Monoterapia con carbamazepina versus fenitoína para la epilepsia: una revisión de datos de participantes individuales
Resumen
Antecedentes
Esta es una actualización de una revisión Cochrane publicada por primera vez en 2002 y actualizada por última vez en 2017. Esta revisión es una de una serie de revisiones Cochrane que estudia las comparaciones por pares de monoterapias.
La epilepsia es una afección neurológica común en la cual las descargas eléctricas anormales del cerebro causan convulsiones recurrentes no provocadas. Se cree que con un tratamiento farmacológico eficaz, hasta el 70% de los pacientes con epilepsia activa tienen la posibilidad de superar las convulsiones y entrar en remisión durante un largo plazo poco después de iniciar el tratamiento farmacológico con un solo fármaco antiepiléptico en forma de monoterapia.
En todo el mundo, la carbamazepina y la fenitoína son fármacos antiepilépticos de amplio espectro de uso común, adecuados para la mayoría de los tipos de crisis epilépticas. La carbamazepina es el tratamiento de primera línea actual para las convulsiones de inicio focal en los Estados Unidos y Europa. La fenitoína ya no se considera un tratamiento de primera línea, debido a las preocupaciones sobre los eventos adversos asociados con su uso, pero el fármaco todavía se utiliza habitualmente en países de ingresos bajos a medios debido a su bajo coste. No se han encontrado diferencias consistentes en la eficacia entre la carbamazepina y la fenitoína en ensayos individuales; sin embargo, los intervalos de confianza generados por estos ensayos son amplios y, por lo tanto, la síntesis de los datos de los ensayos individuales puede mostrar diferencias en la eficacia.
Objetivos
Revisar el tiempo transcurrido hasta el fracaso del tratamiento, la remisión y la primera convulsión con carbamazepina comparada con fenitoína cuando se utilizan como monoterapia en pacientes con convulsiones de inicio focal (focal simple o compleja y secundariamente generalizada), o convulsiones tónico‐clónicas de inicio generalizado (con o sin otros tipos de convulsiones generalizadas).
Métodos de búsqueda
Para la última actualización se realizaron búsquedas en las siguientes bases de datos el 13 de agosto de 2018: el Registro Cochrane de Estudios (Cochrane Register of Studies, CRS Web), que incluye el Registro Especializado Cochrane de Epilepsia (Cochrane Epilepsy's Specialised Register) y el Registro Cochrane Central de Ensayos Controlados (Cochrane Central Register of Controlled Trials, CENTRAL); MEDLINE; el National Institutes of Health Ongoing Trials Register de los EE.UU. (ClinicalTrials.gov) y la World Health Organization International Clinical Trials Registry Platform (ICTRP). Se hicieron búsquedas manuales en revistas relevantes y se estableció contacto con compañías farmacéuticas, investigadores de ensayos originales y expertos en el tema.
Criterios de selección
Ensayos controlados aleatorizados que compararon la monoterapia con carbamazepina o fenitoína en niños o adultos con convulsiones de inicio focal o convulsiones de inicio generalizado (tónico‐clónicas).
Obtención y análisis de los datos
Ésta fue una revisión de datos de pacientes individuales (DPI). El resultado primario fue el tiempo transcurrido hasta el fracaso del tratamiento. Los resultados secundarios fueron el tiempo hasta la primera convulsión después de la asignación al azar, el tiempo hasta la remisión de seis meses, el tiempo hasta la remisión de 12 meses y la incidencia de eventos adversos. Se utilizaron los modelos de regresión de riesgos proporcionales de Cox para obtener estimaciones específicas de los ensayos de los cocientes de riesgos instantáneos (CRI) con intervalos de confianza (IC) del 95% mediante el método de la varianza inversa genérica para obtener los CRI agrupados generales y los IC del 95%.
Resultados principales
Los DPI estuvieron disponibles para 595 participantes de 1102 individuos elegibles, de cuatro de 11 ensayos (es decir, el 54% de los datos potenciales). Para los resultados de remisión, un CRI mayor de 1 indica una ventaja de la fenitoína y para los resultados primera convulsión y retiro, un CRI mayor de 1 indica una ventaja de la carbamazepina. Se consideró que la mayoría de los participantes incluidos en el análisis (78%) presentaban convulsiones de inicio focal al inicio y sólo el 22% presentaban convulsiones de inicio generalizado, por lo tanto, los resultados de esta revisión son principalmente aplicables a los pacientes con convulsiones de inicio focal.
Los resultados primarios de la revisión fueron: tiempo hasta el fracaso del tratamiento por cualquier razón relacionada con el tratamiento (CRI agrupado ajustado según el tipo de convulsión para 546 participantes): 0,94; IC del 95%: 0,70 a 1,26; evidencia de certeza moderada); tiempo hasta el fracaso del tratamiento debido a la falta de eficacia (CRI agrupado ajustado por tipo de convulsión para 546 participantes: 0,99; IC del 95%: 0,69 a 1,41; evidencia de certeza moderada); y ninguno muestra diferencias claras entre los fármacos y el tiempo transcurrido hasta el fracaso del tratamiento debido a eventos adversos (CRI agrupado ajustado por tipo de convulsión para 546 participantes: 1,27; IC del 95%: 0,87 a 1,86; evidencia de certeza moderada), lo que muestra que el fracaso del tratamiento debido a eventos adversos puede ocurrir antes con la carbamazepina que con la fenitoína, pero no se puede descartar una ligera ventaja de la carbamazepina o ninguna diferencia entre los fármacos.
Para los resultados secundarios (CRI agrupados ajustados según el tipo de convulsión), no se encontraron diferencias claras entre la carbamazepina y la fenitoína: tiempo hasta la primera convulsión después de la asignación al azar (582 participantes): 1,15; IC del 95%: 0,94 a 1,40; evidencia de certeza moderada); tiempo hasta la remisión de 12 meses (551 participantes): 1,00; IC del 95%: 0,79 a 1,26; evidencia de certeza moderada) y tiempo hasta la remisión de seis meses (551 participantes): 0,90; IC del 95%: 0,73 a 1,12; evidencia de certeza moderada).
Para todos los resultados, los resultados de los pacientes con convulsiones de inicio focal fueron similares a los resultados generales (evidencia de certeza moderada) y los resultados del subgrupo pequeño de pacientes con convulsiones de inicio generalizado fueron poco precisos, por lo que no se puede descartar una ventaja de alguno de los dos fármacos o ninguna diferencia entre los fármacos (evidencia de certeza baja). También hubo evidencia de que la clasificación errónea del tipo de convulsión puede haber confundido los resultados de esta revisión, en particular para el resultado "tiempo transcurrido hasta el fracaso del tratamiento". La heterogeneidad estuvo presente en el análisis del "tiempo transcurrido hasta la primera convulsión" en los pacientes con convulsiones de inicio generalizado, lo que no se pudo explicar mediante el análisis de subgrupos ni los análisis de sensibilidad.
Se dispuso de información limitada acerca de los eventos adversos en los ensayos y no fue posible comparar las tasas de eventos adversos entre la carbamazepina y la fenitoína. Algunos eventos adversos informados sobre ambos fármacos fueron dolor abdominal, náuseas y vómitos, somnolencia, trastornos motores y cognitivos y efectos secundarios dismórficos (como erupción cutánea).
Conclusiones de los autores
La evidencia de certeza moderada proporcionada por esta revisión sistemática no muestra diferencias entre la carbamazepina y la fenitoína en cuanto a la efectividad (retención) o la eficacia (recurrencia y remisión de las convulsiones) en los pacientes con convulsiones de inicio focal o de inicio generalizado.
Sin embargo, algunos de los ensayos que contribuyeron a los análisis presentaron deficiencias e inconsistencias metodológicas, que pueden haber tenido un impacto en los resultados de esta revisión. Por lo tanto, no se indica que los resultados de esta revisión por sí solos constituyan la base de una elección de tratamiento para un paciente con convulsiones de comienzo reciente. No se encontró evidencia para apoyar o refutar las políticas de tratamiento actuales. Los ensayos futuros se deben diseñar con la mayor calidad posible y tener en cuenta el enmascaramiento, la elección de la población, la clasificación del tipo de convulsión, la duración del seguimiento, la elección de los resultados y el análisis, y la presentación de los resultados.
PICO
Resumen en términos sencillos
Carbamazepina versus fenitoína (administradas como tratamiento farmacológico único) para la epilepsia
Esta es una versión actualizada de la revisión Cochrane publicada anteriormente en el Número 2, 2017 de la Base de Datos Cochrane de Revisiones Sistemáticas (Cochrane Database of Systematic Reviews)
Antecedentes
La epilepsia es un trastorno neurológico común en el cual las convulsiones recurrentes son causadas por descargas eléctricas anormales del cerebro. En esta revisión se estudiaron dos tipos de crisis epilépticas: las convulsiones de inicio generalizado, en las que las descargas eléctricas comienzan en una parte del cerebro y se mueven por todo el cerebro, y las convulsiones de inicio focal, en las que la convulsión se genera y afecta sólo a una parte del cerebro (todo el hemisferio cerebral o parte de un lóbulo del cerebro). En aproximadamente el 70% de los pacientes con epilepsia, las convulsiones de inicio generalizado o de inicio focal se pueden controlar con un único fármaco antiepiléptico. En todo el mundo, la fenitoína y la carbamazepina son fármacos antiepilépticos de uso habitual, aunque la carbamazepina se utiliza con mayor frecuencia en los EE.UU. y Europa debido a las preocupaciones sobre los efectos secundarios asociados con la fenitoína. La fenitoína aún se utiliza en los países de ingresos bajos y medios de África, Asia y América del Sur, debido al bajo coste del medicamento.
Objetivo
Para esta revisión actualizada se examinó la evidencia de 11 ensayos clínicos controlados aleatorizados que compararon la fenitoína y la carbamazepina sobre la base de la efectividad de los fármacos para controlar las convulsiones (es decir, si los pacientes volvieron a presentar convulsiones o tuvieron períodos prolongados sin convulsiones [remisión]), y cuán tolerables fueron los efectos secundarios relacionados con los mismos.
Métodos
Fue posible combinar los datos de 595 pacientes de cuatro de los 11 ensayos y en el caso de los 507 pacientes restantes de siete ensayos no hubo información disponible para su uso en esta revisión. La evidencia está actualizada hasta agosto de 2018.
Resultados clave
Esta revisión de ensayos no encontró diferencias entre estos dos fármacos en los tipos de convulsiones estudiados en cuanto a los resultados de fracaso del tratamiento (retiro del tratamiento por cualquier motivo y también retiro del tratamiento debido a convulsiones continuas o debido a efectos secundarios) y el control de las convulsiones (recurrencia de las convulsiones o logro de un período sin convulsiones [remisión] de seis o 12 meses). Tres cuartas partes de los pacientes reclutados en los cuatro ensayos presentaron convulsiones de inicio focal y sólo una cuarta parte de los pacientes reclutados en los cuatro ensayos presentaron convulsiones de inicio generalizado, por lo que los resultados de esta revisión se aplican principalmente a los pacientes con convulsiones de inicio focal y los resultados son muy limitados para los pacientes con convulsiones de inicio generalizado. Se necesita más información para los pacientes con convulsiones de inicio generalizado.
Algunos efectos secundarios informados por los pacientes que recibieron carbamazepina y los pacientes que recibieron fenitoína fueron dolor abdominal, náuseas, vómitos, cansancio, problemas motores (como coordinación deficiente), problemas cognitivos (mala memoria), erupciones cutáneas y otros problemas cutáneos.
Certeza de la evidencia
La certeza de la evidencia se consideró moderada a baja para el fracaso del tratamiento, moderada para los resultados de remisión y baja para los resultados de convulsiones, ya que es probable que la clasificación errónea del tipo de convulsión influyera en los resultados de la revisión. En dos de los ensayos que proporcionaron datos para esta revisión, el diseño del ensayo permitió que los pacientes y los médicos tratantes supieran qué medicación tomaban. Este diseño puede haber influido en los resultados.
Algunos de los ensayos que contribuyeron con datos a la revisión tuvieron problemas metodológicos, que pueden haber introducido sesgos y resultados inconsistentes en esta revisión, y algunos pacientes mayores de 30 años con convulsiones de inicio generalizado recién diagnosticados pueden haber tenido un diagnóstico erróneo de su tipo de convulsión. Estos problemas pueden haber afectado los resultados de esta revisión y la certeza de la evidencia proporcionada por esta revisión se consideró moderada para los pacientes con convulsiones de inicio focal y de certeza baja para los pacientes con convulsiones de inicio generalizado. No se recomienda utilizar solo los resultados de esta revisión para elegir entre la carbamazepina o la fenitoína para el tratamiento de la epilepsia.
Se indica que todos los ensayos futuros que comparen estos fármacos o cualquier otro fármaco antiepiléptico se diseñen con métodos de alta calidad, y que los tipos de convulsiones de los pacientes incluidos en los ensayos se clasifiquen con mucho cuidado para garantizar que los resultados también sean de alta calidad.
Authors' conclusions
Summary of findings
| Carbamazepine compared with phenytoin for epilepsy | ||||||
| Patient or population: adults and children with new‐onset focal or generalised epilepsy Settings: outpatients Intervention: carbamazepine Comparison: phenytoin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of Participants | Certainty (quality) of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenytoin | Carbamazepine | |||||
| Time to treatment failure (any reason related to treatment) Range of follow‐up:1 day to 4403 days | The median time to treatment failure was 2135 days in the phenytoin group | The median time to treatment failure was 2422 days (307 days longer) in the carbamazepine group | HR 0.94 (0.70 to 1.26)a | 546 (3 studies) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical advantage for carbamazepine There was also no statistically significant difference between drugs in treatment failure due to adverse events (HR 1.27, 95% CI 0.87 to 1.86; P = 0.21; I2 = 3%), or treatment failure due to lack of efficacy: HR 0.99 (95% CI 0.69 to 1.41; P = 0.94; I2 = 0%) |
| Time to treatment failure (any reason related to treatment) Subgroup: focal onset seizures Range of follow‐up: 1 day to 4064 days | The median time to treatment failure was 1300 days in the phenytoin group | The median time to treatment failure was 2422 days (1122 days longer) in the carbamazepine group | HR 0.83 (0.61 to 1.13) | 428 (3 studies) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical advantage for carbamazepine There was also no statistically significant difference between drugs in treatment failure due to adverse events (HR 1.19, 95% CI 0.80 to 1.78; P = 0.38, I2 = 0%), or treatment failure due to lack of efficacy: HR 0.88 (95% CI 0.60 to 1.40, P = 0.52, I2 = 0%) |
| Time to treatment failure (any reason related to treatment) Subgroup: generalised Range of follow‐up:1 day to 4403 days | The 10th percentilec | The 10th percentilec | HR 2.38 (1.04 to 5.47) | 118 (2 studies) | ⊕⊕⊝⊝ lowb,d | HR < 1 indicates a clinical advantage for carbamazepine There was no statistically significant difference between drugs in treatment failure due to adverse events (HR 2.31, 95% CI 0.68 to 7.81; P = 0.18; I2 = 60% , or treatment failure due to lack of efficacy: HR 1.86 (95% CI 0.74 to 4.67; P = 0.19; I2 = 0%) but confidence intervals are wide so we cannot rule out an advantage to either drug or no difference between the drugs |
| *Illustrative risks in the carbamazepine and phenytoin groups are calculated at the median time to treatment failure (i.e. the time to 50% of participants failing or withdrawing from allocated treatment) within each group across all trials. The relative effect (pooled hazard ratio) shows the comparison of 'Time to treatment failure' between the treatment groups. CI: 95% confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High certainty (quality): Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty (quality): Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty (quality): Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very certainty (quality): We are very uncertain about the estimate. | ||||||
| aPooled HR for all participants adjusted for seizure type. All pooled HRs presented calculated with fixed‐effect model. | ||||||
| Carbamazepine compared with phenytoin for epilepsy | ||||||
| Patient or population: adults and children with new‐onset focal or generalised epilepsy Settings: outpatients Intervention: carbamazepine Comparison: phenytoin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of Participants | Certainty (quality) of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenytoin | Carbamazepine | |||||
| Time to first seizure after randomisation Range of follow‐up: 0 days to 4589 days | The median time to first seizure was 124 days in the phenytoin group | The median time to first seizure was 79 days (45 days shorter) in the carbamazepine group | HR 1.15 (0.94 to 1.40) | 582 (4 studies) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical |
| Time to first seizure after randomisation Subgroup: focal onset seizures Range of follow‐up: 0 days to 4589 days | The median time to first seizure was 78 days in the phenytoin group | The median time to first seizure was 62 days (16 days shorter) in the carbamazepine group | HR 1.13 (0.89 to 1.43) | 432 (4 studies) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical |
| Time to first seizure after randomisation Range of follow‐up: 2 days to 4070 days | The median time to first seizure was 323 days in the phenytoin group | The median time to first seizure was 142 days (181 days shorter) in the carbamazepine group | HR 1.19 (0.81 to 1.75) | 150 (3 studies) | ⊕⊕⊝⊝ lowb,c | HR < 1 indicates a clinical |
| Time to achieve 12‐month remission Range of follow‐up: 0 days to 4222 days | The median time to 12‐month remission was 472 days in the phenytoin group | The median time to 12‐month remission was 481 days (9 days longer) in the carbamazepine group | HR 1.00 (0.79 to 1.26) | 551 (3 studies) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical |
| Time to achieve 12‐month remission Range of follow‐up: 0 days to 4222 days | The median time to 12‐month remission was 531 days in the phenytoin group | The median time to 12‐month remission was 515 days (16 days shorter) in the carbamazepine group | HR 1.06 (0.80 to 1.42) | 430 (3 studies) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical |
| Time to achieve 12‐month remission Range of follow‐up: 7 days to 4163 days | The median time to 12‐month remission was 366 days in the phenytoin group | The median time to 12‐month remission was 375 days (9 days longer) in the carbamazepine group | HR 0.88 (0.58 to 1.33) | 121 (2 studies) | ⊕⊕⊝⊝ lowb,d | HR < 1 indicates a clinical |
| *Illustrative risks in the carbamazepine and phenytoin groups are calculated at the median time to first seizure or time to 12‐month remission (i.e. the time to 50% of participants experiencing a first seizure or 12‐months of remission) within each group across all trials. The relative effect (pooled hazard ratio) shows the comparison of 'Time to first seizure' or 'Time to 12‐month remission' between the treatment groups. CI: 95% confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High certainty (quality): Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty (quality): Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty (quality): Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very certainty (quality): We are very uncertain about the estimate. | ||||||
| aPooled HR for all participants adjusted for seizure type. All pooled HRs presented calculated with a fixed‐effect model. | ||||||
Background
This is an updated version of the Cochrane Review previously published in Issue 2, 2017 of the Cochrane Database of Systematic Reviews (Nevitt 2017b).
Description of the condition
Epilepsy is a common neurological condition in which recurrent, unprovoked seizures are caused by abnormal electrical discharges from the brain. Epilepsy is a disorder of many heterogeneous seizure types, with an estimated incidence of 33 to 57 per 100,000 person‐years worldwide (Annegers 1999; Hirtz 2007; MacDonald 2000; Olaffsson 2005; Sander 1996), accounting for approximately 1% of the global burden of disease (Murray 1994). The lifetime risk of epilepsy onset is estimated to be 1300 to 4000 per 100,000 person‐years (Hauser 1993; Juul‐Jenson 1983), and the lifetime prevalence could be as large as 70 million people worldwide (Ngugi 2010). It is believed that with effective drug treatment, up to 70% of individuals with active epilepsy have the potential to become seizure‐free and go into long‐term remission shortly after starting drug therapy (Cockerell 1995; Hauser 1993; Sander 2004), and that around 70% of individuals can achieve seizure freedom using a single antiepileptic drug in monotherapy (Cockerell 1995); current National Institute for Health and Care Excellence (NICE) guidelines recommend that both adults and children with epilepsy should be treated by monotherapy wherever possible (NICE 2012). The remaining 30% of individuals experience refractory or drug‐resistant seizures which often require treatment with combinations of antiepileptic drugs, or alternative treatments such as epilepsy surgery (Kwan 2000).
We study two seizure types in this review: generalised onset seizures (generalised tonic‐clonic seizures with or without other generalised seizure types), in which electrical discharges begin in one part of the brain and move throughout the brain; and focal onset seizures, in which the seizure is generated in and affects only one part of the brain (the whole hemisphere of the brain or part of a lobe of the brain).
Description of the intervention
Carbamazepine and phenytoin are among the most commonly used and earliest drugs licensed for the treatment of epileptic seizures; phenytoin has been used as monotherapy for focal seizures and generalised tonic‐clonic seizures for over 50 years (Gruber 1962) and carbamazepine for over 30 years (Shakir 1980). Current NICE guidelines (NICE 2012) for adults and children recommend carbamazepine as a first‐line treatment for focal onset seizures and as a second‐line treatment for generalised tonic‐clonic seizures if first‐line treatments sodium valproate and lamotrigine are deemed unsuitable; however, there is evidence that carbamazepine may exacerbate some other generalised seizure types such as myoclonic and absence seizures (Liporace 1994; Shields 1983; Snead 1985). Phenytoin is no longer considered a first‐line treatment in the USA and most of Europe, due to concerns over adverse events (Wallace 1997; Wilder 1995), but phenytoin is still used as a first‐line drug in low‐ and middle‐income countries (Ogunrin 2005; Pal 1998).
Both carbamazepine and phenytoin have been shown to have teratogenic effects (disturbances to foetal development) (Bromley 2014; Weston 2016), where the risk is estimated to be two to three times that of the general population (Gladstone 1992; Meador 2008; Morrow 2006; Nulman 1997). Carbamazepine is associated particularly with neural tube defects (Matlow 2012) and phenytoin is associated with foetal hydantoin syndrome (Scheinfeld 2003), low folic acid levels and megaloblastic anaemia (Carl 1992). Both carbamazepine and phenytoin are associated with an allergic rash (Tennis 1997) in 5% to 10% of users, which on rare occasions may be life‐threatening, and phenytoin is also associated with long‐term cosmetic changes including gum hyperplasia, acne and coarsening of the facial features (Mattson 1985; Scheinfeld 2003).
How the intervention might work
Antiepileptic drugs suppress seizures by reducing neuronal excitability. Phenytoin and carbamazepine are broad‐spectrum treatments suitable for many seizure types and both have an anticonvulsant mechanism through blocking ion channels, binding with neurotransmitter receptors or through inhibiting the metabolism or reuptake of neurotransmitters (Ragsdale 1991; Willow 1985) and the modulation of gamma‐aminobutyric acid‐A (GABA‐A) receptors (Granger 1995).
Why it is important to do this review
The aim of this review is to summarise efficacy and tolerability data from existing trials comparing carbamazepine and phenytoin when used as monotherapy treatments. The adverse event profiles of the two drugs are well documented (see example references from Description of the intervention), but no consistent differences in efficacy have been found between the two drugs from a number of randomised controlled trials (RCTs) individually (for example: De Silva 1996; Heller 1995; Mattson 1985; Ramsay 1983). Although no clear difference in efficacy has been found from individual studies, the confidence intervals generated by these studies are wide. We cannot exclude important differences in efficacy, which may be shown by synthesising the data of the individual trials.
There are difficulties in undertaking a systematic review of epilepsy monotherapy trials as the important efficacy outcomes require analysis of time‐to‐event data (for example, time to first seizure after randomisation). Although methods have been developed to synthesise time‐to‐event data using summary information (Parmar 1998; Williamson 2002), the appropriate statistics are not commonly reported in published epilepsy trials (Nolan 2013a; Williamson 2000). Furthermore, although most epilepsy monotherapy trials collect seizure data, there has been no uniformity in the definition and reporting of outcomes. For example, trials may report time to 12‐month remission but not time to first seizure or vice versa, or some trials may define time to first seizure from the date of randomisation while others use the date of achieving maintenance dose. Trial investigators have also adopted differing approaches to the analysis, particularly with respect to the censoring of time‐to‐event data. For these reasons, we performed this review using individual participant data (IPD), which help to overcome these problems. This review is one in a series of Cochrane IPD Reviews investigating pair‐wise monotherapy comparisons (Marson 2000; Nevitt 2018a; Nevitt 2018b; Nevitt 2018c; Nevitt 2018d; Nevitt 2019; Nolan 2013b). These data have also been included in IPD network meta‐analyses of antiepileptic drug monotherapy (Nevitt 2017a; Tudur Smith 2007).
Objectives
To review the time to treatment failure, remission and first seizure with carbamazepine compared with phenytoin when used as monotherapy in people with focal onset seizures (simple or complex focal and secondarily generalised), or generalised onset tonic‐clonic seizures (with or without other generalised seizure types).
Methods
Criteria for considering studies for this review
Types of studies
-
Studies must be randomised controlled trials (RCTs) using either an adequate method of allocation concealment (e.g. sealed opaque envelopes) or a quasi‐randomised method of allocation (e.g. allocation by date of birth).
-
Studies must be of parallel design; cross‐over studies are not an appropriate design for measuring the long‐term outcomes of interest in this review (see Types of outcome measures).
-
Studies must include a comparison of carbamazepine monotherapy with phenytoin monotherapy in individuals with epilepsy; cluster‐randomised studies are therefore not an eligible design.
We included studies regardless of blinding method (unblinded, single‐blind or double‐blind).
Types of participants
-
We included trials recruiting children or adults with focal onset seizures (simple focal, complex focal, or secondarily generalised tonic‐clonic seizures) or generalised onset tonic‐clonic seizures (as a primary generalised seizure type), with or without other generalised seizure types (e.g. absence, myoclonic, etc.).
-
We excluded studies that recruited only individuals with other generalised seizure types, without generalised tonic‐clonic seizures (such as studies recruiting only individuals with a diagnosis of absence seizures or juvenile myoclonic epilepsy, etc.) due to differences in first‐line treatment guidelines (NICE 2012).
-
We included individuals who had a new diagnosis of epilepsy or who had experienced a relapse following antiepileptic monotherapy withdrawal only, due to differences in first‐line treatment guidelines for individuals with drug‐resistant epilepsy (NICE 2012).
Types of interventions
Carbamazepine versus phenytoin (any doses) as monotherapy.
Types of outcome measures
Below is a list of outcomes investigated in this review. Reporting of these outcomes in the original trial report was not an eligibility requirement for this review.
Primary outcomes
Time to treatment failure (retention time). This was a combined outcome reflecting both efficacy and tolerability, as the following may have led to failure of treatment: continued seizures, side effects, non‐compliance or the initiation of additional add‐on treatment. This is an outcome to which the participant makes a contribution and is the primary outcome measure recommended by the Commission on Antiepileptic Drugs of the International League Against Epilepsy (ILAE 1998; ILAE 2006).
We consider time to treatment failure according to three definitions:
-
Time to treatment failure for any treatment‐related reason (continued seizures, side effects, non‐compliance or the initiation of additional add‐on treatment)
-
Time to treatment failure due to adverse events (i.e. side effects)
-
Time to treatment failure due to lack of efficacy (i.e. continued seizures)
Secondary outcomes
-
Time to first seizure post‐randomisation
-
Time to achieve 12‐month remission (seizure‐free period)
-
Time to achieve six‐month remission (seizure‐free period)
-
Adverse events (including those relating to treatment withdrawal)
Search methods for identification of studies
Electronic searches
We conducted searches for the original review in 1999, and subsequently in 2001, 2003, 2005, July 2007, November 2009, November 2011, October 2013, September 2014, and November 2016. For the latest update we searched the following databases, applying no language restrictions:
-
The Cochrane Register of Studies (CRS Web, 13 August 2018), which includes the Cochrane Epilepsy Group’s Specialised Register and the Cochrane Central Register of Controlled Trials (CENTRAL), using the search strategy outlined in Appendix 1.
-
MEDLINE (Ovid, 1946 to August 10 2018), using the search strategy outlined in Appendix 2.
-
ClinicalTrials.gov (13 August 2018), using the search strategy outlined in Appendix 3.
-
World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP, 13 August 2018), using the search strategy outlined in Appendix 4.
Previously we also searched SCOPUS (1823 to 16th September 2014), using the search strategy outlined in Appendix 5, as an alternative to Embase, but this is no longer necessary, because randomised and quasi‐randomised controlled trials in Embase are now included in CENTRAL.
Searching other resources
In addition, we handsearched relevant journals, reviewed the reference lists of retrieved studies to search for additional reports of relevant studies, contacted Novartis (manufacturers of carbamazepine), Parke‐Davis (manufacturers of phenytoin), and experts in the field for information on any ongoing studies, and original investigators of relevant trials found.
Data collection and analysis
Selection of studies
Two review authors (SJN and AGM) independently assessed trials for inclusion, resolving any disagreements by discussion.
Data extraction and management
We requested the following IPD for all trials meeting our inclusion criteria.
Trial methods
-
method of generation of random list
-
method of concealment of randomisation
-
stratification factors
-
blinding methods
Participant covariates
-
gender
-
age
-
seizure types
-
time between first seizure and randomisation
-
number of seizures prior to randomisation (with dates)
-
presence of neurological signs
-
electroencephalographic (EEG) results
-
computerised tomography/magnetic resonance imaging (CT/MRI) results
Follow‐up data
-
treatment allocation
-
date of randomisation
-
dates of follow‐up
-
dates of seizures post‐randomisation or seizure frequency data between follow‐up visits
-
dates of treatment failure and reasons for treatment failure
-
dose
-
dates of dose changes
For each trial for which we did not obtain IPD, we carried out an assessment to see whether any relevant aggregate‐level data had been reported or could be indirectly estimated using the methods of Parmar 1998 and Williamson 2002.
In one study (Mattson 1985), seizure data were provided in terms of the number of seizures recorded between each follow‐up visit rather than specific dates of seizures. To enable us to calculate time‐to‐event outcomes, we applied linear interpolation to approximate dates of seizures between follow‐up visits, assuming a uniform seizure rate. For example, if four seizures were recorded between two visits which occurred on 1st March 1990 and 1st May 1990 (an interval of 61 days), then the date of first seizure would be approximately 13th March 1990 (i.e. 61 days divided by number of seizures plus 1 rounded to the next day, i.e. 13 days). This allowed us to compute an estimate of the time to six‐month remission, 12‐month remission, and the time to first seizure.
We calculated time to six‐month and 12‐month remission from the date of randomisation to the date (or estimated date) the individual had first been free of seizures for six or 12 months respectively. If the person had one or more seizures in the titration period, a six‐month or 12‐month seizure‐free period could also occur between the estimated date of the last seizure in the titration period and the estimated date of the first seizure in the maintenance period.
We calculated time to first seizure from the date of randomisation to the date that their first seizure was estimated to have occurred. If seizure data were missing for a particular visit, we censored these outcomes at the previous visit. We also censored these outcomes if the individual died or if follow‐up ceased prior to the occurrence of the event of interest. These methods had been used in the remaining three trials (De Silva 1996; Heller 1995; Ogunrin 2005) for which outcome data (dates of seizures after randomisation) were provided directly.
In one trial (Ogunrin 2005), all participants completed the 12‐week trial duration without failing treatment or withdrawing from the study. For three trials (De Silva 1996; Heller 1995; Mattson 1985) we extracted dates and reason for treatment failure or withdrawal from trial case report forms for the original review. Two review authors (SJN and CTS) independently extracted data from all case report forms, resolving disagreements by reconsidering the case report forms at conference. For the analysis of time‐to‐event data, we defined an 'event' as either the failure of the allocated treatment because of poor seizure control, adverse events, or both. We also classed non‐compliance with the treatment regimen or the addition of another antiepileptic drug as 'events' for the outcome 'time to treatment failure.' We censored the outcome if treatment was stopped because the individual achieved a period of remission or if the individual was still on allocated treatment at the end of follow‐up.
Assessment of risk of bias in included studies
Two review authors (SJN and CTS) independently assessed all included studies for risks of bias (Higgins 2017), resolving any disagreements by discussion. The domains assessed as being at low, high or unclear risk of bias were random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting, and other potential sources of bias. We took into account all available information for an included study when making 'Risk of bias' judgements, including multiple publications of the study and additional information provided from study authors with IPD.
Measures of treatment effect
We measured all outcomes in this review as time‐to‐event outcomes with the hazard ratio (HR) and 95% confidence interval (CI) used as the measure of treatment effect. We calculated outcomes from IPD provided where possible, or extracted from published trials if possible.
Unit of analysis issues
We did not have any unit of analysis issues. The unit of allocation and analysis was the individual for all included trials, and no trials included in meta‐analysis were of a repeated measures (longitudinal) nature or of a cross‐over design.
Dealing with missing data
For each trial where IPD were supplied, we reproduced information from trial results where possible, and performed the following consistency checks:
-
We cross‐checked trial details against any published report of the trial and contacted original trial authors if we found missing data, errors or inconsistencies;
-
We reviewed the chronological randomisation sequence, and checked the balance of participant characteristics, taking account of factors stratified for in the randomisation procedure.
Assessment of heterogeneity
We assessed heterogeneity statistically using the Q test (P value < 0.10 for significance) and the I2 statistic (Higgins 2003) (greater than 50% indicating considerable heterogeneity), output produced using the generic inverse variance approach in Data and analyses, and visually by inspecting forest plots.
Assessment of reporting biases
Two review authors (SJN and CTS) undertook all full quality and 'Risk of bias' assessments. In theory, a review using IPD should overcome issues of reporting biases, as unpublished data can be provided and unpublished outcomes calculated. Any selective reporting bias detected could be assessed with the ORBIT classification system (Kirkham 2010).
Data synthesis
We carried out our analysis on an intention‐to‐treat basis; i.e. we analysed participants in the group to which they were randomised, irrespective of which treatment they actually received. Therefore, for the time‐to‐event outcomes 'Time to six‐month remission', 'Time to 12‐month remission', and 'Time to first seizure post‐randomisation', we did not censor participants if treatment was withdrawn or failed.
For all outcomes, we investigated the relationship between the time‐to‐event and treatment effect of the antiepileptic drugs. We used Cox proportional hazards regression models to obtain trial‐specific estimates of log (HR), or treatment effect and associated standard errors in Stata Statistical Software, version 14 (Stata 2015). The model assumes that the ratio of hazards (risks) between the two treatment groups is constant over time (i.e. hazards are proportional). We tested this proportional hazards assumption of the Cox regression model for each outcome of each trial by testing the statistical significance of a time‐varying covariate in the model and by visually inspecting survival plots for each outcome of each trial. We evaluated overall estimates of HRs (with 95% confidence intervals (CIs)), using the generic inverse variance method in Data and analyses. We expressed results as a HR and its 95% CI.
By convention, a HR greater than 1 indicates that an event is more likely to occur earlier on carbamazepine than on phenytoin. Hence, for time to treatment failure or time to first seizure, a HR greater than 1 indicates a clinical advantage for phenytoin (e.g. a HR of 1.2 would suggest a 20% increase in risk of treatment failure from carbamazepine compared with phenytoin), and for time to six‐month and 12‐month remission, a HR greater than 1 indicates a clinical advantage for carbamazepine.
Subgroup analysis and investigation of heterogeneity
To examine the potential impact of seizure type on results, we stratified all analyses by seizure type (focal onset versus generalised onset), according to the classification of main seizure type at baseline. We classified focal seizures (simple or complex), and focal secondarily generalised seizures as focal epilepsy.
We classified primarily generalised seizures as generalised epilepsy. We conducted a Chi2 test of interaction between treatment and seizure type. If we found significant statistical heterogeneity to be present, we performed meta‐analysis with a random‐effects model in addition to a fixed‐effect model, presenting the results of both models and performing sensitivity analyses to investigate differences in trial characteristics.
Sensitivity analysis
Misclassification of seizure type is a recognised problem in epilepsy, whereby some people with generalised seizures have been mistakenly classed as having focal onset seizures, and vice versa. There is clinical evidence that individuals with generalised onset seizures are unlikely to have an age of onset greater than 25 to 30 years (Malafosse 1994). Such misclassification affected the results of three reviews in our series of pair‐wise reviews for monotherapy in epilepsy comparing carbamazepine to phenobarbitone, phenytoin and sodium valproate, in which around 30% to 50% of participants analysed may have had their seizure type misclassified as generalised onset (Marson 2000; Nevitt 2017b; Nevitt 2018b). Given the overlap with studies contributing to this review and the other reviews within the series, we suspected that misclassification of seizure type could also be likely in this review, and so we examined the distribution of age at onset for individuals with generalised seizures.
De Silva 1996 was a paediatric study and Mattson 1985 recruited participants with focal seizures only, so there were no participants with new‐onset generalised seizures over the age of 30 in these studies. Twenty‐nine out of 72 individuals (42%) with generalised onset seizures were over the age of 30 in Heller 1995, and six out of 29 individuals (21%) with generalised onset seizures were over the age of 30 in Ogunrin 2005. Therefore out of 150 participants from the four studies providing IPD, 35 (23%) may have been wrongly classified as having new‐onset generalised seizures.
We undertook the following two sensitivity analyses to investigate misclassification for each outcome:
-
We reclassified the 35 individuals with generalised seizure types and age at onset greater than 30 into an 'uncertain seizure type' group.
-
We reclassified the 35 individuals with generalised seizures and age of onset greater than 30 as having focal seizures.
'Summary of findings' tables and certainty of the evidence (GRADE)
For the 2015 update, we added two 'Summary of findings' tables to the review (outcomes in the tables decided before the update started based on clinical relevance).
summary of findings Table for the main comparison reports the primary outcome of 'Time to treatment failure' in the subgroups of participants with focal onset seizures, generalised onset seizures and overall adjusted by seizure type.
summary of findings Table 2 reports the secondary outcomes of 'Time to first seizure' and 'Time to 12‐month remission' in the subgroups of participants with focal onset seizures, generalised onset seizures and overall adjusted by seizure type.
We determined the certainty of the evidence using the GRADE approach, where we downgraded evidence in the presence of high risk of bias in at least one trial, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results and high probability of publication bias. We downgraded evidence by one level if we considered the limitation serious and by two levels for very serious.
Results
Description of studies
Results of the search
For previous versions of the review, we identified 655 records from the databases and search strategies outlined in Electronic searches. We found three further records by handsearching and checking reference lists of included studies. We removed 265 duplicate records and screened 393 records (title and abstract) for inclusion in the review. We excluded 354 records based on title and abstract and assessed 39 full‐text articles for inclusion in the review. We excluded 16 studies (reported in 23 full‐text articles) from the review (see Excluded studies below) and included 11 trials (reported in 16 full‐text articles) in the review (see Included studies below).
For the 2019 update of this review we identified 41 records from the databases.We removed 12 duplicate records and screened 29 records (title and abstract) for inclusion in the review. All 29 records were clearly irrelevant and we excluded them.
See Figure 1 for PRISMA study flow diagram (Moher 2009) for the eligibility screening of all studies identified in searches for all versions of this review (previous searches and the most recent search in August 2018).
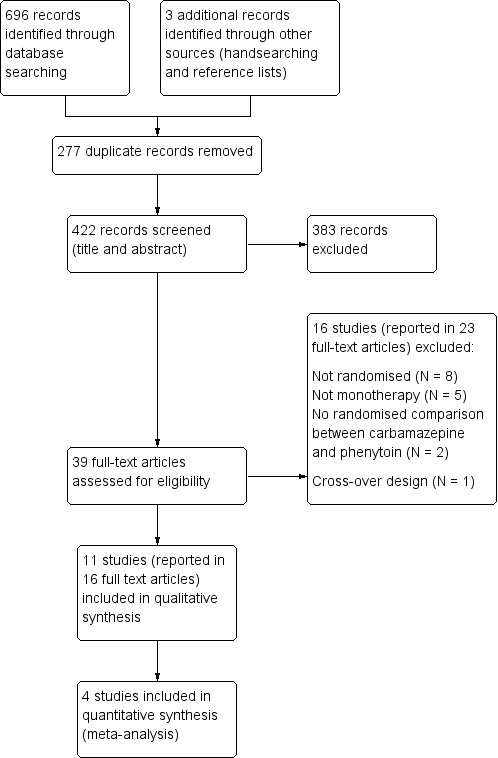
Study flow diagram.
Included studies
We included 11 trials in this review (Callaghan 1985; Czapinski 1997; De Silva 1996; Forsythe 1991; Heller 1995; Mattson 1985; Miura 1993; Ogunrin 2005; Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995). One trial was available in abstract form only (Czapinski 1997).
One trial recruited individuals of all ages (Callaghan 1985), three trials recruited children only (defined as under the age of 16 in De Silva 1996, and under the age of 14 in Forsythe 1991 and Miura 1993); and the remaining seven trials recruited adults only. Three trials defined adults as individuals above the age of 18 (Czapinski 1997; Mattson 1985; Ramsay 1983), one trial classed adults as older than 13 years (Heller 1995), two trials classed adults as older than 14 years (Ogunrin 2005; Ravi Sudhir 1995) and one trials classed adults as older than 15 years (Pulliainen 1994).
Nine trials recruited individuals with focal onset seizures and generalised onset seizures (Callaghan 1985; De Silva 1996; Forsythe 1991; Heller 1995; Miura 1993; Ogunrin 2005; Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995), and two trials recruited individuals with focal onset seizures only (Czapinski 1997; Mattson 1985). Ten trials recruited individuals with new‐onset seizures or previously untreated seizures, or both (Callaghan 1985; Czapinski 1997; De Silva 1996; Forsythe 1991; Heller 1995; Miura 1993; Ogunrin 2005; Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995). One trial recruited "previously untreated or under treated" individuals (Mattson 1985).
Six trials were conducted in Europe (Callaghan 1985; Czapinski 1997; De Silva 1996; Forsythe 1991; Heller 1995; Pulliainen 1994), two in the USA (Mattson 1985; Ramsay 1983), one in Nigeria (Ogunrin 2005), one in India (Ravi Sudhir 1995), and one in Japan (Miura 1993).
Individual participant data (IPD) could not be supplied for seven trials (Callaghan 1985; Czapinski 1997; Forsythe 1991; Miura 1993; Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995), in which 507 individuals had been randomised to either phenytoin or carbamazepine. None of these seven trials reported the specific time‐to‐event outcomes chosen for this systematic review.
Forsythe 1991 presented times at which the allocated drug was withdrawn and the reason for withdrawal in the trial publication for each individual. Hence, we were able to incorporate this trial into the analysis of 'Time to treatment failure’. For each participant, 'withdrawal and time of occurrence by month’ was presented; therefore, to calculate 'Time to treatment failure’ we assumed that, for example, if withdrawal occurred during the fifth month, that withdrawal occurred halfway between the fifth and sixth month (i.e. participants spent 167 full days on treatment before withdrawal).
We could not extract sufficient aggregate data from the trial publication in any other trial, and we therefore could not include them in data synthesis. Full details of outcomes considered and a summary of results in each eligible trial for which IPD were not available can be found in Table 1.
| Trial | Outcomes reported | Summary of results |
| 1. Seizure control: excellent (seizure‐free) | 1. PHT (n = 58); CBZ (n = 59) PHT: 39 (67%); CBZ: 22 (37%) | |
| 1. Proportion achieving 24‐month remission at 3 years 2. Proportion excluded after randomisation due to adverse effects or no efficacy | 1. PHT: 59%; CBZ: 62% 2. PHT: 23%; CBZ: 30% | |
| 1. Cognitive assessments 2. Withdrawals from randomised drug | 1. No significant differences between the two treatment groups on any cognitive tests | |
| 1. Proportion of all randomised participants with seizure recurrence (by seizure type) 2. Proportion of participants with optimum plasma levels with seizure recurrence (by seizure type) | PHT (n = 51); CBZ (n = 66) 1. PHT (focal): 10/31 (32%); PHT (generalised): 7/20 (35%); 2. PHT (focal): 4/17 (24%); PHT (generalised): 1/8 (13%); | |
| 1. Cognitive assessments (visual motor speed, co‐ordination, attention and concentration, verbal and visual‐spatial learning, visual and recognition memory, reasoning, mood, handedness) 2. Harmful side effects | 1. Compared to CBZ, participants on PHT became slower (motor speed of the hand) and their visual memory decreased. There was an equal decrease in negative mood (helplessness, irritability, depression) on PHT and CBZ
| |
| 1. Side effects (major and minor) 3. Laboratory results | 1. Incidence of:
2. Treatment failures among analysed participants: Seizure control (among analysed participants with no major side effects): PHT: 23/27 participants (86%); CBZ: 22/27 participants (82%) 3. Significantly lower mean LDH level at 24 weeks in CBZ participants than PHT participants (P < 0.01). Other laboratory results similar across treatment groups | |
| 1. Cognitive measures (verbal, performance, memory, visual‐motor, perceptomotor organisation, visual organisation, dysfunction) | 1. No significant differences between any tests of cognitive function taken before treatment and after 10 ‐ 12 weeks for both treatment groups |
CBZ = carbamazepine; LDH = lactate dehydrogenase; PHT= phenytoin
IPD were provided by trial authors for the four remaining trials which recruited 595 participants, representing 54% of individuals from 1102 individuals in all eligible trials (De Silva 1996; Heller 1995; Mattson 1985; Ogunrin 2005). Two trials (Mattson 1985; Ogunrin 2005) directly provided computerised data, and the authors of the other two trials (Heller 1995; De Silva 1996) supplied a combination of both computerised and paper‐based (although mostly computerised) data.
Data were available for the following subject characteristics (percentage of 595 participants with data available): seizure type (100%), sex (99%, missing for two participants in Mattson 1985), age at randomisation (98%, data missing for three participants from Mattson 1985 and Heller 1995), drug randomised (99%, data missing for six participants in De Silva 1996), time since first seizure to randomisation (98%, data missing for eight participants from Mattson 1985 and Heller 1995), number of seizures in six months prior to randomisation (93%, data missing for 41 participants, all 37 participants from Ogunrin 2005 and four participants from Mattson 1985 and Heller 1995). See the Characteristics of included studies table and Table 2 for further details.
| Trial | Focal seizures: n (%) | Male participants: | Age at entry (years): Mean (SD), rangeb | Epilepsy duration | Number of seizures in prior 6 months, median (range)d | |||||
| CBZ | PHT | CBZ | PHT | CBZ | PHT | CBZ | PHT | CBZ | PHT | |
| 29 (54%) | 30 (56%) | 30 (56%) | 34 (63%) | 9.2 (3.8) 2 to 15 | 9.5 (3.4) (3 to 16) | 1.7 (2.6), 0 to 12 | 1.0 (2.1) (0 to 14) | 3 (1 to 500) | 3 (1 to 404) | |
| 24 (39%) | 28 (44%) | 30 (49%) | 34 (54%) | 29.3 (14.1) 13 to 69 | 33.5 (14.3) (14 to 72) | 4.4 (7.4), 0.1 to 40 | 3.8 (5.4) (0 to 24) | 2 (1 to 354) | 2 (1 to 575) | |
| 155 (100%) | 165 (100%) | 133 (87%) | 145 (88%) | 42.1 (15.9) 18 to 82 | 40.8 (15.3) (18 to 81) | 5.9 (9.1), 0.5 to 55 | 6.6 (9.1) (0.5 to 59) | 1 (1 to 100) | 1 (1 to 26) | |
| 5 (26%) | 3 (17%) | 12 (63%) | 11 (61%) | 28.2 (5.8) 14 to 38 | 18.8 (2.6) (15 to 26) | NA | NA | 18 (6 to 36) | 12 (6 to 18) | |
n: number of participants; CBZ: carbamazepine; NA: not available; PHT:phenytoin SD: standard deviation
aSex was missing for two participants on CBZ from Mattson 1985.
bAge at randomisation was missing for two participants on CBZ from Mattson 1985 and one participant on CBZ from Heller 1995.
cEpilepsy duration was missing for 41 participants; all 37 participants from Ogunrin 2005, three participants on CBZ from Mattson 1985, one participant on CBZ from Heller 1995.
dNumber of seizures in the prior six months was missing for eight participants, seven participants from Mattson 1985 (three participants on PHT and four on CBZ), one participant on CBZ from Heller 1995.
eRandomised drug missing for six participants in De Silva 1996.
The results of neurological examinations were provided for 326 participants (55%) from three trials (De Silva 1996; Heller 1995; Ogunrin 2005), electroencephalographic (EEG) results were provided for 316 participants (53%) from one trial (Mattson 1985) and computerised tomography/magnetic resonance imaging (CT/MRI) results were provided for 324 participants (54%) in two trials (Mattson 1985; Ogunrin 2005).
Excluded studies
We excluded six studies which were not RCTs (Bird 1966; Kuzuya 1993; Rysz 1994; Sabers 1995; Shorvon 1978; Zeng 2010). We excluded seven trials which did not use carbamazepine and phenytoin in monotherapy (Bittencourt 1993; Canadian Study 1998; Hakami 2012; Kosteljanetz 1979; Rajotte 1967; Simonsen 1976; Troupin 1975). We excluded two trials which did not make a randomised comparison between carbamazepine and phenytoin monotherapy (Kaminow 2003; Shakir 1980), and we excluded one trial which had a cross‐over design (Cereghino 1974). See Characteristics of excluded studies for further details.
Risk of bias in included studies
For further details see Characteristics of included studies, Figure 2 and Figure 3.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
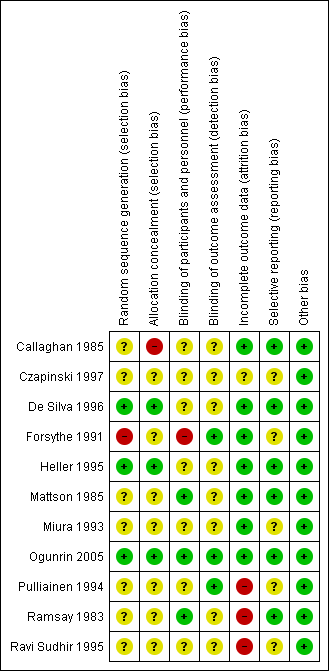
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Trials for which individual participant data (IPD) were provided
Three trials reported adequate methods of randomisation and allocation concealment; two trials used permuted blocks to generate a random list and concealed allocation by using sealed opaque envelopes (De Silva 1996; Heller 1995), and one trial used number tables to generate a random list and concealed allocation by allocating the randomised drug on a different site from where participants were randomised (Ogunrin 2005). We judged all three trials to be at low risk of selection bias. One trial reported only that participants were randomised with stratification for seizure type (Mattson 1985); no further information was provided in the study publication or from the authors about the methods of generating the random list and concealment of allocation, so we rated this trial at unclear risk of selection bias.
Trials for which no IPD were available
Two trials reported inadequate methods of randomisation and allocation concealment; Forsythe 1991 reported a method of quota allocation and did not report how allocation was concealed, and Callaghan 1985 reported a method of randomisation and allocation concealment based on two Latin squares which seems to take into account the drug preference of participants (the “drug of first preference” was selected from the randomisation list on a sequential basis); we judged both of these trials to be at high risk of selection bias. The remaining five trials (Czapinski 1997; Miura 1993; Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995) reported that the participants were "randomised" or "randomly allocated" etc., but did not provide information on the method of generation of the random list or of allocation concealment, and we judged them to be at unclear risk of selection bias.
Blinding
Trials for which IPD were provided
One trial double‐blinded participants and personnel using an additional blank tablet (low risk of performance bias; Mattson 1985), but it is unclear if the outcome assessor was blinded in this trial (unclear risk of detection bias). One trial blinded participants and the outcome assessors who performed cognitive testing (low risk of performance and detection bias), but a research assistant recruiting participants and providing counselling on medication adherence was not blinded (Ogunrin 2005). Two trials were unblinded for “practical and ethical reasons” (De Silva 1996; Heller 1995), but it is unclear whether the outcomes of these trials were influenced by the lack of masking.
Trials for which no IPD were available
One trial double‐blinded participants and personnel using an additional blank tablet (low risk of performance bias; Ramsay 1983), but it is unclear if the outcome assessor was blinded in this trial (unclear risk of detection bias). Two trials single‐blinded the outcome assessor who performed cognitive testing (low risk of detection bias); in one of these trials (Forsythe 1991) the participants and personnel were unblinded (high risk of performance bias), and in the other (Pulliainen 1994) it was unclear if the participants and personnel were blinded or not (unclear risk of performance bias). The remaining four trials (Callaghan 1985; Czapinski 1997; Miura 1993; Ravi Sudhir 1995) did not provide any information on masking of participants, personnel or outcome assessors, so we judged these trials to be at unclear risk of performance bias and detection bias.
Incomplete outcome data
Trials for which IPD were provided
In theory, a review using IPD should overcome issues of attrition bias, as unpublished data can be provided, unpublished outcomes calculated and all randomised participants can be analysed by an intention‐to‐treat approach. All four trials (De Silva 1996; Heller 1995; Mattson 1985; Ogunrin 2005) provided IPD for all randomised individuals and reported the extent of follow‐up for each individual, so we judged all four trials to be at low risk of attrition bias. We queried any missing data with the original study authors. From the information provided by the authors, we deemed the small amount of missing data (Included studies) to be missing at random and that they did not have an effect on our analysis.
Trials for which no IPD were available
Three trials reported attrition rates and analysed all randomised participants using an intention‐to‐treat approach, and we judged them to be at low risk of attrition bias (Callaghan 1985; Forsythe 1991; Miura 1993). One trial reported attrition rates, but it was unclear if all participants were analysed, so we rated this trial at unclear risk of attrition bias (Czapinski 1997). Three studies excluded between 20% and 35% of participants from the final analysis for “non‐compliance”, loss to follow‐up or uncontrolled seizures, and included only those who completed the analysis. This approach is not intention‐to‐treat, so we deemed these three studies to be at high risk of bias (Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995)
Selective reporting
We requested study protocols in all IPD requests, but protocols were not available for any of the 11 included trials, so we made a judgement of the risks of bias based on the information included in the publications, or from the IPD we received (see Characteristics of included studies for more information).
Trials for which IPD were provided
In theory, a review using IPD should overcome issues of reporting biases, as unpublished data can be provided and unpublished outcomes calculated. We acquired sufficient IPD to calculate the four outcomes ('Time to treatment failure', 'Time to six‐month remission','Time to 12‐month remission' and 'Time to first seizure’) for three of the four trials (De Silva 1996; Heller 1995; Mattson 1985). The study duration of Ogunrin 2005 was 12 weeks and all randomised participants completed the study without withdrawing, so we could only calculate 'Time to first seizure' for this study. We judged all four studies to be at low risk of reporting bias.
Trials for which no IPD were available
Seizure outcomes or adverse events, or both, were fully reported in three trials and we judged these trials to be at low risk of reporting bias (Callaghan 1985; Miura 1993; Ramsay 1983). Two trials reported cognitive outcomes and adverse events, but no seizure outcomes (Forsythe 1991; Pulliainen 1994), and one trial reported cognitive outcomes only, but no adverse events or seizure outcomes (Ravi Sudhir 1995); however, as no protocols were available for these three trials, we do not know whether seizure outcomes or recording of adverse events, or both, were planned a priori. One trial was in abstract form only and did not provide sufficient information to assess selective reporting bias (Czapinski 1997). We judged all of these trials to be at unclear risk of reporting bias.
Other potential sources of bias
We did not identify any other potential source of bias in any of the 11 included studies.
Effects of interventions
See: Summary of findings for the main comparison Carbamazepine compared with phenytoin (time to treatment failure); Summary of findings 2 Carbamazepine compared with phenytoin (secondary outcomes)
We have provided a summary of the outcomes reported in trials for which no IPD were available in Table 2.
See Table 3 for details about the number of individuals contributing IPD to each analysis, summary of findings Table for the main comparison for a summary of the results for the primary outcome 'Time to treatment failure' (stratified by seizure type), and summary of findings Table 2 for a summary of results for the secondary outcomes 'Time to first seizure' and 'Time to 12‐month remission'.
| Trial | Number randomised | Time to treatment failure (any reason, adverse events, lack of efficacy) | Time to 12‐month remission | Time to 6‐month remission | Time to first seizure | ||||||||||
| PHT | CBZ | Total | PHT | CBZ | Total | PHT | CBZ | Total | PHT | CBZ | Total | PHT | CBZ | Total | |
| 54 | 54 | 108 | 53 | 53 | 106 | 54 | 54 | 108 | 54 | 54 | 108 | 54 | 54 | 108 | |
| 63 | 61 | 124 | 61 | 60 | 121 | 63 | 61 | 124 | 63 | 61 | 124 | 63 | 61 | 124 | |
| 165 | 155 | 320 | 165 | 154 | 319 | 165 | 154 | 319 | 165 | 154 | 319 | 162 | 151 | 313 | |
| 20 | 23 | 43 | 20 | 23 | 43 | Information not available | Information not available | Information not available | |||||||
| 18 | 19 | 37 | Information not available | Information not available | Information not available | 18 | 19 | 37 | |||||||
| Total | 320 | 312 | 632 | 299 | 290 | 589 | 282 | 269 | 551 | 282 | 269 | 551 | 297 | 285 | 582 |
CBZ = carbamazepine, PHT= phenytoin
aIndividual participant data (IPD) supplied for 114 participants recruited in De Silva 1996; randomised drug not recorded in six participants. Reasons for treatment failure not available for two participants (one randomised to CBZ and one to PHT); these participants are not included in analysis of time to treatment failure.
bReasons for treatment failure not available for three participants (one randomised to CBZ and two to PHT) in Heller 1995; these participants are not included in analysis of time to treatment failure.
cNo follow‐up data after randomisation available for one participant randomised to CBZ in Mattson 1985. Data on seizure recurrence not available for six additional participants (three randomised to CBZ and three to PHT); these participants are not included in the analysis of Time to first seizure.
dIPD for 'Time to treatment failure' available in the study publication of Forsythe 1991. Data for other outcomes not available.
eStudy duration of Ogunrin 2005 is 12 weeks, so six‐ and 12‐month remission of seizures could not be achieved and cannot therefore be calculated. All randomised participants completed the study without withdrawing from treatment, so time to treatment failure cannot be analysed.
Survival curve plots are shown in Figure 4; Figure 5; Figure 6; Figure 7; Figure 8; Figure 9; Figure 10; Figure 11; Figure 12; Figure 13; Figure 14 and Figure 15 .

Time to treatment failure ‐ any reason related to the treatment (CBZ: carbamazepine; PHT: phenytoin)

Time to treatment failure ‐ any reason related to the treatment, by epilepsy type (CBZ: carbamazepine; PHT: phenytoin)

Time to treatment failure due to adverse events (CBZ: carbamazepine; PHT: phenytoin)

Time to treatment failure due to adverse events, by epilepsy type (CBZ: carbamazepine; PHT: phenytoin)

Time to treatment failure due to lack of efficacy (CBZ: carbamazepine; PHT: phenytoin)

Time to treatment failure due to lack of efficacy, by epilepsy type (CBZ: carbamazepine; PHT: phenytoin)

Time to first seizure (CBZ: carbamazepine; PHT: phenytoin)
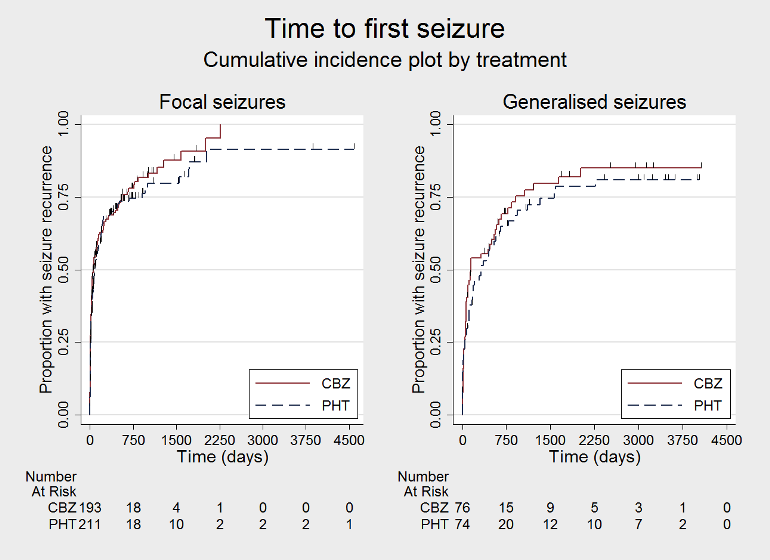
Time to first seizure, by epilepsy type (CBZ: carbamazepine; PHT: phenytoin)
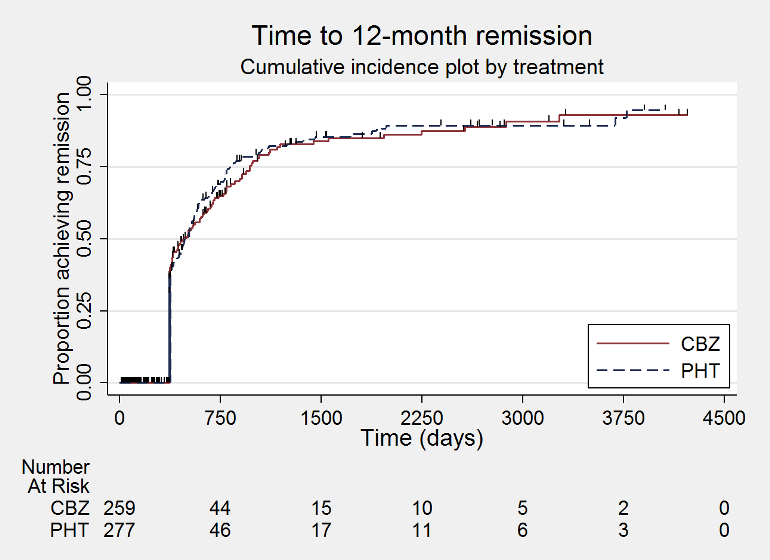
Time to 12 month remission (CBZ: carbamazepine; PHT: phenytoin)

Time to 12 month remission, by epilepsy type (CBZ: carbamazepine; PHT: phenytoin)
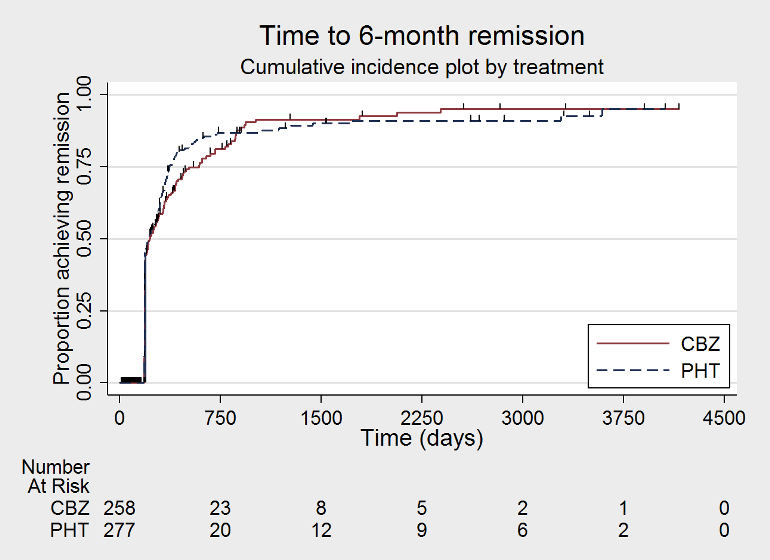
Time to 6 month remission (CBZ: carbamazepine; PHT: phenytoin)
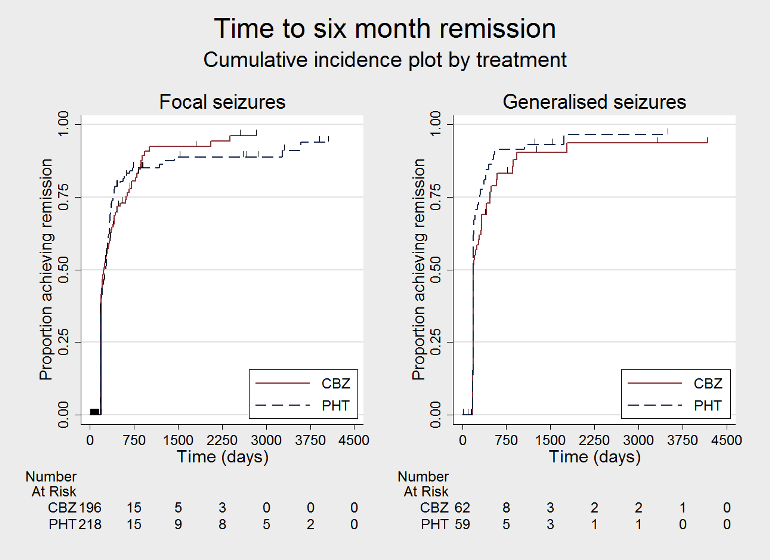
Time to 6 month remission, by epilepsy type (CBZ: carbamazepine; PHT: phenytoin)
We used Stata software version 14 to produce all survival curve plots using data from all trials providing IPD combined (Stata 2015). We note that participants with event times of zero (i.e. those who experienced treatment failure or experienced seizure recurrence on the day of randomisation) are not included in the 'Numbers at risk' on the graphs and that data are not stratified by trial within these survival curve plots. All figures are intended to provide a visual representation of outcomes, extent of follow‐up and visual differences between seizure types. These graphs are not intended to show statistical significance and numerical values may vary compared to the text due to differences in methodology.
We calculated all hazard ratios (HRs) presented below by generic inverse variance meta‐analysis and all HRs presented are calculated with a fixed‐effect model unless otherwise stated. All analyses met the assumption of proportional hazards (addition of time‐varying covariate into the model non‐significant), unless otherwise stated.
Primary outcomes
Time to treatment failure (retention time)
For this outcome, a HR less than one indicates a clinical advantage for carbamazepine.
Time to treatment failure and reason for treatment failure or withdrawal were available for 546 participants from three of the four trials providing IPD: 99% of 558 participants from De Silva 1996, Heller 1995 and Mattson 1985 (see Included studies), and 49.5% of the 1102 participants from the 11 included studies. Although two participants failed treatment (one in each group) in De Silva 1996, a reason for treatment failure was not available and could not be determined from the case notes. Similarly in Heller 1995, for one participant taking carbamazepine, the reason for treatment failure was not available and could not be determined from case notes. Also in Heller 1995, two participants (both on phenytoin) had reasons for treatment failure recorded but no date of treatment failure. We have not included these five participants with missing reasons for treatment failure or treatment failure dates from the two trials in analysis of time to treatment failure. Sufficient IPD were available in the published report for a further 43 participants from one trial (Forsythe 1991). Therefore, 589 participants from four trials were available for the analysis of this outcome (see Table 3). See Table 4 for reasons for premature termination of allocated treatment and how we classified these treatment failures or withdrawals in analysis.
| Reason for early termination | Heller 1995a,b | Totalc | |||||||||
| CBZ | PHT | CBZ | PHT | CBZ | PHT | CBZ | PHT | CBZ | PHT | Total | |
| Adverse events (Event) | 3 | 2 | 4 | 1 | 8 | 1 | 11 | 8 | 26 | 12 | 38 |
| Seizure recurrence (Event) | 12 | 10 | 2 | 1 | 5 | 8 | 3 | 6 | 22 | 25 | 47 |
| Both seizure recurrence and adverse events (Event) | 6 | 5 | 0 | 0 | 4 | 2 | 31 | 33 | 31 | 40 | 81 |
| Non‐compliance/participant choice (Event) | 0 | 0 | 3 | 4 | 0 | 0 | 11 | 26 | 14 | 30 | 44 |
| Participant went into remission (Censored) | 18 | 24 | 0 | 0 | 6 | 14 | 0 | 0 | 24 | 38 | 62 |
| Lost to follow‐up (Censored) | 0 | 0 | 0 | 0 | 0 | 0 | 26 | 19 | 26 | 19 | 45 |
| Death (Censored)d | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 5 | 4 | 5 | 9 |
| Other (Censored)e | 0 | 0 | 0 | 0 | 0 | 0 | 16 | 11 | 16 | 11 | 27 |
| Completed the study (did not withdraw) (Censored) | 14 | 12 | 14 | 14 | 37 | 38 | 53 | 57 | 118 | 121 | 239 |
| Total | 53 | 53 | 23 | 20 | 60 | 63 | 155 | 165 | 281 | 301 | 592 |
n = number of individuals contributing to the outcome 'Time to treatment failure’
aOne participant for Heller 1995 (CBZ) and two for De Silva 1996 (one PHT and one CBZ) have missing reasons for treatment failure.
bTwo participants from Heller 1995 (both PHT) had missing treatment failure times and did not contribute to analysis, but reasons for treatment failure are given.
cAll participants in Ogunrin 2005 completed the study without withdrawing, so this study did not contribute to 'Time to treatment failure'.
dDeath due to reasons not related to the study drug.
eOther reasons from Mattson 1985: participants developed other medical disorders including neurological and psychiatric disorders.
Out of the 592 participants for whom we had reasons for treatment failure or withdrawal (De Silva 1996; Forsythe 1991; Heller 1995; Mattson 1985), 353 participants prematurely withdrew from treatment (60% of total participants): 173 out of 291 participants randomised to carbamazepine (59%), and 180 out of 301 participants randomised to phenytoin (60%).
We deemed 210 participants (59% of total treatment failures) to have withdrawn for reasons related to the trial drug, 103 (60%), on carbamazepine and 107 (59%), on phenytoin, and we classed these reasons as 'events' in analysis. The most common treatment‐related reason for treatment failure was a combination of adverse events and lack of efficacy: 81 withdrawals (39% of total treatment failures), 41 (40% of total treatment failures) on carbamazepine and 40 (37% of total treatment failures) on phenytoin. Non‐compliance with treatment or participant choice was the treatment‐related reason in 21% of total treatment failures, lack of efficacy in 22% of total treatment failures and adverse events in 18% of total treatment failures.
We classed the other 143 reasons (70 on carbamazepine and 73 on phenytoin), which were mostly participants going into remission (43% of other withdrawals) and losses to follow‐up (31% of other withdrawals), to be not related to the treatment and censored these participants in the analysis, in addition to the 239 participants (118 on carbamazepine and 121 on phenytoin) who completed the trial without withdrawing or failing treatment.
Considering 'Time to treatment failure for any reason related to the treatment', the overall pooled HR (for 589 participants in four trials) was 0.99 (95% CI 0.76 to 1.31; P = 0.97; moderate‐certainty evidence; Analysis 1.1), indicating no clear advantage to either drug. No important heterogeneity was present between trials (I2 = 3%).
Considering 'Time to treatment failure due to adverse events' (all other reasons for treatment failure or treatment withdrawal censored in analysis), the overall pooled HR (for 589 participants in four trials) was 1.35 (95% CI 0.93 to 1.95; P = 0.12; moderate‐certainty evidence; Analysis 1.2), suggesting a potential advantage for phenytoin, which is not statistically significant; in other words, treatment failure due to adverse events may occur earlier on carbamazepine than phenytoin but we cannot rule out a slight advantage to carbamazepine or no difference between the drugs.
A moderate amount of heterogeneity was present between trials (I2 = 40%). From visual inspection of the forest plot of Analysis 1.2, the HRs of two trials were around 1.15 to 1.16 (De Silva 1996; Mattson 1985) while the HRs of the other two trials were much larger (HRs of 3.83 and 4.57 respectively) and confidence intervals of the HRs were very wide (Forsythe 1991; Heller 1995). Table 4 shows an imbalance between the drugs between the number of participants failing treatment due to adverse events in Forsythe 1991 and Heller 1995; very few participants on phenytoin failed treatment due to adverse events compared to participants on carbamazepine in these trials. This explains the extreme and imprecise HRs for these two trials and may explain the moderate amount of heterogeneity between trials.
Considering 'Time to treatment failure due to lack of efficacy' (all other reasons for treatment failure or treatment withdrawal censored in analysis), the overall pooled HR (for 589 participants in four trials) was 1.02 (95% CI 0.72 to 1.44; P = 0.92; moderate‐certainty evidence; Analysis 1.3), indicating no clear advantage to either drug. No heterogeneity was present between trials (I2 = 0%).
Subgroup analyses: seizure type (focal versus generalised onset)
Treatment failure data for 43 participants extracted from Forsythe 1991 did not distinguish between epilepsy type (focal onset or generalised onset) and we therefore could not include them in the meta‐analysis stratified by epilepsy type.
Considering 'Time to treatment failure for any reason related to the treatment', for individuals with focal onset seizures (428 participants from three trials), the pooled HR was 0.83 (95% CI 0.61 to 1.13; P = 0.23; I2 = 0%; moderate‐certainty evidence), suggesting a potential advantage for carbamazepine which is not statistically significant. For individuals with generalised onset seizures (118 participants from two trials), the pooled HR was 2.38 (95% CI 1.04 to 5.47; P = 0.04; I2 = 0%; low‐certainty evidence), indicating a statistically significant advantage for phenytoin; in other words, for individuals with generalised seizures, carbamazepine treatment was withdrawn significantly earlier than phenytoin in the two included trials, but the confidence interval around the pooled HR was wide so we are unsure of the magnitude of the advantage to phenytoin. There was statistically significant evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (test of subgroup differences: P = 0.02; I2 = 81.7%; Analysis 1.4).
The overall pooled HR (adjusted by epilepsy type for 546 participants from three trials) was 0.94 (95% CI 0.70 to 1.26; P = 0.68; moderate‐certainty evidence; Analysis 1.4). This result is similar to the unadjusted pooled HR (Analysis 1.1), and conclusions remain unchanged following the exclusion of 43 individuals in the stratified analysis (Forsythe 1991). Heterogeneity present within analysis has increased from I2= 3% to I2= 35%, probably due to the observed interaction between epilepsy type and treatment effect. This is explored further in the section on 'Sensitivity analysis' (see below).
Considering 'Time to treatment failure due to adverse events', for individuals with focal onset seizures (428 participants from three trials), the pooled HR was 1.19 (95% CI 0.80 to 1.78; P = 0.38; I2 = 0%; moderate‐certainty evidence), suggesting a potential advantage for phenytoin which is not statistically significant. For individuals with generalised onset seizures (118 participants from two trials), the pooled HR was 2.31 (95% CI 0.68 to 7.81; P = 0.18; I2 = 60%; low‐certainty evidence), suggesting a potential advantage for phenytoin, but the confidence interval is wide, so we cannot rule out an advantage to carbamazepine or no difference between drugs. There was no evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (test of subgroup differences: P = 0.31; I2 = 2.2%; Analysis 1.5).
There was a large amount of heterogeneity between trials (I2 = 60%) and when we repeated the analysis with a random‐effects model, the confidence interval around the pooled HR becomes even wider, at 2.82 (95% CI 0.37 to 21.32; P = 0.32). This heterogeneity is probably due to the imbalance between the drugs between the number of participants failing treatment due to adverse events; very few participants on phenytoin failed treatment due to adverse events compared to participants on carbamazepine in Heller 1995, while the numbers of participants failing each drug in De Silva 1996 were more balanced (see Table 4).
The overall pooled HR (adjusted by epilepsy type for 546 participants from three trials) was 1.27 (95% CI 0.87 to 1.86; P = 0.21; I2 = 3%; moderate‐certainty evidence; Analysis 1.5). This result is similar to the unadjusted pooled HR (Analysis 1.2), and conclusions remain unchanged following the exclusion of participants from Forsythe 1991.
Considering 'Time to treatment failure due to lack of efficacy', for individuals with focal onset seizures (428 participants from three trials), the pooled HR was 0.88 (95% CI 0.60 to 1.30; P = 0.52; I2 = 0%; moderate‐certainty evidence), suggesting a potential advantage for carbamazepine which is not statistically significant. For individuals with generalised onset seizures (118 participants from two trials), the pooled HR was 1.86 (95% CI 0.74 to 4.67; P = 0.19; I2 = 0%; low‐certainty evidence), suggesting a potential advantage for phenytoin, but the confidence interval is wide, so we cannot rule out an advantage to carbamazepine or no difference between drugs. There was no evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (test of subgroup differences: P = 0.14; I2= 53.2%; Analysis 1.6).
The overall pooled HR (adjusted by epilepsy type for 546 participants from three trials) was 0.99 (95% CI 0.69 to 1.41; P = 0.94; I2 = 0%; moderate‐certainty evidence; Analysis 1.6). This result is similar to the unadjusted pooled HR (Analysis 1.3), and conclusions remain unchanged following the exclusion of participants from Forsythe 1991.
Sensitivity analysis
We conducted sensitivity analyses to investigate misclassification of seizure type, reclassifying 29 individuals from Heller 1995 aged 30 or older with new‐onset generalised seizures to focal onset seizures or to an uncertain seizure type. The results of the two sensitivity analyses are shown in Table 5. It was not possible to estimate a HR and 95% CI for 'Time to treatment failure due to adverse events' or 'Time to treatment failure due to lack of efficacy' for the 29 participants reclassified as an uncertain seizure type, as none of these participants failed phenytoin treatment due to adverse events or failed carbamazepine treatment due to lack of efficacy.
| Outcome | Original analysis | Generalised onset and age at onset > 30 years classified as focal onset | Generalised onset and age at onset > 30 years classified as uncertain seizure type | |||
| Pooled HR (95% CI) fixed‐effect model | Test of subgroup differences | Pooled HR (95% CI) fixed‐effect model | Test of subgroup differences | Pooled HR (95% CI) fixed‐effect model | Test of subgroup differences | |
| Time to treatment failure (for any reason related to treatment)a | F: 0.83 (0.61 to 1.13), I2 = 0% G: 2.38 (1.04 to 5.47), I2 = 0% O: 0.94 (0.70 to 1.26), I2 = 35% | Chi2 = 5.45, df = 1, P = 0.02, I2 = 81.7% | F: 0.88 (0.65 to 1.19), I2 = 0% G: 1.96 (0.81 to 4.78), I2 = 0% O: 0.96 (0.72 to 1.27), I2 = 6% | Chi2 = 2.83, df = 1, P = 0.09, I2 = 64.6% | F: 0.83 (0.61 to 1.13), I2 = 0% G: 1.96 (0.81 to 4.78), I2 = 0% U: 5.23 (0.47 to 58.71), I2 = NA O: 0.93 (0.70 to 1.24), I2 = 7% | Chi2 = 5.24, df = 2, P = 0.07, I2 = 61.8% |
| Time to treatment failure due to adverse eventsa | F: 1.19 (0.80 to 1.78), I2 = 0% G: 2.31 (0.68 to 7.81), I2 = 60% O: 1.27 (0.87 to 1.86), I2 = 3% | Chi2 = 1.02, df = 1, P = 0.31, I2 = 2.2% | F: 1.25 (0.84 to 1.86), I2 = 22% G: 1.72 (0.51 to 5.87), I2 = 0% O: 1.29 (0.88 to 1.88), I2 = 0% | Chi2 = 0.24, df = 1, P = 0.62, I2 = 0% | F: 1.19 (0.80 to 1.78), I2 = 0% G: 1.72 (0.51 to 5.87), I2 = 0% U: Not estimablec O: 1.24 (0.85 to 1.81), I2 = 0% | Chi2 = 0.31, df = 2, P = 0.86, I2 = 0% |
| Time to treatment failure due to lack of efficacya | F: 0.88 (0.60 to 1.30), I2 = 0% G: 1.86 (0.74 to 4.67), I2 = 0% O: 0.99 (0.69 to 1.41), I2 = 0% | Chi2 = 2.14, df = 1, P = 0.14, I2 = 53.2% | F: 0.91 (0.62 to 1.34), I2 = 0% G: 1.81 (0.68 to 4.82), I2 = 0% O: 1.00 (0.70 to 1.43), I2 = 0% | Chi2 = 1.64, df = 1, P = 0.20, I2 = 38.9% | F: 0.88 (0.60 to 1.30), I2 = 0% G: 1.81 (0.68 to 4.82), I2 = 0% U: Not estimablec O: 0.97 (0.68 to 1.40), I2 = 0% | Chi2 = 1.78, df = 2, P = 0.41, I2 = 0% |
| Time to first seizureb | F: 1.13 (0.89 to 1.43), I2 = 0% G: 1.19 (0.81 to 1.75), I2 = 45% O: 1.15 (0.94 to 1.40), I2 = 0% | Chi2 = 0.05, df = 1, P = 0.83, I2 = 0% | F: 1.15 (0.91 to 1.44), I2 = 0% G: 1.19 (0.77 to 1.84), I2 = 53% O: 1.16 (0.94 to 1.42), I2 = 0% | Chi2 = 0.02, df = 1, P = 0.88, I2 = 0% | F: 1.13 (0.89 to 1.43), I2 = 0% G: 1.19 (0.77 to 1.84), I2 = 53% U: 0.82 (0.29 to 2.34), I2 = NA O: 1.13 (0.92 to 1.39), I2 = 0% | Chi2 = 0.41, df = 2, P = 0.82, I2 = 0% |
| Time to 12‐month remissiona | F: 1.06 (0.80 to 1.42), I2 = 0% G: 0.88 (0.58 to 1.33), I2 = 73% O: 1.00 (0.79 to 1.26), I2 = 16% | Chi2 = 0.54, df = 1, P = 0.46, I2 = 0% | F: 1.10 (0.84 to 1.45), I2 = 0% G: 0.69 (0.43 to 1.11), I2 = 0% O: 0.98 (0.77 to 1.24), I2 = 0% | Chi2 = 2.79, df = 1, P = 0.09, I2 = 64.2% | F: 1.06 (0.80 to 1.42), I2 = 0% G: 0.69 (0.43 to 1.11), I2 = 0% U: 1.91 (0.74 to 4.90), I2 = NA O: 0.99 (0.78 to 1.25), I2 = 15% | Chi2 = 4.32, df = 2, P = 0.12, I2 = 53.7% |
| Time to 6‐month remissiona | F: 0.98 (0.75 to 1.27), I2 = 0% G: 0.77 (0.52 to 1.13), I2 = 39% O: 0.90 (0.73 to 1.12), I2 = 0% | Chi2 = 1.03, df = 1, P = 0.31, I2 = 3.2% | F: 0.98 (0.76 to 1.26), I2 = 0% G: 0.59 (0.37 to 0.93), I2 = 0% O: 0.87 (0.70 to 1.09), I2 = 15% | Chi2 = 3.63, df = 1, P = 0.06, I2 = 72.5% | F: 0.98 (0.75 to 1.27), I2 = 0% G: 0.59 (0.37 to 0.93), I2 = 0% U: 1.20 (0.51 to 2.83), I2 = NA O: 0.88 (0.71 to 1.10), I2 = 2% | Chi2 = 4.01, df = 2, P = 0.13, I2 = 50.1% |
Chi2: Chi2 statistic; CI: confidence interval; df: degrees of freedom of Chi2 distribution; F: focal epilepsy; G: generalised epilepsy; HR: Hazard Ratio; O: overall (all participants); U: uncertain epilepsy; P: P value (< 0.05 are classified as statistically significant)
a29 participants reclassified to focal epilepsy or uncertain epilepsy type from Heller 1995.
b35 participants reclassified to focal epilepsy or uncertain epilepsy type from Heller 1995 and Ogunrin 2005.
cHR and 95% CI not estimable as no participants with uncertainty epilepsy type failed carbamazepine treatment due to lack of efficacy or failed phenytoin treatment due to adverse events in Heller 1995.
Considering 'Time to treatment failure for any reason related to the treatment', following reclassification, for the remaining 89 participants with generalised onset seizures the pooled HR was 1.96 (95% CI 0.81 to 4.78; P = 0.14; I2 = 0%), which still indicates a potential advantage for phenytoin, but this advantage is no longer statistically significant. Reclassifying these 29 participants as having new‐onset focal seizures, the pooled HR for 457 participants is 0.88 (95% CI 0.65 to 1.19; P = 0.40; I2 = 0%), indicating a potential slight advantage for carbamazepine, which is not statistically significant.
Overall, following this reclassification for all participants adjusted for epilepsy type, the numerical result was very similar, indicating no clear difference between the drugs (pooled HR 0.96, 95% CI 0.72 to 1.27; P = 0.76). However, following reclassification, heterogeneity in the analysis of all participants was greatly reduced, from I2 = 35% to I2 = 6%, and the observed interaction between seizure type (generalised versus focal onset) and treatment effect is no longer statistically significant (test of subgroup differences: P = 0.09; I2 = 64.6%).
Overall results for all participants adjusted for epilepsy type were similar when the 29 participants were reclassified as of uncertain seizure type, again indicating no clear difference between the drugs (pooled HR 0.93, 95% CI 0.70 to 1.24; P = 0.63). Again following this reclassification, heterogeneity in the analysis of all participants was greatly reduced, from I2 = 35% to I2 = 7%, and the observed interaction between seizure type (generalised versus focal onset) and treatment effect is no longer statistically significant (test of subgroup differences: P = 0.07; I2 = 61.8%).
Considering time to treatment failure due to adverse events, following reclassification, for the remaining 89 participants with generalised onset seizures, conclusions were unchanged suggesting a potential advantage to phenytoin but the confidence interval is wide, so we cannot rule out an advantage to carbamazepine or no difference between drugs (pooled HR 1.72, 95% CI 0.51 to 5.87). Within the subgroup of participants with generalised onset seizures, heterogeneity was reduced from I2 = 60% to I2 = 0% as the reclassification of participants resulted in a more balanced number of participants failing phenytoin or failing carbamazepine treatment in Heller 1995. Reclassifying the 29 participants as having new‐onset focal seizures, the pooled HRs for 457 participants with focal onset seizures and for all participants adjusted for epilepsy type were similar to the original analyses, and conclusions were unchanged (see Table 5).
Considering 'Time to treatment failure due to lack of efficacy', following reclassification, the pooled HRs for the remaining 89 participants with generalised onset seizures, for 457 participants with focal onset seizures and for all participants adjusted for epilepsy type were similar to the original analyses and conclusions were unchanged (see Table 5).
Secondary outcomes
Time to first seizure post‐randomisation
For this outcome, a HR greater than one indicates a clinical advantage for carbamazepine.
Data for 582 participants (99% of 558 randomised participants from De Silva 1996, Heller 1995 and Mattson 1985 (see Included studies), 100% from Ogunrin 2005, and 53% of the 1102 participants from the 11 included studies) from all four trials providing IPD were available for the analysis of this outcome.
Three hundred and eighty‐three out of 582 participants (66%) experienced a recurrence of seizures: 192 out of 297 (64%) on phenytoin and 191 out of 285 on carbamazepine (67%). The overall pooled HR (for 582 participants) was 1.13 (95% CI 0.92 to 1.39; P = 0.23; moderate‐certainty evidence; Analysis 1.7), suggesting a potential advantage for phenytoin, which is not statistically significant; in other words, seizure recurrence may occur earlier on carbamazepine than phenytoin but we cannot rule out a slight advantage to carbamazepine or no difference between the drugs. No important heterogeneity was present between trials (I2 = 34%).
Subgroup analyses: seizure type (focal versus generalised onset)
For the participants with focal onset seizures (432 participants, four trials), the pooled HR was 1.13 (95% CI 0.89 to 1.43; P = 0.32; I2 = 0%, moderate‐certainty evidence), indicating a slight advantage for phenytoin, which is not statistically significant; in other words, seizure recurrence may occur earlier on carbamazepine than on phenytoin, but we cannot rule out a slight advantage to carbamazepine or no difference between drugs.
For the participants with generalised onset seizures (150 participants, three trials), the pooled HR was 1.19 (95% CI 0.81 to 1.75; P = 0.38; I2 = 0%; moderate‐certainty evidence), again indicating a slight advantage for phenytoin, which is not statistically significant. A moderate amount of statistical heterogeneity was present between trials for generalised onset seizures (I2 = 45%). This heterogeneity is explored further in the 'Sensitivity analysis' section (see below).
Overall, the pooled HR (adjusted for epilepsy type for 582 participants, four trials) was 1.15 (95% CI 0.94 to 1.40; P = 0.19), suggesting a slight advantage for phenytoin, which is not statistically significant. There was no evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (test of subgroup differences: P = 0.83; I2 = 0%; Analysis 1.8).
Sensitivity analysis
We conducted sensitivity analyses to investigate misclassification of seizure type, reclassifying 35 individuals from Heller 1995 and Ogunrin 2005 aged 30 or older with new‐onset generalised seizures to focal onset seizures or to an uncertain seizure type. The results of the two sensitivity analyses are shown in Table 5.
Following reclassification, the pooled HRs for the remaining 115 participants with generalised onset seizures, for 457 participants with focal onset seizures and for all participants adjusted for epilepsy type were similar to the original analyses and conclusions were unchanged (see Table 5). Following reclassification, heterogeneity in the subgroup of participants with generalised onset seizures is increased from I2 = 45% to I2 = 53%, so it does not appear that this heterogeneity is due to misclassification of seizure type.
Considering other potential reasons for the heterogeneity in the subgroup of participants with generalised onset seizures, there is a difference in the direction of effects of the three studies, with De Silva 1996 and Ogunrin 2005 showing a potential advantage for phenytoin and Heller 1995 showing a potential advantage for carbamazepine, yet all study‐specific HRs have wide confidence intervals due to small numbers of participants with generalised onset seizures. From correspondence with the study authors, we know that De Silva 1996 and Heller 1995 were conducted under the same protocol and therefore trial characteristics should be homogeneous; the only difference between the two studies is within the age groups recruited (De Silva 1996 recruited children only and Heller 1995 recruited adults only). We therefore performed a further subgroup analysis by adult versus paediatric studies (Ogunrin 2005 also recruited adults only). For 101 adults with generalised onset seizures, the pooled HR was 0.98 (95% CI 0.60 to 1.61; P = 0.94), indicating no clear advantage for either drug, and for 49 children with generalised onset seizures in De Silva 1996 the HR was 1.59 (95% CI 0.86 to 2.94; P = 0.14), indicating a potential advantage for phenytoin, which is not statistically significant. The test for interaction between age groups recruited (adults versus children) and treatment effect was not significant (P = 0.23; I2 = 30.9%). However, participant numbers with generalised onset seizures are quite limited in this review, so we may not have had the power to detect a difference between age groups.
In Ogunrin 2005 there is an indication that the proportional hazards assumption may be violated (see Data synthesis); the P value of time‐varying covariate is 0.02 and visual inspection of the cumulative incidence plot (Figure 16) shows clear crossing of the curves at around 10 days. In other words, up to 10 days seizure recurrence seems to be occurring earlier on phenytoin, but this changes earlier with carbamazepine after 10 days.
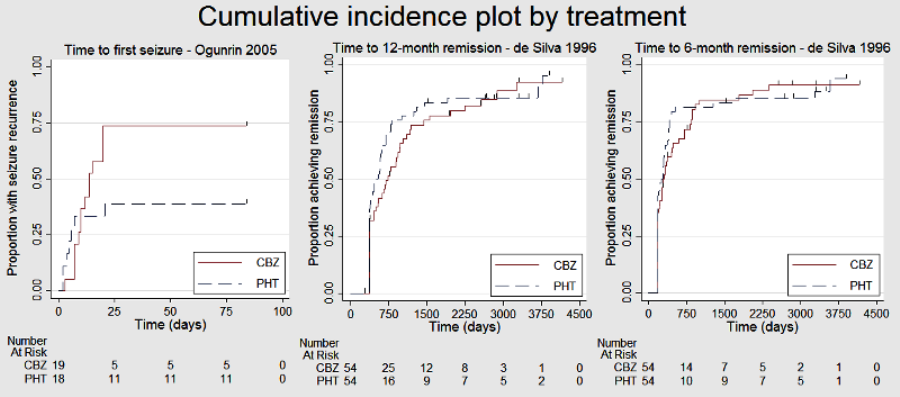
Cumulative incidence plots, proportional hazards assumption checks
As a sensitivity analysis, we fitted a piecewise Cox regression model to investigate any change in treatment effect over time, assuming proportional hazards within each interval. From the visual inspection of Figure 16, the follow‐up period of Ogunrin 2005 is split into two intervals; 0 to 10 days and over 10 days (maximum follow‐up is 84 days). We can estimate separate HRs for each interval as follows:
-
For the interval 0 to 10 days (13 events in 37 participants at risk) the HR is 0.67 (95% CI 0.20 to 2.22; P = 0.51), suggesting a potential advantage for carbamazepine, which is not statistically significant.
-
For intervals over 10 days (eight events in 24 participants at risk) the HR is 3.13 (95% CI 1.09 to 9.09; P = 0.03), suggesting a large statistically significant advantage for phenytoin. Visual inspection of Figure 16 also shows a clear advantage for phenytoin after 10 days.
These results suggest some indication of a change in treatment effect over time, with a slight early advantage for carbamazepine, changing to a large statistically significant advantage for phenytoin later in the study, and support the hypothesis of a change in treatment effect over time for Ogunrin 2005. Ogunrin 2005 is by far the shortest of the studies for which we have IPD (maximum follow‐up was 84 days in Ogunrin 2005 compared to maximum follow‐up of 3995 days in Heller 1995, 4589 days in De Silva 1996 and 1838 days in Mattson 1985), and we did not find statistically significant evidence of a difference between carbamazepine and phenytoin for 'Time to first seizure after randomisation' in any of the three studies with a longer duration (see Analysis 1.7). The apparent large advantage for phenytoin from 10 to 84 days in Ogunrin 2005, may therefore have reduced in size or even changed direction to favour carbamazepine if this study had continued for a longer duration.
Time to achieve 12‐month remission
For this outcome, a HR less than one indicates a clinical advantage for phenytoin.
Data for 551 participants (99% of 558 randomised participants from De Silva 1996, Heller 1995 and Mattson 1985 (see Included studies) and 50% of the 1102 participants from the 11 included studies) from three out of four trials providing IPD were available for the analysis of this outcome. Individuals were followed up for a maximum of 12 weeks in Ogunrin 2005, so could not contribute to this outcome.
Two hundred and eighty‐nine out of 551 participants (52%) achieved 12‐month remission: 155 out of 282 (55%) on phenytoin and 134 out of 269 (50%) on carbamazepine. The overall pooled HR (for 551 participants) was 1.01 (95% CI 0.80 to 1.27; P = 0.95; moderate‐certainty evidence; Analysis 1.9), indicating no clear differences between the drugs. No heterogeneity was present between trials (I2 = 0%).
Subgroup analyses: seizure type (focal versus generalised onset)
For the participants with focal onset seizures (430 participants, three trials), the pooled HR was 1.06 (95% CI 0.80 to 1.42; P = 0.68; I2 = 0%; moderate‐certainty evidence), indicating no clear advantage for either drug. For the participants with generalised onset seizures (121 participants, two trials), the pooled HR was 0.88 (95% CI 0.58 to 1.33; P = 0.54; I2 = 73%; moderate‐certainty evidence), indicating a potential advantage for phenytoin, which is not statistically significant; in other words, 12‐month remission may occur earlier on phenytoin than carbamazepine, but we cannot rule out a slight advantage to carbamazepine or no difference between drugs. There was substantial heterogeneity present between the two trials for individuals with generalised onset seizures (I2 = 73%). When we repeated the analysis with a random‐effects model, the pooled HR was 0.86 (95% CI 0.39 to 1.89; P = 0.70). This heterogeneity is explored further in the 'Sensitivity analysis' section below. There was no evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (test of subgroup differences: P = 0.46; I2 = 0%; Analysis 1.10).
Overall, the pooled HR (adjusted for epilepsy type for 551 participants in three trials) was 1.00 (95% CI 0.79 to 1.26; P = 0.99; I2 = 16%; moderate‐certainty evidence), indicating no clear advantage for either drug.
Sensitivity analysis
We conducted sensitivity analyses to investigate misclassification of seizure type, reclassifying 29 individuals from Heller 1995 aged 30 or older with new‐onset generalised seizures to focal onset seizures or to an uncertain seizure type. The results of the two sensitivity analyses are shown in Table 5.
The pooled HR for 92 participants with generalised onset seizures was 0.69 (95% CI 0.43 to 1.11; P = 0.13; I2 = 0%), showing that all of the heterogeneity in Analysis 1.10 is explained by misclassification of participants with generalised onset seizures. Reclassifying the 29 participants as having new‐onset focal seizures, the pooled HRs for 461 participants with focal onset seizures and for all participants adjusted for epilepsy type were similar to the original analyses and conclusions were unchanged (see Table 5).
In De Silva 1996 there is an indication that the proportional hazards assumption may be violated (see Data synthesis); the P value of time‐varying covariate is 0.051 and visual inspection of the cumulative incidence plot (Figure 16) shows crossing of the curves at around 2500 days. In other words, up to 2500 days, participants on phenytoin seem to be achieving 12‐month remission quicker than those on carbamazepine, but this changes after 2500 days; however, participant numbers are small (15 participants at risk out of 108 randomised), so small changes may be magnified in this case.
As a sensitivity analysis, we fitted a piecewise Cox regression model to investigate any change in treatment effect over time, assuming proportional hazards within each interval. From the visual inspection of Figure 16, the follow‐up period of De Silva 1996 is split into two intervals; 0 to 2500 days and over 2500 days (maximum follow‐up was 4163 days). We can estimate separate HRs for each interval as follows:
-
For the interval 0 to 2500 days (88 events in 108 participants at risk) the HR is 0.78 (95% CI 0.51 to 1.19; P = 0.23), suggesting a potential advantage for phenytoin, which is not statistically significant.
-
For the interval over 2500 days (five events in 15 participants at risk) the HR is 1.59 (95% CI 0.64 to 4.00; P = 0.32), suggesting a potential advantage for carbamazepine, which is not statistically significant.
These results suggest some indication of a change in treatment effect over time, with a potential advantage for phenytoin earlier on in the study, changing to a potential advantage for carbamazepine later in the study. However, CIs of estimates are wide, particularly for the HR after 2500 days, due to small numbers of events and participants at risk, and it is likely that the observed change of direction in effect at around 2500 days is due to small participant numbers after this time.
Time to achieve six‐month remission
For this outcome, a HR less than one indicates a clinical advantage for phenytoin.
Data for 551 participants (99% of 558 randomised participants from De Silva 1996, Heller 1995 and Mattson 1985 (see Included studies) and 50% of the 1102 participants from the 11 included studies) from three out of four trials providing IPD were available for the analysis of this outcome. Individuals were followed up for a maximum of 12 weeks in Ogunrin 2005, so could not contribute to this outcome.
Three hundred and thirty‐eight out of 551 participants (61%) achieved six‐month remission: 179 out of 282 (63%) on phenytoin and 159 out of 269 (59%) on carbamazepine.
The overall pooled HR (for 551 participants) was 0.92 (95% CI 0.74 to 1.14; P = 0.45; moderate‐certainty evidence; Analysis 1.11), indicating no clear differences between the drugs. No heterogeneity was present between trials (I2 = 0%).
Subgroup analyses: seizure type (focal versus generalised onset)
For the participants with focal onset seizures (430 participants, three trials), the pooled HR was 0.98 (95% CI 0.75 to 1.27; P = 0.85; I2 = 0%; moderate‐certainty evidence), indicating no clear advantage for either drug. For the participants with generalised onset seizures (121, two trials), the pooled HR was 0.77 (95% CI 0.52 to 1.13; P = 0.18; I2 = 39%; moderate‐certainty evidence), indicating a potential advantage for phenytoin, which is not statistically significant; in other words, six‐month remission may occur earlier on phenytoin than carbamazepine, but we cannot rule out a slight advantage to carbamazepine or no difference between drugs. There was no evidence of an interaction between epilepsy type (focal onset versus generalised onset) and treatment effect (test of subgroup differences: P = 0.31; I2 = 3.2%; Analysis 1.12).
Overall, the pooled HR (adjusted for epilepsy type for 551 participants, three trials) was 0.90 (95% CI 0.73 to 1.12; P = 0.36; I2 = 0%; moderate‐certainty evidence), indicating a potential advantage for phenytoin, which is not statistically significant; in other words, six‐month remission may occur earlier on phenytoin than carbamazepine, but we cannot rule out a slight advantage to carbamazepine or no difference between drugs.
Sensitivity analysis
We conducted sensitivity analyses to investigate misclassification of seizure type, reclassifying 29 individuals from Heller 1995 aged 30 or older with new‐onset generalised seizures to focal onset seizures or an uncertain seizure type. The results of the two sensitivity analyses are shown in Table 5.
The pooled HR for 92 participants with generalised onset seizures was 0.59 (95% CI 0.37 to 0.93; P = 0.02; I2 = 0%), indicating that for these individuals, six‐month remission occurred significantly earlier on phenytoin than carbamazepine. As observed for 'Time to 12‐month remission', all of the heterogeneity in Analysis 1.12 is explained by misclassification of participants with generalised onset seizures. Reclassifying the 29 participants as having new‐onset focal seizures, the pooled HRs for 461 participants with focal onset seizures and for all participants adjusted for epilepsy type were similar to the original analyses, and conclusions were unchanged (see Table 5).
In De Silva 1996, there is an indication that the proportional hazards assumption may be violated (see Data synthesis); the P value of time‐varying covariate is 0.066 and visual inspection of the cumulative incidence plot (Figure 16) shows crossing of the curves at several points at around 1000 days, 1750 days and 3500 days, suggesting several changes in the direction of treatment effect over time. As in the sensitivity analysis of De Silva 1996 in 'Time to 12‐month remission', after 1000 days participant numbers are small (18 participants at risk out of 108 randomised), so small changes may be magnified in the later stages of study follow‐up.
As a sensitivity analysis, we fitted a piecewise Cox regression model to investigate any change in treatment effect over time, assuming proportional hazards within each interval. From the visual inspection of Figure 16, the follow‐up period of De Silva 1996 is split into three intervals; 0 to 1000 days, 1000 to 1750 days, and over 1750 days (maximum follow‐up is 4163 days). We did not consider an interval of 3500 days to the end of the study, due to very small participant numbers at this time (three participants at risk). We can estimate separate HRs for each interval as follows:
-
For the interval 0 to 1000 days (87 events in 108 participants at risk) the HR is 0.85 (95% CI 0.55 to 1.30; P = 0.44), suggesting a potential advantage for phenytoin, which is not statistically significant.
-
For the interval 1000 to 1750 days (three events in 18 participants at risk) the HR is 0.79 (95% CI 0.24 to 2.70; P = 0.71), again suggesting a potential advantage for phenytoin, which is not statistically significant.
-
For intervals over 1750 days (five events in 14 participants at risk) the HR is 1.32 (95% CI 0.72 to 2.44; P = 0.37), suggesting a potential advantage for carbamazepine, which is not statistically significant.
As above, these results suggest some indication of a change in treatment effect over time, with an advantage for phenytoin earlier on in the study, changing to an advantage for carbamazepine later in the study. However, CIs of estimates are again wide, due to small participant numbers in the later two intervals, so we do not have statistically significant evidence to support the hypothesis of a change in treatment effect over time for De Silva 1996, and conclude that the apparent changes of direction in effect at later stages of the study are likely to be due to small participant numbers.
Adverse events
We extracted all reported information related to adverse events from the study publications. Miura 1993 and Ravi Sudhir 1995 did not report any information on adverse events and we are uncertain without access to protocols if these data were collected (see Selective reporting (reporting bias)). See Table 6 for details of all adverse event data provided in the other nine studies included in this review. In summary, the adverse events reported by two or more studies in this review are as follows:
| Trial | Adverse event dataa | Summary of reported results | |
| Carbamazepine (CBZ) | Phenytoin (PHT) | ||
| All adverse events according to drug (note: no participants withdrew due to adverse events) | CBZ (n = 59): drowsiness (n = 2), rash (n = 3) | PHT (n = 58): gum hypertrophy (n = 2), rash (n = 2), ataxia (n = 2) | |
| “Exclusions” due to adverse events or no efficacy” | Proportion “excluded”: CBZ: 30% (out of 30 randomised to CBZ) | Proportion “excluded”: PHT: 23.3% (out of 30 randomised to PHT) | |
| “Unacceptable” adverse events leading to drug withdrawald | CBZ (n = 54): drowsiness (n = 1), blood dyscrasia (n = 1) | PHT (n = 54): drowsiness (n = 2), skin rash (n = 1), blood dyscrasia (n = 1), hirsutism (n = 1) | |
| Withdrawal due to adverse events (no other adverse event data reported) | 4 participants out of 23 randomised to CBZ withdrew for the following reasons (some withdrew for more than adverse event): slowing of mental function, headache, anorexia, nausea, abdominal pain, fatigue and drowsiness2 | 1 participant out of 20 randomised to PHT withdrew from the study due to depression and anorexia | |
| “Unacceptable” adverse events leading to drug withdrawald | CBZ (n = 61): drowsiness (n = 3), rash (n = 2), headache (n = 1), depression (n = 1) | PHT (n = 63): myalgia (n = 1), irritability (n = 1) | |
| Narrative report of ‘Adverse effects’ and ‘Serious side effects’ | CBZ (n = 155): motor disturbance (ataxia, incoordination, nystagmus, tremor: 33%); dysmorphic and idiosyncratic side effects (gum hypertrophy, hirsutism, acne and rash: 14%); gastrointestinal problems (27%); decreased libido or impotence (13%); No serious side effects | PHT (n = 165); motor disturbance (ataxia, inco‐ordination, nystagmus, tremor: 28%); dysmorphic and idiosyncratic side effects (gum hypertrophy, hirsutism, acne and rash: 22 %); gastrointestinal problems (24%); decreased libido or impotence (11%) 1 serious side effect – 1 participant has confirmed lymphoma, rash improved rapidly following discontinuation of PHT | |
| No adverse events reported | N/A | N/A | |
| Participant reported symptomatic complaints (provided as IPD) | CBZ (n = 19): memory impairment (n = 9) psychomotor retardation (n = 1) inattention (n = 1) transient rash (n = 1) CBZ‐induced cough (n = 1) | PHT (n = 18): memory impairment (n = 7) psychomotor retardation (n = 1) inattention (n = 2) transient rash (n = 1) | |
| Participant‐reported adverse events | 1 participant on CBZ complained of facial skin problems; 1 participant on CBZ complained of tiredness and memory problems | 3 participants on PHT complained of tiredness | |
| Major and minor side effects | CBZ (n = 35): Major side effects: rash (n = 1), pruritus (n = 1), impotence (n = 2), dizziness (n = 1), headaches (n = 1), impaired cognition (n = 1), elevated liver enzymes (n = 1) Mild side effects: nausea (33%), headaches (24%), cognitive impairment (33%), nystagmus (52%), sedation (33%), fine tremor (20%) | PHT (n = 35): Major side effects: rash (n = 4), exfoliative dermatitis (n = 1), impotence (n = 1), dizziness (n = 1), nausea/vomiting (n = 1) Mild side effects: nausea (38%), gingival hyperplasia (12%), headaches (32%), cognitive impairment (15%), nystagmus (40%), sedation (15%), fine tremor (28%) | |
| No adverse events reported | N/A | N/A | |
CBZ = carbamazepine, N/A = not available, PHT= phenytoin
aAdverse event data are recorded as reported narratively in the publications, so exact definition of a symptom may vary. Adverse event data supplied as IPD for Ogunrin 2005. Adverse event data were not requested in original IPD requests (De Silva 1996; Heller 1995; Mattson 1985) but will be for all future IPD requests. For numbers of treatment withdrawals due to adverse events in studies for which IPD were provided (De Silva 1996; Heller 1995; Mattson 1985) see Table 4.
bParticipants may report more than one adverse event.
cCzapinski 1997 is an abstract only, so very little information is reported.
dParticipants may have withdrawn due to adverse event alone or a combination of adverse events and poor efficacy (seizures).
For carbamazepine
-
Gastrointestinal side effects including abdominal pain, nausea and vomiting: (Forsythe 1991; Mattson 1985; Ramsay 1983).
-
Drowsiness/tiredness/fatigue/sedation: (Callaghan 1985; De Silva 1996; Forsythe 1991; Heller 1995; Pulliainen 1994; Ramsay 1983).
-
Rash: (Callaghan 1985; Heller 1995; Mattson 1985; Ogunrin 2005).
-
Decreased libido, or impotence, or both: (Mattson 1985; Ramsay 1983).
-
Headaches: (Forsythe 1991; Heller 1995; Ramsay 1983).
-
Motor disturbance (including ataxia, inco‐ordination, nystagmus, tremor, slowing of mental function, inattention, psychomotor retardation): (Forsythe 1991; Mattson 1985; Ogunrin 2005; Ramsay 1983).
-
Dysmorphic and idiosyncratic side effects (gum hypertrophy, hirsutism, acne, other skin problems): (Mattson 1985; Pulliainen 1994; Ramsay 1983).
-
Cognitive side effects and impairments, including depression and memory problems: (Heller 1995; Ogunrin 2005; Pulliainen 1994; Ramsay 1983).
For phenytoin
-
Gastrointestinal side effects including abdominal pain, nausea and vomiting: (Mattson 1985; Ramsay 1983).
-
Drowsiness/tiredness/fatigue/sedation: (De Silva 1996; Pulliainen 1994; Ramsay 1983).
-
Rash: (Callaghan 1985; De Silva 1996; Mattson 1985; Ogunrin 2005).
-
Decreased libido, or impotence, or both: (Mattson 1985; Ramsay 1983).
-
Motor disturbance (including ataxia, inco‐ordination, nystagmus, tremor, slowing of mental function, inattention, psychomotor retardation): (Callaghan 1985; Mattson 1985; Ogunrin 2005; Ramsay 1983).
-
Dysmorphic and idiosyncratic side effects (gum hypertrophy, hirsutism, acne, other skin problems): (Callaghan 1985; De Silva 1996; Mattson 1985; Ramsay 1983).
-
Cognitive side effects and impairments, including depression and memory problems: (Forsythe 1991; Ogunrin 2005; Ramsay 1983).
Because of the differences in methods of reporting adverse event data across the studies (see Table 6), it is difficult to summarise the 'most common' adverse events overall across the 11 studies, or to deduce whether carbamazepine or phenytoin are most associated with specific adverse events. Adverse event data for individuals were not included in the original IPD requests for earlier versions of this review, but will be sought in all future IPD requests.
Discussion
Summary of main results
The results of this review provide moderate‐certainty evidence that for our primary global effectiveness outcome 'Time to treatment failure for any reason related to treatment' (and also for 'Time to treatment failure due to lack of efficacy'), for all individuals, we found no clear differences between the drugs. The results of this review also provide moderate‐certainty evidence that for all individuals, treatment failure due to adverse events may occur earlier on carbamazepine than phenytoin, but we cannot rule out a slight advantage to carbamazepine or no difference between the drugs.
Considering differences between the subgroups of individuals with focal onset seizures and generalised onset seizures for 'Time to treatment failure for any reason related to treatment', given that subgroup sizes are unbalanced (118 with generalised seizures (22%) and 428 with focal seizures (78%) as classified by the studies) and that results may be confounded by misclassification of seizure type in up to 29 participants, we cannot draw any firm conclusions about an association between treatment and seizure type (i.e. an advantage for carbamazepine for individuals with focal onset seizures and an advantage for phenytoin for individuals with generalised onset seizures). We require more evidence, particularly from individuals with correctly classified generalised onset seizures, to inform this analysis. Furthermore, due to small numbers of participants within the two seizure‐type subgroups failing treatment due to adverse events or due to lack of efficacy, we cannot draw any firm conclusions about any differences between the drugs in the two seizure‐type subgroups for 'Time to treatment failure due to adverse events' or 'Time to treatment failure due to lack of efficacy'.
Similarly for the secondary outcomes 'Time to first seizure', 'Time to 12‐month remission', 'Time to six‐month remission', we found no consistent or statistically significant differences between phenytoin and carbamazepine for participants overall or by seizure type. Evidence for these outcomes was of moderate to low certainty. However, subgroups of participants by seizure type are again unbalanced in size, and misclassification of seizure types may have confounded analyses. More evidence is needed, particularly from individuals with correctly classified generalised seizures, to inform all of the outcomes of this review.
Limited information was available about adverse events in the trials and we could not compare the rates of adverse events between carbamazepine and phenytoin. Some adverse events reported on both drugs were abdominal pain, nausea, and vomiting, drowsiness, motor and cognitive disturbances, dysmorphic side effects (such as rash).
For all outcomes in this review, for all individuals and for the subgroups of participants with focal onset seizures and generalised onset seizures, we therefore encourage caution in the interpretation of the results and we would not encourage basing a choice between these two drugs on the results of this review alone.
Overall completeness and applicability of evidence
We believe our systematic electronic searches identified all relevant evidence for this review. We have gratefully received individual participant data (IPD) for 595 individuals (54% of individuals from all eligible trials) from the authors of four trials (De Silva 1996; Heller 1995; Mattson 1985; Ogunrin 2005), which included a comparison of phenytoin versus carbamazepine for the treatment of epilepsy. However, 484 individuals (44%) from six relevant trials (Callaghan 1985; Czapinski 1997; Miura 1993; Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995) could not be included in any analysis as IPD were not available and outcomes of interest were not reported in the published reports. Sufficient data for 23 individuals (2%) were published in one trial (Forsythe 1991) to contribute to analysis for the primary outcome 'Time to treatment failure', but insufficient data were available to include these individuals in the analyses by seizure type and the analyses of other outcomes. Having to exclude data from half of eligible participants due to lack of IPD and insufficient reporting in study publications is likely to have impacted on the applicability of the evidence, but it is difficult to quantify exactly how large this impact was on the results of this review (see Potential biases in the review process).
Three trials contributing around 80% of the participant data to this review recruited adults only (Heller 1995; Mattson 1985; Ogunrin 2005); the remaining study was a paediatric trial (De Silva 1996). Also, the largest single trial contributing over half of the participant data to this review (Mattson 1985) recruited individuals with focal onset seizures only, so that only around 25% of participants included in this review were experiencing generalised onset seizures. Furthermore, there is evidence within this review to suggest that up to 23% of individuals with new‐onset generalised seizures may have had their seizure type misclassified. For these reasons, the results of this review may not be fully generalisable to children or to individuals with generalised onset seizures, and more evidence is required from participants with generalised seizure types.
Quality of the evidence
The four trials for which IPD were available were generally at low risk of bias (see Figure 3). Three of the trials contributing around half of the participant data to this review described adequate methods of randomisation and allocation concealment (De Silva 1996; Heller 1995; Ogunrin 2005), but the largest single trial, contributing 54% of participant data (Mattson 1985), did not describe the method of randomisation and allocation concealment used, and this information was not available from study authors. We are uncertain whether this lack of information has impacted on the results of this review. Two of the trials providing IPD blinded participants and outcome assessors (Mattson 1985; Ogunrin 2005) and the other two trials (De Silva 1996; Heller 1995) were designed as pragmatic open‐label trials, as masking of treatment would not be “practicable or ethical", would “undermine compliance” and would have “introduced bias due to a very large drop‐out rate.” For the three trials providing treatment failure information, the treatment failure or withdrawal rate in the double‐blinded trial (Mattson 1985) was 40%, and the treatment failure or withdrawal rates were 36% and 24% in De Silva 1996 and Heller 1995 respectively (29.5% treatment failure/withdrawal rate overall in the two open‐label studies, which is statistically significantly lower than the treatment failure/withdrawal rate in the double‐blind study; P = 0.009). It is therefore debatable whether a double‐blind design is the most appropriate for trials of monotherapy in epilepsy of long duration and whether such a design does have an impact upon the dropout rate and therefore on the results of the trial. Further differences between the studies were in the population recruited (age of participants and seizure types). We discuss these differences below in Overall completeness and applicability of evidence.
Trials for which no IPD were available were generally of poorer quality than those for which we had IPD, with two studies describing inadequate methods of randomisation or allocation concealment (Callaghan 1985; Forsythe 1991), three trials presenting incomplete outcome data following exclusion of participants (Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995) and two trials providing very limited information on trial methodology, available only in abstract or summary form (Czapinski 1997; Miura 1993).
Overall, due to the documented methodological issues that may have introduced heterogeneity, biases and imprecision into our meta‐analyses, we rated the evidence provided in this review as of moderate certainty for all individuals and for individuals with focal onset seizures, and of low certainty for individuals with generalised onset seizures according to GRADE criteria (see summary of findings Table for the main comparison; summary of findings Table 2). We would not encourage basing a choice between these two drugs on the results of this review alone.
Potential biases in the review process
We were provided with IPD for 595 out of 1102 eligible participants (54%) from four out of 11 studies included; we conducted all analyses as IPD analyses. Such an approach has many advantages, such as allowing us to standardise definitions of outcomes across trials, and attrition and reporting biases being reduced as we can perform additional analyses and calculate additional outcomes from unpublished data. For the outcomes we used in this review which are of a time‐to‐event nature, an IPD approach is considered to be the ‘gold standard’ approach to analysis (Parmar 1998).
However, despite the advantages of this approach, for reasons out of our control we were not able to obtain IPD for 507 participants from seven eligible studies and except for one study of 43 participants reporting data which could contribute to our primary outcome 'Time to treatment failure' (Forsythe 1991), no aggregate data were available for our outcomes of interest in study publications. We therefore had to exclude around half of eligible participants from our analyses, which may have introduced bias into the review.
From the results reported in these seven studies (see Table 1 for narrative description of the results of each study), only one study showed a statistically significant difference in efficacy between carbamazepine and phenytoin for participants with generalised onset seizures (73% seizure‐free with phenytoin versus 39% seizure‐free with carbamazepine (Callaghan 1985)). There was no difference between treatments for participants with focal onset seizures (P = 0.006). Some significant differences between carbamazepine and phenytoin in terms of specific adverse events and cognitive adverse events were also reported (see Table 1). However, no consistent differences in efficacy or tolerability were reported in these seven studies, so it is unclear whether the exclusion of these studies from our meta‐analysis has impacted upon our results and conclusions. Furthermore, five of the seven studies that we could not include in meta‐analysis were at high risk of bias for at least one methodological aspect (see Figure 3), so inclusion of these data may have introduced bias into our results.
We have evidence from previous reviews conducted by the Cochrane Epilepsy Group (Marson 2000; Nevitt 2017b; Nevitt 2018b) that misclassification of seizure type is an important issue in epilepsy trials. We believe that the results of the original trials and hence the results of this meta‐analysis may have been confounded by classification bias, particularly the 35 individuals from two trials (Heller 1995; Ogunrin 2005) classified with new‐onset generalised seizures over the age of 30 (Malafosse 1994). Sensitivity analysis to investigate potential misclassification of these individuals may impact upon our conclusion for two outcomes ('Time to treatment failure' and 'Time to six‐month remission'), and explains all heterogeneity among individuals with generalised onset seizures for the outcomes 'Time to 12‐month remission' and 'Time to six‐month remission'. Both studies with potentially misclassified participants used the International League Against Epilepsy (ILAE) classification of 1981 (Commission 1981) to classify generalised onset and focal onset seizures. Heller 1995 was initiated before the publication of the revised ILAE classification in 1989 (Commission 1989), so some individuals in Heller 1995 may have been classified correctly according to Commission 1981 but misclassified by the revised Commission 1989. Ogunrin 2005 was initiated around 10 years after the publication of Commission 1989, but this study was conducted in Nigeria, a low‐income country without access to the same facilities as trials conducted in the USA and Europe; seizure types were therefore classified clinically, and electroencephalographs (EEGs)/magnetic resonance imaging (MRI) were not required for diagnosis of epilepsy. Clinical classification may have contributed to potential misclassification in this study.
Finally, we made some assumptions in the statistical methodology used in this review. Firstly, when we received only follow‐up dates and seizure frequencies, we used linear interpolation to estimate seizure times. We are aware that an individual's seizure patterns may be non‐linear; we therefore recommend caution when interpreting the numerical results of the seizure‐related outcomes. We also made an assumption that the treatment effect for each outcome did not change over time (proportional hazards assumption, see Data synthesis). For three of the outcomes, there was evidence that this assumption may have been violated for one of the trials. Sensitivity analysis showed that changes in treatment effect tended to occur in the later stages of the studies when small participant numbers were being followed up, so small changes in treatment effect would be magnified. Furthermore, we are aware that in trials of long duration (e.g. De Silva 1996, Heller 1995 and Mattson 1985 followed up participants for between three and 10 years), the assumption of treatment effect remaining constant over time may not be appropriate. For example, there is likely to be a difference between participants who achieve immediate remission compared with participants who achieve later remission, and we encourage that results should be interpreted with this limitation in mind.
Agreements and disagreements with other studies or reviews
No single trial included in this review has found convincing differences between phenytoin and carbamazepine with respect to seizure control or seizure type. To our knowledge, together with previous versions of this review, this is the only systematic review and meta‐analysis that compares carbamazepine and phenytoin monotherapy for focal onset seizures and generalised onset tonic‐clonic seizures. A network meta‐analysis has been published (Nevitt 2017a), comparing all direct and indirect evidence from carbamazepine, phenytoin and other standard and new antiepileptic drugs licensed for monotherapy. It also found no differences between carbamazepine and phenytoin for the outcomes specified in this review.
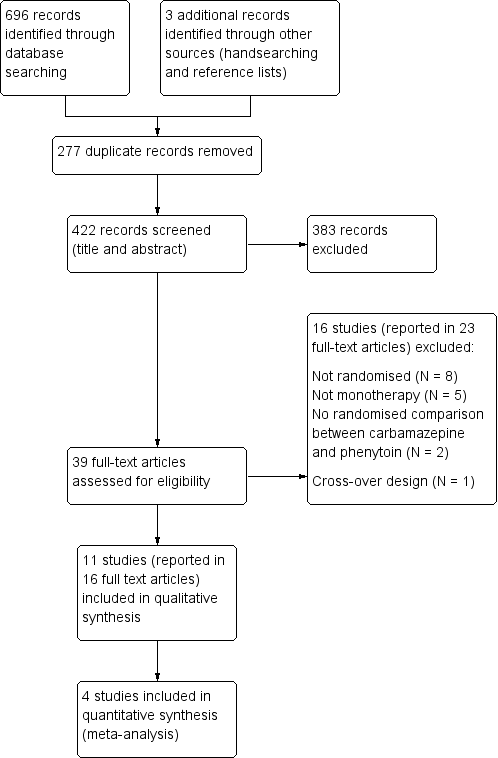
Study flow diagram.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
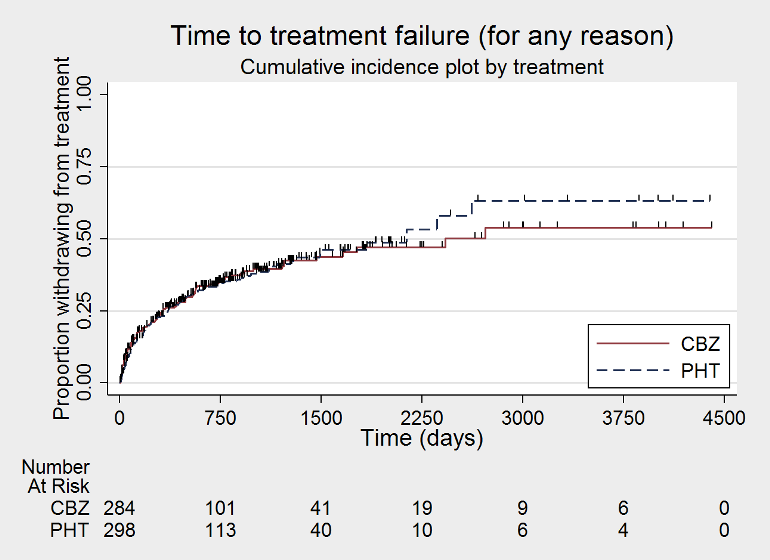
Time to treatment failure ‐ any reason related to the treatment (CBZ: carbamazepine; PHT: phenytoin)

Time to treatment failure ‐ any reason related to the treatment, by epilepsy type (CBZ: carbamazepine; PHT: phenytoin)

Time to treatment failure due to adverse events (CBZ: carbamazepine; PHT: phenytoin)
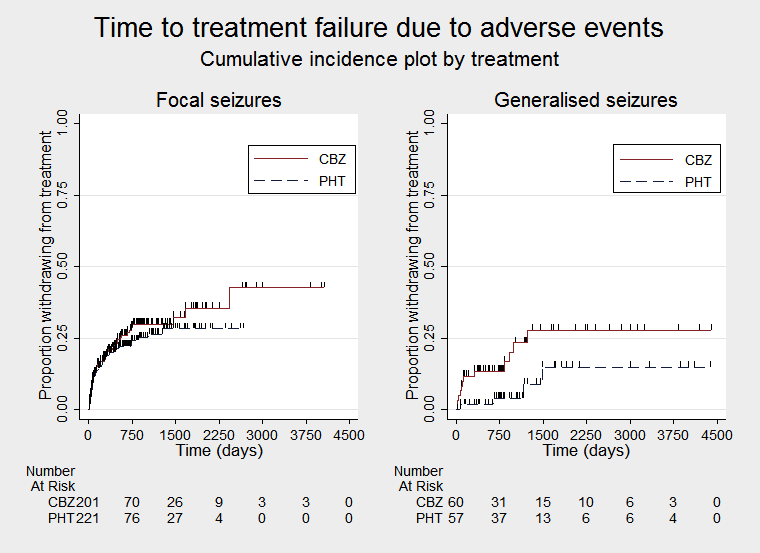
Time to treatment failure due to adverse events, by epilepsy type (CBZ: carbamazepine; PHT: phenytoin)

Time to treatment failure due to lack of efficacy (CBZ: carbamazepine; PHT: phenytoin)

Time to treatment failure due to lack of efficacy, by epilepsy type (CBZ: carbamazepine; PHT: phenytoin)
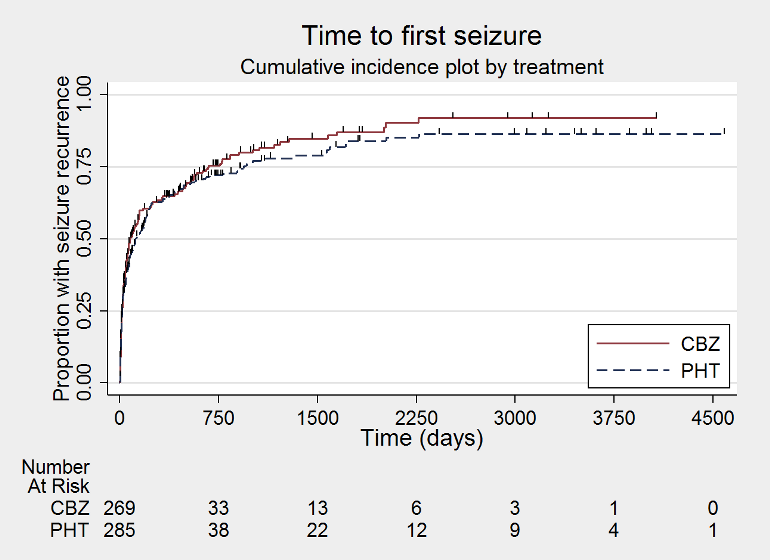
Time to first seizure (CBZ: carbamazepine; PHT: phenytoin)
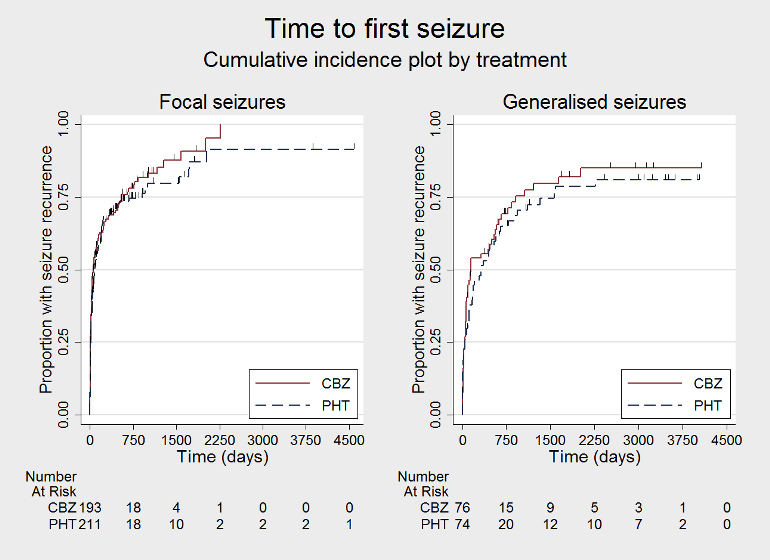
Time to first seizure, by epilepsy type (CBZ: carbamazepine; PHT: phenytoin)
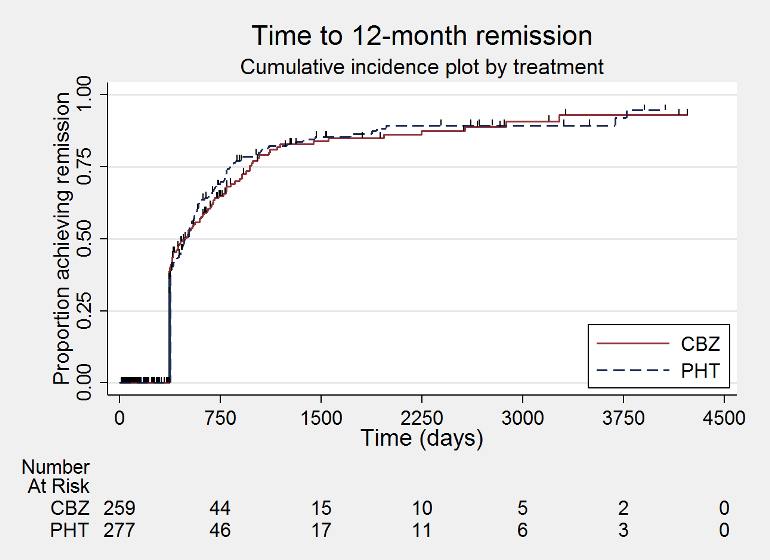
Time to 12 month remission (CBZ: carbamazepine; PHT: phenytoin)

Time to 12 month remission, by epilepsy type (CBZ: carbamazepine; PHT: phenytoin)
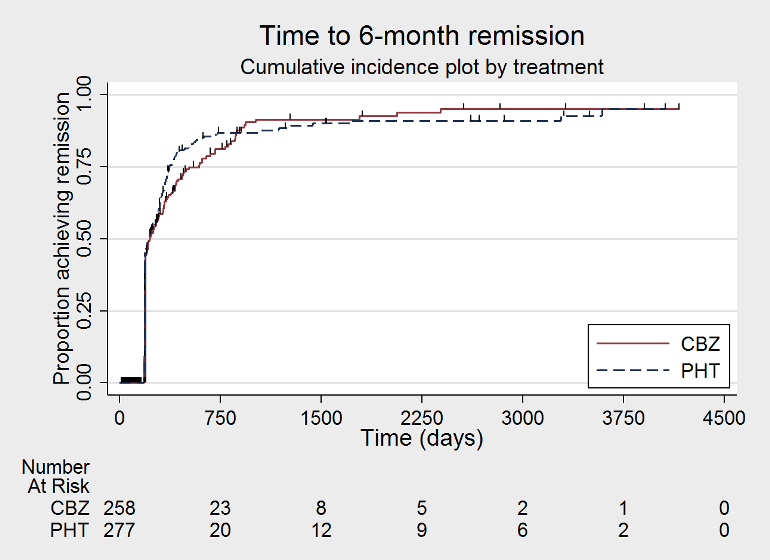
Time to 6 month remission (CBZ: carbamazepine; PHT: phenytoin)
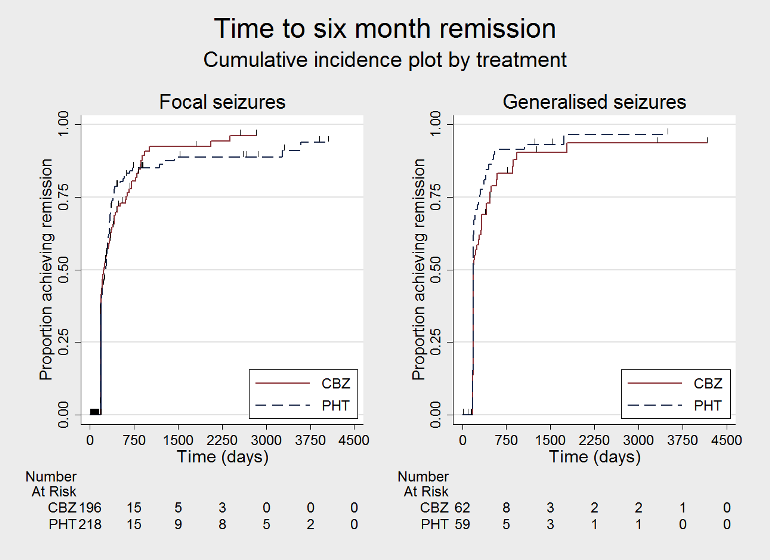
Time to 6 month remission, by epilepsy type (CBZ: carbamazepine; PHT: phenytoin)
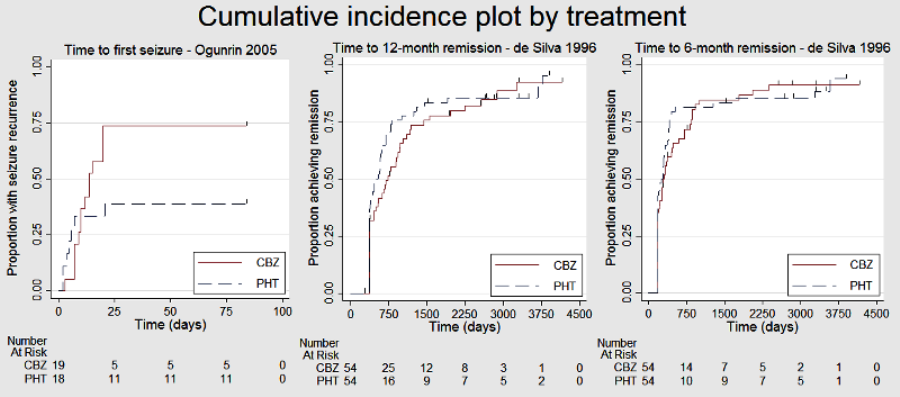
Cumulative incidence plots, proportional hazards assumption checks

Comparison 1 Carbamazepine (CBZ) versus phenytoin (PHT) monotherapy, Outcome 1 Time to treatment failure (any reason related to the treatment).

Comparison 1 Carbamazepine (CBZ) versus phenytoin (PHT) monotherapy, Outcome 2 Time to treatment failure due to adverse events.
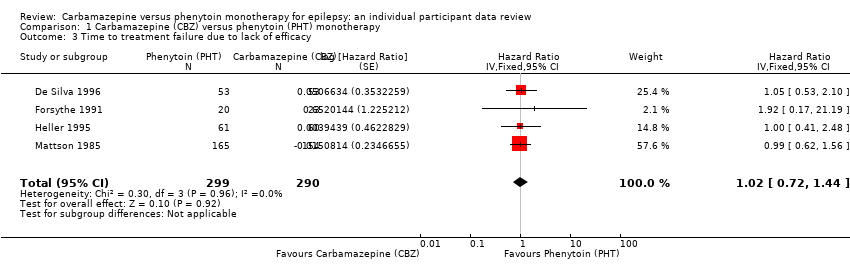
Comparison 1 Carbamazepine (CBZ) versus phenytoin (PHT) monotherapy, Outcome 3 Time to treatment failure due to lack of efficacy.

Comparison 1 Carbamazepine (CBZ) versus phenytoin (PHT) monotherapy, Outcome 4 Time to treatment failure (any reason related to the treatment) ‐ by epilepsy type.

Comparison 1 Carbamazepine (CBZ) versus phenytoin (PHT) monotherapy, Outcome 5 Time to treatment failure due to adverse events ‐ by epilepsy type.
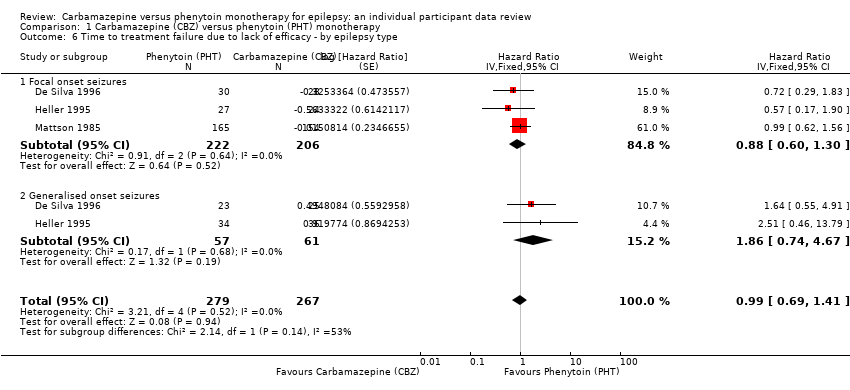
Comparison 1 Carbamazepine (CBZ) versus phenytoin (PHT) monotherapy, Outcome 6 Time to treatment failure due to lack of efficacy ‐ by epilepsy type.
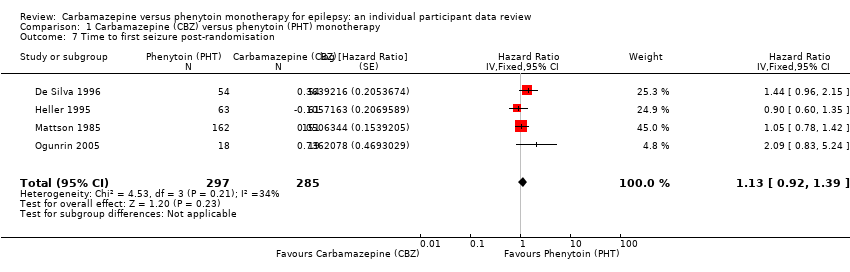
Comparison 1 Carbamazepine (CBZ) versus phenytoin (PHT) monotherapy, Outcome 7 Time to first seizure post‐randomisation.
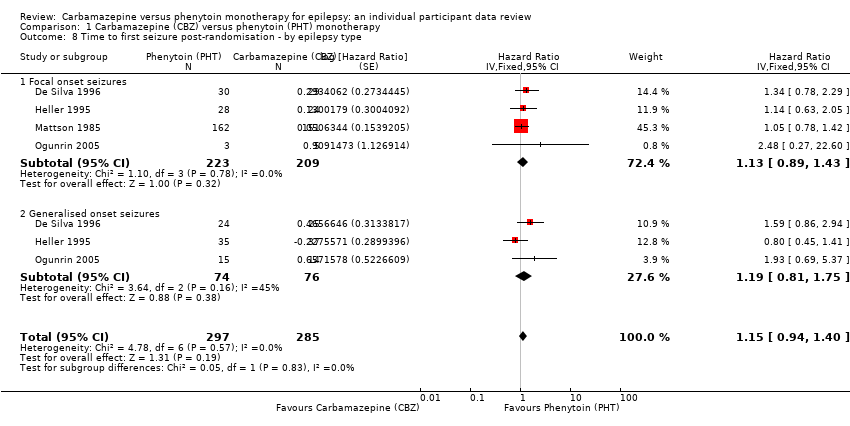
Comparison 1 Carbamazepine (CBZ) versus phenytoin (PHT) monotherapy, Outcome 8 Time to first seizure post‐randomisation ‐ by epilepsy type.

Comparison 1 Carbamazepine (CBZ) versus phenytoin (PHT) monotherapy, Outcome 9 Time to achieve 12‐month remission.
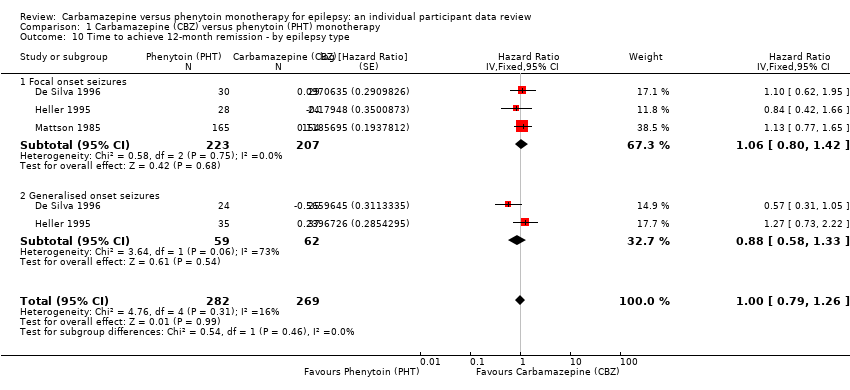
Comparison 1 Carbamazepine (CBZ) versus phenytoin (PHT) monotherapy, Outcome 10 Time to achieve 12‐month remission ‐ by epilepsy type.

Comparison 1 Carbamazepine (CBZ) versus phenytoin (PHT) monotherapy, Outcome 11 Time to achieve six‐month remission.

Comparison 1 Carbamazepine (CBZ) versus phenytoin (PHT) monotherapy, Outcome 12 Time to achieve six‐month remission ‐ by epilepsy type.
| Carbamazepine compared with phenytoin for epilepsy | ||||||
| Patient or population: adults and children with new‐onset focal or generalised epilepsy Settings: outpatients Intervention: carbamazepine Comparison: phenytoin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of Participants | Certainty (quality) of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenytoin | Carbamazepine | |||||
| Time to treatment failure (any reason related to treatment) Range of follow‐up:1 day to 4403 days | The median time to treatment failure was 2135 days in the phenytoin group | The median time to treatment failure was 2422 days (307 days longer) in the carbamazepine group | HR 0.94 (0.70 to 1.26)a | 546 (3 studies) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical advantage for carbamazepine There was also no statistically significant difference between drugs in treatment failure due to adverse events (HR 1.27, 95% CI 0.87 to 1.86; P = 0.21; I2 = 3%), or treatment failure due to lack of efficacy: HR 0.99 (95% CI 0.69 to 1.41; P = 0.94; I2 = 0%) |
| Time to treatment failure (any reason related to treatment) Subgroup: focal onset seizures Range of follow‐up: 1 day to 4064 days | The median time to treatment failure was 1300 days in the phenytoin group | The median time to treatment failure was 2422 days (1122 days longer) in the carbamazepine group | HR 0.83 (0.61 to 1.13) | 428 (3 studies) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical advantage for carbamazepine There was also no statistically significant difference between drugs in treatment failure due to adverse events (HR 1.19, 95% CI 0.80 to 1.78; P = 0.38, I2 = 0%), or treatment failure due to lack of efficacy: HR 0.88 (95% CI 0.60 to 1.40, P = 0.52, I2 = 0%) |
| Time to treatment failure (any reason related to treatment) Subgroup: generalised Range of follow‐up:1 day to 4403 days | The 10th percentilec | The 10th percentilec | HR 2.38 (1.04 to 5.47) | 118 (2 studies) | ⊕⊕⊝⊝ lowb,d | HR < 1 indicates a clinical advantage for carbamazepine There was no statistically significant difference between drugs in treatment failure due to adverse events (HR 2.31, 95% CI 0.68 to 7.81; P = 0.18; I2 = 60% , or treatment failure due to lack of efficacy: HR 1.86 (95% CI 0.74 to 4.67; P = 0.19; I2 = 0%) but confidence intervals are wide so we cannot rule out an advantage to either drug or no difference between the drugs |
| *Illustrative risks in the carbamazepine and phenytoin groups are calculated at the median time to treatment failure (i.e. the time to 50% of participants failing or withdrawing from allocated treatment) within each group across all trials. The relative effect (pooled hazard ratio) shows the comparison of 'Time to treatment failure' between the treatment groups. CI: 95% confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High certainty (quality): Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty (quality): Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty (quality): Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very certainty (quality): We are very uncertain about the estimate. | ||||||
| aPooled HR for all participants adjusted for seizure type. All pooled HRs presented calculated with fixed‐effect model. | ||||||
| Carbamazepine compared with phenytoin for epilepsy | ||||||
| Patient or population: adults and children with new‐onset focal or generalised epilepsy Settings: outpatients Intervention: carbamazepine Comparison: phenytoin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of Participants | Certainty (quality) of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenytoin | Carbamazepine | |||||
| Time to first seizure after randomisation Range of follow‐up: 0 days to 4589 days | The median time to first seizure was 124 days in the phenytoin group | The median time to first seizure was 79 days (45 days shorter) in the carbamazepine group | HR 1.15 (0.94 to 1.40) | 582 (4 studies) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical |
| Time to first seizure after randomisation Subgroup: focal onset seizures Range of follow‐up: 0 days to 4589 days | The median time to first seizure was 78 days in the phenytoin group | The median time to first seizure was 62 days (16 days shorter) in the carbamazepine group | HR 1.13 (0.89 to 1.43) | 432 (4 studies) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical |
| Time to first seizure after randomisation Range of follow‐up: 2 days to 4070 days | The median time to first seizure was 323 days in the phenytoin group | The median time to first seizure was 142 days (181 days shorter) in the carbamazepine group | HR 1.19 (0.81 to 1.75) | 150 (3 studies) | ⊕⊕⊝⊝ lowb,c | HR < 1 indicates a clinical |
| Time to achieve 12‐month remission Range of follow‐up: 0 days to 4222 days | The median time to 12‐month remission was 472 days in the phenytoin group | The median time to 12‐month remission was 481 days (9 days longer) in the carbamazepine group | HR 1.00 (0.79 to 1.26) | 551 (3 studies) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical |
| Time to achieve 12‐month remission Range of follow‐up: 0 days to 4222 days | The median time to 12‐month remission was 531 days in the phenytoin group | The median time to 12‐month remission was 515 days (16 days shorter) in the carbamazepine group | HR 1.06 (0.80 to 1.42) | 430 (3 studies) | ⊕⊕⊕⊝ moderateb | HR < 1 indicates a clinical |
| Time to achieve 12‐month remission Range of follow‐up: 7 days to 4163 days | The median time to 12‐month remission was 366 days in the phenytoin group | The median time to 12‐month remission was 375 days (9 days longer) in the carbamazepine group | HR 0.88 (0.58 to 1.33) | 121 (2 studies) | ⊕⊕⊝⊝ lowb,d | HR < 1 indicates a clinical |
| *Illustrative risks in the carbamazepine and phenytoin groups are calculated at the median time to first seizure or time to 12‐month remission (i.e. the time to 50% of participants experiencing a first seizure or 12‐months of remission) within each group across all trials. The relative effect (pooled hazard ratio) shows the comparison of 'Time to first seizure' or 'Time to 12‐month remission' between the treatment groups. CI: 95% confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High certainty (quality): Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty (quality): Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty (quality): Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very certainty (quality): We are very uncertain about the estimate. | ||||||
| aPooled HR for all participants adjusted for seizure type. All pooled HRs presented calculated with a fixed‐effect model. | ||||||
| Trial | Outcomes reported | Summary of results |
| 1. Seizure control: excellent (seizure‐free) | 1. PHT (n = 58); CBZ (n = 59) PHT: 39 (67%); CBZ: 22 (37%) | |
| 1. Proportion achieving 24‐month remission at 3 years 2. Proportion excluded after randomisation due to adverse effects or no efficacy | 1. PHT: 59%; CBZ: 62% 2. PHT: 23%; CBZ: 30% | |
| 1. Cognitive assessments 2. Withdrawals from randomised drug | 1. No significant differences between the two treatment groups on any cognitive tests | |
| 1. Proportion of all randomised participants with seizure recurrence (by seizure type) 2. Proportion of participants with optimum plasma levels with seizure recurrence (by seizure type) | PHT (n = 51); CBZ (n = 66) 1. PHT (focal): 10/31 (32%); PHT (generalised): 7/20 (35%); 2. PHT (focal): 4/17 (24%); PHT (generalised): 1/8 (13%); | |
| 1. Cognitive assessments (visual motor speed, co‐ordination, attention and concentration, verbal and visual‐spatial learning, visual and recognition memory, reasoning, mood, handedness) 2. Harmful side effects | 1. Compared to CBZ, participants on PHT became slower (motor speed of the hand) and their visual memory decreased. There was an equal decrease in negative mood (helplessness, irritability, depression) on PHT and CBZ
| |
| 1. Side effects (major and minor) 3. Laboratory results | 1. Incidence of:
2. Treatment failures among analysed participants: Seizure control (among analysed participants with no major side effects): PHT: 23/27 participants (86%); CBZ: 22/27 participants (82%) 3. Significantly lower mean LDH level at 24 weeks in CBZ participants than PHT participants (P < 0.01). Other laboratory results similar across treatment groups | |
| 1. Cognitive measures (verbal, performance, memory, visual‐motor, perceptomotor organisation, visual organisation, dysfunction) | 1. No significant differences between any tests of cognitive function taken before treatment and after 10 ‐ 12 weeks for both treatment groups | |
| CBZ = carbamazepine; LDH = lactate dehydrogenase; PHT= phenytoin | ||
| Trial | Focal seizures: n (%) | Male participants: | Age at entry (years): Mean (SD), rangeb | Epilepsy duration | Number of seizures in prior 6 months, median (range)d | |||||
| CBZ | PHT | CBZ | PHT | CBZ | PHT | CBZ | PHT | CBZ | PHT | |
| 29 (54%) | 30 (56%) | 30 (56%) | 34 (63%) | 9.2 (3.8) 2 to 15 | 9.5 (3.4) (3 to 16) | 1.7 (2.6), 0 to 12 | 1.0 (2.1) (0 to 14) | 3 (1 to 500) | 3 (1 to 404) | |
| 24 (39%) | 28 (44%) | 30 (49%) | 34 (54%) | 29.3 (14.1) 13 to 69 | 33.5 (14.3) (14 to 72) | 4.4 (7.4), 0.1 to 40 | 3.8 (5.4) (0 to 24) | 2 (1 to 354) | 2 (1 to 575) | |
| 155 (100%) | 165 (100%) | 133 (87%) | 145 (88%) | 42.1 (15.9) 18 to 82 | 40.8 (15.3) (18 to 81) | 5.9 (9.1), 0.5 to 55 | 6.6 (9.1) (0.5 to 59) | 1 (1 to 100) | 1 (1 to 26) | |
| 5 (26%) | 3 (17%) | 12 (63%) | 11 (61%) | 28.2 (5.8) 14 to 38 | 18.8 (2.6) (15 to 26) | NA | NA | 18 (6 to 36) | 12 (6 to 18) | |
| n: number of participants; CBZ: carbamazepine; NA: not available; PHT:phenytoin SD: standard deviation aSex was missing for two participants on CBZ from Mattson 1985. eRandomised drug missing for six participants in De Silva 1996. | ||||||||||
| Trial | Number randomised | Time to treatment failure (any reason, adverse events, lack of efficacy) | Time to 12‐month remission | Time to 6‐month remission | Time to first seizure | ||||||||||
| PHT | CBZ | Total | PHT | CBZ | Total | PHT | CBZ | Total | PHT | CBZ | Total | PHT | CBZ | Total | |
| 54 | 54 | 108 | 53 | 53 | 106 | 54 | 54 | 108 | 54 | 54 | 108 | 54 | 54 | 108 | |
| 63 | 61 | 124 | 61 | 60 | 121 | 63 | 61 | 124 | 63 | 61 | 124 | 63 | 61 | 124 | |
| 165 | 155 | 320 | 165 | 154 | 319 | 165 | 154 | 319 | 165 | 154 | 319 | 162 | 151 | 313 | |
| 20 | 23 | 43 | 20 | 23 | 43 | Information not available | Information not available | Information not available | |||||||
| 18 | 19 | 37 | Information not available | Information not available | Information not available | 18 | 19 | 37 | |||||||
| Total | 320 | 312 | 632 | 299 | 290 | 589 | 282 | 269 | 551 | 282 | 269 | 551 | 297 | 285 | 582 |
| CBZ = carbamazepine, PHT= phenytoin aIndividual participant data (IPD) supplied for 114 participants recruited in De Silva 1996; randomised drug not recorded in six participants. Reasons for treatment failure not available for two participants (one randomised to CBZ and one to PHT); these participants are not included in analysis of time to treatment failure. | |||||||||||||||
| Reason for early termination | Heller 1995a,b | Totalc | |||||||||
| CBZ | PHT | CBZ | PHT | CBZ | PHT | CBZ | PHT | CBZ | PHT | Total | |
| Adverse events (Event) | 3 | 2 | 4 | 1 | 8 | 1 | 11 | 8 | 26 | 12 | 38 |
| Seizure recurrence (Event) | 12 | 10 | 2 | 1 | 5 | 8 | 3 | 6 | 22 | 25 | 47 |
| Both seizure recurrence and adverse events (Event) | 6 | 5 | 0 | 0 | 4 | 2 | 31 | 33 | 31 | 40 | 81 |
| Non‐compliance/participant choice (Event) | 0 | 0 | 3 | 4 | 0 | 0 | 11 | 26 | 14 | 30 | 44 |
| Participant went into remission (Censored) | 18 | 24 | 0 | 0 | 6 | 14 | 0 | 0 | 24 | 38 | 62 |
| Lost to follow‐up (Censored) | 0 | 0 | 0 | 0 | 0 | 0 | 26 | 19 | 26 | 19 | 45 |
| Death (Censored)d | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 5 | 4 | 5 | 9 |
| Other (Censored)e | 0 | 0 | 0 | 0 | 0 | 0 | 16 | 11 | 16 | 11 | 27 |
| Completed the study (did not withdraw) (Censored) | 14 | 12 | 14 | 14 | 37 | 38 | 53 | 57 | 118 | 121 | 239 |
| Total | 53 | 53 | 23 | 20 | 60 | 63 | 155 | 165 | 281 | 301 | 592 |
| n = number of individuals contributing to the outcome 'Time to treatment failure’ aOne participant for Heller 1995 (CBZ) and two for De Silva 1996 (one PHT and one CBZ) have missing reasons for treatment failure. | |||||||||||
| Outcome | Original analysis | Generalised onset and age at onset > 30 years classified as focal onset | Generalised onset and age at onset > 30 years classified as uncertain seizure type | |||
| Pooled HR (95% CI) fixed‐effect model | Test of subgroup differences | Pooled HR (95% CI) fixed‐effect model | Test of subgroup differences | Pooled HR (95% CI) fixed‐effect model | Test of subgroup differences | |
| Time to treatment failure (for any reason related to treatment)a | F: 0.83 (0.61 to 1.13), I2 = 0% G: 2.38 (1.04 to 5.47), I2 = 0% O: 0.94 (0.70 to 1.26), I2 = 35% | Chi2 = 5.45, df = 1, P = 0.02, I2 = 81.7% | F: 0.88 (0.65 to 1.19), I2 = 0% G: 1.96 (0.81 to 4.78), I2 = 0% O: 0.96 (0.72 to 1.27), I2 = 6% | Chi2 = 2.83, df = 1, P = 0.09, I2 = 64.6% | F: 0.83 (0.61 to 1.13), I2 = 0% G: 1.96 (0.81 to 4.78), I2 = 0% U: 5.23 (0.47 to 58.71), I2 = NA O: 0.93 (0.70 to 1.24), I2 = 7% | Chi2 = 5.24, df = 2, P = 0.07, I2 = 61.8% |
| Time to treatment failure due to adverse eventsa | F: 1.19 (0.80 to 1.78), I2 = 0% G: 2.31 (0.68 to 7.81), I2 = 60% O: 1.27 (0.87 to 1.86), I2 = 3% | Chi2 = 1.02, df = 1, P = 0.31, I2 = 2.2% | F: 1.25 (0.84 to 1.86), I2 = 22% G: 1.72 (0.51 to 5.87), I2 = 0% O: 1.29 (0.88 to 1.88), I2 = 0% | Chi2 = 0.24, df = 1, P = 0.62, I2 = 0% | F: 1.19 (0.80 to 1.78), I2 = 0% G: 1.72 (0.51 to 5.87), I2 = 0% U: Not estimablec O: 1.24 (0.85 to 1.81), I2 = 0% | Chi2 = 0.31, df = 2, P = 0.86, I2 = 0% |
| Time to treatment failure due to lack of efficacya | F: 0.88 (0.60 to 1.30), I2 = 0% G: 1.86 (0.74 to 4.67), I2 = 0% O: 0.99 (0.69 to 1.41), I2 = 0% | Chi2 = 2.14, df = 1, P = 0.14, I2 = 53.2% | F: 0.91 (0.62 to 1.34), I2 = 0% G: 1.81 (0.68 to 4.82), I2 = 0% O: 1.00 (0.70 to 1.43), I2 = 0% | Chi2 = 1.64, df = 1, P = 0.20, I2 = 38.9% | F: 0.88 (0.60 to 1.30), I2 = 0% G: 1.81 (0.68 to 4.82), I2 = 0% U: Not estimablec O: 0.97 (0.68 to 1.40), I2 = 0% | Chi2 = 1.78, df = 2, P = 0.41, I2 = 0% |
| Time to first seizureb | F: 1.13 (0.89 to 1.43), I2 = 0% G: 1.19 (0.81 to 1.75), I2 = 45% O: 1.15 (0.94 to 1.40), I2 = 0% | Chi2 = 0.05, df = 1, P = 0.83, I2 = 0% | F: 1.15 (0.91 to 1.44), I2 = 0% G: 1.19 (0.77 to 1.84), I2 = 53% O: 1.16 (0.94 to 1.42), I2 = 0% | Chi2 = 0.02, df = 1, P = 0.88, I2 = 0% | F: 1.13 (0.89 to 1.43), I2 = 0% G: 1.19 (0.77 to 1.84), I2 = 53% U: 0.82 (0.29 to 2.34), I2 = NA O: 1.13 (0.92 to 1.39), I2 = 0% | Chi2 = 0.41, df = 2, P = 0.82, I2 = 0% |
| Time to 12‐month remissiona | F: 1.06 (0.80 to 1.42), I2 = 0% G: 0.88 (0.58 to 1.33), I2 = 73% O: 1.00 (0.79 to 1.26), I2 = 16% | Chi2 = 0.54, df = 1, P = 0.46, I2 = 0% | F: 1.10 (0.84 to 1.45), I2 = 0% G: 0.69 (0.43 to 1.11), I2 = 0% O: 0.98 (0.77 to 1.24), I2 = 0% | Chi2 = 2.79, df = 1, P = 0.09, I2 = 64.2% | F: 1.06 (0.80 to 1.42), I2 = 0% G: 0.69 (0.43 to 1.11), I2 = 0% U: 1.91 (0.74 to 4.90), I2 = NA O: 0.99 (0.78 to 1.25), I2 = 15% | Chi2 = 4.32, df = 2, P = 0.12, I2 = 53.7% |
| Time to 6‐month remissiona | F: 0.98 (0.75 to 1.27), I2 = 0% G: 0.77 (0.52 to 1.13), I2 = 39% O: 0.90 (0.73 to 1.12), I2 = 0% | Chi2 = 1.03, df = 1, P = 0.31, I2 = 3.2% | F: 0.98 (0.76 to 1.26), I2 = 0% G: 0.59 (0.37 to 0.93), I2 = 0% O: 0.87 (0.70 to 1.09), I2 = 15% | Chi2 = 3.63, df = 1, P = 0.06, I2 = 72.5% | F: 0.98 (0.75 to 1.27), I2 = 0% G: 0.59 (0.37 to 0.93), I2 = 0% U: 1.20 (0.51 to 2.83), I2 = NA O: 0.88 (0.71 to 1.10), I2 = 2% | Chi2 = 4.01, df = 2, P = 0.13, I2 = 50.1% |
| Chi2: Chi2 statistic; CI: confidence interval; df: degrees of freedom of Chi2 distribution; F: focal epilepsy; G: generalised epilepsy; HR: Hazard Ratio; O: overall (all participants); U: uncertain epilepsy; P: P value (< 0.05 are classified as statistically significant) a29 participants reclassified to focal epilepsy or uncertain epilepsy type from Heller 1995. | ||||||
| Trial | Adverse event dataa | Summary of reported results | |
| Carbamazepine (CBZ) | Phenytoin (PHT) | ||
| All adverse events according to drug (note: no participants withdrew due to adverse events) | CBZ (n = 59): drowsiness (n = 2), rash (n = 3) | PHT (n = 58): gum hypertrophy (n = 2), rash (n = 2), ataxia (n = 2) | |
| “Exclusions” due to adverse events or no efficacy” | Proportion “excluded”: CBZ: 30% (out of 30 randomised to CBZ) | Proportion “excluded”: PHT: 23.3% (out of 30 randomised to PHT) | |
| “Unacceptable” adverse events leading to drug withdrawald | CBZ (n = 54): drowsiness (n = 1), blood dyscrasia (n = 1) | PHT (n = 54): drowsiness (n = 2), skin rash (n = 1), blood dyscrasia (n = 1), hirsutism (n = 1) | |
| Withdrawal due to adverse events (no other adverse event data reported) | 4 participants out of 23 randomised to CBZ withdrew for the following reasons (some withdrew for more than adverse event): slowing of mental function, headache, anorexia, nausea, abdominal pain, fatigue and drowsiness2 | 1 participant out of 20 randomised to PHT withdrew from the study due to depression and anorexia | |
| “Unacceptable” adverse events leading to drug withdrawald | CBZ (n = 61): drowsiness (n = 3), rash (n = 2), headache (n = 1), depression (n = 1) | PHT (n = 63): myalgia (n = 1), irritability (n = 1) | |
| Narrative report of ‘Adverse effects’ and ‘Serious side effects’ | CBZ (n = 155): motor disturbance (ataxia, incoordination, nystagmus, tremor: 33%); dysmorphic and idiosyncratic side effects (gum hypertrophy, hirsutism, acne and rash: 14%); gastrointestinal problems (27%); decreased libido or impotence (13%); No serious side effects | PHT (n = 165); motor disturbance (ataxia, inco‐ordination, nystagmus, tremor: 28%); dysmorphic and idiosyncratic side effects (gum hypertrophy, hirsutism, acne and rash: 22 %); gastrointestinal problems (24%); decreased libido or impotence (11%) 1 serious side effect – 1 participant has confirmed lymphoma, rash improved rapidly following discontinuation of PHT | |
| No adverse events reported | N/A | N/A | |
| Participant reported symptomatic complaints (provided as IPD) | CBZ (n = 19): memory impairment (n = 9) psychomotor retardation (n = 1) inattention (n = 1) transient rash (n = 1) CBZ‐induced cough (n = 1) | PHT (n = 18): memory impairment (n = 7) psychomotor retardation (n = 1) inattention (n = 2) transient rash (n = 1) | |
| Participant‐reported adverse events | 1 participant on CBZ complained of facial skin problems; 1 participant on CBZ complained of tiredness and memory problems | 3 participants on PHT complained of tiredness | |
| Major and minor side effects | CBZ (n = 35): Major side effects: rash (n = 1), pruritus (n = 1), impotence (n = 2), dizziness (n = 1), headaches (n = 1), impaired cognition (n = 1), elevated liver enzymes (n = 1) Mild side effects: nausea (33%), headaches (24%), cognitive impairment (33%), nystagmus (52%), sedation (33%), fine tremor (20%) | PHT (n = 35): Major side effects: rash (n = 4), exfoliative dermatitis (n = 1), impotence (n = 1), dizziness (n = 1), nausea/vomiting (n = 1) Mild side effects: nausea (38%), gingival hyperplasia (12%), headaches (32%), cognitive impairment (15%), nystagmus (40%), sedation (15%), fine tremor (28%) | |
| No adverse events reported | N/A | N/A | |
| CBZ = carbamazepine, N/A = not available, PHT= phenytoin aAdverse event data are recorded as reported narratively in the publications, so exact definition of a symptom may vary. Adverse event data supplied as IPD for Ogunrin 2005. Adverse event data were not requested in original IPD requests (De Silva 1996; Heller 1995; Mattson 1985) but will be for all future IPD requests. For numbers of treatment withdrawals due to adverse events in studies for which IPD were provided (De Silva 1996; Heller 1995; Mattson 1985) see Table 4. | |||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Time to treatment failure (any reason related to the treatment) Show forest plot | 4 | 589 | Hazard Ratio (Fixed, 95% CI) | 0.99 [0.76, 1.31] |
| 2 Time to treatment failure due to adverse events Show forest plot | 4 | 589 | Hazard Ratio (Fixed, 95% CI) | 1.35 [0.93, 1.95] |
| 3 Time to treatment failure due to lack of efficacy Show forest plot | 4 | 589 | Hazard Ratio (Fixed, 95% CI) | 1.02 [0.72, 1.44] |
| 4 Time to treatment failure (any reason related to the treatment) ‐ by epilepsy type Show forest plot | 3 | 546 | Hazard Ratio (Fixed, 95% CI) | 0.94 [0.70, 1.26] |
| 4.1 Focal onset seizures | 3 | 428 | Hazard Ratio (Fixed, 95% CI) | 0.83 [0.61, 1.13] |
| 4.2 Generalised onset seizures | 2 | 118 | Hazard Ratio (Fixed, 95% CI) | 2.38 [1.04, 5.47] |
| 5 Time to treatment failure due to adverse events ‐ by epilepsy type Show forest plot | 3 | 546 | Hazard Ratio (Fixed, 95% CI) | 1.27 [0.87, 1.86] |
| 5.1 Generalised onset seizures | 2 | 118 | Hazard Ratio (Fixed, 95% CI) | 2.31 [0.68, 7.81] |
| 5.2 Focal onset seizures | 3 | 428 | Hazard Ratio (Fixed, 95% CI) | 1.19 [0.80, 1.78] |
| 6 Time to treatment failure due to lack of efficacy ‐ by epilepsy type Show forest plot | 3 | 546 | Hazard Ratio (Fixed, 95% CI) | 0.99 [0.69, 1.41] |
| 6.1 Focal onset seizures | 3 | 428 | Hazard Ratio (Fixed, 95% CI) | 0.88 [0.60, 1.30] |
| 6.2 Generalised onset seizures | 2 | 118 | Hazard Ratio (Fixed, 95% CI) | 1.86 [0.74, 4.67] |
| 7 Time to first seizure post‐randomisation Show forest plot | 4 | 582 | Hazard Ratio (Fixed, 95% CI) | 1.13 [0.92, 1.39] |
| 8 Time to first seizure post‐randomisation ‐ by epilepsy type Show forest plot | 4 | 582 | Hazard Ratio (Fixed, 95% CI) | 1.15 [0.94, 1.40] |
| 8.1 Focal onset seizures | 4 | 432 | Hazard Ratio (Fixed, 95% CI) | 1.13 [0.89, 1.43] |
| 8.2 Generalised onset seizures | 3 | 150 | Hazard Ratio (Fixed, 95% CI) | 1.19 [0.81, 1.75] |
| 9 Time to achieve 12‐month remission Show forest plot | 3 | 551 | Hazard Ratio (Fixed, 95% CI) | 1.01 [0.80, 1.27] |
| 10 Time to achieve 12‐month remission ‐ by epilepsy type Show forest plot | 3 | 551 | Hazard Ratio (Fixed, 95% CI) | 1.00 [0.79, 1.26] |
| 10.1 Focal onset seizures | 3 | 430 | Hazard Ratio (Fixed, 95% CI) | 1.06 [0.80, 1.42] |
| 10.2 Generalised onset seizures | 2 | 121 | Hazard Ratio (Fixed, 95% CI) | 0.88 [0.58, 1.33] |
| 11 Time to achieve six‐month remission Show forest plot | 3 | 551 | Hazard Ratio (Fixed, 95% CI) | 0.92 [0.74, 1.14] |
| 12 Time to achieve six‐month remission ‐ by epilepsy type Show forest plot | 3 | 551 | Hazard Ratio (Fixed, 95% CI) | 0.90 [0.73, 1.12] |
| 12.1 Focal onset seizures | 3 | 430 | Hazard Ratio (Fixed, 95% CI) | 0.98 [0.75, 1.27] |
| 12.2 Generalised onset seizures | 2 | 121 | Hazard Ratio (Fixed, 95% CI) | 0.77 [0.52, 1.13] |