مقایسه تاثیر زوکلوپنتیکسول در برابر دارونما در مدیریت درمانی اسکیزوفرنی
چکیده
پیشینه
زوکلوپنتیکسول (zuclopenthixol) یک آنتیسایکوتیک قدیمی است که سه فرمول متمایز دارد (زوکلوپنتیکسول دیهیدروکلراید (zuclopenthixol dihydrochloride)، زوکلوپنتکسول استات (zuclopenthixol acetate) یا آکوفاز (Acuphase) و زوکلوپنتیکسول دکانوات (zuclopenthixol decanoate)). اگرچه استفاده از آن برای سالهای متمادی معمول بوده، هیچ مرور سیستماتیک قبلی در مورد اثربخشی آن در مقایسه با دارونما (placebo) در اسکیزوفرنی (schizophrenia) انجام نشده است.
اهداف
ارزیابی اثربخشی تمام فرمولهای زوکلوپنتیکسول در مقایسه با دارونما در درمان اسکیزوفرنی.
روشهای جستوجو
در 6 نوامبر 2013 و 20 اکتبر 2015، پایگاه ثبت کارآزماییهای گروه اسکیزوفرنی در کاکرین را جستوجو کردیم که مبتنی بر جستوجوهای منظم در پایگاههای MEDLINE؛ EMBASE؛ CINAHL؛ BIOSIS؛ AMED؛ PubMed؛ PsycINFO و پایگاههای ثبت کارآزماییهای بالینی است. منابع همه مطالعات وارد شده را بررسی کرده و برای دستیابی به مطالعات و دادههای مرتبط، با نویسندگان مطالعات وارد شده تماس گرفتیم.
معیارهای انتخاب
همه کارآزماییهای تصادفیسازی و کنترل شدهای را وارد کردیم که زوکلوپنتیکسول را در هر شکلی با دارونما برای درمان اسکیزوفرنی یا سایکوزهای شبه‐اسکیزوفرنی مقایسه کردند.
گردآوری و تجزیهوتحلیل دادهها
بهطور جداگانه، دادهها را استخراج کرده و به صورت متقاطع کنترل کردیم. تعداد کمی مطالعه شناسایی شد، بنابراین همه مطالعات را به صورت متقاطع بررسی کردیم. برای دادههای دو‐حالتی، نسبت خطر (relative risk; RR) اثر‐ثابت و 95% فواصل اطمینان (CI) آنها را محاسبه کردیم. آنالیزها بر اساس قصد درمان (intention‐to‐treat) انجام شدند. در جایی که امکانپذیر بود، پیامدهای پیوسته را به پیامدهای دو‐حالتی تبدیل کردیم. زمانی که این کار ممکن نبود، از تفاوتهای میانگین (MD) برای متغیرهای پیوسته بهره بردیم. خطر سوگیری (bias) را برای مطالعات وارد شده ارزیابی کرده و از رویکرد درجهبندی توصیه، ارزیابی، توسعه و ارزشیابی (Grading of Recommendations Assessment, Development and Evaluation; GRADE) برای تهیه جدول «خلاصهای از یافتهها» استفاده کردیم.
نتایج اصلی
فقط دو مطالعه، شامل 65 شرکتکننده، برای ورود به این مرور واجد شرایط بودند. سطح کیفیت کلی دو مطالعه پائین بود، با حجم نمونه کوچک و منابع قابل توجه سوگیری، بنابراین قادر به استفاده از همه دادهها در مقایسههای خود نبودیم. مطالعات مربوط به سالهای 1968 و 1972 قدیمی بودند، و بعید به نظر میرسید که استانداردهای مدرن مرور همتا (peer review) را گذرانده باشند. ما فقط توانستیم دادههای کوتاه‐مدت و فقط کارآزماییهایی را پیدا کنیم که زوکلوپنتیکسول دیهیدروکلراید را تصادفیسازی کردند. همچنین امیدوار بودیم که دادههایی را برای مقایسه زوکلوپنتیکسول استات در برابر دارونما و زوکلوپنتیکسول دکانوات در برابر دارونما شناسایی کنیم. ما نتوانستیم هیچ مطالعهای را بیابیم که شامل دادههای مربوط به این دو داروی نسبتا پُر‐مصرف باشد.
برای پیامد اولیه مورد نظر، یعنی بهبودی بالینی قابل توجه، یک مطالعه را یافتیم که دادههای قابل استفادهای را ارائه کرد. وضعیت کلی اندازهگیری شده توسط مقیاس درک کلی بالینی (clinical global impression; CGI) هنگامی که توسط روانپزشک یا پرستار ارزیابی شد، رتبهبندیهای متفاوتی را نشان داد. نمرات روانپزشک از اهمیت آماری برخوردار نبود، اما زمانی که توسط کارکنان پرستاری ارزیابی شد، تفاوتی به نفع زوکلوپنتیکسول به اهمیت آماری رسید (1 RCT؛ n = 29؛ RR: 2.57؛ 95% CI؛ 1.06 تا 6.20، دادهها با کیفیت بسیار پائین). همچنین شواهدی مبنی بر افزایش سداتاسیون در افراد درمان شده با زوکلوپنتیکسول در مقایسه با دارونما به دست آمد (1 RCT؛ n = 29؛ RR: 4.67؛ 95% CI؛ 1.23 تا 17.68، دادهها با کیفیت بسیار پائین). دادههای مربوط به «ترک زودهنگام مطالعه» مبهم بودند. هیچ داده قابل استفادهای برای پیامدهایی مانند عود بیماری، وضعیت روانی، مرگومیر، کیفیت زندگی، استفاده از خدمات یا هزینههای اقتصادی درمان در دسترس نبود.
نتیجهگیریهای نویسندگان
این مرور برای افراد مبتلا به اسکیزوفرنی نشان میدهد که زوکلوپنتیکسول دیهیدروکلراید ممکن است به بهبود نشانههای اسکیزوفرنی کمک کند. این مرور شواهدی را از کارآزمایی ارائه میدهد که در صورت مصرف زوکلوپنتیکسول دیهیدروکلراید در مقایسه با دارونما، بیماران ممکن است دچار عوارض جانبی و سداتاسیون شوند. با این حال، شواهد دارای کیفیت بسیار پائین و با برخی منابع مهم سوگیری همراه است. هیچ دادهای در مورد زوکلوپنتیکسول دکانوات یا زوکلوپنتکسول استات وجود ندارد.
دادههای کارآزمایی موجود در مورد اثربخشی مطلق زوکلوپنتیکسول دیهیدروکلراید برای پزشکان، استفاده از آن را تائید میکند، اما ماهیت محدود دادهها و منابع مهم سوگیری نتیجهگیریها را دشوار میسازد. زوکلوپنتیکسول در هر سه شکل خود یک آنتیسایکوتیک شایع است و اینکه دادههای کمی در مورد استفاده از آن وجود دارد، ناامید کننده است.
PICO
خلاصه به زبان ساده
مقایسه تاثیر زوکلوپنتیکسول در برابر دارونما در درمان افراد مبتلا به اسکیزوفرنی
افراد مبتلا به اسکیزوفرنی (schizophrenia) اغلب صداهایی را میشنوند و چیزهایی را میبینند (توهمها (hallucinations)) و باورهای عجیب و غریب دارند (هذیان (delusions)). درمان اصلی این نشانههای اسکیزوفرنی، بر پایه تجویز داروهای آنتیسایکوتیک قرار گرفته است. زوکلوپنتیکسول (zuclopenthixol) یک داروی آنتیسایکوتیک قدیمی است که اولین بار در سال 1962 معرفی شد و دارای سه فرمول متمایز است: زوکلوپنتیکسول دیهیدروکلراید (zuclopenthixol dihydrochloride)، زوکلوپنتکسول استات (zuclopenthixol acetate) (یا آکوفاز (Acuphase)) و زوکلوپنتیکسول دکانوات (zuclopenthixol decanoate). اگرچه استفاده از آن برای سالهای متمادی شایع بوده، هیچ مرور سیستماتیک قبلی در مورد اثربخشی آن در مقایسه با دارونما (placebo) («درمان ساختگی») در درمان اسکیزوفرنی انجام نشده است. با توجه به استفاده گسترده از این دارو، بررسی اثربخشی هر سه فرمول آن مهم است تا متخصصان سلامت، سیاستگذاران و افراد مبتلا به اسکیزوفرنی بتوانند انتخابهای آگاهانهتری داشته باشند.
در سال 2013 به دنبال کارآزماییهای تصادفیسازی و کنترل شدهای بودیم که زوکلوپنتیکسول را با دارونما مقایسه کردند. فقط دو مطالعه را با 65 شرکتکننده پیدا کردیم که میتوانستند در این مرور وارد شوند. کیفیت کلی این مطالعات پائین بود، افراد کمی در آنها حضور داشتند و میزان سوگیری (bias) قابل توجه بود. مطالعات مربوط به سالهای 1968 و 1972 قدیمی بودند، و بعید به نظر میرسید که استانداردهای مدرن مرور همتا (peer review) را پشت سر گذاشته باشند. فقط اطلاعات و دادههای کوتاه‐مدتی به دست آمد که فقط در مورد زوکلوپنتکسول دیهیدروکلراید بودند.
اطلاعات بسیار محدود است، اما نشان میدهد که زوکلوپنتیکسول در مقایسه با دارونما میتواند منجر به بهبود وضعیت عمومی شود. با این حال، خطر بروز عوارض جانبی مانند سداتاسیون و خستگی نیز افزایش مییابد.
با توجه به کیفیت پائین اطلاعات و قدمت دو مطالعه، انجام پژوهش بیشتری مورد نیاز است، به ویژه در مورد زوکلوپنتکسول در مقایسه با داروهای آنتیسایکوتیک جدیدتر.
Authors' conclusions
Summary of findings
| ZUCLOPENTHIXOL DIHYDROCHLORIDE versus PLACEBO for schizophrenia | ||||||
| Patient or population: people with with schizophrenia | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | ZUCLOPENTHIXOL DIHYDROCHLORIDE versus PLACEBO | |||||
| Clinically significant response on global state ‐ as defined by each of the studies, Improvement (CGI) ‐ short term ‐ as rated by nurse | Study population | RR 2.57 | 29 | ⊕⊝⊝⊝ | For this SOF table outcome CGI data as rated by a nurse and a psychiatrist were both available. The nurse data were chosen as it includes both control event and | |
| 286 per 1000 | 734 per 1000 | |||||
| Relapse as defined by the studies | No studies reported these important outcomes | |||||
| Clinically significant response on psychotic symptoms ‐ as defined by each of the studies. | ||||||
| Adverse effects: Sedation | Low | RR 4.67 | 29 | ⊕⊝⊝⊝ | No use of formal rating scales.Several other adverse events were recorded but sedation considered important. | |
| 50 per 1000 | 234 per 1000 | |||||
| Moderate | ||||||
| 150 per 1000 | 701 per 1000 | |||||
| High | ||||||
| 250 per 1000 | 1000 per 1000 | |||||
| Leaving the study early | Study population | RR 0.93 | 29 | ⊕⊝⊝⊝ | ||
| 100 per 1000 | 93 per 1000 | |||||
| Average change in quality of life/satisfaction | No studies reported these important outcomes | |||||
| Significant change in quality of life/satisfaction ‐ as defined by each of the studies | ||||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Risk of bias: rated 'very serious' ‐ randomisation method unclear as describes "randomly assigned". Incomplete outcome data and does not accurately describe losses. Patients blinded but unclear if raters and clinicians are blinded | ||||||
| ZUCLOPENTHIXOL ACETATE versus PLACEBO for schizophrenia | |||||
| Patient or population: people with schizophrenia | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | |
| Assumed risk | Corresponding risk | ||||
| Control | ZUCLOPENTHIXOL ACETATE versus PLACEBO | ||||
| Clinically significant response on global state | No studies reported any of these important outcomes | ||||
| Relapse as defined by the studies | |||||
| Clinically significant response on psychotic symptoms ‐ as defined by each of the studies. | |||||
| Other adverse effects, general and specific | |||||
| Leaving the study early | |||||
| Average change in quality of life/satisfaction | |||||
| Significant change in quality of life/satisfaction ‐ as defined by each of the studies | |||||
| ZUCLOPENTHIXOL DECANOATE versus PLACEBO for schizophrenia | |||||
| Patient or population: people with schizophrenia | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | |
| Assumed risk | Corresponding risk | ||||
| Control | ZUCLOPENTHIXOL DECANOATE versus PLACEBO | ||||
| Clinically significant response on global state | No studies reported any of these important outcomes | ||||
| Relapse as defined by the studies | |||||
| Clinically significant response on psychotic symptoms ‐ as defined by each of the studies | |||||
| Other adverse effects, general and specific | |||||
| Leaving the study early | |||||
| Average change in quality of life/satisfaction | |||||
| Significant change in quality of life/satisfaction ‐ as defined by each of the studies | |||||
Background
Description of the condition
Schizophrenia is a chronic mental health condition that is associated with significant morbidity and mortality. It is characterised by psychotic symptoms, alterations in motivation and volition, neurochemical imbalances and irregularities of emotion. The diagnosis of this condition is based on clinical interview with the patient and observation of their mental state, and comparison of this with diagnostic criteria, such as the ICD‐10 (International Classification of Diseases) (Jablensky 2010).
Lifetime prevalence of schizophrenia is estimated to be 0.33% to 0.66% and the incidence is somewhere between 10 and 22 per 100,000 (Jablensky 1992). Schizophrenia commonly presents in early adulthood or late adolescence. Men have an earlier age of onset than women, and also seem to have a worse prognosis (Picchioni 2007). The incidence of schizophrenia is not uniform across different research sites but it is present and identifiable throughout the world (McGrath 2006).
Schizophrenia is estimated to reduce lifespan by 10 to 15 years, partly due to poor access to health care, stigma and increase in comorbid physical health problems (Van Os 2009). The National Confidential Inquiry (NCI 2012) states schizophrenia leads to an increased risk of suicide compared to the general population and also contributes to an increase in overall mortality. Schizophrenia causes more loss of life‐years than some cancers and physical illnesses (WHO 2004). Given that it is a disabling condition that contributes to a high burden of illness for patient and carers, it is of vital importance to investigate further its causes and effective treatment options.
Description of the intervention
Antipsychotic medication has remained the mainstay of treatment for schizophrenia ever since chlorpromazine was introduced. Zuclopenthixol is one of the earlier antipsychotics (Figure 1), first introduced in 1962, and has remained in use. Clinicians seem to find it convenient to use and patients seem to tolerate it reasonably well (Owens 2012). Zuclopenthixol is commonly known by the name of Clopixol, although this usage in clinical parlance can lead to confusion because of the different formulations that exist. Essentially zuclopenthixol is available in three formulations. Zuclopenthixol dihydrochloride is the tablet form and has a half life of approximately 20 hours (Lundbeck 2011). Zuclopenthixol acetate (Acuphase) is an option if it is anticipated that a patient will be disturbed over a longer time period (NICE 2005) and is also used as the initial medication prior to initiating longer‐acting depot antipsychotic treatments. Acuphase reaches half‐life concentration at 36 hours. The longer‐acting zuclopenthixol decanoate is used as a depot preparation and is convenient in terms of frequency of administering, which can vary between weekly and monthly. Zuclopenthixol decanoate has a half life of 19 days (Lundbeck 2011).

Zuclopenthixol structure
How the intervention might work
Antipsychotic drugs, especially the first generation antipsychotics are hypothesised to work by blocking dopamine receptors in the brain, especially around the mesolimbic dopamine pathway. There are several theories relating to dopamine as the neurotransmitter implicated in symptom causation. As a neurotransmitter, dopamine is involved in four main pathways in the brain. The mesolimbic and mesocortical pathways are related to salience of perceptions, motivation and reward. These pathways are deranged in schizophrenia and are the target of many antipsychotics (Kapur 2005). The nigrostriatal pathway is involved in the control of movement and can be affected by some, especially typical, antipsychotics leading to movement disorders as a common side effect (Miyamoto 2005). The final pathway is the tuberoinfundibular pathway that is involved in prolactin secretion. Hence, many antipsychotics can interfere with prolactin secretion and lead to hyperprolactinaemia (Owens 2012).
Zuclopenthixol belongs to a class of chemical compounds called the thioxanthenes. These all have a common three‐ringed structure and include zuclopenthixol and flupentixol. Zuclopenthixol primarily works by blocking the D2 receptors but has also been shown to have serotonergic‐blocking properties, a high affinity for alpha‐adrenoreceptors and some antihistamine properties (Lundbeck 2011). The side effects of zuclopenthixol use are common to many antipsychotics and include sedation and extra pyramidal side effects. It may also cause some abnormalities of liver function.
Why it is important to do this review
Zuclopenthixol is a widely used drug around the world. There have been numerous studies over the years comparing it with both other drugs and placebos. There are three Cochrane Reviews that have evaluated the efficacy of zuclopenthixol, (Table 1). These past reviews of zuclopenthixol acetate and decanoate did not find placebo data, however, the review of zuclopenthixol dihydrochloride did present some placebo data. Our review enables the placebo data from zuclopenthixol dihydrochloride to be transferred out of the other drug comparisons and sit in its own placebo title covering all three zuclopenthixol formulations. Over the years there has been widespread debate over the ethics of placebo‐controlled studies especially when well known proven treatments exist. Some researchers also argue that placebo trials provide the purest form of evidence when evaluating a particular drug (Vallance 2006). Given the widespread use of this drug, it is essential to know the absolute effects of all three formulations of this commonly used drug so that clinicians, policy makers and recipients of care can make better informed choices.
| Focus of the review | Participants | Reference |
| Zuclopenthixol acetate | acutely ill people with schizophrenia | |
| Zuclopenthixol decanoate | people with schizophrenia | |
| Zuclopenthixol dihydrochloride | people with schizophrenia |
Objectives
To evaluate the effectiveness of all formulations of zuclopenthixol when compared with a placebo in schizophrenia.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials. If a trial was described as 'double blind' but implied randomisation, we intended to include such trials in a sensitivity analysis (see Sensitivity analysis) but we found insufficient numbers of trials. We would have excluded quasi‐randomised studies, such as those allocating by alternate days of the week, but again found no quasi‐randomised trials. If people were given additional treatments along with zuclopenthixol (any formulation), we would have only included data if the adjunctive treatment was evenly distributed between groups and randomised in exactly the same way as the intervention at the start of the trial.
Types of participants
Adults, however defined by authors of studies, with a diagnosis of schizophrenia or schizophreniform disorder, schizoaffective disorder and delusional disorder, again, by any means of diagnosis as defined by authors. We are interested in making sure that information is as relevant to the current care of people with schizophrenia as possible so we intended to clearly highlight the current clinical state (acute, early post‐acute, partial remission, remission) as well as the stage (prodromal, first episode, early illness, persistent) and whether the studies primarily focused on people with particular problems (for example, negative symptoms, treatment‐resistant illnesses). However, the studies we found did not provide this information. We would have excluded children, and people with dementing illnesses, depression and primary problems associated with substance misuse.
If a study had described the participant group as suffering from 'serious mental illnesses' and did not give a particular diagnostic grouping, we would have included these trials assuming that most people would have suffered from schizophrenia, but we found no such trials.
Types of interventions
-
Zuclopenthixol dihydrochloride in any form or at any dose compared with placebo.
-
Zuclopenthixol acetate in any form or at any dose compared with placebo.
-
Zuclopenthixol decanoate in any form or at any dose compared with placebo.
Types of outcome measures
We defined the outcome periods depending on the type of formulation of zuclopenthixol. However, we only found trial data for Zuclopenthicxol dihydrochloride and only for short‐term use. For zuclopenthixol decanoate and dihydrochloride, we had predefined trial duration as short term for those between zero and 12 weeks, medium term as those between 13 to 26 weeks and long term as those longer than 26 weeks. For zuclopenthixol acetate, given that the indications for its use are different and it has a different half life, we considered trials as being short term if the duration was between zero to six hours, medium term from seven to 36 hours, and long term greater than 36 hours.
Primary outcomes
1. Clinically significant response on global state ‐ as defined by each of the studies
Secondary outcomes
1. Death
1.1 Death ‐ not suicide
1.2 Suicide
2. Global state
3.1 Average score in global state
3.2 Average change in global state
3.3 Relapse as defined by the studies
3. Mental state
4.1 Clinically significant response on psychotic symptoms ‐ as defined by each of the studies
4.2 Average score on psychotic symptoms
4.3 Average change in psychotic symptoms
4.4 Clinically significant response on positive symptoms ‐ as defined by each of the studies
4.5 Average score in positive symptoms
4.6 Average change in positive symptoms
4.7 Clinically significant response on negative symptoms ‐ as defined by each of the studies
4.8 Average score on negative symptoms
4.9 Average change in negative symptoms
4. Other adverse effects, general and specific
5. Leaving the study early
6. Service utilisation outcomes
6.1 Hospital admission
6.2 Compulsory hospital admission
6.3 Readmission‐ as defined by each of the studies
7. Quality of life/satisfaction with care for either recipients of care or carers
7.1 Significant change in quality of life/satisfaction ‐ as defined by each of the studies
7.2 Average score in quality of life/satisfaction
7.3 Average change in quality of life/satisfaction
8. Economic outcomes
'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2011) and used GRADE profiler (GRADEPRO) to import data from RevMan 5 (Review Manager) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient‐care and decision making. In our protocol we aimed to select the following main outcomes for inclusion in the 'Summary of findings' table:
-
Clinically significant response on global state ‐ as defined by each of the studies.
-
Relapse as defined by the studies.
-
Clinically significant response on psychotic symptoms ‐ as defined by each of the studies.
-
Other adverse effects, general and specific.
-
Leaving the study early.
-
Average change in quality of life/satisfaction.
-
Significant change in quality of life/satisfaction ‐ as defined by each of the studies.
Search methods for identification of studies
Electronic searches
Cochrane Schizophrenia Group’s Trials Register
On 20 October 2015, the Trials Search Co‐ordinator (TSC) searched the Cochrane Schizophrenia Group’s Study‐Based Register of Trials using the following search strategy:
(*Zuclopenthixol* AND *Placebo*) in Intervention Field of STUDY
In such a study‐based register, searching the major concept retrieves all the synonym keywords and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics.
The Cochrane Schizophrenia Group’s Register of Trials is compiled by systematic searches of major resources (including MEDLINE, EMBASE, AMED, BIOSIS, CINAHL, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group’s Module). There are no language, date, document type, or publication status limitations for inclusion of records into the register.
For previous searches, please see Appendix 1.
Searching other resources
1. Reference searching
We inspected references of all included studies for further relevant studies.
2. Personal contact
We attempted to contact the first author of each included study for information regarding unpublished trials. Please see Results of the search for description.
3. Further searching of trials registers.
We also inspected web sites to see if other trials have been registered. We inspected the United States National Institute of Health website, (http://www.clinicaltrials.gov/). We also inspected the World Health Organization (WHO) international clinical trials registry platform (http://www.who.int/ictrp/en/). No further trials were identified.
Data collection and analysis
Selection of studies
Review author ML independently inspected citations from the searches and identified relevant abstracts. A former member of the review author team, Christine Esbensen (CE) planned to independently re‐inspect a random 20% sample to ensure reliability, however, CE was unable to continue to participate. Only a small number of citations were found, so MJ cross checked them all. No disputes arose. ML obtained and inspected full reports of the abstracts meeting the review criteria and MJ re‐inspected these full reports. No disputes arose.
Data extraction and management
1. Extraction
Review author ML extracted data from all included studies. As only two studies with useable data were identified MJ cross checked the whole sample. No disputes arose.
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if:
-
the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and
-
the measuring instrument has not been written or modified by one of the trialists for that particular trial.
Ideally, the measuring instrument should either be a self‐report or completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly. In 'Description of studies we describe the scales used.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint) which can be difficult in unstable and difficult‐to‐measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if the former were not available. We combined endpoint and change data in the analysis as we preferred to use mean differences (MD) rather than standardised mean differences (SMD) throughout (Higgins 2011a).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to all data before inclusion:
-
standard deviations (SDs) and means are reported in the paper or obtainable from the study authors; One study provided raw data and allowed calculation of the SD. See Results section and for the calculation Table 2 and Table 3;
-
if a scale started from the finite number zero, the SD, when multiplied by two, would be less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution, (Altman 1996);
-
if a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), (Kay 1986)) which can have values from 30 to 210), the calculation described above will be modified to take the scale starting point into account. In these cases skew would be present if 2 SD > (S‐S min), where S is the mean score and 'S min' is the minimum score.
| Individual data | Mean | Sum of mean squares | SD | |||||||||||||||
| CGI severity score clopenthixol | 3 | 4 | 4 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 6 | 6 | 6 | 6 | 6 | 5.07 | 396 | 0.88 |
| Individual data | Mean | Sum of mean squares | SD | |||||||||||||
| CGI Scores Placebo | 4 | 5 | 5 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 5.69 | 426 | 0.63 |
Endpoint scores on scales often have a finite start and end point and these rules can be applied. Skewed data pose less of a problem when looking at means if the sample size is large (> 200) but no large studies were found. If found we would have presented skewed endpoint data from studies of less than 200 participants as other data within the 'Data and analyses' section rather than enter such data into statistical analyses.
When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. We intended to present and enter change data into statistical analyses but none were found.
2.5 Common measure
To facilitate comparison between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month), however, no relevant data were found.
2.6 Conversion of continuous to binary
In one study we converted outcome measures to dichotomous data. This was done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS) (Overall 1962), or the PANSS (Kay 1986), this could be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). However, the study Serafetinides 1972 included data on clinical global impression (CGI) improvement scores. In order to analyse these data the grouping of 'worse or no better' was considered as a negative outcome. 'Better' was considered an improvement and a positive event. Please see Results for details.
2.7 Direction of graph
We entered data in such a way that the area to the left of the line of no effect indicates a favourable outcome for zuclopenthixol. Had keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not un‐improved'), we would have reported data where the left of the line indicates an unfavourable outcome. This was not a problem in this review.
Assessment of risk of bias in included studies
Originally review authors ML and CE planned to work independently to assess risk of bias by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b) to assess trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting. As CE was unable to participate MJ took over this role.
There were no disagreements, but If the raters had disagreed, we would have made the final rating by consensus, with the involvement of another member of the review group. Where inadequate details of randomisation and other characteristics of trials are provided, we attempted to contact the authors of the studies in order to obtain further information, but were unable to find further information.
The level of risk of bias is noted in both the text of the review and in the 'Summary of findings' table.
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). The number needed to treat for an additional beneficial or harmful outcome (NNTB/NNTH) statistic with its confidence intervals is intuitively attractive to clinicians but is problematic both in its accurate calculation in meta‐analyses and interpretation (Hutton 2009). It was not used in this review. For binary data presented in the 'Summary of findings' table, where possible, we calculated illustrative comparative risks. Data on our graphs read, from left to right, making them as clear as possible.
2. Continuous data
For continuous outcomes, we estimated MD between groups. We did not have to calculate effect size measures (SMD).
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, study authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
There were no cluster trials identified, however, had clustering not been accounted for in primary studies, we intended to present data in a table, with a ' * ' symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we would seek to contact first authors of studies to obtain intra‐class correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999).
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC [Design effect = 1 + (m ‐ 1) X ICC] (Donner 2002). If the ICC is not reported, it will be assumed to be 0.1 (Ukoumunne 1999).
If cluster studies had been appropriately analysed taking into account ICCs and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
2. Cross‐over trials
No cross over trials were identified, however a major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). .
3. Studies with multiple treatment groups
Where a study involves more than two treatment arms, if relevant, the additional treatment arms could be presented in comparisons. If data are binary these will be simply added and combined within the two‐by‐two table. If data are continuous, we will combine data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). Where the additional treatment arms are not relevant, we will not use these data. Both of our included studies had other arms but these were not felt to be relevant.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). We decided that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data or use them within analyses. Neither of our included studies lost more than this amount of data. If they had, and if more than 50% of data in one arm of a study had been lost, but the total loss was less than 50%, we would have addressed this within the 'Summary of findings' table/s by down‐rating quality. Finally, we would have downgraded quality within the 'Summary of findings' table/s should loss have been 25% to 50% in total.
2. Binary
If attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we have presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis). Those leaving the study early are all assumed to have the same rates of negative outcome as those who completed, with the exception of the outcomes of death and adverse effects. If any data had been identified for these outcomes, the rate of those who stay in the study ‐ in that particular arm of the trial ‐ would have been used for those who did not. We intended to undertake a sensitivity analysis to test how prone the primary outcomes are to change when data only from people who complete the study to that point are compared with the intention‐to‐treat analysis using the above assumptions. However, we found insufficient data for this analysis.
3. Continuous
3.1 Attrition
If attrition for a continuous outcome was between 0% and 50%, and data only from people who completed the study to that point were reported, we reproduced these data.
3.2 Standard deviations (SDs)
In several data sets SDs were not reported. We first tried to obtain the missing values from the study authors. If these were not available, but an exact standard error (SE) and CIs had been available for group means, and either P value or t value had been available for differences in mean, we could have calculated them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). When only the SE is reported, SDs could be calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a; Higgins 2011c) present detailed formulae for estimating SDs from P values, t or F values, CIs, ranges or other statistics. However, we calculated the SDs according to a validated imputation method which is based on the SDs of the included studies (Furukawa 2006). Please see Results section and for the calculation Table 2 and Table 3. Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. We nevertheless intended to examine the validity of the imputations in a sensitivity analysis but there was insufficient data for this.
3.3 Last observation carried forward
We anticipated that in some studies the method of last observation carried forward (LOCF) would be employed within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, had LOCF data been used in the trial, and if fewer than 50% of the data had been assumed, we would have presented and used these data and indicated that they were the product of LOCF assumptions. No studies using this method were identified.
Assessment of heterogeneity
1. Clinical heterogeneity
Only a small number of studies were identified making heterogeneity analysis difficult. We would have considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We would simply have inspected all studies for clearly outlying people or situations which we had not predicted would arise. If such situations or participant groups had arisen, we would have discussed them fully.
2. Methodological heterogeneity
We would have considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. If such methodological outliers had arisen we would have discussed them.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
Heterogeneity between studies could have been investigated by considering the I2 statistic alongside the Chi2 P value. The I2 statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on the magnitude and direction of effects, and the strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a CI for I2). An I2 statistic estimate greater than or equal to around 50%, accompanied by a statistically significant Chi2 statistic, will be interpreted as evidence of substantial levels of heterogeneity (Deeks 2011). If we had found substantial levels of heterogeneity in the primary outcome, we would have explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity), however, insufficient data were found.
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in section 10.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2011). We tried to locate protocols of the included randomised trials. However, given the age of the studies involved none were identified. If the protocols had been available, we would have compared outcomes in the protocol and in the published report.
2. Funnel plot
We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We intended not to use funnel plots for outcomes where there are 10 or fewer studies, or where all studies are of similar sizes, and hence we have not included any.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model. It puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose to use the random‐effects model for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1 Primary outcomes
As there are three different formulations for Zuclopenthixol, we anticipated there may be different effects from each as they commonly have different indications for use. We intended therefore to undertake a subgroup analysis of the different formulations if there is significant heterogeneity. We found data only on one formulation so were unable to conduct this analysis.
1.2 Clinical state, stage or problem
We undertook this review to provide an overview of the effects of zuclopenthixol for people with schizophrenia in general. In addition, however, we would try to report data on subgroups of people in the same clinical state, stage and with similar problems if possible. Again we were unable to conduct this analysis as insufficient data were found.
2. Investigation of heterogeneity
As very few data were found it was difficult to investigate or analyse further. If inconsistency had been high, this would have been reported. First we would have investigated whether data had been entered correctly. Secondly, if data were correct, we would have visually inspected the graph, and successively removed outlying studies to see if homogeneity was restored. For this review, we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, data would be presented. If not, data would not be pooled and issues would be discussed. We know of no supporting research for this 10% cut off but are investigating use of prediction intervals as an alternative to this unsatisfactory state.
If unanticipated clinical or methodological heterogeneity had been obvious, we would have stated hypotheses regarding these for other reviews or future versions of this review.
Sensitivity analysis
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. However, not all trials clearly state randomising methods, so we have used the data available. Further sensitivity analysis of this is difficult.
2. Assumptions for lost binary data
There were no losses for the binary data. However, if assumptions had been made regarding people lost to follow‐up (see Dealing with missing data), we would have compared the findings of the primary outcomes when we used our assumptions and when we used data only from people who had completed the study to that point. If there had been substantial differences, we would have reported results and discussed them, but would continue to employ our assumption.
No assumptions were made with the SD data. Had assumptions been made regarding missing SD data (see Dealing with missing data), we would have compared the findings of the primary outcomes when we used our assumptions and when we used data only from people who had completed the study to that point. A sensitivity analysis would have been undertaken to test how prone results were to change when completer‐only data only were compared to the imputed data using the above assumption. If there had been a substantial difference, we would have reported results and discussed them.
3. Risk of bias
We planned to analyse the effects of excluding trials that are judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available) allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. However, as we only found two trials this was unnecessary. If the exclusion of trials at high risk of bias had not substantially altered the direction of effect or the precision of the effect estimates, then we would have included data from these trials in the analysis.
4. Imputed values
We planned to undertake a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster randomised trials but we did not find any cluster trials.
5. Fixed and random effects
All data were synthesised using a random‐effects model, however, we also synthesised data for the primary outcome using a fixed‐effect model to evaluate whether this would alter the significance of the results, which It did not. If there had been a difference between the fixed‐effect and random‐effects models then we would have considered whether it is reasonable to conclude that the intervention was more effective in the smaller studies, as the fixed‐effect model gives a greater weight to smaller studies.
Results
Description of studies
For full details please see, Characteristics of included studies and Characteristics of excluded studies.
Results of the search
The search was conducted on 23 January 2013. It yielded 15 records of potential interest. Two records were duplications, so we screened 13 records. There were several reports from Serafetinides. We read these in full and all but one, Serafetinides 1971, were felt to be part of the same study, though this was confusing at times, as the excluded studies by Clark 1970 and Clark 1970a included Seraftinides as a third author. We read the remaining articles in full and judged them to be from one study, the data being used in different ways to look at different outcomes. We read all the Seraftinides group of studies in full to try and identify other pertinent data. We judged a further study, Kordas 1968 was also suitable for inclusion. For details of the decision process see Figure 2.

Processing search results
Both the included studies were old and we attempted to contact both groups of study authors. We could not identify the primary author's current contact details in Kordas 1968, so we contacted the hospital where the study was undertaken but received no response. For Serafetinides 1972, again we could not identify a current contact for the author and sent an email to the University of California's media relations as a general enquiry. They provided us with a copy of the article but were unable to provide any other data. We also contacted Lundbeck who manufacture zuclopenthixol and they stated they had no further data as, given the age of the product, all their data were in the public domain.
Included studies
We were able to include two studies, both randomising zuclopenthixol dihydrochloride compared to placebo. We identified no placebo‐controlled studies of zuclopenthixol acetate or zuclopenthixol decanoate. For full details please see Characteristics of included studies.
1. Design
In Kordas 1968 participants had a washout period and then were rated on behavioural scales. They were allocated into three arms, zuclopenthixol dihydrochloride (Clopenthixol), chlorpromazine and placebo; after four weeks of medication they were re‐rated. The allocation method is unclear. Serafetinides 1972 is a single‐centre four‐armed trial. Patients underwent a washout period and then had a baseline assessment. They were then "randomly assigned" to receive either zuclopenthixol dihydrochloride (Clopenthixol), )chlorpromazine, haloperidol or placebo. After 12 weeks participants were assessed again.
2. Sample sizes
In Kordas 1968 the total number of participants was n = 54, with n = 18 receiving zuclopenthixol dihydrochloride (Clopenthixol), n = 18 receiving chlorpromazine and n = 18 receiving placebo. Serafetinides 1972 has n = 57 patients, with n = 15 receiving zuclopenthixol dihydrochloride (Clopenthixol), n = 14 receiving chlorpromazine, n = 14 receiving haloperidol, and n = 14 receiving placebo.
3. Setting
Kordas 1968 was set in an inpatient unit in Greece. Serafetinides 1972 was set in a single inpatient hospital in the United states of America. They separated men and women into different wings.
4. Participants
All participants in Kordas 1968 had a diagnosis of schizophrenia of at least six years' duration, although no standard diagnostic criteria was identified and no exclusions described. All participants in Serafetinides 1972 had a diagnosis of chronic schizophrenia and again no standard criteria were identified. They excluded those with "organic disease".
5. Interventions
Kordas 1968 had three arms, two of interest, with zuclopenthixol dihydrochloride (Clopenthixol) (oral, dose 50 mg three times a day), and a placebo arm. They also included a chlorpromazine treatment arm. All sets of tablets were identical. In Serafetinides 1972 participants received zuclopenthixol dihydrochloride (oral average dose of 205 mg) or placebo. There were also chlorpromazine and haloperidol treatment arms.
6. Outcomes
In Kordas 1968 the only useable data are reported around side effects, as the behavioural scales are not useable (no mean or SD). They did not report losses. Serafetinides 1972 reported CGI improvement and severity data, as well as side effects and laboratory information. They did not report the SDs for the CGI data but provided the raw scores and allowed calculation of the SD, see Table 2; Table 3 They did provide BPRS, Nurse observation scale for inpatient evaluation, Oklahoma behaviour rating scale and data on the Venables‐O'connor scale but they did not report the SDs for these scales nor the raw scores, and so the data are unusable. The group did report their losses.
6.1 Outcome scales
For information we describe below the outcome scales and their uses in the studies.
6.1.1 Behavioural and inferential scale
This scale has two parts, a behavioural and inferential scale. The behavioural scale has 64 items that are considered more objective and observable, examples include, 'does not talk to patients', 'eats alone', 'attacks self'. The inferential scale contains 49 items considered less objective and includes 'poor memory', 'seems anxious' and 'grandiose feelings'. Each item is scored 0 to 4 with 0 not present and 4 maximum presence (Kordas 1968). This is a rating system that does not seem to have been especially successful. It is quite complicated and the difference between some of the 'objective' and 'subjective' behavioural markers seems crude.
6.1.2 Clinical global impression (CGI) scales
This is a global state scale used to assess both severity of illness and clinical improvement. It is observer‐rated (Guy 1976). The scale compares the condition of the person standardised against other people with the same diagnosis. It uses a seven‐point scoring system with low scores showing decreased severity and overall improvement.
6.1.3 Brief psychiatric rating scale
The BPRS (Overall 1962) is a scale about mental state that is used to assess the severity of abnormal mental state. It is observer‐rated, and different versions have different scales. The most common has 18 items and includes areas such as somatic concern, anxiety and grandiosity. Each item is scored on a seven‐point scale varying from 'not present' to 'extremely severe’ with a corresponding numerical value (1 to 7 or 0 to 6). The patient is then given an overall score.
6.1.4 Nurses' observational scale for inpatient evaluation.
The NOISE scale (Honigfeld 1965) is an observational scale designed to be a quick ward‐based observer rating of behaviour, it is rated on observation for the last three days. It has 30 items including examples such as social competence, social neatness irritability and manifest psychosis. Each item is rated 0 to 4 as never (0) sometimes, often and always (4). It is not as popular as the BPRS or CGI scales but still seems to be relatively commonly used.
6.1.5 Oklahoma behavioural rating scale
This was developed in 1961 (Clark 1961). It consists of nine items such as activity, aggression, communicativeness and toilet behaviour. Each item is rated on a 13‐point scale, from ‐6 to 6, with 0 in the middle: 0 represents typical appropriate behaviour. The plus behaviour is outgoing or overactive and the minus represents withdrawal and negative‐like behaviour.
Seven separate summary scores are then derived from this scale, each providing information different from the other. For example TB, the 'Typical Behaviour' score is the summation of item scores disregarding the direction from zero. It is the extent of deviation of behaviour reflected on the scale and can be referred to as 'gross behavioural pathology score'.Another example is 'Typical Behaviour plus' score which is the summation of item scores which fall on the plus side of zero. Over‐activity and outgoingness were thus analysed separately from their opposite.
This is then presented in tables as TB+ or E+ and comparisons are made before and after treatment. Overall this is a confusing scale that is quite difficult to use which is likely the reason for its unpopularity.
6.1.6 Venables‐O’Connor scale
This is an observational scale that rates patients on 20 items such as hostility, cleanliness or bizarre behaviour. Each item is rated 1 to 5, with 1 being normal and 5 being an extreme case. The definition of the number does change with each item. These scores are then collated to create a form of factor analysis and give scores on various factors such as paranoid and withdrawal factors (Venables 1959). This is a score system that has not remained popular over the years.
Excluded studies
For full details please see Characteristics of excluded studies. We excluded five studies, all as the interventions were not relevant to the review.
There are currently no studies awaiting classification nor any ongoing studies that we are aware of.
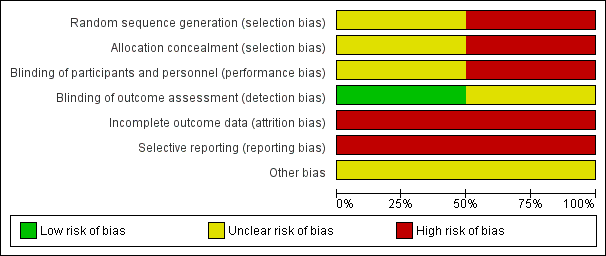
Risk of bias in included studies
For full details please see the risk of bias tables associated with Characteristics of included studies. Given that both studies are of a relatively old design there are some quite significant potential sources of bias. Summaries of risk of bias across the trials can also be viewed in Figure 3 and Figure 4.
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Sequence generation: Kordas 1968 states that 'patients' were allocated to the treatment arms but does not state that this was randomised and gives no description of allocation concealment. We felt this to be a high risk of bias. Serafetinides 1972 states that patients were 'randomly assigned' but gives no information on the method or use of allocation concealment. We judged this trial to have an unclear selection bias. The numbers in the treatment groups in this study are also confusing. The numbers in 'Table 2' in Serafetinides 1972 show 13 in the placebo group. However, the text says that one individual in the placebo group failed to complete. The total number in their table comes to 56 indicating that someone has been lost. We have inferred that this person has come from the placebo group and this group started with 14 participants.
Blinding
Kordas 1968 had an unclear risk of performance and low detection bias. Study authors state that patients and raters were blinded but that medication was distributed by the attending physician: it was unclear what involvement they had. Serafetinides 1972 says that the participants were blinded but makes no comment about the staff. Clinicians could alter the dose of medication and therefore were clearly not blinded. The CGI scores were rated by both nurses and psychiatrists and it is unclear if they were blinded to the treatment. We therefore rated this trial as high risk of bias.
Incomplete outcome data
Both trials had a high risk of attrition bias. In Kordas 1968 the outcome data are incomplete: they have presented variance charts between the different comparison arms but have not provided any actual description of losses. They provide side effect data for the intervention arm but none for the placebo arm. We have assumed this means no significant side effects in the placebo group. Serafetinides 1972 have described their losses and report that four participants left early, three from the chlorpromazine arm and only one from the placebo, and none from the zuclopenthixol dihydrochloride (Clopenthixol) arm. However, when describing side effects they report that one participant taking zuclopenthixol dihydrochloride experienced persistent liver abnormalities which "resulted in withholding medication". The participant underwent a liver biopsy showing steatosis; and did not recover. They do not, however, describe how this participant's outcome was included in the data. They make no reference as to whether they were included in the results or became a loss, or if the medication was withheld for a short period of time and restarted.
Selective reporting
In both studies the risk is high as they have not described any agreed protocol prior to commencement, nor does either study state clear objectives.
Other potential sources of bias
We did not identify other potential sources of bias.
Effects of interventions
See: Summary of findings for the main comparison ZUCLOPENTHIXOL DIHYDROCHLORIDE versus PLACEBO for schizophrenia; Summary of findings 2 ZUCLOPENTHIXOL ACETATE versus PLACEBO for schizophrenia; Summary of findings 3 ZUCLOPENTHIXOL DECANOATE versus PLACEBO for schizophrenia
We identified three different zuclopenthixol preparations and so, potentially three different relevant comparisons with placebo.
1. ZUCLOPENTHIXOL DIHYDROCHLORIDE versus PLACEBO for schizophrenia
We identified two studies that compared zuclopenthixol dihydrochloride (Clopenthixol) with placebo.
1.1 Clinically signficant response
1.1.1 Improvement (CGI) ‐ short term
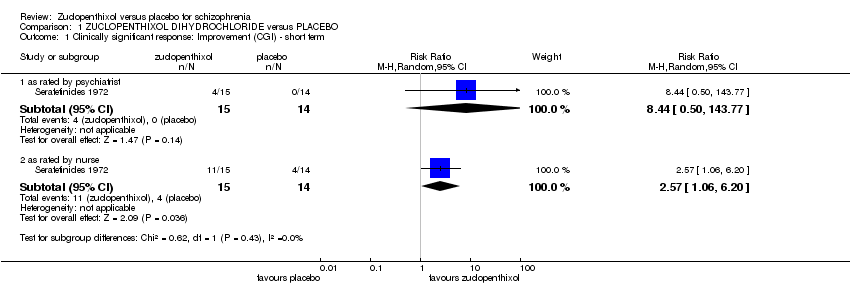
Serafetinides 1972 included data on CGI improvement scores. In order to analyse these data the grouping of worse or no better was considered as a negative outcome. Better was considered an improvement and a positive event. These were rated by both a nurse and a psychiatrist with different scores for each. The psychiatrist rated data (1 RCT n = 29, RR 8.44, 95% CI 0.50 to 143.77) in favour of zuclopenthixol but not significant. The nurse rated data (1 RCT n = 29, RR 2.57 95% CI 1.06 to 6.20) also in favour of zuclopenthixol but statistically significant, Analysis 1.1.
1.1.2 Average score of severity of illness ‐ short term (CGI, high score = poor)
Serafetinides 1972 provided data on CGI severity of illness. The data presented in the paper included the initial and final means for the placebo and treatments arms. Although the SDs were not included they could be calculated from the data presented, see Table 2 and Table 3. Those in the zuclopenthixol group had lower mean scores (1 RCT n = 29, MD ‐0.62, 95% CI ‐1.17 to ‐0.07), Analysis 1.2.
1.2. Other adverse effects, general and specific.
1.2.1 Extrapyramidal effects
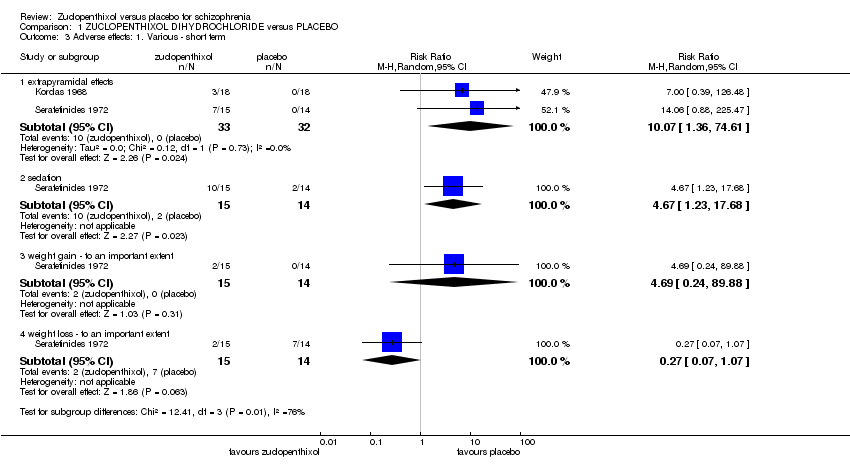
Both studies included some data on extrapyramidal side effects. In Kordas 1968 they described two participants who had extrapyramidal side effects and one who had an oculogyric crisis. Although not explicit in the text we assume that these were three separate participants. The pooled effect indicates zuclopenthixol is more likely to cause side effects (2 RCTs n = 65, RR 10.07, 95% CI 1.36 to 74.61) Analysis 1.3
1.2.2 Sedation
Serafetinides 1972 included some description of sedative effects. They described sedation as 'transient and persistent' but did not define what these mean. These have been combined to give a single category of sedation and shows those taking zuclopenthixol are more likely to become sedated (1 RCT n = 29 RR, 4.67, 95% CI 1.23 to 17.68) Analysis 1.3
1.2.3 Weight change
Again Serafetinides 1972 included some data on weight change in participants. They included data on both losing and gaining weight and in both cases considered a significant change to be a loss or gain of 4.5 kg (10lb) and thus measured as a dichotomous outcome. There were no statistically significant changes for either weight gain or weight loss with zuclopenthixol. (Weight gain: 1 RCT n = 29, RR 4.69, 95%CI 0.24 to 89.88 and weight loss: 1 RCT n = 29, RR 0.27 95% CI 0.07 to 1.07). Analysis 1.3
1.2.4 Abnormal laboratory values
Serafetinides 1972 included data on a variety of abnormal laboratory values. Details of urine analysis looking at the presence of white blood cells (WBC) and sugar were presented and we have pooled these values as urine abnormalities for simplicity (1 RCT n = 29, RR 1.87 95% CI 0.19 to 18.38).
Data were presented for haematological abnormalities of raised WBC (> 12000) eosinophilia (> 5%) and WBC < 4000. These are pooled (1 RCT n = 29, RR 1.87 , 95% CI 0.40 to 8.65). They also reported blood urea nitrogen of > 20 mg per cent but no patients in the zuclopenthixol or placebo arms experienced this.
For data on liver abnormalities raised serum glutamic‐pyruvic transaminase (SGPT) of > 49 units or a total bilirubin of > 1.4 mg per cent were reported. The total pooled effects for liver abnormalities is (1 RCT n = 29, RR 8.44, 95% CI 0.50 to 143.77). The body of text describes that one participant had a persistent raised SGPT and required medication withdrawal and liver biopsy showing steatosis. They then recovered.
Cholesterol data were also presented. They reported no patients in either the placebo or zuclopenthixol arm had a cholesterol > 300 mg per cent; however, one participant taking zuclopenthixol had a cholesterol < 170 mg per cent (1 RCT n = 29, RR 2.81,95% CI 0.12 to 63.83). They also reported data for participants with triglycerides > 4.6 mEq/l (1 RCT n = 29, RR 2.80, 95% CI 0.33 to 23.86).
The final value reported is zinc. One participant in the placebo arm had a zinc level of < 70 mg per cent (1 RCT n = 29, RR 0.31, 95% CI 0.01 to 7.09). One participant in the zuclopenthixol arm had a zinc level of > 120 mg per cent (1 RCT n = 29, RR 2.81, 95% CI 0.12 to 63.83). Analysis 1.4
1.3 Leaving the study early
Kordas 1968 did not describe any losses, nor were raw data sets made available, making it impossible to determine any losses. Serafetinides 1972 described one participant lost in the placebo arm, as they became disturbed and required treatment. It is unclear about losses in the zuclopenthixol treatment arm as they stated that one participant had a persistent raised SGPT and required medication withdrawal and liver biopsy showing steatosis. The participant then recovered but it is unclear if this person resumed the trial treatment or not. They do not give any information about at what point in the study this occurred. We have assumed that this person was lost to follow up as it seems unreasonable to assume that they resumed the same medication after such a clinically significant event. No overall difference in numbers leaving the study early was found (1 RCT n = 29, RR 0.93, 95% CI 0.06 to 13.54). Analysis 1.5.
1.4 Missing outcomes
It is clear from previous trials that not reporting missing outcomes usually tends to favour the intervention arm rather than the control arm. It is disappointing that there were no data found on many of the outcomes of interest we listed in Methods . These include death, mental state, service utilisation outcomes, quality of life and economic outcomes. Given the age and widespread use of zuclopenthixol dihydrochloride it was anticipated that more data would be found. Althouh we were able to glean that some participants had left the study due to a particular reason, it was not clear whether they subsequently resumed the study or not.
We were hoping that there might be additional data to make comparisons of zuclopenthixol acetate versus placebo for schizophrenia and zuclopenthixol decanoate versus placebo for schizophrenia, however we were unable to identify any studies that included data on these two fairly widely used drugs.
Discussion
We found no trials at all investigating the absolute effects of zuclopenthixol acetate (summary of findings Table 2) or decanoate (summary of findings Table 3).
Summary of main results
1. ZUCLOPENTHIXOL DIHYDROCHLORIDE compared to PLACEBO for schizophrenia
Some data were found for zuclopenthixol dihydrochloride vs placebo (summary of findings Table for the main comparison). This was of a very low quality overall. The data are only for short term effects as both studies were of only 12 weeks' duration.
1.1 Clinically significant response on global state CGI improvement scores for nurse follow‐up
The data did show a significant response on global state as rated by nursing staff, however this was not corroborated by the psychiatrists' rating on the same patients, which highlights some perceived differences in improvement/lack of improvement rates (1 RCT, n = 29, RR 2.57, 955 CI 1.06 to 6.20, very low quality data). These data are too limited and unreliable to allow us to draw any clinical conclusion. It is surprising that no useful data exist for this comparison, for this outcome.
1.2 Other adverse events
The data also show an increase in sedation and extrapyramidal side effects for those who were given zuclopenthixol dihydrochloride. However these data were all of very low quality and from a very small number of participants (sedation: 1 RCT, n = 29, RR 4.67, 95% CI 1.23 to 17.68, very low quality data; extrapyramidal effects: 2 RCTs, n = 65, RR 10.07, 95% CI 1.36 to 74.61, very low quality data).
1.3 Missing
That we found no data on ' Death, Mental state, Service utilisation outcomes, Quality of life and Economic outcomes. is disappointing given the well established use of zuclopenthixol dihydrochloride.
2. ZUCLOPENTHIXOL ACETATE versus PLACEBO for schizophrenia
No data were found on any of the outcomes of interest (summary of findings Table 2). There are other non‐placebo comparisons of this preparation (Jayakody 2012) but none comparing it with giving placebo. Recognising that such trials will probably now never take place, this might be a reason for a network meta‐analysis, where comparisons are created by a more indirect route.
3. ZUCLOPENTHIXOL DECANOATE versus PLACEBO for schizophrenia
No data were found on any of the outcomes of interest (summary of findings Table 3). For severe mental illness, depot injections are used in about 25% to 33% of patients and amongst the medications used, zuclopenthixol decanoate is one of the most common (Barnes 2009). In spite of this there are no data to either support its ongoing use nor to indicate that it is not to be used. In the absence of hard data, clinicians appear to be guided in their practice by what has been taught to them over the years and personal experience.
Overall completeness and applicability of evidence
1. Completeness
For zuclopenthixol hydrochloride versus placebo, both included studies had significant amounts of data that were incomplete. Kordas 1968 presented a self‐designed behavioural and inferential scale that is described in detail in Results. They have calculated the variance using an analysis of variance (ANOVA) between different factors comparing placebo and zuclopenthixol arms and claim to have found some sources of this variance to be significant. However, they presented no raw data, only the variance charts which are unusable. They did provide sum of squares, degrees of freedom, mean squares and some F statistics. We looked into alternative methods of calculating SD from this data but none were felt to be appropriate (Schünemann 2011). Given the complexity of this, the review authors discussed the statistical methods and the consensus was that the data were unusable.
Serafetinides 1972 presented a number of different behavioural scales that, again are potentially informative but include too little detail to allow inclusion. They presented BPRS data that included mean and they have stated ACOVA scores and whether regressionwas significant, but they did not provide the P value, just stated 'not significant', without actually defining what value was considered significant. They showed data for the Nurses' observational scale for inpatient evaluation (NOISE). Again this was presented in a table format with final mean values, ACOVA score and regression but no raw data or SDs. The Oklahoma behavioural rating scale and Venables‐O’Connor rating scales were presented in a similar manner with little useable data
The were no data at all found for zuclopenthixol acetate or decanoate.
2. Applicability
There are some difficulties in applying this evidence to modern psychiatric practice. The data included is around 40 years old and includes some data from the USA and Greece. The population studied in both trials was inpatients with chronic schizophrenia. The inpatient population in modern hospitals is likely to be very different from these given changes in working practice. This makes applicability difficult. The included studies were only undertaken over a short period of time. This makes it difficult to apply this evidence to the more chronic setting. The small study sizes also make the applicability and usefulness difficult to interpret. There is no data at all for zuclopenthixol acetate or decanoate which makes it impossible to comment on the applicability.
Quality of the evidence
Overalll the quality of the evidence is very low. We have only been able to identify two studies for zuclopenthixol dihydrochloride which have small sample sizes. Only one study showed useable primary outcome data. We have demonstrated some significant results but these have large confidence intervals and a number of them are only just above the threshold for statistical significance.
Both studies are very old and have significant methodological flaws. We feel it is unlikely that they would pass modern peer review standards. Only one study states that it was randomised but gives no details of how, the other study we assumed to be randomised from the methods description. We have concerns over both studies' blinding of assessors, although they both seemed to blind the participants satisfactorily.
Both studies only present limited outcome data, with very few raw score data sets given, and often vague and confusing data presentation. We have had to make assumptions when handling certain aspects of the data due to the difficulties in the reports. Overall the quality of evidence was low, with significant risks of bias. Please see Figure 3 for risk of bias graph and Figure 4 for risk of bias summary. In practical terms the quality of this evidence is so poor that in reality no meaningful data were found.
The studies we have found are both on Clopenthixol which, as it is given as a tablet, is taken to mean zuclopenthixol dihydrochloride. We have found no useable data on zuclopenthixol acetate or decanoate. This is disappointing given the wide use of all forms of zuclopenthixol.
Potential biases in the review process
The search process only identified a number of small trials. It is possible that other small trials (e.g. conducted for an individual's research projects) may exist and may not have been identified.
Agreements and disagreements with other studies or reviews
Kumar 2005 is a review of zuclopenthixol dihydrochloride and includes a versus‐placebo review. Our findings are consistent with these. They have included a number of the same studies, however, we have felt able to calculate the SDs of the CGI data from Serafetinides 1972 and use this in our review whereas Kumar 2005 did not. An updated version of this review is underway and we have seen an early version of this. Broadly again there is agreement with the versus‐placebo data they have found. Kumar 2005 felt confident to accept that the Kordas 1968 study had no losses of follow up. In Serafetinides 1972 there is a difference in interpretation in the numbers in the placebo group. We have taken the one person lost to be excluded from the final number as the total does not reach 57. This means our placebo arm is 14 and Kumar 2005's is 13. Overall this does not change the significance of the results and likely reflects the poor methodology and recording of the study as previously described.
The data on adverse outcomes is consistent with the product‐characteristic and clinical‐pragmatic experience of using zuclopenthixol.

Zuclopenthixol structure
Processing search results

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Comparison 1 ZUCLOPENTHIXOL DIHYDROCHLORIDE versus PLACEBO, Outcome 1 Clinically significant response: Improvement (CGI) ‐ short term.

Comparison 1 ZUCLOPENTHIXOL DIHYDROCHLORIDE versus PLACEBO, Outcome 2 Global state: Average score of severity of illness ‐ short term (CGI, high score=bad).
Comparison 1 ZUCLOPENTHIXOL DIHYDROCHLORIDE versus PLACEBO, Outcome 3 Adverse effects: 1. Various ‐ short term.

Comparison 1 ZUCLOPENTHIXOL DIHYDROCHLORIDE versus PLACEBO, Outcome 4 Adverse effects: 2. Laboratory values ‐ abnormal ‐ short term.

Comparison 1 ZUCLOPENTHIXOL DIHYDROCHLORIDE versus PLACEBO, Outcome 5 Leaving the study early.
| Excluded Study | Comparison | Existing review |
| Chlorpromazine versus molindone | ||
| Chlorpromazine versus placebo | ||
| Chlorpromazine versus placebo | ||
| Clopenthixol versus risperidone | ||
| D‐fenfluramine versus placebo |
| Methods | Allocation: randomised, full description of methods of randomisation and allocation concealment. |
| Participants | Diagnosis: people with schizophrenia (according to a diagnostic criteria). |
| Interventions | 1. Zuclopenthixol dihydrochloride. N = 150. 2. Placebo. N = 150. |
| Outcomes | Global state: clinically important response to treatment, average score/change of the global state. Mental state: general measurement and specific domains (depressive symptoms, positive symptoms, negative symptoms). Leaving the study early, due to any reason, due to inefficacy of treatment, and due to adverse events. Adverse events: any serious adverse event recorded. Service use: number of hospitalisations, days in hospital. |
| Methods | Allocation: randomised, fully explicit description of methods of randomisation and allocation concealment. |
| Participants | Diagnosis: people with schizophrenia (according to a diagnostic criteria). Acutely disturbed behaviour as described by the study |
| Interventions | 1. Zuclopenthixol acetate either alone or in combination with other medications. n = 150 2. Placebo. n = 150 |
| Outcomes | Global state: clinically important response to treatment, average score/change of the global state. Mental state: general measurement and specific domains (such as depressive symptoms, positive symptoms, negative symptoms) Leaving the study early, due to any reason, due to inefficacy of treatment, and due to adverse events. Adverse events: any serious adverse event recorded. Pharmacological interactions. |
| ZUCLOPENTHIXOL DIHYDROCHLORIDE versus PLACEBO for schizophrenia | ||||||
| Patient or population: people with with schizophrenia | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | ZUCLOPENTHIXOL DIHYDROCHLORIDE versus PLACEBO | |||||
| Clinically significant response on global state ‐ as defined by each of the studies, Improvement (CGI) ‐ short term ‐ as rated by nurse | Study population | RR 2.57 | 29 | ⊕⊝⊝⊝ | For this SOF table outcome CGI data as rated by a nurse and a psychiatrist were both available. The nurse data were chosen as it includes both control event and | |
| 286 per 1000 | 734 per 1000 | |||||
| Relapse as defined by the studies | No studies reported these important outcomes | |||||
| Clinically significant response on psychotic symptoms ‐ as defined by each of the studies. | ||||||
| Adverse effects: Sedation | Low | RR 4.67 | 29 | ⊕⊝⊝⊝ | No use of formal rating scales.Several other adverse events were recorded but sedation considered important. | |
| 50 per 1000 | 234 per 1000 | |||||
| Moderate | ||||||
| 150 per 1000 | 701 per 1000 | |||||
| High | ||||||
| 250 per 1000 | 1000 per 1000 | |||||
| Leaving the study early | Study population | RR 0.93 | 29 | ⊕⊝⊝⊝ | ||
| 100 per 1000 | 93 per 1000 | |||||
| Average change in quality of life/satisfaction | No studies reported these important outcomes | |||||
| Significant change in quality of life/satisfaction ‐ as defined by each of the studies | ||||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Risk of bias: rated 'very serious' ‐ randomisation method unclear as describes "randomly assigned". Incomplete outcome data and does not accurately describe losses. Patients blinded but unclear if raters and clinicians are blinded | ||||||
| ZUCLOPENTHIXOL ACETATE versus PLACEBO for schizophrenia | |||||
| Patient or population: people with schizophrenia | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | |
| Assumed risk | Corresponding risk | ||||
| Control | ZUCLOPENTHIXOL ACETATE versus PLACEBO | ||||
| Clinically significant response on global state | No studies reported any of these important outcomes | ||||
| Relapse as defined by the studies | |||||
| Clinically significant response on psychotic symptoms ‐ as defined by each of the studies. | |||||
| Other adverse effects, general and specific | |||||
| Leaving the study early | |||||
| Average change in quality of life/satisfaction | |||||
| Significant change in quality of life/satisfaction ‐ as defined by each of the studies | |||||
| ZUCLOPENTHIXOL DECANOATE versus PLACEBO for schizophrenia | |||||
| Patient or population: people with schizophrenia | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | |
| Assumed risk | Corresponding risk | ||||
| Control | ZUCLOPENTHIXOL DECANOATE versus PLACEBO | ||||
| Clinically significant response on global state | No studies reported any of these important outcomes | ||||
| Relapse as defined by the studies | |||||
| Clinically significant response on psychotic symptoms ‐ as defined by each of the studies | |||||
| Other adverse effects, general and specific | |||||
| Leaving the study early | |||||
| Average change in quality of life/satisfaction | |||||
| Significant change in quality of life/satisfaction ‐ as defined by each of the studies | |||||
| Focus of the review | Participants | Reference |
| Zuclopenthixol acetate | acutely ill people with schizophrenia | |
| Zuclopenthixol decanoate | people with schizophrenia | |
| Zuclopenthixol dihydrochloride | people with schizophrenia |
| Individual data | Mean | Sum of mean squares | SD | |||||||||||||||
| CGI severity score clopenthixol | 3 | 4 | 4 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 6 | 6 | 6 | 6 | 6 | 5.07 | 396 | 0.88 |
| Individual data | Mean | Sum of mean squares | SD | |||||||||||||
| CGI Scores Placebo | 4 | 5 | 5 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 5.69 | 426 | 0.63 |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinically significant response: Improvement (CGI) ‐ short term Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 as rated by psychiatrist | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 8.44 [0.50, 143.77] |
| 1.2 as rated by nurse | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 2.57 [1.06, 6.20] |
| 2 Global state: Average score of severity of illness ‐ short term (CGI, high score=bad) Show forest plot | 1 | 29 | Mean Difference (IV, Random, 95% CI) | ‐0.62 [‐1.17, ‐0.07] |
| 3 Adverse effects: 1. Various ‐ short term Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 extrapyramidal effects | 2 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 10.07 [1.36, 74.61] |
| 3.2 sedation | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 4.67 [1.23, 17.68] |
| 3.3 weight gain ‐ to an important extent | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 4.69 [0.24, 89.88] |
| 3.4 weight loss ‐ to an important extent | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 0.27 [0.07, 1.07] |
| 4 Adverse effects: 2. Laboratory values ‐ abnormal ‐ short term Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 urine abnormalities | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 1.87 [0.19, 18.38] |
| 4.2 haematological abnormalities | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 1.87 [0.40, 8.65] |
| 4.3 liver abnormalities | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 8.44 [0.50, 143.77] |
| 4.4 cholesterol <170mg per cent | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 2.81 [0.12, 63.83] |
| 4.5 triglycerides | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 2.8 [0.33, 23.86] |
| 4.6 zinc < 70microgram | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.01, 7.09] |
| 4.7 zinc > 120microgram per cent | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 2.81 [0.12, 63.83] |
| 5 Leaving the study early Show forest plot | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.06, 13.54] |