Botulinum toxin type B for cervical dystonia
Abstract
Background
This is an update of a Cochrane review first published in 2004, and previously updated in 2009 (no change in conclusions). Cervical dystonia is a frequent and disabling disorder characterised by painful involuntary head posturing. Botulinum toxin type A (BtA) is usually considered the first line therapy for this condition, although botulinum toxin type B (BtB) is an alternative option.
Objectives
To compare the efficacy, safety and tolerability of botulinum toxin type B (BtB) versus placebo in people with cervical dystonia.
Search methods
We identified studies for inclusion in the review using the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, EMBASE, reference lists of articles and conference proceedings, last run in October 2015. We ran the search from 1977 to 2015. The search was unrestricted by language.
Selection criteria
Double‐blind, parallel, randomised, placebo‐controlled trials (RCTs) of BtB versus placebo in adults with cervical dystonia.
Data collection and analysis
Two independent authors assessed records, selected included studies, extracted data using a paper pro forma and evaluated the risk of bias. We resolved disagreements by consensus or by consulting a third author. We performed one meta‐analysis for the comparison BtB versus placebo. We used random‐effects models when there was heterogeneity and fixed‐effect models when there was no heterogeneity. In addition, we performed pre‐specified subgroup analyses according to BtB doses and BtA previous clinical responsiveness. The primary efficacy outcome was overall improvement on any validated symptomatic rating scale. The primary safety outcome was the number of participants with any adverse event.
Main results
We included four RCTs of moderate overall methodological quality, including 441 participants with cervical dystonia. Three studies excluded participants known to have poorer response to Bt treatment, therefore including an enriched population with a higher probability of benefiting from Bt treatment. None of the trials were independently funded. All RCTs evaluated the effect of a single Bt treatment session using doses between 2500 U and 10,000 U. BtB was associated with an improvement of 14.7% (95% CI 9.8% to 19.5) in the patients' baseline clinical status as assessed by investigators, with reduction of 6.8 points in the Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS‐total score) at week 4 after injection (95% CI 4.54 to 9.01). Mean difference (MD) in TWSTRS‐pain score at week 4 was 2.20 (95% CI 1.25 to 3.15). Overall, both participants and clinicians reported an improvement of subjective clinical status. There were no differences between groups in the withdrawals rate due to adverse events or in the proportion of participants with adverse events. However, BtB‐treated patients had a 7.65 (95% CI 2.75 to 21.32) and a 6.78 (95% CI 2.42 to 19.05) increased risk of treatment‐related dry mouth and dysphagia, respectively. Statistical heterogeneity between studies was low to moderate for most outcomes. All tested dosages were efficacious against placebo without clear‐cut evidence of a dose‐response gradient. However, duration of effect (time until return to baseline TWSTRS‐total score) and risk of dry mouth and dysphagia were greater in the subgroup of participants treated with higher BtB doses. Subgroup analysis showed a higher improvement with BtB among BtA‐non‐responsive participants, although there were no differences in the effect size between the BtA‐responsive and non‐responsive subgroups.
Authors' conclusions
A single BtB‐treatment session is associated with a significant and clinically relevant reduction of cervical dystonia impairment including severity, disability and pain, and is well tolerated, when compared with placebo. However, BtB‐treated patients are at an increased risk of dry mouth and dysphagia. There are no data from RCTs evaluating the effectiveness and safety of repeated BtB injection cycles. There are no RCT data to allow us to draw definitive conclusions on the optimal treatment intervals and doses, usefulness of guidance techniques for injection, and impact on quality of life.
PICO
Plain language summary
Botulinum toxin for people with involuntary posturing of the head
Undesired, uncontrollable, and often painful placement of the head, a disease called cervical dystonia or spasmodic torticollis, is a relatively uncommon condition (affecting 57 to 280 people per million) that can be very disabling and can compromise quality of life. Mostly the cause is unknown and no cure exists. As this is typically a chronic disease, it requires long‐term treatment.
Botulinum toxin (Bt) is a natural powerful chemical produced by a bacterium called Clostridium botulinum, that can cause severe paralysis in animals and humans. It can also be used to treat many conditions, in particular those with involuntary muscle contractions, such as cervical dystonia, by delivering intra‐muscular Bt injections. There are different types of Bt, not all available for therapeutic purposes. Bt type A (BtA) is normally the first‐used treatment in cervical dystonia. However, not all patients respond to BtA injections, and in such situations, treatment with Bt type B (BtB) is of special interest.
This update of a previous Cochrane review aimed to assess the effectiveness (reduction in severity, disability and pain) and safety of BtB in cervical dystonia, in comparison to placebo (a pretend medicine).
We performed a literature search in October 2015 for studies that compared BtB with placebo in people with cervical dystonia.
We found four studies comparing a single BtB treatment session with placebo, including 441 participants in total.
There was moderate‐quality evidence that a single BtB treatment session is efficacious when compared to placebo, improving cervical dystonia symptoms by between 10% and 20%. This clinical benefit applies to people with both a poor and a good response to previous BtA treatments. Both physicians and patients evaluated BtB positively. BtB‐treated patients are, however, at an increased risk of dry mouth and swallowing difficulties.
Further studies are needed to establish the long‐term clinical benefit of BtB treatment, including its impact on quality of life, to evaluate the best treatment intervals and doses, as well as to find out which people with cervical dystonia would benefit the most from BtB treatment.
Authors' conclusions
Summary of findings
| Botulinum Neurotoxin B compared to placebo for cervical dystonia | ||||||
| Patient or population: adults with cervical dystonia | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Botulinum Neurotoxin B | |||||
| Overall cervical dystonia improvement as assessed with TWSTRS: change from baseline to week 4 (range, 0 to 85; more is worst) | ‐7 | ‐7 | The mean change from baseline to week 4 in the BtB group was 6.78 TWSTRS units higher (4.54 higher to 9.01 higher) compared to the placebo group | 316 | ⊕⊕⊕⊝ | |
| Proportion of withdrawals due to adverse events | Study population | RR 0.88 | 440 | ⊕⊕⊝⊝ | ||
| 14 per 1000 | 13 per 1000 | |||||
| Cervical dystonia associated pain: change from baseline to week 4 as assessed with TWSTRS (range, 0 to 20; more is worst) | ‐7 | ‐7 | The mean change from baseline to week 4 in the BtB group was 2.41 TWSTRS units higher (0.82 higher to 4.01 higher) compared to the placebo group | 207 | ⊕⊕⊝⊝ | |
| Subjective change as assessed by the patient at week 4 | ‐7 | ‐7 | The mean change at week 4 in the BtB group was 0.86 standard deviations higher (0.61 higher to 1.1 higher) compared to the placebo group | 316 | ⊕⊕⊕⊕ | |
| Proportion of participants with adverse events | Study population | RR 1.09 | 186 | ⊕⊝⊝⊝ | ||
| 838 per 1000 | 930 per 1000 | |||||
| Adverse events: dry mouth | Study population | RR 7.65 | 438 | ⊕⊕⊕⊕ | ||
| 22 per 1000 | 168 per 1000 | |||||
| Adverse events: dysphagia | Study population | RR 6.78 | 438 | ⊕⊕⊕⊕ | ||
| 22 per 1000 | 148 per 1000 | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Two of 3 studies enrolled an enriched population; none of the included studies had independent funding; blinding of outcome assessment was unclear in all studies 2Three of 4 studies enrolled an enriched population; none of the studies had a clearly stated independent funding; blinding of outcome assessment was unclear in all studies; two out of 4 had an unclear random sequence generation 3The total number of participants included was less than the number generated by a conventional sample size calculation for a single adequately powered trial 4I‐squared of 58% and small overlap between confidence intervals 5Both studies had an enriched population and non‐independent funding; blinding of outcome assessment was unclear in all studies 6I‐squared of 45% and there is a wide variance of point estimates between studies 7 Data were only available as the difference between the BtB and placebo groups | ||||||
Background
This review is an update of a previously published review in the Cochrane Database of Systematic Reviews 2004, Issue 4 (Costa 2004), evaluating the efficacy and safety of Botulinum toxin type B versus placebo in the treatment of cervical dystonia.
Description of the condition
See Table 1 for glossary of terms.
| Term | Definition |
| BtA‐non‐responsive | People who do not experience the expected benefit from treatment with botulinum toxin type A |
| Cervical dystonia or spasmodic toricollis | It is a common movement disorder in which people have abnormal movements or postures of the head and neck that they cannot control. It is frequently accompanied by social embarrassment and pain. |
| Chemodenervation | It is the process by which botulinum toxin causes muscular paralysis. Altought all the anatomical elements necessary for muscular control are intact (i.e. nerve, synapse and muscle), there is a chemical process that disables the transmission of the transmission of the electrical signal from the nerve to the muscle. |
| Dysphagia | A discomfort or difficulty when swallowing. |
| Electromyography | It is an exam that displays the electrical activity of muscles using pieces of metal attached to the skin or inserted into the muscle. |
| Non‐naive | People who have been treated in the past with botulinum toxin. |
| Voluntary action | Movements that we are able to control, start and stop when we want to. |
Dystonia is the third most common movement disorder, after Parkinson’s disease and essential tremor, with an overall prevalence of 164 per million (Steeves 2012). Dystonia syndromes are a group of disabling, painful disorders characterised by involuntary sustained or intermittent muscle contractions causing abnormal, often repetitive, movements or postures of the face, neck, trunk or limbs (Albanese 2013). Dystonic movements are typically patterned or twisting, and are often initiated or worsened by voluntary action (Albanese 2013). These neurological disorders can be classified based on topographic distribution, including focal dystonia (one body region, e.g. cervical dystonia and blepharospasm), segmental dystonia (two or more adjacent regions, e.g. hemifacial spasm), multifocal dystonia (two or more nonadjacent regions), hemidystonia (ipsilateral regions) and generalised dystonia (trunk and two or more other regions) (Albanese 2013; Tarsy 2006).
Focal dystonia is a highly disabling movement disorder, with serious functional and social impairment. Close to half of the patient population quits work by the age of 40 or retires early due to dystonia, and 10 years later, only 25% of patients are working compared to 62% of the general population (Zoons 2012). Moreover, health‐related quality of life is significantly diminished, mainly attributable to depression and anxiety, with scores comparable to people with multiple sclerosis, Parkinson’s disease or stroke (Zoons 2012).
Cervical dystonia, also called spasmodic torticollis, is the most common form of adult‐onset focal dystonia, with estimates from population studies ranging from 57 per million in Europe (ESDE 2000) to as high as 280 per million in the USA (Jankovic 2006). It typically has its onset in the fifth decade (Albanese 2013), and affects more women than men (Defazio 2013). This condition is characterised by abnormal movements of head, neck, and shoulder, resulting in posturing of the head away from its normal central position (Foltz 1959). It may present predominantly with sustained abnormal posture, spasm, jerks, tremor, or a combination of these features. Neck or shoulder pain, or both, occur in more than 70% of patients (Chan 1991; Tarsy 2006).
Cervical dystonia can be classified according to the dominant head position, with the most common type involving horizontal turning, the so‐called rotatory (or simple) torticollis (Chan 1991 ; Albanese 2013). Other common patterns include laterocollis (tilt to one side), retrocollis (tilt upwards resulting in neck extension) and anterocollis (tilt downwards resulting in neck flexion). Complex torticollis, a combination of these abnormal patterns, is frequently found in clinical practice.
The aetiology of most forms of dystonia is still not fully understood, with the exception of early‐onset dystonia, for which a hereditary aetiology is common (Balint 2015). In most cases of focal adult‐onset dystonia, such as cervical dystonia, the pathophysiology is generally considered to result from inhibition of the central nervous system (CNS) at multiple levels (Hallett 1998) resulting in abnormal sensorimotor integration. Cervical dystonia can also be secondary to brain injury, infections of the CNS, drugs (such as levodopa or antipsychotics), toxics, vascular or neoplastic disorders, and may also be psychogenic (i.e. functional) (Albanese 2013). Although most cases of cervical dystonia are currently classified as idiopathic, it should be observed that some may come to be reclassified as inherited, since new gene discoveries are under investigation (Albanese 2013; Balint 2015).
The natural course of cervical dystonia remains unclear though it typically worsens over time. The clinical presentation is seldom progressive to generalised dystonia, although it often extends to contiguous body regions. For most patients, cervical dystonia is a life‐long disorder, with only about 10% undergoing spontaneous remissions (Jahnanshani 1990).
To date, no curative or disease‐modifying treatments are available for cervical dystonia.
Description of the intervention
Botulinum toxin (Bt) is a powerful biological toxin produced by Clostridium botulinum. The active form of botulinum toxin is a di‐chain polypeptide composed of two chains: a heavy chain (100 kDa) and a light chain (50 kDa), and by associating with certain auxiliary proteins (haemagglutinins and non‐haemagglutinins), the toxin forms a non‐covalent multimeric complex of variable size (Simpson 2004). The nontoxic proteins aid the formation of neutralising antibodies, though beyond this their role is unclear (Frevert 2010). Bt binds to peripheral cholinergic nerve terminals of the neuromuscular junction as well as sympathetic ganglionic, parasympathetic ganglionic and postganglionic terminals (Simpson 2004). Bt, after binding to an acceptor protein, is endocytosed at the presynaptic membrane of acetylcholine nerve terminals (Pellizzari 1999). By action of the N‐terminal on the heavy ‐chain, a pore is formed on the endocytic membrane, which permits the release of the light chain into the cytosol. This light chain, which is a zinc protease, performs the key‐action of the botulinum toxin, by cleaving soluble N‐ethylmaleimidesensitive factor attachment receptor proteins (SNARE proteins) (Pellizzari 1999).
SNAREs are docking proteins for acetylcholine vesicles that allow for the release of acetylcholine into the synaptic cleft (Pellizzari 1999). The overall effect of Bt is a local chemodenervation by the temporary blockade of acetylcholine release at cholinergic synapses. Temporary synapses are consequently formed via the process of axonal sprouting (Duchen 1971; Holland 1981; Juzans 1996).
There are seven immunologically distinct botulinum toxin serotypes (labelled A to G). These different Bt serotypes cleave specific SNARE proteins. Serotype A cleaves SNARE protein SNAP 25 located on the inner membrane, and serotype B targets synaptobrevin located on the vesicular membrane (Pellizzari 1999).
Botulinum toxin is injected into the muscles involved in dystonia, with or without guidance by either electromyography (EMG) or ultrasound. As a general rule, the number of muscles injected and the number of injection sites per muscle are tailored to the severity of the case in question and the mass of the muscle, respectively. Within roughly three months after injection of botulinum toxin into skeletal muscle, the nerve terminal resumes exocytosis, and the muscle returns to its baseline clinical function, showing a wearing off response from the Bt injection (Jankovic 2004). Eventually, the muscle paralysis subsides, and this is associated with the formation of new sprouts capable of neurotransmission. Over time, synaptic activity resumes in the original nerve terminals, leading to sprout regression (de Paiva 1999).
Currently there are two commercially available Bt serotypes (BtA and BtB). The following products are commonly available (three BtA and one BtB): onabotulinumtoxinA (Botox®, Allergan Inc., Irvine, CA, USA), abobotulinumtoxinA (Dysport®/Reloxin®/Azzalure®, Ipsen Pharma, Boulogne Billancourt, France), incobotulinumtoxinA (Xeomin®/Bocoture® Merz GmbH, Frankfurt, Germany), and rimabotulinumtoxinB (Myobloc®/Neurobloc®, Solstice Neurosciences Inc., Louisville, KY, USA). Other BtA formulations are available in more restricted markets and are yet to receive a generic name: Prosigne®/Lantox® (Lanzhou Institute of Biological Products, China), PurTox® (Mentor Worldwide LLC, Santa Barbara, CA, USA), and Neuronox® (Medy‐Tox Inc, South Korea) (Walker 2014).
How the intervention might work
The therapeutic potential of all Bt serotypes derives from their ability to inhibit the release of acetylcholine from the presynaptic nerve terminal into the synaptic cleft, causing local chemodenervation (Jankovic 2004). In addition to this, recent research has also suggested that Bt is active at multiple levels, namely sensory nerve terminals, and muscle spindles, which leads to a reduction in sensory input and fewer muscle contractions (Filippi 1993; Matak 2014; Rosales 1996; Rosales 2010).
It has also been suggested that cortical reorganisation may result from changes in the spinal cord, brainstem and central nervous pathways (Palomar 2012). Animal research has shown the presence of supra‐therapeutic levels of Bt by way of retrograde axonal transport and penetration of the central nervous system (Antonucci 2008 ; Boroff 1975). However, Bt has not been shown to penetrate the blood‐brain barrier in humans.
Until recently, SNARE proteins were considered the only target molecules of Bt. Thus, it was widely accepted that the therapeutic and toxic actions of Bt were exclusively mediated by SNARE cleavage preventing the release of synaptic neurotransmitters. However, recent studies have suggested that a number of Bt actions might not be mediated by SNARE cleavage, specifically regarding neuroexocytosis, cell cycle and apoptosis, neuritogenesis and gene expression (Matak 2015). The existence of unknown Bt molecular targets and modulation of unknown signalling pathways is a possibility that may prove to be pharmacologically relevant.
Why it is important to do this review
BtA is the toxin serotype that has been most intensively studied and approved for the treatment of the large number of focal dystonias. BtA is considered the first line therapy for cervical dystonia and has proven to be effective in the symptomatic management of this condition (Albanese 2013). However, not all patients have an adequate clinical response. Primary non‐response to botulinum toxin is seen in cases where the first and subsequent treatment cycles do not elicit a response. Cases of secondary non‐response, however, respond to initial treatment, but over the course of multiple treatment cycles, this effect wanes and is eventually lost. Secondary non‐response is partially explained by the formation of neutralising antibodies, though it is worth noting that there are cases of secondary non‐responders without positive antibody titers (Hanna 1998; Lange 2009) as well as cases with positive titers but with an adequate sensitivity to Bt (Brin 2008; Muller 2009). An estimated 4% to 20% of patients develop neutralising antibodies to the toxin (Brashear 2008; Fabbri 2015), and if secondary non‐responsiveness occurs, it is partially related to the protein load, with higher protein load per dose generating higher antibody titers (Benecke 2012; Frevert 2010).
When clinical non‐response occurs, other Bt serotypes are important treatment options for cervical dystonia (Cullis 2000; Eleopra 1997; Greene 1993). At the present time, BtB is the only approved non‐BtA formulation available for the treatment of cervical dystonia in the United States and in the European Union.
A Cochrane systematic review previously assessed the efficacy and safety of BtB in comparison to placebo in people with cervical dystonia (Costa 2004). This is the second update of that review, having been previously updated in 2009 with no changes to conclusions. The original review concluded that a single injection of BtB was efficacious in comparison to placebo in the treatment of cervical dystonia, with a greater benefit for participants who were BtA non‐responders when compared to BtA‐responders, as assessed by subgroup analysis. Three studies were included in the original review with a total number of 308 participants enrolled.
Since the release of the original review, a new trial has been published (Kaji 2013). Furthermore, Cochrane’s criteria for evaluating studies' risk of bias and evidence quality have evolved and been updated. Therefore, the authors consider it important to update this review.
Objectives
To compare the efficacy, safety and tolerability of botulinum toxin type B (BtB) versus placebo in people with cervical dystonia.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs), blinded, single or multiple dose, parallel‐designed, of any duration, assessing the efficacy or safety, or both, of BtB treatment versus placebo in people with cervical dystonia were eligible for inclusion in this review. We excluded trials in which allocation was not adequately concealed. We excluded non‐parallel study designs, namely cross‐over trials, from this updated version of the review, due to uncertainty about whether this type of study design was appropriate to study people with cervical dystonia, as well as methodological concerns with regards to detection and performance bias.
Types of participants
Adults (i.e. ≥ 18 years of age), in any setting, with a clinical diagnosis made by any physician, specialist or otherwise, of idiopathic cervical dystonia. We allowed trials enrolling participants with any form of cervical dystonia, and additional or more widespread dystonias, for inclusion. Participants could have had prior exposure to BtA or BtB, and could be taking any concomitant medications if on stable regimens.
There were no restrictions regarding the number of participants recruited to trials, or the number of recruitment centres.
Types of interventions
Intramuscular injections of BtB compared to placebo. We allowed all administration schedules and injection techniques, performed with or without guidance by either EMG or echography.
Types of outcome measures
Primary outcomes
Primary efficacy outcome
Overall improvement on any validated symptomatic rating scale, such as Cervical Dystonia Severity Scale (CDSS), Tsui scale, and Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS), measured between weeks 3 and 6.
Primary safety outcome
Number of participants with any adverse event, measured at any point during study follow up.
Secondary outcomes
-
Change in subjective evaluation of clinical status evaluated by both patients and clinicians, as assessed with validated assessment tools such as Patient Subjective Assessment of Change, Patient Global Assessment of Improvement, Patient Evaluation of Global Response (PEGR), Patient and Physician Global Assessment of Change, Investigator Global Assessment of Efficacy (IGAE), and Physician Global Assessment of Change (PGAC), and Visual analogue scale (VAS) for symptom severity, measured between weeks 3 and 6.
-
Changes in pain scores, as assessed with validated assessment tools such as Patient Assessment of Pain, TWSTRS Pain sub‐scale score, and VAS Pain score, measured between weeks 3 and 6.
-
Changes in quality‐ of‐ life assessments, as assessed with validated assessment tools such as Short Form 36 (SF‐36) Quality‐of‐Life questionnaire, measured at any point during study follow up.
-
Number of withdrawals due to adverse events, including adverse events caused by the intervention (type A or type B, or both, adverse drug reactions (ADRs)), and failure of therapy (type F ADRs), measured at any point during study follow up.
-
Number of participants with adverse events of special interest, such as dry mouth, neck weakness, dysphagia, pain at the injection site, voice change, and systemic complaints (e.g. diffuse muscle weakness, malaise, dizziness and headache), measured at any point during study follow up.
-
Duration of effect, assessed by the number of days until need for reinjection or effect waning.
Search methods for identification of studies
For this update, we expanded the search strategy to capture all the search terms for BtB formulations that were available at the time of the search. We designed the search strategy to include other botulinum toxin formulations and other dystonic disorders that were also under revision by this author team.
Electronic searches
In October 2015 we searched the following databases.
-
Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2015, Issue 11);
-
MEDLINE (1977 to October 2015)
-
EMBASE (1977 to October 2015)
We assessed non‐English language papers equally, translated them as necessary and evaluated them for inclusion.
For the identification of studies considered for inclusion in this review, we developed detailed search strategies for each database searched. Please see Appendix 1 for the CENTRAL search strategy, Appendix 2 for the MEDLINE search strategy, and Appendix 3 for the EMBASE search strategy.
We ran the search for the original version of this review in June 2003, based on the search strategy developed for the Movement Disorders Group to identify all papers from 1977, the first year botulinum toxin was used therapeutically in any condition.
Searching other resources
The search strategy also included:
-
searches through reference lists of located trials and review articles concerning botulinum toxin;
-
handsearch of abstracts of international congresses relevant in the fields of movement disorders and botulinum toxins, i.e. American Academy of Neurology, Movement Disorders Society, International Association of Parkinsonism and Related Disorders, and International Neurotoxin Association (1985 to October 2015);
-
personal communication with other researchers in the field;
-
contact with drug manufacturers;
-
whenever necessary, we contacted authors of published trials for further information and unpublished data.
Data collection and analysis
Selection of studies
Two review authors independently screened the studies identified by the search strategy, reading each of the titles and abstracts, excluding studies that were not applicable. If there was no abstract, we opted to retrieve the full text of the study in question.
Two review authors then independently assessed the full‐text articles to see if the studies fulfilled the inclusion criteria. We resolved disagreements by discussion or, if necessary, reached consensus with the participation of a third author.
Data extraction and management
Two authors independently extracted study data onto standardised forms, after which we cross‐checked the forms for accuracy. We resolved disagreements by discussion or, if necessary, arbitration by a third author. We extracted the following data from each study.
-
Participants: inclusion and exclusion criteria, demographics and clinical baseline characteristics, number and reasons for withdrawals, exclusions and loss to follow‐up, if any.
-
Interventions: full description of intervention, duration of treatment period and follow‐up, providers, and co‐interventions, if any.
-
Comparisons: number of randomised participants to each arm, compliance and dropouts, reasons for dropouts, and ability to perform an intention‐to‐treat analysis.
-
Outcomes: definition of outcomes, use of validated measurement tools, time‐point measurements, change from baseline or post‐interventional measures, and missing outcomes, if any.
-
Study design: interventional, randomised, controlled, double‐blind.
Assessment of risk of bias in included studies
We used the recommended Cochrane tool for assessing risk of bias in this review (Higgins 2011a). We added one new criteria, in addition to the seven specific domains of this tool (i.e. random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting and other bias). This extra domain, 'enriched population', was created to evaluate bias originating from either the preferential enrolment of known positive responders to BtA (high risk of bias being arbitrarily defined as >30% of participants being non‐naive to Bt) or the exclusion of known poor responders to BtA.
We also divided the domain 'blinding of outcome assessment' into two categories: subjective and objective assessment. Because the clinical effect of botulinum toxin treatment is easily perceived by most patients, Bt non‐naive patients are likely to recognise the presence or absence of clinical effects, or frequent adverse events, or both, effectively revealing the respective allocation arm. Thus, whenever a study population consisted primarily of non‐naive participants, we took this potential source of bias for subjective outcome assessment into account.
Two independent review authors performed critical assessments for each domain of the risk of bias tool. We resolved disagreements by discussion and, if necessary, reached consensus with the participation of a third author.
Measures of treatment effect
We compared disease symptoms at baseline to disease symptoms in weeks 3 to 6 between BtB and placebo arms. Whenever possible, we extracted continuous outcomes. Where we extracted adequate data from the studies, we pooled these data and used them for comparison. We opted to preferentially use mean differences. When studies investigating the same outcome used different validated rating scales, we calculated a standardised mean difference (SMD). For interpretation of effect sizes with SMDs, we used a rule of thumb to define absence of effect (SMD < 0.2), a small effect (SMD = 0.2 to 0.49), a moderate effect (SMD = 0.5 to 0.79), or a large effect (SMD ≥ 0.80) (Cohen 1988). If necessary for comparison, we dichotomised rating scales using each study author's own criteria for improvement or no improvement. If these criteria were not described, we defined 'improvement' as any beneficial change from baseline, and 'no improvement' as lack of improvement or any deterioration from baseline.
We compared the proportion of participants with adverse events between treatment arms using risk ratios, and performed further analysis for adverse events of special interest reported in the trials.
We planned a meta‐analysis for the duration of effect of BtB formulations (using time‐to‐event data). Where there were no data that could be combined and subjected to such analysis, we undertook a narrative approach to result synthesis.
Unit of analysis issues
Studies with multiple treatment groups
Whenever the included studies had multiple BtB arms with different dosages versus placebo, we combined all the BtB groups to create a single pair‐wise comparison, using the Review Manager (RevMan) 5.3 Calculator (RevMan 2014). This avoided the duplication of the placebo group that would happen if multiple comparisons (e.g. BtB dose 1 versus placebo; BtB dose 2 versus placebo) were included in the meta‐analysis, as well as the loss of information if one dosage group was chosen in detriment of the others. We analysed the importance of dosage in a subgroup analysis.
Dealing with missing data
Where insufficient data were presented in the study report to combine information into the meta‐analysis, we derived the mean value and standard deviation of the outcome measurements using the methods suggested in the Cochrane Handbook, Section 16.1 (Higgins 2011b).
We used the generic inverse variance method when an effect estimate and a valid measure of uncertainty (e.g. standard error (SE), 95% confidence interval (CI) or exact P value) were reported in the study. When two reported groups needed to be combined into a single group, we calculated a pooled standard deviation (SD) estimate (Abrams 2005; Follmann 1992) and used it as the standard deviation for that group.
When change from baseline SD was not reported or not possible to extract, we used alternative methods for imputing SD, namely, those suggested by Cochrane (Cochrane Handbook, Section 16.1.3.2). If a study in this review uses the same scale, degree of error and time period measurements, and SD was available, SD was appropriated from that study (Higgins 2011b). Where not possible to use the aforementioned methods, we used a pooled SD estimate (Abrams 2005; Follmann 1992) instead, assuming a lower degree of accuracy.
Assessment of heterogeneity
Heterogeneity between trial results was tested using a standard chi‐squared test and an I2 statistic was performed to quantify inconsistency across studies (Higgins 2003). When considerable heterogeneity was present (i.e. P < 0.1 or I2 > 50%), we explored the possible causes of heterogeneity by conducting non‐planned subgroup analyses. Where heterogeneity could not readily be explained by the planned and non‐planned exploratory analyses, we incorporated it into a random‐effects (RE) meta‐analysis model.
Assessment of reporting biases
We assessed publication bias through visual inspection of funnel plot asymmetry (Sterne 2001) and Peters’ regression tests (Peters 2006), if more than 10 studies per outcome were available (Sterne 2011).
Data synthesis
We performed statistical analysis with Review Manager (RevMan) version 5.3 (RevMan 2014).
We pooled effect measures by applying the Mantel‐Haenszel method for dichotomous outcomes, and applying the inverse‐variance method for continuous and generalised inverse variance outcomes. We conducted data synthesis using a fixed‐effect model unless considerable heterogeneity was detected, in which case we opted to apply the random‐effects model. We presented all results with 95% CI.
We calculated the number of participants needed to treat for an additional beneficial outcome (NNTB) and for an additional harmful outcome (NNTH) from meta‐analysis estimates, rather than treating data as if they came from a single trial, as the latter approach is more prone to bias, especially when there are significant imbalances between groups within one or more trials in the meta‐analysis (Altman 2002). However, caution is needed in interpreting these findings since they may be misleading because of variation in the event rates in each trial, differences in the outcomes considered, effects of secular trends on disease risk, and differences in clinical setting (Smeeth 1999).
Where data from the study reports could not be combined into a meta‐analysis, we presented a narrative report of result synthesis in the review text.
Subgroup analysis and investigation of heterogeneity
We planned subgroup analysis for the following areas, independently of the presence or not of significant heterogeneity: high (≥ 10000 U) versus medium (> 2500 U to < 10000 U) versus low total treatment dose (≤ 2500 U), all defined arbitrarily; EMG‐guided versus non‐EMG‐ guided injection; and BtA‐responsive versus BtA‐non‐responsive
Results
Description of studies
We included one new study in this update (Kaji 2013, n = 130), adding to the three studies already included in the original review (Brashear 1999; Brin 1999; Lew 1997).
Overall, we included four parallel‐designed studies comparing BtB (different total treatment doses) with placebo in this update, with a total of 441 participants with cervical dystonia.
Results of the search
The search, last run on 26 October 2015, returned 1667 records (189 through CENTRAL, 436 though MEDLINE, 1042 through EMBASE), resulting in 1450 records after removing all duplicates. After title and abstract screening we retrieved twelve full articles. Of these, we excluded a further eight studies, one due to examining the wrong intervention (AN072‐008 1995) and seven due to having the wrong study design (Chinnapongse 2010; Cullis 2000; Dressler 2005; Jacob 2003; Jankovic 2006; Lew 2002; Truong 1997). We did not retrieve any unpublished trials.
We once again included the three studies that had been included in previous versions of this review (Brashear 1999; Brin 1999; Lew 1997) and we included one new study in both the qualitative and quantitative syntheses (Kaji 2013).
See Figure 1 for the Study flow diagram.
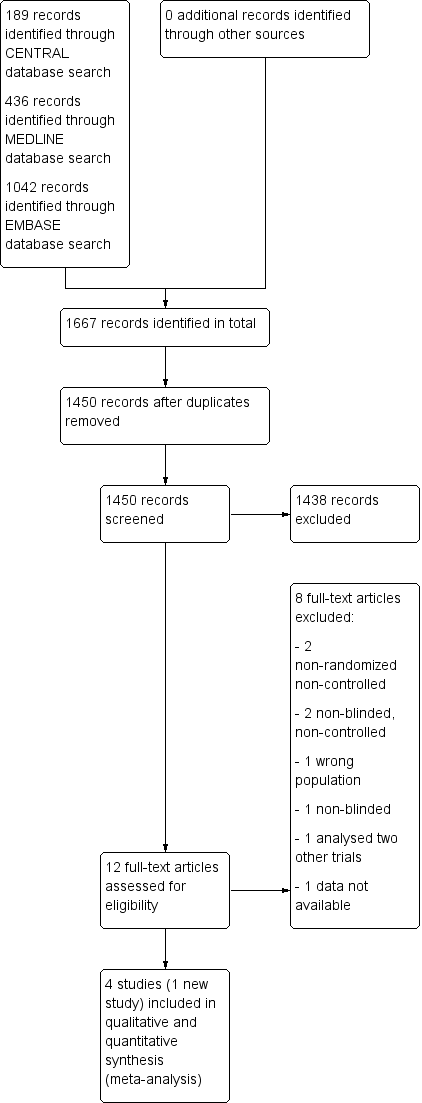
Study flow diagram
Included studies
We have listed all the included studies in this review in the 'Characteristics of included studies' table.
Study participants
The four included studies enrolled a total of 441 adult (aged above 18 years old) participants (57.6% of whom were female (n = 254)). The mean age was 52.9 years across all studies except Lew 1997, where age distribution was not available. Trial size varied from 77 to 133 participants, with all but one study (Brin 1999) enrolling above 100 participants. Three of the included RCTs were multi‐centre studies conducted in the US and published in the late 1990s (Brashear 1999; Brin 1999; Lew 1997), and one was a more recent trial conducted in Japan (Kaji 2013). All trials tested only one injection treatment session and followed participants for 16 weeks.
With respect to baseline characteristics, all studies required participants to have had cervical dystonia for at least one year. The mean duration of cervical dystonia was 7.21 years in the Kaji 2013 trial (ranging from 5.58 years in the 5000 U BtB treatment arm to 8.53 years in the 2500 U BtB treatment arm); the remaining studies did not present the mean duration of disease in the population enrolled. The baseline mean cervical dystonia impairment was moderate to severe in all participants, though well matched between study arms, with TWSTRS total scores ranging from 43.4 to 52.0, and TWSTRS Severity scores from 19.6 to 22.4 (baseline scores not available in Lew 1997, and sub‐scores not reported in Kaji 2013).
Participants’ previous Bt response varied across trials. Lew 1997 and Kaji 2013 trials allowed both BtA‐responsive and BtA‐non‐responsive participants to enter the study; the Brin 1999 trial allowed only BtA‐non‐responders; and the Brashear 1999 trial included only BtA‐responsive participants. Only the Kaji 2013 trial enrolled BtA‐naïve participants (25.4% of total population); in all studies, time since last injection before study entry had to be superior to 16 weeks. All trials except Kaji 2013 excluded clinical forms of cervical dystonia known to perform poorly to botulinum toxin injections, such as pure anterocollis or retrocollis.
The number of withdrawals was small and balanced in all trials. Reasons for withdrawals were given, even though Kaji 2013 did not describe the reasons for each participant withdrawal in detail.
Overall, within studies, participants were well matched between BtB and placebo arms.
Study design and interventions
All studies were designed to evaluate only a single treatment session. Total BtB dosages tested varied between studies. All trials assessed the effect of 10,000 U of BtB (a dose that we have arbitrarily classified as being a high dose). Three studies (Brashear 1999; Kaji 2013; Lew 1997) also included a group treated with 5000 U of BtB (a dose that we have arbitrarily classified as being a medium dose), and two studies (Kaji 2013; Lew 1997) further included a group treated with 2500 U (a dose that we have arbitrarily classified as being a low dose).
Techniques and schema of BtB administration did not vary considerably among the studies. In all the trials, BtB was injected into two to four involved cervical dystonia muscles selected by the investigator, with the use of electromyography left at the discretion of the investigator performing the injection.
All trials were short‐term, with an observational period lasting 16 weeks post‐injection. No re‐injections were allowed.
Three studies (Brashear 1999; Brin 1999; Kaji 2013) assessed efficacy and other primary outcomes using an intent‐to‐treat (ITT) analysis, which included all participants randomised to treatment or, in the case of Kaji 2013, all those to whom treatment was administered. Lew 1997 assessed efficacy and safety outcomes on the per‐protocol (PP) population; this study also used an ITT analysis to assess the duration of effect.
Excluded studies
We have listed all the excluded studies in this review, together with reasons for their exclusion, in the 'Characteristics of excluded studies' table.
One study had been excluded from the original analysis since it was not fully published and relevant data was lacking (AN072‐008 1995). It was a dose‐finding parallel‐designed study comparing a single treatment session of BtB in three different doses (400 U, 1200 U, 2400 U) against placebo, doses currently thought to be insufficient for the great majority of CD patients. The follow‐up period was 16 weeks. Both BtA‐responsive and non‐responsive participants were enrolled, and the primary outcome was change in TWSTRS total score. All three experimental groups had large rates of withdrawals (400 U: 71%, 1200 U: 73%, 2400 U: 48%). For all but two of the participants, who had withdrawn from the study, the reason was the protocol‐defined criteria, ‘lack of response’. We asked the drug company for further information, without success. Our research did not find any additional publications on this trial that could shed any light on this problem, and because we could not rule out selective reporting of results, we decided that this study should remain excluded from our review. Two other parallel‐designed studies comparing different doses of BtB had been excluded from the original review (Cullis 2000; Truong 1997) for lacking a placebo group.
From the updated searches, we excluded a further five studies as one was neither blinded nor placebo‐controlled (Chinnapongse 2010); one was not placebo‐controlled (Jacob 2003); one was a post‐hoc analysis of two trials already included in this review (Lew 2002), and two were non‐randomised, non‐controlled studies focusing on the immunogenicity of BtB (Dressler 2005; Jankovic 2006).
Risk of bias in included studies
The quality assessment of previously included studies was re‐evaluated with the Cochrane Risk of bias tool (current at the time of writing), the results of which can be found in Figure 2 and Figure 3. These assessments were based on the information available in the primary report data.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Overall, no studies were considered to be at low risk of bias across all domains. High risk of bias was attributed only to "enriched population" and "other bias" domains.
Allocation
Two studies (Brashear 1999; Brin 1999) described the process of random sequence generation, in both controlled by an independent organisation; we assessed the other two studies to be at unclear risk of bias for this criterion.
Three studies (Brashear 1999: Brin 1999; Kaji 2013) described an adequate allocation concealment process and were rated as being at a low risk of bias, whereas we assessed the remaining study as being at unclear risk of bias.
Blinding
We evaluated the risk of bias in blinding of participants and personnel involved in the trial to be low for all the studies included in this review, since all trials were described as being double‐blinded and all used vials with identical appearance to mask the intervention employed.
We considered two studies to have adequately blinded investigators measuring objective outcomes (Brashear 1999; Brin 1999); the other two studies did not provide sufficient information to permit judgement, so we rated them as having an unclear risk of bias.
For the assessment of subjective outcomes, we considered all studies to have an unclear risk of bias: Kaji 2013 did not specify the blinding process, and all the other studies enrolled only participants who had previously been treated with botulinum toxin. We considered that studies including only non‐naive participants may introduce bias in patient‐reported assessment of subjective outcomes.
Incomplete outcome data
In Lew 1997, all participants completed the study per protocol. In the remainder, missing outcome data was balanced in numbers across intervention groups and adequate imputation methods were used (ITT). Reasons for missing data were unclear only in Kaji 2013, but we considered the reasons for participant attrition to be unlikely to motivate output imbalances.
Selective reporting
The more clinically relevant outcomes, which are usually evaluated in intervention trials for this condition, were reported in all studies, so we considered them to be at low risk of bias for reporting data. No trial protocol registry was available for any of the four included studies. However, three of these studies were conducted in the 1990's, before trial registration became standard good practice for clinical investigations.
Other potential sources of bias
Enriched population
Brashear 1999 exclusively enrolled BtA‐responsive participants, and was classified as having a high risk of bias for enriched population. All the other studies allowed BtA‐non‐responsive participants.
On the other hand, three studies (Brashear 1999; Brin 1999; Lew 1997) excluded forms of cervical dystonia known to have a poorer clinical response to BtA injection, and were considered to be at a high risk of bias for this domain.
Other Bias
Two trials (Brashear 1999; Brin 1999) declared funding or supply of study vials from industry sources, being rated at a high risk of bias for funding and potential conflicts of interest. Kaji 2013 did not provide a description of funding, but members of a pharmaceutical company were authors of the study, so this trial was also classified as high risk of bias for this domain. Lew 1997 was classified at unclear risk of bias for not stating the source of funding.
Publication bias
We intended to use funnel plots to explore publication bias. However, due to the small number of included studies, the power of this analysis was considered to be inadequate (Sterne 2011).
Effects of interventions
The key results of this review can be found in 'summary of findings Table for the main comparison'.
Botulinum toxin type B versus placebo
Preceding data analysis
Whenever necessary, we used appropriate imputation methods in order to combine the reported data into the meta‐analysis with other studies for which full data were available (see Dealing with missing data). Brashear 1999 and Brin 1999 reported the primary outcome (mean and SD) as total scores for the time point assessed; we obtained change from baseline SD values using pooled SD estimates.
All studies evaluating different BtB dosages (Brashear 1999; Kaji 2013; Lew 1997) presented data separately for each dose, reporting sample sizes, means and SD (when available) for each intervention group. When an overall dose was required to compare to other studies, we combined the reported subgroups using RevMan 5.3 (see Unit of analysis issues) (RevMan 2014). We used the same tool to calculate SD values from SE values presented in Kaji 2013. We conducted sensitivity analyses for every study where imputation methods were applied.
Primary outcomes
1. Overall improvement on any validated symptomatic rating scale for cervical dystonia
The primary outcome in all trials included in this review was change in Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS) (Consky 1994) total or subtotal scores, assessed at week 4 following initial injection of BtB. TWSTRS is currently the most common clinical validated tool to assess and document the status of patients with spasmodic torticollis. The TWSTRS (total score range, 0 to 85) is composite of three sub‐scales that evaluate different features of CD, namely severity (range, 0 to 35), disability (range, 0 to 30) and pain (range, 0 to 20). The higher the score, the greater the level of morbidity. In the absence of a validated value for a clinically meaningful change in TWSTRS total score, we have considered a 10% change from patients' baseline status as a clinically meaningful change.
Three trials (Brashear 1999; Brin 1999; Kaji 2013) reported data as the mean change from baseline in the TWSTRS total‐score, and demonstrated an improvement in participants treated with BtB compared to placebo mean difference (MD) 6.78; 95% CI 4.54 to 9.01; I2= 0%) (Analysis 1.1). This represents an improvement of 14.7% from the participant’s baseline clinical status (46.3 TWSTRS combined score). Lew 1997 was not included in this primary analysis because SD values were not reported. As the study population was not described in detail, lacking important data such as age distribution, duration and severity of disease, we could not impute SD values from similar studies with an acceptable margin of error. However, data from this trial was used to assess NNTB.
The NNTB in TWSTRS total score was three patients (95% CI 2 to 6).
With respect to TWSTRS sub‐scores, BtB was associated with a mean reduction of 2.43 points in TWSTRS Severity (95% CI 1.24 to 3.63; I2= 0%), and of 2.29 points in TWSTRS Disability (95% CI 1.04 to 3.54; I2= 0%). Brashear 1999 was not included in this analysis of TWSTRS sub‐scales as it did not present objective efficacy data for all groups.
1.1. Overall improvement with low vs medium vs high dose of BtB
We carried out a subgroup analysis to assess overall improvement according to BtB dose (see Subgroup analysis and investigation of heterogeneity). All trials tested high BtB dose (10,000 U); three tested medium BtB dose (5000 U), and two tested low BtB dose (2500 U).
All BtB doses were efficacious against placebo (low‐dose: MD 6.95; 95% CI 3.70 to 10.21; I2= 0%; medium‐dose: MD 6.10; 95% CI 3.40 to 8.81; I2= 0%; high‐dose: MD 8.72; 95% CI 6.35 to 11.10; I2= 0%). There was no difference in overall improvement, as assessed with TWSTRS global score, between these dose‐defined subgroups (P= 0.34; I2 = 6.9%) (Analysis 1.2).
1.2 Overall improvement with non‐EMG guided vs EMG‐ guided injections
In all trials, the use of EMG‐guidance was left at the discretion of the investigator. No data were reported concerning participants that did or did not undergo EMG‐guided injection. Thus, it was not possible to perform this planned subgroup analysis.
1.3 Overall improvement in BtA‐responsive vs BtA‐non‐responsive participants
Two trials (Kaji 2013; Lew 1997) enrolled both types of patients. However, we did not include these studies in this analysis because they did not present mean TWSTRS values for each subgroup.
Brashear 1999 included only BtA‐responsive participants, and Brin 1999 included only BtA‐non‐responsive participants. Overall, BtA‐responsive participants improved by 6.22 points (95% CI 1.83 to 10.60), while BtA‐non‐responsive participants improved by 9.0 points in TWSTRS total score (95% CI 4.46 to 13.54). This difference (2.78 points on the TWSTRS total score) between BtA‐responsive and BtA‐non‐responsiveparticipants was not significant (P = 0.39; I2= 0%) (Analysis 1.3).
Lew 1997, which included both types of participants, reported a higher rate of participants classified as 'responders' among participants who were non‐responsive to BtA in comparison to participants who were responsive to BtA (25% versus 66.7% ).
2. Number of participants with any adverse event
2.1 Proportion of participants with adverse events
Adverse events were generally transient and either mild to moderate, or intermittent. They were reported by 90.2% of the participants in the BtB groups, compared to 83.8% of participants in the placebo arm (RR 1.09; 95% CI 0.97 to 1.23; I2= 45%) (Analysis 1.4). This analysis included only two studies (Brashear 1999; Brin 1999) as the others (Kaji 2013; Lew 1997) did not present the total number of participants with adverse events per group.
2.1.1 Proportion of participants with adverse events with low vs medium vs high dose of BtB
Brashear 1999 and Brin 1999 were the studies included for this subgroup analysis. We excluded Kaji 2013 and Lew 1997 because they did not provide data according to the BtB dosages used.
There was no difference in the overall risk of any adverse event between medium (RR 1.07; 95% CI 0.89 to 1.29) or high‐dose (RR 1.09; 95% CI 0.89 to 1.33; I2= 59%) BtB‐treated participants and placebo (Analysis 1.5).
2.1.2 Proportion of participants with adverse events in BtA‐responsive vs BtA‐non‐responsive participants
The overall risk of adverse events reported by Brashear 1999 (with exclusively BtA‐responsive participants) was similar between the BtB and placebo groups (RR 0.97; 95% CI 0.79 to 1.20) (Analysis 1.6). The overall risk of adverse events reported by Brin 1999 (with exclusively BtA‐non‐responsive participants) was higher in the BtB group (RR 1.19; 95% CI 1.03 to 1.37).
Secondary outcomes
1. Change in subjective evaluation of clinical status evaluated by both participants and clinicians
Subjective evaluation of overall improvement by both participants and clinicians was assessed in all trials at week 4 after BtB injection. The trials used two scales to quantify overall improvement: the Global Assessment of Change (GAC) and the Visual Analogue Scale (VAS). GAC ranges from "Very marked worsening" (‐ 4) to "Complete resolution of CD symptoms" (+ 4). VAS (range, 0 mm to 100 mm) assesses the change from baseline in symptom severity, where 0 mm indicates "Much worse", 50 mm: "No change", and 100 mm: "Symptom‐free".
Two of the trials included in quantitative synthesis (Brashear 1999; Brin 1999) reported this outcome using mean change from baseline on the GAC scale whilst the other study (Kaji 2013) reported this outcome as the mean change from baseline on the on the VAS scale, with both dimensions of the outcome (participant and clinician assessments) being reported in all three studies. Overall, both participants and clinicians reported an improvement of subjective clinical status, with a SMD of 0.86 (95% CI 0.61 to 1.10; I2= 0%) (Analysis 1.7) and 0.80 (95% CI 0.55 to 1.04; I2= 0%) (Analysis 1.8) , respectively.
Lew 1997 reported that there was a significant (P = 0.0001) improvement among BtB‐treated participants in both Patient and Investigator Global Assessment ratings. However, since these data were not fully reported, we could not include this trial in the meta‐analysis for this outcome.
All three trials (Brashear 1999; Brin 1999; Kaji 2013) reporting extractable data for subjective assessments were meta‐analysed according to the doses of BtB used, and all doses were associated with a significant benefit when compared to placebo in both participant and clinician subjective assessments (Analysis 1.9 and Analysis 1.10, respectively). There were no differences between the different dose‐based subgroups (low versus medium, versus high‐dose BtB).
Two trials (Brashear 1999; Brin 1999) reported subjective assessment data with regards to BtA‐responsive and BtA‐non‐responsive participants. These data were meta‐analysed, though we found no differences between the different BtA‐responsiveness subgroups (see Analysis 1.11 and Analysis 1.12).
2. Changes in pain scores, as assessed with validated assessment tools
Two trials (Brin 1999; Kaji 2013) provided data on TWSTRS pain sub‐scores (range, 0 to 20), and reported an improvement in participants treated with BtB compared to placebo with a MD of 2.20 (95% CI 1.25 to 3.15; I2= 58%) (Analysis 1.13). We did not include Lew 1997 in this meta‐analysis because they did not report SD values for the overall intervention group, although they did report SD values for dose subgroups. As they did not describe the study population in sufficient detail, lacking important data such as age distribution, duration and severity of disease, we decided not to impute SD values from similar studies as the margin of error would be unknown. Brashear 1999 did not report data for this outcome.
We meta‐analysed three trials (Brin 1999; Kaji 2013; Lew 1997) that reported data as mean change from baseline on the TWSTRS pain sub‐scale, according to the doses of BtB used. These trials were associated with significant benefit when compared to placebo (Analysis 1.14). However, we found no differences between the different dose‐based subgroups (low versus medium, versus high‐dose BtB).
Two trials (Brashear 1999; Brin 1999) reported pain relief data with regards to BtA‐responsive and BtA‐non‐responsive participants. Subgroup analysis did not identify any differences between the different BtA‐responsiveness subgroups (see Analysis 1.15).
3. Changes in quality of life assessments
Lew 1997 assessed quality of life with the Sickness Impact Profile (SIP), a 136‐item questionnaire evaluating quotidian activities, divided into 12 categories: emotional behaviour, social interaction, alertness behaviour, communication, body care and movement, ambulation, mobility, sleep and rest, home management, work, recreation and pastimes, and eating. The results are given in percentages, highest scores representing more disabling status. This trial did not report definitive data , although it does state that scores in the BtB arm did not differ significantly from those in the placebo arm. None of the other studies (Brashear 1999; Brin 1999; Kaji 2013) reported data on this outcome.
4. Number of withdrawals due to adverse events, including adverse events caused by the intervention (type A or type B, or both, adverse drug reactions (ADRs)), and failure of therapy (type F ADRs)
All the included trials reported the number of withdrawals due to adverse events without differences between BtB and placebo (RR 0.88; 95% CI: 0.19 to 4.06; I2= 0%) (Analysis 1.20). For the purpose of this analysis we considered that adverse events may be caused by the intervention (i.e. type A and/or type B ADRs), or lack of efficacy of the treatment (i.e., failure of therapy, a type F ADRs) (Edwards 2000).
The most frequent reason for withdrawal due to adverse events was failure of therapy, which was reported in two participants in Brashear 1999 (one in the BtB arm and the other in the placebo arm) and in two BtB‐treated participants in the Kaji 2013 trial. Withdrawals due to adverse events occurred in one participant in the placebo arm in Brin 1999 and in one BtB‐allocated participant in Brashear 1999. The former participant experienced neck pain, headache, urticaria, eye pain, asthenia and nausea, and the latter died after triple‐vessel coronary artery bypass surgery performed on study day 67, considered unrelated to CD treatment.
5. Number of participants with adverse events of special interest
The most frequent adverse events reported were dry mouth (RR 7.65; 95% CI 2.75 to 21.32; I2= 0%) and dysphagia (RR 6.78; 95% CI 2.42 to 19.05; I2= 0%), which occurred in 17% of participants in the BtB group versus 3% in the placebo group (Analysis 1.21, Analysis 1.22). The NNTH for dry mouth and dysphagia was 7 (95% CI 26 to 2) and 8 (95% CI 32 to 3), respectively.
For all the other adverse events no significant differences were found. Nevertheless, the following adverse events were more frequent with BtB than placebo: injection site pain (RR 1.39; 95% CI 0.73 to 2.66; I2= 0%), nausea (RR 2.06; 95% CI 0.68 to 6.28; I2= 0%), headache (RR 1.90; 95% CI 0.82 to 4.41; I2= 0%), pain (RR 1.15; 95% CI 0.51 to 2.62; I2= 0%), infection (RR 1.14; 95% CI 0.38 to 3.38; I2= 61%) and flu syndrome (RR 1.44; 95% CI 0.23 to 8.92; I2= 67%).
We performed subgroup analysis according to BtB dose for the two most common adverse events, dry mouth and dysphagia. In comparison to placebo, dry mouth was significantly higher among high‐dose BtB‐treated participants (RR 11.47; 95% CI 3.95 to 33.30; I2= 0%), but not among medium and low‐dose BtB‐treated participants in comparison to placebo. However, overall risk of dry mouth was no different between the dose‐defined subgroups (P = 0.18; I2= 41%) (Analysis 1.23). The risk of dysphagia was significantly higher among high‐ (RR 9.19; 95% CI 3.38 to 25.01; I2= 0%) and medium‐dose (RR 5.50; 95% CI 1.25 to 24.17; I2= 0%) BtB‐treated participants, but not among low‐dose BtB‐treated participants in comparison to placebo. However, overall risk of dysphagia was no different between the dose‐defined subgroups (P = 0.85; I2= 0%) (Analysis 1.24).
It is noteworthy that all above mentioned adverse events occurred in more than 10% of BtB‐treated participants.
6. Duration of effect, or number of days until need for reinjection or effect waning
Three trials (Brashear 1999; Brin 1999; Lew 1997) assessed duration of clinical benefit, defined as the time until return to baseline TWSTRS total score. In all trials, the duration of effect thus defined was between 12 and 16 weeks. Data suggested that the change in TWSTRS total score over time was somewhat shorter for the lower doses (2500‐5000 U) than for the higher dose (10,000 U). Since the studies performed only one treatment session, no data was available for long‐term duration of benefit. The newly included study (Kaji 2013) did not assess duration of effect. We did not conduct meta‐analysis due to lack of combinable data (Michiels 2005).
Discussion
Summary of main results
This updated review included four randomised, parallel‐designed, placebo‐controlled trials, enrolling 441 participants with cervical dystonia, of whom 92.5% had been previously treated with BtA. In comparison to placebo, BtB was effective in reducing overall disease impairment, including disease severity, disability and associated pain. An improvement of 14.7% from the participant’s baseline clinical status was found among participants treated with BtB four weeks after a single treatment cycle, reducing by nearly 7 points in TWSTRS‐total score and yielding an NNTB of 3 (for any improvement in TWSTRS‐total score). Subjective assessments by both participants and clinicians also favoured BtB in comparison to placebo. The impact of BtB on other domains of participants' quality of life, such as social functioning or mental health, have not properly been addressed in the included trials.
Overall, there was no difference in rates of adverse events and withdrawals due to adverse events between groups. However, the short duration of the trials, as well as the reduced sample size, precludes strong conclusions with regards to the lack of differences between BtB and placebo. The most common adverse events that were different between the BtB and placebo groups were dry mouth and dysphagia, both considered related to treatment and being about six times more frequent among BtB‐treated patients, with an NNTH of 7 and 8, respectively. No fatalities or serious adverse events were considered related to BtB treatment in any of the trials. Data for special subpopulations, such as children and pregnant women, were not available.
BtB doses
All dosages were efficacious against placebo, but we found no clear‐cut evidence of a dose‐response gradient. It is however noteworthy that these trials were not dose‐response studies and that this conclusion was based on arbitrarily defined dose‐subgroup analyses. On the other hand, higher BtB dosages were associated with a higher risk of dysphagia and dry mouth.
BtA‐responsive versus BtA‐non‐responsive participants
The percentage improvement of disease impairment reported in one trial enrolling exclusively BtA‐non‐responders was higher than that reported in another trial enrolling exclusively BtA‐responders. The reduced sample size precludes strong conclusions with regards to these results, which could be due to several confounding factors, such as methodological differences and population imbalances between the two trials. One further trial, enrolling both types of participants, also suggested a higher efficacy among BtA‐non‐responders. As for adverse events, we found no differences between the groups.
Duration of effect
The effect of BtB lasted approximately 12 to 16 weeks, as assessed by the time needed to return to baseline TWSTRS total scores. Duration of effect thus defined was greater in the subgroup of participants treated with higher BtB doses. Long‐term duration of effect could not be evaluated as all trials evaluated only a single treatment session.
Overall completeness and applicability of evidence
All included trials addressed the primary outcome of our review using the same assessment tool. However, some did not fully report all outcome data, and in some cases results could not be pooled and compared across studies. This limits the amount of data available and, consequently, the confidence in overall conclusions.
Four noteworthy factors challenge the implementation of the evidence in this review. First, there was a limited and considerably heterogeneous regional distribution, with one trial being conducted in Japan and three in the United States. Differences in clinical practice, training of experts, and local guidelines in other regions of the world may present an obstacle to the application of the evidence here demonstrated. Second, sample size across included trials was relatively small and many subgroup analyses for the outcomes of interest present only trends in the results. More studies are needed to provide robust evidence for these trends. Third, the enrolment of enriched populations in clinical trials limits applicability of results into clinical practice, as complex and potentially poorer responders are usually excluded in these trials. The fact that these patients are common in clinical practice further complicates issues of generalisation. Fourth, patients frequently have concomitant medications for their condition, such as muscle relaxants and benzodiazepines. In trials, such medications are reasonably required to be on a stable dose for many weeks to avoid confounding factors. As a result, little is currently known about the impact of these drug regimens with regards to implementation of the evidence in this review.
Quality of the evidence
See Characteristics of included studies, 'Risk of bias' tables and 'Risk of bias' summary tables (Figure 2; Figure 3).
Only two of the included studies adequately described their randomisation and allocation methods, with the remaining two trials being assessed at an unclear risk of bias for these items. All studies were considered appropriately blinded in general; however, only two provided satisfactory descriptions of blinding of objective outcome assessment, and all were considered possibly biased regarding subjective outcome assessment, as all studies predominantly enrolled patients with previous treatment with BtA. This represents major methodological limitations that may have resulted in a biased assessment of the intervention effect, particularly with regards to subjective outcomes, which are highly susceptible to biased estimations, namely pain assessment, subjective assessment by participants and clinicians, and quality of life assessments. Finally, statistical heterogeneity was present for pain and adverse events outcomes which could not be clearly explained by the subgroup analysis performed. However, results from individual studies were all in the same direction.
Some outcomes could not be compared across studies, as some studies lacked reporting of relevant data. Imbalances between baseline characteristics of the participants and incomplete description of the variables meant that we could not confidently impute values for missing data, further reducing the amount of combinable data, and therefore the precision of the results.
The included trials enrolled between 77 and 133 participants, and although individually these trials were underpowered, the pooling of the trials permitted an adequate sample size for the majority of efficacy outcomes. Taken together, we consider that there is moderate quality evidence that a single treatment session of BtB, in certain types of cervical dystonia, is efficacious in reducing disease impairment, including severity, pain and disability. However, the quality of the evidence is low and no robust conclusions can be made regarding safety and tolerability, including withdrawals due to adverse events, as well as regarding continued responsiveness and long‐term efficacy, which are important aspects in a chronic condition such as cervical dystonia.
Potential biases in the review process
Although we followed the methods recommended by Cochrane in order to minimise bias in the review process, certain areas are deserving of attention on the part of readers. Despite having contacted experts in the area, not having searched clinical trial registries opens the current review to two potential problems: firstly, possibly having missed trials and also the possibility of introducing publication bias.
The newly added trial was published in Japanese only. Results tables were presented in English, and important information was extracted from the text by a Japanese collaborator (Dr. Masao Kaneshige). Even though we took steps to minimise this potential source of bias, we cannot ignore its existence.
Agreements and disagreements with other studies or reviews
Overall, the results of this updated review are in agreement with the conclusions of earlier versions. However, we now conclude that no claims can be made regarding a clear‐cut dose‐response relationship for efficacy outcomes. On the other hand, a clear dose‐dependent relationship exists for the treatment‐related adverse events of special interest, such as dysphagia and dry mouth.
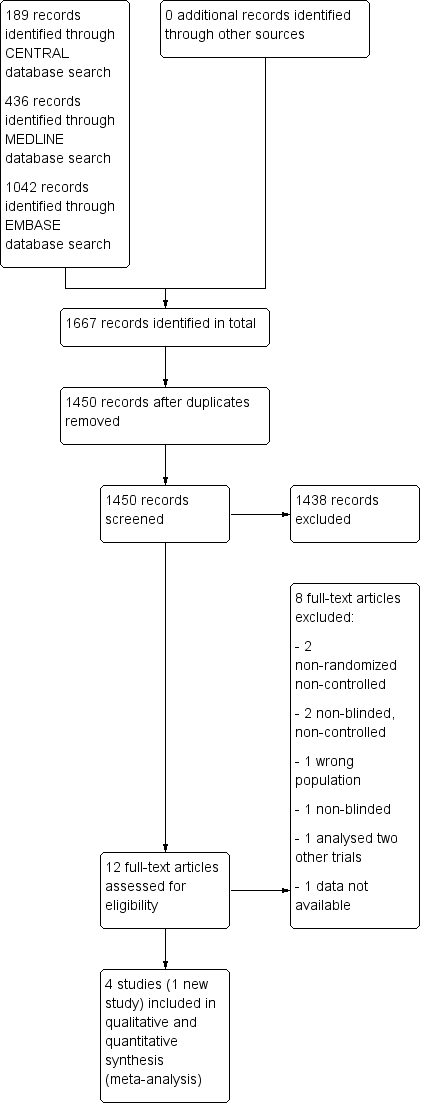
Study flow diagram
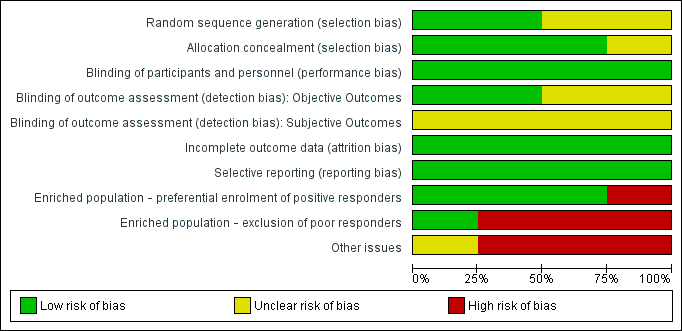
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 1 Overall cervical dystonia improvement as assessed with TWSTRS: change from baseline to week 4.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 2 Overall cervical dystonia improvement as assessed with TWSTRS: change from baseline to week 4 ‐ Doses subgroup analysis.
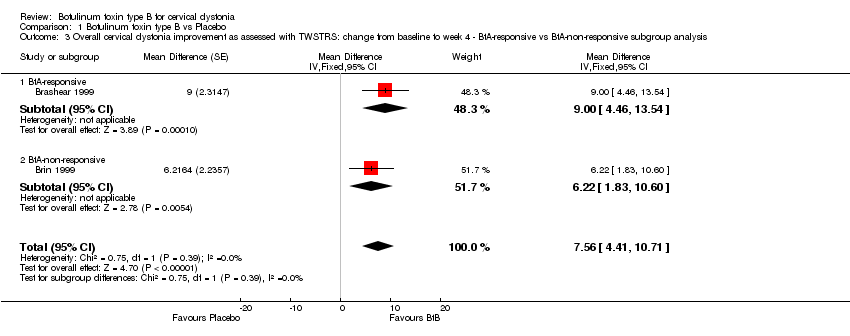
Comparison 1 Botulinum toxin type B vs Placebo, Outcome 3 Overall cervical dystonia improvement as assessed with TWSTRS: change from baseline to week 4 ‐ BtA‐responsive vs BtA‐non‐responsive subgroup analysis.
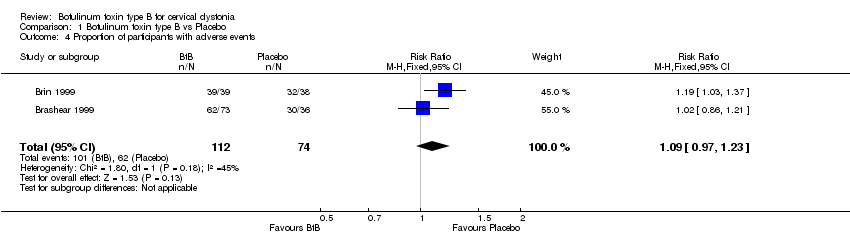
Comparison 1 Botulinum toxin type B vs Placebo, Outcome 4 Proportion of participants with adverse events.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 5 Proportion of participants with adverse events ‐ doses subgroup analysis.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 6 Proportion of participants with adverse events ‐ BtA‐responsive vs BtA‐non‐responsive subgroup analysis.
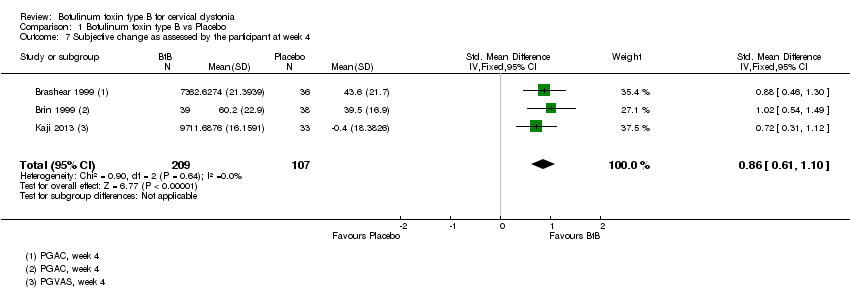
Comparison 1 Botulinum toxin type B vs Placebo, Outcome 7 Subjective change as assessed by the participant at week 4.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 8 Subjective change as assessed by the clinician at week 4.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 9 Subjective change as assessed by the participant at week 4 ‐ doses subgroup analysis.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 10 Subjective change as assessed by the clinician at week 4 ‐ doses subgroup analysis.
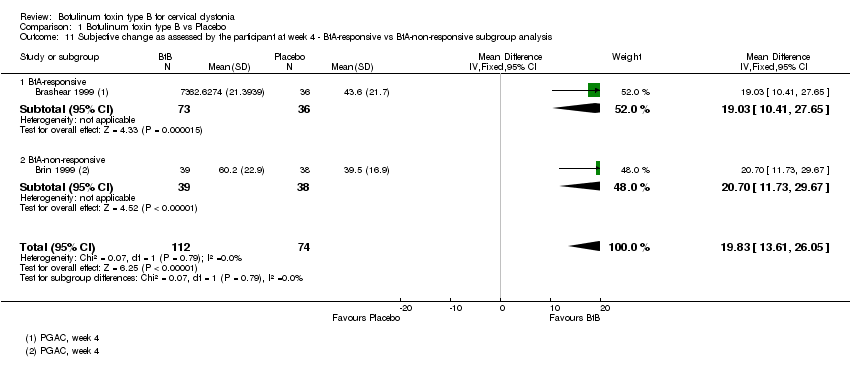
Comparison 1 Botulinum toxin type B vs Placebo, Outcome 11 Subjective change as assessed by the participant at week 4 ‐ BtA‐responsive vs BtA‐non‐responsive subgroup analysis.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 12 Subjective change as assessed by the clinician at week 4 ‐ BtA‐non‐responsive vs ‐responsive subgroup analysis.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 13 Cervical dystonia associated pain: change from baseline to week 4 as assessed with TWSTRS.
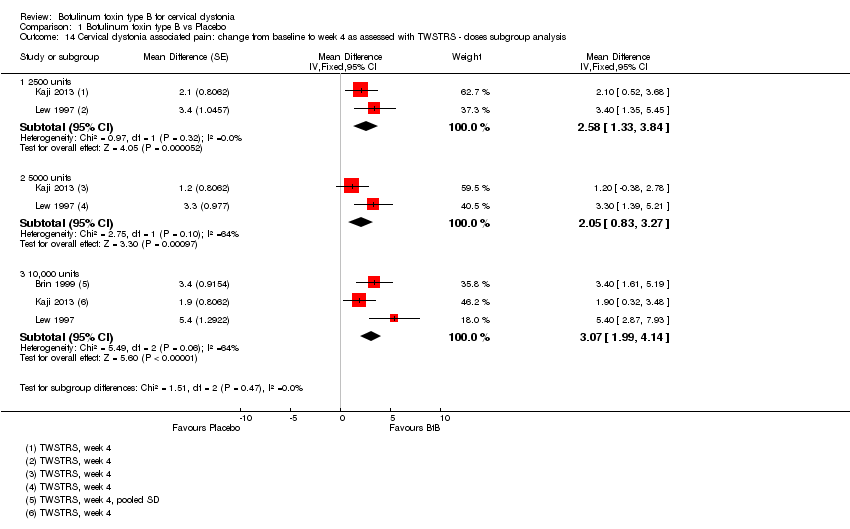
Comparison 1 Botulinum toxin type B vs Placebo, Outcome 14 Cervical dystonia associated pain: change from baseline to week 4 as assessed with TWSTRS ‐ doses subgroup analysis.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 15 Cervical dystonia associated pain: change from baseline to week 4 as assessed with validated scales ‐ BtA‐responsive vs BtA‐non‐responsive subgroup analysis.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 16 Cervical dystonia severity: change from baseline to week 4 as assessed with TWSTRS.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 17 Cervical dystonia severity: change from baseline to week 4 as assessed with TWSTRS ‐ doses subgroup analysis.
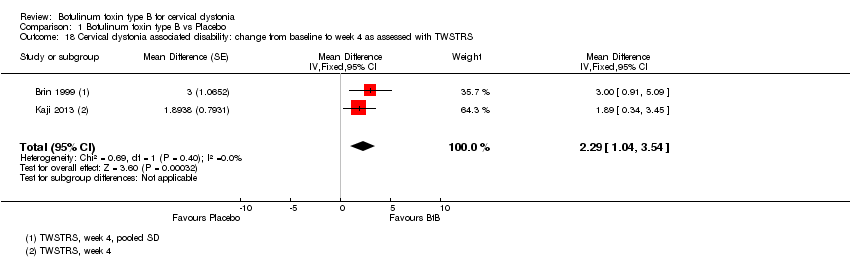
Comparison 1 Botulinum toxin type B vs Placebo, Outcome 18 Cervical dystonia associated disability: change from baseline to week 4 as assessed with TWSTRS.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 19 Cervical dystonia associated disability: change from baseline to week 4 as assessed with TWSTRS ‐ doses subgroup analysis.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 20 Proportion of withdrawals due to adverse events.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 21 Adverse events: dry mouth.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 22 Adverse events: dysphagia.
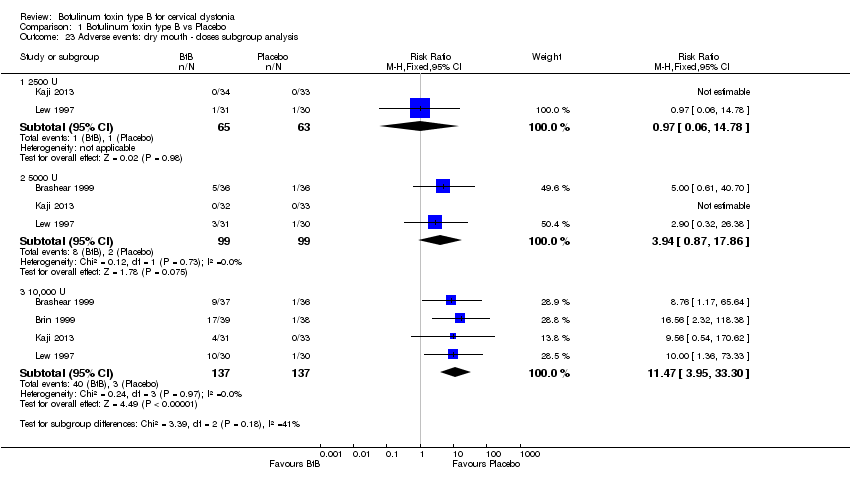
Comparison 1 Botulinum toxin type B vs Placebo, Outcome 23 Adverse events: dry mouth ‐ doses subgroup analysis.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 24 Adverse events: dysphagia ‐ doses subgroup analysis.
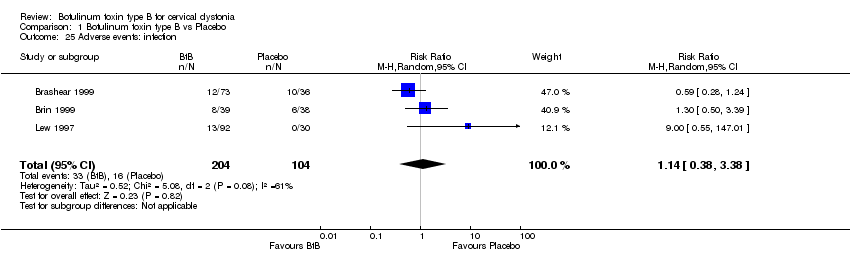
Comparison 1 Botulinum toxin type B vs Placebo, Outcome 25 Adverse events: infection.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 26 Adverse events: neck pain secondary to CD.
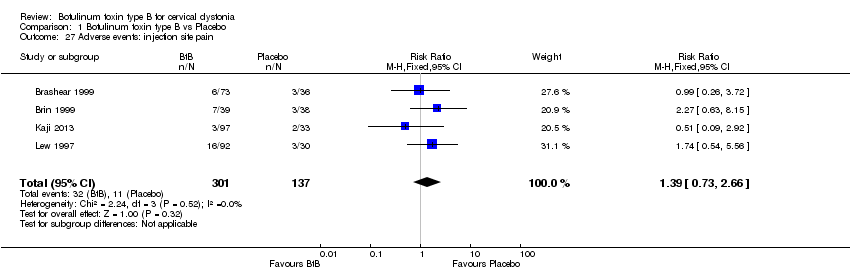
Comparison 1 Botulinum toxin type B vs Placebo, Outcome 27 Adverse events: injection site pain.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 28 Adverse events: nausea.
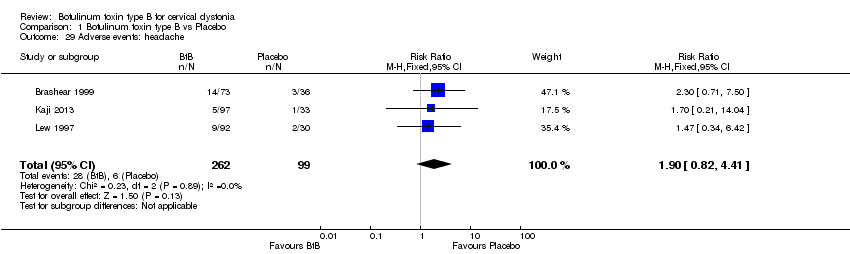
Comparison 1 Botulinum toxin type B vs Placebo, Outcome 29 Adverse events: headache.
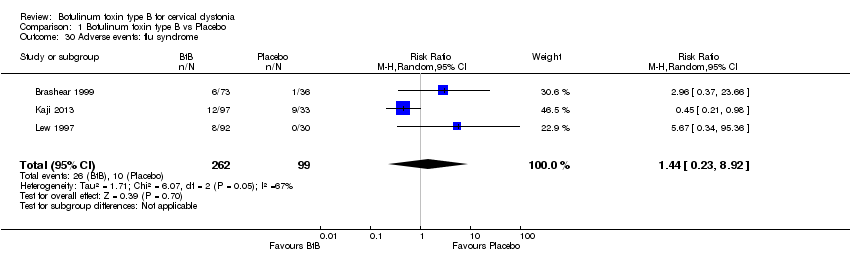
Comparison 1 Botulinum toxin type B vs Placebo, Outcome 30 Adverse events: flu syndrome.

Comparison 1 Botulinum toxin type B vs Placebo, Outcome 31 Adverse events: pain.
| Botulinum Neurotoxin B compared to placebo for cervical dystonia | ||||||
| Patient or population: adults with cervical dystonia | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Botulinum Neurotoxin B | |||||
| Overall cervical dystonia improvement as assessed with TWSTRS: change from baseline to week 4 (range, 0 to 85; more is worst) | ‐7 | ‐7 | The mean change from baseline to week 4 in the BtB group was 6.78 TWSTRS units higher (4.54 higher to 9.01 higher) compared to the placebo group | 316 | ⊕⊕⊕⊝ | |
| Proportion of withdrawals due to adverse events | Study population | RR 0.88 | 440 | ⊕⊕⊝⊝ | ||
| 14 per 1000 | 13 per 1000 | |||||
| Cervical dystonia associated pain: change from baseline to week 4 as assessed with TWSTRS (range, 0 to 20; more is worst) | ‐7 | ‐7 | The mean change from baseline to week 4 in the BtB group was 2.41 TWSTRS units higher (0.82 higher to 4.01 higher) compared to the placebo group | 207 | ⊕⊕⊝⊝ | |
| Subjective change as assessed by the patient at week 4 | ‐7 | ‐7 | The mean change at week 4 in the BtB group was 0.86 standard deviations higher (0.61 higher to 1.1 higher) compared to the placebo group | 316 | ⊕⊕⊕⊕ | |
| Proportion of participants with adverse events | Study population | RR 1.09 | 186 | ⊕⊝⊝⊝ | ||
| 838 per 1000 | 930 per 1000 | |||||
| Adverse events: dry mouth | Study population | RR 7.65 | 438 | ⊕⊕⊕⊕ | ||
| 22 per 1000 | 168 per 1000 | |||||
| Adverse events: dysphagia | Study population | RR 6.78 | 438 | ⊕⊕⊕⊕ | ||
| 22 per 1000 | 148 per 1000 | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Two of 3 studies enrolled an enriched population; none of the included studies had independent funding; blinding of outcome assessment was unclear in all studies 2Three of 4 studies enrolled an enriched population; none of the studies had a clearly stated independent funding; blinding of outcome assessment was unclear in all studies; two out of 4 had an unclear random sequence generation 3The total number of participants included was less than the number generated by a conventional sample size calculation for a single adequately powered trial 4I‐squared of 58% and small overlap between confidence intervals 5Both studies had an enriched population and non‐independent funding; blinding of outcome assessment was unclear in all studies 6I‐squared of 45% and there is a wide variance of point estimates between studies 7 Data were only available as the difference between the BtB and placebo groups | ||||||
| Term | Definition |
| BtA‐non‐responsive | People who do not experience the expected benefit from treatment with botulinum toxin type A |
| Cervical dystonia or spasmodic toricollis | It is a common movement disorder in which people have abnormal movements or postures of the head and neck that they cannot control. It is frequently accompanied by social embarrassment and pain. |
| Chemodenervation | It is the process by which botulinum toxin causes muscular paralysis. Altought all the anatomical elements necessary for muscular control are intact (i.e. nerve, synapse and muscle), there is a chemical process that disables the transmission of the transmission of the electrical signal from the nerve to the muscle. |
| Dysphagia | A discomfort or difficulty when swallowing. |
| Electromyography | It is an exam that displays the electrical activity of muscles using pieces of metal attached to the skin or inserted into the muscle. |
| Non‐naive | People who have been treated in the past with botulinum toxin. |
| Voluntary action | Movements that we are able to control, start and stop when we want to. |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Overall cervical dystonia improvement as assessed with TWSTRS: change from baseline to week 4 Show forest plot | 3 | Mean Difference (Fixed, 95% CI) | 6.78 [4.54, 9.01] | |
| 2 Overall cervical dystonia improvement as assessed with TWSTRS: change from baseline to week 4 ‐ Doses subgroup analysis Show forest plot | 4 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 2.1 2500 units | 2 | Mean Difference (Fixed, 95% CI) | 6.95 [3.70, 10.21] | |
| 2.2 5000 units | 3 | Mean Difference (Fixed, 95% CI) | 6.10 [3.40, 8.81] | |
| 2.3 10,000 units | 4 | Mean Difference (Fixed, 95% CI) | 8.72 [6.35, 11.10] | |
| 3 Overall cervical dystonia improvement as assessed with TWSTRS: change from baseline to week 4 ‐ BtA‐responsive vs BtA‐non‐responsive subgroup analysis Show forest plot | 2 | Mean Difference (Fixed, 95% CI) | 7.56 [4.41, 10.71] | |
| 3.1 BtA‐responsive | 1 | Mean Difference (Fixed, 95% CI) | 9.0 [4.46, 13.54] | |
| 3.2 BtA‐non‐responsive | 1 | Mean Difference (Fixed, 95% CI) | 6.22 [1.83, 10.60] | |
| 4 Proportion of participants with adverse events Show forest plot | 2 | 186 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.97, 1.23] |
| 5 Proportion of participants with adverse events ‐ doses subgroup analysis Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 5000 U | 1 | 72 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.89, 1.29] |
| 5.2 10,000 U | 2 | 150 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.89, 1.33] |
| 6 Proportion of participants with adverse events ‐ BtA‐responsive vs BtA‐non‐responsive subgroup analysis Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 BtA‐responsive | 1 | 73 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.79, 1.20] |
| 6.2 BtA‐non‐responsive | 1 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [1.03, 1.37] |
| 7 Subjective change as assessed by the participant at week 4 Show forest plot | 3 | 316 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.86 [0.61, 1.10] |
| 8 Subjective change as assessed by the clinician at week 4 Show forest plot | 3 | 316 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.80 [0.55, 1.04] |
| 9 Subjective change as assessed by the participant at week 4 ‐ doses subgroup analysis Show forest plot | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 9.1 2500 U | 1 | 67 | Mean Difference (IV, Fixed, 95% CI) | 12.30 [4.21, 20.39] |
| 9.2 5000 U | 2 | 137 | Mean Difference (IV, Fixed, 95% CI) | 9.71 [4.05, 15.37] |
| 9.3 10,000 U | 3 | 214 | Mean Difference (IV, Fixed, 95% CI) | 15.12 [10.32, 19.91] |
| 10 Subjective change as assessed by the clinician at week 4 ‐ doses subgroup analysis Show forest plot | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 10.1 2500 U | 1 | 67 | Mean Difference (IV, Fixed, 95% CI) | 8.60 [1.46, 15.74] |
| 10.2 5000 U | 2 | 137 | Mean Difference (IV, Fixed, 95% CI) | 10.84 [5.65, 16.04] |
| 10.3 10,000 U | 3 | 214 | Mean Difference (IV, Fixed, 95% CI) | 13.22 [9.39, 17.05] |
| 11 Subjective change as assessed by the participant at week 4 ‐ BtA‐responsive vs BtA‐non‐responsive subgroup analysis Show forest plot | 2 | 186 | Mean Difference (IV, Fixed, 95% CI) | 19.83 [13.61, 26.05] |
| 11.1 BtA‐responsive | 1 | 109 | Mean Difference (IV, Fixed, 95% CI) | 19.03 [10.41, 27.65] |
| 11.2 BtA‐non‐responsive | 1 | 77 | Mean Difference (IV, Fixed, 95% CI) | 20.70 [11.73, 29.67] |
| 12 Subjective change as assessed by the clinician at week 4 ‐ BtA‐non‐responsive vs ‐responsive subgroup analysis Show forest plot | 2 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.1 BtA‐responsive | 1 | 109 | Mean Difference (IV, Fixed, 95% CI) | 12.74 [5.83, 19.65] |
| 12.2 BtA‐non‐responsive | 1 | 77 | Mean Difference (IV, Fixed, 95% CI) | 12.70 [7.04, 18.36] |
| 13 Cervical dystonia associated pain: change from baseline to week 4 as assessed with TWSTRS Show forest plot | 2 | Mean Difference (Fixed, 95% CI) | 2.20 [1.25, 3.15] | |
| 14 Cervical dystonia associated pain: change from baseline to week 4 as assessed with TWSTRS ‐ doses subgroup analysis Show forest plot | 3 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 14.1 2500 units | 2 | Mean Difference (Fixed, 95% CI) | 2.58 [1.33, 3.84] | |
| 14.2 5000 units | 2 | Mean Difference (Fixed, 95% CI) | 2.05 [0.83, 3.27] | |
| 14.3 10,000 units | 3 | Mean Difference (Fixed, 95% CI) | 3.07 [1.99, 4.14] | |
| 15 Cervical dystonia associated pain: change from baseline to week 4 as assessed with validated scales ‐ BtA‐responsive vs BtA‐non‐responsive subgroup analysis Show forest plot | 2 | Std. Mean Difference (Fixed, 95% CI) | 0.96 [0.65, 1.27] | |
| 15.1 BtA‐responsive | 1 | Std. Mean Difference (Fixed, 95% CI) | 0.92 [0.50, 1.34] | |
| 15.2 BtA‐non‐responsive | 1 | Std. Mean Difference (Fixed, 95% CI) | 1.01 [0.53, 1.48] | |
| 16 Cervical dystonia severity: change from baseline to week 4 as assessed with TWSTRS Show forest plot | 2 | Mean Difference (Fixed, 95% CI) | 2.43 [1.24, 3.63] | |
| 17 Cervical dystonia severity: change from baseline to week 4 as assessed with TWSTRS ‐ doses subgroup analysis Show forest plot | 3 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 17.1 2500 units | 2 | Mean Difference (Fixed, 95% CI) | 1.9 [0.55, 3.25] | |
| 17.2 5000 units | 2 | Mean Difference (Fixed, 95% CI) | 2.51 [1.25, 3.77] | |
| 17.3 10,000 units | 3 | Mean Difference (Fixed, 95% CI) | 2.96 [1.87, 4.05] | |
| 18 Cervical dystonia associated disability: change from baseline to week 4 as assessed with TWSTRS Show forest plot | 2 | Mean Difference (Fixed, 95% CI) | 2.29 [1.04, 3.54] | |
| 19 Cervical dystonia associated disability: change from baseline to week 4 as assessed with TWSTRS ‐ doses subgroup analysis Show forest plot | 3 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 19.1 2500 units | 2 | Mean Difference (Fixed, 95% CI) | 2.32 [0.86, 3.78] | |
| 19.2 5000 units | 2 | Mean Difference (Fixed, 95% CI) | 2.06 [0.55, 3.57] | |
| 19.3 10,000 units | 3 | Mean Difference (Fixed, 95% CI) | 3.22 [2.09, 4.35] | |
| 20 Proportion of withdrawals due to adverse events Show forest plot | 4 | 440 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.19, 4.06] |
| 21 Adverse events: dry mouth Show forest plot | 4 | 438 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.65 [2.75, 21.32] |
| 22 Adverse events: dysphagia Show forest plot | 4 | 438 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.78 [2.42, 19.05] |
| 23 Adverse events: dry mouth ‐ doses subgroup analysis Show forest plot | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 23.1 2500 U | 2 | 128 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.06, 14.78] |
| 23.2 5000 U | 3 | 198 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.94 [0.87, 17.86] |
| 23.3 10,000 U | 4 | 274 | Risk Ratio (M‐H, Fixed, 95% CI) | 11.47 [3.95, 33.30] |
| 24 Adverse events: dysphagia ‐ doses subgroup analysis Show forest plot | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 24.1 2500 U | 2 | 128 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.79 [0.86, 53.63] |
| 24.2 5000 U | 3 | 198 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.50 [1.25, 24.17] |
| 24.3 10,000 U | 4 | 274 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.19 [3.38, 25.01] |
| 25 Adverse events: infection Show forest plot | 3 | 308 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.38, 3.38] |
| 26 Adverse events: neck pain secondary to CD Show forest plot | 3 | 308 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.67, 1.73] |
| 27 Adverse events: injection site pain Show forest plot | 4 | 438 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.39 [0.73, 2.66] |
| 28 Adverse events: nausea Show forest plot | 2 | 199 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.06 [0.68, 6.28] |
| 29 Adverse events: headache Show forest plot | 3 | 361 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.90 [0.82, 4.41] |
| 30 Adverse events: flu syndrome Show forest plot | 3 | 361 | Risk Ratio (M‐H, Random, 95% CI) | 1.44 [0.23, 8.92] |
| 31 Adverse events: pain Show forest plot | 2 | 231 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.51, 2.62] |