Primaquine at alternative dosing schedules for preventing relapse in people with Plasmodium vivax malaria
Abstract
This is a protocol for a Cochrane Review (Intervention). The objectives are as follows:
To assess the efficacy and safety of alternative primaquine regimens for radical cure ofP. vivax malaria compared to the standard 14 days of primaquine 0.25 mg/kg/day.
Background
Malaria is a potentially life‐threatening disease caused by the Plasmodium parasite, which is transmitted by the bite of an infected female Anopheles mosquito. There are five species of Plasmodium malaria parasites that can cause malaria disease in humans, of these, Plasmodium vivax and Plasmodium falciparum are generally recognized as the most significant threat to human health (WHO 2016a). In 2015, there were an estimated 212 million cases of malaria worldwide, with 429,000 attributable deaths (WHO 2016b). By 2030, the World Health Organization (WHO) aims to reduce malaria case load and mortality by at least 90% (WHO 2016a).
Historically, P. vivax infection was thought to be a milder form of malaria with minimal morbidity, with the greater focus for research on P. falciparum, because of the high number of deaths it causes (Bassat 2016). In recent years the morbidity and mortality of P. vivax have been shown to have been underestimated, with evidence of direct fatality and contribution to mortality in patients who have other co‐morbidities, such as malnutrition, human immunodeficiency virus (HIV), or co‐existing infections (Baird 2013; Bhattacharjee 2013; Rizvi 2013; Singh 2013; Battle 2014; Douglas 2014; Kochar 2014; Arévalo‐Herrera 2015; Baird 2015b). Repeated P. vivax infections through childhood and adulthood also affect personal well‐being, development, and education and can thus negatively impact economic development, both for the individual and the community (Mendis 2001). P. vivax malaria in pregnancy is associated with maternal anaemia, spontaneous abortion, stillbirth, and low birthweight, with especially poor pregnancy outcomes for women with severe infection (McGready 2012; Rijken 2012; Brutus 2013).
Description of the condition
P. vivax infection caused an estimated 13.8 million cases of malaria in 2015 and is responsible for almost half of the global cases of malaria outside of Sub‐Saharan Africa (WHO 2015c). The geographical distribution ofP. vivax malaria is more widespread than any of the other forms of human malaria – around 35% of the world’s population is thought to be at risk, with two‐thirds of cases occurring in South‐East Asia (WHO 2015a). Co‐infection with P. falciparum is also common in many regions (Kumar 2007; Mueller 2009). As malaria control accelerates, the P. vivax proportion in co‐endemic areas tends to rise compared to that of P. falciparum, which highlights the importance and challenge of this infection (John 2012).
P. vivax is also important as many countries progress towards malaria elimination, as it is increasingly recognized as a potential roadblock to eradication (Cibulskis 2015; Bassat 2016). Despite a reduction in the number of cases of P. vivax malaria over the past 20 years, it has several characteristics that enable it to evade control (Newby 2016). The early appearance of gametocytes in the blood, often prior to symptoms of malaria, increases the chance of onward transmission by mosquitoes (WHO 2015a). P. vivax differs to P. falciparum in that as well as having a blood stage schizontal infection, hypnozoites develop in the liver that can be dormant for weeks to months before developing into an infection (Llanos‐Cuentas 2014). It is not known what triggers these relapses. There is difficultly in distinguishing between relapse, recrudescence (subpar treatment of the initial blood stage infection), and reinfection (new infection with P. vivax) (Betuela 2014). A study in Papua New Guinea suggested that relapses cause four‐fifths of P. vivax infections, so are important in sustaining transmission (Robinson 2015). Parasites show high genetic diversity, even in countries that are at malaria elimination stage, where you would expect reduced transmission to result in reduced diversity (Koepfli 2015). P. vivax is likely underestimated worldwide as the dormant liver stage is not detected in routine surveys (Gething 2012). Submicroscopic infections (asymptomatic infection reservoirs) may also lead to underdiagnosis or misdiagnosis. A systematic review showed that across all study sites the polymerase chain reaction (PCR) prevalence of P. vivax was significantly higher than that identified by light microscopy (Cheng 2015). The effect that this may have on P. vivax malaria studies is unclear. There are different strains of P. vivax, which have varying relapse patterns, and this can further complicate matters (White 2016). The Chesson, or tropical, strain is commonly found in South East Asia, Oceania, and parts of the Indian subcontinent and has the shortest relapse interval of about three weeks (if untreated), while the temperate strain may relapse after months (John 2012).
Currently primaquine, an 8‐aminoquinoline, is the only drug available on the market for treating the hypnozoite stage of infection (Ashley 2014). One of the main barriers in P. vivax treatment is the reluctance to use primaquine due to it causing haemolysis in patients with glucose‐6‐phosphate‐dehydrogenase (G6PD) deficiency. G6PD deficiency is the commonest enzyme deficiency worldwide and affects red blood cells, by leading to their premature lysis (Nkhoma 2009). G6PD deficiency is common in countries where P. vivax malaria is endemic, with an estimated population prevalence of 8% (Howes 2012). Within G6PD deficiency there are differing phenotypes, meaning some people may be mildly sensitive to primaquine, while others may be very sensitive and experience life threatening haemolysis (Baird 2015a), which explains the varying responses to primaquine. In many countries where P. vivax is pre‐dominant, locally available testing for G6PD is not available (Baird 2015b). A newer alternative, tafenoquine, another 8‐aminoquinoline, has completed phase III trials and is on track for submission to the Food and Drug Administration (FDA) in 2017 (MMV 2016). Tafenoquine has shown promise in reducing relapses, but there are increased safety concerns in patients with undiagnosed G6PD deficiency compared to primaquine, due to its longer half‐life (Rajapakse 2015).
Description of the intervention
People with P. vivax malaria require treatment with a blood stage antimalarial drug to treat the schizont infection, and a drug to treat the hypnozoite stage (radical cure). The WHO recommends treatment with either chloroquine or an artemisinin‐based combination therapy (ACT) for the blood‐stage infection, followed by treatment with 0.25 to 0.5 mg/kg primaquine for 14 days (WHO 2015b). ACTs and chloroquine have been shown to be effective and comparable in treating the blood stage infection of P. vivax malaria (Gogtay 2013). A previous Cochrane Review showed that primaquine regimes of five days or fewer had similar relapse rates to placebo or no primaquine. Of the comparisons included in the systematic review, a regime of 0.25 mg/kg (15 mg) a day of primaquine for 14 days had the lowest relapse rates of P. vivax infection (Galappaththy 2013). There were no trials at that time that compared higher doses of primaquine at 14 or seven days.
Primaquine was first made available to American soldiers in the 1950s (Baird 2004). Its mechanism and metabolism is not widely understood, but it has a broad spectrum of activity against the Plasmodium parasite. As well as preventing relapse of P. vivax malaria by targeting the latent and developing hypnozoites in the liver, it is also used in malaria prophylaxis (Baird 2003). It is absorbed from the gastrointestinal tract, has a half life of about four to nine hours, and crosses the placenta in pregnancy (Baird 2004). New advancements in studying P. vivax in humanized mice may lead to a greater understanding of the mechanism of action of primaquine (Mikolajczak 2015).
Adverse effects of primaquine include production of methaemoglobin, an oxidated state of haemoglobin which cannot transport oxygen to tissues. Methaemoglobinaemia (an abnormal build‐up of methaemoglobin) can result in cyanosis when levels exceed 10% of the usual haemoglobin level (Vale 2009). As described above, primaquine causes haemolysis in people with G6PD deficiency, which leads to severe intravascular haemolysis and anaemia (Ashley 2014). When taken on an empty stomach it can cause abdominal pain and gastrointestinal upset (Vale 2009). Primaquine cannot be given in pregnancy or early infancy as the G6PD status of the baby is unknown and there would be risk of haemolysis and possible termination if the foetus was G6PD‐deficient. There is currently debate about whether the levels of primaquine in breast milk would be sufficient to cause haemolysis in a G6PD‐deficient baby, but it is not recommended at this time.
How the intervention might work
The WHO advises that 0.25 mg to 0.5 mg/kg of primaquine for 14 days should be used for radical cure of P. vivax malaria in patients over six months old, excluding people with G6PD deficiency and patients who are pregnant or breastfeeding (WHO 2015b). There has been suggestion of failure of the 0.25 mg/kg/14‐day dosing regimen of primaquine for the Chesson strain of P. vivax, which is what was behind the suggestion of the increased dosing of 0.5 mg/kg/day. In the last review, Galappaththy 2013, there were no trials found which compared higher doses of primaquine to the 14‐day regime. The WHO recommends a weekly dose of 0.75 mg/kg for eight weeks for patients with G6PD deficiency but the evidence for this is low quality as there are few high‐quality trials (WHO 2015b).
The 14‐day course of primaquine, which can lead to adherence issues in patients, as well as safety concerns about haemolysis in places where G6PD testing is not available, means that shorter courses of primaquine are desirable. Failure to treat the hypnozoite stage of P. vivax malaria leads to repeated relapses, morbidity, and persistent infection. The logic framework for developing efficacious and safe treatment regimes for P. vivax is illustrated in Figure 1.

Logic framework: treatment outcome pathways in Plasmodium vivax liver hypnozoite infection
It has long been suggested that it may be the total dose of primaquine that is important in treatment of the hypnozoite stage rather than the length of the course (Schmidt 1977). If a higher dose of primaquine could be administered safely over a shorter period of time, this may improve adherence rates, thus reducing relapse rates and morbidity and mortality resulting from P. vivax infection. There are small trials from the 1970s that suggest that shorter, higher dose regimes were as efficacious as the 14‐day courses (Clyde 1977), and there is also evidence of similar efficacy (Saint‐Yves 1977). At the time of the last review (Galappaththy 2013) there were no recent large high‐quality trials that had investigated the use of higher doses given over seven days. We plan to include any such trials in this Cochrane Review.
Why it is important to do this review
The use of primaquine for radical cure of P. vivax malaria continues to pose a therapeutic dilemma for healthcare providers in areas without adequate screening for G6PD status. Clinicians must either chose to give primaquine and risk haemolysis if the patient is G6PD‐deficient, or withhold treatment and accept the complications of ongoing parasite infection and relapses. This is why when clinicians choose to treat with primaquine they prefer a lower dose over a more prolonged period ‐ although this then risks difficulties with adherence and thus reduced efficacy.
From the previous systematic review on primaquine with chloroquine for radical cure (Galappaththy 2013), we know 14 days of 0.25 mg/kg or 15 mg/day (210 mg total dose) is better than shorter regimens of similar daily doses and placebo. A major problem with the radical cure of P. vivax is difficulty with the adherence of the 14‐day course of primaquine, which has led to many countries shortening the regime. Peru was once such example, although a study revealed that patients often still discontinued the therapy after around three days, when they started to feel better (Grietens 2010). A study that compared directly observed therapy (DOT) for 14 days of primaquine, versus non‐DOT primaquine found that the vivax relapse rate was significantly lower in the DOT group (Takeuchi 2010). These problems have led to a more urgent call for shorter treatment regimes. Various trials are investigating regimens that improves dosing and duration of treatment, to improve adherence and reduce the potential for incomplete treatment and development of resistance. As mentioned previously, the significance of the total cumulative primaquine dose given, rather than length of course, is one avenue of investigation. In areas where G6PD screening is present, using higher dosing regimes over shorter time periods, if at least similarly efficacious, could improve adherence and reduce morbidity associated with P. vivax parasitaemia.
WHO guidelines suggest a higher dosing regimen of primaquine for areas with the tropical strain of P. vivax (WHO 2015b), although the previous Cochrane Review, Galappaththy 2013, did not find any trials that assessed this. Therefore investigating the evidence base for this is important. The 2015 WHO guidelines also suggest an alternate dosing regimen of weekly primaquine, which may be safer in patients with G6PD. In the last Cochrane Review, only data from one trial assessed this, so it will be useful to see if there is any further evidence to substantiate this guidance.
In this Cochrane Review, we will exclude comparisons between blood stage drug (chloroquine/ACT) with and without primaquine as the rationale for primaquine use has been sufficiently demonstrated in a previous Cochrane Review (Galappaththy 2013). Similarly, we will not include comparisons that look at different blood stage drugs compared using the same dose of primaquine as an update to an existing Cochrane Review will address this (Gogtay 2013). However, we will stratify our results according to partner drug, as there is increasing evidence that primaquine is metabolized via the CYP2D6 pathway and efficacy may thus be affected if the blood stage antimalarial drug is a CYP2D6 inhibitor (Bennett 2013). This review will exclude comparisons of regimens that do not use the control of 14 days of primaquine at 0.25 mg/kg/day. Also, it will not include comparisons of primaquine regimens of 0.25 mg/kg daily for less than 14 days as Galappaththy 2013 has already assessed these shorter, same daily dose regimens.
Currently there is a lack of consensus among studies as to what the minimum time frame for follow‐up of relapse in P. vivax malaria should be. The WHO guidance on clinical trials in malaria sets out standard follow‐up for blood (or schizontal) stage infection as 28 days after treatment commencement, but has no clear definition on the follow‐up period for radical cure in primaquine studies. It states that "follow up varies from three months to a year in the literature, and should be adapted to regional parasite characteristics" (WHO 2009). In a recent review, John 2012 described relapse of the tropical strain of P. vivax as typically three weeks, but this varies according to blood stage treatment: "three weeks following quinine therapy" and "six to eight weeks following chloroquine" (White 2011). With exposure to primaquine ‐ even if radical cure is not achieved ‐ relapses may be at longer intervals (Sutanto 2013). In the Cochrane Review (Galappaththy 2013), the period of follow‐up was 30 days after starting primaquine treatment. Despite this, the definition of relapse used in the review was the presence of P. vivax parasites more than 28 to 30 days after the full course of primaquine in people living in a non‐endemic area (Murphy 1993; Looareesuwan 1997). Because of the varying lengths of relapse time in P. vivax malaria, as well as the longer schizonticidal half‐life in ACTs, 28 days from treatment commencement may not allow true assessment of radical cure. It also makes assessment of the weekly primaquine regime difficult, as the follow‐up time is before the eight‐week treatment course has finished. In this Cochrane Review we plan to assess parasitaemia at 3, 6, and 12 months follow‐up, in keeping with WHO guidance. We intend to describe the length of follow‐up across studies, and then group them into meaningful lengths of follow‐up, depending on the regimen.
We intend to answer the following questions by comparing the new regimens to the standard regimen of 14 days of primaquine at 0.25 mg/kg (15 mg adult dose).
-
Are higher doses (0.5 mg/kg or 30 mg primaquine/day for 14 days) more efficacious and safe compared to standard therapy (0.25 mg/kg/day for 14 days), in all areas, or only in areas where they are standard treatment (for tropical P. vivax strains in Asia, Pacific)?
-
Are shorter, higher dose regimes (0.5 mg/kg or 30 mg primaquine/day for 7 days) as efficacious and safe compared to standard therapy (0.25 mg/kg/day for 14 days)?
-
Are weekly dosing regimens (0.75 mg/kg or 45 mg/week for 8 weeks) as efficacious and safe compared to standard therapy (0.25 mg/kg/day for 14 days)?
Objectives
To assess the efficacy and safety of alternative primaquine regimens for radical cure ofP. vivax malaria compared to the standard 14 days of primaquine 0.25 mg/kg/day.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials (RCT). We will exclude quasi‐RCTs.
Types of participants
Adults and children with confirmed clinical and parasitological (light microscopy or polymerase chain reaction (PCR), or both) diagnosis of P. vivax malaria. We will include trials that have excluded people with glucose‐6‐phosphate‐dehydrogenase (G6PD) deficiency, and trials that included populations that had not been screened for G6PD deficiency.
Types of interventions
Intervention
Any regimen of either chloroquine or an artemisinin‐based combination therapy (ACT) plus primaquine with any of the following.
-
Higher daily doses for 14 days.
-
Shorter regimens with the same total dose.
-
Using weekly dosing regimens.
Control
Standard regimen of 14 days of primaquine at 0.25 mg/kg (15 mg adult dose) plus either chloroquine or an ACT.
We will include trials that use chloroquine or ACT as the treatment for blood‐borne infection, and we will stratify by the schizonticidal agent.
Types of outcome measures
Primary outcomes
-
P. vivax parasitaemia detected (by light microscopy or polymerase chain reaction (PCR), or both) at 3 months, 6 months, and 12 months follow‐up.
Secondary outcomes
-
P. vivax parasitaemia detected (by light microscopy or polymerase chain reaction (PCR), or both) at one to three months follow‐up.
Adverse effects
-
Serious adverse effects (fatal, life‐threatening, or requiring hospitalization).
-
Adverse effects that result in discontinuation of treatment.
-
Events known to occur with primaquine (cyanosis, leucopenia, methaemoglobinaemia, hypertension, cardiac arrhythmia, abdominal pain, nausea, vomiting, or haemolysis) or those due to a comparator drug used along with primaquine.
-
Anaemia or change in haemoglobin status.
-
Other adverse effects.
Search methods for identification of studies
We will attempt to identify all relevant studies regardless of language or publication status (published, unpublished, in press, or in progress).
Electronic searches
We will search the following databases: the Cochrane Infectious Diseases Group (CIDG) Specialized Register; the Cochrane Central Register of Controlled Trials (CENTRAL), published in the Cochrane Library; MEDLINE (PubMed); Embase (OVID); and LILACS (BIREME), using the search terms detailed in Appendix 1 (Lefebvre 2011). We will also search the World Health Organization (WHO) International Clinical Trials Registry Platform (http://www.who.int/ictrp/search/en/) and ClinicalTrials.gov (https://clinicaltrials.gov/), to identify ongoing trials, using "vivax" and "primaquine" as search terms.
Searching other resources
We will check the reference lists of all studies identified by the above methods for other potentially relevant studies. We will contact researchers working in the field and the World Health Organization (WHO) for unpublished and ongoing trials. We will also search the reference lists and included studies of the Cochrane review by Galappaththy 2013.
Data collection and analysis
Selection of studies
Two review authors will independently screen the titles and abstracts of the search results to identify potentially eligible trials, and will code the articles as either 'retrieve' or 'do not retrieve'. We will obtain the full‐text reports of potentially eligible trials and will assess them for inclusion in the review using a predesigned eligibility form based on the inclusion criteria. We will resolve discrepancies through discussion or, if required, we will consult a third review author. Where necessary we will contact the trial authors for clarification of trial methods. We will list the excluded trials and the reasons for exclusion in a 'Characteristics of excluded studies' table. Where there are multiple reports relating to the same trial, we will include all reports. However, we will only extract data from the most up‐to‐date report that includes the specified outcome. We will detail the trial selection process in a PRISMA diagram.
Data extraction and management
Two review authors will independently extract data from the included trials using a data extraction form, designed for this review, in keeping with Cochrane guidance (Higgins 2011).
For each included trial we plan to extract a minimum of the following data if available.
-
Study design.
-
Endemicity/population demographics.
-
G6PD status of participants (known/unknown).
-
CYP2D6 status (if available).
-
Blood stage antimalarial drug choice.
-
Dose/duration/timing of treatment arms.
-
Supervised or non‐supervised therapy.
-
Duration of follow‐up.
-
Adverse events.
-
Reported outcomes.
We will resolve any differences in data extraction through discussion and consult a third review author if there is any discrepancy. We will enter the extracted data into Review Manager 5 (RevMan 5) (RevMan 2014). We will contact the authors of primary trials in case of any doubts regarding missing data or methodological details of the trial. We will note the limitations in the included studies.
We will group comparisons as illustrated in Table 1.
| Objective | Intervention | Control |
| Are higher doses more effective (0.5 mg/kg or 30 mg primaquine/day for 14 days), in all areas, or only in areas where they are standard treatment (Asia, Pacific)? | Blood‐stage antimalarial drug with primaquine 0.5 mg/kg (30 mg) per day for 14 days (total dose 420 mg) Both intervention and control groups must have received the same treatment: either CQ or ACT for the blood‐borne stage of infection. | Blood‐stage antimalarial drug with primaquine 0.25 mg/kg (15 mg) per day for 14 days (total dose 210 mg) Both intervention and control groups must have received the same treatment: either CQ or ACT for the blood‐borne stage of infection. |
| Are shorter, higher dose regimes of primaquine over 7 days as effective as treatment over 14 days (is the total dose rather than the length of treatment the important factor)? | Blood‐stage antimalarial drug with primaquine 0.5 mg/kg (30 mg) per day for 7 days (total dose 210 mg) Both intervention and control groups must have received the same treatment: either CQ or ACT for the blood‐borne stage of infection. | |
| Are weekly dosing regimens (0.75 mg/kg or 45 mg/week for 8 weeks) as effective? | Blood‐stage antimalarial drug with primaquine 0.75/kg (45 mg) per week for 8 weeks (total dose 360 mg) |
CQ = Chloroquine
ACT = Artemisinin‐based combination therapy
Assessment of risk of bias in included studies
Two review authors will independently assess the risk of bias of each included trial using the Cochrane 'Risk of bias' assessment tool, and discuss any differences of opinion. In the case of missing or unclear information, we will contact the trial authors for clarification. We will summarize the results using Cochrane 'Risk of bias' tables (Higgins 2011).
Measures of treatment effect
For dichotomous data, we will compare interventions using risk ratios to measure treatment effect. Where trial authors present data as odds ratios, we will recalculate the effect. We will define statistical significance as P < 0.05 and for all results we will calculate 95% confidence intervals (CIs). For comparable trials, we will perform meta‐analyses if there is sufficient data.
Unit of analysis issues
For this Cochrane Review, cluster‐randomized designs would be inappropriate for evaluating the research questions. If trials that have used cluster‐randomization meet our inclusion criteria we would expect the results to have been controlled for clustering. If they have not, we will contact the trial authors for an estimate of the intra‐cluster correlation coefficient (ICC) value. We will analyse clustered data using the methods outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
We will split trials that include more than two comparison groups and will analyse them as individual pair‐wise comparisons. If there is a shared control group we will split the control group so that participants are only counted once in the overall meta‐analysis.
Dealing with missing data
We will analyse missing data using available case analysis if we judge the trial to be at low risk of bias for incomplete outcome data. We will attempt to contact trial authors to obtain missing or unclear data. If the missing data render the result uninterpretable, we will exclude the data from meta‐analyses and clearly state the reason for exclusion. If the missing data means that results are interpretable but likely to be at high risk of bias, we may use imputation methods to investigate the impact of the missing data. We will analyse extracted data on an intention‐to‐treat basis where there is no missing data.
Assessment of heterogeneity
We will inspect forest plots for overlapping CIs. We will also apply the Chi² test as a statistical test for the presence of heterogeneity, with a P value of 0.10 used to indicate statistical significance, and we will compute the I² statistic to quantify the percentage of the variability in effect estimates that is due to heterogeneity rather than sampling error (chance). We will investigate possible causes of heterogeneity by subgroup analysis. If substantive heterogeneity persists, which we define as an I² statistic value of greater than 50%, we will use a random‐effects meta‐analysis.
Assessment of reporting biases
We will examine the likelihood of reporting bias using funnel plots provided that there is a sufficient number of included trials.
Data synthesis
We will analyse the data using Review Manager 5 (RevMan 5) (RevMan 2014). We will assess the certainty of the evidence for each outcome measure using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach and we will construct 'Summary of findings' tables using GRADEpro Guideline Development Tool (GDT) (GRADEpro GDT 2014). We will stratify results according to blood stage partner drug. Length of follow‐up will vary with regimes and between studies. We will describe regimes and follow‐up periods and define sensible groupings for follow‐up. If there is sufficient data, we will also perform subgroup analyses according to CYP2D6 status (when available), geographical region/endemicity, length of follow‐up, and directly observed therapy (DOT) or non‐DOT.
Subgroup analysis and investigation of heterogeneity
We will group the analysis by drug regimen. We will describe the interventions and outcomes in all included trials. We will conduct an inventory of length of follow‐up against each drug regimen and then group P. vivax parasitaemia relapse by appropriate groupings for length of follow‐up.
Sensitivity analysis
We will assess the risk of bias that contributed data to the meta‐analyses for the prespecified outcomes with sensitivity analyses against concealment of allocation.
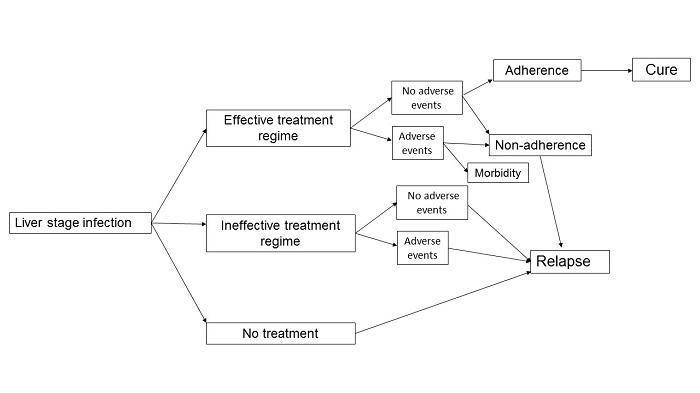
Logic framework: treatment outcome pathways in Plasmodium vivax liver hypnozoite infection
| Objective | Intervention | Control |
| Are higher doses more effective (0.5 mg/kg or 30 mg primaquine/day for 14 days), in all areas, or only in areas where they are standard treatment (Asia, Pacific)? | Blood‐stage antimalarial drug with primaquine 0.5 mg/kg (30 mg) per day for 14 days (total dose 420 mg) Both intervention and control groups must have received the same treatment: either CQ or ACT for the blood‐borne stage of infection. | Blood‐stage antimalarial drug with primaquine 0.25 mg/kg (15 mg) per day for 14 days (total dose 210 mg) Both intervention and control groups must have received the same treatment: either CQ or ACT for the blood‐borne stage of infection. |
| Are shorter, higher dose regimes of primaquine over 7 days as effective as treatment over 14 days (is the total dose rather than the length of treatment the important factor)? | Blood‐stage antimalarial drug with primaquine 0.5 mg/kg (30 mg) per day for 7 days (total dose 210 mg) Both intervention and control groups must have received the same treatment: either CQ or ACT for the blood‐borne stage of infection. | |
| Are weekly dosing regimens (0.75 mg/kg or 45 mg/week for 8 weeks) as effective? | Blood‐stage antimalarial drug with primaquine 0.75/kg (45 mg) per week for 8 weeks (total dose 360 mg) | |
| CQ = Chloroquine ACT = Artemisinin‐based combination therapy | ||