Alemtuzumab para la esclerosis múltiple
Resumen
Antecedentes
La esclerosis múltiple (EM) es una enfermedad desmielinizante, autoinmune, inflamatoria y dependiente de los linfocitos T, que afecta al sistema nervioso central y tiene un curso impredecible. Los tratamientos actuales de la EM se centran en el tratamiento de las exacerbaciones, prevenir las nuevas exacerbaciones y evitar la progresión de la discapacidad. Sin embargo, actualmente no hay un tratamiento eficaz que sea capaz de alcanzar estos objetivos con seguridad y eficacia. Lo anterior ha estimulado el desarrollo y la investigación de fármacos nuevos. Ensayos clínicos recientes indican que alemtuzumab, un anticuerpo monoclonal humanizado contra la superficie de las células CD52, podría ser una opción promisoria para la EM.
Objetivos
Evaluar la seguridad y la efectividad de alemtuzumab administrado solo o en combinación con otros tratamientos para reducir la actividad de la enfermedad en los pacientes con cualquier forma de EM.
Métodos de búsqueda
Se hicieron búsquedas en el registro de ensayos del Grupo Cochrane de Esclerosis Múltiple y Enfermedades Raras del Sistema Nervioso Central (Cochrane Multiple Sclerosis and Rare Diseases of the CNS Group) (30 abril 2015), que contiene ensayos del Registro Cochrane Central de Ensayos Controlados (Cochrane Central Register of Controlled Trials) (CENTRAL), MEDLINE, EMBASE, CINAHL, LILACS y en las bases de datos de registros de ensayos ClinicalTrials.gov y WHO International Clinical Trials Registry Platform. No hubo restricciones de la fuente, fecha de publicación o el idioma.
Criterios de selección
Todos los ensayos clínicos aleatorios (ECA) que incluyeron adultos diagnosticados con cualquier forma de EM según los criterios McDonald y compararon alemtuzumab solo o en combinación con otros fármacos, a cualquier dosis y por cualquier duración, versus placebo u otra farmacoterapia activa o alemtuzumab en otra dosis, régimen o duración. Los resultados primarios complementarios fueron la supervivencia sin recurrencia, la progresión mantenida de la enfermedad y el número de participantes con al menos un evento adverso, incluidos eventos adversos graves.
Obtención y análisis de los datos
Dos autores de la revisión independientes realizaron la selección de los estudios, extrajeron los datos y evaluaron el riesgo de sesgo. Un tercer autor de la revisión comprobó la exactitud del proceso. Se utilizó la herramienta Cochrane del “Riesgo de sesgo” para evaluar el riesgo de sesgo de los estudios incluidos en la revisión. Se utilizó el sistema GRADE para evaluar la calidad del conjunto de pruebas. Para medir el efecto del tratamiento sobre los resultados dicotómicos se utilizó el cociente de riesgos (CR), para el efecto del tratamiento sobre los resultados continuos se utilizó la diferencia de medias (DM), y para los resultados de tiempo hasta el evento se utilizó el cociente de riesgos instantáneos (CRI). Se calcularon los intervalos de confianza (IC) del 95% para estas medidas. Cuando no hubo heterogeneidad se utilizó un modelo de efectos fijos para agrupar los datos.
Resultados principales
Tres ECA (1713 participantes) cumplieron los criterios de selección y se incluyeron en la revisión. Los tres ensayos compararon alemtuzumab versus interferón beta‐1a subcutáneo en pacientes con EM recurrente‐remitente. En los estudios CARE‐MS y CAMMS223 los pacientes no habían recibido tratamiento previamente. El estudio CARE‐MS II incluyó pacientes con al menos una recurrencia mientras recibían tratamiento con interferón beta o acetato de glatiramer. Alemtuzumab se proporcionó durante 12 ó 24 meses; para algunos resultados, el período de seguimiento alcanzó los 36 meses. Los regímenes fueron (a) 12 mg o 24 mg por día administrado por vía intravenosa, una vez al día por cinco días consecutivos al mes cero y 12 o (b) 24 mg por día, por vía intravenosa, una vez al día por tres días consecutivos al mes 12 y 24. Los pacientes del otro brazo de los ensayos recibieron interferón beta‐1a 44 μg subcutáneamente tres veces semanales después del ajuste de la dosis.
A los 24 meses, alemtuzumab 12 mg se asoció con: (a) una mayor supervivencia sin recurrencia (cociente de riesgos instantáneos [CRI] 0,50; IC del 95%: 0,41 a 0,60; 1248 participantes, dos estudios, pruebas de calidad moderada); (b) una mayor supervivencia sin progresión mantenida de la enfermedad (CRI 0,62; IC del 95%: 0,44 a 0,87; 1191 participantes; dos estudios; pruebas de calidad moderada); (c) un número ligeramente mayor de participantes con al menos un evento adverso (CR 1,04; IC del 95%: 1,01 a 1,06; 1248 participantes; dos estudios; pruebas de calidad moderada); (d) un número inferior de participantes con lesiones hiperintensas en T2 nuevas o de mayor tamaño en la imaginología de resonancia magnética (IRM) (CR 0,74; IC del 95%: 0,59 a 0,91; 1238 participantes; dos estudios; I2 = 80%); y (e) un número inferior de abandonos (CR 0,31; IC del 95%: 0,23 a 0,41; 1248 participantes; dos estudios, I2 = 29%; pruebas de baja calidad).
A los 36 meses, alemtuzumab 24 mg se asoció con: (a) mayor supervivencia sin recurrencia (45 versus 17; CRI 0,21; IC del 95%: 0,11 a 0,40; un estudio; 221 participantes); (b) una mayor supervivencia sin progresión mantenida de la enfermedad (CRI 0,33; IC del 95%: 0,16 a 0,69; un estudio; 221 participantes); y (c) ninguna diferencia estadística en la tasa de participantes con al menos un evento adverso. No se encontró ningún estudio que informara de ninguno de los siguientes resultados: tasa de participantes sin actividad clínica de la enfermedad, calidad de vida, fatiga o cambio en los números de lesiones potenciadas en T1 y T2 en la IRM después del tratamiento. No fue posible realizar análisis de subgrupos según el tipo de enfermedad y la discapacidad inicial debido a la falta de datos.
Conclusiones de los autores
En los pacientes con EM recurrente‐remitente, el alemtuzumab 12 mg fue mejor que el interferón beta‐1a subcutáneo para los siguiente resultados evaluados a los 24 meses: supervivencia sin recurrencia, supervivencia sin progresión mantenida de la enfermedad, número de participantes con al menos un evento adverso y número de participantes con lesiones hiperintensas en T2 nuevas o de mayor tamaño en la IRM. La calidad de las pruebas para estos resultados era de baja a moderada. Alemtuzumab 24 mg pareció ser mejor que el interferón beta‐1a subcutáneo para la supervivencia sin recurrencia y la supervivencia sin progresión mantenida de la enfermedad a los 36 meses.
Se necesitan más ensayos clínicos aleatorios para evaluar los efectos de alemtuzumab sobre otras formas de EM y en comparación con otras opciones terapéuticas. Estos estudios nuevos deben evaluar resultados relevantes adicionales como la tasa de participantes sin actividad clínica de la enfermedad, la calidad de vida, la fatiga y los eventos adversos (tasas individuales, eventos adversos graves y eventos adversos a largo plazo). Además, estos estudios nuevos deben evaluar otras dosis y duraciones del ciclo de alemtuzumab.
PICO
Resumen en términos sencillos
Alemtuzumab para la esclerosis múltiple
Antecedentes
La esclerosis múltiple (EM) es una enfermedad crónica del sistema nervioso que afecta a adultos jóvenes y de mediana edad. El daño repetido al recubrimiento de mielina (las membranas que cubren y protegen los nervios) y a otras partes de los nervios puede provocar discapacidad grave. La EM puede estar relacionada con alteraciones del sistema inmunológico. Alemtuzumab es un fármaco biológico (un tipo de anticuerpo) que ya se ha utilizado en otras enfermedades.
Características de los estudios
Se encontraron tres estudios (con 1713 participantes) que cumplieron con los criterios de selección de la revisión. Todos los estudios compararon alemtuzumab versus interferón beta‐1a subcutáneo en pacientes con EM recurrente‐remitente. En dos de los estudios (CARE‐MS y CAMMS223) los participantes recibían tratamiento por primera vez. El tercer estudio (CARE‐MS II) incluyó a participantes con al menos una recurrencia mientras eran tratados con interferón beta o acetato de glatiramer por al menos seis meses.
Resultados clave
La revisión de estos estudios comparativos encontró que, en comparación con interferón beta‐1a subcutáneo, alemtuzumab reduce el riesgo de recurrencia, mejora la función y no parece aumentar el riesgo general de eventos adversos. Además, alemtuzumab reduce el riesgo de lesiones nuevas o de mayor tamaño de EM detectadas mediante imaginología de resonancia magnética (IRM). Sin embargo, hay una falta de información acerca de los efectos del alemtuzumab sobre varios resultados relacionados con el paciente como (a) la calidad de vida, (b) la tasa de cada evento adverso (por separado) y (c) la frecuencia de eventos adversos a largo plazo y los eventos adversos graves.
Calidad de la evidencia
La calidad metodológica general de los estudios incluidos fue de moderada a alta. Sin embargo, debido al escaso número de estudios incluidos y la baja tasa de eventos, la calidad general de las pruebas para los resultados principales se consideró muy baja a moderada. Lo anterior significa que es probable que los estudios nuevos tengan una marcada repercusión sobre la confianza en la estimación del efecto y puedan cambiar la estimación o que no haya seguridad con respecto a la estimación.
Conclusiones de los autores
Summary of findings
| Alemtuzumab 12 mg compared to interferon beta‐1a for multiple sclerosis | ||||||
| Patient or population: patients with multiple sclerosis | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Interferon beta‐1a | Alemtuzumab 12 mg | |||||
| Relapse‐free survival | Not estimated | Not estimated | HR 0.50 (0.41 to 0.60) | 1248 | ⊕⊕⊕⊝ | — |
| Sustained disease progression‐free survival | Not estimated | Not estimated | HR 0.62 (0.44 to 0.87) | 1191 | ⊕⊕⊕⊝ | — |
| Number of participants with at least one adverse event | Study population | RR 1.04 | 1248 | ⊕⊕⊕⊝ | — | |
| 94 per 100 | 98 per 100 | |||||
| Moderate | ||||||
| 94 per 100 | 98 per 100 | |||||
| Change in EDSS score | — | The mean change in EDSS score in the intervention groups was | — | 1199 | ⊕⊝⊝⊝ | — |
| Number of participants with new or enlarging T2‐hyperintense lesions | 69 per 100 | 51 per 100 | RR 0.74 | 1238 | ⊕⊕⊕⊝ | — |
| Dropouts | Study population | RR 0.31 | 1248 | ⊕⊕⊝⊝ | — | |
| 24 per 100 | 8 per 100 | |||||
| Moderate | ||||||
| 24 per 100 | 7 per 100 | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Participants and personnel were not blinded and this outcome could be affected by this fact. | ||||||
Antecedentes
Descripción de la afección
La esclerosis múltiple (EM) es una enfermedad desmielinizante, autoinmune e inflamatoria del sistema nervioso central (cerebro y médula espinal), de la que todavía se desconocen las causas (Coles 1999a; Gray 2004). Es la causa más frecuente de discapacidad neurológica no traumática en adultos jóvenes (Noseworthy 2000). Casi 2 000 000 de personas en el mundo están afectadas por esta afección, que puede deteriorar la calidad de vida de los pacientes de manera significativa y se asocia con costos elevados para los pacientes, sus familias y la sociedad en general (Multiple Sclerosis International Federation 2010).
Se han identificado cuatro tipos de EM: recurrente‐remitente (RR), secundaria‐progresiva (SP), primaria‐progresiva (PP) y progresiva‐recurrente (PR). El curso de la enfermedad es impredecible; mientras algunos pacientes son afectados de forma mínima, otros muestran una progresión rápida de la enfermedad hasta alcanzar una incapacidad física total (Lublin 1996). En la primera forma, la EM se caracteriza por recurrencias y remisiones (RR), pero las secuelas en el tiempo debido a las recurrencias pueden provocar mayor discapacidad (Hawkins 1999). En algunos pacientes la enfermedad es progresiva desde su aparición (PP); otros experimentan períodos de progresión seguidos de recurrencias y remisiones (SP) (Lublin 1996). En otros casos la EM muestra progresión desde la aparición, pero con recurrencias claras (PR).
Descripción de la intervención
Las estrategias terapéuticas para la EM intentan tratar las exacerbaciones, prevenir nuevas exacerbaciones y evitar la progresión de la discapacidad (Filippini 2013). Los tratamientos actuales que modifican la enfermedad reducen la frecuencia de las recurrencias y reducen moderadamente la acumulación de la discapacidad (Coles 2006; Rieckmann 2009). En consecuencia, se necesitan agentes nuevos que controlen eficazmente la enfermedad.
Alemtuzumab (Lemtrada, previamente conocido como Campath‐1H) es un anticuerpo monoclonal humanizado contra la superficie de las células CD52 que se puede encontrar en diversas poblaciones de células, incluidos los linfocitos B y T, los timocitos y los monocitos, pero no en los precursores hematológicos o las células plasmáticas (Gilleece 1993). Sin embargo, todavía se desconoce la función exacta de CD52 (Xia 1991).
En 2001, alemtuzumab se autorizó para la leucemia linfocítica crónica de células B resistente a la fludarabina (FDA 2001; Keating 2002). Desde entonces se ha administrado en algunas otras enfermedades (uso autorizado o sin prescripción), que incluyen la púrpura trombocitopénica inmune, la anemia aplásica, la anemia hemolítica autoinmune, la vasculitis, los trasplantes de células madre hematopoyéticas (como régimen de acondicionamiento) y los trasplantes de órganos (como agente de inducción) (Gomez‐Almaguer 2012; Lockwood 2003; Waldmann 2005; Weissenbacher 2010).
Un estudio publicado en 1999 que incluyó 36 participantes con EM progresiva, informó que las infusiones intravenosas diarias de alemtuzumab (20 mg durante cuatro horas por cinco días) se asociaron con una reducción en las lesiones realzadas por gadolinio en la imaginología de resonancia magnética (IRM) y una reducción de las recurrencias, sin mejoría clínica en la discapacidad (Coles 1999b). Estudios abiertos que incluyeron a pacientes con EM recurrente‐remitente informaron que el fármaco redujo las tasas de recurrencia y discapacidad (Coles 2006; Hirst 2008).
Alemtuzumab ya se autorizó para la EM en la Unión Europea (EMA 2013). La Food and Drug Administration (FDA) de los EE.UU. autorizó el alemtuzumab para el tratamiento de los pacientes con EMRR que han tenido una respuesta insuficiente a dos o más fármacos recomendados para el tratamiento de la EM (FDA 2014).
Las guías de la Association of British Neurologists identificaron al alemtuzumab como con una actividad mayor para prevenir las recurrencias. Sin embargo, debido a los problemas de seguridad, las guías recomendaron este fármaco como tratamiento de segunda línea o en pacientes que progresaron rápidamente a la forma RR (Scolding 2015).
Alemtuzumab está disponible para el tratamiento de la EM en viales de dosis única de 12 mg/1,2 ml (10 mg/mL). La dosis inicial propuesta para la EM es 12 mg diarios por cinco días consecutivos (infusión intravenosa), seguidos de un segundo ciclo de tratamiento de 12 mg/diario por tres días consecutivos. El segundo ciclo de tratamiento se administra 12 meses después del primer ciclo. Inmediatamente antes de la administración de alemtuzumab y durante los tres primeros días de cualquier ciclo de tratamiento se recomienda la premedicación con corticosteroides (FDA 2014). La vida media general del fármaco es aproximadamente 21 días. Alemtuzumab está disponible como un líquido para reconstituirse en una solución para infusión (goteo) en una vena. Una infusión proporciona 12 mg y dura alrededor de cuatro horas.
Alemtuzumab puede producir eventos adversos graves que incluyen otros síndromes autoinmunes que afectan a la tiroides y los glóbulos (trombocitopenia, anemia hemolítica, pancitopenia), así como nefropatías, y además puede aumentar el riesgo de cáncer tiroideo (FDA 2014). En el seguimiento a los cinco años el riesgo acumulativo de enfermedad autoinmune es de aproximadamente el 22%, de enfermedad de Graves del 12%, de púrpura trombocitopénica inmune del 3% y de enfermedad de Goodpasture (glomerulopatía grave) del 0,4% (Cossburn 2011).
Recientemente, la FDA actualizó un resumen general de recomendaciones (Risk Evaluation and Mitigation Strategy Program) acerca de Lemtrada para los pacientes, las farmacias y los profesionales sanitarios (FDA 2015).
De qué manera podría funcionar la intervención
Los estudios de investigación anteriores han indicado que alemtuzumab reduce las células T y B, que pueden ser responsables del daño celular, aunque preserva las células inmunitarias innatas (Rao 2012). También se ha informado un cambio en la composición de los linfocitos que acompaña la reconstitución de los linfocitos (Hill‐Cawthorne 2012).
Por qué es importante realizar esta revisión
Los resultados de los ensayos controlados aleatorios (ECA) de alemtuzumab para la EM son promisorios y se justifica una revisión sistemática de todos los ECA para evaluar la efectividad y la seguridad en la EM.
Objetivos
Evaluar la seguridad y la efectividad de alemtuzumab administrado solo o en combinación con otros tratamientos para reducir la actividad de la enfermedad en los pacientes con cualquier forma de EM.
Métodos
Criterios de inclusión de estudios para esta revisión
Tipos de estudios
Se incluyeron los ensayos clínicos aleatorios (ECA) doble ciego. No se consideraron los ensayos cruzados (crossover).
Tipos de participantes
Se incluyeron adultos diagnosticados con EM según los criterios McDonald (McDonald 2001; Polman 2011), o los criterios Poser (Poser 1983). Se consideraron para inclusión los participantes con cualquier forma de EM (recurrente‐remitente, primaria‐progresiva, secundaria‐progresiva o progresiva‐recurrente).
Tipos de intervenciones
-
Intervención experimental: alemtuzumab solo o en combinación con otros fármacos a cualquier dosis y para cualquier duración del ciclo.
-
Comparador: placebo, otra farmacoterapia activa (es decir, corticosteroides, plasmaféresis, interferones beta, acetato de glatiramer, fingolimod, natalizumab, mitoxantrona, teriflunomida o dimetilfumarato).
Tipos de medida de resultado
Resultados primarios
-
Supervivencia sin recurrencia. La recurrencia se definió como síntomas recién desarrollados o recientemente empeorados de disfunción neurológica, que persisten por más de 24 horas y se confirman objetivamente. Sin embargo, se consideraron criterios menos estrictos y se evaluaron por separado.
-
Supervivencia sin progresión mantenida de la enfermedad, definida como un aumento ≥ 1,0 punto en la puntuación en la Expanded Disability Status Scale (EDSS) (Kurtzke 1983) en participantes con una puntuación inicial ≤ 5,0 o un aumento ≥ 0,5 puntos en participantes con una puntuación inicial ≥ 5,5 puntos confirmado a los seis meses. Un aumento de un punto en la puntuación en la EDSS confirmado a los tres meses de seguimiento se consideró una medida de resultado alternativa de progresión.
-
Número de participantes con al menos un evento adverso, incluyendo los eventos adversos graves.
Todos los resultados primarios se evaluaron después de 12 y 24 meses de seguimiento y al final del período de seguimiento.
Resultados secundarios
-
Número de participantes sin actividad clínica de la enfermedad, definida como ninguna recurrencia y ninguna acumulación mantenida de la discapacidad. La acumulación mantenida de la discapacidad se definió como un aumento de al menos 1,5 puntos en la Expanded Disability Status Scale (EDSS) en los pacientes con una puntuación inicial de 0 y de al menos 1,0 punto en los pacientes con una puntuación inicial de 1,0 o más.
-
Calidad de vida evaluada según la escala Multiple Sclerosis Quality of Life (MSQOL)‐54 (Vickrey 1995) o el Multiple Sclerosis Quality of Life Inventory (MSQLI) (Fischer 1999).
-
Cambio en la discapacidad evaluado según la EDSS (Kurtzke 1983).
-
Fatiga evaluada según la Fatigue Severity Scale o la Fatigue Index Scale (Krupp 1989).
-
Número de participantes con lesiones hiperintensas en T2 nuevas o de mayor tamaño en la imaginología de resonancia magnética (Li 1999).
-
Número de participantes que abandonaron el estudio.
Todos los resultados secundarios se evaluarían después de 12 y 24 meses y al final del período de seguimiento.
Results
Description of studies
Results of the search
The search strategy retrieved 223 references: two in CENTRAL, 128 in MEDLINE, 82 in EMBASE, three in CINAHL, none in PEDro, none in LILACS, four in ClinicalTrials.gov, none in the WHO International Clinical Trials Registry Platform and four from handsearching. We considered a total of 35 references to be potentially eligible. After reading the full text, we included these 35 records. They referred to three RCTs and 32 ancillary reports about these three primary studies. The flow diagram of the process of study identification and selection is presented in Figure 1.

Study flow diagram.
Included studies
The three RCTs included a total of 1713 participants (CAMMS223; CARE‐MS I; CARE‐MS II). All studies were multicentric trials, comparing alemtuzumab versus subcutaneous interferon beta‐1a for patients with relapsing–remitting MS according to the McDonald criteria (McDonald 2001).
Participants were treatment‐naive in the CARE‐MS I and CAMMS223 studies. The CARE‐MS II study included only participants with at least one relapse while being treated with interferon beta or glatiramer for at least six months.
In the CARE‐MS I and CARE‐MS II studies, the interventions were given for 12 months (CARE‐MS I; CARE‐MS II); in the CAMMS223 study, the treatment lasted 24 months (CAMMS223). The following regimens were used in these RCTs:
-
CAMMS223 study, a phase II trial: alemtuzumab (either 12 mg per day or 24 mg per day) was given by intravenous infusion on five consecutive days during the first month and on three consecutive days at months 12 and 24 (CAMMS223).
-
CARE‐MS I (or CAMMS323) study, a phase III trial: alemtuzumab (12 mg per day) was given by intravenous infusion on five consecutive days during the first month and on three consecutive days at month 12 (CARE‐MS I).
-
CARE‐MS II (or CAMMS324) study, a phase III trial: alemtuzumab (either 12 mg per day or 24 mg per day) was given by intravenous infusion on five consecutive days during the first month and on three consecutive days at month 12 (CARE‐MS II).
In all studies, the dose of interferon beta‐1a was 44 μg given subcutaneously three times weekly after dose titration.
Details of these RCTs are available in the table Characteristics of included studies.
Excluded studies
We excluded none of the potentially eligible studies.
Risk of bias in included studies
The risk of bias of each study is detailed in the Characteristics of included studies table. Figure 2 and Figure 3 present the 'Risk of bias' summary along with review authors' judgements about each risk of bias item for each included study. The overall quality of the studies was low since in all of them we categorised at least one of the main domains (generation of allocation sequence, allocation concealment and blinding) as having a high risk of bias.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
We classified all studies as low risk of bias for generation of allocation sequence. The methods were reported in the articles and we judged them to be appropriate.
However, for allocation concealment, we classified one study as having an unclear risk of bias because it did not provide enough information to allow judgement (CAMMS223). We classified the other two studies as having a low risk of bias because they provided an adequate method to ensure allocation concealment (CARE‐MS I; CARE‐MS II).
Blinding
We considered all studies as having a high risk of bias of performance bias (participants and personnel) because both drugs (intervention and comparator) had adverse effects that precluded masking.
We judged the following outcomes separately for detection bias (outcome assessment):
-
EDSS outcome assessment: We classified CARE‐MS I and CARE‐MS II as having a high risk of bias since unmasked raters performed the EDSS assessments. On the other hand, CAMMS223 had a low risk of bias.
-
Efficacy outcomes assessment (except EDSS): We classified all studies as having a low risk of bias.
-
Safety outcomes assessment: CARE‐MS I and CARE‐MS II had a low risk of bias, while CAMMS223 had a high risk of bias.
Incomplete outcome data
One study was at low risk of attrition bias (CAMMS223), one was at high risk because it used an inappropriate modified "intention‐to‐treat" analysis (CARE‐MS I), and one study was at unclear risk of attrition bias (CARE‐MS II)
Selective reporting
We classified two studies as having a low risk of selective reporting bias (CAMMS223; CARE‐MS I). We classified one study as having high risk of bias because the results for some previously planned outcomes were not provided (i.e. quality of life) (CARE‐MS II).
Other potential sources of bias
There were no other known potential sources of bias in the three included trials.
Effects of interventions
Comparison 1: Alemtuzumab 12 mg versus subcutaneous interferon beta‐1a
Primary outcomes
Relapse‐free survival
Alemtuzumab was associated with better relapse‐free survival at 24‐month follow‐up (hazard ratio (HR) 0.50, 95% confidence interval (CI) 0.41 to 0.60; 1248 participants; two studies; moderate quality evidence, I2 = 0%) (Analysis 1.1; Figure 4). This result was consistent when we considered separately naive and previously treated participants.

Forest plot of comparison: 1 Alemtuzumab 12 mg versus interferon beta‐1a, outcome: 1.1 Relapse‐free survival.
Only one study assessed this outcome at 36 months (CAMMS223). This study showed a higher number of participants who relapsed with interferon than with alemtuzumab (45 versus 24; HR 0.31, 95% CI 0.18 to 0.52).
None of the included studies provided data for the 12‐month analysis.
Sustained disease progression‐free survival
Alemtuzumab was associated with a lower number of participants with sustained disease progression‐free survival at both 24‐month (HR 0.62, 95% CI 0.44 to 0.87; 1191 participants; two studies; I2 = 0%) and 36‐month follow‐up (HR 0.25, 95% CI 0.11 to 0.57; 223 participants; one study) (Analysis 1.2; Figure 5). This finding was consistent when we considered a subgroup of previously treated participants. However, for naive participants there was no difference between the interventions.

Forest plot of comparison: 1 Alemtuzumab 12 mg versus interferon beta‐1a, outcome: 1.2 Sustained disease progression‐free survival.
None of the included studies provided data for the 12‐month analysis.
Number of participants with at least one adverse event, including serious adverse events
Alemtuzumab was associated with a higher proportion of participants with at least one adverse event after 24 months (risk ratio (RR) 1.04, 95% CI 1.01 to 1.06; 1248 participants; two studies; I2 = 0%; moderate quality evidence), but not at 36 months (RR 1.00, 95% CI 0.98 to 1.02; 224 participants; one study) (Analysis 1.3; Figure 6).

Forest plot of comparison: 1 Alemtuzumab 12 mg versus interferon beta‐1a, outcome: 1.2 Rate of participants with at least one adverse event.
None of the included studies provided data for the 12‐month analysis.
Secondary outcomes
Number of participants free of clinical disease activity
None of the included studies assessed this outcome.
Quality of life
None of the included studies assessed this outcome.
Change in disability as assessed by the Expanded Disability Status Scale (EDSS)
Alemtuzumab was associated with a significant improvement in EDSS scores after 36 months (mean difference (MD) ‐0.70, 95% CI ‐1.04 to ‐0.36; 223 participants; one study) (CAMMS223). At 24 months, considering both treatment‐naive patients and previously treated patients (who failed after interferon beta or glatiramer treatment), there were no differences in EDSS scores (MD ‐0.20, 95% CI ‐0.60 to 0.20; 1199 participants; two studies; I2 = 88%) (Analysis 1.4). However, when only previously treated patients were assessed, alemtuzumab was associated with better results (MD ‐0.41, 95% CI ‐0.62 to ‐0.20; one study; 628 participants) (CARE‐MS II).
None of the included studies provided data for the 12‐month analysis.
Fatigue as assessed by the Fatigue Severity Scale or the Fatigue Index Scale
None of the included studies assessed this outcome.
Number of participants with new or enlarging T2‐hyperintense lesions on magnetic resonance imaging
Alemtuzumab was associated with a lower rate of participants presenting with new or enlarging lesions, considering both naive and previously treated participants (RR 0.74, 95% CI 0.59 to 0.91; 1238 participants; two studies; I2 = 80%; random‐effects model) (Analysis 1.5).
Number of participants who dropped out
Alemtuzumab was associated with a lower number of dropouts at 24 months (RR 0.31, 95% CI 0.23 to 0.41; 1248 participants; two studies), but not at 36 months (RR 0.81, 95% CI 0.57 to 1.14; 224 participants; one study) (Analysis 1.6).
None of the included studies provided data for the 12‐month analysis.
Comparison 2: Alemtuzumab 24 mg versus subcutaneous interferon beta‐1a
Primary outcomes
Relapse‐free survival
Only one study assessed this outcome at 36 months (CAMMS223). This study showed a higher number of relapses in the interferon group than in the alemtuzumab group (45 versus 17; HR 0.21, 95% CI 0.11 to 0.40) (CAMMS223).
None of the included studies provided data for the 12‐ and 24‐month analyses.
Sustained disease progression‐free survival
Alemtuzumab was associated with a lower number of participants with sustained disease progression‐free survival at 36 months (HR 0.33, 95% CI 0.16 to 0.69; 221 participants; one study) (CAMMS223).
None of the included studies provided data for the 12‐ or 24‐month analyses.
Number of participants with at least one adverse event, including serious adverse events
There were no significant differences between alemtuzumab and subcutaneous interferon beta‐1a at 24 months (RR 1.04, 95% CI 1.00 to 1.07; 391 participants; one study) (CARE‐MS II) or at 36 months (RR 0.99, 95% CI 0.97 to 1.02; 220 participants; one study) (CAMMS223).
None of the included studies provided data for the 12‐month analysis.
Secondary outcomes
Number of participants free of clinical disease activity
None of the included studies assessed this outcome.
Quality of life
None of the included studies assessed this outcome.
Change in disability as assessed by the EDSS
Alemtuzumab was associated with a significant improvement in EDSS scores after 36 months (MD ‐0.83, 95% CI ‐1.17 to ‐0.49; 221 participants; one study) (CAMMS223). None of the included studies provided data for the 12‐ and 24‐month analyses.
Fatigue as assessed by the Fatigue Severity Scale or the Fatigue Index Scale
None of the included studies assessed this outcome.
Number of participants with new or enlarging T2‐hyperintense lesions on magnetic resonance imaging
None of the included studies assessed this outcome.
Number of participants who dropped out
Alemtuzumab was associated with a lower number of dropouts at 24 months (RR 0.27, 95% CI 0.16 to 0.46; 404 participants; one study) (CARE‐MS II), but not at 36 months (RR 0.76, 95% CI 0.53 to 1.09; 221 participants; one study) (CAMMS223).
None of the included studies provided data for the 12‐month analysis.
Discusión
Resumen de los resultados principales
Esta revisión sistemática intentó evaluar los efectos (beneficiosos y perjudiciales) de alemtuzumab en comparación con otra farmacoterapia para cualquier tipo de esclerosis múltiple (EM).
Según los resultados de tres ensayos clínicos aleatorios (ECA), comparado con interferón beta‐1a subcutáneo, alemtuzumab 12 mg se asoció con:
-
mayor supervivencia sin recurrencia a los 24 y 36 meses;
-
un número inferior de participantes con supervivencia sin progresión mantenida de la enfermedad;
-
un número ligeramente mayor de participantes con al menos un evento adverso después de 24 meses;
-
una mejoría mayor en las puntuaciones en la Expanded Disability Status Scale (EDSS) después de 36 meses;
-
una mejoría mayor en las puntuaciones en la EDSS después de 24 meses (en los pacientes previamente tratados con interferón o acetato de glatiramer);
-
un número inferior de participantes con lesiones hiperintensas en T2 nuevas o de mayor tamaño en la imaginología de resonancia magnética;
-
un número inferior de abandonos a los 24 meses, pero no a los 36 meses.
Según los resultados de un ECA, en comparación con interferón beta‐1a subcutáneo, alemtuzumab 24 mg se asoció con:
-
mayor supervivencia sin recurrencia a los 36 meses;
-
un número inferior de participantes con supervivencia sin progresión mantenida de la enfermedad a los 36 meses;
-
ninguna diferencia estadística en el número de participantes con al menos un evento adverso a los 24 y 36 meses;
-
una mejoría mayor en las puntuaciones en la EDSS después de 36 meses;
-
un número inferior de abandonos a los 24 meses, pero no a los 36 meses.
El número mayor de participantes con al menos un evento adverso no se asoció con una tasa mayor de abandonos, probablemente porque la mayoría de estos eventos fueron leves o moderados. Los datos de eventos adversos graves no se proporcionan por separado en ninguno de los estudios incluidos.
Se incluyó el cambio en las puntuaciones en la EDSS como un resultado secundario en lugar de un resultado primario porque los cambios a corto plazo en las puntuaciones en la EDSS pueden no ser un marcador fiable de cambio irreversible en la EM recurrente‐remitente (Healy 2013).
Compleción y aplicabilidad general de las pruebas
Se incluyeron tres ECA que compararon alemtuzumab versus interferón beta‐1a subcutáneo en pacientes con EM recurrente‐remitente. Alemtuzumab se proporcionó durante 12 ó 24 meses y los participantes tuvieron un seguimiento de hasta 36 meses para algunos resultados en uno de los estudios incluidos. Las dosis fueron (a) 12 mg o 24 mg por día por vía intravenosa, una vez al día por cinco días consecutivos al mes cero y 12, o (b) 24 mg por día por vía intravenosa, una vez al día por tres días consecutivos al mes 12 y 24. Los grupos control recibieron interferón beta‐1a, 44 μg subcutáneamente tres veces por semana después del ajuste de la dosis. Por lo tanto, las pruebas disponibles están limitadas a estas intervenciones y pacientes específicos.
Hay una falta de pruebas para los siguientes resultados:
-
número de participantes sin actividad clínica de la enfermedad;
-
calidad de vida;
-
fatiga (por ejemplo, evaluada con la Fatigue Severity Scale o la Fatigue Index Scale).
Hay dos razones probables de esta falta de pruebas: (a) los resultados se propusieron inicialmente en los protocolos de los ensayos pero no estaban disponibles para esta revisión incluso después de establecer contacto con los autores de estos estudios; (b) los resultados no se planificaron originalmente en el estadio de protocolo de los ECA incluidos.
Se debe recalcar que los datos al seguimiento a los 36 meses se basan en un escaso número de participantes, lo que puede aumentar la incertidumbre en estos resultados.
Finalmente, los tres estudios solamente incluyeron a pacientes con EM recurrente‐remitente y no se encontraron pruebas para otras formas de la enfermedad.
Calidad de la evidencia
Como se presentó en el Resumen de los hallazgos para la comparación principal, la calidad del grupo de pruebas obtenido para cada resultado varió de muy baja a moderada.
La calidad general de los ECA fue baja ya que en todos al menos uno de los dominios principales (generación de la secuencia de asignación, ocultación de la asignación y cegamiento) se consideró con alto riesgo de sesgo. En todos los estudios, los participantes y el personal no se cegaron porque los efectos adversos relacionados con cada intervención impiden el enmascaramiento. Además, en dos estudios tampoco se pudo cegar la evaluación de las puntuaciones en la EDSS. Al considerar estos dos hechos, se evaluó por separado el riesgo de sesgo para la EDSS y los eventos adversos.
No se observó heterogeneidad estadísticamente significativa entre los estudios para los resultados primarios complementarios. La calidad de las pruebas para los abandonos estuvo afectada por el escaso número de eventos en los ensayos.
Sesgos potenciales en el proceso de revisión
Para evitar la introducción de sesgo en esta revisión se siguieron estrictamente todas las recomendaciones sobre la búsqueda, la selección de los estudios, la obtención de los datos y el análisis de los datos del Manual Cochrane para revisiones sistemáticas de intervenciones (Higgins 2011).
Los puntos fuertes de esta revisión incluyen una búsqueda amplia de la bibliografía y el uso de análisis de intención de tratar para los datos dicotómicos.
La limitaciones de esta revisión incluyen: (a) no se realizó una evaluación del sesgo de publicación a través del análisis del gráfico en embudo porque se incluyeron menos de diez estudios en el metanálisis y (b) hubo falta de algunos datos de resultados en los ECA incluidos.
Acuerdos y desacuerdos con otros estudios o revisiones
Durante la realización de esta revisión se publicó una revisión sistemática no Cochrane que evaluó todos los tratamientos disponibles para la EM (CADTH 2013). Esta revisión evaluó comparaciones directas e indirectas entre varios fármacos, incluido el alemtuzumab. Los resultados son similares a los de la presente revisión, incluidos los resultados del metanálisis y el riesgo de sesgo de los ECA incluidos.

Study flow diagram.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Forest plot of comparison: 1 Alemtuzumab 12 mg versus interferon beta‐1a, outcome: 1.1 Relapse‐free survival.

Forest plot of comparison: 1 Alemtuzumab 12 mg versus interferon beta‐1a, outcome: 1.2 Sustained disease progression‐free survival.

Forest plot of comparison: 1 Alemtuzumab 12 mg versus interferon beta‐1a, outcome: 1.2 Rate of participants with at least one adverse event.

Comparison 1 Alemtuzumab 12 mg versus interferon beta‐1a, Outcome 1 Relapse‐free survival.

Comparison 1 Alemtuzumab 12 mg versus interferon beta‐1a, Outcome 2 Sustained disease progression‐free survival.

Comparison 1 Alemtuzumab 12 mg versus interferon beta‐1a, Outcome 3 Number of participants with at least one adverse event.

Comparison 1 Alemtuzumab 12 mg versus interferon beta‐1a, Outcome 4 Change in EDSS score.

Comparison 1 Alemtuzumab 12 mg versus interferon beta‐1a, Outcome 5 Number of participants with new or enlarging T2‐hyperintense lesions.
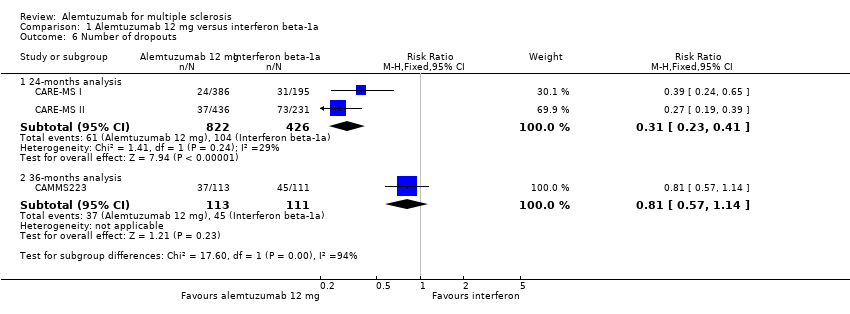
Comparison 1 Alemtuzumab 12 mg versus interferon beta‐1a, Outcome 6 Number of dropouts.
| Alemtuzumab 12 mg compared to interferon beta‐1a for multiple sclerosis | ||||||
| Patient or population: patients with multiple sclerosis | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Interferon beta‐1a | Alemtuzumab 12 mg | |||||
| Relapse‐free survival | Not estimated | Not estimated | HR 0.50 (0.41 to 0.60) | 1248 | ⊕⊕⊕⊝ | — |
| Sustained disease progression‐free survival | Not estimated | Not estimated | HR 0.62 (0.44 to 0.87) | 1191 | ⊕⊕⊕⊝ | — |
| Number of participants with at least one adverse event | Study population | RR 1.04 | 1248 | ⊕⊕⊕⊝ | — | |
| 94 per 100 | 98 per 100 | |||||
| Moderate | ||||||
| 94 per 100 | 98 per 100 | |||||
| Change in EDSS score | — | The mean change in EDSS score in the intervention groups was | — | 1199 | ⊕⊝⊝⊝ | — |
| Number of participants with new or enlarging T2‐hyperintense lesions | 69 per 100 | 51 per 100 | RR 0.74 | 1238 | ⊕⊕⊕⊝ | — |
| Dropouts | Study population | RR 0.31 | 1248 | ⊕⊕⊝⊝ | — | |
| 24 per 100 | 8 per 100 | |||||
| Moderate | ||||||
| 24 per 100 | 7 per 100 | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Participants and personnel were not blinded and this outcome could be affected by this fact. | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Relapse‐free survival Show forest plot | 2 | Hazard Ratio (Fixed, 95% CI) | Subtotals only | |
| 1.1 24‐month analysis | 2 | Hazard Ratio (Fixed, 95% CI) | 0.50 [0.41, 0.60] | |
| 2 Sustained disease progression‐free survival Show forest plot | 3 | Hazard Ratio (Fixed, 95% CI) | Subtotals only | |
| 2.1 24‐month analysis | 2 | Hazard Ratio (Fixed, 95% CI) | 0.62 [0.44, 0.87] | |
| 2.2 36‐month analysis | 1 | Hazard Ratio (Fixed, 95% CI) | 0.25 [0.11, 0.57] | |
| 3 Number of participants with at least one adverse event Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 24‐month analysis | 2 | 1248 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [1.01, 1.06] |
| 3.2 36‐month analysis | 1 | 224 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.98, 1.02] |
| 4 Change in EDSS score Show forest plot | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 24‐month analysis | 2 | 1199 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.60, 0.20] |
| 4.2 36‐month analysis | 1 | 223 | Mean Difference (IV, Random, 95% CI) | ‐0.7 [‐1.04, ‐0.36] |
| 5 Number of participants with new or enlarging T2‐hyperintense lesions Show forest plot | 2 | 1238 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.59, 0.91] |
| 6 Number of dropouts Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 24‐months analysis | 2 | 1248 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.23, 0.41] |
| 6.2 36‐months analysis | 1 | 224 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.57, 1.14] |