Antiemetics for adults for prevention of nausea and vomiting caused by moderately or highly emetogenic chemotherapy: a network meta‐analysis
Abstract
This is a protocol for a Cochrane Review (Intervention). The objectives are as follows:
To compare the effects of antiemetics on the prevention of acute, delayed and overall chemotherapy‐induced nausea and vomiting in adults with solid cancer or haematological malignancies receiving moderately or highly emetogenic chemotherapy and to generate a clinically meaningful treatment ranking according to their safety and efficacy.
Background
This protocol is partly based on suggested wording from the Pain, Palliative and Supportive Care Review Group (PaPaS CRG), the Cochrane Heamatological Malignancies Review Group and the protocol templates for a Cochrane intervention review that compares multiple interventions (Chaimani 2014; Chaimani 2017).
Description of the condition
Many cancer patients, both with solid tumours and with haematological malignancies, suffer from chemotherapy‐induced nausea and vomiting (CINV), which is an important contributor to morbidity, poor performance status and decreased quality of life (Feyer 2011; Jordan 2015). The reported age‐adjusted incidence rate of cancer in the USA in 2010 was 464.6 per 100,000, and the mortality rate was 199.8 per 100,000 persons per year (Howlader 2013). Without appropriate antiemetic therapy, 70% to 80% of cancer patients develop CINV (Feyer 2011). It is classified into five categories, depending on start of CINV in relation to start of chemotherapy and patients’ negative previous experiences (Navari 2016; Tageja 2016).
-
Acute: nausea and vomiting occurring within the first 24 hours of treatment with chemotherapy with a maximal intensity after five to six hours; activated through a peripheral pathway, in which 5‐HT3 receptor activation plays a role.
-
Delayed: nausea and vomiting occurring from 24 hours to 120 hours of treatment with chemotherapy with peaks of intensity between 48 and 72 hours; activated through a central pathway, in which NK1 receptor activation is involved.
-
Breakthrough: nausea and vomiting occurring although appropriate prophylaxis has been administered.
-
Anticipatory: conditioned response to the occurrence of CINV in previous chemotherapy cycles resulting in nausea and vomiting.
-
Refractory: nausea and vomiting recurring in subsequent cycles of chemotherapy, excluding anticipatory CINV.
In this review, we will focus on prevention of acute and delayed CINV.
Several prognostic factors such as younger age, female sex, previous hyperemesis gravidarum or history of vomiting in pregnancy and motion sickness have been found to increase the likelihood of CINV (Di Mattei 2016; Dranitsaris 2017; Furukawa 2014; Hu 2016; Warr 2014); previous alcohol intake reduces the risk of CINV (Hu 2016). CINV remains one of the most distressing symptoms associated with cancer therapy and can lead to dehydration, electrolyte imbalances, malnutrition and metabolic disturbances (Viale 2012). Moreover, CINV is associated with decreased adherence to chemotherapy, which could lead to a decreased response resulting in increased risk of death in cancer patients (Wozniak 1998). Therefore, preventing CINV is an important goal in cancer patients.
According to the Multinational Association of Supportive Care in Cancer (MASCC)/European Society of Medical Oncology (ESMO) and the American Society of Clinical Oncology (ASCO), practice focuses on the emetogenicity of chemotherapeutic agents (minimal, low, moderate, high) and the relative dose of antineoplastic agents used (Basch 2012; Jordan 2017; Roila 2016).
Highly emetogenic chemotherapy includes the following agents or combinations of agents (Basch 2012).
-
Anthracycline/cyclophosphamide combination
-
Carmustine
-
Cisplatin
-
Cyclophosphamide ≥1500 mg/m2
-
Dacarbazine
-
Hexamethylmelamine
-
Mechlorethamine
-
Procarbazine
-
Streptozocin
Moderately emetogenic chemotherapy includes the following agents (Basch 2012).
-
Alemtuzumab
-
Azacitidine
-
Bendamustine
-
Bosutinib
-
Carboplatin
-
Ceritinib
-
Clofarabine
-
Crizotinib
-
Cyclophosphamide 1000 mg/m2
-
Daunorubicin
-
Doxorubicin
-
Epirubicin
-
Idarubicin
-
Ifosfamide
-
Imatinib
-
Irinotecan
-
Oxaliplatin
-
Romidepsin
-
Temozolomide
-
Thiotepa
-
Trabectedin;
-
Temozolomide
-
Vinorelbine
According to the latest MASCC)/ESMO guidelines, carboplatin has a higher emetogenic potential compared to the other moderately emetogenic agents; patients receiving this drug should receive the same prophylaxis as described for patients with highly emetogenic potential (Jordan 2017; Roila 2016).
With appropriate antiemetic prophylaxis, acute and delayed CINV are clinically significantly reduced. In a recent systematic review, Yuan and colleagues found that the complete response rate of patients receiving a NK1 receptor antagonist was significantly higher compared to various control regimens (like 5‐HT3 receptor antagonists plus dexamethasone) with complete response in the acute phase of 85.1% versus 79.6% and complete response of 71.4% versus 58.2% in the delayed phase. According to the authors, the safety profile of NK1 receptor antagonist was comparable to other regimens, with less insomnia but more diarrhoea and hiccups (Yuan 2016).
Description of the intervention
Options for prevention of CINV are 5‐HT3 receptor antagonists (e.g. ondansetron, granisetron, palonosetron) in combination with dexamethasone, or additionally combined with NK1 receptor antagonists (e.g. aprepitant, fosaprepitant, netupitant or rolapitant). Although antiemetic therapy is common in cancer patients at risk for CINV, recommendations in current guidelines are inconsistent. According to ASCO, practice focuses on the emetogenicity of chemotherapeutic agents (minimal, low, moderate, high) and the relative dose of antineoplastic agents used. This guideline recommends 5‐HT3 receptor antagonists plus dexamethasone for patients administered moderately emetogenic chemotherapy (Basch 2012). The latest update of this guideline for patients receiving highly emetogenic chemotherapy (including anthracycline plus cyclophosphamide) recommends a combination of an NK1 receptor antagonist, a 5‐HT3 receptor antagonist, and dexamethasone. The oral combination of palonosetron, netupitant and dexamethasone is one of the specific drugs recommended for these patients (Hesketh 2016).The recommendation in the moderately emetogenic setting is rather difficult. In the latest MASCC/ESMO guideline report, it was acknowledged that carboplatin‐based chemotherapy might have higher risk of nausea and vomiting compared to other drugs in the category moderately emetogenic chemotherapy (Roila 2016). The MASCC/ESMO guideline recommends the same three‐drug combination as the ASCO guideline for patients receiving highly emetogenic chemotherapy (including anthracycline plus cyclophosphamide), but points out that no comparative studies have been published to identify differences in efficacy and toxicity between available NK1 receptor antagonists to recommend one specific drug over another (Roila 2016). In the so called other moderate emetogenic risk group a 5‐HT3 receptor antagonist, and dexamethasone is still standard of care although the National Comprehensive Cancer Network (NCCN) guidelines broaden the indication of an NK1 receptor agonist in this risk category (Ettinger 2017).
Another option for prevention and treatment of CINV‐ or radiotherapy‐induced nausea and vomiting is olanzapine. However, the evidence for efficacy and safety of this drug is not yet clear and is being evaluated in a Cochrane Review (Cochrane protocol already published: Sutherland 2017).
How the intervention might work
For 5‐HT3 receptor antagonists and dexamethasone, solely or in combination with NK1 receptor antagonists, systematic reviews and meta‐analyses have shown that they improve CINV in cancer patients administered especially highly emetogenic chemotherapy including anthracycline‐cyclophosphamide‐based chemotherapy, with inconclusive evidence on effectiveness and rate of adverse events for one drug compared to another (Celio 2013; dos Santos 2013; Hocking 2014; Jin 2012; Jordan 2016; Lee 2013; Popovic 2014).
As the central nervous system, neurotransmitter and their receptors play a critical role in CINV, both 5‐HT3 receptor antagonists and NK1 receptor antagonists inhibit processing of antiemetic signals from the gut to the central nervous system (Janelsins 2013). Both drugs are usually combined with dexamethasone to improve the efficacy. A pilot randomised controlled trial (RCT) including 31 patients receiving cisplatin chemotherapy has shown first the improved efficacy of ondansetron if combined with dexamethasone and a good safety profile (Smith 1991). Another RCT conducted between 1992 and 1994 for patients receiving moderately emetogenic chemotherapy has shown highest efficacy in terms of complete protection of vomiting and nausea and less delayed vomiting and nauseas with the combination of granisetron and dexamethasone compared to granisetron or dexamethasone. No severe adverse events were reported, but constipation and hot flushes were more often found in the granisetron plus dexamethasone arm compared to the single drug arms (Italian Group for Antiemetic Research 1995). Granisetron plus dexamethasone has also shown improved efficacy compared to both single drugs only in patients receiving cisplatin chemotherapy (Heron 1994). Thereafter, a 5‐HT3 receptor antagonist combined with dexamethasone became standard prophylaxis to prevent CINV (Gralla 1999).
As NK1 receptor antagonists inhibit another receptor in the emetic signal activation, they are combined with 5‐HT3 receptor antagonists and dexamethasone for patients receiving cisplatin or other highly emetogenic chemotherapeutic agents (Hesketh 2003; Poli‐Bigelli 2003).
Why it is important to do this review
As mentioned above, the decision making process for prevention of CINV is usually confusing for patients and physicians, as there are no clear recommendations for a consistent approach to the use of antiemetic agents in international guidelines (Hesketh 2016; Roila 2016). Economic arguments are introduced in discussions on the best strategy as direct and indirect costs differ enormously for the various treatment options and could lead to increased healthcare costs (Avritscher 2010; Humphreys 2013). In addition, direct head‐to‐head comparisons of prophylactic options are too sparse to favour one drug or a combined drug‐regimen over another.
The aim of our systematic review and network analysis is to provide a comprehensive overview on the benefits and harms of antiemetic agents for CINV. By systematically identifying all relevant RCTs conducted to date and critically reviewing their reliability and validity considering similar trials in the network analysis, we will overcome statistical limitations of individual studies. The network meta‐analysis will allow a hierarchy of the therapeutic options, in particular, if the benefits of one option compared to another will translate into a clinical important difference. This comprehensive overview is necessary for clinical decision making, it has the potential to have a great impact on international guidelines and clinical pathways. Moreover, it may contribute to a high‐grade decision support for effective therapeutic strategies for the individual person.
The results of this network meta‐analysis will be published in the Cochrane library and presented at national and international expert meetings and conferences (e.g. American Society of Clinical Oncology, Multinational Association of Supportive Care in Cancer). The results of the network analysis have the potential to influence the design of new RCTs on antiemetic agents. As patient‐related outcomes will be evaluated, a direct impact on patient care and treatment might be expected.
Objectives
To compare the effects of antiemetics on the prevention of acute, delayed and overall chemotherapy‐induced nausea and vomiting in adults with solid cancer or haematological malignancies receiving moderately or highly emetogenic chemotherapy and to generate a clinically meaningful treatment ranking according to their safety and efficacy.
Methods
Criteria for considering studies for this review
Types of studies
We will include studies if they are randomised controlled trials (RCTs). We require full journal publication, with the exception of online clinical trial results and summaries of otherwise unpublished clinical trials and abstracts with sufficient data for analysis. In the case of cross‐over trials, only the first period of the trial will be analysed. There will be no limitation with respect to the length of follow‐up.
We will exclude studies that were non‐randomised, case reports and clinical observations.
Types of participants
Studies will include trials involving adult patients according to the definition in the studies (usually ≧ 18 years of age), with a confirmed diagnosis of cancer, irrespective of type and stage of cancer and gender. We will include both patients with solid cancer and patients with haematological malignancies. We will include trials that included patients receiving moderately or highly emetogenic chemotherapy according to the latest Antineoplastic Agents Emetic Risk Classification (Jordan 2017; Roila 2016). As this classification has changed over the years (e.g. anthracycline and cyclophosphamide combination is nowadays classified as highly emetogenic instead of moderately emetogenic), we will use this classification to asses the emetogenic risk of one specific chemotherapeutic agent, irrespective of the emetogenic risk the study authors applied (see section Description of the condition). We will perform separate analyses for populations receiving moderately emetogenic and highly emetogenic chemotherapy, according to the latest definition by MASCC/ESMO (Jordan 2017; Roila 2016). We assume that patients who fulfil the inclusion criteria are equally eligible to be randomised to any of the interventions we plan to compare.
We will exclude trials including participants not receiving the same risk levels of emetogenic chemotherapies, which do not provide subgroup data for each emetogenic risk group. We will exclude trials evaluating participants at risk for radiotherapy‐induced nausea and vomiting. We will exclude trials evaluating participants at risk of vomiting and nausea due to their underlying disease.
Types of interventions
Nowadays, recommended antiemetics for prophylaxis of CINV caused by moderately or highly emetogenic chemotherapy are:
-
5‐Hydroxytryptamine‐3 (5‐HT3) receptor antagonists with dexamethasone, solely, or combined with;
-
Neurokinin‐1 (NK1) receptor antagonist.
Combinations of these interventions at any dose and by any route will be compared to each other in a full network. We will include all RCTs comparing in at least two study arms the intervention of interest, either 5‐Hydroxytryptamine‐3 (5‐HT3) receptor antagonists with dexamethasone or 5‐Hydroxytryptamine‐3 (5‐HT3) receptor antagonists with dexamethasone combined with Neurokinin‐1 (NK1) receptor antagonist) (see Figure 1 for a full network of direct comparisons). In case we identify additional, new drugs belonging to the above mentioned drug classes which are not mentioned in Figure 1, we will include these trials. We will explicitly state in the review that we identified further drugs of interest.

Network of direct comparisons of interest. All drugs and drug combinations are combined with dexamethasone. Strength of the line represents number of RCTs.
We will analyse prophylaxis for cancer patients administered moderately emetogenic or carboplatin‐based chemotherapy or highly emetogenic chemotherapy separately. We assume that any participant that meets the inclusion criteria is, in principle, equally likely to be randomised to any of the eligible interventions. We plan to group interventions by evaluating different drug doses together as one drug of interest, according to the product characteristics.
We will exclude trials evaluating solely treatment of nausea and vomiting, meaning that the drug is not given before administering chemotherapeutic agents to prevent CINV, but once nausea or vomiting appears. Antimetic agents to treat CINV might be the same agents as for prevention of CINV, but to include clinically homogenous trials to answer the research question, we will focus on prophylaxis only.
Comparisons of direct interest
As mentioned above, there is high uncertainty in current guidelines whether to recommend a double or triple combination for cancer patients receiving moderately emetogenic chemotherapy and which triple regimen should be administered to cancer patients receiving highly or carboplatin‐based emetogenic chemotherapy (Basch 2012; Hesketh 2016; Roila 2016). Therefore the following comparisons are of direct interest.
Comparisons in cancer patients receiving moderately emetogenic chemotherapy
-
5‐Hydroxytryptamine‐3 (5‐HT3) receptor antagonists with dexamethasone
versus
-
5‐Hydroxytryptamine‐3 (5‐HT3) receptor antagonists with dexamethasone and
-
Neurokinin‐1 (NK1) receptor antagonist
Comparisons in cancer patients receiving highly emetogenic chemotherapy
-
5‐Hydroxytryptamine‐3 (5‐HT3) receptor antagonists, with dexamethasone and
-
Neurokinin‐1 (NK1) receptor antagonist
versus other specific drug combinations of these three drug classes. Different intervention doses and different routes of administration will be evaluated together, but assessed in subgroup analyses.
Additional interventions to supplement the analysis
In addition to the direct comparisons of interest, we will also include trials analysing one double (5‐Hydroxytryptamine‐3 (5‐HT3) receptor antagonists with dexamethasone) versus another double combination (5‐Hydroxytryptamine‐3 (5‐HT3) receptor antagonists with dexamethasone), or one double versus a triple combination in cancer patients receiving highly emetogenic chemotherapy, to increase the amount of available (indirect) information in the analysis (Ades 2013; Chaimani 2017).
Included trials should be comparable in terms of clinical and methodological criteria to hold for transitivity (Chaimani 2017). Therefore, we will exclude trials evaluating in only one arm an intervention of interest, but in the control arm different drug classes (e.g. metoclopramide). We will exclude these trials as they evaluate drugs which are no longer recommended for primary prophylaxis of CINV in moderate‐ and high‐emetogenic chemotherapy. As these trials might be outdated, the assumption that any participant that meets the inclusion criteria is, in principle, equally likely to be randomised to any of the eligible interventions will not be sustained. The efficacy and safety of cannabinoids is evaluated in the Cochrane review by Smith and colleagues (Smith 2015); cannabinoids are not evaluated in this review.
Types of outcome measures
We will include all trials fitting the inclusion criteria mentioned above, irrespective of reported outcomes. We will estimate the relative ranking of the competing interventions according to each of the following outcomes.
Primary outcomes
-
Complete control of nausea (no nausea and no significant nausea), determined from reports in participants' diaries
-
in the acute phase (first 24 hours of treatment with chemotherapy)
-
in the delayed phase (after 24 to 120 hours of treatment with chemotherapy)
-
overall (0 to 120 hours of treatment with chemotherapy)
-
-
Complete control of vomiting (no vomiting and no use of rescue medications), determined from reports in participants' diaries
-
in the acute phase (first 24 hours of treatment with chemotherapy)
-
in the delayed phase (after 24 to 120 hours of treatment with chemotherapy)
-
overall (0 to 120 hours of treatment with chemotherapy)
-
Secondary outcomes
-
On‐study mortality (deaths occurring up to 30 days after the active study period)
-
Qualiy of life if measured by validated instruments e.g.
-
Functional Living Index–emesis (FLIE) (Lindley 1992)
-
Modified Functional Living Index–emesis (M‐FLIE) (Martin 2003; Martin 2003a)
-
-
Adverse events (with a special focus on neutropenia, febrile neutropenia, infections, local reaction at infusion site, and hiccup)
As there are different underlying mechanisms for anticipatory CINV, which do not respond to prophylactic antiemetics, we will not evaluate this outcome.
Search methods for identification of studies
Electronic searches
We will search the following databases without language restrictions. As at least one intervention arm will include at least one 5‐Hydroxytryptamine‐3 (5‐HT3) receptor antagonist, which has been mentioned first in 1988 for the treatment of chemotherapy‐induced emesis (Carmichael 1988), we will restrict our search from 1988 to present. Only trials that compare at least two of the drug combinations mentioned above are eligible. We will search for all possible comparisons formed by the interventions of interest.
-
The Cochrane Central Register of Controlled Trials (CENTRAL) (via the Cochrane Library, latest issue)
-
MEDLINE (via Ovid, 1988 to present)
-
Embase (via Ovid, 1988 to present)
Medical subject headings (MeSH) or equivalent and text word terms will be used. There will be no language restrictions. Searches will be tailored to individual databases. The search strategy for CENTRAL is in Appendix 1.
Searching other resources
In addition, we will search the following databases/sources.
-
Conference proceedings of annual meetings of the following societies will be searched if they are not included in CENTRAL (1988 to present):
-
American Society of Clinical Oncology (ASCO);
-
European Society of Medical Oncology (ESMO);
-
Multinational Association of Supportive Care in Cancer (MASCC).
-
-
Databases of ongoing trials:
-
-
metaRegister of controlled trials (mRCT) (www.controlled‐trials.com/mrct);
-
clinicaltrials.gov (www.clinicaltrials.gov);
-
WHO International Clinical Trials Registry Platform (ICTRP) (http://apps.who.int/trialsearch/).
-
-
We will check reference lists of reviews and retrieved articles for additional studies and we will perform citation searches on key articles.
-
We will contact experts in the field for unpublished and ongoing trials.
-
We will contact authors where necessary for additional information.
Data collection and analysis
Selection of studies
Two review authors will independently screen the results of the search strategies for eligibility for this review by reading the abstracts using Covidence software (Covidence systematic review software). We will code the abstracts as either 'retrieve' or 'do not retrieve'. In the case of disagreement or if it is unclear whether we should retrieve the abstract or not, we will obtain the full‐ text publication for further discussion. Independent review authors will eliminate studies that clearly do not satisfy the inclusion criteria, and obtain full‐text copies of the remaining studies. Two review authors will read these studies independently to select relevant studies, and in the event of disagreement, a third author will adjudicate. We will not anonymise the studies before assessment. We will include a Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) flow chart in the full review that will show the status of identified studies (Moher 2009) as recommended in Part 2, Section 11.2.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We will include studies in the review irrespective of whether measured outcome data are reported in a ‘usable’ way.
Data extraction and management
Two review authors will extract data using a standardised data extraction form developed in Covidence (Covidence systematic review software). If the authors are unable to reach a consensus, we will consult a third review author for final decision. If required, we will contact the authors of specific studies for supplementary information (Higgins 2011a). After agreement has been reached, we will enter data into Review Manager (RevMan 2014). We will extract the following information.
-
General information: Author, title, source, publication date, country, language, duplicate publications.
-
Quality assessment: Sequence generation, allocation concealment, blinding (participants, personnel, outcome assessors), incomplete outcome data, selective outcome reporting, other sources of bias.
Moreover, we will extract the following information which may act as effect modifier.
-
Study characteristics: Trial design, aims, setting and dates, source of participants, inclusion/exclusion criteria, comparability of groups, subgroup analysis, statistical methods, power calculations, treatment cross‐overs, compliance with assigned treatment, length of follow‐up, time point of randomisation.
-
Participant characteristics: Age, gender, ethnicity, number of participants recruited/allocated/evaluated, participants lost to follow‐up, cancer type and stage, additional diagnoses, type and intensity of antineoplastic therapy, emetogenic risk, other patient‐specific prognostic factors e.g. pregnancy, motion sickness, alcohol intake.
-
Interventions: Type and dosage of antiemetic agents, duration of prophylaxis, duration of follow‐up.
-
Outcomes: Complete control of nausea (acute and delayed phase, overall), complete control of vomiting (acute and delayed phase, overall), on‐study mortality, quality of life, adverse events. Where possible, we will extract data at the arm level, not summary effects.
-
Notes: Sponsorship/funding for trial and notable conflict of interest of review authors.
We will collate multiple reports of the same study, so that each study rather than each report is the unit of interest in the review. We will collect characteristics of the included studies in sufficient detail to populate a table of ‘Characteristics of included studies’ in the full review.
Data on potential effect modifiers
We will extract from each included study data on the following: intervention and population characteristics that may act as effect modifiers:
-
year of publication;
-
type of antineoplastic chemotherapy.
Assessment of risk of bias in included studies
This section is taken from the PaPaS template for protocols.
Two review authors will independently assess risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c) and adapted from those used by the Cochrane Pregnancy and Childbirth Group, with any disagreements resolved by discussion. We will complete a 'Risk of bias' table for each included study using the 'Risk of bias' tool in RevMan (RevMan 2014).
We will assess the following for each study.
-
Random sequence generation (checking for possible selection bias). We will assess the method used to generate the allocation sequence as: low risk of bias (any truly random process, e.g. random number table; computer random number generator); unclear risk of bias (method used to generate sequence not clearly stated). Studies using a non‐random process (e.g. odd or even date of birth; hospital or clinic record number) will be excluded.
-
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions prior to assignment determines whether intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment. We will assess the methods as: low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes); unclear risk of bias (method not clearly stated). Studies that do not conceal allocation (e.g. open list) will be excluded.
-
Blinding of participants and personnel (checking for possible performance bias). We will assess the methods used to blind study participants and personnel from knowledge of which intervention a participant received. We will assess methods as: low risk of bias (study states that it was blinded and describes the method used to achieve blinding, such as identical tablets matched in appearance or smell, or a double‐dummy technique); unclear risk of bias (study states that it was blinded but does not provide an adequate description of how it was achieved). Studies that were not double‐blinded are considered to have high risk of bias.
-
Blinding of outcome assessment (checking for possible detection bias). We will assess the methods used to blind study participants and outcome assessors from knowledge of which intervention a participant received. We will assess the methods as: low risk of bias (study has a clear statement that outcome assessors were unaware of treatment allocation, and ideally describes how this was achieved); unclear risk of bias (study states that outcome assessors were blind to treatment allocation, but lacks a clear statement on how it was achieved). Studies where outcome assessment was not blinded will be considered as having a high risk of bias.
-
Selective reporting (checking for reporting bias). We will assess whether primary and secondary outcome measures were pre‐specified and whether these were consistent with those reported: low risk of bias ( study protocol is available and all of the study’s pre‐specified (primary and secondary) outcomes that are of interest in the review have been reported in the pre‐specified way, or the study protocol is not available but it is clear that the published reports include all expected outcomes, including those that were pre‐specified); unclear risk of bias (insufficient information to permit judgement of ‘low risk’ or ‘high risk'); high risk of bias (not all of the study’s pre‐specified primary outcomes have been reported or one or more primary outcomes is reported using measurements, analysis methods or subsets of the data that were not pre‐specified or one or more reported primary outcomes were not pre‐specified or one or more outcomes of interest in the review are reported incompletely so that they cannot be entered in a meta‐analysis, or the study report fails to include results for a key outcome that would be expected to have been reported for such a study).
-
Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data). We will assess the methods used to deal with incomplete data as: low risk (< 10% of participants did not complete the study and/or used ‘baseline observation carried forward’ analysis); unclear risk of bias (used 'last observation carried forward' analysis); high risk of bias (used 'completer' analysis).
-
Size of study (checking for possible biases confounded by small size). We will assess studies as being at low risk of bias (≥ 200 participants per treatment arm); unclear risk of bias (50 to 199 participants per treatment arm); high risk of bias (< 50 participants per treatment arm).
Measures of treatment effect
Relative treatment effect
We will use intention‐to‐treat data. For binary outcomes, we will use risk ratios (RRs) with 95% confidence intervals (CIs) as the measure of treatment effect. We will calculate continuous outcomes as mean differences (MDs) with 95% CIs. In case different instruments are used to assess effects in continuous outcomes, we will use standardised mean differences (SMD) with 95% CIs.
Relative treatment ranking
We will obtain a treatment hierarchy using P scores (Rücker 2015). P scores allow ranking treatments on a continuous zero to 1 scale in a frequentist network meta‐analysis.
Unit of analysis issues
Studies with multiple treatment groups
As recommended in Chapter 16.5.4 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b), for studies with multiple treatment groups we will combine arms as long as they can be regarded as subtypes of the same intervention.
When arms can not be pooled this way, we will compare each arm with the common comparator separately. For pairwise meta‐analysis, we will split the ‘shared’ group into two or more groups with smaller sample size, and include two or more (reasonably independent) comparisons. For this purpose, for dichotomous outcomes, both the number of events and the total number of participants will be divided up, and for continuous outcomes, the total number of participants will be divided up with unchanged means and standard deviations. For network meta‐analysis, instead of subdividing the common comparator, we will use an approach that accounts for the within‐study correlation between the effect sizes by re‐weighting all comparisons of each multi‐arm study (Rücker 2012; Rücker 2014).
Dealing with missing data
As suggested in Chapter 16 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b), we will take the following steps to deal with missing data.
Whenever possible, we will contact the original investigators to request relevant missing data. If the number of participants evaluated for a given outcome is not reported, we will use the number of participants randomised per treatment arm as the denominator. If only percentages but no absolute number of events are reported for binary outcomes, we will calculate numerators using percentages. If estimates for mean and standard deviations are missing, we will calculate these statistics from reported data whenever possible, using approaches described in Chapter 7.7 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). If standard deviations are missing and we are not able to calculate them from reported data, we will calculate values according to a validated imputation method (Furukawa 2006). If data are not reported numerically but graphically, we will estimate missing data from figures. We will perform sensitivity analyses to assess how sensitive results are to imputing data in some way. We will address in the Discussion section the potential impact of missing data on findings of the review.
Assessment of heterogeneity
Pairwise meta‐analyses
For each direct comparison, we will use visual inspection of the forest plots as well as Cochran’s Q based on a Chi2 statistic and the I2 statistic in order to detect the presence of heterogeneity. We will interpret I2 values according to Chapter 9.5.2 of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011). We will use the P value of the Chi2 test only for describing the extent of heterogeneity and not for determining statistical significance. In addition, we will report Ʈ2, the between‐study variance in random‐effects meta‐analysis.
Network meta‐analysis
A very important pre‐supposition for using network meta‐analysis is to make sure that the network is consistent, meaning that direct and indirect evidence on the same comparisons agree. Inconsistency can be caused by incomparable inclusion and exclusion criteria of the trials in the network.
We will evaluate the assumption of transitivity epidemiologically by comparing the distribution of the potential effect modifiers across the different pairwise comparisons. For each set of studies, grouped by treatment comparison, we will create a table of important clinical and methodological characteristics. We will visually inspect the similarity of these factors, including the inclusion and exclusion criteria of every trial in the network.
To evaluate the presence of inconsistency locally we will use the Bucher method for single loops of evidence (Bucher 1997), as described for example in Dias 2013. For each closed loop, we will calculate the difference between direct and indirect evidence together with its 95% CI. We will use loop‐specific z‐tests to infer about the presence of inconsistency in each loop. We will use graphical representation of estimates of inconsistency together with 95% CIs and will report the percentage of inconsistent loops in the network. It should be noted that in a network of evidence there may be many loops and with multiple testing there is an increased likelihood that we might find an inconsistent loop by chance. Therefore, we will be cautious deriving conclusions from this approach.
To evaluate the presence of inconsistency in the entire network, we will give the generalised heterogeneity statistic Qtotal and the generalised I2 statistic, as described in Schwarzer 2015. We will use the decomp.design command in the R package netmeta (R 2016; netmeta 2016) for decomposition of the heterogeneity statistic into a Q statistic for assessing the heterogeneity between studies with the same design and a Q statistic for assessing the designs inconsistency to identify the amount of heterogeneity/inconsistency within as well as between designs. Furthermore, we will create a net heat plot (Krahn 2013), a graphical tool for locating inconsistency in network meta‐analysis, using the command netheat in the R package netmeta. We will give Qtotal and its components as well as net heat plots based on fixed‐effect and random‐effects models to identify differences between these approaches. For random‐effects models, we will report Ʈ2.
If we find substantive heterogeneity and/or inconsistency, we will explore possible sources by performing pre‐specified sensitivity and subgroup analyses (see below). In addition, we will review the evidence base, reconsider inclusion criteria as well as discuss the potential role of unmeasured effect modifiers to identify further sources.
Assessment of reporting biases
In pairwise comparisons with at least 10 trials, we will examine the presence of small‐study effects graphically by generating funnel plots. We will use linear regression tests (Egger 1997) to test for funnel plot asymmetry. A P value less than 0.1 will be considered significant for this test (Sterne 2011). We will examine the presence of small‐study effects for the primary outcome only. Moreover, we will search study registries, to identify completed, but not published trials.
Data synthesis
Methods for direct treatment comparisons
We will perform analyses according to recommendations provided in Chapter 9 of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011), and we will use the statistical software of Cochrane ‐ Review Manager (RevMan 2014) ‐ for analysis. We will perform separate analyses for cancer patients receiving moderately or highly emetogenic chemotherapy. If applicable, we will use R (R 2016) for additional analyses that can not be done with RevMan.
Pairwise comparisons are part of the network meta‐analysis. However, in order to outline available direct evidence we will provide forest plots for pairwise comparisons with at least 10 trials and if trials are clinically homogenous. We will perform these standard pairwise meta‐analyses using a random‐effects model. We will calculate corresponding 95% CIs for all analyses, and will graphically present the results using forest plots. When trials are clinically too heterogenous to be combined, we will perform only subgroup analyses without calculating an overall estimate.
Methods for indirect and mixed comparisons
Should the data be considered sufficiently similar to be combined, we will perform a network meta‐analysis using the frequentist weighted least squared approach described by Rücker 2012. We will use a random‐effects model, taking into account the correlated treatment effects in multi‐arm studies. We will assume a common estimate for the heterogeneity variance across the different comparisons. To evaluate the extent to which treatments are connected, we will give a network plot for our primary and secondary outcomes. For each comparison, we will give the estimated treatment effect along with its 95% CI. We will graphically present the results using forest plots, with placebo as reference. We will use the R package netmeta (R 2016; netmeta 2016) for statistical analyses.
GRADE
Quality of the evidence
Two review authors will independently rate the quality of each outcome. We will use the GRADE (Grades of Recommendation, Assessment, Development and Evaluation) system to rank the quality of the evidence using the GRADEprofiler Guideline Development Tool software (GRADEpro GDT 2015), and the guidelines provided in Chapter 12.2 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011) and specifically for network meta‐analyses (Puhan 2014).
The GRADE approach uses five considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of the body of evidence for each outcome. The GRADE system uses the following criteria for assigning grade of evidence.
-
High = we are very confident that the true effect lies close to that of the estimate of the effect.
-
Moderate = we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different.
-
Low = our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect.
-
Very low = we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect.
The GRADE system uses the following criteria for assigning a quality level to a body of evidence (Chapter 12, Higgins 2011).
-
High: randomised trials; or double‐upgraded observational studies.
-
Moderate: downgraded randomised trials; or upgraded observational studies.
-
Low: double‐downgraded randomised trials; or observational studies.
-
Very low: triple‐downgraded randomised trials; or downgraded observational studies; or case series/case reports.
We will decrease grade if:
-
serious (‐1) or very serious (‐ 2) limitation to study quality;
-
important inconsistency (‐ 1);
-
some (‐1) or major (‐ 2) uncertainty about directness;
-
imprecise or sparse data (‐ 1);
-
high probability of reporting bias (‐ 1).
'Summary of findings' table
We will include two 'Summary of findings' table(s), one for each of the main comparisons, to present the main findings in a transparent and simple tabular format. In particular, we will include key information concerning the quality of evidence, the magnitude of effect of the interventions examined, and the sum of available data on the outcomes complete control of nausea in the acute and delayed phase, complete control of vomiting in the acute and delayed phase, on‐study mortality, quality of life, and adverse events.
Subgroup analysis and investigation of heterogeneity
We will use the test for interactions to test for subgroup differences. The following subgroup analysis will be conducted, if appropriate:
-
type of chemotherapy (carboplatin versus other moderately emetogenic chemotherapy);
-
drug dosage;
-
route of administration (intravenous (IV) versus oral);
-
cancer type;
-
patient‐specific prognostic factors (e.g. younger age, female sex, alcohol intake, history of vomiting pregnancy, motion sickness).
Sensitivity analysis
To test the robustness of the results, we will conduct fixed‐effect pairwise and network meta‐analyses. We will report the estimates of the fixed‐effect only if they show a difference to the random‐effects model. We will explore the influence of quality components with regard to low and high risk of bias.
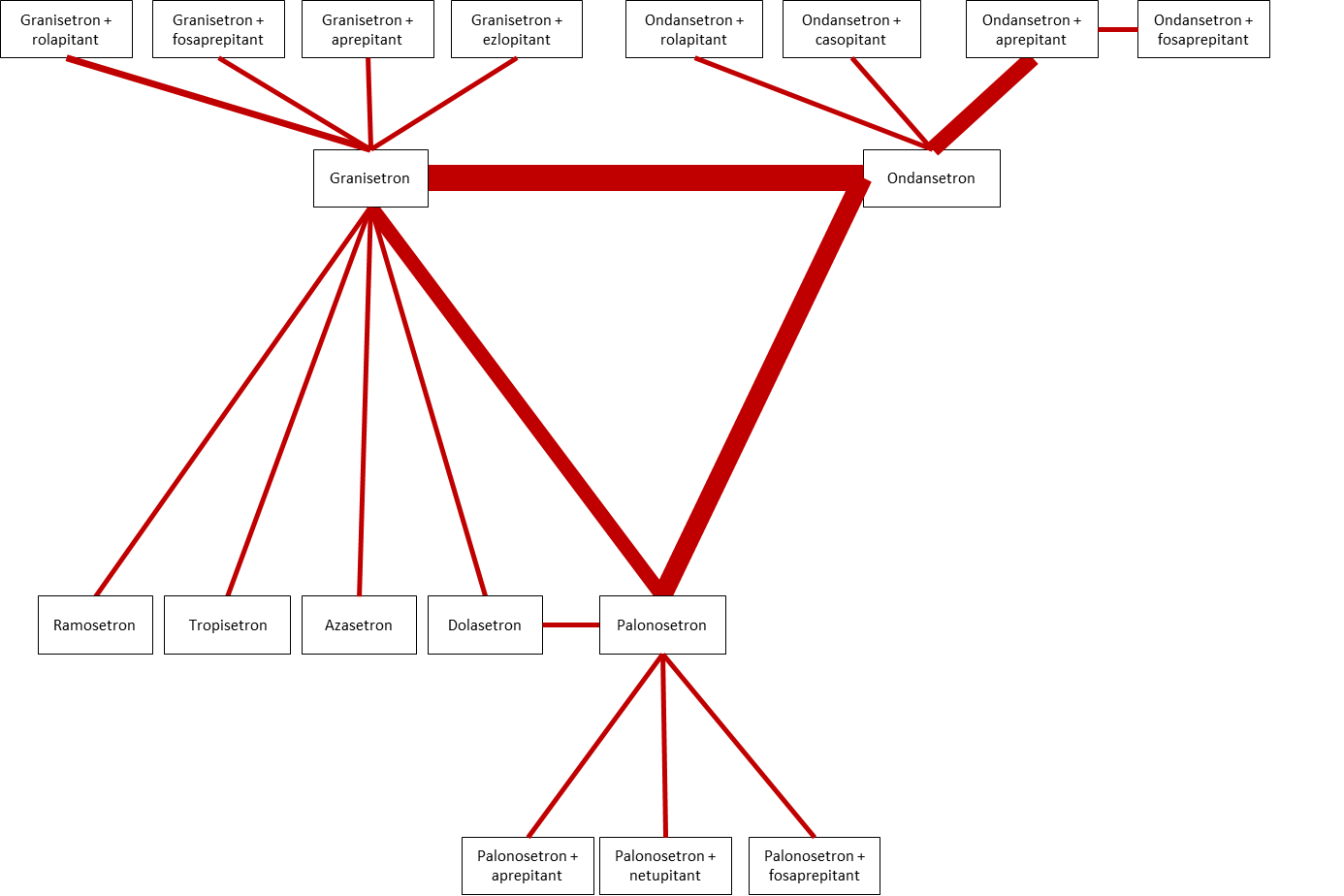
Network of direct comparisons of interest. All drugs and drug combinations are combined with dexamethasone. Strength of the line represents number of RCTs.