Intervenciones para la prevención de la hemorragia digestiva alta en pacientes ingresados a la unidad de cuidados intensivos
Referencias
Referencias de los estudios incluidos en esta revisión
Referencias de los estudios excluidos de esta revisión
Referencias de los estudios en espera de evaluación
Referencias de los estudios en curso
Referencias adicionales
Referencias de otras versiones publicadas de esta revisión
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 10 participants Number analysed: 10 participants Pantoprazole
Placebo
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalances: ‐ | |
| Interventions | Pantoprazole
Placebo
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: ICU Source of funding: ‐ Conflicts of interest: ‐
Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: mentioned only that participants were randomised to treatment. Not enough information on method of randomisation |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information about allocation concealment reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: mentioned only that the trial was double‐blind. No information about the method of blinding |
| Blinding (detection bias) | Low risk | Comment: mentioned that outcome assessment was done blinded to intervention. Presence of 1 or more macroscopic abnormalities (erythema or oedema, erosions, ulcerations, and nasogastric tube lesions) and GI bleeding was assessed, blinded to intervention, at endoscopy by a single experienced gastroenterologist Quote: "assessed, blinded to intervention, at endoscopy by a single experienced gastroenterologist" |
| Blinding (detection bias) | Unclear risk | Comment: The outcome was not addressed in this trial |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: mentioned that outcome assessment was done blinded to intervention |
| Incomplete outcome data (attrition bias) | High risk | Comment: Conference abstract reports about 10 participants from a larger prospective trial. Those who received > 5 doses of pantoprazole or placebo (n = 84) were eligible for the endoscopy substudy, but unclear why data from only 10 participants are reported |
| Selective reporting (reporting bias) | Low risk | Comment: Outcomes reported in Methods section are also reported in Results section |
| Other bias | Unclear risk | Comment: not enough information reported in conference abstract to assess other biases |
| Methods | Open‐label parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 34 participants Number analysed: 34 participants Ranitidine
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: Participants were tracheotomised patients with tetanus. Maximum tetanus severity score was 11 (4 to 16) in the ranitidine group and 10 (6 to 16) in the control group. Groups were similar with respect to age and gender distribution | |
| Interventions | Ranitidine
No prophylaxis
Adherence to regimen: 34 tracheotomised participants who were admitted to medical ICU with tetanus were randomly assigned to ranitidine or control group within 24 hours of tracheal intubation. Six participants who had pneumonia before tracheostomy or had ranitidine before randomisation were excluded. All remaining participants were studied until 48 hours after extubation Duration of trial: ‐ Duration of follow‐up: studied daily until 48 hours after tracheal extubation | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Medical ICU in Department of Medicine, Department of Microbiology, King Edward Memorial Hospital, Parel, Bombay, India Source of funding: Quote: "Study supported, in part, by a grant from Seth GS medical College and KEM Hospital research Society”; Torrent Pharmaceuticals provided ranitidine Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional ethics committee" Informed consent: Quote: "The study was approved by the institutional ethics committee [with] waived informed consent" Comment: not obtained, as mentioned in the trial report Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Trial included patients with tetanus. Trial reports that gram‐negative bacilli were the predominant organisms that caused pneumonia. Participants treated with ranitidine developed pneumonia significantly earlier (median 3 days, range 1 to 5) than participants given control (median 5 days, range 3 to 14 days) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not enough information on method of randomisation reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information on method of allocation concealment reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This trial compared an intervention vs no prophylaxis (blinding was not possible) |
| Blinding (detection bias) | Low risk | Comment: GI bleeding was an objective outcome that was detected as per the definition in the trial protocol |
| Blinding (detection bias) | Low risk | Comment: Pneumonia was an objective outcome that was detected as per the definition in the trial protocol |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All participants who were randomised were included in analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: This trial was supported in part by a grant from Seth GS Medical College and KEM Hospital Research Society. The role of the sponsor in the conduct and reporting of the trial is unclear. No other form of bias is suspected |
| Methods | Double‐blind parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 66 participants Number analysed: 55 participants Pirenzepine
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: baseline imbalances comparable, also in terms of severity of injury/trauma | |
| Interventions | Pirenzepine
Cimetidine
Adherence to regimen: 11 participants withdrew from the trial. However, no reasons are mentioned in the trial report. Also no mention of which interventional group these 11 participants belonged to Duration of trial: ‐ Duration of follow‐up: not clearly mentioned, probably until discharge | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Surgical ICU, Chur, Switzerland Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not enough information reported on method of sequence generation |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information reported on method of allocation concealment |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: This appears to be a double‐dummy placebo‐controlled trial, in which participants from group 1 got a placebo that looks like the intervention in group 2 and vice versa. Thus personnel would have been blinded and likelihood of performance bias is low |
| Blinding (detection bias) | Unclear risk | Comment: not enough information reported on the criteria for diagnosis of upper GI bleeding |
| Blinding (detection bias) | Unclear risk | Comment: The trial did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: This appears to be a double‐dummy placebo‐controlled trial, in which participants from group 1 got a placebo that looks like the intervention in group 2 and vice versa. Thus the likelihood of detection bias seems low |
| Incomplete outcome data (attrition bias) | High risk | Comment: 11 participants withdrew from the trial, but reasons for withdrawal and the group to which they were randomised are not clearly mentioned in the trial report |
| Selective reporting (reporting bias) | Unclear risk | Comment: All intended outcomes were reported but no clear mention of the number of participants in the cimetidine group who had confusion and high K levels |
| Other bias | Low risk | Comment: The trial report is unclear on the source of funding. No other sources of bias detected |
| Methods | Double‐blind parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 146 participants Number analysed: 120 participants Ranitidine
Pantoprazole
Inclusion criteria
Exclusion criteria
Baseline imbalances: We found no statistically significant differences between the 2 groups regarding baseline characteristics, such as age, sex, or APACHE II | |
| Interventions | Ranitidine
Pantoprazole
Adherence to regimen: ‐ Duration of trial: July 2011 to July 2012 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial
Outcomes reported, but not used in the review
| |
| Notes | Setting: ICU, Iran Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Trial was approved by the Ethics Committee of Hamedan University of Medical Sciences. Informed consent: Written informed consent was obtained from legal guardians of participants Clinical trials registration: ‐ Sample size calculation: ‐ Conflicts of interest: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The patients were randomised using online random allocation software (www.allocationsoftware.com)" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "The patients and the attending intensivists responsible for data collection were blinded to the assigned groups" |
| Blinding (detection bias) | Unclear risk | Comment: Trial did not address this outcome |
| Blinding (detection bias) | Low risk | Quote: "Patients underwent chest radiography which was repeated at least twice a week" Comment: Objective outcome measurement unlikely to introduce bias |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "The patients and intensivists responsible for data collection were blinded to the assigned groups" |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All participants were followed up until discharge |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes listed in the Methods section were reported in the Results section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 168 participants Number analysed: 168 participants Cimetidine
Antacids (Maalox)
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: Risk categories and risk factors in the 3 groups were comparable | |
| Interventions | Cimetidine
Antacids (Maalox)
No prophylaxis
Adherence to regimen: 16 participants died, 6 did not comply with therapy, and 9 were transferred to other institutions. Therefore, 31 participants did not complete 10 days of the trial Duration of trial: March 1978 to April 1979 Duration of follow‐up: not clearly mentioned in trial report | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: ICU, University of Rome Source of funding: ‐ Ethics approval: ‐ Informed consent: Quote: "Informed consent was obtained from either the participant or their closest relative" Clinical trials registration: not provided Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “The study was done in a single blind manner, assigning the treatment according to a list of randomised values” Comment: not enough information reported on method of sequence generation |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information reported on method of allocation concealment |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial and personnel were not blinded |
| Blinding (detection bias) | Low risk | Quote: "The observer assessing the occurrence of gastrointestinal bleeding did not know the type of prophylactic measures the patient was receiving" |
| Blinding (detection bias) | Unclear risk | Comment: The trial did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: Unclear whether outcome assessors were blinded for other outcomes |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Unclear risk | Comment: All‐cause mortality in ICU and units of blood transfused were not mentioned separately for each interventional arm. Unclear whether this contributed to reporting bias |
| Other bias | Unclear risk | Comment: source of funding and baseline characteristics unclear |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 43 participants Number analysed: 43 participants Pirenzepine
Famotidine
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: There were no differences between groups that did not receive any prophylaxis (not randomised) and the group that did receive prophylaxis (randomised to pirenzepine and famotidine) | |
| Interventions | Pirenzepine
Famotidine
No prophylaxis
Adherence to regimen: ‐ Duration of trial: October 1988 to November 1991 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in report but not used in review
| |
| Notes | Setting: University Children's Hospital, Department of Pediatric Gastroenterology, Erlangen, Germany Source of funding: ‐ Conficts of interest: ‐ Ethics approval: Quote: "The study was approved by the committee on human research of the University of Erlangen‐Nurnberg" Informed consent: ‐ Clinical trial registration: ‐ Sample size calculation: ‐ Additional notes: Gastric cultures were positive in 95% of participants with mean gastric pH > 4 and in 80% of participants with mean gastric pH < 4. Five and 4 participants in the 2 groups required mechanical ventilation. Candida sp was the predominant species cultured from gastric and tracheal secretions of participants given pirenzepine and famotidine. In the pirenzepine and famotidine groups, an organism cultured from the stomach was grown from the tracheal secretion, 1 to 4 days later, in 6 and 5 participants, respectively | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not enough information reported on method of sequence generation |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information reported on method of allocation concealment |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: unclear on blinding of personnel, although regimens and mode of administration of interventions were similar |
| Blinding (detection bias) | Unclear risk | Comment: The physician who performed endoscopy to detect inflammation or ulceration of the upper GI tract was not blinded to the intervention. There is no clear definition of GI bleeding in the trial. Therefore, unclear on the likelihood of performance or detection bias |
| Blinding (detection bias) | Unclear risk | Comment: no mention of blinding technicians who cultured gastric secretions for pathogenic bacteria, radiologists who interpreted chest X‐rays, or physicians who performed the clinical examination. No clear definition of VAP provided in the trial |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: not enough information reported on blinding of outcome assessors for other outcomes |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were included in the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: Trial compared participants who received prophylaxis vs participants who did not receive prophylaxis. In the intervention group, participants were randomised to receive either pirenzepine or famotidine. The outcome of interest (VAP) was reported separately for participants who were randomised to 2 different arms (pirenzepine and famotidine) |
| Other bias | Unclear risk | Comment: source of funding not clearly mentioned in the trial report. Baseline data on randomised groups unclear |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 304 participants Number analysed: 300 participants Sucralfate
Cimetidine
Control
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "One hundred patients were randomly assigned to each of the treatments. The groups were similar with regard to age, gender, percentage of participants admitted from the emergency room, severity of illness, admission diagnoses, corticosteroid usage and coagulopathy. Patients often had more than one reason for ICU admission. The mean APACHE II scores for the control, sucralfate, and cimetidine groups were 16.5 ± 6.9, 16.8 ± 6.9, and 18.0 ± 8.0, respectively. Approximately one‐third of the patients in each group had APACHE II scores greater than 20.0 Comment: The 3 groups were similar with respect to demographic and other risk factors for stress haemorrhage at the beginning of the trial. The most common diagnosis on admission was pneumonia, which was reported in 89 participants. Bacterial pneumonia (control: 23; sucralfate: 26; cimetidine: 21). Non‐bacterial pneumonia was diagnosed (control: 9; sucralfate: 4; cimetidine: 6). Coagulopathy was present in 16, 14, and 21 participants in the 3 randomised groups | |
| Interventions | Sucralfate
Cimetidine
No prophylaxis
Adherence to regimen: Although 304 participants were randomised, only 300 (100 in each group) were part of the trial because 1 participant died 2 hours after admission, 3 participants were randomised on 2 different occasions, and second admissions were excluded. Results for only 291 participants were available when the trial was finally reviewed by the second committee. The 8 participants who did not agree to endoscopy might have been excluded from the trial denominator (unclear on the 1 remaining participant) Duration of trial: 1 February to 25 November 1992 Duration of follow up: until death or discharge from ICU | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Note: Median time from ICU admission to onset of stress‐related haemorrhage was 5 days
Note: Noscocomial pneumonia occurred after a mean of 6.9 ± 7.2 days in the ICU (median 9 days) Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Henry Ford Hospital and Health Sciences Center, Detroit, Michigan Source of funding: Quote: "Grant support: in part from the Henry Ford Hospital Research and Education Funds to Dr. Ben‐Menachem and to Dr. Fogel" Conflicts of interest: ‐ Ethics approval: Quote: "This protocol was reviewed and approved by the Henry Ford Hospital Institutional Review Board" Informed consent: Quote: "Informed consent was obtained from the patient or from legally authorized representatives when the patient could not provide consent" Clinical trials registration: ‐ Sample size calculation: Quote: "We estimated sample size to provide 80% power to detect a 75% reduction in bleeding rate, that is, a 12% bleeding rate for the control group compared with a 3% rate in either of the two treatment groups. We used an alpha value of 0.05 (two‐tailed) adjusted for the comparison of the control group with each of the prophylaxis groups. As a result of these assumptions, 160 patients were needed in each of the three groups” Additional notes: Mechanical ventilation was used in 65, 72, and 76 participants in the 3 respective groups. Respiratory failure and high dose of corticosteroid were independently associated with increased risk of stress‐related haemorrhage. According to the trial report, 43 participants satisfied the criteria for significant GI bleeding (as per the definition), and 19 of these participants did not have stress‐related bleeding (as determined by oesophagogastroduodenoscopy). Among them, 12 participants (4 controls, 3 receiving sucralfate, and 5 receiving cimetidine) had a normal result of endoscopy, suggesting that the change in haematocrit was due to fluid shifts. Seven participants bled from causes not due to stress ulceration, and 8 participants did not have endoscopy (5 did not give consent, 1 participant with lymphoma and thrombocytopaenia died of multiple‐organ system failure 10 days after the bleeding episode. Three of the 8 participants met criterion 3 of diagnosis Quote: "Two of 20 patients in the control group with coagulopathy had stress‐related haemorrhage. In the cimetidine and sucralfate groups, the incidences were 1 of 22 and 2 of 17, respectively (P > 0.05)" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was by sealed envelope using the permuted block design" |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization was by sealed envelope using the permuted block design" Comment: Method for allocation concealment is clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was a single‐blind trial, and the mode of drug administration could not have allowed blinding. Therefore, risk of performance bias is high |
| Blinding (detection bias) | Low risk | Quote: "The primary study end point was substantial haemorrhage from stress gastritis. Information regarding hematocrit, haemoccult status of stool and nasogastric aspirate, and volume status [were] presented daily to two investigators who were blinded to therapy" Comment: Trial report mentions blinding of outcome assessors for the primary outcome of GI bleed, which was an objective outcome detected as per the definition used in the trial |
| Blinding (detection bias) | Low risk | Comment: Outcome assessors were not blinded. However, this was an objective outcome that was diagnosed as per the definition used in the trial protocol. Therefore, the likelihood of performance or detection bias is low |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Although 304 participants were randomised, only 300 were analysed, as 1 participant died 2 hours after admission, 3 were randomised on 2 different occasions, and second admissions were excluded. Results for only 291 participants were available when the trial was finally reviewed by the second committee. The 8 participants who did not agree to endoscopy might have been excluded from the trial denominator (unclear for the 1 remaining participant). However, intention‐to‐treat analysis was done with 100 participants in each of the 3 groups. Therefore, there was no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes are reported. A subgroup analysis was desirable for participants who received enteral feeds |
| Other bias | Low risk | Comment: This trial was supported in part by Henry Ford Hospital Research and Education Funds. The role of the sponsor in the conduct and reporting of this trial is unclear. No other form of bias is suspected |
| Methods | Stratified double‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 141 participants Number analysed: 141 participants Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Demographic characteristics of both groups, such as age, gender, and severity of illness, which was assessed by the APCHE score, were almost similar. Most participants were given a diagnosis of cardiovascular problems (30 in both groups). Neoplastic disease was more common among participants assigned to the antacid group when compared with the sucralfate group (12 and 5), whereas renal insufficiency and immunodeficiency were more common among participants in the sucralfate group (1 and 6 and 3 and 7, respectively). All other underlying conditions including presence of pneumonia (9 in antacid group and 10 in sucralfate group) on admission were similar between both groups | |
| Interventions | Antacids
Sucralfate
Adherence to regimen: Gastric pH was determined within 24 hours after admission via continuous intragastric monitoring. Participants were stratified into 2 groups according to gastric pH value measured on the first day of admission, which was < 3 (n = 69) or ≥ 3 (n = 72) (continuous intragastric pH in 84 and indicator papers in 57 participants; for 28 in the latter group, continuous pH monitoring was performed later). Participants were later randomised to 2 treatment arms, amongst whom 74 received antacids and 67 received sucralfate Duration of trial: August 1992 to August 1993 Duration of follow‐up: probably until discharge or death | |
| Outcomes | Outcomes sought in review and reported in trial
Note: The mean number of days before the first episode of VAP was 9.2 in the antacid group and 9.3 in the sucralfate group, respectively.
Outcomes sought but not reported in trial
Outcomes reported in report but not used in review
| |
| Notes | Setting: University Hospital Maastricht, Maastricht, The Netherlands Source of funding: Quote: "Supported by grant 28‐2125 from the Praevention Foundation" Conflicts of interest: ‐ Ethics approval: Quote: "The protocol was approved by the institutional review board of the hospital" Informed consent: Quote: "... informed consent was obtained from all participants or if this was not possible because of the clinical condition, from a representative of the family" Clinical trials registration: ‐ Sample size calculation: Quote: "The sample size of both groups of patients was calculated to detect a reduction in the incidence of VAP from an assumed 30% in the antacid group to an expected incidence of 10% in the sucralfate group (α = 0.05 and β = 0.2)" Additional notes: VAP was mainly polymicrobial in nature. Pseudomonas aeruginosa and Staphylococcus aureus were the 2 predominant pathogens cultured from participants with a diagnosis of VAP. The oropharynx was the initial site for colonisation ofEnterobacteriaceae, whereas Pseudomonas aeruginosa and Staphylococcus aureus colonised in the upper respiratory tract first | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were stratified into two groups according to, the gastric pH values measured on the first day of admission which were < 3 (n = 69) or ≥ 3 (n = 72). These patients were later randomised to two treatment arms" |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Sucralfate and antacids were compared in a double blind fashion and analysed according to a prospectively developed plan that used definition adopted before the treatment allocation code was broken" Comment: Allocation concealment might have been in place but not enough information was provided on method of allocation concealment |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "The study had a "double dummy model" design wherein participants receiving sucralfate received an antacid placebo at the same time when antacid was administered to the other group and vice versa" Quote: "Sucralfate and antacids were compared in a double blind fashion and analysed according to a prospectively developed plan that used definition adopted before the treatment allocation code was broken. The attending physician was unaware of the cultural results of the oropharyngeal and gastric samples, or results of pH monitoring" Comment: Personnel (and participants) were unaware of therapy and results |
| Blinding (detection bias) | Unclear risk | Quote: "The study had a "double dummy model" design wherein participants receiving sucralfate received an antacid placebo at the same time when antacid was administered to the other group and vice versa" Comment: The trial by design inherently blinded trial personnel who were assigned to endoscopically detect upper GI bleeding; however, criteria for diagnosis of upper GI bleeding were not clearly reported |
| Blinding (detection bias) | Low risk | Quote: "The study had a "double dummy model" design wherein participants receiving sucralfate received an antacid placebo at the same time when antacid was administered to the other group and vice versa" Comment: The trial by design inherently blinded trial personnel who were assigned to diagnose VAP. Criteria for diagnosis of VAP were clearly reported |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "The study had a "double dummy model" design wherein participants receiving sucralfate received an antacid placebo at the same time when antacid was administered to the other group and vice versa" Comment: The trial by design inherently blinded outcome assessors. Therefore, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: No dropouts were reported, and all randomised participants were part of the final analysis.Therefore, there might not be an attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported and were part of the analysis.. A subgroup analysis for participants who received enteral feeds was desirable |
| Other bias | Low risk | Comment: Trial was supported by grant 28‐2125 from the Praevention Foundation. The role of the sponsor in the conduct and reporting of the trial is unclear |
| Methods | Quasi‐randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 100 participants Number analysed: 100 participants Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There was no statistically significant difference between the sucralfate treated and the antacid treated groups in number of patients, sex" Comment: Average numbers of risk factors per group were also similar; 92.5 and 83 participants had undergone an operation just before trial entry | |
| Interventions | Antacids (Mylanta II)
Sucralfate
Adherence to regimen: Quote: "Of the 52 patients treated with antacids, failure to achieve a pH of 3.5 or greater occurred in 8 patients initially given 30 mL of antacid. Five subsequently required 60 mL/hour and three required 120 mL/hour. All patients receiving antacids maintained a gastric pH of mote than 5" Comment: Iced saline solution lavage was given to all participants with diagnosis of upper GI bleed (by Gastroccult test) Duration of trial: August 1983 to December 1983 Duration of follow up: probably until discharge or death | |
| Outcomes | Outcomes sought in review and reported in trial
Note: antacids 9 and 10 hours after start of the drug and sucralfate; 8, 41, and 45 hours after initiation of prophylaxis Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Queens Hospital Centre, New York, USA Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: No deaths were due to GI bleeding | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: “100 patients admitted to medical and surgical intensive care units were randomised to receive either antacids and sucralfate depending on the year of birth (odd year, sucralfate; even year, antacid)” Comment: This was a quasi‐randomised trial |
| Allocation concealment (selection bias) | High risk | Comment: This was a quasi‐randomised trial, and no information on allocation concealment was reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: no information on blinding reported |
| Blinding (detection bias) | Low risk | Comment: Blinding was not done. However, the outcome was measured as per the definition used in the trial protocol. Moreover, owing to the objective nature of the outcome of interest, the likelihood of detection and performance bias is low |
| Blinding (detection bias) | Unclear risk | Comment: The trial did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no information on blinding or criteria to diagnose other outcomes reported |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were included in the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is unclear. No other source of bias detected |
| Methods | Quasi‐randomised trial | |
| Participants | Baseline characteristics Number randomised: 155 participants Number analysed: 155 participants Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: no statistically significant difference between sucralfate‐treated and antacid‐treated groups in numbers, age, and gender of participants. 130 participants (61 and 69 in each group) had undergone a major operation just before trial entry | |
| Interventions | Antacids (Mylanta or Maalox)
Sucralfate
Adherence to regimen: not clearly mentioned in the trial report Duration of follow‐up: Quote: “Patients were continued in the trial until the onset of gastrointestinal bleeding, until they were discharged from the critical care unit, or until nasogastric suction was discontinued. The patients’ clinical course was followed until they were discharged from the hospital” | |
| Outcomes | Outcomes sought in review and reported in trial
Note: GI bleeding was tested for antacids 9 and 10 hours after initiating prophylaxis, and for sucralfate 8, 41, and 43 hours after initiating prophylaxis Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Long Island Jewish Medical Centre and Queens Hospital Centre Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study protocol was approved by the institutional review boards at Long island Jewish Medical Centre and Queens Hospital Centre" Informed consent: Quote: "Informed consent was obtained from the patient or immediate relative" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: None of the deaths were due to GI bleeding | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Patients were randomly assigned to one of the two treatment regimens according to their date of birth"; "participants born on even days were given antacids and those born on odd days were administered sucralfate" Comment: This was a quasi‐randomised trial |
| Allocation concealment (selection bias) | High risk | Comment: This was a quasi‐randomised trial; no information on method of allocation concealment was reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial; the different interventions and their mode of administration might not have ensured blinding of personnel |
| Blinding (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However, the outcome was measured as per the definition used in the trial protocol. Moreover, owing to the objective nature of the outcome of interest, the likelihood of detection and performance bias was judged as low |
| Blinding (detection bias) | Unclear risk | Comment: The trial did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However, outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is unclear. No other source of bias is suspected |
| Methods | Quasi‐randomised trial | |
| Participants | Baseline characteristics Number randomised: 50 participants Number analysed: 50 participants Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There was no statistically significant difference between the sucralfate treated and the antacid treated groups in number of patients, sex, mean age [...] or number of risk factors per patient" Comment: All participants had undergone aortobifemoral artery Dacron graft placement | |
| Interventions | Antacids (Mylanta II)
Sucralfate
Adherence to regimen: Comment: Iced saline solution lavage was given to all participants with diagnosed upper GI bleeding Duration of trial: August 1983 to December 1984 Duration of follow‐up: not clearly mentioned in the trial report; probably until discharge or death | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in trial but not used in review
| |
| Notes | Settings: Long island Jewish Medical Centre, New Hyde Park, NY 11042 Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: None of the deaths were due to GI bleeding | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "…were randomised to receive either antacid or sucralfate, depending on their year of birth (odd year sucralfate, even year antacid) Comment: quasi‐randomised trial |
| Allocation concealment (selection bias) | High risk | Comment: This was a quasi‐randomised trial; no information on method of allocation concealment was reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial; the different interventions and their mode of administration might not have ensured blinding of personnel |
| Blinding (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However, GI bleeding was an objective outcome that was detected as per the definition used in the trial report |
| Blinding (detection bias) | Unclear risk | Comment: The trial did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: unclear on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were included in the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is unclear. No other source of bias is suspected |
| Methods | Open‐label parallel‐group trial | |
| Participants | Baseline characteristics Number randomised: 83 participants Number analysed: 74 participants Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 groups were almost similar with respect to age, gender, and time from entry into ICU to random selection. However, the sucralfate group had more risk factors for bleeding on admission to the study when compared with the antacid group (respiratory failure was the most common ‐ 34 and 37 participants, respectively). Three participants in the antacid group had coagulopathy vs 1 participant in the antacid group. Two participants in the antacid group were given a diagnosis of pneumonia on admission | |
| Interventions | Antacids (Maalox Therapeutic Concentration)
Sucralfate
Adherence to regimen: Six participants from the antacid group (n = 42) and 3 from the sucralfate group (n = 41) were excluded from analysis for the following reasons: < 12 hours in the study (3 died, 2 enteral feeds, 1 severe diarrhoea, with treatment discontinued), received both interventions, had evidence of GI bleed before intervention, refused participation Duration of trial: January 1984 to September 1985 | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: surgical, medical, or burn intensive care units at San Francisco General Hospital, USA Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The protocol was approved by the University of California, San Fransisco, Hum and Environmental protection committees (approval number 251701‐02)" Informed consent: "obtained from all participants. If the participant was unable to provide informed consent, then the participant's next of kin or legally authorized representative provided consent. If they could not be contacted, then the participant's attending physician was asked to provide permission" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: One participant in each group had significant upper GI bleeding after the trial was completed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no information on sequence generation reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information on allocation concealment reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: Participants and personnel involved in the trial were not blinded |
| Blinding (detection bias) | Low risk | Comment: GI bleeding was an objective outcome that was diagnosed as per the definition used in the trial protocol |
| Blinding (detection bias) | Unclear risk | Comment: Trial did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: unclear whether outcome assessors were blinded. Moreover, criteria for diagnosis of other outcomes of interest not clearly reported |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Not all randomised participants were included in the final analysis (83 were randomised, 74 were included in the analysis). A per‐protocol analysis was done, as 9 participants were involved in the trial for less than 12 hours. There was no imbalance between groups. Therefore, low risk of bias is due to attrition |
| Selective reporting (reporting bias) | Unclear risk | Comment: not enough information reported on outcomes of relevance in the trial |
| Other bias | High risk | Comment: Source of funding is not clearly mentioned in the trial report. No other sources of bias are suspected, but insufficient information is reported in the trial abstract |
| Methods | Quasi‐randomised trial | |
| Participants | Baseline characteristics Number randomised: 51 participants Number analysed: 51 participants Famotidine
Lansoprazole
Inclusion criteria
Exclusion criteria
Baseline imbalances: "There were significantly more males than females in the study. Over 75% of the patients had a Glasgow Coma Scale (GCS) < 9, and median GCS scores were similar between the two groups. All of the patients had at least two risk factors for SRMD, and each treatment group had a similar number of patients with traumatic brain injuries. The median baseline gastric pH was 3.0 for both famotidine and lansoprazole groups" Comment: There were more women in the lansoprazole group than in the famotidine group | |
| Interventions | Famotidine
Lansoprazole
Adherence to regimen: ‐ Duration of trial: August 1999 to April 2005 Duration of follow‐up: “Patients were followed until 24 hours after the discontinuation of SUP, the patient was discharged from the ICU, or if the patient expired, whichever came first" | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review • Duration of pH ≥ 4.0 • Percentage of time gastric residual was < 28 mL | |
| Notes | Setting: The Virginia Commonwealth University (VCU), 1000‐bed, academic, level 1 trauma centre Source of funding: Quote: "This study was funded by TAP Pharmaceuticals" Conflicts of interest: ‐ Ethics approval: Quote: "The Virginia Commonwealth University (VCU) Institutional Review Board approved this study prior to subject enrolment, and this study was conducted in compliance with the Declaration of Helsinki" Informed consent: Quote: "All subjects provided written informed consent prior to study commencement" Clinical trials registration: ‐ Sample size calculation: Quote: "...we assumed that on day 3 of therapy, 85% of the patients receiving lansoprazole would have pH values ≥ 4.0 for 80% of the time compared to only 40% of the patients receiving famotidine. Using these proportions, α = 0.05, β = 0.20, and a two‐way statistical test, approximately 22 patients were needed in each group to show statistical significance" Comment: This was after 30 people admitted to the neurosurgical unit were followed; it was assumed that approximately 40% of them receiving famotidine and 80% receiving lansoprazole maintained gastric pH ≥ 4 80% of the time | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Patients admitted into the unit during odd‐numbered months received famotidine 20 mg IV every 12 hours; patients admitted during even numbered months received lansoprazole" Comment: This was a quasi‐randomised trial in which sequence generation was not done |
| Allocation concealment (selection bias) | High risk | Quote: "Patients admitted into the unit during odd‐numbered months received famotidine 20 mg IV every 12 hours; patients admitted during even numbered months received lansoprazole" Comment: This was a quasi‐randomised trial in which allocation was not concealed |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial; the different interventions and their mode of administration could not have made it possible to blind trial personnel |
| Blinding (detection bias) | Low risk | Comment: This was an unblinded trial in which GI bleeding was detected as per the definition used in the trial protocol |
| Blinding (detection bias) | Unclear risk | Comment: Trial did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: This was an unblinded trial; outcomes of interest were diagnosed as described in the trial protocol |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis. Therefore, no attrition bias is suspected |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: TAP Pharmaceuticals funded the trial. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
| Methods | Double‐blind parallel‐group randomised placebo‐controlled trial | |
| Participants | Baseline characteristics Number randomised: 34 participants Number analysed: 34 participants Ranitidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "All 34 patients were comatose on admission and required ventilatory support"; "No significant differences in demographic characteristics were present between the two treatment groups" Comment: The 2 groups were comparable with respect to mean Glasgow Coma Scale score (mean 8, range 4 to 10; and mean 6.7 range 3 to 10), mean Injury Severity Score (mean 32, range 25 to 41; and mean 30, range 25 to 57), and time from injury to study drug administration | |
| Interventions | Ranitidine
Placebo
Adherence to regimen: Quote: "All 34 patients were comatose on admission and required ventilatory support. Ten patients were withdrawn before completing 72‐hour study period. Five of these patients were in the placebo treatment group and were withdrawn from the trial because of protocol‐defined upper gastrointestinal tract bleeding. Of the remaining four patients,who were from the ranitidine group; one was withdrawn due to death secondary to severe head injury, two were withdrawn because they became combative and removed their NG tubes and pH probes, and the final patient was withdrawn from the trial when steroids were prescribed for an optic nerve injury by the attending physicians. One patient from the placebo group was removed due to withdrawal of NG tube" Comment: 24 participants completed the prescribed 72 hours of the trial, and reasons for the remaining 10 not completing the 72‐hour period are well documented Duration of trial: February 1988 to November 1988 Duration of follow‐up: 48 hours after study withdrawal | |
| Outcomes | Outcomes sought in review and reported in trial
Note: The 5 participants who bled belonged to the placebo group; bleeding occurred before 72 hours into the trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Department of Surgery, University of Louisville, School of Medicine, Louisville, Kentucky, USA Source of funding: Quote: "The study was supported by a grant from Glaxo Inc. Research Institute" Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the Human Studies Committee" Informed consent: Quote: "...informed consent was obtained from each patient's legal representative" Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Within 24 hr of injury, each patient was randomly assigned to receive either 6.25 mg/hr continuous intravenous ranitidine infusion or a saline placebo infusion in accordance with a computer‐generated randomisation scheme" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Unclear risk | Comment: No information on allocation concealment reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "To maintain the integrity of the study blinding and to avoid potential bias, the principal investigator did not have access to the pH data" Comment: This is a double‐blind, placebo‐controlled, parallel‐group study design in which study personnel were blinded |
| Blinding (detection bias) | Low risk | Quote: "To maintain the integrity of the study blinding and to avoid potential bias, the principal investigator did not have access to the pH data" Comment: The trial did investigate the relationship between intragastric pH values and the incidence of bleeding. Moreover, we are unclear whether it was the principal investigator who was also involved in outcome assessment. However, GI bleed was detected as per the study definition, and owing to the objective nature of the outcome, the likelihood of performance and detection bias is low |
| Blinding (detection bias) | Unclear risk | Comment: Trial did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: Blinding for other outcome assessments is unclear. Criteria for diagnosis of other outcomes are not fully described |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "All patients completed at least 8 hours of investigational therapy and were included in the analysis" Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and included in the report |
| Other bias | Low risk | Comment: Trial was supported by a grant from Glaxo Inc. Research Institute; trial authors (number not sure) had affiliation with this company. No other sources of bias are suspected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 62 participants Number analysed: 59 participants Cimetidine
Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 3 groups were almost similar with respect to age, gender, number of participants, and risk factors thought to precipitate GI bleed. Most participants were medical participants with respiratory failure secondary to an intrathoracic process. There was no significant difference between groups with respect to major risk factors for GI bleeding (sepsis, peritonitis, jaundice, hypotension, and trauma). Only acute renal failure was more common with the antacid regimen than with cimetidine and sucralfate (however, the incidence of renal failure did not correlate with upper GI bleed) | |
| Interventions | Cimetidine
Antacids
Sucralfate
Adherence to regimen: sucralfate, H2 receptor antagonists, or antacids were used inadvertently in 3 patients who were excluded from the trial Duration of trial: October 1985 to January 1986 Duration of follow‐up: 24 hours after extubation | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not sought in review
| |
| Notes | Setting: Medical‐Surgical Intensive Care Unit at Akron General Medical Centre, Ohio, USA Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional review board at the Akron General Medical Centre" Informed consent: Quote: "A signed consent was obtained from patients or the next of kin after the potential complications and nature of the procedure were explained" Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Sixty‐two patients were accepted for computerised randomised study between October 1985 and January 1986" |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information on allocation concealment reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and mode of administration of drugs could not have allowed blinding of participants and personnel involved in the trial |
| Blinding (detection bias) | Low risk | Comment: unclear if outcome assessors were blinded. However, GI bleeding was an objective outcome, which was detected as per the definition in the trial protocol |
| Blinding (detection bias) | Unclear risk | Comment: The trial did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: not enough information on criteria for diagnosis of other outcomes described |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "Of the 62 patients originally entered into the study three were excluded because of the inadvertent concomitant use of sucralfate, H2 receptor antagonists or antacids in the study group" Comment: Groups to which these 3 participants belonged are unclear (ITT cannot be performed). These participants were excluded from the final analysis. Because loss to follow‐up was < 10% and appeared to be balanced across groups, this would not have introduced any attrition bias into the trial |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is not clearly mentioned in the trial report. No other form of bias is detected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: ‐ Number analysed: 101 participants Ranitidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: The nature and location of diseases, types of operations, number of preoperative risk factors, and demographic data were comparable in the 2 groups | |
| Interventions | Ranitidine
Placebo
Adherence to regimen: ‐ Duration of trial: July 1988 to December 1989 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes reported in trial and used in review
Outcomes sought but not reported in trial
Outcomes reported, but not used in review
| |
| Notes | Setting: Neurological Intensive Care Unit, Department of Surgery, Queen Mary Hospital, University of Hong Kong Source of funding: University of Hong Kong Research Grant and Lee Wing Tat Research Grant Conflicts of interest: ‐ Ethics approval: The protocol used in our trial was approved by the Ethics Committee of the Faculty of Medicine, at the University of Hong Kong Informed consent: Written consent was obtained from patients or their next of kin Clinical trials registration: ‐ Sample calculation: 49 patients would be required in each arm of the trial with a power of 0.8 and a 0.95 significance level by 2‐tailed test Comments: The endpoint of the trial was the development of symptomatic GD lesions defined as GD bleeding requiring blood transfusions and/or surgery for acute perforated ulcers | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomised in a standard double blind manner" Comment: not enough information on sequence generation reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information on allocation concealment reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "All patients were randomised in a standard double‐blind manner to receive either ranitidine (50 mg) or placebo medication (normal saline) identical in appearance and volume" |
| Blinding (detection bias) | Low risk | Quote: "Final outcomes were also assessed by an independent observer to ascertain whether they were a direct result of the GD lesions" "Endoscopic examination of the GD tract up to the second part of the duodenum was performed in all patients within 12 hours of surgery. Additional bolus doses of sedative and analgesic medications we re given during endoscopy. A nasogastric tube was passed into the stomach after endoscopy; its position was confirmed by radiological means, and it was connected to a bag for free drainage. Aspiration from the tube was performed at 6‐hour intervals and a pH paper was used to measure the pH of the gastric content. The total volume of daily gastric output was recorded" |
| Blinding (detection bias) | Unclear risk | Comment: Trial did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no details on criteria for diagnosis of other outcomes reported |
| Incomplete outcome data (attrition bias) | Low risk | Comment: no missing outcome data. All participants randomised at baseline are also included in analyses of outcomes |
| Selective reporting (reporting bias) | High risk | Comment: Outcome data for blood transfusions were not reported by treatment group, but as totals. Other outcomes were reported completely. More outcomes were reported in the Results section than in the Methods section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 100 participants Number analysed: 100 participants Antacids + cimetidine
Sucralfate
Incusion criteria
Exclusion criteria
Baseline imbalance: Quote: "There were no significant differences in age, burn size, the presence of inhalation injury and requirements for intubation. The groups were compared using severity index based upon age, burn size and the presence of inhalation injury. The severity findings were not different between the groups, indicating the similarity of the participant cohorts" Comment: The 2 groups were comparable at baseline. Participants were admitted for thermal injury (burns). Inhalation injury was found in 22 and 27 participants from the cimetidine + antacid and sucralfate groups. Intubation was required in 29 and 28 participants, respectively | |
| Interventions | Antacids + cimetidine
Sucralfate
Adherence to regimen: 100 participants were randomised to both groups ‐ 50 each. There were 4 protocol violations (2 in each group), leaving 96 participants for evaluation. Continuous enteral feeding began from postburn day 3 onwards for participants who were not able to meet the requirements orally. The duration of intubation was longer in the sucralfate group owing to higher incidence of pneumonia. There were 9 deaths before postburn day 5 ‐ 5 in the acid neutralising group and 4 in the sucralfate group. None of these participants had pneumonia. There were 5 deaths among participants who developed pneumonia after postburn day 5 Duration of trial: March 1990 to December 1992 Duration of follow‐up: probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: US Army Institute of Surgical Research, Fort Sam Huston, Texas Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: Quote: "Consenting participants were randomised in a pair wise fashion" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: There was a higher incidence of pneumonia among participants who were intubated or had inhalation injury and were administered sucralfate. Mixed pneumonia (gram‐positive and gram‐negative pathogens) was most frequent in both groups. The percentage of participants in each group who developed positive sputum cultures was 100 and 98, and for gastric cultures 96 and 83, respectively (for any bacteria) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no information on sequence generation reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information on allocation concealment reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the mode of administration of interventions could not have permitted blinding of participants |
| Blinding (detection bias) | Low risk | Comment: Outcome assessors were not blinded. Only the presence of gross bleeding was considered in the results |
| Blinding (detection bias) | Low risk | Quote: "Chest roentgenograms were reviewed by staff surgeon and radiologist who were unaware of the participants' treatment group" Comment: Outcome assessors for this particular outcome were blinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: Outcome assessors were not blinded. Criteria for diagnosis of other outcomes were not clearly reported |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Not all participants who were randomised were part of the final analysis. Two were excluded from each arm owing to protocol violations. Because the loss to follow‐up of less than 10% is balanced between groups, this would not have introduced any attrition bias into the trial. Results were analysed in the review on the basis of ITT |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed |
| Other bias | Low risk | Comment: Source of funding is unclear. No other source of bias is suspected |
| Methods | Double‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 359 participants Number analysed: 359 participants Omeprazole
Cimetidine
Note: 64 participants in both groups were over 65 years of age Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The two groups were generally well matched, although the mean APACHE II score (indicating severity of critical illness) was higher in the omeprazole‐suspension group" Comment: Coagulopathy was present in 37 and 26 participants, respectively, and acute renal failure was more common in the omeprazole group (47 and 33). The 2 groups were similar to each other with respect to age, gender, baseline gastric pH, and other risk factors known to precipitate GI bleeding in the ICU | |
| Interventions | Omeprazole
Cimetidine
Duration of trial: June 2002 to May 2003 Duration of follow‐up: probably until death or discharge Adherence to regimen: Quote: "A total of 56 patients in the intention‐to‐treat population were excluded from the per‐protocol population (omeprazole, n = 21; cimetidine, n = 35) primarily due to failure to receive dose increases within 12 hours of observing the first of two pH values of 4 (omeprazole, n = 4; cimetidine, n = 25)" "Participation in the trial was discontinued before 14 days in the event of death, discharge from the unit, or extubation (endotracheal or gastric tube)" " ...four patients who were discontinued from the trial while actively bleeding (omeprazole, n = 1; cimetidine, n = 3)" | |
| Outcomes | Outcomes sought in review and reported in trial
Note: Five participants bled during the first 2 days of acid suppression therapy (omeprazole; n = 1; cimetidine; n = 4)
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: 47 ICUs in the United States Source of funding: supported, in part, by Santarus, San Diego, CA Conflicts of interest: ‐ Ethics approval: Quote: "This trial was approved by the institutional review board at each site" Informed consent: Quote: "Before any trial procedures were performed, each patient or his or her authorized legal representative gave written informed consent for trial participation" Clinical trials registration: ‐ Sample size calculation: Quote: "Sample size calculations were based on an assumption of a 12% rate of upper GI bleeding in cimetidine‐treated patients and a 6% rate in omeprazole suspension–treated patients. To establish the non‐inferiority of omeprazole suspension with 90% power at the one‐sided α = 0.25 level, 142 patients were required in each treatment group. Assuming that 20% of randomised patients would not satisfy per‐protocol requirements, enrolment of 178 patients per group was planned" Additional notes: An additional 75 participants had overt upper GI bleeding but did not meet the primary endpoint of clinically significant upper GI bleeding as defined by the trial protocol. Moreover all participants who met the endpoint criteria of clinically significant upper GI bleeding were in the per‐protocol population. Four of the 17 participants who had clinically significant bleeding died (omeprazole: n = 2; cimetidine: n = 2), but bleeding was not the cause of death for any of these participants | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Patients, trial site personnel, and the sponsor were blinded to patient treatment assignment" "Within each site, patients were randomly assigned to receive immediate‐release omeprazole oral suspension (Zegerid, Santarus, San Diego, CA) and continuous intravenous infusion of placebo or continuous intravenous cimetidine and placebo oral suspension for up to 14 days" Comment: The presence of placebo in both arms might have ensured that participants and personnel were blinded |
| Blinding (detection bias) | Low risk | Quote: "Patients, trial site personnel, and the sponsor were blinded to patient treatment assignment" Comment: The presence of placebo in both arms ensured that participants and personnel and outcome assessors were blinded. Moreover, GI bleeding was an objective outcome that was detected as per the trial protocol, so the likelihood of performance and detection bias is low |
| Blinding (detection bias) | Low risk | Quote: "Patients, trial site personnel, and the sponsor were blinded to patient treatment assignment" Comment: The presence of placebo in both arms ensured that participants and personnel and outcome assessors were blinded. Moreover, nosocomial pneumonia was an objective outcome that was detected as per the trial protocol, so the likelihood of performance and detection bias is low |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Patients, trial site personnel, and the sponsor were blinded to patient treatment assignment" Comment: The presence of placebo in both arms ensured that participants and personnel were blinded. Moreover, other outcomes of interest were objective in nature and were diagnosed according to the trial protocol, so the likelihood of performance and detection bias was low |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "The intent‐to‐treat population included all randomised patients. The per‐protocol population was used for the analysis of the primary efficacy end point; the intent‐to‐treat population was also used for the primary end point and for all other analyses" Comment: ITT analysis was performed for all outcomes of interest, so the likelihood of attrition bias was low |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes intended in the trial were analysed and reported. However, there is no mention of the presence or absence of any adverse events |
| Other bias | Unclear risk | Comment: One of the trial interventions, omeprazole oral suspension, was sponsored by Zegerid, Santarus, San Diego, CA, which is also the sponsor of the trial. Analysis of data was performed by Santarus, the sponsor of the trial. Also the analysis used was a non‐inferiority analysis, to compare rates of upper GI bleeding in the 2 treatment groups |
| Methods | Multi‐centre blinded randomised placebo‐controlled trial | |
| Participants | Baseline characteristics Number randomised: 1200 participants Number analysed: 1200 participants Ranitidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalance: Quote: "Demographic and baseline physiologic characteristics were similar in the two groups" Comment: Mean and SD for APACHE II scores were 24.7 +/‐ 7.1 and 24.6 +/‐ 7.3 in the ranitidine and sucralfate groups. The main reasons for admission were medical: elective surgery or emergency surgery | |
| Interventions | Ranitidine
Sucralfate
Adherence to regimen: Quote: "No patient received active drug instead of the assigned placebo, or vice versa. Of the scheduled doses of ranitidine and sucralfate, 94.2% and 91.7%, respectively, were administered. Among patients who missed doses, the mean number of doses missed was 2.3 (median, 3; interquartile range, 2 to 3) for ranitidine and 2.9 (median, 4; interquartile range, 1 to 4) for sucralfate" Duration of trial: October 1992 to May 1996 Duration of follow‐up: probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial:
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: 16 ICUs: McMaster University, Hamilton, The University of Toronto, Toronto, The University of Western Ontario, London, Dalhousie University, Halifax, Memorial University, St. John’s, Newf , Queen’s University, Kingston. University of British Columbia, University of Ottawa, Ottawa, University of Alberta, Edmonton Vancouver General Hospital, Vancouver, Royal Alexandra Hospital, Edmonton, Grey Nun’s Hospital, Edmonton, Winnipeg Health Sciences Center, Winnipeg, Man., Toronto Hospital, General Division, Toronto; Wellesley Hospital, Toronto London Health Sciences Center (Victoria Campus), London, Ont., London Health Sciences Center (University Campus), London, Ont., St. Joseph’s Health Center, London, Ont., St. Joseph’s Hospital, Hamilton, Ont., Henderson Hospital, Hamilton, Ont., Kingston General Hospital, Kingston,Ont., Ottawa Civic Hospital, Ottawa, Ont., Health Sciences Center, St. John’s, Newf, Victoria General Hospital, Halifax, N.S. Source of funding: Quote: "Supported by the Medical Research Council of Canada and Hoechst Marion Roussel. Drugs were supplied by Glaxo Wellcome, Baxter, and Hoechst Marion Roussel. Dr. Cook is a Career Scientist of the Ontario Ministry of Health" Conflicts of interest: ‐ Ethics approval: Quote: "The protocol was approved by the institutional review board of all participating enters ..." Informed consent: Quote: "...and the patients or their proxies gave informed consent" Clinical trials registration: ‐ Sample size calculation: Quote: "On the basis of data published through 1991, when our study was designed, we anticipated a 25 percent incidence of pneumonia and identified a 25 percent reduction in the risk of pneumonia associated with sucralfate as being plausible and clinically important. This led to the calculation of a sample size of 1200 patients as necessary to give the study 75 percent power to detect such a difference, assuming a two‐sided significance test at the 0.05 level. We analysed all patients in the groups to which they were randomly assigned, according to the intention‐to‐treat principle" Additional notes: Gram‐negative bacilli and gram‐positive cocci were the main isolates from endotracheal aspirates from patients with ventilator‐associated pneumonia | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to study groups in blocks of six, with stratification according to centre, by means of a computer generated random‐number table prepared at the McMaster University Methods Center and managed by the ICU study pharmacist at each site" |
| Allocation concealment (selection bias) | Low risk | Quote: "...managed by the ICU study pharmacist at each site who administered the coded drugs" Comment: Method to obtain allocation concealment is clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "The patients, research nurses, and all ICU care givers were unaware of the treatment assignments for the duration of the study. Therefore, clinicians did not monitor gastric pH" "The radiologists, outcome adjudicators, all investigators, and the study statistician were also blinded until all events had been adjudicated and the analyses completed" Comment: Blinding was done, so the likelihood of performance bias and detection bias is low |
| Blinding (detection bias) | Low risk | Quote: "The patients, research nurses, and all ICU care givers were unaware of the treatment assignments for the duration of the study. Therefore, clinicians did not monitor gastric pH" "The radiologists, outcome adjudicators, all investigators, and the study statistician were also blinded until all events had been adjudicated and the analyses completed" Comment: Blinding was done. Moreover the outcome of interest was objective in nature, so the likelihood of performance bias and detection bias is low |
| Blinding (detection bias) | Low risk | Quote: "The patients, research nurses, and all ICU care givers were unaware of the treatment assignments for the duration of the study. Therefore, clinicians did not monitor gastric pH" "The radiologists, outcome adjudicators, all investigators, and the study statistician were also blinded until all events had been adjudicated and the analyses completed" Comment: Blinding was done. Moreover the outcome of interest was objective in nature, so the likelihood of performance bias and detection bias is low |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "The patients, research nurses, and all ICU care givers were unaware of the treatment assignments for the duration of the study..." Comment: Blinding was done. Moreover all other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All participants who were randomised to the 2 groups were analysed |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported |
| Other bias | Low risk | Comment: Hoechst Marion Roussel supported the trial and also sponsored sucralfate needed for administering to study participants. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 52 participants Number analysed: 52 participants Ranitidine
Sucralfate
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: There were no major differences in demographic profiles between study groups Comment: Participants were from medical and surgical units, in equal proportions across the 3 groups. However, the underlying reason for admission is not mentioned in the trial report | |
| Interventions | Ranitidine
Sucralfate
No prophylaxis
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: All India Institute of Medical Sciences (AIIMS), New Delhi, India Source of funding: Quote: "Supported by the Medical Research Council of Canada and Hoechst Marion Roussel. Drugs were supplied by Glaxo Wellcome, Baxter, and Hoechst Marion Roussel. Dr. Cook is a Career Scientist of the Ontario Ministry of Health" Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional Notes: Gram‐negative organisms were found in the gastric culture of 18 ranitidine participants and 6 sucralfate participants, and the control group had no pathogenic organisms. Pseudomonas was the most common organism in the gastric and BAL cultures of the ranitidine and sucralfate groups | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the mode of administration of interventions could not have permitted blinding of participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: not clearly mentioned in the trial report. However owing to the objective nature of the outcome, which was detected as per the trial protocol, the likelihood of performance and detection bias is low |
| Blinding (detection bias) | Unclear risk | Comment: Trial did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: No other outcomes of interest were assessed in this trial |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | High risk | Comment: unclear on source of funding. Baseline imbalance on the numbers randomised to the 3 groups as the no prophylaxis arm had only 7 participants compared with 21 in the sucralfate arm and 24 in the ranitidine arm |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 182 participants Number analysed: 181 participants Nasojejunal nutrition
Nasogastric nutrition
Inclusion criteria
Exclusion criteria
Baseline imbalances: Baseline characteristics of the groups were similar | |
| Interventions | Nasojejunal nutrition
Nasogastric nutrition
Adherence to treatment: ‐ Duration of trial: ‐ Duration of follow‐up: 28 days | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Medical‐Surgical ICUs, Alfred Hospital, Melbourne, Australia Sponsorship source: Cook Critical Care provided the frictional nasojejunal tubes Conflicts of interest: no conflicts of interest disclosed Comments: Other recommended treatment includes ranitidine Ethics approval: All sites received institutional human research ethics committee approval Informed consent: Prior written informed consent was obtained from the patient’s surrogate decision‐maker Clinical trials registration: ‐ Sample size calculation: 180‐patient sample size provided 80% power with 2‐sided significance of 0.05 to detect a 12% difference in mean energy delivery | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "They were randomly assigned to early nasojejunal nutrition or naso‐gastric nutrition using a computer‐generated randomisation schedule with variable block size" |
| Allocation concealment (selection bias) | Low risk | Quote: "Treatment assignments were concealed before randomisation using a centralized voice‐recognition phone system" |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "There was no blinding after randomisation" Comment: high for personnel; indifferent for patients |
| Blinding (detection bias) | Low risk | Quote: "gastrointestinal haemorrhage ([was measured as] defined as major or minor based on definitions from a previous trial)" Comment: detailed description provided in supplementary material to the trial. Outcome measurement unlikely to introduce bias to the trial |
| Blinding (detection bias) | Low risk | Quote: "chest radiograph images" Comment: Outcome measurement was objective and therefore was unlikely to introduce bias |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "The collected information was provided to a blinded adjudication panel of two intensivists and one respiratory physician for each patient. To maintain blinding, the radiograph images had the enteral tube shadows in the mediastinum and upper abdomen obscured. Panel members individually adjudicated the diagnosis of VAP using objective criteria (Supplemental Digital Appendix A, Supplemental Digital Content 1, http://links.lww. com/CCM/A454). Patients were formally categorized as meeting the VAP outcome if at least two of the three adjudicators determined the pres‐ ence of VAP and this occurred after the patient had been mechanically ventilated for >72 hrs. A Data Monitoring Committee regularly viewed data on safety aspects with blinding maintained and did not recommend study cessation" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "Data were analyzed according to the intention‐to‐treat principle with no imputation of missing values" Comment: No incomplete outcome data were suspected. All participants enrolled at baseline were treated and analysed |
| Selective reporting (reporting bias) | Low risk | Comment: All prespecified outcomes listed in the Methods section were reported in the Results section. No selective outcome reporting was suspected |
| Other bias | Low risk | Comment: No other sources of bias were suspected |
| Methods | Randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: ‐ Number analysed: ‐ Ranitidine
Sucralfate
Omeprazole
Inclusion criteria
Exclusion criteria
Baseline imbalances: Groups were similar to each other with respect to demographic characteristics. APACHE ll scores were 55.7 ± 23.2, 54.1 ± 21.7, and 56.1 ± 23.7 in the ranitidine, sucralfate, and omeprazole groups, respectively. Pneumonia was present in 2, 3, and 3 participants in the 3 groups | |
| Interventions | Ranitidine
Sucralfate
Omeprazole
Adherence to regimen: ‐ Duration of trial: February 1997 to April 1998 Duration of treatment: Duration of treatment that was established was based on clinical evaluation, control of risk factors, clinical improvement, and full nutritional support, oral or enteral Duration of follow‐up: during the period that participants were in the ICU. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Intensive Care Unit, Hospital São Domingos, Sao Luis do Maranhão, Brasil Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were randomly assigned for one of the three groups when they were admitted for the intensive unit care, they used a simple technique of randomisation, and sealed envelopes" |
| Allocation concealment (selection bias) | Unclear risk | Comment: Trial authors described that they used sealed envelopes. It was not stated whether they were numbered and opaque |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: not enough information reported |
| Blinding (detection bias) | Unclear risk | Comment: not enough information reported on criteria for assessment of upper GI bleeding |
| Blinding (detection bias) | Unclear risk | Comment: not enough information reported on criteria for assessment of pneumonia |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear; however all other outcomes of interest were objective outcomes. Therefore the likelihood of performance and detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were analysed |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes described in the Methods section were analysed in the Results section |
| Other bias | Low risk | Comment: no other source of bias detected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 130 participants Number analysed: 130 participants Sucralfate
Antacid and/or H2 receptor antagonists
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The two treatment groups were similar in terms of demographic characteristics and severity of illness on admission to the study" "The distribution of underlying diseases, indications for intubation and surgical procedures according to the site were similar in both the groups" "Thirty two of the 61 patients in the sucralfate group and twenty nine of the 69 participants in the antacid H2 group had infiltrates at baseline evaluation (n = 38), adult respiratory distress syndrome (n = 8), or congestive heart failure (n = 15), making it difficult to diagnose new infiltrates on a chest film" Comment: similar distribution with respect to demographic and baseline risk factors. However, a total of 35 participants (sucralfate: n = 16; antacid and/or H2: 19) had pneumonia on admission and/or had infiltrates (as mentioned above) | |
| Interventions | Sucralfate
Antacid and/or H2 receptor antagonists
Adherence to regimen: Quote: "Six patients who were initially assigned to sucralfate but subsequently assigned to antacids or H2 blockers by the treating physician were included in the sucralfate group (for analysis), no patients were switched from antacids or H2 blockers to sucralfate. Four of the six patients who were crossed over had evidence of upper GI tract bleeding, the other two who were switched to antacids 5 and 11 days after randomisation had no evidence of such bleeding. Two of the six patients had pneumonia after the crossover occurred but were included in the sucralfate group" "Four patients in the sucralfate group and 17 in the antacid‐H2 group underwent tracheostomy" Comment: ITT was performed for the review and not per‐protocol analysis Duration of trial: April 1986 to February 1987 Duration of follow‐up: until 48 hours after extubation | |
| Outcomes | Outcomes sought in review and reported in trial
Note: Pneumonia developed after 9.6 (4.6) days in sucralfate group and after 9.6 (6.3) days in antacid H2 group
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: surgical, medical, or coronary intensive care units, Boston City Hospital Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study protocol was reviewed and approved by the hospital institutional review board for human studies" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Quantitative data were extracted for the 2 randomised groups alone (sucralfate and antacid ‐ H2 receptor groups). Data were not extracted separately for participants treated with antacid or H2 receptor alone, as this would break the randomisation. Of bacteria isolated from the tracheal aspirates of participants treated with pneumonia, gram‐negative bacilli were predominant; Pseudomonas aeruginosa in the sucralfate group; Enterobacteriaceae in antacid‐H2 group | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the mode of administration of the interventions would not have permitted blinding of participants |
| Blinding (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded, but GI bleeding was an objective outcome that was detected as per the definition in the trial protocol |
| Blinding (detection bias) | Low risk | Quote: "Chest roentgenograms were interpreted by at least one of us, who had no knowledge on the patient's treatment group after randomisation" Comment: Blinding was done. Moreover, pneumonia was an objective outcome that was detected as per the definition in the trial protocol |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded. Moreover the outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Intention‐to‐treat analysis was performed wherein all participants who were initially randomised to each of the 2 study groups were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes have been reported |
| Other bias | Low risk | Comment: Source of funding is not clearly mentioned in the trial report. No other form of bias was detected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 60 participants Number analysed: 60 participants Sucralfate
Ranitidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Participants in the ranitidine group were relatively older. The acute physiology and chronic health evaluation (APCHE II) score was 11.6 (1.3) for sucralfate and 12.4 (1.5) for ranitidine Admission diagnosis: More participants in the sucralfate group had both CNS injury (11/30) and trauma (8/30), and CNS injury and respiratory failure were the most common diagnoses on admission among participants in the ranitidine group (8/30) and (7/30) | |
| Interventions | Sucralfate
Ranitidine
Adherence to regimen: All participants were subjected to treatment within 6 hours after ICU admission.Enteral nutrition was given (exact numbers and interventional groups not specified) Duration of trial: ‐ Duration of follow‐up: probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial
Note: GI bleeding in 2 participants (day 3 in the participant from the sucralfate group and day 5 in the participant from the ranitidine group) Outcomes sought but not reported in trial
Outcomes reported in trial but not sought in review
| |
| Notes | Setting: ICU, Manchester Royal Infirmary, Manchester, UK Source of funding: ‐ Ethics approval: Quote: "The protocol for the study was approved by the hospital ethics committee" Informed consent: Quote: "Informed consent was obtained from the patient's next of kin" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Retrograde colonisation from stomach to oropharynx was found in 6 participants in the sucralfate group and 14 participants in group B. From the oropharynx to the trachea, colonisation occurred in 5 of the 6 participants in group A and in 12 of the 14 participants in group B. Colonisation occurred relatively later in sucralfate‐treated participants. More pneumonia cases were reported in group B, and the pathogens were mainly similar in both groups (mainly gram‐negative bacilli) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The patients were allocated by the use of a random sampling table into two groups" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering the study interventions would not have made it possible to blind the study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Quote: "Endoscopic evolution of the gastric and the duodenal mucosa was performed within 24 hours of admission by an operator who was blinded to the randomisation and was repeated on day 4 if the participant was still receiving mechanical ventilation" Comment: Endoscopy was performed if there was fresh blood (< 50 mL) in the nasogastric aspirate, but the endoscopist was blinded. Moreover, the outcome of interest was objective in nature |
| Blinding (detection bias) | Low risk | Comment: no mention of blinding of radiologists, lab technicians, or physicians. However, pneumonia was diagnosed as per the study definition and due to the objective nature of the outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: not clear on blinding assessors for other outcomes of interest. However given the objective nature of the outcomes, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All 60 randomised participants were included in the final analysis. Therefore there is low risk of attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported. No mention of any adverse events |
| Other bias | Low risk | Comment: Source of funding is not clearly mentioned in the trial report. No other sources of bias are suspected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 26 participants Number analysed: 26 participants Sucralfate
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Participants in the placebo group were relatively older. Acute Pysiology and Chronic Health Evaluation II (APCHEII) scores were 12.5 (1.7) and 14.7 (2.2), respectively. Trauma was the most common cause for admission (6 in sucralfate group and 4 in the placebo group) | |
| Interventions | Sucralfate
Placebo
Adherence to regimen: Treatment began within 8 hours of ICU admission. Endoscopy performed on day 3 in the placebo group revealed acute ulcerations in 5 participants, as a result of which they were withdrawn and put on sucralfate 2 g every 8 hours. On day 6, 2 more patients from placebo group were switched over to sucralfate owing to acute gastric ulceration. Mucosal deterioration and acute ulceration were significant in the placebo group. The study was halted after recruitment of 26 participants for ethical reasons Duration of trial: ‐ Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Note: GI bleeding occurred in 1 participant from placebo group on 20th day of the trial Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Department of Surgery, Critical Care Unit, Department of Microbiology, Manchester Royal Infirmary, Manchester, UK Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The protocol for the study was approved by the hospital ethics committee" Informed consent: Quote: "...informed consent was obtained from the patient's next of kin" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Oropharyngeal retrograde colonisation occurred in 1 participant in the sucralfate group and in 2 participants in the placebo group. Tracheal colonisation occurred in 2 participants from the sucralfate group and in 1 participant from the placebo group. However only the latter developed pneumonia. Mortality in ICU was attributed to multiple organ failure | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were allocated by the use of a random sampling table into two groups..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the trial report |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: This was a placebo‐controlled trial, and the mode of administering the interventions was similar. However, unclear whether personnel and participants were blinded |
| Blinding (detection bias) | Low risk | Quote: "Endoscopic evolution of the gastric and the duodenal mucosa was performed within 24 hours of admission by an operator who was blinded to the randomisation code and was repeated on day 3, 6 and 10 if the participant was still receiving mechanical ventilation" Comment: Interim gastroscopy was performed if there was fresh blood in the nasogastric aspirate, but the endoscopist was blinded. However, the outcome of interest was objective in nature |
| Blinding (detection bias) | Low risk | Comment: no mention of blinding of radiologists, lab technicians, or physicians. However, pneumonia was diagnosed as per the study definition and due to the objective nature of the outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: no clear mention of blinding of outcome assessors. However owing to the objective nature of the outcomes, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All participants randomised were included in the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes are reported, although unclear on adverse events |
| Other bias | Low risk | Comment: Source of funding is not clearly mentioned in the trial report. No other sources of bias are suspected |
| Methods | Double‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 140 participants Number analysed: 140 participants Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The patients who were randomly assigned to receive antacid or sucralfate stress ulcer prophylaxis were similar in most risk factors for postoperative complications but they differed in the percentage of patients with a history of treatment for chronic obstructive pulmonary disease (COPD). Significantly more patients with a clinical diagnosis of COPD preoperatively were randomly assigned to receive sucralfate (COPD was found to be an independent risk factor for development of pneumonia)" Comment: Baseline age and gender distributions are not mentioned; however the distribution of all risk factors except COPD is normal across the 2 groups. Most participants from both groups were admitted for general surgery of the abdomen (23 in antacid group and 27 in sucralfate group, respectively) | |
| Interventions | Antacid
Sucralfate
Adherence to regimen: Quote: "One hundred twenty‐two participants received 3 or more days of stress ulcer prophylaxis with the study drugs, while only 25 received 7 or more days.Many patients were treated with intravenous H2‐receptor blockers after 3 days postoperatively, which was when the study drugs were generally discontinued" Duration of trial: June 1990 to April 1992 Duration of follow‐up: Quote: "Daily assessment for gastrointestinal tract bleeding, infections, and other postoperative complications was continued until discharge from the hospital or death for all 140 patients" | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: The Surgical Service, Veterans Affairs Medical Center, Iowa City, Iowa Source of funding: Quote: "This study was funded through a Veterans Affairs Merit Review grant." Conflicts of interest: ‐ Ethics approval: Quote: "...approved for human subjects by the appropriate committees of the University of Iowa College of Medicine and the Iowa City Veterans Affairs Medical Center" Informed consent: Quote: "Patients were contacted before their elective surgical procedures, and less than 10% of the patients asked to participate refused to do so. Patients who declined entry into the study were not monitored further" Clinical trials registration: ‐ Sample size calculation: Quote: "A biostatistician calculated that an 80% likelihood of detection of a 50% improvement in the expected postoperative pneumonia rate of 15% to 20% would require entry of more than 300 patients into each arm of the study. With pilot work showing that more than 50% of patients in the surgical intensive care unit harboured pathogens in their gastric contents, we determined that approximately 70 patients in each arm would give an 80% likelihood of detecting an effect by the stress ulcer prophylactic agent on the gastric microbial flora" Comment: Sample size was calculated for the primary endpoint of pneumonia Additional notes: Study endpoints of pneumonia and GI bleeding were diagnosed postoperatively. Many participants received H2 antagonists 3 days postoperatively; study drugs were discontinued by then. Pulmonary colonisation with gastric micro‐organism occurred in 16% of antacid‐treated and 15% of sucralfate‐treated participants | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was performed in blocks of 20 patients before the study began, to ensure that both treatment groups would be evenly represented throughout the 2 years of patient accrual" |
| Allocation concealment (selection bias) | Low risk | Quote: "In keeping with the double‐blind design, the treatment group assignment of each patient was known only to the pharmacy service until all the clinical and microbial data collection was complete" Comment: Allocation concealment was done |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "The sham agents were designed to be similar in colour and viscosity to the active agents, and free of acid‐neutralizing properties" Comment: This was a double‐blind study in which participants and trial personnel appear to be blinded. Therefore the likelihood of performance bias is low |
| Blinding (detection bias) | Low risk | Quote: "The sham agents were designed to be similar in colour and viscosity to the active agents, and free of acid‐neutralizing properties" Comment: This was a double‐blind study in which participants and trial personnel appear to be blinded. Moreover the outcome of GI bleed was detected as per the definition in the study protocol owing to the objective nature of the outcome |
| Blinding (detection bias) | Low risk | Quote: "Pneumonia was diagnosed by consensus of 2 investigators not involved with the clinical Care”; "The sham agents were designed to be similar in colour and viscosity to the active agents, and free of acid‐neutralizing properties" Comment: Blinding of outcome assessors was done. Moreover the outcome of pneumonia was detected as per the definition in the study protocol owing to the objective nature of the outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "The sham agents were designed to be similar in colour and viscosity to the active agents, and free of acid‐neutralizing properties" Comment: Blinding of outcome assessors was done. Moreover because of the objective nature of the outcomes, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were analysed for the outcomes of interest |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes have been reported |
| Other bias | Low risk | Comment: Funding was through a Veterans Affairs Merit Review grant. The role of the sponsor in the conduct and reporting of the trial is unclear. No other bias was detected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 616 participants Number analysed: 278 participants Sucralfate
Cimetidine bolus
Cimetidine continuous
Inclusion criteria
Exclusion criteria
Baseline imbalances: After exclusion, 99 participants received sucralfate, 114 received cimetidine (bolus), and 65 received cimetidine as continuous infusion. Blunt trauma was the most common cause for admission in 72% and penetrating trauma in 28% (86% of these were gunshot wounds; 14% were stab wounds). The APACHE ll scores were 14 (5) (mean, SD) for the sucralfate group, 14 (8) (mean, SD) for the cimetidine bolus group, and 13 (6) (mean, SD) for the cimetidine infusion group | |
| Interventions | Sucralfate
Cimetidine bolus
Cimetidine continuous
Adherence to regimen: 616 participants were initially randomised to 3 treatment groups. Data on 338 participants were excluded from analysis for the following reasons:
The remaining 278 participants were part of the study until discharge or death Duration of trial: January 1990 to April 1991 Duration of follow‐up: Quote: "Evaluable patients were studied until discharge or death" | |
| Outcomes | Outcomes sought in review and reported in trial
Note: Pneumonia developed 5.6 (1.6) (mean, SD) days after injury
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Department of Surgery and Clinical Pharmacy, Presley Regional Trauma Centre, University of Tennessee, Memphis Source of funding: Quote: "Supported in part by educational grant from Smith Kline Beecham Inc" Conflicts of interest: Ethics approval: Quote: "The protocol was approved by the Institutional Review Board of The University of Tennesse, Memphis" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: Quote: "Power analysis was made using a significance level of 0.5 a power of 0.8 and a two sided alternative hypothesis" Additional notes: In the bacterial isolates of 81 participants, Staphylococcus aureus was the most predominant followed by Haemophilus influenzae. H2 receptor antagonists were combined to form a common interventional arm vs sucralfate, as the review does not aim to investigate | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to one of the three prophylaxis groups by random number table" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial. Moreover, the mode of administration of study drugs would not have permitted blinding |
| Blinding (detection bias) | Low risk | Comment: GI bleeding was an objective outcome that was diagnosed as per the definition in the study protocol |
| Blinding (detection bias) | Low risk | Comment: Pneumonia was an objective outcome that was diagnosed as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | High risk | Comment: Yes, around 55% of all participants who were randomised were excluded from the final analysis because of less time spent in the ICU (< 48 hours), adverse reactions, younger than 18 years of age (which was clearly an exclusion criterion), and other protocol violations. Although a per‐protocol analysis was done, there appears to be an imbalance between groups with respect to the number of participants available for analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes are clearly mentioned in the study report |
| Other bias | Low risk | Comment: The study was supported in part by an educational grant from Smith Kline Beecham Inc. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: participants Number analysed: participants Enteral plus parenteral nutrition
Enteral nutrition
Parenteral nutrition
Inclusion criteria
Exclusion criteria
Baseline imbalances: There was no significant difference among the 3 groups in terms of age, sex, weight, and serum‐albumin at baseline (P > 0.05) | |
| Interventions | Enteral plus parenteral nutrition
Enteral nutrition
Parenteral nutrition
Adherence to regimen: ‐ Duration of trial: January 2009 to May 2012 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Neurological Intensive Care Unit Source of funding: Natural Science Foundation of Shandong Province (Y2008C35) and Technology Supporting Program of Qingdao (12‐1‐3‐5‐(1)‐nsh) Conflicts of interest: ‐ Ethics approval: This study had been approved by the local institutional review board and the Human Ethics Committee of the Affiliated Hospital of Qingdao University Informed consent: Informed consent had been provided by patients’ guardians Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "patients were assigned to EN group, PN group and EN+PN groups randomly according to the sequence of their assigned hospital record number" Comment: method of randomisation not adequate |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: no information about blinding reported |
| Blinding (detection bias) | Unclear risk | Comment: No definition of criteria was used to diagnose upper GI bleeding as reported |
| Blinding (detection bias) | Unclear risk | Comment: No definition of criteria was used to diagnose nosocomial pneumonia was reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no information reported |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: insufficient information to allow judgement; once 120 patients were enrolled, but no information on how many patients fitted eligibility criteria |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes listed in the Methods section are also reported in the Results section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 120 Number analysed: 120 Early onset drug treatment
Late onset drug treatment
Enteral nutrition
Inclusion criteria:
Exclusion criteria
Baseline imbalances: no baseline difference in sex, age, BMI, and causes of brain injury | |
| Interventions | Early‐onset drug treatment
Late‐onset drug treatment
Enteral nutrition
Adherence to regimen: ‐ Duration of follow‐up: ‐ Duration of trial: May 2011 to May 2013 | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought in review and not reported in trial
Outcomes reported in trial, but not used in review
| |
| Notes | Setting: Department of Nursing, First People’s Hospital of Yunnan Province, Kunming, Yunnan Province, China Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: randomisation by random number tables |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: no information reported |
| Blinding (detection bias) | Unclear risk | Comment: no definition of criteria used to diagnose upper GI bleeding as reported |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no information reported |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All participants were observed and analysed after intervention |
| Selective reporting (reporting bias) | Low risk | Comment: All observed results were reported, including stress ulcer bleeding rate in group A; the correlation between gastric pH and ulcer healing time in group A; incidence of stress ulcer bleeding rate by different gastric pH and different blood sugar level in group C; clinical therapeutic effects |
| Other bias | Low risk | Comment: no other sources of bias suspected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: not clearly mentioned in study abstract Number analysed: 189 participants Overall
Inclusion criteria Study design, inclusion and exclusion criteria similar to Martin 1993
Exclusion criteria
Baseline imbalances: not clearly mentioned in study abstract | |
| Interventions | Pantoprazole
Pantoprazole
Pantoprazole
Pantoprazole
Cimetidine
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: ‐ Adherence to regimen: ‐ Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Quote: "Overall patents with a mean APSII score of 16.8 or greater tended to die while those with 15.33 or less, tended to survive" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in study abstract |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in study abstract |
| Blinding of participants and personnel (performance bias) | High risk | Comment: unblinded, open trial |
| Blinding (detection bias) | Low risk | Comment: unblinded, open trial. The definition for detecting GI bleeding is not clearly mentioned in the study abstract. However, GI bleeding was an objective outcome |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unblinded, open trial. However owing to the objective nature of the outcomes of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Unclear risk | Quote: "189 patients received at least 1 dose of medication and had sufficient data points to be included in the analysis" Comment: unclear on how many were randomised initially to the different arms. Therefore, unclear on attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes appear to be reported |
| Other bias | Low risk | Comment: Source of funding remains unclear. No other sources of bias are suspected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 94 participants Number analysed: 79 participants Proton pump inhibitors
H2 receptor antagonists
Inclusion criteria
Exclusion criteria
Baseline imbalances: no significant difference (P > 0.05) between groups regarding demographics | |
| Interventions | Proton pump inhibitors
H2 receptor antagonists
Adherence to treatment: ‐ Duration of follow‐up: ‐ Duration of trial: ‐ | |
| Outcomes | Outcomes reported in trial and used in review
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: General ICU, University of Szeged, Hungary Sponsorship source: ‐ Conflicts of interest: ‐ Comments: conference abstract Ethics approval: Quote: "After ethics committee approval ..." Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: Quote: "However, the completion of the study on the planned 198 patients is required to come to the final conclusions" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no details reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: no details reported, but lack of blinding unlikely to introduce bias to the outcome and outcome measures |
| Blinding (detection bias) | High risk | Comment: no details reported, and no diagnostic criteria described |
| Blinding (detection bias) | Low risk | Comment: Pneumonia was an objective outcome that was diagnosed as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no details reported |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: no incomplete reporting of outcome data. Patient flow was reported transparently; attrition was unlikely to have introduced bias to the results |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes listed are reported in the Results section. Conference abstract |
| Other bias | Unclear risk | Comment: no other sources of bias suspected, but not enough details reported in conference abstract |
| Methods | Double‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 36 participants Number analysed: 36 participants Placebo
Antacids (Mylanta)
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 3 groups were comparable except for a significantly higher number of participants with chronic obstructive pulmonary disease in the antacid group as compared with the control group. They were also similar with respect to the mean number of risk factors per individual in any of the groups | |
| Interventions | Placebo
Antacid
Cimetidine
Adherence to regimen: 6 participants were unable to complete the protocol: 5 from the antacid group (4 had severe diarrhoea, and 1 had severe hypophosphataemia and hypomagnesaemia) and 1 from the control group (severe diarrhoea). However, intention to treat was performed by study authors Duration of trial: December 1978 to May 1980 Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial
Note: Only data on overt bleeding were extracted, as they were considered to be clinically significant
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Department of Medicine, University of Virginia School of Medicine, Charlottesville, VA 22908 Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The protocol was approved by the Human Investigation Committee of the University of Virginia in February 1978" Informed consent: Quote: "Informed consent was obtained from the patients or from a member of their immediate family" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: According to the study report, no participant died of haemorrhage and overt bleeding ceased when participants were administered cimetidine and antacids | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Quote: "If a participant developed severe diarrhoea, an independent referee broke the treatment code and substituted alternagel in those participants receiving Mylanta ll" Comment: Study must have had some sort of allocation concealment to make the above statement materialise. However, method used for allocation concealment is not clear |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "If a participant developed severe diarrhoea, an independent referee broke the treatment code and substituted Alternagel in those participants receiving Mylanta ll and the investigators were not informed of the outcomes of the referees intervention" Comment: Investigators might have been blinded in the rest of the study too because this was a placebo‐controlled trial; each treatment arm (with the actual intervention) received a placebo form of the other drug (with which it was being compared) and vice versa |
| Blinding (detection bias) | Low risk | Comment: This was a placebo‐controlled trial; each treatment arm (with the actual intervention) received a placebo form of the other drug (with which it was being compared) and vice versa. Moreover, this was an objective outcome detected as per the definition provided in the study |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: This was a placebo‐controlled trial; each treatment arm (with the actual intervention) received a placebo form of the other drug (with which it was being compared) and vice versa. Moreover, owing to the objective nature of the outcome of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were accounted for in the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on source of funding. No other form of bias detected |
| Methods | Double‐blind randomised placebo‐controlled trial | |
| Participants | Baseline characteristics Number randomised: unclear on how many were initially randomised. Study report mentions that 531 participants were eligible for the trial, of whom 221 participants completed the trial with reasons for excluding of the remaining participants Number analysed: 221 participants Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The drug and placebo groups were similar for demographic features and risk factors.The number of patients with each risk factor known to predispose to stress ulceration and the mean number of risk factors per patient were similar in both groups such as minor operative procedure, respiratory failure, renal failure, sepsis, shock, trauma, coma and liver failure" | |
| Interventions | Cimetidine
Placebo
Adherence to regimen: Quote: "Of the remaining 531 eligible patients 221 completed the trial (41%). The remainder were excluded because of inability to obtain early consent (n = 69), refusal to enter the study (n = 83), death within the first 24 hours (n = 48), accidental omission by house staff (n = 86), and miscellaneous reasons (n = 24)" Comment: not sure when randomisation took place and exact numbers of participants who were initially randomised Duration of trial: 21 months Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial
Note: Data were not provided separately for overt and occult bleeds. Of the 70% of participants who were in the study for 1 to 3 days, 9 of the 11 placebo and 5 of the 6 cimetidine participants bled
Outcomes sought but not reported in trial
Outcomes reported in report but not used in review
| |
| Notes | Setting: Kingston General Hospita, Ontario, Canada Source of funding: Quote: "This project was supported by Smith Kline and French Canada Ltd" Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the Institutional Review Board and Grant agency at the Misnistry of Healthof Chez Republic" Informed consent: Quote: "The study was approved by the committee on human research, Department of Medicine, Queen's University" Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Patients were randomised in a double blind method to receive injections of either cimetidine 300 mg in 20 mL normal saline or placebo prepared in an identical manner, given intravenously every 6 hours" Comment: This was a placebo‐controlled trial, and participants and personnel were blinded |
| Blinding (detection bias) | Low risk | Quote: "Patients were randomised in a double blind method to receive injections of either cimetidine 300 mg in 20 mL normal saline or placebo prepared in an identical manner, given intravenously every 6 hours" Comment: This was a placebo‐controlled trial, and GI bleeding was detected as per the definition in the study owing to the objective nature of the outcome of interest |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: This was a placebo‐controlled trial, and participants and personnel were blinded. Moreover owing to the objective nature of the outcomes of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Unclear risk | Quote: "Of the remaining 531 eligible patients 221 completed the trial (41%). The remainder were excluded because of inability to obtain early consent (n = 69), refusal to enter the study (n = 83), death within the first 24 hours (n = 48), accidental omission by house staff (n = 86), and miscellaneous reasons (n = 24)" Comment: not sure when randomisation took place and exact numbers of participants who were initially randomised. Therefore unclear on attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: This project was supported by Smith Kline and French Canada Ltd. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias suspected |
| Methods | Double‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 50 participants Number analysed: 50 participants Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Both groups were comparable with respect to age, gender, and underlying disorders | |
| Interventions | Cimetidine
Placebo
Adherence to regimen: no dropouts mentioned Duration of trial: July 1977 to March 1979 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: Departments of Medicine and Surgery, Medical College, Virginia, USA Source of funding: Quote: "The work was supported by Smith Kline and French Laboratories.clearly mentioned in the study report" Ethics approval: Quote: "The study was approved by the committee on the conduct of clinical research.Medical College of Virginia, Virginia commonwealth university" Informed consent: Quote: "Informed consent was obtained from the patients closest relative or legal agent and later from the patient if sufficient neurologic recovery ensued" Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were randomly given coded medication in a standard double blind fashion" Comment: Allocation concealment might have been done |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Patients were randomly given coded medication in a standard double blind fashion" Comment: This was a placebo‐controlled double‐blind trial, so study personnel would have been blinded |
| Blinding (detection bias) | Low risk | Comment: not clearly mentioned, but this was a placebo‐controlled double‐blind trial, and GI bleeding was an objective outcome that was detected as per the study definition |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: not clearly mentioned, but this was a placebo‐controlled double‐blind trial, and outcomes of interest were objective in nature and were detected as per the study definition |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes stated were reported in the Results |
| Other bias | Low risk | Quote: "The work was supported by Smith Kline and French Laboratories" Comment: The role of the sponsor in the conduct and reporting of the trial is unclear. No other source of bias is suspected |
| Methods | Double‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 827 participants Number analysed: 158 participants Ranitidine
Pirenzipine
Placebo
Notes: Age not given for all randomised participants, but only for those who were in mechanical ventilation for longer than 48 hours Inclusion criteria
Exclusion criteria
Baseline imbalances: Groups were similar with respect to age and APACHE ll score | |
| Interventions | Ranitidine
Pirenzipine
Placebo
Adherence to regimen: All 827 randomised patients completed the trial. However, only 158 participants who were on mechanical ventilation for ≥ 48 hours were part of the analysis, as the primary outcome was pneumonia rate Duration of trial: December 1991 to December 1992 Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: Department of Surgery, Division of General and Trauma Surgery, Johann Wolfgang Goethe University, Frankfurt am Main, Germany Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: approved by the Ethics Committee of Johann Wolfgang Goethe University, Frankfurt/Main Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: Quote: "Assuming a pneumonia rate of 25% in ranitidine treated patients and a pneumonia rate of 15% in placebo treated patients, the complete sample size was calculated to be at least 100 patients (alpha = 0.05 and beta = 0.20)" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A complete and balanced randomisation schedule was generated by the institute of biomathematics of the university of Frankfurt" Comment: method adopted to obtain random sequence generation clearly mentioned in the study report |
| Allocation concealment (selection bias) | Low risk | Quote: "At the time of entering the ICU patients were assigned to a consecutive study number, the application of a blinded drug regimen was started" "...the drugs were prepared in advance by a staff who were not involved in the treatment of patients and identified by the above mentioned consecutive study number i.e. the code was located exclusively with this group during the trial'' Comment: Randomisation code was used exclusively with staff who were not involved in the treatment of patients, and these staff were responsible for preparing the 3 interventions through a similar mechanism (dissolving each in 100 mL of 5% glucose) to avoid detection during treatment |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Blinding of drugs (placebo, ranitidine, pirenzipine) was achieved by dissolving them in glucose (100 mL; 5% Braun, Melsungen, Germany) resulting in an equal infusion volume for each patient. Drugs were prepared by staff who were not involved in the treatment of patients and identified by the above mentioned consecutive study number"; "All blinded drugs (only identifiable by their consecutive study number) were given to the patients by a staff nurse in the ICU" Comment: This was a placebo‐controlled nature of the study design in which the 3 interventions were prepared in a similar manner (dissolving each in 100 mL of 5% glucose) to ensure adequate blinding because of which the details of interventions received were not known. Participants and personnel were blinded, ensuring no performance bias |
| Blinding (detection bias) | Low risk | Quote: "Data documentation for each patient was performed prospectively and daily by a different staff not involved in the treatment of patients in the randomised assignment of the drugs" Comment: not clear whether they were involved in outcome assessment. Moreover, GI bleeding was detected as per the definition in the study protocol and was an objective outcome |
| Blinding (detection bias) | Low risk | Quote: "Data documentation for each patient was performed prospectively and daily by a different staff not involved in the treatment of patients in the randomised assignment of the drugs" Comment: not clear whether they were involved in outcome assessment. Moreover, pneumonia was detected as per the definition in the study protocol and was an objective outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Data documentation for each patient was performed prospectively and daily by a different staff not involved in the treatment of patients in the randomised assignment of the drugs" Comment: not clear whether they were involved in outcome assessment. However, outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | High risk | Comment: 741 patients randomised, but only 158 completed 48 hours of mechanical ventilation and were reported in the trial |
| Selective reporting (reporting bias) | Low risk | Comment: Data on all randomised participants were not included in the results. Only outcomes for participants who were on mechanical ventilation for longer than 48 hours are reported. Of 827 randomised patients, a total of 158 were mechanically ventilated for ≥ 48 hours. Data on the remaining 669 patients were not available. However, this does not account for reporting bias, as the study intended to report its primary outcome of pneumonia only for participants who completed ≥ 48 hours on mechanical ventilation. All intended outcomes were reported only for participants who spent ≥ 48 hours in the ICU |
| Other bias | Low risk | Comment: unclear on the source of funding. No other sources of bias suspected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 100 participants Number analysed: 100 participants Antacids
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "Fifty one of the 100 participants entered into the study were randomised to receive antacid prophylaxis. Six major risk factors were analysed for each participant in each group. These risk factors were respiratory failure, extra abdominal sepsis, peritonitis, jaundice, renal failure and hypotension. There were 110 risk factors in 51 participants in the antacid group, a mean of 2.2 risk factors per participant. There were 115 risk factors in 49 participants in the group not receiving antacid prophylaxis, a mean of 2.4 risk factors per participant. All participants in respiratory failure were treated with constant‐volume ventilators. The severity of respiratory failure was judged by the alveolar‐ atrial oxygen. The mean highest value was 363+/‐ 25 mm Hg (SEM) in the participants receiving antacid and 324+/‐ 29 mm Hg in those not receiving Antacid prophylaxis (p > 0.05). The difference in mean blood urea nitrogen values (96+/‐ 10 mm/dl in the Antacid group vs. 90+/‐ 10 mm Hg in those not receiving antacid prophylaxis) was not statistically significant between the two groups. The extent of jaundice was also similar in the two groups" Comment: Both groups were similar with respect to their baseline characteristics. The major risk factor was respiratory failure | |
| Interventions | Antacids
No prophylaxis
Adherence to regimen: Quote: "The study was ended when oral feedings were begun and the nasogastric tube was removed, or the participant was discharged from the intensive care unit" Comment: In 2 participant who had diarrhoea, antacid was stopped, and in 2 other participants, a different antacid was administered. In 1 participant from antacid group with elevated magnesium levels, antacid was changed to a preparation that did not contain magnesium Duration of trial: March 1972 to March 1977 Notes duration of treatment: Mean duration for the entire study of 100 participants was 2.9 ± 0.29 days (SEM). Study was terminated when oral feedings were begun and the nasogastric tube was removed, or when the participant was discharged from the ICU Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcome (outcomes are not categorised as primary and secondary, but from the study report we feel GI Bleeding was the primary outcome)
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: Respiratory‐Surgical Intensive Care Unit of the Beth Israel Hospital, 300 Brookline Avenue, Boston, MA, 02215 Source of funding: Quote: "we are indebted to Stuart Pharmaceuticals, Division of I.C.I United States, Inc., Wilmington, DE (manufacturers of Mylanta II), whose generous contribution in part made this study possible…" Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the Committee on Cliniccal Investigations, New procedures and new forums of therapy of the Beth Israel Hospital" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "…the patients were randomised by a table of random numbers" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information on allocation concealment reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering the study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | High risk | Comment: no clear definition for the diagnosis of upper GI bleeding or blinding of outcome assessors reported |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the analysis. Therefore there is no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported in the study |
| Other bias | Low risk | Quote: "we are indebted to Stuart Pharmaceuticals, Division of I.C.I United States ,Inc., Wilmington, DE (manufacturers of Mylanta II), whose generous contribution in part made this study possible…" Comment: The above mentioned organisation also manufactured the antacid used as intervention in this study, which is suggestive of potential conflict of interest. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 210 participants Number analysed: 210 participants Tepterone
Ranitidine
Rabeprazole
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "Prevalence of hypertension, diabetes, renal insufficiency (serum creatinine level of more than 2.0 mg/dl), and smoking history were similar. The rates of operative characteristics such as CABG, valve replacement, and aortic surgery were also similar. There were also no differences regarding CPB duration, postoperative maximum creatine kinase (CK)‐MB levels" Comment: Participants had undergone an open heart surgery | |
| Interventions | Tepterone
Ranitidine
Rabenprazole
Adherence to regimen: Quote: "There was hematemesis complications in 4 patients during postoperative days 4 and 6 in each of groups I and II. Otherwise, those with active ulcers were completely healed by PPI administered for 2 weeks, and confirmed by GFS. These patients required a blood transfusion and all had emergency GFS. In contrast, in group III, no patients developed upper GI bleeding" Notes concomitant medication: Quote: "Systemic heparinization (10,000 U/day) was delayed until the second postoperative day. Patients undergoing CABG or aortic surgery had aspirin (81 mg/day) and those with valve replacement were administered Warfarin (2–3 mg/day) from the first postoperative day. Even though there was no complaints in relation to the upper GI system, gastric fibroscopy was used in all patients postoperatively during days 5 to 7" Concomitant medications: as mentioned in concurrent medication Duration of trial: January 2001 to December 2003 Duration of treatment: ‐ Duration of follow‐up: not clearly mentioned in the study report. Most probably until discharge or death (although mortality is not reported) | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: Department of Cardiovascular Surgery, Nihon University School of Medicine, Tokyo, Japan Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The Ethics committee of Nihon University School of Medicine approved the current trial" Informed Consent: Quote: "All patients gave written informed consent" Clinical trials registration: ‐ Sample Size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: This was not a placebo‐controlled trial, and the study is unclear on blinding of study personnel and participants. Therefore, it is unclear on performance bias |
| Blinding (detection bias) | High risk | Comment: unclear on blinding of outcome assessors, and there was no clear definition for detecting clinically significant upper GI bleeding. Therefore high risk of performance and detection bias |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However owing to the objective nature of the outcome of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis. Therefore there is no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported in the study |
| Other bias | Low risk | Comment: unclear on the source of funding. No other bias detected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 120 participants Number analysed: 120 participants Naloxone
Naloxone + pantoprazole
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalances: no significant difference in primary disease, sex, age, and course of disease between study group and control group | |
| Interventions | Naloxene
Naloxene + pantoprazole
Adherence to regimen: ‐ Duration of trial: May 2009 to May 2014 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: ICU Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: Signed informed consent was required by all participants Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: insufficient information to allow judgment |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to allow judgment |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: insufficient information to allow judgment |
| Blinding (detection bias) | High risk | Comment: no details about criteria for diagnosis of upper GI bleeding or blinding of outcome assessors reported |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: insufficient information to allow judgement |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: insufficient information to allow judgement |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes reported in Methods section also reported in Results |
| Other bias | Unclear risk | Comment: insufficient information to allow judgement |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 86 participants Number analysed: 64 participants Sucralfate
Antacid
Inclusion criteria
Exclusion criteria
Baseline imbalances: None of the differences was statistically significant | |
| Interventions | Sucralfate
Antacid
Adherence to regimen: ‐ Duration of trial: April 1985 to March 1986 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcome sought in review but not reported in trial
Outcomes reported in trial, but not used in review
| |
| Notes | Setting: Medical Intensive Care Unit, Department of Medicine, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand Sponsorship source: Siam Pharmaceutical Company, Bangkok Ethics approval: ‐ Informed consent: Failure to receive informed consent is mentioned under exclusion criteria Clinical trials registration: ‐ Sample size calculation: ‐ Comments: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "108 subjects were divided into acute and chronic cases of respiratory failure and were allocated by means of a table of random numbers to receive either sucralfate or antacid" |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: no details about blinding reported, but lack of blinding unlikely to introduce bias to findings |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding (detection bias) | Low risk | Comment: GI bleeding was an objective outcome that was diagnosed as per the definition in the study protocol. Therefore the likelihood of performance or detection bias is low |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no details reported |
| Incomplete outcome data (attrition bias) | High risk | Comment: 16 patients were excluded from analysis in the sucralfate group and 6 from the antacid group, without a described reason |
| Selective reporting (reporting bias) | Low risk | Comment: no selective reporting of outcomes suspected; all outcomes listed in the Methods section and objectives are also reported in the Results section |
| Other bias | Low risk | Comment: funded by pharmaceutical industry. No other sources of bias suspected |
| Methods | Single‐center randomised placebo‐controlled trial | |
| Participants | Baseline characteristics Number randomised: 323 participants Number analysed: 287 participants Omeprazole
Famotidine
Sucralfate
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "At randomisation no statistically significant difference was found among the four groups in demographic and baseline physiological characters. The distribution of risk factors such as participants who had trauma, surgery or coagulopathy at ICU admission were also similar in all groups" Comment: no significant difference between the 2 groups with respect to demographic and baseline risk factors. Trauma was the main reason for admission. Coagulopaty was diagnosed in 31 participants at baseline (7, 10, 6, and 8 in each of the 4 groups, respectively) | |
| Interventions | Omeprazole
Famotidine
Sucralfate
Placebo
Notes for placebo intervention: A placebo group was included in the protocol design because at study initiation and during the study period, no drug was approved for prophylaxis of stress‐related upper gastrointestinal bleeding by the FDA. The European Medicines Evaluation Agency required a placebo comparison group for registration of studies in this indication Adherence to regimen: Quote: "Of the 323 randomised patients 36 were subsequently excluded from analysis (1 patient died suddenly within 2 hours after randomisation, 18 underwent mechanical ventilation < 48 hours and 16 were not assessable because of missing important data" Thus only 287 patients could be analysed. No drug‐related adverse events were seen during the whole study, possibly related adverse events were rare, and in no patient was discontinuation of therapy necessary. Duration of trial: February 2000 to June 2002 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Traumatological Hospital Brno, a Czech republic ministry of health teaching and research facility Source of funding: Quote: "The study was supported by a grant of IGA MZ CB ND 5932‐3/2000" Conflicts of interest: Ethics approval: Quote: "The study was approved by the Institutional Review Board and Grant agency at the Misnistry of Healthof Chez Republic" Informed consent: Quote: "participants not able or willing to give informed consent were eluded from the study" Clinical trials registration: not provided in the study report Sample size calculation: not mentioned in the study report Additional notes: GI bleeding occurred more commonly in participants with coagulopathies (3/31, 10% vs 4/245, 2%, P = 0.006). The most common pathogens in participants with pneumonia were Staphylococcus andStreptococcus. ICU stay and mortality were greater in participants with investigated complications of GI bleed and pneumonia but were not influenced by prophylaxis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to study group by means of computer generated random number table to generate a random treatment list" |
| Allocation concealment (selection bias) | Low risk | Quote: "Treatment regimens were included in opaque sealed numbered envelopes and the envelope with the lowest number was always used for the consecutive patient" Comment: method adopted to allocation concealment clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: "Blinding of the drug administered intravenously was achieved by dissolving them in 100 mL of saline solution drug were prepared by staff not involved in the study according to the randomisation. All blinded drugs were identifiable by their study number and were given to the patients by the study nurse" Comment: Blinding is mentioned only for groups that received intravenous medications (omeprazole, famotidine, and placebo), not sucralfate. Unclear whether this would have led to performance bias |
| Blinding (detection bias) | Low risk | Comment: mentioned that outcome assessment was done blinded to intervention. The presence of 1 or more macroscopic abnormalities (erythema or oedema, erosions, ulcerations, and naso‐gastric tube lesions) and GI bleeding was assessed, blinded to intervention, at endoscopy by a single experienced gastroenterologist Quote: "assessed, blinded to intervention, at endoscopy by a single experienced gastroenterologist" |
| Blinding (detection bias) | Low risk | Quote: "Chest radiographs were interpreted by two independent radiologists" Comment: Blinding of outcome assessors was done. Pneumonia was an objective outcome detected as per the study protocol |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Of the 323 randomised patients, 36 were subsequently excluded from analysis. Groups to which they were initially randomised remain unclear though. Thus only 287 participants were available for analysis. However, the number of dropouts and their characteristics were comparable in all 4 arms. Therefore there is no likelihood of risk of attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported in the study |
| Other bias | Low risk | Comment: Study was funded by a grant of IGA MZ CB ND 5932‐3/2000. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias were detected |
| Methods | Parallel‐group quasi‐randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 104 participants Number analysed: 104 participants Sucralfate
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 groups were largely similar according to demographic characteristics, underlying diseases, and diagnoses at admission. However, there were slight imbalances with regard to age, gender, polytrauma, and acute respiratory failure | |
| Interventions | Sucralfate
Cimetidine
Adherence to treatment: ‐ Duration of trial: May 1986 to January 1989 Duration of follow‐up (days): ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought in review, but not reported in trial
Outcomes reported in trial but not sought in review
| |
| Notes | Setting: ICU, Department of Hospital Epidemiology, Medical Biometry and Anaesthesiology, University Hospital Freiburg, Germany Sponsorship source: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Comment: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "and tube‐fed patients were excluded. Patients were assigned to stress ulcer prophylaxis with either sucralfate or cimetidine by setting up groups of 10 patients who received one prophylactic regimen followed by the next 10 patients receiving the other regimen. Patients received either intravenous" Judgement comment: quasi‐randomisation. Patients were assigned to stress ulcer prophylaxis with either sucralfate or cimetidine by setting up groups of 10 patients who received 1 prophylactic regimen followed by the next 10 patients receiving the other regimen |
| Allocation concealment (selection bias) | Unclear risk | Judgement comment: Provided the sequence was consecutive, patients were known in each group |
| Blinding of participants and personnel (performance bias) | Low risk | Judgement comment: no blinding reported, but lack of blinding is unlikely to introduce bias. Outcome measures and outcomes are objectively measured |
| Blinding (detection bias) | Low risk | Quote: "Macroscopically visible blood in gastric aspirates was taken for evidence of stress bleeding" Comment: outcome measured objectively |
| Blinding (detection bias) | Low risk | Comment: criteria for the diagnosis of pneumonia clearly reported in the trial |
| Blinding of outcome assessment (detection bias) | High risk | Quote: "The chest x‐ray was interpreted by a radiologist who was not aware of the particular prophylactic regimen" Judgement comment: but investigators and personnel were unblinded for other criteria for pneumonia and for other outcomes |
| Incomplete outcome data (attrition bias) | Low risk | Judgement comment: All outcomes have included all patients. No incomplete reporting of data suspected. Attrition was low and was reported transparently |
| Selective reporting (reporting bias) | Low risk | Judgement comment: no incomplete reporting of outcomes suspected. All outcomes listed in the Methods section are also reported in the Results section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
| Methods | Multi‐centre double‐blind randomised placebo‐controlled trial | |
| Participants | Baseline characteristics Number randomised: 87 participants Number analysed: 87 participants Cimetidine
Placebo
Inclusion criteria Participants admitted to ICUs were eligible for entry into the trial if they had at least
Exclusion criteria
Baseline imbalances: Quote: "Patient demographics and risk factors for bleeding on entry into the study show that the treatment groups were comparable" | |
| Interventions | Cimetidine
Placebo
Adherence to regimen: Quote: "The average time in the study was longer for patients treated with cimetidine (mean, 83 hours ± 53) than for patients treated with placebo (mean, 53 hours ± 41). This difference was because more patients treated with placebo bled and left the study early" "Two patients being treated with cimetidine experienced adverse events for which they were withdrawn from the study: one developed moderate nausea and vomiting and one developed a change in mental status. In both cases, the adverse experiences were reversible. No patients treated with placebo were withdrawn from treatment, but adverse experiences attributed to placebo treatment included one case of mental confusion" Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: The study was a multi‐centric study involving 14 centres; University of Kentucky, Lexington, KY; Boston City Hospital, Boston, King Drew Medical Center, Los Angeles, CA; Winthrop University Hospital, Mineola, NY; Ellis Hospital, Schenectady, NY; Detroit Rec. Hospital, Detroit, MI; Mt. Sinai Medical Center, New York, Hartford Hospital, Hartford, CT; St. Thomas Hospital, Akron, OH; Cleveland Metro General Hospital, Cleveland, OH; University of Oklahoma Health Science Center, Oklahoma City, OK; Ravenswood Hospital, Chicago, IL; Brookdale Hospital, Brooklyn, NY; University of Texas Health Science Center, San Antonio, TX Source of funding: Study appears to have been funded by Smith Kline & French Laboratories, Philadelphia, PA, although this is not clearly mentioned in the study report Conflicts of interest: Ethics approval: Quote: "The study was approved by all institutional review boards" Comment: This study was approved by the ethics committees at all 14 centres Informed consent: Quote: " ...and all patients (or their legal guardians) provided written, informed consent" Comment: For participating in the trial, informed consent was obtained from participants or their legal guardians Clinical trials registration: not provided in the study report Sample size calculation: not clearly mentioned in the study report Additional Notes:Staphylococcus aureus was the organism found to have caused pneumonia. One participant treated with cimetidine had melena, but the upper GI source could not be detected, so this participant was not categorised as having an upper GI bleed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not enough information about sequence generation reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information about allocation concealment reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: This was a placebo‐controlled trial; unclear about whether participants and study personnel were blinded |
| Blinding (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded. GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) | High risk | Comment: Outcome assessors were not blinded. Study had no clear mention of the definition to detect pneumonia. Moreover 14% of participants had pneumonia on entry into the study. Lack of blinding could have influenced detection of this condition, as it is mentioned that the physician did not attribute pneumonia to the study medication |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: Outcomes of interest were in part objective in nature; for other outcomes, the definition is not described and no blinding is in place |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "All patients who received study drugs were included in the denominator for analysis" Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Lead author is affiliated with Smith Kline & French Laboratories, Philadelphia, PA. Clear conflict of interest. No other sources of bias suspected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 75 participants Number analysed: 75 participants Famotidine
Sucralfate
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: Age, gender, and clinical features of participants in the 3 groups were comparable. Participants were people who required ICU care after head injury (mainly because if road side accident) | |
| Interventions | Famotidine
Sucralfate
No prophylaxis
Adherence to regimen: Quote: "Out of 56 patients who had upper GI bleeding, 39 started bleeding within 3 days of head injury" Comment: Only 1 patient in the famotidine group required escape treatment as compared with 3 in the sucralfate group and 8 in the no prophylaxis group. The exact reason for escape of treatment is not mentioned but could be attributed to untimely death or upper GI bleed that was clinically significant as per the study protocol. The 56 participants with upper GI bleed are inclusive of all grades of bleeding according to the study protocol. Interventions were discontinued if at least 3 consecutive positive occult blood readings were detected Duration of trial: ‐ Duration of follow‐up: 15 days | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Note: Among the 56 participants who bled, 39 of them started to bleed within 3 days of head injury. Data on occult bleeding were not used in the review Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Neurosurgery Unit of Medical College and Hospital, Ludhiana, India Source of funding: ‐ Ethics approval: Quote: "After approval from Hospital Ethics Committee, the study was conducted in 75 consecutive patients" Comment: The ethics committee of the concerned institution had approved the study Informed consent: Quote: "Since informed consent could not be taken directly from patients, it was taken from the next of kin" Comment: Informed consent was obtained Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: According to the study report, data on endoscopy could not be analysed because of small numbers of participants who underwent the procedure. Occult bleeding occurred in 38 participants (no prophylaxis: 11, famotidine: 12, and sucralfate: 15) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and 1 arm received no prophylaxis; this would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded. However GI bleeding was an objective outcome that was detected as per the definition in the study |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded. All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis in the study report |
| Selective reporting (reporting bias) | Low risk | Comment: The intended outcome of GI bleeding was reported in the study report |
| Other bias | Low risk | Comment: unclear on the source of funding. No other form of bias detected |
| Methods | Parallel‐group quasi‐randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 148 participants Number analysed: 137 participants Pantoprazole
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalance: ‐ | |
| Interventions | Pantoprazole
Sucralfate
Adherence to regimen: ‐ Duration of trial: early 2010 to mid‐2011 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: ICU, Infectious Diseases and Tropical Medicine Research Center, Isfahan University of Medical Sciences, Isfahan, Iran Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Study was approved by the medical universities ethics committee at Isfahan University of Medical Sciences Informed consent: Quote: "after they agreed to cooperate" Comment: included in the trial report Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Patients were consecutively assigned to each group. The first patient was selected randomly" Comment: no randomisation, quasi‐randomised study |
| Allocation concealment (selection bias) | High risk | Comment: consecutive pattern of allocation |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: no blinding reported, but lack of blinding is unlikely to introduce bias to outcome measures or outcomes |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding (detection bias) | Low risk | Quote: "Ventilator‑associated pneumonia is defined as a group of pneumonias that occur 48 hours after the patient is ventilated if the patient did not have primary signs of the infection at the time of arriving ICU. VAP is one of the most prevalent nosocomial infections and pneumonia is causes 27% of infections in ICU" Comment: Criteria for diagnosis of the outcome were reported objectively |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no blinding of outcome assessors described |
| Incomplete outcome data (attrition bias) | High risk | Comment: "11 patients were excluded for different reasons like not using the drug, changing the drug by the physician, and so on. Three of them were from the pantoprazole and 8 of them were from sucralfate group" |
| Selective reporting (reporting bias) | Unclear risk | Comment: no outcomes of interest described in the Methods section; assessment of selective outcome reporting difficult |
| Other bias | Low risk | Comment: no other sources of bias suspected |
| Methods | Randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 263 participants Number analysed: 249 participants Antacids
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The patients were randomised to the four different groups were fairly homogenous and were exposed to essentially the same risk factors" Comment: no clear mention of gender, age, and type of risk factor distribution in the study report. In all participants, a variety of illness classified II or greater by therapeutic intervention scoring system (TISS) were diagnosed | |
| Interventions | Antacids bolus
Antacids continuous
Cimetidine bolus
Cimetidine continuous
Adherence to regimen: Quote: "...14 patients initially admitted to the study were excluded for failure to follow protocol", "Any patient who developed severe diarrhoea was given alternagel instead of Mylanta" Comments: not sure which groups the 14 participants belonged to. Nine participants needed to be switched to alternagel Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Hurley Medical Centre, Michigan, USA Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Mean blood transfusion due to gastrointestinal haemorrhage was 1335 mL (range: 0 to 4500 mL). This is not mentioned separately for both groups. Irreversible haemorrhagic shock was the cause of death in 2 cimetidine participants | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: Given the mode of administration and dosing regimens, it would not have been possible to blind study personnel. Therefore unclear whether this would have caused any performance bias |
| Blinding (detection bias) | Low risk | Comment: unclear whether the outcome assessor was blinded. GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "...14 patients initially admitted to the study were excluded for failure to follow protocol" Comment: unclear on which groups the 14 participants were initially randomised to. But this accounts to be around 5% of total participants and is uniformly distributed across groups |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on the source of funding. No additional biases suspected |
| Methods | Randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 401 participants Number analysed: 298 participants Overall
Inclusion criteria
Exclusion criteria
Baseline imbalances: Most participants were referred for cardiac surgical treatment. Groups were similar in demographic characteristics such as age and race. The sucralfate group had the fewest women. Ten participants assigned to receive meciadanol had a history of peptic ulcer disease (3 with a history of bleeding); 14 with sucralfate had a history of peptic ulcer disease (5 with a history of bleeding), and 13 assigned to receive antacid had a history of peptic ulcer disease (4 with bleeding) | |
| Interventions | Bioflavonoid (Maciadanol)
Sucralfate
Antacids (Maalox, magnesium aluminium hydroxide gel)
Adherence to regimen: Overall 401 patients had entered the study and 298 had completed the study. Those who did not complete the study could not be evaluated or were excluded because of protocol error (receiving other antacids or antiulcer drugs), by physician request, or by their own request. Participants who remained in SICU for less than 24 hours were also excluded from the study. 85 participants completed the maciadanol arm, 100 the sucralfate arm, and 113 the antacid arm Duration of trial: 16 months Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Surgical Intensive Care Unit of Johns Hopkins Medical Institutions, Baltimore, and Francis Scott Key Medical Centre, Baltimore Source of funding: Zyma, S. A., Nyon, Switzerland Conflicts of interest: Ethics approval: Quote: "The study was approved by the institutional review board of Johns Hopkins Medical Center and the Francis Scott Key Medical Center" Informed consent: mentioned in the study report Clinical trials registration: ‐ Sample size calculation: Quote: "The power calculations resulting from study sample size were one tailed with an alpha of 0.05 and a power of 80.0 per cent..." | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomised by a table of random numbers..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering the study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Quote: "All data were collected by an observer unaware of the results" Comment: not clear how this would have contributed to blinding. GI bleeding was detected as per the definition in the study. However, owing to the objective nature of the outcome of interest, the likelihood of performance and detection bias is low |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "All data were collected by an observer unaware of the results" Comment: unclear on blinding of outcome assessors. However, all other outcome data were objective in nature |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: Overall 401 patients had entered the study and 298 had completed the study. Study did a per‐protocol analysis for outcomes. Participants in the Maciadanol arm were 24% less than in the antacid arm. Unclear whether this could have influenced outcomes |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported in the study report |
| Other bias | Low risk | Comment: Source of funding is mentioned. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
| Methods | Double‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 30 participants Number analysed: 30 participants Overall
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "... groups were comparable in terms of age, gender and degree of consciousness" Comment: Participants were people who were admitted to the neurosurgical ICU | |
| Interventions | Ranitidine + placebo
Ranitidine + pirenzepine
Adherence to regimen: no dropouts. There does not seem to be any change in dosage, nor were are any adverse events mentioned Duration of trial: ‐ Duration of follow‐up: no information given. It seems that there was no further follow‐up after the end of the study | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Neurosurgical ICU, Cologne, Germany Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent : ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: Study mentions that it is double‐blinded, but it does not describe clearly who was blinded or how this was done. However, because this is a placebo‐controlled trial, we assume that participants and study personnel were the ones blinded |
| Blinding (detection bias) | Low risk | Comment: Study mentions that it is double‐blinded, but it does not describe clearly who was blinded or how this was done. Moreover GI bleeding was an objective outcome that was detected as per the study definition |
| Blinding (detection bias) | High risk | Comment: Study mentions that it is double‐blinded, but it does not describe clearly who was blinded or how this was done. The definition for VAP was not clearly mentioned in the study report |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: Study mentions that it is double‐blinded, but it does not describe clearly who was blinded or how this was done. Moreover outcomes were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on the source of funding. No additional biases were suspected |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 53 participants Number analysed: 53 participants Ranitidine
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 groups were comparable with respect to gestational age, birth weight, Apgar score, cord blood pH, and gender. Participants were preterm and full‐term infants | |
| Interventions | Ranitidine
No prophylaxis
Adherence to regimen: Of 53 participants who were randomised, only 48 were analysed as there were 5 dropouts due to early death (2 participants at gestational age < 33 weeks and 1 participant at gestational age ≥ 33 weeks) and oesophageal atresia (n = 2). 3 infants belonged to the ranitidine group and 2 belonged to the control group Duration of trial: 10‐Month period Duration of treatment : 4 days; study was discontinued when significant results of the first block of 20 preterm infants randomised were available for interpretation Duration of follow‐up: Routine follow‐ups were made at 7 days, then afterwards weekly up to 4 weeks | |
| Outcomes | Outcomes sought in review and reported in trial
Note: The intervention was given for 4 days only, whereas endoscopic findings were done at 3 and 6 days (even after treatment), the results of which could not be extracted separately
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Department of Neonatal Intensive Care, University Hospital, Tampere, Finland Source of funding: Quote: "Supported in part by the Finnish Foundation of Pediatric Research" Conflicts of interest: ‐ Ethics approval: Quote: "The study protocol was approved by the Ethics Committee of the Tampere University Hospital" Informed consent: Quote: "The parents received both oral and written information on the study; their informed consent was obtained" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Randomisation of participants was performed after infants were stratified into 2 groups (infants at less than and more than 33 weeks' gestational age) because infants usually less than 33 weeks are ventilated more often. Thus balance was achieved with respect to gestational age for both groups. Study was discontinued after significant results for the first 20 preterm infants (the first block) were available.When the study was stopped, results for a second block of 10 preterm infants and a third block of 7 preterm infants were available, along with results for a block of 16 and 20 mature infants | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The first randomisation block included 20 envelopes in random order for both groups (gestational age < 33 weeks and gestational age ≥ 33 weeks). Followed by two randomisation blocks of 10 envelops for more preterm infants. Babies closer to term were randomised in one block with 20 envelops" |
| Allocation concealment (selection bias) | Low risk | Quote: "The first randomisation block included 20 envelopes in random order for both groups (gestational age < 33 weeks and gestational age ≥ 33 weeks). Followed by two randomisation blocks of 10 envelops for more preterm infants. Babies closer to term were randomised in one block with 20 envelops" Comment: Method adopted to conceal allocation is clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Only the sequence number of the patient were written in the patients records by the nurse responsible for medication; thus the attending physicians the endoscopist and the pathologist remained blinded as to the treatment group" Comment: Study personnel were blinded to treatments. Moreover outcomes of interest were objective in nature |
| Blinding (detection bias) | Low risk | Quote: "Only the sequence number of the patient [was] written in the patient records by the nurse responsible for medication; thus the attending physicians, the endoscopist, and the pathologist remained blinded as to the treatment group" Comment: The endoscopist was blinded to whether or not the participant received prophylaxis. Moreover the outcome measured was objective in nature |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Only the sequence number of the patient [was] written in the patient records by the nurse responsible for medication; thus the attending physicians, the endoscopist, and the pathologist remained blinded as to the treatment group" Comment: Outcome assessors were blinded. Moreover owing to the objective nature of the outcomes of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 5 babies were not part of the analysis (3 in treatment and 2 in control groups). A sensitivity analysis was done in which it was assumed that the 3 dropout neonates randomised to the treatment group had mucosal abnormalities, and the 2 dropout neonates randomised to the control group had no mucosal abnormalities. Results of this assumption still indicate that ranitidine was effective in preventing gastric mucosal lesions in infants under stress |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported and analysed |
| Other bias | Low risk | Comment: The Finnish Foundation of Pediatric Research funded the study. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 40 participants Number analysed: 38 participants Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Mechanical ventilation was used in 2 times more patients in the placebo group | |
| Interventions | Cimetidine
Placebo
Adherence to treatment: ‐ Duration of trial: ‐ Duration of follow‐up (days): ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcome sought in review and not reported in trial
Outcomes reported in trial and not sought in review
| |
| Notes | Setting: PICU, Department of Pediatrics, Pediatric intensive Care Unit, Hopital Sainte‐Justine, University of Montreal, Canada Sponsorship source: Smith Kline & French Laboratories Conflicts of interest: ‐ Ethics approval: Study was approved by the Ethics Committee of Hopital Sainte‐Justine Informed consent: Written informed consent was obtained from a parent or guardian of each patient Clinical trials registration: ‐ Sample size calculation: We calculated a necessary sample size of 37 participants | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Allocation of treatment followed a double‐blind pattern, according to a list set up by the Smith Kline & French Laboratories from a table of random numbers" Judgement comment: list of random number set up by study sponsor. Some risk of bias suspected |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Allocation of treatment followed a double‐blind pattern, according to a list set up by the Smith Kline & French Laboratories from a table of random numbers" Judgement comment: no details about allocation concealment reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: "double‐blind pattern". Procedures for blinding not explicitly reported, but lack of blinding is unlikely to introduce bias to the outcomes or outcome measures |
| Blinding (detection bias) | Low risk | Quote: "UGIB noted from the nasogastric tube, which had been inserted in each patient. Outcome was assessed by the appearance of UGIB and by gastric fluid pH. Massive UGIB was defined as red or brown haemorrhage from the nasogastric tube associated with a decrease in arterial blood pressure of >20 mm Hg or with an acute decrease in haemoglobin level of >2 gm/dl; obvious UGIB was defined as red or brown haemorrhage from the nasogastric tube without a decrease in arterial blood pressure or in haemoglobin level of >2 gm/dl; slight UGIB was defined as minimal bleeding appearing only at the aspiration of gastric fluid" Comment: criteria for diagnosis of this outcome reported objectively |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no details about blinding of outcome assessors reported |
| Incomplete outcome data (attrition bias) | Low risk | Comment: no incomplete outcome data suspected. All participants randomised at baseline were included in analyses |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes listed in the Methods section (GI bleeding) were also reported in the Results section |
| Other bias | High risk | Comment: Industry participates in providing drugs and conducting the trial. Mechanical ventilation (risk factor for GI bleeding) was used more often in placebo group |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 84 participants Number analysed: 84 participants Ranitidine
Sucralfate
Inclusion criteria
Notes: not clear whether any at all, 1, or more of these risk factors is required to be classified as high risk Exclusion criteria
Baseline imbalance: Both groups were comparable with respect to age, underlying disorders, and factors predisposing to development of stress ulcers | |
| Interventions | Ranitidine
Sucralfate
Adherence to regimen: no dropouts mentioned; however, after bleeding was detected, 1 participant in the sucralfate group was switched over to ranitidine. 2 individuals in the ranitidine group who had bleeding were switched to the sucralfate group, 1 to pirenzepine; in 3, enteral feeding was started Duration of trial: 18 months Duration of treatment: until participant was discharged from ICU or when acute problem improved, so that bleeding prophylaxis was no longer deemed necessary Duration of follow‐up: until patient was discharged from ICU or prophylaxis was no longer required (unclear how this was determined) | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: ICU Medical University Hospital, Wien, Austria Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Positive bacterial cultures were obtained from bronchial secretions of (43.3% of 74 bronchial secretions in participants receiving ranitidine and 18.6% of 59 bronchial secretions in those receiving sucralfate). Only 30 participants in the ranitidine group and 25 participants in the sucralfate group needed mechanical ventilation. The duration of this was 8.6 ± 4.6 in the ranitidine group and 8.9 ± 6.1 in the sucralfate group | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: GI bleeding was detected as per the definition in the study protocol, and the the nature of the outcome of interest is objective |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature, so the likelihood of performance bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants completed the trial and were included in the final analysis. No treatment withdrawals and no trial group changes |
| Selective reporting (reporting bias) | High risk | Quote: "There was no difference in side effects between the two medication groups" Comment: Adverse reactions due to the interventions are not clearly mentioned in the study report. This would have caused reporting bias in the study report. All other intended outcomes are reported |
| Other bias | Low risk | Comment: no mention of the source of funding. No additional biases were detected |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 32 participants Number analysed: 32 participants Sucralfate
Ranitidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "Male: female ratios and mean age of the two treatment groups appear not quite similar,but we could not detect any significant difference" Comment: All participants were long‐term ventilated persons | |
| Interventions | Sucralfate
Ranitidine
Adherence to regimen: ‐ Duration of trial: ‐ Note duration of treatment: Quote: "The study was terminated when the patient was dismissed from the intensive care unit or died" Duration of follow‐up: not clearly mentioned in the study report | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Department of Medicine, University of Vienna, Austria Source of funding: ‐ Conflicts of interest: Ethics approval: Quote: "The study was approved by the local ethical committee" Informed consent: Quote: "...and informed consent was obtained from the next of kin after the potential complications of the procedures were explained" Comment: Consent was obtained Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Quote: "During mechanical ventilation, upper gastrointestinal bleeding developed in three sucralfate‐ and seven ranitidine‐treated patients (18.7 versus 43.7 percent, p < 0.05). Until the end of the study, bleeding developed in only three sucralfate‐ but nine ranitidine treated patients (18.7 versus 56.2 percent, p ˜ 0.05)" Comment: Data for 9 participants (in ranitidine) were taken for analysis because it was felt that this occurred when participants were still in the ICU. One death in each group was attributed to GI bleed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: ":.. 32 mechanically ventilated patients were randomly assigned, according to a preset table, to receive either ranitidine or sucralfate" Comment: not clear on how the sequence was generated in the "preset table" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial. Moreover the different modes of administering study drug would not have made it possible to blind participants. Therefore the likelihood of performance bias is high |
| Blinding (detection bias) | High risk | Comment: unclear whether outcome assessors were blinded to this outcome. The definition of this outcome was not clearly described in the study report |
| Blinding (detection bias) | Low risk | Quote: ”Analysis of chest radiographs was performed by a radiologist, who was not aware of the radiologist, who was not aware of the clinical status or the therapy of the patients” Comment: Outcome assessors for nosocomial pneumonia were blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded to other outcomes of interest. However, outcomes were objective in nature. Therefore the likelihood of performance and detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported in the study |
| Other bias | Low risk | Comment: unclear on the source of funding. No other sources of bias suspected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 57 participants Number analysed: 57 participants Cimetidine
Famotidine
Ranitidine
Antacids
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There were no statistically significant difference in age distribution or sex distribution among the four treatment groups" Comment: all participants about to undergo elective coronary artery bypass graft surgery | |
| Interventions | Cimetidine
Famotidine
Ranitidine
Antacids (Mylanta II)
Adherence to regimen: All participants received the drugs at least 12 hours before surgery and continued to receive them postoperatively. All participants used NG tube postoperatively Duration of trial: ‐ Duration of follow‐up: until discharge from hospital | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: cardiac surgery at the Medical Centre of Delware Adherence to regimen: ‐ Source of funding: ‐ Ethics approval: ‐ Informed consent: Quote: "A consent form was signed by all patient before surgery" Comment: Consent was obtained Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: H2 receptor antagonists were combined to form a common interventional arm vs antacids, as the review did not aim to investigate intraclass effects among included interventions. Blood transfusions were done but were not attributed to GI bleeding | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: All other outcomes were objective in nature, which was detected as per the definition in the study protocol. Therefore the likelihood of performance and detection bias is low |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: All randomised participants were accounted in the final analysis |
| Selective reporting (reporting bias) | Unclear risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on source of funding. No other known form of bias detected |
| Methods | Double‐blind placebo‐controlled trial | |
| Participants | Baseline characteristics Number randomised: 31 participants Number analysed: 31 participants Ranitidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Comment: All participants are admitted with severe head injury | |
| Interventions | Ranitidine
Placebo
Adherence to regimen: Comment: 1 participant died owing to the underlying cause of severe head injury on day 2, 1 participant removed pH electrode on day 3, and 5 participants in the placebo group had upper GI bleeding Duration of trial: ‐ Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: University of Louisville, Louisville, KY Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: This was a double‐blind placebo‐controlled trial. Therefore the likelihood of performance bias is low |
| Blinding (detection bias) | Low risk | Comment: not clearly mentioned in the study report. GI bleeding was an objective outcome that was detected as per the definition in the study |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: not clearly mentioned in the study report. However, the outcome was an objective outcome. Therefore, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | High risk | Comment: unclear on source of funding and baseline characteristics of participants |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 60 participants Number analysed: 60 participants Esomeprazole
Famotidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: no significant difference for sex, age, GCS, AP‐II, intracranial pressure, and operation time between these 2 groups | |
| Interventions | Esomeprazole
Famotidine
Adherence to regimen: ‐ Duration of trial: March 2007 to March 2010 Duration of follow‐up: 30 days | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial
Outcome reported in trial but not used in review
| |
| Notes | Setting: Neurosurgical ICU, Division of Gastroenterology and Hepatology, Department of Internal Medicine, Far Eastern Memorial Hospital, New Taipei, Taiwan Source of funding: Quote: "This study was performed in the Far Eastern Memorial Hospital and was supported by the research grant FEMH‐94‐C‐016 from the Far Eastern Memorial Hospital" Conflicts of interest: Quote: "All contributing authors declare no conflicts of interest" Ethics approval: Quote: "This study was approved by the Research Ethics Review Committee of the Far Eastern Memorial Hospital" Informed consent: Quote: "After explaining the study purpose and obtaining written consent from their family members" Clinical trials registration: ‐ Sample size calculation: ‐ Sponsorship source: supported by the research grant FEMH‐94‐ C‐016 from the Far Eastern Memorial Hospital | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were randomly allocated to two groups" Comment: not enough details reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: no details on blinding reported, but lack of blinding is unlikely to introduce bias to the outcomes or outcome measures |
| Blinding (detection bias) | Low risk | Quote: "[We] defined upper gastrointestinal bleeding as tarry stool, haematemesis, drainage of more than 60 mL coffee Comment: Outcome was measured objectively |
| Blinding (detection bias) | Low risk | Quote: "We defined ventilator‐associated pneumonia as pneumonia occurring after 48 hours of ventilator use that fulfils three or more of the following four criteria: (1) presence of persistent (> 48 hours) or new onset infiltration in CXR; (2) positive sputum smear (with < 10 epithelial cells per low‐power field, 100×, and > 25 white blood cells per low‐power field or presence of polymorphonuclear cells with phagocytosis); (3) fever with body temperature > 38.3°C; and (4) leukocytosis > 12 × 10⁹/L" Comment: Outcome was measured objectively |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no details reported |
| Incomplete outcome data (attrition bias) | Low risk | Comment: no incomplete outcome data; all participants randomised at baseline were included in analyses |
| Selective reporting (reporting bias) | Low risk | Comment: no incomplete reporting of outcomes suspected. All outcomes listed in the Methods section were also reported in the Results section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 70 participants Number analysed: 67 participants Ranitidine
Omeprazole
Inclusion criteria
Acute Physiologic and Chronic Health Evaluation (APACHE II) scores were calculated at baseline Exclusion criteria
Baseline imbalances: Quote: "There were no statistically significant differences between the ranitidine‐treated and the omeprazole‐treated groups for age, gender, race, or APACHE II scores. Ranitidine subjects had significantly more risk factors at baseline. There were no significant differences in the number of patients who required mechanical ventilation: ranitidine, 26/35 (72%) and omeprazole 16/32 (50%)" Comment: More participants in the ranitidine group had major neurological insults or trauma. 7 and 5 participants in both groups had coagulopathy at baseline | |
| Interventions | Ranitidine
Omeprazole
Adherence to regimen: "Seventy patients formed the study group. Thirty five were randomised to the ranitidine treatment group, 35 received omeprazole. Three omeprazole subjects were excluded from data analysis because of errors in randomisation or enrolment criteria protocol violations, resulting in 32 omeprazole‐treated patients included in the final analysis" Duration of trial: over a 10‐month period Duration of follow up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: New Hanover Regional Medical Center and the Coastal AHEC, Wilmington, North Carolina, and Department of Medicine, School of Medicine, University of North Carolina at Chapel Hill Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "This study was approved by the Institutional Review Board" Informed consent: Quote: "...informed consent was obtained from each patient or their legally authorized representative" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Mortality was related to increased APACHE II scores. Endoscopy was performed on 27 participants (ranitidine 15 and omeprazole 15) and stress ulcers were detected in all but 2 participants (from each group). The source of bleeding in these participants could not be determined, but it was presumed to be due to stress ulcerations | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. GI bleeding was detected as per the definition in the study protocol |
| Blinding (detection bias) | High risk | Comment: unclear on blinding of outcome assessors. The definition of nosocomial pneumonia is not clearly mentioned in the study report |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However, owing to the objective nature of the outcomes of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comments: Three participants (from the omeprazole group) were not part of the final analysis for legitimate reasons such as errors in randomisation or enrolment criteria protocol violations. However, we did an intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | High risk | Comment: Source of funding was not clearly mentioned in the study report. Baseline differences between groups were detected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 120 participants Number analysed: 120 participants Lansoprazole
Other medications
Inclusion criteria
Exclusion criteria
Baseline imbalances: no difference in gender, age, and mean number of comorbidities between the 2 groups. Use of ulcerogenic medications was similar between the 2 groups. There was no significant difference between the 2 groups in GCS scores, initial APACHE II score in the medical or surgical ICUs, ventilator‐dependent days before starting to be weaned, rapid shallow index (a weaning parameter), and baseline albumin and haemoglobin levels before weaning | |
| Interventions | Lansoprazole
Other medications
Adherence to treatment: ‐ Duration of trial: June 2009 ‐ February 2012 Duration of follow‐up: 30 days | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Respiratory Care Center, Division of Gastroenterology and Hepatology, Far Eastern Memorial Hospital, New Taipei City, Taiwan Sponsorship source: Quote: "This study was supported by a grant from Far Eastern Memorial Hospital (FEMH) (FEMH‐2008‐C‐043)" Conflicts of interest: ‐ Ethics approval: Quote: "The protocol was approved by the Research Ethics Review Committee of the Far Eastern Memorial Hospital" Informed consent: Quote: "After obtaining written consent from their families..." Clinical trials registration: ClinicalTrials.gov ID: NCT00708149, Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "They were randomly allocated into two groups using blocked randomisation" |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details reported. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: "non‐double‐blind" Comment: Performance bias is a possibility, even if outcomes are objective |
| Blinding (detection bias) | Low risk | Comment: outcome measured objectively as defined in the trial report |
| Blinding (detection bias) | Low risk | Comment: outcome measured objectively as defined in the trial report |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote: "randomised, non‐double‐blind" Comment: no blinding in place, lack of blinding unlikely to influence outcome measures and outcomes |
| Incomplete outcome data (attrition bias) | Low risk | Comment: no incomplete reporting suspected; all participants randomised at baseline are also included in analyses |
| Selective reporting (reporting bias) | Low risk | Quote: "(ClinicalTrials.gov ID: NCT00708149). Outcome measures. The primary end point of our study was apparent UGI bleeding 2,20 within 2 weeks of enrolment, which was defined as follows: (1) a 'coffee ground' substance from the NG aspirate 60 mL; (2) fresh blood from the NG tube; or (3) passage of tarry stool. Secondary end points included clinically significant UGI bleeding 2,20 (definition: UGI bleeding with haemoglobin level decrease !2 gm/dL or in need of a blood transfusion of > 2 units), successful weaning rate, ventilator‐associated pneumonia (definition: clinical pulmonary infection score 21 > 6, a scoring system composed of body temperature, white blood cell count, purulent secretions, diffuse or localized pulmonary infiltrate in CXR, progression of infiltrate, and culture of secretions), and a 30‐day survival rate" Comment: All outcomes reported in the Methods section are also reported in the Results |
| Other bias | Low risk | Comment: no other sources of bias suspected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 165 participants Number analysed: 140 participants Overall
Inclusion criteria
Exclusion criteria
Baseline imbalances: Groups were similar when compared using indexes (Theraputic Interventiobn Scoring System, Physiological Stability Index, Multi Organ System Failure Scoring System, Zinner and Tryba). Detailed baseline characteristics for each group are not provided in the study report | |
| Interventions | No prophylaxis
Almagate
Ranitidine
Sucralfate
Adherence to regimen: 165 randomised, 25 "excluded because of protocol reasons"; 1 patient in the Amalgamate group had watery diarrhoea and treatment was switched to ranitidine. In 6 patients from the control group (no prophylaxis) who developed GI bleed, 2 were given Amalgamate, 2 were given sucralfate, and 2 were given amalgamate Duration of trial: June 1986 to June 1988 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought in review but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Pediatric ICU, LaPaz Children’s Hospital, Madrid, Spain Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional review board" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: One of the 7 participants in the control group with GI bleeding did not receive any treatment; the others were given the following: amalgamate to 2 participants, ranitidine to 2 participants, and sucralfate to 2 participants. The last participant needed ranitidine too to decrease the intensity of bleeding. In the antacid group, 1 child died of haemorrhage, and the other was given ranitidine and sucralfate to contain the bleeding. In the ranitidine group, 1 participant improved within the first 24 hours, without receiving any other drug. In the second participant, the intensity of bleeding decreased when amalgamate was added, and the third participant died. In the sucralfate group, the statue of the lone participant with haemorrhage improved without any addition of another drug It is mentioned that there was no incidence of nosocomial pneumonia, but under "side effects", the study reports 5 incidences of the same and goes on to say that there was a difference between groups with respect to this outcome. The incidence in each group remains unclear | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: no blinding of outcome assessors reported, but GI bleeding was detected as per the definition in the study protocol |
| Blinding (detection bias) | High risk | Comment: no blinding of outcome assessors reported, and definition for detecting pneumonia was not clearly mentioned in the study protocol. Moreover the outcome was not clearly reported in the study |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: The outcome of interest was objective in nature, so the likelihood of performance and detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Although it mentions that 165 children were randomised and 25 were excluded because of various protocol violations, it is not clear to which of the 4 study groups these 25 children belonged. A per‐protocol analysis was done, and there appears to be no imbalance between groups with respect to the number of participants available for analysis. Therefore, the likelihood of bias due to attrition is low |
| Selective reporting (reporting bias) | High risk | Comment: A high mortality rate of 38.4% is found in patients with important (major) upper GI bleeding, but counts for each intervention are not given separately. Data on macroscopic upper GI bleeding, slight (microscopic) upper GI bleeding, and mortality are not reported for each intervention separately but are reported for the entire study. Data for pneumonia are not clear |
| Other bias | Low risk | Comment: Study is unclear on source of funding. No other known source of bias |
| Methods | Double‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 182 participants Number analysed: 182 participants Cimetidine
Antacids
Placebo
No prophylaxis
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalance: "All 4 groups were similar in age, sex, clinical severity of illness and mortality (25%)" | |
| Interventions | Cimetidine
Antacids
Placebo
No prophylaxis
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: Johns Hopkins University, Baltimore, Maryland, USA Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Cimetidine and placebo helped reduce the incidence of GI bleeding in participants with respiratory illness | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: This was a double‐blind placebo‐controlled trial. Study personnel were blinded.Therefore the likelihood of performance bias is low |
| Blinding (detection bias) | Low risk | Comment: This was a double‐blind placebo‐controlled trial, and GI bleed was an objective outcome that was diagnosed as per the definition in the study protocol |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: No other outcome of interest was mentioned in this study |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on source of funding. No other sources of bias suspected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 90 participants Number analysed: 90 participants Ranitidine
Pantoprazole
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalances: ‐ | |
| Interventions | Ranitidine
Pantoprazole
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: ICU Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not enough detail reported. Quote: "randomly divided" |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to allow judgement |
| Blinding of participants and personnel (performance bias) | High risk | Comment: no information about blinding reported; no definitions for diagnosis of outcomes reported |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no incomplete reporting of data suspected |
| Incomplete outcome data (attrition bias) | Low risk | Comment: no incomplete reporting of data suspected |
| Selective reporting (reporting bias) | Unclear risk | Comment: no selective reporting of outcomes suspected. Outcome reported in the Methods section is also reported in the Results section |
| Other bias | Unclear risk | Comment: not enough detail reported in conference abstract to assess risk of other biases |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 75 participants Number analysed: 75 participants Antacids
No prophylaxis
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalances: ‐ | |
| Interventions | Antacids
No prophylaxis
Adherence to regimen: The first 25 participants received either antacids or no prophylaxis. The trial was discontinued when H2 receptor antagonists became available. Of the 50 remaining participants, 10 received metiamide and 16 received cimetidine (after case reports of agranulocytosis by metiamide). The remaining 24 got no prophylaxis Duration of trial: January 1975 to July 1976 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Liver Unit, King's College Hospital and Medical School, Denmark Hill, London SES Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Only data for the comparison of antacid with no prophylaxis were extracted for the review, as it was felt that the second part of the trial, which compared H2 receptor antagonist vs no prophylaxis, was not a properly randomised trial | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the trial was stopped and H2 receptors administered instead of antacids |
| Blinding (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors, but the definition for diagnosis of GI bleeding is mentioned in the study report |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors, but outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | High risk | Comment: Data are not reported separately for participants who received cimetidine and metiamide. Mortality data are clubbed for both groups of controls (those compared with antacids and H2 receptor antagonists) |
| Other bias | High risk | Comment: unclear on source of funding and baseline characteristics of participants |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 158 participants Number analysed: 145 participants Overall
Inclusion criteria
Exclusion criteria
Baseline imbalances: 48 participants had 'primitive pneumonia' on admission, 14 had tracheobronchitis, and bacterial colonisation was noted in 36 participants, respectively (not clear to which intervention these participants belonged) | |
| Interventions | Antacids
Sucralfate
Adherence to regimen: Quote: "A single blind randomisation was performed within 12 hours of admission according to a simultaneous and independent dual randomisation. The first randomisation involved mechanical SSD versus no‐SSD. The second randomisation involved ulcer prophylaxis with aluminium hydroxide versus sucralfate. At the end, 4 random classes were defined" "158 of them were randomly selected on the probability of intubation for more than 3 days, 13 were then excluded because of death (n = 5) or extubation (n = 8) before day 3" Duration of trial: 14 months Duration of follow‐up: not clearly mentioned in the study report | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: Hospital Nord, CHRU St. Etienne, France Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: "A single blind randomisation was performed within 12 hours of admission according to a simultaneous and independent dual randomisation" Comment: not clear who were blinded. Therefore, unclear on the likelihood of performance bias |
| Blinding (detection bias) | Unclear risk | Quote: "A single blind randomisation was performed within 12 hours of admission according to a simultaneous and independent dual randomisation" Comment: unclear on blinding of outcome assessors. The definition for detecting GI bleeding is not clearly mentioned in the study report |
| Blinding (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. Nosocomial pneumonia was detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Of the 158 randomised participants, only 145 were part of the final analysis. There were 13 dropouts for the reasons mentioned above. The group to which they were randomised is not clearly mentioned in the study report, and an intention‐to‐treat analysis was not done.However, the dropouts accounted for less than 10% of randomised participants and appear to be equally distributed (given the double randomisation design of the study). Therefore, the likelihood of attrition bias is low |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | High risk | Comment: source of funding and baseline imbalances of antacid and sucralfate groups unclear |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 98 participants Number analysed: 98 participants Ranitidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 groups were similar with respect to gender distribution, age, admission APACHE II scores, Injury Severity Score, Revised Trauma Score, and history of smoking. Overall, patterns of injury were also similar in both groups | |
| Interventions | Ranitidine
Sucralfate
Adherence to regimen: All 98 participants were admitted for a minimum of 72 hours in ICU Duration of trial: April 1991 to October 1993 Duration of follow‐up: up to 2 weeks | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: Harborview Medical Center, 325 Ninth Avenue ZA ‐ 16, Seattle, WA 98104 Source of funding: Quote: "Supported in part by grant C#R49/CCR002570" Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the University of Washington Human Subjects Review Board" Informed consent: Quote: "Informed consent was obtained from each patient or patient representative within 24 hours of study enrolment" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Of the 12 participants who were classified as having gross bleeding, 7 in ranitidine and 5 in sucralfate groups had 'coffee‐ground' aspirates | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However, GI bleeding was an objective outcome that was detected as per the definition in study protocol |
| Blinding (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However, nosocomial pneumonia was an objective outcome that was detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However, all other outcomes of interest were objective in nature. Therefore, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants completed the trial and were included in the final analysis. There are no treatment withdrawals and no trial group changes. Therefore, there is no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Trial is supported by a grant from the Centers for Disease and Control and Prevention CDC#R49/CCR002570. The role of the sponsor in the conduct and reporting of the trial is unclear. No other source of bias detected |
| Methods | Quasi‐randomised trial | |
| Participants | Baseline characteristics Number randomised: 77 participants Number analysed: 77 participants Antacids
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: no difference between antacid and cimetidine groups in age and gender. The main reasons for admission were head injury and orthopaedic injury | |
| Interventions | Antacids
Cimetidine
Adherence to regimen: 49 participants in both groups maintained a gastric pH ≥ 4 (29 in antacid and 20 in cimetidine). Fifteen participants required increase in dosage, as they were not able to maintain a gastric pH ≥ 4 at the initial dose (6 in antacid and 7 in cimetidine). For 4 participants who were on cimetidine, an additional antacid administration was required. Nine participants failed to maintain a pH ≥ 4 despite maximum dose as per the study protocol (2 in antacid and 7 in cimetidine) Duration of trial: January 1979 to August 1979 Duration of follow‐up: Quote: "Patients were observed throughout their hospitalisation for GI bleeding and if it developed they were readmitted to the study and to intensive care unit" | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Louisville General Hospital surgical intensive care unit Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: It is mentioned that 19 participants had pneumonia (7 in antacid and 12 in cimetidine), but it was not an outcome intended to be reported in the study. Not sure if this was present on admission | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "patients were assigned to either antacid or cimetidine treatment group according to odd or even date of admission" Comment: This was a quasi‐randomised trial, so sequence generation was not done |
| Allocation concealment (selection bias) | High risk | Quote: "patients were assigned to either antacid or cimetidine treatment group according to odd or even date of admission" Comment: This was a quasi‐randomised trial in which allocation was not concealed |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: Blinding was not done ,and GI bleeding was an objective outcome that was detected as per the definition in the study objectives |
| Blinding (detection bias) | Unclear risk | Comment: Blinding was not done, and there was no intention to report pneumonia in the study objectives. It was reported when it was diagnosed in participants and was the main cause of sepsis in participants. Unclear whether pneumonia was present on admission |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature; because of this, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All participants were included in the final analysis. There was no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported. Pneumonia was reported although it was not intended. However this was an outcome of interest for the review. Therefore it could have caused reporting bias |
| Other bias | Low risk | Comment: Source of funding is unclear from the study report. No other sources of bias are suspected |
| Methods | Double‐blind randomised, double‐dummy trial | |
| Participants | Baseline characteristics Number randomised: 127 participants Number analysed: 127 participants Misoprostol
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The groups were clinically equivalent at entry with respect to age, gender race, risk factors such as hypotension, sepsis, coagulopathy, renal failure, hepatic failure, cardiac failure, adult respiratory distress syndrome (ARDS), gastric and duodenal lesions" Comment: no significant difference between the 2 groups with respect to demographic and baseline risk factors. Most participants were given a diagnosis of hypotension at baseline. Coagulopatyhy was present in 15 and 14 participants in both groups. Only 15 and 19 participants in both groups were free of any haemorrhagic gastric lesions at baseline | |
| Interventions | Misoprostol
Cimetidine
Adherence to regimen: Quote: "Patients meeting the above criteria for bleeding underwent an endoscopic evaluation within 12 hours and were removed from the study if a upper GI bleeding source was confirmed. All patients still in the study at 72 hours underwent a follow‐up endoscopy to determine whether the condition of the gastric or duodenal mucosa had changed. When possible, patients also underwent an endoscopy on exit from the study. If a patient underwent more than one follow‐up endoscopy, the score that represented the most severe damage was used" Duration of trial: July 1986 to January 1988. Duration of follow up: Quote: "All patients still in the study at 72 hours underwent a follow‐up endoscopy to determine whether the condition of the gastric or duodenal mucosa had changed" Notes duration of treatment: Patients were studied until 1 of 3 events occurred:
| |
| Outcomes | Outcomes sought in review and reported in trial
Note: GI bleeding developed 3 to 14 days after the first dose of study medication
Outcomes sought but not reported in trial
Outcomes reported in the trial but not used in review
| |
| Notes | Setting: 25 medical centres in United States: Medical University of South Carolina, Charleston, South Carolina; Our Lady of Mercy Center, Bronx, New York; Cook County Hospital, Chicago, Illinois; Medical Center of Central George, Macon, Georgia; VA Medical Center, Dayton, Ohio, St. Francis Medical Center, Trenton, New Jersey; Maine Medical Center, Portland, Maine; VA Medical Center, Detroit, Michigan; Buffalo General Hospital, Buffalo, New York; University of South Alabama, Mobile, Alabama; Butterworth Hospital, Grand Rapids, Michigan; Indiana University Medical Center, Indianapolis, Indiana; Brackenridge Hospital, Austin,Texas; Meharry Medical Center, NashvilleTennessee; 6196 Eagle Crest Drive, Huntington Beach, California; Humana Hospital‐University, Louisville, Kentucky; Hershey Medical Center, Hershey, Pennsylvania; University Hospital, Columbia, Missouri; Denver General Hospital, Denver, Colorado; Truman Medical Center, Kansas City, Missouri; VA Medical Center, Long Beach, California; Chicago Medical School, North Chicago, Illinois; St. Louis University Medical Center, St. Louis, Missouri; Buffalo VA Medical Center, Buffalo, New York; University of Chicago, Chicago, Illinois Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "At each of the 25 institutions, the protocol was approved by the institutional review board" Informed consent: Quote: "Written informed consent was obtained from the patient or surrogate before study entry" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Mortality was significantly associated with adult respiratory distress syndrome (ARDS), at baseline or at subsequent development, upper GI haemorrhage and additional organ system failure. 87 participants were given diagnosis of haemorrhagic lesions, and 10 participants met the criteria of upper GI haemorrhage as per the study definition | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: This was a double‐blind 'double‐dummy study' in which placebo was administered to both groups. There personnel involved were blinded, and so the likelihood of performance bias is low |
| Blinding (detection bias) | Low risk | Comment: This was a double‐blind 'double‐dummy study' in which placebo was administered to both groups. GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded. However, owing to the objective nature of the outcome of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants completed the trial and were included in the final analysis. There are no treatment withdrawals and no trial group changes. Therefore there is no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is unclear. No other sources of bias are suspected |
| Methods | Multi‐centre double‐blind randomised placebo‐controlled trial | |
| Participants | Baseline characteristics Number randomised: 131 participants Number analysed: 131 participants Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 treatment groups were similar in terms of demographic and clinical characteristics at baseline, including age, sex, race, type and number of risk factors for bleeding, and nasogastric pH. Most participants had major surgery (40 in cimetidine and 49 in placebo groups). Pneumonia was diagnosed at baseline in 9 and 5 participants, respectively | |
| Interventions | Cimetidine
Placebo
Adherence to regimen: Patients with renal failure were given 25 mg/h. Patients with pH below 4 on two different occasions (1 hour apart) 100 mg/h (if renally impaired 50 mg/h) Duration of trial: September 1988 to March 1989 Duration of follow up: In participants who developed GI bleeding, transfusion monitoring was continued for an additional 24‐hour period and a chest radiograph was taken, 48 hours after the medications were discontinued | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: multi‐centre study (20 institutions) Source of funding: Smith Kline Beecham Pharmaceuticals Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional review board" Clinical trials registration: ‐ Sample size calculation: Quote: "The study was designed as a multi centred trial with 200 randomised participants. The sample size was derived from the assumption that the population failure rate for placebo infused patients would be 20% vs. 5% for cimetidine infused patients. Thus, 100 patients per group (200 patients total) would be required to provide a power of 90%, with a two sided type 1 error of 0.05" Comment: Study was designed to include 200 participants but was terminated after enrolment of 131 participants because of the statically significant reduction in bleeding among participants treated with cimetidine Additional notes: Enteral feeding was given to 5 participants (4 in cimetidine and 1 in placebo groups, and among these a participant from the cimetidine group had protocol defined GI bleeding). An intention to treat was done for the incidence of nosocomial pneumonia (although it was present at baseline in 9 and 5 participants, respectively) as the study definition required new and persistent infiltrates on the chest radiograph. Mortality was not attributed to upper GI haemorrhage | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Low risk | Quote: "All the patients received a one time, 50 mL loading dose of coded medication (cimetidine or placebo) in 5% dextrose in water that was infused over a 20 minute period. This loading dose was followed immediately by a continuous infusion of coded medication (cimetidine or placebo) in 5% dextrose in water at a rate of approximately 10.4 mL/hour, using an infusion pump" Comment: Allocation concealment might have been done since the medication is coded |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "All the patients received a one time, 50 mL loading dose of coded medication (cimetidine or placebo) in 5% dextrose in water that was infused over a 20 minute period" Comments: This was a placebo‐controlled study in which medications administered were coded and participants and personnel involved in the trial were blinded |
| Blinding (detection bias) | Low risk | Quote: "To prevent bias in the interpretation of endpoints and safety data each institution designated one investigator to monitor pH and a second investigator, blinded to the pH determinations, to monitor signs and symptoms of upper GI haemorrhage, pneumonia and other safety parameters" Comment: This was a placebo‐controlled study in which medications administered were coded and the outcome assessor who monitored tsigns and symptoms of GI bleeding was unaware of the gastric pH value of respective participants. Moreover GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) | Low risk | Quote: "To prevent bias in the interpretation of endpoints and safety data each institution designated one investigator to monitor pH and a second investigator, blinded to the pH determinations, to monitor signs and symptoms of upper GI haemorrhage, pneumonia and other safety parameters" Comment: This was a placebo‐controlled study in which medications administered were coded and the outcome assessor who monitored signs and symptoms for pneumonia was unaware of the gastric pH value of respective participants. Moreover, nosocomial pneumonia was an objective outcome detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: This was a placebo‐controlled trial in which medications administered were coded. This would have ensured blinding for other outcome assessments. Moreover, outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were included in the final analysis. There are no treatment withdrawals and no trial group changes |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Smith Kline Beecham Pharmaceuticals funded part of the study. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
| Methods | Multi‐centre double‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 167 participants Number analysed: 167 participants Ranitidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "No statistically significant difference was present between treatment groups with regard to any demographic variables" Comment: The imbalance between the 2 groups was only with respect to number of people on mechanical ventilation at study entry. 65 in placebo group and 80 in the ranitidine group. The other baseline characteristics were comparable. Nosocomial pneumonia was present on entry in 2 participants in both groups, respectively. Prothrombin time > upper limit was present in 31/77 and 28/83 participants, respectively | |
| Interventions | Ranitidine
Placebo
Adherence to regimen: If upper GI bleeding was detected according to the definition, then participant was withdrawn from the study. All participants adhered to the prescribed study regimen Duration of trial: January 1990 to September 1991 Duration of follow‐up: probably until discharge or untimely death | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
If any of the preceding variables were present, the following 4 questions were addressed to establish the diagnosis of stress ulcer GI bleeding:
If the answer to any of the preceding 4 questions was "yes", the participant was considered to have GI bleeding Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported in report but not used in review
| |
| Notes | Setting: 10 ICUs from across the United States of America Source of funding: Quote: "This study was supported in part by a research grant from Glaxo Pharmaceuticals" Ethics approval: Quote: "The study was approved by the institutional review boards of all participating sites." Comment: ethics approval obtained from all 10 participating sites Informed consent: Quote: "Informed consent was obtained from patient or a legally authorized representative" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Two participants who were diagnosed with pneumonia at baseline were excluded from the denominator of participants who subsequently developed nosocomial pneumonia during the study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were then randomised to treatment with 6.25 mg/hour continuous ranitidine or placebo infusion according to a computer generated randomisation scheme" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: This is a placebo‐controlled trial in which both the intervention and the control were administered at the same rate as per a computer‐generated randomisation scheme, which suggests that participants and study personnel were blinded. Therefore the likelihood of performance bias is low |
| Blinding (detection bias) | Low risk | Comment: This was a placebo‐controlled trial, and GI bleeding was an objective outcome that was detected as per the definition in the study report |
| Blinding (detection bias) | Low risk | Comment: This is a placebo‐controlled trial, and nosocomial pneumonia was an objective outcome detected as per the definition in the study report |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: This is a placebo‐controlled trial, and all other outcomes of interest were objective in nature. Therefore the likelihood of performance or detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants completed the trial and were included in the final analysis. There are no treatment withdrawals and no trial group changes. Therefore the likelihood of attrition bias is low |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: This study was supported in part by a research grant from Glaxo Pharmaceuticals, and some of the equipment used was provided by this organisation. The role of the sponsor in the conduct and reporting of the trial is unclear. No other form of bias was detected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 31 participants Number analysed: 31 participants Ranitidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: no significant difference between the 2 groups with respect to demographic and baseline risk factors. Most participants were admitted post surgery | |
| Interventions | Ranitidine
Sucralfate
Adherence to regimen: All participants appear to have adhered to the regimen to which they were randomised Duration of trial: ‐ Duration of follow‐up: probably until discharge or untimely death of the participant | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcin report but not used in review
| |
| Notes | Setting: Department of Intensive Care Unit, Karadeniz Teknik University, Trabzon, Turkey Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Only 3 participants treated with ranitidine and 1 with sucralfate had secondary pneumonia due to some bacterial agent isolated from the stomach. Colonistion of the oropharynx and tracheostomy were more common in participants treated with ranitidine | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and blinding of study personnel or participants was not possible owing to the different modes of administration of study drugs |
| Blinding (detection bias) | Unclear risk | Comment: The definition for detecting GI bleeding was not clearly mentioned in the study report, and the study is unclear on blinding of outcome assessors |
| Blinding (detection bias) | Low risk | Comment: The definition for detecting secondary pneumonia was clearly mentioned in the study report. Still, the study is unclear on blinding of outcome assessors |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants completed the trial and were included in the final analysis.There are no treatment withdrawals and no trial group changes. |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported |
| Other bias | Low risk | Comment: No mention of the source of funding. No additional biases were detected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 313 Number analysed: 311 Esomeprazole
Famotidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 treatment groups were similar with respect to baseline demographic characteristics, history of ulcers, cardiac disease, percutaneous coronary stenting, baseline haemoglobin and serum creatinine levels, and use of enoxaparin or thrombolytics | |
| Interventions | Esomeprazole
Famotidine
Adherence to regimen: Compliance was assessed by pill count. Good compliance with study drugs (≥ 90% ): 100% in famotidine group and 98.8% in esomeprazole group Duration of trial: July 2008 to September 2010 Duration of follow‐up: minimum of 4 weeks and maximum of 52 weeks | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial
Outcomes reported in trial, but not used in review
| |
| Notes | Setting: acute medical wards, cardiac care unit, and intensive care unit, Department of Medicine and Geriatric, Ruttonjee Hospital, Hong Kong Sponsorship source: Cardiac Research Fund, Ruttonjee Hospital Conflict of interest: Quote: "Potential competing interests: None" Ethics approval: Quote: "The study protocol was approved by the Ethics Committee of the Hong Kong East Cluster" Informed consent: Quote: "All patients gave their written, informed consent" Clinical trials registration: This study was registered at http://www.clinicaltrials.gov (Identifier NCT00683111) Sample size calculation: Yes, described under statistical analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Ward clerks at the four acute medical wards, the cardiac ward, and the intensive care ward generated 50 treatment codes labelled A and 50 codes labelled B. These were sealed in identical, blinded envelopes that were shuffled randomly" |
| Allocation concealment (selection bias) | Low risk | Quote: "Ward clerks at the four acute medical wards, the cardiac ward, and the intensive care ward generated 50 treatment codes labelled A and 50 codes labelled B. These were sealed in identical, blinded envelopes that were shuffled randomly. Th e investigators drew a blinded envelope randomly, and the pharmacist dispensed the repackaged medi‐ cation. The investigators and patients were blinded to the treatment‐group assignments" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "The investigators drew a blinded envelope randomly, and the pharmacist dispensed the repackaged medication. The investigators and patients were blinded to the treatment‐group assignments. The treatment codes were released aft er approval of the completion of the study by the Ethics Committee" |
| Blinding (detection bias) | Low risk | Quote: "overt bleeding of gastroduodenal origin (confirmed by means of upper endoscopy), overt upper GIB of unknown origin, bleeding of occult gastrointestinal origin (confirmed by means of upper gastrointestinal endoscopy), obstruction, or perforation" Comment: objective criteria for the measurement of GI bleeding reported |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "The investigators drew a blinded envelope randomly, and the pharmacist dispensed the repackaged medication. The investigators and patients were blinded to the treatment‐group assignments. The treatment codes were released after approval of the completion of the study by the Ethics Committee" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "No patients were lost to follow‐up. Premature termination occurred in 34 (20.9%) and 31 (20.9%) patients in the esomeprazole and famotidine groups, respectively. No patient in the esomeprazole group and three (2.1%) patients in the famotidine group refused to continue the study. In the famotidine group, one patient with significant dyspepsia withdrew consent, while the remaining two patients did not give specific reasons" Comment: Flow chart of participant flow is included, and no incomplete reporting of outcome data is suspected |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes that are listed in the Methods section are also reported in the Results section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 86 participants Number analysed: 86 participants Ranitidine
Antacids
Inclusion criteria
Exclusion criteria
Baseline imbalances: Groups were similar with respect to age, gender, and admission diagnosis. Acute and chronic respiratory failure were the most common causes for admission | |
| Interventions | Ranitidine
Antacids
Adherence to regimen: Quote: "Eightysix patients were randomised, 42 received ranitidine, 44 received antacids (malox: 42 and amphojel:3), 38 receiving ranitidine completed 48 hours of study while 23 continued up to 72 hours. Of the patients receiving antacids, 39 completed 48 hours of study, while 25 continued up to 76 hours" Duration of trial: ‐ Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Department of Adult Intensive Care, Royal Alexandria Hospitals, Edmonton, and the Division of Critical Care Medicine, University of Alberta, Edmonton, Alberta, Canada Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Using a computer generated table of numbers, patients were assigned by restricted randomisation...” |
| Allocation concealment (selection bias) | Low risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants.Therefore, high risk of performance bias |
| Blinding (detection bias) | High risk | Comment: There was no clear definition for detecting clinically significant upper GI bleeding, and it is unclear whether the unblinded nature of the study influenced this outcome, which was otherwise objective in nature |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature. Therefore the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Although 1 participant withdrew from each group during the course of the study, all randomised participants were part of the final analysis.Therefore there was no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes have been reported |
| Other bias | Low risk | Comment: no mention of the source of funding. No additional biases were detected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 78 participants Number analysed: 78 participants (for outcomes of interest to this review) Cimetidine bolus
Cimetidine continuous
Sucralfate
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: Baseline characteristics for participants who met the criteria for early withdrawal from the study (for reasons mentioned below) are not mentioned. Nearly 40 participants were withdrawn from the study | |
| Interventions | Cimetidine bolus
Cimetidine continuous
Sucralfate
No prophylaxis
Adherence to regimen: Quote: "Patients who met criteria for early withdrawal (< 3 days) were not included in the final analysis" Comment: Withdrawal was mainly due to GI bleeding, gastric colonisation on entry, extubation, mortality, and inability to aspirate GI secretions and change to tube feedings Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial (none of them were the primary outcomes in the study)
Note to 1: Only those that occurred within the first 3 days and caused withdrawal were reported Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: Department of Critical Care Medicine, Saint Vincent Hospital, Worcester, Massachusetts, and Department of Surgery, New England Medical Centre, Boston, Massachusetts Source of funding: Smith Kline Beecham, Inc., Philadelphia, Pennsylvania Informed consent: ‐ Ethics approval: Quote: "The study was approved by the institutional review board" Clinical trials registration: ‐ Sample size calculation: ‐ Comment: Power analysis has been done at the end of the study to detect a correlation of less than 0.4 Additional notes: Cimetidine arms were combined to form a common interventional arm as the review did not aim to investigate efficacy on the basis of dose or mode of administration in the same drug | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "All other patients were randomised (table of random numbers) into one of the three groups" Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Comment: This is an unblinded trial, and GI bleeding was detected as per the definition in the trial protocol |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: This is an unblinded trial, and all other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 40 randomised participants were withdrawn from the study (more than 50%) for protocol violations and were excluded from the final analysis. However, relevant data (for GI bleed, ICU mortality) could be obtained from the study |
| Selective reporting (reporting bias) | High risk | Comment: The duration of intubation,hospital mortality and participants requiring antibiotics are mentioned only for participants who were part of the final analysis, and this excluded earlier withdrawals. This accounts for selective reporting |
| Other bias | High risk | Comment: This study was supported by a grant from Smith Kline Beecham, and some of the equipment used was received from this organisation. However, the role of the sponsor in the conduct and reporting of the trial is unclear. Baseline characteristics for 40 participants (excluded owing to early withdrawal) are not mentioned in the study report |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 30 participants Number analysed: 30 participants Pantoprazole
Famotidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: ‐ | |
| Interventions | Pantoprazole
Famotidine
Adherence to regimen: no loss to follow‐up; all finished treatment Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge from the hospital | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcome
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: ‐ Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: not clearly mentioned in the study report. However, this was not a placebo‐controlled trial, and modes of interventions were different. Therefore it might not have been possible to blind personnel, so the likelihood of performance bias is low |
| Blinding (detection bias) | High risk | Comment: not clearly mentioned in the study report. However, GI bleeding was an objective outcome, but the definition was not clearly mentioned in the study report |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: No other outcomes of interest were part of this study |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes of interest were analysed and reported |
| Other bias | Low risk | Comment: unclear on the source of funding. No other source of bias detected |
| Methods | Double‐blind randomised placebo‐controlled trial | |
| Participants | Baseline characteristics Number randomised: 39 participants Number analysed: 39 participants Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "The treatment groups were similar in the number of patients, sex and severity of illness as determined by the primary diagnosis. The cimetidine group tended to be older, but this difference was not statistically significant. Both treatment arms were also similar in the initial pretreatment endoscopic classification" Comment: The 2 groups appear to be similar in their demographic and other characteristics, suggesting no imbalance between the 2 at baseline. Mosg in both groups were diagnosed with cardiopulmonary disease | |
| Interventions | Cimetidine
Placebo
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge from the hospital | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported but not used in review
| |
| Notes | Setting: Gastroenterology Service, Department of Medicine, Walter Reed Army Medical Center, Washington, DC Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "This protocol was approved by the Clinical Investigation and Human Use Committees of the Walter Reed Army Medical Center" Informed consent: Quote: "Each patient or guardian gave informed consent" Clinical trials registration: not provided in the study report Sample size calculation: ‐ Additional notes: On initial endoscopy, it was found that there were endoscopic signs of bleeding in 14 of 29 participants with mucosal abnormalities. 3 participants from the placebo arm developed new signs of bleeding during the study, when serial endoscopy was done. One death in the placebo group was attributed to upper GI bleed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: “The group not receiving cimetidine received an intravenous injection of placebo every 6 hours to ensure the double blind nature of the study” Comment: This suggests that participants and personnel were blinded to the interventions |
| Blinding (detection bias) | Low risk | Quote: "All endoscopic examinations were done by a single investigator and witnessed by a second investigator who observed the procedure through a lecturescope. During the endoscopy, findings were discussed and agreed on by both investigators before an entry was made on the report form. Both investigators were uninformed as to the patient’s treatment group" Comment: Outcome assessors were blinded. The definition for detecting GI bleed was not clearly mentioned. However, it was an objective outcome detected through endoscopy. Therefore the likelihood of performance or detection bias is low |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported and analysed in the study |
| Other bias | Low risk | Comment: unclear on the source of funding. No additional biases were detected |
| Methods | Multi‐centre randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: not clear Number analysed: 58 participants Omeprazole
Ranitidine
Inclusion criteria
Exclusion criteria: not clearly mentioned in the study report, most probably participants who did not satisfy the inclusion criteria Baseline imbalances: Quote: "APACHE II, ISS scores and other variables were comparable in both the groups" Comment: The 3 groups appeared to be comparable at baseline | |
| Interventions | Omeprazole
Ranitidine
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: University Hospitals and Clinics, Department of Surgery, 1 Hospital Drive, 65212, USA Source of funding: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: This was a poster presented at Society of Critical Care Medicine; 27th Educational and Scientific Symposium; San Antonio, Texas, USA; February 4 to 8 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | High risk | Comment: no clear mention of blinding of outcome assessors. Moreover, there is no clear definition to detect GI bleeding |
| Blinding (detection bias) | High risk | Comment: no clear mention of blinding of outcome assessors. Moreover, there is no clear definition to detect pneumonia |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: No other outcomes of interest were reported |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: Report says there were 58 evaluable participants; it does not specify the number randomised |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on source of funding. No other source of bias suspected |
| Methods | Open‐label quasi‐randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 92 participants Number analysed: 83 participants Sucralfate
Ranitidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There were no significant differences between the two groups in demographic data, trauma score, revised trauma score, Injury severity score and APACHE II score" | |
| Interventions | Sucralfate
Ranitidine
Adherence to regimen: Of the 92 participants, 9 were subsequently excluded for protocol breaks: 4 patients because they did not meet age criteria, 3 patients because admitting chest radiographic films were abnormal, and 2 patients because of inadvertent extubation Duration of trial: January 1989 to August 1991 Duration of follow‐up: All patients were followed until hospital discharge or death | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: SICU, Grant Medical Centre, Columbus, Ohio, USA Source of funding: Quote: "Supported in part by a Roche Hospital Pharmacy Research Grant" Conflicts of interest: ‐ Ethics approval: Study was approved and monitored by the Institutional Review Board Informed consent: Consent for therapy was considered consent for study inclusion, and specific informed consent was waived by the Institutional Review Board Clinical trials registration: ‐ Sample size calculation: Quote: "The present study was designed to produce a power of 90% with a 30% difference in pneumonia rates between the groups" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Randomisation occurred by the use of computer generated random number table. Odd numbered patients received ranitidine 50 mg every 6 hours and even numbered patients received sucralfate 1 g dissolved in 15 mL of sterile water ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: Different modes of administering study drugs and absence of placebo would not have made it possible to blind the study |
| Blinding (detection bias) | Low risk | Comment: Study report is unclear on blinding of outcome assessors. However GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) | Low risk | Comment: Study report is unclear on blinding of outcome assessors. However, nosocomial pneumonia was detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: Study report is unclear on blinding of outcome assessors. However, all other outcomes were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "Nine patients were subsequently excluded for protocol breaks: four patients because they did not meet the age criteria, three patients because admitting chest radiographic films were abnormal and two patients had inadvertent extubation. These patients had been evenly distributed between study groups and none of the excluded patients developed pneumonia" Comment: Although 92 participants were enrolled in the study, only 83 were analysed. The interventional arms to which these 9 participants were randomised are not clearly mentioned in the study report, but it does say that participants were evenly distributed between study groups, and none of them developed pneumonia. Therefore the likelihood of attrition bias is minimal |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were part of the final analysis |
| Other bias | Low risk | Comment: Study was supported in part by a Roche Hospital Pharmacy Research Grant. However, the role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias suspected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 143 Number analysed: 126 Antacid
No intervention
Inclusion criteria Critically ill with 1 or more of the following risk factors for acute erosive gastritis:
Exclusion criteria
Baseline imbalances: ‐ | |
| Interventions | Antacid
No intervention
Adherence to regimen: ‐ Duration of follow‐up: ‐ Duration of trial: January 1981 to June 1983 | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought in review and not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: ICU, Department of Surgery, University Hospital and the Department of Mathematics University of Saskatchewan Sponsorship source: none Conflicts of interest: ‐ Comments: 28% of patients in the control group and 32% of patients in the treatment group were in the study for 1 day Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were assigned randomly to control or treatment group using a table of random numbers, for groups of four" |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: no details reported, but lack of blinding is unlikely to bias outcome measures and outcomes |
| Blinding (detection bias) | Low risk | Quote: "The pH of the gastric aspirate was tested every 2 hours using phenaphthazine paper (Nitrazine, Squibb; or Colorphast. E. Merck), and observed and tested for blood every 4 hours using cumene hydroperoxide; 3,3', 5,5 '‐tetramethylbenzidine (Ames Lab‐stix). Upper GI bleeding was quantitated in the following manner: microscopic, defined as a small ( + ), moderate ( ++ ), or large ( +++) chemical reaction; moderate, defined as less than 200 mL of blood visible at any time; or severe, defined as more than 200 mL of blood, or as moderate bleeding recurring at least three times in 6 hours, or as moderate bleeding with worsening vital signs during 6 hours of observation" Comment: objective criteria for the diagnosis of GI bleeding reported |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no details reported, but lack of blinding is unlikely to bias outcome measures and outcomes |
| Incomplete outcome data (attrition bias) | Unclear risk | Quote: "Of 143 eligible patients, nine control patients and eight treatment patients were excluded because of an insufficient number of gastric pH measurements (six patients), missing protocol sheets (five patients), randomisation error (four patients), or clotting abnormalities (two patients)" Comment: Of 143 eligible patients 17 were excluded for various reasons unrelated to treatment |
| Selective reporting (reporting bias) | Unclear risk | Comment: no selective outcome reporting suspected. All outcomes listed in the Methods section were reported in the Results section |
| Other bias | Low risk | Comment: no other sources of bias suspected |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 44 Number analysed: 38 (for the primary outcome of GI bleed), 44 (for all other outcomes of importance to the review) Cimetidine
Antacid
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 2 groups were almost similar with respect to age, gender, and number of participants. Most participants were diagnosed with respiratory failure or sepsis, and this was equally distributed in both groups | |
| Interventions | Cimetidine
Antacids
Adherence to regimen: Two patients in the cimetidine group were removed from the protocol (n = 23) owing to refusal of endoscopy and protocol error, respectively. From the antacid group (n = 21), 5 patients were removed; 4 could not be endoscoped (2 refused endoscopy and 2 developed cardiac complications), and 1 patient with severe ileus (developed nausea and vomiting). Amphogel was substituted for Mylanta II in patients with severe diarrhoea and renal failure Duration of trial: ‐ Duration of follow‐up: not mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes were not classified as primary or secondary in the study report; however outcomes reported were: Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: McGill University, Sir Mortimer B. Davis‐Jewish Hospital, Montreal, Quebec, Canada Source of funding: Smith Kline and French Canada Ltd., and Park Davis Canada Ltd. Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the ethics committee on clinical investigations of the Sir Mortimer B.Davis‐Jewish General Hospital" Informed consent: Quote: "A signed consent was obtained from patients or their immediate relations" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Trial reports that NG aspirates were frequently positive for occult bleeds and were not a useful tool for determining clinically significant upper GI bleeding | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Quote: "After approximately 72 hours all patients were gastroscoped by a single endoscopist who had no knowledge about which therapy the patient was on…" Comment: GI bleeding was an objective outcome that was detected as per the definition in the study protocol; the outcome assessor was blinded |
| Blinding (detection bias) | Unclear risk | Comment: This was not an outcome of interest in this study |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However, all other outcomes were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 6 participants were excluded from analysis of the primary outcome of diagnosing GI bleed endoscopically (as endoscopy could not be done on these participants) for reasons mentioned under "adherence to the regimen". An intention‐to‐treat analysis was done for other outcomes of interest |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were reported and analysed |
| Other bias | Low risk | Comment: Study was funded by Smith Kline and French Canada Ltd., and Park Davis Canada Ltd., and some of the equipment used was received from this organisation. The role of the sponsor in the conduct and reporting of the trial is unclear |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 41 participants Number analysed: 41 participants Ranitidine
Omeprazole (bolus)
Omeprazole (infusion)
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There was no difference between the groups with regard to sex, age, ethnic origin, the presence of droperidol in the premedication and the drugs used after the operation" Comments: The 4 groups of participants who were scheduled for CABG appear to be similar to each other with respect to demographic characteristics and medications given | |
| Interventions | Ranitidine
Omeprazole (bolus)
Omeprazole (infusion)
Placebo
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study record. Most probably until discharge | |
| Outcomes | Outcomes sought in review and reported in trial (none of these were primary outcomes for this study)
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Department of Anaesthetics and Department of Surgery, Royal Postgraduate Medical School, Hammersmith Hospital, London W12ONN, UK Source of funding: not clearly mentioned in the study report. However, it is mentioned that Astra Clinical Research Unit, Edinburgh, supplied the drugs and supported the trial Conflicts of interest: ‐ Ethics approval: Quote: "41 patients who were scheduled for CABG were entered into the study, which was approved by the local ethics committee" Informed consent: Quote: "Informed consent was obtained from each patient on the eve of the operation" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: The omeprazole arms were combined to form a common interventional arm as the review did not aim to investigate efficacy on the basis of dose or mode of administration of the same drug | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were assigned to one of the four treatment groups from a random list" Comment: Random sequence generation is not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Patients were assigned to one of the four treatment groups from a random list" Comment: unclear on how allocation was concealed |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "The personnel collecting the aspirates did not know which treatment the patient received" Comment: This was a placebo‐controlled trial. Blinding of personnel was done for the primary outcomes of measuring gastric pH and volume of gastric secretion. The likelihood of performance bias is low |
| Blinding (detection bias) | High risk | Comment: not clear whether outcome assessors were blinded. The definition for diagnosing GI bleed, which was an objective outcome, was not clearly mentioned in the study report |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: not clear whether outcome assessors were blinded. Moreover, the outcome of interest was objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: It is mentioned that Astra Clinical Research Unit, Edinburgh, supplied the drugs and supported the trial. But it is unclear whether they had any influence on the results of the trial. No other sources of bias detected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: Unclear Number analysed: 50 participants Ranitidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There were no significant differences between the groups with respect to age, sex, distribution of underlying diseases, the severity of illness, prophylactic antibiotic therapy, and gastric pH at admission" Comment: no significant difference between the 2 groups with respect to demographic and baseline risk factors. Laparotomy was the most common cause for admission in both groups. The APCHE II score was 14.21 ± 5.44 and 13.34 ± 6.03 in the ranitidine and sucralfate groups, respectively | |
| Interventions | Ranitidine
Sucralfate
Adherence to regimen: Quote: "Seven patients were extubated and one patient died before four days of observation and could not be analysed for the development of late onset pneumonia. 42 patients observed for more than four days" Comment: Of the initial number of participants who were randomised, only those who were eventually intubated for longer than 24 hours were part of the study analysis Duration of trial: ‐ Duration of follow‐up: Patients were followed up for a period of 7 days with daily chest radiograph, complete blood count with differential, serum electrolytes, and gastric pH measurements | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
15 early‐onset and late‐onset cases of pneumonia were diagnosed if they occurred during the first 4 days of or 4 days after initiation of mechanical ventilation, respectively. Patients observed for longer than 4 days and were evaluated for the development of late‐onset pneumonia Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Department of Anaesthesia and Intensive Care, Vardhman Mahavir Medical College and Safdarjang Hospital, New Delhi Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study protocol was approved by the institutional ethics committee" Informed consent: Quote: "...informed consent was obtained from the patients or, if this was not possible because of the clinical condition, from a relative of the family" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Of the 25 participants who developed pneumonia, 11 (44%) had the source traced to gastric colonisation (10 in ranitidine and 1 in sucralfate group). Klebsiella species was the most commonly isolated (gastric and tracheal aspirates). Late‐onset pneumonia was more common in the ranitidine group than in the sucralfate group (10 and 2; P = 0.001), and there was no significant difference in early‐onset pneumonia between the 2 groups (5 and 8; P = 0.098) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was done using a computer generated random number table" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Comment: no clear mention of blinding the outcome assessor to this outcome. But GI bleeding was an objective outcome detected as per the definition in the study report |
| Blinding (detection bias) | Low risk | Quote: "Chest radiographs were interpreted by a radiologist who had no knowledge of the patients’ treatment group after randomisation" Comment: VAP was detected as per the definition in the study report, and the radiologist was blinded to the interventions |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: no clear mention of blinding outcome assessors. However, outcomes were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: Although 50 participants in each treatment arm were evaluated, we are not sure of the number of participants who were initially randomised to each of these arms |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: The source of funding is not mentioned. No additional biases were detected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised:75 participants Number analysed: 75 participants Cimetidine
Antacids
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "Of the 38 participants in the cimetidine group, 19 were male and 19 were female, and the mean age was 62 years. Of the 37 participants in the antacid group 22 were male and 15 were female participants. and their mean age was 63 years. The principle diagnosis of both the groups were almost similar (most of them had intra abdominal diseases ; antacid group (7) and cimetidine group (10). There was no statistically significant difference between the two groups with respect to risk factors such as major operative procedures, respiratory failure, sepsis, peritonitis, multiple trauma, renal failure, hypotension and jaundice" "Before administration of any medication, the initial gastric pH values were below 3.5 in 16 of the cimetidine treated participates and 16 of the antacid treated participants. The guaiac test was initially positive in 11 participants in cimetidine group and 9 in antacid group. None of these 20 participants had gross evidence of GI bleeding and were included in the study" Comment: Both of these groups were similar with respect to their baseline characteristics | |
| Interventions | Antacids
Cimetidine
Adherence to regimen: Quote: "In 38 participants treated with cimetidine, failure to achieve a pH of 3.5 or greater on one or more occasions occurred in 18 participants who were initially given 300 mg intravenously every six hours and in nine given a dose of 300 mg every four hours. Seven of eight participants who required 400 mg of cimetidine every four hours had gastric pH values below 3.5 on one or more occasions even at that dosage" "In 37 participants treated with antacids, failure to achieve a pH of 3.5 or greater occurred in nine participants initially given 30 mL of antacid, six given 60 mL, and one given 120 mL. However, during 1259 hourly administrations of 30 mL of antacids, the gastric pH remained below 3.5 only 21 times (1.7 percent)" "Patients in whom cimetidine "failed" (who had GI bleed, n = 7), were continued on cimetidine, and antacids were added at an initial dosage of 30 ml/hour and patients who bled with antacids would receive cimetidine also" "The study was ended when oral feedings were begun and the nasogastric tube was removed, or the participant was discharged from the intensive care unit" Duration of trial: January 1978 to March 1979 Duration of follow up: not mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes are not categorised as primary and secondary, but from the study report, we feel GI bleeding was the primary outcome Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Departments of Anaesthesia and Surgery, Harvard Medical School, and the Beth Israel Hospital Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the Committee on Cliniccal Investigations, New Procedures and New Forums of Therapy of the Beth Israel Hospital" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: In 1 participant from the cimetidine group, GI bleeding was the cause of death | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "…randomised by the table of random numbers to receive either cimetidine or antacids" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded. Definition of GI bleeding described in the study report |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is not mentioned. No additional biases were detected |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 375 participants Number analysed: 244 participants Antacids
Ranitidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "At randomisation, no statistically significant difference was found among the three groups in terms of age (P = 0.058), sex (P > 0.2), APACHE II scores (P > 0.2), Glasgow coma scores (P > 0.2), and other underlying characteristics such as pneumonia on admission, participants receiving antibiotic therapy or enteral nutrition" Comment: The 3 groups were similar. Among the participants from surgical ICU, 30, 28, and 33 participants in the antacid, ranitidine, and sucralfate groups were requiring emergency surgery. Most participants from surgical ICU were diagnosed with trauma requiring some form of intervention. Among participants from the medical ICU, most had some pulmonary disease; 9, 7, and 6 participants were diagnosed with pneumonia on admission to each of the respective groups | |
| Interventions | Antacid
Ranitidine
Sucralfate
Adherence to regimen: Quote: "375 were randomly assigned to a treatment group and 258 were eventually intubated for more than 24 hours. Fourteen were un assessable because of missing data (4, 3, and 7 patients in the antacid, ranitidine, and sucralfate groups, respectively). Thus, 244 patients could be analysed. Of these 244 patients, 81 received antacid, 80 received ranitidine, and 83 received sucralfate.The protocol had to be interrupted before extubation in 23 (28%) of the patients in the antacid group, 19 (24%) of the patients in the ranitidine group, and 17 (20%) in the sucralfate group. Renal insufficiency developed in 5, 8, and 6 patients in the antacid, ranitidine, and sucralfate groups, respectively. In the antacid group, 6 patients developed diarrhoea or ileus, which was attributed to the treatment. In the ranitidine group, one patient developed leukopaenia and another patient developed a rash. Removal of the nasogastric tube, withdrawal of supportive care, or discharge from the hospital was the other reason for premature protocol interruption. Five patients in the antacid group, 5 patients in the ranitidine group, and 8 patients in the sucralfate group had these characteristics. In addition, protocol violation prompted interruption of treatment in 7 patients in the antacid group, 4 patients in the ranitidine group, and 3 patients in the sucralfate group. For patients in whom the protocol was interrupted, the total number of assessable days before interruption was not statistically different among the three groups (P > 0.2)" Comment: It is also mentioned that physicians had to modify the anti‐stress ulcer prophylaxis regimen in 1, 2, and 3 participants in the antacid, ranitidine, and sucralfate groups, respectively, due to GI bleed Duration of trial: January 1989 to January 1991 Duration of follow up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Note: Early‐onset pneumonia developed in 9, 8, and 7 participants in antacid, ranitidine, and sucralfate groups, respectively. Late‐onset pneumonia developed in 11, 14, and 4 participants in the antacid, ranitidine, and sucralfate groups, respectively.Three of the 4 cases of late‐onset pneumonia in the sucralfate group were observed on day 5 Note: In the antacid group, GI bleeding developed on third day for 2 and on day 18 for 1 participant. In the ranitidine group; it was diagnosed on the second day for 2 participants and on days 3, 4, and 6 for the remaining 3 participants. In the sucralfate group, it was detected on the second day for three and on days 3, 5,8,12, and 23 for the remaining participants. Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Division Autonome de Medecine Preventive Hospitaliere, Centre Hospitalier Universitaire Vaudois, CH‐1011 Lausanne, Switzerland Source of funding: by Merck and Co. Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Mortality was attributed to pneumonia in 4 participants (1 in the antacid and 3 in ranitidine groups, respectively, whereas it was attributed to GI haemorrhage in 1 participant from the sucralfate group. Staphylococcus aureus, Streptococcus pneumoniae, and Haemophilus influenzae, alone or in combination, accounted for more than half of early‐onset pneumonia cases (54%), whereas gram‐negative bacilli were most commonly isolated in late‐onset pneumonia | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was done using a random permutable table to generate a random treatment list" |
| Allocation concealment (selection bias) | Low risk | Quote: "Treatment regimens were included in opaque, sealed envelopes" Comment: Method adopted to obtain allocation concealment is clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Comment: There is no clear mention of blinding outcome assessors. However, GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) | Low risk | Quote: "They were interpreted by a pneumologist who had knowledge of all relevant data except for the patient's stress ulcer prophylactic regimen, gastric pH, or colonization data" Comment: VAP was detected as per the definition in the study protocol by an outcome assessor who was blinded to the above mentioned participant data |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: There is no clear mention of blinding outcome assessors. However, all other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Of the initial 375 randomised participants, only 258 were part of the analysis, as only these participants were in the trial for longer than 24 hours, as this was criterion was necessary to measure the outcomes of interest. Data on 14 participants were missing, and they seem to be well balanced across the 3 groups |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported in the study |
| Other bias | Low risk | Comment: The study was funded by Merck and Co. However, the role of the sponsor in the conduct and reporting of the trial is unclear. No other form of bias was detected |
| Methods | Open label, randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 97 participants Number analysed: 40 participants Ranitidine
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "No significant differences between treatment and control groups were detected in any of the clinical and therapeutic characteristics among both the groups. However, hypotension was more frequent in the control group (12 vs. 5)" Comment: Both groups were comparable with respect to age and gender distribution, and risk factors such as disseminated intravascular coagulation in addition to severe intracranial lesion and respiratory failure. Severe head injury was the main primary diagnosis in both groups | |
| Interventions | Ranitidine
No prophylaxis
Adherence to regimen: For treatment of the 97 eligible participants, only 40 completed the trial and were available for analysis (19 in the treatment arm and 21 in the control arm) The remainder were excluded for the following reasons:
In the treatment group, 5 (26%) participants remained on the original ranitidine dosage of 50 mg every 8 hours throughout the study, 5 (26%) required a dosage increase to 50 mg every 6 hours, and 9 (47%) needed additional antacids Duration of trial: August 1984 to September 1986 Duration of follow up: up to 7 days after study completion | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Department of Internal Medicine and Surgical Intensive Care Unit, University Hospital Basel, Switzerland Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the ethical committee of the Basel University Hospital" Informed consent: Quote: "Informed consent was obtained from each patient's legal guardian" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Data are provided on number of deaths during the study (n = 1 in the ranitidine group), which is not similar to all‐cause mortality in ICU | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "The need to monitor gastric pH to determine intensification of stress lesion prophylaxis in the treatment group prevented double‐blind study design” Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Quote: “The need to monitor gastric pH to determine intensification of stress lesion prophylaxis in the treatment group prevented double‐blind study design” Comment: Outcome assessors were not blinded. However, GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: It is mentioned that of the 97 eligible participants, only 40 completed the trial and were available for analysis. The remaining 43 participants were excluded for reasons mentioned under "Adherence to the regimen". Therefore, a protocol analysis was done to measure the outcomes of interest, and the number of participants appears to be balanced between groups. Therefore, the likelihood of this affecting the outcomes of interest is minimal |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: The source of funding is not clearly mentioned in the study report. No additional biases were detected |
| Methods | Randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: ‐ Number analysed: ‐ Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: ‐ | |
| Interventions | Cimetidine
Placebo
Adherence to regimen
Duration of trial: March 1977 to June 1978 Duration of follow up: until death or discharge from hospital | |
| Outcomes | Outcome sought in review and reported in trial
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: Department of Surgery, Marburg, Germany Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The definite protocol was submitted to the local ethical committee...". Comment: Approval from the ethics committee was sought. Moreover, the trial was constantly monitored by an executive group Informed consent: Informed consent was sought from participants, as it was a criterion for inclusion/exclusion from the trial Clinical trials registration: ‐ Sample size calculation: Although the trial was planned as a double‐blind trial with a fixed sample size (100 participants) of people admitted to ICU, it was executed as a sequential single‐blind study only in 1 subgroup of participants (polytrauma) and was ended before the planned termination date for ethical reasons | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: Participants were randomised in blocks of 4. The method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: "The study was executed as a single blind sequential trial (a deviation from the planned double blind method) due to ethical reasons" Comment: unclear on who was blinded and how it was executed |
| Blinding (detection bias) | Low risk | Comment: GI bleeding was detected as per the definition in the study protocol, not blinding the outcome assessor to this objective outcome would not have caused detection or performance bias |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | High risk | Comment: Data for only 1 strata of participants (polytrauma) are available, as the strata were terminated as mentioned above in "Adherence to regimen". This appears to be an incomplete report |
| Selective reporting (reporting bias) | High risk | Comment: Data for only 1 strata of participants (polytrauma) are available, as the strata were terminated as mentioned above in "Adherence to regimen" |
| Other bias | Low risk | Comment: The source of funding is not clearly mentioned in the study report. No additional biases were detected |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 94 participants Number analysed:73 participants Total parenteral nutrition (TNP)
TPN + sucralfate
TPN + ranitidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Groups were similar with respect to age and gender. The 2 main reasons for admission were respiratory disease (n = 26) and multiple injuries (n = 14). There is no clear mention of the distribution of clinical features across study groups | |
| Interventions | TPN
TPN + sucralfate
TPN + ranitidine
Adherence to regimen: Quote: "24 participant were withdrawn from the study before the sixth day on protocol with the following reasons for withdrawal: weaned from mechanical ventilation before the sixth day (n = 10), death (n = 8), acute upper GI haemorrhage (n = 5, 2 stress induced gastroduodenal ulcers, 2 chronic duodenal ulcers, 1 stomach cancer) and early tolerance to enteral feedings (n = 1)" Comment: The interventional arms to which these participants were initially randomised are not clearly mentioned in the study report Duration of trial: December 1988 to January 1990 Duration of follow up: not mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: ICU and gastroenterology service, Hospital del Pino, Las Palmas de Gran Canaria, Canary Islands, Spain Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional review board" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Commets: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Commets: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Quote: ”All endoscopic examinations, except a few cases performed on emergency basis was done by a single investigator uninformed as to the treatment group” Comment: Outcome assessors were mostly blinded and GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on the blinding of outcome assessors. However, all other outcomes were objective in nature |
| Incomplete outcome data (attrition bias) | High risk | Comment: Of the 97 participants, 24 were withdrawn from the study, as they did not complete a minimum of 6 days in the ICU as required by the protocol of this study. The interventions to which they were originally randomised were not clear from the study report. A per‐protocol analysis was done for the outcomes of interest, but there appears to be an imbalance between groups with respect to the final number of participants available for analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Unclear risk | Comment: Source of funding is not clearly stated. Baseline characteristics (clinical) are not clearly mentioned for each group. No additional biases were detected |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 114 participants Numner analysed:114 participants Cimetidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There was no significant difference in mean age, sex ratio, or number of patients admitted to medical or surgical services" Comment: The 2 groups were similar with respect to their baseline characteristics | |
| Interventions | Cimetidine
Sucralfate
Adherence to regimen: 114 participants who met the inclusion criteria were enrolled into the study. 25 (22%; 12 participants from cimetidine group and 13 participants from sucralfate group) participants were withdrawn from the study for the following reasons:
The remaining 89 participants constituted the study group Duration of trial: January 2009 to September 2009 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Note: Other than the CDC and prevention criteria, each participant was required to have 2 chest roentgenograms showing persistent infiltrates, with agreement by intensivist and radiologist Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported in report but not used in review
| |
| Notes | Setting: Medical and Surgical Intensive Care Unit, Springfield, Mass, Tufts University Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional review board" Informed consent: Quote: "Informed consent was obtained from each patient or guardian" Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ”Patients were randomised according to computer generated numbers to receive either cimetidine or sucralfate” |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report. |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Comment: Outcome assessors were not blinded, but GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) | Low risk | Quote: "The radiologist and the intensivist who assessed the chest roentgenograms for diagnosing pneumonia were both blinded to the treatment that the participant was receiving" Comment: Nosocomial pneumonia was detected as per the definition in the study protocol by an outcome assessor who was blinded to participant data |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on the blinding of outcome assessors. However, outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "...with 10 mL of water. Twenty‐five patients (22%) were withdrawn from the study for the following reasons: two because of extubation within 48 hours of enrolment and discharge from the ICU, two because the nasogastric tube was removed (the nasogastric tube was required to instil sucralfate into the stomach and monitor bleeding), three at the request of the patient or guardian, eight because they died within 48 hours, four because of inadvertent medication change, three because of documented cases of aspiration by a witness, three because of adverse reactions from medications (two patients in the cimetidine group had confusion and neutropaenia, and one patient in the sucralfate group could not tolerate the nasogastric tube clamped). The patients who were withdrawn were equally distributed between the two treatment groups. The remaining 89 patients constituted" Comment: no incomplete reporting of outcomes suspected |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is not clearly mentioned in the study design. No other sources of bias suspected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 214 participants Number analysed: 214 participants Pantoprazole
Placebo
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalances: ‐ | |
| Interventions | Pantoprazole
Placebo
Adherence to regimen: ‐ Duration of trial: January 2014 to January 2015 Duration of follow‐up: 90 days | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: ICU Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not enough details reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "double blind fashion" Comment: no details on blinding reported. Lack of blinding is unlikely to introduce bias to outcome measures or outcomes |
| Blinding (detection bias) | Low risk | Quote: "(haematemesis, bloody gastric aspirate, melaena or haematochezia), clinically significant bleeding (overt bleeding accompanied by a drop in mean arterial pressure > 20mmHg, or reduction in haemoglobin > 20g/L, or need for surgical intervention)" Comment: criteria for diagnosis of GI bleeding described. |
| Blinding (detection bias) | Unclear risk | Comment: no criteria for diagnosis of VAP described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no information about blinding of outcome assessors described |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: not enough information reported to assess incomplete outcome data. Conference abstract |
| Selective reporting (reporting bias) | Low risk | Comment: no incomplete reporting of outcomes suspected. All outcomes listed in the Methods section were also reported in the Results section briefly |
| Other bias | Unclear risk | Comment: no other sources of bias suspected, but very little information reported overall |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Overall
Inclusion criteria
Exclusion criteria: ‐ Baseline imbalances: ‐ | |
| Interventions | Pantoprazole
Placebo
Adherence to regimen: ‐ Duration of trial: January 2014 to January 2015 Duration of follow‐up: 90 days | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought in review but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: ICU Sponsorship source: ‐ Ethics approval: ‐ Conflicts of interest: ‐ Informed consent: ‐ Study protocol: ‐ Sample size calculation: ‐ Additinal notes: conference abstract | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned" Comment: not enough details reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "double blind fashion" Comment: no details on blinding reported. Lack of blinding is unlikely to introduce bias to outcome measures or outcomes |
| Blinding (detection bias) | Low risk | Quote: "(hematemesis, bloody gastric aspirate, melena or hematochezia), clinically significant bleeding (overt bleeding accompanied by a drop in mean arterial pressure > 20 mmHg, or reduction in haemoglobin > 20 g/L, or need for surgical intervention)" Comment: outcome measured objectively |
| Blinding (detection bias) | Unclear risk | Comment: no criteria for diagnosis of VAP described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no information about blinding of outcome assessors described |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: not enough information reported to assess incomplete outcome data. Conference abstract |
| Selective reporting (reporting bias) | Low risk | Comment: no incomplete reporting of outcomes suspected. All outcomes listed in the Methods section were also reported in the Results section briefly |
| Other bias | Unclear risk | Comment: no other sources of bias suspected, but too little information reported in this conference abstract to assess other biases |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 89 participants Number analysed: 89 participants Antacids
Cimetidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: ‐ | |
| Interventions | Antacids
Cimetidine
Sucralfate
Adherence to regimen: Quote: "Patients received cimetidine or antacids had the doses of each drug increased if the pH failed to reach 4 on any two consecutive 2‐ hours sampling patients receiving antacids alone at maximal doses whose intragastric pH < 4 received combination therapy to maintain intragastric pH > 4 and therefore were discontinued from the study" "Upper GI bleeding requiring blood transfusions were seen in two patients, one receiving cimetidine and one receiving sucralfate. These two participants were eliminated from the study and were endoscopically shown to have diffuse erosive gastritis" Duration of trial: December 1988 to January 1990 Duration of follow up: not mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes were not classified as primary and secondary, but main outcomes in the study report are as follows; Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Surgical Intensive Care Unit, Rhode Island Hospital/Brown University, Providence, USA Source of funding: Smith Kline Beecham Pharmacuticals Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the Rhode Island Hospital Human welfare protection committee" Informed consent: Quote: "Informed consent was obtained from each participant before induction into the study" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: The most prevalent organisms isolated from NG tube were Candida albicans, Enterococcus organisms, and β‐haemolytic streptococci; they were identical across all 3 groups | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | High risk | Comment: unclear whether outcome assessors were blinded, and definition of GI bleeding not clearly mentioned in the study report |
| Blinding (detection bias) | High risk | Comment: unclear whether outcome assessors were blinded, and definition of pneumonia was not clearly mentioned in the study report |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is not clearly mentioned in the study design. No other source of bias suspected |
| Methods | Randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 51 participants Number analysed: 51 participants Antacids + ranitidine
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Groups were similar in demographic characteristics. Antacid + Ranitidine group had an APACHE II score of 14.7 ± 4.9, and sucralfate group had a score of 13.2 ± 5.1 More participants in the sucralfate group were diagnosed with polytrauma (n = 16 vs 4 in the sucralfate group) | |
| Interventions | Antacids + ranitidine
Sucralfate
Adherence to regimen: no change in dose/regimen mentioned. No information about dropouts Duration of trial: January 1990 to June 1991 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Hospital de Bellvitge‐Princeps d’Espanya Source of funding: grant from the heath ministry Conflicts of interest: ‐ Ethics approval: Study was approved by the clinical investigations committee at the Hospital Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Two episodes of mild upper GI bleeding occurred in each group; this was not of clinical significance | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding (detection bias) | Low risk | Comment: Study mentions that 2 outcome assessors were blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on blinding of assessors for other outcomes. However owing to the objective nature of the outcomes of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | High risk | Comment: All‐cause mortality in the ICU was not separately mentioned for each group. However, all other intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: This study was funded by the ministry of health. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias suspected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 60 participants Number analysed: 46 participants Antacids (Mylanta II)
Prostaglandin ((15)‐R‐15‐methyl prostaglandin E2)
Inclusion criteria
Exclusion criteria
Baseline imbalances: no significant differences between the 2 groups in baseline characteristics such as age, sex, and risk factors. Most participants were diagnosed with intra‐abdominal disease (antacids: 7 and prostaglandin: 10) | |
| Interventions | Antacids (Mylanta II)
Prostaglandin ((15)‐R‐15‐methyl prostaglandin E2)
Adherence to regimen: 14 patients were excluded after randomisation for reasons mentioned above under "Exclusion criteria". 11 of the 12 patients in whom prostaglandin E2 prophylaxis failed (upper GI bleeding) were switched to antacid regimen. One of the 11 patients had "double" failure because antacid also failed Duration of trial: October 1980 to August 1982 Duration of follow up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes were not classified as primary or secondary in the study report; however outcomes reported were as follows. Outcomes sought in review and reported in trial
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Department of Surgery and Medicine Harvard Medical School and Beth Israel Hospital, Boston, Massachusetts Source of funding: Quote: "This study was supported in part by funds from Upjohn University, Kalamazoo, Michigan and the Charls Diana Research Institures, Beth Israel Hospital, Boston" Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the Committee on Clinical Investigation, New Procedures and new Forms of Therapy of the Beth Israel Hospital" Comment: mentioned for only 1 centre Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Patients were selected by a table of random numbers” Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Comment: Outcome assessor was not blinded. GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: Outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 14 patients were excluded from analysis after randomisation for various reasons mentioned in "adherence to the regimen". The interventional arms to which these participants were randomised remain unclear. A per‐protocol analysis was performed, and there appears to be a balance between groups in terms of the number of participants available for final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: This study was supported in part by a research grant from Upjohn University, Kalamazoo, Michigan, and the Charls Diana Research Institures, Beth Israel Hospital. The role of the sponsor in the conduct and reporting of the trial is unclear |
| Methods | Double‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 129 participants Number analysed: 129 participants Ranitidine
Omeprazole
Inclusion criteria
Exclusion criteria
Baseline imbalances: Patients’ age in group A ranged from 5 to 85 years with mean age of 49.19 years. Group B patients were in the age range of 5 to 95 years with mean age of 52.41 years. In the omeprazole group. There were 32 (52.5%) males and 29 (47.5%) females. These numbers were 35 (51.5%) males and 33 (48.5%) females in the ranitidine group. The 2 groups were similar with respect to the baseline risk factors responsible for GI bleeding | |
| Interventions | Ranitidine
Omeprazole
Adherence to regimen: 128 participants were randomised to receive 2 interventions; Ranitidine + Placebo (68) and Omeprazole + Placebo (61). 12 participants died in the first group while 3 died in the second group Duration of trial: June 2000 to January 2001 Duration of follow up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Department of Internal Medicine and Intensive Care Unit, Imam Hossein Hospital, Shahid Beheshti University, M.C., Tehran, Iran Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was approved by the institutional ethics committee..." Informed consent: Quote: "Written consents signed by the patients or their family members were obtained for participation in the study" Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: In the ranitidine group, 4 in 14 participants who had overt bleeding had clinically important GI bleeding. This was 1 in 3 participants from omeprazole group | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Using the table numbers randomly, all ICU beds were divided into two groups of A and B" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Comment: This was a randomised double‐blind placebo‐controlled trial, and outcome assessors appeared to be blinded. GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) | Low risk | Comment: This was a randomised double‐blind placebo‐controlled trial, and outcome assessors appeared to be blinded. VAP was an objective outcome detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: This was a randomised double‐blind controlled trial, and outcome assessors seem to be blinded |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding not known. No other known source of bias |
| Methods | Multi‐centre open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 202 participants Number analysed: 202 participants Pantoprazole (40 mg per 24 hours)
Pantoprazole (40 mg per 12 hours)
Pantoprazole (80 mg per 24 hours)
Pantoprazole (80 mg per 12 hours)
Pantoprazole (80 mg per 8 hours)
Cimetidine
Inclusion criteria
Exclusion criteria
Baseline characteristics: Groups were similar with respect to age, gender, and race. Trauma was the main cause for admission, and coagulopathy was present in 2 participants from the pantoprazole group | |
| Interventions | Pantoprazole
Cimetidine
Adherence to regimen: Quote: "Subjects received study medication within 24 hours of the precipitating stress event and treatment was to be continued for at least 48 hours and up to 7 days. Patients were considered as completers if they received the study medication as described and participated in the study for at least 48 hours" Of 202 participants, 144 remained NPO, while 58 participants were switched over to enterally feed, mainly on day 2 (48 participants) "32 patients (16%) prematurely discontinued from the study. The most frequent reason for premature discontinuation was removal or inability to maintain tolerate NG/OG tube. Other reasons for premature discontinuation from the study included adverse events (3 in total, 2 in pantoprazole and 1 in cimetidine) or legal guardian request" Duration of trial: June 2000 to September 2001 Duration of follow up: up to 30 days | |
| Outcomes | Outcomes sought in review and reported in trial (none of these were primary outcomes of interest for this study)
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: 14 ICU centres across the United States Source of funding: Quote: "Supported by Wyeth Pharmaceuticals, Collegeville, PA" Ethics approval: Quote: "The study conducted according to declaration of Helsinki and its amendments. and was approved by the independent ethics committee or institutional review board at each ICU centre" Informed consent: Quote: "Written informed consent was obtained from all patients or their legal representatives before enrolment" Clinical trials registration: ‐ Sample size calculation: The sample size of 30 patients per group was chosen based on a common standard deviation of 24% for the primary end point (pH response associated with each treatment group), and there was approximately 80% power to detect a mean difference of 18% for treatment group comparisons Additonal notes:H influenzae was one of the most common bacterial isolates among participants who developed pneumonia. The pantoprazole arms were combined to form a common interventional arm vs the H2 receptor antagonist as the review did not aim to investigate efficacy on the basis of dose or mode of administration in the same drug | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...the study centre called the central randomisation centre to obtain the treatment assignment..." |
| Allocation concealment (selection bias) | Low risk | Quote: "...the study centre called the central randomisation centre to obtain the treatment assignment..." Comment: Allocation was probably concealed |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: "One physician per patient remained blinded to the patients treatment assignment and pH data to assess patient’s safety” Comment: Each patient had a physician in charge, who was blinded. Unclear on how this was possible, as it is not a placebo‐controlled trial, and all other personnel were aware of the intervention |
| Blinding (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. VAP was detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However, owing to the objective nature of the outcomes of interest, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants completed the trial and were included in the final analysis. There were no treatment withdrawals and no trial group changes |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Study was supported by Wyeth Pharmaceuticals, Collegeville, PA. The role of the sponsor in the conduct and reporting of the trial is unclear |
| Methods | Randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 30 participants Number analysed: 30 participants Sucralfate
Ranitidine
Inclusion criteria
Exclusion criteria
Baseline characteristics: Quote: "The study sample was homogeneous in terms of age and sex distribution, duration of treatment, severity of illness, inclusion criteria, and basic intensive care regimens" | |
| Interventions | Sucralfate
Ranitidine
Adherence to regimen: ‐ Duration of trial: ‐ Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial ‐ Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Surgical ICUs at a university hospital in Germany Source of funding: ‐ Ethics approval: Quote: "The study was approved by the Ethics Committee of the University of Düsseldorf" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additonal notes: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: no information reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: no information reported |
| Blinding (detection bias) | Unclear risk | Comment: no information reported |
| Blinding (detection bias) | Unclear risk | Comment: This was not an outcome of the study |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: This was not an outcome of the study |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All participants randomised were included in the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes described in the Methods section were included in the Results section |
| Other bias | Low risk | Comment: no other source of bias suspected. Source of funding unclear |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 144 participants Number analysed: 123 participants Antacids
Cimetidine
Inclusion criteria ‐ Exclusion criteria
Baseline imbalances: Quote: "No significant difference exists between the groups in relation to the risk factors such as abdominal trauma, cardiovascular disease, respiratory failure, sepsis, neurologic injury, orthopedic injury, vascular injury, renal failure, hepatic failure, alcohol or drug abuse, metastatic carcinoma, hypotension and history of peptic ulcer. Similarly, the two groups are quite comparable in age and sex ratio" Comment: There was no significant difference between the 2 groups with respect to demographic and baseline risk factors. Nine and 6 participants in both groups had a history of ulcers | |
| Interventions | Antacids
Cimetidine
Adherence to regimen: Quote: "One hundred forty‐four patients were included in this study. Fifty‐eight patients were randomised to the antacid treatment group and 65 patients were randomised to the cimetidine therapy group. Forty‐six patients were not included in these results because they met protocol criteria for less than 24 hours. Twenty‐one patients required no therapy because of persistent pH ≥ 4. Forty‐eight (74%) cimetidine recipients had the expected elevation of pH ≥ 4. Seventeen (26%) failed despite maximum dosage of cimetidine. All failures with cimetidine had antacids added and responded successfully. No failure of antacid therapy was seen in this study and therefore no patients crossed over from the antacid therapy group into the cimetidine therapy group.The twenty‐one patients who maintained a gastric pH ≥ 4 required no prophylaxis and were not further dealt with further in this paper. All studies continued until the patient no longer required gastric decompression or was discharged from the intensive care unit as a result of improvement or death" Comment: also stated that all patients responded to antacid therapy "Forty participants responded to 30 cc every hour to maintain gastric pH at the desired level. Only three patients required 120 cc per hour to maintain this level. In the group receiving cimetidine, 34% responded to 300 mg every six hours. An additional 40% of the participants responded to administration at a dosage level of every four or every three hours" Duration of trial: October 1978 to July 1979 Duration of follow up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial (none of these were primary outcomes of the study)
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Harborview Medical Center Surgical Intensive Care Unit (SICU), 325 Ninth Avenue, Seattle, WA 98104 Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: It is mentioned that diarrhoea occurred in 5 of the 75 participants treated with antacids. This included 58 antacid‐treated participants and 17 participants in whom cimetidine "failed" as per the study protocol. These data are unclear on the incidence in the original denominator, so could not be analysed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The randomisation process was based on a random number table in a blinded fashion" |
| Allocation concealment (selection bias) | Unclear risk | Comment: Not clearly mentioned in the study report. However, the baseline there is no imbalance in baseline characteristics, indicating low risk of selection bias |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Day‐to‐day clinical management was performed by an attending physician independent of the study protocol" Comment: Personnel were blinded |
| Blinding (detection bias) | Low risk | Comment: no clear mention of blinding of outcome assessors. GI bleeding was an objective outcome detected as per the definition in the study protocol |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: A per‐protocol analysis was performed to measure the outcomes of interest. Only 123 of 144 participants were accounted for in the analysis. Groups to which these 67 participants were randomised remains unclear. However, there is no imbalance between groups with respect to the number of participants available for final analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes stated were analysed and reported |
| Other bias | Low risk | Comment: source of funding not clear. No other known form of bias detected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 27 participants Number analysed: 27 participants Pantoprazole I
Pantoprazole II
Ranitidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: No statistical differences in age, sex, and basal pH and SOFA were found between treatment groups | |
| Interventions | Pantoprazole I
Pantoprazole II
Ranitidine
Adherence to regimen: ‐ Duration of trial: April 2010 to August 2011 Duration of follow‐up: 2 days | |
| Outcomes | Outcomes sought in review and reported in trial ‐ Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: ICU, Department of Pharmacotherapy, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Study protocol was approved by our institutional ethics committee Informed consent: Written consent form was obtained from each patient’s closest family member Clinical trials registration: This trial is registered in www.anzctr.org.au Sample size calculation: ‐ Additional notes: | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to 3 study groups according to a computer‐generated table of random numbers" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not enough information reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: insufficient information to allow judgement, but the outcome is unlikely to be influenced by performance bias |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: insufficient information to allow judgement, but the outcome is unlikely to be influenced by detection bias, provided it was a laboratory measurement. |
| Incomplete outcome data (attrition bias) | Low risk | Comment: no incomplete outcome data suspected. All patients randomised at baseline were also included in the analyses |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes listed in the Methods section were also reported in the Results |
| Other bias | Low risk | Comment: no other sources of bias suspected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 24 participants Number analysed: 20 participants Ranitidine
Pantoprazole
Inclusion criteria
Exclusion criteria
Baseline imbalances: Participants were comparable with respect to age, gender, and other clinical characteristics including the presence of H pylori, APACHE II score was 12 +/‐ 7 and 16 +/‐ 4 for ranitidine and pantoprazole groups | |
| Interventions | Ranitidine
Pantoprazole
Adherence to regimen: Quote: "Four patients, 3 from ranitidine group, were excluded for technical reasons” Duration of trial: ‐ Duration of treatment: ‐ Duration of follow‐up: not clearly mentioned in the trial report. Probably until discharge or an untimely event of death | |
| Outcomes | Outcomes sought in review and reported in trial (none of these were the primary outcomes of the trial)
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: State University of Campinas (UNICAMP), Campinas, São Paulo, Brazil Source of funding: Quote: “Support funds for research and development were provided by FAEPEX. Fundação de Apoio ao Ensino, à Pesquisa e à Extensão” Conflicts of interest: ‐ Ethics approval: Quote: "The present study was approved by the ethics committee of the State University of Campinas, Brazil (no. 035/2003, dated February 18, 2003)" Informed consent: Quote: "The informed consent form was signed by a relative of each patient included in the study, as required by Resolution no. 196/96 of the National Health Council, Brazilian Ministry of Health, concerning research involving human beings" Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and different modes of administering the study interventions would not have made it possible to blind study personnel and participants.Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: This was an open‐label trial, and outcome assessors were not blinded. However GI bleeding was an objective outcome that had to be detected as per the study protocol |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: This was an open‐label trial, and outcome assessors were not blinded. However the outcome of interest was an objective outcome, so the likelihood of detection bias is low. |
| Incomplete outcome data (attrition bias) | High risk | Quote “Four patients, 3 from ranitidine group, were excluded for technical reasons” Comment: Nearly 16% of randomised participants were excluded from analysis. An intention to treat analysis was not performed.This would have contributed to attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes have been reported |
| Other bias | Low risk | Comment: Funding was provided by FAEPEX. Fundação de Apoio ao Ensino, à Pesquisa e à Extensão.However, the role of the sponsor in the conduct and reporting of the trial is unclear |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 424 participants Number analysed: 242 participants Sucralfate
Antacids
Ranitidine
Inclusion criteria
Exclusion criteria
Baseline imbalances: Participants were similar in the different groups with respect to age, gender, race, median Injury Severity Score (ISS), Trauma Score (TS), Glasgow Coma Scale Score, and APCHE II (Acute Physiology and Chronic Health Evaluation) Score and Habits (Tobacco and Caffine consumption). The only significant baseline difference observed in the 11 variables recorded for participants in the 3 drug treatment groups were median alcohol level on admission (P = 0.045) and self‐reported use of alcohol (P = 0.007) | |
| Interventions | Sucralfate
Antacids
Ranitidine
Adherence to regimen: Between November 1990 and May 1994, 424 patients were randomised to 1 of 3 treatment regimens. One hundred eighty‐two patients were discontinued from study participation, mainly because of the following occurring within 24 hours of study entry:
A patient was considered to have discontinued the study if gastrointestinal haemorrhage occurred before study day 7, if diet by mouth or by gastric feeding was begun before day 7, or if the study drug was discontinued or changed. A patient was considered to have completed the study if the patient developed pneumonia > 24 hours after study entry, completed study day 7, was extubated for 48 hours before study day 7, or died. Data from patients who died before study day 7 but who completed at least 24 hours on the study were included in the analysis. All patients randomised to the study were followed‐up until hospital discharge for the outcome variables of laboratory, roentgenogram, and clinical signs of pneumonia, GI bleeding, or death. Forty of the 242 patients completed all 7 study days without getting pneumonia > 24 hours after study entry or dropping out of the study Duration of trial: November 1990 to May 1994 Duration of follow up: Quote: "Mean follow up for intention to treat was 24.3 days (median, 16 days)" "The mean follow‐up time for patients in the drug treatment groups was 27.3 days, with a range of 1 to 257 days (median, 19 days)" Participants were followed up until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial Outcomes are not categorised as primary and secondary
Note: Greatest incidence of pneumonia developed within 7 days into the study Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Carolinas Medical Center, Charlotte, North Carolina , USA Source of funding: Quote: "This project was supported by the Health Services Foundation of Carolinas Medical Center” Conflicts of interest: ‐ Ethics approval: Quote: "The study protocol was reviewed and approved by the institutional review board at Carolinas Medical Center" Informed consent: Quote: "10 participants were excluded because consent was not signed" Clinical trials registration: ‐ Sample size calculation: Sample size analysis showed that 80 participants were needed in each treatment arm (assuming an alpha level of 0.05 and a power of 80%). This assumes a pneumonia rate of 40%, which is supported by previous studies in intensive care unit patients, and also assumes that one of the treatments would reduce the rate to 20%, enough to be meaningful clinically; therefore, our goal was to recruit 240 assessable patients Additional notes: Gram‐negative micro‐organisms were the most common tracheal isolates both in participants with and without pneumonia, and 13% of participants who developed pneumonia had retrograde colonisation from stomach or trachea | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Patients were randomised using a computer‐generated random number table to one of the following stress ulcer prophylaxis regimens...” Comment: Method adopted to obtain random sequence generation is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded. GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded. Pneumonia was an objective outcome that was detected as per the definition in the study protocol |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 424 patients were randomised to 1 of 3 treatment regimens. One hundred eighty‐two patients were discontinued from study participation for reasons mentioned under 'adherence of regimens'. Intention to treat was done for the outcomes of pneumonia and mortality only. All participants who were randomised to the 2 groups were not analysed for all outcomes, and a per‐protocol analysis was done. The number of participants available for analysis appears to be balanced between groups. Therefore, the likelihood of bias due to attrition is low |
| Selective reporting (reporting bias) | High risk | Comment: All intended outcomes were analysed and reported, but intention to treat was done for the outcomes of pneumonia and mortality only |
| Other bias | Low risk | Comment: Source of funding was Health Services Foundation of Carolinas Medical Center. The role of the sponsor in the conduct and reporting of the trial is unclear. No other known source of bias |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised:100 Number analysed: 100 Cimetidine
Antacids
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There were no statically significant differences among the three treatment groups on basic or laboratory parameters" Comment: The 3 groups were similar with respect to demographic data | |
| Interventions | Cimetidine
Antacids
Sucralfate
Adherence to regimen: Quote: "... nausea and vomiting led to the discontinuation of antacid in four patients. The interval between administration of antacid doses had to be extended to four hours in 18 patients following the removal of their stomach tubes because they refused more frequent doses. Moreover, antacids could not be administered every two hours during the night in patients without a stomach tube since it was unreasonable to awaken them repeatedly for this purpose. Nausea or vomiting occurred in nine patients receiving sucralfate following the removal of the stomach tubes. This necessitated discontinuing the drug in one Duration of trial: September 1982 to December 1983 Duration of follow up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Department of Anaesthesiology and Biometry Hannover School of Medicine, Hannover, Federal Republic of Germany Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The study was carried out from September 1982 to December 1983 with institutional approval and in accordance with the guidelines of the German Drug Law" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants |
| Blinding (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded. However, GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: All other outcomes of interest were objective in nature |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on the source of funding. No other form of bias detected |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 100 participants Number analysed: 100 (for all outcomes except for pneumonia, reasons mentioned below) Antacid
Sucralfate
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "Both the groups were comparable on basic clinical or laboratory parameters" Comment: Both groups were comparable with respect to the distribution of demographic characteristics and risk factors | |
| Interventions | Antacids
Sucralfate
Adherence to regimen: Antacid therapy was discontinued in 3 participants (vomiting after extubation (n = 2) and alkalosis on the second day (n = 1)) Duration of trial: July 1984 to November 1986 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Department of Anaesthesiology, Hannover School of Medicine, Hannover, Federal Republic of Germany Source of funding: Quote: “This study was supported by a grant from E. Merck” Ethics approval: Quote: "...with the approval of the local ethical committee and in accordance with the guidelines of the German Drug Law" Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded. GI bleeding was detected as per the definition in the study protocol. Therefore, the likelihood of detection bias is low |
| Blinding (detection bias) | Low risk | Quote: "The diagnosis of pneumonia was done by a physician who was unaware of the object of the study” Comment: There was clear definition for diagnosing pneumonia, and the outcome assessor was blinded to the interventions. Therefore, the likelihood of detection bias is low |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However, outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Quote: “For analysis of pulmonary infections, 39 participants were withdrawn from the study because of thoracic trauma or pneumonia at the time of admission to ICU (18 from antacid arm and 21 from sucralfate arm)” For all other outcomes, all 100 randomised participants were part of the analysis Comment: Excluding these participants is justified as it could not have led to the true estimate of the outcome (incidence of nosocomial pneumonia) due to the intervention |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Study was supported by a grant from E. Merck. The role of the sponsor in the conduct and reporting of the trial is unclear. No other form of bias detected |
| Methods | Randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 400 participants Number analysed: 400 participants Ranitidine
Pirenzepine
Inclusion criteria
Exclusion criteria
Baseline imbalances: no significant differences | |
| Interventions | Ranitidine
Pirenzepine
Adherence to regimen: ‐ Duration of trial: October 1984 to November 1986 Duration of follow‐up: unclear, but might be until discharge from ICU | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Anaesthesiology Department, Hannover and Bochum, Germany Source of funding: ‐ Conflicts of interest: ‐ Ethics Approval: Study was conducted according to the drug law. No further information Informed Consent: ‐ Clinical Trials Registration: ‐ Sample Size Calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded. GI bleeding was an objective outcome that was detected as per the definition in the study protocol |
| Blinding (detection bias) | Low risk | Comment: unclear whether outcome assessors were blinded. Pneumonia was an objective outcome that was detected as per the definition in the study protocol. Outcome assessor was blinded to study aims |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: Outcome assessor was blinded to study aims. Therefore, low risk of bias |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All randomised participants were part of the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes have been reported |
| Other bias | Low risk | Comment: No additional biases were detected |
| Methods | Double‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 34 participants Number analysed: 28 participants Cimetidine
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote "All the factors including the distribution of patients according to sex, age, nature of intensive care unit were almost same within the two groups except for the mean risk factors (it was 2.6 per person in the cimetidine group and 1.9 in the placebo group)" Comment: More people in the cimetidine group had 3 or more risk factors when compared with the placebo group | |
| Interventions | Cimetidine
Placebo
Adherence to regimen: 34 participants entered the study. While 28 completed the trial, 6 people dropped out for the following reasons: 1 participant died on the second day of the study from sepsis, 1 participant developed exanthema on the second day after which all medication was stopped, 1 participant was inadvertently given open cimetidine, 1 participant had bleeding duodenal ulcer proven endoscopically at the end of the study, 1 participant developed anuria, and 1 participant proved to have previous gastric surgery Duration of trial: ‐ Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported in trial but not used in review
| |
| Notes | Setting: Rotterdam University Hospital, Netherlands Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "The study was performed double blind. The investigators were not involved in the treatment of the patients" Comment: This was a placebo‐controlled trial, and study personnel and investigators were blinded. Therefore, the likelihood of performance bias is low |
| Blinding (detection bias) | Low risk | Quote: "The study was performed double blind. The investigators were not involved in the treatment of the patients" Comment: GI bleeding was detected as per the definition in the study protocol and the investigators were blinded. Therefore, the likelihood of detection or performance bias is low |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "The study was performed double blind. The investigators were not involved in the treatment of the patients" Comment: Study was performed as a double‐blinded randomised controlled trial, and the outcomes of interest were objective in nature. Therefore, the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Not all randomised participants were part of the final analysed. 6 participants were excluded for reasons mentioned under "Adherence to the regimen". Moreover, it is unclear to which group these participants belonged, and an intention‐to‐treat analysis was not done. A per‐protocol analysis was performed for the outcomes of interest, and the number of participants available for analysis appears to be balanced between groups. Therefore, the likelihood of attrition bias is low |
| Selective reporting (reporting bias) | High risk | Comment: All intended outcomes were analysed and reported. All‐cause mortality in ICU was not specifically reported for each group |
| Other bias | Low risk | Comment: unclear on the source of funding. No additional biases were detected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 90 participants Number analysed: 58 participants Prostaglandin
Placebo
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "29 participants in both the groups were similar with regard to sex distribution, age and number and nature of risk factors" Comment: no significant difference between the 2 groups with respect to demographic and baseline risk factors | |
| Interventions | Prostaglandin
Placebo
Adherence to regimen: Quote: "The study lasted 3 ‐ 7 days. Treatment was discontinued if enteral feeding was given within the first three days, if gastric tube was removed or participant underwent surgery or had GI bleeding" Of 32 participants, 8 participants had gastrectomy performed within first 3 days; in 1 participant, no chromium labelling was performed, 6 participants were discharged within first 3 days from ICU 3 participants showed non‐compliance with entry criteria, 12 participants received enteral feeds or underwent gastric tube removal within first 3 days, and 9 participants died within the first 3 days Duration of trial: November 1981 to September 1983 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Departments of Internal Medicine and Surgery, University Hospital, Dijkzigt, Rotterdam, The Netherlands Source of funding: Quote: "The study was supported, in part, by a grant–in‐aid from Upjohn Inc., Kalamazoo, MI" Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Patients were randomly assigned to receive placebo or prostaglandin therapy in a double blind fashion" Comment: This was a placebo‐controlled trial where most likely participants and study personnel were blinded to the interventions. Therefore, the likelihood of performance bias is low |
| Blinding (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However, GI bleeding was an objective outcome that was detected as per the definition in the study protocol. Therefore the likelihood of detection bias is low |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear on blinding of outcome assessors. However, the outcome of interest was objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Not all randomised participants were part of the final analysis. 90 were randomised, and only 58 were part of the study. However, this was a per‐protocol analysis, and the numbers were well balanced between groups. Therefore there is no serious attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported in the study |
| Other bias | Low risk | Comment: Upjohn Inc., Kalamazoo, MI, partially funded the study. The role of the sponsor in the conduct and reporting of the trial is unclear. No other sources of bias are suspected |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 100 Number analysed: 100 Bowel stimulation protocol
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: none | |
| Interventions | Bowel stimulation protocol
No prophylaxis
| |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought in review and not reported in trial
Outcomes reported in trial and not used in review
| |
| Notes | Setting: Department of ICU, the Second Hospital Affiliated to Tianjin Medical University, Tianjin, China Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Comment: mentioned in the study report that study authors sought ethics approval Informed consent: Comment: mentioned in the study report that study authors sought for informed consent Clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: Patients were randomly divided into an intervention group and a control group, with 50 patients in each group, but the detailed methods were not mentioned |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: lack of blinding of participants and healthcare providers, but outcomes were measured objectively |
| Blinding (detection bias) | Low risk | Comment: Outcome was measured objectively, and no detection bias is suspected |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no information reported |
| Incomplete outcome data (attrition bias) | Low risk | Comment: no missing data detected |
| Selective reporting (reporting bias) | Low risk | Comment: All outcomes described in Methods were reported in Table 2 |
| Other bias | Unclear risk | Comment: Detailed diagnosis of GI dysfunction was not reported ‐ probably differential diagnostic activity between intervention group and control group |
| Methods | Parallel‐group randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: ‐ Number analysed: ‐ Famotidine
Pantoprazole
Inclusion criteria: each patient admitted to ICU/CCM Exclusion criteria: ‐ Baseline imbalances: Both groups had similar baseline characteristics including risk factors for SRMB (2.7 vs 2.7, P = 0.50); however, the pantoprazole group had higher APACHE‐II scores (23.9 vs 20.1; :P < 0.01) | |
| Interventions | Famotidine
Pantoprazole
Adherence to regimen: ‐ Duration of trial: December 2012 to April 2013 Duration of follow‐up: until discharge | |
| Outcomes | Outcomes sought in review and reported in trial
Outcomes sought in review and not reported in trial
Outcomes reported in trial, but not used in review ‐ | |
| Notes | Setting: ICU/CCU, Kingsbrook Jewish Medical Center, Brooklyn, NY 11203, USA, Arnold & Marie Schwartz College of Pharmacy and Health Sciences, Long Island University, Brooklyn, NY 11201, USA Sponsorship source: ‐ Conflicts of interest: ‐ Comments: 2 conference abstracts reporting on 1 study Ethical approval: Quote: "Expedited IRB approval was granted" Informed consent: Quote: "Informed consent was not required" Sample size calculation: ‐ Clinical trials registration: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomized" Comment: not enough details reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no information reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: no information on blinding reported. Lack of blinding is unlikely to introduce bias to outcome measures and outcomes |
| Blinding (detection bias) | Unclear risk | Comment: not enough details described |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: no information on blinding of outcome assessors reported. Lack of blinding might potentially introduce bias |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: conference abstracts. Not enough information reported to assess incomplete outcome data |
| Selective reporting (reporting bias) | Unclear risk | Comment: Secondary outcomes listed in the Methods sections of the two abstracts differ slightly. Not enough information reported to assess selective outcome reporting |
| Other bias | Unclear risk | Comment: not enough information reported to assess other sources of bias |
| Methods | Quasi‐randomised trial | |
| Participants | Baseline characteristics Number randomised: 77 participants Number analysed: 77 participants Antacid
Cimetidine (300 mg every 4 hours)
Cimetidine (300 mg every 6 hours)
Cimetidine (400 mg every 4 hours)
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "A modification of Theraputic intervention scoring system (TISS) was used to grade severity of illness for patient selection and group comparison" Comment: Average TISS score was 3 for antacids and 3.16 for combined cimetidine groups | |
| Interventions | Antacids
Cimetidine
Adherence to regimen: Quote: "The pH control in the antacid group was achieved with an average dose of 30 mL of antacid every two hours (range 15 to 120 mL/hour). Only one patient required more than 60 mL/hour for pH control. The duration of cimetidine treatment was similar in the three groups regardless of the efficacy of pH control" Comment: According to the study report, pH was controlled in 14 cimetidine participants. The rest might have switched over to antacids according to the study protocol (but this is not clearly mentioned in the study report) Duration of trial: February 1979 to August 1979 Duration of follow up: Quote: "All patients were followed up until death, discharge from ICU or institution of enteral feedings" | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: Departments of Surgery, South‐Western Medical School, University of Texas Health Science Centre, Dallas Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Informed consent: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: Cimetidine groups were combined to form a common interventional arm vs antacids, as the review did not aim to investigate efficacy among different routes of administration of the same intervention | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote: "Patients entering the study were randomised by hospital unit number into one of the four groups…" Comment: This was a quasi–randomised trial, and sequence was not generated |
| Allocation concealment (selection bias) | High risk | Quote: "Patients entering the study were randomised by hospital unit number into one of the four groups…" Comment: This was a quasi–randomised trial, and allocation was not concealed |
| Blinding of participants and personnel (performance bias) | High risk | Comment: Tthis was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: unclear on the blinding of outcome assessors. However, GI bleeding was an objective outcome that was detected as per the study definition, so the likelihood of performance or detection bias is low |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: Outcomes of interest were objective in nature, so the likelihood of performance or detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: All quasi‐randomised participants were part of the final analysis, so there is no attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: unclear on source of funding. No other form of bias detected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 200 participants Number analysed: 160 participants Sucralfate
Rantidine
Omeprazole
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: no significant difference between groups with respect to age, gender, or primary disease (sepsis and bronchitis were the most common primary diseases among participants) | |
| Interventions | Sucralfate
Ranitidine
Omeprazole
No prophylaxis
Adherence to regimen: Quote: "Administration of the drugs were unblinded and were begun within 6 hours of PICU admission" "40 patients were excluded from study according to the exclusion criteria" Duration of trial: August 2000 to February 2002 Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: PICU of Faculty of Medicine, Cukurova University, Adana, Turkey Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: ‐ Clinical trials registration: ‐ Sample size calculation: ‐ Additional notes: According to the study, hospital mortality was defined as deaths occurring in the PICU, and hospital stay was defined as days spent in the PICU. Pseudomonas aeruginosa and Klebsiella pneumoniae were the most common organisms cultured in participants with the diagnosis of ventilator‐associated pneumonia | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomised to one of the following stress ulcer prophylaxis regimens by using a computer generated random number table" Comment: The method adopted to generate a random sequence is clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in study report |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "Administration of drugs were unblinded..." Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Therefore, high risk of performance bias |
| Blinding (detection bias) | Unclear risk | Comment: not clear whether outcome assessors were blinded. Moreover, the definition for diagnosing GI bleeding, which is an objective outcome, is not clearly mentioned in the study report. Not clear whether this would have caused any detection bias |
| Blinding (detection bias) | Low risk | Comment: not clear whether outcome assessors were blinded. But ventilator‐associated pneumonia was assessed as per the definition in the study report. Therefore, the likelihood of performance or detection bias is low |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: not clear whether outcome assessors were blinded. However, the other outcomes of interest were objective in nature, so the likelihood of performance or detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "...200 patients were randomised to one of the 4 treatment regimens. 40 patients were excluded from study according to the exclusion criteria" Comment: A per‐protocol analysis was done, and only those participants who completed ≥ 48 hours in the study were part of the final analysis. There is also a balance between groups with respect to the number of participants analysed. Therefore, the likelihood of attrition bias is low |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes have been analysed and reported |
| Other bias | Low risk | Comment: unclear on the source of funding. No additional biases were detected |
| Methods | Open‐label randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 300 participants (the report says that 40 additional participants were entered into the randomised trial, not sure if they were randomised) Number analysed: 300 participants Cimetidine
Antacids
No prophylaxis
Inclusion criteria
Exclusion criteria
Baseline imbalances: The 3 groups were similar with respect to age, gender, and medical conditions. Most participants were admitted for cardiac or general surgery | |
| Interventions | Cimetidine
Antacids
No prophylaxis
Adherence to regimen: 33 participants who met the inclusion criteria were randomised (100 in each treatment arm). 40 additional participants entered into the study but were removed for the following reasons: protocol errors or request from physician (n = 31), no reason mentioned (n = 9) Duration of trial: December 1977 to December 1979 Duration of follow‐up: not clearly mentioned in the study report. Probably until death or discharge | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcome
Secondary outcomes
Outcomes sought but not reported in trial
Outcomes reported but not used in review
| |
| Notes | Setting: surgical intensive care unit of the Johns Hopkins Hospital and the intensive care unit of the Baltimore City Hospitals Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: study approved by the joint committee on clinical investigation of John Hopkins Medical Institutions Informed consent: Quote: "Informed consent was obtained from all participants" clinical trials registration: ‐ Sample size calculation: ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | High risk | Comment: This was not a placebo‐controlled trial, and the different modes of administering study interventions would not have made it possible to blind study personnel and participants. Moreover, 2 arm received no prophylaxis. Therefore, high risk of performance bias |
| Blinding (detection bias) | Low risk | Comment: It is unclear whether outcome assessors were blinded. GI bleeding was an objective outcome detected as per the definition in the study protocol. Therefore, the likelihood of detection bias is low |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: It is unclear whether outcome assessors were blinded. Moreover, outcomes of interest were objective in nature. Therefore the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "Three hundred participants entered the inclusion criteria and were randomised to three treatment groups. Forty additional participants were entered into RCT but were removed from the protocol. Thirty one of these participants were excluded because of protocol errors or because of request by physician. These were evenly distributed between the treatment groups" Comment: The reason for exclusion of the remaining 9 participants is not clear. The study arms of these 40 participants are also not clearly mentioned in the study report. However, there appears to be no imbalance between study groups with respect to the number of participants available for analysis. Therefore the likelihood of bias due to attrition is low |
| Selective reporting (reporting bias) | Low risk | Comment: All intended outcomes were analysed and reported |
| Other bias | Low risk | Comment: Source of funding is unclear. No other known source of bias |
| Methods | Single‐blind randomised controlled trial | |
| Participants | Baseline characteristics Number randomised: 371 Number analysed: 281 (for primary outcome) Misoprostol
Antacid
Inclusion criteria
Exclusion criteria
Baseline imbalances: Quote: "There were no statistical differences between the two treatment groups with respect to age, sex, or race. They were also comparable in mean height, weight, and vital signs on admission, and these did not differ with respect to study site. Initial gastric lesion scores were 0 or 1 in 88% of the patients, and initial duodenal lesion scores were 0 or 1 in 96% of the patients, with no significant differences between the two treatment groups (p = 0.141 and 0.848, respectively)" Comment: The 2 groups were similar with respect to demographic and baseline risk factors | |
| Interventions | Misoprostol
Antacid
Adherence to regimen: Quote: "Comparable numbers of patients completed the 14 days of treatment or were released earlier, having met the dietary requirement. By the eighth day, only 16% of the misoprostol group and 21% of the antacid group remained in the ICU (p = not significant). Total time of nasogastric medication administration was also somewhat shorter in the misoprostol group (655 patient days vs. 731 patient days in the antacid group), but again these differences were not statistically significant.", "141 in Misoprostol and 140 in Antacid were only analysed as they met the following four criteria for other evaluations 1. completed at least three days in the study, 2. took at least 80% of the assigned medication, 3. did not withdraw from the study except for side effects of the medication, 4. had sufficient follow up endoscopy information to permit outcome evaluation" Comment: 46 participants on misoprostol and 44 participants on antacid regimens were not unavailable for analysis of the primary outcome of GI bleeding Duration of trial: ‐ Duration of follow‐up: ‐ | |
| Outcomes | Outcomes sought in review and reported in trial Primary outcomes
Secondary outcomes
Outcomes sought but not reported in trial report
Outcomes reported in report but not used in review
| |
| Notes | Setting: 16 university‐associated medical centres: King/Drew Medical Center, Los Angeles, University of Minnesota Hospitals, Minneapolis, MN, Georgetown University Hospital, Washington, DC, Denver General Hospital, Denver, Health Science Center, Brooklyn, NY: Cook County Hospital, Chicago, IL, Minneapolis VA Medical Center, Minneapolis, Hines VA Hospital, Milton S. Hershey Medical Center, VA Medical Center, Fresno, CA, W.P, Temple University Hospital, Philadelphia, PA, University of California, Irvine, CA, VA Medical Center, San Francisco, CA, University Texas Health Science Center, Dallas, TX, Johns Hopkins Medical Institutions, Baltimore Source of funding: ‐ Conflicts of interest: ‐ Ethics approval: Quote: "The Institutional Review Board at each of the study sites approved the protocol" Informed consent: Quote: "Each patient gave written informed consent" Clinical trials registration: ‐ Sample size calculation: Quote: "The protocol was designed to require a minimum of 270 fully evaluable patients to complete the study (135 in each group). This sample size is sufficient to detect differences of 20% or more between two treatment groups (p = 0.05; power = 0.90) with two‐sided tests of significance. That is, this study size should detect a clinically significant difference between misoprostol and antacid that is greater than 20%" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: not clearly mentioned in the study report |
| Allocation concealment (selection bias) | Unclear risk | Comment: nNot clearly mentioned in the study report |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Acceptable candidates were randomly assigned to each group and they received either tablets containing 200 mcg of misoprostol every four hours plus placebo liquid antacid every two hours, or placebo tablets every four hours plus magnesium‐aluminum hydroxide liquid antacid every two hours. Tablets were dissolved in 20 mL of water and administered six times daily through a nasogastric tube, or were given orally if no tube was in place" Comment: This was a 'double‐dummy' placebo‐controlled trial, and participants and study personnel appear to be blinded. Therefore, the likelihood of performance bias is low |
| Blinding (detection bias) | Low risk | Comment: This was a 'double‐dummy' placebo‐controlled trial. Moreover, GI bleeding was an objective outcome that was detected as per the definition in the study protocol. Therefore, there is no likelihood of performance bias in this study |
| Blinding (detection bias) | Unclear risk | Comment: Study did not address this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: This was a 'double‐dummy' placebo‐controlled trial. However, outcomes of interest were objective in nature, so the likelihood of detection bias is low |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Not all 371 randomised participants were part of the final analysis. The study was designed in such a way that only those participants who met certain criteria for evaluation of the primary outcome (as described under 'adherence to the regimen') were included in the study report (141 participants in misoprostol and 140 participants in antacid groups). A per‐protocol analysis was done. However, the participants for whom outcomes were reported were well balanced between groups. Therefore, the likelihood of bias due to attrition is low |
| Selective reporting (reporting bias) | High risk | Comment: Study says that all randomised participants were analysed for safety of the drug, but this was true only for the adverse event of diarrhoea and not for other adverse events. The reasons for excluding some of the participants is not clearly mentioned in the study report |
| Other bias | Low risk | Comment: unclear on the source of funding. No other form of bias detected |
ADH: antidiuretic hormone.
APACHE: Acute Physiologic Assessment and Chronic Health Evaluation.
APS: acute physiology score.
BAL: bronchoalveolar lavage.
BMI: body mass index.
bw: body weight.
CABG: coronary artery bypass graft.
CNS: central nervous system.
CXR: chest X‐ray.
GCS: Glasgow Coma Scale.
GD: gastroduodenal.
GI: gastrointestinal.
ICU: intensive care unit.
IMED: infusion pump.
IQR: interquartile ratio.
ITT: intention‐to‐treat.
IV: intravenous.
IVAC: infusion pump.
NG: nasogastric.
NPO: nothing by mouth.
NSAID: non‐steroidal anti‐inflammatory drug.
OR: odds ratio.
PaO2: partial pressure of oxygen in arterial blood.
PCT: procalcitonin
PICU: paediatric intensive care unit.
PSB: protected specimen brush.
SD: standard deviation.
SE: standard error.
SICU: surgical intensive care unit.
SOFA: sepsis‐related organ failure assessment score.
SRMD: stress‐related mucosal disease.
SUP: stress‐ ulcer prophylaxis
TISS: therapeutic intervention scoring system.
VAP: ventilator‐associated pneumonia.
WBC: white blood cell.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) | |
| Wrong study population | |
| The review does not deal with comparing different drugs in the same class (H2 blocker antagonists) | |
| Wrong study design | |
| Wrong study design | |
| Wrong study design | |
| Wrong study population | |
| Wrong study population | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) | |
| The review does not deal with comparing different drugs in the same class (H2 blocker antagonists) | |
| Wrong study design | |
| Wrong indication | |
| Wrong setting, no ICU setting | |
| Wrong setting | |
| Wrong study design | |
| Wrong indication/study aim (prevention of gastritis) (conference abstract) | |
| Wrong study design | |
| The review does not deal with comparing different drugs in the same class (proton pump inhibitors) | |
| Wrong intervention and comparison | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) | |
| Wrong population | |
| Wrong study design | |
| Wrong intervention and study aim | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) | |
| Wrong study design | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) | |
| Wrong study design | |
| Wrong population | |
| Wrong setting | |
| The review does not deal with comparing different drugs in the same class (nutrition regimen) | |
| Wrong population | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) | |
| Wrong study design | |
| Wrong intervention and comparison | |
| Wrong study design | |
| Wrong study design | |
| Wrong intervention and comparison | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) | |
| The review does not deal with comparing different drugs in the same class (proton pump inhibitors) | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) | |
| Wrong study population | |
| Wrong indication/study aim (prevention of gastritis) | |
| Wrong study population | |
| Wrong study population | |
| Wrong study design | |
| Wrong study population | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) | |
| Wrong population (patients on continuous venovenous hemofiltration) | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonists) | |
| The review does not deal with comparing different drugs in the same class (proton pump inhibitor) | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) | |
| The review does not deal with comparing different drugs in the same class (nutrition regimen) | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) | |
| Wrong indication (management of gastrointestinal bleeding) | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) | |
| The review does not deal with comparing different drugs in the same class (omeprazole) | |
| Wrong study design | |
| Wrong study population | |
| Wrong study design | |
| Wrong study population | |
| The review does not deal with comparing different drugs in the same class (omeprazole) | |
| Wrong setting | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) | |
| The review does not deal with comparing different drugs in the same class (H2 receptor antagonist) | |
| Wrong setting, no ICU setting | |
| Wrong study population |
ICU: intensive care unit.
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | Randomised controlled trial |
| Participants | People admitted to surgical ICU |
| Interventions | ‐ |
| Outcomes | ‐ |
| Notes | Full text could not be obtained |
| Methods | Randomised controlled trial |
| Participants | People admitted to surgical ICU |
| Interventions | ‐ |
| Outcomes | ‐ |
| Notes | Full text could not be obtained |
ICU: intensive care unit.
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Proton pump inhibitors vs histamine‐2 receptor blockers for ulcer prophylaxis therapy in the intensive care unit |
| Methods | Multi‐centre open‐label cluster‐randomised cross‐over study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: proton pump inhibitors Control: H2 receptor antagonists |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | April 2016 |
| Contact information | Dr. Paul Young, Wellington Hospital, Riddiford Street, Newtown, Wellington 6021, New Zealand; [email protected] |
| Notes | anzctr.org.au/Trial/Registration/TrialReview.aspx?id=370438 |
| Trial name or title | Stress ulcer prophylaxis in the intensive care unit |
| Methods | Parallel‐group double‐blind randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: 4 mL pantoprazole IV Control: placebo |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | August 2016 |
| Contact information | Morten Hylander; [email protected] |
| Notes | clinicaltrialsregister.eu/ctr‐search/search?query=EUCTR2015‐000318‐24‐DK |
| Trial name or title | Omeprazole treatment for prophylaxis of gastrointestinal bleeding and gastro‐oesophageal reflux in critically ill children [Tratamiento con omeprazole para la profilaxis de la hemorragia digestiva y el reflujo gastroesofágico en niños críticos] |
| Methods | ‐ |
| Participants | Infants and toddlers, children, adolescents, under 18 |
| Interventions | Intervention: 0.4 mL omeprazole IV Control: 10 mL ranitidine IV |
| Outcomes | Primary outcomes:
Secondary outcomes: ‐ |
| Starting date | February 2008 |
| Contact information | ‐ |
| Notes | clinicaltrialsregister.eu/ctr‐search/search?query=EUCTR2007‐006102‐19‐ES |
| Trial name or title | Ranitidine and pantoprazole in prevention of stress ulcer |
| Methods | Not stated |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: pantoprazole 40 mg infusion once a day until end of patient admission Control: ranitidine 50 mg infusion twice a day until end of patient admission |
| Outcomes | Primary outcome
Secondary outcome
|
| Starting date | 20 February 2011 |
| Contact information | Farshid Rahimi Bashar, Mahdiye Street, Hamedan University Of Medical Sciences, Hamedan Iran, Islamic Republic of Telephone: 00988118276295; [email protected] |
| Notes | ‐ |
| Trial name or title | A placebo‐controlled double‐blind randomised feasibility trial of desmopressin (DDAVP) in critical illness prior to procedures |
| Methods | Placebo‐controlled double‐blind randomised study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: desmopressin Control: ‐ |
| Outcomes | Primary outcome
Secondary outcome ‐ |
| Starting date | November 2016 |
| Contact information | ‐ |
| Notes | clinicaltrialsregister.eu/ctr‐search/search?query=EUCTR2016‐001126‐33‐GB |
| Trial name or title | Stress ulcer prophylaxis with a proton pump inhibitor vs placebo in critically ill patients (SUP‐ICU trial): study protocol for a randomised controlled trial |
| Methods | Investigator‐initiated pragmatic international multi‐centre randomised blinded parallel‐group study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: pantoprazole 40 mg IV (pantoprazole; Actavis, Gentofte, Denmark) Control: placebo, given once daily IV, from randomisation until ICU discharge or death for a maximum of 90 days |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | January 2016 |
| Contact information | Mette Krag, Department of Intensive Care 4131, Copenhagen University Hospital, Rigshospitalet; [email protected] |
| Notes | ‐ |
| Trial name or title | Gastric pH in critically ill patients |
| Methods | Double‐blind parallel‐group randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: esomeprazole 40 mg once daily Control: ranitidine 50 mg every hour |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | July 2004 |
| Contact information | Christian Madl, MD, Medical University of Vienna |
| Notes | clinicaltrials.gov/ct2/show/record/NCT00590928 |
| Trial name or title | A clinico‐bacteriological study and effect of stress ulcer prophylaxis on occurrence of ventilator associated pneumonia |
| Methods | Parallel‐group open‐label randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: ranitidine 50 mg IV 8‐hourly for entire duration of ICU stay Control: sucralfate 1 g via NG tube 6‐hourly for entire duration of ICU stay |
| Outcomes | Primary outcome
Secondary outcome ‐ |
| Starting date | March 2005 |
| Contact information | Rajiv Singla, MD, Maulana Azad Medical College and Lok Nayak Hospital, Delhi, India |
| Notes | clinicaltrials.gov/ct2/show/record/NCT00702871 |
| Trial name or title | Stress ulcer prophylaxis of intravenous esomeprazole in Chinese seriously ill patients (SUP) |
| Methods | Double‐blind parallel‐group randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: IV esomeprazole 30 min intermittent infusions given for maximum 14 days Control: IV cimetidine 30 min bolus infusion followed by IV cimetidine continuous infusion given for maximum 14 days |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | July 2014 |
| Contact information | Xinyu Qin, Professor and Chairman, Department of General Surgery, Zhongshan, Hospital, Fudan University |
| Notes | clinicaltrials.gov/ct2/show/record/NCT02157376 |
| Trial name or title | Re‐evaluating the inhibition of stress erosions: gastrointestinal bleeding prophylaxis In ICU (REVISE) |
| Methods | Quadruple blind parallel‐group randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: pantoprazole 40 mg in 50 mL 0.9% normal saline intravenously once daily Control: placebo 50 mL of 0.9% normal saline intravenously once daily |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | May 2015 |
| Contact information | ‐ |
| Notes | clinicaltrials.gov/ct2/show/record/NCT02290327 |
| Trial name or title | Sup‐Icu RENal (SIREN) ‐ a subanalysis of the prospective SUP (stress ulcer prophylaxis)‐ICU trial on the risk of GI bleeding in ICU patients receiving renal replacement therapy |
| Methods | Parallel‐group triple‐blind randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: pantoprazole IV Control: 0.9% saline IV |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | February 2016 |
| Contact information | Joerg Schefold; [email protected] |
| Notes | clinicaltrials.gov/ct2/show/record/NCT02718261 |
| Trial name or title | Effects of enteral nutrition on stress ulcer haemorrhage ‐ multi‐center randomised controlled trial |
| Methods | Open‐label parallel‐group randomised controlled study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: enteral nutrition + proton pump inhibitor Control: enteral nutrition only |
| Outcomes | Primary outcome
Secondary outcome ‐ |
| Starting date | August 2016 |
| Contact information | |
| Notes | clinicaltrials.gov/ct2/show/record/NCT03098537 |
APACHE: Acute Physiologic Assessment and Chronic Health Evaluation.
aPTT: activated partial thromboplastin time.
CDI: Clostridium difficile infection.
CT: computed tomography.
DDAVP: desmopressin.
EN: enteral nutrition.
ESRD: end‐stage renal disease.
GI: gastrointestinal.
H2RA: histamine receptor‐2 antagonist.
hCG: human chorionic gonadotropin.
HIV: human immunodeficiency virus.
ICU: intensive care unit.
INR: international normalised ratio.
IV: intravenous.
NG: nasogastric.
NSAIDs: non‐steroidal anti‐inflammatory drugs.
OG: orogastric.
PLT: platelet blood test.
PPI: proton pump inhibitor.
PT: prothrombin time.
RBC: red blood cell.
RRT: rapid resolution therapy.
SAR: serious adverse reaction.
SDM: substitute decision‐maker.
TIA: transient ischaemic attack.
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 30 | 3132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.39, 0.57] |
| Analysis 1.1  Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 H2 receptor antagonists vs placebo or no prophylaxis | 24 | 1844 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.37, 0.59] |
| 1.2 Proton pump inhibitors vs placebo or no prophylaxis | 3 | 159 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.13, 2.59] |
| 1.3 Prostagladin analogues vs placebo or no prophylaxis | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.22, 4.55] |
| 1.4 Anticholinergics vs placebo or no prophylaxis | 2 | 103 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.30, 2.49] |
| 1.5 Antacids vs placebo or no prophylaxis | 7 | 515 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.23, 0.63] |
| 1.6 Sucralfate vs placebo or no prophylaxis | 7 | 453 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.25, 0.87] |
| 2 Nosocomial pneumonia Show forest plot | 9 | 1331 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.90, 1.48] |
| Analysis 1.2  Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia. | ||||
| 2.1 H2 receptor antagonists vs placebo or no prophylaxis | 8 | 788 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.77, 1.46] |
| 2.2 Proton pump inhibitors vs placebo or no prophylaxis | 2 | 149 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.59, 2.17] |
| 2.3 Anticholinergics vs placebo or no prophylaxis | 1 | 72 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.43, 2.59] |
| 2.4 Sucralfate vs placebo or no prophylaxis | 4 | 322 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.59 [0.84, 3.01] |
| 3 All‐cause mortality in ICU Show forest plot | 19 | 2159 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.90, 1.34] |
| Analysis 1.3  Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU. | ||||
| 3.1 H2 receptor antagonists vs placebo or no prophylaxis | 14 | 1209 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.89, 1.53] |
| 3.2 Proton pump inhibitors vs placebo or no prophylaxis | 3 | 180 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.46, 2.38] |
| 3.3 Prostagladin analogues vs placebo or no prophylaxis | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.48, 2.74] |
| 3.4 Anticholinergics vs placebo or no prophylaxis | 2 | 103 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.59, 2.56] |
| 3.5 Antacids vs placebo or no prophylaxis | 2 | 250 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.58, 1.79] |
| 3.6 Sucralfate vs placebo or no prophylaxis | 5 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.58, 1.51] |
| 4 All‐cause mortality in hospital Show forest plot | 5 | 857 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.85, 1.55] |
| Analysis 1.4  Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 4 All‐cause mortality in hospital. | ||||
| 4.1 H2 receptor antagonists vs placebo or no prophylaxis | 4 | 387 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.71, 1.83] |
| 4.2 Proton pump inhibitors vs placebo or no prophylaxis | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.42, 3.22] |
| 4.3 Antacids vs placebo or no prophylaxis | 1 | 126 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.79, 2.64] |
| 4.4 Sucralfate vs placebo or no prophylaxis | 2 | 244 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.53, 1.68] |
| 5 Duration of ICU stay Show forest plot | 2 | 447 | Mean Difference (IV, Fixed, 95% CI) | 0.24 [‐1.13, 1.61] |
| Analysis 1.5 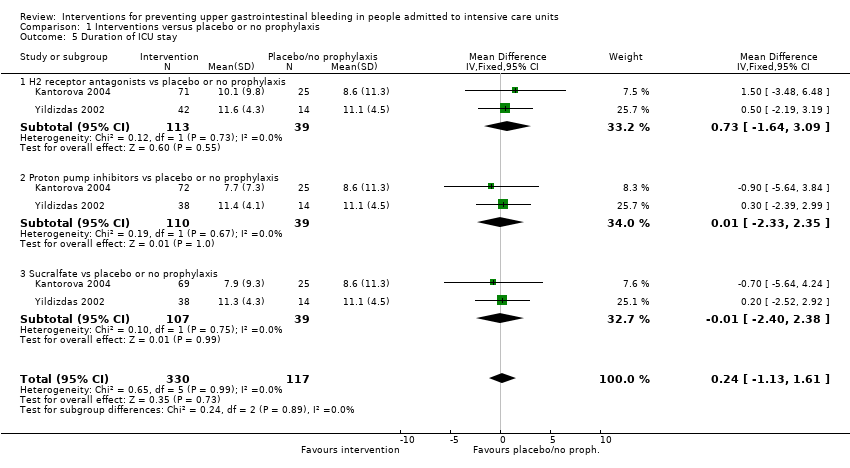 Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 5 Duration of ICU stay. | ||||
| 5.1 H2 receptor antagonists vs placebo or no prophylaxis | 2 | 152 | Mean Difference (IV, Fixed, 95% CI) | 0.73 [‐1.64, 3.09] |
| 5.2 Proton pump inhibitors vs placebo or no prophylaxis | 2 | 149 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐2.33, 2.35] |
| 5.3 Sucralfate vs placebo or no prophylaxis | 2 | 146 | Mean Difference (IV, Fixed, 95% CI) | ‐0.01 [‐2.40, 2.38] |
| 6 Duration of intubation Show forest plot | 2 | 447 | Mean Difference (IV, Fixed, 95% CI) | 0.87 [‐0.58, 2.31] |
| Analysis 1.6  Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 6 Duration of intubation. | ||||
| 6.1 H2 receptor antagonists vs placebo or no prophylaxis | 2 | 152 | Mean Difference (IV, Fixed, 95% CI) | 0.78 [‐1.72, 3.29] |
| 6.2 Proton pump inhibitors vs placebo or no prophylaxis | 2 | 149 | Mean Difference (IV, Fixed, 95% CI) | 0.36 [‐2.18, 2.90] |
| 6.3 Sucralfate vs placebo or no prophylaxis | 2 | 146 | Mean Difference (IV, Fixed, 95% CI) | 1.43 [‐1.04, 3.89] |
| 7 Number of participants requiring blood transfusions Show forest plot | 9 | 981 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.41, 0.97] |
| Analysis 1.7  Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 7 Number of participants requiring blood transfusions. | ||||
| 7.1 H2 receptor antagonists vs placebo or no prophylaxis | 7 | 605 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.35, 0.94] |
| 7.2 Antacids vs placebo or no prophylaxis | 2 | 226 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.30, 2.96] |
| 7.3 Sucralfate vs placebo or no prophylaxis | 1 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.13, 4.34] |
| 8 Units of blood transfused Show forest plot | 2 | 309 | Mean Difference (IV, Random, 95% CI) | 0.09 [‐0.99, 1.17] |
| Analysis 1.8  Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 8 Units of blood transfused. | ||||
| 8.1 H2 receptor antagonists vs placebo or no prophylaxis | 2 | 159 | Mean Difference (IV, Random, 95% CI) | ‐1.73 [‐6.37, 2.90] |
| 8.2 Sucralfate vs placebo or no prophylaxis | 1 | 150 | Mean Difference (IV, Random, 95% CI) | 0.80 [0.25, 1.35] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 24 | 2149 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.36, 0.70] |
| Analysis 2.1 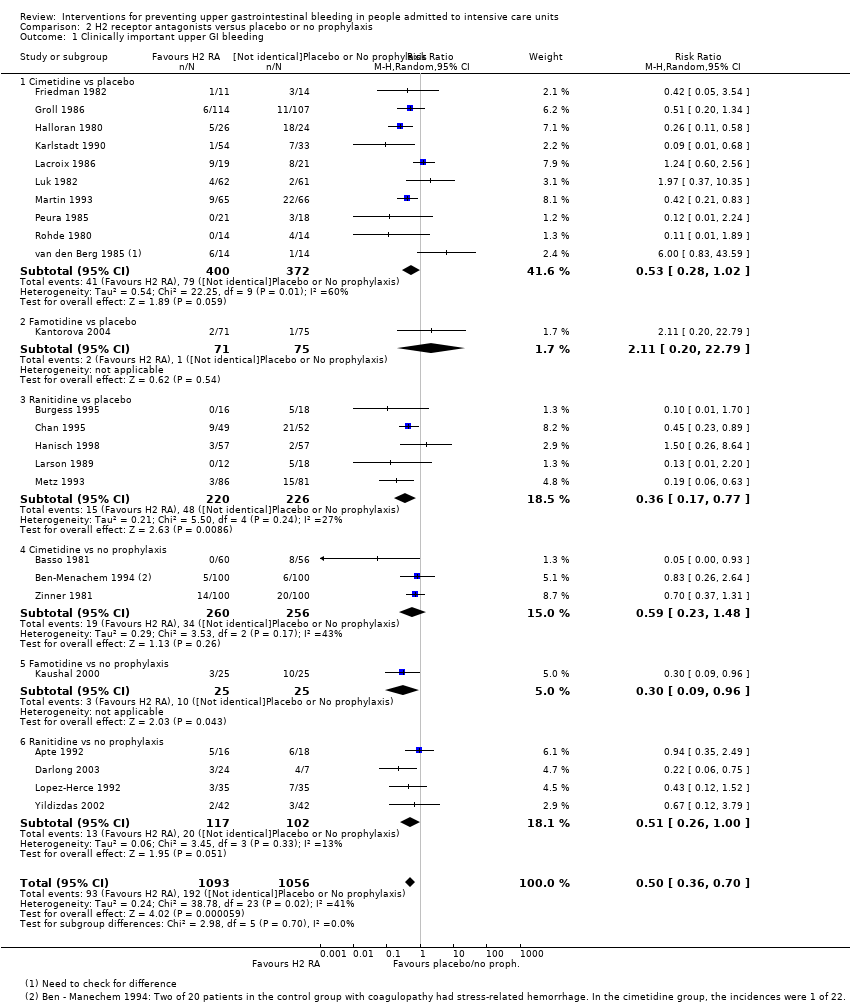 Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Cimetidine vs placebo | 10 | 772 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.28, 1.02] |
| 1.2 Famotidine vs placebo | 1 | 146 | Risk Ratio (M‐H, Random, 95% CI) | 2.11 [0.20, 22.79] |
| 1.3 Ranitidine vs placebo | 5 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.36 [0.17, 0.77] |
| 1.4 Cimetidine vs no prophylaxis | 3 | 516 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.23, 1.48] |
| 1.5 Famotidine vs no prophylaxis | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.3 [0.09, 0.96] |
| 1.6 Ranitidine vs no prophylaxis | 4 | 219 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.26, 1.00] |
| 2 Nosocomial pneumonia Show forest plot | 8 | 945 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.85, 1.48] |
| Analysis 2.2  Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia. | ||||
| 2.1 Cimetidine vs placebo | 2 | 204 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.06, 2.00] |
| 2.2 Famotidine vs placebo | 1 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.48 [0.49, 4.45] |
| 2.3 Ranitidine vs placebo | 2 | 277 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.47, 1.31] |
| 2.4 Cimetidine vs no prophylaxis | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.17 [0.86, 5.47] |
| 2.5 Ranitidine vs no prophylaxis | 2 | 118 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.93, 1.90] |
| 3 All‐cause mortality in ICU Show forest plot | 14 | 1428 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.88, 1.42] |
| Analysis 2.3  Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU. | ||||
| 3.1 Cimetidine vs placebo | 4 | 478 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.66, 1.68] |
| 3.2 Famotidine vs placebo | 1 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.55, 3.16] |
| 3.3 Ranitidine vs placebo | 2 | 148 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.31, 1.54] |
| 3.4 Cimetidine vs no prophylaxis | 2 | 400 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.61, 1.63] |
| 3.5 Famotidine vs no prophylaxis | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.59, 2.64] |
| 3.6 Ranitidine vs no prophylaxis | 4 | 206 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [0.97, 2.58] |
| 4 All‐cause mortality in hospital Show forest plot | 4 | 487 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.79, 1.70] |
| Analysis 2.4  Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 4 All‐cause mortality in hospital. | ||||
| 4.1 Famotidine vs placebo | 1 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.43, 1.86] |
| 4.2 Ranitidine vs placebo | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.47] |
| 4.3 Cimetidine vs no prophylaxis | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.47 [0.88, 2.46] |
| 4.4 Ranitidine vs no prophylaxis | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.33, 2.53] |
| 5 Duration of ICU stay Show forest plot | 2 | 230 | Mean Difference (IV, Fixed, 95% CI) | 0.73 [‐0.92, 2.38] |
| Analysis 2.5  Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 5 Duration of ICU stay. | ||||
| 5.1 Famotidine vs placebo | 1 | 146 | Mean Difference (IV, Fixed, 95% CI) | 1.5 [‐1.93, 4.93] |
| 5.2 Ranitidine vs no prophylaxis | 1 | 84 | Mean Difference (IV, Fixed, 95% CI) | 0.5 [‐1.38, 2.38] |
| 6 Duration of intubation Show forest plot | 2 | 230 | Mean Difference (IV, Fixed, 95% CI) | 0.79 [‐0.95, 2.54] |
| Analysis 2.6  Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 6 Duration of intubation. | ||||
| 6.1 Famotidine vs placebo | 1 | 146 | Mean Difference (IV, Fixed, 95% CI) | 1.20 [‐1.86, 4.26] |
| 6.2 Ranitidine vs no prophylaxis | 1 | 84 | Mean Difference (IV, Fixed, 95% CI) | 0.60 [‐1.52, 2.72] |
| 7 Number of participants requiring blood transfusions Show forest plot | 7 | 655 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.36, 0.95] |
| Analysis 2.7  Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 7 Number of participants requiring blood transfusions. | ||||
| 7.1 Cimetidine vs placebo | 3 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.19, 0.79] |
| 7.2 Ranitidine vs placebo | 2 | 148 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.24, 4.60] |
| 7.3 Cimetidine vs no prophylaxis | 2 | 400 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.35, 1.71] |
| 8 Units of blood transfused Show forest plot | 2 | 209 | Mean Difference (IV, Fixed, 95% CI) | 0.33 [‐0.04, 0.70] |
| Analysis 2.8  Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 8 Units of blood transfused. | ||||
| 8.1 Cimetidine vs placebo | 1 | 9 | Mean Difference (IV, Fixed, 95% CI) | ‐4.35 [‐7.35, ‐1.35] |
| 8.2 Cimetidine vs no prophylaxis | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 0.40 [0.03, 0.77] |
| 9 Adverse events of interventions Show forest plot | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 2.9  Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 9 Adverse events of interventions. | ||||
| 9.1 Diarrhoea | 2 | 225 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.57, 2.96] |
| 9.2 Thrombocytopenia | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.12, 72.77] |
| 9.3 Hypophosphatemia | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.75 [0.17, 84.02] |
| 9.4 Mental confusion | 5 | 657 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.01 [1.10, 3.65] |
| 9.5 Nausea and vomiting | 2 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.09, 2.35] |
| 9.6 Increased creatinine levels | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.64, 1.69] |
| 9.7 Erythema | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 71.22] |
| 9.8 Pancreatitis | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.01, 6.66] |
| 9.9 Chest infection | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.74 [0.92, 3.30] |
| 9.10 Delirium | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.59 [0.11, 59.93] |
| 9.11 Hallucinations | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.59 [0.11, 59.93] |
| 9.12 Severe bleeding | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.47] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 3 | 237 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.18, 2.22] |
| Analysis 3.1 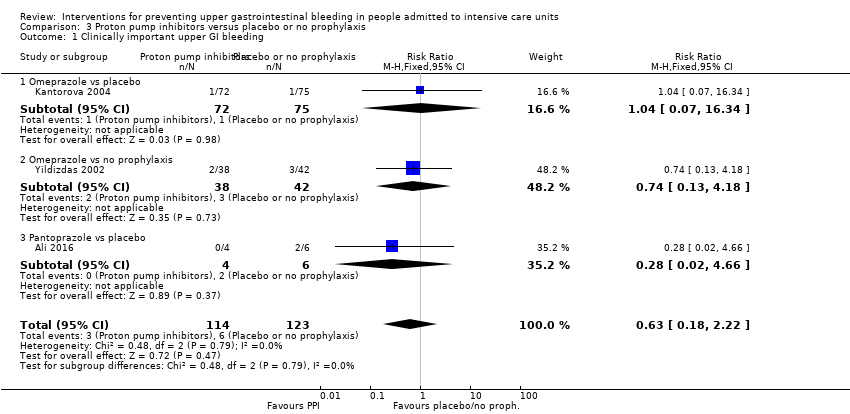 Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Omeprazole vs placebo | 1 | 147 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.07, 16.34] |
| 1.2 Omeprazole vs no prophylaxis | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.13, 4.18] |
| 1.3 Pantoprazole vs placebo | 1 | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.02, 4.66] |
| 2 Nosocomial pneumonia Show forest plot | 2 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.77, 1.98] |
| Analysis 3.2  Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia. | ||||
| 2.1 Omeprazole vs placebo | 1 | 147 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.57, 4.86] |
| 2.2 Omeprazole vs no prophylaxis | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.66, 1.84] |
| 3 All‐cause mortality in ICU Show forest plot | 3 | 258 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.60, 1.99] |
| Analysis 3.3 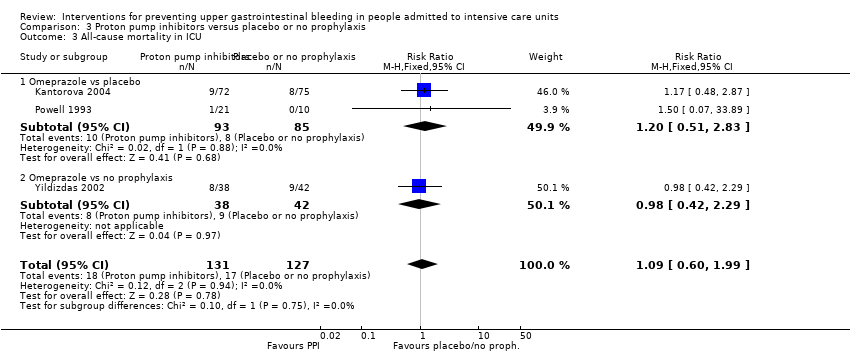 Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU. | ||||
| 3.1 Omeprazole vs placebo | 2 | 178 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.51, 2.83] |
| 3.2 Omeprazole vs no prophylaxis | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.42, 2.29] |
| 4 All‐cause mortality in hospital Show forest plot | 1 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.54, 2.13] |
| Analysis 3.4 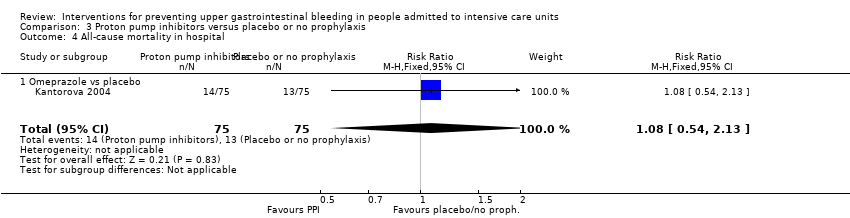 Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 4 All‐cause mortality in hospital. | ||||
| 4.1 Omeprazole vs placebo | 1 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.54, 2.13] |
| 5 Duration of ICU stay Show forest plot | 2 | 227 | Mean Difference (IV, Fixed, 95% CI) | ‐0.03 [‐1.63, 1.58] |
| Analysis 3.5  Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 5 Duration of ICU stay. | ||||
| 5.1 Omeprazole vs placebo | 1 | 147 | Mean Difference (IV, Fixed, 95% CI) | ‐0.90 [‐3.96, 2.16] |
| 5.2 Omeprazole vs no prophylaxis | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐1.58, 2.18] |
| 6 Duration of intubation Show forest plot | 2 | 227 | Mean Difference (IV, Fixed, 95% CI) | 0.36 [‐1.43, 2.15] |
| Analysis 3.6  Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 6 Duration of intubation. | ||||
| 6.1 Omeprazole vs placebo | 1 | 147 | Mean Difference (IV, Fixed, 95% CI) | 0.5 [‐2.72, 3.72] |
| 6.2 Omeprazole vs no prophylaxis | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐1.85, 2.45] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 4.1  Comparison 4 Proton pump inhibitors + sucralfate versus no prophylaxis, Outcome 1 Clinically important upper GI bleeding. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 5.1  Comparison 5 Prostaglandin analogues versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Prostaglandin analogues vs placebo | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality in ICU Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 5.2  Comparison 5 Prostaglandin analogues versus placebo or no prophylaxis, Outcome 2 All‐cause mortality in ICU. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 2 | 131 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.36, 2.51] |
| Analysis 6.1  Comparison 6 Anticholinergics versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Pirenzepine vs placebo | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.94 [0.34, 11.13] |
| 1.2 Pirenzepin + ranitidine vs placebo + ranitidine | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.17, 2.07] |
| 2 Nosocomial pneumonia Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.2  Comparison 6 Anticholinergics versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia. | ||||
| 2.1 Pirenzepine vs placebo | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 All‐cause mortality in ICU Show forest plot | 2 | 131 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.66, 2.30] |
| Analysis 6.3  Comparison 6 Anticholinergics versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU. | ||||
| 3.1 Pirenzepine vs placebo | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.65, 2.60] |
| 3.2 Pirenzepine + ranitidine vs placebo + ranitidine | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.24, 4.18] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 8 | 774 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.25, 0.99] |
| Analysis 7.1  Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Antacids vs placebo | 2 | 145 | Risk Ratio (M‐H, Random, 95% CI) | 2.04 [0.72, 5.79] |
| 1.2 Antacids vs no prophylaxis | 6 | 629 | Risk Ratio (M‐H, Random, 95% CI) | 0.35 [0.20, 0.60] |
| 2 All‐cause mortality in ICU Show forest plot | 2 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.53, 1.96] |
| Analysis 7.2  Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 2 All‐cause mortality in ICU. | ||||
| 2.1 Antacids vs no prophylaxis | 2 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.53, 1.96] |
| 3 All‐cause mortality in hospital Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 7.3 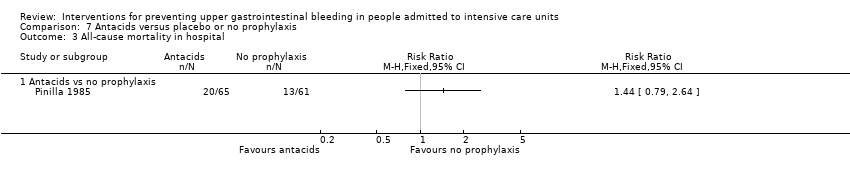 Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in hospital. | ||||
| 3.1 Antacids vs no prophylaxis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Number of participants requiring blood transfusions Show forest plot | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 7.4  Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 4 Number of participants requiring blood transfusions. | ||||
| 4.1 Antacids vs no prophylaxis | 2 | 226 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.30, 2.96] |
| 5 Adverse events of interventions Show forest plot | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 7.5  Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 5 Adverse events of interventions. | ||||
| 5.1 Diarrhoea | 4 | 395 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.56 [1.83, 6.94] |
| 5.2 Hypomagnesaemia | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.75 [0.17, 84.02] |
| 5.3 Hypophosphataemia | 2 | 225 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.48 [1.81, 16.61] |
| 5.4 Hypermagnesaemia | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.73 [0.36, 127.02] |
| 5.5 Nausea and vomiting | 3 | 370 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.39 [0.86, 6.64] |
| 5.6 Mental confusion | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [0.61, 2.67] |
| 5.7 Creatinine increase | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.73, 1.87] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 7 | 598 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.32, 0.88] |
| Analysis 8.1  Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Sucralfate vs placebo | 2 | 170 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.30, 6.62] |
| 1.2 Sucralfate vs no prophylaxis | 5 | 428 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.26, 0.80] |
| 2 Nosocomial pneumonia Show forest plot | 4 | 450 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.86, 2.04] |
| Analysis 8.2 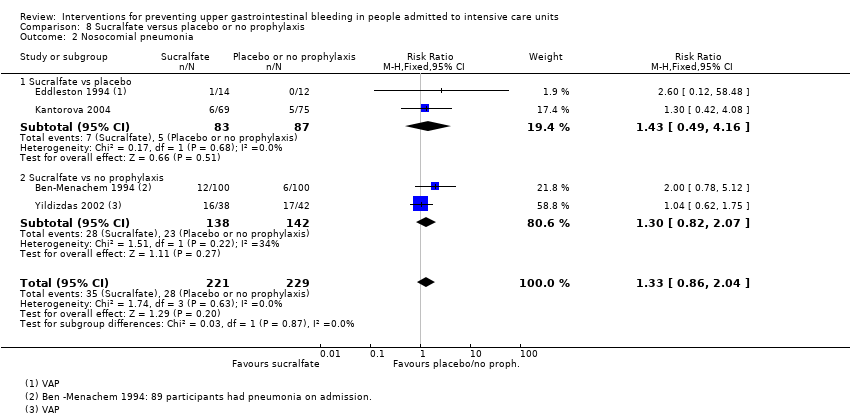 Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia. | ||||
| 2.1 Sucralfate vs placebo | 2 | 170 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.49, 4.16] |
| 2.2 Sucralfate vs no prophylaxis | 2 | 280 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.82, 2.07] |
| 3 All‐cause mortality in ICU Show forest plot | 5 | 500 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.66, 1.43] |
| Analysis 8.3  Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU. | ||||
| 3.1 Sucralfate vs placebo | 2 | 170 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.48, 1.80] |
| 3.2 Sucralfate vs no prophylaxis | 3 | 330 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.62, 1.60] |
| 4 All‐cause mortality in hospital Show forest plot | 2 | 344 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.62, 1.52] |
| Analysis 8.4  Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 4 All‐cause mortality in hospital. | ||||
| 4.1 Sucralfate vs placebo | 1 | 144 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.54, 2.18] |
| 4.2 Sucralfate vs no prophylaxis | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.49, 1.62] |
| 5 Duration of ICU stay Show forest plot | 2 | 224 | Mean Difference (IV, Fixed, 95% CI) | ‐0.02 [‐1.70, 1.65] |
| Analysis 8.5  Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 5 Duration of ICU stay. | ||||
| 5.1 Sucralfate vs placebo | 1 | 144 | Mean Difference (IV, Fixed, 95% CI) | ‐0.70 [‐4.07, 2.67] |
| 5.2 Sucralfate vs no prophylaxis | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐1.73, 2.13] |
| 6 Duration of intubation Show forest plot | 2 | 224 | Mean Difference (IV, Fixed, 95% CI) | 1.42 [‐0.27, 3.10] |
| Analysis 8.6  Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 6 Duration of intubation. | ||||
| 6.1 Sucralfate vs placebo | 1 | 144 | Mean Difference (IV, Fixed, 95% CI) | 0.80 [‐2.20, 3.80] |
| 6.2 Sucralfate vs no prophylaxis | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 1.70 [‐0.34, 3.74] |
| 7 Number of participants requiring blood transfusions Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 8.7  Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 7 Number of participants requiring blood transfusions. | ||||
| 7.1 Sucralfate vs no prophylaxis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Units of blood transfused Show forest plot | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 0.8 [0.32, 1.28] |
| Analysis 8.8  Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 8 Units of blood transfused. | ||||
| 8.1 Sucralfate vs no prophylaxis | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 0.8 [0.32, 1.28] |
| 9 Adverse events of interventions Show forest plot | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.50, 161.13] |
| Analysis 8.9  Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 9 Adverse events of interventions. | ||||
| 9.1 Nausea / Vomiting | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.50, 161.13] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 13 | 1636 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.90 [1.83, 4.58] |
| Analysis 9.1 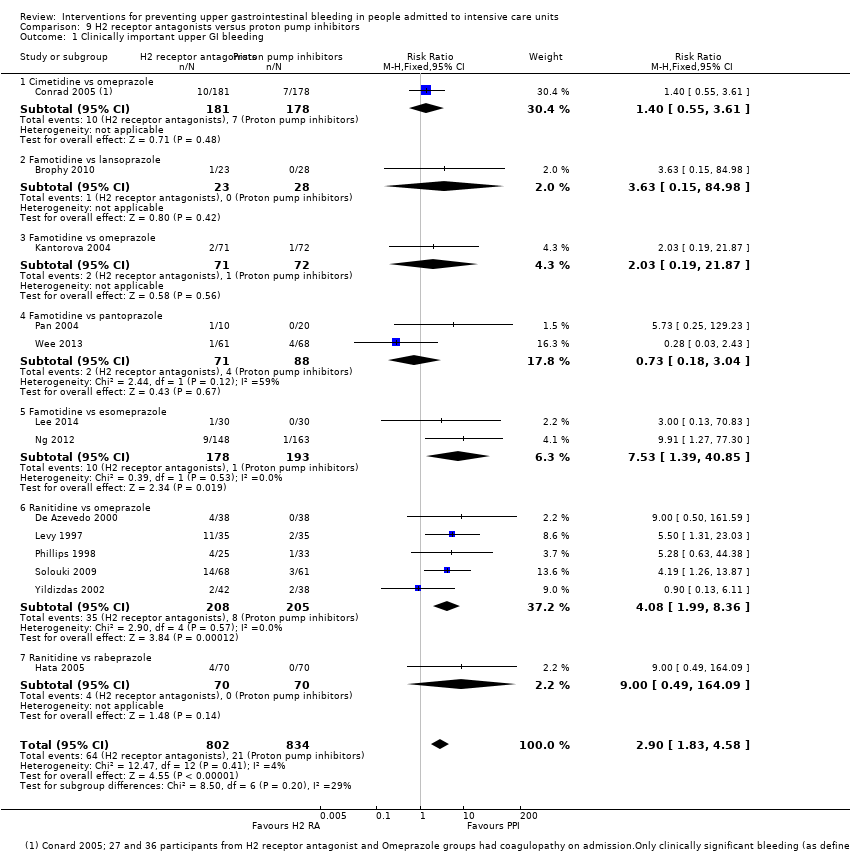 Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Cimetidine vs omeprazole | 1 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.55, 3.61] |
| 1.2 Famotidine vs lansoprazole | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.63 [0.15, 84.98] |
| 1.3 Famotidine vs omeprazole | 1 | 143 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.03 [0.19, 21.87] |
| 1.4 Famotidine vs pantoprazole | 2 | 159 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.18, 3.04] |
| 1.5 Famotidine vs esomeprazole | 2 | 371 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.53 [1.39, 40.85] |
| 1.6 Ranitidine vs omeprazole | 5 | 413 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.08 [1.99, 8.36] |
| 1.7 Ranitidine vs rabeprazole | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.49, 164.09] |
| 2 Nosocomial pneumonia Show forest plot | 10 | 1256 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.77, 1.35] |
| Analysis 9.2  Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 2 Nosocomial pneumonia. | ||||
| 2.1 Cimetidine vs omeprazole | 1 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.45, 1.54] |
| 2.2 Cimetidine vs pantoprazole | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.28, 2.91] |
| 2.3 Famotidine vs esomeprazole | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 15.26] |
| 2.4 Famotidine vs omeprazole | 1 | 143 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.34, 2.32] |
| 2.5 Ranitidine vs omeprazole | 5 | 413 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.80, 1.75] |
| 2.6 H2 receptor antagonists (not defined) vs proton pump inhibitors (not defined) | 1 | 79 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.47, 2.26] |
| 3 All‐cause mortality in ICU Show forest plot | 12 | 1564 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.78, 1.19] |
| Analysis 9.3 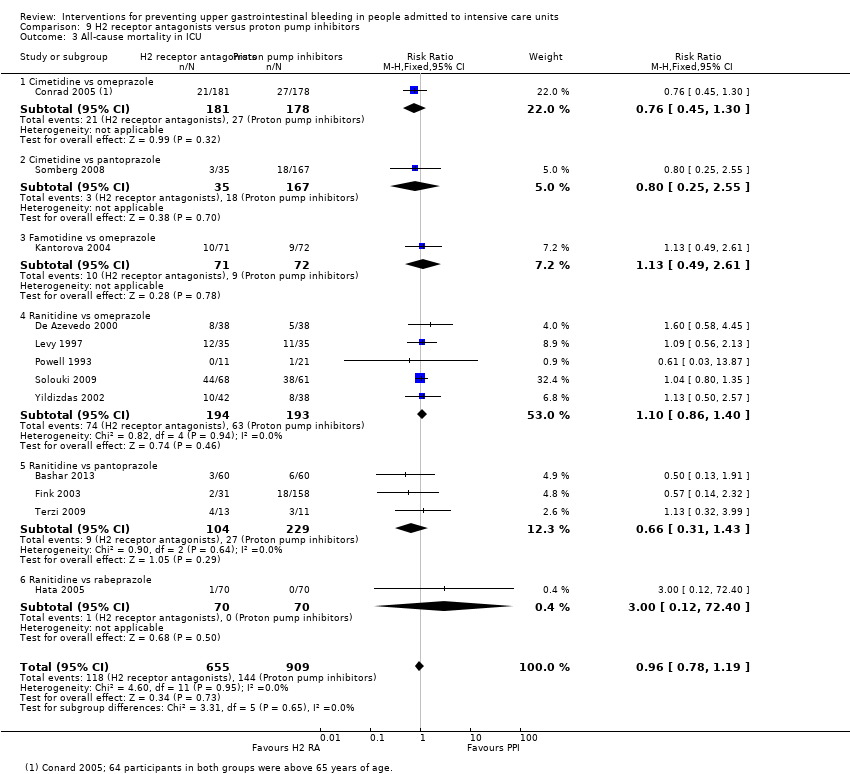 Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 3 All‐cause mortality in ICU. | ||||
| 3.1 Cimetidine vs omeprazole | 1 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.45, 1.30] |
| 3.2 Cimetidine vs pantoprazole | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.25, 2.55] |
| 3.3 Famotidine vs omeprazole | 1 | 143 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.49, 2.61] |
| 3.4 Ranitidine vs omeprazole | 5 | 387 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.86, 1.40] |
| 3.5 Ranitidine vs pantoprazole | 3 | 333 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.31, 1.43] |
| 3.6 Ranitidine vs rabeprazole | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.12, 72.40] |
| 4 All‐cause mortality in hospital Show forest plot | 2 | 454 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.37, 1.43] |
| Analysis 9.4  Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 4 All‐cause mortality in hospital. | ||||
| 4.1 Famotidine vs esomeprazole | 1 | 311 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.04, 3.49] |
| 4.2 Famotidine vs omeprazole | 1 | 143 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.39, 1.63] |
| 5 Duration of ICU stay Show forest plot | 5 | 482 | Mean Difference (IV, Fixed, 95% CI) | 0.14 [‐1.14, 1.41] |
| Analysis 9.5  Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 5 Duration of ICU stay. | ||||
| 5.1 Famotidine vs esomeprazole | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐6.51, 5.91] |
| 5.2 Famotidine vs omeprazole | 1 | 143 | Mean Difference (IV, Fixed, 95% CI) | 2.40 [‐0.44, 5.24] |
| 5.3 Ranitidine vs omeprazole | 3 | 279 | Mean Difference (IV, Fixed, 95% CI) | ‐0.44 [‐1.90, 1.02] |
| 6 Duration of intubation Show forest plot | 5 | 542 | Mean Difference (IV, Fixed, 95% CI) | ‐0.35 [‐1.48, 0.78] |
| Analysis 9.6  Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 6 Duration of intubation. | ||||
| 6.1 Famotidine vs omeprazole | 1 | 143 | Mean Difference (IV, Fixed, 95% CI) | 0.70 [‐2.24, 3.64] |
| 6.2 Ranitidine vs omeprazole | 3 | 279 | Mean Difference (IV, Fixed, 95% CI) | ‐0.78 [‐2.24, 0.67] |
| 6.3 Ranitidine vs pantoprazole | 1 | 120 | Mean Difference (IV, Fixed, 95% CI) | 0.07 [‐2.18, 2.32] |
| 7 Number of participants requiring blood transfusions Show forest plot | 3 | 575 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.98 [0.75, 5.21] |
| Analysis 9.7  Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 7 Number of participants requiring blood transfusions. | ||||
| 7.1 Cimetidine vs omeprazole | 1 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.29, 3.34] |
| 7.2 Ranitidine vs omeprazole | 1 | 76 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.25, 100.80] |
| 7.3 Ranitidine vs rabeprazole | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.49, 164.09] |
| 8 Adverse events of interventions Show forest plot | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 9.8  Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 8 Adverse events of interventions. | ||||
| 8.1 Pyrexia | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.05, 19.03] |
| 8.2 Thrombocytopaenia | 2 | 253 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.64 [0.65, 20.46] |
| 8.3 Neuroleptic malignant syndrome | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.4 Cholestatic jaundice | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.5 Abnormal liver function test | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.6 Pruritus | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.7 Phlebitis | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.8 Major CV events | 1 | 311 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.26, 2.43] |
| 8.9 Abdominal distension and vomiting | 1 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.62, 2.14] |
| 8.10 Hypomagnesaemia | 1 | 129 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.16, 1.13] |
| 8.11 Nausea and vomiting | 1 | 129 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.13, 1.77] |
| 8.12 Diarrhoea | 1 | 129 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.16, 7.67] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 16 | 1700 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.67, 1.36] |
| Analysis 10.1  Comparison 10 H2 receptor antagonists versus antacids, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Cimetidine vs antacids | 11 | 1155 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.65, 1.78] |
| 1.2 Cimetidine + pirenzepine vs antacid + pirenzepine | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.68] |
| 1.3 Ranitidine vs antacids | 4 | 479 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.42, 1.23] |
| 2 Nosocomial pneumonia Show forest plot | 4 | 581 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.81, 1.36] |
| Analysis 10.2  Comparison 10 H2 receptor antagonists versus antacids, Outcome 2 Nosocomial pneumonia. | ||||
| 2.1 Cimetidine vs antacids | 2 | 136 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.70, 2.19] |
| 2.2 Ranitidine vs antacids | 2 | 445 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.75, 1.34] |
| 3 All‐cause mortality in ICU Show forest plot | 11 | 1321 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.66, 1.55] |
| Analysis 10.3  Comparison 10 H2 receptor antagonists versus antacids, Outcome 3 All‐cause mortality in ICU. | ||||
| 3.1 Cimetidine vs antacids | 8 | 885 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.69, 1.59] |
| 3.2 Cimetidine + pirenzepine vs antacid + pirenzepine | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.37, 4.25] |
| 3.3 Ranitidine vs antacids | 2 | 370 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.14, 8.97] |
| 4 All‐cause mortality in hospital Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 10.4  Comparison 10 H2 receptor antagonists versus antacids, Outcome 4 All‐cause mortality in hospital. | ||||
| 4.1 Ranitidine vs antacids | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Duration of intubation Show forest plot | 3 | 121 | Mean Difference (IV, Random, 95% CI) | ‐0.81 [‐3.85, 2.23] |
| Analysis 10.5  Comparison 10 H2 receptor antagonists versus antacids, Outcome 5 Duration of intubation. | ||||
| 5.1 Cimetidine vs antacids | 3 | 121 | Mean Difference (IV, Random, 95% CI) | ‐0.81 [‐3.85, 2.23] |
| 6 Number of participants requiring blood transfusions Show forest plot | 6 | 744 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.49 [1.35, 4.62] |
| Analysis 10.6  Comparison 10 H2 receptor antagonists versus antacids, Outcome 6 Number of participants requiring blood transfusions. | ||||
| 6.1 Cimetidine vs antacids | 5 | 583 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.47 [1.32, 4.63] |
| 6.2 Ranitidine vs antacids | 1 | 161 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.04 [0.13, 73.46] |
| 7 Adverse events of interventions Show forest plot | 12 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 10.7  Comparison 10 H2 receptor antagonists versus antacids, Outcome 7 Adverse events of interventions. | ||||
| 7.1 Diarrhoea | 6 | 777 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.13, 0.43] |
| 7.2 Thrombocytopaenia | 4 | 452 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.93, 2.09] |
| 7.3 Nausea and vomiting | 4 | 380 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.19, 1.10] |
| 7.4 Hypophosphataemia | 2 | 108 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.04, 1.30] |
| 7.5 Hypomagnesaemia | 1 | 22 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.02, 7.39] |
| 7.6 Increase in creatinine | 2 | 286 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.56, 1.28] |
| 7.7 Mental confusion | 4 | 476 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.77, 2.07] |
| 7.8 Hypermagnesaemia | 2 | 115 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.17, 2.03] |
| 7.9 Rash/Erythema | 2 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.02 [0.32, 28.53] |
| 7.10 Alkalosis | 1 | 75 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.73] |
| 7.11 Dryness of mouth | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.15 [0.26, 103.33] |
| 7.12 Leucopaenia | 1 | 161 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.04 [0.13, 73.46] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 24 | 3316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.87, 1.41] |
| Analysis 11.1  Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Cimetidine vs sucralfate | 7 | 873 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.37 [0.87, 2.14] |
| 1.2 Famotidine vs sucralfate | 2 | 190 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.21, 1.78] |
| 1.3 Ranitidine vs sucralfate | 14 | 2186 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.76, 1.39] |
| 1.4 Cimetidine + pirenzepine vs sucralfate + pirenzepine | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.15 [0.26, 103.33] |
| 2 Nosocomial pneumonia Show forest plot | 17 | 3041 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [1.07, 1.40] |
| Analysis 11.2 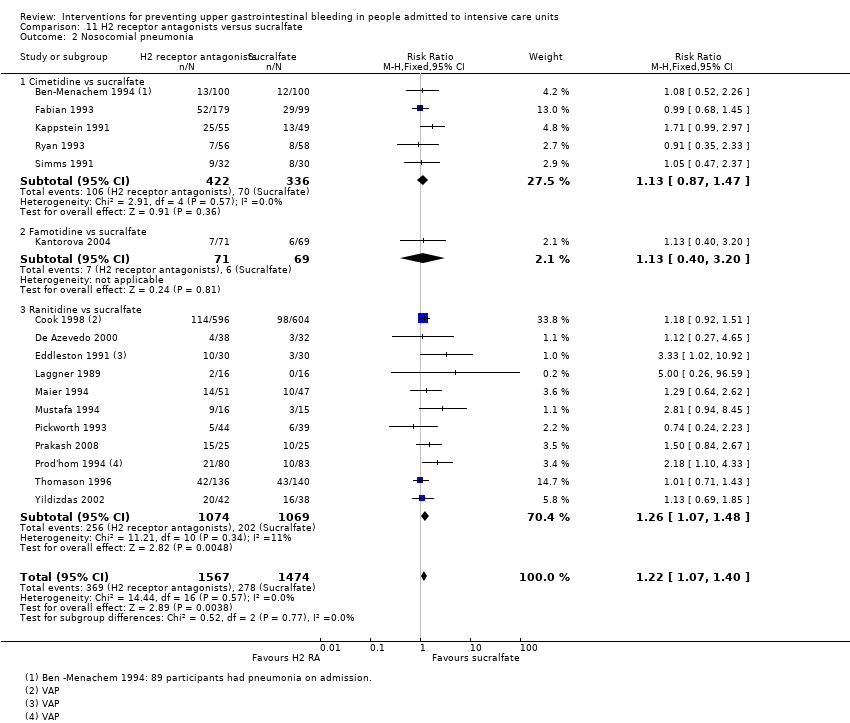 Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 2 Nosocomial pneumonia. | ||||
| 2.1 Cimetidine vs sucralfate | 5 | 758 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.87, 1.47] |
| 2.2 Famotidine vs sucralfate | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.40, 3.20] |
| 2.3 Ranitidine vs sucralfate | 11 | 2143 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [1.07, 1.48] |
| 3 All‐cause mortality in ICU Show forest plot | 21 | 3178 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.95, 1.24] |
| Analysis 11.3 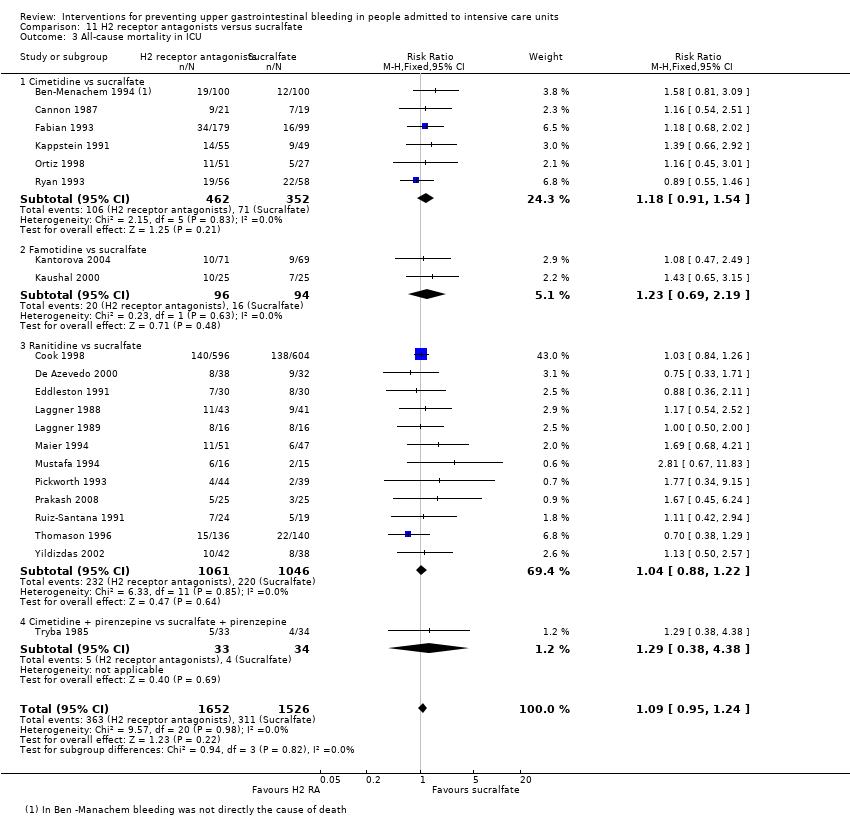 Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 3 All‐cause mortality in ICU. | ||||
| 3.1 Cimetidine vs sucralfate | 6 | 814 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.91, 1.54] |
| 3.2 Famotidine vs sucralfate | 2 | 190 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.69, 2.19] |
| 3.3 Ranitidine vs sucralfate | 12 | 2107 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.88, 1.22] |
| 3.4 Cimetidine + pirenzepine vs sucralfate + pirenzepine | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.38, 4.38] |
| 4 All‐cause mortality in hospital Show forest plot | 4 | 717 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.86, 1.50] |
| Analysis 11.4  Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 4 All‐cause mortality in hospital. | ||||
| 4.1 Cimetidine vs sucralfate | 2 | 413 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.28 [0.86, 1.92] |
| 4.2 Ranitidine vs sucralfate | 1 | 164 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.71, 1.74] |
| 4.3 Famotidine vs sucralfate | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.40, 1.71] |
| 5 Duration of intubation Show forest plot | 10 | 1751 | Mean Difference (IV, Random, 95% CI) | 0.22 [‐1.55, 2.00] |
| Analysis 11.5  Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 5 Duration of intubation. | ||||
| 5.1 Cimetidine vs sucralfate | 2 | 97 | Mean Difference (IV, Random, 95% CI) | 0.58 [‐1.71, 2.87] |
| 5.2 Famotidine vs sucralfate | 1 | 140 | Mean Difference (IV, Random, 95% CI) | 0.40 [‐2.30, 3.10] |
| 5.3 Ranitidine vs sucralfate | 7 | 1514 | Mean Difference (IV, Random, 95% CI) | 0.15 [‐2.12, 2.43] |
| 6 Duration of ICU stay Show forest plot | 6 | 1791 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐1.92, 1.95] |
| Analysis 11.6  Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 6 Duration of ICU stay. | ||||
| 6.1 Cimetidine vs sucralfate | 1 | 213 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐3.05, 3.05] |
| 6.2 Famotidine vs sucralfate | 1 | 140 | Mean Difference (IV, Random, 95% CI) | 2.20 [‐0.96, 5.36] |
| 6.3 Ranitidine vs sucralfate | 4 | 1438 | Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐2.70, 1.84] |
| 7 Number of participants requiring blood transfusion Show forest plot | 9 | 1095 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.70, 2.23] |
| Analysis 11.7 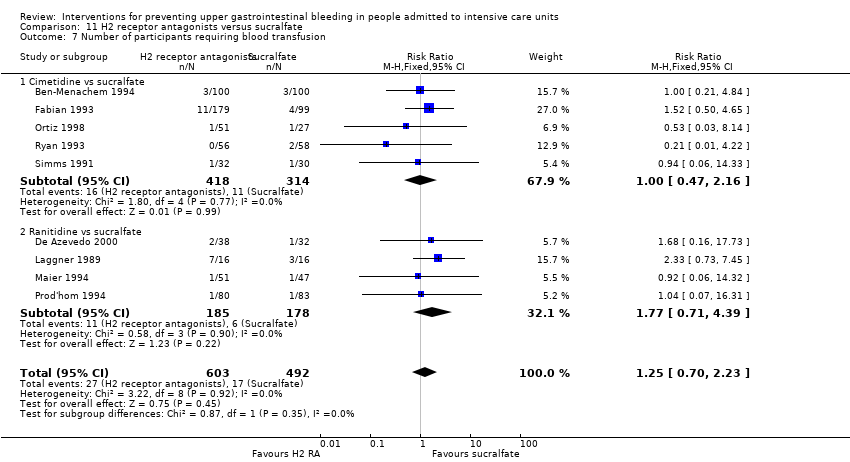 Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 7 Number of participants requiring blood transfusion. | ||||
| 7.1 Cimetidine vs sucralfate | 5 | 732 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.47, 2.16] |
| 7.2 Ranitidine vs sucralfate | 4 | 363 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.77 [0.71, 4.39] |
| 8 Units of blood transfused Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| Analysis 11.8  Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 8 Units of blood transfused. | ||||
| 8.1 Cimetidine vs sucralfate | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Adverse events of interventions Show forest plot | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 11.9  Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 9 Adverse events of interventions. | ||||
| 9.1 Thrombocytopaenia | 2 | 240 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.72 [0.56, 39.47] |
| 9.2 Nausea and vomiting | 2 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.07 [0.01, 0.54] |
| 9.3 Hypermagnesaemia | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.71 [0.31, 23.93] |
| 9.4 Rash/Erythema | 2 | 233 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.06 [0.32, 28.87] |
| 9.5 Confusion | 3 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.48 [0.77, 26.00] |
| 9.6 Neutropaenia | 1 | 114 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.18 [0.25, 105.47] |
| 9.7 Dryness of mouth | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.15 [0.26, 103.33] |
| 9.8 Leucopaenia | 1 | 163 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.11 [0.13, 75.26] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 3 | 556 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.37 [0.58, 3.26] |
| Analysis 12.1 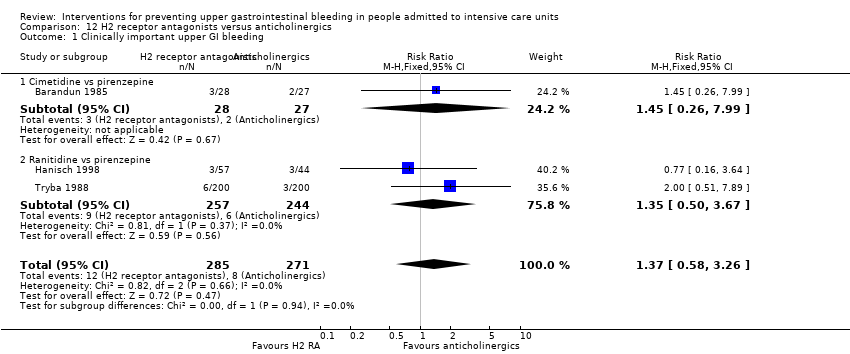 Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Cimetidine vs pirenzepine | 1 | 55 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.45 [0.26, 7.99] |
| 1.2 Ranitidine vs pirenzepine | 2 | 501 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.50, 3.67] |
| 2 Nosocomial pneumonia Show forest plot | 3 | 544 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.50, 1.84] |
| Analysis 12.2  Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 2 Nosocomial pneumonia. | ||||
| 2.1 Famotidine vs pirenzepine | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.42] |
| 2.2 Ranitidine vs pirenzepine | 2 | 501 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.53, 2.01] |
| 3 All‐cause mortality in ICU Show forest plot | 2 | 501 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.21, 3.87] |
| Analysis 12.3  Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 3 All‐cause mortality in ICU. | ||||
| 3.1 Ranitidine vs pirenzepine | 2 | 501 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.21, 3.87] |
| 4 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 12.4  Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 4 Number of participants requiring blood transfusion. | ||||
| 4.1 Ranitidine vs pirenzepine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse events of interventions Show forest plot | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 12.5  Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 5 Adverse events of interventions. | ||||
| 5.1 Tachycardia | 1 | 55 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 1.90] |
| 5.2 High temperature | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.21, 1.32] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 13.1  Comparison 13 H2 receptor antagonists versus prostaglandin analogues, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Cimetidine vs misoprostol | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality in ICU Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 13.2  Comparison 13 H2 receptor antagonists versus prostaglandin analogues, Outcome 2 All‐cause mortality in ICU. | ||||
| 2.1 Cimetidine vs misoprostol | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 14.1  Comparison 14 H2 receptor antagonists versus teprenone, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Ranitidine vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality in ICU Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 14.2  Comparison 14 H2 receptor antagonists versus teprenone, Outcome 2 All‐cause mortality in ICU. | ||||
| 2.1 Ranitidine vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 14.3  Comparison 14 H2 receptor antagonists versus teprenone, Outcome 3 Number of participants requiring blood transfusion. | ||||
| 3.1 Ranitidine vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 2 | 230 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.06, 0.95] |
| Analysis 15.1  Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Cimetidine + antacids vs sucralfate | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.70] |
| 1.2 Cimetidine or ranitidine + antacids vs sucralfate | 1 | 130 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.06, 1.41] |
| 2 Nosocomial pneumonia Show forest plot | 3 | 281 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.51, 2.32] |
| Analysis 15.2  Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 2 Nosocomial pneumonia. | ||||
| 2.1 Cimetidine + antacids vs sucralfate | 1 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.26, 1.07] |
| 2.2 Ranitidine + antacids vs sucralfate | 1 | 51 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.64, 2.53] |
| 2.3 Cimetidine or ranitidine + antacids vs sucralfate | 1 | 130 | Risk Ratio (M‐H, Random, 95% CI) | 2.02 [0.89, 4.58] |
| 3 All‐cause mortality in ICU Show forest plot | 2 | 230 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.92, 2.05] |
| Analysis 15.3  Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 3 All‐cause mortality in ICU. | ||||
| 3.1 Cimetidine + antacids vs sucralfate | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.46, 2.19] |
| 3.2 Cimetidine or ranitidine + antacids vs sucralfate | 1 | 130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.57 [0.99, 2.50] |
| 4 Duration of ICU stay Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| Analysis 15.4  Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 4 Duration of ICU stay. | ||||
| 5 Duration of intubation Show forest plot | 2 | 230 | Mean Difference (IV, Random, 95% CI) | ‐1.24 [‐13.82, 11.33] |
| Analysis 15.5  Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 5 Duration of intubation. | ||||
| 5.1 Cimetidine + antacids vs sucralfate | 1 | 100 | Mean Difference (IV, Random, 95% CI) | ‐8.8 [‐20.11, 2.51] |
| 5.2 Cimetidine or ranitidine + antacids vs sucralfate | 1 | 130 | Mean Difference (IV, Random, 95% CI) | 4.20 [‐0.54, 8.94] |
| 6 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 15.6 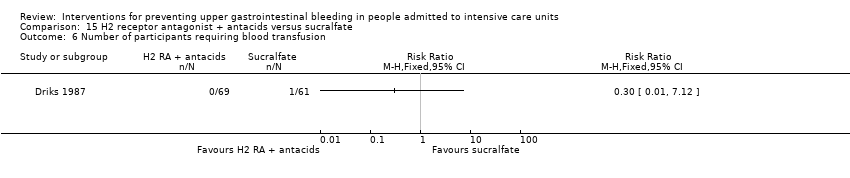 Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 6 Number of participants requiring blood transfusion. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 16.1  Comparison 16 Proton pump inhibitors versus teprenone, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Proton pump inhibitors vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality in ICU Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 16.2  Comparison 16 Proton pump inhibitors versus teprenone, Outcome 2 All‐cause mortality in ICU. | ||||
| 2.1 Proton pump inhibitors vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 16.3  Comparison 16 Proton pump inhibitors versus teprenone, Outcome 3 Number of participants requiring blood transfusion. | ||||
| 3.1 Proton pump inhibitors vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 17.1  Comparison 17 Proton pump inhibitor plus naloxone versus naloxone, Outcome 1 Clinically important upper GI bleeding. | ||||
| 2 All‐cause mortality in hospital Show forest plot | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 17.2  Comparison 17 Proton pump inhibitor plus naloxone versus naloxone, Outcome 2 All‐cause mortality in hospital. | ||||
| 3 Adverse events ‐ gastrointestinal discomfort Show forest plot | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 17.3  Comparison 17 Proton pump inhibitor plus naloxone versus naloxone, Outcome 3 Adverse events ‐ gastrointestinal discomfort. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 18.1  Comparison 18 Proton pump inhibitors versus other medication (not defined), Outcome 1 Clinically important upper GI bleeding. | ||||
| 2 Nosocomial pneumonia Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 18.2  Comparison 18 Proton pump inhibitors versus other medication (not defined), Outcome 2 Nosocomial pneumonia. | ||||
| 3 All‐cause mortality in hospital Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 18.3  Comparison 18 Proton pump inhibitors versus other medication (not defined), Outcome 3 All‐cause mortality in hospital. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 16 | 1772 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.72, 1.39] |
| Analysis 19.1  Comparison 19 Antacids versus sucralfate, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Antacids vs sucralfate | 15 | 1705 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.69, 1.35] |
| 1.2 Antacid + pirenzepine vs sucralfate + pirenzepine | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.15 [0.26, 103.33] |
| 2 Nosocomial pneumonia Show forest plot | 7 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 19.2  Comparison 19 Antacids versus sucralfate, Outcome 2 Nosocomial pneumonia. | ||||
| 2.1 Antacids vs sucralfate | 7 | 996 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.84, 1.30] |
| 3 All‐cause mortality in ICU Show forest plot | 11 | 1249 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.93, 1.40] |
| Analysis 19.3  Comparison 19 Antacids versus sucralfate, Outcome 3 All‐cause mortality in ICU. | ||||
| 3.1 Antacid vs sucralfate | 10 | 1182 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.94, 1.41] |
| 3.2 Antacid + pirenzepine vs sucralfate + pirenzepine | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.28, 3.78] |
| 4 All‐cause mortality in hospital Show forest plot | 3 | 450 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.69, 1.39] |
| Analysis 19.4  Comparison 19 Antacids versus sucralfate, Outcome 4 All‐cause mortality in hospital. | ||||
| 5 Duration of ICU stay Show forest plot | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| Analysis 19.5  Comparison 19 Antacids versus sucralfate, Outcome 5 Duration of ICU stay. | ||||
| 5.1 Antacids vs sucralfate | 2 | 227 | Mean Difference (IV, Fixed, 95% CI) | ‐2.50 [‐6.61, 1.61] |
| 6 Duration of intubation Show forest plot | 4 | Std. Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| Analysis 19.6  Comparison 19 Antacids versus sucralfate, Outcome 6 Duration of intubation. | ||||
| 6.1 Antacids vs sucralfate | 4 | 281 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.18 [‐0.41, 0.06] |
| 7 Number of participants requiring blood transfusion Show forest plot | 6 | 667 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.40, 1.34] |
| Analysis 19.7  Comparison 19 Antacids versus sucralfate, Outcome 7 Number of participants requiring blood transfusion. | ||||
| 8 Adverse events of interventions Show forest plot | 9 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 19.8  Comparison 19 Antacids versus sucralfate, Outcome 8 Adverse events of interventions. | ||||
| 8.1 Diarrhoea | 6 | 599 | Risk Ratio (M‐H, Fixed, 95% CI) | 12.40 [3.88, 39.64] |
| 8.2 Hypermagnesaemia | 4 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.72 [1.24, 17.95] |
| 8.3 Nausea and vomiting | 3 | 223 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.28, 1.41] |
| 8.4 Thrombocytopaenia | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.26, 97.70] |
| 8.5 Severe alkalosis | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 71.92] |
| 8.6 Allergic reactions | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.01, 4.06] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 2 | 329 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.12, 0.91] |
| Analysis 20.1  Comparison 20 Antacids versus prostaglandin analogues, Outcome 1 Clinically important upper GI bleeding. | ||||
| 2 All‐cause mortality in ICU Show forest plot | 2 | 417 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.42, 1.67] |
| Analysis 20.2  Comparison 20 Antacids versus prostaglandin analogues, Outcome 2 All‐cause mortality in ICU. | ||||
| 3 Adverse events of interventions Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 20.3  Comparison 20 Antacids versus prostaglandin analogues, Outcome 3 Adverse events of interventions. | ||||
| 3.1 Diarrhoea | 1 | 368 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.64, 1.33] |
| 3.2 Elevated serum bicarbonate | 1 | 338 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.21 [1.27, 3.87] |
| 3.3 Phospate levels < 2.5 mg/dL | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.66 [1.01, 2.73] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 21.1  Comparison 21 Antacids versus bioflavonoids, Outcome 1 Clinically important upper GI bleeding. | ||||
| 2 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 21.2  Comparison 21 Antacids versus bioflavonoids, Outcome 2 Number of participants requiring blood transfusion. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 22.1  Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 Sucralfate vs omeprazole | 3 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.58 [0.77, 8.63] |
| 2 Nosocomial pneumonia Show forest plot | 4 | 424 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.41, 1.09] |
| Analysis 22.2  Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 2 Nosocomial pneumonia. | ||||
| 2.1 Sucralfate vs omeprazole | 3 | 287 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.57, 1.36] |
| 2.2 Sucralfate vs pantoprazole | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.20, 0.75] |
| 3 All‐cause mortality in ICU Show forest plot | 4 | 424 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.68, 1.68] |
| Analysis 22.3  Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 3 All‐cause mortality in ICU. | ||||
| 3.1 Sucralfate vs omeprazole | 3 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.75, 2.11] |
| 3.2 Sucralfate vs pantoprazole | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.25, 1.68] |
| 4 All‐cause mortality in hospital Show forest plot | 2 | 278 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.46, 1.37] |
| Analysis 22.4  Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 4 All‐cause mortality in hospital. | ||||
| 4.1 Sucralfate vs omeprazole | 1 | 141 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.49, 1.91] |
| 4.2 Sucralfate vs pantoprazole | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.21, 1.45] |
| 5 Duration of ICU stay Show forest plot | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| Analysis 22.5  Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 5 Duration of ICU stay. | ||||
| 5.1 Sucralfate vs omeprazole | 2 | 217 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐1.68, 1.70] |
| 6 Duration of intubation Show forest plot | 3 | 354 | Mean Difference (IV, Fixed, 95% CI) | ‐0.16 [‐1.61, 1.28] |
| Analysis 22.6  Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 6 Duration of intubation. | ||||
| 6.1 Sucralfate vs omeprazole | 2 | 217 | Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐1.56, 1.60] |
| 6.2 Sucralfate vs pantoprazole | 1 | 137 | Mean Difference (IV, Fixed, 95% CI) | ‐1.10 [‐4.69, 2.49] |
| 7 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 22.7  Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 7 Number of participants requiring blood transfusion. | ||||
| 7.1 Sucralfate vs omeprazole | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.91 [0.29, 118.78] |
| 8 Adverse events of interventions Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 22.8  Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 8 Adverse events of interventions. | ||||
| 8.1 Fever | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.70, 0.94] |
| 8.2 Leucocytosis | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.55, 0.80] |
| 8.3 Sudden purulent sputum | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.38, 1.86] |
| 8.4 Sudden cough or aggravation of coughing | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.23 [0.07, 0.79] |
| 8.5 Dyspnoea | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.54, 0.87] |
| 8.6 Rales or bronchial sounds | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.19, 0.51] |
| 8.7 Aggravation of blood gas exchange | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.49, 1.18] |
| 8.8 Change in sputum quality | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.23 [0.13, 0.40] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 23.1  Comparison 23 Sucralfate versus bioflavonoids, Outcome 1 Clinically important upper GI bleeding. | ||||
| 2 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 23.2  Comparison 23 Sucralfate versus bioflavonoids, Outcome 2 Number of participants requiring blood transfusion. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 24.1 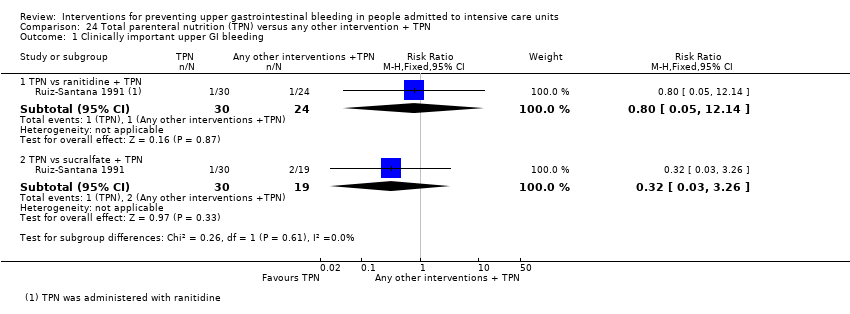 Comparison 24 Total parenteral nutrition (TPN) versus any other intervention + TPN, Outcome 1 Clinically important upper GI bleeding. | ||||
| 1.1 TPN vs ranitidine + TPN | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.8 [0.05, 12.14] |
| 1.2 TPN vs sucralfate + TPN | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.03, 3.26] |
| 2 All‐cause mortality in ICU Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 24.2  Comparison 24 Total parenteral nutrition (TPN) versus any other intervention + TPN, Outcome 2 All‐cause mortality in ICU. | ||||
| 2.1 TPN vs ranitidine + TPN | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.41, 3.09] |
| 2.2 TPN vs sucralfate + TPN | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.26, 1.52] |
| 3 Duration of intubation Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| Analysis 24.3  Comparison 24 Total parenteral nutrition (TPN) versus any other intervention + TPN, Outcome 3 Duration of intubation. | ||||
| 3.1 TPN vs ranitidine + TPN | 1 | 54 | Mean Difference (IV, Fixed, 95% CI) | ‐2.0 [‐9.53, 5.53] |
| 3.2 TPN vs sucralfate + TPN | 1 | 49 | Mean Difference (IV, Fixed, 95% CI) | 3.0 [‐1.50, 7.50] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 25.1  Comparison 25 Bowel stimulation versus no prophylaxis, Outcome 1 Clinically important upper GI bleeding. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 26.1  Comparison 26 Nasojejunal nutrition versus nasogastric nutrition, Outcome 1 Clinically important upper GI bleeding. | ||||
| 2 Nosocomial pneumonia Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 26.2  Comparison 26 Nasojejunal nutrition versus nasogastric nutrition, Outcome 2 Nosocomial pneumonia. | ||||
| 3 All‐cause mortality in hospital Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 26.3  Comparison 26 Nasojejunal nutrition versus nasogastric nutrition, Outcome 3 All‐cause mortality in hospital. | ||||
| 4 Adverse events of interventions Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 26.4  Comparison 26 Nasojejunal nutrition versus nasogastric nutrition, Outcome 4 Adverse events of interventions. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Nosocomial pneumonia Show forest plot | 1 | 120 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.44, 1.40] |
| Analysis 27.1  Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 1 Nosocomial pneumonia. | ||||
| 1.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.29, 1.25] |
| 1.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.47, 3.33] |
| 2 All‐cause mortality in hospital Show forest plot | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.06, 0.60] |
| Analysis 27.2  Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 2 All‐cause mortality in hospital. | ||||
| 2.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.05, 1.30] |
| 2.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.03, 0.74] |
| 3 Duration of ICU stay Show forest plot | 1 | 120 | Mean Difference (IV, Fixed, 95% CI) | ‐5.98 [‐8.81, ‐3.16] |
| Analysis 27.3  Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 3 Duration of ICU stay. | ||||
| 3.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐3.81 [‐7.59, ‐0.03] |
| 3.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐8.72 [‐12.97, ‐4.47] |
| 4 Duration of intubation Show forest plot | 1 | 120 | Mean Difference (IV, Fixed, 95% CI) | ‐7.37 [‐9.29, ‐5.45] |
| Analysis 27.4 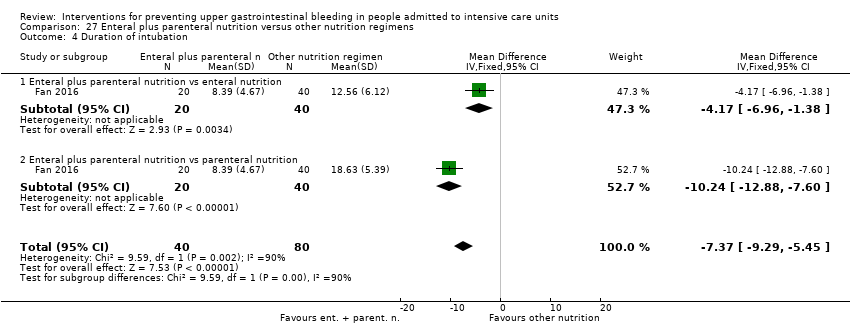 Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 4 Duration of intubation. | ||||
| 4.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐4.17 [‐6.96, ‐1.38] |
| 4.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐10.24 [‐12.88, ‐7.60] |
| 5 Adverse events ‐ stress ulcer Show forest plot | 1 | 120 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.36, 1.33] |
| Analysis 27.5  Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 5 Adverse events ‐ stress ulcer. | ||||
| 5.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.38, 3.45] |
| 5.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.23, 1.20] |
| 6 Adverse events ‐ diarrhoea Show forest plot | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.17, 1.02] |
| Analysis 27.6 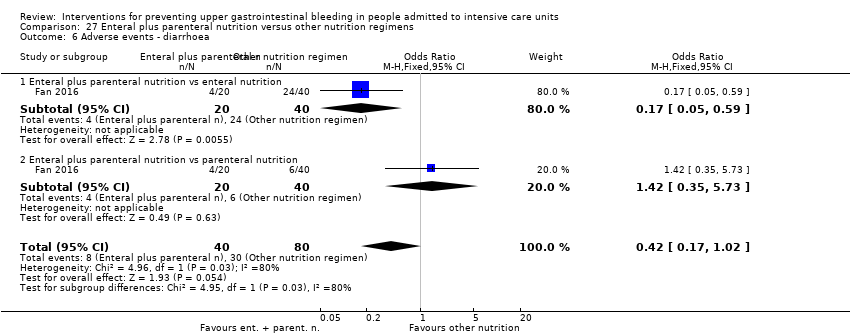 Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 6 Adverse events ‐ diarrhoea. | ||||
| 6.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.05, 0.59] |
| 6.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.35, 5.73] |
| 7 Adverse events ‐ pyaemia Show forest plot | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.37, 2.09] |
| Analysis 27.7  Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 7 Adverse events ‐ pyaemia. | ||||
| 7.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.11 [0.87, 19.41] |
| 7.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.11, 1.21] |
| 8 Adverse events ‐ intracranial infection Show forest plot | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.15, 1.25] |
| Analysis 27.8 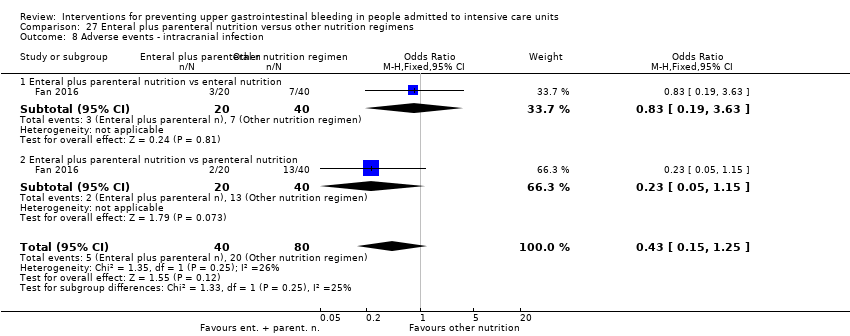 Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 8 Adverse events ‐ intracranial infection. | ||||
| 8.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.19, 3.63] |
| 8.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.05, 1.15] |
| 9 Adverse events ‐ hypoproteinaemia Show forest plot | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.04, 0.27] |
| Analysis 27.9  Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 9 Adverse events ‐ hypoproteinaemia. | ||||
| 9.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.06, 0.72] |
| 9.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.04 [0.01, 0.19] |

PRISMA flow chart of included studies.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
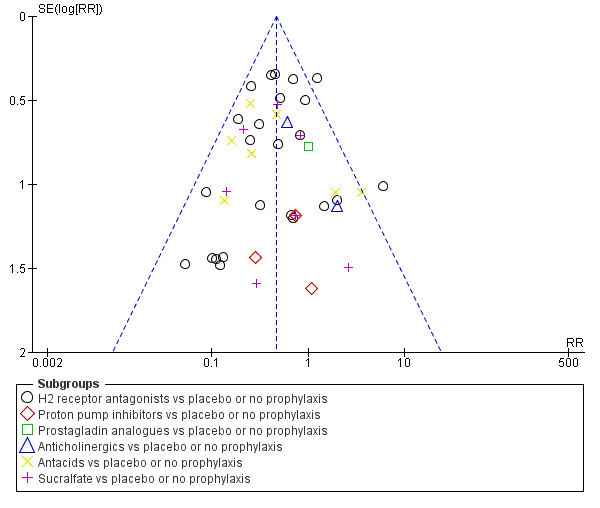
Funnel plot of comparison: 1 Interventions versus placebo or no prophylaxis, outcome: 1.1 Clinically important upper GI bleeding.
Funnel plot of comparison: 2 H2 receptor antagonists versus placebo or no prophylaxis, outcome: 2.1 Clinically important upper GI bleeding.

Funnel plot of comparison: 9 H2 receptor antagonists versus proton pump inhibitors, outcome: 9.1 Clinically important upper GI bleeding.
Funnel plot of comparison: 10 H2 receptor antagonists versus antacids, outcome: 10.1 Clinically important upper GI bleeding.
Funnel plot of comparison: 11 H2 receptor antagonists versus sucralfate, outcome: 11.1 Clinically important upper GI bleeding.

Funnel plot of comparison: 19 Antacids versus sucralfate, outcome: 19.1 Clinically important upper GI bleeding.

Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.

Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia.

Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU.

Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 4 All‐cause mortality in hospital.

Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 5 Duration of ICU stay.
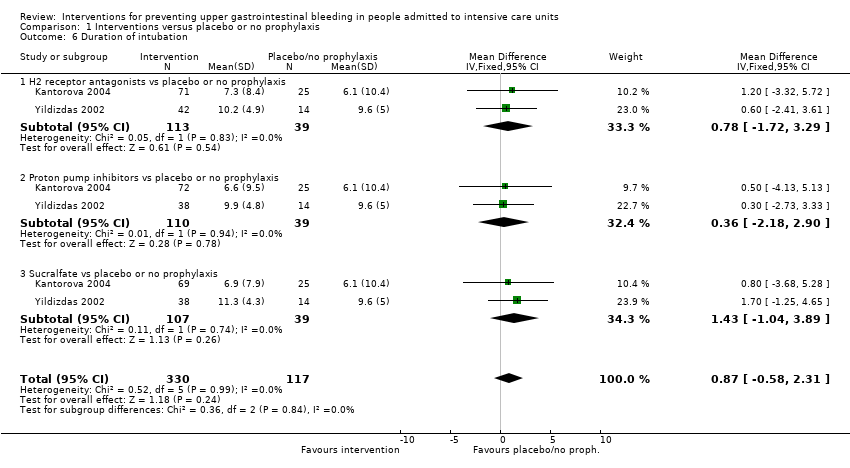
Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 6 Duration of intubation.

Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 7 Number of participants requiring blood transfusions.
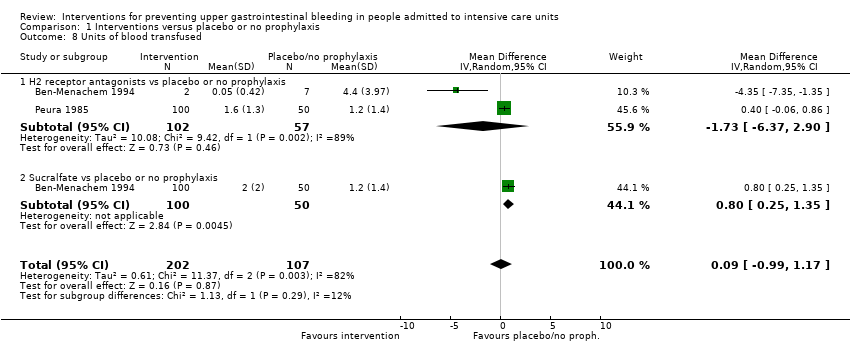
Comparison 1 Interventions versus placebo or no prophylaxis, Outcome 8 Units of blood transfused.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia.
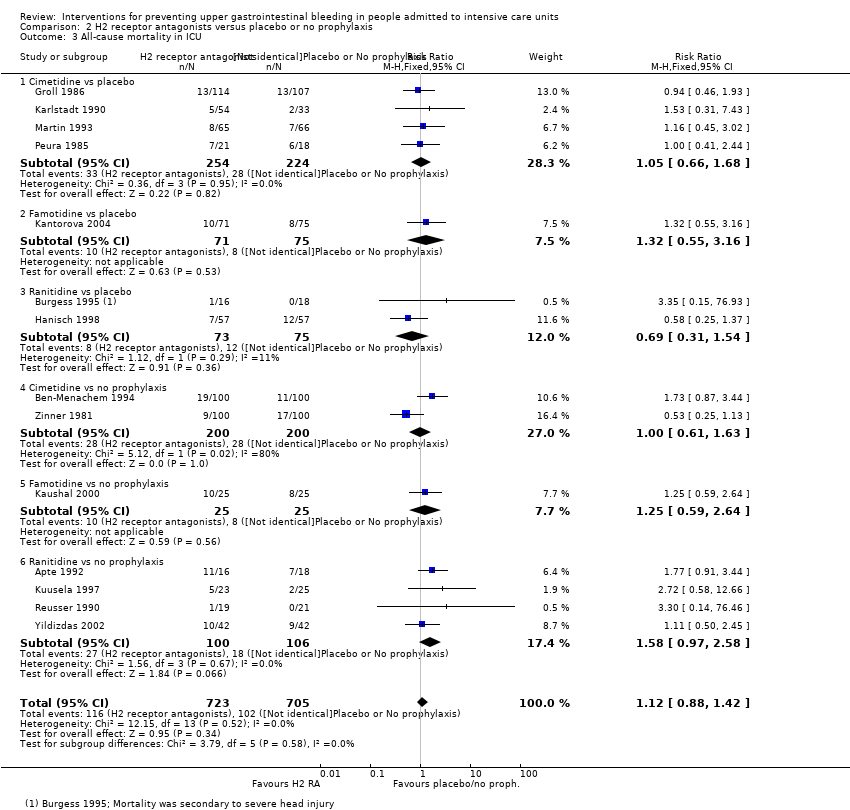
Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU.
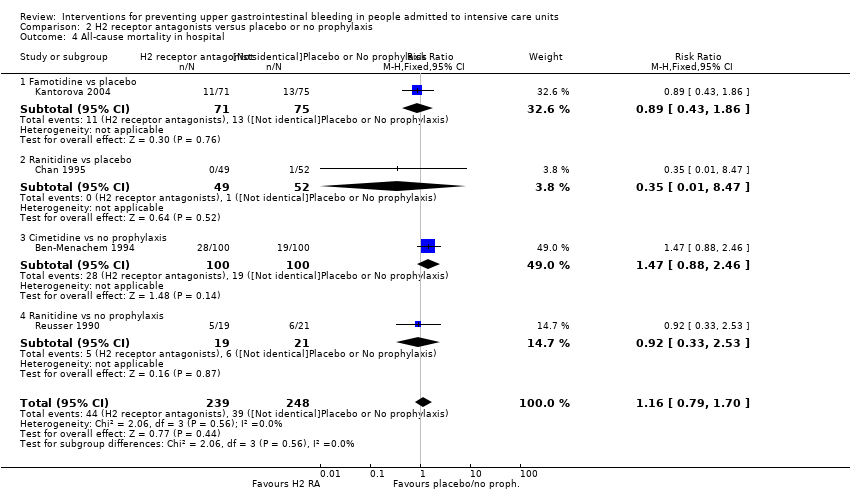
Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 4 All‐cause mortality in hospital.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 5 Duration of ICU stay.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 6 Duration of intubation.
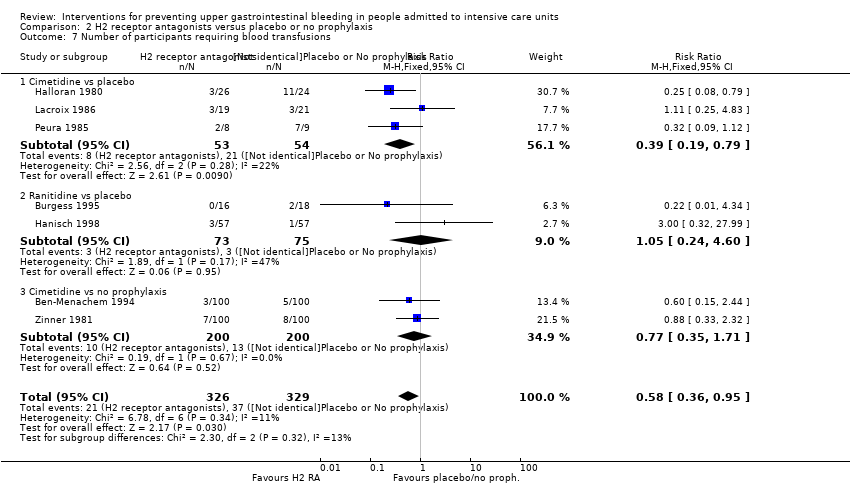
Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 7 Number of participants requiring blood transfusions.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 8 Units of blood transfused.

Comparison 2 H2 receptor antagonists versus placebo or no prophylaxis, Outcome 9 Adverse events of interventions.

Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
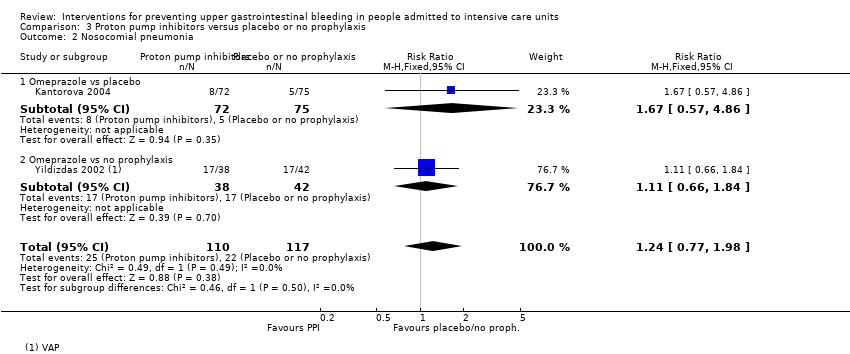
Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia.

Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU.

Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 4 All‐cause mortality in hospital.

Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 5 Duration of ICU stay.
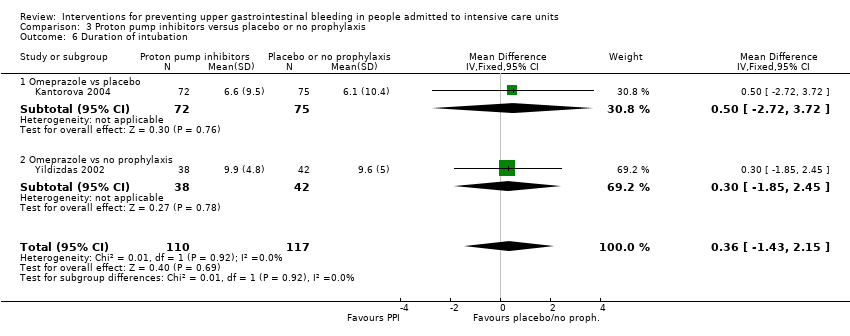
Comparison 3 Proton pump inhibitors versus placebo or no prophylaxis, Outcome 6 Duration of intubation.

Comparison 4 Proton pump inhibitors + sucralfate versus no prophylaxis, Outcome 1 Clinically important upper GI bleeding.

Comparison 5 Prostaglandin analogues versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
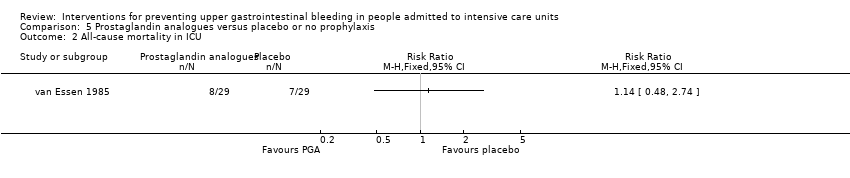
Comparison 5 Prostaglandin analogues versus placebo or no prophylaxis, Outcome 2 All‐cause mortality in ICU.

Comparison 6 Anticholinergics versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.

Comparison 6 Anticholinergics versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia.

Comparison 6 Anticholinergics versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU.
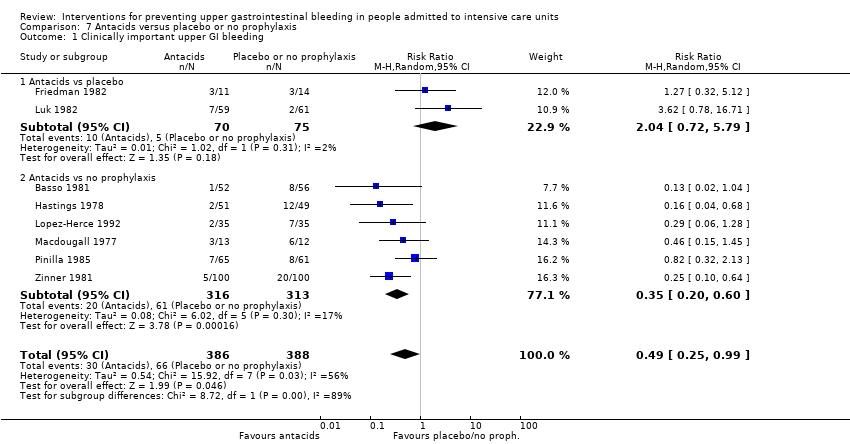
Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
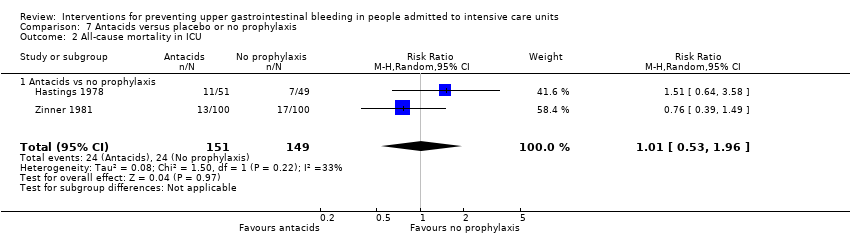
Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 2 All‐cause mortality in ICU.

Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in hospital.

Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 4 Number of participants requiring blood transfusions.

Comparison 7 Antacids versus placebo or no prophylaxis, Outcome 5 Adverse events of interventions.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 1 Clinically important upper GI bleeding.
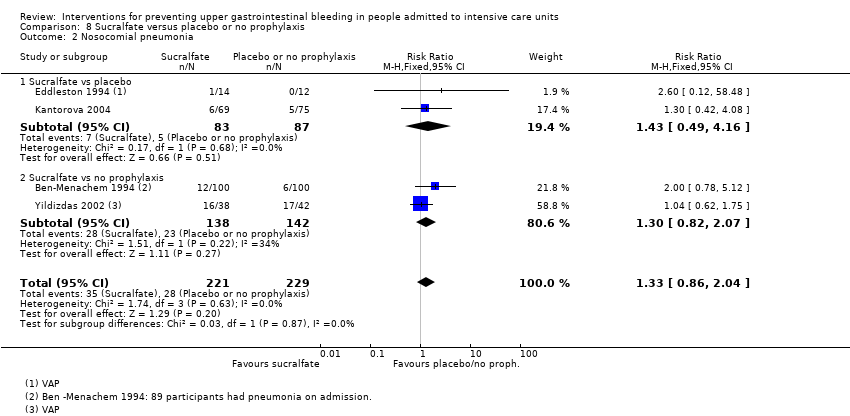
Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 2 Nosocomial pneumonia.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 3 All‐cause mortality in ICU.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 4 All‐cause mortality in hospital.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 5 Duration of ICU stay.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 6 Duration of intubation.
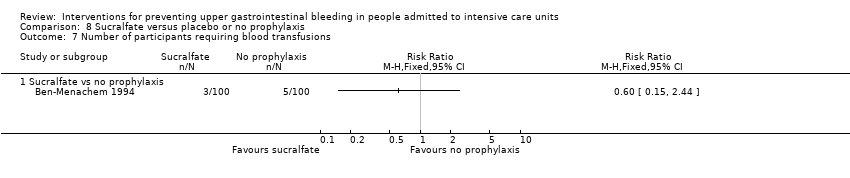
Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 7 Number of participants requiring blood transfusions.
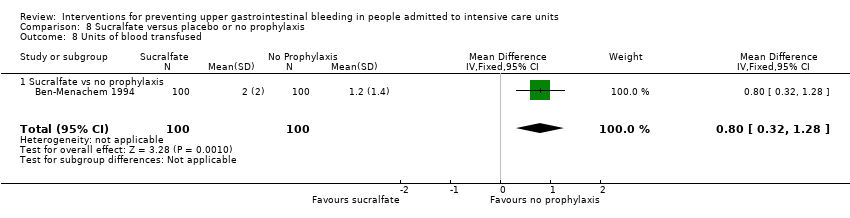
Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 8 Units of blood transfused.

Comparison 8 Sucralfate versus placebo or no prophylaxis, Outcome 9 Adverse events of interventions.

Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 1 Clinically important upper GI bleeding.

Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 2 Nosocomial pneumonia.
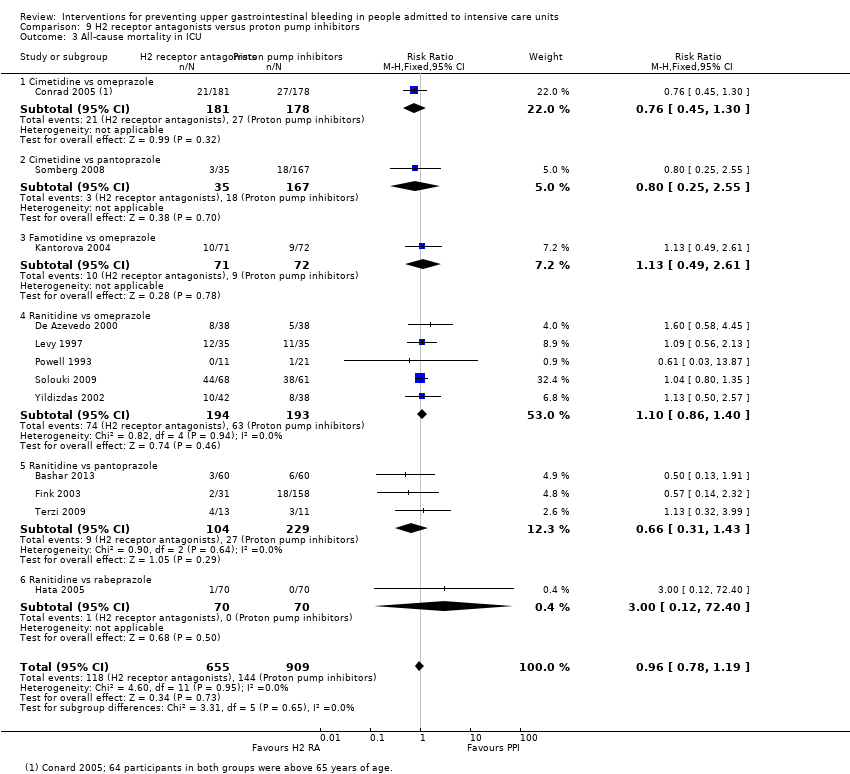
Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 3 All‐cause mortality in ICU.

Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 4 All‐cause mortality in hospital.

Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 5 Duration of ICU stay.
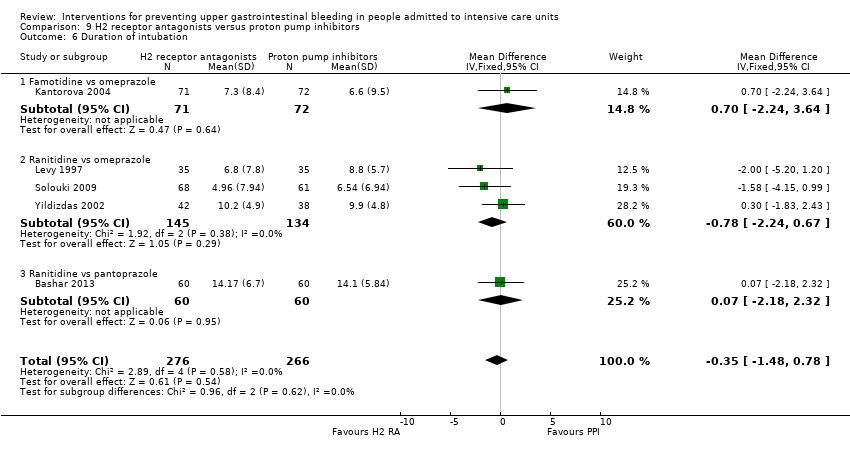
Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 6 Duration of intubation.
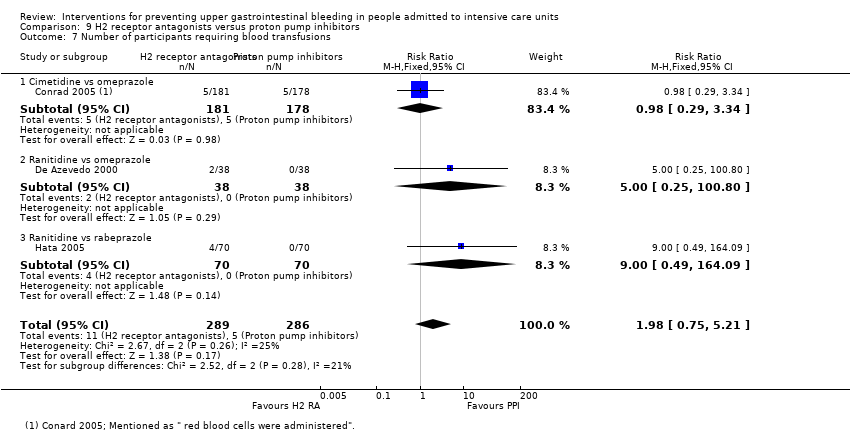
Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 7 Number of participants requiring blood transfusions.

Comparison 9 H2 receptor antagonists versus proton pump inhibitors, Outcome 8 Adverse events of interventions.

Comparison 10 H2 receptor antagonists versus antacids, Outcome 1 Clinically important upper GI bleeding.

Comparison 10 H2 receptor antagonists versus antacids, Outcome 2 Nosocomial pneumonia.

Comparison 10 H2 receptor antagonists versus antacids, Outcome 3 All‐cause mortality in ICU.
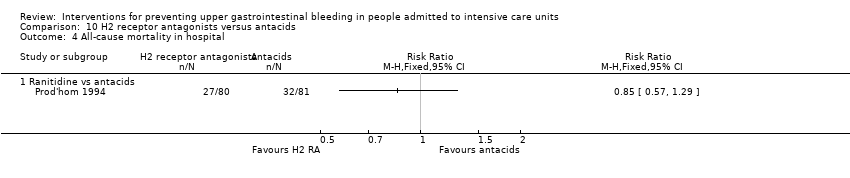
Comparison 10 H2 receptor antagonists versus antacids, Outcome 4 All‐cause mortality in hospital.

Comparison 10 H2 receptor antagonists versus antacids, Outcome 5 Duration of intubation.

Comparison 10 H2 receptor antagonists versus antacids, Outcome 6 Number of participants requiring blood transfusions.

Comparison 10 H2 receptor antagonists versus antacids, Outcome 7 Adverse events of interventions.
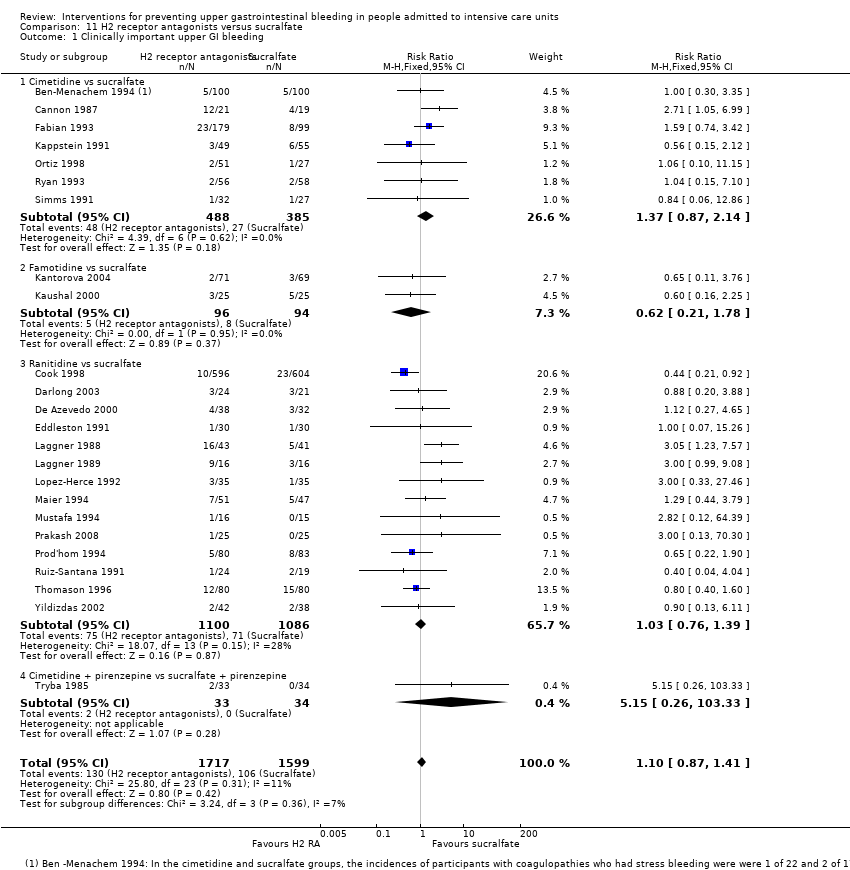
Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 1 Clinically important upper GI bleeding.
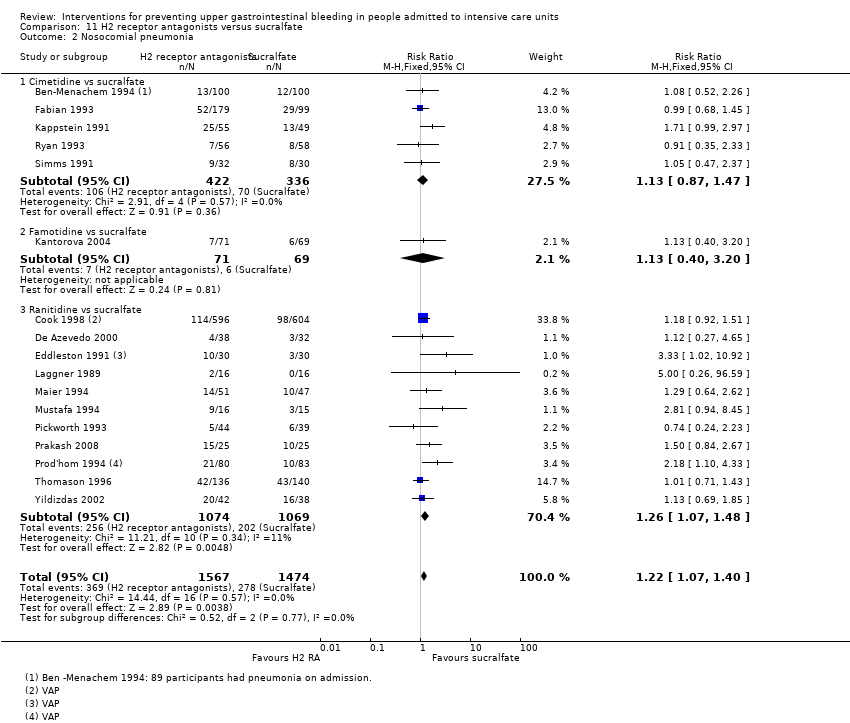
Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 2 Nosocomial pneumonia.

Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 3 All‐cause mortality in ICU.

Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 4 All‐cause mortality in hospital.

Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 5 Duration of intubation.

Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 6 Duration of ICU stay.

Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 7 Number of participants requiring blood transfusion.

Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 8 Units of blood transfused.

Comparison 11 H2 receptor antagonists versus sucralfate, Outcome 9 Adverse events of interventions.

Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 1 Clinically important upper GI bleeding.
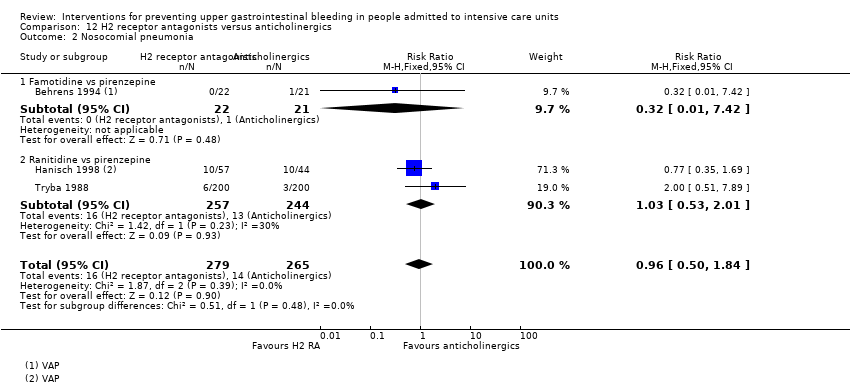
Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 2 Nosocomial pneumonia.

Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 3 All‐cause mortality in ICU.

Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 4 Number of participants requiring blood transfusion.
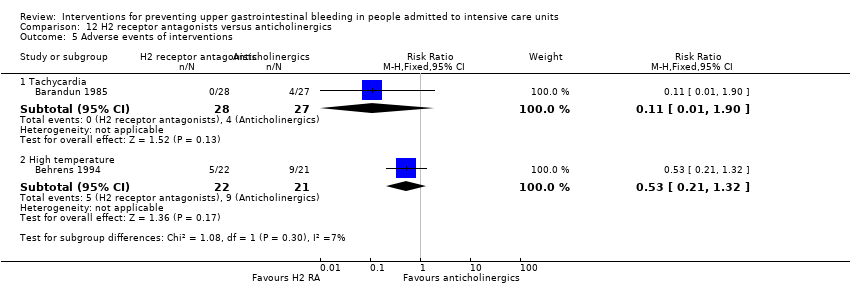
Comparison 12 H2 receptor antagonists versus anticholinergics, Outcome 5 Adverse events of interventions.

Comparison 13 H2 receptor antagonists versus prostaglandin analogues, Outcome 1 Clinically important upper GI bleeding.

Comparison 13 H2 receptor antagonists versus prostaglandin analogues, Outcome 2 All‐cause mortality in ICU.

Comparison 14 H2 receptor antagonists versus teprenone, Outcome 1 Clinically important upper GI bleeding.

Comparison 14 H2 receptor antagonists versus teprenone, Outcome 2 All‐cause mortality in ICU.

Comparison 14 H2 receptor antagonists versus teprenone, Outcome 3 Number of participants requiring blood transfusion.

Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 1 Clinically important upper GI bleeding.

Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 2 Nosocomial pneumonia.

Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 3 All‐cause mortality in ICU.

Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 4 Duration of ICU stay.

Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 5 Duration of intubation.

Comparison 15 H2 receptor antagonist + antacids versus sucralfate, Outcome 6 Number of participants requiring blood transfusion.

Comparison 16 Proton pump inhibitors versus teprenone, Outcome 1 Clinically important upper GI bleeding.

Comparison 16 Proton pump inhibitors versus teprenone, Outcome 2 All‐cause mortality in ICU.

Comparison 16 Proton pump inhibitors versus teprenone, Outcome 3 Number of participants requiring blood transfusion.
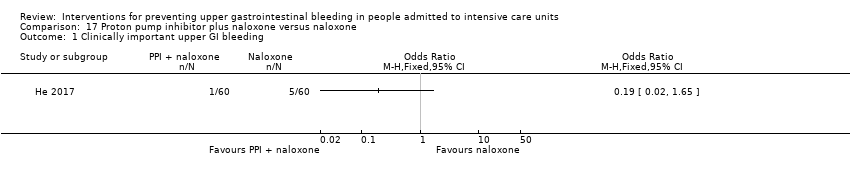
Comparison 17 Proton pump inhibitor plus naloxone versus naloxone, Outcome 1 Clinically important upper GI bleeding.
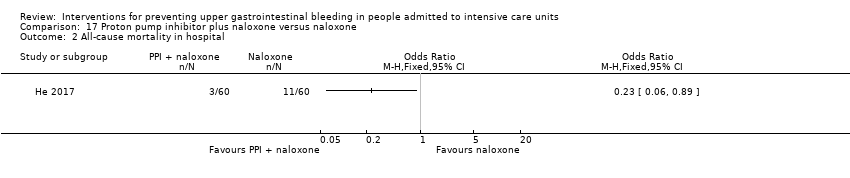
Comparison 17 Proton pump inhibitor plus naloxone versus naloxone, Outcome 2 All‐cause mortality in hospital.

Comparison 17 Proton pump inhibitor plus naloxone versus naloxone, Outcome 3 Adverse events ‐ gastrointestinal discomfort.
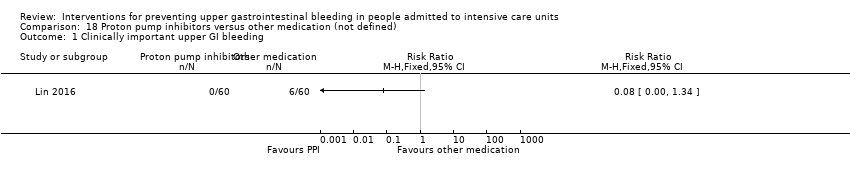
Comparison 18 Proton pump inhibitors versus other medication (not defined), Outcome 1 Clinically important upper GI bleeding.

Comparison 18 Proton pump inhibitors versus other medication (not defined), Outcome 2 Nosocomial pneumonia.

Comparison 18 Proton pump inhibitors versus other medication (not defined), Outcome 3 All‐cause mortality in hospital.

Comparison 19 Antacids versus sucralfate, Outcome 1 Clinically important upper GI bleeding.
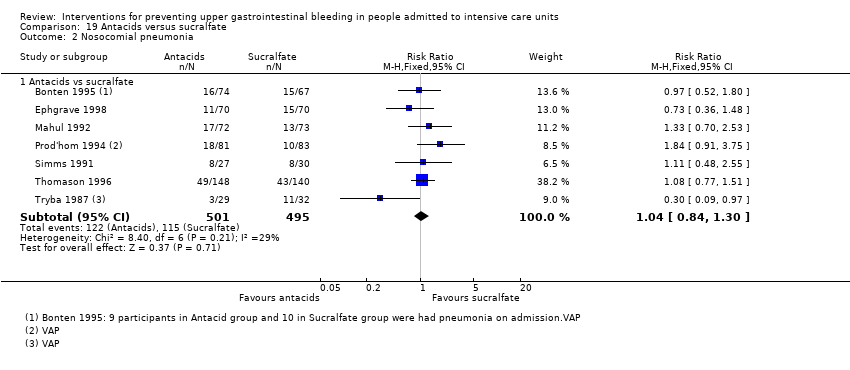
Comparison 19 Antacids versus sucralfate, Outcome 2 Nosocomial pneumonia.

Comparison 19 Antacids versus sucralfate, Outcome 3 All‐cause mortality in ICU.

Comparison 19 Antacids versus sucralfate, Outcome 4 All‐cause mortality in hospital.

Comparison 19 Antacids versus sucralfate, Outcome 5 Duration of ICU stay.

Comparison 19 Antacids versus sucralfate, Outcome 6 Duration of intubation.

Comparison 19 Antacids versus sucralfate, Outcome 7 Number of participants requiring blood transfusion.

Comparison 19 Antacids versus sucralfate, Outcome 8 Adverse events of interventions.

Comparison 20 Antacids versus prostaglandin analogues, Outcome 1 Clinically important upper GI bleeding.

Comparison 20 Antacids versus prostaglandin analogues, Outcome 2 All‐cause mortality in ICU.

Comparison 20 Antacids versus prostaglandin analogues, Outcome 3 Adverse events of interventions.
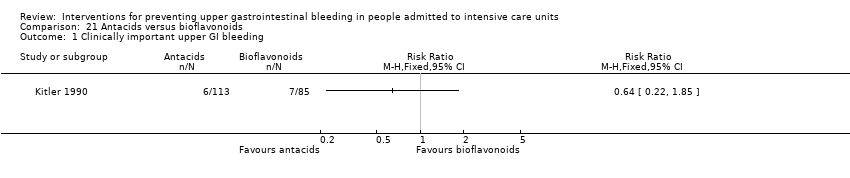
Comparison 21 Antacids versus bioflavonoids, Outcome 1 Clinically important upper GI bleeding.
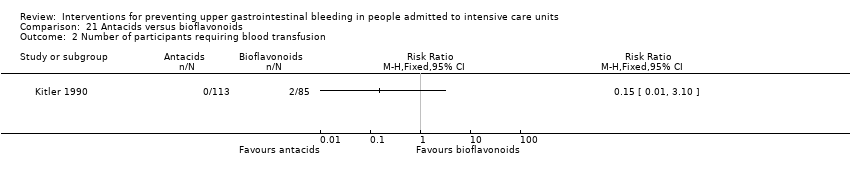
Comparison 21 Antacids versus bioflavonoids, Outcome 2 Number of participants requiring blood transfusion.

Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 1 Clinically important upper GI bleeding.
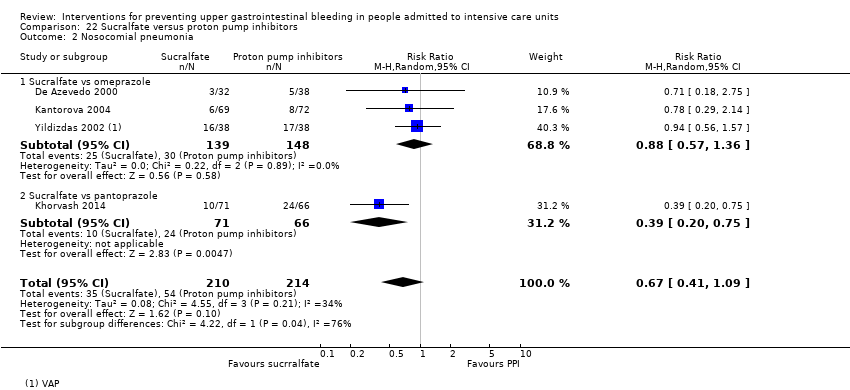
Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 2 Nosocomial pneumonia.
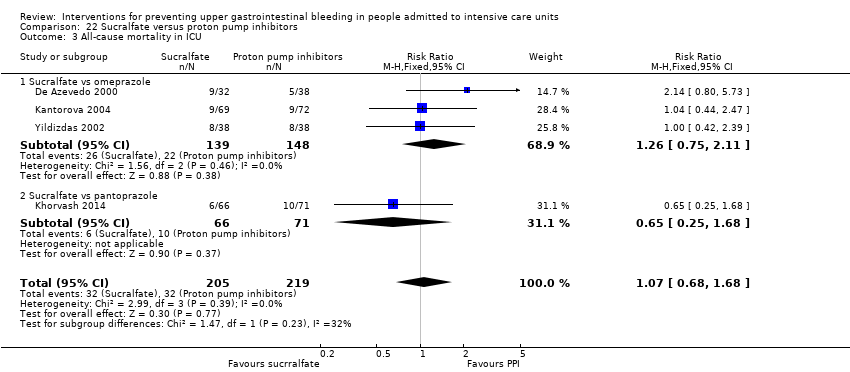
Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 3 All‐cause mortality in ICU.
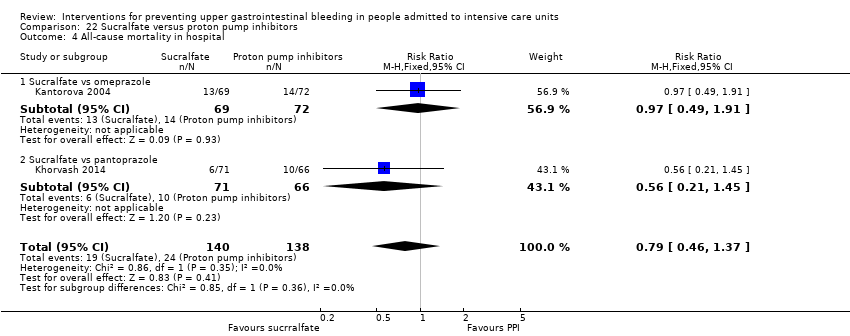
Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 4 All‐cause mortality in hospital.

Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 5 Duration of ICU stay.

Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 6 Duration of intubation.
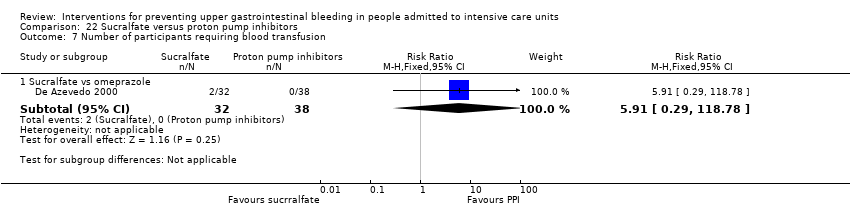
Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 7 Number of participants requiring blood transfusion.

Comparison 22 Sucralfate versus proton pump inhibitors, Outcome 8 Adverse events of interventions.

Comparison 23 Sucralfate versus bioflavonoids, Outcome 1 Clinically important upper GI bleeding.

Comparison 23 Sucralfate versus bioflavonoids, Outcome 2 Number of participants requiring blood transfusion.

Comparison 24 Total parenteral nutrition (TPN) versus any other intervention + TPN, Outcome 1 Clinically important upper GI bleeding.

Comparison 24 Total parenteral nutrition (TPN) versus any other intervention + TPN, Outcome 2 All‐cause mortality in ICU.

Comparison 24 Total parenteral nutrition (TPN) versus any other intervention + TPN, Outcome 3 Duration of intubation.

Comparison 25 Bowel stimulation versus no prophylaxis, Outcome 1 Clinically important upper GI bleeding.

Comparison 26 Nasojejunal nutrition versus nasogastric nutrition, Outcome 1 Clinically important upper GI bleeding.

Comparison 26 Nasojejunal nutrition versus nasogastric nutrition, Outcome 2 Nosocomial pneumonia.
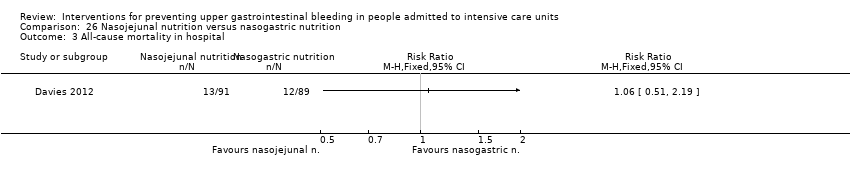
Comparison 26 Nasojejunal nutrition versus nasogastric nutrition, Outcome 3 All‐cause mortality in hospital.

Comparison 26 Nasojejunal nutrition versus nasogastric nutrition, Outcome 4 Adverse events of interventions.
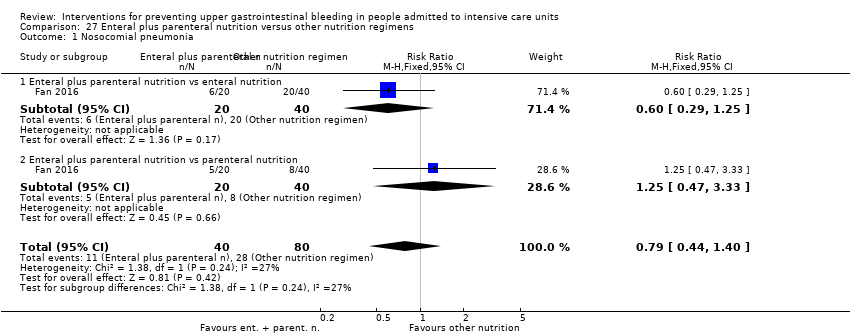
Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 1 Nosocomial pneumonia.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 2 All‐cause mortality in hospital.
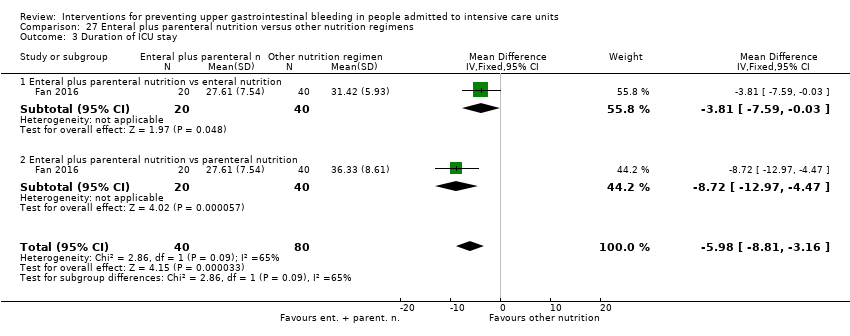
Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 3 Duration of ICU stay.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 4 Duration of intubation.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 5 Adverse events ‐ stress ulcer.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 6 Adverse events ‐ diarrhoea.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 7 Adverse events ‐ pyaemia.
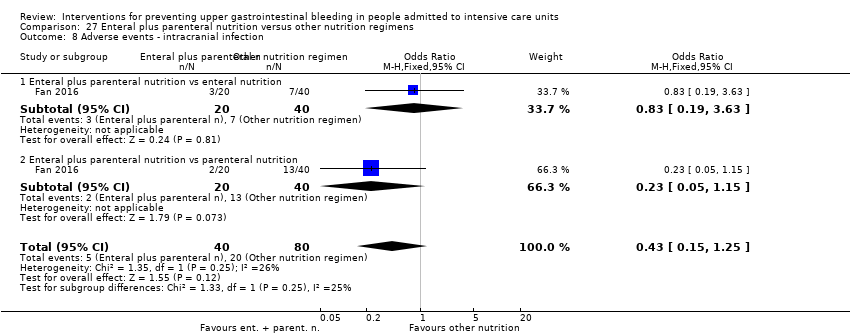
Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 8 Adverse events ‐ intracranial infection.

Comparison 27 Enteral plus parenteral nutrition versus other nutrition regimens, Outcome 9 Adverse events ‐ hypoproteinaemia.
| Any intervention compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with placebo or no prophylaxis | Risk with Interventions | |||||
| Clinically important upper GI bleeding Follow‐up: 15 days† | Study population | RR 0.47 | 3207 | ⊕⊕⊕⊝ | ||
| 188 per 1000 | 88 per 1000 | |||||
| Nosocomial pneumonia Follow‐up: 48 hours after extubation‡ | Study population | RR 1.15 | 1331 | ⊕⊕⊝⊝ | ||
| 143 per 1000 | 164 per 1000 | |||||
| All‐cause mortality in ICU Follow‐up: 4 weeks§ | Study population | RR 1.10 | 2159 | ⊕⊕⊝⊝ | ||
| 152 per 1000 | 168 per 1000 | |||||
| Duration of ICU stay Follow‐up: not reported | Mean duration of ICU stay ranged from 8.6 to 11.1 days | MD 0.24 days higher | ‐ | 447 | ⊕⊕⊝⊝ | |
| Number of participants requiring blood transfusion Follow‐up: 48 hours after discharge‡ | Study population | RR 0.63 | 981 | ⊕⊕⊕⊝ | ||
| 96 per 1000 | 60 per 1000 | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). †Duration of follow‐up reported in four studies. §Duration of follow‐up reported in five studies. | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in nine studies, high risk of detection bias in five studies, high risk of attrition bias in four studies, high risk of reporting bias in five studies, and high risk of other biases in four studies. bDowngraded by one level for imprecision because effect estimate and 95% CI were compatible with benefit and harm. cDowngraded by one level for risk of bias because of high risk of performance bias in three studies, high risk of detection bias in one study, and high risk of attrition bias in two studies. dDowngraded by one level for risk of bias because of high risk of performance bias in seven studies and high risk of attrition bias in two studies. eDowngraded by one level for risk of bias because of high risk of performance bias in one study. fDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in two studies, high risk of attrition bias in one study, high risk of reporting bias in one study, and high risk of other biases in one study. | ||||||
| H2 receptor antagonists compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with placebo or no prophylaxis | Risk with H2 receptor antagonists | |||||
| Clinically important upper GI bleeding Follow‐up: 15 days/weeks† | Study population | RR 0.50 | 2149 | ⊕⊕⊕⊝ | ||
| 182 per 1000 | 91 per 1000 | |||||
| Nosocomial pneumonia Follow‐up: 48 hours after extubation‡ | Study population | RR 1.12 | 945 | ⊕⊕⊝⊝ | ||
| 146 per 1000 | 164 per 1000 | |||||
| All‐cause mortality in ICU Follow‐up: 4 weeks§ | Study population | RR 1.12 | 1428 | ⊕⊕⊝⊝ | ||
| 145 per 1000 | 162 per 1000 | |||||
| Duration of ICU stay Follow‐up: not reported | Mean duration of ICU stay ranged from 8.6 to 11.1 days | MD 0.73 days higher | ‐ | 230 | ⊕⊕⊝⊝ | |
| Number of participants requiring blood transfusions Follow‐up: 48 hours after extubationǁ | Study population | RR 0.58 | 655 | ⊕⊕⊕⊝ | ||
| 112 per 1000 | 65 per 1000 | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ‡Duration of follow‐up reported in two studies. §Duration of follow‐up reported in five studies. ǁDuration of follow‐up reported in one study. | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in eight studies, high risk of attrition bias in two studies, high risk of reporting bias in four studies, and high risk of other biases in three studies. bDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm. cDowngraded by one level for risk of bias because of high risk performance bias in three studies and high risk of attrition bias in one study. dDowngraded by one level for risk of bias because of high risk of performance bias in three studies and high risk of attrition bias in one study. eDowngraded by one level for risk of bias because of high risk of performance bias in one study. fDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in two studies, high risk of attrition bias in one study, and high risk of other biases in one study. | ||||||
| Proton pump inhibitors compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with placebo or no prophylaxis | Risk with proton pump inhibitors | |||||
| Clinically important upper GI bleeding Follow‐up: not reported | Study population | RR 0.63 | 237 | ⊕⊕⊝⊝ | ||
| 49 per 1000 | 31 per 1000 | |||||
| Nosocomial pneumonia Follow‐up: not reported | Study population | RR 1.24 | 227 | ⊕⊕⊝⊝ | ||
| 188 per 1000 | 233 per 1000 | |||||
| All‐cause mortality in ICU Follow‐up: not reported | Study population | RR 1.09 | 258 | ⊕⊕⊝⊝ | ||
| 134 per 1000 | 146 per 1000 | |||||
| Duration of ICU stay Follow‐up: not reported | Mean duration of ICU stay ranged from 8.6 to 11.1 days | MD 0.03 days lower | ‐ | 227 | ⊕⊕⊝⊝ | |
| Number of participants requiring blood transfusion | Not reported | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm. bDowngraded by one level for risk of bias because of high risk of performance bias in one study and high risk of attrition bias in one study. cDowngraded by one level for risk of bias because of high risk of performance bias in one study. | ||||||
| Antacids compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with placebo or no prophylaxis | Risk with antacids | |||||
| Clinically important upper GI bleeding Follow‐up: not reported | Study population | RR 0.49 | 774 | ⊕⊕⊝⊝ | ||
| 170 per 1000 | 83 per 1000 | |||||
| Nosocomial pneumonia | Not reported | |||||
| All‐cause mortality in ICU Follow‐up: not reported | Study population | RR 1.01 | 300 | ⊕⊕⊝⊝ | ||
| 161 per 1000 | 163 per 1000 | |||||
| Duration of ICU stay | Not reported | |||||
| Number of participants requiring blood transfusions Follow‐up: not reported | Study population | RR 0.94 | 226 | ⊕⊕⊝⊝ | ||
| 45 per 1000 | 43 per 1000 | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDowngraded by one level for inconsistency because of moderate heterogeneity; I² = 56%. bDowngraded by one level for risk of bias because of high risk of performance bias in five studies, high risk of detection bias in one study, high risk of reporting bias in two studies, and high risk of other biases in one study. cDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm. dDowngraded by one level for risk of bias because of high risk of performance bias in two studies. eDowngraded by one level for risk of bias because of high risk of performance bias in one study. | ||||||
| Sucralfate compared with placebo or no prophylaxis for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with placebo or no prophylaxis | Risk with sucralfate | |||||
| Clinically important upper GI bleeding Follow‐up: 15 days† | Study population | RR 0.53 | 598 | ⊕⊕⊕⊝ | ||
| 108 per 1000 | 57 per 1000 | |||||
| Nosocomial pneumonia Follow‐up: not reported | Study population | RR 1.33 | 450 | ⊕⊕⊝⊝ | ||
| 122 per 1000 | 163 per 1000 | |||||
| All‐cause mortality in ICU Follow‐up: 15 days† | Study population | RR 0.97 | 500 | ⊕⊕⊝⊝ | ||
| 165 per 1000 | 160 per 1000 | |||||
| Duration of ICU stay Follow‐up: not reported | Mean duration of ICU stay ranged from 8.6 to 11.1 days | MD 0.02 days lower | ‐ | 224 | ⊕⊕⊝⊝ | |
| Number of participants requiring blood transfusion Follow‐up: not reported | Study population | RR 0.60 | 200 | ⊕⊕⊝⊝ | ||
| 50 per 1000 | 30 per 1000 | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDowngraded by one level for risk of bias because of high risk of performance bias in five studies, high risk of reporting bias in one study, and high risk of other biases in one study. bDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm. cDowngraded by one level for risk of bias because of high risk of performance bias in two studies. dDowngraded by one level for risk of bias because of high risk of performance bias in three studies. eDowngraded by one level for risk of bias because of high risk of performance bias in one study. | ||||||
| H2 receptor antagonists compared with proton pump inhibitors for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with proton pump inhibitors | Risk with H2 receptor antagonists | |||||
| Clinically important upper GI bleeding Follow‐up: not reported | Study population | RR 2.90 | 1636 | ⊕⊕⊝⊝ | ||
| 25 per 1000 | 73 per 1000 | |||||
| Nosocomial pneumonia Follow‐up: 30 days† | Study population | RR 1.02 | 1256 | ⊕⊕⊝⊝ | ||
| 123 per 1000 | 126 per 1000 | |||||
| All‐cause mortality in ICU Follow‐up: 30 days† | Study population | RR 0.96 | 1564 | ⊕⊕⊝⊝ | ||
| 158 per 1000 | 152 per 1000 | |||||
| Duration of ICU stay Follow‐up: not reported | Mean duration of ICU stay ranged from 7.7 to 23.6 days | MD 0.14 days higher | ‐ | 482 | ⊕⊕⊝⊝ | |
| Number of participants requiring blood transfusion Follow‐up: not reported | Study population | RR 1.98 | 575 | ⊕⊕⊕⊝ | ||
| 17 per 1000 | 35 per 1000 | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDowngraded by one level for inconsistency because of substantial heterogeneity; I² = 59%. bDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in five studies, high risk of detection bias in two studies, and high risk of other biases in one study. cDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm. dDowngraded by one level for risk of bias because of high risk of performance bias in four studies, high risk of detection bias in two studies, and high risk of other biases in one study. eDowngraded by one level for risk of bias because of high risk of performance bias in five studies, high risk of attrition bias in one study, and high risk of other biases in one study. fDowngraded by one level for risk of bias because of high risk of performance bias in three studies, high risk of attrition bias in one study, and high risk of other biases in one study. | ||||||
| H2 receptor antagonists compared with antacids for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with antacids | Risk with H2 receptor antagonists | |||||
| Clinically important upper GI bleeding Follow‐up: 25 days† | Study population | RR 0.96 | 1700 | ⊕⊕⊝⊝ | ||
| 86 per 1000 | 82 per 1000 | |||||
| Nosocomial pneumonia Follow‐up: 25 days‡ | Study population | RR 1.05 | 581 | ⊕⊕⊝⊝ | ||
| 280 per 1000 | 294 per 1000 | |||||
| All‐cause mortality in ICU Follow‐up: 25 days§ | Study population | RR 1.01 | 1321 | ⊕⊝⊝⊝ | ||
| 163 per 1000 | 165 per 1000 | |||||
| Duration of ICU stay | Not reported | |||||
| Number of participants requiring blood transfusion Follow‐up: not reported | Study population | RR 2.49 | 744 | ⊕⊕⊕⊝ | ||
| 30 per 1000 | 75 per 1000 | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ‡Duration of follow‐up reported in one study. §Duration of follow‐up reported in three studies. | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm. bDowngraded by one level for risk of bias because of high risk of selection bias in two studies, high risk of performance bias in 12 studies, high risk of detection bias in two studies, and high risk of reporting bias in two studies. cDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in four studies, high risk of detection bias in one study, and high risk of reporting bias in one study. dDowngraded by one level for inconsistency because of moderate heterogeneity; I² = 53%. eDowngraded by one level for risk of bias because of high risk of selection bias in two studies, high risk of performance bias in nine studies, and high risk of reporting bias in one study. fDowngraded by one level for risk of bias because of high risk of selection bias in one study and high risk of performance bias in four studies. | ||||||
| H2 receptor antagonists compared with sucralfate for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with sucralfate | Risk with H2 receptor antagonists | |||||
| Clinically important upper GI bleeding Follow‐up: 15 days† | Study population | RR 1.10 | 3316 | ⊕⊕⊝⊝ | ||
| 66 per 1000 | 73 per 1000 | |||||
| Nosocomial pneumonia Follow‐up: 25 days‡ | Study population | RR 1.22 | 3041 | ⊕⊕⊕⊝ | ||
| 189 per 1000 | 230 per 1000 | |||||
| All‐cause mortality in ICU Follow‐up: 25 days§ | Study population | RR 1.09 | 3178 | ⊕⊕⊝⊝ | ||
| 204 per 1000 | 222 per 1000 | |||||
| Duration of ICU stay Follow‐up: 2 weeks | Mean duration of ICU stay ranged from 7.9 to 13.7 days | MD 0.01 days higher | ‐ | 1791 | ⊕⊝⊝⊝ | |
| Number of participants requiring blood transfusion Follow‐up: until death or dischargeǁ | Study population | RR 1.25 | 1095 | ⊕⊕⊝⊝ | ||
| 35 per 1000 | 43 per 1000 | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). †Duration of follow‐up reported in five studies. ‡Duration of follow‐up reported in three studies. §Duration of follow‐up reported in six studies. ǁDuration of follow‐up reported in one study. | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm. bDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in 20 studies, high risk of detection bias in two studies, high risk of attrition bias in two studies, high risk of reporting bias in four studies, and high risk of other biases in two studies. cDowngraded by one level for risk of bias because of high risk of selection bias in two studies, high risk of performance bias in 12 studies, high risk of detection bias in one study, high risk of attrition bias in one study, and high risk of reporting bias in two studies. dDowngraded by one level for risk of bias because of high risk of selection bias in two studies, high risk of performance bias in 16 studies, high risk of detection bias in one study, high risk of attrition bias in two studies, high risk of reporting bias in three studies, and high risk of other biases in one study. eDowngraded by one level for inconsistency because of considerable heterogeneity; I² = 82%. fDowngraded by one level for risk of bias because of high risk of performance bias in four studies and high risk of attrition bias in one study. gDowngraded by one level for risk of bias because of high risk of performance bias in eight studies, high risk of attrition bias in one study, high risk of reporting bias in one study, and high risk of other biases in one study. | ||||||
| Antacids compared with sucralfate for preventing upper gastrointestinal bleeding in people admitted to intensive care units | ||||||
| Patient or population: people admitted to intensive care units | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with sucralfate | Risk with antacids | |||||
| Clinically important upper GI bleeding Follow‐up: 21 days† | Study population | RR 1.00 | 1772 | ⊕⊕⊝⊝ | ||
| 66 per 1000 | 66 per 1000 | |||||
| Nosocomial pneumonia Follow‐up: 25 days‡ | Study population | RR 1.04 | 996 | ⊕⊕⊝⊝ | ||
| 232 per 1000 | 242 per 1000 | |||||
| All‐cause mortality in ICU Follow‐up: 25 days† | Study population | RR 1.15 | 1249 | ⊕⊕⊝⊝ | ||
| 206 per 1000 | 237 per 1000 | |||||
| Duration of ICU stay Follow‐up: not reported | Mean duration of ICU stay ranged from 10.4 to 16.8 days | MD 2.5 days lower | ‐ | 227 | ⊕⊕⊝⊝ | |
| Number of participants requiring blood transfusion Follow‐up: until discharge or onset of GI bleeding§ | Study population | RR 0.73 | 667 | ⊕⊕⊝⊝ | ||
| 52 per 1000 | 38 per 1000 | |||||
| Serious adverse events | Not reported | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ‡Duration of follow‐up reported in two studies. §Duration of follow‐up reported in one study. | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDowngraded by one level for imprecision because 95% CI was compatible with benefit and harm. bDowngraded by one level for risk of bias because of high risk of selection bias in three studies, high risk of performance bias in 12 studies, high risk of detection bias in one study, high risk of attrition bias in one study, high risk of reporting bias in two studies, and high risk of other biases in two studies. cDowngraded by one level for risk of bias because of high risk of performance bias in four studies, high risk of detection bias in one study, high risk of reporting bias in one study, and high risk of other biases in one study. dDowngraded by one level for risk of bias because of high risk of selection bias in three studies, high risk of performance bias in eight studies, high risk of attrition bias in one study, high risk of reporting bias in one study, and high risk of other biases in one study. eDowngraded by one level for risk of bias because of high risk of attrition bias in one study. fDowngraded by one level for risk of bias because of high risk of selection bias in one study, high risk of performance bias in six studies, and high risk of other biases in one study. | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 30 | 3132 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.39, 0.57] |
| 1.1 H2 receptor antagonists vs placebo or no prophylaxis | 24 | 1844 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.37, 0.59] |
| 1.2 Proton pump inhibitors vs placebo or no prophylaxis | 3 | 159 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.13, 2.59] |
| 1.3 Prostagladin analogues vs placebo or no prophylaxis | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.22, 4.55] |
| 1.4 Anticholinergics vs placebo or no prophylaxis | 2 | 103 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.87 [0.30, 2.49] |
| 1.5 Antacids vs placebo or no prophylaxis | 7 | 515 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.23, 0.63] |
| 1.6 Sucralfate vs placebo or no prophylaxis | 7 | 453 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.25, 0.87] |
| 2 Nosocomial pneumonia Show forest plot | 9 | 1331 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.90, 1.48] |
| 2.1 H2 receptor antagonists vs placebo or no prophylaxis | 8 | 788 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.77, 1.46] |
| 2.2 Proton pump inhibitors vs placebo or no prophylaxis | 2 | 149 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.59, 2.17] |
| 2.3 Anticholinergics vs placebo or no prophylaxis | 1 | 72 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.43, 2.59] |
| 2.4 Sucralfate vs placebo or no prophylaxis | 4 | 322 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.59 [0.84, 3.01] |
| 3 All‐cause mortality in ICU Show forest plot | 19 | 2159 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.90, 1.34] |
| 3.1 H2 receptor antagonists vs placebo or no prophylaxis | 14 | 1209 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.89, 1.53] |
| 3.2 Proton pump inhibitors vs placebo or no prophylaxis | 3 | 180 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.46, 2.38] |
| 3.3 Prostagladin analogues vs placebo or no prophylaxis | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.48, 2.74] |
| 3.4 Anticholinergics vs placebo or no prophylaxis | 2 | 103 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.59, 2.56] |
| 3.5 Antacids vs placebo or no prophylaxis | 2 | 250 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.58, 1.79] |
| 3.6 Sucralfate vs placebo or no prophylaxis | 5 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.58, 1.51] |
| 4 All‐cause mortality in hospital Show forest plot | 5 | 857 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.85, 1.55] |
| 4.1 H2 receptor antagonists vs placebo or no prophylaxis | 4 | 387 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.71, 1.83] |
| 4.2 Proton pump inhibitors vs placebo or no prophylaxis | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.42, 3.22] |
| 4.3 Antacids vs placebo or no prophylaxis | 1 | 126 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.79, 2.64] |
| 4.4 Sucralfate vs placebo or no prophylaxis | 2 | 244 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.53, 1.68] |
| 5 Duration of ICU stay Show forest plot | 2 | 447 | Mean Difference (IV, Fixed, 95% CI) | 0.24 [‐1.13, 1.61] |
| 5.1 H2 receptor antagonists vs placebo or no prophylaxis | 2 | 152 | Mean Difference (IV, Fixed, 95% CI) | 0.73 [‐1.64, 3.09] |
| 5.2 Proton pump inhibitors vs placebo or no prophylaxis | 2 | 149 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐2.33, 2.35] |
| 5.3 Sucralfate vs placebo or no prophylaxis | 2 | 146 | Mean Difference (IV, Fixed, 95% CI) | ‐0.01 [‐2.40, 2.38] |
| 6 Duration of intubation Show forest plot | 2 | 447 | Mean Difference (IV, Fixed, 95% CI) | 0.87 [‐0.58, 2.31] |
| 6.1 H2 receptor antagonists vs placebo or no prophylaxis | 2 | 152 | Mean Difference (IV, Fixed, 95% CI) | 0.78 [‐1.72, 3.29] |
| 6.2 Proton pump inhibitors vs placebo or no prophylaxis | 2 | 149 | Mean Difference (IV, Fixed, 95% CI) | 0.36 [‐2.18, 2.90] |
| 6.3 Sucralfate vs placebo or no prophylaxis | 2 | 146 | Mean Difference (IV, Fixed, 95% CI) | 1.43 [‐1.04, 3.89] |
| 7 Number of participants requiring blood transfusions Show forest plot | 9 | 981 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.41, 0.97] |
| 7.1 H2 receptor antagonists vs placebo or no prophylaxis | 7 | 605 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.57 [0.35, 0.94] |
| 7.2 Antacids vs placebo or no prophylaxis | 2 | 226 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.30, 2.96] |
| 7.3 Sucralfate vs placebo or no prophylaxis | 1 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.13, 4.34] |
| 8 Units of blood transfused Show forest plot | 2 | 309 | Mean Difference (IV, Random, 95% CI) | 0.09 [‐0.99, 1.17] |
| 8.1 H2 receptor antagonists vs placebo or no prophylaxis | 2 | 159 | Mean Difference (IV, Random, 95% CI) | ‐1.73 [‐6.37, 2.90] |
| 8.2 Sucralfate vs placebo or no prophylaxis | 1 | 150 | Mean Difference (IV, Random, 95% CI) | 0.80 [0.25, 1.35] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 24 | 2149 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.36, 0.70] |
| 1.1 Cimetidine vs placebo | 10 | 772 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.28, 1.02] |
| 1.2 Famotidine vs placebo | 1 | 146 | Risk Ratio (M‐H, Random, 95% CI) | 2.11 [0.20, 22.79] |
| 1.3 Ranitidine vs placebo | 5 | 446 | Risk Ratio (M‐H, Random, 95% CI) | 0.36 [0.17, 0.77] |
| 1.4 Cimetidine vs no prophylaxis | 3 | 516 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.23, 1.48] |
| 1.5 Famotidine vs no prophylaxis | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.3 [0.09, 0.96] |
| 1.6 Ranitidine vs no prophylaxis | 4 | 219 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.26, 1.00] |
| 2 Nosocomial pneumonia Show forest plot | 8 | 945 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.85, 1.48] |
| 2.1 Cimetidine vs placebo | 2 | 204 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.06, 2.00] |
| 2.2 Famotidine vs placebo | 1 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.48 [0.49, 4.45] |
| 2.3 Ranitidine vs placebo | 2 | 277 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.47, 1.31] |
| 2.4 Cimetidine vs no prophylaxis | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.17 [0.86, 5.47] |
| 2.5 Ranitidine vs no prophylaxis | 2 | 118 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.93, 1.90] |
| 3 All‐cause mortality in ICU Show forest plot | 14 | 1428 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.88, 1.42] |
| 3.1 Cimetidine vs placebo | 4 | 478 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.66, 1.68] |
| 3.2 Famotidine vs placebo | 1 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.55, 3.16] |
| 3.3 Ranitidine vs placebo | 2 | 148 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.31, 1.54] |
| 3.4 Cimetidine vs no prophylaxis | 2 | 400 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.61, 1.63] |
| 3.5 Famotidine vs no prophylaxis | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.59, 2.64] |
| 3.6 Ranitidine vs no prophylaxis | 4 | 206 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [0.97, 2.58] |
| 4 All‐cause mortality in hospital Show forest plot | 4 | 487 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.79, 1.70] |
| 4.1 Famotidine vs placebo | 1 | 146 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.43, 1.86] |
| 4.2 Ranitidine vs placebo | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.47] |
| 4.3 Cimetidine vs no prophylaxis | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.47 [0.88, 2.46] |
| 4.4 Ranitidine vs no prophylaxis | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.33, 2.53] |
| 5 Duration of ICU stay Show forest plot | 2 | 230 | Mean Difference (IV, Fixed, 95% CI) | 0.73 [‐0.92, 2.38] |
| 5.1 Famotidine vs placebo | 1 | 146 | Mean Difference (IV, Fixed, 95% CI) | 1.5 [‐1.93, 4.93] |
| 5.2 Ranitidine vs no prophylaxis | 1 | 84 | Mean Difference (IV, Fixed, 95% CI) | 0.5 [‐1.38, 2.38] |
| 6 Duration of intubation Show forest plot | 2 | 230 | Mean Difference (IV, Fixed, 95% CI) | 0.79 [‐0.95, 2.54] |
| 6.1 Famotidine vs placebo | 1 | 146 | Mean Difference (IV, Fixed, 95% CI) | 1.20 [‐1.86, 4.26] |
| 6.2 Ranitidine vs no prophylaxis | 1 | 84 | Mean Difference (IV, Fixed, 95% CI) | 0.60 [‐1.52, 2.72] |
| 7 Number of participants requiring blood transfusions Show forest plot | 7 | 655 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.36, 0.95] |
| 7.1 Cimetidine vs placebo | 3 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.39 [0.19, 0.79] |
| 7.2 Ranitidine vs placebo | 2 | 148 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.24, 4.60] |
| 7.3 Cimetidine vs no prophylaxis | 2 | 400 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.35, 1.71] |
| 8 Units of blood transfused Show forest plot | 2 | 209 | Mean Difference (IV, Fixed, 95% CI) | 0.33 [‐0.04, 0.70] |
| 8.1 Cimetidine vs placebo | 1 | 9 | Mean Difference (IV, Fixed, 95% CI) | ‐4.35 [‐7.35, ‐1.35] |
| 8.2 Cimetidine vs no prophylaxis | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 0.40 [0.03, 0.77] |
| 9 Adverse events of interventions Show forest plot | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 Diarrhoea | 2 | 225 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.57, 2.96] |
| 9.2 Thrombocytopenia | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.12, 72.77] |
| 9.3 Hypophosphatemia | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.75 [0.17, 84.02] |
| 9.4 Mental confusion | 5 | 657 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.01 [1.10, 3.65] |
| 9.5 Nausea and vomiting | 2 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.09, 2.35] |
| 9.6 Increased creatinine levels | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.64, 1.69] |
| 9.7 Erythema | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 71.22] |
| 9.8 Pancreatitis | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.01, 6.66] |
| 9.9 Chest infection | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.74 [0.92, 3.30] |
| 9.10 Delirium | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.59 [0.11, 59.93] |
| 9.11 Hallucinations | 1 | 39 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.59 [0.11, 59.93] |
| 9.12 Severe bleeding | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.47] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 3 | 237 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.18, 2.22] |
| 1.1 Omeprazole vs placebo | 1 | 147 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.07, 16.34] |
| 1.2 Omeprazole vs no prophylaxis | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.13, 4.18] |
| 1.3 Pantoprazole vs placebo | 1 | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.02, 4.66] |
| 2 Nosocomial pneumonia Show forest plot | 2 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.77, 1.98] |
| 2.1 Omeprazole vs placebo | 1 | 147 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.57, 4.86] |
| 2.2 Omeprazole vs no prophylaxis | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.66, 1.84] |
| 3 All‐cause mortality in ICU Show forest plot | 3 | 258 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.60, 1.99] |
| 3.1 Omeprazole vs placebo | 2 | 178 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.51, 2.83] |
| 3.2 Omeprazole vs no prophylaxis | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.42, 2.29] |
| 4 All‐cause mortality in hospital Show forest plot | 1 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.54, 2.13] |
| 4.1 Omeprazole vs placebo | 1 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.54, 2.13] |
| 5 Duration of ICU stay Show forest plot | 2 | 227 | Mean Difference (IV, Fixed, 95% CI) | ‐0.03 [‐1.63, 1.58] |
| 5.1 Omeprazole vs placebo | 1 | 147 | Mean Difference (IV, Fixed, 95% CI) | ‐0.90 [‐3.96, 2.16] |
| 5.2 Omeprazole vs no prophylaxis | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐1.58, 2.18] |
| 6 Duration of intubation Show forest plot | 2 | 227 | Mean Difference (IV, Fixed, 95% CI) | 0.36 [‐1.43, 2.15] |
| 6.1 Omeprazole vs placebo | 1 | 147 | Mean Difference (IV, Fixed, 95% CI) | 0.5 [‐2.72, 3.72] |
| 6.2 Omeprazole vs no prophylaxis | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 0.30 [‐1.85, 2.45] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Prostaglandin analogues vs placebo | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality in ICU Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 2 | 131 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.36, 2.51] |
| 1.1 Pirenzepine vs placebo | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.94 [0.34, 11.13] |
| 1.2 Pirenzepin + ranitidine vs placebo + ranitidine | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.17, 2.07] |
| 2 Nosocomial pneumonia Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Pirenzepine vs placebo | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 All‐cause mortality in ICU Show forest plot | 2 | 131 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.66, 2.30] |
| 3.1 Pirenzepine vs placebo | 1 | 101 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.65, 2.60] |
| 3.2 Pirenzepine + ranitidine vs placebo + ranitidine | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.24, 4.18] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 8 | 774 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.25, 0.99] |
| 1.1 Antacids vs placebo | 2 | 145 | Risk Ratio (M‐H, Random, 95% CI) | 2.04 [0.72, 5.79] |
| 1.2 Antacids vs no prophylaxis | 6 | 629 | Risk Ratio (M‐H, Random, 95% CI) | 0.35 [0.20, 0.60] |
| 2 All‐cause mortality in ICU Show forest plot | 2 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.53, 1.96] |
| 2.1 Antacids vs no prophylaxis | 2 | 300 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.53, 1.96] |
| 3 All‐cause mortality in hospital Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Antacids vs no prophylaxis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Number of participants requiring blood transfusions Show forest plot | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Antacids vs no prophylaxis | 2 | 226 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.30, 2.96] |
| 5 Adverse events of interventions Show forest plot | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Diarrhoea | 4 | 395 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.56 [1.83, 6.94] |
| 5.2 Hypomagnesaemia | 1 | 25 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.75 [0.17, 84.02] |
| 5.3 Hypophosphataemia | 2 | 225 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.48 [1.81, 16.61] |
| 5.4 Hypermagnesaemia | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.73 [0.36, 127.02] |
| 5.5 Nausea and vomiting | 3 | 370 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.39 [0.86, 6.64] |
| 5.6 Mental confusion | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [0.61, 2.67] |
| 5.7 Creatinine increase | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.73, 1.87] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 7 | 598 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.32, 0.88] |
| 1.1 Sucralfate vs placebo | 2 | 170 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.30, 6.62] |
| 1.2 Sucralfate vs no prophylaxis | 5 | 428 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.26, 0.80] |
| 2 Nosocomial pneumonia Show forest plot | 4 | 450 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.86, 2.04] |
| 2.1 Sucralfate vs placebo | 2 | 170 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.49, 4.16] |
| 2.2 Sucralfate vs no prophylaxis | 2 | 280 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.82, 2.07] |
| 3 All‐cause mortality in ICU Show forest plot | 5 | 500 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.66, 1.43] |
| 3.1 Sucralfate vs placebo | 2 | 170 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.48, 1.80] |
| 3.2 Sucralfate vs no prophylaxis | 3 | 330 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.62, 1.60] |
| 4 All‐cause mortality in hospital Show forest plot | 2 | 344 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.62, 1.52] |
| 4.1 Sucralfate vs placebo | 1 | 144 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.54, 2.18] |
| 4.2 Sucralfate vs no prophylaxis | 1 | 200 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.49, 1.62] |
| 5 Duration of ICU stay Show forest plot | 2 | 224 | Mean Difference (IV, Fixed, 95% CI) | ‐0.02 [‐1.70, 1.65] |
| 5.1 Sucralfate vs placebo | 1 | 144 | Mean Difference (IV, Fixed, 95% CI) | ‐0.70 [‐4.07, 2.67] |
| 5.2 Sucralfate vs no prophylaxis | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 0.20 [‐1.73, 2.13] |
| 6 Duration of intubation Show forest plot | 2 | 224 | Mean Difference (IV, Fixed, 95% CI) | 1.42 [‐0.27, 3.10] |
| 6.1 Sucralfate vs placebo | 1 | 144 | Mean Difference (IV, Fixed, 95% CI) | 0.80 [‐2.20, 3.80] |
| 6.2 Sucralfate vs no prophylaxis | 1 | 80 | Mean Difference (IV, Fixed, 95% CI) | 1.70 [‐0.34, 3.74] |
| 7 Number of participants requiring blood transfusions Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1 Sucralfate vs no prophylaxis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Units of blood transfused Show forest plot | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 0.8 [0.32, 1.28] |
| 8.1 Sucralfate vs no prophylaxis | 1 | 200 | Mean Difference (IV, Fixed, 95% CI) | 0.8 [0.32, 1.28] |
| 9 Adverse events of interventions Show forest plot | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.50, 161.13] |
| 9.1 Nausea / Vomiting | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.50, 161.13] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 13 | 1636 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.90 [1.83, 4.58] |
| 1.1 Cimetidine vs omeprazole | 1 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.55, 3.61] |
| 1.2 Famotidine vs lansoprazole | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.63 [0.15, 84.98] |
| 1.3 Famotidine vs omeprazole | 1 | 143 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.03 [0.19, 21.87] |
| 1.4 Famotidine vs pantoprazole | 2 | 159 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.18, 3.04] |
| 1.5 Famotidine vs esomeprazole | 2 | 371 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.53 [1.39, 40.85] |
| 1.6 Ranitidine vs omeprazole | 5 | 413 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.08 [1.99, 8.36] |
| 1.7 Ranitidine vs rabeprazole | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.49, 164.09] |
| 2 Nosocomial pneumonia Show forest plot | 10 | 1256 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.77, 1.35] |
| 2.1 Cimetidine vs omeprazole | 1 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.45, 1.54] |
| 2.2 Cimetidine vs pantoprazole | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.28, 2.91] |
| 2.3 Famotidine vs esomeprazole | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.07, 15.26] |
| 2.4 Famotidine vs omeprazole | 1 | 143 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.89 [0.34, 2.32] |
| 2.5 Ranitidine vs omeprazole | 5 | 413 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.80, 1.75] |
| 2.6 H2 receptor antagonists (not defined) vs proton pump inhibitors (not defined) | 1 | 79 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.47, 2.26] |
| 3 All‐cause mortality in ICU Show forest plot | 12 | 1564 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.78, 1.19] |
| 3.1 Cimetidine vs omeprazole | 1 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.45, 1.30] |
| 3.2 Cimetidine vs pantoprazole | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.25, 2.55] |
| 3.3 Famotidine vs omeprazole | 1 | 143 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.49, 2.61] |
| 3.4 Ranitidine vs omeprazole | 5 | 387 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.86, 1.40] |
| 3.5 Ranitidine vs pantoprazole | 3 | 333 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.31, 1.43] |
| 3.6 Ranitidine vs rabeprazole | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.12, 72.40] |
| 4 All‐cause mortality in hospital Show forest plot | 2 | 454 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.37, 1.43] |
| 4.1 Famotidine vs esomeprazole | 1 | 311 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.04, 3.49] |
| 4.2 Famotidine vs omeprazole | 1 | 143 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.39, 1.63] |
| 5 Duration of ICU stay Show forest plot | 5 | 482 | Mean Difference (IV, Fixed, 95% CI) | 0.14 [‐1.14, 1.41] |
| 5.1 Famotidine vs esomeprazole | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐6.51, 5.91] |
| 5.2 Famotidine vs omeprazole | 1 | 143 | Mean Difference (IV, Fixed, 95% CI) | 2.40 [‐0.44, 5.24] |
| 5.3 Ranitidine vs omeprazole | 3 | 279 | Mean Difference (IV, Fixed, 95% CI) | ‐0.44 [‐1.90, 1.02] |
| 6 Duration of intubation Show forest plot | 5 | 542 | Mean Difference (IV, Fixed, 95% CI) | ‐0.35 [‐1.48, 0.78] |
| 6.1 Famotidine vs omeprazole | 1 | 143 | Mean Difference (IV, Fixed, 95% CI) | 0.70 [‐2.24, 3.64] |
| 6.2 Ranitidine vs omeprazole | 3 | 279 | Mean Difference (IV, Fixed, 95% CI) | ‐0.78 [‐2.24, 0.67] |
| 6.3 Ranitidine vs pantoprazole | 1 | 120 | Mean Difference (IV, Fixed, 95% CI) | 0.07 [‐2.18, 2.32] |
| 7 Number of participants requiring blood transfusions Show forest plot | 3 | 575 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.98 [0.75, 5.21] |
| 7.1 Cimetidine vs omeprazole | 1 | 359 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.29, 3.34] |
| 7.2 Ranitidine vs omeprazole | 1 | 76 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.25, 100.80] |
| 7.3 Ranitidine vs rabeprazole | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.0 [0.49, 164.09] |
| 8 Adverse events of interventions Show forest plot | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Pyrexia | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.05, 19.03] |
| 8.2 Thrombocytopaenia | 2 | 253 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.64 [0.65, 20.46] |
| 8.3 Neuroleptic malignant syndrome | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.4 Cholestatic jaundice | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.5 Abnormal liver function test | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.6 Pruritus | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.7 Phlebitis | 1 | 202 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [0.06, 37.42] |
| 8.8 Major CV events | 1 | 311 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.26, 2.43] |
| 8.9 Abdominal distension and vomiting | 1 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.62, 2.14] |
| 8.10 Hypomagnesaemia | 1 | 129 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.16, 1.13] |
| 8.11 Nausea and vomiting | 1 | 129 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.48 [0.13, 1.77] |
| 8.12 Diarrhoea | 1 | 129 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.16, 7.67] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 16 | 1700 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.67, 1.36] |
| 1.1 Cimetidine vs antacids | 11 | 1155 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.65, 1.78] |
| 1.2 Cimetidine + pirenzepine vs antacid + pirenzepine | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.68] |
| 1.3 Ranitidine vs antacids | 4 | 479 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.42, 1.23] |
| 2 Nosocomial pneumonia Show forest plot | 4 | 581 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.81, 1.36] |
| 2.1 Cimetidine vs antacids | 2 | 136 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.70, 2.19] |
| 2.2 Ranitidine vs antacids | 2 | 445 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.75, 1.34] |
| 3 All‐cause mortality in ICU Show forest plot | 11 | 1321 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.66, 1.55] |
| 3.1 Cimetidine vs antacids | 8 | 885 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.69, 1.59] |
| 3.2 Cimetidine + pirenzepine vs antacid + pirenzepine | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.37, 4.25] |
| 3.3 Ranitidine vs antacids | 2 | 370 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.14, 8.97] |
| 4 All‐cause mortality in hospital Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Ranitidine vs antacids | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Duration of intubation Show forest plot | 3 | 121 | Mean Difference (IV, Random, 95% CI) | ‐0.81 [‐3.85, 2.23] |
| 5.1 Cimetidine vs antacids | 3 | 121 | Mean Difference (IV, Random, 95% CI) | ‐0.81 [‐3.85, 2.23] |
| 6 Number of participants requiring blood transfusions Show forest plot | 6 | 744 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.49 [1.35, 4.62] |
| 6.1 Cimetidine vs antacids | 5 | 583 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.47 [1.32, 4.63] |
| 6.2 Ranitidine vs antacids | 1 | 161 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.04 [0.13, 73.46] |
| 7 Adverse events of interventions Show forest plot | 12 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Diarrhoea | 6 | 777 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.13, 0.43] |
| 7.2 Thrombocytopaenia | 4 | 452 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.93, 2.09] |
| 7.3 Nausea and vomiting | 4 | 380 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.19, 1.10] |
| 7.4 Hypophosphataemia | 2 | 108 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.04, 1.30] |
| 7.5 Hypomagnesaemia | 1 | 22 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.02, 7.39] |
| 7.6 Increase in creatinine | 2 | 286 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.56, 1.28] |
| 7.7 Mental confusion | 4 | 476 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.77, 2.07] |
| 7.8 Hypermagnesaemia | 2 | 115 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.17, 2.03] |
| 7.9 Rash/Erythema | 2 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.02 [0.32, 28.53] |
| 7.10 Alkalosis | 1 | 75 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.73] |
| 7.11 Dryness of mouth | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.15 [0.26, 103.33] |
| 7.12 Leucopaenia | 1 | 161 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.04 [0.13, 73.46] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 24 | 3316 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.87, 1.41] |
| 1.1 Cimetidine vs sucralfate | 7 | 873 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.37 [0.87, 2.14] |
| 1.2 Famotidine vs sucralfate | 2 | 190 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.21, 1.78] |
| 1.3 Ranitidine vs sucralfate | 14 | 2186 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.76, 1.39] |
| 1.4 Cimetidine + pirenzepine vs sucralfate + pirenzepine | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.15 [0.26, 103.33] |
| 2 Nosocomial pneumonia Show forest plot | 17 | 3041 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [1.07, 1.40] |
| 2.1 Cimetidine vs sucralfate | 5 | 758 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.87, 1.47] |
| 2.2 Famotidine vs sucralfate | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.40, 3.20] |
| 2.3 Ranitidine vs sucralfate | 11 | 2143 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [1.07, 1.48] |
| 3 All‐cause mortality in ICU Show forest plot | 21 | 3178 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.95, 1.24] |
| 3.1 Cimetidine vs sucralfate | 6 | 814 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.91, 1.54] |
| 3.2 Famotidine vs sucralfate | 2 | 190 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.23 [0.69, 2.19] |
| 3.3 Ranitidine vs sucralfate | 12 | 2107 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.88, 1.22] |
| 3.4 Cimetidine + pirenzepine vs sucralfate + pirenzepine | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.38, 4.38] |
| 4 All‐cause mortality in hospital Show forest plot | 4 | 717 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.86, 1.50] |
| 4.1 Cimetidine vs sucralfate | 2 | 413 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.28 [0.86, 1.92] |
| 4.2 Ranitidine vs sucralfate | 1 | 164 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [0.71, 1.74] |
| 4.3 Famotidine vs sucralfate | 1 | 140 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.40, 1.71] |
| 5 Duration of intubation Show forest plot | 10 | 1751 | Mean Difference (IV, Random, 95% CI) | 0.22 [‐1.55, 2.00] |
| 5.1 Cimetidine vs sucralfate | 2 | 97 | Mean Difference (IV, Random, 95% CI) | 0.58 [‐1.71, 2.87] |
| 5.2 Famotidine vs sucralfate | 1 | 140 | Mean Difference (IV, Random, 95% CI) | 0.40 [‐2.30, 3.10] |
| 5.3 Ranitidine vs sucralfate | 7 | 1514 | Mean Difference (IV, Random, 95% CI) | 0.15 [‐2.12, 2.43] |
| 6 Duration of ICU stay Show forest plot | 6 | 1791 | Mean Difference (IV, Random, 95% CI) | 0.01 [‐1.92, 1.95] |
| 6.1 Cimetidine vs sucralfate | 1 | 213 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐3.05, 3.05] |
| 6.2 Famotidine vs sucralfate | 1 | 140 | Mean Difference (IV, Random, 95% CI) | 2.20 [‐0.96, 5.36] |
| 6.3 Ranitidine vs sucralfate | 4 | 1438 | Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐2.70, 1.84] |
| 7 Number of participants requiring blood transfusion Show forest plot | 9 | 1095 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.70, 2.23] |
| 7.1 Cimetidine vs sucralfate | 5 | 732 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.47, 2.16] |
| 7.2 Ranitidine vs sucralfate | 4 | 363 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.77 [0.71, 4.39] |
| 8 Units of blood transfused Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 8.1 Cimetidine vs sucralfate | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Adverse events of interventions Show forest plot | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 Thrombocytopaenia | 2 | 240 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.72 [0.56, 39.47] |
| 9.2 Nausea and vomiting | 2 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.07 [0.01, 0.54] |
| 9.3 Hypermagnesaemia | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.71 [0.31, 23.93] |
| 9.4 Rash/Erythema | 2 | 233 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.06 [0.32, 28.87] |
| 9.5 Confusion | 3 | 382 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.48 [0.77, 26.00] |
| 9.6 Neutropaenia | 1 | 114 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.18 [0.25, 105.47] |
| 9.7 Dryness of mouth | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.15 [0.26, 103.33] |
| 9.8 Leucopaenia | 1 | 163 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.11 [0.13, 75.26] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 3 | 556 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.37 [0.58, 3.26] |
| 1.1 Cimetidine vs pirenzepine | 1 | 55 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.45 [0.26, 7.99] |
| 1.2 Ranitidine vs pirenzepine | 2 | 501 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.35 [0.50, 3.67] |
| 2 Nosocomial pneumonia Show forest plot | 3 | 544 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.50, 1.84] |
| 2.1 Famotidine vs pirenzepine | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.42] |
| 2.2 Ranitidine vs pirenzepine | 2 | 501 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.53, 2.01] |
| 3 All‐cause mortality in ICU Show forest plot | 2 | 501 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.21, 3.87] |
| 3.1 Ranitidine vs pirenzepine | 2 | 501 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.21, 3.87] |
| 4 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Ranitidine vs pirenzepine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse events of interventions Show forest plot | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Tachycardia | 1 | 55 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.01, 1.90] |
| 5.2 High temperature | 1 | 43 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.21, 1.32] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Cimetidine vs misoprostol | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality in ICU Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Cimetidine vs misoprostol | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Ranitidine vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality in ICU Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Ranitidine vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Ranitidine vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 2 | 230 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.06, 0.95] |
| 1.1 Cimetidine + antacids vs sucralfate | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.70] |
| 1.2 Cimetidine or ranitidine + antacids vs sucralfate | 1 | 130 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.06, 1.41] |
| 2 Nosocomial pneumonia Show forest plot | 3 | 281 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.51, 2.32] |
| 2.1 Cimetidine + antacids vs sucralfate | 1 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.26, 1.07] |
| 2.2 Ranitidine + antacids vs sucralfate | 1 | 51 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.64, 2.53] |
| 2.3 Cimetidine or ranitidine + antacids vs sucralfate | 1 | 130 | Risk Ratio (M‐H, Random, 95% CI) | 2.02 [0.89, 4.58] |
| 3 All‐cause mortality in ICU Show forest plot | 2 | 230 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.92, 2.05] |
| 3.1 Cimetidine + antacids vs sucralfate | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.46, 2.19] |
| 3.2 Cimetidine or ranitidine + antacids vs sucralfate | 1 | 130 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.57 [0.99, 2.50] |
| 4 Duration of ICU stay Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5 Duration of intubation Show forest plot | 2 | 230 | Mean Difference (IV, Random, 95% CI) | ‐1.24 [‐13.82, 11.33] |
| 5.1 Cimetidine + antacids vs sucralfate | 1 | 100 | Mean Difference (IV, Random, 95% CI) | ‐8.8 [‐20.11, 2.51] |
| 5.2 Cimetidine or ranitidine + antacids vs sucralfate | 1 | 130 | Mean Difference (IV, Random, 95% CI) | 4.20 [‐0.54, 8.94] |
| 6 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Proton pump inhibitors vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 All‐cause mortality in ICU Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Proton pump inhibitors vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Proton pump inhibitors vs teprenone | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 All‐cause mortality in hospital Show forest plot | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Adverse events ‐ gastrointestinal discomfort Show forest plot | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Nosocomial pneumonia Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 All‐cause mortality in hospital Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 16 | 1772 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.72, 1.39] |
| 1.1 Antacids vs sucralfate | 15 | 1705 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.69, 1.35] |
| 1.2 Antacid + pirenzepine vs sucralfate + pirenzepine | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.15 [0.26, 103.33] |
| 2 Nosocomial pneumonia Show forest plot | 7 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Antacids vs sucralfate | 7 | 996 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.84, 1.30] |
| 3 All‐cause mortality in ICU Show forest plot | 11 | 1249 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.93, 1.40] |
| 3.1 Antacid vs sucralfate | 10 | 1182 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.94, 1.41] |
| 3.2 Antacid + pirenzepine vs sucralfate + pirenzepine | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.28, 3.78] |
| 4 All‐cause mortality in hospital Show forest plot | 3 | 450 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.69, 1.39] |
| 5 Duration of ICU stay Show forest plot | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Antacids vs sucralfate | 2 | 227 | Mean Difference (IV, Fixed, 95% CI) | ‐2.50 [‐6.61, 1.61] |
| 6 Duration of intubation Show forest plot | 4 | Std. Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 Antacids vs sucralfate | 4 | 281 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.18 [‐0.41, 0.06] |
| 7 Number of participants requiring blood transfusion Show forest plot | 6 | 667 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.40, 1.34] |
| 8 Adverse events of interventions Show forest plot | 9 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Diarrhoea | 6 | 599 | Risk Ratio (M‐H, Fixed, 95% CI) | 12.40 [3.88, 39.64] |
| 8.2 Hypermagnesaemia | 4 | 317 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.72 [1.24, 17.95] |
| 8.3 Nausea and vomiting | 3 | 223 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.28, 1.41] |
| 8.4 Thrombocytopaenia | 1 | 38 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.26, 97.70] |
| 8.5 Severe alkalosis | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 71.92] |
| 8.6 Allergic reactions | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.01, 4.06] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 2 | 329 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.12, 0.91] |
| 2 All‐cause mortality in ICU Show forest plot | 2 | 417 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.42, 1.67] |
| 3 Adverse events of interventions Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Diarrhoea | 1 | 368 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.92 [0.64, 1.33] |
| 3.2 Elevated serum bicarbonate | 1 | 338 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.21 [1.27, 3.87] |
| 3.3 Phospate levels < 2.5 mg/dL | 1 | 276 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.66 [1.01, 2.73] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Sucralfate vs omeprazole | 3 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.58 [0.77, 8.63] |
| 2 Nosocomial pneumonia Show forest plot | 4 | 424 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.41, 1.09] |
| 2.1 Sucralfate vs omeprazole | 3 | 287 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.57, 1.36] |
| 2.2 Sucralfate vs pantoprazole | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.20, 0.75] |
| 3 All‐cause mortality in ICU Show forest plot | 4 | 424 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.68, 1.68] |
| 3.1 Sucralfate vs omeprazole | 3 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.26 [0.75, 2.11] |
| 3.2 Sucralfate vs pantoprazole | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.25, 1.68] |
| 4 All‐cause mortality in hospital Show forest plot | 2 | 278 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.46, 1.37] |
| 4.1 Sucralfate vs omeprazole | 1 | 141 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.49, 1.91] |
| 4.2 Sucralfate vs pantoprazole | 1 | 137 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.21, 1.45] |
| 5 Duration of ICU stay Show forest plot | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5.1 Sucralfate vs omeprazole | 2 | 217 | Mean Difference (IV, Fixed, 95% CI) | 0.01 [‐1.68, 1.70] |
| 6 Duration of intubation Show forest plot | 3 | 354 | Mean Difference (IV, Fixed, 95% CI) | ‐0.16 [‐1.61, 1.28] |
| 6.1 Sucralfate vs omeprazole | 2 | 217 | Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐1.56, 1.60] |
| 6.2 Sucralfate vs pantoprazole | 1 | 137 | Mean Difference (IV, Fixed, 95% CI) | ‐1.10 [‐4.69, 2.49] |
| 7 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Sucralfate vs omeprazole | 1 | 70 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.91 [0.29, 118.78] |
| 8 Adverse events of interventions Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Fever | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.70, 0.94] |
| 8.2 Leucocytosis | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.55, 0.80] |
| 8.3 Sudden purulent sputum | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.38, 1.86] |
| 8.4 Sudden cough or aggravation of coughing | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.23 [0.07, 0.79] |
| 8.5 Dyspnoea | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.54, 0.87] |
| 8.6 Rales or bronchial sounds | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.19, 0.51] |
| 8.7 Aggravation of blood gas exchange | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.49, 1.18] |
| 8.8 Change in sputum quality | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.23 [0.13, 0.40] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Number of participants requiring blood transfusion Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 TPN vs ranitidine + TPN | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.8 [0.05, 12.14] |
| 1.2 TPN vs sucralfate + TPN | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.03, 3.26] |
| 2 All‐cause mortality in ICU Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 TPN vs ranitidine + TPN | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.41, 3.09] |
| 2.2 TPN vs sucralfate + TPN | 1 | 49 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.26, 1.52] |
| 3 Duration of intubation Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 TPN vs ranitidine + TPN | 1 | 54 | Mean Difference (IV, Fixed, 95% CI) | ‐2.0 [‐9.53, 5.53] |
| 3.2 TPN vs sucralfate + TPN | 1 | 49 | Mean Difference (IV, Fixed, 95% CI) | 3.0 [‐1.50, 7.50] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically important upper GI bleeding Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Nosocomial pneumonia Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 All‐cause mortality in hospital Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Adverse events of interventions Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Nosocomial pneumonia Show forest plot | 1 | 120 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.44, 1.40] |
| 1.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.29, 1.25] |
| 1.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.47, 3.33] |
| 2 All‐cause mortality in hospital Show forest plot | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.06, 0.60] |
| 2.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.05, 1.30] |
| 2.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.03, 0.74] |
| 3 Duration of ICU stay Show forest plot | 1 | 120 | Mean Difference (IV, Fixed, 95% CI) | ‐5.98 [‐8.81, ‐3.16] |
| 3.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐3.81 [‐7.59, ‐0.03] |
| 3.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐8.72 [‐12.97, ‐4.47] |
| 4 Duration of intubation Show forest plot | 1 | 120 | Mean Difference (IV, Fixed, 95% CI) | ‐7.37 [‐9.29, ‐5.45] |
| 4.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐4.17 [‐6.96, ‐1.38] |
| 4.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Mean Difference (IV, Fixed, 95% CI) | ‐10.24 [‐12.88, ‐7.60] |
| 5 Adverse events ‐ stress ulcer Show forest plot | 1 | 120 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.36, 1.33] |
| 5.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.38, 3.45] |
| 5.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.23, 1.20] |
| 6 Adverse events ‐ diarrhoea Show forest plot | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.17, 1.02] |
| 6.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.05, 0.59] |
| 6.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.35, 5.73] |
| 7 Adverse events ‐ pyaemia Show forest plot | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.37, 2.09] |
| 7.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.11 [0.87, 19.41] |
| 7.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.11, 1.21] |
| 8 Adverse events ‐ intracranial infection Show forest plot | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.15, 1.25] |
| 8.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.19, 3.63] |
| 8.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.05, 1.15] |
| 9 Adverse events ‐ hypoproteinaemia Show forest plot | 1 | 120 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.11 [0.04, 0.27] |
| 9.1 Enteral plus parenteral nutrition vs enteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.06, 0.72] |
| 9.2 Enteral plus parenteral nutrition vs parenteral nutrition | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.04 [0.01, 0.19] |