Sulthiame add‐on therapy for epilepsy
Abstract
Background
This is an updated version of the Cochrane Review previously published in the Cochrane Database of Systematic Reviews 2015, Issue 10. Epilepsy is a common neurological condition, characterised by recurrent seizures. Most people respond to conventional antiepileptic drugs, however, around 30% will continue to experience seizures, despite treatment with multiple antiepileptic drugs. Sulthiame, also known as sultiame, is a widely used antiepileptic drug in Europe and Israel. We present a summary of the evidence for the use of sulthiame as add‐on therapy in epilepsy.
Objectives
To assess the efficacy and tolerability of sulthiame as add‐on therapy for people with epilepsy of any aetiology compared with placebo or another antiepileptic drug.
Search methods
For the latest update, we searched the Cochrane Register of Studies (CRS Web), which includes the Cochrane Epilepsy Group's Specialized Register and CENTRAL (17 January 2019), MEDLINE Ovid (1946 to January 16, 2019), ClinicalTrials.gov and the WHO ICTRP Search Portal (17 January 2019). We imposed no language restrictions. We contacted the manufacturers of sulthiame, and researchers in the field to seek any ongoing or unpublished studies.
Selection criteria
Randomised controlled trials of add‐on sulthiame, with any level of blinding (single, double or unblinded) in people of any age, with epilepsy of any aetiology.
Data collection and analysis
Two review authors independently selected trials for inclusion, and extracted relevant data. We assessed these outcomes: (1) 50% or greater reduction in seizure frequency between baseline and end of follow‐up; (2) complete cessation of seizures during follow‐up; (3) mean seizure frequency; (4) time‐to‐treatment withdrawal; (5) adverse effects; and (6) quality of life. We used intention‐to‐treat for primary analyses. We presented results as risk ratios (RR) with 95% confidence intervals (CIs). However, due to the paucity of trials, we mainly conducted a narrative analysis.
Main results
We included one placebo‐controlled trial that recruited 37 infants with newly diagnosed West syndrome. This trial was funded by DESITIN Pharma, Germany. During the study, sulthiame was given as an add‐on therapy to pyridoxine. No data were reported for the outcomes: 50% or greater reduction in seizure frequency between baseline and end of follow‐up; mean seizure frequency; or quality of life. For complete cessation of seizures during a nine‐day follow‐up period for add‐on sulthiame versus placebo, the RR was 11.14 (95% CI 0.67 to 184.47; very low‐certainty evidence), however, this difference was not shown to be statistically significant (P = 0.09). The number of infants experiencing one or more adverse events was not significantly different between the two treatment groups (RR 0.85, 95% CI 0.44 to 1.64; very low‐certainty evidence; P = 0.63). Somnolence was more prevalent amongst infants randomised to add‐on sulthiame compared to placebo, but again, the difference was not statistically significant (RR 3.40, 95% CI 0.42 to 27.59; very low‐certainty evidence; P = 0.25). We were unable to conduct meaningful analysis of time‐to‐treatment withdrawal and adverse effects due to incomplete data.
Authors' conclusions
Sulthiame may lead to a cessation of seizures when used as an add‐on therapy to pyridoxine in infants with West syndrome, however, we are very uncertain about the reliability of this finding. The included study was small and had a significant risk of bias, largely due to the lack of details regarding blinding and the incomplete reporting of outcomes. Both issues negatively impacted the certainty of the evidence. No conclusions can be drawn about the occurrence of adverse effects, change in quality of life, or mean reduction in seizure frequency. No evidence exists for the use of sulthiame as an add‐on therapy in people with epilepsy outside West syndrome.
Large, multi‐centre randomised controlled trials are needed to inform clinical practice, if sulthiame is to be used as an add‐on therapy for epilepsy.
PICO
Plain language summary
Sulthiame as add‐on therapy for epilepsy
Review question
A team of Cochrane researchers investigated how well sulthiame worked when it was used as an add‐on antiepileptic medicine (medicines that reduce seizures) in people with any type of epilepsy.
Background
Epilepsy is a common neurological (brain) condition that is characterised by repeated seizures. Most people respond well to conventional antiepileptic medicines, however, about 30% continue to have seizures. These people are said to have drug‐resistant epilepsy.
Sulthiame is an antiepileptic drug that is used widely in some European countries and in Israel. Sometimes it is used as an additional (add‐on) antiepileptic medicine for people with epilepsy, alongside an existing antiepileptic medicine.
Main results
Randomised controlled trials produce the most reliable evidence for medicines. The team searched the medical literature for randomised controlled trials that compared sulthiame as an add‐on therapy to add‐on placebo (an inactive, dummy drug), or another antiepileptic medicine.
The researchers found one relevant trial that included 37 infants, aged from three to 15 months, who had a diagnosis of West syndrome, a type of epilepsy. This trial was funded by DESITIN Pharma (Germany). All infants were started on an antiepileptic medicine, pyridoxine, three days before they added sulthiame or placebo. The infants' parents did not know which add‐on therapy their children received. The trial lasted for nine days.
Very uncertain evidence from the trial suggests that sulthiame may stop seizures in people with West syndrome whose seizures do not stop with pyridoxine. Thirty per cent more infants had their seizures stop when they received add‐on sulthiame (6/20 participants) compared to add‐on placebo (0/17 participants). This difference was not statistically significant, mainly because there were so few infants included in the trial.
The same number of infants experienced one or more adverse effects in both groups (9 in each). More infants experienced somnolence (drowsiness) when they received add‐on sulthiame (4/20), compared to those who received add‐on placebo (1/17), but again, this was not statistically significant.
The small number of infants in the trial, and its short duration, means that we are not confident that the results are reliable.
Further randomised controlled trials are required before meaningful conclusions can be drawn about how well sulthiame works as an add‐on therapy in West syndrome and other types of epilepsy, and to establish whether it produces any serious unwanted or harmful effects.
The evidence is current to January 2019.
Authors' conclusions
Summary of findings
| Sulthiame add‐on compared to placebo for epilepsy | ||||||
| Patient or population: patients between 3 to 15 months of age with West syndrome | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with placebo | Risk with Sulthiame add‐on | |||||
| 50% or greater reduction in seizure frequency | Outcome was not reported by Debus 2004 | |||||
| Complete cessation of seizures during follow‐up Follow‐up: 9 days | Study population | RR 11.14 | 37 | ⊕⊝⊝⊝ | ||
| 0 per 1,000 | 0 per 1000 | |||||
| Mean seizure frequency | Outcome was not reported by Debus 2004 | |||||
| Time‐to‐treatment withdrawal | Outcome was not reported by Debus 2004 | |||||
| Adverse effects: total number of participants experiencing one or more adverse effects Follow‐up: 9 days | Study population | RR 0.85 | 37 | ⊕⊝⊝⊝ | ||
| 529 per 1,000 | 450 per 1000 | |||||
| Adverse effects: somnolence Follow‐up: 9 days | Study population | RR 3.40 | 37 | ⊕⊝⊝⊝ | ||
| 59 per 1,000 | 200 per 1000 | |||||
| Quality of life | Outcome was not reported by Debus 2004 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded once for risk of bias: Debus 2004 did not describe how blinding was achieved or maintained, and we suspected reporting bias for the adverse effect outcomes. bDowngraded twice for imprecision: small study population, therefore, the number of events failed to suffice optimal information size. cEvidence was not upgraded for effect size: normally, the evidence would be upgraded twice when a RR is greater than 5.00, however, due to the concerns regarding risk of bias and small sample size, we were unable to upgrade the evidence. dEvidence was not upgraded for effect size: normally, the evidence would be upgraded once when a RR is greater than 2.00, however, due to the concerns regarding risk of bias and small sample size, we were unable to upgrade the evidence. | ||||||
Background
This review is an update of the previously published review in the Cochrane Database of Systematic Reviews (Milburn‐McNulty 2015).
Description of the condition
Epilepsy is a common neurological condition that is characterised by recurrent seizures. It has an estimated worldwide prevalence of between four and 10 per 1000 of the general population (WHO 2019). The majority of people will respond well to conventional antiepileptic drugs (AED (International League Against Epilepsy 1997)). However, around 30% will not achieve remission, despite trying numerous antiepileptic drugs, often in combination (Sander 1993; Schmidt 1995; Brodie 1996). In an attempt to improve outcomes for these drug‐resistant people, a number of newer potential antiepileptic drugs have been assessed over the past 20 to 30 years. One such drug is sulthiame.
Description of the intervention
Sulthiame (also known as sultiame) is used widely as an antiepileptic drug in some European countries and in Israel (Gross‐Selbeck 2001; Koepp 2002; Engler 2003; Ben‐Zeev 2004; Chahem 2007; Swiderska 2011). It is usually taken in tablet form, with doses taken two to three times per day. When used as monotherapy, sulthiame has been reported to reduce the occurrence of seizures and reduce electroencephalographic (EEG) discharges in people with benign epilepsy of childhood with centrotemporal spikes (Rating 2000; Bast 2003; Ben‐Zeev 2004; Wirrell 2008), benign focal epilepsy of childhood (Engler 2003; Ben‐Zeev 2004), symptomatic, localisation‐related epilepsy, juvenile myoclonic epilepsy (Ben‐Zeev 2004), and adults with drug‐resistant epilepsy and learning disabilities (Koepp 2002). In addition, sulthiame as an add‐on therapy has been reported to reduce seizure activity in people with drug‐resistant epilepsy (Livingston 1967; Chahem 2007; Miyajima 2009).
Reported adverse effects of sulthiame include: deterioration of reading ability, memory, attention skills, and mathematical ability (Wirrell 2008), mixed respiratory and metabolic acidosis (a condition characterised by an abnormally low arterial blood pH (Weissbach 2010)), and crystalluria (the excretion of crystals in the urine (Go 2005)).
How the intervention might work
Sulthiame is a sulphonamide, which may exert antiepileptic activity by producing a modest intracellular acidosis in central neurons via its action as a carbonic anhydrase inhibitor, thereby reducing the frequency of action potentials and epileptiform bursts (Leniger 2002).
Why it is important to do this review
A summary of the best available evidence about the efficacy and tolerability of sulthiame as an add‐on therapy for people with epilepsy is required to inform the most appropriate clinical use of this drug and to inform decisions about the further assessment of this drug.
Objectives
To assess the efficacy and tolerability of sulthiame as add‐on therapy for people with epilepsy of any aetiology compared with placebo or another antiepileptic drug.
Methods
Criteria for considering studies for this review
Types of studies
-
Randomised controlled trials (RCTs)
-
Double, single, or unblinded trials
-
Placebo‐controlled trials
-
Parallel group or cross‐over studies
Types of participants
-
Participants with drug‐resistant epilepsy (defined as epilepsy in which seizure control is not adequately managed with one or more antiepileptic drugs)
-
Participants of any age
-
Participants with epilepsy of any aetiology
Types of interventions
-
For the active treatment group, sulthiame as an adjunct to the participant's AED regimen
-
For the control group, placebo or another AED added to the participant's AED regimen
Types of outcome measures
Primary outcomes
-
50% or greater reduction in seizure frequency. We selected this outcome as it is commonly reported in studies assessing the efficacy of AEDs.
Secondary outcomes
-
Complete cessation of seizures during follow‐up
-
Mean seizure frequency
-
Time‐to‐treatment withdrawal; reflective of both intolerable adverse effects and lack of efficacy
-
Any reported adverse effects, such as, but not limited to, deterioration in cognitive ability, crystalluria, or respiratory and metabolic acidosis
-
Quality of life; an overall improvement or deterioration
Search methods for identification of studies
Electronic searches
We ran searches for the original review in January 2012, and subsequent searches on 28 August 2012, 3 June 2014, 11 August 2015, and 22 November 2017. For the latest update, we searched the following databases on 17 January 2019:
-
The Cochrane Register of Studies (CRS Web; includes the Cochrane Epilepsy Group Specialized Register and the Cochrane Central Register of Controlled Trials (CENTRAL)), using the search strategy outlined in Appendix 1;
-
MEDLINE Ovid (1946 to 16 January 2019), using the search strategy outlined in Appendix 2;
-
ClinicalTrials.gov, using the search strategy outlined in Appendix 3;
-
The WHO International Clinical Trials Registry Platform (ICTRP), using the search strategy outlined in Appendix 4.
We also searched SCOPUS (1823 to 3 June 2014), as an alternative to Embase, using the search strategy outlined in Appendix 5, but this is no longer necessary, because randomised and quasi‐randomised controlled trials from Embase are now included in CENTRAL.
We did not impose any language restrictions.
Searching other resources
We checked the reference lists of retrieved reports for additional reports of relevant studies. We also contacted the manufacturers of sulthiame for information about ongoing or unpublished studies.
Data collection and analysis
Selection of studies
Two review authors (RB and KMM) independently assessed studies for inclusion. They resolved disagreements by discussion.
Data extraction and management
We extracted data from the trial and assessed the design of the trial and demographic makeup of the participants, in addition to the outcomes listed in the Types of outcome measures section. Two review authors (RB and KMM) independently assessed studies, and resolved disagreements by discussion. The types of data extracted are listed below.
Trial design
-
Method of randomisation
-
Method of concealment
-
Duration of baseline period
-
Duration of treatment period
-
Duration of 'wash‐out' period for cross‐over studies
-
Dose of sulthiame
-
Description of adverse effects
-
Description of withdrawals and drop‐outs
Demographic information
-
Number of participants in treatment group
-
Number of participants in control group
-
Age
-
Sex
-
Type of seizures and epilepsy
-
Mean baseline seizures frequency
-
AED(s) on which participants were already established
Assessment of risk of bias in included studies
Two review authors (RB and KMM) independently assessed the risk of bias associated with each study's methodology, using the trial design factors outlined in the Data extraction and management section. Risk of bias was evaluated according to the Cochrane 'Risk of bias' tool, as detailed in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). The Cochrane 'Risk of bias' tool comprises seven parameters: (1) random sequence generation, (2) allocation concealment, (3) incomplete outcome data, (4) selective reporting, (5) blinding of participants and personnel, (6) blinding of outcome assessors, and (7) other bias. For each entry, they awarded a judgement (low risk of bias, high risk of bias, or unclear risk of bias), supported by an agreed review author comment. They resolved disagreements by discussion.
Measures of treatment effect
For binary data, we expressed relative treatment effects as risk ratios (RR) with 95% confidence intervals (CI); for continuous data, we had planned to use mean difference (MD) with 95% CI. When we compared data where one group had zero events, we calculated the RR by adding 0.5 to each value in the contingency table, as advised in section 9.2.2.2 of Chapter 9 of the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2011). We considered that a P value of less than 0.05 demonstrated statistical significance.
Unit of analysis issues
The inclusion of cross‐over studies in meta‐analyses introduces unit of analysis issues because each participant contributes data to both treatment groups. We had planned to extract data from the first treatment period of any eligible cross‐over study, had any been identified. Essentially, we would have regarded the first treatment period as a parallel study, thus preventing data from the same participant from being considered twice, whilst simultaneously avoiding any issues of carry‐over effect.
We did not include any cross‐over studies in this current update, hence there were no unit of analysis issues.
Dealing with missing data
We performed intention‐to‐treat analysis, and assumed treatment withdrawal to be due to either lack of efficacy or intolerable adverse effects. We had planned to calculate any missing statistics from the raw data where possible.
Assessment of heterogeneity
We had planned to assess methodological heterogeneity by comparing each trial for aspects outlined in the trial design section of Data extraction and management. We had planned to assess clinical heterogeneity by comparing each trial for aspects outlined in the demographic information section of Data extraction and management. We had planned to assess statistical heterogeneity using the I² statistic, and the following parameters as a guideline:
-
0% to 40%: might not be important;
-
30% to 60%: may represent moderate heterogeneity;
-
50% to 90%: may represent substantial heterogeneity;
-
75% to 100%: represents considerable heterogeneity.
Assessment of reporting biases
We reported bias according to Chapter 10 of theCochrane Handbook for Systematic Reviews of Interventions (Sterne 2011). If we had identified sufficient RCTs, we had planned to make a funnel plot to help identify publication bias, and investigate any visual asymmetry by exploratory analysis. We attempted to obtain source data for any studies included in the analysis in order to assess any non‐reported outcomes.
Data synthesis
We had planned to analyse data in a meta‐analysis using a fixed‐effect model in Review Manager 5 (RevMan 2014), provided this was clinically appropriate, and provided that we found no substantial heterogeneity. If we had found evidence of substantial heterogeneity, we would have explored the factors for heterogeneity. If substantial heterogeneity could not be readily explained, we would have used a random‐effects model to perform meta‐analysis. We used an intention‐to‐treat approach for the primary analysis, in which all participants were included in the treatment groups to which they were allocated, regardless of whether or not they received the treatment. A P value of < 0.05 qualified statistical significance.
Subgroup analysis and investigation of heterogeneity
We had planned to assess the effects of sulthiame in participants with focal epilepsy and participants with generalised epilepsy separately.
Sensitivity analysis
We had planned to assess the influence on results of studies of poor methodological quality by undertaking analyses with and without these studies.
Summarising and interpreting results
We used the GRADE approach, as outlined in the GRADE Handbook (Schünemann 2013), to interpret findings, and GRADEpro GDT software (which imports data from Review Manager 5 software (GRADEpro 2015)), to create a 'Summary of findings' table for the following outcomes: 50% or greater reduction in seizure frequency, complete cessation of seizures during follow‐up, mean seizure frequency, time‐to‐treatment withdrawal, total number of participants experiencing one or more adverse effects, total number of participants experiencing somnolence, and quality of life.
Results
Description of studies
Results of the search
Figure 1 summarises the results of the searches and the process of screening and selecting studies for inclusion in the review. Our search identified a total of 107 papers and trials register records. We removed 61 duplicate records, leaving 46 records to be screened. After reviewing the titles and abstracts, we excluded 23 records as it was clear that they were not randomised controlled add‐on studies comparing sulthiame (sultiame) with placebo or active control in epilepsy. Further evaluation at the full‐text stage of the remaining items is presented below and in the tables Characteristics of included studies and Characteristics of excluded studies.

Study flow diagram
Included studies
One study met our inclusion criteria (Debus 2004). It was a randomised, double‐blind, placebo‐controlled, parallel group study. Infants with newly diagnosed West syndrome, which had to include the features of infantile spasms, and either hypsarrhythmia or hemihypsarrhythmia, were recruited into the study. Infants with a history of epilepsy before the diagnosis of West syndrome were excluded, unless treated with phenytoin or phenobarbitone, both of which are reported to be ineffective in West syndrome (Hrachovy 1991). Thirty‐seven infants were included in the study; 20 received sulthiame and 17 received placebo.
The text publication reported that the infants had a mean age of 7.7 months (range: 3.5 to 15 months). The author, however, provided previously unpublished data that conflicted with this. During correspondence, the author informed us that the intervention group had consisted of 11 boys and nine girls, and that their mean (range) age had been 7.5 months (two to 15 months). According to the correspondence, the control group consisted of seven boys and 10 girls and their mean age (range) was 6.1 months (three to 13 months). Notably, the minimum age of infants for both groups, provided during correspondence, was lower than the minimum age stated in the text publication (two months compared 3.5 months, respectively). We were unable to verify which mean age and age range was correct.
The aetiological make up of the groups was: idiopathic cause – four (20%) in the intervention group, three (18%) in the control group; premature birth – five (25%) in the intervention group, three (18%) in the control group; tuberous sclerosis – three (15%) in the intervention group, four (24%) in the control group; malformations – two (10%) in the intervention group, two (12%) in the control group; birth asphyxia – two (10%) in the intervention group, one (6%) in the control group; trisomy 21 – one (5%) in the intervention group, one (6%) in the control group; congenital heart defect – one (5%) in the intervention group, one (6%) in the control group; porencephaly – none in the intervention group, one (6%) in the control group; encephalopathy – none in the intervention group, one (6%) in the control group; unclear – two (10%) in the intervention group, none in the control group.
After three days of baseline pyridoxine (150 to 300 mg/kg/day), the infants were randomised to receive sulthiame 5 mg/kg/day or placebo. After a further three days, non‐responders had the dose of sulthiame or placebo doubled. At the end of day nine, the study medication was disclosed following an electroencephalograph, which had to include a period of sleep. Baseline seizure frequency was not reported. The author of the study confirmed that this was not measured because the positive endpoint of the study was the absence of seizures, regardless of seizure frequency before the start of the medication. The study does state that there was no significant difference in baseline seizure frequency between the intervention and control groups. As it was not possible to perform a meta‐analysis, we presented our findings as a narrative analysis.
This trial was funded by DESITIN Pharma, in Germany.
Excluded studies
We excluded a further 19 records at the full‐text screening phase. Amongst them were seven RCTs: six studied sulthiame as a monotherapy in epilepsy (Li 2000; Rating 2000; Bast 2003; Basnec 2005; ISRCTN66730162; Borggraefe 2013), and one compared sulthiame with placebo as a monotherapy in healthy participants with no history of epilepsy, measuring axonal excitability of cortical neurons as a primary outcome (Siniatchkin 2006). We excluded three studies as they were not RCTs (Ingram 1963; Griffiths 1964; Livingston 1967), and another study was excluded as it included participants with and without epilepsy (Moffat 1970). There were multiple references that were linked to single studies, which accounts for the larger number of records compared to the number of excluded studies. Please refer to Characteristics of excluded studies for further details
Risk of bias in included studies
The rating for each domain and the respective reasoning can be found in the 'Risk of bias' table within the 'Characteristics of included studies' table, and is summarised in Figure 2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study
Allocation
The Debus 2004 study stated that the infants were randomised to either the intervention or control group, but did not report the method of randomisation. When contacted, the author of the study provided information about the previously unpublished method of randomisation for the purpose of this Cochrane Review. Infants were allocated a number between one and six after a pharmacist had rolled a die. Odd numbers were assigned to sulthiame, even numbers were assigned to placebo. We judged this to be an adequate method for random sequence generation and awarded low risk of bias.
The study author also provided previously unpublished details regarding allocation concealment. Sealed envelopes with participant identification numbers written on the outside, and a letter containing the allocation hidden on the inside, were sent by the pharmacy with correspondingly numbered medication boxes. As a result, allocation was effectively concealed, and we judged the study to be at low risk of bias for allocation concealment.
Blinding
The study report stated that the treatment was started in a double‐blind fashion, but did not provide details of the method or effectiveness of the blinding process beyond the description given for allocation concealment. Without the use of identical tablets for placebo and sulthiame, it is possible that the infants' parents or study personnel could have become aware of, or guessed the infant's group allocation. Therefore, it was unclear how blinding was maintained for the duration of the treatment period. Consequently, we awarded unclear risk of bias for performance and detection bias.
Incomplete outcome data
Four infants did not complete the study period. An intention‐to‐treat analysis, including these infants, was reported, therefore we awarded low risk of bias for attrition bias.
Selective reporting
The study report stated that four infants withdrew from the study (three from the intervention group and one from the control group). The time‐to‐treatment withdrawal for one infant in the intervention group was reported as being day six, but no timing was given for the remaining three infants. The study author provided previously unpublished data for two of these.
Adverse effects were also reported incompletely in this study. The total number of infants experiencing adverse effects in each group was reported (nine in the intervention group, nine in the control group), in addition to the number of infants experiencing somnolence (four in the treatment group, one in the control group). The remaining adverse effects (vomiting, restlessness, loss of appetite, diarrhoea, and abdominal pain) were reported for their occurrence in the study population as a whole, but the specific number of infants in each group who experienced each of these adverse effects was not reported. The author informed us that these data were not recorded. For this reason, we judged that the study was at high risk of reporting bias.
Other potential sources of bias
Other bias arose from the lack of baseline demographic data. Consequently, we were unable to determine whether there were any baseline imbalances between the two treatment groups. In addition, the author of the study gave us a contradictory statement regarding the baseline period, stating that seizure frequency was not measured at baseline. However, the study publication reported that there was no significant difference in baseline seizure frequency between treatment groups. As a result, we awarded the study unclear risk of other bias.
Effects of interventions
The results are summarised in summary of findings Table for the main comparison.
A 50% or greater reduction in seizure frequency
No data were reported for the primary outcome.
Complete cessation of seizures during follow‐up
The study reported that six infants in the intervention group and none of the infants in the control group showed a complete response, meaning cessation of infantile spasms and the disappearance of hypsarrhythmia on EEG testing. The aetiology of the responding infants was: idiopathic aetiology (n = 3), premature birth (n = 1), trisomy 21 (n = 1), and congenital heart defect (n = 1). Overall, the risk ratio (RR) for sulthiame compared with placebo was 11.14 (95% confidence interval (CI) 0.67 to 184.47; Analysis 1.1). Although the effect was statistically insignificant (P = 0.09), the RR was notably very large.
Mean seizure frequency
No data were reported for this outcome.
Time‐to‐treatment withdrawal
The study reported that one infant was withdrawn from the intervention group due to withdrawal of parental consent, precipitated by excessive somnolence. The publication did not state when this happened, however, the study author provided previously unpublished data and specified that this withdrawal occurred on day six. The study also reported that one infant was withdrawn from the control group because they had erroneously and openly received sulthiame during the study period, but did not report the time of treatment withdrawal. When asked, the study author stated that it was not clear when this occurred. It was also not clear whose decision it was to withdraw this infant, the parent's or the investigator's.
The study publication reported that a further two infants were withdrawn from treatment, but did not report the reasons for this, or the time of withdrawal. The study author provided previously unpublished data which informed us that the infants had both been allocated to the intervention group, and were withdrawn from treatment on days five and six, both due to withdrawal of parental consent. Overall, more infants were withdrawn from the treatment with add‐on sulthiame group compared to the add‐on placebo group.
Adverse effects
The study reported that a total of nine infants in the sulthiame intervention group (45%), and nine infants in the placebo control group (53%), experienced one or more adverse effects (RR 0.85, 95% CI 0.44 to 1.64; P = 0.63; Analysis 1.2). The study authors reported somnolence in four (20%) of the infants in the intervention group, and one (6%) of the infants in the control group (RR 3.40, 95% CI 0.42 to 27.59; P = 0.25; Analysis 1.3). The study authors reported the remaining adverse effects, stating that they were distributed equally amongst each group, without providing specific figures. Across the study population there was vomiting in 14 infants (38%), restlessness in six infants (16%), loss of appetite in two infants (5.5%), diarrhoea in one infant (3%), and abdominal pain in one infant (3%).
Quality of life
No data were reported for this outcome.
Discussion
Summary of main results
We included one trial in this review (Debus 2004). This trial assessed the effect of sulthiame as an add‐on therapy for West syndrome, after a baseline treatment with pyridoxine for three days. The inclusion criteria were infants with a new diagnosis of West syndrome, exhibiting infantile spasms, and hypsarrhythmia or hemihypsarrhythmia. The exclusion criteria were a prior diagnosis of epilepsy, unless treated with phenytoin or phenobarbitone, however, it was not clear how many infants entered the trial having been treated with either of these antiepileptic drugs, or whether the dose was reduced or discontinued prior to the study.
The study recruited 37 infants, 20 in the intervention group and 17 in the control group. Six infants from the treatment group and none from the control group became seizure‐free during the study. Although this outcome did suggest that add‐on sulthiame is more effective for seizure control, the outcome did not reach statistical significance. Adverse effects were incompletely reported. Overall, there was no significant difference between the total number of adverse effects reported in each group, or the number of infants experiencing somnolence in each group. Time‐to‐treatment withdrawal was incompletely reported, and we could not conduct any meaningful analysis on the data reported. No data were reported for the outcomes: 50% or greater reduction in seizure frequency, mean seizure frequency, or quality of life.
Notably, although no data were reported for the primary outcome, 50% or greater reduction in seizure frequency, as specified in the review protocol, we did not have any concerns about the lack of reporting. This outcome is not considered clinically meaningful in West syndrome, and is subsequently not routinely reported in these studies. Two of the five secondary outcomes , treatment withdrawal and adverse effects, however, were incompletely reported and provided more reason for concern. Specifically, the author was unable to confirm the time to withdrawal for the placebo‐randomised infant. Additionally, the author could not provide any further data regarding the incidence rate of adverse effects, stating that the data had not been recorded. We therefore judged that, overall, the risk of reporting bias was high in this study.
Further to this, the risk of bias for randomisation and allocation concealment was unclear from the published details. The study author, however, subsequently provided us with methodological details that demonstrated adequate methods. We consequently changed our judgement to low risk of selection bias. We did not receive any more details regarding the methods of blinding, therefore, we continued to award unclear risk of bias. Similarly, we judged that the study was at unclear risk of other bias due to the limited baseline data. We did, however, rate that there was low attrition bias due to the intention‐to‐treat population used.
The results of the study suggest that sulthiame, compared to placebo, may increase the incidence of complete cessation of seizures for infants with West syndrome when used as an add‐on therapy to pyridoxine, however, we are very uncertain about the reliability of this finding. Due to the small sample size, short treatment duration, and the significant risk of bias described above, we have serious concerns about the certainty of the evidence presented. Notably, none of the reported outcomes reached statistical significance. No conclusions can be drawn regarding the occurrence of individual adverse effects or quality of life, as these two measures were either incompletely reported, or not reported at all.
Overall completeness and applicability of evidence
We were unable to conduct a meta‐analysis as originally intended, due to the lack of eligible studies and the subsequent inclusion of a single study. This meant that we could not conduct any subgroup or sensitivity analyses either.
There were further limitations, specifically concerning the data provided by the single included study. First, the study did not report data for our primary outcome (50% or greater reduction in seizure frequency), or for two of the planned secondary outcomes (mean seizure frequency, and quality of life). As a result, we were unable to reach any conclusions for these outcomes.
The second issue regards the generalisability of the data for the outcomes that were reported. Most notably, the included study only investigated the use of sulthiame in infants with West syndrome. As a result, the findings of this review are limited to this specific type of epilepsy, and cannot be generalised to all epilepsy syndromes. It is also important to note that the infants included in the Debus 2004 study did not, by definition, have drug‐resistant epilepsy. In this study, sulthiame was used as an add‐on to pyridoxine, a form of Vitamin B6, not in addition to another anti‐epileptic drug. Hence, the review does provide some insight into the use of sulthiame as an add‐on therapy, but not in the context of drug‐resistant epilepsy. As a result, the findings of the review might have limited applications in clinical practice.
All of the participants in the included study were younger than 15 months. We have no data concerning the effect of sulthiame in older children or adults. Therefore, the review findings restricted specifically to infants with West syndrome.
Moreover, the study comprised a very small sample size population (37 infants) and is thus, likely to be statistically underpowered. Consequently, it is not appropriate to assume that the effect estimated in this review will necessarily be generalisable to all infants with West syndrome. Therefore, it is evident that the evidence for this review is severely lacking in completeness.
Quality of the evidence
We judged the single study included in this review to be at high risk of bias. This mainly resulted from the incomplete reporting of several of the intended outcomes. The study publication lacked methodological details, which the study author confirmed during correspondence. However, the study author failed to provide details on the method by which blinding was achieved and maintained. Accordingly, we downgraded the certainty of evidence for all outcomes once for risk of bias, and twice more due to the insufficient number of events reported. This was largely as a consequence of the small sample size. As a result, we GRADE‐assessed the evidence for all three reported outcomes as being of very low certainty.
Potential biases in the review process
For the purposes of this review, we defined drug‐resistant epilepsy as epilepsy in which seizure control had not been adequately managed by one or more antiepileptic drugs. The definition of drug‐resistant epilepsy used here is not consistent with that endorsed by the International League Against Epilepsy (ILAE), which specifies that drug‐resistant epilepsy should refer to the failure of adequate trials of two tolerated and appropriately chosen and used antiepileptic drug schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom (Kwan 2010).
Although currently, none of the studies screened satisfied either definition of drug‐resistant epilepsy, the definition used for the purposes of this review should be reviewed before conducting future updates, to ensure that it remains consistent with current recommendations. Importantly, the definition of drug‐resistant epilepsy used here has direct implications for which studies are eligible for inclusion, and consequently, could alter the conclusions reached.
Agreements and disagreements with other studies or reviews
The conclusions of the present review update remain consistent with the conclusions of the previously published versions of this review, largely because we found no additional eligible studies (Milburn‐McNulty 2011; Milburn‐McNulty 2013; Milburn‐McNulty 2015). Independent of this review and the included study, few reports exist regarding the use of sulthiame as an add‐on therapy. We were able to identify two retrospective studies, which despite being ineligible for this review, remain informative about sulthiame as an add‐on therapy for epilepsy (Swiderska 2011; Caraballo 2018).
The most recent of the two studies investigated the use of add‐on sulthiame in participants, aged 4 to 16 years (mean age 9 years) with drug‐resistant Lennox‐Gastaut syndrome, a paediatric epilepsy characterised by multiple seizure types (Caraballo 2018). Eligible participants had failed treatment with four or more previous anti‐epileptic drugs, prior to commencing treatment with sulthiame. During the study, the medical records of 44 participants were reviewed and retrospectively analysed. Participants had received sulthiame 5 to 30 mg/kg/day in addition to their current anti‐epileptic treatment (median two concomitant anti‐epileptic drugs), over a treatment period of 12 to 60 months (mean treatment period 20 months). Twenty‐seven of the 44 participants (61%) involved in the study attained a 50% or greater reduction in seizure frequency, one of whom was rendered seizure‐free. Adverse effects were reported by ten of the 44 participants and included: hyperpnoea (n = 4), dyspnoea (n = 4), nausea (n = 1), drowsiness (n = 1), headache (n = 4), decreased appetite (n = 2), allergic skin rash (n = 2), and irritability (n = 2). The instances of hyperpnoea and dyspnoea responded to adjustments in the dosage of sulthiame. All reported adverse effects were mild and transient in nature, and in none of the reported cases did adverse effects lead to discontinuation of treatment.
The earlier study by Swiderska 2011 also reviewed the medical records of paediatric participants (aged 2 to 17 years; mean age 10.7 years) with drug‐resistant epilepsy (defined as people who failed to respond to treatment with two or more previous anti‐epileptic drugs). The medical records of 20 participants were analysed as part of the review. Participants received a median dose of sulthiame 8.2 mg/kg/day in addition to their anti‐epileptic drugs (range 1 to 3 anti‐epileptic drugs; mean 1.2 anti‐epileptic drugs) over a treatment period ranging form two to 37 months (mean treatment period 18 months). At three months, 14 of 20 participants (70%) reported a 50% or greater reduction in seizure frequency, however, at subsequent follow‐up points (11, 18, and 20 months), only four participants (20%) responded; three of whom were rendered seizure‐free. Seven participants reported one or more adverse effects, which included drowsiness (n = 2), cognitive slowing (n = 1), hypersalivation (n = 1), breathlessness (n = 1), tachypnoea (n = 1), and diarrhoea (n = 1). The adverse effects reported were largely mild in severity and responsive to reductions in dosage. In only two instances did participants withdraw from treatment as a result of adverse effects (one due to cognitive impairment, and one due to excessive drowsiness).
Both retrospective studies highlighted that sulthiame was effective when used as an add‐on treatment for epilepsy, similar to that implied by the findings of this current review. Notably, the studies evaluated the use of sulthiame over much longer treatment durations than the treatment period used by Debus 2004 (6 days). The efficacy noted after much longer treatment periods in these retrospective studies suggests that the therapeutic effect, reported with regards to seizure freedom, within the short treatment period used by Debus 2004, could potentially be maintained over much longer treatment durations. The good tolerability profile of sulthiame was also emphasised by both retrospective studies, with the majority of associated adverse effects being mild in severity. Correspondingly, in this review, and as reported by Debus 2004, an equal number of participants reported one or more adverse effects in both the sulthiame add‐on treatment group and in the placebo group, implying that sulthiame is well‐tolerated.
As a result, the conclusions of this review, which included data from one randomised controlled trial, appear to be fairly consistent with those of the retrospective studies, despite the significant differences in methodology (Caraballo 2018; Swiderska 2011).

Study flow diagram

Risk of bias summary: review authors' judgements about each risk of bias item for each included study

Comparison 1 Sulthiame add‐on versus placebo, Outcome 1 Complete cessation of seizures.
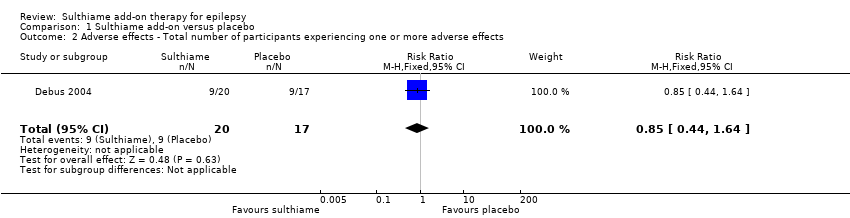
Comparison 1 Sulthiame add‐on versus placebo, Outcome 2 Adverse effects ‐ Total number of participants experiencing one or more adverse effects.

Comparison 1 Sulthiame add‐on versus placebo, Outcome 3 Adverse effects ‐ Somnolence.
| Sulthiame add‐on compared to placebo for epilepsy | ||||||
| Patient or population: patients between 3 to 15 months of age with West syndrome | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with placebo | Risk with Sulthiame add‐on | |||||
| 50% or greater reduction in seizure frequency | Outcome was not reported by Debus 2004 | |||||
| Complete cessation of seizures during follow‐up Follow‐up: 9 days | Study population | RR 11.14 | 37 | ⊕⊝⊝⊝ | ||
| 0 per 1,000 | 0 per 1000 | |||||
| Mean seizure frequency | Outcome was not reported by Debus 2004 | |||||
| Time‐to‐treatment withdrawal | Outcome was not reported by Debus 2004 | |||||
| Adverse effects: total number of participants experiencing one or more adverse effects Follow‐up: 9 days | Study population | RR 0.85 | 37 | ⊕⊝⊝⊝ | ||
| 529 per 1,000 | 450 per 1000 | |||||
| Adverse effects: somnolence Follow‐up: 9 days | Study population | RR 3.40 | 37 | ⊕⊝⊝⊝ | ||
| 59 per 1,000 | 200 per 1000 | |||||
| Quality of life | Outcome was not reported by Debus 2004 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded once for risk of bias: Debus 2004 did not describe how blinding was achieved or maintained, and we suspected reporting bias for the adverse effect outcomes. bDowngraded twice for imprecision: small study population, therefore, the number of events failed to suffice optimal information size. cEvidence was not upgraded for effect size: normally, the evidence would be upgraded twice when a RR is greater than 5.00, however, due to the concerns regarding risk of bias and small sample size, we were unable to upgrade the evidence. dEvidence was not upgraded for effect size: normally, the evidence would be upgraded once when a RR is greater than 2.00, however, due to the concerns regarding risk of bias and small sample size, we were unable to upgrade the evidence. | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Complete cessation of seizures Show forest plot | 1 | 37 | Risk Ratio (M‐H, Fixed, 95% CI) | 11.14 [0.67, 184.47] |
| 2 Adverse effects ‐ Total number of participants experiencing one or more adverse effects Show forest plot | 1 | 37 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.44, 1.64] |
| 3 Adverse effects ‐ Somnolence Show forest plot | 1 | 37 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.4 [0.42, 27.59] |