Antiemetics for adults for prevention of nausea and vomiting caused by moderately or highly emetogenic chemotherapy: a network meta‐analysis
Referencias
References to studies included in this review
References to studies excluded from this review
References to studies awaiting assessment
References to ongoing studies
Additional references
References to other published versions of this review
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Study characteristics | ||
| Methods | Randomised, stratified, parallel‐group, active‐comparator, phase 3 trial with 3 arms
Enrolment period: July 2000 to December 2001
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: subjects were followed 5 days for efficacy endpoints and 15 days for safety endpoints | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD (range), years: 53.4 ± 13.7 (palonosetron 0.25 mg), 50.6 ± 14.1 (palonosetron 0.75 mg), 50.9 ± 14.2 (ondansetron 32 mg) Gender: male + female Tumour/cancer type: malignant tumour (ovarian cancer, lung cancer, Hodgkin lymphoma, gastric cancer, breast cancer) Chemotherapy regimen: cisplatin, cyclophosphamide, dacarbazine Country/continent: North America, Europe (76 centres) | |
| Interventions | Experimental: arm A: palonosetron 0.25 mg palonosetron 0.25 mg i.v. + dexamethasone 20 mg i.v. Experimental: arm B: palonosetron 0.75 mg palonosetron 0.75 mg i.v. + dexamethasone 20 mg i.v. Experimental: arm C: ondansetron 32 mg ondansetron 32 mg i.v. + dexamethasone 20 mg i.v. | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind, double‐dummy ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind, double‐dummy ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both participants and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both participants and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the intent‐to‐treat (ITT) cohort included all randomized patients who received chemotherapy and study drug (n = 667)" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the safety cohort (safety analysis) included all patients who received study drug and had at least one safety assessment after treatment (n = 673)" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, cross‐over, placebo‐controlled trial with 2 arms
Study period: January 2015 to June 2015
Masking: single‐blind Baseline patient characteristics: n.r. Follow‐up: yes, time period not mentioned | |
| Participants | Inclusion criteria
Exclusion criteria: n.r. Median age (range), years: 45 (38 to 56) Gender: 8 males + 7 females Tumour/cancer type: non‐Hodgkin lymphoma (NHL) Chemotherapy regimen: ESHAP chemotherapy regimen (etoposide 40 mg/m²/d as a 1‐h i.v. infusion from Day 1 to Day 4; cisplatin 25 mg/m²/d as a continuous infusion from Day 1 to Day 4; solumedrol 500 mg/d as a 15‐min i.v. infusion from Day 1 to Day 5; cytarabine 2 g/m² given as a 2‐h i.v. infusion on Day 5) Country: Egypt (single centre) | |
| Interventions | Cross‐over trial Experimental: arm A: aprepitant 125/80 mg, then placebo Day 1: aprepitant 125 mg + ondansetron 8 mg + dexamethasone 8 mg Days 2 to 3: aprepitant 80 mg + ondansetron 8 mg + dexamethasone 8 mg Days 4 to 5: ondansetron 8 mg + dexamethasone 8 mg with cross‐over to the opposite treatment in the third and fourth cycles Experimental: arm B: placebo. then aprepitant 125/80 mg Days 1 to 3: placebo + ondansetron 8 mg + dexamethasone 8 mg Days 4 to 5: ondansetron 8 mg + dexamethasone 8 mg with cross‐over to the opposite treatment in the third and fourth cycles | |
| Outcomes | Primary endpoint
Secondary endpoint
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "treatment group assignments were made by block randomization" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... single‐blinded ..." and "... prospective placebo‐controlled cross‐over study ..." |
| Blinding of participants and personnel (performance bias) | High risk | Comment: personnel were not blinded |
| Blinding of outcome assessment (detection bias) | High risk | Comment: personnel were not blinded towards the intervention, which might influence subjective outcome assessments |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "twelve of the 15 patients who entered the study were fully evaluable and completed 4 cycles of ESHAP chemotherapy regimen" |
| Selective reporting (reporting bias) | Low risk | Comment: outcome measures were described in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, cross‐sectional trial with 2 arms
Recruitment period: n.r.
Masking: n.r. Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria: n.r. Exclusion criteria: n.r. Median age (range), years: 58 (38 to 72) Gender: male (n = 56, 93%) + female (n = 4, 7%) Tumour/cancer type: non‐small cell lung cancer Chemotherapy regimen: cisplatin (75 mg/m², Day 1) and docetaxel (75 mg/m², Day 1) Country: Turkey | |
| Interventions | Experimental: arm A: aprepitant 125/80 aprepitant (125 mg on Day 1, 80 mg on Days 2 and 3) + p.o. ondansetron 4 mg (4 days, beginning on Day 1 of chemotherapy) + dexamethasone (8 mg on Day 1, 4 mg on Days 2 and 3) Control: arm B p.o. ondansetron 4 mg (4 days, beginning on Day 1 of chemotherapy) + dexamethasone (8 mg on Day 1, 4 mg on Days 2 and 3) | |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "... patients were prospectively randomized ..." Comment: method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: blinding not reported |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: not reported |
| Selective reporting (reporting bias) | Low risk | Comment: outcome measure was reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐ controlled, cross‐over, phase 3 trial with 2 arms
Study period: December 2007 to February 2011
Masking: double‐blind (participants, care provider) Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: 33 years (16 to 62 years) Gender: male Tumour/cancer type: germ cell tumour Chemotherapy regimen: 2 identical courses of standard 5‐day cisplatin‐based chemotherapy regimen Country: United States (multi‐centre, 6) | |
| Interventions | Cross‐over study Experimental: arm A: aprepitant 125/80, then placebo Days 1 to 2: dexamethasone 20 mg + 5‐HT₃ RA Day 3: aprepitant 125 mg + 5‐HT₃ RA Days 4 to 5: aprepitant 80 mg + 5‐HT₃ RA Days 6 to 7: aprepitant 80 mg + dexamethasone 4 mg twice per day, then received matched placebo PO daily on Days 3 through 7 during study Cycle 2 Experimental: arm B: placebo, then aprepitant 125/80 Days 1 to 2: dexamethasone 20 mg + 5‐HT₃ RA Day 3: placebo + 5‐HT₃ RA Days 4 to 5: placebo + 5‐HT₃ RA Days 6 to 7: placebo + dexamethasone 8 mg twice per day | |
| Outcomes | Primary outcome
Secondary outcome(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not reported |
| Allocation concealment (selection bias) | Low risk | Quote: "treatment group assignments were made by personnel not otherwise involved with the study to ensure in‐house blinding" |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: NCT00572572 reported that both participants and care providers were blinded |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: NCT00572572 reported that both participants and care providers were blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "sixty of the 69 patients who entered the study were fully evaluable and completed at least 5 days of both cycles" |
| Selective reporting (reporting bias) | Low risk | Comment: all of the outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, controlled study with 2 arms
Recruitment period: January 2013 to March 2014
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 61.7 ± 11.7 in aprepitant group, 65.4 ± 10.0 in fosaprepitant group Gender: male (74) + female (19) Tumour/cancer type: malignant tumour (lung cancer, gastric cancer, oesophageal cancer, head and neck cancer) Chemotherapy regimen: CDDP (60 mg/m² or higher) Country: Japan (single centre) | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg + palonosetron 0.75 mg or granisetron 3 mg or azasetron 10 mg + dexamethasone 6.6 to 9.9 mg Days 2 to 4: aprepitant 80 mg + dexamethasone 3.3 to 6.6 mg Day 5: aprepitant 80 mg Experimental: arm B: fosaprepitant Day 1: fosaprepitant meglumine 150 mg + palonosetron 0.75 mg or granisetron 3 mg or azasetron 10 mg + dexamethasone 6.6 to 9.9 mg Days 2 to 4: dexamethasone 3.3 to 6.6 mg | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: no allocation concealment reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all randomised patients were included in the analysis |
| Selective reporting (reporting bias) | Unclear risk | Comment: in the results section, palonosetron, granisetron, and azasetron were not separately reported |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, open‐label trial
Study period: n.r.
Masking: open‐label Median follow‐up: n.r. ITT analysis: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: n.r. Gender: female Tumour/cancer type: breast cancer Chemotherapy regimen: AC, TC, TCH regimens Country: United States (Texas) | |
| Interventions | Experimental: arm A: palonosetron, dexamethasone, and fosaprepitant Experimental: arm B: ondansetron, dexamethasone, fosaprepitant | |
| Outcomes | Primary outcome measures
Secondary outcome measures
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Comment: outcome assessors (participants) not blinded to intervention |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: outcome robust to blinding |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: conference abstract, not fully evaluable |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: conference abstract, not fully evaluable |
| Selective reporting (reporting bias) | Unclear risk | Comment: conference abstract, not fully evaluable |
| Other bias | Unclear risk | Comment: conference abstract, not fully evaluable |
| Study characteristics | ||
| Methods | Randomised, parallel, comparative, phase 2 study with 2 arms
Study period: September 2011 to August 2013
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria: n.r. Mean years ± SD: 66.46 ± 9.81 in aprepitant group, 63.48 ± 10.23 in control group Gender: male (64) + female (49) Tumour/cancer type: colorectal cancer Chemotherapy regimen: oxaliplatin‐based or irinotecan‐based MEC (FOLFOX, XELOX, or FOLFIRI) Country: Japan (18 institutions, multi‐centre) | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg p.o. + 5‐HT₃ RAs + dexamethasone 6.6 mg i.v. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 4 mg p.o. Control: arm B Day 1: 5‐HT₃ RAs + dexamethasone 9.9 mg i.v. Days 2 to 3: dexamethasone 8 mg p.o. | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... randomly assigned to the aprepitant (5‑HT3 RA + reduced‑dose dexamethasone + aprepitant) or standard (5‑HT3 + dexamethasone) regimen group according to a computer‑generated, blinded allocation schedule" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis. |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although this was an open‐label study, we assume that knowledge of both patient and personnel had no influence on objective outcomes (e.g. neutropenia) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "... in total, 113 patients were included in the full analysis set" |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all of the included patients were evaluated for objective outcomes |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes have been reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, dose‐ranging, phase 2 trial with 6 arms
Enrolment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: 57 (22 to 83) in arm A, 58.5 (33 to 88) in arm B, 57.4 (20 to 85) in arm C, 59.2 (26 to 82) in arm D, 57.9 (22 to 84) in arm E, 57.9 (28 to 88) in arm F Gender: male (287) + female (436) Tumour/cancer type: solid malignancy (breast, NSCLC, colon or rectum, ovary, other) Chemotherapy regimen: cyclophosphamide from 500 mg/m² to 1500 mg/m² if given with other MEC, or from 750 mg/m² to 1500 mg/m² if given alone or with agents that had minimal or low emetogenic potential; oxaliplatin ≥ 85 mg/m², doxorubicin ≥ 60 mg/m², epirubicin ≥ 90 mg/m², irinotecan (dosed as part of an irinotecan, leucovorin, 5‐fluorouracil (FOLFIRI) regimen), or carboplatin at an area under the curve (AUC) ≥ 5 Country: 99 centres in 24 countries | |
| Interventions | Control: arm A Day 1: 30 min before MEC (casopitant placebo + ondansetron 8 mg p.o. + dexamethasone 8 mg i.v.), 8 h later (ondansetron 8 mg p.o.) Days 2 to 3: casopitant placebo + ondansetron 8 mg p.o. b.i.d. Primary treatment: arm B Day 1: 30 min before MEC (casopitant 50 mg + ondansetron 8 mg p.o. + dexamethasone 8 mg i.v.), 8 h later (ondansetron 8 mg p.o.) Days 2 to 3: casopitant 50 mg + ondansetron 8 mg p.o. b.i.d. Primary treatment: arm C Day 1: 30 min before MEC (casopitant 100 mg + ondansetron 8 mg p.o. + dexamethasone 8 mg i.v.), 8 h later (ondansetron 8 mg p.o.) Days 2 to 3: casopitant 100 mg + ondansetron 8 mg p.o. b.i.d. Primary treatment: arm D Day 1: 30 min before MEC (casopitant 150 mg + ondansetron 8 mg p.o. + dexamethasone 8 mg i.v.), 8 h later (ondansetron 8 mg p.o.) Days 2 to 3: casopitant 150 mg + ondansetron 8 mg p.o. b.i.d. Exploratory: arm E Day 1: 30 min before MEC (casopitant 150 mg + ondansetron 8 mg p.o. + dexamethasone 8 mg i.v.), 8 h later (ondansetron 8 mg p.o.) Days 2 to 3: casopitant placebo + ondansetron 8 mg p.o. b.i.d. Exploratory: arm F Day 1: 30 min before MEC (casopitant 150 mg + ondansetron 16 mg p.o. + dexamethasone 8 mg i.v.), 8 h later (ondansetron placebo) Days 2 to 3: casopitant 150 mg + ondansetron 16 mg q.a.m., ondansetron placebo q.p.m. | |
| Outcomes | Primary endpoint(s)
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomized to 1 of 6 treatment groups (Table 1) by a centralized, automated randomization system and were stratified by sex and chemotherapy treatment (taxane‐based or non taxane‐based)" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. neutropenia) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 723 patients have been included in the efficacy analysis and primary outcomes have been assessed in the intent‐to‐treat (ITT) population |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the safety population consisted of the 711 patients who received at least 1 dose of study medication in any treatment cycle" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes have been reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, comparative, phase 2 study with 2 arms
Enrolment period: n.r.
Masking: open‐label Baseline patient characteristics: reported Follow‐up: patients were followed for a total of 6 days, starting from first day of chemotherapy | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 49 (21 to 70) in ondansetron + aprepitant arm, 53 (30 to 68) in ondansetron arm Gender: male (55) + female (43) Tumour/cancer type: AML, MDS, CML Chemotherapy regimen: cytarabine‐based chemotherapy at a dose ≥1g/m²/d for at least 3 days Country: n.r. | |
| Interventions | Experimental: arm A: aprepitant + ondansetron aprepitant (APREP) 125 mg p.o. plus ondansetron 8 mg i.v. 30 min before cytarabine followed by ondansetron 24 mg i.v. continuous infusion daily until 6 to 12 hours and aprepitant 80 mg p.o. daily until 1 day after the end of last chemotherapy Experimental: arm B: ondansetron Ondansetron (OND) 8 mg i.v. 30 min before cytarabine followed by ondansetron 24 mg i.v. continuous infusion daily until 6 to 12 h after the end of last chemotherapy infusion | |
| Outcomes | Primary endpoint
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although patients and personnel were not blinded, we assume no risk of bias for objective outcomes |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "ninety‐eight patients were registered in the study, 49 to each arm. Among them 83 were evaluable for efficacy, 42 in the OND arm and 41 in the OND + APREP arm" |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients who had received a single dose of study drug were included for safety analysis (N = 87) |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised trial with 2 arms
Enrolment period: n.r.
Masking: single‐blind (patients) Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria: n.r. Exclusion criteria: n.r. Mean/median age, years: n.r. Gender: male (3) + female (9) Tumour/cancer type: n.r. Chemotherapy regimen: combination therapy with Cy > 600 mg or IFO > 1 g/m² Country: Belgium | |
| Interventions | Experimental: arm A: ondansetron 8 mg ondansetron + 10 mg dexamethasone Experimental: arm B: granisetron 3 mg granisetron + 10 mg dexamethasone | |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "treatment allocation was randomized in blocks of four" Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "the patients were kept blind ..." |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding of personnel not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: even though patients were blinded, it is unclear whether personnel were blinded, which could have an impact on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 12 patients (9 F) enrolled for 53 treatments were included in this study |
| Selective reporting (reporting bias) | Unclear risk | Comment: conference abstract, not evaluable |
| Other bias | Unclear risk | Comment: conference abstract, not evaluable |
| Study characteristics | ||
| Methods | Randomised, parallel‐group, controlled trial with 2 arms
Study period: May 2004 to January 2009
Masking: quadruple‐blind (participant, care provider, investigator, outcomes assessor) Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: 46 ± 12.9 in aprepitant group, 46 ± 13 in placebo group Gender: male (28) + female (12) Tumour/cancer type: n.r. Chemotherapy regimen: cyclophosphamide‐containing regimen before transplant Country: United States (single centre) | |
| Interventions | Experimental: arm A: aprepitant aprepitant: loading dose of 125‐mg capsule once a day for 1 day, then maintenance dose of 80‐mg capsule daily through Day +4 of bone marrow transplant dexamethasone: for cyclophosphamide total body irradiation (CyTBI) patients: dexamethasone study drug 1 capsule p.o. daily, 1 h before chemotherapy, with aprepitant on total body irradiation (TBI) and cyclophosphamide chemotherapy days; for busulfan cyclophosphamide (BuCy) patients: dexamethasone 1 capsule orally once daily, discontinued after last dose of chemotherapy ondansetron: for CyTBI patients: ondansetron 8 mg orally every 12 h, beginning 1 h before first TBI dose and discontinued after last dose; then ondansetron 8 mg i.v. every 12 h × 4 doses, beginning 30 min before first cyclophosphamide chemotherapy; for BuCy patients: ondansetron 8 mg p.o. every 6 h, beginning 1 h before first busulfan dose and discontinued after last busulfan dose is given; then ondansetron 8 mg i.v. every 12 h × 4 doses, beginning 30 min before first cyclophosphamide chemotherapy Experimental: arm B: placebo placebo comparator: sugar pill: loading dose of 125‐mg capsule once a day for 1 day, then maintenance dose of 80‐mg capsule daily through Day +4 of bone marrow transplant dexamethasone and ondansetron: same doses and routine as arm A | |
| Outcomes | Primary outcome measures
Secondary outcome measures
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: NCT00248547 reported that masking was quadruple (participant, care provider, investigator, outcomes assessor) |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: NCT00248547 reported that masking was quadruple (participant, care provider, investigator, outcomes assessor) |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all randomised patients were assessed for the primary outcome |
| Selective reporting (reporting bias) | High risk | Comment: primary outcome reported; "effects on nausea, appetite and taste changes" and "pharmacokinetic interaction" are missing |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, prospective, placebo‐controlled, parallel, pilot study with 2 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: 46 (19 to 60) in aprepitant group, 46 (19 to 63) in placebo group Gender: male (28) + female (12) Tumour/cancer type: acute lymphoblastic leukemia, acute myelogenous leukemia, chronic lymphocytic leukemia, chronic myelogenous leukemia, non‐Hodgkin lymphoma, myelodysplastic syndrome, myelofibrosis, Waldenstrom’s macroglobulinemia Chemotherapy regimen: very high doses of cyclophosphamide Country: United States (single centre) | |
| Interventions | Experimental: arm A: aprepitant aprepitant 125 mg on Day 1 and 80 mg through Day +4 + ondansetron given per institutional guidelines on each day of chemotherapy or radiation as described in appendix A + dexamethasone 12 mg p.o. for 1 dose given on Day 1, and 8 mg p.o. once daily given on subsequent days Experimental: arm B: placebo placebo + ondansetron + dexamethasone | |
| Outcomes | Primay objective
Secondary objectives
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "a randomization list was generated using a permuted block randomization with a random block size of either 2 or 4" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "the medications were administered on the same schedule to effectively blind the patients and healthcare staff" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "the medications were administered on the same schedule to effectively blind the patients and healthcare staff" |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the safety analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel‐group trial with 4 treatment arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: Days 6 to 8 for adverse events | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 55 ± 16 (group I), 53 ± 14 (group II), 54 ± 14 (group III), 54 ± 13 (group IV) Gender (male:female): 58:42 (group I), 50:50 (group II), 61:39 (group III), 60:40 (group IV) Tumour/cancer type: solid malignancy (lung cancer, gastrointestinal cancer, head and neck cancer, genitourinary cancer, other) Chemotherapy regimen: cisplatin‐based chemotherapy at a dose ≥ 70 mg/m² Country: n.r. (multi‐centre) | |
| Interventions | Experimental: treatment group I Day 1: placebo (evening pre‐cisplatin), granisetron (10 µg/kg) + dexamethasone (20 mg placebo) + placebo (treatment pre‐cisplatin) Days 2 to 5: placebo Experimental: treatment group II Day 1: placebo (evening pre‐cisplatin), granisetron (10 µg/kg) + dexamethasone (20 mg placebo) + MK‐869 (400 mg placebo) (treatment pre‐cisplatin) Days 2 to 5: MK‐869 (300 mg placebo) Experimental: treatment group III Day 1: MK‐869 (400 mg placebo) (evening pre‐cisplatin), placebo + dexamethasone (20 mg placebo) + MK‐869 (400 mg placebo) (treatment pre‐cisplatin) Days 2 to 5: MK‐869 (300 mg placebo) Experimental: treatment group IV Day 1: placebo (evening pre‐cisplatin), placebo + dexamethasone (20 mg placebo) + MK‐869 (400 mg placebo) (treatment pre‐cisplatin) Days 2 to 5: MK‐869 (300 mg placebo) | |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... computer‐generated, randomized allocation schedule ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. hiccups, laboratory parameters) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the statistical analysis approach for efficacy was intent‐to‐treat (all patients that had data after cisplatin administration were included). The incidence of emesis in the acute and delayed periods as well as the use of rescue medication in both periods was evaluated" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "all 350 patients who received study medication were included in the analysis of safety" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes measures were described in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled trial with 3 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: Days 19 to 29 post cisplatin for follow‐up examination and laboratory tests | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 56.0 ± 13.0 (aprepitant 125/80), 58.4 ± 13.4 (aprepitant 40/25 mg), 53.7 ± 13.2 (placebo) Gender: male + female Tumour/cancer type: solid tumour of respiratory, urogenital, and other origin Chemotherapy regimen: cisplatin at a dose ≥ 70 mg/m² Country: 21 sites in the United States and 29 sites outside the United States (multi‐centre) | |
| Interventions | Experimental: arm A: aprepitant 125/80 mg + standard therapy Day 1: aprepitant 125 mg + ondansetron 32 mg i.v. + dexamethasone 20 mg p.o. Days 2 to 5: aprepitant 80 mg + dexamethasone 8 mg p.o. Experimental: arm B:aprepitant 40/25 mg + standard therapy Day 1: aprepitant 40 mg + ondansetron 32 mg i.v.+ dexamethasone 20 mg p.o. Days 2 to 5: aprepitant 25 mg + dexamethasone 8 mg p.o. Standard: arm C: placebo + standard therapy Day 1: placebo + ondansetron 32 mg i.v. + dexamethasone 20 mg p.o. Days 2 to 5: placebo + dexamethasone 8 mg p.o. ***Aprepitant 375/250 mg + standard therapy (this arm was replaced by arm B) Day 1: aprepitant 375 mg + ondansetron 32 mg i.v. + dexamethasone 20 mg p.o. Days 2 to 5: aprepitant 250 mg + dexamethasone 8 mg p.o. | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
Exploratory endpoints
| |
| Notes |
***Aprepitant 375/250‐mg dose regimen in the current study was discontinued and was replaced with an aprepitant 40/25‐mg dose regimen, and a new randomisation schedule was generated after the dose group adjustment | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... were assigned to 1 of 3 treatment groups according to a computer‐generated randomization schedule" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. hiccup, study mortality) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "... all analyses were performed using an intent‐to treat approach ..." |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the safety analyses are based on data from all patients both before and after the dose‐group adjustment" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised controlled trial with 2 arms
Enrolment period: February 2003 to August 2003
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: ramosetron group 54.53 ± 10.16, granisetron group 53.97 ± 10.50 Gender: male + female Tumour/cancer type: solid malignancy (head and neck, cervix, lung, ovary, stomach‐oesophagus, urinary bladder, testis, other) Chemotherapy regimen: cisplatin at dose ≥ 70 mg/m² Country: Thailand (single centre) | |
| Interventions | Experimental: arm A: ramosetron
Active control: arm B: granisetron
| |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not reported |
| Allocation concealment (selection bias) | Low risk | Quote: "... one nurse prepared for the study drug using identical syringes. The study drug was then sent to another nurse confirming that the study drug was stable and identical in appearance. Then a syringe containing the study drug was handed directly to the investigator. The study drug code was sealed and not opened until all evaluations had been finalized after the completion of treatment" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: adverse events were recorded for all participants |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, cross‐over trial with 2 arms
Recruitment period: March 1997 to December 1997
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria: n.r. Exclusion criteria
Median age (range), years: 52 (42 to 67) Gender: male + female Tumour/cancer type: solid tumours (stomach, lung, head and neck, ovary, metastasis of unknown origin) Chemotherapy regimen: cisplatin (≥ 50 mg/m²) Country: Korea | |
| Interventions | Cross‐over study Experimental: arm A tropisetron + dexamethasone Experimental: arm B ondansetron + dexamethasone | |
| Outcomes | Primary endpoint
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: cannot be determined due to language barrier |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label trial |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label trial |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: cannot be determined due to language barrier |
| Selective reporting (reporting bias) | Unclear risk | Comment: cannot be judged due to language barrier |
| Other bias | Unclear risk | Comment: cannot be judged due to language barrier |
| Study characteristics | ||
| Methods | Randomised, cross‐over trial with 3 arms
Recruitment period: March 1996 to May 1998
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 48 (22 to 74) Gender: male (86.5%) + female (13.5 %) Tumour/cancer type: solid malignancy (nasopharynx, oral cavity, hypopharynx, larynx, ear) Chemotherapy regimen: cisplatin at a dose of 100 mg/m² on Day 1 and 5‐fluorouracil (5‐FU) 1000 mg/m² on Days 1 to 3, repeated every 21 days Country: Hong Kong, China | |
| Interventions | Cross‐over study: patients were randomised to receive 1 of the 3 5‐HT₃ antagonists in the first cycle; treatment was crossed over to the other 2 5‐HT₃ antagonists in the second and third cycles Experimental: arm A: granisetron granisetron 3 mg i.v. + dexamethasone 20 mg i.v. Experimental: arm B: tropisetron tropisetron 5 mg i.v. + dexamethasone 20 mg i.v. Experimental: arm C: ondansetron ondansetron 8 mg i.v. + dexamethasone 20 mg i.v. before cisplatin, followed by 2 p.o. doses of 8 mg at 4 and 8 hours after the start of chemotherapy on Day 1 | |
| Outcomes | Primary endpoint
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomization was performed using a computer‐generated code" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the antiemetic efficacy analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, controlled, parallel‐group, placebo‐controlled trial with 2 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: 62.1 (39 to 71) in placebo group, 57.4 (40 to 69) in aprepitant group Gender: male (19) + female (11) Tumour/cancer type: multiple myeloma Chemotherapy regimen: melphalan Country: Germany (single centre) | |
| Interventions | Experimental: arm a: aprepitant aprepitant (125 mg) (1 h infusion of 100 mg m‐2) + granisetron (Kevatril) 2 mg once daily on Days 1 to 4 + dexamethasone 8 mg once daily on Day 1 and 4 mg once daily on Days 2 and 3 Experimental: arm B: placebo matched placebo + granisetron (Kevatril) 2 mg once daily on Days 1 to 4 + dexamethasone 8 mg once daily on Day 1 and 4 mg once daily on Days 2 and 3 | |
| Outcomes | Primary endpoint
Secondary objectives
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... patients were randomized 1:1 to placebo and aprepitant" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the efficacy analysis |
| Selective reporting (reporting bias) | High risk | Comment: safety is mentioned in the introduction but no results are provided |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel, stratified, comparative phase 3 trial with 3 arms
Study period: May 2000 to December 2001
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: subjects were followed for 15 days | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 53.3 ± 13.1 in palonosetron 0.25 mg group, 55.2 ± 13.1 in palonosetron 0.75 mg group, 53.6 ± 12.7 in dolasetron group Gender: male (102) + female (467) Tumour/cancer type: malignant disease Chemotherapy regimen: any dose of carboplatin, epirubicin, idarubicin, ifosfamide, irinotecan, or mitoxantrone; or methotrexate > 250 mg/m², cyclophosphamide < 1500 mg/m², cyclophosphamide < 1500 mg/m², doxorubicin > 25 mg/m², or cisplatin ≤ 50 mg/m² Country: 61 centres in North America (United States and Mexico) | |
| Interventions | Experimental: arm A: palonosetron 0.25 mg palonosetron 0.25 mg + dexamethasone 20 mg (or, if unavailable, a single dose of p.o. dexamethasone 20 mg or i.v. methylprednisolone 125 mg) Experimental: arm B: palonosetron 0.75 mg palonosetron 0.75 mg + dexamethasone 20 mg (or, if unavailable, a single dose of p.o. dexamethasone 20 mg or i.v. methylprednisolone 125 mg) Experimental: arm C: dolasetron dolasetron 100 mg + dexamethasone 20 mg (or, if unavailable, a single dose of p.o. dexamethasone 20 mg or i.v. methylprednisolone 125 mg) | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomized using an interactive voice response system across all study sites according to specific procedures" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. on study mortality) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "... 569 patients were included in the ITT cohort analysis" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the safety cohort included all treated patients who had at least one safety assessment after treatment with study drugs" and "the safety cohort comprised 582 patients" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes have been reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised study with 2 arms
Enrolment period: November 2010 to March 2012
Masking: n.r. Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: 67.8 (41 to 85) in azasetron group, 68.0 (44 to 83) in granisetron group Gender: male (80) + female (25) Tumour/cancer type: lung cancer Chemotherapy regimen: carboplatin (area under the concentration vs time curve of 5 to 6) in combination with paclitaxel (200 mg/m²), etoposide (100 mg/m²), gemcitabine (1000 mg/m²), or pemetrexed (500 mg/m²) Country: Japan (single centre) | |
| Interventions | Experimental: arm A: azasetron Day 1: p.o. azasetron 10 mg + i.v. dexamethasone 12 mg Days 2 to 3: p.o. dexamethasone 8 mg/d Experimental: arm B: granisetron Day 1: i.v. granisetron 3 mg + i.v. dexamethasone 12 mg Days 2 to 3: p.o. dexamethasone 8 mg/d | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomization was carried out by using the random number table" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: patients were possibly not blinded; therefore we do not know if there is potential for risk of bias |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although blinding was not reported, we assume that for objective outcomes, there would be no potential for risk of bias |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all participants have been included in the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: safety outcomes were reported for all participants |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, phase 3 trial with 2 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria: n.r. Mean/median age (range), years: n.r. Gender: n.r. Tumor/cancer type: n.r. Chemotherapy regimen: cyclophosphamide i.v. chemotherapy (3 g/m²) Country: Italy (single centre) | |
| Interventions | Experimental: arm A aprepitant + palonosetron + dexamethasone Experimental: arm B placebo + palonosetron + dexamethasone | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blinded ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blinded ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the analysis was performed according to the intention‐to‐treat principle" |
| Selective reporting (reporting bias) | Unclear risk | Comment: conference abstract, not evaluable |
| Other bias | Unclear risk | Comment: not evaluable |
| Study characteristics | ||
| Methods | Randomised, controlled trial with 3 arms
Recruitment period: n.r.
Masking: n.r. Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria: n.r. Exclusion criteria: n.r. Mean/median age (range), years: n.r. Gender: n.r. Tumour/cancer type: osteosarcoma Chemotherapy regimen: cisplatin (120 mg/m², 48‐h CI) followed by doxorubicin (75 mg/m², 24‐h CI); then, in the second cycle, delivered 3 weeks later, ifosfamide 15 g/m² (120‐h CI) Country: n.r. | |
| Interventions | Experimental: arm A: granisetron granisetron 2 mg/m² + dexamethasone 8 mg/m² Experimental: arm B: tropisetron tropisetron 5.3 mg/m² + dexamethasone 8 mg/m² Experimental: arm C: ondansetron ondansetron 3.3 mg/m² + dexamethasone 8 mg/m² | |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: not reported |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: not reported |
| Selective reporting (reporting bias) | Unclear risk | Comment: conference abstract, not evaluable |
| Other bias | Unclear risk | Comment: conference abstract, not evaluable |
| Study characteristics | ||
| Methods | Randomised, comparative trial with 3 arms
Recruitment period: September 1997 to September 1998
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria: n.r. Mean age, years: 45 (oral ondansetron), 46 (oral granisetron), 49 (intravenous ondansetron) Gender: male + female Tumour/cancer type: n.r. Chemotherapy regimen: STAMP V, TBI/VP/CY, TANC, Bu/Cy, BEAM, BCNU/VP/CY, ICE, Carboplatin/VP, Carboplatin/MTZ/CY, MMT, Thiotepa/CY, TBI/CY Country: United States (single centre) | |
| Interventions | Experimental: arm A: oral ondansetron ondansetron 8 mg p.o. (every 8 hours) + dexamethasone 10 mg i.v. (once daily) Experimental: arm B: oral granisetron granisetron 1 mg p.o. (every 12 hours each day) + dexamethasone 10 mg i.v. (once daily) Experimental: arm C: intravenous ondansetron ondansetron 32 mg i.v. (single daily dose) + dexamethasone 10 mg i.v. (once daily) | |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the randomization scheme was determined by the study’s biostatistician based on a permuted block design (K = 6)" |
| Allocation concealment (selection bias) | Low risk | Quote: "the treatment allocation scheme was maintained by the study pharmacist, who assumed responsibility for blinded drug distribution" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "... the trial was analysed according to intention to‐treat" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, prospective, cross‐over, comparative study with 2 arms
Enrolment period: January 2011 to January 2012
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 57.5 (36 to 76) Gender: female (38) Tumour/cancer type: gynaecological cancer (endometrial cancer, cervical cancer, ovarian or tubal cancer, double endometrial and ovarian cancer) Chemotherapy regimen: TC regimen (paclitaxel and carboplatin) Country: Japan (single centre) | |
| Interventions | Cross‐over study Experimental: arm A: palonosetron Day 1: aprepitant 125 mg p.o. + palonosetron 0.75 mg i.v. + dexamethasone 20 mg i.v. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 4 mg p.o. Experimental: arm B: granisetron Day 1: aprepitant 125 mg p.o. + granisetron 3 mg i.v. + dexamethasone 20 mg i.v. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 4 mg p.o. | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the group assignment was performed by simple randomization using a table of random numbers and patients were informed of which group (arm A or B) they were assigned" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... non‐blinded ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... non‐blinded ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the intent‐to‐treat population included 19 patients who received palonosetron on Day 1 (Arm A) and 19 patients who received granisetron on Day 1 (Arm B)" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, cross‐over, self‐control study with 2 arms
Recruitment period: n.r.
Masking: n.r. Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria: n.r. Exclusion criteria: n.r. Mean/median age (range), years: n.r. Gender: n.r. Tumour/cancer type: n.r. Chemotherapy regimen: cisplatin‐based chemotherapy Country: n.r. | |
| Interventions | Cross‐over study Experimental: arm A: palonosetron Day 1: palonosetron 0. 25 mg i.v. + dexamethasone 10 mg i.v. Day 2: dexamethasone 10 mg i.v. Day 3: palonosetron 0. 25 mg i.v. + dexamethasone 10 mg i.v. Experimental: arm B: granisetron Days 1 to 3: granisetron 3 mg i.v. + dexamethasone 10 mg i.v. | |
| Outcomes | not reported | |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: blinding not reported |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: no outcomes reported |
| Selective reporting (reporting bias) | Unclear risk | Comment: conference abstract, not evaluable |
| Other bias | Unclear risk | Comment: conference abstract, not evaluable |
| Study characteristics | ||
| Methods | Randomised, controlled trial with 3 arms
Recruitment period: 5 November 2007 to 30 September 2009
Masking: double‐blind Baseline patient characteristics: n.r. Follow‐up: patients were followed for 5 days for efficacy endpoints and for 8 days for safety endpoints | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 48 (ondansetron); 49 (granisetron); 47 (palonosetron) Gender: male + female Tumour/cancer type: malignant disease Chemotherapy regimen
Country: India (multi‐centre) | |
| Interventions | Experimental: arm A: ondansetron Day 1: ondansetron 8 mg i.v. + dexamethasone 16 mg i.v. Day 2: ondansetron 8 mg i.v. + dexamethasone 4 mg i.v. Day 3: dexamethasone 4 mg i.v. Experimental: arm B: granisetron Day 1: granisetron 3 mg i.v. + dexamethasone 16 mg i.v. Day 2: granisetron 3 mg i.v. + dexamethasone 4 mg i.v. Day 3: dexamethasone 4 mg i.v. Experimental: arm C: palonosetron Day 1: palonosetron 0.75 mg i.v. + dexamethasone 16 mg i.v. Days 2 to 3: dexamethasone 4 mg i.v. Day 3: dexamethasone 4 mg i.v. | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "all study personnel and patients were blinded to the treatment assignment for the duration of the study and the nursing staffs injecting the drugs were prohibited from divulging any information on drug assignment even to the doctors giving the chemotherapeutic drugs" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "all study personnel and patients were blinded to the treatment assignment for the duration of the study and the nursing staffs injecting the drugs were prohibited from divulging any information on drug assignment even to the doctors giving the chemotherapeutic drugs" |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all participants were included for the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all participants were included in reporting on adverse events and deaths |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, phase 3 trial with 3 arms
Recruitment period: 6 November 2006 to 9 October 2007
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Age (range), years: 57 (20 to 84) in 3‐day i.v. + p.o. casopitant group, 59 (18 to 80) in single‐dose p.o. casopitant group, 59 (20 to 82) in control group Gender: male + female Tumour/cancer type: malignant solid tumour (non‐small cell lung, head and neck, small cell lung, ovary, cervix, bladder, other) Chemotherapy regimen: cisplatin, gemcitabine, vinorelbine, fluorouracil, etoposide, paclitaxel, cyclophosphamide, doxorubicin, docetaxel Country: 22 countries (77 centres) | |
| Interventions | Experimental: arm A: 3‐day i.v. + p.o. casopitant Day 1: casopitant 90 mg i.v. + casopitant placebo p.o. + ondansetron 32 mg i.v. + dexamethasone 12 mg p.o. + dexamethasone placebo p.o. Days 2 to 3: AM (dexamethasone 8 mg p.o. + casopitant 50 mg p.o.), PM (dexamethasone placebo p.o.) Day 4: AM (dexamethasone 8 mg p.o.), PM (dexamethasone placebo p.o.) Experimental: arm B: single‐dose oral casopitant Day 1: casopitant 150 mg p.o. + casopitant placebo i.v. + ondansetron 32 mg i.v. + dexamethasone 12 mg p.o. + dexamethasone placebo p.o. Days 2 to 3: AM (dexamethasone 8 mg p.o. + casopitant placebo p.o.), PM (dexamethasone 8 mg p.o.) Day 4: AM (dexamethasone 8 mg p.o.), PM (dexamethasone 8 mg p.o.) Control: arm C Day 1: casopitant placebo p.o. + casopitant placebo i.v. + ondansetron 32 mg i.v. + dexamethasone 20 mg p.o. Days 2 to 3: AM (dexamethasone 8 mg p.o. + casopitant placebo p.o.), PM (dexamethasone 8 mg p.o.) Day 4: AM (dexamethasone 8 mg p.o.), PM (dexamethasone 8 mg p.o.) | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were then randomly assigned, through a second telephone call ..." and "randomisation was performed centrally at the study level ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "these supplies were blinded to the pharmacist, the investigator, and the patient" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "these supplies were blinded to the pharmacist, the investigator, and the patient" |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. neutropenia, on‐study mortality) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "efficacy analysis were done in the modified intention‐to‐treat population" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "safety analyses were done with all patients who received placebo or study drug" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel‐group, phase 3 trial with 2 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 56 (19 to 86) in i.v. fosaprepitant group, 57 (19 to 82) in oral aprepitant group Gender: male + female Tumour/cancer type: solid tumour (lung cancer, gastrointestinal cancer, reproductive or genitourinary cancer, miscellaneous or site unspecified, renal and urinary tract cancer, breast cancer, lymphoma, hepatic and biliary cancer, endocrine cancer) Chemotherapy regimen: cisplatin at a dose ≥ 70 mg/m² Country/continent: North America, South America, Europe, Asia‑Pacific, Africa | |
| Interventions | Experimental: arm A: i.v. fosaprepitant Day 1: 150 mg fosaprepitant dimeglumine + 32 mg i.v. ondansetron + 12 mg p.o. dexamethasone Day 2: 8 mg p.o. dexamethasone Days 3 to 4: 8 mg p.o. dexamethasone twice a day Experimental: arm B: p.o. aprepitant 125/80 Day 1: 125 mg p.o. aprepitant + 32 mg i.v. ondansetron + 12 mg p.o. dexamethasone Days 2 to 3: 80 mg p.o. aprepitant + 8 mg p.o. dexamethasone Day 4: 8 mg p.o. dexamethasone | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: patients were stratified by sex and were randomly assigned to treatment groups according to a computer‐generated, blinded allocation schedule |
| Allocation concealment (selection bias) | Low risk | Comment: to ensure in‐house blinding, treatment group assignments were made by personnel not otherwise involved with the study |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‑blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‑blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. infusion site reaction) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included from the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all included patients were checked for adverse events |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, phase 3 trial with 2 arms
Enrolment period: July 2011 to June 2012
Masking: double‐blind Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: n.r. Gender: n.r. Tumour/cancer type: malignant solid tumour Chemotherapy regimen: HEC containing ≥ 50 mg/m² cisplatin (CDDP) Country: Japan | |
| Interventions | Experimental: arm A: palonosetron Day 1: aprepitant 125 mg p.o. + palonosetron 0.75 mg i.v. + dexamethasone 9.9 mg i.v. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 6.6 mg i.v. Day 4: dexamethasone 6.6 mg i.v. Experimental: arm B: granisetron Day 1: aprepitant 125 mg p.o. + granisetron 1 mg i.v. + dexamethasone 9.9 mg i.v. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 6.6 mg i.v. Day 4: dexamethasone 6.6 mg i.v. | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomization was done centrally using the minimization method with stratification with respect to center, age (< 60 versus ≥ 60 years), gender, and cisplatin dose (< 70 versus ≥ 70 mg/m2)" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "at each center, one pharmacist was pre‐designated who was notified of treatment label ... All other staffs (physicians, study pharmacists, and nurses involved in the study) as well as patients were kept blinded to the assigned treatments" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "at each center, one pharmacist was pre‐designated who was notified of treatment label ... All other staffs (physicians, study pharmacists, and nurses involved in the study) as well as patients were kept blinded to the assigned treatments" |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "...our study designated study pharmacists at each center who were blinded (masked) to treatment allocation and who evaluated efficacy end points for each patient every day based on diary data and interview" |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included for safety analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all pre‐specified outcomes were reported |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised study with 2 arms
Enrolment period: July 1997 to February 1999
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 59 (20 to 91) in ondansetron group, 62.5 (25 to 84) in granisetron group Gender: male (15) + female (46) Tumour/cancer type: breast, lymphoma, multiple myeloma, other Chemotherapy regimen: moderately emetogenic chemotherapy (carboplatin ≥ 300 mg/m², cisplatin ≥ 20 to < 50 mg/m², cyclophosphamide, p.o. ≥ 100 mg/m², cyclophosphamide, i.v. ≥ 500 to < 1000 mg/m², dacarbazine ≥ 350 to < 500 mg/m², daunorubicin ≥ 45 mg/m², doxorubicin ≥ 40 mg/m² as a single agent or ≥ 25 mg/m² combined with other chemotherapeutic agents, idarubicin ≥ 12 mg/m², ifosfamide ≥ 1000 mg/m², methotrexate ≥ 250 mg/m², mitoxantrone ≥ 12 mg/m²) Country: n.r. (multi‐centre) | |
| Interventions | Experimental: arm A: ondansetron p.o. single‐dose ondansetron 16 mg + dexamethasone 12 mg Experimental: arm B: granisetron p.o. granisetron 1 mg + dexamethasone 12 mg | |
| Outcomes | Primary objective
Secondary objective(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "sixty‐five patients (75% women) took part in the study, but only 61 were included in the analysis (Table 2)" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, pilot, placebo‐controlled, comparative trial with 3 arms
Patient evaluation period: June 2005 to May 2007
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Age ± SD, years: 59.6 ± 10.7 (palonosetron + 3‐day aprepitant), 58.3 ± 10.5 (palonosetron + 1‐day aprepitant), 56.1 ± 12.6 (palonosetron + placebo) Gender: male + female Tumour/cancer type: solid malignancy (breast cancer, lung cancer, head and neck cancer, other) Chemotherapy regimen
Country: United States (single institute) | |
| Interventions | Experimental: arm A: palonosetron + 3‐day aprepitant Day 1: aprepitant 125 mg p.o. + palonosetron 0.25 mg i.v. + dexamethasone 12 mg p.o. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 8 mg p.o. Day 4: dexamethasone 8 mg p.o. Experimental: arm B: palonosetron + 1‐day aprepitant Day 1: aprepitant 125 mg p.o. + palonosetron 0.25 mg i.v. + dexamethasone 12 mg p.o. Days 2 to 3: matching placebo + dexamethasone 8 mg p.o. Day 4: dexamethasone 8 mg p.o. Experimental: arm C: palonosetron + placebo Day 1: placebo + palonosetron 0.25 mg i.v. + dexamethasone 18 mg p.o. Days 2 to 4: dexamethasone 8 mg p.o. | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included for efficacy analysis |
| Incomplete outcome data (attrition bias) | Unclear risk | Quote: "there were no reports of serious adverse events that were related to study medication" Probably for all patients |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Unclear risk | Comment: the study was temporarily halted, and the protocol was amended with removal of arm C. Descriptive statistics for patients in arm C were not included in the report |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, parallel‐group, phase 3 controlled trial with 4 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Median age, years: 51 in single p.o. dose group, 51 in 3‐day p.o. group, 53 in 3‐day i.v./p.o. group, 52 in control group Gender: male (40) + female (1893) Tumour/cancer type: breast cancer and other Chemotherapy regimen: anthracycline and cyclophosphamide (AC)‐based regimen Country: 196 centres in 32 countries | |
| Interventions | Experimental: arm A: casopitant single oral dosage Day 1: casopitant 150 mg p.o. + casopitant placebo i.v. + ondansetron 8 mg p.o. twice daily + dexamethasone 8 mg i.v. Days 2 to 3: casopitant placebo p.o. + ondansetron 8 mg p.o. twice daily Experimental: arm B: casopitant 3‐day oral dosage Day 1: casopitant 150 mg p.o. + casopitant placebo i.v. + ondansetron 8 mg p.o. twice daily + dexamethasone 8 mg i.v. Days 2 to 3: casopitant 50 mg p.o. + ondansetron 8 mg p.o. twice daily Experimental: arm C: casopitant 3‐day intravenous/oral dosage Day 1: casopitant placebo p.o. + casopitant 90 mg i.v. + ondansetron 8 mg p.o. twice daily + dexamethasone 8 mg i.v. Days 2 to 3: casopitant 50 mg p.o. + ondansetron 8 mg p.o. twice daily Control: arm D Day 1: casopitant placebo p.o. + casopitant placebo i.v. + ondansetron 8 mg p.o. twice daily + dexamethasone 8 mg i.v. Days 2 to 3: casopitant placebo p.o. + ondansetron 8 mg p.o. twice daily | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "after the initial screening visit, investigators used a Randomisation and Medication system to register patients for the trial ..." Comment: method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. study mortality, infusion/injection site reaction) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "of the 1,933 patients randomly assigned to the study, a modified intent to treat (mITT) population (n=1,917) was used for the efficacy analysis" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the safety population (n=1,920) included all patients randomly assigned to a study arm who received any investigational drug (casopitant or casopitant placebo)"; and "there was a single death in each study arm, none of which were attributed to the investigational drug by the investigator" |
| Selective reporting (reporting bias) | Unclear risk | Comment: most outcome measures were reported in the results section. Results were submitted to study registry, then were cancelled. Satisfaction and impact on daily life were not reported |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, phase 2 trial with 2 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: 66 (36 to 81) in ezlopitant group, 62 (40 to 76) in placebo group Gender: male + female Tumour/cancer type: solid tumour (lung cancer, ovary cancer, oesophagus/oropharynx cancer, other/unspecified) Chemotherapy regimen: cisplatin at a dose ≥ 100 mg/m² Country: n.r. (10 institutions, multi‐centre) | |
| Interventions | Experimental: arm A: Gran/Dex + CJ‐11,974 granisetron 10 μg/kg i.v. 30 min before cisplatin + dexamethasone 20 mg i.v. 30 min before cisplatin + CJ‐11,974 100 mg orally 30 min before and 12 h after cisplatin, and twice daily on Days 2 through 5 after cisplatin Experimental: arm B: Gran/Dex + placebo granisetron 10 μg/kg i.v. 30 min before cisplatin + dexamethasone 20 mg i.v. 30 min before cisplatin + identical‐appearing placebo capsules orally 30 min before and 12 h after cisplatin, and twice daily on Days 2 through 5 after cisplatin | |
| Outcomes | Primary efficacy endpoint
Secondary efficacy endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "three (two from group 1 and one from group 2) of the 61 total patients who received the study drug were not included in efficacy analyses because assessments were not recorded..." |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "all 61 patients treated on the study were considered assessable for adverse events. Safety results are listed in Table 4" |
| Selective reporting (reporting bias) | Low risk | Comment: all efficacy measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled trial with 2 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range) ± SD, years: 59 (18 to 84) ± 12, aprepitant 125/80 regimen; 58 (19 to 83) ± 12, placebo group Gender: male + female Tumour/cancer type: solid malignancy (respiratory cancer, urogenital cancer, others) Chemotherapy regimen: cisplatin ≥ 70 mg/m² Country: 15 centres in United States, 14 centres in other countries | |
| Interventions | Experimental: arm A: aprepitant 125/80 Day 1: p.o. aprepitant 125 mg + i.v. ondansetron 32 mg + p.o. dexamethasone 12 mg Days 2 to 3: p.o. aprepitant 80 mg + p.o. dexamethasone 8 mg Day 4: p.o. dexamethasone 8 mg Standard therapy: arm B Day 1: i.v. ondansetron 32 mg + p.o. dexamethasone 20 mg Days 2 to 4: p.o. dexamethasone 8 mg twice per day | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... computer‐generated random assignment schedule ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: a modified intent‐to‐treat approach, which included all patients who received cisplatin, took study drug, and had at least 1 post‐treatment assessment, was used to analyse the data |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "all patients who received cisplatin and at least one dose of study drug were included in the statistical analyses for safety" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, active‐controlled, parallel‐group, phase 3 study with 2 arms
Study period: 2008 March 10 to 13 April 2009
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (SD), years: 61.3 (11.03) in casopitant group, 61.3 (10.77) in placebo group Gender: male (392) + female (318) Tumour/cancer type: colorectal cancer Chemotherapy regimen: oxaliplatin doses between 85 and 130 mg/m² Country: 89 centres (hospitals or outpatient clinics) in 11 countries (multi‐centre) | |
| Interventions | Experimental: arm A: casopitant casopitant + ondansetron + dexamethasone Control: arm B: placebo placebo + ondansetron + dexamethasone | |
| Outcomes | Primary outcome
Secondary outcome(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. neutropenia) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the efficacy endpoints were analyzed for the modified intention to treat (MITT) population which comprised the subset of the intention to treat (ITT) population who received any investigational product and had oxaliplatin administered" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the safety population included all subjects who received any investigational product" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section and in the study registry |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel‐group, phase 2 trial with 5 arms
Recruitment period: 2008
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age, years: 55 (PALO), 55 (NEPA100), 55 (NEPA200), 53 (NEPA300), 55.5 (APR + OND) Gender: male (386) + female (291) Tumour/cancer type: solid tumour (lung/respiratory, head and neck, ovarian, other urogenital, gastric, other GI, breast, other) Chemotherapy regimen: cisplatin‐based chemotherapy at a dose ≥ 50 mg/m² alone or in combination with other chemotherapy agents Country: 29 sites in Russia, 15 sites in Ukraine | |
| Interventions | Experimental: arm A: PALO Day 1: p.o. palonosetron 0.50 mg + p.o. dexamethasone 20 mg + placebo Days 2 to 4: p.o. dexamethasone 8 mg b.i.d. Experimental: arm B: NEPA100 Day 1: p.o. netupitant 100 mg + p.o. palonosetron 0.50 mg + p.o. dexamethasone 12 mg Days 2 to 4: p.o. dexamethasone 4 mg b.i.d. Experimental: arm C: NEPA200 Day 1: p.o. netupitant 200 mg + p.o. palonosetron 0.50 mg + p.o. dexamethasone 12 mg Days 2 to 4: p.o. dexamethasone 4 mg b.i.d. Experimental: arm D: NEPA300 Day 1: p.o. netupitant 300 mg + p.o. palonosetron 0.50 mg + p.o. dexamethasone 12 mg Days 2 to 4: p.o. dexamethasone 4 mg b.i.d. Experimental: arm E: APR + OND Day 1: p.o. aprepitant 125 mg + i.v. ondansetron 32 mg + p.o. dexamethasone 12 mg Days 2 to 3: p.o. aprepitant 80 mg in morning + p.o. dexamethasone 4 mg b.i.d. Day 4: p.o. dexamethasone 4 mg b.i.d. | |
| Outcomes | Primary efficacy endpoint
Secondary efficacy endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. hiccups, study mortality) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "15 patients did not receive study treatment and were not included in the safety population and 677 (98%) patients were included in the full analysis set" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "15 patients did not receive study treatment and were not included in the safety population and 677 (98%) patients were included in the full analysis set"; and "one patient (NEPA100) died during the study due to multiple organ failure. His death was not considered related to study medication" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel, comparative, active‐control trial with 2 arms
Recruitment period: January 2006 to December 2007
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 51 (29 to 73) in ramosetron + dexamethasone group, 51 (22 to 74) in granisetron + dexamethasone group Gender: male (110) + female (175) Tumour/cancer type: solid malignancy (breast, lung, nasopharynx, mouth, rectum, liver, bladder, stomach, oesophagus, testis, brain, other) Chemotherapy regimen: cisplatin, doxorubicin, epirubicin, or oxaliplatin Country: Taiwan (4 centres) | |
| Interventions | Experimental: arm A: ramosetron + dexamethasone ramosetron 0.3 mg + dexamethasone 20 mg Experimental: arm B: granisetron + dexamethasone granisetron 3 mg + dexamethasone 20 mg | |
| Outcomes | Primary endpoint
Secondary endpoint(s) To be evaluated during first, second, third, and fourth 6‐h durations and total 24‐h period after the start of chemotherapy
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: Both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included for the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: safety data were reported for all randomised patients who received the study drug |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, phase 3 trial with 2 arms
Study period: August 2009 to April 2010
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: follow‐up on Days 19 to 29 | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years : 53.1 ± 10.1 (aprepitant regimen), 53.6 ± 10.6 (placebo group) Median age, years: 56 (aprepitant regimen), 54 (placebo group) Gender: male + female Tumour/cancer type: solid tumour (lung cancer, nasopharyngeal cancer, gastrointestinal cancer, reproductive cancer, breast cancer, lymphoma, other) Chemotherapy regimen: cisplatin (≥ 70 mg/m²) Country: China (16 independent centres) | |
| Interventions | Experimental: arm A: aprepitant regimen Day 1: aprepitant 125 mg p.o. + granisetron 3 mg i.v. + dexamethasone 6 mg p.o. Day 2: aprepitant 80 mg p.o. + dexamethasone 3.75 mg p.o. Day 3: aprepitant 80 mg p.o. + dexamethasone 3.75 mg p.o. Day 4: dexamethasone 3.75 mg p.o. Standard: arm A Day 1: placebo + granisetron 3 mg i.v. + dexamethasone 10.5 mg p.o. Day 2: placebo + dexamethasone 7.5 mg p.o. Day 3: placebo + dexamethasone 7.5 mg p.o. Day 4: dexamethasone 7.5 mg p.o. | |
| Outcomes | Primary endpoint
Secondary endpoints ·
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: computer‐generated randomisation |
| Allocation concealment (selection bias) | Low risk | Quote: "... computer‐generated blinded allocation ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "all patients treated with cisplatin or aprepitant who underwent one or more posttreatment assessments were included in the modified intent‐to‐treat analysis" |
| Incomplete outcome data (attrition bias) | Low risk | Comment: safety data were reported for all randomised patients who received the study drug |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, cross‐over study with 2 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: n.r. Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria: n.r. Mean age, years: 53.5 Gender: male (10) + female (24) Tumour/cancer type: cervical cancer and head and neck cancers were predominant Chemotherapy regimen: cisplatin‐based Country: n.r. | |
| Interventions | Cross‐over study Experimental: arm A: ondansetron Day 1: ondansetron 12 mg i.v. + dexamethasone 8 mg i.v. Days 2 to 5: ondansetron 8 mg p.o. b.d. + dexamethasone 4 mg p.o. b.d. Experimental: arm B: granisetron Day 1: granisetron 12 mg i.v. + dexamethasone 8 mg i.v. Days 2 to 5: granisetron 1 mg p.o. b.d. + dexamethasone 4 mg p.o. b.d. | |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised study but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote. "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote. "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the patient‐reported outcome analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, cross‐over, phase 2 trial with 2 arms
Recruitment period: November 2010 to August 2013
Masking: open‐label Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 65 (30 to 75) in aprepitant + granisetron + dexamethasone group, 64 (33 to 77) in palonosetron + dexamethasone group Gender: male + female Tumour/cancer type: advanced or recurrent oesophageal or gastric cancer Chemotherapy regimen
Country: Japan (single centre) | |
| Interventions | Cross‐over trial Experimental: arm A: aprepitant + granisetron + dexamethasone, then palonosetron + dexamethasone 1 h before start of treatment with cisplatin: 125 mg aprepitant (administered p.o.) + 3 mg granisetron (administered i.v.) + 6.6 mg dexamethasone (administered i.v.) after 24 h and 48 h: 80 mg aprepitant (administered p.o.) + 4 mg dexamethasone (administered p.o.) during second cycle, study treatments were crossed over, that is, aprepitant + granisetron + dexamethasone group received palonosetron + dexamethasone Experimental: arm B: palonosetron + dexamethasone, then aprepitant + granisetron + dexamethasone before treatment with cisplatin: 0.75 mg palonosetron (administered i.v.) + 13.2 mg dexamethasone (administered i.v.) after 24 h and 48 h: 8 mg dexamethasone (administered p.o.) during second cycle, study treatments were crossed over, that is, palonosetron + dexamethasone group received aprepitant + granisetron + dexamethasone | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients scheduled to receive chemotherapy who provided informed consent were assigned randomly to receive AGD or PD in a 1: 1 ratio" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open ‐no one is blinded ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open ‐no one is blinded ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although this was an open‐label study, both patients and personnel had no influence on objective outcomes (e.g. hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "effectiveness and safety were evaluated in the remaining 84 patients ..." |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "effectiveness and safety were evaluated in the remaining 84 patients ..." |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were described in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel‐group, phase 2 study with 2 arms
Enrolment period: n.r.
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 67 (34 to 84) in aprepitant group, 66 (44 to 81) in control group Gender: male (110) + female (24) Tumour/cancer type: adenocarcinoma, squamous cell carcinoma, other Chemotherapy regimen: carboplatin + paclitaxel, carboplatin + paclitaxel + bevacizumab, carboplatin + pemetrexed, carboplatin + pemetrexed + bevacizumab Country: Japan (multi‐centre) | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg + first‐generation 5‐HT₃ antagonist + dexamethasone 8 mg Days 2 to 3: aprepitant 80 mg + dexamethasone 8 mg Control: arm B Day 1: first‐generation 5‐HT₃ antagonist + dexamethasone 8 mg Days 2 to 3: dexamethasone 8 mg | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomization was done centrally using a computer program and stratified by sex and non‐platinum agent" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although this was an open‐label study, both patients and personnel had no influence on objective outcomes (e.g. hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 1 patient discontinued due to anaphylactic shock, and remaining 133 patients were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "all 134 patients (including one patient who could not complete chemotherapy because of anaphylactic shock due to paclitaxel) were included in the safety analysis" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Unclear risk | Quote: "additional antiemetic agents and other supportive treatments were administered at the discretion of the treating physicians" |
| Study characteristics | ||
| Methods | Randomised, prospective, cross‐over study with 2 arms
Enrolment period: n.r.
Masking: open‐label Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Median age, years: 58.3 in males, 49.5 in females Gender: male (14) + female (33) Tumour/cancer type: solid tumour (breast, lung, melanoma, other) Chemotherapy regimen: non‐cisplatin‐containing chemotherapy (CNF, CMF, CEF, VAC, carboplatin‐containing, DTIC‐containing, epirubicin‐containing, MTX‐5‐FU, MMM) Country: n.r. | |
| Interventions | Cross‐over study Experimental: arm A: ondansetron 8 mg ondansetron + 10 mg dexamethasone (loading dose of ondansetron was followed by 8 mg ondansetron given orally twice at 8‐h intervals) Experimental: arm B: tropisetron 5 mg tropisetron + 10 mg dexamethasone | |
| Outcomes | control of vomiting during first 24 h was scored as
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "thirty‐nine patients were evaluable for cross‐over analysis" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel, phase 3 study with 2 arms comparison of netupitant + palonosetron + dexamethasone vs aprepitant + palonosetron + dexamethasone Enrolment period: July 2011 to September 2012
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 57 ± 10 in NEPA group, 58 ± 11 in APR + PALO group Gender: male (106) + female (90) Tumour/cancer type: solid malignancy (lung/respiratory, gynaecological, head and neck, breast, other) Chemotherapy regimen: carboplatin Country: multi‐national, multi‐centre | |
| Interventions | Experimental: arm A: netupitant + palonosetron + dexamethasone oral netupitant/palonosetron (300 mg/0.50 mg) hard capsule (on Day 1) with oral dexamethasone before each scheduled chemotherapy cycle Active comparator: arm B: aprepitant + palonosetron + dexamethasone oral aprepitant hard capsule 125 mg (on Day 1) + 80 mg daily (for the following 2 days) and oral palonosetron soft capsule 0.50 mg (on Day 1) given with oral dexamethasone at each scheduled chemotherapy cycle | |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomly allocated in a 3:1 ratio to receive one of the following treatments ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all included patients have been analysed in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised trial with 2 arms
Enrolment period: September 1994 to April 1996
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age, years: 43.5 in granisetron group, 43 in ondansetron group Gender: n.r. Tumour/cancer type: breast cancer Chemotherapy regimen: 1500 mg cyclophosphamide/m²/d, 125 mg thiotepa/m²/d, 200 mg carboplatin/m²/d Country: n.r. | |
| Interventions | Experimental: arm A: granisetron granisetron as a 0.5‐mg i.v. bolus 30 min before chemotherapy followed by continuous infusion of 0.04 mg/h (1 mg/d) for 7 days + 10 mg dexamethasone/d i.v. for 7 days Experimental: arm B: ondansetron ondansetron as 8‐mg i.v. bolus 30 min before chemotherapy followed by continuous infusion of 1 mg/h (24 mg/d) for 7 days + 10 mg dexamethasone/d i.v. for 7 days | |
| Outcomes | Primary endpoint
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... computer generated a list whereby each patient was assigned to either arm 1 or arm 2 as they were enrolled on the trial" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "both patient and treatment team remained blinded to the identity of the study drug throughout the patient's hospitalization" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "both patient and treatment team remained blinded to the identity of the study drug throughout the patient's hospitalization" |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote. "of the 48 patients enrolled, 45 were evaluable" |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included for assessed adverse events Comment: not reported |
| Selective reporting (reporting bias) | Low risk | Comment: outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised trial with 2 arms
Enrolment period: August 2015 to September 2017
Masking: single‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (SD), years: 59.4 (12.0) in ramosetron group, 60.3 (11.8) in palonosetron group Gender: 37.2% (62.8% male) female in ramosetron group, 38.7% female (61.3% male) in palonosetron group Tumour/cancer type: solid tumours (lung and thymus, breast, head and neck, gynaecological and genitourinary, gastrointestinal, others) Chemotherapy regimen: individual highly emetogenic chemotherapies: 71.5% in ramosetron group, 72.5% in palonosetron group received cisplatin Country: Korea, multi‐centre | |
| Interventions | Experimental: arm A: ramosetron aprepitant (Day 1, 125 mg p.o. 1 h before chemotherapy; Days 2 to 3, 80 mg p.o.), ramosetron (Day 1, 0.3 mg i.v. 30 min before chemotherapy), and dexamethasone (Day 1, 12 mg p.o. or i.v. 30 min before chemotherapy; Days 2 to 4, 8 mg p.o.) Experimental: arm B: palonosetron aprepitant (Day 1, 125 mg p.o. 1 h before chemotherapy; Days 2 to 3, 80 mg p.o.), palonosetron (Day 1, 0.25 mg i.v. 30 min before chemotherapy), and dexamethasone (Day 1, 12 mg p.o. or i.v. 30 min before chemotherapy; Days 2 to 4, 8 mg p.o.) | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "stratified block randomization was conducted with a 1:1 ratio between groups, randomly mixing block sizes of 2 and 4, considering (1) chemotherapeutic regimen (cisplatin vs.non‐cisplatin), (2) treatment schedule (single‐day vs. multiple‐ day), and (3) sex (male vs. female), as stratification factors"; "patients were assigned according to a pre‐defined randomization sequence created by an independent investigator with no clinical involvement in the trial"
|
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Single‐blinded; participants were not aware of treatment |
| Blinding of participants and personnel (performance bias) | High risk | Physicians were aware of treatment |
| Blinding of outcome assessment (detection bias) | Low risk | Outcome assessors (participants) were blinded to intervention |
| Blinding of outcome assessment (detection bias) | Low risk | Outcome was robust to blinding |
| Incomplete outcome data (attrition bias) | High risk | Modified ITT was analysed (participants who received at least 1 treatment) |
| Incomplete outcome data (attrition bias) | High risk | Safety population (ITT) analysed, but not all participants received intervention |
| Selective reporting (reporting bias) | Low risk | No reasons for any concerns detected |
| Other bias | Low risk | No other sources of bias detected |
| Study characteristics | ||
| Methods | Randomised, cross‐over study with 2 arms
Enrolment period: n.r.
Masking: open‐label Baseline patient characteristics: not reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: n.r. Tumour/cancer type: head and neck cancer Chemotherapy regimen: moderately emetogenic cancer chemotherapeutic drugs (i.v. docetaxel 60 mg/m², i.v. carboplatin 300 mg/m², and i.v. 5‐fluorouracil 600 mg/m²) Country: India (single centre) | |
| Interventions | Cross‐over study Experimental: arm A: palonosetron palonosetron 0.25 mg i.v. + dexamethasone 16 mg i.v. (half hour before chemotherapy) Experimental: arm B: ondansetron ondansetron 16 mg i.v. + dexamethasone 16 mg i.v. (half hour before chemotherapy) | |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all 30 patients have been included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, prospective study with 2 arms
Enrolment period: n.r.
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range): 52 (36 to 70) in PDA group, 51 (34 to 69) in OD group Gender: male (52) + female (8) Tumour/cancer type: head and neck cancer (squamous cell carcinoma of head and neck) Chemotherapy regimen: docetaxel 60 mg/m² intravenously (i.v.), carboplatin 300 mg/m² i.v., and 5‐FU (5‐fluorouracil) 600 mg/m² i.v. Country: India (single centre) | |
| Interventions | Experimental: arm A: aprepitant + palonosetron + dexamethasone (PDA) Day 1: p.o. aprepitant 125 mg + palonosetron 0.25 mg i.v. + dexamethasone 12 mg i.v. Days 2 to 3: capsule aprepitant 80 mg o.d. + tablet dexamethasone 8 mg b.d. Control: arm B: ondansetron + dexamethasone (OD) Day 1: ondansetron 16 mg i.v. + dexamethasone 12 mg i.v. + ondansetron 8 mg b.d. (after chemotherapy) Days 2 to 3: ondansetron 8 mg b.d. + dexamethasone 8 mg b.d. | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the efficacy analysis |
| Selective reporting (reporting bias) | High risk | Comment: the result of safety analysis was not reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, prospective, phase 3 trial with 2 arms
Enrolment period: June 2011 to September 2012
Masking: single‐blind (participant) Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 58.9 ± 10.4 in ramosetron group, 59.0 ± 11.6 in ondansetron group Gender: male + female Tumour/cancer type: solid tumour (lung cancer, lymphoma, stomach cancer, head and neck cancer, breast cancer, oesophagus cancer, hepatobiliary and pancreas cancer, other) Chemotherapy regimen: highly emetogenic chemotherapeutic agents (NCCN Guideline v1.0 2011 antiemesis) Country: Korea (17 institutions, multi‐centre) | |
| Interventions | Experimental: arm A: ramosetron Day 1: ramosetron 0.3 mg i.v. + aprepitant 125 mg p.o. + dexamethasone 12 mg p.o. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 8 mg p.o. Day 4: dexamethasone 8 mg p.o. Experimental: arm B: ondansetron Day 1: ondansetron 16 mg i.v. + aprepitant 125 mg p.o. + dexamethasone 12 mg p.o. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 8 mg p.o. Day 4: dexamethasone 8 mg p.o. | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were assigned to the RAD or OAD groups (1:1 ratio) according to a stratified block randomization table" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... single‐blind ..." Comment: patients were blinded |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... single‐blind ..." Comment: only patients were blinded |
| Blinding of outcome assessment (detection bias) | High risk | Comment: although patients were blinded, unblinded personnel might have an influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although this was a single‐blind study, both patients and personnel had no influence on objective outcomes (e.g. study mortality) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "... an m‐ITTpopulation of 299 was subjected to the efficacy analysis ..." |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "three deaths of OAD patients during the study were considered unrelated to the medication" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, active‐comparator, phase 4 trial (MK‐0869‐225) with 2 arms
Study period: 2012 December 28 to 4 August 2014
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD (range), years: 59.7 ±11.4 (23 to 84) in aprepitant group, 60.9 ± 11.5 (28 to 85) in control group Gender: male (263) + female (217) Tumour/cancer type: solid malignancy (gastrointestinal, lung, gynaecological, other) Chemotherapy regimen: carboplatin, oxaliplatin, irinotecan‐based Country: South Korea (multi‐centre, 20 sites) | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg capsule p.o. + ondansetron 16 mg i.v. + dexamethasone 12 mg p.o. Days 2 to 3: aprepitant 80 mg capsule p.o. + placebo for ondansetron 8 mg p.o. twice daily (b.i.d.) Control: arm B Day 1: aprepitant placebo capsule p.o. + ondansetron 16 mg i.v. + dexamethasone 20 mg p.o. Days 2 to 3: aprepitant placebo capsule p.o. + ondansetron 8 mg p.o. b.i.d. | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
Exploratory endpoint
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "eligible patients were randomised (1:1) to receive either a 3‐day aprepitant or control regimen" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. neutropenia, hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "of the 494 randomized subjects, 480 were included in the modified intent‐to‐treat population" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the all‐patients‐as treated (APaT) population was used for safety analyses" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, cross‐over trial with 2 arms
Enrolment period: 1 April 2011 to 31 March 2013
Masking: single‐blind Baseline patient characteristics: reported Follow‐up: patients were followed up for 10 days during each course for efficacy and safety endpoints | |
| Participants | Inclusion criteria
Exclusion criteria
Age (range), years: 36.1 (15 to 65) in palonosetron arm, 50.6 (18 to 70) in granisetron arm Gender: male + female Tumour/cancer type: osteosarcoma, malignant fibrous histiocytoma, synovial sarcoma, leiomyosarcoma, rhabdomyosarcoma, dedifferentiated liposarcoma, myxoid liposarcoma, clear cell sarcoma Chemotherapy regimen: cisplatin + doxorubicin, ifosfamide + doxorubicin/ifosfamide + etoposide Country: Japan | |
| Interventions | Cross‐over study Experimental: arm A: palonosetron Day 1: p.o. 125 mg aprepitant + i.v. 0.75 mg palonosetron + i.v. 6.6 mg dexamethasone Days 2 to 5: 80 mg aprepitant + 6.6 mg dexamethasone Experimental: arm B: granisetron Day 1: p.o. 125 mg aprepitant + i.v. 3 mg × 2 granisetron + i.v. 6.6 mg dexamethasone Days 2 to 5: 80 mg aprepitant + 3 mg × 2 granisetron + 6.6 mg dexamethasone | |
| Outcomes | Primary endpoints
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "a single randomization method was used to assign eligible patients to the palonosetron or granisetron arm" |
| Allocation concealment (selection bias) | Unclear risk | Commen: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "all participants were blinded to the antiemetic treatment assignments for the duration of the study" |
| Blinding of participants and personnel (performance bias) | High risk | Comment: only patients were blinded to the study intervention |
| Blinding of outcome assessment (detection bias) | High risk | Comment: although patients were blinded, unblinded personnel might have an influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although this was a single‐blind study, we assume that both patients and personnel had no influence on objective outcomes (e.g. hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "all patients were eligible for efficacy analysis ..." |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "safety was assessed for all patients who received treatment" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, cross‐over study with 2 arms
Study period: April 2013 to November 2014
Masking: single‐blind Baseline patient characteristics: yes Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
All patients provided written informed consent before entering the study Mean/median age, years: 80% ≥ 50 years Gender: 37% male Tumour/cancer type: colorectal, breast, other Chemotherapy regimen: MEC (oxaliplatin base, irinotecan base, other) Country: n.r. | |
| Interventions | Experimental: arm A: fosaprepitant Day 1: fosaprepitant 150 mg + granisetron 3 mg + dexamethasone 4.95 mg Experimental: arm B: palonosetron Day 1: i.v. palonosetron 0.75 mg + dexamethasone 9.9 mg | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: "... minimization method ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: single‐blind |
| Blinding of participants and personnel (performance bias) | High risk | Comment: single‐blind only |
| Blinding of outcome assessment (detection bias) | High risk | Comment: single‐blind study; knowledge of treatment may have affected outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: single‐blind study; we assume that knowledge of treatment would not influence objective outcomes |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the total of 35 patients and 70 therapies was available for analysis" and "we analyzed the per‐protocol cohort including all patients who received the study medication and completed the follow‐up period" Comment: 2 participants did not start study treatment; 2 withdrew because they could not complete chemotherapy |
| Incomplete outcome data (attrition bias) | Low risk | Comment: adverse events reported for all patients who completed chemotherapy and study treatment |
| Selective reporting (reporting bias) | Low risk | Comment: outcomes only for the overall phase have been reported, as no significant difference was found in any other evaluation points |
| Other bias | Unclear risk | Comment: participants did not record the incidence and severity of CINV daily, but only on Days 2 and 5 |
| Study characteristics | ||
| Methods | Randomised, cross‐over trial with 2 arms
Patient hospitalisation period: March 1998 to June 1999
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: recorded daily for 7 days after the start of chemotherapy | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 61.2 (26 to 81) in granisetron‐ramosetron group, 58.7 (33 to 77) in ramosetron‐granisetron group Gender: male + female Tumour/cancer type: solid tumour (gastric cancer, oesophageal cancer) Chemotherapy regimen: 2 courses of combined chemotherapy (including ≥ 60 mg/m² cisplatin) Country: Japan | |
| Interventions | Cross‐over study Experimental: arm A: granisetron‐ramosetron granisetron 3 mg i.v. during treatment phase 1, followed by ramosetron 0.3 mg i.v. during treatment phase 2 + methylprednisolone sodium (Solumedrol) 250 mg i.v. immediately before and 6 h after chemotherapy Experimental: arm B: ramosetron‐granisetron ramosetron 0.3 mg i.v. during treatment phase 1, followed by granisetron 3 mg i.v. during treatment phase 2 + methylprednisolone sodium (Solumedrol) 250 mg i.v. immediately before and 6 h after chemotherapy | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Low risk | Quote: "the study drug code was sealed, preserved at the registration center and not opened until all evaluations had been finalized, after the completion of treatment phase 2" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: "clinical response was evaluated in the remaining 30 patients" |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all included patients recorded adverse events |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, controlled, prospective, parallel‐group trial with 2 arms
Enrolment period: April 2013 to February 2015
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age, years: 70 (57 to 90) in aprepitant group, 73 (43 to 84) in control group Gender: male (57) + female (23) Tumour/cancer type: non‐small cell lung cancer Chemotherapy regimen: carboplatin + paclitaxel, carboplatin + paclitaxel + bevacizumab, carboplatin + pemetrexed, carboplatin + pemetrexed + bevacizumab, carboplatin + S‐1 Country: Japan (multi‐centre) | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg + palonosetron 0.75 mg + dexamethasone 8 mg Days 2 to 3: aprepitant 80 mg + dexamethasone 8 mg Control: arm B Day 1: palonosetron 0.75 mg + dexamethasone 8 mg Days 2 to 3: dexamethasone 8 mg | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomization was performed centrally by computer software and stratified by sex, age, and non‐platinum chemotherapy agent" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although this was an open‐label study, both patients and personnel had no influence on objective outcomes (e.g. neutropenia, hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "one patient withdrew consent before chemotherapy, and 80 patients (41 in the aprepitant group and 39 in the control group) were assessed for efficacy and safety" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "one patient withdrew consent before chemotherapy, and 80 patients (41 in the aprepitant group and 39 in the control group) were assessed for efficacy and safety" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, cross‐over trial with 2 arms
Recruitment period: n.r.
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 61 (32 to 77) in arm A, 58.5 (30 to 71) in arm B Gender: male (29) + female (10) Tumour/cancer type: solid tumour (lung cancer, head and neck tumour, unknown tumour, oesophageal cancer, gastric cancer, malignant lymphoma) Chemotherapy regimen: cisplatin (≥ 50 mg/m²), ifosfamide (≥ 3000 mg/m²), mitomycin (6 mg/m²), fluorouracil (1000 mg/m²), etoposide (80 mg/m²) Country: Korea (multi‐centre) | |
| Interventions | Cross‐over study Experimental: arm A first ondansetron + dexamethasone, then tropisetron + dexamethasone Experimental: arm B first tropisetron + dexamethasone, then ondansetron + dexamethasone | |
| Outcomes | not readable | |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: not evaluable due to language barrier |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: for objective outcomes, we assume that not blinding participants and personnel would not influence risk of bias |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: not evaluable because of language barrier |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: not evaluable because of language barrier |
| Selective reporting (reporting bias) | Unclear risk | Comment: not evaluable because of language barrier |
| Other bias | Unclear risk | Comment: not evaluable because of language barrier |
| Study characteristics | ||
| Methods | Randomised, prospective study with 2 arms
Recruitment period: June 2015 to February 2018
Masking: n.r. Baseline patient characteristics: reported Follow‐up: patients were followed on Days 6 to 8 and on Days 19 to 21 | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 51.74 ± 7.082 in aprepitant group, 47.46 ± 8.180 in standard group Gender: female Tumour/cancer type: breast cancer Chemotherapy regimen: anthracycline (30 mg/m²/d for pirarubicin or 45 mg/m²/d for epirubicin) and cyclophosphamide Country: Mongolia, China (single centre) | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg p.o. + tropisetron 5 mg i.v. + dexamethasone 6 mg p.o. Day 2: aprepitant 80 mg p.o. + tropisetron 5 mg i.v. + dexamethasone 3.75 mg p.o. Day 3: aprepitant 80 mg p.o. + dexamethasone 3.75 mg p.o. Day 4: dexamethasone 3.75 mg p.o. Standard: arm B Day 1: tropisetron 5 mg i.v. + dexamethasone 10.5 mg p.o. Day 2: tropisetron 5 mg i.v. + dexamethasone 7.5 mg p.o. Days 3 to 4: dexamethasone 7.5 mg p.o. | |
| Outcomes | Primary endpoints
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised study but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: blinding was not reported; therefore it is possible that knowledge of allocated treatment could have posed a risk of bias |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: not reported |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel‐design study with 2 arms
Enrolment period: November 2010 to October 2012
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 54.5 ± 11.9 in control group, 62.6 ± 12.7 in aprepitant group Gender: n.r. Tumour/cancer type: ovarian cancer, endometrial cancer Chemotherapy regimen: paclitaxel and carboplatin (TC) Country: Japan (single centre) | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg p.o. + first‐generation 5‐HT₃ antagonist 3 mg + dexamethasone 8 or 16 mg i.v. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 8 or 4 mg p.o. Control: arm B Day 1: first‐generation 5‐HT₃ antagonist 3 mg + dexamethasone 8 or 4 mg i.v. Days 2 to 3: dexamethasone 8 or 16 mg p.o. | |
| Outcomes | Primary outcome
Secondary outcome(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Low risk | Quote: "twenty‐three eligible patients were divided randomly into two groups (A and B) using sealed opaque envelopes" |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all 23 patients were included in patient‐reported efficacy analysis |
| Selective reporting (reporting bias) | High risk | Quote: "safety was evaluated using general laboratory tests" Comment: no results regarding the safety analysis were reported |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, open‐label study
Study period: n.r.
Masking: open‐label Baseline patient characteristics: n.r. Median follow‐up: n.r. ITT analysis: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: n.r. Gender: n.r. Tumour/cancer type: n.r. Chemotherapy regimen: cisplatin‐based, or combination of cyclophosphamide and anthracyclines Country: Egypt | |
| Interventions | Experimental: arm A: granisetron with dexamethasone Experimental: arm B: palonosetron with dexamethasone | |
| Outcomes | Primary outcome measures
Secondary outcome measures: adverse events | |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Comment: outcome assessors (participants) not blinded to intervention |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: outcome robust to blinding |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: conference abstract, not fully evaluable |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: conference abstract, not fully evaluable |
| Selective reporting (reporting bias) | Unclear risk | Comment: conference abstract, not fully evaluable |
| Other bias | Unclear risk | Comment: conference abstract, not fully evaluable |
| Study characteristics | ||
| Methods | Randomised study with 2 arms
Enrolment period: n.r.
Masking: n.r. Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria: n.r. Exclusion criteria: n.r. Mean/median age, years: n.r. Gender: n.r. Tumour/cancer type: n.r. Chemotherapy regimen: n.r. Country: Japan | |
| Interventions | Experimental: arm A: aprepitant aprepitant + palonosetron + dexamethasone Control: arm B palonosetron + dexamethasone | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: blinding not reported |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding not reported; however, this should not affect objective outcomes |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | All patients were included in the analysis |
| Selective reporting (reporting bias) | Low risk | Comment: not reported |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised trial with 2 arms
Enrolment period: December 2012 to October 2014
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age, years: 54 (27 to 82) in granisetron group, 54 (30 to 74) in palonosetron group Gender: female Tumour/cancer type: breast cancer Chemotherapy regimen: anthracycline plus cyclophosphamide (AC) regimen Country: Japan | |
| Interventions | Experimental: arm A: palonosetron fosaprepitant + palonosetron 0.75 mg + dexamethasone Experimental: arm B: granisetron fosaprepitant + granisetron 1 mg + dexamethasone | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised study but method of randomisation not described |
| Allocation concealment (selection bias) | Low risk | Quote: "concealment: central registration" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 326 patients were evaluable |
| Selective reporting (reporting bias) | High risk | Comment: safety was planned but not reported; complete response for nausea in acute and overall phases not reported |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, phase 2 trial with 3 arms
Recruitment period: n.r.
Masking: n.r. Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria: n.r. Exclusion criteria: n.r. Mean/median age (range), years: n.r. Gender: n.r. Tumour/cancer type: acute myeloid leukemia, myelodysplastic syndrome Chemotherapy regimen: MD‐HD‐CHEMO with HDAC‐containing regimens Country: n.r. | |
| Interventions | Experimental: arm A: ONDA 1 to 5 ONDA 8 mg i.v. bolus, then 24 mg i.v., continuous infusion on Day 1 through Day 5 and for 12 h after Ara C infusion ends Experimental: arm B: PALO 1 to 5 PALO 0.25 mg i.v. bolus over 30 seconds on Day 1 through Day 5 of Ara C infusion Experimental: arm C: PALO 1, 3, 5 PALO 0.25 mg i.v. bolus over 30 seconds on Days 1, 3, and 5 of Ara C infusion All patients received Solumedrol 40 mg i.v. before Ara C | |
| Outcomes | Primary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "open randomized comparative trial" |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "open randomized comparative trial" |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 95 patients were evaluable |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Unclear risk | Comment: conference abstract, not evaluable |
| Study characteristics | ||
| Methods | Randomised, prospective, phase 2 study with 2 arms
Study period: January 2011 to April 2014
Masking: n.r. Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria: n.r. Mean/median age, years: n.r. Gender: n.r. Tumour/cancer type: lung cancer Chemotherapy regimen: MEC Country: n.r. | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg p.o. + palonosetron 0.75 mg i.v. + dexamethasone 4.95 mg i.v. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 4 mg p.o. Experimental: arm B Day 1: granisetron 3 mg i.v. + dexamethasone 9.9 mg i.v. Days 2 to 3: dexamethasone 8 mg p.o. | |
| Outcomes | Primary endpoint
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: not reported |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the efficacy analysis |
| Selective reporting (reporting bias) | Unclear risk | Comment: conference abstract only |
| Other bias | Unclear risk | Comment: conference abstract, not evaluable |
| Study characteristics | ||
| Methods | Randomised, parallel‐design study with 2 arms
Enrolment period: January to December 2015
Masking: single‐blind Baseline patient characteristics: sex and age reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (SD), years: 47.8 (14.4) Gender: 63.5% female, 36.5% male Tumour/cancer type: Hodgkin lymphoma Chemotherapy regimen: Adriamycin, Bleomycin, Vinblastine, Dacarbazine Country: Iraq (single centre) | |
| Interventions | Experimental: arm A oral aprepitant on Days 1 to 3 (Day 1, 125 mg 1 h before chemotherapy; Days 2 to 3, 80 mg), ondansetron on Day 1 (Day 1, 32 mg i.v. infusion over 15 min at 30 to 60 min before chemotherapy), oral dexamethasone on Days 1 to 4 (Day 1, 12 mg 30 min before chemotherapy; Days 2 to 4, 8 mg in the morning) Control: arm B ondansetron on Days 1 to 4 (Day 1, 32 mg i.v. infusion over 15 min at 30 to 60 min before chemotherapy; Days 2 to 4, 8 mg p.o. twice daily), p.o. dexamethasone on Days 1 to 4 (Day 1, 20 mg 20 min before chemotherapy; Days 2 to 4, 8 mg twice daily) | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "computer‐generated, random allocation schedule" |
| Allocation concealment (selection bias) | Low risk | Quote: "computer‐generated, random allocation schedule" |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: single‐blinded; participants not aware of assigned treatment |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "the selection of both groups was known by the researcher after taking the agreement of physician responsible for patients' treatment" |
| Blinding of outcome assessment (detection bias) | Low risk | Outcome assessors (participants) blinded to intervention |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: mean scores reported, but unclear whether data on all participants were included in analysis |
| Selective reporting (reporting bias) | Low risk | Comment: no reasons for any concerns detected |
| Other bias | Low risk | Comment: no reasons for any concerns detected |
| Study characteristics | ||
| Methods | Randomised, cross‐over trial with 2 arms
Recruitment period: March 2009 to April 2010
Masking: single‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria:
Exclusion criteria
Median age (range), years: 51 (36 to 73) in azasetron first group, 50 (42 to 68) in granisetron NK first group Gender: female Tumour/cancer type: breast cancer Chemotherapy regimen: FEC100 (5‐FU (500 mg/m²), epirubicin hydrochloride (100 mg/m²), and cyclophosphamide hydrate (500 mg/m²) on Day 1, every 3 weeks) Country: Japan (single centre) | |
| Interventions | Cross‐over study Experimental: arm A: azasetron first azasetron 10 mg mixed with dexamethasone and intravenously infused over 30 min before administration of FEC100 regimen granisetron hydrochloride 2 mg and dexamethasone 8 mg p.o. administered for 3 days after FEC100 regimen Experimental: arm B: granisetron NK first granisetron NK 3 mg mixed with dexamethasone and intravenously infused over 30 min before administration of FEC100 regimen granisetron hydrochloride 2 mg and dexamethasone 8 mg p.o. administered for 3 days after FEC100 regimen | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... single‐blind ..." Comment: patients were blinded |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... single‐blind ..." Comment: personnel were not blinded |
| Blinding of outcome assessment (detection bias) | High risk | Comment: although patients were blinded, unblinded personnel might have had an influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although personnel were not blinded, we assume that this had no effect on objective outcomes |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in analysis of adverse events |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised study with 2 arms
Study period: January 2011 to August 2011
Masking: open‐label Baseline patient characteristics: yes Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: female Tumour/cancer type: malignant neoplasm Chemotherapy regimen: HEC Country: United States (single centre) | |
| Interventions | Experimental: arm A: palonosetron palonosetron 0.25 mg Day 1; aprepitant 125 mg Day 1, 80 mg Days 2 to 3; dexamethasone 12 mg Day 1, 8 mg Days 2 to 4 Experimental: arm B: ondansetron ondansetron 24 mg Day 1; aprepitant 125 mg Day 1, 80 mg Days 2 to 3; dexamethasone 12 mg Day 1, 8 mg Days 2 to 4 | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the study results tab |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel‐group study with 2 arms
Study period: April 2011 to March 2014
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age, years: 64.1 in aprepitant group, 64.2 in control group Gender: male (252) + female (161) Tumour/cancer type: colorectal cancer Chemotherapy regimen: oxaliplatin‐based chemotherapy (FOLFOX, XELOX, or SOX regimen including oxaliplatin at ≥ 85 mg/m²) Country: 25 hospitals in Japan (multi‐centre) | |
| Interventions | Experimental: arm A: aprepitant or fosaprepitant Day 1: p.o. aprepitant 125 mg + i.v. 5‐HT₃ receptor antagonist + dexamethasone 6.6 mg Days 2 to 3: aprepitant 80 mg + p.o. dexamethasone 2 mg twice daily Day 1: i.v. fosaprepitant 150 mg + 5‐HT₃ receptor antagonist + dexamethasone 6.6 mg Day 2: p.o. dexamethasone 2 mg twice daily Day 3: p.o. dexamethasone 4 mg twice daily Control: arm B Day 1: i.v. 5‐HT₃ receptor antagonist + dexamethasone 9.9 mg Days 2 to 3: p.o. dexamethasone (4 mg) twice daily | |
| Outcomes | Primary outcome
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the allocation of patients was performed using the minimisation method with age, sex, chemotherapy‐naive/non‐naive status, regimen (FOLFOX, XELOX or SOX) and study centre as the adjustment factors" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although this was an open‐label study, both patients and personnel had no influence on objective outcomes (e.g. hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all 413 patients have been included in the full analysis set (Fig. 1) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "tolerability analyses included 398 patients who received oxaliplatin‐based chemotherapy in the first cycle" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Prospective, stratified, randomised, cross‐over, comparative trial with 2 arms
Recruitment period: n.r.
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria: n.r. Median age (range), years: 53 (35 to 75) in palonosetron‐first group, 53 (40 to 71) in granisetron‐first group Gender: female Tumour/cancer type: breast cancer Chemotherapy regimen AC treatment, adriamycin (60 mg/m²) and cyclophosphamide (600 mg/m²) EC treatment, epirubicin (90 mg/m²) and cyclophosphamide (600 mg/m²) FEC treatment, 5‐fluorouracil (500 mg/m²), epirubicin (90 mg/m²), and cyclophosphamide (500 mg/m²) Country: Japan (single centre) | |
| Interventions | Cross‐over study Experimental: arm A: palonosetron‐first Day 1: aprepitant p.o. 125 mg + palonosetron i.v. 0.75 mg + dexamethasone i.v. 13.2 mg Days 2 to 3: aprepitant p.o. 80 mg + dexamethasone p.o. 8 mg Day 4: dexamethasone p.o. 8 mg Experimental: arm A: granisetron‐first Day 1: aprepitant p.o. 125 mg + granisetron i.v. 3 mg + dexamethasone i.v. 13.2 mg Days 2 to 3: aprepitant p.o. 80 mg + dexamethasone p.o. 8 mg Day 4: dexamethasone p.o. 8 mg | |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... stratified randomization ...", "... using a table of random numbers ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... non‑blinded ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... non‑blinded ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were described in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, phase 2 study with 2 arms
Enrolment period: August 2011 to March 2013
Masking: n.r. Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria: n.r. Exclusion criteria: n.r. Mean/median age, years: n.r. Gender: n.r. Tumour/cancer type: colorectal cancer Chemotherapy regimen: standard MEC regimen Country: Japan (multi‐centre) | |
| Interventions | Experimental: arm A: aprepitant aprepitant + palonosetron + dexamethasone Control: arm B palonosetron + dexamethasone | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: blinding not reported |
| Incomplete outcome data (attrition bias) | High risk | Quote: "fifteen patients were excluded because of insufficient diary and protocol violation. Finally, we analyzed 45 patients (26 male, 19 female) for this study" Comment: the percentage of excluded patients was 25% of the initial included population |
| Selective reporting (reporting bias) | Unclear risk | Comment: only complete response was reported in detail; other secondary outcomes were not reported with statistical figures, as no significant differences were observed between the 2 groups. Conference abstract only, not evaluable |
| Other bias | Unclear risk | Comment: conference abstract, not evaluable |
| Study characteristics | ||
| Methods | Randomised, parallel‐group, phase 3, placebo‐controlled trial with 2 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD (range), years: 54 ± 13 (18 to 82) in aprepitant regimen group, 53 ± 14 (18 to 81) in standard therapy regimen group Gender: male + female Tumour/cancer type: solid tumours (respiratory, urogenital, eyes/ears/nose/throat, other) Chemotherapy regimen: chemotherapy regimen that included cisplatin 70 mg/m² Country: Argentina, Brazil, Chile, Colombia, Guatemala, Mexico, Peru, Venezuela (18 centres) | |
| Interventions | Experimental: arm A: aprepitant 125/80 mg Day 1: p.o. aprepitant 125 mg + i.v. ondansetron 32 mg + p.o. dexamethasone 12 mg Days 2 to 3: p.o. aprepitant 80 mg + p.o. dexamethasone 8 mg Day 4: p.o. dexamethasone 8 mg Standard: arm B Day 1: i.v. ondansetron 32 mg + p.o. dexamethasone 20 mg Days 2 to 4: p.o. dexamethasone 8 mg twice daily | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... computer‐generated randomization ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: a modified intent‐to‐treat approach was used to analyse efficacy data |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, prospective, parallel‐group, phase 3 trial with 3 arms
Study period: June 2006 to February 2009
Masking: double‐blind (participant, investigator) Baseline patient characteristics: n.r. Follow‐up: after completion of study treatment, patients are followed at approximately 30 days | |
| Participants | Inclusion criteria
Exclusion criteria: n.r. Mean/median age (range), years: MEC: APF530 250 mg: 60.3 (SD 12.5); APF530 500 mg: 59.1 (SD 13.3); palonosetron 0.25 mg: 60.4 (12.8); HEC: APF530 250 mg: 53.0 (SD 12.7); APF530 500 mg: 52.8 (SD 11.9); palonosetron 0.25 mg: 54.5 (SD 12.8) Gender: male + female Tumour/cancer type: solid tumour (lung cancer, breast cancer, ovarian cancer, lymphoma) Chemotherapy regimen: cyclophosphamide + doxorubicin or epirubicin (78% of MEC participants and 75% of HEC participants) Country: United States (52 study locations, multi‐centre) | |
| Interventions | Active comparator: arm A Day 1 of chemotherapy course 1: palonosetron hydrochloride i.v. + placebo subcutaneously (SC) + dexamethasone i.v. patients in high‐risk (level 5) stratum also receive oral dexamethasone on Days 2 to 4 of all treatment courses Experimental: arm B Day 1 of chemotherapy course 1: APF530 SC + placebo i.v. + dexamethasone i.v. Day 1 of chemotherapy courses 2 to 4: APF530 SC + dexamethasone i.v. patients in high‐risk (level 5) stratum also receive oral dexamethasone as in arm A Experimental: arm C Day 1 of chemotherapy course 1: APF530 SC at higher dose + placebo i.v. + dexamethasone i.v. Day 1 of chemotherapy courses 2 to 4: APF530 SC (at same higher dose) + dexamethasone i.v. patients in high‐risk (level 5) stratum also receive oral dexamethasone as in arm A | |
| Outcomes | Primary outcome measures
Secondary outcome measures
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomized 1:1:1 to ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. injection site reaction) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "efficacy analyses were performed separately for ASCO‐derived MEC and HEC strata and were based on a modified intent‐to‐treat (mITT) population" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the safety population in this reanalysis comprised all 1395 patients who were randomized and received study drug ..." |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, prospective, parallel‐group, controlled trial with 2 arms
Study period: January 2007 to December 2008
Masking: double‐blind (participant, investigator) Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 57.1 ± 11.8 in aprepitant group, 55.9 ± 12.6 in control group Gender: male (196) + female (652) Tumour/cancer type: solid malignancy (breast cancer, colorectal cancer, lung cancer, ovarian cancer) Chemotherapy regimen: non‐AC (anthracycline (doxorubicin and epirubicin) and cyclophosphamide), AC (anthracycline (doxorubicin and epirubicin) and cyclophosphamide) Countries: USA, Mexico, Canada, Chile, Brazil, Peru, Colombia, Panama, Hong Kong, Australia, South Africa, France, Germany, Israel, Russia | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg p.o. 1 h before chemotherapy + ondansetron 8 mg p.o. 30 to 60 min before chemotherapy; 8 mg p.o. 8 h after first dose + dexamethasone 12 mg p.o. 30 min before chemotherapy Days 2 to 3: aprepitant 80 mg p.o. + ondansetron placebo p.o. b.i.d. Control: arm B: placebo Day 1: aprepitant placebo p.o. 1 h before chemotherapy + ondansetron 8 mg p.o. 30 to 60 min before chemotherapy; 8 mg p.o. 8 h after first dose + dexamethasone 20 mg p.o. 30 min before chemotherapy Days 2 to 3: aprepitant placebo p.o. + ondansetron 8 mg p.o. b.i.d. | |
| Outcomes | Primary outcome measure
Secondary outcome measure
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... computer‐generated, random, blinded allocation schedule ..." |
| Allocation concealment (selection bias) | Low risk | Comment: allocation was blinded |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind (participant, investigator) |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind (participant, investigator) |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. neutropenia) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the analysis of efficacy was based on the full analysis set population, which included those patients who received MEC, took a dose of study drug, and completed at least one posttreatment efficacy assessment" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "all patients who received at least one dose of study drug were included in the safety analyses" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel‐group, placebo‐controlled, dose‐ranging,phase 2 trial with 5 arms
Study period: September 2006 to March 2008
Masking: double‐blind (participant, investigator) Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 55 (22 to 86) in rolapitant 9‐mg group, 53 (26 to 76) in rolapitant 22.5‐mg group, 57 (19 to 79) in rolapitant 90‐mg group, 56 (20 to 75) in rolapitant 180‐mg group, 54 (18 to 77) in control group Gender: male (244) + female (210) Tumor/cancer type: n.r. Chemotherapy regimen: HEC (≥ 70 mg/m² cisplatin‐based chemotherapy) Country: 75 sites in 21 countries | |
| Interventions | Experimental: arm A: rolapitant 9 mg Day 1: rolapitant 9 mg administered approximately 2 h before first dose of chemotherapeutic agent on Day 1 of Cycle 1 + i.v. ondansetron 32 mg 0.5 h before initiation of chemotherapy + oral dexamethasone 20 mg 0.5 h before initiation of chemotherapy Days 2 to 4: dexamethasone 8 mg twice daily Experimental: arm B: rolapitant 22.5 mg Day 1: rolapitant 22.5 mg administered approximately 2 h before first dose of chemotherapeutic agent on Day 1 of Cycle 1 + i.v. ondansetron 32 mg 0.5 h before initiation of chemotherapy + oral dexamethasone 20 mg 0.5 h before initiation of chemotherapy Days 2 to 4: dexamethasone 8 mg twice daily Experimental: arm C: rolapitant 90 mg Day 1: rolapitant 90 mg administered approximately 2 h before first dose of chemotherapeutic agent on Day 1 of Cycle 1 + i.v. ondansetron 32 mg 0.5 h before initiation of chemotherapy + oral dexamethasone 20 mg 0.5 h before initiation of chemotherapy Days 2 to 4: dexamethasone 8 mg twice daily Experimental: arm D: rolapitant 180 mg Day 1: rolapitant 180 mg administered approximately 2 h before first dose of chemotherapeutic agent on Day 1 of Cycle 1 + i.v. ondansetron 32 mg 0.5 h before initiation of chemotherapy + oral dexamethasone 20 mg 0.5 h before initiation of chemotherapy Days 2 to 4: dexamethasone 8 mg twice daily Control: arm E Day 1: placebo + i.v. ondansetron 32 mg 0.5 h before initiation of chemotherapy + oral dexamethasone 20 mg 0.5 h before initiation of chemotherapy Days 2 to 4: dexamethasone 8 mg twice daily | |
| Outcomes | Primary outcome measure
Secondary outcome measures
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind (participant, investigator) |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind (participant, investigator) |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. febrile neutropenia, neutropenia) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included for the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included for the safety analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, active‐controlled, parallel‐group, phase 3 trial with 2 arms
Study period: February 2012 to May 2014
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 57.0 ± 10.08 in rolapitant group, 57.7 ± 11.15 in placebo group Gender: male (304) + female (222) Tumour/cancer type: solid tumour (breast, colon or rectum, head and neck, lung, ovary, stomach, and other tumours) Chemotherapy regimen: cisplatin‐based chemotherapy (≥ 60 mg/m²) Country: United States (multi‐centre) | |
| Interventions | Experimental: arm A: rolapitant Day 1: oral dose of rolapitant 180 mg (equivalent to 200 mg rolapitant hydrochloride monohydrate) 1 to 2 h before administration of chemotherapy + granisetron (10 µg/kg i.v.) + dexamethasone (20 mg p.o.) Days 2 to 4: dexamethasone (8 mg p.o.) to be administered orally b.i.d. Control: arm B Day 1: placebo + granisetron (10 µg/kg i.v.) + dexamethasone (20 mg p.o.) Days 2 to 4: dexamethasone (8 mg p.o.) to be administered orally b.i.d. | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "for randomisation of patients, we used an interactive web‐based randomisation system (IWRS) at cycle 1" |
| Allocation concealment (selection bias) | Low risk | Quote: "an independent group not involved with study implementation created a randomisation schedule for study drug labelling" |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind (participant, investigator) |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind (participant, investigator) |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. febrile neutropenia, neutropenia) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: analysis included modified ITT population |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the safety population included all patients who were randomly allocated to a treatment group and who received at least one dose of study drug" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel‐group, active‐controlled, phase 3 trial with 2 arms
Study period: February 2012 to March 2014
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 58.5 ± 10.05 in rolapitant group, 58.5 ± 9.25 in placebo group Gender: male (369) + female (175) Tumour/cancer type: solid tumour (breast, colon or rectum, head and neck, lung, ovary, stomach, and other tumours) Chemotherapy regimen: cisplatin‐based chemotherapy (≥ 60 mg/m²) Country: United States (multi‐centre) | |
| Interventions | Experimental: arm A: rolapitant Day 1: oral dose of rolapitant 180 mg (equivalent to 200 mg rolapitant hydrochloride monohydrate) 1 to 2 h before administration of chemotherapy + granisetron (10 µg/kg i.v.) + dexamethasone (20 mg p.o.) Days 2 to 4: dexamethasone (8 mg p.o.) to be administered orally b.i.d. Control: arm B Day 1: placebo + granisetron (10 µg/kg i.v.) + dexamethasone (20 mg p.o.) Days 2 to 4: dexamethasone (8 mg p.o.) to be administered orally b.i.d. | |
| Outcomes | Primary endpoints
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "for randomisation of patients, we used an interactive web‐based randomisation system (IWRS) at cycle 1" |
| Allocation concealment (selection bias) | Low risk | Quote: "an independent group not involved with study implementation created a randomisation schedule for study drug labelling" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "double‐blind (participant, investigator)" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "double‐blind (participant, investigator)" |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention; therefore they probably had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. febrile neutropenia, neutropenia) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: analysis included modified ITT population |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the safety population included all patients who were randomly allocated to a treatment group and who received at least one dose of study drug" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Double‐blind, multi‐centre, randomised trial with 2 arms
Recruitment period: December 1992 to July 1994
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age (range), years: 61 Gender: male + female Tumour/cancer type: solid tumours Chemotherapy regimen: cisplatin at doses ≥ 50 mg/m², used alone or in combination with other antineoplastic agents Country: Italy (multi‐centre) | |
| Interventions | Experimental: arm A: ondansetron ondansetron 8 mg i.v. diluted in 50 mL normal saline and administered 15 min, 30 min before chemotherapy + dexamethasone 20 mg i.v. added to 5‐HT₃ antagonist and administered 15 min, 45 min before chemotherapy Experimental: arm B: granisetron granisetron 3 mg i.v. diluted in 50 mL normal saline and administered 15 min, 30 min before chemotherapy + dexamethasone 20 mg i.v. added to 5‐HT₃ antagonist and administered 15 min, 45 min before chemotherapy | |
| Outcomes | Primary endpoint
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "using a centralized list of computer‐generated random permuted blocks of 10 patients for each centre ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind study |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind study |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "a total of 973 patients took part in the study and 966 of them were evaluated for efficacy according to the intention‐to‐treat principle" Comment: some patients missing without reasons provided; however, we judged this number to be small |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 967 participants were included for adverse events assessment; this number represents nearly the entire enrolled cohort |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, active‐controlled, parallel‐group, phase 3 trial with 2 arms
Study period: April 2011 to November 2012
Masking: quadruple‐blind (participant, care provider, investigator, outcomes assessor) Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
The following inclusion criteria must be checked before inclusion at each cycle of the multiple‐cycle extension
Exclusion criteria
The following exclusion criteria must be checked before inclusion at each cycle of the multiple‐cycle extension
Mean age (SD), years: 53.7 (10.66) in netupitant + palonosetron group, 54.1 (10.65) in palonosetron group Gender: male + female Tumour/cancer type: breast cancer Chemotherapy regimen: anthracycline and cyclophosphamide‐containing chemotherapy Country: United States (28), Argentina (9), Belarus (6), Brazil (12), Bulgaria (12), Croatia (9), Germany (11), Hungary (8), India (13), Italy (5), Mexico (5), Poland (10), Romania (10), Russian Federation (22), Ukraine (12) (172 centres) | |
| Interventions | Experimental: arm A: NEPA + dexamethasone Day 1: oral netupitant/palonosetron (300 mg/0.50 mg) hard capsule with oral dexamethasone before each scheduled chemotherapy cycle Experimental: arm B: palonosetron + dexamethasone Day 1: oral palonosetron 0.50 mg (Aloxi) with oral dexamethasone before each scheduled chemotherapy cycle | |
| Outcomes | Primary outcome measure
Secondary outcome measures
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "quadruple (participant, care provider, investigator, outcomes assessor)" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "quadruple (participant, care provider, investigator, outcomes assessor)" |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: patients, personnel, investigator, and outcome assessor were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all randomised patients were included in the ITT population |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, phase 3 study with 2 arms
Recruitment period: June 2010 to March 2015
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years (median, range): fosaprepitant group: 48 (25 to 74); placebo group: 47 (26 to 77) Gender: female Tumour/cancer type: cervical cancer Chemotherapy regimen: cisplatin 40 mg/m² Country: 8 centres in 4 countries (Germany, Australia, Norway, Denmark) | |
| Interventions | Experimental: arm A: fosaprepitant fosaprepitant + palonosetron + dexamethasone Experimental: arm B: placebo placebo + palonosetron + dexamethasone | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the randomisation numbers were generated by an independent research staff member using a web randomisation sequence generator" |
| Allocation concealment (selection bias) | Unclear risk | Quote: "the blocks, in sealed envelopes, were provided to the pharmacists who were not masked to treatment at the study centres, and sealed blinding envelopes unique to each randomisation number were provided to the centres."; "To avoid treatments being visually distinguishable, study drug and placebo were provided in identically wrapped and labelled infusion bags of the same volume" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. neutrophil count decreased) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "... the 234 patients receiving study medication represented the modified intention‐to‐treat population (figure 1) and were all included in the analysis of the primary and secondary outcomes and in the safety analysis" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "... the 234 patients receiving study medication represented the modified intention‐to‐treat population (figure 1) and were all included in the analysis of the primary and secondary outcomes and in the safety analysis" |
| Selective reporting (reporting bias) | Low risk | Comment: study authors mentioned explicitly that FLIE results are not reported in this article |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel‐group, comparative, phase 3, active‐comparator trial with 2 arms
Study period: July 2006 to May 2007
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 58.4 ± 10.4 in palonosetron arm, 58 ± 10.5 in granisetron arm Gender: male + female Tumour/cancer type: solid tumour (non‐small cell lung cancer, small cell lung cancer, breast cancer, others) Chemotherapy regimen: cisplatin, doxorubicin + cyclophosphamide/epirubicin + cyclophosphamide Country: Japan (75 centres) | |
| Interventions | Experimental: arm A: palonosetron i.v. injection of 0.75 mg palonosetron (5 mL), then granisetron placebo before administration of highly emetogenic chemotherapy, concomitantly administered with corticosteroids Experimental: arm B granisetron i.v. injection of palonosetron placebo, then 40 μg/kg granisetron hydrochloride before administration of highly emetogenic chemotherapy, concomitantly administered with corticosteroids | |
| Outcomes | Primary outcome measure
Secondary outcome measures
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomisation was done centrally by computer ..."; "a non‐deterministic minimisation method with a stochastic‐biased coin was applied to the randomisation of patients" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. study mortality) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the modified ITT cohort was used for the primary efficacy analysis" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "... including one death caused by bleeding from pulmonary carcinoma in the granisetron group..." |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, parallel, phase 3 trial with 2 arms
Recruitment period: August 2009 to December 2009
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 62 (26 to 86) in fosaprepitant group, 63 (25 to 85) in placebo group Gender: male (257) + female (90) Tumour/cancer type: solid tumour (respiratory, genitourinary, digestive, head and neck, other) Chemotherapy regimen: chemotherapy including cisplatin (≥ 70 mg/m²) Country: Japan (68 institutions) | |
| Interventions | Experimental: arm A: fosaprepitant Day 1: i.v. fosaprepitant (150 mg) + granisetron (40 μg/kg) + dexamethasone phosphate (10 mg) Day 2: dexamethasone phosphate (4 mg) Day 3: dexamethasone phosphate (8 mg) Experimental: arm B: placebo Day 1: placebo + granisetron (40 μg/kg body weight) + dexamethasone phosphate (20 mg) Days 2 to 3: dexamethasone phosphate (8 mg) | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. study mortality, neutropenia) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the efficacy analysis was carried out on the modified intention‐to‐treat (ITT) population" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "therefore, 340 patients (167 in the standard arm and 173 in the fosaprepitant arm) were assessable in the modified ITT analysis, and 344 patients were included in the safety analysis" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, comparative, interventional, parallel, phase 3 trial with 2 arms
Recruitment period: 2012 to 2015
Masking: double‐blind Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age (range), years: n.r. Gender: female Tumour/cancer type: breast cancer Chemotherapy regimen: anthracycline and cyclophosphamide‐containing regimens (AC) Country: Japan (11 institutions) | |
| Interventions | Experimental: arm A: palonosetron Day 1: aprepitant (125 mg) + palonosetron (0.75 mg) + dexamethasone (9.9 mg) Days 2 to 3: aprepitant (80 mg) Experimental: arm B: granisetron Day 1: aprepitant (125 mg) + granisetron (40 μg/kg) + dexamethasone (9.9 mg) Days 2 to 3: aprepitant (80 mg) | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included for patient‐reported outcomes analysis |
| Selective reporting (reporting bias) | High risk | Comment: only complete response and quality of life were reported in the results section |
| Other bias | Unclear risk | Comment: conference abstract, not evaluable |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, phase 3 trial with 2 arms
Recruitment period: July 2005 to January 2012
Masking: double‐blind Baseline patient characteristics: n.r. Follow‐up: Days 5 to 7 | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 58.3 (27 to 72) in aprepitant arm, 57.9 (35 to 72) in placebo arm Gender: male + female Tumour/cancer type: multiple myeloma Chemotherapy regimen: high‐dose melphalan therapy Country: Heidelberg University Hospital, Heidelberg, Germany (single centre) | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg + granisetron 2 mg + dexamethasone 4 mg Days 2 to 3: aprepitant 80 mg + granisetron 2 mg + dexamethasone 2 mg Day 4: aprepitant 80 mg + granisetron 2 mg Experimental: arm B: placebo Day 1: placebo 125 mg + granisetron 2 mg + dexamethasone 8 mg Days 2 to 3: placebo 80 mg + granisetron 2 mg + dexamethasone 4 mg Day 4: placebo 80 mg + granisetron 2 mg | |
| Outcomes | Primary endpoint
Secondary endpoint
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... computer‐generated randomization list ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "362 patients underwent ITT analysis out of 363 randomly assigned patients" |
| Incomplete outcome data (attrition bias) | Low risk | Comment: ITT data set used for safety analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel‐group trial with 2 arms
Enrolment period: patients were enrolled from 2 January 2004 and were followed up until 30 September 2004
Masking: double‐blind, with sponsor blinding Baseline patient characteristics: reported Follow‐up: follow‐up visit on Days 19 to 29 | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD (range), years: 59 ± 11 (20 to 79) in aprepitant regimen, 58 ±11 (23 to 82) in control regimen Gender: male + female Tumour/cancer type: solid tumour (respiratory, urogenital, gastrointestinal, eyes/ears/nose/throat, other) Chemotherapy regimen: chemotherapy including cisplatin ≥ 70 mg/m²in Cycle 1 Country: 56 investigator sites in Europe, North America, South America, and Korea | |
| Interventions | Experimental: arm A: aprepitant 125/80 mg Day 1: p.o. aprepitant 125 mg + i.v. ondansetron 32 mg + p.o. dexamethasone 12 mg Days 2 to 3: p.o. aprepitant 80 mg + p.o. ondansetron placebo twice daily + p.o. dexamethasone 8 mg in the morning and placebo in the evening Day 4: p.o. ondansetron placebo twice daily + p.o. dexamethasone 8 mg in the morning and placebo in the evening Control: arm B Day 1: aprepitant placebo + i.v. ondansetron 32 mg + p.o. dexamethasone 20 mg Days 2 to 3: aprepitant placebo + p.o. ondansetron 8 mg twice daily + p.o. dexamethasone 8 mg twice daily Day 4: p.o. ondansetron 8 mg twice daily + p.o. dexamethasone 8 mg twice daily | |
| Outcomes | Primary efficacy endpoint
Secondary endpoints
Exploratory endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients received either the aprepitant or the control regimen in a 1:1 ratio according to a sponsor‐supplied, computer‐generated, random allocation schedule" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind, parallel‐group trial with sponsor blinding ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind, parallel‐group trial with sponsor blinding ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. febrile neutropenia, hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the efficacy analyses used a modified intention‐to‐treat (mITT) population ..." |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included for the tolerability analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were described in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, phase 3 study with 2 arms
Enrolment period: n.r.
Masking: double‐blind Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria: n.r. Mean/median age, years: n.r. Gender: n.r. Tumour/cancer type: n.r. Chemotherapy regimen: MEC (cyclophosphamide, doxorubicin, epirubicin, carboplatin, irinotecan, daunorubicin, or cytarabine) Country: n.r. | |
| Interventions | Experimental: arm A: rolapitant rolapitant + granisetron + dexamethasone Experimental: arm B: placebo placebo + granisetron + dexamethasone | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...were randomized 1:1 to either ..." Comment: sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: one thousand three hundred forty‐four evaluable patients were included for efficacy analysis |
| Selective reporting (reporting bias) | Unclear risk | Comment: conference abstract only, not evaluable |
| Other bias | Unclear risk | Comment: conference abstract only, not evaluable |
| Study characteristics | ||
| Methods | Randomised, prospective, double‐dummy, parallel‐group, phase 3 trial with 2 arms
Study period: March 2014 to May 2015
Masking: quadruple‐blind (participant, care provider, investigator, outcomes assessor) Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 55.7 ± 11.75 in granisetron group, 55.6 ± 11.94 in ondansetron group Gender: male + female Tumour/cancer type: malignant disease Chemotherapy regimen: anthracycline + cyclophosphamide Country: United States (77 centres) | |
| Interventions | Experimental: arm A: fosaprepitant + granisetron + dexamethasone Day 1: single SC dose of APF530 500 mg (10 mg granisetron) + placebo for ondansetron i.v. + fosaprepitant 150 mg i.v. + dexamethasone 12 mg i.v. Day 2: dexamethasone 8 mg p.o. QD Days 3 to 4: dexamethasone 8 mg p.o. b.i.d. Experimental: arm B: fosaprepitant + ondansetron + dexamethasone Day 1: single i.v. dose of ondansetron 0.15 mg/kg (to maximum of 16 mg) + placebo for APF530 SC + fosaprepitant 150 mg i.v. + dexamethasone 12 mg i.v. Day 2: dexamethasone 8 mg p.o. QD Days 3 to 4: dexamethasone 8 mg p.o. b.i.d. | |
| Outcomes | Primary outcome measure
Secondary outcome measures
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "masking: quadruple (Participant, Care Provider, Investigator, Outcomes Assessor)" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "masking: quadruple (Participant, Care Provider, Investigator, Outcomes Assessor)" |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. injection‐site reactions, neutropenia) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "efficacy analyses were performed on the modified intent‐to‐treat (mITT) population, comprising all randomized patients who received study drug and an HEC regimen and had post baseline efficacy data" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "safety analyses were based on the safety population, comprising all randomized patients who received study drug" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, parallel‐group, active‐controlled, phase 3 study with 2 arms
Study period: 5 March to 6 September 2013
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 58 (22 to 86) in rolapitant group, 56 (22 to 88) in active control group Gender: male (265) + female (1067) Tumour/cancer type: malignant solid tumour (breast, colon or rectum, head and neck, lung, ovary, stomach, other tumours) Chemotherapy regimen: MEC including ≥ 1 of the following agents: cyclophosphamide i.v. (< 1500 mg/m²), doxorubicin, epirubicin, carboplatin, idarubicin, ifosfamide, irinotecan, daunorubicin, cytarabine i.v. (> 1 g/m²) Country: 170 cancer centres in 23 countries (multi‐centre) | |
| Interventions | Experimental: arm A: rolapitant Day 1: rolapitant (200 mg p.o.) + granisetron (2 mg p.o.) + dexamethasone (20 mg p.o.) Days 2 to 3: granisetron (2 mg p.o.) administered orally Control: arm B Day 1: placebo + granisetron (2 mg p.o.) + dexamethasone (20 mg p.o.) Days 2 to 3: granisetron (2 mg p.o.) administered orally | |
| Outcomes | Primary outcome
Secondary outcome(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "for randomisation of patients, we used an interactive web‐based randomisation system (IWRS) at cycle 1" |
| Allocation concealment (selection bias) | Low risk | Quote: "for masking, a double‐blind technique was used. Placebo capsules were identical in appearance to rolapitant capsules. Through out the study, neither the patient nor the investigators and assessors knew which treatment the patient was receiving."; "... an independent group with no further role in study implementation ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. neutropenia, febrile neutropenia) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "therefore, in cycle 1, the modified intention‐to‐treat population comprised 666 patients in each treatment group" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the safety population consisted of all patients who received at least one dose of study drug" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, cross‐over, active‐controlled, phase 4 study with 2 arms
Enrolment period: 17 August 2012 to 14 February 2014
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age± SD, years: 58.88 ± 10.53 in granisetron and palonosetron group, 60.41 ± 10.06 in palonosetron and granisetron group Gender: male (117) + female (71) Tumour/cancer type: n.r. Chemotherapy regimen: MEC, FOLFIRI, or FOLFOX Country: 6 tertiary referral hospitals in Korea (multi‐centre) | |
| Interventions | Cross‐over study Experimental: arm A: granisetron and palonosetron transdermal granisetron (1 GTDS patch, 7 days) in the first cycle, palonosetron (i.v. 0.25 mg/d, 1 day) in the second cycle before receiving MEC in 2 consecutive cycles prophylactic dexamethasone (i.v. 10 mg) within 30 min before chemotherapy on Day 1 Experimental: arm B: palonosetron and granisetron palonosetron in the first cycle and GTDS in the second cycle prophylactic dexamethasone (i.v. 10 mg) within 30 min before chemotherapy on Day 1 | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding should not affect risk of bias of objective outcomes |
| Incomplete outcome data (attrition bias) | High risk | Quote: "three hundred thirty‐three cycles were included in the per protocol analysis ‐ 165 cycles for the GTDS and 168 cycles for the palonosetron were analyzed (Fig. 2)" Comment: per‐protocol analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: safety assessed for all completed cycles |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, prospective, comparative clinical trial with 2 arms
Enrolment period: October 2013 to March 2015
Masking: open‐label Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 41.2 ± 4.57 in aprepitant group, 39.6 ± 6.85 in control group Gender: male (74) + female (34) Tumour/cancer type: B‐ or T‐cell non‐Hodgkin lymphoma Chemotherapy regimen: cyclophosphamide (750 mg/m²), epirubicin (60 mg/m²), vincristine (1.4 mg/m²) Country: China (single centre) | |
| Interventions | Experimental: arm A: aprepitant Day 1: p.o. aprepitant 125 mg + i.v. ondansetron 24 mg + p.o. prednisone 100 mg Days 2 to 3: p.o. aprepitant 80 mg + prednisone 100 mg once daily Days 4 to 5: p.o. prednisone 100 mg once daily Control: arm B Day 1: i.v. ondansetron 24 mg + p.o. prednisone 100 mg Days 2 to 5: p.o. prednisone 100 mg once daily | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were assigned to one of the two treatment groups, according to a computer‐generated random assignment schedule" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards intervention and therefore might influence subjective outcomes analysis |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although it was an open‐label study, both patients and personnel had no influence on objective outcomes (e.g. hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 101 patients have been included in the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: safety data reported for all 101 patients |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised trial with 2 arms
Recruitment period: n.r. Enrolled/randomised patient number: n.r. Masking: double‐blind Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria: n.r. Exclusion criteria: n.r. Mean/median age (range), years: n.r. Gender: n.r. Tumour/cancer type: n.r. Chemotherapy regimen: cisplatin Country: n.r. | |
| Interventions | Experimental: arm A: granisetron granisetron + dexamethasone (8 mg i.v., then treatment 4 mg t.d.s. for 3 days afterwards) Experimental: arm B: ondansetron ondansetron + dexamethasone (8 mg i.v., then treatment 4 mg t.d.s. for 3 days afterwards) | |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised study but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: not reported |
| Selective reporting (reporting bias) | Unclear risk | Comment: not evaluable, conference abstract only |
| Other bias | Unclear risk | Conference abstract, not evaluable |
| Study characteristics | ||
| Methods | Randomised, cross‐over, controlled trial with 2 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: 56 (37 to 74) for all patients Gender: male (9) + female (12) Tumour/cancer type: n.r. Chemotherapy regimen: highly emetogenic chemotherapy, including cisplatin, mean dose 74 mg/m² Country: UK | |
| Interventions | Cross‐over study Experimental: arm A: ondansetron ondansetron 8 mg + i.v. bolus dexamethasone 8 mg immediately before chemotherapy and p.o. dexamethasone 4 mg 3 times a day on Days 2 to 4 Experimental: arm B: granisetron granisetron 3 mg by infusion + i.v. bolus dexamethasone 8 mg immediately before chemotherapy and p.o. dexamethasone 4 mg 3 times a day on Days 2 to 4 | |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... random allocation using a Latin square design in sets of four" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | Comment: outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomized, comparative, phase 3 trial with 2 arms
Registration period: September 2004 to July 2008
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 50 (20 to 75) in aprepitant 125/80 mg group, 51 (19 to 79) in placebo group Gender: male + female Tumour/cancer type: malignant disease (non‐Hodgkin lymphoma, AML, multiple myeloma, ALL, Hodgkin lymphoma, CML, myelodysplastic syndrome, myeloproliferative disorder, chronic lymphocytic leukaemia, myelofibrosis) Chemotherapy regimen: high‐dose cyclophosphamide preparative regimens Country: United States (single centre) | |
| Interventions | Experimental: arm A: aprepitant 125/80 mg aprepitant 125 mg p.o. Day 1, then 80 mg daily during preparative regimen + 3 days dexamethasone 7.5 mg i.v. daily during preparative regimen + 1 day ondansetron 8 mg p.o. q8h daily during preparative regimen + 1 day Experimental: arm B: placebo placebo p.o. daily during preparative regimen + 3 days dexamethasone 10 mg i.v. daily during preparative regimen + 1 day ondansetron 8 mg p.o. q8h daily during preparative regimen + 1 day | |
| Outcomes | Primary endpoints
Secondary endpoints
Additional analyses
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Comment: patients who received the study drug were included in the intent‐to‐treat analysis |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "five patients died in the aprepitant arm due to sepsis (3 patients), toxic epidermal necrolysis and sepsis (1 patient), and veno‐occlusive disease of the liver (1 patient), whereas 2 patients died in the control arm due to viral pneumonia/encephalitis (1 patient) and fungal pneumonia (1 patient) within 30 days" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Multi‐centre, randomised, double‐blind, placebo‐controlled, parallel‐group, phase 2 study with 3 arms
Registration period: September 2016 to November 2017
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 67 (36 to 79) in placebo group, 66 (41 to 76) in fosnetupitant 81‐mg group, 67 (37 to 78) in fosnetupitant 235‐mg group Gender: 24.2% female (75.8% male) in placebo group, 25.1% female (74.9% male) in fosnetupitant 81‐mg group, 24.1% female (75.9% male) in fosnetupitant 235‐mg group Tumour/cancer type: confirmed malignant solid tumour Chemotherapy regimen: cisplatin‐based, cisplatin at a dose ≥ 70 mg/m² Country: Japan (multi‐centre) | |
| Interventions | Experimental: arm A: fosnetupitant 81 mg Fosnetupitant (81 mg intravenously, approximately 60 min before administration of cisplatin and infused for 30 min on Day 1), palonosetron (0.75 mg intravenously, approximately 60 min before administration of cisplatin and infused for 30 min on Day 1), dexamethasone (Day 1, 60 min before administration of cisplatin, 9.9 mg; Days 2 to 4, 6.6 mg administered intravenously in the morning) Experimental: arm B: fosnetupitant 235 mg Fosnetupitant (235 mg intravenously, approximately 60 min before administration of cisplatin and infused for 30 min on Day 1), palonosetron (0.75 mg intravenously, approximately 60 min before administration of cisplatin and infused for 30 min on Day 1), dexamethasone (Day 1, 60 min before administration of cisplatin, 9.9 mg; Days 2 to 4, 6.6 mg administered intravenously in the morning) Control: arm C: placebo Placebo, palonosetron (0.75 mg intravenously, approximately 60 min before administration of cisplatin and infused for 30 min on Day 1), dexamethasone (Day 1, 60 min before administration of cisplatin,13.2 mg; Days 2 to 4, 6.6 mg administered intravenously in the morning) | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Minimisation method used for random allocation, stratified by sex and age class (age < 55 years vs age ≥ 55 years) |
| Allocation concealment (selection bias) | Low risk | Quote: "treatment assignment was masked from all patients, investigators, and study personnel except for the pharmacists who were preparing the study drugs at the institutions, who were prohibited from divulging any information regarding drug assignment" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "treatment assignment was masked from all patients, investigators, and study personnel except for the pharmacists who were preparing the study drugs at the institutions, who were prohibited from divulging any information regarding drug assignment" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "treatment assignment was masked from all patients, investigators, and study personnel except for the pharmacists who were preparing the study drugs at the institutions, who were prohibited from divulging any information regarding drug assignment" |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "treatment assignment was masked from all patients, investigators, and study personnel except for the pharmacists who were preparing the study drugs at the institutions, who were prohibited from divulging any information regarding drug assignment" |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "treatment assignment was masked from all patients, investigators, and study personnel except for the pharmacists who were preparing the study drugs at the institutions, who were prohibited from divulging any information regarding drug assignment" |
| Incomplete outcome data (attrition bias) | Low risk | Data available for nearly all participants (194/197 in placebo group, 195/199 in fosnetu 81‐mg group, 195/198 in fosnetu 235‐mg group) |
| Incomplete outcome data (attrition bias) | Low risk | All participants who received ≥ 1 dose included in safety analysis |
| Selective reporting (reporting bias) | Low risk | No reason for any concern detected |
| Other bias | Low risk | No other bias detected |
| Study characteristics | ||
| Methods | Randomised, prospective, phase 2 study with 2 arms
Enrolment period: November 2010 to March 2014
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age, years: 58.5 in aprepitant group, 57.7 in control group Gender: female Tumour/cancer type: gynaecological cancer Chemotherapy regimen: carboplatin target area under the curve of 5 and 175 mg/m² paclitaxel (TC) therapy Country: Japan (single centre) | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg + palonosetron 0.75 mg + dexamethasone 6.6 mg Days 2 to 3: aprepitant 80 mg + dexamethasone 8 mg Control: arm B Day 1: palonosetron 0.75 mg + dexamethasone 13.2 mg Days 2 to 3: dexamethasone 8 mg | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "... open‐label ..." |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although participants were not blinded, we assume that this does not affect objective outcomes |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all 78 patients were included in the efficacy analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: side effects reported as percentage of all patients |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled trial with 2 arms
Recruitment period: June 2010 to June 2012
Masking: double‐blind (attending nurse and patient) Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years : 58.11 ± 8.84 (experimental group), 56.52 ± 8.25 (control group) Gender: 64 male + 32 female Tumour/cancer type: lymphoma (38) and myeloma (58) Chemotherapy regimen: BEAM, BEAC for lymphoma patients and high‐dose melphalan, BBM for myeloma patients Country: Sweden (single centre) | |
| Interventions | Experimental: arm A 6 mg of betamethasone (T Betapred 0.5 mg, 12 tablets daily) + T Navobane (tropisetron) (5 mg) + aprepitant (Emend), started 1 h before first HDCT dose for SCT and administered daily until 7 days after the end of chemotherapy Control: arm B 6 mg of betamethasone (T Betapred 0.5 mg, 12 tablets daily) + T Navobane (tropisetron) (5 mg) + placebo, started 1 h before first HDCT dose for SCT and administered daily until 7 days after the end of chemotherapy | |
| Outcomes | Primary efficacy endpoint
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation was not reported |
| Allocation concealment (selection bias) | Low risk | Quote: "a random assignment to the experimental (EXP) or control (CTR) group was performed by research nurses not participating in any other way in the study" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "study drug or placebo was unknown to the attending nurse and the patient in the study" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "study drug or placebo was unknown to the attending nurse and the patient in the study" |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the analysis is made on an intention‐to‐treat basis" |
| Selective reporting (reporting bias) | Unclear risk | Comment: adverse events mentioned as not differing, no proportions provided. All other outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, parallel, comparative, phase 2 trial with 3 arms
Study start date: August 2005
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years : 63.3 ± 9.4 (aprepitant 40 ⁄ 25 + standard therapy), 60.5 ± 9.7 (aprepitant 125 ⁄ 80 mg + standard therapy), 62.2 ± 9.8 (standard therapy) Gender: male + female Tumour/cancer type: solid malignant tumour Chemotherapy regimen: cisplatin at a dose ≥ 70 mg/m² Country: Japan (9 facilities) | |
| Interventions | Experimental: arm A: aprepitant 40 ⁄ 25 + standard therapy Day 1: aprepitant 40 mg + dexamethasone 8 mg + granisetron 40 µg/kg Days 2 to 3: aprepitant 25 mg + dexamethasone 6 mg Days 4 to 5: aprepitant 25 mg Experimental: arm B: aprepitant 125 ⁄ 80 mg + standard therapy Day 1: aprepitant 125 mg + dexamethasone 6 mg + granisetron 40 µg/kg Days 2 to 3: aprepitant 80 mg + dexamethasone 4 mg Days 4 to 5: aprepitant 80 mg Standard: arm C Day 1: placebo + dexamethasone 12 mg + granisetron 40 µg/kg Days 2 to 3: placebo + dexamethasone 8 mg Days 4 to 5: placebo | |
| Outcomes | Primary endpoint
Secondary endpoints
Both primary and secondary endpoints were assessed in the overall phase (Days 1 to 5), the acute phase (Day 1), and the delayed phase (Days 2 to 5) | |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "treatment assignment (dynamic allocation) was performed using a minimization method for balancing four factors (sex, presence or absence of at least one emetogenic antitumour agent used in combination with cisplatin, presence or absence of previous treatment with cisplatin, and institution) between the treatment and control groups" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. study mortality) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "of these, 449 patients were included in the safety analysis set, 439 subjects were included in the FAS" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "all 453 enrolled subjects were included in the safety analysis" Quote: "serious adverse events led to the death of one patient in the standard therapy group and one in the 125 ⁄80 mg group. The former died of febrile neutropenia, acute respiratory distress syndrome (ARDS) and septic shock, and the latter died of cardiac failure" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, phase 2 study with 2 arms
Study period: January 2011 to September 2012
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age, years: 53 (36 to 67) in aprepitant group, 59 (33 to 69) in placebo group Gender: female Tumour/cancer type: ovarian cancer (early/advanced), endometrial cancer, other, ascites or peritoneal dissemination Chemotherapy regimen: carboplatin + intravenous cytotoxic anti‐tumour drugs such as paclitaxel and pemetrexed; carboplatin + paclitaxel; carboplatin + liposomal doxorubicin; irinotecan + fluorouracil, bevacizumab, or cetuximab Country: Japan (multi‐centre) | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg p.o. + granisetron 1 mg i.v. + dexamethasone 12 mg i.v. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 4 mg i.v. Experimental: arm B: placebo Day 1: placebo 0 mg p.o. + granisetron 1 mg i.v. + dexamethasone 20 mg i.v. Days 2 to 3: placebo 0 mg p.o. + dexamethasone 8 mg i.v. | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... randomly assigned to the aprepitant group or placebo group according to a computer‐generated, blinded allocation schedule" |
| Allocation concealment (selection bias) | Unclear risk | Comment: precise allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "of these, 91 patients were included in the full analysis set" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "safety was evaluated in all the 92 subjects who were assigned to treatment, including the patient who discontinued chemotherapy due to a hypersensitivity reaction" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, prospective, single‐centre, comparative, phase 3 trial
Study period: October 2010 to January 2013
Masking: open‐label Baseline patient characteristics: reported Median follow‐up: n.r. ITT analysis: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age, years: 68 Gender: 22 men and 13 women Tumour/cancer type: NSCLC, SCLC, BC Chemotherapy regimen: HEC or MEC; not further specified Country: Japan (single centre) | |
| Interventions |
| |
| Outcomes | Primary outcome measure
Secondary outcome measures
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stratified and 1:1 randomised; method not further described |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) | High risk | Open‐label |
| Blinding of participants and personnel (performance bias) | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Outcome assessors (participants) not blinded to intervention |
| Incomplete outcome data (attrition bias) | Low risk | Data available for all randomised participants |
| Selective reporting (reporting bias) | Unclear risk | Wrong UMIN ID provided in paper; UMIN ID 000005268 is a clinical trial to evaluate effect of spectacle lens that reduces myopia progression |
| Other bias | Low risk | No other sources of bias detected |
| Study characteristics | ||
| Methods | Randomised, parallel‐group, placebo‐controlled, phase 3 trial with 2 arms
Recruitment period: October 2002 to December 2003
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: 53.1 (aprepitant regimen), 52.1 (control regimen) Gender: male (2) + female (864) Tumour/cancer type: breast cancer Chemotherapy regimen: anthracycline plus cyclophosphamide‐based chemotherapy regimen (the following agents were administered alone or in combination: i.v. cyclophosphamide 750 to 1500 mg/m² (± 5%); i.v. cyclophosphamide 500 to 1500 mg/m² (± 5%) and i.v. doxorubicin ≤ 60 mg/m² (± 5%); i.v. cyclophosphamide 500 to 1500 mg/m² (± 5%) and i.v. epirubicin ≤ 100 mg/m² (± 5%). Other chemotherapeutic agents, Hesketh level 2 or lower, could be added to the above chemotherapeutic regimens) Country: 95 centres in the United States, Germany, Austria, Canada, Hong Kong, Hungary, Spain, United Kingdom, Italy, Australia, and Greece | |
| Interventions | Experimental: arm A: aprepitant 125/80 mg Day 1: p.o. aprepitant 125 mg + p.o. ondansetron 8 mg (30 to 60 min before chemotherapy and again 8 h after chemotherapy) + p.o. dexamethasone 12 mg Days 2 to 3: p.o. aprepitant 80 mg Control: arm B: placebo Day 1: placebo + p.o. ondansetron 8 mg (30 to 60 min before chemotherapy and again 8 h after chemotherapy) + p.o. dexamethasone 20 mg Days 2 to 3: placebo + p.o. ondansetron 8 mg | |
| Outcomes | Primary endpoint
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... using a computer‐generated allocation schedule with a block size of four" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "... modified intent‐to‐treat analysis was conducted, including all patients who received chemotherapy, took the study drug, and had at least one post‐treatment assessment" |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included in the safety analysis |
| Selective reporting (reporting bias) | Low risk | Comment: outcome measure was reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised study with 2 arms
Enrolment period: n.r.
Masking: double‐blind Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria: n.r. Mean/median age, years: n.r. Gender: female Tumour/cancer type: breast cancer Chemotherapy regimen: MEC Country: n.r. | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg p.o. + ondansetron 8 mg p.o. b.i.d. + dexamethasone 12 mg p.o. Days 2 to 3: aprepitant 80 mg p.o. Control: arm B: ondansetron Day 1: ondansetron 8 mg p.o. b.i.d. + dexamethasone 20 mg p.o. Days 2 to 3: ondansetron 8 mg p.o. b.i.d. | |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: breast cancer patients were also included in the modified intent‐to‐treat population |
| Selective reporting (reporting bias) | Unclear risk | Comment: conference abstract only, not evaluable |
| Other bias | Unclear risk | Comment: conference abstract, not evaluable |
| Study characteristics | ||
| Methods | Randomised, active‐comparator, parallel‐group, phase 3 superiority trial (PN031) with 2 arms
Enrolment period: 30 October 2012 to 03 November 2014
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 60.0 ± 11.8 in fosaprepitant group, 59.1 ± 12.3 in control group Gender: male (409) + female (591) Tumour/cancer type: malignant disease (lung, breast, colorectal, gynaecological, gastrointestinal, head and neck, other) Chemotherapy regimen: MEC agents except for the combination of anthracycline and cyclophosphamide Country: 125 sites across 30 countries | |
| Interventions | Experimental: arm A: fosaprepitant Day 1: fosaprepitant 150 mg intravenous (i.v.) infusion, ~ 30 minutes before chemotherapy + dexamethasone 12 mg orally (p.o.) ~ 30 minutes before chemotherapy + ondansetron 16 mg total dose: 8 mg p.o. ~ 30 to 60 minutes before chemotherapy, followed by 8 mg p.o. 8 hours after first dose + dexamethasone placebo, p.o. ~ 30 minutes before chemotherapy Days 2 to 3: ondansetron placebo, p.o. every 12 hours Control: arm B: fosaprepitant Day 1: fosaprepitant placebo, 150 mL i.v. infusion, ~ 30 minutes before chemotherapy + dexamethasone 20 mg, p.o. ~ 30 minutes before chemotherapy + ondansetron 16 mg total dose: 8 mg p.o. ~ 30 to 60 minutes before chemotherapy; followed by 8 mg p.o. 8 hours after first dose Days 2 to 3: ondansetron 8 mg p.o. every 12 hours | |
| Outcomes | Primary endpoint(s)
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "subjects were randomized (1:1) to the single‐dose fosaprepitant or control regimen via an interactive voice response system/interactive web response system, and stratified based on sex" |
| Allocation concealment (selection bias) | Low risk | Quote: "study medications were supplied in a blinded manner as fosaprepitant/placebo i.v. bags, ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. neutropenia) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the ITT and ASaT populations comprised 1000 and 1001 subjects, respectively (Figure 1)" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the ITT and ASaT populations comprised 1000 and 1001 subjects, respectively (Figure 1)" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, prospective, pilot study with 2 arms
Study period: January 2011 to July 2011
Masking: open‐label Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 52.9 ± 12.7 in ondansetron group, 50.9 ± 9.2 in palonosetron group Gender: female Tumour/cancer type: breast cancer (N = 39), lymphoma (N = 1) Chemotherapy regimen: AC (doxorubicin/cyclophosphamide), AC plus bevacizumab, ABVD Country: USA (single centre) | |
| Interventions | Experimental: arm A: palonosetron Day 1: aprepitant 125 mg p.o. + palonosetron 0.25 mg i.v. + dexamethasone 12 mg p.o. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 8 mg p.o. Day 4: dexamethasone 8 mg p.o. Experimental: arm B: ondansetron Day 1: aprepitant 125 mg p.o. + ondansetron 24 mg p.o. Days 2 to 3: aprepitant 80 mg p.o. + dexamethasone 8 mg p.o. Day 4: dexamethasone 8 mg p.o. | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... randomized to one of two treatments according to a permuted block‐design with block sizes of 2, 4, or 6" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of participants and personnel (performance bias) | High risk | Comment: open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Comment: patients and personnel were not blinded towards the intervention and therefore might influence subjective outcomes analysis |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "an intention to‐treat approach was used to evaluate patients" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, cross‐over trial with 2 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria: n.r. Median age (range), years: 46 (29 to 71) in granisetron, 46 (30 to 73) in ondansetron Gender: male (4) + female (36) Tumour/cancer type: solid tumour (breast, ovarian, lung, other) Chemotherapy regimen: cisplatin ≥ 50 mg m–2 or cyclophosphamide ≥ 500 mg m–2 Country: n.r. | |
| Interventions | Cross‐over study Experimental: arm A: granisetron granisetron 3 mg i.v. + dexamethasone 10 mg i.v. Experimental: arm B: ondansetron patients with previous treatment failure continued treatment with ondansetron 8 mg i.v. + dexamethasone 10 mg i.v. | |
| Outcomes | Primary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but the method of randomisation was not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment was not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 40 patients were included in the efficacy analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised trial with 2 arms
Recruitment period: March 2014 to March 2017
Masking: open‐label Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean ± SD, years: 39.8 ± 8.6 in aprepitant group, 41.5 ± 9.4 in control group Gender: male (54) + female (51) Tumour/cancer type: bone or soft tissue sarcoma Chemotherapy regimen: 30 mg/m² on Days 1 and 2 for doxorubicin and 3 g/m² on Days 1 to 3 for ifosfamide Country: China | |
| Interventions | Experimental: arm A: aprepitant Day 1: 125 mg p.o. aprepitant + 0.25 mg i.v. palonosetron + 5 mg i.v. dexamethasone Days 2 to 3: 80 mg p.o. aprepitant + 5 mg i.v. dexamethasone Control: arm B Day 1: 0.25 mg i.v. palonosetron + 10 mg i.v. dexamethasone Days 2 to 3: 10 mg i.v. dexamethasone | |
| Outcomes | Primary endpoints
Secondary endpoint
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "... using a computer‐generated allocation schedule with a block size of four" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "we have no placebo, and the administration of dexamethasone is different in the two groups, so it is difficult for us to make this trial double blinded" |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: "we have no placebo, and the administration of dexamethasone is different in the two groups, so it is difficult for us to make this trial double blinded" |
| Blinding of outcome assessment (detection bias) | High risk | Comment: study was not blinded; knowledge of treatment may affect outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: although the study was not blinded, we assume that knowledge of treatment had no effect on outcome assessment for objective outcomes |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "105 patients (51 patients in the aprepitant group and 54 patients in the control group) were included in the efficacy analyses"; "three patients were excluded from both the efficacy and safety analyses because they did not receive at least 1 day’s dose of study drug"; modified ITT analysis |
| Incomplete outcome data (attrition bias) | Low risk | Comment: "three patients were excluded from both the efficacy and safety analyses because they did not receive at least 1 day’s dose of study drug"; modified ITT analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled, parallel‐group trial with 2 arms
Study period: April 2011 to December 2013
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (range), years: 59 (26 to 77) in aprepitant group, 59 (24 to 79) in placebo group Gender: female Tumour/cancer type: gynaecological cancer (ovarian cancer, endometrial cancer, cervical cancer, peritoneal cancer, tubal cancer) Chemotherapy regimen: TC combination chemotherapy (paclitaxel and carboplatin) Country: Japan (9 institutes, multi‐centre) | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg p.o. + granisetron/ondansetron ¼ mg + dexamethasone 20 mg i.v. Days 2 to 3: aprepitant 80 mg p.o. Experimental: arm B: placebo Day 1: placebo 0 mg p.o. + granisetron/ondansetron ¼ mg + dexamethasone 20 mg i.v. | |
| Outcomes | Primary endpoint
Secondary endpoint(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomised trial but method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "efficacy analyses were conducted with the full analysis set, which was defined as all randomized patients who received at least one dose of the study drugs" |
| Incomplete outcome data (attrition bias) | High risk | Quote: "safety was evaluated in all patients taking the study drugs", but "adverse events obviously caused by paclitaxel and/or carboplatin (e.g. alopecia, neutropenia) were excluded, and only toxicities not related to the study drugs were recorded" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, active‐control, parallel‐group, phase 3 trial with 2 arms
Recruitment period: November 2014 to July 2015
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: yes | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 55 (20 to 79) in fosaprepitant group, 53 (18 to 74) in aprepitant group Gender: male (326) + female (319) Tumour/cancer type: n.r. Chemotherapy regimen: HEC (according to NCCN Clinical Practice Guidelines in Oncology: Antiemesis version 1.2014) Country: China (21 centres) | |
| Interventions | Experimental: arm A: fosaprepitant Day 1: fosaprepitant 150 mg i.v. + granisetron 3 mg i.v. + dexamethasone 6 mg p.o. or i.v. Day 2: dexamethasone 3.75 mg p.o. Day 3: dexamethasone 3.75 mg p.o. every 12 h Day 4: dexamethasone 3.75 mg p.o. every 12 h Experimental: arm B: aprepitant Day 1: aprepitant 125 mg p.o. + granisetron 3 mg i.v. + dexamethasone 6 mg p.o. or i.v. Day 2: aprepitant 80 mg p.o. + dexamethasone 3.75 mg p.o. Day 3: aprepitant 80 mg p.o. + dexamethasone 3.75 mg p.o. Day 4: dexamethasone 3.75 mg p.o. | |
| Outcomes | Primary efficacy endpoint
Secondary efficacy endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomized in a 1:1 ratio using a central randomization system ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. hiccups) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "assessments of efficacy, tolerability and safety variables were performed for 5 days after the start of chemotherapy (0–120 hr), including the acute and delayed phases" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "a total of 645 patients were included in the safety study" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, placebo‐controlled study with 2 arms
Enrolment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Median age (range), years: 46.5 (32 to 66) in aprepitant group, 48.5 (26 to 68) in standard group Gender: female (124) Tumour/cancer type: invasive ductal carcinoma, invasive lobular carcinoma, other Chemotherapy regimen: doxorubicin 60 mg/m² + cyclophosphamide 600 mg/m² Country: Hong Kong, China (single centre) | |
| Interventions | Experimental: arm A: aprepitant Day 1: aprepitant 125 mg, ondansetron 8 mg, dexamethasone 12 mg, before chemotherapy and ondansetron 8 mg 8 h later Days 2 to 3: aprepitant 80 QD Standard: arm B: ondansetron Day 1: ondansetron 8 mg and dexamethasone 20 mg before chemotherapy and ondansetron 8 mg 8 h later Days 2 to 3: ondansetron 8 mg b.i.d. | |
| Outcomes | Primary objective
Secondary objective(s)
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were assigned to 1 of 2 anti‐emetic regimens according to an in‐house blinding and allocation schedule of random numbers" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. neutropenia, febrile neutropenia) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "thus, data from 124 evaluable patients (62 in each arm) was available for analysis. The modified intention‐to‐treat (mITT) approach was used for all efficacy analyses" |
| Incomplete outcome data (attrition bias) | Low risk | Comment: all patients were included for safety analysis |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised, single initial cycle, parallel‐group, international, phase 3 trial with 2 arms
Recruitment period: n.r.
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age ± SD, years: 54.6 ± 9.63 in NEPA group, 54.5 ± 10.24 in APR/GRAN group Gender: male (589) + female (240) Tumour/cancer type: solid tumour (lung, head and neck, and other) Chemotherapy regimen: cisplatin‐based chemotherapy at a dose ≥ 50 mg/m² alone or in combination with other chemotherapy agents Country: 30 sites in China, 5 sites in Taiwan, 3 sites in Thailand, 8 sites in Korea (46 centres) | |
| Interventions | Experimental: arm A: NEPA Day 1: NEPA (300 mg netupitant and 0.5 mg palonosetron) + dexamethasone 12 mg Days 2 to 4: dexamethasone 8 mg daily Experimental: arm B: APR/GRAN Day 1: aprepitant 125 mg + 3 mg i.v. granisetron + dexamethasone 12 mg Days 2 to 3: aprepitant 80 mg daily + dexamethasone 8 mg daily Day 4: dexamethasone 8 mg daily | |
| Outcomes | Primary efficacy endpoint
Secondary efficacy endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were stratified by gender and randomly assigned (1:1) to receive either NEPA or APR/GRAN treatment ..." |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "... double‐blind ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: both patients and personnel were blinded towards the intervention and thus had no influence on outcome assessment (e.g. study mortality) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the full analysis set (FAS) population (efficacy analyses) was defined as all patients who were randomized and received protocol‐required cisplatin and study treatment ...": of 334 randomised, 328 were included in FAS |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "the safety analysis population consisted of all patients who received study treatment" |
| Selective reporting (reporting bias) | Low risk | Comment: all outcome measures were reported in the results section |
| Other bias | Low risk | Comment: no information to suggest other sources of bias |
| Study characteristics | ||
| Methods | Randomised study with 2 arms
Recruitment period: n.r.
Masking: n.r. Baseline patient characteristics: n.r. Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria: n.r. Mean/median age, years: n.r. Gender: n.r. Tumour/cancer type: locally advanced or metastatic lung cancer Chemotherapy regimen: cisplatin‐based combination chemotherapy Country: China | |
| Interventions | Experimental: arm A: aprepitant aprepitant + palonosetron + dexamethasone Experimental: arm B: placebo placebo + palonosetron + dexamethasone | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "random sequence generation not reported" |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: blinding not reported; therefore we do not know if this was a risk of bias |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: unclear whether all randomised patients were included for efficacy analysis |
| Selective reporting (reporting bias) | Unclear risk | Comment: conference abstract, not evaluable |
| Other bias | Unclear risk | Comment: conference abstract, not evaluable |
| Study characteristics | ||
| Methods | Randomised study with 2 arms
Recruitment period: October 2014 to November 2015
Masking: double‐blind Baseline patient characteristics: reported Follow‐up: n.r. | |
| Participants | Inclusion criteria
Exclusion criteria
Mean age (SD), years: 55.88 (10.37) in fosaprepitant group, 55.88 (10.19) in aprepitant group Gender: 40.50% female (59.50% male) in fosaprepitant group, 40.87% female (59.13% male) in aprepitant group Tumour/cancer type: solid malignant tumour Chemotherapy regimen: single day of cisplatin (dosage ≥ 50 mg/m² and infusion time ≤ 3 hours) Country: China | |
| Interventions | Experimental: arm A: fosaprepitant fosaprepitant (Day 1, 150 mg i.v.) + palonosetron (0.25 mg i.v.) + dexamethasone (6 mg on Day 1 followed by 3.75 mg on Day 2 and 3.75 mg p.o. every 12 hours on Days 3 to 4) + aprepitant simulating agents (placebo, scheduled as received in aprepitant group) Experimental: arm B: aprepitant aprepitant (Day 1, 125 mg, p.o.; Days 2 to 3, 80 mg p.o.) + palonosetron + dexamethasone (6 mg on Day 1 followed 3.75 mg on Days 2 to 4, with dexamethasone simulation agent 3.75 mg p.o. on Days 3 to 4), fosaprepitant simulating agent (placebo, scheduled as received in fosaprepitant group) | |
| Outcomes | Primary endpoint
Secondary endpoints
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "computer‐generated, blinded allocation schedule. Patients firstly stratified by gender and then based on whether the first administration of chemotherapy or not and the emetogenic potential of anticancer agents (excluding cisplatin) were randomized to different treatment groups" |
| Allocation concealment (selection bias) | Low risk | Computer‐generated, blinded allocation schedule |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "both patients and researchers were blinded to the therapeutic grouping" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "both patients and researchers were blinded to the therapeutic grouping" |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "both patients and researchers were blinded to the therapeutic grouping" |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "both patients and researchers were blinded to the therapeutic grouping" |
| Incomplete outcome data (attrition bias) | Low risk | Data available for nearly all participants (1/322 participants in fosa group and 3/326 participants from apre group excluded because no study drug received) |
| Incomplete outcome data (attrition bias) | Low risk | All participants who received at least 1 dose included in safety analysis |
| Selective reporting (reporting bias) | Low risk | No reasons for any concern detected |
| Other bias | Low risk | No other sources of bias detected |
5‐HT₃: serotonin.
5‐HT₃ RA: 5‐HT₃ receptor antagonist.
ABVD: doxorubicin (Adriamycin), bleomycin, vinblastine (Velbe), dacarbazine (DTIC).
AC: doxorubicin, cyclophosphamide.
ALT: alanine aminotransferase.
AML: acute myeloid leukemia.
APF: granisetron.
AST: aspartate aminotransferase.
AUC: area under the curve.
BBM: berbamine.
BCNU: carmustine.
BEAM: carmustine, etoposide, cytarabine, and melphalan.
BEAC: carmustine, etoposide, cytarabine, cyclophosphamide.
β‐hCG: β‐subunit of human chorionic gonadotropin.
b.i.d.: twice daily.
Bu: busulfan.
CDDP: cisplatin.
CEF: cyclophosphamide, epirubicin, 5‐fluorouracil.
CINV: chemotherapy‐induced nausea and vomiting.
CNF: cyclophosphamide, novantrone, and 5‐fluorouracil.
CNS: central nervous system.
CMF: cyclophosphamide, mitoxantrone, and 5‐fluorouracil.
CML: chronic myeloid leukemia.
CR: complete response.
CrCl: creatinine clearance.
CTCAE: Common Terminology Criteria for Adverse Events.
Cy: cyclophosphamide.
CyTBI: cyclophosphamide total body irradiation.
EC: epirubicin, cyclophosphamide.
ECG: electrocardiogram.
ECOG: Eastern Cooperative Oncology Group.
ESHAP: multi‐day cisplatin along with etoposide, methylprednisolone, high‐dose cytarabine.
FAC: 5‐fluorouracil (5‐FU) + AC.
FEC: fluorouracil, epirubicin, cyclophosphamide.
FLIE: Functional Living Index‐Emesis.
FOLFOX: 5‐fluorouracil + leucovorin + oxaliplatin.
g/m²: gram per square meter.
GTDS: granisetron transdermal delivery system.
h: hour.
hCG: human chorionic gonadotropin.
HDCT: high‐dose chemotherapy.
HEC: highly emetogenic chemotherapy.
HSCT: haematopoietic stem cell transplantation.
IFO: ifosfamide.
ITT: intention‐to‐treat.
IU/L: international units per litre.
i.v.: intravenous.
L: litre.
MEC: moderately emetogenic chemotherapy.
μg: microgram.
mg: milligram.
mg/dL: milligrams per decilitre.
mg/m²: milligram per square meter.
min: minutes.
mITT: modified intention‐to‐treat.
MMT: paclitaxel.
msec: millisecond.
MTZ: mitoxantrone.
NCCN: National Comprehensive Cancer Network.
NSCLC: non‐small cell lung carcinoma.
NK₁: neurokinin‐1.
n.r.: not reported.
PBSC: peripheral blood stem cell.
p.o.: per os (orally).
PRN: medicine as required.
q8h: every 8 hours.
QAM: in the morning.
QD: once a day.
QoL: quality of life.
QPM: in the evening.
SC: subcutaneously.
SCT: stem cell transplantation.
SOX: S‐1, oxaliplatin.
SPAMP V: cyclophosphamide, thiotepa, carboplatin.
TAC: docetaxel, doxorubicin, cyclophosphamide.
TANC: paclitaxel, mitoxantrone, carboplatin.
TBI: total body irradiation.
TC: paclitaxel, carboplatin.
ULN: upper limit of normal.
VAS: visual analogue scale.
VP: etoposide.
vs: versus.
WBC: white blood cell count.
XELOX: capecitabine, oxaliplatin.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| non‐randomised study | |
| antiblastic therapy used as treatment regimen | |
| non‐randomised study | |
| application of dexamethasone has not been reported | |
| dexamethasone has been used in only 1 arm | |
| cost‐benefit analysis | |
| no use of 5‐hydroxytryptamine‐3 (5‐HT₃) receptor antagonists in first 2 arms, along with neurokinin‐1 (NK₁) receptor antagonist | |
| application of dexamethasone has not been reported | |
| a letter | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| objectives of this article are (1) to assess the pharmacokinetics of aprepitant in cancer patients undergoing HSCT, and (2) to examine the potential drug–drug interaction between aprepitant and cyclophosphamide | |
| application of dexamethasone has not been reported | |
| chemotherapy regimen is not clearly reported | |
| non‐randomised study | |
| use of 5‐hydroxytryptamine‐3 (5‐HT₃) receptor antagonist has been reported in only 1 arm | |
| non‐randomised study | |
| no information given regarding randomisation, no comparator against ondansetron | |
| docetaxel belongs to low emetogenic chemotherapy regimen | |
| dexamethasone was permitted as rescue medication | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| dexamethasone has been used in only 1 arm | |
| correspondence | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| no use of dexamethasone reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| comparison of identical dose of Zudan and Zofran; both are ondansetron | |
| no information available regarding randomisation | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| retrospective subset analysis | |
| cost‐effectiveness study | |
| application of dexamethasone has not been reported | |
| use of metoclopramide in 1 arm | |
| on Days 2 to 4 after chemotherapy, all patients received oral metoclopramide + intramuscular dexamethasone as antiemetic prophylaxis for delayed emesis | |
| use of metoclopramide | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| concurrent chemoradiotherapy | |
| chemotherapy regimen is not clearly reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| detailed chemotherapy regimens within the HEC group have not been reported | |
| addition of lorazepam in all treatment arms | |
| use of 5‐hydroxytryptamine‐3 (5‐HT₃) receptor antagonist has not been reported | |
| chemotherapy regimen is not clearly mentioned | |
| application of dexamethasone has not been reported | |
| comparison of ondansetron, prochlorperazine, and dexamethasone in 3 individual arms | |
| dolasetron (dol) vs ondansetron (ond) with and without dexamethasone (dex) to evaluate additive effects of i.v. DEX with each drug | |
| no information available regarding randomisation | |
| pharmacokinetic study | |
| addition of lorazepam in all treatment arms | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| patients were randomly assigned to receive granisetron alone (arm 1) or granisetron, dexamethasone, and prochlorperazine (arm 2) | |
| treatment arms include granisetron + dexamethasone vs granisetron alone | |
| efficacy determination of dronabinol alone and in combination with ondansetron vs ondansetron alone | |
| application of dexamethasone has not been reported in Cycle 1 | |
| study type: pooled analysis of different trials | |
| no information given regarding randomisation | |
| cost‐effectiveness study | |
| application of dexamethasone has not been reported | |
| review | |
| comparison of different preparations of the same drug | |
| 5‐HT₃ not defined and use of both fosaprepitant and aprepitant in 1 group reported | |
| 5‐HT₃ not defined and use of both fosaprepitant and aprepitant in 1 group reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| dexamethasone has been reported in only 1 group | |
| ondansetron or saline injection has been given to patients | |
| pharmacokinetic and dose‐finding study | |
| application of dexamethasone has not been reported | |
| non‐randomised study | |
| application of dexamethasone has not been reported | |
| no information given regarding randomisation | |
| application of dexamethasone has not been reported | |
| dexamethasone or methylprednisolone was permitted as a prophylactic component of pre‐therapy | |
| dexamethasone has been reported in only 1 group | |
| no information given regarding randomisation | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| dose‐finding study | |
| no individual data have been provided for HEC and MEC regimens | |
| application of dexamethasone has not been reported | |
| 5 weeks of fractionated radiotherapy and concomitant weekly cisplatin have been used | |
| non‐randomised study; historical control group | |
| chemotherapy regimen is not clearly reported | |
| application of dexamethasone and chemotherapy regimen have not been reported | |
| application of dexamethasone has not been reported | |
| a letter | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| pharmacogenomics study | |
| no information given regarding randomisation | |
| non‐randomised study and no comparator has been used | |
| no individual data have been provided for HEC and MEC regimens | |
| application of dexamethasone has not been reported | |
| dose‐finding study | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| concurrent radiochemotherapy | |
| application of dexamethasone and chemotherapy regimen have not been reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| no use of dexamethasone has been reported | |
| pharmacogenomics study | |
| application of dexamethasone has not been reported | |
| retrospective study | |
| pharmacological study | |
| pharmacological study | |
| pharmacological study | |
| pharmacological study | |
| no use of 5‐hydroxytryptamine‐3 (5‐HT₃) receptor antagonist has been reported in 2 of 3 arms | |
| metoclopramide used as part of the antiemetic regimen | |
| pharmacological study | |
| application of dexamethasone has not been reported | |
| use of dexamethasone has not been reported | |
| no clear distinction in presenting results for granisetron and ondansetron | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| non‐randomised study | |
| application of dexamethasone and chemotherapy regimen have not been reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| phase 1, pharmacokinetic study | |
| use of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| application of dexamethasone has not been reported | |
| phase 1, pharmacokinetic study |
Characteristics of studies awaiting classification [ordered by study ID]
| Methods | Randomised, cross‐over study with 2 arms
Study period: April 2017 to March 2020
Masking: open‐label Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: histologically or cytologically confirmed malignant tumour Chemotherapy regimen: cisplatin, doxorubicin ≥ 60 mg/m², epirubicin > 90 mg/m², ifosfamide ≥ 2 g/m² Country: China |
| Interventions | Cross‐over study Experimental: arm A Cycle 1: palonosetron 0.25 mg on Day 1, Day 3 + dexamethasone 10 mg on Day 1, 5 mg on Days 2 to 5 Cycle 2: tropisetron 5 mg on Days 1 to 3/5 + dexamethasone 10 mg on Day 1, 5 mg on Days 2 to 5 Experimental: arm B Cycle 1: tropisetron 5 mg on Days 1 to 3/5 + dexamethasone 10 mg on Day 1, 5 mg on Days 2 to 5 Cycle 2: palonosetron 0.25 mg on Day 1, Day 3 + dexamethasone 10 mg on Day 1, 5 mg on Days 2 to 5 |
| Outcomes | Primary outcome
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, interventional, parallel study with 2 arms
Recruitment period: November 2017 to April 2018
Masking: open‐label Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: malignancy Chemotherapy regimen: moderate or low emetogenic potential chemotherapeutic agents except for carboplatin Country: India |
| Interventions | Experimental: arm A: granisetron Day 1: i.v. injections of granisetron 1 mg + i.v. dexamethasone 8 mg given 30 min before administration of moderate emetogenic and mild emetogenic chemotherapy agents Days 2 to 5: p.o. granisetron 1 mg once daily before food Control: arm B: ondansetron Day 1: i.v. injections of ondansetron 8 mg + i.v. dexamethasone 8 mg given 30 min before administration of moderate emetogenic chemotherapy agents and low chemotherapeutic agents Days 2 to 5: p.o. ondansetron 8 mg once daily before food |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, phase 2, placebo‐ and active‐controlled study with 3 arms
Masking: double‐blind Baseline patient characteristics: reported |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: solid tumours Chemotherapy regimen: cisplatin‐based chemotherapy Country: 47 centres in 18 countries (multi‐centre) |
| Interventions | Experimental: arm A casopitant 50 mg + ondansetron + dexamethasone Experimental: arm B casopitant 100 mg + ondansetron + dexamethasone Experimental: arm C casopitant 150 mg + ondansetron + dexamethasone |
| Outcomes | Primary outcome
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, placebo‐controlled, dose‐ranging study Recruitment period: February 2005 to n.r.
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: solid malignancy Chemotherapy regimen: MEC Country: Czech Republic, Germany, Hungary, Ireland, Spain, United Kingdom |
| Interventions | oral neurokinin₁ receptor antagonist, GW679769, administered as 50 mg, 100 mg, and 150 mg oral tablets in combination with ondansetron hydrochloride and dexamethasone |
| Outcomes | Primary endpoints
Secondary objectives
|
| Notes |
|
| Methods | Randomised, placebo‐controlled study with 2 arms
Recruitment period: June 2005 to n.r.
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: multiple myeloma Chemotherapy regimen: high‐dose melphalan Country: Germany (single centre) |
| Interventions | Experimental: arm A aprepitant + granisetron + dexamethasone Experimental: arm B placebo + granisetron + dexamethasone |
| Outcomes | Primary outcome
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, parallel‐group, cross‐over study with 3 arms
Recruitment period: September 2009 to n.r.
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: histologically or cytologically confirmed malignant disease Chemotherapy regimen
Country: n.r. |
| Interventions | Cross‐over study Experimental: arm A palonosetron 0.25 mg + dexamethasone 8 mg Experimental: arm B palonosetron 0.50 mg + dexamethasone 8 mg Experimental: arm C palonosetron 0.75 mg + dexamethasone 8 mg |
| Outcomes | Primary endpoint
Secondary endpoints
Additional secondary outcomes
|
| Notes |
|
| Methods | Randomised, phase 3, active‐controlled study Masking: double‐blind Baseline patient characteristics: reported |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: n.r. Chemotherapy regimen: MEC Country: 196 centres in 32 countries (multi‐centre) |
| Interventions | casopitant + ondansetron + dexamethasone |
| Outcomes | Primary endpoints
Secondary endpoints for Cycle 1
|
| Notes |
|
| Methods | Randomised, parallel‐group study Recruitment period: November 2006 to n.r.
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: solid malignancy Chemotherapy regimen
Country: France, Germamy |
| Interventions | aprepitant, fosaprepitant, ondansetron, dexamethasone |
| Outcomes | Primary endpoints
Secondary objectives
|
| Notes |
|
| Methods | Randomised, active‐controlled, parallel‐group study Recruitment period: March 2008 to n.r.
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: solid malignancy Chemotherapy regimen: cisplatin Country: Denmark, Germany, Hungary, Italy, Lithuania, Netherlands, Poland, Portugal, Spain, Sweden |
| Interventions | single dose of intravenous MK‐0517 |
| Outcomes | Primary endpoints
Secondary objectives
|
| Notes |
|
| Methods | Randomised, parallel‐group study Recruitment period: February 2008 to n.r.
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: colorectal cancer Chemotherapy regimen: oxaliplatin at a dose between 85 mg/m² and 130 mg/m² Country: Bulgaria, Czech Republic, Germany, Hungary. Italy, Slovakia |
| Interventions | casopitant + ondansetron + dexamethasone |
| Outcomes | Primary endpoint
Secondary objectives
|
| Notes |
|
| Methods | Randomised, parallel‐group study Recruitment period: April 2008 to n.r.
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: malignancy Chemotherapy regimen: cisplatin Country: Italy |
| Interventions | aprepitant |
| Outcomes | Primary endpoint
Secondary objectives
|
| Notes |
|
| Methods | Randomised, phase 3, active‐controlled, parallel‐group study with 2 arms
Recruitment period: April 2011 to November 2012 Sample size: 726 Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: solid tumours Chemotherapy regimen: MEC Countries: Argentina, Brazil, Bulgaria, Croatia, Germany, Hungary, India, Italy, Mexico, Poland, Romania, Russian Federation, Ukraine, United States |
| Interventions | Experimental: arm A netupitant + palonosetron + dexamethasone Experimental: arm B palonosetron + dexamethasone |
| Outcomes | Primary endpoint
Secondary endpoints
|
| Notes |
|
| Methods | Randomised, phase 3, placebo‐controlled study Recruitment period: February 2010 to n.r.
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: multiple myeloma, Hodgkin lymphoma, non‐Hodgkin lymphoma Chemotherapy regimen: cyclophosphamide i.v. chemotherapy (3 g/m²) Country: Italy |
| Interventions | aprepitant |
| Outcomes | Primary endpoint
Secondary objective
|
| Notes |
|
| Methods | Randomised, phase 3, double‐blind, active‐control study Recruitment period: July 2011 to September 2012 Sample size: 309 Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: malignant tumours Chemotherapy regimen HEC: cisplatin, mechlorethamine, streptozocin, cyclophosphamide ≥ 1500 mg/m², carmustine, dacarbazine MEC: any i.v. dose of oxaliplatin, carboplatin, epirubicin, idarubicin, ifosfamide, irinotecan, daunorubicin, or doxorubicin; i.v. cyclophosphamide (< 1500 mg/m²), i.v. cytarabine (> 1 g/m²); azacitidine, alemtuzumab, bendamustine, or clofarabine Countries: Bulgaria, Czech Republic, Germany, Hungary, India, Poland, Russian Federation, Serbia, Ukraine, United States |
| Interventions | Experimental: arm A netupitant + palonosetron + dexamethasone Experimental: arm B aprepitant + palonosetron + dexamethasone |
| Outcomes | Primary objective
Secondary objectives
|
| Notes |
|
| Methods | Randomised, phase 3, active‐controlled study Recruitment period: December 2015 to n.r.
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: solid tumour malignancy Chemotherapy regimen
Country: Austria, Croatia, Czech Republic, Germany, Israel, Italy, Poland, Serbia, South Africa, Spain, Ukraine, United States |
| Interventions | pro‐netupitant (260 mg) + palonosetron (0.25 mg) + dexamethasone |
| Outcomes | Primary endpoints
Secondary endpoints
|
| Notes |
|
| Methods | Randomised, interventional, phase 3 study with 2 arms
Target sample size: 100 Masking: n.r. Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: minimum 20 years Gender: male and female Tumour/cancer type: not specified Chemotherapy regimen: AC/EC Country: Japan |
| Interventions | Experimental: arm A Fosnetupitant 235 mg i.v. before start of chemotherapy Active control: arm B Fosaprepitant 150 mg i.v. before start of chemotherapy |
| Outcomes | Primary outcome
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, comparative study with 2 arms
Recruitment period: n.r.
Masking: n.r. Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria: n.r. Exclusion criteria: n.r. Mean/median age, years: n.r. Gender: n.r. Tumour/cancer type: n.r. Chemotherapy regimen: moderately emetogenic chemotherapy regimen Country: n.r. |
| Interventions | Experimental: arm A ondansetron Experimental: arm B tropisetron |
| Outcomes | n.r. |
| Notes |
|
| Methods | Randomised, phase 2, parallel‐assignment study Study start date: February 2005 Study completion date: not provided
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: solid malignancy Chemotherapy regimen: n.r. Country: Argentina, Austria, Belgium, Chile, Croatia, Czech Republic, Hong Kong, Hungary, Italy, Mexico, Netherlands, Pakistan, Peru, Philippines, Poland, Romania, Singapore, Slovakia, Taiwan |
| Interventions | Drug: aprepitant, ondansetron, GW679769, dexamethasone Study arms: not provided |
| Outcomes | Primary outcome
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, phase 2 study with 2 arms
Study period: May 2010 to December 2012
Masking: triple (participant, care provider, investigator) Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: lymphoproliferative disease Chemotherapy regimen: myeloablative chemotherapy + autologous stem cell transplantation Country: Sweden |
| Interventions | Experimental: arm A aprepitant given orally 125 mg the first day, then 80 mg daily during the chemotherapy course + 1/dexamethasone 6 mg daily during chemotherapy days + 2/tropisetron (Navoban) 5 mg daily during chemotherapy and 2 days after Control: arm B placebo + 1/dexamethasone 6 mg daily during chemotherapy days + 2/tropisetron (Navoban) 5 mg daily during chemotherapy and 2 days after |
| Outcomes | Primary outcome
Secondary outcome
|
| Notes |
|
| Methods | Randomised, placebo‐controlled, cross‐over study with 2 arms Recruitment period: April 2015 to February 2018
Masking: quadruple (participant, care provider, investigator, outcomes assessor) Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: non‐small cell lung cancer Chemotherapy regimen: carboplatin‐based combination chemotherapy Country: United States |
| Interventions | Cross‐over study Experimental: arm A fosaprepitant (Emend) for injection; 150 mg was administered, 1 time, i.v. on Day 1 only, as an infusion with a duration of 30 minutes Experimental: arm B saline placebo intravenously on Day 1 of first chemotherapy cycle |
| Outcomes | Primary outcome
Secondary outcomes
|
| Notes |
|
| Methods | Randomised study with 2 arms
Recruitment period: 19 April 2006 to 1 April 2010
Masking: open‐label Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: gastrointestinal malignancy Chemotherapy regimen: oxaliplatin‐containing regimen in combination with 5‐fluorouracil (combinations such as fluorouracil, oxaliplatin, and leucovorin calcium (FOLFOX), FOLFOX + bevacizumab, FOLFOX + cetuximab) Country: United States |
| Interventions | Experimental: arm A dolasetron mesylate p.o. or i.v., dexamethasone p.o. or i.v., and aprepitant p.o. 1 day before chemotherapy; dexamethasone p.o. and aprepitant p.o. on Days 2 and 3 after chemotherapy begins during Course 2 to 3 Experimental: arm B dolasetron mesylate and dexamethasone as in arm A and placebo p.o. 1 day before chemotherapy; dexamethasone p.o. and placebo p.o. on Days 2 and 3 after chemotherapy begins during Courses 2 to 3 |
| Outcomes | Primary outcome
Secondary outcome
|
| Notes |
|
| Methods | Randomised, interventional, phase 2, parallel study with 2 arms
Estimated enrolled patients: 91, terminated per principal investigator's request after enrolment of 37 participants Masking: open‐label Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: male + female Tumour/cancer type: sarcoma Chemotherapy regimen: doxorubicin + ifosfamide (AI) or ifosfamide (AI) and vincristine (VAI) is indicated Country: United States |
| Interventions | Experimental: arm A: rolapitant i.v. dexamethasone daily and ondansetron i.v. on Days 1 to 5, and p.o. rolapitant hydrochloride on Day 1 treatment repeats every 21 days for 6 cycles in the absence of disease progression or unacceptable toxicity Experimental: arm B: fosaprepitant i.v. dexamethasone and i.v. ondansetron on Days 1 to 5, and i.v. fosaprepitant dimeglumine over 30 min on Day 1 Cycle 2. Treatment repeats every 21 days for 6 cycles in the absence of disease progression or unacceptable toxicity |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, phase 3b study with 2 arms
Study period: 16 March 2018 to 19 September 2018
Masking: quadruple (participant, care provider, investigator, outcomes assessor) Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: female Tumour/cancer type: breast cancer Chemotherapy regimen: anthracycline + cyclophosphamide Country: United States, Georgia (multi‐centre) |
| Interventions | Experimental: arm A i.v. fosnetupitant/ palonosetron (260 mg/0.25 mg) fixed‐dose combination, administered as a 30‐min infusion of a 50‐mL solution on Day 1 of each cycle p.o. dexamethasone will be administered on Day 1 of each cycle (12 mg) Control: arm B p.o. netupitant/palonosetron (300 mg/0.50 mg) fixed‐dose combination on Day 1 of each cycle p.o. dexamethasone will be administered on Day 1 of each cycle (12 mg) |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, multi‐centre, parallel‐group, active‐controlled, phase 3 study Recruitment period
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: 18 to 99 Gender: male + female Tumour/cancer type: n.r. Chemotherapy regimen: HEC Country: n.r. (multi‐centre) |
| Interventions |
|
| Outcomes |
|
| Notes |
|
| Methods | Randomised study with 2 arms
Recruitment period: n.r.
Masking: n.r. Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria: n.r. Exclusion criteria: n.r. Mean/median age, years: n.r. Gender: n.r. Tumour/cancer type: HIV‐related non‐Hodgkin lymphoma (HIV‐NHL) Chemotherapy regimen: moderately emetogenic chemotherapy regimen Country: n.r. |
| Interventions | Experimental: arm A granisetron Experimental: arm B ondansetron |
| Outcomes | n.r. |
| Notes |
|
| Methods | Randomised, interventional, phase 2 study with 2 arms
Recruitment period: October 2010 to n.r.
Masking: open‐label Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: head and neck cancer Chemotherapy regimen: cisplatin ≥ 60 mg/m² Country: Japan |
| Interventions | Cross‐over study Experimental: arm A: granisetron aprepitant was administered p.o. at 125 mg/body 1 h or 1 h and a half before cisplatin administration. On Days 2 and 3, aprepitant was administrated p.o. at 80 mg/body granisetron 40 μg/body and dexamethasone 12.3 mg/body were administered i.v. 30 min before cisplatin administration. On Days 2 and 3, granisetron 40 μg/body and dexamethasone 6.6 mg/body were administered Experimental: arm B: palonosetron aprepitant was administered p.o. at 125 mg/body 1 h or 1 h and a half before cisplatin administration. On Days 2 and 3, aprepitant was administrated p.o. at 80 mg/body palonosetron 0.75 mg/body and dexamethasone 12.3 mg/body were administered i.v. 30 min before cisplatin administration. On Days 2 and 3, dexamethasone 6.6 mg/body was administered |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, phase 3, active‐controlled study with 2 arms
Study period: May 2011 to June 2012
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: solid malignancy (lung cancer, gastric cancer, oesophagus cancer, cervical cancer, endometrial cancer, head and neck cancer, etc.) Chemotherapy regimen: cisplatin ≥ 50 mg/m² Country: Japan |
| Interventions | Experimental: arm A: granisetron granisetron 1 mg + dexamethasone (Days 1 to 4) + aprepitant (Days 1 to 3) Experimental: arm B: palonosetron palonosetron 0.75 mg + dexamethasone (Days 1 to 4) + aprepitant (Days 1 to 3) |
| Outcomes | Primary outcome
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, phase 2 study with 2 arms
Recruitment period: January 2011 to January 2013
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: female Tumour/cancer type: haematology and clinical oncology Chemotherapy regimen: carboplatin‐ or irinotecan‐ (> 150 mg/m²) based regimens Country: Japan |
| Interventions | Experimental: arm A Day 1, aprepitant 125 mg, granisetron 1 mg, and dexamethasone 12 mg before chemotherapy; Days 2 through 3, aprepitant 80 mg QD and dexamethasone 4 mg QD Control: arm B Day 1, granisetron 1 mg and dexamethasone 20 mg before chemotherapy; Days 2 through 3, dexamethasone 8 mg QD |
| Outcomes | Primary outcome
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, cross‐over, active‐controlled study with 2 arms Recruitment period: February 2012 to n.r.
Masking: open‐label Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: advanced/recurrent gastric cancer or colorectal cancer Chemotherapy regimen: FOLFILI, CPT‐11 monotherapy, FOLFOX, or XELOX regimen Country: Japan |
| Interventions | Cross‐over study Experimental: arm A
Experimental: arm B
|
| Outcomes | Primary outcomes
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, active‐controlled, cross‐over study with 2 arms
Recruitment period: August 2012 to n.r.
Masking: single‐blind (participants were blinded) Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: female Tumour/cancer type: gynaecological cancer Chemotherapy regimen: carboplatin (> AUC 5) Country: Japan |
| Interventions | Cross‐over study Experimental: arm A NK₁ receptor antagonist (fosaprepitant) + 5‐HT₃ receptor antagonist + dexamethasone Experimental: arm B palonosetron + dexamethasone |
| Outcomes | Primary outcome
|
| Notes |
|
| Methods | Randomised, interventional, phase 3 study with 2 arms
Recruitment period: November 2012 to n.r.
Masking: double‐blind Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: breast cancer Chemotherapy regimen: AC/EC/FAC/FEC chemotherapy Country: Japan |
| Interventions | Experimental: arm A: granisetron granisetron + dexamethasone Experimental: arm B: palonosetron palonosetron + dexamethasone |
| Outcomes | Primary outcome
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, active‐controlled, phase 2 study with 2 arms
Recruitment period: April 2014 to n.r.
Masking: open‐label Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: lung cancer Chemotherapy regimen: carboplatin Country: Japan |
| Interventions | Experimental: arm A aprepitant + palonosetron + dexamethasone Experimental: arm B palonosetron + dexamethasone |
| Outcomes | n.r. |
| Notes |
|
| Methods | Randomised study with 2 arms
Recruitment period: May 2013 to October 2015
Masking: open‐label Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: male + female Tumour/cancer type: n.r. Chemotherapy regimen: carboplatin (CBDCA)‐based moderately emetogenic chemotherapy (MEC) Country: Japan |
| Interventions | Experimental: arm A aprepitant (Day 1: 125 mg, Days 2 to 3: 80 mg) + granisetron 3 mg + dexamethasone (Day 1: 6.6 mg i.v., Days 2 to 3: 2 mg POX2) Experimental: arm B palonosetron 0.75 mg + dexamethasone (Day 1: 9.9 mg i.v., Days 2 to 3: 4 mg POX2) |
| Outcomes | Primary outcome
Secondary outcomes
|
| Notes |
|
| Methods | Randomised, phase 2 study with 2 arms
Recruitment period: November 2011 to n.r.
Masking: open‐label Baseline patient characteristics: n.r. |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: n.r. Gender: female Tumour/cancer type: gynaecological cancer Chemotherapy regimen: carboplatin (area under the concentration curve of 5) and paclitaxel at 175 mg/m² Country: Japan |
| Interventions | Experimental: arm A p.o. administration of 125 mg aprepitant 90 min before administration of chemotherapy drug on Day 1 and of 80 mg on Days 2 and 3, and 0.75 mg + palonosetron administered i.v. on Day 1 + 6 mg dexamethasone administered i.v. on Day 1 and 4 mg dexamethasone administered p.o. on Days 2 and 3 Experimental: arm B 0.75 mg palonosetron administered i.v. on Day 1 + 6 mg dexamethasone administered i.v. on Day 1 and 4 mg dexamethasone administered p.o. on Days 2 and 3 |
| Outcomes | Primary outcome
|
| Notes |
|
5‐FU: 5‐fluorouracil.
5‐HT₃: serotonin (5‐hydroxytryptamine).
AC: doxorubicin, cyclophosphamide.
ALT: alanine aminotransferase.
AST: aspartate aminotransferase.
CNS: central nervous system.
Cr: creatinine.
CPT‐11: camptothecin‐11.
CTCAE: Common Terminology Criteria for Adverse Events.
d: day (e.g., d1, d3).
EC: epirubicin.
ECG: electrocardiogram.
ECOG: Eastern Cooperative Oncology Group.
ESMO: European Society for Medical Oncology.
FAC: 5‐fluorouracil (5FU) + AC.
FEC: fluorouracil, epirubicin, cyclophosphamide.
FLIE: Functional Living Index‐Emesis.
FOLFILI: folinic acid, fluorouracil, and irinotecan.
FOLFOX: folinic acid, fluorouracil, and oxaliplatin.
g/L: gram per litre.
g/m²: gram per square meter.
h: hour.
Hb: haemoglobin.
HBV: hepatitis B virus.
HCV: hepatitis C virus.
HEC: highly emetogenic chemotherapy.
HIV: human immunodeficiency virus.
IU: international unit.
i.v.: intravenous.
L: litre.
MASCC: Annual Meeting on Supportive Cancer Care.
MEC: moderately emetogenic chemotherapy.
mg: milligram.
mL/min: millilitre per minute.
msec: millisecond.
NCI: National Cancer Institute.
NK₁: neurokinin‐1.
n.r.: not reported.
NSCLC: non‐small cell lung carcinoma.
PLT: platelets.
p.o.: oral.
QD: once a day.
QTc: QT interval corrected for heart rate.
SOX: S‐1 plus oxaliplatin.
TBIL: total bilirubin.
ULN: upper limit of normal.
VAS: visual analogue scale.
VP‐16: etoposide.
VM‐26: teniposide.
vs: versus.
WBC: white blood cell count.
XELOX: capecitabine, oxaliplatin.
Characteristics of ongoing studies [ordered by study ID]
| Study name | A randomized, open, parallel controlled phase II clinical study comparing the efficacy and safety of dexamethasone, palonosetron, or aprepitant in the control of acute and delayed vomiting in non‐small cell lung cancer patients receiving multiple moderately emetogenic chemotherapy regimens |
| Methods | Randomised, interventional, active‐controlled study with 2 arms
Target sample size: 100 Masking: open‐label Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: both Tumour/cancer type: non‐small cell lung cancer Chemotherapy regimen: not reported, moderately emetogenic chemotherapy Country: China |
| Interventions | Experimental: arm A: aprepitant/palonosetron/dexamethasone Intervention details not reported Experimental: arm B: palonosetron/dexamethasone Intervention details not reported |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | 15 August 2019 |
| Contact information | Research and public contact: Zhang Lemeng (Hunan Cancer Hospital, 283 Tongzipo Road, Yuelu District, Changsha, Hu'nan, China, email: [email protected]) |
| Notes |
|
| Study name | Comparison between the effect of Triplet Aprepitant/Dexamethasone/Ondansetron vs doublet Dexamethasone/Ondansetron for prevention of moderately emetogenic chemotherapy: placebo‐controlled double blind, randomised clinical trial of efficacy |
| Methods | Randomised, interventional, active‐controlled study with 2 arms
Target sample size: 90 (actual sample size reached: 160) Masking: double‐blind Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: not available Tumour/cancer type: not available Chemotherapy regimen: not available, moderately emetogenic chemotherapy Country: Iran |
| Interventions | Experimental: arm A: aprepitant/dexamethasone/ondansetron p.o. administration of 125 mg aprepitant, i.v. injection of 12 mg dexamethasone and 8 mg ondansetron on Day 1, followed by 80 mg Abitant on Days 2 and 3 Experimental: arm B: dexamethasone/ondansetron i.v. injection of 12 mg dexamethasone and 8 mg ondansetron on Day 1 |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | 21 December 2018 |
| Contact information | Research and public contact: Mania Rajabzadeh Kheradmardi (Shahid Beheshti University of Medical Sciences, Jorjani Center of Radiation Oncology, Imam Husein Hospital, Shahid Madani Ave, Nezam Abad Town, Tehran, Iran, email: [email protected]) |
| Notes |
|
| Study name | A randomized, double‐blind, double‐dummy, parallel group, international multi center study assessing the efficacy and safety of a netupitant‐palonosetron Fixed Dose Combination (FDC) compared to an extemporary combination of granisetron and aprepitant on the prevention of highly emetogenic chemotherapy‐induced nausea and vomiting in patients with cancer |
| Methods | Randomised, interventional, phase 3 study with 2 arms
Target sample size: 832 Masking: double‐blind Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: male + female Tumour/cancer type: solid tumour malignancy Chemotherapy regimen: cisplatin‐based chemotherapy regimen Country: multi‐national (7 centres) |
| Interventions | Experimental: arm A p.o. administration of NETU‐PALO FDC (containing 300 mg netupitant and 0.5 mg palonosetron) on Day 1 (with adjusted dexamethasone regimen: 12 mg on Day 1 + 8 mg daily from Day 2 to Day 4) Active control: arm B p.o. aprepitant 125 mg (on Day 1) + 80 mg daily (on Days 2 and day 3) and 3 mg i.v. granisetron on Day 1 (with adjusted dexamethasone regimen: 12 mg on Day 1 + 8 mg daily from Day 2 to Day 4) |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | 15 January 2015 |
| Contact information | Younyoung Cho, Konkuk University Medical Center |
| Notes |
|
| Study name | Effectiveness and quality of life analysis of palonosetron against ondansetron combined with dexamethasone and fosaprepitant in prevention of acute and delayed emesis associated to chemotherapy moderately and highly emetogenic in breast cancer |
| Methods | Randomised, interventional, parallel study with 2 arms
Estimated enrolled patients: 560 Masking: open‐label Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: female Tumour/cancer type: breast cancer Chemotherapy regimen: anthracyclines combined with cyclophosphamide or carboplatin combined with docetaxel or docetaxel combined with cyclophosphamide Country: Mexico |
| Interventions | Experimental: arm A: palonosetron early emesis: palonosetron 0.25 mg i.v. + dexamethasone 12 mg i.v. + fosaprepitant 150 mg i.v. delayed emesis: dexamethasone 8 mg p.o. on Days 2, 3, and 4 Experimental: arm B: ondansetron early emesis: ondansetron 16 mg i.v. + dexamethasone 12 mg i.v. + fosaprepitant 150 mg i.v. delayed emesis: metoclopramide 10 mg p.o. every 6 hours + dexamethasone 8 mg p.o. every 24 hours |
| Outcomes | Primary outcome
Secondary outcome
|
| Starting date | 5 November 2015 |
| Contact information | Contact: Claudia H. Arce Salinas, MD; phone: 56280400 ext 12065; email: [email protected] Contact: Juan P González Serrano, BD; phone: 5519480352; email: [email protected] |
| Notes |
|
| Study name | Study of oral neurokinin‐1 antagonist, aprepitant for the prevention of nausea and vomiting in patients receiving chemotherapy with irinotecan alone or combination of irinotecan plus cisplatin for unresectable gastric cancer |
| Methods | Randomised, cross‐over, interventional, placebo‐controlled study with 2 arms
Target sample size: 80 Masking: double‐blind Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: male + female Tumour/cancer type: gastric cancer Chemotherapy regimen: irinotecan alone or combination of irinotecan plus cisplatin Country: Japan |
| Interventions | Cross‐over study Experimental: arm A: aprepitant aprepitant: 125 mg p.o./d on Day 1 followed by 80 mg p.o./d on Days 2 and 3 Experimental: arm B: placebo placebo: placebo capsules p.o. on Days 1 to 3 |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | 1 August 2010 |
| Contact information | Research contact: Hiroto Miwa (Division of Upper Gastroenterology, Department of Internal Medicine; Hyogo College of Medicine; 1‐1 Mukogawa‐cho, Nishinomiya, Hyogo, Japan) Public contact: Junji Tanaka (Division of Upper Gastroenterology, Department of Internal Medicine; Hyogo College of Medicine; 1‐1 Mukogawa‐cho, Nishinomiya, Hyogo, Japan) |
| Notes |
|
| Study name | Effect of oral neurokinin‐1 antagonist, aprepitant for chemotherapy‐induced nausea and vomiting in patients with gynecologic cancer receiving carboplatin/paclitaxel chemotherapy |
| Methods | Randomised, interventional, parallel, active‐controlled study with 2 arms
Target sample size: 60 Masking: open‐label Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: female Tumour/cancer type: gynaecological cancer Chemotherapy regimen: carboplatin/paclitaxel Country: Japan |
| Interventions | Experimental: arm A: aprepitant aprepitant 125 mg p.o. on Day 1 Experimental: arm B granisetron 3 mg i.v. on Day 1 |
| Outcomes | Primary outcome
|
| Starting date | 29 November 2010 |
| Contact information | Research contact: Hiroshi Tsujioka (Department of Gynecology; Fukuoka University Hospital; 7‐45‐1 Nanakuma, Jonan‐ku, Fukuoka, Japan) |
| Notes |
|
| Study name | Aprepitant for nausea, vomiting with the TC therapy of the gynecology cancer patient or the DC therapy, fosaprepitant, granisetron, protective efficacy of the dexamethasone combination therapy |
| Methods | Randomised, interventional, active‐controlled study with 2 arms
Target sample size: 140 Masking: open‐label Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: female Tumour/cancer type: uterine cancer, ovarian cancer Chemotherapy regimen: paclitaxel/carboplatin or docetaxel/carboplatin Country: Japan |
| Interventions | Experimental: arm A: aprepitant/fosaprepitant aprepitant on Days 1 to 3 (or fosaprepitant on Day 1) Experimental: arm B granisetron on Day 1 |
| Outcomes | Primary outcome
|
| Starting date | 1 May 2011 |
| Contact information | Research contact: Hideaki Masuzaki (Department of Obstetrics and Gynecology; Nagasaki University (graduate school); 1‐7‐1 Sakamoto, Nagasaki, Japan) Public contact: Shuhei Abe (Department of Obstetrics and Gynecology; Nagasaki University (graduate school); 1‐7‐1 Sakamoto, Nagasaki, Japan; email: [email protected]) |
| Notes |
|
| Study name | Randomized phase II study of aprepitant in patients with colorectal cancer receiving FOLFOX, FOLFIRI, or XELOX chemotherapy regimen |
| Methods | Randomised, phase 2, parallel, interventional controlled study with 2 arms
Target sample size: 100 Masking: n.r. Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: male + female Tumour/cancer type: colon and rectal cancer Chemotherapy regimen: FOLFOX, FOLFIRI, or XELOX chemotherapy regimen Country: Japan |
| Interventions | Experimental: arm A: aprepitant aprepitant 125 mg p.o. on Day 1 Experimental: arm B 5‐HT₃ receptor antagonist i.v. on Day 1 |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | 1 November 2011 |
| Contact information | Contact 1: Shoji Natsugoe (Department of Digestive Surgery, Breast and Thyroid Surgery; Kagoshima University Graduate School of Medical and Dental Sciences; 8‐35‐1 Sakuragaoka, Kagoshima, Japan; telephone: 099‐275‐5358) Contact 2: Sumiya Ishigami (Department of Digestive Surgery, Breast and Thyroid Surgery; Kagoshima University Graduate School of Medical and Dental Sciences; 8‐35‐1 Sakuragaoka, Kagoshima, Japan; telephone: 099‐275‐5360) |
| Notes |
|
| Study name | Multicenter double‐blind randomized comparative parallel study with concomitant therapy of 3 drugs, aprepitant + dexamethasone+palonosetron or aprepitant + dexamethasone+ granisetron, for prevention of nausea/vomiting in breast cancer patients receiving AC therapy |
| Methods | Randomised, interventional, controlled study with 2 arms
Target sample size: 660 Masking: double blind Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: female Tumour/cancer type: breast cancer Chemotherapy regimen: doxorubicin hydrochloride (adriamycin) and cyclophosphamide Country: Japan |
| Interventions | Experimental: arm A: granisetron aprepitant (Day 1: 125 mg, Days 2 to 3: 80 mg) + dexamethasone (Day 1: 9.9 mg) + granisetron (Day 1: 40 (microgram)/kg) Experimental: arm B: palonosetron aprepitant (Day 1: 125 mg, Days 2 to 3: 80 mg) + dexamethasone (Day 1: 9.9 mg) + palonosetron (Day 1: 0.75 mg) |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | 1 June 2012 |
| Contact information | Research and public contact: Mitsue Saito (Department of Breast Oncology; Juntendo University Hospital; 3‐1‐3, Hongo, Bunkyo‐ku, Tokyo 113‐8421; email: [email protected]) |
| Notes |
|
| Study name | Effect of aprepitant for nausea and vomiting during paclitaxel + carboplatin (TC) therapy |
| Methods | Randomised, cross‐over, interventional study with 2 arms
Target sample size: 50 Masking: open‐label Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: female Tumour/cancer type: gynaecological malignancy Chemotherapy regimen: paclitaxel + carboplatin (TC) regimen Country: Japan |
| Interventions | Cross‐over study Experimental: arm A first course 5‐HT₃ receptor antagonist + dexamethasone i.v. Experimental: arm B first course 5‐HT₃ receptor antagonist + dexamethasone i.v. + aprepitant p.o. |
| Outcomes | Objective
|
| Starting date | 18 January 2011 |
| Contact information | Research contact: Masahide Ohmichi; Department of Obstetrics and Gynecology; Osaka Medical College; 2‐7, Daigaku‐machi, Takatsuki, Osaka; email: m‐[email protected]‐med.ac.jp Public contact: Masanori Kanemura; Department of Obstetrics and Gynecology; Osaka Medical College; 2‐7, Daigaku‐machi, Takatsuki, Osaka; email: [email protected]‐med.ac.jp |
| Notes |
|
| Study name | Palonosetron versus granisetron in combination with fosaprepitant and dexamethasone for TC therapy in patients with gynecologic cancer |
| Methods | Randomised, factorial, active‐controlled study with 2 arms
Target sample size: 240 Masking: open‐label Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: female Tumour/cancer type: gynaecological cancer Chemotherapy regimen: paclitaxel + carboplatin (TC) regimen Country: Japan |
| Interventions | Experimental: arm A: palonosetron fosaprepitant + palonosetron + dexamethasone Experimental: arm B: granisetron fosaprepitant + granisetron + dexamethasone |
| Outcomes | Primary outcome
|
| Starting date | 10 June 2018 |
| Contact information | Research and contact person: Soshi Kusunoki; Department of Obstetrics and Gynecology; Faculty of Medicine, Juntendo University; Bunkyoku, Tokyo, Japan; email: [email protected] |
| Notes |
|
| Study name | To establish of optimal antiemetic therapy for trastuzumab deruxtecan therapy‐induced nausea and vomiting in patients with breast cancer: an open‐label, randomized pilot study |
| Methods | Randomised, interventional, controlled study with 2 arms
Target sample size: 40 Masking: open‐label Baseline patient characteristics: not available |
| Participants | Inclusion criteria
Exclusion criteria
Mean/median age, years: not available Gender: female Tumour/cancer type: breast cancer Chemotherapy regimen: trastuzumab deruxtecan Country: Japan |
| Interventions | Experimental: arm A granisetron + dexamethasone + aprepitant (fosaprepitant) Experimental: arm B granisetron + dexamethasone |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | 1 July 2020 |
| Contact information | Research and public contact: Hirotoshi Iihara (Gifu University Hospital, Department of Pharmacy, 500‐1194 1‐1 Yanagido, Gifu, email: dai0920@gifu‐u.ac.jp) |
| Notes |
|
AC: doxorubicin, cyclophosphamide.
ALT: alanine transaminase.
AST: aspartate aminotransferase.
CINV: chemotherapy‐induced nausea and vomiting.
ECG: electrocardiogram.
ECOG: Eastern Cooperative Oncology Group.
dL: decilitre.
FOLFILI: folinic acid, fluorouracil, and irinotecan.
FOLFOX: folinic acid, fluorouracil, and oxaliplatin.
g: gram.
i.v.: intravenous.
kg: kilogram.
min: minute.
mL: millilitre.
NK₁: neurokinin‐1.
p.o.: oral.
QoL: quality of life.
ULN: upper limit of normal.
VAS: visual analogue scale.
WBC: white blood cell count.
XELOX: capecitabine, oxaliplatin.

Study flow diagram.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Network graph for the outcome complete control of vomiting in the overall phase (HEC).
A line connects any two treatments when there is at least one study comparing the two treatments. Line width: number of patients.
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

League table for the outcome complete control of vomiting in the overall phase (HEC). Network estimates with 95% CIs are given. Descending P score shows ranking of treatment options. Statistically significant results are marked in yellow. Global approach to check inconsistency/heterogeneity: Q‐statistics, I².
No. of studies: 34. No. of treatments: 14. No. of pair‐wise comparisons: 36. No. of designs: 21.
Qtotal = 23.95, df = 22, P = 0.35/Qwithin = 17.60, df = 13, P = 0.17/Qbetween = 6.34, df = 9, P = 0.71; I² = 8.1%, Tau² = 0.0005.
Treatment effects + 95% CIs (risk ratios, random‐effects model).
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

Exemplary network meta‐analysis forest plot for the outcome complete control of vomiting during the overall phase (HEC) (random‐effects model).
Aprepitant + granisetron was used as exemplary reference treatment. Ranking of treatments is ordered by P score (descending).
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.
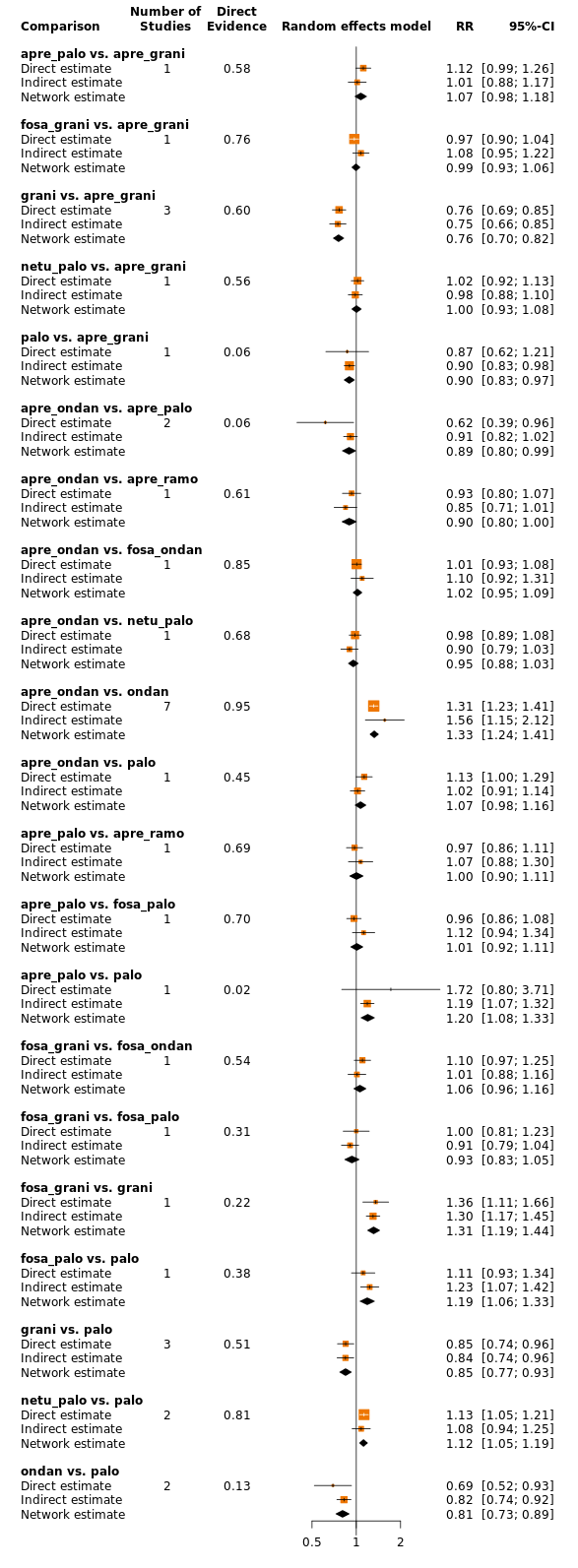
Comparison of direct and indirect evidence (in closed loops) for the outcome complete control of vomiting in the overall phase (HEC). CI: confidence interval; RR: risk ratio.
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

Network graph for the outcome serious adverse events (HEC).
A line connects any 2 treatments when there is at least 1 study comparing the 2 treatments. Line width: number of patients.
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.
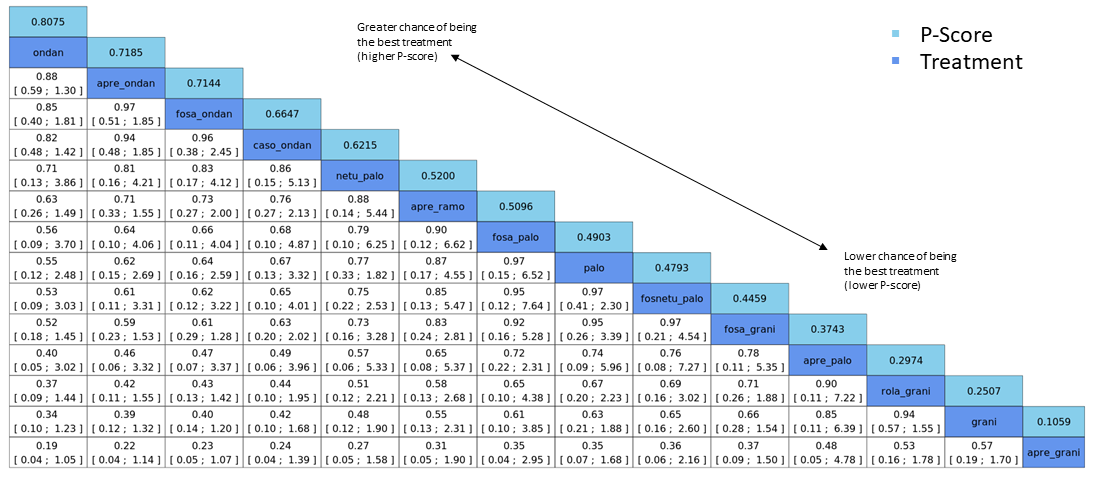
League table for the outcome serious adverse events (HEC). Network estimates with 95% CIs are given. Descending P score shows ranking of treatment options. Statistically significant results are marked in yellow. Global approach to check inconsistency/heterogeneity: Q‐statistics, I².
No. of studies: 20. No. of treatments: 12. No. of pair‐wise comparisons: 22. No. of designs: 13.
Qtotal = 20.26, df = 9, P = 0.016/Qwithin = 16.39, df = 7, P = 0.022/Qbetween = 3.87, df = 2, P = 0.14; I² = 55.6%, Tau² = 0.1057.
Treatment effects + 95% CIs (risk ratios, random‐effects model).
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

Exemplary network meta‐analysis forest plot for the outcome serious adverse events (HEC) (random‐effects model).
Aprepitant + granisetron was used as exemplary reference treatment. Ranking of treatments is ordered by P score (descending).
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

Comparison of direct and indirect evidence (in closed loops) for the outcome serious adverse events (HEC). CI: confidence interval; RR: risk ratio.
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

Exemplary ranking plot representing simultaneously the efficacy (x‐axis, CR during the overall phase) and the acceptability (y‐axis, SAEs) of all antiemetic regimens for patients receiving highly emetogenic chemotherapy.
Only antiemetic regimens for which data for both endpoints (CR during the overall phase and SAEs) were available are represented in the ranking plot.
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

League table with network estimates (RR with 95% CIs) of all treatment combinations for efficacy (CR during the overall phase) and acceptability (SAEs) (HEC).
Treatments are presented in alphabetical order. For efficacy, RRs > 1 favour the first treatment in alphabetical order. For safety, RRs < 1 favour the first treatment in alphabetical order.
n.a.: no data were available for this comparison
Statistically significant results are marked bold.
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.
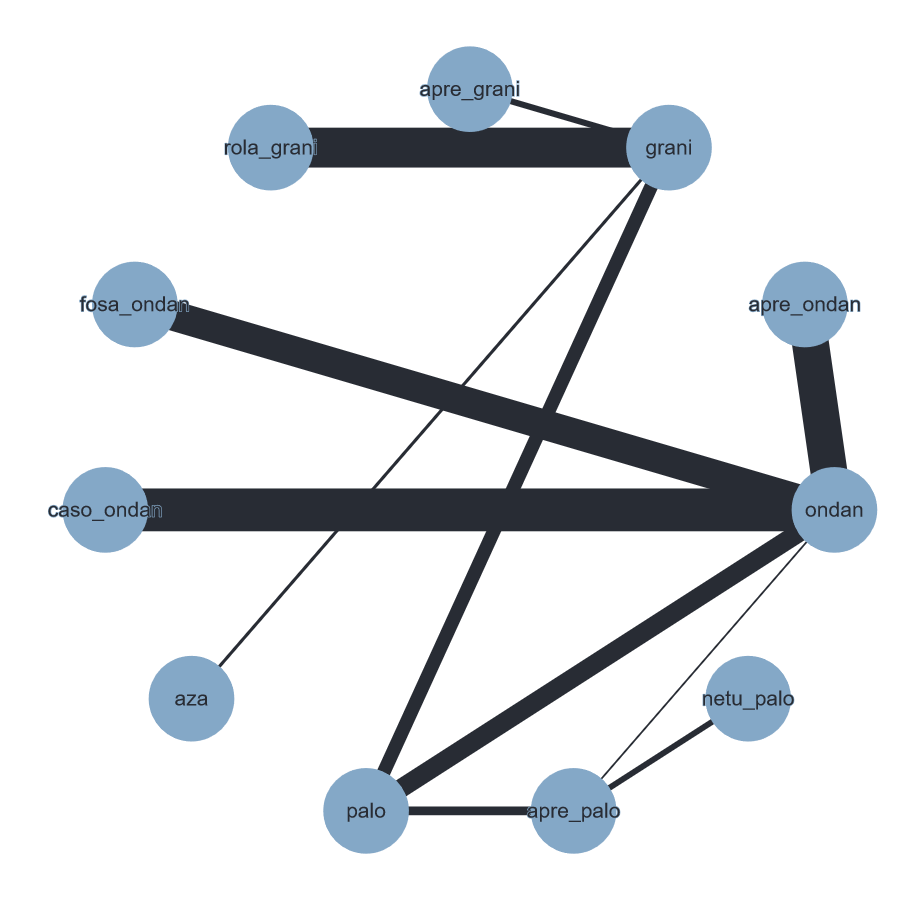
Network graph for the outcome complete control of vomiting during the overall phase (MEC).
A line connects any 2 treatments when there is at least 1 study comparing the 2 treatments. Line width: number of patients.
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

League table for the outcome complete control of vomiting during the overall phase (MEC). Network estimates with 95% CIs are given. Descending P score shows ranking of treatment options. Statistically significant results are marked in yellow. Global approach to check inconsistency/heterogeneity: Q‐statistics, I².
No. of studies: 22. No. of treatments: 11. No. of pair‐wise comparisons: 22. No. of designs: 11.
Qtotal = 13.90, df = 12, P = 0.31/Qwithin = 13.80, df = 11, P = 0.24/Qbetween = 0.09, df = 1, P = 0.76; I² = 13.7%, Tau² = 0.0018.
Treatment effects + 95% CIs (risk ratios, random‐effects model); RR > 1 favours the upper treatment/treatment on the left.
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

Exemplary network meta‐analysis forest plot for the outcome complete control of vomiting during the overall phase (MEC) (random‐effects model).
Granisetron was used as exemplary reference treatment. Ranking of treatments is ordered by P score (descending).
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

Comparison of direct and indirect evidence (in closed loops) for the outcome complete control of vomiting during the overall phase (MEC). CI: confidence interval; RR: risk ratio.
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

Network graph for the outcome serious adverse events (MEC).
A line connects any 2 treatments when there is at least 1 study comparing the 2 treatments. Line width: number of patients.
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

League table for the outcome serious adverse events (MEC). Network estimates with 95% CIs are given. Descending P score shows ranking of treatment options. Statistically significant results are marked in yellow. Global approach to check inconsistency/heterogeneity: Q‐statistics, I².
No. of studies: 4. No. of treatments: 5. No. of pair‐wise comparisons: 4. No. of designs: 4.
Heterogeneity/inconsistency: Q = 0, df = 0, P = not available; I² = not available, Tau² = not available.
Treatment effects + 95% CIs (risk ratios, random‐effects model).
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

Exemplary network meta‐analysis forest plot for the outcome serious adverse events (MEC) (random‐effects model).
Ondansetron was used as exemplary reference treatment. Ranking of treatments is ordered by P score (descending).
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

Exemplary ranking plot representing simultaneously the efficacy (x‐axis, CR during overall phase) and the acceptability (y‐axis, SAEs) of all antiemetic regimens for patients receiving moderately emetogenic chemotherapy.
Only antiemetic regimens for which data for both endpoints (CR during the overall phase and SAEs) were available are represented in the ranking plot.
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.

League table with network estimates (RR with 95% CIs) of all treatment combinations for efficacy (CR during the overall phase) and acceptability (SAEs) (MEC).
Treatments are presented in alphabetical order. For efficacy, RRs > 1 favour the first treatment in alphabetical order. For safety, RRs < 1 favour the first treatment in alphabetical order.
n.a.: no data were available for this comparison.
Statistically significant results are marked bold.
A list of treatment abbreviations is provided in Table 1; all treatments included a corticosteroid.
| Efficacy | ||||||
|---|---|---|---|---|---|---|
| Antiemetics for adults for prevention of nausea and vomiting caused by highly emetogenic chemotherapy | ||||||
| Patient or population: adult cancer patients at risk for CINV caused by highly emetogenic chemotherapy Settings: inpatient and outpatient care Intervention: neurokinin‐1 (NK₁) receptor antagonist and 5‐hydroxytryptamine‐3 (5‐HT₃) receptor antagonists + corticosteroid Comparison: aprepitant (NK₁) combined with granisetron (5‐HT₃) + corticosteroid Outcome: complete control of vomiting during the overall phase (0 to 120 h of treatment with chemotherapy) RR < 1 indicates an advantage for the intervention Combinations of these interventions at any dose and by any route as mentioned above have been compared to one another in a full network | ||||||
| Interventions (corticosteroids included in all regimens)a | Illustrative comparative risks* (95% CI) | Risk ratio | No. of participants | Certainty of the evidence | Comments | |
| Assumed risk with aprepitant + granisetron | Corresponding risk with the intervention | |||||
|
|
| |||||
| fosnetupitant + palonosetron | 704 of 1000 | 810 of 1000 (683 to 944) | RR 1.15 | 21,642 (39) | ⊕⊕⊕⊝ moderateb | Fosnetupitant + palonosetron probably increases complete response in the overall phase when compared with aprepitant + granisetron |
| aprepitant + palonosetron | 704 of 1000 | 753 of 1000 (690 to 831) | RR 1.07 | 21,642 (39) | ⊕⊕⊝⊝ lowb,c | Aprepitant + palonosetron may result in a slight increase in complete response in the overall phase when compared with aprepitant + granisetron |
| aprepitant + ramosetron | 704 of 1000 | 753 of 1000 (669 to 852) | RR 1.07 | 21,642 (39) | ⊕⊕⊝⊝ lowb,c | Aprepitant + ramosetron may result in a slight increase in complete response in the overall phase when compared with aprepitant + granisetron |
| fosaprepitant + palonosetron | 704 of 1000 | 746 of 1000 (676 to 838) | RR 1.06 | 21,642 (39) | ⊕⊕⊝⊝ lowb,c | Fosaprepitant + palonosetron may result in a slight increase in complete response in the overall phase when compared with aprepitant + granisetron |
| netupitant + palonosetron | 704 of 1000 | 704 of 1000 (655 to 760) | RR 1.00 | 21,642 (39) | ⊕⊕⊕⊕ high | Netupitant + palonosetron has little to no impact on complete response in the overall phase when compared with aprepitant + granisetron |
| fosaprepitant + granisetron | 704 of 1000 | 697 of 1000 (655 to 746) | RR 0.99 | 21,642 (39) | ⊕⊕⊕⊕ high | Fosaprepitant + granisetron has little to no impact on complete response in the overall phase when compared with aprepitant + granisetron |
| aprepitant + ondansetron | 704 of 1000 | 676 of 1000 (620 to 739) | RR 0.96 | 21,642 (39) | ⊕⊕⊝⊝ lowb,c | Aprepitant + ondansetron may result in a slight decrease in complete response in the overall phase when compared with aprepitant + granisetron |
| fosaprepitant + ondansetron | 704 of 1000 | 662 of 1000 (598 to 732) | RR 0.94 | 21,642 (39) | ⊕⊕⊝⊝ lowb,c | Fosaprepitant + ondansetron may result in a slight decrease in complete response in the overall phase when compared with aprepitant + granisetron |
| casopitant + ondansetron | 704 of 1000 | 634 of 1000 (556 to 725) | RR 0.90 | 21,642 (39) | ⊕⊕⊝⊝ lowb,c | Aprepitant + ondansetron may decrease complete response in the overall phase when compared with aprepitant + granisetron |
| rolapitant + granisetron | 704 of 1000 | 627 of 1000 (549 to 711) | RR 0.89 | 21,642 (39) | ⊕⊕⊕⊝ moderateb | Rolapitant + granisetron probably decreases complete response in the overall phase when compared with aprepitant + granisetron |
| rolapitant + ondansetron | 704 of 1000 | 598 of 1000 (458 to 788) | RR 0.85 (0.65 to 1.12) | 21,642 (39) | ⊕⊕⊝⊝ lowc,d | Rolapitant + ondansetron may decrease complete response in the overall phase when compared with aprepitant + granisetron |
| *Basis for the assumed risk is actual event rates reported for the main comparator summed across studies: 1312 of 1863 (70.4%) participants treated with aprepitant + granisetron achieved complete response during the overall phase (aprepitant + granisetron was used in 7 studies reporting the outcome). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| aEither dexamethasone or methylprednisolone was used in all treatment regimens. bDowngraded once for serious imprecision because 95% CIs cross unity. cDowngraded once for serious study limitations due to high risk of bias. dDowngraded once for serious imprecision due to wide confidence intervals. | ||||||
| Safety | ||||||
|---|---|---|---|---|---|---|
| Antiemetics for adults for prevention of nausea and vomiting caused by highly emetogenic chemotherapy | ||||||
| Patient or population: adult cancer patients at risk for CINV caused by highly emetogenic chemotherapy Settings: inpatient and outpatient care Intervention: neurokinin‐1 (NK₁) receptor antagonist and 5‐hydroxytryptamine‐3 (5‐HT₃) receptor antagonists + corticosteroid Comparison: aprepitant (NK₁) combined with granisetron (5‐HT₃) + corticosteroid Outcome: serious adverse events RR < 1 indicates an advantage for the intervention Combinations of these interventions at any dose and by any route as mentioned above have been compared to one another in a full network | ||||||
| Interventions (corticosteroids included in all regimens)a | Illustrative comparative risks* (95% CI) | Risk ratio | No. of participants | Certainty of the evidence | Comments | |
| Assumed risk with aprepitant + granisetron | Corresponding risk with the intervention | |||||
| aprepitant + ondansetron | 35 of 1000 | 8 of 1000 (1 to 40) | RR 0.22 | 16,065 (23) | ⊕⊝⊝⊝ very lowb,c,d | Evidence is very uncertain about the effect of aprepitant + ondansetron on risk of serious adverse events when compared to aprepitant + granisetron |
| fosaprepitant + ondansetron | 35 of 1000 | 8 of 1000 (2 to 37) | RR 0.23 | 16,065 (23) | ⊕⊕⊝⊝ lowb,c | Fosaprepitant + ondansetron may decrease the risk of serious adverse events slightly when compared to aprepitant + granisetron |
| casopitant + ondansetron | 35 of 1000 | 8 of 1000 (1 to 49) | RR 0.24 | 16,065 (23) | ⊕⊕⊝⊝ lowb,c | Casopitant + ondansetron may decrease the risk of serious adverse events slightly when compared to aprepitant + granisetron |
| netupitant + palonosetron | 35 of 1000 | 9 of 1000 (2 to 55) | RR 0.27 | 16,065 (23) | ⊕⊕⊝⊝ lowb,c | Netupitant + palonosetron may decrease the risk of serious adverse events slightly when compared to aprepitant + granisetron |
| aprepitant + ramosetron | 35 of 1000 | 11 of 1000 (2 to 67) | RR 0.31 | 16,065 (23) | ⊕⊝⊝⊝ very lowb,c,d | Evidence is very uncertain about the effect of aprepitant plus ramosetron on risk of serious adverse events when compared to aprepitant + granisetron |
| fosaprepitant + palonosetron | 35 of 1000 | 12 of 1000 (1 to 103) | RR 0.35 | 16,065 (23) | ⊕⊝⊝⊝ very lowb,e | Evidence is very uncertain about the effect of fosaprepitant + palonosetron on risk of serious adverse events when compared to aprepitant + granisetron |
| fosnetupitant + palonosetron | 35 of 1000 | 13 of 1000 (2 to 76) | RR 0.36 | 16,065 (23) | ⊕⊝⊝⊝ very lowb,e | Evidence is very uncertain about the effect of fosnetupitant + palonosetron on risk of serious adverse events when compared to aprepitant + granisetron |
| fosaprepitant + granisetron | 35 of 1000 | 13 of 1000 (3 to 53) | RR 0.37 | 16,065 (23) | ⊕⊕⊝⊝ lowb,c | Fosaprepitant + granisetron may decrease the risk of serious adverse events slightly when compared to aprepitant + granisetron |
| aprepitant + palonosetron | 35 of 1000 | 17 of 1000 (2 to 167) | RR 0.48 | 16,065 (23) | ⊕⊝⊝⊝ very lowb,d,e | Evidence is very uncertain about the effect of aprepitant + palonosetron on risk of serious adverse events when compared to aprepitant + granisetron |
| rolapitant + granisetron | 35 of 1000 | 20 of 1000 (7 to 60) | RR 0.57 | 16,065 (23) | ⊕⊕⊝⊝ lowb,c | Rolapitant + granisetron may decrease the risk of serious adverse events slightly when compared to aprepitant + granisetron |
| *Basis for the assumed risk is actual event rates reported for the main comparator summed across studies: 20 of 573 (3.5%) participants treated with aprepitant + granisetron experienced at least 1 SAE (aprepitant + granisetron was used in 2 studies reporting the outcome, with follow‐up of up to 29 days). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| aEither dexamethasone or methylprednisolone was used in all treatment regimens. bDowngraded once for moderate inconsistency. cDowngraded once for serious imprecision because 95% CIs cross unity and confidence intervals are wide. dDowngraded once for serious study limitations due to high risk of bias. eDowngraded twice for very serious imprecision because 95% CIs cross unity and confidence intervals are very wide, suggesting high possibility of harm. | ||||||
| Efficacy | ||||||
|---|---|---|---|---|---|---|
| Antiemetics for adults for prevention of nausea and vomiting caused by moderately emetogenic chemotherapy | ||||||
| Patient or population: adult cancer patients at risk for CINV caused by moderately emetogenic chemotherapy Settings: inpatient and outpatient care Intervention
Comparison: granisetron (5‐HT₃) + corticosteroid Outcome: complete control of vomiting during the overall phase (0 to 120 h of treatment with chemotherapy) RR < 1 indicates an advantage for the intervention Combinations of these interventions at any dose and by any route as mentioned above have been compared to one another in a full network | ||||||
| Interventions (corticosteroids included in all regimens)a | Illustrative comparative risks* (95% CI) | Risk ratio | No. of participants | Certainty of the evidence | Comments | |
| Assumed risk with granisetron | Corresponding risk with the intervention | |||||
| aprepitant + palonosetron | 555 of 1000 | 716 of 1000 (555 to 921) | RR 1.29 | 7800 (22) | ⊕⊕⊝⊝ lowb,c | Aprepitant + palonosetron may increase complete response in the overall phase when compared to granisetron |
| netupitant + palonosetron | 555 of 1000 | 694 of 1000 (510 to 944) | RR 1.25 | 7800 (22) | ⊕⊕⊝⊝ lowb,d | Netupitant + palonosetron may increase complete response in the overall phase when compared to granisetron |
| rolapitant + granisetron | 555 of 1000 | 660 of 1000 (588 to 738) | RR 1.19 | 7800 (22) | ⊕⊕⊕⊕ high | Rolapitant + granisetron results in an increase in complete response in the overall phase when compared to granisetron |
| palonosetron | 555 of 1000 | 588 of 1000 (472 to 733) | RR 1.06 | 7800 (22) | ⊕⊕⊝⊝ lowb,d | Palonosetron may or may not increase complete response in the overall phase when compared to granisetron |
| aprepitant + granisetron | 555 of 1000 | 577 of 1000 (483 to 694) | RR 1.06 | 7800 (22) | ⊕⊕⊝⊝ lowb,d | Aprepitant + palonosetron may or may not increase complete response in the overall phase when compared to granisetron |
| azasetron | 555 of 1000 | 561 of 1000 (422 to 738) | RR 1.01 | 7800 (22) | ⊕⊕⊝⊝ lowb,e | Azasetron may result in little to no difference in complete response in the overall phase when compared to granisetron |
| fosaprepitant + ondansetron | 555 of 1000 | 500 of 1000 (366 to 677) | RR 0.90 | 7800 (22) | ⊕⊕⊝⊝ lowb,d | Fosaprepitant + ondansetron may decrease complete response in the overall phase when compared to granisetron |
| aprepitant + ondansetron | 555 of 1000 | 477 of 1000 (355 to 649) | RR 0.86 | 7800 (22) | ⊕⊕⊝⊝ lowb,d | Aprepitant + ondansetron may decrease complete response in the overall phase when compared to granisetron |
| casopitant + ondansetron | 555 of 1000 | 461 of 1000 (344 to 622) | RR 0.83 (0.62 to 1.12) | 7800 (22) | ⊕⊕⊝⊝ lowb,d | Casopitant + ondansetron may decrease complete response in the overall phase when compared to granisetron |
| ondansetron | 555 of 1000 | 433 of 1000 (327 to 577) | RR 0.78 | 7800 (22) | ⊕⊕⊝⊝ lowb,d | Ondansetron may decrease complete response in the overall phase when compared to granisetron |
| *Basis for the assumed risk is actual event rates reported for the main comparator summed across studies: 623 of 1123 (55.5%) participants treated with granisetron achieved complete response during the overall phase (granisetron was used in 5 studies reporting the outcome). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| aEither dexamethasone or methylprednisolone was used in all treatment regimens. bDowngraded once for serious study limitations due to high risk of bias. cDowngraded once for serious imprecision because 95% CIs included zero effect line. dDowngraded once for serious imprecision because 95% CIs cross unity. eDowngraded once for serious imprecision due to wide confidence intervals. | ||||||
| Safety | ||||||
|---|---|---|---|---|---|---|
| Antiemetics for adults for prevention of nausea and vomiting caused by moderately emetogenic chemotherapy | ||||||
| Patient or population: adult cancer patients at risk for CINV caused by moderately emetogenic chemotherapy Settings: inpatient and outpatient care Intervention
Comparison: granisetron (5‐HT₃) + corticosteroid Outcome: serious adverse events RR < 1 indicates an advantage for the intervention Combinations of these interventions at any dose and by any route as mentioned above have been compared to one another in a full network | ||||||
| Interventions (corticosteroids included in all regimens)a | Illustrative comparative risks* (95% CI) | Risk ratio | No. of participants | Quality of the evidence | Comments | |
| Assumed risk with granisetron | Corresponding risk with the intervention | |||||
| rolapitant + granisetron | 153 of 1000 | 176 of 1000 (135 to 230) | RR 1.15 | 1344 (1) | ⊕⊕⊝⊝ lowb | Rolapitant + granisetron may increase the risk of serious adverse events slightly when compared to granisetron |
| *Basis for the assumed risk is actual event rates reported for the main comparator summed across studies: 103 of 674 (10.3%) participants treated with granisetron experienced at least 1 SAE (granisetron was used in 1 study reporting the outcome; time frame for reporting safety data was not described). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| aEither dexamethasone or methylprednisolone was used in all treatment regimens. bDowngraded twice for very serious imprecision because 95% CIs cross unity, confidence intervals are wide, and information size is small. | ||||||
| Drug combinations | Treatment regimena | Abbreviation | Used in HECb setting | Used in MECc setting |
|---|---|---|---|---|
| NK₁ receptor antagonists and 5‐HT₃ receptor antagonists + corticosteroid | aprepitant with granisetron | apre_grani | X | X |
| aprepitant with ondansetron | apre_ondan | X | X | |
| aprepitant with palonosetron | apre_palo | X | X | |
| aprepitant with ramosetron | apre_ramo | X |
| |
| aprepitant with tropisetron | apre_tropi | X |
| |
| casopitant with ondansetron | caso_ondan | X | X | |
| fosaprepitant with granisetron | fosa_grani | X | X | |
| ezlopitant with granisetron | ezlo_grani | X |
| |
| fosaprepitant with ondansetron | fosa_ondan | X | X | |
| fosaprepitant with palonosetron | fosa_palo | X |
| |
| fosnetupitant with palonosetron | fosnetu_palo | X |
| |
| netupitant with palonosetron | netu_palo | X | X | |
| rolapitant with granisetron | rola_grani | X | X | |
| rolapitant with ondansetron | rola_ondan | X |
| |
| 5‐HT₃ receptor antagonists+ corticosteroid | azasetron | aza | X | X |
| dolasetron | dola |
| X | |
| granisetron | grani | X | X | |
| ondansetron | ondan | X | X | |
| palonosetron | palo | X | X | |
| ramosetron | ramo | X | X | |
| tropisetron | tropi | X | X | |
| aAll treatment regimens also include a corticosteroid. bHighly emetogenic chemotherapy. cModerately emetogenic chemotherapy. | ||||
| Outcome | Definition | Unit of outcome measurement | Referred to as/abbreviation | Prioritisation |
|---|---|---|---|---|
| Complete control of nausea | No nausea and no significant nausea, as defined on a study levela Assessed for:
| Binary; participants with complete control of nausea | No nausea | Overall phase prioritised for GRADE assessment |
| Complete control of vomiting | No vomiting and no use of rescue medications Assessed for:
| Binary; participants with complete control of vomiting | Complete response (CR) | Delayed and overall phases prioritised for GRADE assessment Overall phase chosen as most important efficacy outcome |
| Quality of life | No impairment in quality of life during active study period | Binary; participants with no impairment in quality of life | QoL | Prioritised for GRADE assessment |
| On‐study mortality | Deaths occurring from randomisation up to 30 days after the active study period | Binary; participants who died | OSM | Prioritised for GRADE assessment |
| Adverse events | As defined on a study level; during active study period | Binary; participants with at least 1 event | AEs | ‐ |
| Serious adverse events | As defined on a study level; during active study period | Binary; participants with at least 1 event | SAEs | Prioritised for GRADE assessment Chosen as most crucial safety outcome |
| Neutropenia | As defined on a study level; during active study period | Binary; participants with at least 1 event | ‐ | ‐ |
| Febrile neutropenia | As defined on a study level; during active study period | Binary; participants with at least 1 event | ‐ | ‐ |
| Infection | As defined on a study level; during active study period | Binary; participants with at least 1 event | ‐ | ‐ |
| Local reaction at infusion site | As defined on a study level; during active study period | Binary; participants with at least 1 event | ‐ | Prioritised for GRADE assessment |
| Hiccup | As defined on a study level; during active study period | Binary; participants with at least 1 event | ‐ | ‐ |
| aStandardised tools are typically used to assess degree of nausea and vomiting (Wood 2011). No nausea and no significant nausea were defined on a study level and typically refer to pre‐defined cutoffs, e.g. in Rapoport 2015 (a) or Schwartzberg 2015, nausea was assessed on a visual analogue scale (VAS; 0 to 100 mm; 0 = no nausea, 100 = severe nausea; < 5 mm = no nausea, < 25 mm = no significant nausea). No significant nausea is typically more subjective because of the wider range on the scale and is therefore less objective, especially in an open‐label study design. To increase comparability of studies and minimise biased results, we were therefore interested in patients with no nausea. | ||||
| Antiemetics for adults for prevention of nausea and vomiting caused by highly emetogenic chemotherapy | ||||||
| Patient or population: adult cancer patients at risk for CINV caused by highly emetogenic chemotherapy Settings: inpatient and outpatient care Intervention: neurokinin‐1 (NK₁) receptor antagonist and 5‐hydroxytryptamine‐3 (5‐HT₃) receptor antagonists + corticosteroid Comparison: aprepitant (NK₁) combined with granisetron (5‐HT₃) + corticosteroid Outcome: complete control of nausea during the overall phase (0 to 120 h of treatment with chemotherapy) RR < 1 indicates an advantage for the intervention Combinations of these interventions at any dose and by any route as mentioned above have been compared to one another in a full network | ||||||
| Interventions (corticosteroids included in all regimens)a | Illustrative comparative risks* (95% CI) | Risk ratio | No. of participants | Certainty of the evidence | Comments | |
| Assumed risk with aprepitant + granisetron | Corresponding risk with the intervention | |||||
| fosaprepitant + palonosetron | 896 of 1000 | NE of 1000 (NE to NE) | RR 1.46 | 14,588 (22) | ⊕⊕⊕⊝ moderateb | Fosaprepitant + palonosetron probably results in a large increase in complete control of nausea in the overall phase when compared with aprepitant + granisetron |
| fosnetupitant + palonosetron | 896 of 1000 | NE of 1000 (851 to NE) | RR 1.21 | 14,588 (22) | ⊕⊕⊕⊝ moderatec | Fosnetupitant + palonosetron probably increases complete control of nausea in the overall phase when compared with aprepitant + granisetron |
| ezlopitant + granisetron | 896 of 1000 | NE of 1000 (554 to NE) | RR 1.31 | 14,588 (22) | ⊕⊕⊝⊝ lowd | Ezlopitant + granisetron may increase complete control of nausea in the overall phase when compared with aprepitant + granisetron |
| rolapitant + granisetron | 896 of 1000 | NE of 1000 (860 to NE) | RR 1.12 | 14,588 (22) | ⊕⊕⊕⊝ moderatec | Rolapitant + granisetron probably increases complete control of nausea in the overall phase when compared with aprepitant + granisetron |
| fosaprepitant + granisetron | 896 of 1000 | 914 of 1000 (780 to NE) | RR 1.02 | 14,588 (22) | ⊕⊕⊕⊕ high | Fosaprepitant + granisetron has little to no effect on complete control of nausea in the overall phase when compared with aprepitant + granisetron |
| rolapitant + ondansetron | 896 of 1000 | 860 of 1000 (591 to NE) | RR 0.96 | 14,588 (22) | ⊕⊕⊕⊝ moderatec | Rolapitant + ondansetron probably decreases complete control of nausea slightly in the overall phase when compared with aprepitant + granisetron |
| netupitant + palonosetron | 896 of 1000 | 860 of 1000 (753 to 986) | RR 0.96 | 14,588 (22) | ⊕⊕⊕⊕ high | Netupitant + palonosetron has little to no effect on complete control of nausea in the overall phase when compared with aprepitant + granisetron |
| fosaprepitant + ondansetron | 896 of 1000 | 806 of 1000 (645 to NE) | RR 0.90 | 14,588 (22) | ⊕⊕⊕⊝ moderatec | Fosaprepitant + ondansetron probably decreases complete control of nausea slightly in the overall phase when compared with aprepitant + granisetron |
| aprepitant + ondansetron | 896 of 1000 | 780 of 1000 (609 to NE) | RR 0.87 | 14,588 (22) | ⊕⊕⊕⊝ moderatec | Aprepitant + ondansetron probably decreases complete control of nausea in the overall phase when compared with aprepitant + granisetron |
| casopitant + ondansetron | 896 of 1000 | 717 of 1000 (538 to 950) | RR 0.80 | 14,588 (22) | ⊕⊕⊕⊝ moderatec | Casopitant + ondansetron probably decreases complete control of nausea in the overall phase when compared with aprepitant + granisetron |
| *Basis for the assumed risk is actual event rates reported for the main comparator summed across studies: 412 of 460 (89.6%) participants treated with aprepitant + granisetron experienced no nausea during the overall phase (aprepitant + granisetron was used in 5 studies reporting the outcome). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| aEither dexamethasone or methylprednisolone was used in all treatment regimens. bDowngraded once for serious study limitations due to high risk of bias. cDowngraded once for serious imprecision because 95% CIs cross unity and confidence intervals are wide. dDowngraded twice for very serious imprecision because wide confidence intervals suggest both a potentially substantial harm and benefit for the intervention. | ||||||
| Antiemetics for adults for prevention of nausea and vomiting caused by highly emetogenic chemotherapy | ||||||
| Patient or population: adult cancer patients at risk for CINV caused by highly emetogenic chemotherapy Settings: inpatient and outpatient care Intervention: neurokinin‐1 (NK₁) receptor antagonist and 5‐hydroxytryptamine‐3 (5‐HT₃) receptor antagonists + corticosteroid Comparison: aprepitant (NK₁) combined with granisetron (5‐HT₃) + corticosteroid Outcome: complete control of vomiting during the delayed phase (24 to 120 h of treatment with chemotherapy) RR > 1 indicates an advantage for the intervention Combinations of these interventions at any dose and by any route as mentioned above have been compared to one another in a full network | ||||||
| Interventions (corticosteroids included in all regimens)a | Illustrative comparative risks* (95% CI) | Risk ratio | No. of participants | Certainty of the evidence | Comments | |
|---|---|---|---|---|---|---|
| Assumed risk aprepitant + granisetron | Corresponding risk with the intervention | |||||
| fosnetupitant + palonosetron | 694 of 1000 | 784 of 1000 (632 to 972) | RR 1.13 | 21,563 (37) | ⊕⊕⊝⊝ lowb,c | Fosnetupitant + palonosetron may increase complete control of vomiting in the delayed phase when compared to aprepitant + granisetron |
| fosaprepitant + palonosetron | 694 of 1000 | 736 of 1000 (632 to 854) | RR 1.06 | 21,563 (37) | ⊕⊝⊝⊝ very lowb,c,d | Evidence is very uncertain about the effect of fosaprepitant + palonosetron on complete control of vomiting in the delayed phase when compared to aprepitant + granisetron |
| aprepitant + palonosetron | 694 of 1000 | 722 of 1000 (652 to 798) | RR 1.04 | 21,563 (37) | ⊕⊝⊝⊝ very lowb,c,d | Evidence is very uncertain about the effect of aprepitant + palonosetron on complete control of vomiting in the delayed phase when compared to aprepitant + granisetron |
| aprepitant + ramosetron | 694 of 1000 | 722 of 1000 (625 to 1.21) | RR 1.04 | 21,563 (37) | ⊕⊝⊝⊝ very lowb,c,d | Evidence is very uncertain about the effect of aprepitant + ramosetron on complete control of vomiting in the delayed phase when compared to aprepitant + granisetron |
| fosaprepitant + granisetron | 694 of 1000 | 701 of 1000 (632 to 770) | RR 1.01 | 21,563 (37) | ⊕⊕⊕⊝ moderateb | Fosaprepitant + granisetron probably has little to no effect on complete control of vomiting in the delayed phase when compared to aprepitant + granisetron |
| netupitant + palonosetron | 694 of 1000 | 687 of 1000 (618 to 763) | RR 0.99 | 21,563 (37) | ⊕⊕⊕⊝ moderateb | Netupitant + palonosetron probably has little to no effect on complete control of vomiting in the delayed phase when compared to aprepitant + granisetron |
| aprepitant + ondansetron | 694 of 1000 | 645 of 1000 (576 to 722) | RR 0.93 | 21,563 (37) | ⊕⊝⊝⊝ very lowb,c,d | Evidence is very uncertain about the effect of aprepitant + ondansetron on complete control of vomiting in the delayed phase when compared to aprepitant + granisetron |
| rolapitant + granisetron | 694 of 1000 | 632 of 1000 (541 to 736) | RR 0.91 | 21,563 (37) | ⊕⊕⊝⊝ lowb,c | Rolapitant + granisetron may decrease complete control of vomiting in the delayed phase when compared to aprepitant + granisetron |
| fosaprepitant + ondansetron | 694 of 1000 | 632 of 1000 (548 to 722) | RR 0.91 | 21,563 (37) | ⊕⊕⊝⊝ lowb,c | Fosaprepitant + ondansetron may decrease complete control of vomiting in the delayed phase when compared to aprepitant + granisetron |
| casopitant + ondansetron | 694 of 1000 | 618 of 1000 (507 to 756) | RR 0.89 | 21,563 (37) | ⊕⊕⊝⊝ lowb,c | Casopitant + ondansetron may decrease complete control of vomiting in the delayed phase when compared to aprepitant + granisetron |
| rolapitant + ondansetron | 694 of 1000 | 583 of 1000 (437 to 784) | RR 0.84 (0.63 to 1.13) | 21,563 (37) | ⊕⊝⊝⊝ very lowb,c,d | Evidence is very uncertain about the effect of rolapitant + ondansetron on complete control of vomiting in the delayed phase when compared to aprepitant + granisetron |
| *Basis for the assumed risk is actual event rates reported for the main comparator summed across studies: 1537 of 2215 (69.4%) participants treated with aprepitant + granisetron achieved complete response during the delayed phase (aprepitant + granisetron was used in 10 studies reporting the outcome). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| aEither dexamethasone or methylprednisolone was used in all treatment regimens. bDowngraded once for moderate inconsistency. cDowngraded once for serious imprecision because 95% CIs cross unity and confidence intervals are wide. dDowngraded once for serious study limitations due to high risk of bias. | ||||||
| Antiemetics for adults for prevention of nausea and vomiting caused by highly emetogenic chemotherapy | ||||||
| Patient or population: adult cancer patients at risk for CINV caused by highly emetogenic chemotherapy Settings: inpatient and outpatient care Intervention: neurokinin‐1 (NK₁) receptor antagonist and 5‐hydroxytryptamine‐3 (5‐HT₃) receptor antagonists + corticosteroid Comparison: aprepitant (NK₁) combined with granisetron (5‐HT₃) + corticosteroid Outcome: no impairment in quality of life RR <1 indicates an advantage for the intervention Combinations of these interventions at any dose and by any route as mentioned above have been compared to one another in a full network | ||||||
| Interventions (corticosteroids included in all regimens)a | Illustrative comparative risks* (95% CI) | Risk ratio (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk with aprepitant + granisetron | Corresponding risk with the intervention | |||||
| rolapitant + ondansetron | 714 of 1000 | 893 of 1000 (486 to 1649) | RR 1.25 | 7894 (14) | ⊕⊝⊝⊝ very lowb,c | Evidence is uncertain about the effect of rolapitant + ondansetron on quality of life when compared to aprepitant + granisetron |
| netupitant + palonosetron | 714 of 1000 | 764 of 1000 (585 to 1007) | RR 1.07 | 7894 (14) | ⊕⊝⊝⊝ very lowb,d | Evidence is uncertain about the effect of netupitant + palonosetron on quality of life when compared to aprepitant + granisetron |
| casopitant + ondansetron | 714 of 1000 | 743 of 1000 (421 to 1307) | RR 1.04 | 7894 (14) | ⊕⊝⊝⊝ very lowb,c | Evidence is uncertain about the effect of casopitant + ondansetron on quality of life when compared to aprepitant + granisetron |
| rolapitant + granisetron | 714 of 1000 | 693 of 1000 (521 to 921) | RR 0.97 | 7894 (14) | ⊕⊝⊝⊝ very lowb,d | Evidence is uncertain about the effect of rolapitant + granisetron on quality of life when compared to aprepitant + granisetron |
| aprepitant + ondansetron | 714 of 1000 | 657 of 1000 (393 to 1100) | RR 0.92 | 7894 (14) | ⊕⊝⊝⊝ very lowb,c,e | Evidence is uncertain about the effect of aprepitant + ondansetron on quality of life when compared to aprepitant + granisetron |
| *Basis for the assumed risk is actual event rates reported for the main comparator summed across studies: 569 of 797 participants treated with aprepitant + granisetron experienced no impact on QoL (aprepitant + granisetron was used in 3 studies reporting the outcome, follow‐up on Day 6). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| aEither dexamethasone or methylprednisolone was used in all treatment regimens. bDowngraded twice for high inconsistency within the network. cDowngraded twice for very serious imprecision because 95% CIs cross unity and confidence intervals are very wide, suggesting high benefit for the comparator. dDowngraded once for serious imprecision because 95% CIs cross unity and confidence intervals are wide. eDowngraded once for serious study limitations due to high risk of bias. | ||||||
| Antiemetics for adults for prevention of nausea and vomiting caused by highly emetogenic chemotherapy | ||||||
| Patient or population: adult cancer patients at risk for CINV caused by highly emetogenic chemotherapy Settings: inpatient and outpatient care Intervention: neurokinin‐1 (NK₁) receptor antagonist and 5‐hydroxytryptamine‐3 (5‐HT₃) receptor antagonists + corticosteroid Comparison: aprepitant (NK₁) combined with granisetron (5‐HT₃) + corticosteroid Outcome: on‐study mortality RR < 1 indicates an advantage for the intervention Combinations of these interventions at any dose and by any route as mentioned above have been compared to one another in a full network | ||||||
| Interventions (corticosteroids included in all regimens)a | Illustrative comparative risks* (95% CI) | Risk ratio (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk with aprepitant + granisetron | Corresponding risk with the intervention | |||||
| netupitant + palonosetron | 8 of 1000 | 2 of 1000 (0 to 19) | RR 0.29 | 8030 (16) | ⊕⊕⊝⊝ lowb | Netupitant + palonosetron may have little to no effect on on‐study mortality when compared with aprepitant + granisetron |
| aprepitant + ondansetron | 8 of 1000 | 5 of 1000 (1 to 35) | RR 0.57 | 8030 (16) | ⊕⊕⊝⊝ lowb | Aprepitant + ondansetron may have little to no effect on on‐study mortality when compared with aprepitant + granisetron |
| casopitant + ondansetron | 8 of 1000 | 5 of 1000 (1 to 44) | RR 0.65 | 8030 (16) | ⊕⊕⊝⊝ lowb | Casopitant + ondansetron may have little to no effect on on‐study mortality when compared with aprepitant + granisetron |
| rolapitant + granisetron | 8 of 1000 | 5 of 1000 (1 to 33) | RR 0.66 | 8030 (16) | ⊕⊕⊝⊝ lowb | Rolapitant + granisetron may have little to no effect on on‐study mortality when compared with aprepitant + granisetron |
| rolapitant + ondansetron | 8 of 1000 | 12 of 1000 (1 to 216) | RR 1.56 | 8030 (16) | ⊕⊕⊝⊝ lowb | Rolapitant + ondansetron may have little to no effect on on‐study mortality when compared with aprepitant + granisetron |
| *Basis for the assumed risk is actual event rates reported for the main comparator summed across studies: 7 of 844 (0.08%) participants treated with aprepitant + granisetron died during the study (aprepitant + granisetron was used in 4 studies reporting the outcome, with follow‐up of up to 29 days). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| aEither dexamethasone or methylprednisolone was used in all treatment regimens. bDowngraded twice for very serious imprecision due to few events and very wide confidence intervals, suggesting potential benefit and harm for the comparator. | ||||||
| Antiemetics for adults for prevention of nausea and vomiting caused by moderately emetogenic chemotherapy | ||||||
| Patient or population: adult cancer patients at risk for CINV caused by moderately emetogenic chemotherapy Settings: inpatient and outpatient care Intervention:
Comparison: granisetron (5‐HT₃) + corticosteroid Outcome: complete control of nausea during the overall phase (0 to 120 h of treatment with chemotherapy) RR < 1 indicates an advantage for the intervention. Combinations of these interventions at any dose and by any route as mentioned above have been compared to one another in a full network | ||||||
| Interventions (corticosteroids included in all regimens)a | Illustrative comparative risks* (95% CI) | Risk ratio (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk with granisetron | Corresponding risk with the intervention | |||||
|
|
| |||||
| aprepitant + granisetron | 419 of 1000 | 570 of 1000 (365 to 897) | RR 1.36 | 1423 (2) | ⊕⊕⊝⊝ lowb | Aprepitant + granisetron may increase complete control of nausea in the overall phase when compared with granisetron |
| rolapitant + granisetron | 419 of 1000 | 453 of 1000 (402 to 511) | RR 1.08 | 1423 (2) | ⊕⊕⊝⊝ lowc | Rolapitant + granisetron may increase complete control of nausea in the overall phase slightly when compared with granisetron |
| *Basis for the assumed risk is actual event rates reported for the main comparator summed across studies: 298 of 712 (41.9%) participants treated with granisetron experienced no nausea during the overall phase (granisetron was used in 2 studies reporting the outcome). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| aEither dexamethasone or methylprednisolone was used in all treatment regimens. bDowngraded twice for very serious imprecision because 95% CIs cross unity, information size is small, and confidence intervals are very wide, suggesting benefit and harm for the comparator. cDowngraded twice for very serious imprecision because 95% CIs cross unity, confidence intervals are wide, and information size is small. | ||||||
| Antiemetics for adults for prevention of nausea and vomiting caused by moderately emetogenic chemotherapy | ||||||
| Patient or population: adult cancer patients at risk for CINV caused by moderately emetogenic chemotherapy Settings: inpatient and outpatient care Intervention:
Comparison: granisetron (5‐HT₃) + corticosteroid Outcome: complete control of vomiting during the delayed phase (24 to 120 h of treatment with chemotherapy) RR > 1 indicates an advantage for the intervention Combinations of these interventions at any dose and by any route as mentioned above have been compared to one another in a full network | ||||||
| Interventions (corticosteroids included in all regimens)a | Illustrative comparative risks* (95% CI) | Risk ratio | No. of participants | Certainty of the evidence | Comments | |
|---|---|---|---|---|---|---|
| Assumed risk with granisetron | Corresponding risk with the intervention | |||||
|
|
| |||||
| aprepitant + palonosetron | 641 of 1000 | 823 of 1000 (712 to 962) | RR 1.29 | 8421 (21) | ⊕⊕⊕⊝ moderateb | Aprepitant + palonosetron likely results in a large increase of complete control of vomiting during the delayed phase when compared to granisetron |
| rolapitant + granisetron | 641 of 1000 | 744 of 1000 (679 to 814) | RR 1.16 | 8421 (21) | ⊕⊕⊕⊕ high | Rolapitant + granisetron increases complete control of vomiting during the delayed phase when compared to granisetron |
| palonosetron | 641 of 1000 | 679 of 1000 (622 to 750) | RR 1.06 | 8421 (21) | ⊕⊕⊝⊝ lowb,c | Palonosetron may increase complete control of vomiting during the delayed phase slightly when compared to granisetron, but the evidence is uncertain |
| aprepitant + granisetron | 641 of 1000 | 667 of 1000 (564 to 788) | RR 1.04 | 8421 (21) | ⊕⊕⊝⊝ lowb,c | Palonosetron may or may not increase complete control of vomiting during the delayed phase slightly when compared to granisetron, but the evidence is uncertain |
| azasetron | 641 of 1000 | 647 of 1000 (494 to 846) | RR 1.01 | 8421 (21) | ⊕⊕⊝⊝ lowb,d | Azasetron may result in little to no difference in complete control of vomiting during the delayed phase slightly when compared to granisetron, but the evidence is uncertain |
| fosaprepitant + ondansetron | 641 of 1000 | 596 of 1000 (551 to 718) | RR 0.98 | 8421 (21) | ⊕⊕⊕⊝ moderateb | Fosaprepitant + ondansetron probably results in little to no difference in complete control of vomiting during the delayed phase slightly when compared to granisetron |
| aprepitant + ondansetron | 641 of 1000 | 596 of 1000 (526 to 679) | RR 0.93 | 8421 (21) | ⊕⊕⊝⊝ lowb,c | Aprepitant + ondansetron may decrease complete control of vomiting during the delayed phase slightly when compared to granisetron, but the evidence is uncertain |
| casopitant + ondansetron | 641 of 1000 | 570 of 1000 (506 to 647) | RR 0.89 | 8421 (21) | ⊕⊕⊝⊝ lowb,d | Casopitant + ondansetron may decrease complete control of vomiting during the delayed phase when compared to granisetron, but the evidence is uncertain |
| ondansetron | 641 of 1000 | 551 of 1000 (493 to 609) | RR 0.86 | 8421 (21) | ⊕⊕⊕⊝ moderateb | Ondansetron probably decreases complete control of vomiting during the delayed phase when compared to granisetron |
| *Basis for the assumed risk is actual event rates reported for the main comparator summed across studies: 953 of 1486 (64.1%) participants treated with granisetron achieved complete response during the delayed phase (granisetron was used in 7 studies reporting the outcome). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| aEither dexamethasone or methylprednisolone was used in all treatment regimens. bDowngraded once for serious study limitations due to high risk of bias. cDowngraded once for serious imprecision because 95% CIs cross unity and include potential advantages and disadvantages. dDowngraded once for serious imprecision due to wide confidence intervals. | ||||||
| Antiemetics for adults for prevention of nausea and vomiting caused by moderately emetogenic chemotherapy | ||||||
| Patient or population: adult cancer patients at risk for CINV caused by moderately emetogenic chemotherapy Settings: inpatient and outpatient care Intervention
Comparison: granisetron (5‐HT₃) + corticosteroid Outcome: no impairment in quality of life RR < 1 indicates an advantage for the intervention. Combinations of these interventions at any dose and by any route as mentioned above have been compared to one another in a full network. | ||||||
| Interventions (corticosteroids included in all regimens)a | Illustrative comparative risks* (95% CI) | Risk ratio (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk with granisetron | Corresponding risk with the intervention
| |||||
| rolapitant + granisetron | 674 of 1000 | 620 of 1000 (580 to 667) | RR 0.92 | 1212 (1) | ⊕⊕⊕⊝ moderateb | Rolapitant + granisetron probably decreases quality of life slightly when compared to granisetron |
| *Basis for the assumed risk is actual event rates reported for the main comparator summed across studies: 409 of 607 (67.4%) participants treated with granisetron experienced no impact on QoL (granisetron was used in 1 study reporting the outcome, follow‐up on Day 6). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| aEither dexamethasone or methylprednisolone was used in all treatment regimens. bDowngraded once for serious imprecision for the small sample size. | ||||||
| Antiemetics for adults for prevention of nausea and vomiting caused by moderately emetogenic chemotherapy | ||||||
| Patient or population: adult cancer patients at risk for CINV caused by moderately emetogenic chemotherapy Settings: inpatient and outpatient care Intervention
Comparison: granisetron (5‐HT₃) + corticosteroid Outcome: on‐study mortality RR < 1 indicates an advantage for the intervention. Combinations of these interventions at any dose and by any route as mentioned above have been compared to one another in a full network. | ||||||
| Interventions (corticosteroids included in all regimens)a | Illustrative comparative risks* (95% CI) | Risk ratio (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk with granisetron
| Corresponding risk with the intervention | |||||
| rolapitant + granisetron | 6 of 1000 | 18 of 1000 (6 to 56) | RR 3.00 | 1369 (1) | ⊕⊕⊝⊝ lowb | Rolapitant + granisetron may make little to no difference in on‐study mortality when compared to granisetron |
| *Basis for the assumed risk is actual event rates reported for the main comparator summed across studies: 4 of 685 (0.6%) participants treated with granisetron died during the study (granisetron was used in 1 study reporting the outcome, time frame for reporting safety data was not described). The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the risk ratio of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
| aEither dexamethasone or methylprednisolone was used in all treatment regimens. bDowngraded twice for very serious imprecision because 95% CIs cross unity and because of the small information size. | ||||||