Antagonistas opiáceos Mu para la disfunción intestinal inducida por opiáceos en pacientes con cáncer y en pacientes que reciben cuidados paliativos
Resumen
Antecedentes
La disfunción intestinal inducida por opiáceos (DIIO) se caracteriza por estreñimiento, evacuación incompleta, timpanismo abdominal y reflujo gástrico. Es uno de los principales eventos adversos del tratamiento para el dolor en el cáncer y en los cuidados paliativos, y da lugar a una mayor morbilidad y a una reducción en la calidad de vida.
Ésta es una actualización de dos revisiones Cochrane. Una se publicó en 2011, Número 1, sobre laxantes y metilnaltrexona para el tratamiento del estreñimiento en pacientes que reciben cuidados paliativos, se actualizó en 2015 y excluyó la metilnaltrexona. La otra se publicó en 2008, Número 4, sobre antagonistas opiáceos Mu (AOM) para la DIIO. En esta revisión actualizada solo se incluyeron los ensayos sobre los AOM (incluida la metilnaltrexona) para la DIIO en los pacientes con cáncer y en los pacientes que reciben cuidados paliativos.
Objetivos
Evaluar la efectividad y la seguridad de los AOM para la DIIO en los pacientes con cáncer y los pacientes que reciben cuidados paliativos.
Métodos de búsqueda
Se hicieron búsquedas en el Registro Cochrane Central de Ensayos Controlados (Cochrane Central Register of Controlled Trials), MEDLINE, Embase, CINAHL y en Web of Science hasta agosto 2017. También se buscó en los registros de ensayos clínicos y en los sitios web de agencias reguladoras. Se estableció contacto con los fabricantes de AOM para identificar datos adicionales.
Criterios de selección
Se incluyeron los ensayos controlados aleatorios (ECA) que evaluaron la efectividad y la seguridad de los AOM para la DIIO en los pacientes con cáncer y en los pacientes en estadio paliativo, independientemente del tipo de enfermedad terminal que presentaran.
Obtención y análisis de los datos
Dos autores de la revisión , evaluaron el riesgo de sesgo y extrajeron los datos. Se consideró adecuado combinar los datos de los ensayos si hubo homogeneidad suficiente entre los ensayos. Los resultados primarios fueron laxación, repercusión sobre el alivio del dolor y eventos adversos. La repercusión sobre el alivio del dolor fue un resultado primario porque un posible efecto adverso de los AOM es la reducción de la analgesia de los opiáceos. Se evaluó la evidencia sobre estos resultados usando GRADE.
Resultados principales
Se identificaron cuatro nuevos ensayos para esta actualización, lo que lleva el número total incluido en esta revisión a ocho. En los ensayos se asignaron al azar 1022 hombres y mujeres con cáncer independientemente del estadio, o en un estadio de atención paliativa de cualquier enfermedad. Los AOM evaluados fueron la naldemedina y la naloxona orales (solas o en combinación con oxicodona) y la metilnaltrexona subcutánea. Los ensayos compararon AOM con placebo o con una intervención activa administrada en diferentes dosis o en combinación con otros fármacos. El ensayo de naldemedina y los dos de naloxona en combinación con oxicodona se realizaron en pacientes con cáncer, independientemente del estadio de la enfermedad. El ensayo sobre naloxona sola se realizó en pacientes con cáncer avanzado. Los cuatro ensayos sobre metilnaltrexona se realizaron en pacientes en cuidados paliativos, donde la mayoría presentaba cáncer. Todos los ensayos fueron vulnerables a los sesgos; cuatro tuvieron alto riesgo de sesgo ya que incluyeron una muestra de menos de 50 participantes por brazo.
En el ensayo de naldemedina en comparación con placebo en 225 participantes, hubo más laxaciones espontáneas durante las dos semanas de tratamiento en el grupo de intervención (cociente de riesgos [CR] 1,93; intervalos de confianza [IC] del 95%: 1,36 a 2,74; evidencia de calidad moderada). En comparación con dosis mayores, las dosis menores dieron lugar a menos laxaciones espontáneas (0,1 mg versus 0,2 mg: CR 0,73; IC del 95%: 0,55 a 0,95; 0,1 mg versus 0,4 mg: CR 0,69; IC del 95%: 0,53 a 0,89; evidencia de calidad moderada). Hubo evidencia de calidad moderada de que la naldemedina no tuvo efectos sobre la abstinencia de los opiáceos. Hubo cinco eventos adversos graves. Todos ocurrieron en pacientes que tomaban naldemedina (evidencia de baja calidad). Se produjo un aumento en otros eventos adversos (no graves) en los grupos de naldemedina (CR 1,36; IC del 95%: 1,04 a 1,79; evidencia de calidad moderada). El evento adverso más común fue la diarrea.
Los ensayos sobre naloxona administrada sola o en combinación con oxicodona (un opiáceo) en comparación con oxicodona sola no evaluó la respuesta de laxación durante las dos primeras semanas de administración. Hubo evidencia de muy baja calidad para la naloxona sola, y evidencia de calidad moderada para la oxicodona/naloxona, de que estos fármacos no tuvieron efecto sobre la analgesia. Hubo evidencia de baja calidad de que la oxicodona/naloxona no aumentaron el riesgo de eventos adversos y evidencia de calidad moderada de que no aumentaron el riesgo de eventos adversos graves.
En el análisis combinado de dos ensayos con 287 participantes, se encontró que la metilnaltrexona comparada con placebo indujo más laxaciones en el transcurso de 24 horas (CR 2,77; IC del 95%: 1,91 a 4,04; I² = 0%; evidencia de calidad moderada). En el análisis combinado se encontró que la metilnaltrexona indujo más respuestas de laxación en dos semanas (CR 9,98; IC del 95%: 4,96 a 20,09; I² = 0%; evidencia de calidad moderada). La proporción de participantes que tuvieron una respuesta de laxación sin necesidad de rescate en el transcurso de 24 horas desde la primera dosis fue del 59,1% en los brazos de metilnaltrexona y del 19,1% en el brazo placebo. Hubo evidencia de calidad moderada de que no se afectó la tasa de abstinencia de los opiáceos. La metilnaltrexona no aumentó la probabilidad de eventos adversos graves; hubo menos en el brazo de intervención (CR 0,59; IC del 95%: 0,38 a 0,93; I² = 0%; evidencia de calidad moderada). No hubo diferencias en la proporción de participantes que presentaron un evento adverso (CR 1,17; IC del 95%: 0,94 a 1,45; I² = 74%; evidencia de baja calidad). La metilnaltrexona aumentó la probabilidad de dolor abdominal y flatulencia.
Dos ensayos compararon diferentes regímenes de metilnaltrexona, dosis mayores versus dosis inferiores. Para la laxación temprana, hubo evidencia de baja calidad de ninguna diferencia clara entre las dosis en cuanto a la analgesia y los eventos adversos. Ambos ensayos midieron la respuesta de laxación en el transcurso de 24 horas desde la primera dosis (ensayo uno: CR 0,82; IC del 95%: 0,41 a 1,66; ensayo dos: CR 1,07; IC del 95%: 0,81 a 1,42).
Conclusiones de los autores
En esta actualización las conclusiones para la naldemedina son nuevas. Hay evidencia de calidad moderada que indica que, administrada por vía oral, la naldemedina mejora la función intestinal a las dos semanas en los pacientes con cáncer y DIIO, pero aumenta el riesgo de eventos adversos. No han cambiado las conclusiones para la naloxona y la metilnaltrexona. Los ensayos sobre naloxona no evaluaron la laxación a las 24 horas ni en dos semanas. Hay evidencia de calidad moderada de que la metilnaltrexona mejora la función intestinal en los pacientes que reciben cuidados paliativos a corto plazo y a las dos semanas, y evidencia de baja calidad de que no aumenta los eventos adversos. Se necesitan más ensayos que incluyan evaluaciones adicionales de los eventos adversos. En ninguno de los ensayos actuales se evaluaron los efectos en los niños.
PICO
Resumen en términos sencillos
Antagonistas opiáceos Mu para la disfunción intestinal debido a los opiáceos en pacientes con cáncer y en pacientes que reciben cuidados paliativos
Antecedentes
Los opiáceos (fármacos similares a morfina) se utilizan para tratar el dolor intenso. Lamentablemente, causan efectos secundarios. La disfunción intestinal inducida por opiáceos (DIIO) es un término utilizado para describir el estreñimiento, la evacuación incompleta de los intestinos, el timpanismo abdominal y el aumento del reflujo (retroceso) de los contenidos del estómago. La DIIO puede ser tan grave como para que un paciente decida limitar el tratamiento con opiáceos para mejorar la función intestinal. La DIIO es frecuente en los pacientes con cáncer y en los pacientes que reciben cuidados paliativos (atención a pacientes con una enfermedad terminal cuando ya no es posible la curación). Los laxantes suelen ser el tratamiento de primera elección para la DIIO. Estos fármacos no siempre funcionan. Los antagonistas opiáceos Mu (AOM) son fármacos específicos para la DIIO. Las guías clínicas pueden recomendarlos cuando fracasan los laxantes.
Características de los ensayos
El objetivo de esta revisión actualizada fue determinar lo que se conoce acerca de la efectividad y la seguridad de los AOM para el tratamiento de la DIIO en pacientes con cáncer y pacientes que reciben cuidados paliativos, en los que han fracasado los laxantes. Un efecto secundario posible de los AOM es la reducción del alivio del dolor producido por los opiáceos; por lo tanto, se examinó su repercusión sobre el alivio del dolor. Solo se incluyeron ensayos controlados aleatorios (ECA), que son ensayos clínicos bien diseñados que aportan la evidencia más fiable. Los ECA debían evaluar un AOM como los fármacos naldemedina, metilnaltrexona y naloxona. Los grupos de comparación de los ensayos podrían ser placebo (una sustancia sin efecto activo conocido), atención habitual u otro tratamiento como un tipo diferente de AOM.
Resultados clave
La búsqueda hasta agosto de 2017 encontró ocho ensayos con 1022 adultos. Los AOM evaluados en los pacientes con cáncer fueron naldemedina y naloxona orales tomadas en combinación con un tratamiento con opiáceos (para el dolor). La metilnaltrexona administrada mediante inyección se evaluó en cuidados paliativos, donde la mayoría de los participantes presentaban cáncer avanzado.
Los resultados fueron la naldemedina o metilnaltrexona en comparación con placebo. La naloxona se comparó con placebo o con tratamiento con opiáceos solamente.
La calidad general de la evidencia de estos estudios se calificó de muy baja a moderada. "Muy baja" indica que no hay seguridad acerca de los resultados. "Alta" significa que existe mucha confianza en los resultados. Hubo problemas con el diseño de los estudios, como el reporte insuficiente de los métodos del ensayo.
Evacuaciones intestinales en el transcurso de 24 horas y de hasta dos semanas
Hubo evidencia de calidad moderada de que la naldemedina aumentó las evacuaciones intestinales en el transcurso de hasta dos semanas. Los ensayos no midieron los efectos de la naloxona sobre las evacuaciones intestinales a las dos semanas. Metilnaltrexona aumentó las evacuaciones intestinales o laxaciones (heces más blandas) en el transcurso de 24 horas y de hasta dos semanas (evidencia de calidad moderada).
Alivio del dolor
Hubo evidencia de calidad moderada de que la naldemedina no cambió el alivio del dolor. Los ensayos no midieron la intensidad del dolor con naldemedina. Hubo evidencia muy de baja calidad de que la naloxona administrada sola no cambió el alivio del dolor. Hubo evidencia de calidad moderada de que la naloxona administrada con un tratamiento con opiáceos no cambió el alivio del dolor. Hubo evidencia de calidad moderada a baja de que la metilnaltrexona no cambió el alivio del dolor.
Riesgo de efectos secundarios graves (hospitalización, potencialmente mortales o mortales) y otros efectos secundarios
Hubo evidencia de baja calidad de que la naldemedina y la naloxona en combinación con un tratamiento con opiáceos no aumentaron el riesgo de efectos secundarios graves. Para la naldemedina, hubo cinco efectos secundarios graves en el ensayo, aunque ninguno se describió como relacionado con el fármaco de estudio. La metilnaltrexona probablemente no aumentó el riesgo de efectos secundarios graves (evidencia de calidad moderada).
Hubo evidencia de calidad moderada de que la naldemedina aumentó el riesgo de efectos secundarios. Hubo evidencia de calidad moderada de que la naloxona administrada con oxicodona (un analgésico opiáceo) no aumentó el riesgo de efectos secundarios. Hubo evidencia de baja calidad de que la metilnaltrexona no aumentó el riesgo general de un efecto secundario. La metilnaltrexona aumentó el riesgo de dolor abdominal y flatulencia.
Conclusión
Hubo evidencia de calidad moderada para sugerir que la naldemedina mejoró la función intestinal en el transcurso de dos semanas en los pacientes adultos con cáncer y DIIO, pero aumentó el riesgo de efectos secundarios; y que la metilnaltrexona mejoró la función intestinal en los pacientes que reciben cuidados paliativos, y evidencia de baja calidad de que la metilnaltrexona no aumentó los efectos secundarios. Los resultados de esta revisión deben interpretarse con precaución, ya que no se obtuvieron de evidencia de alta calidad. No se identificaron estudios en niños.
Conclusiones de los autores
Summary of findings
| Naldemedine compared to placebo for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Settings: cancer care Intervention: naldemedine Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Naldemedine | |||||
| Laxation response within 24 hours of dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14a | 375 per 1000 | 724 per 1000 | RR 1.93 (1.36 to 2.74) NNTB 2.88 (2.04 to 4.92) | 225 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: opioid withdrawalc | — | — | 0.1 mg: MD ‐0.13 (‐0.57 to 0.31); 0.2 mg: MD ‐0.40 (‐0.87 to 0.07); 0.4 mg: MD ‐0.02 (‐0.45 to 0.41) | 225 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: pain intensity | — | — | — | — | — | Not reported |
| Serious adverse eventsa | — | — | 5 SAEs occurred, all in naldemedine group. | 225 (1 study) | ⊕⊕⊝⊝ | — |
| Adverse eventsa | 518 per 1000 | 704 per 1000 (539 to 927) | RR 1.36 (1.04 to 1.79) | 225 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; NNTB: number needed to treat for an additional beneficial outcome; RR: risk ratio; SAE: serious adverse events. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by clinician or self‐report and in the case of adverse events using severity grades according to the Common Terminology Criteria for Adverse Events. | ||||||
| Lower‐dose naldemedine compared to higher‐dose naldemedine for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Setting: cancer care Intervention: lower dose naldemedine 0.1 mg daily Comparison: higher dose naldemedine 0.2 mg or 0.4 mg daily | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Higher dose 0.2 mg/0.4 mg daily | Lower dose 0.1 mg daily | |||||
| Laxation response within 24 hours of dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14a | 0.1 mg vs 0.2 mg: 776 per 1000 0.1 mg vs 0.4 mg: 821 per 1000 | 0.1 mg vs 0.2 mg: 564 per 1000 (430 to 739) 0.1 mg vs 0.4 mg: 564 per 1000 (433 to 733) | 0.1 mg vs 0.2 mg: RR 0.73 (0.55 to 0.95) 0.1 mg vs 0.4 mg: RR 0.69 (0.53 to 0.89) | 226 (1 study) 0.1 mg vs 0.2 mg: n = 113 0.1 mg vs 0.4 mg: n = 111 | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: opioid withdrawal | — | — | — | — | — | Not reported |
| Effect on analgesia: pain intensity | — | — | — | — | — | Not reported |
| Serious adverse events | — | — | — | — | — | Not reported |
| Adverse events | — | — | — | — | — | Not reported |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by self‐report. | ||||||
| Naloxone compared with placebo for cancer and people receiving palliative care with opioid‐induced bowel dysfunction | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Settings: cancer care Intervention: naloxone Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Naloxone | |||||
| Laxation response within 24 hours of a dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14 | — | — | — | — | — | Not reported |
| Effect on analgesia: opioid withdrawal | — | — | — | — | — | Not reported |
| Effect on analgesia: pain intensitya | — | — | No statistical difference in pain experienced when taking placebo or naloxone. Full data, including pre‐cross‐over results, were not provided. | 17 (1 study) | ⊕⊝⊝⊝ | — |
| Serious adverse events | — | — | — | — | — | Not reported |
| Adverse events | — | — | — | — | — | Not reported |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured using 4‐point scale (0 = no pain, 3 = severe pain). | ||||||
| Oxycodone/naloxone prolonged release tablets compared with oxycodone prolonged‐released tablets for opioid‐induced bowel dysfunction | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Settings: cancer care Intervention: oxycodone/naloxone prolonged‐release tablets Comparison: oxycodone prolonged‐released tablets | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Oxycodone | Oxycodone/naloxone | |||||
| Laxation response within 24 hours of dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14 | — | — | — | — | — | Not reported |
| Effect on analgesia: opioid withdrawalc | — | — | Intervention group: mean 6.64 (SD 5.97) comparison group: mean 7.29 (SD 4.59) at 7 days | 184 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: pain intensitya | — | — | Intervention group: mean 3.50 (SD 1.88) and comparison group: mean 3.52 (SD 1.80) at 4 weeks | 184 (1 study) | ⊕⊕⊕⊝ Moderateb | Another study, Dupoiron 2017 also found outcome to be similar between trial arms, but did not provide any data. |
| Serious adverse events | 43 per 1000 | 87 per 1000 (27 to 279) | RR 2.00 (95% CI 0.62 to 6.41) | 184 (1 study) | ⊕⊕⊝⊝ Lowb,d | — |
| Adverse events | 754 per 1000 | 815 per 1000 (709 to 935) | RR 1.08 (95% CI 0.94 to 1.24) | 234 (2 studies) | ⊕⊕⊕⊝ Moderateb | — |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; SD: standard deviation. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured using the Brief Pain Inventory‐Short Form. | ||||||
| Methylnaltrexone compared to placebo for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Setting: palliative care Intervention: methylnaltrexone Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Risk with placebo | Risk with methylnaltrexone | |||||
| Laxation response within 24 hours of dosea | 195 per 1000 | 568 per 1000 | RR 2.77 (1.91 to 4.04) | 287 | ⊕⊕⊕⊝ Moderateb | — |
| Laxation response between day 1 and day 14 (specifically within 4 hours after 4 or more of the 7 doses)a | 52 per 1000 | 517 per 1000 | RR 9.98 (4.96 to 20.09) | 305 | ⊕⊕⊕⊝ Moderate b,c | — |
| Effect on analgesia: opioid withdrawald | Study 1: day 1: MD 0.00 (‐0.46 to 0.46); day 14: MD 0.10 (‐0.63 to 0.83) Study 2: median change to day 2 = 0 in both trials arms | 236 (2 studies) | ⊕⊕⊕⊝ Moderateb | ‐ | ||
| Effect on analgesia: pain intensitye | Study 1: at 4 hours (methylnaltrexone 0.15 mg/kg: MD ‐0.76 (‐1.47 to 0.05); methylnaltrexone 0.3 mg/kg: MD ‐0.25 (‐0.91 to 0.41) Study 2: at day 1 and 14 (day 1: MD 0.20 (‐0.62 to 1.02); day 14: MD ‐0.70 (‐1.52 to 0.12) | 287 (2 studies) | ⊕⊕⊝⊝ Lowb,f | Another study, Bull 2015, found similar pain intensity experienced in trial arms, full data not provided. | ||
| Serious adverse events | 238 per 1000 | 142 per 1000 | RR 0.59 (0.38 to 0.93) | 364 | ⊕⊕⊕⊝ Moderateb | — |
| Adverse events | 700 per 1000 | 815 per 1000 | RR 1.17 (CI 0.94 to 1.45) | 518 | ⊕⊕⊝⊝ Lowb,g | Heterogeneity was substantial (74%). It was explained in sensitivity analysis by omitting the trial at a high risk of bias because of small sizes. The effect estimate was reduced. The direction of effect not changed. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by self‐report or clinician report. dMeasured using the modified Himmelsbach Opioid Withdrawal Scale. | ||||||
| Lower dose methylnaltrexone compared to higher dose for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Setting: palliative care Intervention 1: lower‐dose methylnaltrexone (study 1: 3 doses, 1 week, 1 mg; study 2: 1 dose, 0.15 mg/kg) Intervention 2: higher‐dose methylnaltrexone (study 1: 3 doses, 1 week, 5‐12.5 mg; study 2: 1 dose, 0.30 mg/kg) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Higher dose | Lower dose | |||||
| Laxation response within 24 hours of first dosea | Study 1: 609 per 1000 Study 2: 639 per 1000 | Study 1: 499 per 1000 (250 to 100) Study 2: 681 per 1000 (515 to 904) | Study 1: RR 0.82 (0.41 to 1.66) Study 2: RR 1.07 (0.81 to 1.42) | 135 (2 studies) Study 1: n = 33 Study 2: n = 102 | ⊕⊕⊝⊝ Lowb | Unable to combine study data as methylnaltrexone low and higher doses differed per trial |
| Laxation responsea | At 3 days: 706 per 1000 | At 3 days: 332 per 1000 | At 3 days: RR 0.47 (0.18 to 1.25) | 33 participants (1 study) | ⊕⊕⊝⊝ Lowb | Unable to combine study data as methylnaltrexone low and higher doses differed per trial |
| At 5 days: 688 per 1000 | At 5 days: 144 per 1000 | At 3 days: RR 0.21 (0.03 to 1.31) | ||||
| Effect on analgesia: opioid withdrawalc | — | — | MD ‐0.04 (‐0.73 to 0.65) | 102 participants (1 study) | ⊕⊕⊝⊝ Lowb | Another study,Portenoy 2008, also found outcome to be similar between trial arms, but did not provide any data |
| Effect on analgesia: pain intensityd | — | — | MD ‐0.51 (‐1.49 to 0.47) | 102 participants (1 study) | ⊕⊕⊝⊝ Lowb | Another study, Portenoy 2008, also found outcome to be similar between trial arms, but did not provide any data |
| Serious adverse event | — | — | — | — | Not reported | |
| Adverse event | Study 1: 1000 per 1000 Study 2: 800 per 1000 | Study 1: 1000 per 1000 (1000 to 1000) Study 2: 723 per 1000 (580 to 902) | Study 1: RR 1.00 (1.00 to 1.00) Study 2: RR 0.90 (0.73 to 1.13) | 135 (2 studies) Study 1: n = 33 Study 2: n = 102 | ⊕⊕⊝⊝ Lowb | Unable to combine study data as methylnaltrexone low and higher doses differed per trial |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by clinician or self‐report. bDowngraded by two levels for study limitations: one for unclear risk of bias (reporting bias) and one for small sample size (high risk of bias). cMeasured using the modified Himmelsbach Opioid Withdrawal Scale. dMeasured by participant‐rated scale 0‐10. | ||||||
Antecedentes
Esta revisión es una actualización (parcial) de dos revisiones publicadas anteriormente en la Base de Datos Cochrane de Revisiones Sistemáticas (Cochrane Database of Systematic Reviews).
-
Antagonistas opiáceos Mu para la disfunción intestinal inducida por opiáceos, 2008, Número 2 (McNicol 2008).
-
Laxantes o metilnaltrexona para el tratamiento del estreñimiento en pacientes que reciben cuidados paliativos, 2011, Número 1 (Candy 2011).
Esta actualización de la revisión presentó los resultados sobre la efectividad y la seguridad de los antagonistas opiáceos Mu (AOM) para la disfunción intestinal inducida por opiáceos (DIIO) en pacientes con cáncer y en pacientes que reciben cuidados paliativos. Se ha publicado una revisión actualizada sobre los laxantes para el tratamiento del estreñimiento en pacientes que reciben cuidados paliativos (Candy 2015). Se prevé que la evidencia sobre la efectividad de los AOM para la DIIO en otras poblaciones, como por ejemplo, los pacientes con dolor crónico no maligno, se evalúe en una revisión Cochrane posterior.
Descripción de la afección
Los opiáceos como el sulfato de morfina, la oxicodona y el fentanil son analgésicos potentes. Son recomendados por la Organización Mundial de la Salud (OMS) y están en las guías clínicas para el tratamiento del dolor moderado a intenso del cáncer y en otras poblaciones como los pacientes que necesitan cuidados paliativos (WHO 2016). Se utilizan ampliamente, aunque a nivel global hay una gran variación que indica una subutilización de los opiáceos para el tratamiento del dolor en algunos lugares (Manjiani 2014).
Sin embargo, los opiáceos se asocian con eventos adversos. El más frecuente e incapacitante es la disfunción intestinal, que puede ser suficientemente grave para que el paciente limite el uso de los opiáceos (Cook 2008). Los opiáceos, independientemente del método de administración (oral, parenteral, transdérmico), interfieren con la motilidad propulsiva gastrointestinal (Leppert 2010). Los opiáceos aumentan la absorción de los líquidos en el intestino y disminuyen la secreción epitelial. Retrasan el vaciamiento gástrico y disminuyen el peristaltismo intestinal.
La DIIO se ha descrito como "Un cambio, cuando se inicia el tratamiento con opiáceos, de los hábitos intestinales iniciales, que se caracteriza por cualquiera de los siguientes: reducción de la frecuencia de evacuaciones intestinales (convencionalmente menos de tres por semana), desarrollo o empeoramiento del esfuerzo para las evacuaciones intestinales, sensación de evacuación rectal incompleta o mayor consistencia de las heces" (Kumar 2012). Esta afección puede incluso dar lugar a la impactación de las heces (Camilleri 2014). Además del estreñimiento, la DIIO describe una constelación de síntomas incluidos timpanismo abdominal, distensión abdominal, reflujo gástrico, retortijones abdominales, sensación de sequedad en la boca, plenitud epigástrica, náuseas y vómitos (Leppert 2015; Pappagallo 2001). Puede causar angustia psicológica y agitación en los pacientes en fase terminal. La DIIO aumenta el uso de los servicios sanitarios, en ocasiones requiere de hospitalización y puede reducir extraordinariamente la calidad de vida ya comprometida. Puede provocar que los pacientes no traten suficientemente el dolor (Pizzi 2012); sin embargo, debido a que la dosis que produce estreñimiento puede ser solo el 25% de la requerida para una analgesia adecuada, la reducción de la dosis no es una opción apropiada para el tratamiento de la DIIO (Ketwaroo 2013).
La incidencia calculada de la DIIO en las poblaciones ingresadas en cuidados paliativos y en los pacientes con enfermedad avanzada es elevada, entre el 65% y el 90% (Panchal 2007; Sykes 1998). Aunque estos cálculos son relativamente antiguos, no existe evidencia que indique que este ya no sea el caso.
Descripción de la intervención
El tratamiento preventivo recomendado de la DIIO en los cuidados paliativos y en las enfermedades avanzadas es la administración de un laxante estimulante y un ablandador de heces, además de medidas generales como aumentar la ingesta de alimentos, una dieta con alto contenido de fibra, ingestión de líquidos, aumentar la actividad física y privacidad durante la defecación (NICE 2012). Sin embargo, estas medidas no siempre son efectivas; en los pacientes que reciben opiáceos, se calcula que más del 80% se mantienen estreñidos a pesar del uso regular de los laxantes (Coyne 2014; Diego 2011).
Los AOM como la metilnaltrexona, la naloxona y el naloxegol, están diseñados específicamente para dirigirse a la fisiopatología de la DIIO y "neutralizar" el efecto de constipación de los opiáceos. La metilnaltrexona está autorizada para el tratamiento del estreñimiento inducido por opiáceos en los cuidados paliativos en más de 50 países (Bader 2013). En las guías clínicas, cuando se considera la metilnaltrexona, se describe que actúa al aumentar la acción de los laxantes o como una opción cuando fracasan los laxantes (European Association of Palliative Care, Caraceni 2012), y solo se debe utilizar bajo la supervisión de un médico especialista en cuidados paliativos (Scottish Palliative Care Guidelines 2014). Es importante señalar que el National Institute for Health and Care Excellence (NICE) no puede recomendar el uso de la metilnaltrexona en el National Health Service (NHS) del Reino Unido para el tratamiento de la DIIO en los pacientes con enfermedades avanzadas que reciben cuidados paliativos porque el fabricante no presentó evidencia al respecto (NICE 2013).
De qué manera podría funcionar la intervención
Los opiáceos median sus efectos gastrointestinales y analgésicos a través de las mismas subclases de receptores opiáceos en el cuerpo humano: mu, kappa y delta. No se comprende completamente cómo participa cada tipo de receptor en la DIIO (Neefjes 2014). El efecto opiáceo periférico sobre los receptores opiáceos Mu en la pared intestinal puede desempeñar una función importante en la DIIO (Leppert 2010). La coordinación de la motilidad se interrumpe mediante la activación de los receptores opiáceos Mu que inhiben las vías neurales excitadoras e inhibidoras en el sistema nervioso entérico.
Un enfoque para la disociación del efecto analgésico de los opiáceos es separar la actividad central del opiáceo de su actividad periférica (Wang 2013). Lo anterior se puede lograr con un antagonista de los receptores opiáceos que actúe de manera periférica, con una capacidad limitada para cruzar la barrera hematoencefálica y que, por lo tanto, no interfiera con la analgesia (Brown 1985). Alternativamente, se puede lograr mediante la administración de una preparación que tenga un metabolismo de un "primer paso" por el hígado, por lo que no ingresa a la circulación sistémica.
Hay varios antagonistas Mu en uso y otros en desarrollo. La naloxona está comercialmente disponible; actúa centralmente pero tiene un efecto terapéutico estrecho con ciertas dosis, lo que revierte la analgesia aconsejable (Camilleri 2011). Experimenta un metabolismo de primer paso extenso y en la dosis correcta no revierte el efecto analgésico de los opiáceos. Se administra por vía oral. El desarrollo de una preparación de liberación prolongada de naloxona para cubrir la mayor parte posible de los intestinos delgado y grueso cuando se utiliza con oxicodona, ha dado lugar a estudios adicionales sobre el compuesto (Camilleri 2011). Hay otros preparados que no cruzan la barrera hematoencefálica, que incluyen alvimopan, metilnaltrexona, naloxegol y naldemedina. Alvimopan tiene una afinidad alta por los receptores opiáceos periféricos. Solo se recomienda para el uso a corto plazo, por ejemplo después de la cirugía, debido a la posibilidad de eventos miocárdicos (Merck 2015). Está contraindicado en los pacientes con enfermedades avanzadas (Leppert 2015). La metilnaltrexona es menos liposoluble que la naloxona y, por lo tanto, es menos probable que cruce la barrera hematoencefálica. En la actualidad solo está disponible en preparaciones subcutáneas. El naloxegol, que se administra por vía oral, tiene una fracción de polietilenglicol que limita su capacidad de cruzar la barrera hematoencefálica (Pritchard 2015). La naldemedina es un derivado de la naloxona. Hay detalles publicados limitados sobre sus mecanismos de acción, y en la actualidad está en evaluación en ensayos de fase III en pacientes con cáncer.
Por qué es importante realizar esta revisión
Hay revisiones de los AOM para la DIIO en diferentes poblaciones (p.ej. Ford 2013). Sin embargo, es importante evaluar su efectividad específicamente en el cáncer y en los pacientes de atención paliativa (Bader 2012; Clark 2014). Lo anterior se debe a las diferencias inherentes en estos grupos que pueden repercutir, probablemente de forma negativa, sobre el efecto de un AOM. La repercusión puede diferir debido a la fisiopatología multifactorial del estreñimiento en los pacientes con cáncer y enfermedades avanzadas (Leppert 2010). Lo anterior puede incluir anomalías estructurales como la obstrucción intestinal; los tumores pelvianos; la fibrosis por radiación; o los trastornos metabólicos como la deshidratación, la hipercalcemia y la hipopotasemia. Asimismo, puede incluir trastornos neurológicos. También pueden existir problemas generales que aumentan el riesgo y que complican el tratamiento de la DIIO como la edad avanzada, la depresión, la sedación con fármacos, la quimioterapia, los tratamientos múltiples y la falta de privacidad de los enfermos hospitalizados para la evacuación intestinal. Según progresa la enfermedad, los pacientes pueden presentar mayor debilidad, menos actividad, pérdida del apetito y, con el tiempo, insuficiencia de múltiples órganos, todo lo cual puede repercutir sobre la función intestinal (Bader 2012). Además, debido a estos factores los pacientes con cáncer, y en particular los pacientes en un estadio de cuidados paliativos, pueden tener un riesgo mayor que otras poblaciones menos enfermas de presentar eventos adversos con un AOM.
Objetivos
Evaluar la efectividad y la seguridad de los AOM para la DIIO en los pacientes con cáncer y en los pacientes que reciben cuidados paliativos.
Métodos
Criterios de inclusión de estudios para esta revisión
Tipos de estudios
Se incluyeron los ensayos controlados aleatorios (ECA) doble ciego que investigaron la eficacia de los AOM para la DIIO. No se incluyeron las fases de extensión abiertas de los ensayos ni los análisis post hoc de los ensayos porque tienen un mayor riesgo de sesgo. No se aplicaron restricciones de idioma. Se exigió la publicación completa en revistas. Además de los ensayos presentados como publicación completa en revistas, se incluyó cualquier resumen de los resultados de un ensayo clínico en línea de datos de ensayos por otra parte no publicados en relación con el ensayo publicado.
Tipos de participantes
Los ensayos elegibles incluyeron participantes de cualquier edad o sexo que:
-
fueran pacientes con cáncer o pacientes en un estadio paliativo independientemente de la enfermedad y la edad, o ambos;
-
estuvieran, todos o la mayoría de los pacientes (más del 95%), en un régimen estable con opiáceos y presentaran DIIO que no se había resuelto con la ingestión de laxantes.
Se incluyeron los ensayos con poblaciones de participantes en las que no todos cumplieran con estos criterios de elegibilidad, siempre que al menos el 50% de la muestra fueran pacientes con cáncer o pacientes que recibían cuidados paliativos o en un estadio avanzado de su enfermedad, o en los que se proporcionaran análisis de subgrupos de cualquiera de estos grupos de participantes.
No se incluyeron los ensayos en cualquier población si los AOM para la disfunción intestinal se administraron por un íleo posoperatorio asociado (detención del peristaltismo intestinal). Lo anterior se debió a que este íleo no es causado principalmente por los opiáceos (Marderstein 2008). También se excluyeron los ensayos en voluntarios sanos, participantes con estreñimiento debido al uso inadecuado de fármacos y participantes con estreñimiento debido a una obstrucción intestinal.
Tipos de intervenciones
Se incluyeron los ensayos de intervenciones que evaluaron los antagonistas opiáceos de los receptores Mu que actúan de manera periférica o sistémica, y que administraron cualquier dosis y por cualquiera vía. Se incluyeron, por ejemplo, metilnaltrexona y naloxona.
La intervención de comparación de interés fue un AOM diferente, una intervención farmacológica o no farmacológica alternativa, placebo o ningún tratamiento.
Tipos de medida de resultado
Resultados primarios
Los resultados primarios de interés fueron la eficacia con respecto a la respuesta de laxación y la seguridad.
-
Eficacia:
-
la respuesta de laxación en las primeras 24 horas y entre los días uno y 14 después de la primera dosis. La respuesta de laxación se podría haber medido con una escala validada como el Bowel Function Index (BFI) de tres ítems sobre la facilidad de la defecación o la sensación personal de vaciado intestinal incompleto;
-
efecto sobre la analgesia. Lo anterior se podría haber medido como necesidad de analgésicos, síntomas de abstinencia de los opiáceos e intensidad del dolor. Este fue un resultado primario porque los AOM pueden tener un efecto adverso sobre el alivio del dolor.
-
-
Seguridad:
-
eventos adversos graves;
-
número y tipo de eventos adversos.
-
Resultados secundarios
-
Número de participantes que abandonaron debido a eventos adversos.
-
Otras medidas de respuesta de laxación, como el tiempo de tránsito intestinal y el alivio en un punto temporal más allá del día 14.
-
Alivio de otros síntomas asociados con el estreñimiento como dolor abdominal y pérdida del apetito.
-
Uso de medicación de rescate para la laxación. Es la necesidad de medicación adicional porque el alivio del estreñimiento no ha ocurrido en un tiempo aceptable, por ejemplo, en el transcurso de cuatro horas después de la administración de la intervención con metilnaltrexona subcutánea . La medicación de rescate puede ser en forma de un supositorio laxante o un enema.
-
Calidad de vida, satisfacción de los participantes con las evacuaciones intestinales y preferencia de los participantes.
Métodos de búsqueda para la identificación de los estudios
Búsquedas electrónicas
For this update, we searched five databases.
-
CENTRAL (CRSO) 2007 to issue 7 of 12 2017.
-
MEDLINE and MEDLINE in process (Ovid) 2007 to 28 August 2017.
-
Embase (Ovid) 2007 to week 35 2017.
-
CINAHL (EBSCO) 1982 to August 2017.
-
Web of Science (SCI‐Expanded and CPCI‐S) 1945 to 28 August 2017.
The search strategies are listed in Appendix 1.
Búsqueda de otros recursos
We searched three clinical trials registries to March 2016.
-
The metaRegister of controlled trials (mRCT) (www.controlled‐trials.com/mrct).
-
ClinicalTrials. gov (clinicaltrials.gov).
-
The WHO International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/).
We searched two regulatory agency websites, US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), for drug reports.
We searched pharmaceutical companies' websites of known manufacturers of MOAs to identify trial data.
We searched two pharmaceutical company trials registers:
-
AstraZeneca Clinical Trials (www.astrazenecaclinicaltrials.com);
-
GlaxoSmithKline Clinical Trial Register (www.gsk‐clinicalstudyregister.com).
We checked references lists of included trials and any identified systematic reviews. We also undertook a forward citation search of all included trials. We checked conference proceedings of the National Cancer Research Institute (NCRI) Cancer Conference and the European Association of Palliative Care (EAPC). We contacted authors of any identified relevant conference abstracts to ask for full details of their trials.
We wrote to pharmaceutical companies of known manufacturers of MOAs to obtain any trial data not available in peer‐review publications; these were AstraZeneca, Mundipharma GmbH, Progenics, Shionogi, and Valeant. For this purpose, we adapted a letter developed by authors of a previous Cochrane Review; see Appendix 2 for a copy of this letter.
Obtención y análisis de los datos
Selección de los estudios
Two review authors (BC, LJ) independently screened the citations identified in the database searches. Where it was unclear or likely that the studies fulfilled our inclusion criteria, we retrieved the full‐text articles. If disagreements on eligibility had occurred, we would have resolved them by discussion, or if persistent, by a third review author (PS). If necessary for further clarification such as if it was unclear whether the trial identified was completed and whether their findings were available, we sought contact with the author or sponsor.
Extracción y manejo de los datos
We extracted data (as detailed in Types of outcome measures) from each trial. One review author (BC) extracted the data and another review author checked them (LJ/VV). We resolved disagreements by discussion, or if persistent, we would have involved a third review author (PS).
Evaluación del riesgo de sesgo de los estudios incluidos
Two review authors (BC, VV) independently assessed risk of bias for each trial using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions, resolving any disagreements by discussion (Higgins 2011). We completed a 'Risk of bias' table for each included trial. We assessed the following.
-
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (any truly random process: random number table; computer random number generator); and unclear risk of bias (method used to generate sequence not clearly stated). We excluded studies using a non‐random process, which were therefore at high risk of bias (odd or even date of birth; hospital or clinic record number).
-
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions before assignment determines whether the intervention allocation could have been foreseen in advance of, or during, recruitment, or changed after assignment. We assessed the methods as: low risk of bias (telephone or central randomisation; consecutively numbered, sealed, opaque envelopes); and unclear risk of bias if the method was not clearly stated. We excluded trials that did not conceal allocation, which were therefore at high risk of bias (open list).
-
Blinding of participants and personnel (performance bias and detection bias). We assessed the methods used to blind trial participants and personnel (performance bias) and outcome assessors (detection bias) from knowledge after trial assignment of which intervention a participant received. We assessed the methods as: low risk of bias if the trial stated that it was blinded and described the method used to achieve blinding: identical tablets, matched in appearance and smell; and unclear risk of bias if the trial stated that it was blinded but did not provide an adequate description of how blinding was achieved. We judged a trial as high risk if blinding was attempted but it was likely that the blinding could have been broken and that the outcome was likely to be influenced by lack of blinding. We did not include any trial that was reported as not being double blinded.
-
Incomplete outcome data (attrition bias). We assessed whether there was attrition bias due to the amount, nature, or handling of incomplete outcome data. We judged the trial as having low risk of attrition bias if there were no missing outcome data or the reasons for missing data were unlikely to be related to true outcome, or missing data and reasons for it were similar across trial arms, or the missing data had been imputed using appropriate methods. We judged the trial as high risk if the reason for missing outcome data was likely to be related to the outcome, with either imbalance across trial arms in numbers of reasons for missing data and if an inappropriate application of simple imputation was potentially used. We judged the trial as unclear risk if there was insufficient reporting of attrition to permit judgement of low or high risk.
-
Selective outcome reporting (checking if there was a selection of a subset of the original variables recorded on the basis of the results). We assessed selective outcome reporting, if a protocol was available, by comparing outcomes in the protocol and published report. If they were the same we assessed it as low risk in this domain, if they differed, we considered it as high risk. If a protocol was not available, then we compared the outcomes listed in the methods section of an article with the outcomes for which results were reported. If they differed, we considered the trial as high risk. If a protocol was not available and even though the outcomes listed in the methods section and the results section were the same, we considered the trial as having an unclear risk of bias in this domain. Since not all trials have a protocol available, we expected to find a number of trials in this review to be at unclear risk.
-
Sample size (checking for possible biases confounded by small sample size). Small trials have been shown to overestimate treatment effects, probably because the conduct of small trials is more likely to be less rigorous, allowing critical criteria to be compromised (Zhang 2013). We considered trials to be at low risk of bias if they had 200 participants or more per treatment arm, at unclear risk if they had 50 to 199 participants per treatment arm, and at high risk if they had fewer than 50 participants per treatment arm.
We incorporated the results of the 'Risk of bias' assessment into the review through systematic narrative description and commentary about each item.
Medidas del efecto del tratamiento
We reported trial results organised by type of MOA and comparator evaluated. We measured treatment effects using dichotomous data, an ordinal rating scale, or qualitative evidence. For cross‐over trials, we only generated, as appropriate, a risk ratio (RR) or mean difference (MD) for pre‐cross‐over results.
Dichotomous data
For dichotomous data, we generated RRs and their 95% confidence intervals (CIs). For primary outcomes, we calculated numbers needed to treat (NNT) using the 'treat‐as‐one‐trial' method. To indicate direction of effect, we presented results as either number needed to treat for an additional beneficial outcome (NNTB) or number needed to treat for an additional harmful outcome (NNTH) to indicate direction of effect.
Continuous data
We assessed effects measures for ordinal data as continuous data. We generated the MD for continuous and ordinal data where the data were provided as a mean and standard deviation (SD).
If baseline data were reported preintervention and postintervention, we reported means or proportions for both intervention and control groups and calculated the change from baseline.
If limitations in the trial data prevented reporting a RR or if continuous data, an MD, we reported the results with caution due to lack of transparency of the evidence.
Qualitative evidence
If there had been any qualitative data in the included trials, we planned to extract them in consultation with the Cochrane Qualitative and Implementation Methods Group. Such qualitative data may aim to capture the participant's views on the value of the intervention.
Cuestiones relativas a la unidad de análisis
In our handling of each trial analytic, we considered issues that may have impacted on findings. For these we took guidance from the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). These were:
-
groups of participants randomised together with the same intervention (e.g. cluster‐randomised trials);
-
participants receiving more than one intervention (e.g. cross‐over trials);
-
multiple observations for the same outcomes (such as repeated measures).
Manejo de los datos faltantes
Given the nature of this field, there was a significant amount of missing data as a result of trial attrition due to the death of the participant.
We planned to contact trial authors if we had found data to be missing. For trials using continuous outcomes in which SDs were not reported, and no information was available from the authors, we calculated the SDs using the standard error of the mean (SEM).
Evaluación de la heterogeneidad
We assessed statistical heterogeneity using the I² statistic. The I² statistic is a reliable and robust test to quantify heterogeneity, since it does not depend on the number of trials or on the between‐trial variance. I² measures the extent of inconsistency among trials' results, and can be interpreted as the proportion of total variation in trial estimates that is due to heterogeneity rather than sampling error. We considered an I² value of greater than 50% to indicate substantial heterogeneity (Deeks 2006). Where possible, we undertook subanalyses or sensitivity analyses in an attempt to explain heterogeneity.
Evaluación de los sesgos de notificación
To reduce the risk of reporting bias, we undertook comprehensive database and registry searches, including searches of clinical trial registers and drug regulatory agency websites. We also searched websites of, and wrote to, pharmaceutical companies that are known manufacturers of MOAs to identify trial data.
Owing to an insufficient number of included studies (fewer than 10), as appropriate a test power was not ensured; we did not create funnel plots or conduct Egger's test for funnel plot asymmetry (Egger 1997; Sterne 2011). In applying in combined analysis as appropriate random‐effects estimates of the intervention effect, we decided not to exclude small studies, as this might have led to an inappropriate reduction of studies in a field that is just emerging. Nevertheless, in case of small‐study effects, we cautiously considered sample size when grading and discussing the evidence for each outcome (Roberts 2015). We expect that in updates of the review, when more studies have been published, we will be able to explore reporting biases further by comparing fixed‐effect and random‐effects estimates or L'Abbé plots as a visual method of assessing differences in results of individual studies.
Síntesis de los datos
Where trial data were of sufficient quality and sufficiently similar (in diagnostic criteria, intervention, outcome measure, length of follow‐up, and type of analysis), we combined data in a meta‐analysis to provide a pooled effect estimate. We used a fixed‐effect model in the first instance. If we found no statistical heterogeneity, we used a random‐effects model to check the robustness of the fixed‐effect model. If there was substantial (over 50%) statistical heterogeneity, we reported the random‐effects model only. Where this occurred, we stated we used the random‐effects model.
Análisis de subgrupos e investigación de la heterogeneidad
Where heterogeneity was identified in a meta‐analysis, we undertook subgroup and sensitivity analysis to investigate its possible sources. Subgroup analysis explores whether the overall effect varied with different trial populations, and with the nature and content of the interventions. In this update, we planned the following subgroup analysis:
-
studies of participants with advanced disease or in palliative care, as impact of MOAs may differ in such participants than those at an earlier stage of cancer.
Análisis de sensibilidad
If sufficient trials were available, we sought to perform, in a meta‐analysis, sensitivity analyses to explore the influence of:
-
publication status by excluding unpublished trials;
-
trial quality by excluding trials that had a high risk of bias;
-
use of appropriate measures/validated measures of outcome by excluding trials that did not use appropriate/validated measures.
We presented in a table for ease of comparisons such investigations of heterogeneity.
Quality of evidence
Two review authors (BC, VV) independently rated the quality of the primary outcomes. We used the GRADE system to rank the quality of the evidence using the guidelines provided in Chapter 12.2 of the CochraneHandbook for Systematic Reviews of Interventions (Higgins 2011).
The GRADE system uses the following criteria for assigning a quality level to a body of evidence (Chapter 12, Higgins 2011).
-
High: randomised trials; or double‐upgraded observational studies.
-
Moderate: downgraded randomised trials; or upgraded observational studies.
-
Low: double‐downgraded randomised trials; or observational studies.
-
Very low: triple‐downgraded randomised trials; or downgraded observational studies; or case series/case reports.
The GRADE approach uses five considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the quality of the body of evidence for each outcome. The GRADE system uses the following criteria for assigning grade of evidence.
-
High: we are very confident that the true effect lies close to that of the estimate of the effect.
-
Moderate: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different.
-
Low: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect.
-
Very low: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect.
Factors that may decrease the quality level of a body of evidence are:
-
limitations in the design and implementation of available studies suggesting high likelihood of bias;
-
indirectness of evidence (indirect population, intervention, control, outcomes);
-
unexplained heterogeneity or inconsistency of results (including problems with subgroup analyses);
-
imprecision of results (wide CIs);
-
high probability of publication bias (0.7854 to 1.1359).
Factors that may increase the quality level of a body of evidence are:
-
large magnitude of effect;
-
all plausible confounding would reduce a demonstrated effect or suggest a spurious effect when results show no effect;
-
dose‐response gradient.
We decreased the grade rating by one (‐1) or two (‐2) (up to a maximum of ‐3 to 'very low') if we identified:
-
serious (‐1) or very serious (‐2) limitation to study quality;
-
important inconsistency (‐1);
-
some (‐1) or major (‐2) uncertainty about directness;
-
imprecise or sparse data (‐1);
-
high probability of reporting bias (‐1).
In certain circumstances, we adjusted the overall rating for a particular outcome as recommended by GRADE guidelines (Guyatt 2013a). For example, we considered whether there were so few data that the results were highly susceptible to the random play of chance, or if a study used last observation carried forward imputation in circumstances where there were substantial differences in adverse event withdrawals, one would have no confidence in the result and would need to downgrade the quality of the evidence by three levels to very low quality (Guyatt 2013b). In other circumstances, we would not downgrade for imprecision if CIs were wide, if the outcome threshold according to how much harm would be acceptable given a benefit or vice versa.
'Summary of findings' table
We included 'Summary of findings' tables to present the main findings in a transparent and simple tabular format. We have summarised the level of overall quality of evidence on all primary outcomes in the 'Summary of findings' tables. This does not include quality evaluations on the individual types of adverse events. This decision was made as we were not judging quality for all types of adverse events using GRADE; we were only judging those adverse events that were most commonly reported. We included key information concerning the quality of evidence, the magnitude of effect of the interventions examined, and the sum of available data on the outcomes laxation response (within 24 hours; between day one and day 14), effect on analgesia (pain intensity, opioid withdrawal), serious adverse events, and number of adverse events.
Results
Description of studies
Results of the search
In this update, we first searched for evidence on 24 September 2014. This search was run without restricting the search terms to only those relating to cancer and palliative care populations. We reran our search to 29 August 2017 updating search terms and restricting to those relating to cancer and palliative care populations. We identified 633 unique citations. See Figure 1 for the flowchart of the screening process.
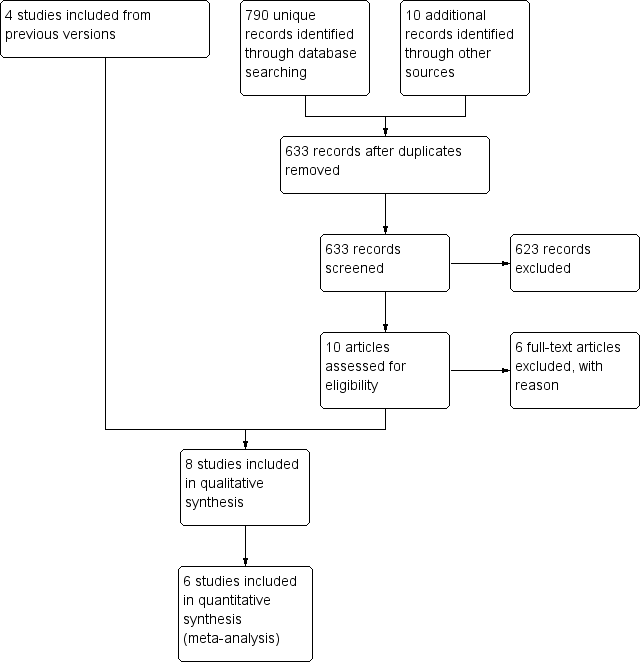
Study flow diagram.
Included studies
We included seven published RCTs of 976 participants (Ahmedzai 2012; Bull 2015; Katakami 2017; Portenoy 2008; Slatkin 2009; Sykes 1996; Thomas 2008). We included one RCT in a general population with a subset analysis in 46 people with cancer (Dupoiron 2017). We include four trials (Portenoy 2008; Slatkin 2009; Sykes 1996; Thomas 2008) that were identified in the earlier Cochrane Reviews (Candy 2011; McNicol 2008) for which this review forms a partial update. For four trials, we also identified regulatory (FDA and the EMA) assessments of the manufactures' Clinical Study Reports (Ahmedzai 2012; Portenoy 2008; Slatkin 2009; Thomas 2008) (these reports are referenced under Ahmedzai 2012 for oxycodone/naloxone, and Slatkin 2009 for methylnaltrexone).
Seven trials were multi‐centre parallel RCTs. The other was a single‐centre cross‐over controlled trial (Sykes 1996). All trials were sponsored by a pharmaceutical company, apart from Sykes 1996. Included trial populations were from North America (Bull 2015; Portenoy 2008; Slatkin 2009; Thomas 2008), Japan (Katakami 2017), South Korea (Katakami 2017) and the UK (Sykes 1996). Two trials involved sites in multiple countries. In one this included sites in Australia, Czech Republic, France, Germany, Hungary, Israel, the Netherlands, Poland, and the UK (Ahmedzai 2012), and in the other France, Germany, Poland, and the UK (Dupoiron 2017).
Three trials evaluated participants with chronic cancer pain who were not described as being at an advanced disease stage (Ahmedzai 2012; Dupoiron 2017; Katakami 2017). Where specified, the healthcare setting was a clinic (Ahmedzai 2012; Dupoiron 2017).
The five other trials evaluated effects in participants with an advanced disease including cancer, and other conditions such as AIDS or circulatory disease. Although in all these trials the majority had a primary diagnosis of cancer. Three of these trials were based in multiple care settings including inpatients and outpatients of a hospice or hospital, and long‐term care facilities (Bull 2015; Slatkin 2009; Thomas 2008). Another was hospice based only (Sykes 1996), and the other did not report the setting (Portenoy 2008).
In all trials, according to inclusion criteria, at baseline all or the majority (over 95%) of participants were on a stable opioid regimen, had OIBD, and were taking laxatives. Six trials specified that the indication for opioids was pain (Ahmedzai 2012; Dupoiron 2017; Katakami 2017; Portenoy 2008; Slatkin 2009; Thomas 2008). The other trials did not state an indication. All studies were on adults. All trials reported laxative use at baseline. For all it was either the need to take regular laxatives was part of the inclusion criteria, or it was stated that all or the majority (90% or greater) used regular laxatives.
In four trials, the intervention of interest was subcutaneous methylnaltrexone (Bull 2015; Portenoy 2008; Slatkin 2009; Thomas 2008). Three trials tested oral naloxone; in one naloxone only (Sykes 1996) and in two oxycodone (an opioid) in combination with naloxone (Ahmedzai 2012; Dupoiron 2017). The other trial evaluated oral naldemedine (Katakami 2017). We identified no trials in cancer or palliative care populations that evaluated naloxegol or another MOA.
Three of the trials involved multiple trial arms (Katakami 2017; Portenoy 2008; Slatkin 2009), the others were two armed. The interventions were either compared with a placebo or with the active intervention administered at different doses or in combination with other drugs. Outcomes on laxation were measured as self‐report or clinician report, for instance on rescue‐free laxation (Bull 2015; Katakami 2017; Portenoy 2008; Slatkin 2009; Thomas 2008), or by using a validated scale such as the BFI (Ahmedzai 2012; Dupoiron 2017), Patient Assessment of Constipation Symptoms (PAC‐SYM), and the Global Clinical Impression of Change (GCIC) (Slatkin 2009). One trial also used small bowel transit time using a lactulose and hydrogen breath test (Sykes 1996). Further details of these trials are shown in the Characteristics of included studies table.
Five trials involved a subsequent open‐label extension phase (Bull 2015; Portenoy 2008; Slatkin 2009; Sykes 1996; Thomas 2008). We did not report results on effectiveness from open‐label extension as the participants were no longer blinded.
Excluded studies
We excluded five trials because they did not include participants with cancer or at the palliative stage of a disease in their sample. These trials are listed in the Characteristics of excluded studies table.
Ongoing studies
We also identified 11 trials known to have been started but results as yet are not published (Dimitroulis 2014; JAPIC‐CTI‐132340; NCT00135577; NCT00331045/00101998; NCT02745353; NCT02839889; NCT01438567; NCT02321397; NCT02574819; Neefjes 2014; Peppin 2013). Four of these trials are evaluating methylnaltrexone (Dimitroulis 2014; NCT02574819; Neefjes 2014; Peppin 2013), one naldemedine (JAPIC‐CTI‐132340), one oxycodone/naloxone (NCT01438567; NCT02321397), two naloxegol (NCT02745353; NCT02839889), and two alvimopan (NCT00135577; NCT00331045/00101998). Further details of these are in the Characteristics of ongoing studies table.
Studies awaiting classification
We are awaiting classification for one trial on naloxegol (Webster 2013). We are unsure until we receive details from the authors or funders whether the trial fulfils our inclusion criteria. See Characteristics of studies awaiting classification for further details.
Risk of bias in included studies
All trials were vulnerable to a number of biases, most commonly reporting bias and small sample sizes. See Figure 2; Figure 3.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
The method of randomisation sequence generation was described adequately in four trials (Ahmedzai 2012; Katakami 2017; Slatkin 2009; Thomas 2008). In the other four trials this was inclear as they did not provide any details. The method of concealment of allocation was described adequately in two trials (Ahmedzai 2012; Slatkin 2009). In the other six trials this was unclear as they did not provide any details.
Blinding
Three trials were at a low risk of performance bias and detection bias (Katakami 2017; Slatkin 2009; Thomas 2008). In the other trials, it was unclear as they provided no details on who was blinded or how blinding was conducted.
Incomplete outcome data
The risk of attrition bias was low in most trials apart from one trial where it was unclear how many had dropped out of the subgroup of people with cancer (Dupoiron 2017).
Selective reporting
The risk of selective reporting was unclear in seven trials as there were no published protocols. One of these trials did not state a primary outcome (Sykes 1996).The eighth study had low risk of bias as it had a protocol (Dupoiron 2017).
Other potential sources of bias
We assessed sample size. Four trials were at a high risk of biased results as they involved fewer than 50 participants in at least one trial arm (Dupoiron 2017; Portenoy 2008; Slatkin 2009; Sykes 1996). In one of these trials, only a subset of their study sample was relevant to this review (Dupoiron 2017). Also in one of these trials while there were fewer than 50 participants in one of their treatments arms, as we combined in our exploration of the impact of MOAs their two treatment groups in comparison with placebo this risk was no longer apparent (Slatkin 2009).
All the other trials were at an unclear risk of bias as they involved treatment arms with between 50 and 199 participants.
Effects of interventions
See: Summary of findings for the main comparison Naldemedine compared to placebo for opioid‐induced bowel dysfunction in cancer and people receiving palliative care; Summary of findings 2 Lower‐dose naldemedine compared to higher‐dose naldemedine for opioid‐induced bowel dysfunction in cancer and people receiving palliative care; Summary of findings 3 Naloxone compared with placebo for cancer and people receiving palliative care with opioid‐induced bowel dysfunction; Summary of findings 4 Oxycodone/naloxone prolonged release tablets compared with oxycodone prolonged‐released tablets for opioid‐induced bowel dysfunction; Summary of findings 5 Methylnaltrexone compared to placebo for opioid‐induced bowel dysfunction in cancer and people receiving palliative care; Summary of findings 6 Lower‐dose methylnaltrexone compared to higher‐dose methylnaltrexone for opioid‐induced bowel dysfunction in cancer and people receiving palliative care
The trials varied in population (participants with any disease at a palliative stage or people with cancer irrespective of disease stage), intervention, and how they reported the outcomes. This limited the number of combined analyses. Subgroup and sensitivity analyses were limited because of the small number of trials included in any combined analysis. We have standardised the results where possible and reported the findings in the trials as fully as possible.
Naldemedine
One trial of 225 participants with cancer evaluated the effectiveness of two weeks of treatment with naldemedine compared with placebo and at different doses in people with cancer irrespective of disease stage (Katakami 2017). Doses of naldemedine were 0.1 mg, 0.2 mg, or 0.4 mg daily. Overall, we rated the quality of the evidence on laxation, effect on analgesia, and adverse events as moderate. See summary of findings Table for the main comparison.
Primary outcomes
Laxation response
The study did not measure laxation response within the first 24 hours following the first drug dose and participant evaluation of improvement in bowel function.
In comparison with placebo, naldemedine increased the number of participants who had spontaneous bowel movements over the two‐week treatment phase (RR 1.93, 95% CI 1.36 to 2.74. NNTB 3, 95% CI 2 to 5). We judged the quality of evidence for laxation response within two weeks to be moderate. We downgraded the quality of evidence by one level for limitations to the study design. This was because of unclear risk of bias (reporting bias).
The proportion of participants who had a rescue‐free laxation response over the two weeks differed by dose of naldemedine, with the higher dose resulting in more laxations (0.1 mg: 56.4%; 0.2 mg: 77.6%; 0.4 mg: 82.1%). All these were clear improvements when compared with placebo, which had a laxation responder rate of 37.5% (0.1 mg: P = 0.0464; 0.2 mg: P = 0.001; and 0.4 mg: P = 0.001). There was a dose difference in laxation response. This was in comparison with higher doses (0.2 mg and 0.4 mg) the dose of 0.1 mg daily resulted in fewer spontaneous bowel movements (0.1 mg versus 0.2 mg: RR 0.73, 95% CI 0.55 to 0.95; 0.1 mg versus 0.4 mg: RR 0.69, 95% CI 0.53 to 0.89). There was no clear difference between the dose of 0.2 mg daily compared to 0.4 mg daily in bowel movements (RR 0.95, 95% CI 0.79 to 1.14). We judged the quality of evidence on dose response to be moderate. We downgraded the quality of evidence by one level for limitations to the study design. This was because of unclear risk of bias (reporting bias).
Effect on analgesia
Naldemedine had no effect on analgesia compared with placebo in that there was no noticeable increase in opiate withdrawal over two weeks (0.1 mg: MD ‐0.13, 95% CI ‐0.57 to 0.31; 0.2 mg: MD ‐0.40, 95% ‐0.87 to 0.07; 0.4 mg: MD ‐ 0.02, 95% CI ‐0.45 to 0.41). The study did not measure the effect on analgesia using pain intensity. We judged the quality of evidence for effect on analgesia (opioid withdrawal) to be moderate. We downgraded the quality of evidence by one level for limitations to the study design. This was because of unclear risk of bias (reporting bias).
Serious adverse events
There were five serious adverse events. All events occurred in the naldemedine group. One participant experienced a gastrointestinal bleed (taking naldemedine 0.1 mg); one participant each experienced pneumonia, anaemia, or asthenia. One participant died due to bile duct cancer. The investigator considered the death unrelated to the study drug. Judgments on whether the other events were related to the study drug were not reported. Four of the serious adverse events occurred in the highest dose (0.4 mg). We judged the quality of evidence for serious adverse events to be low. We downgraded the quality of evidence by two levels, one for limitations to the study design and one for imprecision. This was because of unclear risk of bias (reporting bias) and a limited number of events.
Number and type of adverse events
There was a clear difference in the proportion of participants in the intervention arm compared to participants in the placebo arm who experienced an adverse event (RR 1.36, 95% CI 1.04 to 1.79). We judged the quality of evidence that naldemedine increased the risk of an adverse event to be moderate. We downgraded the quality of evidence by one level for limitations to the study design. This was because of unclear risk of bias (reporting bias).
The most common adverse event was diarrhoea. There was no clear difference in the proportion of participants in the intervention arm compared to participants in the placebo arm who experienced diarrhoea (RR 1.58, 95% CI 0.97 to 2.57). For eight of the participants receiving naldemedine, the diarrhoea was moderate and in another participant, it was severe. Diarrhoea in participants in the placebo group was mild. Other adverse events reported/measured included white blood cell count, abdominal pain, nausea, and vomiting (see Table 1).
| Adverse event | Naldemedine (%) | Placebo (%) |
| Diarrhoea | 67 (39) | 14 (25) |
| Decreased WBC count | 9 (5) | 3 (5) |
| Abdominal pain | 6 (4) | 0 (0) |
| Vomiting | 5 (3) | 0 (0) |
| Bone marrow failure | 3 (2) | 2 (4) |
| Decreased appetite | 6 (4) | 1 (2) |
| Nasopharyngitis | 4 (2) | 1 (2) |
| Nausea | 4 (2) | 4 (7) |
| Rash | 3 (2) | 2 (4) |
| Decreased platelet count | 3 (2) | 0 (0) |
| Decreased total protein | 7 (4) | 1 (2) |
| Glucose in urine | 4 (2) | 1 (2) |
| Abnormal haematology test | 2 (1) | 0 (0) |
| Decreased RBC count | 4 (2) | 0 (0) |
| Hypertension | 2 (1) | 0 (0) |
| Increased blood alkaline phosphatase | 4 (2) | 1 (2) |
| Increased blood lactate dehydrogenase | 2 (1) | 1 (2) |
| Increased blood pressure | 2 (1) | 0 (0) |
| Increased blood urea | 4 (2) | 1 (2) |
| Increased WBC count | 1 (2) | 2 (4) |
| Protein present in urine | 5 (3) | 0 (0) |
| Upper abdominal pain | 3 (2) | 1 (2) |
RBC: red blood cell; WBC: white blood cell.
All comparisons were not statistically significant.
Secondary outcomes
Number of participants who dropped out due to adverse events
There was no clear difference in the proportion of participants who dropped out due to an adverse event between participants taking naldemedine and participants taking placebo (RR 2.68, 95% CI 0.34 to 20.98).
Other measures of laxation response
The trial did not report other measures of laxation response.
Relief of other constipation‐associated symptoms
The trial did not report relief of other constipation‐associated symptoms.
Use of rescue medication for laxation
The trial did not measure use of 'rescue' medication for laxation.
Quality of life, satisfaction with bowel movements, and participant preference
The trial did not report quality of life, satisfaction with bowel movements, and participant preference.
Naloxone
One cross‐over trial evaluated the effectiveness of oral naloxone compared with placebo in 17 participants with advanced cancer (Sykes 1996). The participants received two days on either placebo or naloxone followed (without washout) by another two days on the trial agent that was not received on day one and two. Naloxone was given four‐hourly for a total daily dose of 0.5%, 1%, 2%, 5%, 10%, or 20% of the total daily dose of morphine. Overall, we rated the quality of the evidence on effect on analgesia as very low (laxation and adverse events not reported). See summary of findings Table 3.
Primary outcomes
Laxation response
The trial did not measure laxation response within the first 24 hours or between days one and 14 after first dose.
Effect on analgesia
The authors stated there was no evidence of opioid withdrawal or difference in pain experienced between the comparisons. There was no full data, including pre‐cross‐over results provided. We judged the quality of evidence for effect on analgesia to be very low. We downgraded by three levels. This was because evidence is from one study with small sample size, the study was of cross‐over design with no washout between drug cross‐over, and because of unclear risk of bias (reporting bias).
Serious adverse events
There were no serious adverse events.
Number and type of adverse events
The trial did not provide the overall number of adverse events experienced by the participants.
Secondary outcomes
Number of participants who dropped out due to adverse events
Four participants withdrew from the trial. One participant withdrew because of severe diarrhoea caused by the lactulose taken as part of the test on bowel function, one participant because of general deterioration in health while taking naloxone (although not thought to be a causal relationship), one participant because of diarrhoea experienced while receiving the placebo, and one participant withdrew because of nausea after two doses of naloxone at the 10% level (5 mg in this case).
Other measures of laxation response
On the second day following treatment the trial measured small bowel transit times (SBTTs). This was by lactulose‐hydrogen breath tests to detect the release of hydrogen resulting from breakdown of lactulose by colonic bacteria. They found no clear difference in SBTTs between naloxone and placebo groups. The investigators did not provide full data, including pre‐cross‐over results. They used no other measures of laxation response
Relief of other constipation‐associated symptoms
The trial did not report other constipation‐associated symptoms.
Use of rescue medication for laxation
The trial did not report use of rescue medication for laxation.
Quality of life, participant satisfaction with bowel movements, and participant preference
The trial did not report quality of life, participant satisfaction with bowel movements, and participant preference.
Oxycodone/naloxone prolonged‐release tablets versus oxycodone prolonged‐release tablets
Two trials of 231 participants with cancer evaluated oxycodone/naloxone prolonged‐release tablets (OXN PR) compared with oxycodone prolonged‐release (OXY PR) tablets (Ahmedzai 2012; Dupoiron 2017).
One trial of 185 participants with cancer of any stage evaluated the effectiveness of four weeks of treatment (Ahmedzai 2012). The drug dose in both trials arms was 120 mg daily. In addition to the published trial paper we also reviewed FDA and EMA drug reports; these documents did not add any additional data (for references to these reports see Ahmedzai 2012). The other trial evaluated the effectiveness of five weeks of treatment in a general population. We included the trial's subset data on the 46 people with cancer (Dupoiron 2017). The dose of oxycodone/naloxone was up to daily maximum oxycodone 160 mg/naloxone 80 mg.
Overall, we rated the quality of the evidence on effect on analgesia and adverse events as moderate (effect on laxation not reported) (summary of findings Table 4).
Primary outcomes
Laxation response
The trials did not report laxation response within the first 24 hours and between days one and 14 after first dose.
Effect on analgesia
In one trial using the Brief Pain Inventory‐Short Form at four weeks of treatment, pain scores were similar between the trial arms (OXN PR: mean 3.50, SD 1.88; OXY PR: mean 3.52, SD 1.80) (Ahmedzai 2012). In the other trial, pain scores remained at a low level throughout the study and were comparable between groups (Dupoiron 2017). Neither trial provided full data. We judged the quality of evidence on effect on analgesia in regards to pain intensity as moderate. We downgraded the quality of the evidence by one level because of study limitations (unclear risk of reporting bias).
Serious adverse events
In one trial 18 participants died during the trial, nine in each trial arm (Ahmedzai 2012), in the other trial one person died in the OXN PR arm and three in the OXY PR arm (Dupoiron 2017). None of the deaths were attributed to the trial drugs.
In one trial, there were 12 serious adverse events attributed to the study medication, eight in the OXN PR arm and four in the OXY PR arm (Ahmedzai 2012). There was no clear difference between trial arms in proportion of participants experiencing a serious adverse event (RR 2.00, 95% CI 0.62 to 6.41). The authors did not describe what these events were. In the other trial, there were three serious adverse events in the OXN PR arm and five in the OXY PR arm (Dupoiron 2017). None were attributed to the study medication.
We judged the quality of evidence for serious adverse events to be low. We downgraded the quality of evidence by one level because of study limitations (unclear risk of bias (reporting bias)) and one level for imprecision because of wide CIs.
Number and type of adverse events
In combined analysis of the two trials (234 participants), there was no clear difference between OXN PR in comparison with OXY PR in the proportion of particants experiencing an adverse event (RR 1.08; 95% CO 0.94 to 1.24]. I² = 0%; Analysis 4.1).
In both trials, a common adverse event was gastrointestinal symptoms. In one study, there was no clear difference in the number of such events per trial arm (RR 1.21, 95% CI 0.81 to 1.83) (Ahmedzai 2012); in the other trial, there were two events of gastritis in the OXN PR arm and none in the OXY PR arm (Dupoiron 2017). Also in this trial, there were two events of hypercholesterolaemia and hypertriglyceridaemia in the OXN PR arm and none in the other arm (Dupoiron 2017).
We judged the quality of evidence for number of adverse events to be moderate. We downgraded the quality of evidence by one level because of study limitations (unclear risk of bias (reporting bias).
Secondary outcomes
Number of participants who dropped out due to adverse events
There was no clear difference in the proportion of participants who dropped out of the trial in the OXN PR arm compared to the OXY PR arm (20/92 with OXN PR versus 12/92 OXY PR; RR 1.67, 95% CI 0.87 to 3.21) (Ahmedzai 2012). The other trial did not report the number who dropped out due to adverse events in the subset of people with cancer (Dupoiron 2017).
Other measures of laxation response
One trial measured laxation responses from baseline to four weeks (Ahmedzai 2012). In the assessment using the BFI‐Short Form, the trial reported no significant difference in changes between the trial arms in bowel function (MD ‐0.02, 95% CI ‐0.65 to 0.61). Using the PAC‐SYM on participants' assessment of bowel symptoms there was a difference between the trial arms favouring intervention group in total symptom score (MD ‐5.10, 95% CI ‐8.08 to ‐2.12), and in frequency of symptoms (MD ‐0.56, 95% CI ‐0.94 to ‐0.18). The trial did not report per arm the proportion of participants who had a laxation at each of the time points over the four weeks.
Laxation response was the primary outcome in one trial at five weeks (Dupoiron 2017). At this time point, the researchers found a clinically meaningful and treatment difference in BFI in favour of OXN PR arm (14.0, SD 8.1; P = 0.047; full data not provided).
Neither trial reported other measures of laxation response.
Relief of other constipation‐associated symptoms
One study assessed constipation‐associated symptoms using the PAC‐SYM scale, which primarily focuses on bowel‐related symptoms in addition to those of bloat, stomach cramps, and abdominal pain. However, only the total symptom score was provided (Ahmedzai 2012). Therefore, neither trial reported useful data on relief of other constipation‐associated symptoms.
Use of rescue medication for laxation
The use of laxatives (oral bisacodyl) at the end of follow‐up was not significantly different between the trial arms (MD ‐6.59, 95% CI ‐16.61 to 3.43) (Ahmedzai 2012). The authors reported the need for rescue medication was "generally low"' in both treatment groups; they reported in terms of frequency and dose that the difference in use was not significantly different. At the end of follow‐up in the other trial, the mean daily doses of rescue medication in the OXN PR arm was 0.6 (SD 1.1) and in the OXY PR arm 1.4 (SD 2.3) (Dupoiron 2017).
Quality of life, participant satisfaction with bowel movements, and participant preference
One trial used two measures to assess quality of life; the European QoL EQ‐5D instrument and the European Organization for Research and Treatment of Cancer QoL Questionnaire‐Core 30 (EORTC QLQ‐C30) (Ahmedzai 2012). Using the EQ‐5D they found no clear difference between the trial arms (MD 0.01, 95% CI ‐0.11 to 0.13). They did not provide full results at follow‐up for the EORTC QLQ‐C30 scale. The trial did not assess overall satisfaction and participant preference. The other trial did not measure any of these outcomes (Dupoiron 2017).
Methylnaltrexone
Three trials of 518 participants with advanced disease evaluated the effectiveness of subcutaneous methylnaltrexone compared to placebo (Bull 2015; Slatkin 2009; Thomas 2008). In addition to the three published trial papers, we also reviewed FDA and EMA drug reports, which did not identify any additional data (for references to these see Slatkin 2009).
One trial involved a single dose of either methylnaltrexone 0.15 mg/kg or 0.30 mg/kg (Slatkin 2009). The other two trials administered methylnaltrexone every other day for two weeks. One trial administered methylnaltrexone 0.15 mg/kg of bodyweight (Thomas 2008), and the other trial, with the aim of improving ease of administration, administered methylnaltrexone 8 mg to participants whose bodyweight was between 38 kg and 62 kg, or methylnaltrexone 12 mg if they weighed more than 62 kg (Bull 2015). Overall, we rated the quality of the evidence on laxation, effect on analgesia, and adverse events as moderate. See summary of findings Table 5.
Primary outcomes
Laxation response
In combined analysis of two trials (287 participants), there was a clear difference favouring methylnaltrexone in comparison with placebo in rescue‐free laxation within 24 hours of the first dose (RR 2.77, 95% CI 1.91 to 4.04. I² = 0%; Analysis 1.1) (Slatkin 2009; Thomas 2008). The NNTB was 3 (95% CI 2 to 3). The proportion of participants who had a rescue‐free laxation response with methylnaltrexone within 24 hours of the first dose of treatment was 59.1% and in the placebo arm was 19.5%. We judged the quality of evidence for laxation within 24 hours of the first dose to be moderate. We downgraded by one level for study limitations (unclear risk of bias (reporting bias)).
Two trials measured the laxation response over two weeks (305 participants) (Bull 2015; Thomas 2008). In combined analysis, more participants in the methylnaltrexone group compared to participants in the placebo group had a rescue‐free laxation within four hours of at least four of the maximum seven doses (RR 9.98, 95% CI 4.96 to 20.09. I² = 0%; Analysis 1.2) (Bull 2015; Thomas 2008). The NNTB was 2 (95% CI 2 to 2). The proportion of participants who had a rescue‐free laxation response with methylnaltrexone (within four hours of at least four of the maximum seven doses) was 52.6% and in the placebo arm was 5.3%. We judged the quality of evidence for laxation response over two weeks to be moderate. We downgraded by one level for study limitations (unclear risk of bias (reporting bias)). As the effect size was large, we did not downgrade for imprecision because of wide CIs.
Effect on analgesia
Two trials assessed opioid withdrawal symptoms (Slatkin 2009; Thomas 2008). In one trial, they stated that the median change from baseline to day two in score using the Himmelsbach Opioid Withdrawal Scale in both trials arms was 0 (Slatkin 2009). In the other trial at day one and 14, there was no clear difference between the trial arms (day one: MD 0.00, 95% CI ‐0.46 to 0.46; day 14: MD 0.10, 95% CI ‐0.63 to 0.83). We judged the quality of evidence for effect on analgesia in regards to opioid withdrawal to be moderate. We downgraded the quality of evidence by one level for study limitations because of unclear risk of bias (reporting bias).
In one trial, there was a reduction from baseline to four‐hour follow‐up in pain intensity in the methylnaltrexone 0.15 mg/kg group compared to placebo, but not in the methylnaltrexone 0.3 mg/kg group compared to placebo (0.15 mg/kg: MD ‐0.76, 95% CI ‐1.47 to ‐0.05; 0.3 mg/kg: MD ‐0.25, 95% CI ‐0.91 to 0.41) (Slatkin 2009). In the other two trials, participants had similar pain scores in both trial arms at follow‐up (Bull 2015; Thomas 2008). The dose of methylnaltrexone in the trial by Thomas 2008 was 0.15 mg/kg and by Bull 2015 was either 8 mg or 12 mg dependent on participant's bodyweight. In the trial by Thomas 2008, there was no clear difference in score between the trial arms at day one and day 14 (day one: MD 0.20, 95% CI ‐0.62 to 1.02; day 14: MD ‐0.70, 95% CI ‐1.52 to 0.12). The other trial did not provide full data (Bull 2015). We judged the quality of evidence for effect on analgesia in regards to pain intensity to be low. We downgraded the quality of evidence by two levels, one for study limitations (unclear risk of bias (reporting bias)), and one for inconsistency because of differing estimates of effect.
Serious adverse events
In combined analysis of two trials, with 364 participants, the proportion of participants experiencing a serious adverse event was lower in the methylnaltrexone arm (25/179 in methylnaltrexone arm versus 44/185 in placebo arm; RR 0.59, 95% CI 0.38 to 0.93; I² = 0%; Analysis 2.1) (Bull 2015; Thomas 2008). In Thomas 2008, the type of serious adverse events in the 11 participants who were receiving methylnaltrexone were: aneurysm ruptured, respiratory arrest, exacerbation of dyspnoea, suicidal ideation, aggression, malignant neoplasm progression, concomitant disease progression, myocardial ischaemia, aggravation of coronary artery disease, and aggravation of congestive heart failure. Bull 2015 did not describe the types of serious adverse events. In both trials, the investigators considered all serious adverse events as either not related or unlikely to be related to the trial drug. One trial did not report serious adverse events occurring during the randomised phase (Slatkin 2009), although during the open‐label phase three participants experienced such an event. One participant had flushing, one participant had delirium possibly related to methylnaltrexone, and one participant had severe diarrhoea and subsequent dehydration and cardiovascular collapse considered to be related to methylnaltrexone. We judged the quality of evidence for risk of a serious adverse event to be moderate. We downgraded the quality of evidence by one level for study limitations. This was because of unclear risk of bias (reporting bias).
Number and type of adverse events
In combined analysis of the three trials, with 518 participants, the proportion of participants experiencing adverse events was not clearly different between the trial arms (RR 1.17, 95% CI 0.94 to 1.45; random‐effects model; I² = 74% suggesting substantial heterogeneity between trials; Analysis 3.1) (Bull 2015; Slatkin 2009; Thomas 2008). We considered subgroup and sensitivity analyses to explain the heterogeneity. Sensitivity analysis demonstrated that the primary meta‐analysis was robust. Omitting the trial at high risk of bias because of small sample size (Slatkin 2009) resulted in a smaller estimate of effect. It did not change the overall result or conclusions. See Table 2. We judged the quality of evidence for adverse events to be low. It was downgraded by two levels; one for study limitations (unclear risk of reporting bias) and one because of inconsistency due to substantial statistical heterogeneity between the trials, this was upgraded since sensitivity analysis accounted for heterogeneity.
| Methylnaltrexone vs placeboa | |
| AEs | RR 1.07, 95% CI 0.96 to 1.19 |
| AE of abdominal pain | RR 2.15, 95% CI 1.28 to 3.62 |
| AE of nausea | RR 0.87, 95% CI 0.46 to 1.65 |
| AE of vomiting | RR 0.70, 95% CI 0.33 to 1.47 |
aomitting trial of high risk of bias.
AE: adverse event; CI: confidence intervals; RR: risk ratio.
Two of the trials reported some of the adverse events as severe (Slatkin 2009; Thomas 2008). One reported that 19/102 participants experienced a severe adverse event that was possibly related to the intervention drug; most commonly this event was abdominal pain (Slatkin 2009). The other trial did not describe what the events were and whether they were considered related to methylnaltrexone, but more participants in the placebo group experienced severe adverse events than in the intervention group (5/63 (8%) with methylnaltrexone versus 9/71 (13%) with placebo).
All three trials reported that participants in both trial arms experienced abdominal pain, flatulence, nausea, and vomiting (Bull 2015; Slatkin 2009; Thomas 2008). In combined analysis, participants in the intervention arm compared to participants in the placebo arm were more likely to experience abdominal pain (RR 2.39, 95% CI 1.07 to 5.34; random‐effects model; I² = 65% suggesting substantial heterogeneity between trials). Likewise, in combined analysis participants in the intervention arm compared to participants in the placebo arm were more likely to experience flatulence (RR 2.09, 95% CI 1.07 to 4.08; I² = 0%). In combined analyses, there was no clear difference between the trial arms in the proportion who experienced nausea (RR 0.97, 95% CI 0.89 to 1.06; random‐effects model; I² = 63% suggesting substantial heterogeneity between trials) or vomiting (RR 0.99, 95% CI 0.92 to 1.08; random‐effects model; I² = 67% suggesting substantial heterogeneity between trials). Sensitivity analyses demonstrated that the primary meta‐analyses were robust with regards to risk of experiencing abdominal pain, nausea, and vomiting. Omitting the trial at high risk of bias because of small sample size resulted in a smaller estimate of effect (Slatkin 2009). It did not change the overall result or conclusions. See Table 2.
Two trials also reported that participants experienced diarrhoea (Bull 2015; Thomas 2008), dizziness (Slatkin 2009; Thomas 2008), peripheral oedema (Bull 2015; Thomas 2008), and restlessness (Slatkin 2009; Thomas 2008). In combined analysis, there was no clear difference between trials in the proportion of participants experiencing these adverse events. See Table 3. Two trials also reported on falls (Bull 2015; Thomas 2008), and somnolence (Slatkin 2009; Thomas 2008). For these adverse events in combined analysis, there was also no significant difference between trial arms. See Table 3.
| Adverse event | RR (95% CI) | I²statistic on heterogeneity |
| Abdominal pain | 2.39 (1.07 to 5.34) | 65% |
| Diarrhoea | 1.02 (0.93 to 1.11) | 51% |
| Dizziness | 4.09 (0.99 to 16.83) | 0% |
| Falls | 1.02 (0.89 to 1.16) | 84% |
| Flatulence | 2.09 (1.07 to 4.08) | 0% |
| Nausea | 0.97 (0.89 to 1.06) | 63% |
| Peripheral oedema | 1.01 (0.50 to 2.03) | 0% |
| Restlessness | 0.83 (0.32 to 2.12) | 0% |
| Somnolence | 1.00 (0.93 to 1.08) | 73% |
| Vomiting | 0.99 (0.92 to 1.08) | 67% |
CI: confidence interval; RR: risk ratio.
Only one trial reported other adverse events. See Table 4 for the proportion of participants per trial and per arm that reported these events.
| Adverse event | Methylnaltrexone (%) | Placebo (%) |
| Abdominal distensiona | 1 (2) | 6 (8) |
| Abdominal tendernessa | 1 (2) | 4 (6) |
| Astheniaa | 4 (6) | 4 (6) |
| Anxietyb | 5 (4.9) | 0 (0) |
| Arthralgiab | 3 (2.9) | 1 (1.9) |
| Back painc | 9 (7.8) | 3 (2.9) |
| Confusional statec | 7 (6.0) | 9 (7.9) |
| Dehydrationa | 2 (3) | 4 (6) |
| Fatigueb | 4 (3.9) | 1 (1.9) |
| Hypotensiona | 0 (0) | 4 (6) |
| Increased body temperaturea | 5 (8) | 2 (3) |
| Lethergya | 4 (6) | 4 (6) |
| Malignant‐neoplasm progressiona | 7 (11) | 9 (13) |
| Pain exacerbationb | 8 (8) | 2 (4) |
| Rhinorrhoeab | 6 (5.9) | 1 (1) |
| Sweating increasedb | 8 (7.8) | 4 (7.7) |
| Tachycardiaa | 1 (1) | 4 (6) |
aReported in trial by Thomas 2008.
bReported in trial by Slatkin 2009.
cReported in trial by Bull 2015.
Secondary outcomes
Number of participants who dropped out due to adverse events
Two trials reported that there were dropouts due to adverse events. In combined analysis, there was no significant difference in the proportion of participants who dropped out between the trial arms (RR 1.22, 95% CI 0.54 to 2.76; I² = 0%; Analysis 3.2) (Bull 2015; Thomas 2008). The other trial reported no dropouts due to adverse events.
Other measures of laxation response
In both trials of two‐week treatments there was a shorter time to laxation in the methylnaltrexone group that persisted for each of the seven treatment doses (for all comparisons: P < 0.005 (Bull 2015); P < 0.002 (Thomas 2008)). In combined analysis of all three trials, there was a difference favouring methylnaltrexone in comparison with placebo in rescue‐free laxation within four hours of the first dose (placebo or methylnaltrexone) (RR 3.87, 95% CI 2.83 to 5.28; I² = 0%; Analysis 1.3) (Bull 2015; Slatkin 2009; Thomas 2008). The proportion of participants in total who had a laxation response within four hours of the first (or second) dose of treatment ranged from 61.4% in the methylnaltrexone groups and 16.0% in the placebo groups. The single‐dose trial using the GCIC found that the proportion of participants who reported an improvement in constipation distress at four hours significantly favoured the methylnaltrexone arm (irrespective of dose) compared to placebo (RR 1.84, 95% CI 1.23 to 2.75) (Slatkin 2009). The trial by Bull 2015 measured the mean number of rescue‐free laxation responses within 24 hours after dosing in the first and second week: at both time points participants receiving methylnaltrexone had significantly more laxations than participants receiving placebo (first week: MD 1.90, 95% CI 0.55 to 3.25; second week: MD 1.20, 95% CI 0.15 to 2.25).
Two trials reported clinician ratings of bowel status using the GCIC ratings were assessed in two trials (287 participants) (Slatkin 2009; Thomas 2008). In combined analysis, clinician ratings that bowel status had improved at week one were greater in those in the interventions groups (RR 2.37, 95% CI 1.66 to 3.38; I² = 25%; Analysis 1.7).
All three trials reported that time to first rescue‐free laxation favoured methylnaltrexone (median times in the intervention group versus placebo: 0.8 hours versus 23.6 hours (Bull 2015), 1.1 hours versus > 24 hours (Slatkin 2009), 6.3 hours versus > 48 hours (Thomas 2008)).
Relief of other constipation‐associated symptoms
None of the trials measured the relief of other constipation‐associated symptoms such as bloating, abdominal distention, gastric reflux, abdominal cramping, dry mouth, epigastric fullness, nausea, and vomiting, although some of these symptoms were reported as adverse events.
Use of rescue medication for laxation
Two trials recorded the need for rescue laxatives. In both trials, more participants in the placebo group needed rescue laxatives than participants in the intervention groups.
In one trial, fewer participants used rescue laxatives in the methylnaltrexone group than in the placebo group (31/116 with methylnaltrexone versus 46/114 with placebo; RR 0.66, 95% CI 0.46 to 0.96) (Bull 2015). It was also recorded in the FDA report on the trial by Thomas 2008 that there was an increased use in enemas in both trial arms. This was greater in the placebo group (23.8% with methylnaltrexone versus 35.2% with placebo).
Quality of life, overall satisfaction with bowel movements, and participant preference
None of the trials measured quality of life or participant preference. Two trials (287 participants) assessed participant ratings of bowel status using the GCIC ratings (Slatkin 2009; Thomas 2008). In combined analysis, participant ratings that bowel status had improved at one week were significantly greater in the methylnaltrexone groups (RR 2.32, 95% CI 1.64 to 3.27; I² = 0%; Analysis 1.6). Both trials measured constipation distress at day one (RR 1.87, 95% CI 1.34 to 2.59; I² = 25%).
Methylnaltrexone dose response
Two trials with 135 participants with advanced disease evaluated different dosing regimens of methylnaltrexone (Portenoy 2008; Slatkin 2009). One trial, irrespective of bodyweight, explored fixed doses of methylnaltrexone 1 mg, 5 mg, 12.5 mg, or 20 mg in 33 participants (Portenoy 2008). Because of the limited number of participants in the trials, we provided outcomes for participants taking 1 mg compared to participants of 5 mg or greater. The drug was administered on alternate days over one week. The other trial compared one dose on different dose‐ranging schedules of 0.15 mg/kg for 47 participants with 0.3 mg/kg for 55 participants (Slatkin 2009). We did not consider combining the data because the dosing schedules differed. Overall, we rated the quality of evidence on laxation, effect on analgesia, and adverse events to be low. See summary of findings Table 6.
Primary outcomes
Laxation response
Both trials measured laxation response within 24 hours of the first dose of methylnaltrexone. There was no clear difference between participants taking a higher dose to participants on a lower dose (RR 0.82, 95% CI 0.41 to 1.66 (Portenoy 2008); RR 1.07, 95% CI 0.81 to 1.42 (Slatkin 2009)). We judged the quality of evidence for laxation response within 24 hours as low. We downgraded the quality of evidence by two levels, because of study limitations. This was because of unclear risk of bias (reporting bias) and small sample sizes (high risk of bias).
One trial reported outcomes after dosing at day three and five (Portenoy 2008). Laxation response within 24 hours did not clearly differ in participants receiving 1 mg compared to participants receiving 5 mg or greater on day three or on day five (day three: RR 0.47, 95% CI 0.18 to 1.25; day five: RR 0.21, 95% CI 0.03 to 1.31). The other trial did not measure response beyond one day (Slatkin 2009). We judged the quality of evidence for laxation response at day three and five to be low. We downgraded the quality of evidence by two levels for study limitations. This was because of unclear risk of bias (reporting bias) and results from a single study with a small sample size (high risk of bias).
Effect on analgesia
In both trials, pain intensity did not change from baseline and was similar in both trial arms receiving the different doses of methylnaltrexone (Portenoy 2008; Slatkin 2009). In one trial, the mean change in pain from baseline to four‐hour evaluation was not clearly different (MD ‐0.51, 95% CI ‐1.49 to 0.47) (Slatkin 2009); the other trial did not provide full results. Both trials reported that the use of methylnaltrexone did not result in opioid withdrawal (Portenoy 2008; Slatkin 2009). In one trial, there was a no clear difference in the mean change from baseline to four‐hour evaluation (MD ‐0.04, 95% CI ‐0.73 to 0.65) (Slatkin 2009); the other trial did not provide full results. We judged the quality of evidence for effect on analgesia to be low. We downgraded the quality of evidence by two levels for study limitations. This was because of unclear risk of bias (reporting bias) and small sample sizes (high risk of bias).
Serious adverse events
In one trial, 15 participants experienced a serious adverse event; however, risk of serious adverse events were not reported per trial arm (Portenoy 2008). The events were lymphadenectomy, febrile neutropenia, depressed level of consciousness, suicide attempt, and delirium. All were considered unrelated to study medication. In the other trial, no serious adverse events occurred during the randomised trial phase, although during the open‐label phase three participants experienced such an event of which one had severe diarrhoea and subsequent dehydration and cardiovascular collapse (Slatkin 2009). These were considered to be related to the drug. We did not judge the quality of evidence for serious adverse events as they did not report incidence per trial arm.
Number and type of adverse events
In the fixed‐dose trial, all participants experienced an adverse event (Portenoy 2008). In the other trial, there was no clear difference in dose arms in the occurrence of an adverse event (RR 0.90, 95% CI 0.73 to 1.13) (Slatkin 2009). The most common adverse event in both trials and per trial arm was abdominal pain. We judged the quality of evidence for adverse events to be low. We downgraded the quality of evidence by two levels for study limitations because of unclear risk of bias (reporting bias) and small sample size (high risk of bias).
Secondary outcomes
Number of participants who dropped out due to adverse events
In one trial, one participant discontinued the trial (during the double‐blind treatment phase) because of an adverse event (Portenoy 2008). This was an 84‐year old man who withdrew due to syncope (taking methylnaltrexone 12.5 mg). The event was transient and resolved without sequelae; the investigators assessed that it was related to the medication. This trial also reported that in the open‐label phase, after receiving three doses, a 20‐year old man was withdrawn from the trial due to abdominal cramping that was considered as probably related to the trial medication. In the other trial, none of the participants discontinued because of an adverse event (Slatkin 2009).
Other measures of laxation response
In the single‐dose trial of 154 participants there was no clear difference in the proportion of participants who had a rescue‐free laxation within four hours of receiving methylnaltrexone between participants in the trial arm who received 0.15 mg/kg with participants who received 0.3 mg/kg (RR 1.06, 95% CI 0.77 to 1.46) (Slatkin 2009). In this trial, the proportion of participants who reported an improvement in constipation distress within four hours was similar (30/47 (64.4%) with methylnaltrexone 0.15 mg/kg versus 35/55 (63.5%) with methylnaltrexone 0.3 mg/kg), as was global improvement (27/47 (58.7%) with methylnaltrexone 0.15 mg/kg versus 32/55 (58.8%) with methylnaltrexone 0.3 mg/kg). In the fixed dose trial, 10% (1/10) of participants taking methylnaltrexone 1 mg had a laxation within four hours compared to 43% (3/7) taking methylnaltrexone 5 mg, 60% (6/10) taking methylnaltrexone 12.5 mg, and 33% (2/6) taking methylnaltrexone 20 mg (Portenoy 2008). Laxation response within four hours did not significantly differ in participants receiving 1 mg compared to participants receiving 5 mg or greater (RR 0.21, 95% CI 0.03 to 1.41).
One trial reported outcomes at four hours after dosing at day three and five (Portenoy 2008). Laxation response within four hours did not significantly differ in participants receiving 1 mg compared to participants receiving 5 mg or greater on day three or day five (day three: RR 0.34, 95% CI 0.10 to 1.22; day five: RR 0.09, 95% CI 0.01 to 1.38).
In one trial, the median time to rescue‐free laxation was 1.10 hours in the methylnaltrexone 0.15 mg/kg group and 0.8 hours in the methylnaltrexone 0.3 mg/kg group (Slatkin 2009). In the other trial, for the lowest dose of 1 mg it was more than 48 hours whereas for the higher doses the first laxation was at 1.72 with methylnaltrexone 5 mg, 0.48 with methylnaltrexone 12.5 mg, and 6.75 hours with methylnaltrexone 20 mg (Portenoy 2008).
One of the two‐week trials reported that there was a similar proportion of watery bowel movements in the methylnaltrexone arm compared with the placebo arm (16% (after 28 of 176 drug trial doses) with methylnaltrexone) versus 17% (after 8 out of 48 doses) with placebo) (Thomas 2008). In the single dose trial in participants who had a laxation within four hours of dosing, eight out of the 29 receiving methylnaltrexone had at least one watery laxation compared to none of the seven participants receiving placebo (Slatkin 2009).
Two trials (287 participants) measured self‐report of constipation distress (Slatkin 2009; Thomas 2008). In both trials, distress was reduced more at 24 hours in the methylnaltrexone group than in placebo group (combined analysis: RR 1.87, 95% CI 1.34 to 2.59; I² = 0%; Analysis 1.5).
One trial reported bowel status using the Clinical Global Impression of Change (Slatkin 2009). There was improvement at the end of the double‐blind phase for methylnaltrexone 0.15 mg/kg (58.7%) and methylnaltrexone 0.3 mg/kg (58.8%).
Relief of other constipation‐associated symptoms
None of the trials measured the relief of other constipation‐associated symptoms, although some of these symptoms, such as abdominal pain and nausea, were recorded as adverse events.
Use of rescue medication for laxation
One trial reported that participants in the trial arm receiving the lowest dose (methylnaltrexone 1 mg) required a rescue laxative approximately twice as often as those in the higher dose groups (Portenoy 2008). The other trial did not compare use of rescue medication (Slatkin 2009).
Quality of life, satisfaction with bowel movements, and participant preference
None of the trials assessed the impact of treatment on quality of life or participant preference (Slatkin 2009; Thomas 2008). One trial assessed participants' level of satisfaction with the trial medication using a 7‐point scale (Portenoy 2008). They did not report the level of satisfaction but reported no difference in satisfaction between the trial arms.
Discusión
Resumen de los resultados principales
Esta actualización de la revisión intentó establecer la efectividad y la seguridad de los AOM para la DIIO en los pacientes con cáncer y en los pacientes que reciben cuidados paliativos. Tres de los ocho ECA exploraron los resultados en poblaciones con cáncer, independientemente del estadio de la enfermedad; uno comparó naldemedina oral con placebo, dos compararon oxicodona/naloxona oral de liberación prolongada con oxicodona sola. La naloxona oral sola comparada con placebo se evaluó en pacientes con cáncer avanzado. Los otros cuatro ensayos compararon metilnaltrexona subcutánea con placebo o diferentes regímenes de metilnaltrexona en poblaciones de atención paliativa, en las que la mayoría de los participantes tenían cáncer avanzado. Todos evaluaron el efecto de los AOM en poblaciones en las que todos o más del 90% de los participantes recibieron regímenes regulares de laxantes. Cuatro ensayos tuvieron alto riesgo de sesgo debido a los tamaños de la muestra pequeños (menos de 50 participantes por brazo del ensayo), todos tuvieron riesgo incierto de sesgo de selección por el informe deficiente de la ocultación de la asignación o la generación de la secuencia aleatoria (o ambas), y siete tuvieron riesgo incierto de sesgo de informe porque no proporcionaron el protocolo. Solo se proporcionaron datos de todos los resultados primarios de interés para la metilnaltrexona comparada con placebo. En un ensayo, solo un subgrupo de la muestra fue elegible para esta revisión. Se utilizaron los criterios GRADE sobre la calidad de la evidencia para evaluar los resultados primarios. Las evaluaciones mediante GRADE difirieron según la intervención del ensayo; en general, fue moderada para la naldemedina y la metilnaltrexona. Para la naloxona como adyuvante fue moderada, y para naloxona tomada sola fue muy baja. Sin embargo, estas evaluaciones para la naloxona solo se refirieron al efecto sobre la analgesia y los eventos adversos y no para la laxación, para la que no se proporcionaron datos.
Para la naldemedina, se encontró evidencia de calidad moderada de que el fármaco aumentó claramente el número de laxaciones espontáneas en el transcurso de dos semanas en pacientes con cáncer. También se identificó una relación dosis‐respuesta con las dosis mayores, que claramente aumentó durante este tiempo el número de laxaciones espontáneas (evidencia de calidad moderada). Las dosis mayores fueron 0,2 mg y 0,4 mg diarios en comparación con la dosis de 0,1 mg diarios. La evidencia de que la naldemedina no tuvo repercusión sobre la analgesia en cuanto a los síntomas de abstinencia de los opiáceos fue de calidad moderada. Hubo cinco eventos adversos graves que ocurrieron en participantes que tomaban naldemedina y ninguno en el grupo placebo. Esta evidencia se consideró de baja calidad. La calidad de la evidencia de que hubo un aumento de los eventos adversos se consideró moderada. El evento adverso más común fue la diarrea.
La evidencia sobre la naloxona fue limitada. Ni el ensayo de naloxona sola en pacientes con cáncer avanzado ni los dos que la evaluaron en combinación con oxicodona en pacientes con cáncer evaluaron la respuesta de laxación a las 24 horas o en el transcurso dos semanas y, para algunas evaluaciones (p.ej. el efecto sobre la analgesia), los estudios no proporcionaron datos completos. El ensayo de naloxona sola no informó eventos adversos.
Hubo evidencia moderada a partir del análisis combinado de que en los pacientes que recibieron metilnaltrexona como atención paliativa mejoró la laxación; hasta el 59,1% de los participantes tuvieron una laxación en el transcurso de 24 horas de la primera dosis, mientras que en el grupo placebo ocurrió en el 19,5% (CR 2,77; IC del 95%: 1,91 a 4,04; NNTB 3; IC del 95%: 2 a 3). Un ensayo demostró claramente que menos participantes del brazo de metilnaltrexona requirieron laxantes de rescate en comparación con placebo. También hubo evidencia de calidad moderada de un análisis combinado sobre la respuesta de laxación en dos semanas; hasta el 52,6% de los participantes tuvieron una respuesta de laxación sin rescate (en el transcurso de cuatro horas de al menos cuatro de un máximo de siete dosis), mientras que en el grupo placebo fue del 5,3% (CR 9,98; IC del 95%: 4,96 a 20,09; NNTB 2 (IC del 95%: 2 a 3). La evidencia de la repercusión de metilnaltrexona sobre la analgesia en cuanto a los síntomas de abstinencia de los opiáceos se consideró moderada, y con respecto a la intensidad del dolor se consideró baja. Hubo evidencia de calidad moderada de que el fármaco no aumentó el riesgo de eventos adversos graves. Hubo evidencia de baja calidad de que la metilnaltrexona aumentó el riesgo de eventos adversos en cuanto al dolor abdominal y la flatulencia. Ninguno de los eventos adversos fue grave. Sin embargo, en un ensayo, en una fase posterior de un estudio abierto (cuando los investigadores y los participantes conocían qué tratamiento se administraba), un participante presentó diarrea grave, deshidratación posterior y colapso cardiovascular que se consideraron relacionados con el fármaco.
Se encontró evidencia de baja calidad de que no hubo repercusión de las diferentes dosis de metilnaltrexona sobre la respuesta de laxación, la analgesia ni los eventos adversos. No fue posible evaluar la calidad con respecto al riesgo de eventos adversos graves. Sin embargo, es importante destacar que, durante uno de los ensayos, 15 participantes presentaron un evento adverso grave. Los investigadores no proporcionaron detalles sobre cuáles fueron los eventos ni si se consideraron relacionados con el fármaco del ensayo. Todos los participantes experimentaron un evento adverso en este ensayo.
Compleción y aplicabilidad general de las pruebas
Se buscó evidencia de los ensayos, mucho más allá de los artículos publicados. Cuando estuvieron disponibles, se obtuvieron los documentos regulatorios, aunque proporcionaron pocos datos nuevos.
Los hallazgos de la revisión fueron limitados. Los ensayos fueron pocos, lo que limitó los análisis combinados. En algunos análisis hubo heterogeneidad estadística entre los ensayos. Lo anterior se relacionó con los efectos adversos de la metilnaltrexona en comparación con placebo. Los análisis de sensibilidad demostraron que los metanálisis primarios fueron consistentes con respecto al riesgo de eventos adversos generales, así como los eventos adversos individuales dolor abdominal, náuseas y vómitos. La omisión del ensayo con alto riesgo de sesgo debido al tamaño de la muestra pequeño (Slatkin 2009) dio lugar a que todos los análisis produjeran una estimación más pequeña del efecto, pero no cambió la dirección del efecto.
La evidencia sobre la naldemedina provino solo de un ensayo. Se conoce al menos otro ensayo finalizado, pero que no se publicado completamente; cuando esté disponible la evidencia de este ensayo podría informar mejor la evidencia sobre este fármaco.
En los ensayos de naloxona, no todos los resultados primarios de interés fueron el centro de los estudios de investigación. El ensayo sobre naloxona sola midió uno de los tres resultados primarios de interés, específicamente el efecto sobre la analgesia, pero no el efecto sobre la laxación ni los eventos adversos. Sin embargo, los dos ensayos de naloxona en combinación con oxicodona midieron la laxación en puntos temporales posteriores (cuatro y cinco semanas). En los estudios se encontró una mejoría en la función intestinal en cuatro de cada cinco evaluaciones.
Se podría argumentar que el grupo de evidencia es más fuerte para metilnaltrexona, ya que se obtuvo a partir de más estudios y con un número total de participantes mayor que el de los ensayos de otros AOM. Es importante reflejar que esta evidencia es solo de calidad moderada, y en uno de los cuatro ensayos de metilnaltrexona la evaluación de la repercusión del fármaco se puede haber afectado porque los participantes del brazo placebo recibieron dosis mayores de opiáceos que los del brazo de metilnaltrexona. El presente análisis fue más limitado para la dosis‐respuesta de la metilnaltrexona porque no fue posible combinar los estudios debido a los diferentes regímenes de dosis.
Los resultados de los participantes con respecto a la satisfacción y la preferencia se evaluaron de manera deficiente. Sólo un ensayo evaluó la calidad de vida. (Ahmedzai 2012). El ensayo no encontró diferencias en la calidad de vida entre los participantes del grupo de oxicodona/naloxona y los participantes del grupo de oxicodona sola. También hay otros resultados que no se evaluaron en estos ensayos, en los que estudios de investigación anteriores indicaban la necesidad de exploración adicional. Por ejemplo, si los AOM, en particular la metilnaltrexona, aumentan la supervivencia del cáncer (Janku 2015).
No se encontraron ensayos completados sobre naloxegol que cumplieran los criterios de inclusión, que en 2014 fue aprobado por la FDA para su uso en la DIIO en pacientes sin cáncer. Sin embargo, se está en espera de aclaración de los autores o patrocinadores de un ensayo, ya que no está claro si cumple los criterios de inclusión (Webster 2013), y hay dos ensayos potencialmente relevantes en curso (NCT02745353; NCT02839889).
Las evaluaciones sobre el desarrollo de nuevos AOM para la DIIO, su efectividad y su seguridad son un campo de investigación activo. Se señalaron 11 ensayos en poblaciones de pacientes con cáncer o pacientes que recibieron cuidados paliativos (o ambos) que estaban en curso o se completaron, pero los resultados publicados aún no estaban disponibles en el momento de publicar esta revisión (Dimitroulis 2014; JAPIC‐CTI‐132340; NCT00135577; NCT00331045/00101998; NCT01438567; NCT02321397; NCT02574819; NCT02745353; NCT02839889; Neefjes 2014; Peppin 2013).
Calidad de la evidencia
Los ocho ensayos fueron vulnerables a los sesgos. Cuatro tuvieron alto riesgo de sesgo ya que incluyeron una muestra de menos de 50 participantes por brazo del ensayo. La calidad de la evidencia para los resultados primarios de interés se evaluó mediante los criterios GRADE.
Para la naldemedina y para la metilnaltrexona la calidad general de la evidencia para mejorar la respuesta de laxación (en comparación con placebo) fue moderada. Lo anterior significa que hay certeza moderada en la estimación del efecto y que es probable que el verdadero efecto esté próximo a la estimación del efecto, pero hay alguna posibilidad de que sea significativamente diferente. La base de la evidencia para evaluar la calidad se podría considerar más fuerte para la metilnaltrexona que para la naldemedina porque se basa en varios ensayos con un total de 518 participantes, en lugar de un ensayo sobre naldemedina con 225 participantes. Sin embargo, mientras que en los ensayos de metilnaltrexona se utilizaron los datos de 518 participantes para las evaluaciones de los eventos adversos, en las comparaciones sobre la respuesta de laxación se utilizaron los datos de 287 participantes.
La calidad de la evidencia de que los AOM evaluados no tuvieron repercusión sobre el efecto de la analgesia con respecto a la naldemedina se consideró moderada (para la abstinencia de los opiáceos, no informaron la intensidad del dolor), y baja a moderada con respecto a la metilnaltrexona. La calidad de la evidencia de que los AOM aumentaron el riesgo de eventos adversos graves fue baja para la naldemedina y moderada para la metilnaltrexona. En cuanto a los eventos adversos no graves, hubo evidencia de calidad moderada de que la naldemedina aumentó el riesgo de eventos adversos y evidencia de baja calidad de que la metilnaltrexona no aumentó el riesgo. La calidad general de la evidencia de que no hubo diferencias en los resultados primarios de interés entre las dosis mayores comparadas con las dosis menores de metilnaltrexona fue baja. La calidad de la evidencia sobre naloxona (en comparación con placebo) fue muy baja con respecto a ningún efecto sobre la analgesia (no se informaron los eventos adversos ni la respuesta de laxación). La calidad de la evidencia sobre la naloxona en combinación con oxicodona fue moderada, y no tuvo repercusión sobre la analgesia ni sobre el riesgo de eventos adversos (no se informó la respuesta temprana de laxación).
Sesgos potenciales en el proceso de revisión
Se buscó de forma exhaustiva la evidencia de los ensayos, incluidas cinco bases de datos de referencias. Se buscaron los datos de ensayos no publicados en las bases de datos de organismos farmacéuticos y reguladores. Sin embargo, hay guías limitadas sobre cómo buscar datos no publicados y la búsqueda en los sitios web de organismos reguladores no es sencilla.
La inclusión se limitó a los ensayos que especificaron que los participantes presentaban cáncer o estaban en cuidados paliativos, independientemente del estadio de la enfermedad. Es probable que lo anterior haya dado lugar a la pérdida de datos, ya que los ensayos excluidos podrían haber incluido pacientes con estas características, pero los artículos de los ensayos no proporcionaron estos detalles "más finos".
Se incluyeron ensayos con limitaciones metodológicas. Además, hay un posible problema debido a los efectos de arrastre en el ensayo con diseño cruzado (Sykes 1996), y el análisis combinado estuvo limitado por el número de ensayos disponibles.
Acuerdos y desacuerdos con otros estudios o revisiones
Esta revisión sistemática Cochrane examinó específicamente la evidencia sobre los AOM para la DIIO en los pacientes con cáncer y atención paliativa. Los resultados sobre metilnaltrexona en los cuidados paliativos fueron similares a los de la revisión Cochrane anterior relevante que incluyó dos de los cuatro ensayos sobre metilnaltrexona incluidos en la presente revisión (Candy 2011).
Hay revisiones que han evaluado el efecto de los AOM para la DIIO en poblaciones generales, aunque no son revisiones Cochrane recientes (publicadas desde 2014). Una revisión con una población más amplia identificó 14 ensayos (Ford 2013). Además de cuatro de los ensayos incluidos en esta revisión, esta incluyó ensayos sobre estreñimiento inducido por metadona y ensayos que incluyeron participantes que recibieron opiáceos para el dolor crónico no maligno. En su metanálisis de 14 ensayos con 4101 participantes encontraron que los AOM metilnaltrexona, naloxona y alvimopan fueron superiores a placebo para el tratamiento del estreñimiento inducido por opiáceos. Sin embargo, los números de eventos adversos fueron significativamente mayores. Las revisiones realizadas por Mehta 2016 y Siemens 2015 encontraron resultados positivos sobre la respuesta de laxación de la metilnaltrexona y otros AOM comparados con placebo, evaluados en poblaciones generales para tratar el estreñimiento inducido por opiáceos.

Study flow diagram.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
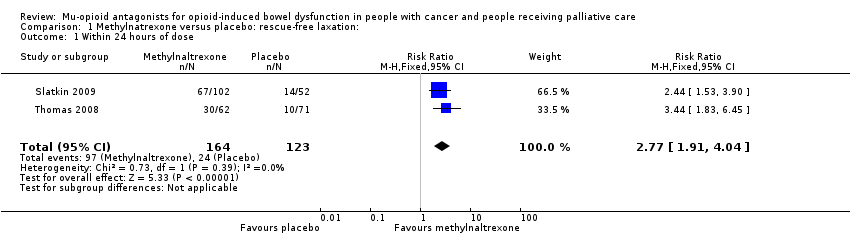
Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 1 Within 24 hours of dose.

Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 2 Within 4 hours after 4 of the 7 doses.
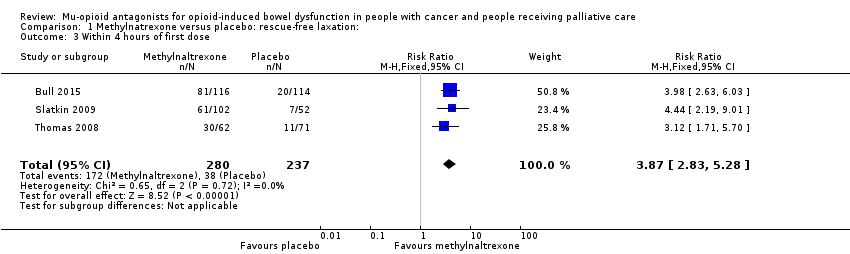
Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 3 Within 4 hours of first dose.

Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 4 Within 4 hours after 1 or 2 doses of the first 4 doses.

Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 5 Improvement in constipation distress at day 1.
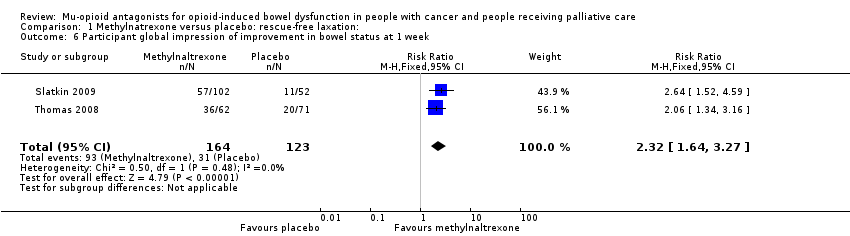
Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 6 Participant global impression of improvement in bowel status at 1 week.
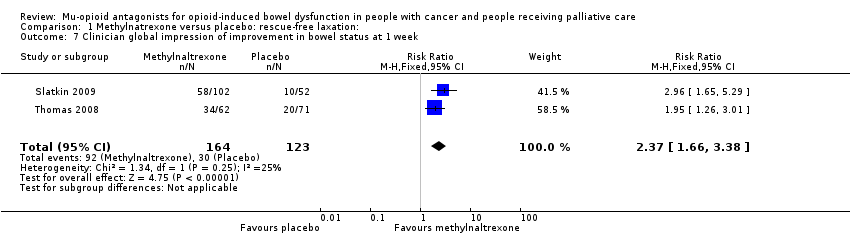
Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 7 Clinician global impression of improvement in bowel status at 1 week.

Comparison 2 Methylnaltrexone versus placebo: serious adverse event, Outcome 1 Serious adverse event.

Comparison 3 Methylnaltrexone versus placebo: adverse event, Outcome 1 Adverse events.

Comparison 3 Methylnaltrexone versus placebo: adverse event, Outcome 2 Dropouts due to adverse event.

Comparison 4 Oxycodone/naloxone prolonged‐release tablets versus oxycodone prolonged‐release: adverse event, Outcome 1 Adverse events.
| Naldemedine compared to placebo for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Settings: cancer care Intervention: naldemedine Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Naldemedine | |||||
| Laxation response within 24 hours of dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14a | 375 per 1000 | 724 per 1000 | RR 1.93 (1.36 to 2.74) NNTB 2.88 (2.04 to 4.92) | 225 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: opioid withdrawalc | — | — | 0.1 mg: MD ‐0.13 (‐0.57 to 0.31); 0.2 mg: MD ‐0.40 (‐0.87 to 0.07); 0.4 mg: MD ‐0.02 (‐0.45 to 0.41) | 225 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: pain intensity | — | — | — | — | — | Not reported |
| Serious adverse eventsa | — | — | 5 SAEs occurred, all in naldemedine group. | 225 (1 study) | ⊕⊕⊝⊝ | — |
| Adverse eventsa | 518 per 1000 | 704 per 1000 (539 to 927) | RR 1.36 (1.04 to 1.79) | 225 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; NNTB: number needed to treat for an additional beneficial outcome; RR: risk ratio; SAE: serious adverse events. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by clinician or self‐report and in the case of adverse events using severity grades according to the Common Terminology Criteria for Adverse Events. | ||||||
| Lower‐dose naldemedine compared to higher‐dose naldemedine for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Setting: cancer care Intervention: lower dose naldemedine 0.1 mg daily Comparison: higher dose naldemedine 0.2 mg or 0.4 mg daily | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Higher dose 0.2 mg/0.4 mg daily | Lower dose 0.1 mg daily | |||||
| Laxation response within 24 hours of dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14a | 0.1 mg vs 0.2 mg: 776 per 1000 0.1 mg vs 0.4 mg: 821 per 1000 | 0.1 mg vs 0.2 mg: 564 per 1000 (430 to 739) 0.1 mg vs 0.4 mg: 564 per 1000 (433 to 733) | 0.1 mg vs 0.2 mg: RR 0.73 (0.55 to 0.95) 0.1 mg vs 0.4 mg: RR 0.69 (0.53 to 0.89) | 226 (1 study) 0.1 mg vs 0.2 mg: n = 113 0.1 mg vs 0.4 mg: n = 111 | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: opioid withdrawal | — | — | — | — | — | Not reported |
| Effect on analgesia: pain intensity | — | — | — | — | — | Not reported |
| Serious adverse events | — | — | — | — | — | Not reported |
| Adverse events | — | — | — | — | — | Not reported |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by self‐report. | ||||||
| Naloxone compared with placebo for cancer and people receiving palliative care with opioid‐induced bowel dysfunction | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Settings: cancer care Intervention: naloxone Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Naloxone | |||||
| Laxation response within 24 hours of a dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14 | — | — | — | — | — | Not reported |
| Effect on analgesia: opioid withdrawal | — | — | — | — | — | Not reported |
| Effect on analgesia: pain intensitya | — | — | No statistical difference in pain experienced when taking placebo or naloxone. Full data, including pre‐cross‐over results, were not provided. | 17 (1 study) | ⊕⊝⊝⊝ | — |
| Serious adverse events | — | — | — | — | — | Not reported |
| Adverse events | — | — | — | — | — | Not reported |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured using 4‐point scale (0 = no pain, 3 = severe pain). | ||||||
| Oxycodone/naloxone prolonged release tablets compared with oxycodone prolonged‐released tablets for opioid‐induced bowel dysfunction | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Settings: cancer care Intervention: oxycodone/naloxone prolonged‐release tablets Comparison: oxycodone prolonged‐released tablets | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Oxycodone | Oxycodone/naloxone | |||||
| Laxation response within 24 hours of dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14 | — | — | — | — | — | Not reported |
| Effect on analgesia: opioid withdrawalc | — | — | Intervention group: mean 6.64 (SD 5.97) comparison group: mean 7.29 (SD 4.59) at 7 days | 184 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: pain intensitya | — | — | Intervention group: mean 3.50 (SD 1.88) and comparison group: mean 3.52 (SD 1.80) at 4 weeks | 184 (1 study) | ⊕⊕⊕⊝ Moderateb | Another study, Dupoiron 2017 also found outcome to be similar between trial arms, but did not provide any data. |
| Serious adverse events | 43 per 1000 | 87 per 1000 (27 to 279) | RR 2.00 (95% CI 0.62 to 6.41) | 184 (1 study) | ⊕⊕⊝⊝ Lowb,d | — |
| Adverse events | 754 per 1000 | 815 per 1000 (709 to 935) | RR 1.08 (95% CI 0.94 to 1.24) | 234 (2 studies) | ⊕⊕⊕⊝ Moderateb | — |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; SD: standard deviation. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured using the Brief Pain Inventory‐Short Form. | ||||||
| Methylnaltrexone compared to placebo for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Setting: palliative care Intervention: methylnaltrexone Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Risk with placebo | Risk with methylnaltrexone | |||||
| Laxation response within 24 hours of dosea | 195 per 1000 | 568 per 1000 | RR 2.77 (1.91 to 4.04) | 287 | ⊕⊕⊕⊝ Moderateb | — |
| Laxation response between day 1 and day 14 (specifically within 4 hours after 4 or more of the 7 doses)a | 52 per 1000 | 517 per 1000 | RR 9.98 (4.96 to 20.09) | 305 | ⊕⊕⊕⊝ Moderate b,c | — |
| Effect on analgesia: opioid withdrawald | Study 1: day 1: MD 0.00 (‐0.46 to 0.46); day 14: MD 0.10 (‐0.63 to 0.83) Study 2: median change to day 2 = 0 in both trials arms | 236 (2 studies) | ⊕⊕⊕⊝ Moderateb | ‐ | ||
| Effect on analgesia: pain intensitye | Study 1: at 4 hours (methylnaltrexone 0.15 mg/kg: MD ‐0.76 (‐1.47 to 0.05); methylnaltrexone 0.3 mg/kg: MD ‐0.25 (‐0.91 to 0.41) Study 2: at day 1 and 14 (day 1: MD 0.20 (‐0.62 to 1.02); day 14: MD ‐0.70 (‐1.52 to 0.12) | 287 (2 studies) | ⊕⊕⊝⊝ Lowb,f | Another study, Bull 2015, found similar pain intensity experienced in trial arms, full data not provided. | ||
| Serious adverse events | 238 per 1000 | 142 per 1000 | RR 0.59 (0.38 to 0.93) | 364 | ⊕⊕⊕⊝ Moderateb | — |
| Adverse events | 700 per 1000 | 815 per 1000 | RR 1.17 (CI 0.94 to 1.45) | 518 | ⊕⊕⊝⊝ Lowb,g | Heterogeneity was substantial (74%). It was explained in sensitivity analysis by omitting the trial at a high risk of bias because of small sizes. The effect estimate was reduced. The direction of effect not changed. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by self‐report or clinician report. dMeasured using the modified Himmelsbach Opioid Withdrawal Scale. | ||||||
| Lower dose methylnaltrexone compared to higher dose for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Setting: palliative care Intervention 1: lower‐dose methylnaltrexone (study 1: 3 doses, 1 week, 1 mg; study 2: 1 dose, 0.15 mg/kg) Intervention 2: higher‐dose methylnaltrexone (study 1: 3 doses, 1 week, 5‐12.5 mg; study 2: 1 dose, 0.30 mg/kg) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Higher dose | Lower dose | |||||
| Laxation response within 24 hours of first dosea | Study 1: 609 per 1000 Study 2: 639 per 1000 | Study 1: 499 per 1000 (250 to 100) Study 2: 681 per 1000 (515 to 904) | Study 1: RR 0.82 (0.41 to 1.66) Study 2: RR 1.07 (0.81 to 1.42) | 135 (2 studies) Study 1: n = 33 Study 2: n = 102 | ⊕⊕⊝⊝ Lowb | Unable to combine study data as methylnaltrexone low and higher doses differed per trial |
| Laxation responsea | At 3 days: 706 per 1000 | At 3 days: 332 per 1000 | At 3 days: RR 0.47 (0.18 to 1.25) | 33 participants (1 study) | ⊕⊕⊝⊝ Lowb | Unable to combine study data as methylnaltrexone low and higher doses differed per trial |
| At 5 days: 688 per 1000 | At 5 days: 144 per 1000 | At 3 days: RR 0.21 (0.03 to 1.31) | ||||
| Effect on analgesia: opioid withdrawalc | — | — | MD ‐0.04 (‐0.73 to 0.65) | 102 participants (1 study) | ⊕⊕⊝⊝ Lowb | Another study,Portenoy 2008, also found outcome to be similar between trial arms, but did not provide any data |
| Effect on analgesia: pain intensityd | — | — | MD ‐0.51 (‐1.49 to 0.47) | 102 participants (1 study) | ⊕⊕⊝⊝ Lowb | Another study, Portenoy 2008, also found outcome to be similar between trial arms, but did not provide any data |
| Serious adverse event | — | — | — | — | Not reported | |
| Adverse event | Study 1: 1000 per 1000 Study 2: 800 per 1000 | Study 1: 1000 per 1000 (1000 to 1000) Study 2: 723 per 1000 (580 to 902) | Study 1: RR 1.00 (1.00 to 1.00) Study 2: RR 0.90 (0.73 to 1.13) | 135 (2 studies) Study 1: n = 33 Study 2: n = 102 | ⊕⊕⊝⊝ Lowb | Unable to combine study data as methylnaltrexone low and higher doses differed per trial |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by clinician or self‐report. bDowngraded by two levels for study limitations: one for unclear risk of bias (reporting bias) and one for small sample size (high risk of bias). cMeasured using the modified Himmelsbach Opioid Withdrawal Scale. dMeasured by participant‐rated scale 0‐10. | ||||||
| Adverse event | Naldemedine (%) | Placebo (%) |
| Diarrhoea | 67 (39) | 14 (25) |
| Decreased WBC count | 9 (5) | 3 (5) |
| Abdominal pain | 6 (4) | 0 (0) |
| Vomiting | 5 (3) | 0 (0) |
| Bone marrow failure | 3 (2) | 2 (4) |
| Decreased appetite | 6 (4) | 1 (2) |
| Nasopharyngitis | 4 (2) | 1 (2) |
| Nausea | 4 (2) | 4 (7) |
| Rash | 3 (2) | 2 (4) |
| Decreased platelet count | 3 (2) | 0 (0) |
| Decreased total protein | 7 (4) | 1 (2) |
| Glucose in urine | 4 (2) | 1 (2) |
| Abnormal haematology test | 2 (1) | 0 (0) |
| Decreased RBC count | 4 (2) | 0 (0) |
| Hypertension | 2 (1) | 0 (0) |
| Increased blood alkaline phosphatase | 4 (2) | 1 (2) |
| Increased blood lactate dehydrogenase | 2 (1) | 1 (2) |
| Increased blood pressure | 2 (1) | 0 (0) |
| Increased blood urea | 4 (2) | 1 (2) |
| Increased WBC count | 1 (2) | 2 (4) |
| Protein present in urine | 5 (3) | 0 (0) |
| Upper abdominal pain | 3 (2) | 1 (2) |
| RBC: red blood cell; WBC: white blood cell. All comparisons were not statistically significant. | ||
| Methylnaltrexone vs placeboa | |
| AEs | RR 1.07, 95% CI 0.96 to 1.19 |
| AE of abdominal pain | RR 2.15, 95% CI 1.28 to 3.62 |
| AE of nausea | RR 0.87, 95% CI 0.46 to 1.65 |
| AE of vomiting | RR 0.70, 95% CI 0.33 to 1.47 |
| aomitting trial of high risk of bias. AE: adverse event; CI: confidence intervals; RR: risk ratio. | |
| Adverse event | RR (95% CI) | I²statistic on heterogeneity |
| Abdominal pain | 2.39 (1.07 to 5.34) | 65% |
| Diarrhoea | 1.02 (0.93 to 1.11) | 51% |
| Dizziness | 4.09 (0.99 to 16.83) | 0% |
| Falls | 1.02 (0.89 to 1.16) | 84% |
| Flatulence | 2.09 (1.07 to 4.08) | 0% |
| Nausea | 0.97 (0.89 to 1.06) | 63% |
| Peripheral oedema | 1.01 (0.50 to 2.03) | 0% |
| Restlessness | 0.83 (0.32 to 2.12) | 0% |
| Somnolence | 1.00 (0.93 to 1.08) | 73% |
| Vomiting | 0.99 (0.92 to 1.08) | 67% |
| CI: confidence interval; RR: risk ratio. | ||
| Adverse event | Methylnaltrexone (%) | Placebo (%) |
| Abdominal distensiona | 1 (2) | 6 (8) |
| Abdominal tendernessa | 1 (2) | 4 (6) |
| Astheniaa | 4 (6) | 4 (6) |
| Anxietyb | 5 (4.9) | 0 (0) |
| Arthralgiab | 3 (2.9) | 1 (1.9) |
| Back painc | 9 (7.8) | 3 (2.9) |
| Confusional statec | 7 (6.0) | 9 (7.9) |
| Dehydrationa | 2 (3) | 4 (6) |
| Fatigueb | 4 (3.9) | 1 (1.9) |
| Hypotensiona | 0 (0) | 4 (6) |
| Increased body temperaturea | 5 (8) | 2 (3) |
| Lethergya | 4 (6) | 4 (6) |
| Malignant‐neoplasm progressiona | 7 (11) | 9 (13) |
| Pain exacerbationb | 8 (8) | 2 (4) |
| Rhinorrhoeab | 6 (5.9) | 1 (1) |
| Sweating increasedb | 8 (7.8) | 4 (7.7) |
| Tachycardiaa | 1 (1) | 4 (6) |
| aReported in trial by Thomas 2008. bReported in trial by Slatkin 2009. cReported in trial by Bull 2015. | ||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Within 24 hours of dose Show forest plot | 2 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.77 [1.91, 4.04] |
| 2 Within 4 hours after 4 of the 7 doses Show forest plot | 2 | 305 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.98 [4.96, 20.09] |
| 3 Within 4 hours of first dose Show forest plot | 3 | 517 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.87 [2.83, 5.28] |
| 4 Within 4 hours after 1 or 2 doses of the first 4 doses Show forest plot | 2 | 363 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.89 [4.46, 10.66] |
| 5 Improvement in constipation distress at day 1 Show forest plot | 2 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.87 [1.34, 2.59] |
| 6 Participant global impression of improvement in bowel status at 1 week Show forest plot | 2 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.32 [1.64, 3.27] |
| 7 Clinician global impression of improvement in bowel status at 1 week Show forest plot | 2 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.37 [1.66, 3.38] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Serious adverse event Show forest plot | 2 | 364 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.38, 0.93] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse events Show forest plot | 3 | 518 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.94, 1.45] |
| 2 Dropouts due to adverse event Show forest plot | 2 | 363 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.54, 2.76] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse events Show forest plot | 2 | 234 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.94, 1.24] |