Vitamin D supplementation for chronic liver diseases in adults
Abstract
Background
Vitamin D deficiency is often reported in people with chronic liver diseases. Improving vitamin D status could therefore be beneficial for people with chronic liver diseases.
Objectives
To assess the beneficial and harmful effects of vitamin D supplementation in adults with chronic liver diseases.
Search methods
We searched the Cochrane Hepato‐Biliary Group Controlled Trials Register, CENTRAL, MEDLINE Ovid, Embase Ovid, LILACS, Science Citation Index Expanded, and Conference Proceedings Citation Index‐Science. We also searched ClinicalTrials.gov and the World Health Organization International Clinical Trials Registry Platform. We scanned bibliographies of relevant publications and enquired experts and pharmaceutical companies as to additional trials. All searches were up to November 2020.
Selection criteria
Randomised clinical trials that compared vitamin D at any dose, duration, and route of administration versus placebo or no intervention in adults with chronic liver diseases. Vitamin D could have been administered as supplemental vitamin D (vitamin D3 (cholecalciferol) or vitamin D2 (ergocalciferol)), or an active form of vitamin D (1α‐hydroxyvitamin D (alfacalcidol), 25‐hydroxyvitamin D (calcidiol), or 1,25‐dihydroxyvitamin D (calcitriol)).
Data collection and analysis
We used standard methodological procedures expected by Cochrane. We used GRADE to assess the certainty of evidence.
Main results
We included 27 randomised clinical trials with 1979 adult participants. This review update added 12 trials with 945 participants. We assessed all trials at high risk of bias. All trials had a parallel‐group design. Eleven trials were conducted in high‐income countries and 16 trials in middle‐income countries. Ten trials included participants with chronic hepatitis C, five trials participants with liver cirrhosis, 11 trials participants with non‐alcoholic fatty liver disease, and one trial liver transplant recipients. All of the included trials reported the baseline vitamin D status of participants. Participants in nine trials had baseline serum 25‐hydroxyvitamin D levels at or above vitamin D adequacy (20 ng/mL), whilst participants in the remaining 18 trials were vitamin D insufficient (less than 20 ng/mL). Twenty‐four trials administered vitamin D orally, two trials intramuscularly, and one trial intramuscularly and orally. In all 27 trials, the mean duration of vitamin D supplementation was 6 months, and the mean follow‐up of participants from randomisation was 7 months. Twenty trials (1592 participants; 44% women; mean age 48 years) tested vitamin D3 (cholecalciferol); three trials (156 participants; 28% women; mean age 54 years) tested vitamin D2; four trials (291 participants; 60% women; mean age 52 years) tested 1,25‐dihydroxyvitamin D; and one trial (18 participants; 0% women; mean age 52 years) tested 25‐hydroxyvitamin D. One trial did not report the form of vitamin D. Twelve trials used a placebo, whilst the other 15 trials used no intervention in the control group. Fourteen trials appeared to be free of vested interest. Eleven trials did not provide any information on clinical trial support or sponsorship. Two trials were funded by industry.
We are very uncertain regarding the effect of vitamin D versus placebo or no intervention on all‐cause mortality (risk ratio (RR) 0.86, 95% confidence interval (CI) 0.51 to 1.45; 27 trials; 1979 participants). The mean follow‐up was 7 months (range 1 to 18 months). We are very uncertain regarding the effect of vitamin D versus placebo or no intervention on liver‐related mortality (RR 1.62, 95% CI 0.08 to 34.66; 1 trial; 18 participants) (follow‐up: 12 months); serious adverse events such as hypercalcaemia (RR 5.00, 95% CI 0.25 to 100.8; 1 trial; 76 participants); myocardial infarction (RR 0.75, 95% CI 0.08 to 6.81; 2 trials; 86 participants); thyroiditis (RR 0.33, 95% CI 0.01 to 7.91; 1 trial; 68 participants); circular haemorrhoidal prolapse (RR 3.00, 95% CI 0.14 to 65.9; 1 trial; 20 participants); bronchopneumonia (RR 0.33, 95% CI 0.02 to 7.32; 1 trial 20 participants); and non‐serious adverse events. The certainty of evidence for all outcomes is very low.
We found no data on liver‐related morbidity such as gastrointestinal bleeding, hepatic encephalopathy, hepatorenal syndrome, ascites, or liver cancer. There were also no data on health‐related quality of life.
The evidence is also very uncertain regarding the effect of vitamin D versus placebo or no intervention on rapid, early, and sustained virological response in people with chronic hepatitis C.
Authors' conclusions
Given the high risk of bias and insufficient power of the included trials and the very low certainty of the available evidence, vitamin D supplementation versus placebo or no intervention may increase or reduce all‐cause mortality, liver‐related mortality, serious adverse events, or non‐serious adverse events in adults with chronic liver diseases. There is a lack of data on liver‐related morbidity and health‐related quality of life. Further evidence on clinically important outcomes analysed in this review is needed.
PICO
Plain language summary
Vitamin D supplementation for chronic liver diseases
Review question
Is vitamin D supplementation beneficial or harmful for adults with chronic liver diseases?
Background
The available evidence on vitamin D and chronic liver diseases in adults is inconclusive. The aim of this systematic review (a summary of results of available healthcare trials) was to analyse the benefits and harms of the different forms of vitamin D in people with chronic liver diseases.
Study characteristics
Twenty‐seven trials with 1979 adult participants provided data for this review. This review update added 12 trials with 945 participants. The 1979 trial participants were randomly assigned to vitamin D compared with placebo (dummy pill) or no treatment. Eleven trials were conducted in high‐income countries, and 16 trials in middle‐income countries. The age range of the participants was 28 years to 61 years, and on average 44% were women. Ten trials included people with chronic hepatitis C, five trials people with liver cirrhosis, 11 trials people with non‐alcoholic fatty liver disease, and one trial liver transplant recipients. There were no trials including people with chronic hepatitis B or inherited liver diseases. All of the included trials reported the baseline vitamin D status of participants. Vitamin D administration lasted on average six months, and most trials used the cholecalciferol (vitamin D3) form.
Funding
Fourteen trials appeared to be free of vested interest that could bias the trial results. Eleven trials may not have been free of vested interest, as they did not provide any information on clinical trial support or sponsorship. Two trials were funded by industry. We found no difference between trials without industry support compared to trials at risk of industry support in our analysis.
Key results
There is not enough evidence to determine whether vitamin D has beneficial or harmful effects, or has little to no effect on chronic liver diseases in adults. There were too few participants in the individual trials as well as in our evidence synthesis. The trials were at high risk of bias so we lack fair assessments of the benefits and harms of vitamin D in this population. Neither benefits nor harms of vitamin D supplementation in people with chronic liver diseases can be excluded. There were no trials including people with chronic hepatitis B and inherited liver diseases.
Quality of the evidence
We judged all trials to be at high risk of bias (that is an underestimation or overestimation of the true intervention effect). The certainty of evidence is very low.
Currentness of evidence
The evidence is current to November 2020.
Authors' conclusions
Summary of findings
| Vitamin D compared with placebo or no intervention for chronic liver diseases in adults | ||||||
| Patient or population: people with chronic liver diseases | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
|---|---|---|---|---|---|---|
| Risk with placebo or no intervention | Risk with vitamin D | |||||
| All‐cause mortality Follow‐up: mean 7 months (1 to 18 months) | Study population | RR 0.86 | 1979 | ⊕⊝⊝⊝ | ||
| 21 per 1000 | 18 per 1000 | |||||
| Liver‐related mortality Follow‐up: 12 months | Study population | RR 1.62 | 18 | ⊕⊝⊝⊝ | No information was available to calculate absolute effects. | |
| ‐ | ‐ | |||||
| Serious adverse events Follow‐up: mean 10.5 months (6 to 12 months) | Study population | ‐ | ‐ | ⊕⊝⊝⊝ | ||
| Several serious adverse events were reported: hypercalcaemia (RR 5.00, 95% CI 0.25 to 100.8; 1 trial; 76 participants); myocardial infarction (RR 0.75, 95% CI 0.08 to 6.81; 2 trials; 86 participants); thyroiditis (RR 0.33, 95% CI 0.01 to 7.91; 1 trial; 68 participants); circular haemorrhoidal prolapse (RR 3.00, 95% CI 0.14 to 65.9; 1 trial; 20 participants); bronchopneumonia (RR 0.33, 95% CI 0.02 to 7.32; 1 trial; 20 participants). | ||||||
| Liver‐related morbidity | Study population | ‐ | (0 RCTs) | ‐ | ||
| ‐ | ‐ | |||||
| Health‐related quality of life | Study population | ‐ | (0 RCTs) | ‐ | ||
| ‐ | ‐ | |||||
| Non‐serious adverse events Follow‐up: mean 7 months (3 to 12 months) | Study population | ‐ | ‐ | ⊕⊝⊝⊝ | ||
| 1 trial reported 1 single non‐serious adverse event, and another trial reported 16 single non‐serious adverse events, for a total of 17 types of non‐serious adverse events. | ||||||
| Failure of sustained virological response Follow‐up: mean 16 months (6 to 18 months) | Study population | RR 0.65 | 630 | ⊕⊝⊝⊝ | ||
| 484 per 1000 | 315 per 1000 | |||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Downgraded because of risk of bias (1 level) (all trials were at high risk of bias); and imprecision (2 levels) (few events, and the optimal information size of 63,116 participants (based on a proportion of 2% in the control group, a relative risk reduction of 20%, an alpha of 1.25%, and a beta of 10%) was not met; wide CI which included both benefits and harms). | ||||||
Background
Vitamin D is either synthesised in the skin (vitamin D3 (cholecalciferol)) or is obtained from dietary sources (vitamin D3 or vitamin D2 (ergocalciferol)). Vitamin D3 and D2 do not have biological activity. Both forms are metabolised in the liver to 25‐hydroxyvitamin D (calcidiol) and in the kidneys to the biologically active form known as 1,25‐dihydroxyvitamin D (calcitriol), which functions as a steroid‐like hormone (Wesley Pike 2005). The effects of 1,25‐dihydroxyvitamin D are mediated by its binding to vitamin D receptors in the cells (Wesley Pike 2005). Renal production of 1,25‐dihydroxyvitamin D is regulated by parathyroid hormone levels, by serum calcium and phosphorus levels, and by the phosphaturic hormone fibroblast growth factor‐23 (Kovesdy 2013).
Description of the condition
Vitamin D status is determined by the measurement of the serum 25‐hydroxyvitamin D level (Lips 2004; Dawson‐Hughes 2005; Bischoff‐Ferrari 2009). A number of methods are used to measure vitamin D status (radioimmunoassay; high‐performance/pressure liquid chromatography (HPLC); liquid chromatography‐tandem mass spectrometry (LC‐MS/MS); and more recently chemiluminescent immunoassay (CLIA)) (Atef 2018). The accuracy of these methods varies significantly. HPLC and LC‐MS/MS can measure vitamin D2 and D3 independently and are considered as the gold standard (Hollis 2008).
Optimal sun exposure and dietary intake are related to optimal vitamin D status. The US Institute of Medicine recommended target serum 25‐hydroxyvitamin D levels of 20 ng/mL (50 nmol/L) (IOM 2011). Based on the systematic review prepared by the US Institute of Medicine, there are insufficient data to determine the safe upper limit of serum 25‐hydroxyvitamin D levels (IOM 2011). However, serum 25‐hydroxyvitamin D concentrations above 50 ng/mL (125 nmol/L) are considered potentially harmful (IOM 2011). The International Osteoporosis Foundation and the Endocrine Society Task Force recommend a target serum 25‐hydroxyvitamin D level of 30 ng/mL (75 nmol/L) (Dawson‐Hughes 2010; Holick 2011).
The worldwide prevalence of suboptimal vitamin D status is estimated to be high (Lips 2010; Van Schoor 2011; Hilger 2014). The major causes of vitamin D deficiency are insufficient exposure to sunlight, decreased dietary intake, skin pigmentation, obesity, and advanced age (Lips 2006; Holick 2007; Tsiaras 2011; SACN 2016). One systematic review of prospective and intervention studies that assessed the effect of vitamin D status on non‐skeletal outcomes suggested that low vitamin D status in a wide spectrum of diseases may be a marker of ill health (Autier 2014).
Vitamin D undergoes important biotransformation in the liver. The liver also plays a critical role in the inactivation of vitamin D. Because vitamin D is metabolised by the liver, abnormal vitamin D metabolism might be expected to be associated with chronic liver diseases. Vitamin D deficiency has been frequently reported in people with chronic liver diseases (Arteh 2010; Malham 2011; Kitson 2012; Lim 2012; Stokes 2013; Skaaby 2014). There is evidence that low vitamin D status is associated with increased mortality in chronic liver diseases (Putz‐Bankuti 2012; Wang 2013; Stokes 2014; Finkelmeier 2015; Paternostro 2017).
Description of the intervention
Vitamin D can be administered as supplemental vitamin D (vitamin D3 (cholecalciferol) or vitamin D2 (ergocalciferol)) or as an active form of vitamin D (1α‐hydroxyvitamin D (alfacalcidol), 25‐hydroxyvitamin D (calcidiol), or 1,25‐dihydroxyvitamin D (calcitriol)). Vitamin D supplementation prevents osteoporosis and osteomalacia (Lips 2006). It is speculated that vitamin D supplementation may confer benefits beyond the skeletal system, including chronic liver diseases (Davis 2007; Kitson 2012; Han 2013; Elangovan 2017).
How the intervention might work
Vitamin D supplementation may have beneficial effects on bone disorders in people with chronic liver diseases (Guañabens 2010; Luxon 2011). Vitamin D supplementation has also been suggested as a potential therapeutic in people with chronic hepatitis B infection (Farnik 2013; Mahamid 2013); chronic hepatitis C infection (Petta 2010; Gutierrez 2011; Bitetto 2012; Cacopardo 2012; Cholongitas 2012; Luong 2012); autoimmune hepatitis (Luong 2013a); non‐alcoholic fatty liver disease (Geier 2011; Eliades 2013; Kwok 2013; Eliades 2015); primary biliary cirrhosis (Li 2013; Luong 2013b); alcoholic cirrhosis (Trépo 2013; Konstantakis 2016); and hepatocellular carcinoma (Chiang 2011; Lange 2013). It is currently unclear how vitamin D exerts its postulated beneficial effects apart from possibly correcting vitamin D serum levels to something seemingly more normal (Zittermann 2014).
Why it is important to do this review
Observational studies reported a high prevalence of vitamin D insufficiency across a spectrum of chronic liver diseases (Arteh 2010; Lim 2012; Han 2013; Finkelmeier 2014). However, the available evidence on the benefits and harms of vitamin D supplementation in people with chronic liver diseases is insufficient and inconsistent. Meta‐analyses of observational studies and interventional trials in people with chronic hepatitis B or C virus infection and non‐alcoholic fatty liver disease found contradictory results (Villar 2013; Kitson 2014: Mosannen 2017; Tabrizi 2017; Kim 2018; Hariri 2019; Hu 2019; Mansour‐Ghanaei 2019; Sharifi 2019). Results of our previous systematic reviews indicate that vitamin D3 supplementation may potentially prolong life span in adults from the general population (Bjelakovic 2014a), but this observation has been effectively contradicted by recent large randomised clinical trials (Scragg 2017; Manson 2019), and vitamin D does not seem to have an effect on cancer occurrence and cardiovascular diseases (Bjelakovic 2014b; Scragg 2018; Manson 2019; Bischoff‐Ferrari 2020).
Objectives
To assess the beneficial and harmful effects of vitamin D supplementation in adults with chronic liver diseases.
Methods
Criteria for considering studies for this review
Types of studies
Randomised clinical trials, irrespective of blinding, publication status, or language.
Types of participants
Adults (aged 18 years or over) diagnosed with a chronic liver disease (alcoholic, non‐alcoholic fatty liver disease, post‐hepatitis B and C, cholestatic, inherited, and autoimmune diseases).
Types of interventions
Experimental
Vitamin D at any dose and for any duration, administered as monotherapy or in combination with calcium. The route of administration could be enteral (orally) or parenteral. Vitamin D could be administered as supplemental vitamin D (vitamin D3 (cholecalciferol) or vitamin D2 (ergocalciferol)) or as an active form of vitamin D (1α‐hydroxyvitamin D (alfacalcidol), 25‐hydroxyvitamin D (calcidiol), or 1,25‐dihydroxyvitamin D (calcitriol)).
Control
Placebo (identical in appearance and smell) or no intervention.
Concomitant interventions were allowed if used equally in all intervention groups.
Types of outcome measures
Primary outcomes
-
All‐cause mortality.
-
Liver‐related mortality.
-
Serious adverse events. Depending on the availability of data, we attempted to classify adverse events as serious or non‐serious. Serious adverse events were defined as any outward medical occurrence that was life‐threatening; resulted in death, or persistent or significant disability; or any medical event that may have jeopardised the person; or required intervention to prevent it (ICH‐GCP 1997). We considered all other adverse events as non‐serious (see Secondary outcomes below).
Secondary outcomes
-
Liver‐related morbidity (gastrointestinal bleeding, hepatic encephalopathy, hepatorenal syndrome, ascites, liver cancer).
-
Health‐related quality of life (any valid continuous outcome scale used by the trialists).
-
Non‐serious adverse events.
-
Failure of virological response at week four (without rapid virological response), at week 12 (without early virological response), and at six months after treatment (sustained virological response) (e.g. without clearance of hepatitis B virus DNA (HBV‐DNA) or hepatitis C virus ribonucleic acid (HCV‐RNA) from serum).
-
Acute cellular rejection in liver transplant recipients.
-
Vitamin D status.
-
Bone mineral density.
-
Biochemical indices (aspartate aminotransferase, alanine aminotransferase, alkaline phosphatases, gamma‐glutamyl transpeptidase, albumin, bilirubin, triglyceride, cholesterol, calcium, glucose, phosphorus, adiponectin, insulin, parathyroid hormone, C‐reactive protein).
Covariates, effect modifiers, and confounders
We recorded any possible covariates, effect modifiers, and confounders such as dosage and form of vitamin D, dosing schedule, duration of supplementation, duration of follow‐up, mean age, risk of bias, calcium co‐administration, other medications, compliance, and attrition.
Timing of outcome measurement
We applied no restrictions regarding duration of the intervention or length of follow‐up. We assessed outcome data at the end of the trial follow‐up period.
Search methods for identification of studies
Electronic searches
We searched the Cochrane Hepato‐Biliary Group Controlled Trials Register (maintained and searched internally by the Cochrane Hepato‐Biliary Group Information Specialist via the Cochrane Register of Studies Web; 24 November 2020), the Cochrane Central Register of Controlled Trials (CENTRAL; 24 November 2020) in the Cochrane Library, MEDLINE Ovid (1946 to 24 November 2020), Embase Ovid (1974 to 24 November 2020), LILACS (Latin American and Caribbean Health Science Information database) (BIREME; 1982 to 24 November 2020), Science Citation Index Expanded (Web of Science, 1900 to 24 November 2020), and Conference Proceedings Citation Index‐Science (Web of Science; 1990 to 24 November 2020). The search strategies with the time spans of the searches are provided in Appendix 1.
We also searched ClinicalTrials.gov (www.clinicaltrials.gov/) and the World Health Organization International Clinical Trials Registry Platform (www.who.int/ictrp/en/). There were no language limitations.
Searching other resources
We contacted experts and the main manufacturers of vitamin D to enquire as to unpublished randomised trials. We identified additional trials by searching the reference lists of the included trials and systematic reviews, meta‐analyses, and health technology assessment reports.
Data collection and analysis
One review author (MB) performed the electronic searches. Two review authors (GB and DN) independently participated in the manual searches and identified trials eligible for inclusion from the search results.
Selection of studies
Two review authors (MB and GB) independently scanned the abstract, title, or both of every record retrieved to identify studies for further assessment. We investigated all potentially relevant articles as full text. One review author (GB) listed the excluded studies along with the reasons for their exclusion. When a discrepancy occurred in the trial selection, we consulted one review author (CG) to reach consensus. If resolving disagreement was not possible, we added the article to those 'awaiting assessment', and contacted the trial authors for clarification. We also contacted trial authors when information required to make an assessment was not found in the published trial reports. Inter‐rater agreement for trial selection was measured using the Kappa statistic (Cohen 1960). Agreement between the review authors was very good (Kappa = 0.85). We included an adapted PRISMA flow diagram of study selection (Moher 2009).
Data extraction and management
For studies that fulfilled the inclusion criteria, three review authors (GB, DN, and MB) independently extracted the relevant population, intervention characteristics, and risk of bias components using standard data extraction templates. We identified any duplicate publications. Disagreements were resolved by discussion or by consultation with another review author (CG) when required.
Dealing with duplicate publications and companion papers
In the case of duplicate publications and companion papers of a primary study, we maximised our yield of information by simultaneous evaluation of all available data.
Assessment of risk of bias in included studies
Two review authors (GB and DN) independently assessed the risk of bias of each included trial according to the recommendations in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021), and methodological studies (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008; Savović 2012a; Savović 2012b; Savović 2018b). We used the following definitions in our risk of bias assessment.
Allocation sequence generation
-
Low risk of bias: study authors performed sequence generation using computer random number generation or a random number table. Drawing lots, tossing a coin, shuffling cards, and throwing dice were adequate if performed by an independent person not otherwise involved in the study.
-
Unclear risk of bias: method of sequence generation not mentioned.
-
High risk of bias: sequence generation method was not random.
Allocation concealment
-
Low risk of bias: the participant allocations could not have been foreseen in advance of, or during, enrolment. A central and independent randomisation unit controlled allocation. Investigators were unaware of allocation sequence (e.g. if allocation sequence was hidden in sequentially numbered, opaque, and sealed envelopes).
-
Unclear risk of bias: the method used to conceal allocation is not mentioned so that intervention allocations may have been foreseen before, or during, enrolment.
-
High risk of bias: it is likely that the investigators who assigned the participants knew the allocation sequence.
Blinding of participants and personnel
-
Low risk of bias: any of the following: no blinding or incomplete blinding, but we judged that the outcome was not likely to be influenced by lack of blinding; or blinding of participants and key study personnel ensured, and it was unlikely that the blinding could have been broken.
-
Unclear risk of bias: any of the following: insufficient information to permit judgement of 'low risk' or 'high risk'; or the trial did not address this outcome.
-
High risk of bias: any of the following: no blinding or incomplete blinding, and the outcome was likely to be influenced by lack of blinding; or blinding of key study participants and personnel attempted, but it was likely that the blinding could have been broken, and the outcome was likely to be influenced by lack of blinding.
Blinded outcome assessment
-
Low risk of bias: any of the following: no blinding of outcome assessment, but we judged that the outcome measurement was not likely to be influenced by lack of blinding; or blinding of outcome assessment ensured, and it was unlikely that the blinding could have been broken.
-
Unclear risk of bias: any of the following: insufficient information to permit judgement of 'low risk' or 'high risk'; or the trial did not address this outcome.
-
High risk of bias: any of the following: no blinding of outcome assessment, and the outcome measurement was likely to be influenced by lack of blinding; or blinding of outcome assessment, but it was likely that the blinding could have been broken, and the outcome measurement was likely to be influenced by lack of blinding.
Incomplete outcome data
-
Low risk of bias: missing data were unlikely to make treatment effects depart from plausible values. The study used sufficient methods, such as multiple imputation, to handle missing data.
-
Unclear risk of bias: there was insufficient information to assess whether missing data in combination with the method used to handle missing data were likely to induce bias on the results.
-
High risk of bias: the results were likely to be biased due to missing data.
Selective outcome reporting
-
Low risk of bias: the trial reported all predefined outcomes. If the original trial protocol was available, the outcomes should have been those called for in that protocol. If the trial protocol was obtained from a trial registry (e.g. www.clinicaltrials.gov), the outcomes sought should have been those enumerated in the original protocol if the trial protocol was registered before or at the time that the trial was begun. If the trial protocol was registered after the trial had begun, we did not consider those outcomes to be reliable.
-
Unclear risk of bias: the study authors did not report all predefined outcomes fully, or it was unclear whether the study authors recorded data on these outcomes.
-
High risk of bias: the study authors did not report one or more of the predefined outcomes.
Other bias
-
Low risk of bias: the trial appeared to be free of other components (e.g. academic bias) that could put it at risk of bias.
-
Unclear risk of bias: the trial may or may not have been free of other components that could put it at risk of bias.
-
High risk of bias: there were other factors in the trial that could put it at risk of bias (e.g. authors had conducted trials on the same topic).
Overall risk of bias
We judged a trial to be at overall low risk of bias if we assessed the trial at low risk of bias for all of the above domains. We judged a trial to be at high risk of bias if we assessed the trial as having an unclear risk of bias or a high risk of bias in one or more of the risk of bias domains.
Measures of treatment effect
Dichotomous outcomes
For dichotomous outcomes, we calculated and presented risk ratios (RR) with 95% confidence intervals (CI). We planned to calculate and present Peto's odds ratio for rare events such as all‐cause mortality and liver‐related mortality. As there were no differences between the results with Peto's odds ratio and the RR for these two outcomes, we presented the results with RR (Deeks 2021).
Continuous outcomes
For continuous outcomes, we calculated and presented mean differences (MD) with 95% CI.
In the case of time‐to‐event data, we planned to plot and meta‐analyse estimates of hazard ratios (HR) and 95% CIs as presented in the study reports using the generic inverse‐variance method in Review Manager 5 (Review Manager 2020).
Unit of analysis issues
The unit of analysis was the participant as randomised to the intervention group of a clinical trial. In trials with one experimental and one control parallel‐group design, we compared the experimental intervention group versus the control group. In trials with parallel‐group design with more than two intervention groups, we compared the combined vitamin D groups versus the placebo or no intervention group.
For cross‐over trials, we planned to include the relevant data from the first trial period to avoid residual effects from the treatment (Higgins 2011; Higgins 2021). In order to avoid repeated observations on trial participants, we recorded all time points for these observations, but we used the trial data at the longest follow‐up for analysis (Higgins 2011; Higgins 2021).
We planned to include cluster‐randomised trials and assess risk of bias as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011; Higgins 2021).
Dealing with missing data
We attempted to obtain relevant missing data from study authors whenever we lacked important numerical data, such as number of screened or randomised participants, or if there was a lack of data regarding the performance of intention‐to‐treat (ITT) analyses, or data on as‐treated or per‐protocol participant analyses, which prevented us from performing our analyses appropriately. We investigated attrition rates (e.g. dropouts, losses to follow‐up, and withdrawals) and critically appraised issues of missing data (e.g. last‐observation‐carried‐forward and imputation methods).
Regarding the primary outcomes, we included trial participants with incomplete or missing data in sensitivity analyses by imputing them according to the following scenarios (Hollis 1999).
-
Extreme‐case analysis favouring the experimental intervention (best‐worse‐case scenario): none of the dropouts/participants lost from the experimental arm, but all the dropouts/participants lost from the control arm experienced the outcome, including all randomised participants in the denominator.
-
Extreme‐case analysis favouring the control intervention (worst‐best‐case scenario): all dropouts/participants lost from the experimental arm, but none from the control arm experienced the outcome, including all randomised participants in the denominator.
Assessment of heterogeneity
We identified heterogeneity by visual inspection of the forest plots, and by using a standard Chi2 test and a significance level of α = 0.1 (Higgins 2002; Higgins 2003).
We interpreted the I2 statistic as follows (Higgins 2021):
-
0% to 40%: might not be important;
-
30% to 60%: may represent moderate heterogeneity;
-
50% to 90%: may represent substantial heterogeneity;
-
75% to 100%: considerable heterogeneity.
For heterogeneity adjustment of the required information size in the Trials Sequential Analysis, we used diversity (D 2 ), as the I2 statistic used for this purpose consistently underestimates the required information size (Wetterslev 2009).
When we found considerable heterogeneity, we attempted to determine the potential reasons for it by examining the individual trial and subgroup characteristics.
Assessment of reporting biases
To assess the potential existence of publication bias, we planned to use a funnel plot in an exploratory data analysis of the outcome all‐cause mortality, if 10 or more trials were included (Higgins 2021). There are several explanations for the asymmetry of a funnel plot, including true heterogeneity of effect with respect to trial size, poor methodological design of small trials, and publication bias.
We performed adjusted rank correlation, Begg 1994, and a regression asymmetry test, Egger 1997, for detection of bias. We considered a P value of less than 0.10 as significant in these analyses.
Data synthesis
Meta‐analysis
We performed statistical analyses according to the guidelines described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2021).
For the statistical analyses, we used Review Manager 5 (Review Manager 2020), Trial Sequential Analysis version 0.9.5.10 beta (TSA 2017), Stata 8.2 (StataCorp 2005), and SigmaStat 3.0 (Sigma Stat 2003). We analysed the data using both fixed‐effect (DeMets 1987), and random‐effects (DerSimonian 1986), models for meta‐analyses. We presented the results of the random‐effects model analyses. If there were statistically significant discrepancies in the results (e.g. one model giving a significant intervention effect, and the other model giving no significant intervention effect), we presented both models, but considered the more conservative point estimate of the two as the most informative (Jakobsen 2014a). The more conservative point estimate is the estimate closest to one (for dichotomous outcomes) or zero effect (for continuous outcomes). If the two‐point estimates were equal, we used the estimate with the widest CI as our main result of the two analyses (Jakobsen 2014a). For dichotomous outcomes, we calculated RR, and for continuous outcomes we calculated MD or standardised mean difference (SMD) for health‐related quality of life. For all association measures, we used 95% CIs. We performed the analyses using the ITT principle, that is including all randomised participants irrespective of completeness of data. Participants with missing data were included in the analyses using a carry forward of the last observed response. Accordingly, participants who had been lost to follow‐up were counted as being alive.
We compared the intervention effects in subgroups of trials using the method described by Borenstein and colleagues (Borenstein 2009), and implemented it in Review Manager 5 analyses.
Subgroup analysis and investigation of heterogeneity
We planned to conduct a subgroup analysis comparing trials at low risk of bias to trials at unclear or high risk of bias in order to assess the risk of bias to intervention effects (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008; Savović 2012a; Savović 2012b; Savović 2018b). Given that all trials were at high risk of bias, we were not able to conduct this subgroup analysis.
We conducted the following subgroup analyses.
-
According to the aetiology of the chronic liver disease, as vitamin D may have a different effect on the outcome all‐cause mortality in people with chronic liver disease of different aetiology (e.g. non‐alcoholic fatty liver disease, chronic hepatitis C, liver cirrhosis, liver transplant recipients):
-
people with non‐alcoholic fatty liver disease compared to people with chronic hepatitis C;
-
people with non‐alcoholic fatty liver disease compared to people with liver cirrhosis;
-
people with non‐alcoholic fatty liver disease compared to liver transplant recipients.
-
-
According to vested interests. Trials at low risk of vested interests compared to trials at unclear or high risk of vested interests (Lundh 2017).
-
According to vitamin D status at entry (vitamin D sufficient compared to vitamin D insufficient participants). As some participants in some trials had baseline 25‐hydroxyvitamin D levels at or above vitamin D adequacy (20 ng/mL serum), whilst some participants in other trials were vitamin D insufficient (less than 20 ng/mL serum), we conducted this post hoc subgroup analysis.
-
According to the different forms of vitamin D used for supplementation, as vitamin D form may have a different effect on the outcome all‐cause mortality:
-
vitamin D3 compared with placebo or no intervention;
-
vitamin D2 compared with placebo or no intervention;
-
25‐dihydroxyvitamin D compared with placebo or no intervention;
-
1,25‐dihydroxyvitamin D compared with placebo or no intervention.
-
Sensitivity analysis
In addition to the sensitivity analyses described in Dealing with missing data, we used Trial Sequential Analysis as a sensitivity analysis to assess imprecision.
Trial Sequential Analysis
We controlled apparently significant beneficial and harmful intervention effects (potential type I errors) and neutral intervention effects (potential type II errors) with Trial Sequential Analysis to evaluate if these effects could be caused by random errors (Brok 2008; Wetterslev 2008; Brok 2009; Thorlund 2009; Wetterslev 2009; Thorlund 2011; Thorlund 2017; TSA 2017; Wetterslev 2017). The underlying assumption of Trial Sequential Analysis is that testing for significance may be performed each time a new trial is added to the meta‐analysis. We added the trials according to the year of publication, and if more than one trial was published in a year, we added trials alphabetically according to the last name of the first author.
We used Trial Sequential Analysis because cumulative meta‐analyses are at risk of producing random errors due to sparse data and repetitive testing of the accumulating data (Wetterslev 2008). To control for random errors, we calculated the required information size (i.e. the number of participants needed in a meta‐analysis to detect or reject a certain intervention effect) (Wetterslev 2008). The required information size calculation should account for the diversity, present in the meta‐analysis (Wetterslev 2008; Wetterslev 2009). We assessed the diversity‐adjusted required information size (DARIS) for the three primary and the first four secondary outcomes presented in the summary of findings Table 1, by adjusting for multiplicity, using a P value of 0.125, a risk of type II error of 10%, and the observed diversity of the included trials in the random‐effects model meta‐analysis (Jakobsen 2014a). For dichotomous outcomes, we used the proportion in the control group in the meta‐analysis and a relative risk reduction of 20%. For the continuous outcome health‐related quality of life, we would have used the standard deviation (SD) divided by 2 as the minimal relevant difference plus the SD of the difference for calculating the DARIS.
We constructed trial sequential monitoring boundaries for benefit, harm, or futility, based on the DARIS (Thorlund 2017). These boundaries determined the statistical inference one may draw regarding the cumulative meta‐analysis that has not reached the required information size. If the cumulative Z‐curve crosses the trial sequential monitoring boundary for benefit or harm before the diversity‐adjusted required information size is reached, firm evidence may be established, and further trials may be superfluous. In contrast, if the boundary is not surpassed, it is most likely necessary to continue doing trials to detect or reject a certain intervention effect. This can be determined by assessing if the cumulative Z‐curve crosses the trial sequential monitoring boundaries for futility. A more detailed description of Trial Sequential Analysis can be found at www.ctu.dk/tsa/ (Thorlund 2017), and in Wetterslev 2017.
In Trial Sequential Analysis, imprecision is downgraded two levels if the accrued number of participants is below 50% of the DARIS, and one level if it is between 50% and 100% of DARIS. We did not downgrade if the cumulative Z‐curve crossed the monitoring boundaries for benefit, harm, or futility, or if DARIS was reached.
See also Dealing with missing data.
Summary of findings and assessment of the certainty of the evidence
We created summary of findings tables using GRADEpro GDT (GRADEpro GDT). We used the GRADE approach to assess the quality of a body of evidence, that is the extent of certainty on which one can be confident that an estimate of effect or association reflects the item being assessed. The certainty of a body of evidence considers within‐study risk of bias, directness of the evidence (population, intervention, control, outcomes), unexplained heterogeneity or inconsistency of results (including problems with subgroup analyses), imprecision of results (wide CIs, optimal information size criterion), and risk of publication bias (Balshem 2011; Guyatt 2011a; Guyatt 2011b; Guyatt 2011c; Guyatt 2011d; Guyatt 2011e; Guyatt 2011f; Guyatt 2011g; Guyatt 2011h; Guyatt 2013a; Guyatt 2013b; Guyatt 2013c; Guyatt 2013d; Mustafa 2013; Schünemann 2013; Guyatt 2017). We presented the following outcomes: all‐cause mortality, liver‐related mortality, serious adverse events, liver‐related morbidity, health‐related quality of life, non‐serious adverse events, and failure of sustained virological response. After each outcome, we provided the mean and range of follow‐up, or end of follow‐up when there was only one trial that provided data.
These grades of certainty are defined as follows.
-
High certainty: we are very confident that the true effect lies close to that of the estimate of the effect.
-
Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different.
-
Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect.
-
Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect.
Results
Description of studies
In the previous version of this review, we included 15 randomised trials (described in 19 references) with 1034 participants providing data for analyses (Bjelakovic 2017). As described below, our updated searches resulted in the inclusion of an additional 12 randomised trials.
Results of the search
We identified 4672 references of possible interest through the updated electronic searches of the Cochrane Hepato‐Biliary Group Controlled Trials Register (134 records); the Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library (1192 records); MEDLINE Ovid (468 records); Embase Ovid (1900 records); LILACS (28 records); and Science Citation Index Expanded and Conference Proceedings Citation Index‐Science (950 records). We identified an additional two ongoing trials through searching databases of ongoing trials, and four records from reference lists. We excluded 961 duplicates and 3683 clearly irrelevant references through reading of abstracts. Accordingly, we retrieved 34 references for further assessment. Of these, we excluded 19 references because they were not randomised trials, and three references because they did not fulfil our inclusion criteria.
Consequently, we included 12 new randomised trials (described in 12 references) in this updated review version; a total of 27 trials (31 references) with 1979 participants provided data for our analyses (Figure 1).
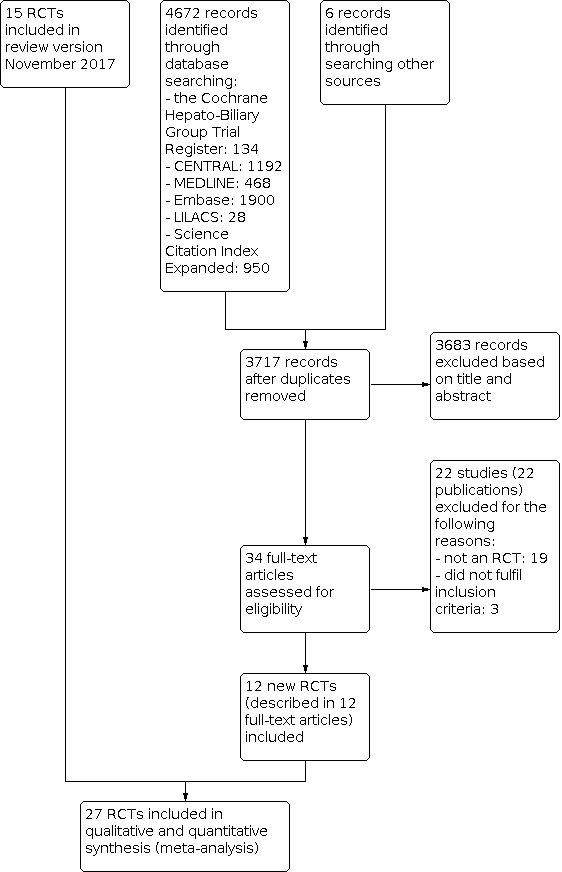
Study flow diagram.
Included studies
For details of the included studies, see Characteristics of included studies; Table 1; Table 2; Table 3.
| Study ID | Protocol | Design | Groups | Bias | Blinding | Participants | Women | Mean |
|---|---|---|---|---|---|---|---|---|
| Yes | Parallel group | 2 | High | NI | 72 | 44 | 47 | |
| No | Parallel group | 2 | High | NI | 115 | 50 | 64 | |
| Yes | Parallel group | 2 | High | PL | 65 | 35 | 59 | |
| Yes | Parallel group | 2 | High | NI | 60 | 40 | 41 | |
| No | Parallel group | 2 | High | PL | 60 | ‐ | ‐ | |
| Yes | Parallel group | 2 | High | PL | 106 | 59 | 45 | |
| No | Parallel group | 2 | High | NI | 101 | 25 | 40 | |
| Yes | Parallel group | 2 | High | PL | 60 | 52 | 48 | |
| Yes | Parallel group | 2 | High | PL | 20 | ‐ | 44 | |
| Yes | Parallel group | 2 | High | NI | 82 | 100 | 34 | |
| No | Parallel group | 2 | High | PL | 109 | 36 | 28 | |
| Yes | Parallel group | 2 | High | NI | 148 | 49 | 52 | |
| No | Parallel group | 2 | High | NI | 101 | 24 | 45 | |
| Yes | Parallel group | 2 | High | PL | 80 | 46 | 52 | |
| Yes | Parallel group | 2 | High | PL | 58 | 38 | 50 | |
| Yes | Parallel group | 3 | High | PL | 120 | 38 | 41 | |
| No | Parallel group | 3 | High | NI | 18 | 0 | 61 | |
| No | Parallel group | 2 | High | NI | 50 | 58 | 47 | |
| Yes | Parallel group | 2 | High | PL | 36 | 25 | 61 | |
| No | Parallel group | 2 | High | NI | 81 | 32 | 38 | |
| No | Parallel group | 2 | High | PL | 60 | 51 | 60 | |
| No | Parallel group | 2 | High | NI | 76 | 66 | 61 | |
| No | Parallel group | 2 | High | NI | 34 | 100 | 56 | |
| Yes | Parallel group | 2 | High | NI | 40 | 50 | 42 | |
| Yes | Parallel group | 2 | High | NI | 68 | 13 | 42 | |
| No | Parallel group | 3 | High | PL | 75 | 17 | 48 | |
| No | Parallel group | 2 | High | NI | 84 | 49 | 59 |
n: number of participants
NI: no intervention
PL: placebo
| Study ID | Participants | Outcome measures | Sponsor | Country |
|---|---|---|---|---|
| Chronic hepatitis C genotype 1 | Sustained virological response | No information | Israel | |
| Chronic hepatitis C genotype 1 | Sustained virological response | No information | Japan | |
| NAFLD | Liver steatosis, liver function | No | Italy | |
| Chronic hepatitis C genotype 1, 4 | Sustained virological response | No | India | |
| NAFLD | Biochemical indices, HOMA, FibroScan measurement | No information | Thailand | |
| NAFLD | Biochemical indices | No | Iran | |
| Chronic hepatitis C genotype 4 | Sustained virological response | No information | Egypt | |
| NAFLD | Liver steatosis, liver function | No | Iran | |
| NAFLD (NASH) | Liver steatosis, liver function | Yes | Switzerland | |
| NAFLD | Serum 25‐hydroxyvitamin D, adiponectin, HOMA‐IR, liver enzymes, and change in grade of NAFLD | No | Iran | |
| NAFLD | Body weight, BMI, insulin resistance, dyslipidaemia, hepatic enzymes, CRP, and adiponectin | No information | Pakistan | |
| Chronic hepatitis C genotype 1, 2, 3 | Sustained virological response | No information | Republic of Korea | |
| Liver cirrhosis | Mortality | No information | India | |
| Chronic hepatitis C | Serum levels of T‐helper cells associated cytokines | No | Thailand | |
| Chronic hepatitis C | Serum fibrotic markers | No | Thailand | |
| NAFLD | Liver function, body fat | No | Iran | |
| Liver cirrhosis | Bone mineral density | Yes | USA | |
| Chronic hepatitis C genotype 2 or 3 | Sustained virological response | No information | Israel | |
| Liver cirrhosis | Vitamin D status, liver function | No | Austria | |
| NAFLD | Insulin resistance and serum ALT | No | India | |
| NAFLD | Liver function, insulin resistance index | No | Iran | |
| Liver cirrhosis | Bone mineral density | No information | Japan | |
| Primary biliary cirrhosis | Bone mineral density | No information | Japan | |
| NAFLD | Biochemical indices, liver steatosis | No information | Iran | |
| Chronic hepatitis C genotype 1, 2, 3, 4 | Early virological response | No | Iran | |
| Liver transplant recipients | Acute cellular rejection rate | No | China | |
| Chronic hepatitis C genotype 1 | Sustained virological response | No information | Japan |
ALT: alanine aminotransferase
BMI: body mass index
CRP: C‐reactive protein
HOMA‐IR: homeostatic model assessment for insulin resistance
NAFLD: non‐alcoholic fatty liver disease
NASH: non‐alcoholic steatohepatitis
| Study ID | Vitamin | Calcium | Route | Regimen | Treatment | Follow‐up | Co‐intervention | |||
|---|---|---|---|---|---|---|---|---|---|---|
| D3 | D2 | 25(OH)D | 1,25(OH)2D | |||||||
| 2000 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 48 | 72 | PEG‐IFN, RBV | |
| 2000 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 16 | 24 | PEG‐IFN, RBV, SP | |
| 2000 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 24 | 24 | ‐ | |
| 2000 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 48 | 48 | PEG‐IFN, RBV | |
| ‐ | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 20 | 20 | ||
| 50,000 | ‐ | ‐ | 0.25 | ‐ | Orally | Weekly and daily | 12 | 12 | ||
| 2143 | ‐ | ‐ | ‐ | ‐ | Orally | Weekly | 48 | 72 | PEG‐IFN, RBV | |
| 7143 | ‐ | ‐ | ‐ | ‐ | Orally | Weekly | 10 | 10 | ‐ | |
| 2100 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 48 | 48 | ||
| 600,000 | ‐ | ‐ | ‐ | ‐ | Intramuscularly | Single dose | Single dose | 4 | Vitamin E 400 IU/day | |
| 50,000 | ‐ | ‐ | ‐ | ‐ | Orally | Weekly | 12 | 12 | ||
| 800 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 24, 48 | 48, 72 | PEG‐IFN, RBV | |
| 300,000; 800 | ‐ | ‐ | ‐ | 1000 | Intramuscularly and orally | Single dose; daily | 24 | 24 | ||
| ‐ | 60,000; 80,000; 100,000 | ‐ | ‐ | ‐ | Orally | Weekly | 6 | 6 | ||
| ‐ | 60,000; 80,000; 100,000 | ‐ | ‐ | ‐ | Orally | Weekly | 6 | 6 | ||
| 1000 | ‐ | ‐ | ‐ | 500 | Orally | Daily | 10 | 12 | ‐ | |
| ‐ | 17,857 | 2400 | ‐ | ‐ | Orally | Daily | 52 | 52 | ‐ | |
| 2000 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 24 | 48 | PEG‐IFN, RBV | |
| 2800 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 8 | 8 | ‐ | |
| 600,000 | ‐ | ‐ | ‐ | ‐ | Intramuscularly | Single dose | Single dose | 24 | ||
| 3571 | ‐ | ‐ | ‐ | ‐ | Orally | Twice a week | 16 | 16 | ‐ | |
| ‐ | ‐ | ‐ | 1 | ‐ | Orally | Daily | 52 | 52 | ‐ | |
| ‐ | ‐ | ‐ | 1 | ‐ | Orally | Daily | 52 | 52 | ‐ | |
| 50,000 | ‐ | ‐ | ‐ | ‐ | Orally | Weekly | 12 | 72 | Lifestyle modification | |
| 1600 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 12 | 12 | PEG‐IFN, RBV | |
| ‐ | ‐ | ‐ | 0.25 | 1000 | Orally | Daily | 4 | 4 | ‐ | |
| 1000 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 16 | 24 | PEG‐IFN, RBV | |
1,25(OH)2D: calcitriol
25(OH)D: calcidiol
IU: international unit
PEG‐IFN: pegylated‐interferon
RBV: ribavirin
SP: simeprevir
All 27 included trials used a parallel‐group design, with two (Shiomi 1999a; Shiomi 1999b; Abu‐Mouch 2011; Nimer 2012; Sharifi 2014; Yokoyama 2014; Esmat 2015; Atsukawa 2016; Barchetta 2016; Boonyagard 2016; Foroughi 2016; Pilz 2016: Vosoghinia 2016: Jha 2017; Komolmit 2017a; Komolmit 2017b; Sakpal 2017; Behera 2018; Geier 2018; Hosseini 2018; Taghvaei 2018; Hussain 2019; Jeong 2019) or three intervention groups (Mobarhan 1984; Xing 2013; Lorvand Amiri 2016; Dabbaghmanesh 2018). The trials were published from 1984 to 2019 (Table 1).
The trials were conducted in Africa (Esmat 2015), Asia (Shiomi 1999a; Shiomi 1999b; Abu‐Mouch 2011; Nimer 2012; Xing 2013; Sharifi 2014; Yokoyama 2014; Atsukawa 2016, Foroughi 2016; Lorvand Amiri 2016; Vosoghinia 2016; Boonyagard 2016; Jha 2017; Komolmit 2017a; Komolmit 2017b; Sakpal 2017; Behera 2018; Dabbaghmanesh 2018; Hosseini 2018; Taghvaei 2018; Hussain 2019; Jeong 2019); Europe (Barchetta 2016; Pilz 2016; Geier 2018), and North America (Mobarhan 1984). Eleven trials were conducted in high‐income countries (Mobarhan 1984; Shiomi 1999a; Shiomi 1999b; Abu‐Mouch 2011; Nimer 2012; Yokoyama 2014; Atsukawa 2016; Barchetta 2016; Pilz 2016; Geier 2018; Jeong 2019), and 16 trials were conducted in middle‐income countries (Table 2) (Xing 2013; Sharifi 2014; Esmat 2015; Boonyagard 2016; Foroughi 2016; Lorvand Amiri 2016; Vosoghinia 2016; Jha 2017; Komolmit 2017a; Komolmit 2017b; Sakpal 2017; Behera 2018; Dabbaghmanesh 2018; Hosseini 2018; Taghvaei 2018; Hussain 2019).
Participants
A total of 1979 participants were randomly assigned in the 27 trials. The number of participants in each trial ranged from 18 to 148 (median 84). The mean age of participants was 48 years (range 28 years to 61 years). The mean proportion of women was 44% (Table 1).
Ten trials included participants with chronic hepatitis C (Abu‐Mouch 2011; Nimer 2012; Yokoyama 2014; Esmat 2015; Atsukawa 2016; Vosoghinia 2016; Komolmit 2017a; Komolmit 2017b; Behera 2018; Jeong 2019); five trials participants with liver cirrhosis (Mobarhan 1984; Shiomi 1999a; Shiomi 1999b; Pilz 2016; Jha 2017); 11 trials participants with non‐alcoholic fatty liver disease (Sharifi 2014; Barchetta 2016; Boonyagard 2016; Foroughi 2016; Lorvand Amiri 2016; Sakpal 2017; Dabbaghmanesh 2018; Geier 2018; Hosseini 2018; Taghvaei 2018; Hussain 2019); and one trial liver transplant recipients (Table 2) (Xing 2013).
All of the included trials reported the baseline vitamin D status of participants based on serum 25‐hydroxyvitamin D levels. Participants in nine trials had baseline 25‐hydroxyvitamin D levels at or above vitamin D adequacy (20 ng/mL) (Abu‐Mouch 2011; Nimer 2012; Yokoyama 2014; Atsukawa 2016; Foroughi 2016; Vosoghinia 2016; Behera 2018; Geier 2018; Komolmit 2017a). Participants in the remaining 18 trials had baseline 25‐hydroxyvitamin D levels considered to be vitamin D insufficient (less than 20 ng/mL) (Mobarhan 1984; Shiomi 1999a; Shiomi 1999b; Xing 2013; Sharifi 2014; Esmat 2015; Barchetta 2016; Boonyagard 2016; Lorvand Amiri 2016; Pilz 2016; Jha 2017; Komolmit 2017b; Sakpal 2017; Dabbaghmanesh 2018; Hosseini 2018; Taghvaei 2018; Hussain 2019; Jeong 2019).
Experimental interventions
One trial did not report form and dose of vitamin D (Boonyagard 2016). One trial with three intervention groups administered 1,25‐dihydroxyvitamin D combined with calcium gluconate in one intervention group, calcium gluconate alone in another intervention group, and placebo in a third group (Xing 2013). We thus compared the 1,25‐dihydroxyvitamin D plus calcium gluconate group versus the calcium gluconate group and placebo group combined. Another trial with three intervention groups used vitamin D3 singly in one intervention group, vitamin D3 combined with calcium carbonate in another intervention group, and placebo in a third group (Table 3) (Lorvand Amiri 2016). We thus compared the first two groups together versus the placebo group. One trial with three intervention groups administered 25‐dihydroxyvitamin D in one intervention group, vitamin D2 in another intervention group, and no intervention in a third group (Mobarhan 1984). We compared vitamin D groups together versus the no intervention group. Another trial with three intervention groups administered 1,25‐dihydroxyvitamin D in one intervention group, vitamin D3 in another intervention group, and placebo in a third group (Dabbaghmanesh 2018). We compared the vitamin D groups together versus the placebo group.
Vitamin D3 (cholecalciferol)
Vitamin D was administered as vitamin D3 (cholecalciferol) in 20 trials (1592 participants; 44% women; mean age 48 years) (Abu‐Mouch 2011; Nimer 2012; Sharifi 2014; Yokoyama 2014; Esmat 2015; Atsukawa 2016; Barchetta 2016; Foroughi 2016; Lorvand Amiri 2016; Pilz 2016; Vosoghinia 2016; Jha 2017; Sakpal 2017; Behera 2018; Dabbaghmanesh 2018; Geier 2018; Hosseini 2018; Taghvaei 2018; Hussain 2019; Jeong 2019). Vitamin D3 was tested orally in 24 trials. Two trials administered vitamin D3 intramuscularly (Sakpal 2017; Hosseini 2018), and one trial administered vitamin D3 intramuscularly and orally (Jha 2017). Vitamin D3 was administered daily in 11 trials (Abu‐Mouch 2011; Nimer 2012; Yokoyama 2014; Atsukawa 2016; Barchetta 2016; Lorvand Amiri 2016; Pilz 2016; Vosoghinia 2016; Behera 2018; Geier 2018; Jeong 2019); weekly in five trials (Esmat 2015; Foroughi 2016; Dabbaghmanesh 2018; Taghvaei 2018; Hussain 2019); twice a week in one trial (Sharifi 2014); in a single dose in two trials (Sakpal 2017; Hosseini 2018); and in a single dose and daily in one trial (Jha 2017). Mean daily dose of vitamin D3 was 2791 international units (IU). The duration of supplementation in trials using vitamin D3 was 8 to 48 weeks (mean 24 weeks). The length of the follow‐up period was from 8 to 72 weeks (mean 28 weeks) (Table 3).
Vitamin D2 (ergocalciferol)
Vitamin D was administered as vitamin D2 (ergocalciferol) in three trials (156 participants; 28% women; mean age 54 years) (Mobarhan 1984; Komolmit 2017a; Komolmit 2017b). Vitamin D2 was tested in a dose of 50,000 IU orally, two or three times weekly for one year in one trial (Mobarhan 1984), and 60,000 to 100,000 IU orally weekly in two trials (Komolmit 2017a; Komolmit 2017b). Mean daily dose of vitamin D2 was 11,429 IU. The duration of supplementation and follow‐up in trials using vitamin D2 was 6 to 52 weeks (mean 21 weeks). The length of the follow‐up period was from 6 to 52 weeks (mean 21 weeks) (Table 3).
1,25‐dihydroxyvitamin D (calcitriol)
Vitamin D was administered as 1,25‐dihydroxyvitamin D in four trials (291 participants; 60% women; mean age 52 years) (Shiomi 1999a; Shiomi 1999b; Xing 2013; Dabbaghmanesh 2018). 1,25‐dihydroxyvitamin D was tested singly, orally, and daily in two trials (Shiomi 1999a; Shiomi 1999b). One trial administered 1,25‐dihydroxyvitamin D combined with calcium (Xing 2013). One trial with a parallel‐group design and three arms tested 1,25‐dihydroxyvitamin D and vitamin D3 in separate arms (Dabbaghmanesh 2018). The dose of 1,25‐dihydroxyvitamin D was 1.0 μg in two trials (Shiomi 1999a; Shiomi 1999b), and 0.25 μg in two trials (Xing 2013; Dabbaghmanesh 2018). Mean daily dose of 1,25‐dihydroxyvitamin D was 0.625 μg. The duration of supplementation and follow‐up in trials using 1,25‐dihydroxyvitamin D was four to 52 weeks (mean 30 weeks) (Table 3).
25‐hydroxyvitamin D (calcidiol)
Vitamin D was administered as 25‐hydroxyvitamin D in one trial (18 participants; 0% women; mean age 52 years) (Mobarhan 1984). 25‐hydroxyvitamin D was tested at a dose of 800 IU/day to 2000 IU/day, orally, for one year (Table 3).
Control interventions
Twelve trials used a placebo in the control group (Xing 2013; Sharifi 2014; Barchetta 2016; Boonyagard 2016; Foroughi 2016; Lorvand Amiri 2016; Pilz 2016; Komolmit 2017a; Komolmit 2017b; Dabbaghmanesh 2018; Geier 2018; Hussain 2019), whilst the remaining 15 trials used no intervention in the control group (Table 1) (Mobarhan 1984; Shiomi 1999a; Shiomi 1999b; Abu‐Mouch 2011; Nimer 2012; Yokoyama 2014; Esmat 2015; Atsukawa 2016; Vosoghinia 2016; Jha 2017; Sakpal 2017; Behera 2018; Hosseini 2018; Taghvaei 2018; Jeong 2019).
Co‐interventions
Seven trials used pegylated‐interferon and ribavirin combined with vitamin D3 in the intervention groups versus pegylated‐interferon and ribavirin in the control group (Abu‐Mouch 2011; Nimer 2012; Yokoyama 2014; Esmat 2015; Vosoghinia 2016; Behera 2018; Jeong 2019). One trial used pegylated‐interferon, ribavirin, and simeprevir (direct‐acting antiviral agent) combined with vitamin D3 in the intervention group versus pegylated‐interferon, ribavirin, and simeprevir in the control group (Atsukawa 2016). One trial supplemented all participants with vitamin E 400 IU (Hosseini 2018). One trial in people with non‐alcoholic fatty liver disease used lifestyle modification (Taghvaei 2018).
Follow‐up
The mean follow‐up period in all 27 trials was 7 months (range 1 to 18 months).
Excluded studies
For details of the excluded studies, see Characteristics of excluded studies.
Risk of bias in included studies
We assessed all trials at high risk of bias (had unclear or high risk of bias in one or more domains assessed) (Figure 2; Figure 3; Table 1). We did not use the test for funnel plot asymmetry because only four trials were included in the meta‐analysis. The adjusted‐rank correlation test (P = 0.34) and a regression asymmetry test (P = 0.48) found no significant evidence of bias.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Fifteen trials described the generation of allocation sequence adequately (Abu‐Mouch 2011; Nimer 2012; Sharifi 2014; Atsukawa 2016; Barchetta 2016; Foroughi 2016; Lorvand Amiri 2016; Pilz 2016; Komolmit 2017a; Komolmit 2017b; Behera 2018; Dabbaghmanesh 2018; Hosseini 2018; Taghvaei 2018; Hussain 2019). The remaining 12 trials were described as being randomised, but the method used for sequence generation was not described or was described insufficiently.
Twelve trials described the method used to conceal allocation adequately (Shiomi 1999a; Abu‐Mouch 2011; Nimer 2012; Sharifi 2014; Esmat 2015; Barchetta 2016; Lorvand Amiri 2016; Pilz 2016; Vosoghinia 2016; Komolmit 2017a; Komolmit 2017b; Geier 2018). The remaining 15 trials were described as being randomised, but the method used for allocation concealment was not described or was described insufficiently.
Blinding
Eight trials performed and adequately described blinding of participants and personnel (Sharifi 2014; Esmat 2015; Barchetta 2016; Lorvand Amiri 2016; Pilz 2016; Dabbaghmanesh 2018; Komolmit 2017a; Komolmit 2017b). Eleven trials did not blind participants and personnel (Mobarhan 1984; Shiomi 1999a; Shiomi 1999b; Abu‐Mouch 2011; Nimer 2012; Yokoyama 2014; Atsukawa 2016; Vosoghinia 2016; Jha 2017; Sakpal 2017; Behera 2018), whilst in eight trials the method used for blinding of participants and personnel was not described or was described insufficiently (Xing 2013; Boonyagard 2016; Foroughi 2016; Geier 2018; Hosseini 2018; Taghvaei 2018; Hussain 2019; Jeong 2019).
Seven trials performed and adequately described blinding of outcome assessors (Sharifi 2014; Esmat 2015; Barchetta 2016; Pilz 2016; Komolmit 2017a; Komolmit 2017b; Dabbaghmanesh 2018). In the remaining 19 trials the method for blinding of outcome assessors was not described or was described insufficiently.
Incomplete outcome data
Twenty trials adequately addressed incomplete outcome data (Mobarhan 1984; Shiomi 1999a; Shiomi 1999b; Abu‐Mouch 2011; Nimer 2012; Xing 2013; Yokoyama 2014; Sharifi 2014; Foroughi 2016; Lorvand Amiri 2016; Pilz 2016; Vosoghinia 2016; Jha 2017; Komolmit 2017a; Komolmit 2017b; Behera 2018; Dabbaghmanesh 2018; Geier 2018; Hosseini 2018; Taghvaei 2018). In seven trials information was insufficient to permit an assessment of whether missing data in combination with the method used to handle missing data was likely to induce bias on the effect estimate (Esmat 2015; Atsukawa 2016; Barchetta 2016; Boonyagard 2016; Sakpal 2017; Hussain 2019; Jeong 2019).
Selective reporting
Thirteen trials reported the outcomes stated in their respective protocols (Mobarhan 1984; Abu‐Mouch 2011; Barchetta 2016; Foroughi 2016; Lorvand Amiri 2016; Pilz 2016; Vosoghinia 2016; Komolmit 2017a; Komolmit 2017b; Behera 2018; Geier 2018; Hosseini 2018; Taghvaei 2018). In 11 trials it was unclear whether all predefined and clinically relevant and reasonably expected outcomes had been reported (Nimer 2012; Xing 2013; Yokoyama 2014; Sharifi 2014; Esmat 2015; Atsukawa 2016; Boonyagard 2016; Jha 2017; Sakpal 2017; Dabbaghmanesh 2018; Hussain 2019). The authors of three trials did not fully report all predefined outcomes (Shiomi 1999a; Shiomi 1999b; Jeong 2019).
Other potential sources of bias
We did not identify any clear signs of academic bias, small‐trial bias, or other potential sources of bias in 13 trials (Mobarhan 1984; Abu‐Mouch 2011; Nimer 2012; Xing 2013; Sharifi 2014; Yokoyama 2014; Esmat 2015; Atsukawa 2016; Barchetta 2016; Dabbaghmanesh 2018; Hosseini 2018; Jha 2017; Hussain 2019). The remaining 14 trials may or may not have been free of other issues that could put them at risk of bias (Shiomi 1999a; Shiomi 1999b; Boonyagard 2016; Foroughi 2016; Lorvand Amiri 2016; Pilz 2016; Vosoghinia 2016; Komolmit 2017a; Komolmit 2017b; Sakpal 2017; Behera 2018; Geier 2018; Taghvaei 2018; Jeong 2019).
Effects of interventions
Primary outcomes
All‐cause mortality
We are very uncertain about the effect of vitamin D versus placebo or no intervention on all‐cause mortality (risk ratio (RR) 0.86, 95% confidence interval (CI) 0.51 to 1.45; I2 = 0%; 27 trials; 1979 participants; Analysis 1.1; very low‐certainty evidence). We are very uncertain about the effect of vitamin D versus placebo or no intervention on all‐cause mortality in people with non‐alcoholic fatty liver disease (no data reported; 11 trials; 803 participants); chronic hepatitis C (RR 0.33, 95% CI 0.04 to 3.13; I2 = 0%; 10 trials; 836 participants); liver cirrhosis (RR 0.91, 95% CI 0.53 to 1.55; I2 = 0%; 5 trials; 265 participants); or liver transplant recipients (no data reported; 1 trial; 75 participants) (Analysis 1.1; summary of findings Table 1). The certainty of evidence is very low. The mean follow‐up was 7 months (range 1 to 18 months).
Subgroup analysis for overall risk of bias
All trials were at high risk of bias, therefore we did not conduct subgroup analysis.
Subgroup analysis for vested interest
Thirteen trials appeared to be free of vested interest. Twelve trials did not provide any information on clinical trial support or sponsorship. Two trials were funded by industry. The test for subgroup differences showed no significant differences in the effect of vitamin D on all‐cause mortality in trials funded by industry (RR 2.69, 95% CI 0.15 to 48.64; 38 participants; 2 trials) and in trials without vested interest (RR 0.83, 95% CI 0.48 to 1.41; I2 = 0%; 1941 participants; 25 trials) (Analysis 1.2).
Subgroup analysis according to vitamin D status at entry
The test for subgroup differences showed insignificant differences in the effect of vitamin D versus placebo or no intervention on all‐cause mortality in participants with normal vitamin D status (RR 0.33, 95% CI 0.04 to 3.13; I2 = 0%; 8 trials; 549 participants; Analysis 1.3) and with low vitamin D status (RR 0.91, 95% CI 0.53 to 1.55; I2 = 0%; 19 trials; 1430 participants; Analysis 1.3).
Subgroup analysis according to form of vitamin D
The test for subgroup differences showed insignificant differences in the effect of different forms of vitamin D versus placebo or no intervention on all‐cause mortality: vitamin D3 (RR 0.83, 95% CI 0.48 to 1.41; I2 = 0%; 20 trials; 1578 participants); vitamin D2 (RR 3.00, 95% CI 0.15 to 61.74; 1 trial; 150 participants); 25‐hydroxyvitamin D (RR 3.00, 95% CI 0.15 to 61.74; 1 trial; 150 participants); and 1,25 dihydroxyvitamin D (4 zero‐event trials; 291 participants) (Analysis 1.4).
Sensitivity analysis for attrition bias
The authors of three trials did not report the exact numbers of participants with missing outcomes in the intervention and control groups (Boonyagard 2016; Jha 2017; Sakpal 2017). There were no losses to follow‐up in 10 trials (Shiomi 1999a; Shiomi 1999b; Abu‐Mouch 2011; Nimer 2012; Xing 2013; Foroughi 2016; Komolmit 2017a; Komolmit 2017b; Behera 2018; Taghvaei 2018). In the remaining 14 included trials, the authors reported the exact numbers of participants with missing outcomes in the intervention and control groups. A total of 65/663 (9.8%) participants had missing outcomes in the vitamin D groups versus 65/572 (11.4%) participants in the control groups.
Best‐worst‐case scenario sensitivity analysis
When we assumed that all participants lost to follow‐up in the experimental intervention group survived, and all those with missing outcomes in the control group died, vitamin D supplementation significantly decreased mortality (RR 0.14, 95% CI 0.06 to 0.30; P < 0.001; I2 = 0%; 1737 participants; 24 trials; Analysis 1.5).
Worst‐best‐case scenario sensitivity analysis
When we assumed that all participants lost to follow‐up in the experimental intervention group died, and all those with missing outcomes in the control group survived, vitamin D supplementation significantly increased mortality (RR 7.95, 95% CI 3.55 to 17.77; P < 0.001; I2 = 0%; 1737 participants; 24 trials; Analysis 1.5).
Sensitivity analysis for imprecision
Trial Sequential Analysis was performed based on a mortality proportion in the control group of 2%, a relative risk reduction of 20% in the experimental intervention group, a type I error of 1.25%, and type II error of 10% (90% power). There was no diversity. The required information size was 63,116 participants. The cumulative Z‐curve did not cross the trial sequential monitoring boundary for benefit or harm after the 27th trial. The trial sequential monitoring boundary was ignored due to little information use (3.14%) (Figure 4). We downgraded imprecision two levels with Trial Sequential Analysis for this outcome, which was in agreement with our GRADE assessment.

All‐cause mortality.
Trial Sequential Analysis was performed based on a mortality in the control group of 2%, a relative risk reduction of 20% in the experimental intervention group, a type I error of 1.25%, and a type II error of 10% (90% power). There was no diversity. The required information size was 63,116 participants. The cumulative Z‐curve (blue line) did not cross the trial sequential monitoring boundary for benefit or harm after the 27th trial. The trial sequential monitoring boundaries were ignored due to little information (3.14%). The blue line represents the cumulative Z‐score of the meta‐analysis. The green dotted lines represent the conventional statistical boundaries.
Liver‐related mortality
The evidence of vitamin D versus placebo or no intervention on the effect of vitamin D on liver‐related mortality is very uncertain (RR 1.62, 95% CI 0.08 to 34.66; 1 trial; 18 participants; very low‐certainty evidence; Analysis 1.6; summary of findings Table 1). The follow‐up was 12 months.
Subgroup analysis according to vitamin D status at entry
Only one trial including participants with low vitamin D status reported liver‐related mortality, making subgroup analysis impossible.
Sensitivity analysis for imprecision
Because of few data, we could not conduct Trial Sequential Analysis, which would only have revealed a similar need to downgrade for imprecision. We downgraded our GRADE assessment two levels for imprecision.
Serious adverse events
The evidence of vitamin D (calcitriol) versus placebo or no intervention is very uncertain on the effect of vitamin D on the risk of hypercalcaemia (RR 5.00, 95% CI 0.25 to 100.8; 1 trial; 76 participants; very low‐certainty evidence; Analysis 1.7); myocardial infarction (RR 0.75, 95% CI 0.08 to 6.81; 2 trials; 86 participants; very low‐certainty evidence; Analysis 1.7); thyroiditis (RR 0.33, 95% CI 0.01 to 7.91; 1 trial; 68 participants; very low‐certainty evidence; Analysis 1.7); circular haemorrhoidal prolapse (RR 3.00, 95% CI 0.14 to 65.90; 1 trial; 20 participants; very low‐certainty evidence; Analysis 1.7); and bronchopneumonia (RR 0.33, 95% CI 0.02 to 7.32; 1 trial; 20 participants; very low‐certainty evidence; Analysis 1.7; summary of findings Table 1). The mean follow‐up was 10.5 months.
Sensitivity analysis for imprecision
Because of few data, we could not conduct Trial Sequential Analysis, which would only have revealed a similar need to downgrade imprecision. We downgraded our GRADE assessment two levels for imprecision.
Secondary outcomes
Liver‐related morbidity
We found no data on liver‐related morbidity.
Health‐related quality of life
We found no data on health‐related quality of life.
Non‐serious adverse events
The evidence is very uncertain as to whether vitamin D3 increases or decreases the risks of glossitis (RR 3.70, 95% CI 0.16 to 87.58; 1 trial; 65 participants; Analysis 1.10); depression (RR 3.00, 95% CI 0.14 to 65.90; 1 trial; 20 participants; Analysis 1.10); lower back pain (RR 3.00, 95% CI 0.14 to 65.90; 1 trial; 20 participants; Analysis 1.10); abdominal bloating (RR 0.33, 95% CI 0.02 to 7.32; 1 trial; 20 participants; Analysis 1.10); cold (RR 0.33, 95% CI 0.02 to 7.32; 1 trial; 20 participants; Analysis 1.10); constipation (RR 0.33, 95% CI 0.02 to 7.32; 1 trial; 20 participants; Analysis 1.10); sore throat (RR 0.33, 95% CI 0.02 to 7.32; 1 trial; 20 participants; Analysis 1.10); sour taste in mouth (RR 0.33, 95% CI 0.02 to 7.32; one trial; 20 participants; Analysis 1.10); contused lacerated wound (RR 0.33, 95% CI 0.02 to 7.32; 1 trial; 20 participants; Analysis 1.10); multiple white matter lesions (RR 0.33, 95% CI 0.02 to 7.32; 1 trial; 20 participants; Analysis 1.10); gastro‐oesophageal reflux (RR 3.00, 95% CI 0.14 to 65.90; 1 trial; 20 participants; Analysis 1.10); abdominal menstrual cramps (RR 3.00, 95% CI 0.14 to 65.90; 1 trial; 20 participants; Analysis 1.10); tubular colon adenoma (RR 3.00, 95% CI 0.14 to 65.90; 1 trial; 20 participants; Analysis 1.10); gastric motility disturbance (RR 3.00, 95% CI 0.14 to 65.90; 1 trial; 20 participants; Analysis 1.10); irritable bowel syndrome (RR 5.00, 95% CI 0.27 to 92.62; 1 trial; 20 participants; Analysis 1.10); knee pain (RR 3.00, 95% CI 0.14 to 65.90; 1 trial; 20 participants; Analysis 1.10); and severe allergy (RR 5.09, 95% CI 0.25 to 103.64; 1 trial; 109 participants; Analysis 1.10) due to the overall rating of very low certainty of evidence (summary of findings Table 1). The mean follow‐up was seven months.
Several non‐serious adverse events were reported in people with chronic hepatitis C treated with a combination of vitamin D and pegylated‐interferon and ribavirin. These were similar in both vitamin D and control groups and consistent with typical interferon‐ribavirin‐induced systemic symptoms such as nausea, headache, insomnia, chills, myalgia, pyrexia, pruritus, mild neutropenia, mild thrombocytopenia, mild neutropenia, and mild anaemia (Abu‐Mouch 2011; Nimer 2012; Yokoyama 2014; Esmat 2015; Atsukawa 2016; Behera 2018; Jeong 2019).
Failure of virological response
Failure of rapid virological response (at week four) in people with chronic viral hepatitis C
Vitamin D3 versus placebo may increase or have no effect on rapid virological response in people with chronic hepatitis C, but the evidence is very uncertain (RR 0.75, 95% CI 0.60 to 0.95; P = 0.02; I2 = 0%; 3 trials; 247 participants; very low‐certainty evidence; Analysis 1.11). The mean follow‐up was 16 months.
Sensitivity analysis for imprecision
Trial Sequential Analysis was conducted based on a failure of rapid virological response in the control group of 53%, a relative risk reduction (RRR) of 20% in the intervention group, a type I error of 1.25%, and type II error of 10% (90% power). There was no diversity. The required information size was 1269 participants. The cumulative Z‐curve crossed the conventional monitoring boundary for benefit, but did not cross the trial sequential monitoring boundaries for benefit, futility or harm (Figure 5). We downgraded imprecision two levels with Trial Sequential Analysis, for this outcome, which was in agreement with our GRADE assessment.

Rapid virological response.
Trial Sequential Analysis was performed based on a failure of rapid virological response in the control group of 53%, a relative risk reduction (RRR) of 20% in the intervention group, a type I error of 1.25%, and a type II error of 10% (90% power). There was no diversity. The required information size was 1269 participants. The cumulative Z‐curve (blue line) crossed the conventional monitoring boundary for benefit but did not cross the trial sequential monitoring boundary for benefit (red down‐sloping line). The blue line represents the cumulative Z‐score of the meta‐analysis. The green dotted lines represent the conventional statistical boundaries. The red inward‐sloping lines represent the trial sequential monitoring boundaries.
Failure of early virological response (at week 12) in people with chronic viral hepatitis C
Vitamin D3 versus placebo may increase or have no effect on early virological response in people with chronic hepatitis C, but the evidence is very uncertain (RR 0.33, 95% CI 0.11 to 1.00; P = 0.05; I2= 75%; 4 trials; 315 participants; very low‐certainty evidence; Analysis 1.12). The mean follow‐up was 13 months.
Sensitivity analysis for imprecision
Trial Sequential Analysis was performed based on a failure of early virological response in the control group of 34%, a relative risk reduction of 20% in the intervention group, a type I error of 1.25%, and type II error of 10% (90% power). The diversity was 88%. The required information size was 21,306 participants. The cumulative Z‐curve (blue line) crossed the conventional monitoring boundary for benefit. The trial sequential monitoring boundary was ignored because of little information use (1.48%) (Figure 6). We downgraded two levels for imprecision with Trial Sequential Analysis for this outcome, which was in agreement with our GRADE assessment.

Early virological response.
Trial Sequential Analysis was performed based on failure of early virological response in the control group of 34%, a relative risk reduction of 20% in the intervention group, a type I error of 1.25%, and a type II error of 10% (90% power). The diversity was 88%. The required information size was 21,306 participants. The cumulative Z‐curve (blue line) crossed the conventional monitoring boundary for benefit. The trial sequential monitoring boundary was ignored due to little information (1.48%). The blue line represents the cumulative Z‐score of the meta‐analysis. The green lines represent the conventional statistical boundaries.
Failure of sustained virological response (at six months after treatment) in people with chronic viral hepatitis C
Vitamin D3 may increase or have no effect on sustained virological response in people with chronic hepatitis C, but the evidence is very uncertain (RR 0.65, 95% CI 0.42 to 1.01; I2 = 76%; 7 trials; 630 participants; very low‐certainty evidence; Analysis 1.13; summary of findings Table 1). The mean follow‐up was 16 months.
Sensitivity analysis for imprecision
Trial Sequential Analysis was performed based on a failure of sustained virological response in the control group of 48%, a relative risk reduction of 20% in the intervention group, a type I error of 1.25%, and type II error of 10% (90% power). The diversity was 80%. The required information size was 7570 participants (Figure 7). We downgraded two levels for imprecision with Trial Sequential Analysis for this outcome, which was in agreement with our GRADE assessment.
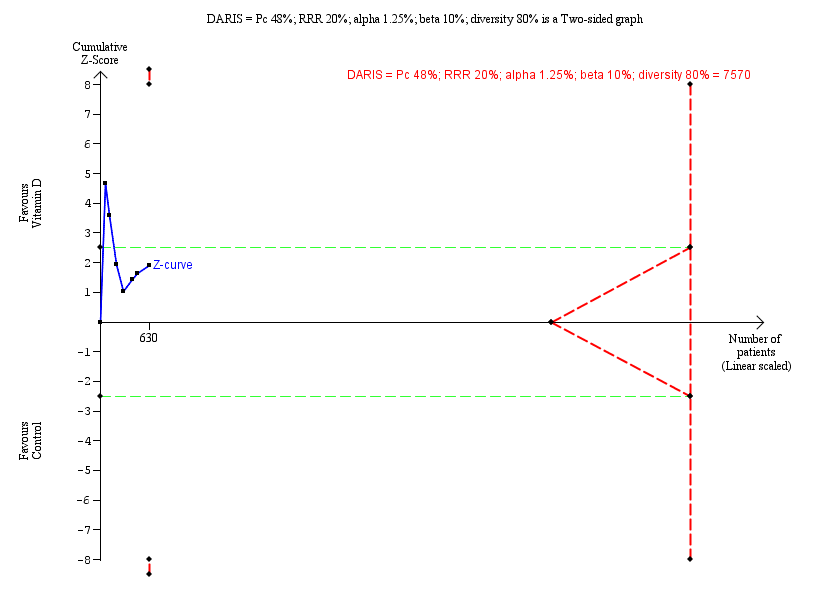
Sustained virological response.
Trial Sequential Analysis was performed based on failure of sustained virological response in the control group of 48%, a relative risk reduction (RRR) of 20% in the intervention group, a type I error of 1.25%, and a type II error of 10% (90% power). Diversity was 80%. The required information size was 7570 participants. The cumulative Z‐curve (blue line) crossed the conventional monitoring boundary for benefit. However, it did not cross any of the monitoring boundaries for benefit, harm, or futility. The blue line represents the cumulative Z‐score of the meta‐analysis. The green lines represent the conventional statistical boundaries. The red inward‐sloping lines represent the trial sequential monitoring boundaries for benefit and harm.
Acute cellular rejection in liver transplant recipients
The evidence is very uncertain on the effect of 1,25‐dihydroxyvitamin D on acute cellular rejection in liver transplant recipients, which may decrease or increase (RR 0.33, 95% CI 0.04 to 2.62; 1 trial; 75 participants; very low‐certainty evidence; Analysis 1.14). The follow‐up was one week.
Vitamin D status
Vitamin D supplementation versus placebo seems to increase vitamin D status of participants, but the evidence is very uncertain (MD 18.49 ng/mL, 95% CI 14.52 to 22.47; I2 = 93%; 15 trials; 1078 participants; very low‐certainty evidence; Analysis 1.15). The mean follow‐up was six months.
Bone mineral density
Vitamin D seems to show an effect on bone mineral density in people with alcoholic liver cirrhosis, but the evidence is very uncertain (MD 0.15 ng/mL, 95% CI 0.04 to 0.26; 1 trial; 18 participants; very low‐certainty evidence; Analysis 1.16). Follow‐up was 12 months (Mobarhan 1984). Two other trials reported bone mineral density, but we could not use the data in analysis (Shiomi 1999a; Shiomi 1999b).
Biochemical indices
Worse prognosis if value result is higher than the normal range
The evidence is very uncertain on the effect of vitamin D on serum activity of aspartate aminotransferase (MD ‐1.75 IU/L, 95% CI ‐5.41 to 1.91; I2 = 82%; 12 trials; 774 participants; Analysis 1.17); serum activity of alanine aminotransferase (MD ‐2.30 IU/L, 95% CI ‐7.60 to 3.00; I2 = 86%; 13 trials; 855 participants; Analysis 1.18); serum activity of alkaline phosphatases (MD ‐0.95 IU/L, 95% CI ‐15.10 to 13.20; I2 = 52%; 6 trials; 344 participants; Analysis 1.19); serum activity of gamma‐glutamyl transpeptidase (MD ‐2.69 IU/L, 95% CI ‐5.26 to ‐0.11; I2 = 0%; 4 trials; 227 participants; Analysis 1.20); serum concentration of bilirubin (MD 0.32 mg/dL, 95% CI 0.00 to 0.63; I2 = 29%; 3 trials; 74 participants; Analysis 1.21); serum concentration of triglyceride (MD 11.27 mg/dL, 95% CI ‐10.99 to 33.53; I2 = 87%; 5 trials; 460 participants; Analysis 1.22); serum concentration of cholesterol (MD 3.51 mg/dL, 95% CI ‐2.83 to 9.85; I2 = 0%; 4 trials; 400 participants; Analysis 1.23); and serum concentration of low‐density lipoprotein (LDL) cholesterol (MD ‐0.97 mg/dL, 95% CI ‐8.70 to 6.76; I2 = 60%; 4 trials; 400 participants; Analysis 1.24).
Worse prognosis if value result is lower than the normal range
The evidence is very uncertain on the effect of vitamin D on serum concentration of albumin (MD ‐1.18 g/L, 95% CI ‐2.96 to 0.59; I2 = 0%; 3 trials; 74 participants; Analysis 1.25) and serum concentration of high‐density lipoprotein (HDL) cholesterol (MD 1.14 mg/dL, 95% CI ‐0.64 to 2.92; I2 = 0%; 4 trials; 400 participants; Analysis 1.26).
Worse prognosis if value result is lower or higher than the normal range
The evidence is very uncertain on the effect of vitamin D on serum concentration of calcium (MD 0.04 mg/dL, 95% CI ‐0.12 to 0.19; I2 = 46%; 7 trials; 423 participants; Analysis 1.27); serum concentration of glucose (MD 1.44 mg/dL, 95% CI ‐5.05 to 7.94; I2 = 85%; 6 trials; 469 participants; Analysis 1.28); serum concentration of phosphorus (MD 0.17 mg/dL, 95% CI ‐0.16 to 0.50; I2 = 53%; 4 trials; 307 participants; Analysis 1.29); serum concentration of adiponectin (MD 1.02 µg/mL, 95% CI ‐0.27 to 2.30; I2 = 62%; 4 trials; 276 participants; Analysis 1.30); serum concentration of insulin (MD 0.03 mIU/mL, 95% CI ‐1.15 to 1.21; I2 = 0%; 6 trials; 428 participants; Analysis 1.31); serum concentration of parathyroid hormone (MD ‐15.18 pg/mL, 95% CI ‐38.54 to 8.18, 2 trials; 118 participants; Analysis 1.32); and serum concentration of C‐reactive protein (MD ‐0.50 mg/L, 95% CI ‐0.93 to ‐0.07; I2 = 86%; 4 trials; 254 participants; Analysis 1.33).
Summary of findings
We have presented our findings for the following outcomes in summary of findings Table 1: all‐cause mortality (mean follow‐up of nine months); liver‐related mortality (mean follow‐up of 12 months); serious adverse events (mean follow‐up of 10.5 months); liver‐related morbidity (no trials); health‐related quality of life (no trials); non‐serious adverse events (mean follow‐up of seven months); failure of sustained virological response (mean follow‐up of 16 months). We downgraded the certainty of the evidence for the outcomes for which data were available to very low. For the outcomes all‐cause mortality, liver‐related mortality, serious adverse events, and non‐serious adverse events, we downgraded the evidence because of risk of bias and imprecision; and for sustained virological response, we downgraded the evidence because of risk of bias, imprecision, inconsistency, and indirectness.
Discussion
Summary of main results
Compared to the previous version of this review (Bjelakovic 2017), the number of trials included in the current review has expanded with the addition of 12 new trials (44%), adding another 945 participants (48%). The current review thus includes 27 randomised clinical trials with 1979 participants. However, our results remain largely the same. The evidence is very uncertain regarding the effect of vitamin D supplements in the form of vitamin D3, vitamin D2, 1,25‐dihydroxyvitamin D, or 25‐dihydroxyvitamin D on all‐cause mortality, liver‐related mortality, and serious and non‐serious adverse events in people with chronic liver diseases. The trials did not present data on liver‐related morbidity such as gastrointestinal bleeding, hepatic encephalopathy, hepatorenal syndrome, ascites, or liver cancer. There were no data on health‐related quality of life. It is very uncertain if vitamin D increases the number of people with sustained virological response or decreases the number of people with acute cellular rejection in liver transplant recipients. Analyses of three trials in people with chronic hepatitis C suggested that vitamin D3 might be beneficial in increasing the number of people with rapid virological response, but the evidence is very uncertain. Vitamin D status of participants with chronic liver diseases seems to increase after supplementation with vitamin D. Vitamin D may or may not have an effect on biochemical indices, but the evidence for all biochemical indices is very uncertain.
The results of our systematic review should be interpreted with great caution because all the included trials were assessed at high risk of bias. The number of people and the trials that provided outcome data were insufficient, which adds to the risk of both type I and type II errors (Keus 2010; Wetterslev 2017). Our sensitivity analysis with Trial Sequential Analysis revealed that there was insufficient information to reach robust conclusions. In this second edition of our review, we defined what the minimal relevant difference for our continuous outcome would be if data are published. Moreover, type 1 and 2 values, diversity, control group proportions, and plausible relative risk reduction will all affect the DARIS calculated, especially as these have changed since the previous version of the review. This is in order to control risks of type 1 and type 2 errors, but will increase the requirement for trial participants. The latter may be seen as an obstacle by many.
Although vitamin D deficiency is considered to be common in people with chronic liver diseases (Chen 2014; Iruzubieta 2014; Elangovan 2017), we found no convincing evidence that vitamin D supplementation might have therapeutic impact in these individuals; however, as highlighted, the evidence is very uncertain.
Overall completeness and applicability of evidence
We included all eligible randomised clinical trials up to November 2020. We found a large number of randomised trials with a small number of participants. We found significant statistical heterogeneity in some of our analyses, such as biochemical indices. This decreases the precision and power of our analyses (Turner 2013; Higgins 2021). Our analyses revealed that outcome reporting was missing in approximately 10% of trial participants. Accordingly, our 'best‐worst‐case' and 'worst‐best‐case' analyses on all‐cause mortality revealed that our results were compatible with both a large beneficial effect and a large detrimental effect of vitamin D. Although these extreme sensitivity analyses are unlikely scenarios, they reveal how missing numbers of participants can substantially change findings from showing great benefit into showing a null effect, or possibly even a harmful effect. We therefore advise critical evaluation of the evidence.
Quality of the evidence
This review followed the overall plan of our published, peer‐reviewed Cochrane protocol (Bjelakovic 2015), some parts of which we revised to enhance clarity for the reader (See Differences between protocol and review). We conducted a thorough review in accordance with Cochrane methodology (Higgins 2011; Higgins 2021), and implemented findings of methodological studies (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008; Savović 2012a; Savović 2012b; Lundh 2017; Savović 2018a).
We repeatedly searched several databases and contacted authors of trials and industry producing vitamin D supplements, therefore we believe it is unlikely that we have overlooked important randomised clinical trials. As stated below, we may have missed trials only reported to regulatory authorities. However, such trials are often neutral or negative (Schroll 2013). We found no evidence of publication bias (Johnson 2007). However, only about every second trial is reported (Gluud 2008), so we cannot exclude reporting biases.
We used GRADEpro GDT to construct a summary of findings table (summary of findings Table 1) (GRADEpro GDT). We calculated the optimal information size when rating imprecision with Trial Sequential Analysis. The GRADE assessments showed that the certainty of evidence was very low for all‐cause mortality, liver‐related mortality, serious adverse events (hypercalcaemia, myocardial infarction, thyroiditis, circular haemorrhoidal prolapse, and bronchopneumonia), liver‐related morbidity, health‐related quality of life, non‐serious adverse events, and failure of sustained virological response. All included trials were at high risk of bias.
In some of our analyses (i.e. biochemical indices) heterogeneity was substantial. This was due to the fact that biochemical indices were measured in people with different aetiology of chronic liver diseases. We also assessed the certainty of the evidence using the GRADE approach based on risk of attrition bias for imprecision, significant between‐trial heterogeneity for inconsistency, and design errors for indirectness. We also conducted Trial Sequential Analysis based on the estimation of the DARIS to avoid an undue risk of random errors in a cumulative meta‐analysis and to prevent premature statements of superiority of vitamin D or of lack of effect (Brok 2008; Wetterslev 2008; Brok 2009; Thorlund 2009; Wetterslev 2009; Thorlund 2011; Thorlund 2017; TSA 2017; Wetterslev 2017). We compared the results of imprecision with GRADE and with Trial Sequential Analysis. The results did not differ, which supports previous studies (Castellini 2018; Gartlehner 2019).
Potential biases in the review process
Certain limitations of this review warrant consideration. As with all systematic reviews, our findings and interpretations are limited by the certainty and quantity of the available evidence on the effects of vitamin D on chronic liver diseases. Despite extensive speculations in the literature and a number of epidemiological studies that claimed possible beneficial effects of vitamin D in people with chronic liver diseases, only a few randomised clinical trials assessed such effects. The duration of supplementation and duration of follow‐up were short in some included trials, which may make it difficult to detect any effects, beneficial or harmful. We assessed all 27 included trials at high risk of bias. Instead of reporting clinical outcomes, most of the trials based their analyses on surrogate outcomes. Such outcomes may be clinically meaningless if they have not been properly validated against clinical outcomes (Gluud 2007; Jakobsen 2017). Many of the included trials were not adequately powered. These factors corrupt the validity of our results (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008; Savović 2012a; Savović 2012b). Adverse events were insufficiently reported. It has been noted that adverse events are very often neglected in randomised trials (Ioannidis 2009). In a number of trials in people with chronic hepatitis C, vitamin D was administered in combination with pegylated‐interferon and ribavirin, which made it difficult to judge the beneficial or harmful effects of vitamin D, or to judge which intervention one should assign any of the observed adverse events. Significant between‐trial heterogeneity was present in some of our meta‐analyses. This may emphasise the inconsistency of our findings and may additionally question some of these findings.
Most of the included trials used vitamin D3; three trials tested vitamin D2; four trials tested 1,25‐dihydroxyvitamin D; and one trial tested 25‐dihydroxyvitamin D.
We did not search the files of regulatory agencies such as the US Food and Drug Administration and European Medicines Agency, which may have biased our selection of trials (Schroll 2013; Boesen 2021). We did not conduct searches for observational studies on harms, which may have biased our findings towards benefits of the interventions with our less focus on harms (Storebø 2015; Storebø 2018).
Different types of bias could have influenced the results of our meta‐analyses including selective reporting of some results in trial publications (Chan 2004; Williamson 2005; Furukawa 2007). Outcome reporting in the included trials was insufficient and inconsistent. There are several possible explanations for selective reporting of outcomes in randomised clinical trials. Trials in which the outcome was not reported may not have measured our outcomes of interest. Researchers may not have reported unexpected results or results may have not satisfied sponsors (Lesser 2007). Pharmaceutical companies provided vitamin D in two of the 27 included trials. This number may in actuality be higher because this information was not available in 11 trials. It could be that researchers have selectively reported outcomes, which may also allude to a publication bias. We are well aware of the difficulties in collecting data on outcomes in clinical trials that focus on safety and efficacy evaluations. The worst result of outcome reporting bias and suppression of some significant or non‐significant findings could be the use of harmful interventions. The results of meta‐analyses may underestimate the true effects of interventions when there is exaggerated outcome reporting bias. One would wish that the results of randomised clinical trials were reported in greater detail (Nordic Trial Alliance 2015). In some of the trials, instead of full reporting, we found partial or qualitative reporting. The huge human efforts of investigators and the high cost of randomised clinical trials should be justified with more rigour in their reporting. In spite of the large investment in the reviewed trials, a number of questions remain unanswered.
Other types of bias, such as academic bias, bias from trials with deficiencies in the trial design (Schulz 1995; Moher 1998; Kjaergard 2001), and small‐trial bias, Siersma 2007, could possibly have influenced our results. Meta‐analysis of randomised trials increases the power and precision of the estimated intervention effect, but this effect may be influenced by systematic errors or random errors and can lead to a report of false significant results (Gluud 2006; Wetterslev 2008). It is probable that the results of our meta‐analysis were influenced by random errors and systematic errors.
A number of design errors may have influenced our results, the first of which is abuse of surrogate outcomes. In most of the included trials, study authors used non‐validated surrogate outcomes such as biochemical indices and liver steatosis, assuming that normal levels are beneficial. The ideal primary outcome in a randomised clinical trial is the outcome relevant to the person's quality of life or course of disease. Relying on non‐validated potential surrogate outcomes is potentially dangerous when assessing new therapies (Gluud 2007; Garattini 2016). We lack validated surrogate outcome measures in hepatology. Some trials included in this review examined early, rapid, or sustained virological response as a surrogate outcome for successful treatment. However, improved early, rapid, or sustained virological response does not definitively mean significant improvement in clinical outcomes (Gluud 2007; Jakobsen 2017). The use of new interventions in hepatology should not be justified unless these have been confirmed beneficial on clinical outcomes (Gluud 2006; Jakobsen 2017). These issues could be resolved with the development and application of agreed‐upon sets of outcomes, known as core outcome sets (www.comet-initiative.org). The increase in the number of hepato‐biliary randomised trials will never be considered a sufficient valuable source for data if aspects of trial design, such as sample size, completeness of data reporting, duration of follow‐up, and bias risk, are not improved.
Agreements and disagreements with other studies or reviews
Efforts in evaluating the benefits and harms of vitamin D supplementation in people with chronic liver diseases resulted in the absence of evidence or potentially neutral results. It is likely that vitamin D deficiency is not a pathogenetic mechanism contributing to liver damage. There is also the possibility that vitamin D deficiency is the consequence but not the cause of chronic liver diseases. Inflammatory processes involved in the pathogenesis of chronic liver diseases, as well as other chronic diseases, reduce serum vitamin D levels, which can explain their low vitamin D status (Autier 2014). Lifestyle, race, and genetic variations could also be related to vitamin D status (Skaaby 2016). Vitamin D supplementation apparently had no effect on all‐cause mortality, but we are unable to exclude meaningful benefits or harms. This result may be due to the fact that the included randomised clinical trials focused on a group of people with well‐compensated liver diseases at low risk of mortality.
Five trials in the current review included people with liver cirrhosis. Given the very low certainty of the evidence, we could not determine if vitamin D supplementation may decrease mortality in people with liver cirrhosis. This finding is contrary to earlier claims in the literature that vitamin D deficiency was associated with increased mortality in people with advanced cirrhosis (Putz‐Bankuti 2012; Wang 2013; Stokes 2014; Finkelmeier 2015; Paternostro 2017). It seems that vitamin D status in people with liver cirrhosis is not only related to liver dysfunction (Lim 2012). In earlier years, it was thought that people with cholestatic liver disease were more likely to be vitamin D deficient. Today, it is evident that people with liver cirrhosis, non‐alcoholic fatty liver disease, and chronic hepatitis C are also at risk for low vitamin D levels. Vitamin D deficiency in the last group of people is likely to be multifactorial in aetiology including decreased intake and absorption, altered activity of hepatic 25‐hydroxylase, and insufficient exposure to sunlight (Lim 2012). Trials including people with liver cirrhosis reported data on biochemical indices after vitamin D supplementation. There seemed to be no significant differences between supplemented and control groups in most of the recorded values, but again we cannot be sure.
Our review did not confirm implications that vitamin D supplementation can be beneficial as an adjuvant to other drugs such as interferon or ribavirin (Luong 2012). Meta‐analysis of 10 trials that included participants with chronic hepatitis C suggests that vitamin D may benefit rapid, early, and sustained virological response, but our findings are very uncertain. One study suggested noffect of vitamin D supplementation in people with advanced chronic hepatitis C (Corey 2012). Oliveira and colleagues observed no association between vitamin D and the degree of liver fibrosis in people with chronic hepatitis C (Oliveira 2017). Our results are contrary to the result of a meta‐analysis that found a positive relationship between high vitamin D status and sustained virological response in people with hepatitis C virus infection (Villar 2013). Another meta‐analysis observed that additional use of vitamin D has a positive effect on sustained virological response of people with chronic hepatitis C (Kim 2018). A further meta‐analysis, by Kitson and colleagues, found that baseline vitamin D status was not associated with sustained virological response in people with chronic hepatitis C (Kitson 2014). However, because of paucity of data, we warn that our results may be deeply influenced by systematic and random errors. We found no randomised trials that tested vitamin D supplementation in people with chronic hepatitis B. Farnik and colleagues found that low vitamin D levels were associated with increased hepatitis B virus replication in people with chronic hepatitis B (Farnik 2013), whilst Mahamid and colleagues showed a correlation between normal vitamin D levels and spontaneous hepatitis B surface antigen clearance from serum (Mahamid 2013). Hoan and colleagues observed vitamin D deficiency in the majority of hepatitis B‐infected people (Hoan 2016). A recently published systematic review and meta‐analysis found that vitamin D levels were lower in people with chronic hepatitis B than in healthy controls (Hu 2019). However, whether vitamin D deficiency is the cause or a consequence of chronic hepatitis is still unknown (Bitetto 2011). We cannot judge if or to what extent the results reached in the mentioned publications are reliable or not, as we have not evaluated them methodologically.
Non‐alcoholic fatty liver disease has become the most common form of chronic liver disease in high‐income countries (Sayiner 2016; Younossi 2016). There is a growing interest in exploring the relationship between vitamin D deficiency and severity of non‐alcoholic fatty liver disease. Eleven trials included in our review administered vitamin D to participants with non‐alcoholic fatty liver disease. We were unable to extract data on clinically important outcomes from these trials. We found no significant effect of vitamin D on surrogate outcomes such as liver function tests. Our results are in accordance with the results of three recent meta‐analyses (Tabrizi 2017; Mansour‐Ghanaei 2019; Guo 2020), which also found no significant effect of vitamin D supplementation on biochemical indices in people with non‐alcoholic fatty liver disease. Two meta‐analyses of case‐control and cross‐sectional studies found that people with non‐alcoholic fatty liver disease were more likely to be vitamin D deficient than people in the control groups, suggesting that vitamin D may play a role in the development of non‐alcoholic fatty liver disease (Eliades 2013; Wang 2015). A recent systematic review of six randomised clinical trials observed improved lipid profile and inflammatory mediators after vitamin D supplementation (Hariri 2019). A systematic review and meta‐analysis of observational studies showed that vitamin D status may not be associated with non‐alcoholic fatty liver disease histologic severity (Jaruvongvanich 2017). We also found that vitamin D supplementation may not be beneficial in this population.
The evidence on the effect of vitamin D supplementation on liver‐related morbidity and health‐related quality of life is still insufficient.
Although three included randomised trials analysed the influence of vitamin D supplementation on bone mineral density in people with liver cirrhosis, we were able to use the data from only one trial (Mobarhan 1984). One systematic review and meta‐analysis concluded that vitamin D supplementation for osteoporosis prevention in community‐dwelling adults without specific risk factors for vitamin D deficiency seemed to be inappropriate (Reid 2014). Another systematic review and meta‐analysis suggested that vitamin D supplementation did not prevent fractures, falls, and increase bone density in adults (Bolland 2018). Similarly, another updated systematic evidence review for the US Preventive Service Task Force found no benefit from vitamin D supplementation for the prevention of cancer and cardiovascular disease (Fortmann 2013). Bolland and colleagues found that vitamin D did not reduce skeletal, vascular, or cancer outcomes (Bolland 2014). Recently completed population‐based randomised clinical trials as well as meta‐analyses of randomised clinical trials found no effect of vitamin D supplementation on cancer occurrence and cardiovascular diseases (Scragg 2017; Scragg 2018; Barbarawi 2019; Keum 2019; Manson 2019; Bischoff‐Ferrari 2020). Interestingly, evidence from a Cochrane Review, Bjelakovic 2014b, a meta‐analysis, Keum 2019, and a large randomised clinical trial, Zhang 2019, suggest that vitamin D may reduce cancer mortality in the general population. A recent systematic review and meta‐analysis of randomised clinical trials found no association between vitamin D supplementation and mortality in critically ill patients (Peng 2020).
It seems that health claims are again ahead of the evidence. The great enthusiasm for vitamin D as a cure for a myriad of diseases, reinforced by observational studies showing that healthy people have higher vitamin D status, has not been supported by evidence obtained from randomised clinical trials. It is very likely that low vitamin D status is not the cause but rather the consequence of chronic diseases (Grey 2010; Guallar 2010; Harvey 2012; Kupferschmidt 2012; Autier 2014). We now have some evidence that vitamin D status is a biomarker of health status (Skaaby 2016). It is likely that less healthy people are obese, less active, and more sunlight‐deprived than healthier people, and therefore have lower vitamin D status (Lucas 2005; Bolland 2006; Grey 2010; Autier 2016; Skaaby 2016). It seems that the cautionary tale of antioxidant supplements is reiterated (Garattini 2016). The current evidence still does not support the use of vitamin D supplementation to prevent or cure chronic liver diseases. Results of ongoing randomised clinical trials may help us further in resolving the vitamin D enigma. The current evidence suggests that it is prudent to get vitamin D from sun exposure and a balanced diet.

Study flow diagram.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

All‐cause mortality.
Trial Sequential Analysis was performed based on a mortality in the control group of 2%, a relative risk reduction of 20% in the experimental intervention group, a type I error of 1.25%, and a type II error of 10% (90% power). There was no diversity. The required information size was 63,116 participants. The cumulative Z‐curve (blue line) did not cross the trial sequential monitoring boundary for benefit or harm after the 27th trial. The trial sequential monitoring boundaries were ignored due to little information (3.14%). The blue line represents the cumulative Z‐score of the meta‐analysis. The green dotted lines represent the conventional statistical boundaries.

Rapid virological response.
Trial Sequential Analysis was performed based on a failure of rapid virological response in the control group of 53%, a relative risk reduction (RRR) of 20% in the intervention group, a type I error of 1.25%, and a type II error of 10% (90% power). There was no diversity. The required information size was 1269 participants. The cumulative Z‐curve (blue line) crossed the conventional monitoring boundary for benefit but did not cross the trial sequential monitoring boundary for benefit (red down‐sloping line). The blue line represents the cumulative Z‐score of the meta‐analysis. The green dotted lines represent the conventional statistical boundaries. The red inward‐sloping lines represent the trial sequential monitoring boundaries.
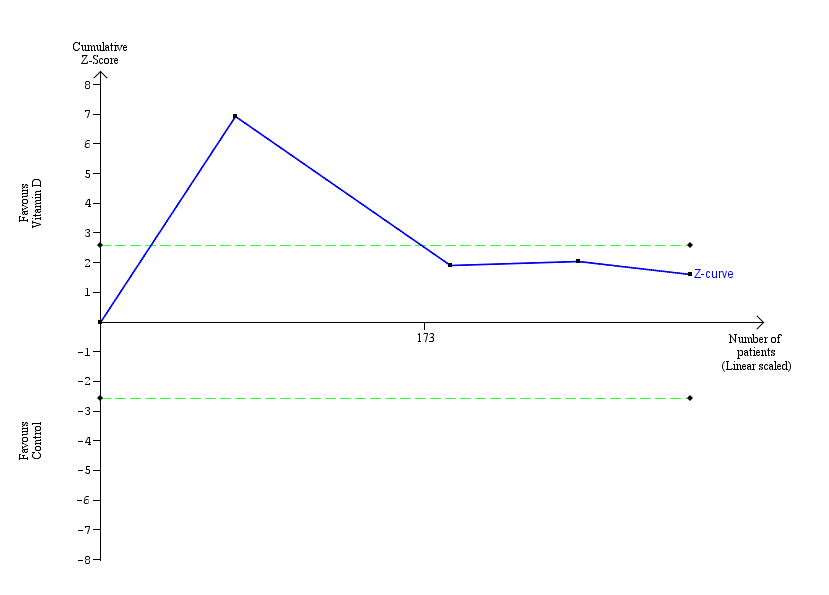
Early virological response.
Trial Sequential Analysis was performed based on failure of early virological response in the control group of 34%, a relative risk reduction of 20% in the intervention group, a type I error of 1.25%, and a type II error of 10% (90% power). The diversity was 88%. The required information size was 21,306 participants. The cumulative Z‐curve (blue line) crossed the conventional monitoring boundary for benefit. The trial sequential monitoring boundary was ignored due to little information (1.48%). The blue line represents the cumulative Z‐score of the meta‐analysis. The green lines represent the conventional statistical boundaries.

Sustained virological response.
Trial Sequential Analysis was performed based on failure of sustained virological response in the control group of 48%, a relative risk reduction (RRR) of 20% in the intervention group, a type I error of 1.25%, and a type II error of 10% (90% power). Diversity was 80%. The required information size was 7570 participants. The cumulative Z‐curve (blue line) crossed the conventional monitoring boundary for benefit. However, it did not cross any of the monitoring boundaries for benefit, harm, or futility. The blue line represents the cumulative Z‐score of the meta‐analysis. The green lines represent the conventional statistical boundaries. The red inward‐sloping lines represent the trial sequential monitoring boundaries for benefit and harm.

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 1: 1.1 All‐cause mortality

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 2: 1.1 All‐cause mortality according to vested interest

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 3: All‐cause mortality according to vitamin D status at entry

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 4: All‐cause mortality according to form of vitamin D

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 5: All‐cause mortality (best‐worst‐case and worst‐best‐case scenarios)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 6: Liver‐related mortality

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 7: Serious adverse events

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 8: Liver‐related morbidity

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 9: Health‐related quality of life

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 10: Non‐serious adverse events

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 11: Failure of rapid virological response

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 12: Failure of early virological response

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 13: Failure of sustained virological response

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 14: Acute cellular rejection in liver transplant recipients

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 15: Vitamin D status (ng/mL)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 16: Bone mineral density (g/cm)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 17: Aspartate aminotransferase (IU/L)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 18: Alanine aminotransferase (IU/L

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 19: Alkaline phosphatases (IU/L)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 20: Gamma‐glutamyl transpeptidase (IU/L)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 21: Bilirubin (mg/dL)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 22: Triglyceride (mg/dL)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 23: Cholesterol (mg/dL)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 24: LDL cholesterol (mg/dL)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 25: Albumin (g/L)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 26: HDL cholesterol (mg/dL)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 27: Calcium (mg/dL)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 28: Glucose (mg/dL)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 29: Phosphorus (mg/dL)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 30: Adiponectin (µg/mL)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 31: Insulin (mIU/mL)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 32: Parathyroid hormone (pg/mL)

Comparison 1: Vitamin D versus placebo or no intervention, Outcome 33: C‐reactive protein (mg/L)
| Vitamin D compared with placebo or no intervention for chronic liver diseases in adults | ||||||
| Patient or population: people with chronic liver diseases | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
|---|---|---|---|---|---|---|
| Risk with placebo or no intervention | Risk with vitamin D | |||||
| All‐cause mortality Follow‐up: mean 7 months (1 to 18 months) | Study population | RR 0.86 | 1979 | ⊕⊝⊝⊝ | ||
| 21 per 1000 | 18 per 1000 | |||||
| Liver‐related mortality Follow‐up: 12 months | Study population | RR 1.62 | 18 | ⊕⊝⊝⊝ | No information was available to calculate absolute effects. | |
| ‐ | ‐ | |||||
| Serious adverse events Follow‐up: mean 10.5 months (6 to 12 months) | Study population | ‐ | ‐ | ⊕⊝⊝⊝ | ||
| Several serious adverse events were reported: hypercalcaemia (RR 5.00, 95% CI 0.25 to 100.8; 1 trial; 76 participants); myocardial infarction (RR 0.75, 95% CI 0.08 to 6.81; 2 trials; 86 participants); thyroiditis (RR 0.33, 95% CI 0.01 to 7.91; 1 trial; 68 participants); circular haemorrhoidal prolapse (RR 3.00, 95% CI 0.14 to 65.9; 1 trial; 20 participants); bronchopneumonia (RR 0.33, 95% CI 0.02 to 7.32; 1 trial; 20 participants). | ||||||
| Liver‐related morbidity | Study population | ‐ | (0 RCTs) | ‐ | ||
| ‐ | ‐ | |||||
| Health‐related quality of life | Study population | ‐ | (0 RCTs) | ‐ | ||
| ‐ | ‐ | |||||
| Non‐serious adverse events Follow‐up: mean 7 months (3 to 12 months) | Study population | ‐ | ‐ | ⊕⊝⊝⊝ | ||
| 1 trial reported 1 single non‐serious adverse event, and another trial reported 16 single non‐serious adverse events, for a total of 17 types of non‐serious adverse events. | ||||||
| Failure of sustained virological response Follow‐up: mean 16 months (6 to 18 months) | Study population | RR 0.65 | 630 | ⊕⊝⊝⊝ | ||
| 484 per 1000 | 315 per 1000 | |||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Downgraded because of risk of bias (1 level) (all trials were at high risk of bias); and imprecision (2 levels) (few events, and the optimal information size of 63,116 participants (based on a proportion of 2% in the control group, a relative risk reduction of 20%, an alpha of 1.25%, and a beta of 10%) was not met; wide CI which included both benefits and harms). | ||||||
| Study ID | Protocol | Design | Groups | Bias | Blinding | Participants | Women | Mean |
|---|---|---|---|---|---|---|---|---|
| Yes | Parallel group | 2 | High | NI | 72 | 44 | 47 | |
| No | Parallel group | 2 | High | NI | 115 | 50 | 64 | |
| Yes | Parallel group | 2 | High | PL | 65 | 35 | 59 | |
| Yes | Parallel group | 2 | High | NI | 60 | 40 | 41 | |
| No | Parallel group | 2 | High | PL | 60 | ‐ | ‐ | |
| Yes | Parallel group | 2 | High | PL | 106 | 59 | 45 | |
| No | Parallel group | 2 | High | NI | 101 | 25 | 40 | |
| Yes | Parallel group | 2 | High | PL | 60 | 52 | 48 | |
| Yes | Parallel group | 2 | High | PL | 20 | ‐ | 44 | |
| Yes | Parallel group | 2 | High | NI | 82 | 100 | 34 | |
| No | Parallel group | 2 | High | PL | 109 | 36 | 28 | |
| Yes | Parallel group | 2 | High | NI | 148 | 49 | 52 | |
| No | Parallel group | 2 | High | NI | 101 | 24 | 45 | |
| Yes | Parallel group | 2 | High | PL | 80 | 46 | 52 | |
| Yes | Parallel group | 2 | High | PL | 58 | 38 | 50 | |
| Yes | Parallel group | 3 | High | PL | 120 | 38 | 41 | |
| No | Parallel group | 3 | High | NI | 18 | 0 | 61 | |
| No | Parallel group | 2 | High | NI | 50 | 58 | 47 | |
| Yes | Parallel group | 2 | High | PL | 36 | 25 | 61 | |
| No | Parallel group | 2 | High | NI | 81 | 32 | 38 | |
| No | Parallel group | 2 | High | PL | 60 | 51 | 60 | |
| No | Parallel group | 2 | High | NI | 76 | 66 | 61 | |
| No | Parallel group | 2 | High | NI | 34 | 100 | 56 | |
| Yes | Parallel group | 2 | High | NI | 40 | 50 | 42 | |
| Yes | Parallel group | 2 | High | NI | 68 | 13 | 42 | |
| No | Parallel group | 3 | High | PL | 75 | 17 | 48 | |
| No | Parallel group | 2 | High | NI | 84 | 49 | 59 | |
| n: number of participants | ||||||||
| Study ID | Participants | Outcome measures | Sponsor | Country |
|---|---|---|---|---|
| Chronic hepatitis C genotype 1 | Sustained virological response | No information | Israel | |
| Chronic hepatitis C genotype 1 | Sustained virological response | No information | Japan | |
| NAFLD | Liver steatosis, liver function | No | Italy | |
| Chronic hepatitis C genotype 1, 4 | Sustained virological response | No | India | |
| NAFLD | Biochemical indices, HOMA, FibroScan measurement | No information | Thailand | |
| NAFLD | Biochemical indices | No | Iran | |
| Chronic hepatitis C genotype 4 | Sustained virological response | No information | Egypt | |
| NAFLD | Liver steatosis, liver function | No | Iran | |
| NAFLD (NASH) | Liver steatosis, liver function | Yes | Switzerland | |
| NAFLD | Serum 25‐hydroxyvitamin D, adiponectin, HOMA‐IR, liver enzymes, and change in grade of NAFLD | No | Iran | |
| NAFLD | Body weight, BMI, insulin resistance, dyslipidaemia, hepatic enzymes, CRP, and adiponectin | No information | Pakistan | |
| Chronic hepatitis C genotype 1, 2, 3 | Sustained virological response | No information | Republic of Korea | |
| Liver cirrhosis | Mortality | No information | India | |
| Chronic hepatitis C | Serum levels of T‐helper cells associated cytokines | No | Thailand | |
| Chronic hepatitis C | Serum fibrotic markers | No | Thailand | |
| NAFLD | Liver function, body fat | No | Iran | |
| Liver cirrhosis | Bone mineral density | Yes | USA | |
| Chronic hepatitis C genotype 2 or 3 | Sustained virological response | No information | Israel | |
| Liver cirrhosis | Vitamin D status, liver function | No | Austria | |
| NAFLD | Insulin resistance and serum ALT | No | India | |
| NAFLD | Liver function, insulin resistance index | No | Iran | |
| Liver cirrhosis | Bone mineral density | No information | Japan | |
| Primary biliary cirrhosis | Bone mineral density | No information | Japan | |
| NAFLD | Biochemical indices, liver steatosis | No information | Iran | |
| Chronic hepatitis C genotype 1, 2, 3, 4 | Early virological response | No | Iran | |
| Liver transplant recipients | Acute cellular rejection rate | No | China | |
| Chronic hepatitis C genotype 1 | Sustained virological response | No information | Japan | |
| ALT: alanine aminotransferase | ||||
| Study ID | Vitamin | Calcium | Route | Regimen | Treatment | Follow‐up | Co‐intervention | |||
|---|---|---|---|---|---|---|---|---|---|---|
| D3 | D2 | 25(OH)D | 1,25(OH)2D | |||||||
| 2000 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 48 | 72 | PEG‐IFN, RBV | |
| 2000 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 16 | 24 | PEG‐IFN, RBV, SP | |
| 2000 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 24 | 24 | ‐ | |
| 2000 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 48 | 48 | PEG‐IFN, RBV | |
| ‐ | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 20 | 20 | ||
| 50,000 | ‐ | ‐ | 0.25 | ‐ | Orally | Weekly and daily | 12 | 12 | ||
| 2143 | ‐ | ‐ | ‐ | ‐ | Orally | Weekly | 48 | 72 | PEG‐IFN, RBV | |
| 7143 | ‐ | ‐ | ‐ | ‐ | Orally | Weekly | 10 | 10 | ‐ | |
| 2100 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 48 | 48 | ||
| 600,000 | ‐ | ‐ | ‐ | ‐ | Intramuscularly | Single dose | Single dose | 4 | Vitamin E 400 IU/day | |
| 50,000 | ‐ | ‐ | ‐ | ‐ | Orally | Weekly | 12 | 12 | ||
| 800 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 24, 48 | 48, 72 | PEG‐IFN, RBV | |
| 300,000; 800 | ‐ | ‐ | ‐ | 1000 | Intramuscularly and orally | Single dose; daily | 24 | 24 | ||
| ‐ | 60,000; 80,000; 100,000 | ‐ | ‐ | ‐ | Orally | Weekly | 6 | 6 | ||
| ‐ | 60,000; 80,000; 100,000 | ‐ | ‐ | ‐ | Orally | Weekly | 6 | 6 | ||
| 1000 | ‐ | ‐ | ‐ | 500 | Orally | Daily | 10 | 12 | ‐ | |
| ‐ | 17,857 | 2400 | ‐ | ‐ | Orally | Daily | 52 | 52 | ‐ | |
| 2000 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 24 | 48 | PEG‐IFN, RBV | |
| 2800 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 8 | 8 | ‐ | |
| 600,000 | ‐ | ‐ | ‐ | ‐ | Intramuscularly | Single dose | Single dose | 24 | ||
| 3571 | ‐ | ‐ | ‐ | ‐ | Orally | Twice a week | 16 | 16 | ‐ | |
| ‐ | ‐ | ‐ | 1 | ‐ | Orally | Daily | 52 | 52 | ‐ | |
| ‐ | ‐ | ‐ | 1 | ‐ | Orally | Daily | 52 | 52 | ‐ | |
| 50,000 | ‐ | ‐ | ‐ | ‐ | Orally | Weekly | 12 | 72 | Lifestyle modification | |
| 1600 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 12 | 12 | PEG‐IFN, RBV | |
| ‐ | ‐ | ‐ | 0.25 | 1000 | Orally | Daily | 4 | 4 | ‐ | |
| 1000 | ‐ | ‐ | ‐ | ‐ | Orally | Daily | 16 | 24 | PEG‐IFN, RBV | |
| 1,25(OH)2D: calcitriol | ||||||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1.1 1.1 All‐cause mortality Show forest plot | 27 | 1979 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.51, 1.45] |
| 1.1.1 Non‐alcoholic fatty liver disease | 11 | 803 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.1.2 Chronic hepatitis C | 10 | 836 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.04, 3.13] |
| 1.1.3 Liver cirrhosis | 5 | 265 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.53, 1.55] |
| 1.1.4 Liver transplant recipients | 1 | 75 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.2 1.1 All‐cause mortality according to vested interest Show forest plot | 27 | 1979 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.51, 1.45] |
| 1.2.1 Trials with vested interest | 2 | 38 | Risk Ratio (M‐H, Random, 95% CI) | 2.69 [0.15, 48.64] |
| 1.2.2 Trials without vested interest | 25 | 1941 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.48, 1.41] |
| 1.3 All‐cause mortality according to vitamin D status at entry Show forest plot | 27 | 1979 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.51, 1.45] |
| 1.3.1 Normal vitamin D status | 8 | 549 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.04, 3.13] |
| 1.3.2 Low vitamin D status | 19 | 1430 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.53, 1.55] |
| 1.4 All‐cause mortality according to form of vitamin D Show forest plot | 26 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.4.1 Vitamin D 3 | 20 | 1578 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.48, 1.41] |
| 1.4.2 Vitamin D 2 | 3 | 150 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.15, 61.74] |
| 1.4.3 1,25‐dihydroxyvitamin D | 4 | 291 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.4.4 25‐hydroxyvitamin D | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.15, 61.74] |
| 1.5 All‐cause mortality (best‐worst‐case and worst‐best‐case scenarios) Show forest plot | 24 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.5.1 Best‐worst‐case scenario | 24 | 1737 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.06, 0.30] |
| 1.5.2 Worst‐best‐case scenario | 24 | 1737 | Risk Ratio (M‐H, Random, 95% CI) | 7.95 [3.55, 17.77] |
| 1.6 Liver‐related mortality Show forest plot | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | 1.62 [0.08, 34.66] |
| 1.7 Serious adverse events Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.7.1 Hypercalcaemia | 1 | 76 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.25, 100.80] |
| 1.7.2 Myocardial infarction | 2 | 86 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.08, 6.81] |
| 1.7.3 Thyroiditis | 1 | 68 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.91] |
| 1.7.4 Circular haemorrhoidal prolapse | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.14, 65.90] |
| 1.7.5 Bronchopneumonia | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.32] |
| 1.8 Liver‐related morbidity Show forest plot | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.9 Health‐related quality of life Show forest plot | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.10 Non‐serious adverse events Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.10.1 Glossitis | 1 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 3.70 [0.16, 87.58] |
| 1.10.2 Depression | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.14, 65.90] |
| 1.10.3 Lower back pain | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.14, 65.90] |
| 1.10.4 Abdominal bloating | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.32] |
| 1.10.5 Cold | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.32] |
| 1.10.6 Constipation | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.32] |
| 1.10.7 Sore throat | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.32] |
| 1.10.8 Sour taste in mouth | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.32] |
| 1.10.9 Contused lacerated wound | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.32] |
| 1.10.10 Multiple white matter lesions | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.32] |
| 1.10.11 Gastro‐oesophageal reflux | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.14, 65.90] |
| 1.10.12 Abdominal menstrual cramps | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.14, 65.90] |
| 1.10.13 Tubular colon adenoma | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.14, 65.90] |
| 1.10.14 Gastric motility disturbance | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.14, 65.90] |
| 1.10.15 Irritable bowel syndrome | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.27, 92.62] |
| 1.10.16 Knee pain | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.14, 65.90] |
| 1.10.17 Severe allergy | 1 | 109 | Risk Ratio (M‐H, Random, 95% CI) | 5.09 [0.25, 103.64] |
| 1.11 Failure of rapid virological response Show forest plot | 3 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.60, 0.95] |
| 1.12 Failure of early virological response Show forest plot | 4 | 315 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.11, 1.00] |
| 1.13 Failure of sustained virological response Show forest plot | 7 | 630 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.42, 1.01] |
| 1.14 Acute cellular rejection in liver transplant recipients Show forest plot | 1 | 75 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.04, 2.62] |
| 1.15 Vitamin D status (ng/mL) Show forest plot | 15 | 1078 | Mean Difference (IV, Random, 95% CI) | 18.49 [14.52, 22.47] |
| 1.16 Bone mineral density (g/cm) Show forest plot | 1 | 18 | Mean Difference (IV, Random, 95% CI) | 0.15 [0.04, 0.26] |
| 1.17 Aspartate aminotransferase (IU/L) Show forest plot | 12 | 774 | Mean Difference (IV, Random, 95% CI) | ‐1.75 [‐5.41, 1.91] |
| 1.18 Alanine aminotransferase (IU/L Show forest plot | 13 | 855 | Mean Difference (IV, Random, 95% CI) | ‐2.30 [‐7.60, 3.00] |
| 1.19 Alkaline phosphatases (IU/L) Show forest plot | 6 | 344 | Mean Difference (IV, Random, 95% CI) | ‐0.95 [‐15.10, 13.20] |
| 1.20 Gamma‐glutamyl transpeptidase (IU/L) Show forest plot | 4 | 227 | Mean Difference (IV, Random, 95% CI) | ‐2.69 [‐5.26, ‐0.11] |
| 1.21 Bilirubin (mg/dL) Show forest plot | 3 | 74 | Mean Difference (IV, Random, 95% CI) | 0.32 [0.00, 0.63] |
| 1.22 Triglyceride (mg/dL) Show forest plot | 5 | 460 | Mean Difference (IV, Random, 95% CI) | 11.27 [‐10.99, 33.53] |
| 1.23 Cholesterol (mg/dL) Show forest plot | 4 | 400 | Mean Difference (IV, Random, 95% CI) | 3.51 [‐2.83, 9.85] |
| 1.24 LDL cholesterol (mg/dL) Show forest plot | 4 | 400 | Mean Difference (IV, Random, 95% CI) | ‐0.97 [‐8.70, 6.76] |
| 1.25 Albumin (g/L) Show forest plot | 3 | 74 | Mean Difference (IV, Random, 95% CI) | ‐1.18 [‐2.96, 0.59] |
| 1.26 HDL cholesterol (mg/dL) Show forest plot | 4 | 400 | Mean Difference (IV, Random, 95% CI) | 1.14 [‐0.64, 2.92] |
| 1.27 Calcium (mg/dL) Show forest plot | 7 | 423 | Mean Difference (IV, Random, 95% CI) | 0.04 [‐0.12, 0.19] |
| 1.28 Glucose (mg/dL) Show forest plot | 6 | 469 | Mean Difference (IV, Random, 95% CI) | 1.44 [‐5.05, 7.94] |
| 1.29 Phosphorus (mg/dL) Show forest plot | 4 | 307 | Mean Difference (IV, Random, 95% CI) | 0.17 [‐0.16, 0.50] |
| 1.30 Adiponectin (µg/mL) Show forest plot | 4 | 276 | Mean Difference (IV, Random, 95% CI) | 1.02 [‐0.27, 2.30] |
| 1.31 Insulin (mIU/mL) Show forest plot | 6 | 428 | Mean Difference (IV, Random, 95% CI) | 0.03 [‐1.15, 1.21] |
| 1.32 Parathyroid hormone (pg/mL) Show forest plot | 2 | 118 | Mean Difference (IV, Random, 95% CI) | ‐15.18 [‐38.54, 8.18] |
| 1.33 C‐reactive protein (mg/L) Show forest plot | 4 | 254 | Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐0.93, ‐0.07] |