Terapija hormonom rasta u osoba s talasemijom
Abstract
Background
Thalassaemia is a recessively‐inherited blood disorder that leads to anaemia of varying severity. In those affected by the more severe forms, regular blood transfusions are required which may lead to iron overload. Accumulated iron from blood transfusions may be deposited in vital organs including the heart, liver and endocrine organs such as the pituitary glands which can affect growth hormone production. Growth hormone deficiency is one of the factors that can lead to short stature, a common complication in people with thalassaemia. Growth hormone replacement therapy has been used in children with thalassaemia who have short stature and growth hormone deficiency.
Objectives
To assess the benefits and safety of growth hormone therapy in people with thalassaemia.
Search methods
We searched the Cochrane Haemoglobinopathies Trials Register, compiled from electronic database searches and handsearching of journals and conference abstract books. We also searched the reference lists of relevant articles, reviews and clinical trial registries. Our database and trial registry searches are current to 10 August 2017 and 08 August 2017, respectively.
Selection criteria
Randomised and quasi‐randomised controlled trials comparing the use of growth hormone therapy to placebo or standard care in people with thalassaemia of any type or severity.
Data collection and analysis
Two authors independently selected trials for inclusion. Data extraction and assessment of risk of bias were also conducted independently by two authors. The quality of the evidence was assessed using GRADE criteria.
Main results
One parallel trial conducted in Turkey was included. The trial recruited 20 children with homozygous beta thalassaemia who had short stature; 10 children received growth hormone therapy administered subcutaneously on a daily basis at a dose of 0.7 IU/kg per week and 10 children received standard care. The overall risk of bias in this trial was low except for the selection criteria and attrition bias which were unclear. The quality of the evidence for all major outcomes was moderate, the main concern was imprecision of the estimates due to the small sample size leading to wide confidence intervals. Final height (cm) (the review's pre‐specified primary outcome) and change in height were not assessed in the included trial. The trial reported no clear difference between groups in height standard deviation (SD) score after one year, mean difference (MD) ‐0.09 (95% confidence interval (CI) ‐0.33 to 0.15 (moderate quality evidence). However, modest improvements appeared to be observed in the following key outcomes in children receiving growth hormone therapy compared to control (moderate quality evidence): change between baseline and final visit in height SD score, MD 0.26 (95% CI 0.13 to 0.39); height velocity, MD 2.28 cm/year (95% CI 1.76 to 2.80); height velocity SD score, MD 3.31 (95% CI 2.43 to 4.19); and change in height velocity SD score between baseline and final visit, MD 3.41 (95% CI 2.45 to 4.37). No adverse effects of treatment were reported in either group; however, while there was no clear difference between groups in the oral glucose tolerance test at one year, fasting blood glucose was significantly higher in the growth hormone therapy group compared to control, although both results were still within the normal range, MD 6.67 mg/dL (95% CI 2.66 to 10.68). There were no data beyond the one‐year trial period.
Authors' conclusions
A small single trial contributed evidence of moderate quality that the use of growth hormone for a year may improve height velocity of children with thalassaemia although height SD score in the treatment group was similar to the control group. There are no randomised controlled trials in adults or trials that address the use of growth hormone therapy over a longer period and assess its effect on final height and quality of life. The optimal dosage of growth hormone and the ideal time to start this therapy remain uncertain. Large well‐designed randomised controlled trials over a longer period with sufficient duration of follow up are needed.
PICO
Laički sažetak
Terapija hormonom rasta u osoba s talasemijom
Istraživačko pitanje
Ovaj Cochraneov sustavni pregled literature analizirao je dokaze iz istraživanja o liječenju osoba s talasemijom pomoću hormona rasta.
Dosadašnje spoznaje
Talasemija je nasljedni krvni poremećaj koji uzrokuje anemiju različite težine. Osobe koje imaju teže oblike talasemije trebaju redovite transfuzije krvi od ranog djetinjstva što rezultira viškom željeza koje se nakuplja u vitalnim organima poput srca, jetre i žlijezda koje luče hormone (endokrine žlijezde). Jedna od tih žlijezda je žlijezda hipofiza koja luči hormon rasta koji regulira rast i funkciju ljudskog tijela. Ako je proizvodnja hormona rasta narušena taloženjem željeza, pogođena djeca ne mogu narasti u punom kapacitetu.
Nizak rast je vrlo uobičajen među ljudima s talasemijom. Može biti uzrokovan različitih čimbenicima, uključujući probleme s hormonom rasta ili drugim hormonima, nedovoljne transfuzije krvi ili lošu prehranu. Sintetički hormon rasta je jedan od načina liječenja niskog rasta u talasemiji, osobito u djece s poremećajem stvaranja hormona rasta. To obično uključuje injekcije hormona rasta koje se primjenjuju pod kožu (supkutano) nekoliko puta tjedno tijekom određenog vremenskog razdoblja. Međutim, nejasno je da li primjena sintetičkog hormona rasta osobama s talasemijom pruža bilo kakve trajne ili jasne prednosti.
Datum pretraživanja literature
Dokazi se temelje na literaturi objavljenoj do 8. kolovoza 2017.
Obilježja uključenih istraživanja
Pronađena je samo jedna studija. Studija je uključila 20 djece s beta talasemijom koji su znatno niži nego što bi trebali biti na temelju grafikona rasta. Deset djece nasumično je odabrano za svakodnevno liječenje hormonom rasta uz svoje uobičajeno (standardni) liječenje, a ostalo desetoro djece je primalo samo njihovo uobičajeno liječenje. Visina djece i krvne pretrage su praćeni svaka tri mjeseca. Istraživanje je provedeno tijekom jedne godine.
Ključni rezultati
Brzina rasta je stopa porasta djeteta, a računa se mjerenjem razlike visina u određenom periodu vremena (obično kao godišnji porast izražen u cm). U ovom pregledu, djeca koja su primala hormon rasta za godinu dana su imala veću brzinu rasta (u prosjeku godišnje 2.28 cm više) u odnosu na onu koja nisu primala hormon rasta. Drugim riječima, djeca na tretmanu hormonom rasta rasla su malo brže u odnosu na onu koja ga nisu primala. Visina djeteta također se može pratiti na temelju standardnih grafikona populacije. Koristeći ovo mjerenje, na kraju godine, djeca liječena s hormonom rasta imala su slične rezultate kao i ona koja nisu primala hormon rasta. Nitko od djece nije imao nuspojave. Iako između skupina nakon godinu dana nije bilo jasne razlike u testovima tolerancije oralne glukoze, djeca koja su primala hormon rasta imala su veće razine glukoze u krvi natašte, ali još uvijek unutar normalnih granica. Istraživanje nema podatke o promjeni visine nakon jednogodišnjeg razdoblja, stoga nije poznato da li je hormon rasta na bilo koji način utjecao na konačnu visinu.
Nisu rađena istraživanja koja bi pratila primjenu hormona rasta u osoba sa talasemijom tijekom dužeg razdoblja, pri različitim dozama ili u različitim dobnim grupama; niti je bilo istraživanja učinka terapije hormonom rasta na konačnu visinu ili kvalitetu života.
Kvaliteta dokaza
Sve u svemu, smatra se da su dokazi ishoda opisanog istraživanja (kratkoročni rast i nuspojave) umjerenog kvaliteta s najvećom zamjerkom na mali broj sudionika.
Zaključak
Temeljem jednog malog istraživanja, umjerene kvalitete dokaza, primjena hormona rasta može malo poboljšati neke mjere rasta. Međutim, nema podataka o utjecaju na konačnu visinu ili kvalitetu života. Potrebno je više istraživanja da bi se donio jasan zaključak o korisnim učincima i rizicima primjene hormona rasta u osoba s talasemijom.
Authors' conclusions
Summary of findings
| Growth hormone for people with thalassaemia | ||||||
| Patient or population: people with thalassaemia (any age) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with control | Risk with growth hormone | |||||
| Final height and change in height | The included trial did not assess either of these outcomes. | |||||
| Adverse effects Oral glucose tolerance test (mg/dL) (at one year) | The mean oral glucose tolerance test was 336.56 mg/dL. | MD 0.03 lower (17.45 lower to 17.39 higher). | ‐ | 20 | ⊕⊕⊕⊝ | Fasting blood glucose levels in the growth hormone group were significantly higher than in the control group but both were still within the normal range. |
| Height SDS | The mean height SDS was ‐2.85. | MD 0.09 lower | ‐ | 20 | ⊕⊕⊕⊝ | |
| Change in height SDS (difference between baseline and final visit at one year) | The change in mean height SDS was ‐0.05. | MD 0.26 higher | ‐ | 20 | ⊕⊕⊕⊝ | |
| Height velocity (cm/year) | The mean height velocity was 3.99 cm/year. | MD 2.28 higher | ‐ | 20 | ⊕⊕⊕⊝ | |
| Height velocity SDS | The mean height velocity SDS was ‐1.56. | MD 3.31 higher | ‐ | 20 | ⊕⊕⊕⊝ | |
| Change in height velocity SDS (difference between baseline and final visit at one year) | The change in mean height velocity SDS was 1.76. | MD 3.41 higher | ‐ | 20 | ⊕⊕⊕⊝ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Data contributed by a single trial with small sample and 95% CI is wide. | ||||||
Background
Please refer to the glossary for an explanation of terms (Appendix 1).
Description of the condition
The thalassemias are a group of autosomally recessive inherited conditions characterised by the absence or reduced synthesis of one of the two polypeptide chains (alpha (α) or beta (β)) that form the normal adult human haemoglobin molecule (haemoglobin A, α₂β₂) leading to reduced haemoglobin in red cells and anaemia (Higgs 2008). The thalassaemia syndromes are named according to the globin chain affected or the abnormal haemoglobin involved; mutations of the α globin gene cause α thalassaemia, while the β globin gene defects give rise to β thalassaemia (Peters 2012). It has been estimated that about 1.5% of the global population (80 to 90 million people) are carriers of β thalassaemia, with approximately 60,000 symptomatic individuals born annually (Galanello 2010). Similarly, α thalassaemia occurs at high frequencies throughout tropical and subtropical regions of the world (Harteveld 2010).
The thalassemias can be classified according to phenotype (clinical severity) or genotype (type of mutation) in which clinical presentation can be highly variable ranging from asymptomatic carriers to transfusion‐dependent thalassaemia (Peters 2012). The more clinically severe forms of thalassaemia affect multiple systems, where the manifestations are either caused by the condition itself or by the complications from various treatments such as frequent blood transfusions. These individuals usually present within the first two years of life with severe anaemia and if untreated or poorly transfused, they suffer from growth retardation, poor musculature, hepatosplenomegaly, leg ulcers, development of masses from extramedullary hematopoiesis and skeletal changes due to bone marrow expansion (Galanello 2010). Regular blood transfusions will improve growth and development, reduce hepatosplenomegaly as well as bone deformities, but can lead to complications of iron overload such as cardiomyopathy, liver cirrhosis and endocrinopathies (Peters 2012).
An association between iron overload and target‐endocrine gland toxicity has been established from biological, clinical and radiological studies in people with β thalassaemia (Belhoul 2012; Taher 2009; Wood 2011). These endocrine complications may manifest as short stature, hypogonadism, delayed puberty or secondary amenorrhoea, impaired glucose tolerance or diabetes, hypothyroidism, hypoparathyroidism or osteopenia or osteoporosis (Toumba 2007). Growth hormone deficiency (GHD) has been recognised as one of the endocrine complications among this population as the anterior pituitary is particularly sensitive to free radical oxidative stress secondary to iron overload (De Sanctis 2002).
Studies in many people with thalassaemia who are of short stature have shown dysfunction of the growth hormone releasing hormone‐growth hormone‐insulin‐like growth factor 1 (GHRH‐GH‐IGF‐1) axis. Growth hormone reserve, which is defined biochemically by the peak serum concentration after stimulation with a known secretagogue, was reported to be normal or reduced with a wide variability (8% to 80%) in people with thalassaemia who are of short stature due to defects in the pituitary gland or hypothalamus or both (Delvecchio 2010). The reported prevalence of GHD or insulin‐like growth factor 1 (IGF‐I) deficiency, or both, in adults with thalassaemia varies from 8% to 44 % (Soliman 2013), whilst a study of 94 adults with thalassaemia revealed that 21 (22.3%) had severe GHD and 18 (19.1%) had partial GHD (Scacchi 2007). It is estimated that between 8% and 50% of short children with thalassaemia have GHD (Scacchi 2007). In this population, GH neurosecretory dysfunction (in which pulsatile GH secretion is abnormal despite normal GH response to provocative stimulation test) has also been described (Chatterjee 1993; Katzos 1995; Roth 1997; Shehadeh 1990).
The major concerns with GHD in children surround their growth and height attainment; and among people with thalassaemia, GHD has been recognised as a cause for growth and maturational delay (Soliman 2015). In adults, GHD has been associated with: an adverse lipid profile; increased cardiovascular and cerebrovascular events; and decreased bone mineral density, muscle strength, exercise capacity, cognitive function and quality of life (QoL) (de Boer 1995; Rosen 1990). Although predominantly seen in those with thalassaemia major, GHD may affect those with thalassaemia intermedia where it manifests with a less severe form of anaemia (Karamifar 2006).
Short stature is highly prevalent among individuals with thalassaemia, with a rate ranging from 8% to 75% (Low 2005); and it is frequently disproportionate with a reduction in the upper to lower segment ratio. Short stature usually becomes noticeable from the age of five to six years in males and from eight years in females; and in those who are well‐transfused and well‐chelated, it may be observed after the first 10 years of life. With the delay or attenuation of the pubertal growth spurt, growth failure becomes more evident leading to a reduction in final height (Delvecchio 2010). In this population, growth plate fusion is usually delayed until the end of the second decade of life.
The pathogenesis of short stature in thalassaemia is multifactorial and may be attributed to chronic anaemia and hypoxia, iron overload, chronic liver disease, nutritional deficiency, bone dysplasia due to desferrioxamine toxicity and endocrinopathies (hypogonadism, delayed puberty, hypothyroidism and GH‐IGF‐1 axis deregulations) (Anita 2003; Delvecchio 2010; Noetzli 2012; Soliman 1999). In an international multicentre study conducted in a large series of children and adolescents with β thalassaemia major, short stature was present in 31.1% of males and 30.5% of females, higher than the prevalence of biochemically‐proven GHD, as seen in 7.9% of males and 8.8% of females (De Sanctis 2004). Growth issues in people with thalassaemia should be addressed with measures such as ensuring optimal transfusion and chelation therapies, treating nutritional deficiencies and prompt diagnosis and treatment of endocrinopathies such as hypothyroidism, abnormal glucose homeostasis, pubertal delay and GHD (Cappelini 2014). As many of these aspects (such as proper transfusion regimens and chelation therapies) have been addressed, attempts to improve linear growth has increasingly included the used of GH replacement therapy.
Description of the intervention
Growth hormone (also known as somatotropin or somatropin) is a polypeptide hormone synthesized, stored, and secreted by somatotropic cells in the anterior pituitary gland. Until 1985, GH for replacement therapy in children with GHD was extracted from cadaveric pituitary glands, but due to the possibility of transmitting Creutzfeldt‐Jakob disease, this form of GH was withdrawn worldwide and replaced with recombinant human GH (Pfaffle 2015). Recombinant DNA technology offers a safe and economical method for the production of recombinant human GH in various heterologous systems, without the risk of transfer of human pathogens. Advancement in recombinant DNA technology has allowed for the expression of proteins in host cells, such as Escherichia coli (E coli). This process involves the insertion of the human GH gene into plasmids of E coli bacteria, culture of the recombinant bacterial cells, followed by, extraction of the human GH produced by these bacteria from the extracellular media (Ghasemi 2004; Rezaei 2012). Recombinant human GH is identical to the natural structure and function of human GH. For the purpose of this review, GH therapy will thence refer to recombinant human GH.
Recombinant human GH is licensed for use in people with short stature associated with GHD, Turner syndrome, Prader‐Willi syndrome, chronic renal insufficiency, short stature homeobox‐containing gene deficiency and being born small for gestational age in which statistically significantly larger height standard deviation score (SDS) values were reported (Takeda 2010). Recombinant human GH is measured in international units (IU) and mg. The dose commonly used in children for treating of GHD is 0.1 IU/kg/day, administered via subcutaneous injection at bedtime (Arcasoy 1999; Rappaport 1997; Saggese 2001; Wu 2003). Bedtime dosing is designed to mimic the metabolic effects of GH secretion in normal individuals as closely as possible (Jørgensen 1990).
A literature review of a number of studies evaluated the efficacy of recombinant human GH in children and adolescents with thalassaemia major, but some of the studies were noted to be non‐homogenous or had relatively small sample sizes (Delvecchio 2010). The review authors propose that individuals with thalassaemia may benefit from a short course of treatment with recombinant human GH, while more prolonged treatment should be reserved for adolescents with psychological problems due to short stature (Delvecchio 2010). In individuals with thalassaemia, GH therapy may improve bone mineralization (Soliman 1998). Evidence on various management options to prevent or reduce the severity of thalassaemia‐related bone disease has been synthesised in a further Cochrane Review (Bhardwaj 2013).
There are concerns on treating individuals with GH therapy due to the risk of developing diabetes mellitus (Yuen 2013), for which people with thalassaemia are already at risk, as well as the potential effects on lipid metabolism (Low 2005). Contrasting results have been reported for the long‐term adverse events of GH therapy such as increased mortality from cardiovascular events, bone tumour or haemorrhagic stroke (Carel 2012; Poidvin 2014; Savendahl 2012). Lastly, the cost of GH therapy should be taken into account where its cost‐effectiveness has been estimated at a willingness‐to‐pay threshold of GBP 20,000 to 30,000 per quality‐adjusted life‐year (QALY) gained (Takeda 2010).
How the intervention might work
The biological effects of GH on somatic growth and tissue regeneration have been inextricably linked with the actions of IGF‐1, where their interdependent roles control normal growth during childhood and maintain tissue integrity during aging (Woelfle 2003). Both GH and IGF‐1 are fundamental in achieving a normal longitudinal bone growth and mass during the postnatal period and, in association with sex steroids, play a major role in bone growth and development (Bouillon 2000).
In general, the goals of GH therapy differ somewhat in children and adults. In children the goals are to promote linear growth, restore body composition, and improve QoL; whereas in adults, the goals are to restore normal body composition, improve muscle and cardiac function, normalize serum lipid concentrations and improve QoL (Vance 1999). For individuals with thalassaemia, GH therapy has mainly been used to address their poor growth. With improvement in transfusion and chelation therapy nowadays, GH therapy has an increasing role in addressing short stature amongst people with thalassaemia as some of these individuals have reduced GH reserve or GH neurosecretory dysfunction. Therapy to replace GH aims to improve their growth velocity and eventually to improve their final height.
Administration of recombinant human GH may aim to address a state of deficiency or to boost an already 'normal' level. It should be noted that some children with thalassaemia major had a significant, but lower IGF‐1 response after GH administration, compared with the IGF‐1 response in children with GHD (Soliman 1998a) and supra‐physiological doses of GH may be required to obtain therapeutic response due to partial GH insensitivity (Soliman 2009). In children with β thalassaemia major, growth failure in the presence of normal GH reserve and low serum IGF‐1 concentrations suggests a state of partial GH insensitivity at the post‐receptor level. This partial GH insensitivity can be overcome by supra‐physiological doses of exogenous GH given at 0.14 IU/kg/day subcutaneously. Prolonged treatment with GH, however, may not improve final height (Cavallo 2005; Katzos 2000; Low 1995; Low 1998) and supra‐physiological doses of GH might increase the risk of inducing diabetes and hypertension (Soliman 1996; Soliman 1998a). The overall safety profile of recombinant human GH continues to be favourable, but careful monitoring for the presence of certain conditions such as malignancy, is pertinent both during and after therapy (Bell 2010).
Why it is important to do this review
The improvement in therapeutic approaches such as frequent blood transfusion and chelation therapies has greatly increased the life expectancy of these individuals, hence researchers are focusing on the impact of endocrinological alterations on QoL (Scacchi 2007). Short stature has been associated with poorer QoL whereas increase in height standard deviation score (SDS) due to GH treatment has been associated with an increase in QoL (Geisler 2012). As such, GH therapy may have an increasing role to play in improving height as well as other parameters that translate into the overall well‐being and improved QoL in people with thalassaemia. As there is no systematic review to date, we aim to provide an overall picture of the benefits and harms of GH therapy in people with thalassaemia to inform practice and research.
Objectives
To assess the risks and benefits of GH therapy in children and adults with thalassaemia.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs) and quasi‐RCTs where the groups should be comparable at baseline. Cross‐over trials will be excluded.
Types of participants
We included trials of people (of any age) with thalassaemia (major and intermedia). All types of thalassaemia (including α thalassaemia and β thalassaemia) are eligible for inclusion. The participants' care might be in any setting (including primary, secondary or tertiary care) and they might or might not have received regular blood transfusions.
Types of interventions
Trials were eligible for inclusion if they compared the use of biosynthetic human GH (somatropin), marketed under any brand name, to either placebo or no treatment for a minimum of six months. Apart from the difference in the active intervention of interest, all participants in the same trial should have received a standardised management and follow‐up plan for thalassaemia and related problems.
Types of outcome measures
Primary outcomes
-
Final height
-
height (cm)
-
height SDS
-
height SDS relative to expected height based on mid‐parental height
-
-
Adverse effects (such as but not limited to benign intracranial hypertension, slipped capital femoral epiphyses, effects on glucose metabolism in both non‐diabetic as well as diabetic patients and incidence of malignant disease, haemorrhagic stroke or cardiovascular events)
-
Satisfaction (measured by validated questionnaires e.g. (Leiberman 1993; Rubin 2011))
-
participants
-
parents or caregivers
-
Secondary outcomes
-
Short‐term growth
-
change in height (cm) over trial period
-
change in height SDS over trial period
-
height velocity (expressed as change in height over treatment period, measured during the trial period, or height velocity SDS)
-
-
Bone mineralization (bone density scores) including final scores and change in scores over the trial period
-
QoL (measured using a validated scale or specific measures of physical or social function) where final QoL and change in QoL over the trial period were included
-
Costs of care per year (costs per individual per year)
-
Serum insulin‐like growth factor (IGF‐1) (post hoc change)
We had planned to accept outcomes not defined a priori but which were considered clinically important after discussion among review team members, with justifications stated under 'Differences between protocol and review'. This did not occur.
Search methods for identification of studies
We searched for all relevant published and unpublished trials without restrictions on language, year or publication status.
Electronic searches
We identified relevant studies from the Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register using the terms: (thalassaemia OR (haemoglobinopathies AND general)) AND growth.
The Haemoglobinopathies Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and weekly searches of MEDLINE. Unpublished work is identified by searching the abstract books of five major conferences: the European Haematology Association conference; the American Society of Hematology conference; the British Society for Haematology Annual Scientific Meeting; the Caribbean Health Research Council Meetings; and the National Sickle Cell Disease Program Annual Meeting. For full details of all searching activities for the register, please see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group website.
Date of the most recent search: 10 August 2017.
We also searched the following trial registries:
1. ISRCTN registry (www.isrctn.com; searched 08 August 2017);
2. US National Institutes of Health Ongoing Trials Register Clinicaltrials.gov (www.clinicaltrials.gov; searched 08 August 2017);
3. WHO International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch; searched 08 August 2017).
For details of our search strategies, please see the appendices (Appendix 2).
Searching other resources
Reference lists
We checked the bibliographies of included studies for further references to relevant trials, although none were identified. We did not undertake any grey literature searches.
Data collection and analysis
We followed standard Cochrane methods as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a).
Selection of studies
Following the recommendations in the Cochrane Handbook for Systematic Reviews of Interventions, we undertook screening and selection in two stages (Higgins 2011b). In stage one, three pairs of review authors (Team 1: MKT, PM; Team 2: CFN, AR; Team 3: SLT, NML) each screened one third of the combined, de‐duplicated records retrieved from the first round of the searches. Within each pair the authors undertook the screening independently by inspecting the titles and abstracts and excluding trials that were clearly not relevant, leaving a shortlist of articles for further assessment. In stage two, two authors (CFN and PM) further assessed the shortlist of trials to determine whether these should be included in the meta‐analysis, excluded or placed under 'Studies awaiting assessment'. We extracted trial‐related information on a dedicated data collection form, and recorded the reasons for excluding any trials. The two authors (CFN and PM) discussed any differences in their decisions in each of the two stages mentioned above leading to a consensus; with the involvement of an arbiter if necessary (Stage 1: JYH; Stage 2: MKT).
We planned to accept published and unpublished trials, both in full article and abstract forms, as long as there was sufficient information in the report to enable a meaningful risk of bias assessment and the extraction of outcome data. Both identified trials were published as full papers. If required, we would have contacted the authors of relevant trials to obtain further information if there was critical information missing from the published report; however, it was not necessary to do this for the single included trial. Had we not been able to obtain sufficient information from a potentially eligible trial, we would have listed the trial as 'Awaiting classification' until sufficient information was available.
Data extraction and management
We followed the recommendations in the relevant chapter of the Cochrane Handbook for Systematic Reviews of Interventions with regards to data extraction and management (Higgins 2011b). Two review authors (CFN and SLT) independently extracted and coded all data for the short‐listed trials using a data collection form designed for this review.
We extracted the following data:
-
characteristics of the trial: methods (randomised, quasi‐randomised, non‐randomised); setting (hospital or community); year of publication; characteristics of population; intervention; comparator treatments; and outcome measures. If a trial was found not to fulfil our inclusion criteria, we stated the reason for exclusion (and later transcribed the reasons for exclusion onto the 'Characteristics of excluded studies' table in the review) and did not extract further data;
-
risk of bias profile as detailed below in Assessment of risk of bias in included studies;
-
outcome data: we extracted outcomes reported in the single included trial, and recorded the outcome data in a way suitable for future meta‐analysis (which was not conducted for this review as only one trial was included). We planned to document any trials that did not report key outcomes that were expected to be reported, namely, the primary outcomes as defined in our review, and would have assigned the trial as having high risk of bias in selective outcome reporting (see Assessment of reporting biases for detail). We would have contacted the trial authors to request for further data if necessary.
Had relevant data been available, we would have separated outcomes measured at different time points, for instance, height or height velocity (continuous) or adverse events (dichotomous) measured at three months or less (short term), three to six months (medium term) or beyond six months (long term). We would have reported the results of the trials individually if pooled analyses were not possible.
We resolved any disagreements by discussion with the aim of reaching a consensus, or, if this was not possible, we involved an arbiter (MKT). If any of the review authors had been investigators on a potentially relevant trial, they would not extract data from that trial.
Assessment of risk of bias in included studies
Two authors (SLT and NML) independently assessed the included trial for risk of bias according to six major criteria as stated in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c).
-
Sequence generation
-
Allocation concealment
-
Blinding (assessed separately for clinical and laboratory outcomes if both types of outcomes were available) for:
-
participants and personnel, and
-
outcome assessor
-
-
Incomplete outcome data
-
Selective outcome reporting
-
Other issues (e.g. extreme baseline imbalance)
A detailed description on each of the risk of bias criteria is provided in the appendices (Appendix 3).
We accorded a judgment of low risk, high risk or unclear risk, with justifications on each criterion and completed a risk of bias table for the included trial based on the information from the article. We discussed any disagreement among the review authors and involved a third author (CFN) if necessary. We also presented our assessment of the risk of bias using the risk of bias graph and risk of bias summary.
Measures of treatment effect
We reported the outcome estimates following the recommendations in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011).
For dichotomous or categorical data (presence of adverse events), we planned to report primarily using relative risk (RR), as well as risk difference (RD) with number needed to treat to benefit (NNTB) or number needed to harm (NNTH) regardless of statistical significance, in accordance with the recommendations in the Cochrane Handbook for Systematic Reviews of Interventions, Chapter 9 and Chapter 12 (Higgins 2011d; Schünemann 2011b). We would have used 95% confidence intervals (CIs) for adverse effects, unless there were more than five adverse effects, in which case we would have used 99% CIs. If the adverse events were reported as the total number of adverse events of a particular type, rather than the number of participants with the adverse events, we would have converted them to the number of participants with the adverse events, and reported them as dichotomous or categorical data as mentioned above. If the information reported were insufficient to us to convert to dichotomous data, we would have contacted the author for further information.
For continuous data such as height (including all units of measurements), short‐term growth, bone mineralisation, QoL, participant or parental satisfaction, we used the mean difference (MD) with 95% CIs. We would have used the standardised MD (SMD) with 95% CIs if several included trials had used different measurement scales. Due to the highly skewed distribution of cost variables, as a measure of variability, we would have reported the distribution of costs per patient per year, instead of SDs and considered both direct costs (medical and non‐medical costs) and indirect costs under this section.
Unit of analysis issues
For trials with multiple treatment groups (e.g. different dosages or regimens of GH), we planned to adjust the data by following the methods stated in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011e). Specifically, if only two arms were relevant to this review, we would only include the relevant arms. If there were more than two relevant arms, we would set up separate pair‐wise comparisons, for example, GH regimen A versus GH regimen B (Comparison 1), GH regimen A versus control (Comparison 2) and GH regimen B versus control (Comparison 3). In such cases, number of participants in the control group will not be totalled up to avoid multiple‐counting.
We did not plan to include cross‐over trials, as we anticipated potential issues caused by the period effect, namely, differential impacts of growth hormone administration at different period of the illness or study.
We did not encounter any cluster RCTs. Had we included any cluster‐RCTs, we would have assessed whether any adjustment had been made for the effects of clustering using the appropriate analysis methods such as the Generalized Estimating Equation (GEE) modelling; for example, those in which assignment of intervention and control group is made at the hospital or clinic, rather than the individual level. If no adjustment had been made, we would have performed adjustment by calculating the design effect based on a fairly large assumed intra‐cluster correlation (ICC) of 0.10, which has been shown to be a generally realistic estimate from trials on implementation research (Campbell 2001). If the unit of analysis had not been stated in the trial, we would have inspected the width of the standard error (SE) or 95% CI of the estimated treatment effects. If we found an inappropriately small SE or a narrow 95% CI, we would have asked the authors of the trial to provide information on the unit of analysis.
Dealing with missing data
We followed the recommendations in the Cochrane Handbook for Systematic Reviews of Interventions when dealing with possible missing data (Higgins 2011e). Although there were no dropouts reported in the included trial, for future reviews, we will specifically determine the dropout rates from each trial, and assess the number of participants who are initially randomised against the total number analysed to determine whether the intention‐to‐treat principle is followed. Trials with a substantial dropout rate with no reasonable explanation or markedly different dropout rates between the assigned groups, will be assigned as having a high risk of bias for the criterion of incomplete outcome data. If we consider the extent of missing data to be critical to the final estimates in any future meta‐analysis, we will contact the authors of the individual trials to request further information.
If necessary, we would have performed a sensitivity analysis to assess how the overall results were affected with and without the inclusion of trials with a high risk of attrition bias from incomplete outcome data.
For this initial version of the review, we contacted the corresponding author with regards to additional information about the trial and the participants. We received a response stating that the lead author is unable to provide data beyond what was available from the published article.
Assessment of heterogeneity
If we are able to include more than one trial in future, we will assessed heterogeneity by visually inspecting the forest plots and using the I² statistic to quantify the proportion of inconsistency in the results among the included trials (Higgins 2003). We will use the following values of the I² statistic as cut‐offs as our working guide for reporting heterogeneity where any I² value of 50% or more indicates substantial heterogeneity:
-
below 30% ‐ might not be important;
-
30% to 60% ‐ moderate heterogeneity;
-
50% to 90% ‐ substantial heterogeneity; and
-
75% to 100% ‐ considerable heterogeneity.
Assessment of reporting biases
We matched the pre‐specified outcomes of the trial as published in the trial registry protocol or the methods section of the trial report (if the former was not available) with the outcomes reported in the results. Additionally, we assessed whether certain key outcomes, notably, our primary outcome of height, was included in the trials. For trials that failed to report all pre‐specified outcomes or key outcomes, we would accord them a high risk of bias in the domain of selective outcome reporting.
For publication bias, we planned to use the funnel plot as a screening tool if there were sufficient number of trials (more than 10) reporting the same outcome, as the funnel plot is only useful with a minimum number of 10 included trials. If there was a significant asymmetry of the funnel plot suggesting possible publication bias, we would include a statement in our results with a corresponding note of caution in our discussion, i.e. asymmetric funnel plots do not necessarily indicate a publication bias (Sterne 2011).
Data synthesis
We presented and analysed the data from the single included trial which were relevant to the primary and secondary outcomes of this review by using the Review Manager software (RevMan 2014). If more than one trial is included in the future, we plan to perform meta‐analyses using the Review Manager software with a fixed‐effect model. However, if there is substantial heterogeneity, as indicated by an I² statistic value of 50% or above with no plausible explanation for the observed heterogeneity to enable any subgroup analysis, and if we still consider meta‐analysis to be appropriate, we will combine the data using a random‐effects analysis as the primary analysis, which adjusts for the degree of heterogeneity in arriving at the SEs of the trial‐specific estimates. If, however, we are unable to undertake a meta‐analysis, we will report the findings of the included trials narratively (Deeks 2011).
Subgroup analysis and investigation of heterogeneity
We planned to perform the following subgroup analyses if data had been available:
-
participants with thalassaemia major versus thalassaemia intermedia;
-
participants in different age groups (children up to 18 years of age versus adults above 18 years of age);
-
participants with documented GH deficiency versus those without;
-
participants with documented hypogonadotrophic hypogonadism versus those without.
We planned to undertake these subgroup analyses a priori for both the primary and secondary outcomes regardless of the presence or absence of heterogeneity to assess the effects of GH treatment in different groups of participants. In future updates of the review, if there are inadequate data to group the participants and allow subgroup analysis, we will contacted the author for more information ‐ if the information remained inadequate, we will include a corresponding explanation on our inability to perform subgroup analysis due to missing information.
Sensitivity analysis
As recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011), we planned to perform sensitivity analyses for the primary outcomes (and any secondary outcomes with a sufficient number of studies included) to assess the impact of excluding studies with high risks in:
-
selection bias (in either one or both criteria of random sequence generation and allocation concealment);
-
attrition bias (incomplete outcome data).
Separate sensitivity analyses for each of the two categories of bias would have been performed had relevant studies been available. We planned to limit the sensitivity analyses based on risk of bias to selection and attrition biases because our major outcomes, namely, growth‐related outcomes are objective outcomes, and we considered these two risk of bias domains to be most likely to influence the results.
Summary of findings table
Following the recommendations of the Cochrane Handbook for Systematic Reviews of Interventions, we included a 'Summary of findings' (SOF) table presenting seven major outcomes from our review (Schünemann 2011a). We followed the grading of recommendations assessment, development and evaluation (GRADE) approach and used the web‐based GRADEpro software (http://gdt.guidelinedevelopment.org). We included the following information in the SOF table.
-
Trial characteristics
-
participant population: participants with thalassaemia (any age)
-
setting: any
-
intervention: GH therapy
-
comparator: placebo or a different GH therapy regimen
-
-
Outcomes (in the order of priority subject to the availability of data)
-
final height
-
adverse effects
-
change in height
-
change in height SDS
-
height velocity
-
QoL
-
cost
-
We used the mean baseline risk (the total number of events in the control group divided by the total number of participants in the control group) to represent the assumed risk. We assessed the overall quality of the body of evidence gathered using the eight GRADE considerations (trial limitations, consistency of effect, imprecision, indirectness and publication bias, large effect, plausible confounding and dose response relationship) (Schünemann 2011b). The GRADE system classifies the quality of evidence as high, moderate, low, and very low, with the following explanations:
-
high quality of evidence: further research is very unlikely to change our confidence in the estimate of effect;
-
moderate quality of evidence: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate;
-
low quality of evidence: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate;
-
very low quality of evidence: we are very uncertain about the estimate.
Results
Description of studies
Results of the search
We identified 57 citations from the combined searches, including three duplicates. After screening their titles and abstracts for relevance, we short‐listed two articles. After inspecting the full‐text of the two articles in detail, we included only one trial in our review (Arcasoy 1999). The PRISMA flow diagram is shown in Figure 1.

Study flow diagram.
Included studies
We found only one trial which met the inclusion criteria (Arcasoy 1999). Full details of the trial are presented in the tables (Characteristics of included studies).
Trial design
The trial was an RCT conducted in Turkey which lasted one year. The researchers aimed to study the GH reserve in children with thalassaemia, their growth response to GH therapy and the possible side‐effects of GH therapy. This trial was sponsored in part by Pharmacia‐Upjohn.
Participants
A total of 20 children with homozygous β thalassaemia participated in the trial, but the trial authors did not state if these children had thalassaemia major or intermedia; although it was reported that they received regular blood transfusions to keep pre‐transfusion haemoglobin values above 9 to 10 g/dL. The children were of short stature and the trial's inclusion criteria stated a height below ‐2 SD for age, height velocity below 25th percentile and bone age delay of more than two years. Participants' age ranged from 6 to 16 years and all were pre‐pubertal. There were 10 children in the intervention group (six boys and four girls) and 10 children (all boys) in the control group. Otherwise, there were no significant differences in the baseline characteristics (such as height parameters, age, levels of their haemoglobin, serum IGF‐1, ferritin and bone age) between the intervention and control groups.
The trial authors noted that the participants' basal IGF‐1 levels were significantly lower than age‐matched norms (below the first percentile). Prior to randomisation, all the participants first underwent tests to assess their GH responses to pharmacological and physiological stimuli. Response to pharmacologic stimuli and IGF‐1 generation were found to be severely depressed in most participants, despite their ability to maintain physiologic pulsatile secretion and GH response to recombinant GH therapy, highlighting the significant discordance between GH response to pharmacologic and physiological stimuli. The results of these tests and the proportion of children diagnosed with GH deficiency were not reported separately for the intervention and the control group, with the exception for baseline serum IGF‐1 where no significant difference was found between groups.
Interventions
In the intervention group, 10 children received recombinant GH (Genotropin, Pharmacia) which was administered subcutaneously on a daily basis at a dose of 0.7 IU/kg per week for a duration of 12 months in addition to standard treatment. The control group (n = 10) received standard treatment for their condition; no placebo was used in this trial.
Outcomes
All children were followed up at three‐monthly intervals with auxological measurements (height, height SDS, height velocity, height velocity SDS, body weight) and laboratory parameters which included fasting blood glucose, oral glucose tolerance test, plasma zinc and thyroid function tests (serum T4, T3, TSH). Bone age measurement was repeated at the end of the trial period.
Excluded studies
We excluded one trial since the intervention was hormonal treatment, but not in the form of GH therapy (El Beshlawy 2008). See the 'Characteristics of excluded studies' table.
Ongoing studies
We did not identify any ongoing trials.
Studies awaiting classification
There are no trials awaiting classification.
Risk of bias in included studies
The risk of bias assessment relates to the single included trial (Arcasoy 1999); the results are summarised below.
Allocation
Sequence generation
The trial was described as randomised but the methods of sequence generation were not stated; therefore, we judged this criterion to have an unclear risk of bias.
Allocation concealment
The authors stated that randomisation sequence was concealed in "closed" envelopes, but it was not stated if the envelopes were opaque or not. Hence, a judgement of unclear risk of bias was made.
Blinding
The outcomes reported by the trial were all objective measurements such as height parameters and laboratory results. The trial did not specifically report any blinding of the participants and personnel, but blinding appeared highly unlikely as the intervention (GH) was administered subcutaneously whereas the control group received no placebo intervention. However, we considered that it was highly unlikely that the lack of blinding would affect the growth outcomes as the growth of this group of children was not known to be readily influenced by any form of known co‐intervention. Therefore, we judged that there was a low risk of performance bias.
It was not stated who the assessors of the growth outcomes were, and whether the assessors were blinded to the allocation status of the participants. However, we considered this as unlikely to influence the outcomes, which were objectively measured such as growth parameters and laboratory results. Detection bias was judged to be of low risk.
Incomplete outcome data
The authors stated that all 20 children were followed up until the trial was completed, although it was unclear whether all data were available for analysis for all participants at all periods of the measurement. Therefore, we judged this criterion to have an unclear risk of bias.
Selective reporting
The main outcomes defined in the review methodology (growth response and side effects) were reported in sufficient detail. In terms of growth response, the height and height velocity measurements and SDSs for these parameters were reported in means and SDs at the end of the trial period as defined in the methodology. The side effects which were reported, such as effects on glucose metabolism and thyroid function, were reasonable given the relatively short period of the trial. The risk of selective outcome reporting in this trial was low.
Other potential sources of bias
There was no significant difference in the baseline characteristics of both the intervention and control groups except for their gender, which was probably attributed to the small number of trial participants. There were 10 males in the control group and six boys and four girls in the intervention group; all were pre‐pubertal during enrolment. Height velocity measured in absolute values (cm/year) is influenced by gender during pubertal years, for example girls have an earlier take‐off age and reach their peak height velocity earlier than boys (Abbassi 1998). In contrast, height SDS and height velocity SDS were established on gender‐specific charts based on population norms and the interpretation of these scores will not be influenced by the gender imbalance seen in both groups. Therefore, we considered the improvement in the height velocity (cm/year) seen in the GH‐treated children unlikely to be related to the gender differences as their height velocity SDS had similarly improved.
We noted that the authors had acknowledged that the trial was partially supported by Pharmacia Upjohn, who we believe provided the trial drugs (GH). We screened for other source of bias including any evidence of fraud or publication bias but found no such bias. Overall, we judged this criterion to have an unclear risk of bias.
Effects of interventions
See: Summary of findings for the main comparison Growth hormone for people with thalassaemia
The major outcomes along with their corresponding quality of evidence (rated using the GRADE approach) are presented in the summary of findings table (summary of findings Table for the main comparison). We graded the evidence for all outcomes as moderate in quality (downgraded one level due to small sample size leading to imprecision).
Growth hormone therapy versus no growth hormone or standard care
Primary outcomes
1. Final height
The included trial did not assess this outcome (Arcasoy 1999).
2. Adverse effects
The single included trial reported that there were no adverse effects such as the development of glucose intolerance or thyroid dysfunction in any participant in either group (total participants = 20). However, the paper did report data for the oral glucose tolerance test and for fasting blood glucose at one year. There was no difference between groups in the oral glucose tolerance test at one year MD ‐0.03 mg/dL (95% CI ‐17.45 to 17.39) (moderate quality evidence) (Analysis 1.1), but at the same time point fasting blood glucose was significantly higher in the GH group than in the control group, MD 6.67 mg/dL (95% CI 2.66 to 10.68) (moderate quality evidence) (Analysis 1.2).
3. Participant or parental satisfaction
The included trial did not assess this outcome (Arcasoy 1999).
Secondary outcomes
1. Short‐term growth
a. Change in height (cm) over trial period
The included trial did not assess this outcome (Arcasoy 1999).
b. Change in height SDS over trial period
Based on the single included trial (n = 20) (Arcasoy 1999), there was no significant difference in the height SDS after 12 months between participants who received GH and those who did not, MD ‐ 0.09 (95% CI ‐0.33 to 0.15) (Analysis 1.3); this was judged to be moderate quality of evidence, downgraded one level for imprecision. Height SDS in the intervention group improved significantly more from baseline than in the control group, MD 0.26 (95% CI 0.13 to 0.39) (Analysis 1.4); again evidence was judged to be of moderate quality, downgraded one level for imprecision.
c. Height velocity
The height velocity (cm/year) measured at the end of the trial showed that the group who received GH had a significantly higher height velocity compared to the control group, MD 2.28 cm/year (95% CI 1.76 to 2.80) (Analysis 1.5), evidence was judged to be of moderate quality, downgraded one level for imprecision. In addition, height velocity SDS at one year was significantly higher in the intervention group when compared to the control group, MD 3.31 (95% CI 2.43 to 4.19) (Analysis 1.6); moderate quality of evidence which was downgraded one level for imprecision. Lastly, the change from baseline in height velocity SDS at one year similarly showed that participants in the intervention group who received GH improved significantly more than the control group, MD 3.41 (95% CI 2.45 to 4.37) (Analysis 1.7), again with moderate quality of evidence which was downgraded one level for imprecision.
2. Bone mineralisation (bone density scores)
The included trial did not assess this outcome (Arcasoy 1999).
3. QoL
The included trial did not assess this outcome (Arcasoy 1999).
4. Costs of care per year
The included trial did not assess this outcome (Arcasoy 1999).
5. Serum insulin‐like growth hormone (IGF‐1)
At the end of the trial period, serum IGF‐1 in the intervention group was significantly higher compared to the control group, MD 19.55 ng/mL (95% CI 2.03 to 37.07) (Analysis 1.8). The authors reported that the improved value of serum IGF‐1 in the intervention group; however, this remained below the fifth percentile (Arcasoy 1999).
Discussion
Summary of main results
Results from the single trial included in this review showed that in children with thalassaemia who have problems achieving their target height, the use of GH therapy for one year modestly improved height velocity and height velocity SDS in comparison to those who did not receive GH therapy. Despite the improvement in height SDS being marginally greater in the intervention group, there was no difference between their height SDS when compared to the control group at the end of the one‐year period. None of the participants in either the intervention or the control group reported any adverse event such as the development of glucose intolerance or thyroid dysfunction during the trial period; fasting blood sugar in both groups also remained within the normal range despite being notably higher in the GH group at the end of the trial. After one year, the serum IGF‐1 in the intervention group was significantly higher compared to the control group; but the improvement was modest as the improved value was still below the fifth percentile. Bone age between both groups was not significantly different at the end of the trial period.
Overall completeness and applicability of evidence
After a comprehensive search, we could only identify a single small RCT involving 20 participants which was eligible for inclusion in the review (Arcasoy 1999). The trial was conducted in Turkey and it remains unclear if the results can be generalised to people with thalassaemia from other parts of the world. The GH status of the participants at baseline was not stated clearly, and this posed some uncertainties on the applicability of the findings to those with and without proven GH deficiency. This trial did address some of the outcomes of interest to our review such as short‐term growth and adverse events (Arcasoy 1999). As there were no data beyond the one‐year trial period, we are uncertain if the GH therapy received will have any effect on the participants' future height velocity, height SDS and final height. It also remains unclear if a longer intervention period would continuously improve height velocity and height SDS and eventually final height. We could not find any trial eligible for inclusion in this review which addressed the optimal dosage and duration of GH therapy. Additionally, there were some issues in the applicability of the evidence for both genders, as the control group in the included trial only consisted of boys.
Quality of the evidence
The quality of the evidence for all major outcomes in this review was moderate, as there was a serious concern with the imprecision of the estimates due to the small sample size which translated into wide confidence intervals (summary of findings Table for the main comparison). As this review is based on a single small trial, the inclusion of additional trials in the future may alter the conclusions. Additionally, the single included trial was sponsored in part by a pharmaceutical company; however, while we note a concern here, we decided not to rate the trial as high risk for other potential sources of bias since there is a lack of clear evidence on the association of industry‐sponsorship to the overall risk of biases as assessed within the Cochrane risk‐of‐bias domains (other than publication bias which is assessed separately) (Lundh 2017).
Potential biases in the review process
Although there were no adverse effects reported in both groups in the included trial, the finding was limited by the small number of participants and the short duration of the trial, especially for the intervention in question which usually is administered over the long term.
Agreements and disagreements with other studies or reviews
We are aware of a review on the effects of recombinant GH on height velocity in people with thalassaemia major (Delvecchio 2010). This review presented the results of the RCT included in this review (Arcasoy 1999) along with 16 observational studies. Due to the limitation of observational data, the authors found the real efficacy of GH to be debatable and suggested that GH therapy may be most beneficial in promoting growth during the first year of treatment, but that long‐term treatment seemed ineffective in improving final height (Delvecchio 2010). Our review based on the single RCT found that significant improvement in height velocity was noted after a year of GH treatment, but no conclusions could be drawn beyond that. Guidelines published by the Thalassaemia Internation Federation recommend GH treatment in people with thalassaemia who have growth problems and GH deficiency (Cappelini 2014); however, the optimal dose and duration are not stated. The International Network on Endocrine Complications in Thalassemia (I‐CET) propose GH therapy for these children at a dose of 0.025 to 0.05 mg/kg/day (0.5 to 1.0 units/kg/wk) given subcutaneously daily (at night) till the child reaches near final height with careful monitoring for side effects (De Sanctis 2013).

Study flow diagram.

Comparison 1 Growth hormone versus control, Outcome 1 Oral glucose tolerance test sum (mg/dL).
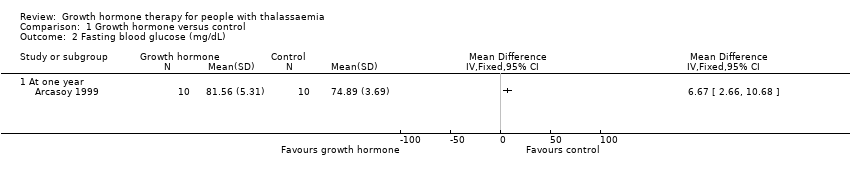
Comparison 1 Growth hormone versus control, Outcome 2 Fasting blood glucose (mg/dL).

Comparison 1 Growth hormone versus control, Outcome 3 Height SD score.

Comparison 1 Growth hormone versus control, Outcome 4 Change from baseline in height SD score.
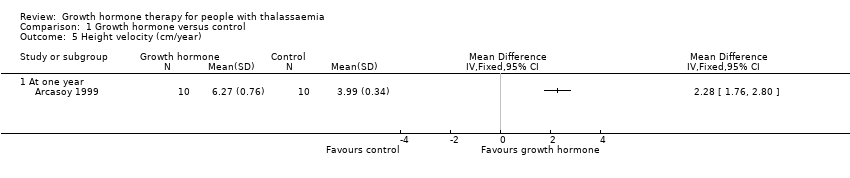
Comparison 1 Growth hormone versus control, Outcome 5 Height velocity (cm/year).

Comparison 1 Growth hormone versus control, Outcome 6 Height velocity SD score.
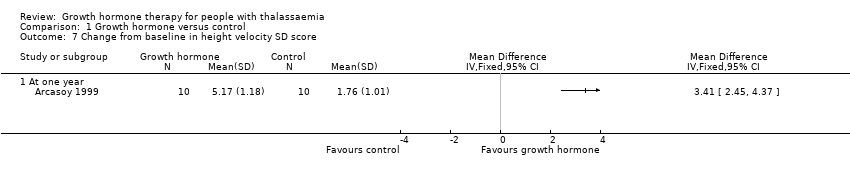
Comparison 1 Growth hormone versus control, Outcome 7 Change from baseline in height velocity SD score.

Comparison 1 Growth hormone versus control, Outcome 8 Serum insulin‐like growth hormone (IGF‐1) (ng/mL).
| Growth hormone for people with thalassaemia | ||||||
| Patient or population: people with thalassaemia (any age) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with control | Risk with growth hormone | |||||
| Final height and change in height | The included trial did not assess either of these outcomes. | |||||
| Adverse effects Oral glucose tolerance test (mg/dL) (at one year) | The mean oral glucose tolerance test was 336.56 mg/dL. | MD 0.03 lower (17.45 lower to 17.39 higher). | ‐ | 20 | ⊕⊕⊕⊝ | Fasting blood glucose levels in the growth hormone group were significantly higher than in the control group but both were still within the normal range. |
| Height SDS | The mean height SDS was ‐2.85. | MD 0.09 lower | ‐ | 20 | ⊕⊕⊕⊝ | |
| Change in height SDS (difference between baseline and final visit at one year) | The change in mean height SDS was ‐0.05. | MD 0.26 higher | ‐ | 20 | ⊕⊕⊕⊝ | |
| Height velocity (cm/year) | The mean height velocity was 3.99 cm/year. | MD 2.28 higher | ‐ | 20 | ⊕⊕⊕⊝ | |
| Height velocity SDS | The mean height velocity SDS was ‐1.56. | MD 3.31 higher | ‐ | 20 | ⊕⊕⊕⊝ | |
| Change in height velocity SDS (difference between baseline and final visit at one year) | The change in mean height velocity SDS was 1.76. | MD 3.41 higher | ‐ | 20 | ⊕⊕⊕⊝ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Data contributed by a single trial with small sample and 95% CI is wide. | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Oral glucose tolerance test sum (mg/dL) Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 At one year | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Fasting blood glucose (mg/dL) Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 At one year | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Height SD score Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 At one year | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Change from baseline in height SD score Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 At one year | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Height velocity (cm/year) Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5.1 At one year | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Height velocity SD score Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 6.1 At one year | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Change from baseline in height velocity SD score Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 7.1 At one year | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Serum insulin‐like growth hormone (IGF‐1) (ng/mL) Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 8.1 At one year | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |