Intervenciones para la micosis fungoide
Referencias
References to studies included in this review
References to studies excluded from this review
References to studies awaiting assessment
References to ongoing studies
Additional references
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | This was a randomised, open‐label, cross‐over trial, which lasted 12 months. | |
| Participants | The study recruited 16 participants (10 were in the PUVA‐first group; 6 were in the ECP‐first group) with plaque‐stage (1B ⁄ T2, Bunn Lamberg 1B) MF and a peripheral blood T‐cell clone (detected by polymerase chain reaction (PCR)‐single‐strand conformational polymorphism (SSCP) methodology), but with no evidence of lymph node involvement. Demographics of the included participants
Exclusion criteria of the trial
| |
| Interventions |
| |
| Outcomes | Outcomes of the trial
| |
| Notes | No funding body was declared. This study was conducted at Skin Tumour Clinic, St John's Institute of Dermatology, United Kingdom. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A statistician generated the randomisation. |
| Allocation concealment (selection bias) | Unclear risk | It was unclear whether used envelopes were sealed and opaque. |
| Blinding of participants and personnel (performance bias) | Unclear risk | This was not possible because of different types of interventions. |
| Blinding of outcome assessment (detection bias) | Unclear risk | There were insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) | High risk | 8/16 participants (50%) were lost to follow up: 3/10 participants in the PUVA‐first group and 5/6 participants in the ECP‐first group. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | High risk | It was unclear if concomitant medication was permitted. |
| Methods | This was a randomised, double‐blind, parallel‐group trial, which lasted 4 months. | |
| Participants | The study recruited 4 participants (3 in the intervention group and 1 in the control group) with histologically‐proven MF plaque stage 1B MF (T2N0M0). Demographics of the included participants
Exclusion criteria of the trial These were not reported. | |
| Interventions |
| |
| Outcomes | Outcomes of the trial
| |
| Notes | The funding body was 3M Health Care Limited supplied Aldara. This study was conducted in the United Kindom (1 centre). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. |
| Blinding of participants and personnel (performance bias) | Low risk | The study was described as double‐blind. |
| Blinding of outcome assessment (detection bias) | Unclear risk | There were insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) | Low risk | No dropouts were reported. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | High risk | The study had a very small sample size (n = 4): 3 participants in the intervention group, and 1 participant in the placebo group. |
| Methods | This was a randomised, open‐label, parallel‐group trial, which lasted 16 weeks. | |
| Participants | The study recruited 58 participants (15 in the low‐dose group with 6.5 mg/m² daily versus 43 in the high‐dose group, which consisted of 28 participants who had 300 mg/m² daily and 15 participants who had 650 mg/m² daily) with histologically‐confirmed mycosis fungoides:
Demographics of the included participants
Exclusion criteria of the trial
| |
| Interventions |
| |
| Outcomes | Outcomes of the trial
| |
| Notes | The high‐dose was reduced twice (from 650 to > 500, and from 500 to > 300) during the trial; there was separate assessments of the high‐dose and "optimal" dose groups. 11/15 participants in the low‐dose group crossed over to high‐dose therapy after 8 weeks of treatment. Randomisation discontinued during the trial after interim analysis and was reinstalled after consideration by the U.S. Food and Drug Administration (FDA). The dropout rate for withdrawals was 72.4%. Dr Duvic was funded by research grants from Ligand Pharmaceuticals, San Diego California, USA (R21‐CA74117); from the National Institutes of Health, Bethesda, Maryland; and from the MD Anderson Cancer Centre (CA16672‐22). This study was conducted in 18 CTCL clinics at academic referral centres in the USA, Canada, Australia, and Europe. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Blinding of participants and personnel (performance bias) | High risk | "Blinding was not possible because of the number of capsules given." |
| Blinding of outcome assessment (detection bias) | Low risk | "The physician was blinded to CA response because it was calculated from the case report form." |
| Incomplete outcome data (attrition bias) | High risk | The dropout rate for withdrawals was 72.4%. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | High risk | The initial dose in the intervention group was reduced from 650 mg/m²/day to 500 mg/m²/day to 300 mg/m²/day due to adverse reactions. The study discontinued randomisation. |
| Methods | This was a randomised, double‐blind, parallel‐group trial, which lasted 24 weeks. | |
| Participants | The study recruited 89 participants (43 in the intervention group and 46 in the control) with histologically‐confirmed MF manifested as patches with or without plaques (stage I), but without enlarged nodes, visceral involvement, or generalised erythroderma. Demographics of the included participants
Exclusion criteria of the trial
| |
| Interventions |
| |
| Outcomes | Outcomes of the trial
| |
| Notes | Funding came from BioCryst Pharmaceuticals, Inc (Birmingham, Alabama, USA). This study was conducted in 10 tertiary care centres in the USA. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | No specific information was given. The data were managed by a third party (Quintiles Inc). |
| Allocation concealment (selection bias) | Low risk | No specific information was given. The data were managed by a third party (Quintiles Inc). |
| Blinding of participants and personnel (performance bias) | Low risk | Only the sponsor was able to un‐blind in case of withdrawal. |
| Blinding of outcome assessment (detection bias) | Low risk | Only the sponsor was able to un=‐blind in case of withdrawal. |
| Incomplete outcome data (attrition bias) | High risk | ITT analysis was carried out and last observation carried forward. 24/89 participants were lost to follow up: 14/43 (33%) in the intervention group and 11/46 (24%) in the placebo group. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | Low risk | None were found. |
| Methods | This was a randomised, open‐label, parallel‐group trial, which lasted 24 weeks. | |
| Participants | The study recruited 43 participants (20 in the high‐dose group and 23 in the low‐dose group) with histologically‐proven mycosis fungoides stages IB and IIA, with lymph node biopsies negative for MF involvement. Demographics of the included participants
Exclusion criteria of the trial These were not reported. | |
| Interventions |
| |
| Outcomes | Outcomes of the trial
| |
| Notes | Data were abstracted from the manuscript sent by the corresponding author; the sample size was smaller than planned according to the author. Dose reduction was necessary in 14/39 participants because of hyperlipidaemic side‐effects, although antilipidaemic therapy was prescribed for each participant. This study was conducted in 12 tertiary care centres in the USA. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Blinding of participants and personnel (performance bias) | High risk | This was an open‐label trial. |
| Blinding of outcome assessment (detection bias) | Unclear risk | No separate outcome assessor was mentioned. |
| Incomplete outcome data (attrition bias) | Low risk | ITT analysis was carried out: 4 participants (10%) dropped out after randomisation without receiving a single treatment; 1 person (5%) in the 300 mg/day bexarotene group dropped out for "other" reason. |
| Selective reporting (reporting bias) | Low risk | We contacted the corresponding author for additional outcome data, and we had an email response. We were sent a manuscript of unpublished data, and we had further confirmation by email that all outcomes were reported. |
| Other bias | High risk | The study had a smaller sample size than planned; dose reduction was necessary in 14/39 participants due to hypertriglyceridaemia, although preventive antilipidaemic therapy was prescribed for each participant. |
| Methods | This was a randomised, open‐label, parallel‐group trial. | |
| Participants | The study recruited 103 participants (52 in the combined‐therapy group and 51 in the conservative‐therapy group) with histologically‐proven MF of all stages. Demographics of the included participants
Exclusion criteria of the trial
| |
| Interventions |
a) oral methotrexate (20 mg/m² p.o. twice weekly for stage IVB participants) b) PUVA (oral methoxsalen 0.6 mg/kg body weight followed by UVA light therapy 3 x/week) c) electron‐beam therapy (as described in the combined‐therapy group) combined with methotrexate (as described above) d) systemic chemotherapy (as described in the combined‐therapy group) | |
| Outcomes | Outcomes of the trial
| |
| Notes | The funding body was not declared. This study was conducted in 7 secondary/tertiary care centres in the USA. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified block randomisation was undertaken. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Blinding of participants and personnel (performance bias) | Unclear risk | This was not possible because of different interventions used. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Incomplete outcome data (attrition bias) | Unclear risk | ITT analysis and last observation carried forward was carried out. There were 6/52 (12%) dropouts in the combined‐therapy group (2 refused to receive treatment; 1 withdrew because of congestive heart failure; 1 withdrew because of residual cutaneous disease; and 2 refused treatment after clinical response) and 2/51 (4%) in conservative‐treatment group (no reasons were stated). |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, and the author requested original data from their former employer, but the data were not available so far. |
| Other bias | Unclear risk | It was unclear if previous treatment was stopped. |
| Methods | This was a randomised, parallel‐group trial, which lasted 6 months. | |
| Participants | The study recruited 71 participants (35 in the low‐dose group and 36 in the high‐dose group) with histologically‐proven mycosis fungoides type with ≥ 20% of lymphocytes within the skin biopsy stain positively for CD25 by immunohistochemistry. Further inclusion criteria was as follows: stage Ib‐III CTCL (CTCL Cooperative Group staging) recurred or persisted after ≥ 4 previous treatments for CTCL (excluding topical or systemic corticosteroids) or stage IVa CTCL participants who failed at least 1 previous therapy study consideration, and Eastern Cooperative Oncology Group Performance Status of 0, 1, or 2. Lymph node involvement was no greater than LN₂, and no CTCL involvement of bone marrow. Demographics of the included participants
Exclusion criteria of the trial
| |
| Interventions |
| |
| Outcomes | Outcomes of the trial
| |
| Notes | Only 42% of all randomised participants received 8 courses of treatment as planned. There was no comparison reported between both treatment groups regarding QoL from baseline to the end of the study (only subgroup analyses of responders vs non‐responders). The funding body was Seragen, Inc (a wholly‐owned subsidiary of Ligand Pharmaceuticals Inc, San Diego, CA). This study was conducted in 20 centres across the USA. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was stratified by stage of CTCL for multicentre trial. It was likely to have been carried out by a third party and concealed, although this was not formally stated. |
| Allocation concealment (selection bias) | Unclear risk | Randomisation was stratified by stage of CTCL for multicentre trial. However, it was unclear whether randomisation was concealed. |
| Blinding of participants and personnel (performance bias) | High risk | This was unlikely to be blinded, since the drug was diluted to a certain minimum concentration in both arms and administered by a pump device for 15 to 60 minutes. |
| Blinding of outcome assessment (detection bias) | Low risk | "All responses were verified by an independent panel of physicians [the Data End Point Review Committee]." |
| Incomplete outcome data (attrition bias) | High risk | 41/71 (58%) participants dropped out. Discontinuation was due to adverse events (11/35 (31%) participants in the 9 µg/kg/day group vs 15/36 (42%) in the 18 µg/kg/day group) and treatment failure (6/35 (17%) in the 9 µg/kg/day group vs 2/36 (6%) in the 18 µg/kg/day group). |
| Selective reporting (reporting bias) | High risk | The quality of life assessment was compared between responders and non‐responders instead of comparing treatment groups. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | Unclear risk | There were insufficient information to permit judgement. |
| Methods | This was a randomised, double‐blind (verified by author contact), within‐participant trial, which lasted 6 weeks. | |
| Participants | The study recruited 12 participants (with 1 lesion per treatment): men or non‐pregnant women aged 18 to 70 with stable patch or plaque phase MF of at least 4 months' duration. Demographics of the included participants
Exclusion criteria of the trial These were not reported. | |
| Interventions |
| |
| Outcomes | Outcomes of the trial
| |
| Notes | The study was supported in part by the USA's Department of Energy Merit Review. Funding came from the Department of Veterans Affairs (Dr Wood) and Vimrx Inc. Disclosure: Dr Rook has been a consultant to Hy BioPharma Inc. This study was conducted in 4 tertiary care centres in the USA. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information and had an email response: The corresponding author no longer had access to data from the former employer. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information and had an email response: The corresponding author no longer had access to data from the former employer. |
| Blinding of participants and personnel (performance bias) | Low risk | The study was described in the publication as "double‐blind" and "open‐label". The email response from the author confirmed that the study was double‐blind. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study did not provide information about this. We sought information and had an email response: The corresponding author no longer had access to data from the former employer. |
| Incomplete outcome data (attrition bias) | Low risk | There were no dropouts. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We sought information and had an email response: The corresponding author no longer had access to data from the former employer. |
| Other bias | Unclear risk | It was unclear if previous treatment was stopped and if concomitant medication was permitted. |
| Methods | This was a randomised, open‐label, parallel‐group trial, which lasted 48 weeks. | |
| Participants | The study recruited 82 participants (40 in the IFN‐α + PUVA group and 42 in the IFN‐α + acitretin group) with small‐ to medium‐sized pleomorphic T‐cell lymphoma or mycosis fungoides stage I or II. The principle investigator (Stadler) stated on author contact that all participants had histologically‐proven mycosis fungoides. Demographics of the included participants
Exclusion criteria of the trial These were not reported. | |
| Interventions |
| |
| Outcomes | Outcomes of the trial
| |
| Notes | The funding body was not declared. This study was conducted in 21 tertiary care centres in Germany, Austria, and Switzerland. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was by a central institution/third party (Estimate GmbH, Augsburg/Germany) and stratified by pretreatment. |
| Allocation concealment (selection bias) | Low risk | Randomisation was by central institution/third party (Estimate GmbH, Augsburg/Germany) and stratified by pretreatment. |
| Blinding of participants and personnel (performance bias) | Unclear risk | This was not possible because of different interventions (PUVA vs capsules). |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Incomplete outcome data (attrition bias) | High risk | The primary analysis was per‐protocol. ITT analysis was also carried out for comparison between study groups regarding complete remission: 16/98 (16%) participants dropped out (6 participants did not receive any treatment; 6 participants had insufficient data monitored; and 4 participants had wrong staging at enrolment); there was no distribution between groups reported. 40/49 participants in the PUVA group and 42/49 participants in the acitretin group were evaluable. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | Unclear risk | It was unclear if previous treatment was stopped and if concomitant medication was permitted. |
| Methods | This was a randomised parallel‐group trial, which lasted 52 weeks. | |
| Participants | The study recruited 124 participants with cutaneous T‐cell lymphoma (stages lA to IIA) ‐ type mycosis fungoides or small to medium cellular pleomorphic type. The principle investigator (Stadler) stated on author contact that all participants had histologically‐proven mycosis fungoides. Demographics of the included participants
Exclusion criteria of the trial These were not reported. | |
| Interventions |
| |
| Outcomes | Outcomes of the trial
| |
| Notes | The funding body was not declared. The disclosure in Stadler 2006 stated: "No significant financial relationships to disclose." This study was conducted in 26 tertiary care centres in Germany and Switzerland. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Blinding of participants and personnel (performance bias) | High risk | Blinding was unlikely since no placebo injections were described. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Incomplete outcome data (attrition bias) | High risk | 31/124 (25%) randomised participants were not evaluable, and no reasons for this were stated. |
| Selective reporting (reporting bias) | High risk | The only outcome reported was complete remission; there was no report on adverse effects. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | Unclear risk | It was unclear if previous treatment was stopped and if concomitant medication was permitted. |
| Methods | This was a randomised, double‐blind, parallel‐group trial, which lasted 12 months. | |
| Participants | The study recruited 16 participants (8 in the intervention group and 8 in the control group) with histologically‐proven MF van Scott stage II to IV. Demographics of the included participants
Exclusion criteria of the trial These were not reported. | |
| Interventions |
| |
| Outcomes | Outcomes of the trial
| |
| Notes | The funding body was Landsforeningen til kraeftens bekaempelse (a grant came from the National Institution for Cancer Prevention of Danish Cancer Society). This study was conducted in the Department of Dermatology, University of Aarhus, Denmark. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Allocation concealment (selection bias) | High risk | The corresponding trial author confirmed in an email response that the randomisation list was open. |
| Blinding of participants and personnel (performance bias) | Low risk | This was a double‐blind study. |
| Blinding of outcome assessment (detection bias) | Low risk | The corresponding trial author confirmed in an email response that the outcome assessors were blinded. |
| Incomplete outcome data (attrition bias) | Unclear risk | No losses to follow up were reported, but the number of participants randomised was not stated. |
| Selective reporting (reporting bias) | Low risk | This was unknown. We contacted the corresponding author for additional outcome data, and the author responded with a completed data extraction form. |
| Other bias | High risk | Concomitant treatment was permitted. |
| Methods | This was a randomised, double‐blind, within‐participant trial, which lasted 4 weeks. | |
| Participants | The study recruited 6 participants (2 lesions per treatment) with plaque phase MF, MFCG stage nomenclature of 1979 stage IA (T1, Nx, T0, M0), stage IB (T2, Nx, T0, M0), or stage IIA (T2, N1, T0, M0) Demographics of the included participants
Exclusion criteria of the trial
| |
| Interventions |
| |
| Outcomes | Outcomes of the trial
| |
| Notes | The funding body was not declared. This study was conducted in a tertiary care centre in the USA. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Lesions were allocated by a random code; no further information was given; information was sought, but we received no response. |
| Allocation concealment (selection bias) | Unclear risk | Information was sought, but we received no response. |
| Blinding of participants and personnel (performance bias) | Low risk | The trial was described as double‐blind for the first part, which we data extracted. |
| Blinding of outcome assessment (detection bias) | Unclear risk | No separate outcome assessor was described. |
| Incomplete outcome data (attrition bias) | Low risk | ITT analysis was carried out. There were no dropouts. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | Unclear risk | There were insufficient information to permit judgement. |
| Methods | This was a randomised, double‐blind, parallel‐group trial, which lasted 8 weeks. | |
| Participants | The study recruited 12 participants (9 from the intervention group and 3 from the control group) with early plaque or patch stage MF (stage IA or IB), with no evidence of physical examination on lymphadenopathy or organomegaly. Demographics of the included participants
Exclusion criteria of the trial
| |
| Interventions |
a) IFN‐α 2MU in superficial dermis 3 times weekly; b) betamethasone dipropionate ointment 0.05% twice daily; or c) no treatment.
a) placebo (buffered glycine serum human albumin) in superficial dermis 3 times weekly; b) betamethasone dipropionate ointment 0.05% twice daily; or c) no treatment. | |
| Outcomes | Outcomes of the trial
| |
| Notes | Partial improvement was reported as mean difference in size of lesions without the possibility to identify participants' improvement according to our defined secondary outcome. Lesions in the IFN‐α group generally improved better than in the placebo group, possibly due to a systemic effect of IFN‐α as discussed by the authors. The IFN‐α was supplied by Schering Corp. This study was conducted in a tertiary care centre in Pittsburgh, USA. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Blinding of participants and personnel (performance bias) | Low risk | This trial had a double‐blind setting. |
| Blinding of outcome assessment (detection bias) | Low risk | Histopathologic features of biopsies were assessed without knowledge of the treatment or group. |
| Incomplete outcome data (attrition bias) | Low risk | ITT analysis was carried out. There were no dropouts. |
| Selective reporting (reporting bias) | High risk | Only the mean difference in the decrease of the lesions were reported; no incidence of partial remission (i.e. > 50% reduction of disease) was reported. The corresponding author was contacted for additional outcome data but did not respond within 4 weeks. |
| Other bias | High risk | The groups were unequal: There was a higher proportion of stage 1B, more men, longer duration of skin disease, and longer time since diagnosis in intervention group |
| Methods | This was a randomised, open‐label, parallel‐group trial, which lasted 24 weeks. | |
| Participants | The study recruited 29 participants (12 in the intervention group and 17 in the control group) with mycosis fungoides stage IA to IIA. Demographics of the included participants
Exclusion criteria of the trial
| |
| Interventions |
| |
| Outcomes | Outcomes of the trial
| |
| Notes | The funding body was Ministerio de Ciencia y Tecnologia (BIO2000‐0275‐C02 ⁄01‐⁄02, SAF2001‐0060, SAF2005‐00221), Comunidad Autonoma de Madrid (CAM 08.1 ⁄0011 ⁄2001.1), and the Ministerio de Sanidad y Consumo (FISP05 ⁄1710, FIS 01‐0035, G03 ⁄179, PI051623) RETICS, Spain. The author, MBW, was supported by FISP05 ⁄1710, and LT was supported by grants from the CNIO and the Higher Education Authority of Ireland, St James Hospital, Dublin. Participants were categorised to responders and non‐responders instead of treatment groups. Some information was taken from the clinicaltrials.gov website (NCT00630903). The main primary aim of the study was to examine the gene expression profiles of primary skin biopsies from these participants. This study was conducted in 9 tertiary care hospitals in Madrid, Spain. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Blinding of participants and personnel (performance bias) | High risk | This was taken from the previous version of the NCT00630903 protocol: The study was described as open‐label. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Incomplete outcome data (attrition bias) | Low risk | ITT analysis was carried out. There were no dropouts. |
| Selective reporting (reporting bias) | High risk | Participants were characterised and divided into responders and non‐responders instead of treatment groups. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | High risk | Some information was taken from protocol NCT00630903 (www.clinicaltrials.gov). The study was described as terminated due to accrual. |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| There was no relevant end point according to the protocol report. | |
| There was no relevant end point according to the protocol report. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. | |
| There was not enough information to confirm inclusion criteria, and we had no reaction when we attempted to contact the corresponding author. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. | |
| There was not enough information to confirm inclusion criteria, and we had no reply when we attempted to contact the corresponding author. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. | |
| There was not enough information to confirm inclusion criteria, and we had no reply when we attempted to contact the corresponding author. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. | |
| There was not enough information to abstract data from the publication. | |
| An email response confirmed that the study was not completed. (The Principal Investigator passed away). | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. | |
| There was not enough information to confirm inclusion criteria, and we had no reply when we attempted to contact the corresponding author. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. | |
| This study was not a randomised controlled trial. | |
| There was not enough information to confirm inclusion criteria, and we had no reply when we attempted to contact the corresponding author. | |
| This was scientific fraud (see Grant 2009). | |
| This was a report of an ongoing trial; there was not enough information to confirm inclusion criteria, and we had no reply when we attempted to contact the corresponding author. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. | |
| There was no relevant end point according to the protocol report. | |
| This study explicitly excluded MF. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. This was identified by Molin 1979 and retrieved as a reference in the adverse event search. | |
| There was not enough information to confirm inclusion criteria, and we had no reply when we attempted to contact the corresponding author. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. | |
| Less than 90% of enrolled participants had Alibert‐Bazin‐type MF, and no subgroup analysis was available. |
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | This is a further report about data from Olsen 2001. This is a randomised, parallel‐group trial, which lasted 6 months. |
| Participants | The study recruited 71 participants (35 in the low‐dose group and 36 in the high‐dose group) with histologically‐proven mycosis fungoides type with ≥ 20% of lymphocytes within the skin biopsy stain positively for CD25 by immunohistochemistry. Further inclusion criteria was as follows: stage Ib‐III CTCL (CTCL Co‐operative Group staging) recurred or persisted after ≥ 4 previous treatments for CTCL (excluding topical or systemic corticosteroids) or stage IVa CTCL participants who failed at least 1 previous therapy study consideration, and Eastern Cooperative Oncology Group Performance Status of 0, 1, or 2. Lymph node involvement was no greater than LN₂, and no CTCL involvement of bone marrow. Demographics of the included participants
Exclusion criteria of the trial
|
| Interventions |
|
| Outcomes | Outcomes of the trial
|
| Notes | This was a meeting abstract; the study also provides data for the excluded trial by Prince 2010. This study was conducted in 20 centres across the USA. |
| Methods | This was a randomised non‐inferiority trial, which lasted 12 months. |
| Participants | This study recruited 260 stage I‐IIA mycosis fungoides participants. |
| Interventions |
|
| Outcomes | Outcomes of the trial
|
| Notes | This was a meeting abstract; please see also NCT00168064 in the 'Characteristics of ongoing studies' section. |
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | A Randomized, Open‐Label Phase III Trial to Evaluate the Efficacy and Safety of Bexarotene (Targretin) Capsules Combined With PUVA, Compared to PUVA Treatment Alone in Patients With Mycosis Fungoides |
| Methods | This is a randomised, open‐label, multicentre study. Participants are stratified according to participating centre, age (60 and under vs over 60), and stage of disease (IB vs IIA). Participants are randomised to 1 of 2 treatment arms. Participants are followed every 8 weeks until the first documented progression or relapse. Projected accrual A total of 145 participants will be accrued for this study within 25 months. |
| Participants | Participants aged 18 years and older are eligible for this study. Both genders are eligible for this study. Healthy volunteers are not accepted. Disease characteristics
Participant characteristics
Prior concurrent therapy
|
| Interventions |
|
| Outcomes | Primary outcomes of the trial
Secondary outcomes of the trial
|
| Starting date | January 2003 |
| Contact information | Sponsors and collaborators
Investigators
|
| Notes | Other Study ID Numbers: CDR0000271933, EORTC‐21011 |
| Trial name or title | A Phase I/II Open Label, Multi‐Center Study For The Evaluation Of CPG 7909 In Patients With Stage IB To IVA Cutaneous T‐Cell Lymphoma |
| Methods | This is a phase I, open‐label, multicentre, dose‐escalation study followed by a randomised phase II study. Participants are followed every 4 weeks. Projected accrual A total of 3 to 56 participants (3 to 36 for phase I and 20 [10 per treatment |
| Participants | Participants aged 18 years and older are eligible for this study. Both genders are eligible for this study. Healthy volunteers are not accepted. Disease characteristics
Participant characteristics
Prior concurrent therapy
|
| Interventions |
|
| Outcomes | Primary outcomes of the trial
Secondary outcomes of the trial
|
| Starting date | July 2004 |
| Contact information | Sponsors and collaborators
Investigators
|
| Notes | ‐ |
| Trial name or title | A Phase II Pivotal Trial to Evaluate the Safety and Efficacy of Nitrogen Mustard (NM) 0.02% Ointment Formulations in Patients With Stage I or IIA Mycosis Fungoides (MF) |
| Methods | This is a phase II, randomised, double‐blind, parallel‐group safety/efficacy study with an estimated enrolment of 250 participants. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
|
| Interventions |
"All affected areas (lesions) are to be treated once daily for 12 months with NM 0.02% PG or NM 0.02% AP ointment." |
| Outcomes | Primary outcomes of the trial
Secondary outcomes of the trial
|
| Starting date | May 2006 |
| Contact information | Sponsors and collaborators Yaupon Therapeutics Investiogators Study Director: Stuart Lessin, MD, Fox Chase Cancer Center |
| Notes | Please see also Lessin 2011 in the 'Studies awaiting classification' section. |
| Trial name or title | Phase IV Randomized Study Of Two Dose Levels Of Targretin˜ Capsules In Patients With Refractory Cutaneous T‐Cell Lymphoma |
| Methods | This is a phase IV, randomised, open‐label, parallel‐group safety/efficacy study with an estimated enrolment of 60 participants. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
|
| Interventions |
|
| Outcomes | Primary outcomes of the trial
Secondary outcomes of the trial
|
| Starting date | December 2009 |
| Contact information | Sponsors and Collaborators
Investigators
|
| Notes | ‐ |
| Trial name or title | A Phase III Study of Lenalidomide Maintenance After Debulking With Gemcitabine or Liposomal Doxorubicin +/‐ Radiotherapy in Patients With Advanced Cutaneous T‐Cell Lymphoma Not Previously Treated With Intravenous Chemotherapy |
| Methods | This is a randomised, phase III, open‐label trial studying observation to see how well it works compared with lenalidomide in treating participants who are in complete or partial response after receiving previous gemcitabine hydrochloride or doxorubicin hydrochloride liposome for stage IIB, stage III, or stage IV cutaneous T‐cell lymphoma or stage IIB, stage III, or stage IV mycosis fungoides/Sézary syndrome with an estimated enrolment of 105 participants. |
| Participants | Disease characteristics
NOTE: *These recommended regimens can be altered according to local institutional policies. In case of drug intolerance, the study regimen can be switched from one regimen to the other. NOTE: **Local low‐dose/energy‐ionizing radiation therapy allowed as part of the debulking process to treat lesions that do not respond after 3 courses of debulking chemotherapy.
Participant characteristics
Prior concurrent therapy
|
| Interventions |
After completion of study treatment, participants are followed at 4 weeks and then every 12 weeks thereafter. |
| Outcomes | Primary outcomes of the trial
Secondary outcomes of the trial
|
| Starting date | July 2010 |
| Contact information | Sponsors and collaborators
Investigators
|
| Notes | ‐ |
| Trial name or title | A Multicenter, Open‐label, Randomized, Phase I/II Study Evaluating the Safety and Efficacy of Low‐dose (12 Gy) Total Skin Electron Beam Therapy (TSEBT) Combined With Vorinostat Versus Low‐dose TSEBT Monotherapy in Mycosis Fungoides (MF) |
| Methods | This is a multicentre, open‐label, parallel‐group safety/efficacy study with an estimated enrolment of 60 participants. |
| Participants | Inclusion criteria of the trial A participant will be eligible for inclusion only if all of the following criteria apply:
Exclusion criteria of the trial A participant will not be eligible for inclusion if any of the following criteria apply:
|
| Interventions |
|
| Outcomes | Primary outcomes of the trial
Secondary outcomes of the trial
|
| Starting date | December 2010 |
| Contact information | Sponsors and collaborators
Investigators
Contact: Cameron Harrison, (650) 721‐7186, [email protected] |
| Notes | ‐ |
| Trial name or title | Vorinostat (Zolinza®) in Combination With (Velcade®) Versus Vorinostat Alone in Refractory or Recurrent Advanced CTCL: A Randomized Phase III Study |
| Methods | This is a randomised, phase III open‐label trial studying how well vorinostat works when given alone compared with vorinostat given together with bortezomib in treating participants with refractory or recurrent stage IIB, stage III, or stage IV cutaneous T‐cell lymphoma with estimated enrolment of 189 participants. |
| Participants | Disease characteristics
Participant characteristics
Prior concurrent therapy
|
| Interventions |
Blood and tissue samples are collected periodically for translational research to provide insight into disease mechanism and identify biomarkers useful for prediction of treatment response. After completion of study treatment, participants are followed up at 4 weeks and then every 3 months until disease progression. |
| Outcomes | Primary outcomes of the trial
Secondary outcomes of the trial
|
| Starting date | January 2012 |
| Contact information | Sponsors and collaborators
Investigators
|
| Notes | ‐ |
| Trial name or title | A Randomized, Double‐Blind, Placebo‐Controlled, Dose‐Escalating Phase 1b Study to Assess the Safety, Pharmacodynamics and Pharmacokinetics of SHP 141, A Histone Deacetylase Inhibitor, Administered Topically Up to 28 Days to Patients With Stage IA, IB or IIA Cutaneous T‐Cell Lymphoma |
| Methods | This is a phase I, randomised, placebo‐controlled, double‐blind, parallel‐group safety/efficacy study with an estimated enrolment of 48 participants. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
|
| Interventions |
|
| Outcomes | Primary outcomes of the trial
|
| Starting date | September 2011 |
| Contact information | Sponsors and Collaborators
Investigators
|
| Notes | ‐ |
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse effects Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.1  Comparison 1 Topical peldesine versus placebo, Outcome 1 Adverse effects. | ||||
| 1.1 Pruritus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Rash | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 1.2  Comparison 1 Topical peldesine versus placebo, Outcome 2 Clearance. | ||||
| 3 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 1.3  Comparison 1 Topical peldesine versus placebo, Outcome 3 Improvement. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 2.1  Comparison 2 Topical imiquimod versus placebo, Outcome 1 Clearance. | ||||
| 2 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 2.2  Comparison 2 Topical imiquimod versus placebo, Outcome 2 Improvement. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 3.1  Comparison 3 Topical hypericin versus placebo, Outcome 1 Improvement. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse effects Show forest plot | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 4.1  Comparison 4 IFN‐α versus placebo, Outcome 1 Adverse effects. | ||||
| 1.1 Erythema | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Fever | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Myalgia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Chills or weakness | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Nausea, arthralgia, and malaise | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Clearance Show forest plot | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 4.2  Comparison 4 IFN‐α versus placebo, Outcome 2 Clearance. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clearance Show forest plot | 2 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.87, 1.31] |
| Analysis 5.1  Comparison 5 IFN‐α + PUVA versus PUVA alone, Outcome 1 Clearance. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse effects Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.1  Comparison 6 Denileukin diftitox high versus low dose, Outcome 1 Adverse effects. | ||||
| 1.1 Constitutional symptoms | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Grade 3 to 4 constitutional symptoms | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Infections | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Grade 3 to 4 infections | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Gastrointestinal syndromes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 Grade 3 to 4 gastrointestinal syndromes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.7 CNS syndromes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.8 Grade 3 to 4 CNS syndromes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.9 Rash | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.10 Grade 3 to 4 rash | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.11 Vascular leak syndrome | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.12 Grade 3 to 4 vascular leak syndrome | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.13 Thrombotic events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.14 Grade 3 to 4 thrombotic events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.15 Cardiopulmonary events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.16 Grade 3 to 4 cardiopulmonary events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.17 Acute infusion related events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.18 Grade 3 to 4 acute infusion related events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.19 Grade 3 to 4 laboratory abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 6.2  Comparison 6 Denileukin diftitox high versus low dose, Outcome 2 Clearance. | ||||
| 3 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 6.3  Comparison 6 Denileukin diftitox high versus low dose, Outcome 3 Improvement. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse effects Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 7.1  Comparison 7 Bexarotene high versus low dose, Outcome 1 Adverse effects. | ||||
| 1.1 Photosensitivity | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Nausea | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Constipation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Fatigue | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Pruritus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 Arthralgia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.7 Nasopharyngitis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.8 Headache | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.9 Insomnia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.10 Free T4 abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.11 Cholesterol abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.12 Triglycerid abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.13 SGOT/SGPT abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 7.2  Comparison 7 Bexarotene high versus low dose, Outcome 2 Clearance. | ||||
| 3 Relapse Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 7.3  Comparison 7 Bexarotene high versus low dose, Outcome 3 Relapse. | ||||
| 4 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 7.4  Comparison 7 Bexarotene high versus low dose, Outcome 4 Improvement. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 8.1  Comparison 8 Extracorporal photopheresis versus PUVA, Outcome 1 Clearance. | ||||
| 2 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 8.2  Comparison 8 Extracorporal photopheresis versus PUVA, Outcome 2 Improvement. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse effects Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 9.1  Comparison 9 Combined therapy versus conservative therapy, Outcome 1 Adverse effects. | ||||
| 1.1 Hospitalization | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Fatal myocardial infarction | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Cutaneous toxicity from electron beam therapy | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Acute non‐lymphocytic leukaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Non‐melanoma skin cancer | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 Unspecified | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 9.2  Comparison 9 Combined therapy versus conservative therapy, Outcome 2 Clearance. | ||||
| 3 Relapse Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 9.3  Comparison 9 Combined therapy versus conservative therapy, Outcome 3 Relapse. | ||||
| 4 Survival rate Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 9.4  Comparison 9 Combined therapy versus conservative therapy, Outcome 4 Survival rate. | ||||
| 5 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 9.5  Comparison 9 Combined therapy versus conservative therapy, Outcome 5 Improvement. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse effects Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 10.1  Comparison 10 IFN‐α + PUVA versus IFN‐α + acitretin, Outcome 1 Adverse effects. | ||||
| 1.1 Grade I to II adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Grade III adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Adverse events requiring treatment discontinuation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Flu‐like symptoms | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Dryness/redness of skin or hair loss | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 Neurological disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.7 Psychiatric disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.8 Gastrointestinal disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.9 Elevated liver or biliary tract enzymes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.10 Elevated triglycerides | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.11 Anemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.12 Leukopenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.13 Impotentia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.14 Redness and infiltration at application site | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 10.2  Comparison 10 IFN‐α + PUVA versus IFN‐α + acitretin, Outcome 2 Clearance. | ||||
| 3 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 10.3  Comparison 10 IFN‐α + PUVA versus IFN‐α + acitretin, Outcome 3 Improvement. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 11.1  Comparison 11 Topical nitrogen mustard with active transfer factor versus topical nitrogen mustard with inactivated transfer factor, Outcome 1 Clearance. | ||||
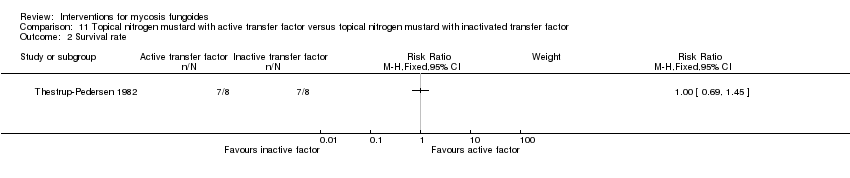
| 2 Survival rate Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 11.2  Comparison 11 Topical nitrogen mustard with active transfer factor versus topical nitrogen mustard with inactivated transfer factor, Outcome 2 Survival rate. | ||||
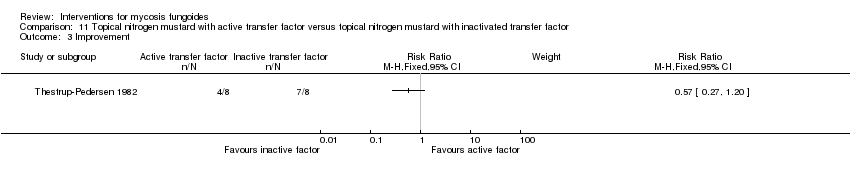
| 3 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 11.3  Comparison 11 Topical nitrogen mustard with active transfer factor versus topical nitrogen mustard with inactivated transfer factor, Outcome 3 Improvement. | ||||

Study flow chart

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
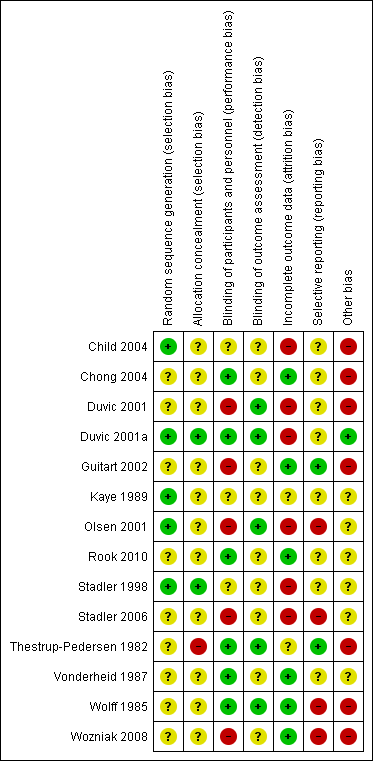
'Risk of bias' summary: review authors' judgements about each 'Risk of bias' item for each included study.

Forest plot of comparison: 5 IFN‐α + PUVA versus PUVA alone, outcome: 5.1 Clearance.

Comparison 1 Topical peldesine versus placebo, Outcome 1 Adverse effects.

Comparison 1 Topical peldesine versus placebo, Outcome 2 Clearance.
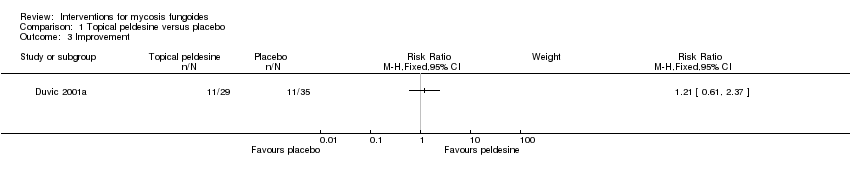
Comparison 1 Topical peldesine versus placebo, Outcome 3 Improvement.
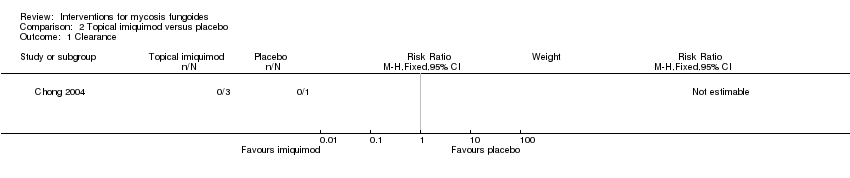
Comparison 2 Topical imiquimod versus placebo, Outcome 1 Clearance.

Comparison 2 Topical imiquimod versus placebo, Outcome 2 Improvement.

Comparison 3 Topical hypericin versus placebo, Outcome 1 Improvement.

Comparison 4 IFN‐α versus placebo, Outcome 1 Adverse effects.
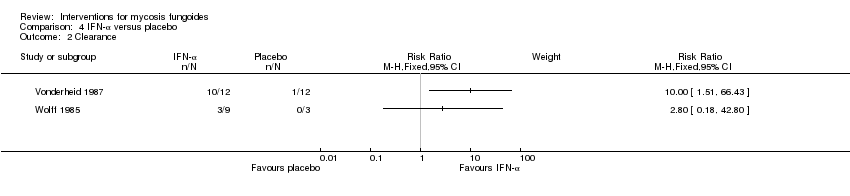
Comparison 4 IFN‐α versus placebo, Outcome 2 Clearance.
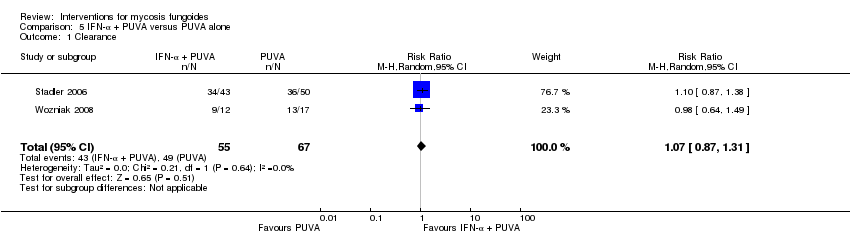
Comparison 5 IFN‐α + PUVA versus PUVA alone, Outcome 1 Clearance.

Comparison 6 Denileukin diftitox high versus low dose, Outcome 1 Adverse effects.

Comparison 6 Denileukin diftitox high versus low dose, Outcome 2 Clearance.
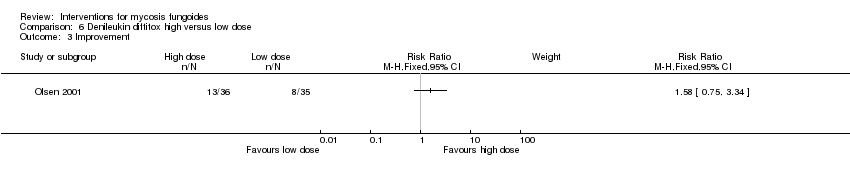
Comparison 6 Denileukin diftitox high versus low dose, Outcome 3 Improvement.
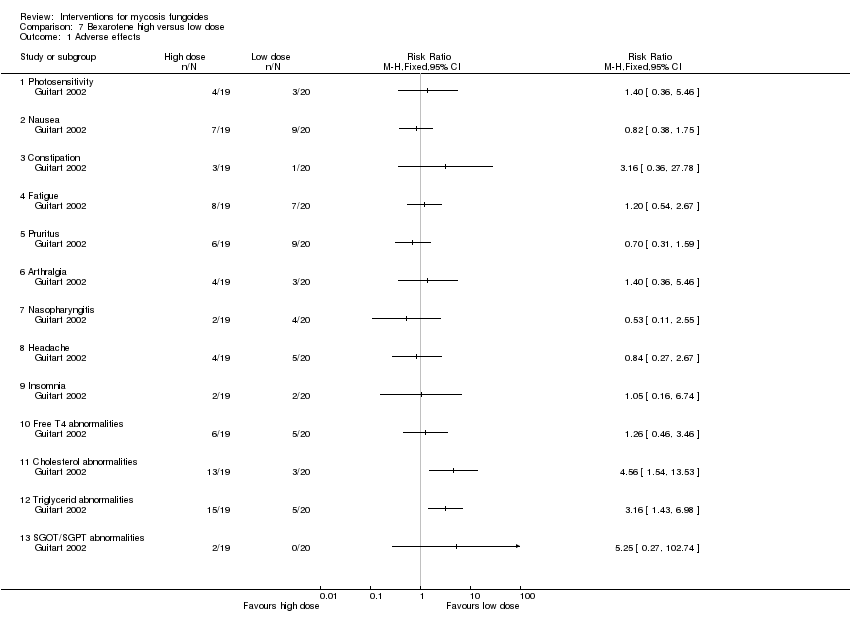
Comparison 7 Bexarotene high versus low dose, Outcome 1 Adverse effects.

Comparison 7 Bexarotene high versus low dose, Outcome 2 Clearance.

Comparison 7 Bexarotene high versus low dose, Outcome 3 Relapse.
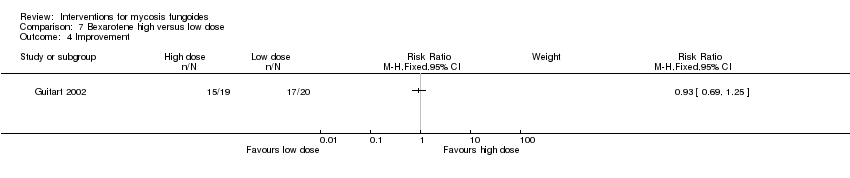
Comparison 7 Bexarotene high versus low dose, Outcome 4 Improvement.
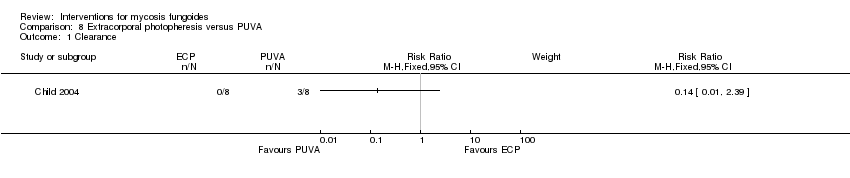
Comparison 8 Extracorporal photopheresis versus PUVA, Outcome 1 Clearance.
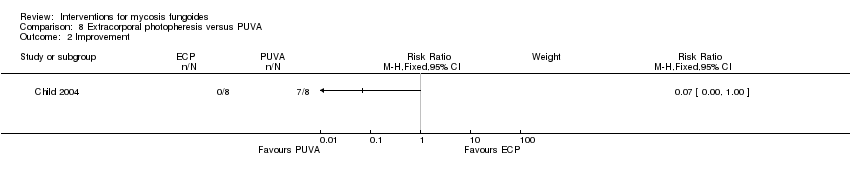
Comparison 8 Extracorporal photopheresis versus PUVA, Outcome 2 Improvement.
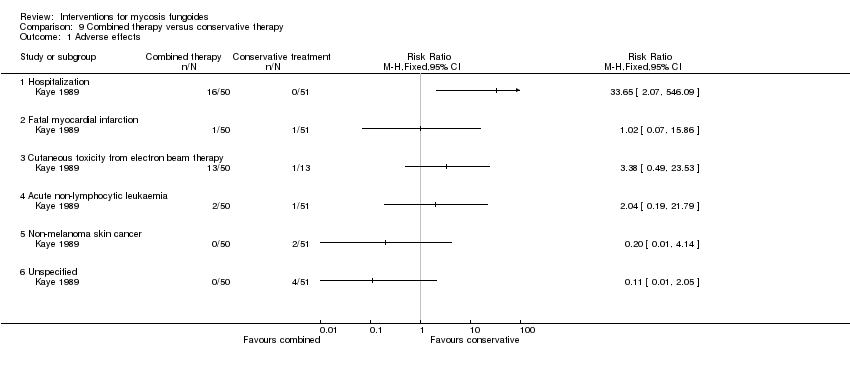
Comparison 9 Combined therapy versus conservative therapy, Outcome 1 Adverse effects.

Comparison 9 Combined therapy versus conservative therapy, Outcome 2 Clearance.

Comparison 9 Combined therapy versus conservative therapy, Outcome 3 Relapse.
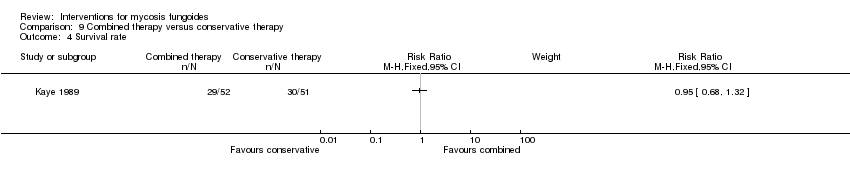
Comparison 9 Combined therapy versus conservative therapy, Outcome 4 Survival rate.

Comparison 9 Combined therapy versus conservative therapy, Outcome 5 Improvement.

Comparison 10 IFN‐α + PUVA versus IFN‐α + acitretin, Outcome 1 Adverse effects.

Comparison 10 IFN‐α + PUVA versus IFN‐α + acitretin, Outcome 2 Clearance.

Comparison 10 IFN‐α + PUVA versus IFN‐α + acitretin, Outcome 3 Improvement.

Comparison 11 Topical nitrogen mustard with active transfer factor versus topical nitrogen mustard with inactivated transfer factor, Outcome 1 Clearance.
Comparison 11 Topical nitrogen mustard with active transfer factor versus topical nitrogen mustard with inactivated transfer factor, Outcome 2 Survival rate.
Comparison 11 Topical nitrogen mustard with active transfer factor versus topical nitrogen mustard with inactivated transfer factor, Outcome 3 Improvement.
| 2007 MF and Sézary syndrome | 1979 CTCL | Disease‐specific survival rates in % (Agar 2010) | |||
| 5 year | 10 year | ||||
| IA | T1 | IA | T1 | 98 | 95 |
| N0 | N0 | ||||
| M0 | M0 | ||||
| B0‐1 | ‐ | ||||
| IB | T2 | IB | T2 | 89 | 77 |
| N0 | N0 | ||||
| M0 | M0 | ||||
| 0‐1 | ‐ | ||||
| IIA | T1‐2 | IIA | T1‐2 | 89 | 67 |
| N1‐2 | N1 | ||||
| M0 | M0 | ||||
| B0‐1 | ‐ | ||||
| IIB | T3 | IIB | T3 | 56 | 42 |
| N0‐2 | N0,1 | ||||
| M0 | M0 | ||||
| B0‐1 | ‐ | ||||
| III | T4 | III | T4 | ||
| N0‐2 | N0,1 | ||||
| M0 | M0 | ||||
| B0‐1 | ‐ | ||||
| IIIA | T4 | 54 | 45 | ||
| N0‐2 | |||||
| M0 | |||||
| B0 | |||||
| IIIB | T4 | 48 | 45 | ||
| N0‐2 | |||||
| M0 | |||||
| B1 | |||||
| IVA1 | T1‐4 | IVA | T1‐4 | 41 | 20 |
| N0‐2 | |||||
| M0 | N2‐3 ‐ | ||||
| B2 | |||||
| IVA2 | T1‐4 | M0 | 23 | 20 | |
| N3 | |||||
| M0 | ‐ | ||||
| B0‐2 | |||||
| IVB | T1‐4 | IVB | T1‐4 | 18 | not reached |
| N0‐3 | N0‐3 | ||||
| M1 | M1 | ||||
| B0‐2 | ‐ | ||||
| General medical terms | Explanation |
|
| A solid elevated area on the skin that is more broad than it is high |
|
| Any new and abnormal growth |
|
| Cutaneous T‐ and B‐cell lymphoma that primarily affect the skin |
|
| Group of skin‐directed T‐cell neoplasms with diverse clinical and histological features and prognosis |
|
| Group of skin‐directed B‐cell neoplasms with diverse clinical and histological features and prognosis |
|
| Group of neoplasms derived from the natural killer cells (NK‐cells) with diverse clinical and histological features and prognosis |
|
| Clinically‐aggressive neoplasm with a high incidence of cutaneous involvement and risk of leukaemic dissemination |
|
| Death of the cells in the damaged area of skin |
|
| A type of lymphocyte (white cell) |
| Adverse effects | |
|
| Death of myocardial tissue due to blocked blood supply |
|
| Serious disease with high production of non‐lymphatic leucocytes |
|
| The alanine transaminase is a liver enzyme (SGPT) |
|
| Low count of red blood cells |
|
| Low count of red blood cells with low amount of haemoglobin, the red molecule that transports oxygen within the blood vessels |
|
| Very acute generalsied reaction with flush/urticaria, bad breathing, lowered blood pressure, nausea |
|
| Joint pain |
|
| The aspartate transaminase is an enzyme mainly present in the liver but also in the blood, muscle cells, and bones (SGOT) |
|
| Lack of energy or physical weakness or both |
|
| Structural or functional disease of the cardiac muscle |
|
| Adverse effect where the heart and the lung are involved |
|
| Shivering attack |
|
| Adverse effect where the central nervous system (brain, spinal cord) is involved |
|
| Altered reactivity to a specific antigen leading to cutaneous alterations |
|
| Cutaneous adverse effect of an agent used in therapeutic dosages |
|
| No stool |
|
| Inflammation and detachment of the skin |
|
| Many fluid stools |
|
| Bad breathing |
|
| Red dot or area on the skin |
|
| To be exhausted |
|
| Symptoms which are often seen with influenza, such as fever, chills, and muscular pain |
|
| Adverse effects affecting the digestive system (oesophagus, stomach, bowel) |
|
| Pathological increased loss of hair |
|
| Capacity of a substance to have damaging effects on the liver |
|
| Confinement of a patient in a hospital |
|
| Elevated levels of cholesterol in the blood |
|
| Elevated levels of lipids in the blood |
|
| Low blood pressure |
|
| Low function of thyroid gland |
|
| Inability to engage in sexual intercourse |
|
| Not being able to sleep |
|
| The lactate dehydrogenase is an enzyme which helps to produce energy in the body when oxygen is absent |
|
| Low count of white blood cells |
|
| To feel ill |
|
| Inflammation of mucosa |
|
| Muscle pain |
|
| Inflammation of the inner nose and the throat |
|
| About to vomit while feeling sick |
|
| Abnormal state of the nervous system or nerves |
|
| Skin cancer which does not originate from melanocytes |
|
| Enhanced responsibility to light or ultraviolet light |
|
| Itching of the skin |
|
| Dermatitis resulting from overexposure to sources of radiant energy |
|
| A lot of red dots on the skin |
|
| Liver enzymes, see also AST and ALT |
|
| Enzyme of the thyroid gland |
|
| Low count of thrombocytes |
|
| Blood coagulates within blood vessels |
|
| Lipid |
|
| When the blood vessels are not tight anymore and parts of the blood leak out in the surrounding tissue |
|
| Blood vessels widening |
| Acronym | Description (letters used for acronym in capitals) |
| ADF | Arbeitsgemeinschaft Dermatologische Forschung |
| BCNU | Carmustine, a nitrogen mustard related alkylating agent |
| CENTRAL | Cochrane Central Register of Controlled Trials |
| CI | Confidence interval |
| CTCL | Cutaneous T‐Cell Lymphoma |
| DDG | German Dermatologic Society |
| EORTC | European Organization of Research and Treatment of Cancer |
| ISCL | International Society for Cutaneous Lymphoma (ISCL) |
| ITT | Intention‐to‐treat |
| LILACS | Latin American and Caribbean Health Science Information database |
| MF | Mycosis Fungoides |
| PICOS | Participants, Interventions, Controls, Outcomes and Study |
| PRISMA | Preferred Reporting Items of Systematic Reviews and Meta‐Analyses |
| RCT | Randomised‐Controlled Trial |
| RR | Risk Ratio |
| TSEB | Total Skin Electron Beam |
| TNMB | Tumour, lymph Node, Metastasis and Blood |
| UK | United Kingdom |
| US | Unites States of America |
| USCLC | United States Cutaneous Lymphoma Consortium |
| WHO | World Health Organization |
| 2007 MF and Sézary syndrome | 1979 CTCL | ||
| T: Skin | |||
| T0 | N.E. | Clinically and/or histopathologically suspicious lesions | |
| T1 | Limited patches, papules, and/or plaques covering < 10% of the skin surface. May further stratify into T1a (patch only) versus T1b (plaque ± patch) | Limited plaques, papules, or eczematous patches | |
| T2 | Patches, papules, or plaques covering ≥ 10% of the skin surface. May further stratify into T2a (patch only) versus T2b (plaque ± patch) | Generalised plaques, papules, or erythematous | |
| T3 | 1 or more tumours ( ≥ 1 cm diameter) | Tumors, 1 or more | |
| T4 | Confluence of erythema covering ≥ 80% body surface area | Generalised erythroderma | |
| N: Node | |||
| N0 | No clinically abnormal peripheral lymph nodes; biopsy not required | No clinically abnormal peripheral lymph nodes palpable, | |
| N1 | Clinically abnormal peripheral lymph nodes, | Palpable Clinically abnormal peripheral lymph nodes, histopathology | |
| N1a | Clone negative | ‐ | |
| N1b | Clone positive | ‐ | |
| N2 | Clinically abnormal peripheral lymph nodes, | No clinically abnormal peripheral lymph nodes, | |
| N2a | Clone negative | ‐ | |
| N2b | Clone positive | ‐ | |
| N3 | Clinically abnormal peripheral lymph nodes, | Palpable clinically abnormal peripheral lymph nodes, pathology | |
| N3x | Clinically abnormal peripheral lymph nodes, no histologic confirmation | ‐ | |
| M: Visceral | |||
| M0 | No visceral organ involvement | No visceral organ involvement | |
| M1 | Visceral involvement (must have pathology confirmation and organ involved should be specified) | Visceral involvement (must have pathology | |
| B: Blood | |||
| B0 | Absence of significant blood involvement: Less than 5% of peripheral blood lymphocytes are atypical (Sézary) cells | Atypical circulating cells not present (less than 5%) | |
| B0a | Clone negative | ‐ | |
| B0b | Clone positive | ‐ | |
| B1 | Low blood tumour burden: More than 5% of peripheral blood lymphocytes are atypical (Sézary) cells but do not meet the criteria of B2 | Atypical circulating cells present (more than 5%), record total | |
| B1a | Clone negative | ‐ | |
| B1b | Clone positive | ‐ | |
| B2 | High blood tumour burden: ≥ 1000/ µL Sézary cells with positive clone | ‐ | |
| For the 'N' category histopathologic grading is necessary with the new 2007 TNMB classification. It may be either done via the Dutch System or the NCI/VA classification system (Sausville 1988; Scheffer 1980). | |||
| Study | Participants randomised | First intervention | Second intervention | Reported outcomes | baseline risk | Significant differences in outcome |
| 89 participants stage I (IA: 45, IB:44) | topical peldesine 1% given for 24 weeks | placebo cream given for 24 weeks | common adverse effects clearance survival rates improvement | common adverse effects (rash): 3% with placebo cream | common adverse effects (rash): The relative risk of 7.24 (95% CI 0.92 to 56.76) showed a higher risk for peldesine (Fisher test P≤0.041). | |
| 4 participants plaque stage IB | topical imiquimod 5% given for 16 weeks | placebo cream given for 16 weeks | common adverse effects clearance (assessed 16 weeks after end of intervention) improvement (assessed 16 weeks after end of intervention) rare adverse effects | none | ||
| 12 participants patch or plaque stage, lesional comparison | hypericin 0.05‐0.25% in combination with visible light given for 6 weeks | placebo cream in combination with visible light given for 6 weeks | common adverse effects survival rates improvement | improvement: 8% with placebo cream | improvement: The relative risk of 7.00 (95% CI 1.01 to 48.54) favoured hypericin (Fisher test P≤0.028). | |
| 6 participants stage IA to IIA (IA: 1; IB: 1; IIA: 4), lesional comparison | injections of interferon‐α 2b given for 4 weeks | injections of isotonic sterile water given for 4 weeks | common adverse effects (assessed after 3 weeks of intervention) clearance (assessed 4 weeks after end of intervention) | common adverse effects (mild erythema): 0% with injections of sterile water clearance: 8% injections of sterile water | common adverse effects (mild erythema): The relative risk of 11.00 (95% CI 0.74 to 163.49) favoured interferon‐α (Fisher test P≤0.016). clearance: The RR 10.00 (95% CI 1.51 to 66.43) favoured interferon‐α (Fisher test P≤0.0007). | |
| 12 participants stage IA (7) to IB (5) | injections of interferon‐α given for 4 weeks | injections of buffered glycine serum human albumin given for 4 weeks | common adverse effects clearance | common adverse effects (mild fever): 0% with injections of buffered glycine serum human albumin | common adverse effects (mild fever): The relative risk of 11.00 (95% CI 0.70‐173.66) showed a higher risk for interferon‐α (Fisher test P≤0.03). | |
| 124 participants stage lA to IIA | injections of interferon‐α in combination with PUVA given for up to 52 weeks | PUVA alone given for up to 52 weeks | clearance | none | ||
| 29 participants stage lA to IIA (IA: 14; IB: 6; IIA: 9) | injections of interferon‐α in combination with PUVA given for 24 weeks | PUVA alone given for 24 weeks | clearance relapse disease‐free interval | none | ||
| 71 participants stage IB‐IVA (IB:16, IIA: 10, IIB: 19, III: 11, IVA:15) | denileukin diftitox intravenous infusion 9µg/kg/day given for up to 6 months | denileukin diftitox intravenous infusion 18µg/kg/day given for up to 6 months | quality of life common adverse effects clearance survival rates (assessed 90 days after end of intervention) improvement rare adverse effects | none | ||
| 58 participants stage I to IIA (IA: 17, IB: 34, IIA: 6, IIB: 1) | bexarotene capsules dosed 300 to 650mg/m²/day given for 16 weeks | bexarotene capsules dosed 6.5mg/m²/day given for 16 weeks | quality of life common adverse effects clearance relapse survival rates (assessed 4 weeks after end of intervention) improvement rare adverse effects | no calculations, due to methodological problems | ||
| 43 participants stage IB (36) and IIA (7) | bexarotene 300mg/day in combination with PUVA and fenofibrate 54mg/day given for 24 weeks | bexarotene 150mg/day in combination with PUVA and fenofibrate 54mg/day given for 24 weeks | common adverse effects clearance relapse (assessed 6 months after end of intervention) disease‐free interval survival rates improvement | common adverse effects (cholesterol levels): 15% with bexarotene 150mg/day common adverse effects (triglyceride levels):25% with bexarotene 150mg/day | common adverse effects (cholesterol levels): The relative risk of 4.56 (5% CI 1.54 to 13.53) showed a higher risk for bexarotene 300mg/day (Fisher test P≤0.002). common adverse effects (triglyceride levels): The RR 3.16 (95% CI 1.93 to 6.98) showed a higher risk for bexarotene 300mg/day (Fisher test P≤0.002). | |
| 8 participants plaque stage (Bunn Lamberg 1B) | extracorporeal photopheresis given for 6 months | PUVA given for 3 months | common adverse effects clearance survival rates (assessed 2‐21 months after end of intervention) improvement | improvement:0% with extracorporeal photopheresis | improvement: The relative risk of 0.07 (95% CI 0.00 to 1.00) favoured PUVA (Fisher test P≤0.002). | |
| 103 participants of all stages (IA: 6, IB: 16, IIA: 9, IIB: 12; III: 2, IVA: 42, IVB: 16) | "combined therapy" (electron‐beam radiation and parenteral chemotherapy) given for 8 to 12 weeks | "conservative treatment" (topical treatment with mechlorethamine supported by a stepwise escalation of the therapy according to stage of disease) duration of intervention not reported | common adverse effects clearance relapse disease‐free interval survival rates (assessed more than 5 years after end of intervention) improvement | common adverse effects (hospitalisation): 0% with "conservative treatment" clearance: 18% with "conservative treatment" improvement: 65% with "conservative treatment" | common adverse effects (hospitalizations): The relative risk of 33.65 (95% CI 2.07 to 546.09) showed a higher risk for the "combined therapy" (Fisher test P≤0.0001). clearance: The relative risk of 2.18 (95% CI 1.10 to 4.33) favoured the "combined therapy" (Fisher test P≤0.03). improvement: The relative risk of 1.40 (95% CI 1.12 to 1.74) favoured the "combined therapy" (Fisher test P≤0.003). | |
| 82 participants stage I and II (IA: 36; IB: 28; IIA: 10; IIB: 8) | injections of interferon‐α in combination with PUVA given for up to 48 weeks | injections of interferon‐α in combination with acitretin given for up to 48 weeks | common adverse effects clearance improvement | common adverse effects (adverse effects grade III): 10% IFN‐α in combination with PUVA common adverse effects (requiring discontinuation): 5% IFN‐α in combination with PUVA common adverse effects (neurological disorders): 8% IFN‐α in combination with PUVA clearance: 38% with IFN‐α in combination with acitretin | common adverse effects (adverse effects grade III): The relative risk of 3.10 (95% CI 1.10 to 8.70) showed a higher risk for interferon‐α in combination with acitretin (Fisher test P≤0.03). common adverse effects (requiring discontinuation): The relative risk of 4.29 (95% CI 0.99 to 18.63) showed a higher risk for interferon‐α in combination with acitretin (Fisher test P≤0.049). common adverse effects (neurological disorders): The relative risk of 3.49 (95% CI 1.05 to 11.60) showed a higher risk for interferon‐α in combination with acitretin (Fisher test P≤0.04). clearance: The relative risk of 0.54 (95% CI 0.35 to 0.84) favoured interferon‐α in combination with PUVA (Fisher test P≤0.005). | |
| 16 participants van Scott stage II‐IV (II: 14; III: 1; IV: 1) | topically applied nitrogen mustard with active transfer factor given for 1 year | topically applied nitrogen mustard with inactivated transfer factor given for 1 year | common adverse effects clearance (assessed 1 year after end of intervention) survival rates (assessed 1 year after end of intervention) improvement (assessed 1 year after end of intervention) | clearance: 0% with topically applied nitrogen mustard with active transfer factor | clearance: The relative risk of 0.09 (95% CI 0.01 to 1.41) favoured topically applied nitrogen mustard with inactivated transfer factor (Fisher test P≤0.03). |
| Intervention | Rare severe adverse effects |
| PUVA | transient lymphomatoid papulosis (Aronsson 1982) |
| extracorporeal photopheresis | sarcoma (Korpusik 2008) |
| imiquimod | no reported SAEs found |
| electron beam | total epilation (Braverman 1987, Desai 1988), |
| cyclophosphamide, doxorubicin, etoposide, vincristine | haematologic toxicity: febrile neutropenia/staphylococcal bacteraemia/disseminated herpes infection/pneumocystis carinii pneumonia/neurologic toxicity grade 3/decreased left ventricular ejection fraction (Akpek 1999) |
| active transfer factor | no reported SAEs found |
| methotrexate | severe edematous erythroderma/denudation on the trunk and extremities/Interstitial pulmonary fibrosis (Zackheim 1996) |
| interferon‐alpha | acrocyanosis (Campo‐Voegeli 1998) |
| bexarotene | neutropenia/non‐ST‐elevation myocardial infarction/elevated liver enzymes (Abbott 2009) |
| peldesine (BCX‐34) | no reported SAEs found |
| denileukin diftitox (ONTAK) | lethal vascular leak syndrome with rhabdomyolysis (Avarbock 2008) |
| nitrogen mustard | urticaria and anaphylactoid reaction (Daughters 1973, Grunnet 1976, Sanchez 1977) |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse effects Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Pruritus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Rash | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse effects Show forest plot | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Erythema | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Fever | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Myalgia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Chills or weakness | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Nausea, arthralgia, and malaise | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Clearance Show forest plot | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clearance Show forest plot | 2 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.87, 1.31] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse effects Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Constitutional symptoms | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Grade 3 to 4 constitutional symptoms | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Infections | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Grade 3 to 4 infections | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Gastrointestinal syndromes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 Grade 3 to 4 gastrointestinal syndromes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.7 CNS syndromes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.8 Grade 3 to 4 CNS syndromes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.9 Rash | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.10 Grade 3 to 4 rash | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.11 Vascular leak syndrome | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.12 Grade 3 to 4 vascular leak syndrome | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.13 Thrombotic events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.14 Grade 3 to 4 thrombotic events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.15 Cardiopulmonary events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.16 Grade 3 to 4 cardiopulmonary events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.17 Acute infusion related events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.18 Grade 3 to 4 acute infusion related events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.19 Grade 3 to 4 laboratory abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse effects Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Photosensitivity | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Nausea | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Constipation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Fatigue | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Pruritus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 Arthralgia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.7 Nasopharyngitis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.8 Headache | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.9 Insomnia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.10 Free T4 abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.11 Cholesterol abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.12 Triglycerid abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.13 SGOT/SGPT abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Relapse Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse effects Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Hospitalization | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Fatal myocardial infarction | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Cutaneous toxicity from electron beam therapy | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Acute non‐lymphocytic leukaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Non‐melanoma skin cancer | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 Unspecified | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Relapse Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4 Survival rate Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse effects Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Grade I to II adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Grade III adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Adverse events requiring treatment discontinuation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Flu‐like symptoms | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Dryness/redness of skin or hair loss | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 Neurological disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.7 Psychiatric disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.8 Gastrointestinal disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.9 Elevated liver or biliary tract enzymes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.10 Elevated triglycerides | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.11 Anemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.12 Leukopenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.13 Impotentia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.14 Redness and infiltration at application site | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clearance Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2 Survival rate Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Improvement Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |