Farmacoterapia para el deterioro cognitivo crónico en el traumatismo craneoencefálico
Resumen
Antecedentes
El traumatismo craneoencefálico (TCE) es una causa importante de discapacidad crónica. Es la causa principal de discapacidad antes de los 40 años de edad en todo el mundo y da lugar a discapacidad grave en alrededor de 150 a 200 000 000 de personas por año. Además de los problemas del estado de ánimo y conductuales, la cognición (en particular la memoria, la atención y la función ejecutiva) habitualmente se deteriora a causa del TCE. Los problemas cognitivos después del TCE son uno de los factores más importantes para determinar el bienestar subjetivo y la calidad de vida de los pacientes. Los fármacos se utilizan ampliamente en un intento por mejorar las funciones cognitivas. Aunque se han examinado los agentes colinérgicos en el TCE, todavía no ha habido una revisión sistemática o metanálisis del efecto sobre los problemas cognitivos crónicos de todos los agentes farmacológicos de acción central.
Objetivos
Evaluar los efectos de los agentes farmacológicos de acción central para el tratamiento del deterioro cognitivo crónico posterior al traumatismo craneoencefálico en adultos.
Métodos de búsqueda
Se hicieron búsquedas en ALOIS— en el registro especializado del Grupo Cochrane de Demencia y Trastornos Cognitivos (Cochrane Dementia and Cognitive Improvement Group's Specialised Register) — el 16 noviembre 2013, 23 febrero 2013, 20 enero 2014 y el 30 diciembre 2014, utilizando los términos: traumatic OR TBI OR "brain injury" OR "brain injuries" OR TBIs OR "axonal injury" OR "axonal injuries". ALOIS contiene los registros de los ensayos clínicos identificados de las búsquedas mensuales de varias bases de datos principales de atención sanitaria, numerosos registros de ensayos y fuentes de literatura gris. Se hicieron búsquedas suplementarias en MEDLINE, EMBASE, PsycINFO, The Cochrane Library, CINAHL, LILACs, ClinicalTrials.gov, en el World Health Organization (WHO) Portal (ICTRP) y en Web of Science con actas de congresos.
Criterios de selección
Se incluyeron los ensayos controlados aleatorios (ECA) que evaluaron la efectividad de cualquier agente farmacológico de acción central que afecta a uno o más de los principales sistemas neurotransmisores en pacientes con traumatismo craneoencefálico crónico; y con un mínimo de 12 meses entre la lesión y el ingreso al ensayo.
Obtención y análisis de los datos
Dos autores de la revisión examinaron los títulos y resúmenes de las citas obtenidas a partir de la búsqueda. Se recuperaron los artículos relevantes para realizar una evaluación adicional. Se realizó una búsqueda bibliográfica de los artículos pertinentes. Los datos se extrajeron mediante una herramienta estandarizada, y se incluyeron los datos sobre la incidencia de efectos adversos. Cuando fue necesario, se solicitaron datos no publicados adicionales de los autores de los estudios. Un único autor evaluó el riesgo de sesgo.
Resultados principales
Sólo cuatro estudios cumplieron los criterios de inclusión, con un total de 274 participantes. Se investigaron cuatro agentes farmacológicos: modafinilo (51 participantes); (−)‐OSU6162, un estabilizador de la monoamina (12 participantes de los cuales seis presentaron un TCE); atomoxetina (60 participantes); y rivastigmina (157 participantes). No fue posible realizar un metanálisis debido al escaso número y la heterogeneidad de los estudios.
Todos los estudios examinaron el rendimiento cognitivo y la mayoría de las subpruebas psicométricas no mostraron diferencias entre el tratamiento y placebo (n = 274, pruebas de muy baja calidad). El (−)‐OSU6162 demostró superioridad sobre placebo en tres medidas, aunque un desempeño marcadamente inferior en otras. La rivastigmina fue mejor que placebo en una medida primaria y en un único resultado cognitivo en un análisis secundario de un subgrupo con deterioro de la memoria más grave al inicio. El estudio del modafinilo evaluó la mejoría clínica global (n = 51; pruebas de baja calidad) y no encontró diferencias entre el tratamiento y placebo. La seguridad, medida según los eventos adversos, se informó en todos los estudios (n = 274; pruebas de muy baja calidad), y se informaron significativamente más náuseas en los participantes que recibieron rivastigmina en comparación con placebo. No hubo otras diferencias en seguridad entre el tratamiento y el placebo. Ningún estudio informó muertes.
Conclusiones de los autores
No hay pruebas suficientes para determinar si el tratamiento farmacológico es efectivo en el deterioro cognitivo crónico en pacientes con TCE. Aunque hay un hallazgo positivo para la rivastigmina en una medida primaria, en todas las otras medidas primarias no fue mejor que placebo. Los hallazgos positivos para el (−)‐OSU6162 se deben interpretar de forma cautelosa debido a que el estudio fue pequeño (n = 6). Para el modafinilo y la atomoxetina no se encontraron efectos positivos. Los cuatro fármacos parecen tolerarse relativamente bien, aunque las pruebas son escasas.
PICO
Resumen en términos sencillos
Farmacoterapias para el deterioro cognitivo crónico en el traumatismo craneoencefálico
Antecedentes: El traumatismo craneoencefálico (TCE) es una causa principal de discapacidad a largo plazo en todo el mundo. La discapacidad a menudo se relaciona con deterioro cognitivo crónico como cambios en la memoria, la atención y la solución de problemas.
Método: Se examinaron los ensayos controlados aleatorios que investigaron la eficacia de cualquiera de los fármacos utilizados habitualmente para tratar el deterioro cognitivo después del TCE. Se incluyeron solamente los estudios que comenzaron el tratamiento al menos 12 meses después de la lesión; en ese momento el deterioro cognitivo habitualmente es estable.
Resultados: Se identificaron sólo cuatro ensayos para la inclusión. Los ensayos investigaron cuatro fármacos diferentes: modafinilo; el fármaco experimental (−)‐OSU6162; atomoxetina; y rivastigmina, en comparación con placebo. En la mayoría de las medidas no hubo diferencias entre el tratamiento y placebo. Además, la calidad de las pruebas se evaluó como muy baja.
El fármaco experimental denominado (−)‐OSU6162 fue mejor que placebo en tres medidas cognitivas, aunque este estudio fue pequeño y solamente incluyó seis participantes con TCE. No se encontró que el modafinilo, la atomoxetina y la rivastigmina fueran mejores que placebo. No se encontraron diferencias entre el modafinilo y placebo en la evaluación de la mejoría clínica global. En comparación con placebo, más participantes que recibieron modafinilo y menos que recibieron rivastigmina abandonaron los ensayos. Más pacientes que recibieron modafinilo, atomoxetina y rivastigmina experimentaron efectos adversos que los que recibieron placebo, aunque es probable que la diferencia se deba al azar. Solamente las náuseas fueron estadísticamente más probables en los pacientes que recibieron rivastigmina. En el estudio de (−)‐OSU6162, un participante de tres que recibieron placebo experimentó efectos adversos que requirieron una reducción de la dosis, y no se informaron abandonos. Ningún estudio informó muertes.
Conclusión: Según las pruebas actuales, no es posible establecer recomendaciones a favor o en contra de la farmacoterapia para el deterioro cognitivo crónico en el TCE.
Authors' conclusions
Summary of findings
| Modafanil, (−)‐OSU6162, atomoxetine or rivastigmine compared to placebo for chronic cognitive impairment in traumatic brain injury | |||||
| Patient or population: Participants with chronic cognitive impairment in traumatic brain injury | |||||
| Outcomes | Effect of drug treatment for people with cognitive impairment in traumatic brain injury | Relative effect | No of Participants | Quality of the evidence | Comments |
| Cognitive performance on psychometric tests | The majority of sub‐tests showed no difference between treatment and placebo. Superiority over placebo was shown in one measure in Silver 2006 and several measures in Johansson 2012 and Johansson 2015. However, interpretation of these findings are cautioned. | See comment. | 274 (4 studies) | ⊕⊝⊝⊝ very low1,2,3 | Data synthesis was not possible due to the heterogeneity of studies. |
| Clinical global improvement | A single study reported no difference between treatment and placebo. | See comment. | 51 (1 study) | ⊕⊕⊝⊝ low1,3 | Data synthesis was not possible as only one study reported a measure on clinical global improvement. |
| Acceptability | No differences between treatment and placebo were found. | See comment. | 274(4 studies) | ⊕⊝⊝⊝ very low1,2,3 | Data synthesis was not possible due to the heterogeneity of studies. |
| Safety | More nausea was reported in participants receiving rivastigmine than placebo (Silver 2006). No other differences were found. | See comment. | 274 (4 studies) | ⊕⊝⊝⊝ very low1,2,3 | Data synthesis was not possible due to the heterogeneity of studies. |
| Mortality | No deaths were reported by any study. | Not estimable. | 274 (4 studies) | ‐ | |
| GRADE Working Group grades of evidence | |||||
| 1 downgraded one level due to serious indirectness, as two of four studies did not investigate cognitive impairment as a primary outcome 2 downgraded one level due to serious inconsistency, due to wide variance of point estimates. 3 downgraded one level due to serious imprecision, as the total population size was less than 400. | |||||
Background
Description of the condition
Traumatic brain injury (TBI) can result from closed‐head injury, penetrating‐head injury or a combination thereof. Penetrating‐head injury can affect any region of the brain while closed‐head injury, which includes rapid acceleration‐deceleration injury, can result in focal lesions but always causes diffuse damage. Diffuse damage consists of a combination of one or more of diffuse axonal injury (DAI), hypoxic brain damage, vascular injury, and swelling (Adams 1991), which frequently involve the white matter of the frontal lobes, the corpus callosum and the corona radiata (Gentry 1988). TBI varies widely in its severity. The commonest form, mild TBI, by definition, is associated with altered mental state at the time of injury; either no or brief (under 30 minutes) loss of consciousness; Glasgow Coma Scale score 13 to 15 after 30 minutes; and post‐traumatic amnesia less than 24 hours (Kay 1993). Mild TBI therefore encompasses a broad spectrum of severities, from feeling momentarily dazed following the trauma to 30 minutes loss of consciousness requiring hospital admission. In severe TBI the loss of consciousness or post‐traumatic amnesia can persist for one week or longer, sometimes even weeks to months (Silver 2008).
TBI is a major cause of chronic disability, even following mild injury (Thornhill 2000). Worldwide, it is the leading cause of disability in the under 40s, resulting in severe disability in some 150 to 200 million people per annum (Fleminger 2005). Neuropsychiatric sequelae of varying severity (which include problems with attention, arousal, concentration, executive functioning, memory impairment, personality change, affective disorders, anxiety, psychosis, aggression and irritability), occur in over two‐thirds of survivors while the vulnerability for such complications can persist for decades (Koponen 2002; Himanen 2006; Silver 2008). The majority of those with mild TBI show significant improvement of their symptoms over the first 6 months (Bernstein 1999); but if symptoms have not resolved by 6 to 12 months, they tend to persist (Brown 1994). Those with moderate to severe TBI show significant improvement of cognitive function over the first year following injury (Tabaddor 1984), but improvement appears to plateau by the end of year two (Whyte 2013).
In addition to mood and behavioural problems, memory, attention and executive function are commonly impaired by TBI (Lippert‐Gruner 2006), and prove difficult to remedy (Salazar 2000). Selective attention, the attending to chosen information at the expense of other stimuli, is mediated bottom‐up by the ascending reticular activating system and top‐down by the prefrontal, parietal and limbic cortices (Aharon‐Peretz 2007). Those with TBI demonstrate slowed reaction times, poor sustained attention, difficulty in ignoring task‐irrelevant information and perform poorly on tests of divided attention (Aharon‐Peretz 2007). The Central Executive System (CES) component of working memory appears to be particularly vulnerable in TBI, and impairment is associated with other symptoms of the dysexecutive syndrome (McDowell 1997). Executive dysfunction may partly explain problems with recall of long‐term episodic and semantic memory, due to an ineffective search strategy, though damage in the medial temporal lobes or diencephalic structures will cause anterograde amnesia that is characterised by impaired recall and recognition. Closed‐head injury tends to cause cognitive impairment in multiple domains whereas penetrating injuries can result in a focal cognitive deficit.
The UK's 'National Service Framework for Long Term Conditions' (DH) has highlighted the need for comprehensive interdisciplinary care that improves people's quality of life and carers' distress. Neurocognitive problems following TBI are one of the most important factors in determining people's subjective well‐being and their quality of life (Teasdale 2005). Hence, the focus is on improving neurocognitive functions, which may include the use of pharmacological approaches in addition to traditional neurorehabilitation.
Description of the intervention
The interventions to be studied are all widely‐available, centrally acting pharmacological agents that affect one or more of the major neurotransmitter systems (dopaminergic, serotonergic, GABAergic, glutamatergic and cholinergic) that are thought to underpin cognitive functions.
How the intervention might work
The neurotransmitter changes caused by traumatic brain injury have been far more extensively researched in the acute phase of brain injury than in chronic TBI (Mysiw 1997). During the acute phase the location of the injury results in different patterns of altered neurotransmitter function (Van Woerkom 1977). However, there are reasons to suspect that pharmacological manipulation of neurotransmitter systems in chronic TBI could enhance cognitive function.
Animal neurotransmitter studies have shown dopamine (DA) is found in high concentrations in the dorsolateral prefrontal cortex and anterior cingulate, areas known to be intimately involved in working memory, attention and executive cognitive functioning (Previc 1999). The noradrenergic (NA) neurones originate in the locus coeruleus and lateral tegmental area and project into the cerebellum and hippocampus as well as diffusely through the cerebral cortex. Both NA and DA are known to play significant roles in arousal, motivation, learning and memory (Cooper 1991a; Cooper 1991b).
Severe TBI (acute Glasgow coma scale (GCS) ≤ 8) with diffuse axonal injury is associated with reduced turnover of 5‐hydroxy tryptophan (5HT). The serotonergic system is implicated in the function of the reticular activating system (RAS) and hippocampus, involved in alertness and memory respectively (Van Woerkom 1990). The serotonergic system, which projects from the midline raphe to the limbic system, neostriatum, thalamus and cerebellar and cerebral cortices, is involved in arousal, memory, mood and learning (Cooper 1991c).
There are two main cholinergic systems. The basal forebrain cholinergic complex includes the nucleus basalis of Meynert, septal nuclei and the nucleus of the diagonal band. A second system arises from the pedunculopontine and laterodorsal tegmental nuclei and these project to the thalamus, cerebellum and ascending reticular formation. The cholinergic system therefore helps maintain arousal and has been found to increase neuronal responsiveness to sensory input (Heilman 1993). An increase in cholinergic function is necessary for tasks requiring sustained and divided attention, maintaining vigilance, neural plasticity required for laying down memory and recognition memory (Tenovuo 2006). It has been suggested that following TBI there is a perturbation of cholinergic systems (Hayes 1989). After an acute period of increased cholinergic activity, a long‐standing hypocholinergic state develops. Structures in the medial temporal lobes, such as the hippocampus and limbic system that are rich in cholinergic neurons, are highly susceptible to traumatic and hypoxic injury (Arciniegas 1999).
Why it is important to do this review
Whilst Poole and Agrawal examined the role for cholinergic agents in TBI (Poole 2008), there has not yet been a systematic review or meta‐analysis of all commonly used psychotropic drugs in chronic cognitive impairment. Given the lack of knowledge of chronic changes in neurotransmitter systems following TBI the authors believed that it was sensible to study all psychotropic drugs that modulate the main neurotransmitter systems (noradrenaline, dopamine, serotonin, glutamate, acetylcholine).
Objectives
To assess the effects of centrally acting pharmacological agents for treatment of chronic cognitive impairment subsequent to traumatic brain injury in adults.
Methods
Criteria for considering studies for this review
Types of studies
As outlined in the protocol (Poole 2011), we only included randomised controlled trials (RCTs) or cross‐over design studies assessing the effectiveness of any one centrally acting pharmacological agent that affects one or more of the main neurotransmitter systems in people with chronic traumatic brain injury (12 months or longer since the injury). We chose twelve months as a suitable time point because the majority of spontaneous biological improvement in neurocognitive functions takes place within this period (Brooks 1976); and only a few people show significant spontaneous recovery after 12 months (Jennett & Bond 1975). However, we acknowledge that any chosen time point is somewhat arbitrary. We only included trials if treatment with the pharmacological agent was compared with a placebo control group, and required that included trials showed evidence of concealment of allocation to treatment and control groups.
Types of participants
Human adults who have sustained mild to severe traumatic brain injury (TBI) resulting in chronic cognitive impairment.
Types of interventions
1) Antidepressants
2) Antipsychotics
3) Dopamine agonists
4) Cholinergic agents
5) Psychostimulants
The authors reviewed trials investigating all centrally acting pharmacological agents that modulate the main neurotransmitter systems.
We did not review combinations of pharmacological agents because pharmacodynamic and kinetic factors would obscure the results.
We required that the intervention must be instigated 12 months or more after the injury.
Types of outcome measures
We required all outcome measures to be valid measures of the domain in question (that is tests of memory must be validated as such). However, we expected a wide variety of measures to be employed across studies and so we did not require that each one has been validated in the TBI population as this was anticipated to limit the number of studies that could be included. We required outcome measures to have demonstrated a high test‐retest reliability (> 0.8) in studies assessing their psychometric properties.
Primary outcomes
• Global severity of cognitive impairment
• Memory performance on psychometric tests
• Performance on neuropsychological tests that evaluate other aspects of cognitive functioning
• Scores on screening measures assessing cognitive function or general severity of cognitive impairment (e.g. mini–mental state examination (MMSE), Cambridge cognitive assessment (CAMCOG), Alzheimer disease assessment scale‐cognitive (ADAS‐Cog) or clinical dementia rating (CDR) scale
• Clinical global impression of change
Secondary outcomes
• Acceptability of treatment (as measured by withdrawal from trial)
• Safety (as measured by incidence of adverse effects)
• Mortality
• Subjective benefit (as measured by a validated tool)
Search methods for identification of studies
Electronic searches
We searched ALOIS (www.medicine.ox.ac.uk/alois)—the Cochrane Dementia and Cognitive Improvement Group’s Specialised Register—on 16 November 2011, 23 February 2013, 20 January 2014 and 30 December 2014. The search terms used were: traumatic OR TBI OR "brain injury" OR "brain injuries" OR TBIs OR "axonal injury" OR "axonal injuries".
ALOIS is maintained by the Trials Search Co‐ordinator of the Cochrane Dementia and Cognitive Improvement Group and contains studies in the areas of dementia prevention, dementia treatment and cognitive enhancement in healthy people. The studies are identified from:
-
Monthly searches of a number of major healthcare databases: MEDLINE (Ovid SP), EMBASE (Ovid SP), CINAHL, (EBSCOhost), PsycINFO (Ovid SP) and LILACS (BIREME)
-
Monthly searches of a number of trial registers: ISRCTN; UMIN (Japan's Trial Register); the WHO portal (which covers ClinicalTrials.gov; ISRCTN; the Chinese Clinical Trials Register; the German Clinical Trials Register; the Iranian Registry of Clinical Trials; and the Netherlands National Trials Register; plus others)
-
Quarterly search of The Cochrane Library’s Central Register of Controlled Trials (CENTRAL)
-
Six‐monthly searches of a number of grey literature sources: ISI Web of Knowledge Conference Proceedings; Index to Theses; Australasian Digital Theses
To view a list of all sources searched for ALOIS see About ALOIS on the ALOIS website.
Details of the search strategies used for the retrieval of reports of trials from the healthcare databases, CENTRAL and conference proceedings can be viewed in the ‘Methods used in reviews’ section within the editorial information about the Dementia and Cognitive Improvement Group.
We performed additional searches in many of the sources listed above to cover the timeframe from the last searches performed for ALOIS to ensure that the search for the review was as up to date and as comprehensive as possible. The search strategies used can be seen in Appendix 1.
The searches retrieved a total of 9200 results. After a first‐assess and a de‐duplication, 676 references were left to further assess for possible inclusion or exclusion within the review.
Searching other resources
The authors handsearched the bibliographies of all relevant papers identified for any additional citations and also consulted experts and pharmaceutical companies to check for any omissions from our identified studies.
Data collection and analysis
Selection of studies
Two authors (DD and NP) examined titles and abstracts of citations obtained from the search, and obviously irrelevant articles were discarded. An article was retrieved for further assessment in the presence of any suggestion that the article described a relevant randomised controlled trial.
Two authors (DD and NP) independently assessed the retrieved articles for inclusion in the review according to the criteria above. Disagreements were resolved by discussion.
Data extraction and management
Two authors (DD and NP) extracted data. If any differences emerged then agreement was reached by consensus.
The authors extracted data from the published reports using standardised tools. In addition to summary statistics for each trial and each outcome we extracted details on the number and types of participants, the interventions, randomisation and blinding. For continuous data we extracted the mean change from baseline, the standard error of the mean change, and the number of participants for each treatment group at each assessment. For binary data we sought the numbers in each treatment group and the numbers experiencing the outcome of interest. We requested additional data from the original study authors if this data was unavailable in the published paper.
We defined the baseline assessment as the latest available assessment prior to randomisation, but no longer than two months before.
We sought data for each outcome measure and on every person assessed.
In studies where a cross‐over design was used, we only extracted data from the first treatment phase after randomisation.
Assessment of risk of bias in included studies
The same review author (DD) assessed the methodological quality of all studies included in the review. The author used the Cochrane 'Risk of bias' tool as described in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), to assess the methods used by each study to control six potential sources of bias (sequence generation; allocation concealment; blinding of participants, personnel and outcome assessors; incomplete outcome data; selective outcome reporting; and other sources of bias). Overall quality of evidence was assessed using the GRADE approach (Schünemann 2011).
The authors gave particular emphasis to allocation concealment, as empirical research has shown that a lack of adequate allocation concealment is associated with bias. Trials with unclear concealment measures have been shown to yield more pronounced estimates of treatment effects than trials that have taken adequate measures to conceal allocation schedules, but this is less pronounced than for inadequately concealed trials (Chalmers 1983; Schulz 1995). Thus trials were included if they were of low or unclear risk, whilst those assessed as high risk were not.
Measures of treatment effect
The authors expressed the treatment effect for continuous outcomes, as the mean difference. For dichotomous outcomes, we expressed risk ratios which for clinicians are easier to interpret than odds ratios. We did not report treatment effects in terms of number needed to treat for an additional beneficial outcome (NNTB) as the data extracted gave mean changes from baseline only and could not be interpreted in terms of treatment response.
Unit of analysis issues
The authors only included data from the first treatment phase after randomisation in studies of a cross‐over design.
Dealing with missing data
The authors sought intention‐to‐treat data but, if unavailable in the publications, we included 'on‐treatment' data or the data for those who completed the trial.
Assessment of heterogeneity
The authors did not conduct an assessment of heterogeneity as the three studies identified were clinically heterogeneous and results were not pooled.
Assessment of reporting biases
The authors minimised the potential for publication bias by contacting study authors, pharmaceutical companies and experts to assist in the identification of unpublished studies. Furthermore, we contacted authors of identified studies to enquire about duplication of studies and outcome reporting bias.
We did not create a funnel plot as an insufficient number of studies were identified.
Data synthesis
We deemed data synthesis to be inappropriate due to the small number of studies identified, which also differed in interventions and outcome measures.
Subgroup analysis and investigation of heterogeneity
We did not assess for statistical heterogeneity as data synthesis was not possible.
Sensitivity analysis
We did not conduct sensitivity analyses as we did not conduct data synthesis.
Results
Description of studies
Results of the search
The results of the search are summarised in Figure 1. A total of 9200 records were identified after removal of duplicates and first assessment. One additional paper was identified after contacting a study author (Johansson 2015). A total of 676 papers were reviewed for suitability for inclusion, of which four studies met the criteria for inclusion. Two cross‐over studies could not be included as they did not report separate phase one outcome data, which the authors were unable to provide when contacted (Speech 1993; Tenovuo 2009). One study did not include a placebo control group (Johansson 2015). One study investigating rivastigmine was identified to be ongoing, and is described in the Characteristics of ongoing studies table.

Study flow diagram.
Included studies
The four studies meeting inclusion criteria are described in the Characteristics of included studies tables. One study investigated modafinil (Jha 2008), a wake‐promoting agent with effects on multiple neurotransmitter systems, including histaminergic, serotonergic, and glutaminergic activity that may be secondary to stimulation of catecholamine systems (Minzenberg 2008). The second investigated (−)‐OSU6162 (Johansson 2012), a monoamine stabiliser agent with dopaminergic (Lahti 2007), and serotonergic effects (Burstein 2011; Carlsson 2011). The third investigated atomoxetine (Ripley 2014), a noradrenaline reuptake inhibitor (Zerbe 1985). Lastly, Silver 2006 investigated rivastigmine which is an acetylcholinesterase and butyrylcholinesterase inhibitor (Giacobini 2002). Three studies (Jha 2008, Ripley 2014 and Silver 2006) were conducted in the United States, whilst Johansson 2012 was conducted in Sweden.
Jha 2008 is a single centre randomised placebo‐controlled crossover trial, examining the efficacy of modafinil in the treatment of fatigue or excessive day‐time sleepiness, which included secondary measures assessing global and cognitive function. Fifty‐one men and women aged 16 to 65 were recruited from an inpatient rehabilitation unit. For inclusion, participants were required to suffer from fatigue or excessive daytime sleepiness that compromised functioning. Although the mean baseline ImPACT (Immediate Post Concussion Assessment Cognitive Testing) score was provided for the group this is difficult to interpret. The ImPACT is a neuropsychological test which compares the subject against aged‐matched normative data. It would therefore be important to know the baseline for each individual to ascertain the degree of cognitive impairment at baseline. Relevant exclusion criteria were: the presence of a neurological or neuropsychiatric diagnosis that would impair evaluation of the intervention; other diagnoses that may cause excessive daytime sleepiness; concurrent medication use; clinically significant systemic medical conditions; epilepsy; cardiovascular disease; severe hepatic or renal impairment; psychiatric or behavioural disturbance that would impair evaluation of the intervention; pregnancy. In the treatment arm, modafinil was titrated to 200 mg twice daily orally over a two‐week period and continued for a further eight weeks. Participants in the placebo arm were administered tablets at the same frequency as for modafinil. After a four‐week washout period participants were switched to the opposite arm. The primary outcome measures of the study related to fatigue and daytime sleepiness. Relevant cognitive or global outcome measures included the Medical Outcome Study 12‐Item Short Form Survey (SF‐12), ImPACT, and the Conners' Continuous Performance Test II (CPT‐II). The measures were administered at baseline, week four and week ten of treatment (repeated at week four and ten following cross‐over). Only results from the first treatment phase were included in this review. We contacted the study authors who provided unpublished phase one outcome data for cognitive outcomes and adverse effects.
Johansson 2012 is a single centre cross‐over randomised placebo controlled study, examining the efficacy of (−)‐OSU6162 on long‐term mental fatigue following TBI or stroke. Twelve men and women aged 30 to 65 were recruited through a local Swedish newspaper advertisement or from the Department of Neurology at Sahlgrenska University Hospital in Gothenburg. Participants were included if they were at least one year post‐TBI or stroke, but not more than 10 years, and had suffered pathological mental fatigue for at least one year. Participants were required to have otherwise recovered from neurological symptoms, have a Glasgow Outcome Scale (Extended) (GOS‐E) score indicating at least moderate disability or better (> 5), and have completed education to high school equivalent and speak Swedish. Participants were excluded if they were deemed to suffer significant cognitive impairment, as measured by scores more than two standard deviations worse on the cognitive tests used by the study, or in the case of significant neurological or psychiatric comorbidity, substance misuse, or pregnancy. Participants receiving (−)‐OSU6162 were administered 15 mg twice daily in week one, 30 mg twice daily in week two, and 45 mg twice daily in weeks three and four. Dose variation was possible if a participant had responded at a lower dose and then experienced decreased efficacy or adverse effects at a higher dose, therefore permitting a reduction to the previous dose. The process of administration in the placebo arm was not explicitly described. The primary outcome measure of the study assessed fatigue. The relevant secondary cognitive outcome measures were the Trail Making Tests A,B,C & D, WAIS‐III digit symbol coding, WAIS‐III digit span and the FAS verbal fluency test. These were administered at baseline and four weeks. A week 8 assessment was conducted following the crossover, the results of which are not relevant to this review. We contacted the study authors, who provided unpublished outcome data specific to TBI participants in phase one of the trial.
Ripley 2014 is a single centre randomised placebo‐controlled cross‐over trial, examining the efficacy of atomoxetine in the treatment of attention impairment. Sixty men and women aged 18 to 65 were recruited by posting flyers to people who had received inpatient rehabilitation following TBI, or through clinicians handing leaflets to people with TBI in the community. Flyers were also distributed to various brain injury organisations. For inclusion, participants were required to have a history of moderate to severe TBI, defined by a Glasgow Coma Scale score of 12 or less, post‐traumatic amnesia which lasted more than 24 hours or radiographic evidence of intracranial trauma. Telephone screening was conducted to administer the Adult ADHD Self‐Report Scale (ASRS‐v1.1) and the Cognitive Failures Questionnaire (CFQ). The Other CFQ was also administered if a primary carer or significant other was available. A score of four or higher on the ASRS, 0.35 or higher on the CFQ, or 0.42 or higher on the Other CFQ was required for inclusion. Exclusion criteria were: non‐English speaking participants; current seizure history; cardiovascular disease; history of psychiatric illness requiring hospital treatment; use of monoamine oxidase inhibitors; severe renal or hepatic impairment; pregnancy or lactating. After baseline assessment all participants received a placebo run‐in for two weeks. In the treatment arm, atomoxetine 40 mg was administered twice a day for two weeks. Participants in the placebo arm were administered tablets of identical appearance according to the same schedule. After a two‐week placebo washout period participants were switched to the opposite arm for a further two weeks. The primary outcome measure of the study was the Power of Attention factor of the Cognitive Drug Research (CDR) Computerized Cognitive Assessment System. The CDR is a computer‐controlled battery of tests that provides factor scores for Power of Attention (POA), Continuity of Attention (COA), Quality of Working Memory (QWM), Quality of Episodic Memory (QEM), and Speed of Memory (SOM). Secondary measures in the study included COA, QEM and SOM factor scores of the CDR. Further secondary measures were the Efficiency of Attention score calculated from the Power of Attention score, Stroop Colour and Word Test, the Adult ADHD Self‐Report Scale and Neurobehavioral Functioning Inventory scores. Only results from the first treatment phase were included in this review. The study authors provided unpublished phase one outcome data for cognitive outcomes and adverse effects on request.
Silver 2006 is a multicentre randomised placebo controlled trial examining the efficacy of rivastigmine on cognitive function. One‐hundred and fifty men and women aged 18 to 50 were recruited across 19 sites. Participants with cognitive impairment following non‐penetrating traumatic brain injury were included. At baseline, participants were required to have a difference of at least one standard deviation between current estimated intelligence measured by the Wechsler Adult Intelligence Scale, third edition (WAIS‐III) and current verbal attention or verbal memory functioning measured by the Cambridge Neuropsychological Test Automated Battery Rapid Visual Information Processing A subtest (CANTAB RVIP A) or the Hopkins Verbal Learning Test (HVLT) total trials one to three. Relevant exclusion criteria were: medical, psychiatric or substance use disorders that could impair evaluation of the intervention; acute or severe pulmonary or cardiovascular disease; primary neurodegenerative disorders; epilepsy; medication known to affect cognitive functioning in TBI; history of major brain surgery or penetrating brain injury. Participants receiving rivastigmine started at 1.5 mg twice daily for four weeks, after which the dose was increased to 3 mg twice daily. If not tolerated due to adverse side effects, the dose could either be reduced to 4.5 mg or 3 mg daily (also in divided doses). A matching placebo was administered according to the same schedule as for rivastigmine. Primary outcome measures were the CANTAB RVIP A and the HVLT. These were administered at baseline and weeks four, eight and twelve. Secondary outcome measures were only administered at baseline and week twelve, and included; Controlled Oral Word Association; WAIS‐III Digit span; WAIS‐III Letter Number Sequencing; and Trail Making Tests Parts A and B. We contacted the study authors for additional data, which was unavailable. Therefore, outcomes for the Neurobehavioral Functioning Inventory (NFI), Diener Satisfaction with Life Scale, and Clinical Global Impression of Change (CGI) could not be reported.
Excluded studies
We excluded two cross‐over studies as neither reported phase 1 outcome data, which the authors were unable to provide when contacted (Speech 1993; Tenovuo 2009). We excluded three other studies as the time since TBI was not clearly stated (León‐Carrión 2000; Plenger 1996; Schneider 1999). One study (Johansson 2015) was excluded as the control group did not receive a placebo.
Risk of bias in included studies
Judgements for risks of bias are detailed in the Characteristics of included studies section and summarised in Figure 2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Jha 2008 and Ripley 2014 described adequate procedures to reduce selection bias and were rated as low risk. Johansson 2012 did not describe the randomisation process, other than reporting that an external system was employed; therefore we rated the study as unclear risk for random sequence generation, and low risk of allocation concealment due to the use of an external process. A limited description of the randomisation and concealment process was provided by Silver 2006; therefore we rated the risks of bias as unclear.
Blinding
All studies were assessed to be of low risk of bias.
Incomplete outcome data
We assessed risk of attrition bias as high in Jha 2008 as not all participants who were randomised were included in the analysis and reference was not made to intention‐to‐treat analysis. We assessed Johansson 2012, Ripley 2014 and Silver 2006 as low risk.
Selective reporting
Outcomes were reported as planned in the protocol by Jha 2008, Johansson 2012, and Ripley 2014 , and therefore we assessed reporting bias as low risk.
Silver 2006 varied from the protocol in terms of inclusion criteria and outcome measurement time‐scales. The published protocol registered with ClinicalTrials.gov planned to include participants aged 18 to 65, while the published study states the inclusion criteria as 18 to 50 years. Of more concern, a different cognitive battery was used in the study to that proposed in the protocol to determine inclusion into the trial. The protocol included subjects one standard deviation or more below the mean on the California Verbal Learning test, Tower of London test, Verbal Memory Learning test and the Test Battery for Attentional Performance. However, the completed study included subjects who performed below one standard deviation on the Verbal subtest on the Wechsler Adult Intelligence Scale (WAIS) and attention or verbal memory as assessed by the Cambridge Neuropsychological Test Automated Battery CANTAB), Rapid Visual Information processing (RVIP), or the Hopkins Verbal learning Test (HVLT). Also, the protocol planned for treatment for 20 weeks, as opposed to 12 weeks in the completed study. Both these discrepancies appear to have been balanced across both arms of the study: we therefore assessed the risk of bias as low.
Other potential sources of bias
Only Ripley 2014 and Silver 2006 investigated treatment of cognitive impairment as a primary outcome measure, with Jha 2008 and Johansson 2012 primarily investigating treatment of fatigue. Limited information on the severity of TBI was provided by the studies, and similarly limited data on baseline cognitive impairment at inclusion were provided. These factors may have introduced bias through variation in the recruitment or selection of participants, and consequently assessment of outcome.
Effects of interventions
Each of the reported studies included measures relating to one of the primary outcomes of this review: namely, cognitive function as assessed by standardised psychometric tests. Of the four studies, only Ripley 2014 and Silver 2006 reported cognitive function as a primary outcome measures. Primary outcome measures for Jha 2008 were for fatigue; and for Johansson 2012, mental fatigue. Reported secondary measures included safety and acceptability data. Due to the small numbers, we have reported outcomes separately for each study. Synthesis of data was not possible, with sub‐tests of psychometric batteries favouring both treatment and placebo arms, as shown in summary of findings Table for the main comparison.
In Jha 2008 unpublished data for relevant cognitive outcomes were provided by the study authors. These data were generated by the Statistical Package for the Social Sciences (SPSS), rather than the original statistician’s Statistical Analysis Software (SAS) syntax for the mixed model used in the published paper. Consequently this resulted in different mean scores compared to the original study, but not to the significance of the findings. A single significant difference at week 4 was found in ImPACT visual motor speed composite score, which was not attributable treatment, as there was a positive change in the placebo arm, whilst the score in the modafinil arm remained essentially unchanged (Analysis 1.5). At week 10, no significant differences between modafinil and placebo were found on all relevant measures, including the SF‐12 mental, SF‐12 physical, ImPACT verbal memory composite, ImPACT visual memory composite, ImPACT visual motor speed composite, ImPACT reaction time composite, CCPT‐II number of omissions or CCPT‐II number of commissions (Analysis 2.1 to Analysis 2.8).
The Modafinil group experienced more adverse events than those taking placebo. However, there were no significant differences in the effect estimates (Analysis 7.1 to Analysis 7.10). Two participants in the treatment arm left the trial early versus none in the control group. Again, this was not statistically significant (Analysis 6.1). No deaths were reported during the study.
In Ripley 2014 atomoxetine was found not to be significantly different from placebo on all cognitive test (Analysis 4.1 to Analysis 4.5). There was no difference in acceptability, with no drop outs reported during the first treatment phase (Analysis 6.4). Numerically more side effects were reported with atomoxetine, but there were no significant differences in effects estimates (Analysis 9.1 to Analysis 9.10). These analyses accord with the findings of the original paper (Ripley 2014), in which no statistical difference between atomoxetine and placebo was found, when pre‐ and post cross‐over phases were combined in the analysis.
In Silver 2006 rivastigmine was found not to be significantly different from placebo on all cognitive tests (Analysis 5.1 to Analysis 5.5, and Analysis 5.7 to Analysis 5.12) apart from an effect estimate favouring rivastigmine according to the CANTAB RVIP, mean latency (−44.54 milliseconds, 95% CI −88.62 to −0.46 Analysis 5.6). However, Silver 2006 did not report a significant difference for this measure (P = 0.129), which may be explained by the use of a different statistical test. On a secondary analysis of a subgroup of participants with greater cognitive impairment at baseline, as measured by at least 25% impairment on the HVLT, significant superiority of rivastigmine over placebo was found on the CANTAB RVIP mean latency test at weeks four and 12 (reported by the study in graphical format only). Numerical data other than a P value of < 0.05 were not reported, and were not available when requested from the study authors.
Placebo was numerically slightly less acceptable than rivastigmine (Analysis 6.2). Rivastigmine was numerically less safe than placebo as measured by adverse effects, with a higher rate of nausea in participants given rivastigmine the only significant difference compared to placebo (19/80, 23.8% versus 6/77, 7.8%, risk ratio 3.05, 95% CI 1.29 to 7.22 Analysis 10.6). No deaths were reported during the study.
In Johansson 2012 data for phase 1 TBI participants were provided by the study authors. Effect estimates showed significantly higher scores for (−)‐OSU6162 than placebo for Trail Making Test A (−9.20 seconds, 95% CI −12.19 to −6.21 Analysis 3.1), Trail Making Test B (−6.20 seconds, 95% CI, −7.81 to −4.59 Analysis 3.2) and WAIS‐III Digit symbol coding (8.60, 95% CI 6.47 to 10.73 Analysis 3.5) showed significantly higher scores for (−)‐OSU6162 than placebo. Trail Making Test D strongly favoured placebo (53.80 seconds, 95% CI 36.76 to 70.24 Analysis 3.4). No significant differences in effect estimates were found for Trail Making Test C, WAIS‐III digit span, and FAS verbal fluency (Analysis 3.3, Analysis 3.6 & Analysis 3.7). One participant required a dose reduction due to adverse effects in the placebo arm. There were no drop‐outs (TBI participants). No reference was made to deaths.
Discussion
Summary of main results
The search has found only seven RCTs (including six cross‐over trials) investigating the pharmacological treatment of chronic cognitive impairment at least 12 months after TBI, with five trials providing data suitable for inclusion. Two cross‐over trials that otherwise met criteria for inclusion were not included as phase one outcome data were unavailable. One trial was rejected as it did not include a placebo control group.
The pharmacological agents investigated were modafinil, rivastigmine, atomoxetine, and (−)‐OSU6162. Superiority over placebo for (−)‐OSU6162 was demonstrated in Trail Making Tests A, B and WAIS‐III digit symbol coding, but (−)‐OSU6162 was also inferior to placebo in Trail Making Test D (Johansson 2012). Superiority over placebo was not demonstrated for modafinil (Jha 2008); or atomoxetine (Ripley 2014). Superiority of rivastigmine over placebo was demonstrated by only one primary measure (CANTAB RVIP, mean latency) according to the effect estimate by this review. Interpretation of this finding is cautioned as Silver 2006 did not report a statistically significant difference, which may be attributed to a different statistical test used. In addition the confidence interval for the effect estimate is wide and approaches statistical non‐significance. Separately on a secondary analysis of a subgroup of participants assessed to have greater cognitive impairment, significant superiority of rivastigmine was demonstrated on a single measure (Silver 2006).
Overall completeness and applicability of evidence
No strong RCT evidence to support the pharmacological treatment of chronic cognitive impairment in TBI has been found by this review. However, when considering the significant disability associated with chronic TBI, pharmacological treatments may still be considered as part of a comprehensive management strategy for chronic cognitive impairment, including non‐pharmacological approaches for which there is reasonable evidence (Lu 2012).
Quality of the evidence
Since only four studies investigating different agents with heterogenous outcome measures met the criteria for inclusion, synthesis of outcomes was not possible. Only two studies concerned the treatment of cognitive impairment as a primary measure (Ripley 2014;Silver 2006), whilst the other two examined the impact on cognitive function as a secondary outcome, with primary outcomes concerned with the treatment of mental fatigue thereby limiting generalisability (Jha 2008; Johansson 2012). That Johansson 2012 also excluded participants with significant cognitive impairment further limits generalisability. The small study size of Johansson 2012, with only six TBI participants may have increased the risk of erroneous results. This study also assessed a relatively short period of treatment (four weeks). The majority of the data reported in this review for Jha 2008, Johansson 2012, and Ripley 2014 were also unpublished. Overall the studies were largely assessed to be at low or unclear risk of bias.
Using the GRADE approach, we rated the quality of evidence for cognitive performance on psychometric tests, acceptability and safety as very low. Quality was downgraded one level for each rating, due to: serious indirectness, as two of four studies did not investigate cognitive impairment as a primary outcome; serious inconsistency, due to wide variance of point estimates; and serious imprecision, as the total population size was less than 400. Quality of evidence for clinical global improvement was rated as low, downgraded due to serious indirectness, as the relevant study did not investigate cognitive impairment as a primary outcome; and serious imprecision, as the population size was less than 400.
Potential biases in the review process
Strengths of this review are that a comprehensive search strategy was employed, which together with a bibliographic search is likely to have identified all relevant studies. The title and abstract search and review of full‐text papers was conducted independently by two study authors (DD & NP) to reduce inclusion bias. A limitation was that a single‐author assessment of bias was conducted. A weakness of this review is that phase 1 data from two cross‐over studies were not available from the respective authors (Speech 1993;Tenovuo 2009), and therefore could not be included, reducing the number of included studies. However, only Tenovuo 2009 reported statistically significant results on two measures, which suggests the findings of this review would not have been substantially altered.
Agreements and disagreements with other studies or reviews
The findings of this review reflect those of Poole 2008 which found little high quality evidence for the pharmacological treatment of cognitive impairment in TBI; whilst no definitive evidence was found, the review concluded that there was suggestive evidence for cholinesterase inhibitors and choline. Another earlier review by Griffin 2003 concluded that there was only preliminary evidence for further investigation of cholinergic agents. Since Poole 2008 which reviewed the RCT by Silver 2006, only one further RCT relevant to this review has been published (Jha 2008). Methodological differences between this review and that by Poole 2008 and Griffin 2003 is that it is limited to chronic cognitive impairment, but is also broader by including drugs other than cholinergic agents.

Study flow diagram.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
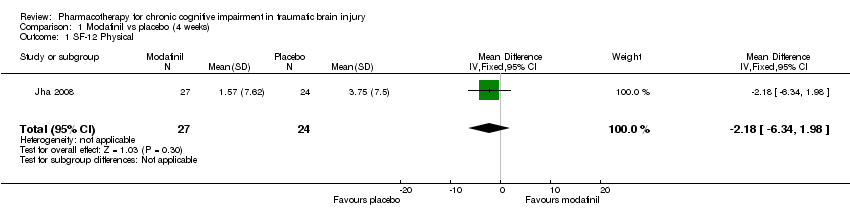
Comparison 1 Modafinil vs placebo (4 weeks), Outcome 1 SF‐12 Physical.

Comparison 1 Modafinil vs placebo (4 weeks), Outcome 2 SF‐12 Mental.

Comparison 1 Modafinil vs placebo (4 weeks), Outcome 3 ImPACT verbal memory composite.
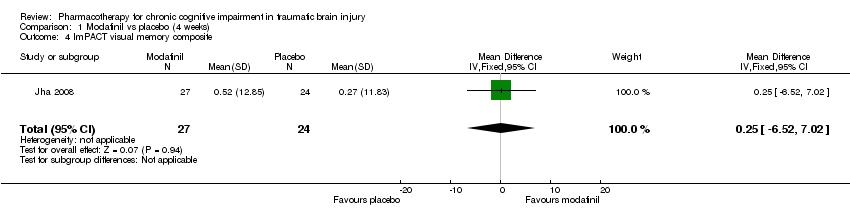
Comparison 1 Modafinil vs placebo (4 weeks), Outcome 4 ImPACT visual memory composite.

Comparison 1 Modafinil vs placebo (4 weeks), Outcome 5 ImPACT visual motor speed composite.
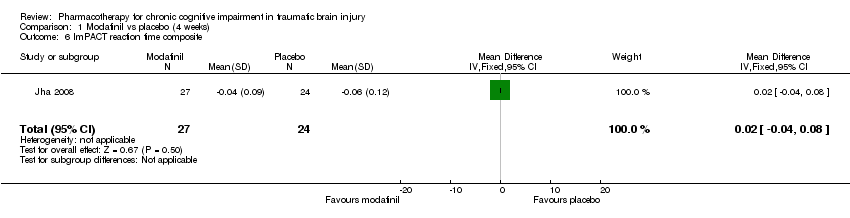
Comparison 1 Modafinil vs placebo (4 weeks), Outcome 6 ImPACT reaction time composite.

Comparison 1 Modafinil vs placebo (4 weeks), Outcome 7 CCPT‐II No. of omissions.
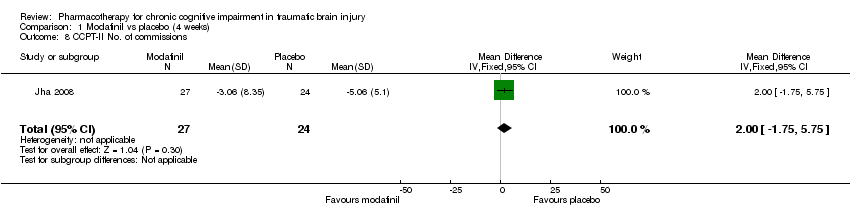
Comparison 1 Modafinil vs placebo (4 weeks), Outcome 8 CCPT‐II No. of commissions.

Comparison 2 Modafinil vs placebo (10 weeks), Outcome 1 SF‐12 Physical.

Comparison 2 Modafinil vs placebo (10 weeks), Outcome 2 SF‐12 Mental.

Comparison 2 Modafinil vs placebo (10 weeks), Outcome 3 ImPACT verbal memory composite.
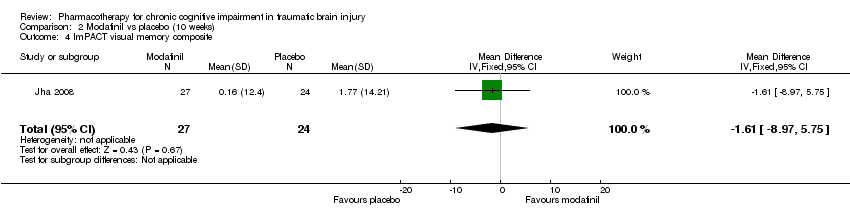
Comparison 2 Modafinil vs placebo (10 weeks), Outcome 4 ImPACT visual memory composite.

Comparison 2 Modafinil vs placebo (10 weeks), Outcome 5 ImPACT visual motor speed composite.

Comparison 2 Modafinil vs placebo (10 weeks), Outcome 6 ImPACT reaction time composite .
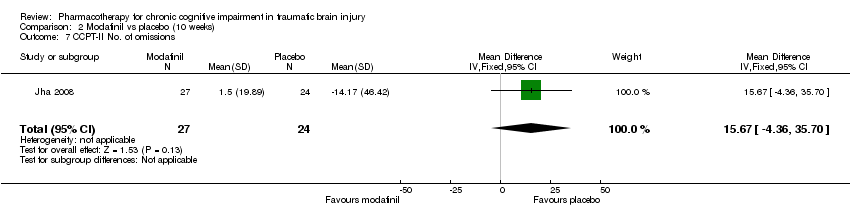
Comparison 2 Modafinil vs placebo (10 weeks), Outcome 7 CCPT‐II No. of omissions.

Comparison 2 Modafinil vs placebo (10 weeks), Outcome 8 CCPT‐II No. of commissions.
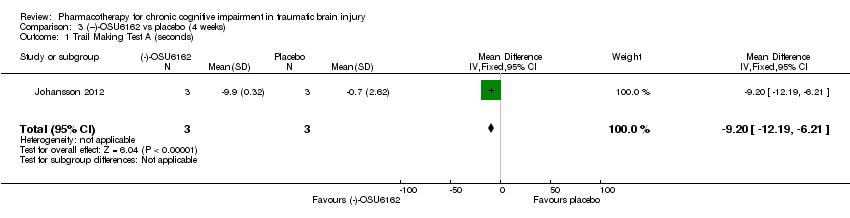
Comparison 3 (−)‐OSU6162 vs placebo (4 weeks), Outcome 1 Trail Making Test A (seconds).

Comparison 3 (−)‐OSU6162 vs placebo (4 weeks), Outcome 2 Trail Making Test B (seconds).
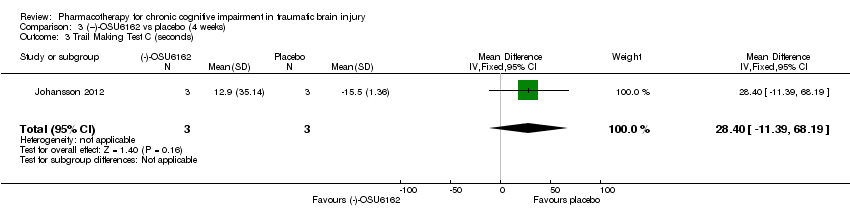
Comparison 3 (−)‐OSU6162 vs placebo (4 weeks), Outcome 3 Trail Making Test C (seconds).

Comparison 3 (−)‐OSU6162 vs placebo (4 weeks), Outcome 4 Trail Making Test D (seconds).

Comparison 3 (−)‐OSU6162 vs placebo (4 weeks), Outcome 5 WAIS‐III Digit Symbol Coding.
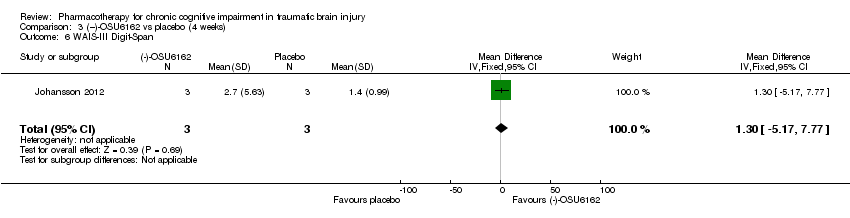
Comparison 3 (−)‐OSU6162 vs placebo (4 weeks), Outcome 6 WAIS‐III Digit‐Span.
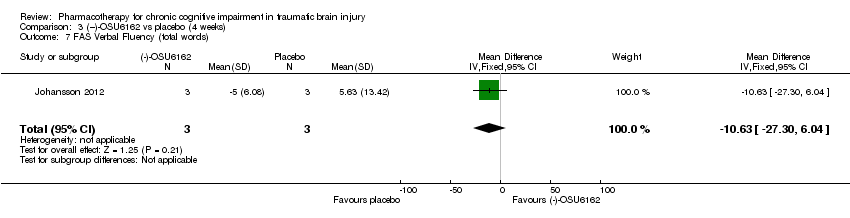
Comparison 3 (−)‐OSU6162 vs placebo (4 weeks), Outcome 7 FAS Verbal Fluency (total words).
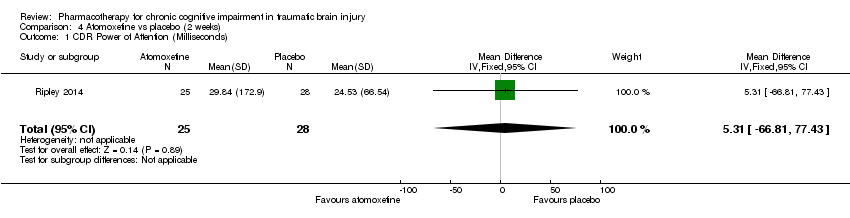
Comparison 4 Atomoxetine vs placebo (2 weeks), Outcome 1 CDR Power of Attention (Milliseconds).
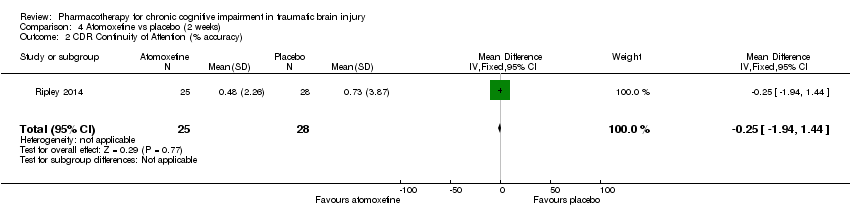
Comparison 4 Atomoxetine vs placebo (2 weeks), Outcome 2 CDR Continuity of Attention (% accuracy).

Comparison 4 Atomoxetine vs placebo (2 weeks), Outcome 3 CDR Efficiency (COA/POA, % accuracy/millisecond).
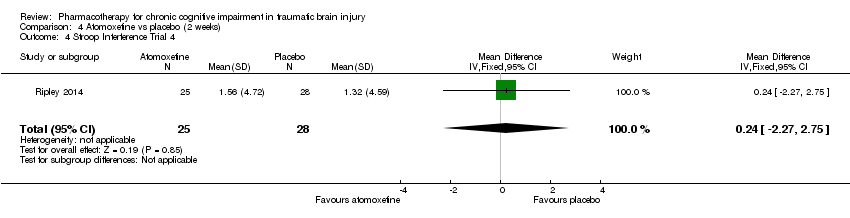
Comparison 4 Atomoxetine vs placebo (2 weeks), Outcome 4 Stroop Interference Trial 4.

Comparison 4 Atomoxetine vs placebo (2 weeks), Outcome 5 Adult ADHD Self‐Report Scale (ASRS‐v1.1.).

Comparison 5 Rivastigmine vs placebo (12 weeks), Outcome 1 HVLT‐total word recall.
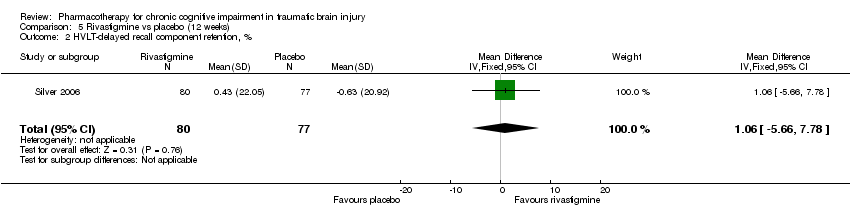
Comparison 5 Rivastigmine vs placebo (12 weeks), Outcome 2 HVLT‐delayed recall component retention, %.

Comparison 5 Rivastigmine vs placebo (12 weeks), Outcome 3 HVLT–recognition discriminant index.

Comparison 5 Rivastigmine vs placebo (12 weeks), Outcome 4 CANTAB RVIP’A.

Comparison 5 Rivastigmine vs placebo (12 weeks), Outcome 5 CANTAB–SWM, total errors.

Comparison 5 Rivastigmine vs placebo (12 weeks), Outcome 6 CANTAB RVIP, mean latency, ms.

Comparison 5 Rivastigmine vs placebo (12 weeks), Outcome 7 CANTAB‐RT, simple reaction time, ms.

Comparison 5 Rivastigmine vs placebo (12 weeks), Outcome 8 CANTAB‐PAL, total errors.
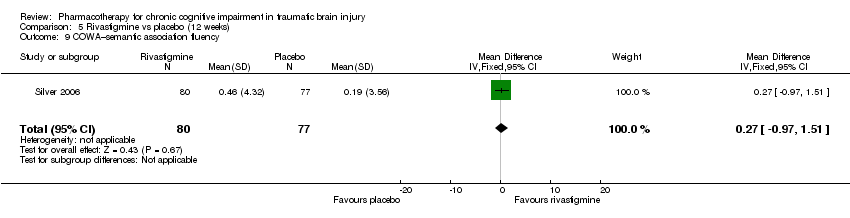
Comparison 5 Rivastigmine vs placebo (12 weeks), Outcome 9 COWA–semantic association fluency.
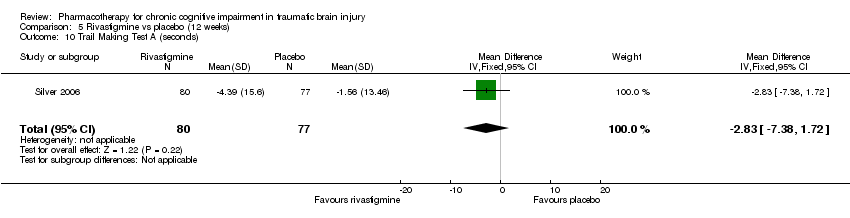
Comparison 5 Rivastigmine vs placebo (12 weeks), Outcome 10 Trail Making Test A (seconds).
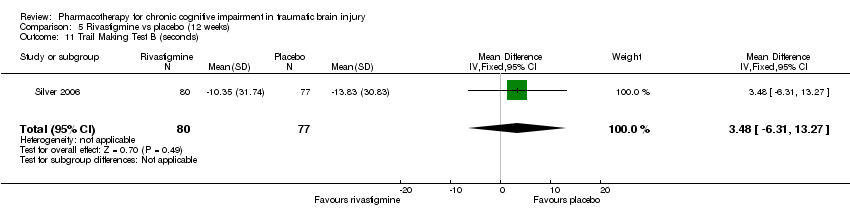
Comparison 5 Rivastigmine vs placebo (12 weeks), Outcome 11 Trail Making Test B (seconds).

Comparison 5 Rivastigmine vs placebo (12 weeks), Outcome 12 WAIS‐III‐DS scaled score.

Comparison 6 Acceptability of treatment, Outcome 1 Modafinil vs placebo.

Comparison 6 Acceptability of treatment, Outcome 2 Rivastigmine vs placebo.

Comparison 6 Acceptability of treatment, Outcome 3 (‐)‐OSU6162 vs placebo.

Comparison 6 Acceptability of treatment, Outcome 4 Atomoxetine vs placebo.

Comparison 7 Modafinil vs placebo adverse events, Outcome 1 Dizziness.

Comparison 7 Modafinil vs placebo adverse events, Outcome 2 Dysgeusia.

Comparison 7 Modafinil vs placebo adverse events, Outcome 3 Dyspepsia.

Comparison 7 Modafinil vs placebo adverse events, Outcome 4 Fatigue.

Comparison 7 Modafinil vs placebo adverse events, Outcome 5 Insomnia.

Comparison 7 Modafinil vs placebo adverse events, Outcome 6 Memory impairment.
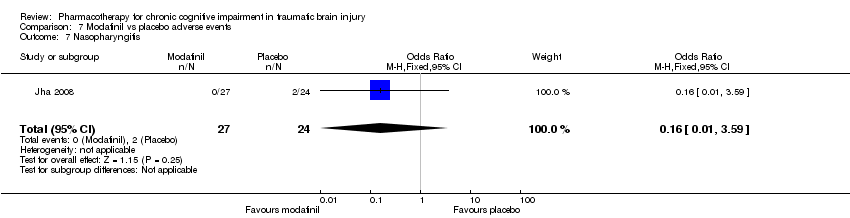
Comparison 7 Modafinil vs placebo adverse events, Outcome 7 Nasopharyngitis.

Comparison 7 Modafinil vs placebo adverse events, Outcome 8 Nausea.

Comparison 7 Modafinil vs placebo adverse events, Outcome 9 Weight loss.
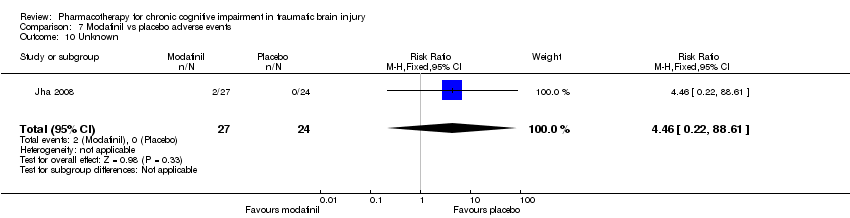
Comparison 7 Modafinil vs placebo adverse events, Outcome 10 Unknown.

Comparison 8 (−)‐OSU6162 vs placebo adverse events, Outcome 1 Nausea.
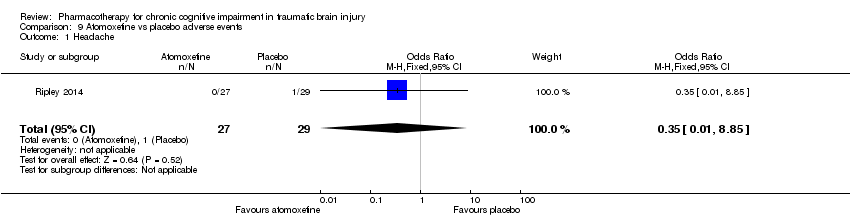
Comparison 9 Atomoxetine vs placebo adverse events, Outcome 1 Headache.
Comparison 9 Atomoxetine vs placebo adverse events, Outcome 2 Dry mouth.

Comparison 9 Atomoxetine vs placebo adverse events, Outcome 3 Globus pharyngeus.

Comparison 9 Atomoxetine vs placebo adverse events, Outcome 4 Hypertension.

Comparison 9 Atomoxetine vs placebo adverse events, Outcome 5 Insomnia.

Comparison 9 Atomoxetine vs placebo adverse events, Outcome 6 Irritable bowl.

Comparison 9 Atomoxetine vs placebo adverse events, Outcome 7 Loss of appetite.

Comparison 9 Atomoxetine vs placebo adverse events, Outcome 8 Nasal congestion.

Comparison 9 Atomoxetine vs placebo adverse events, Outcome 9 Shoulder pain.
Comparison 9 Atomoxetine vs placebo adverse events, Outcome 10 Urinary retention.
Comparison 10 Rivastigmine vs placebo adverse events, Outcome 1 Anxiety.

Comparison 10 Rivastigmine vs placebo adverse events, Outcome 2 Arthralgia.
Comparison 10 Rivastigmine vs placebo adverse events, Outcome 3 Diarrhoea.

Comparison 10 Rivastigmine vs placebo adverse events, Outcome 4 Dizziness.

Comparison 10 Rivastigmine vs placebo adverse events, Outcome 5 Headache.

Comparison 10 Rivastigmine vs placebo adverse events, Outcome 6 Nausea.

Comparison 10 Rivastigmine vs placebo adverse events, Outcome 7 Upper respiratory tract infection.
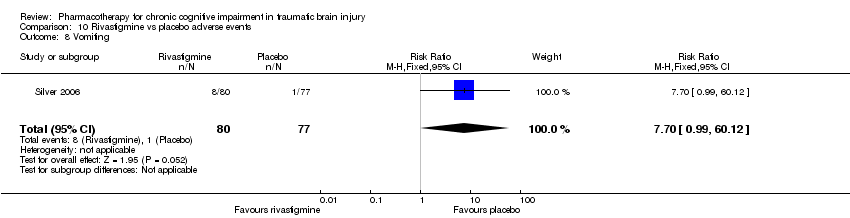
Comparison 10 Rivastigmine vs placebo adverse events, Outcome 8 Vomiting.
| Modafanil, (−)‐OSU6162, atomoxetine or rivastigmine compared to placebo for chronic cognitive impairment in traumatic brain injury | |||||
| Patient or population: Participants with chronic cognitive impairment in traumatic brain injury | |||||
| Outcomes | Effect of drug treatment for people with cognitive impairment in traumatic brain injury | Relative effect | No of Participants | Quality of the evidence | Comments |
| Cognitive performance on psychometric tests | The majority of sub‐tests showed no difference between treatment and placebo. Superiority over placebo was shown in one measure in Silver 2006 and several measures in Johansson 2012 and Johansson 2015. However, interpretation of these findings are cautioned. | See comment. | 274 (4 studies) | ⊕⊝⊝⊝ very low1,2,3 | Data synthesis was not possible due to the heterogeneity of studies. |
| Clinical global improvement | A single study reported no difference between treatment and placebo. | See comment. | 51 (1 study) | ⊕⊕⊝⊝ low1,3 | Data synthesis was not possible as only one study reported a measure on clinical global improvement. |
| Acceptability | No differences between treatment and placebo were found. | See comment. | 274(4 studies) | ⊕⊝⊝⊝ very low1,2,3 | Data synthesis was not possible due to the heterogeneity of studies. |
| Safety | More nausea was reported in participants receiving rivastigmine than placebo (Silver 2006). No other differences were found. | See comment. | 274 (4 studies) | ⊕⊝⊝⊝ very low1,2,3 | Data synthesis was not possible due to the heterogeneity of studies. |
| Mortality | No deaths were reported by any study. | Not estimable. | 274 (4 studies) | ‐ | |
| GRADE Working Group grades of evidence | |||||
| 1 downgraded one level due to serious indirectness, as two of four studies did not investigate cognitive impairment as a primary outcome 2 downgraded one level due to serious inconsistency, due to wide variance of point estimates. 3 downgraded one level due to serious imprecision, as the total population size was less than 400. | |||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 SF‐12 Physical Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐2.18 [‐6.34, 1.98] |
| 2 SF‐12 Mental Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 1.95 [‐2.84, 6.74] |
| 3 ImPACT verbal memory composite Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 1.42 [‐4.42, 7.26] |
| 4 ImPACT visual memory composite Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 0.25 [‐6.52, 7.02] |
| 5 ImPACT visual motor speed composite Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐3.45 [‐6.48, ‐0.42] |
| 6 ImPACT reaction time composite Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐0.04, 0.08] |
| 7 CCPT‐II No. of omissions Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 16.57 [‐3.59, 36.73] |
| 8 CCPT‐II No. of commissions Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 2.00 [‐1.75, 5.75] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 SF‐12 Physical Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐0.53 [‐5.46, 4.40] |
| 2 SF‐12 Mental Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 0.41 [‐4.28, 5.10] |
| 3 ImPACT verbal memory composite Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 4.11 [‐1.37, 9.59] |
| 4 ImPACT visual memory composite Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐1.61 [‐8.97, 5.75] |
| 5 ImPACT visual motor speed composite Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐1.44 [‐4.61, 1.73] |
| 6 ImPACT reaction time composite Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐0.03 [‐0.08, 0.02] |
| 7 CCPT‐II No. of omissions Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 15.67 [‐4.36, 35.70] |
| 8 CCPT‐II No. of commissions Show forest plot | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 2.96 [‐0.47, 6.39] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Trail Making Test A (seconds) Show forest plot | 1 | 6 | Mean Difference (IV, Fixed, 95% CI) | ‐9.20 [‐12.19, ‐6.21] |
| 2 Trail Making Test B (seconds) Show forest plot | 1 | 6 | Mean Difference (IV, Fixed, 95% CI) | ‐6.20 [‐7.81, ‐4.59] |
| 3 Trail Making Test C (seconds) Show forest plot | 1 | 6 | Mean Difference (IV, Fixed, 95% CI) | 28.4 [‐11.39, 68.19] |
| 4 Trail Making Test D (seconds) Show forest plot | 1 | 6 | Mean Difference (IV, Fixed, 95% CI) | 53.5 [36.76, 70.24] |
| 5 WAIS‐III Digit Symbol Coding Show forest plot | 1 | 6 | Mean Difference (IV, Fixed, 95% CI) | 8.6 [6.47, 10.73] |
| 6 WAIS‐III Digit‐Span Show forest plot | 1 | 6 | Mean Difference (IV, Fixed, 95% CI) | 1.30 [‐5.17, 7.77] |
| 7 FAS Verbal Fluency (total words) Show forest plot | 1 | 6 | Mean Difference (IV, Fixed, 95% CI) | ‐10.63 [‐27.30, 6.04] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 CDR Power of Attention (Milliseconds) Show forest plot | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | 5.31 [‐66.81, 77.43] |
| 2 CDR Continuity of Attention (% accuracy) Show forest plot | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐0.25 [‐1.94, 1.44] |
| 3 CDR Efficiency (COA/POA, % accuracy/millisecond) Show forest plot | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | 0.76 [‐3.10, 4.62] |
| 4 Stroop Interference Trial 4 Show forest plot | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | 0.24 [‐2.27, 2.75] |
| 5 Adult ADHD Self‐Report Scale (ASRS‐v1.1.) Show forest plot | 1 | 53 | Mean Difference (IV, Fixed, 95% CI) | ‐1.19 [‐5.63, 3.25] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 HVLT‐total word recall Show forest plot | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | 0.21 [‐1.14, 1.56] |
| 2 HVLT‐delayed recall component retention, % Show forest plot | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | 1.06 [‐5.66, 7.78] |
| 3 HVLT–recognition discriminant index Show forest plot | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | ‐0.06 [‐0.78, 0.66] |
| 4 CANTAB RVIP’A Show forest plot | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | ‐0.01 [‐0.03, 0.00] |
| 5 CANTAB–SWM, total errors Show forest plot | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | 1.05 [‐5.85, 7.95] |
| 6 CANTAB RVIP, mean latency, ms Show forest plot | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | ‐44.54 [‐88.62, ‐0.46] |
| 7 CANTAB‐RT, simple reaction time, ms Show forest plot | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | ‐19.81 [‐59.23, 19.61] |
| 8 CANTAB‐PAL, total errors Show forest plot | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | ‐3.16 [‐10.75, 4.43] |
| 9 COWA–semantic association fluency Show forest plot | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | 0.27 [‐0.97, 1.51] |
| 10 Trail Making Test A (seconds) Show forest plot | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | ‐2.83 [‐7.38, 1.72] |
| 11 Trail Making Test B (seconds) Show forest plot | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | 3.48 [‐6.31, 13.27] |
| 12 WAIS‐III‐DS scaled score Show forest plot | 1 | 157 | Mean Difference (IV, Fixed, 95% CI) | 0.22 [‐0.33, 0.77] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Modafinil vs placebo Show forest plot | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.46 [0.22, 88.61] |
| 2 Rivastigmine vs placebo Show forest plot | 1 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.35, 1.59] |
| 3 (‐)‐OSU6162 vs placebo Show forest plot | 1 | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Atomoxetine vs placebo Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Dizziness Show forest plot | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.56 [0.43, 29.66] |
| 2 Dysgeusia Show forest plot | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.46 [0.22, 88.61] |
| 3 Dyspepsia Show forest plot | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.46 [0.22, 88.61] |
| 4 Fatigue Show forest plot | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.44 [0.56, 35.41] |
| 5 Insomnia Show forest plot | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.67 [0.59, 11.99] |
| 6 Memory impairment Show forest plot | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.46 [0.22, 88.61] |
| 7 Nasopharyngitis Show forest plot | 1 | 51 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.16 [0.01, 3.59] |
| 8 Nausea Show forest plot | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.67 [0.30, 23.96] |
| 9 Weight loss Show forest plot | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.46 [0.22, 88.61] |
| 10 Unknown Show forest plot | 1 | 51 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.46 [0.22, 88.61] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Nausea Show forest plot | 1 | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.01, 8.62] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Headache Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.85] |
| 2 Dry mouth Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.78 [0.27, 126.14] |
| 3 Globus pharyngeus Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.34 [0.13, 85.56] |
| 4 Hypertension Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.34 [0.13, 85.56] |
| 5 Insomnia Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.78 [0.27, 126.14] |
| 6 Irritable bowl Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.34 [0.13, 85.56] |
| 7 Loss of appetite Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.34 [0.13, 85.56] |
| 8 Nasal congestion Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.34 [0.13, 85.56] |
| 9 Shoulder pain Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.34 [0.13, 85.56] |
| 10 Urinary retention Show forest plot | 1 | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.34 [0.13, 85.56] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Anxiety Show forest plot | 1 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.67 [0.47, 158.32] |
| 2 Arthralgia Show forest plot | 1 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.93 [0.36, 10.21] |
| 3 Diarrhoea Show forest plot | 1 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.41 [0.48, 12.03] |
| 4 Dizziness Show forest plot | 1 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.7 [0.99, 60.12] |
| 5 Headache Show forest plot | 1 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.35, 2.10] |
| 6 Nausea Show forest plot | 1 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.05 [1.29, 7.22] |
| 7 Upper respiratory tract infection Show forest plot | 1 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.17 [0.70, 6.74] |
| 8 Vomiting Show forest plot | 1 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.7 [0.99, 60.12] |