Салицилат‐содержащие местнораздражающие средства при острой и хронической мышечно‐скелетной боли у взрослых
Abstract
Background
Rubefacients containing salicylates cause irritation of the skin and are believed to relieve various musculoskeletal pains. They are available on prescription, and are common components in over‐the‐counter remedies. This is an update of a review of rubefacients for acute and chronic pain, originally published in 2009, which found limited evidence for efficacy.
Objectives
To assess the efficacy and safety of topically applied salicylates in acute and chronic musculoskeletal pain in adults.
Search methods
We searched CENTRAL, MEDLINE, and EMBASE, from inception to 22 August 2014, together with the Oxford Pain Relief Database, two clinical trial registries, and the reference lists of included studies and relevant reviews.
Selection criteria
Randomised, double‐blind, placebo‐ or active‐controlled clinical trials of topical rubefacients containing salicylates to treat musculoskeletal pain in adults, with at least 10 participants per treatment arm, and reporting outcomes at close to 7 (minimum 3, maximum 10) days for acute conditions and 14 (minimum 7) days or longer for chronic conditions.
Data collection and analysis
Two review authors independently assessed trials for inclusion and risk of bias, and extracted data. We calculated risk ratio (RR) and number needed to treat to benefit or harm (NNT or NNH) with 95% confidence intervals (CI) using a fixed‐effect model. We analysed acute and chronic conditions separately.
Main results
New searches for this update identified one new study that satisfied our inclusion criteria, although it contributed information only for withdrawals. Six placebo‐ and one active‐controlled studies (560 and 137 participants, respectively) in acute pain, and seven placebo‐ and three active‐controlled studies (489 and 182 participants, respectively) in chronic pain were included in the review. All studies were potentially at risk of bias, and there were substantial differences between studies in terms of the participants (for example the level of baseline pain), the treatments (different salicylates combined with various other potentially active ingredients), and the methods (for example the outcomes reported). Not all of the studies contributed usable information for all of the outcomes sought.
For the primary outcome of clinical success at seven days in acute conditions (mostly sprains, strains, and acute low back pain), the RR was 1.9 (95% CI 1.5 to 2.5) and the NNT was 3.2 (2.4 to 4.9) for salicylates compared with placebo, but this result was not robust (very low quality evidence). Using a random‐effects model for analysis the RR was 2.7 (1.05 to 7.0). For the same outcome in chronic conditions (mostly osteoarthritis, bursitis, and chronic back pain), the RR was 1.6 (1.2 to 2.0) and the NNT was 6.2 (4.0 to 13) (very low quality evidence). This result was not substantially changed using a random‐effects model for analysis. In both categories there were a number of factors might have influenced the results but sensitivity analysis was limited because of the small number of studies and participants.
For both acute and chronic painful conditions any evidence of efficacy came from the older, smaller studies, while the larger, more recent studies showed no effect.
Adverse events were more common with salicylate than with placebo but most of the events occurred in only two studies. There was no difference when these studies were removed from the analysis (very low quality evidence). Local adverse events (at the application site) were again more common with salicylate but were nearly all in one study (in which salicylate was combined with another irritant). There was no difference when this study was removed (very low quality evidence).
There were insufficient data to draw conclusions against active controls.
Authors' conclusions
The evidence does not support the use of topical rubefacients containing salicylates for acute injuries or chronic conditions. They seem to be relatively well tolerated in the short‐term, based on limited data. The amount and quality of the available data mean that uncertainty remains about the effects of salicylate‐containing rubefacients.
PICOs
Резюме на простом языке
Топические местнораздражающие средства при острой и хронической мышечно‐скелетной боли у взрослых
Это обновление обзора о местнораздражающих средствах при острой и хронической боли, впервые опубликованного в 2009 году, которое включает одно новое исследование.
Топические (местные) лекарства это лекарства, которые наносят на поверхности тела, такие как кожа, для лечения болезней/недомоганий. Топические (местные) продукты/препараты могут быть в форме крема, пены, геля, лосьона и мази. Топические (местные) препараты/продукты могут включать в себя широкий спектр лекарственных средств.
Местнораздражающие средства это лекарства, которые вызывают раздражение и покраснение кожи за счет усиления кровотока. Считают, что они облегчают боль при различных состояниях мышечно‐скелетной системы, и доступны как по рецепту, так и без него. Салицилаты ‐ часто используемые местнораздражающие средства.
Этот обзор рассмотрел доказательства пользы топических местнораздражающих средств, содержащих салицилаты, в рандомизированных и двойных‐слепых исследованиях. Эти исследования проводили у людей с острыми состояниями, причиняющими боль, такими как растяжения и вывихи, или с состояниями хронической боли, такими как остеоартрит. Мы хотели узнать, помогают ли топические салицилат‐содержащие местнораздражающие средства при боли.
Доказательства для топических салицилат‐содержащих местнораздражающих средств ограничены качеством, валидностью и размером доступных исследований. Доказательства эффективности при состояниях острой и хронической боли были из более старых и небольших исследований, тогда как более крупные и недавние исследования показали отсутствие эффекта. Нет надежных доказательств, что топические салицилат‐содержащие местнораздражающие средства полезны в облегчении боли.
Authors' conclusions
Summary of findings
| Salicylate‐containing topical rubefacients compared with topical placebo for acute and chronic painful conditions | ||||||
| Patient or population: adults with strains or sprains (acute) or osteoarthritis or low back pain (chronic) Settings: community Intervention: salicylate‐containing topical rubefacient Comparison: topical placebo | ||||||
| Outcomes | Probable outcome with | Probable outcome with | RR NNT, NNTp, or NNH | No of studies, participants | Quality of the evidence | Comments |
| Clinical success (eg 50% reduction in pain) Acute conditions | 640 in 1000 | 335 in 1000 | RR 1.9 (1.5 to 2.5) NNT 3.2 (2.4 to 4.9) | 4 studies 324 participants | ⊕⊝⊝⊝ | Most recent, largest study showed no effect Note NNT cannot be trusted because of low numbers and poor quality studies |
| Clinical success (eg 50% reduction in pain) Chronic conditions | 447 in 1000 | 284 in 1000 | RR 1.6 (1.2 to 2.0) NNT 6.2 (4.0 to 13) | 6 studies 455 participants | ⊕⊝⊝⊝ | Most recent, largest studies showed no effect Note NNT cannot be trusted because of low numbers and poor quality studies |
| Adverse events ‐ any adverse events Acute and chronic conditions combined | 152 in 1000 | 94 in 1000 | RR 1.6 (1.2 to 2.0) NNH 17 (9.9 to 58) | 11 studies 984 participants | ⊕⊕⊝⊝ | Inadequate reporting of adverse events is common Acute and chronic conditions combined |
| Adverse events ‐ local adverse events Acute and chronic conditions combined | 56 in 1000 | 24 in 1000 | RR 2.2 (1.1 to 4.1) NNH 31 (16 to 300) | 10 studies 869 participants | ⊕⊝⊝⊝ | Small numbers of events Acute and chronic conditions combined |
| Withdrawals ‐ lack of efficacy Acute and chronic conditions combined | 24 in 1000 | 72 in 1000 | RR 0.4 (0.2 to 0.9) NNTp 21 (12 to 120) | 5 studies 501 participants | ⊕⊝⊝⊝ | Small numbers of events Acute and chronic conditions combined |
| Withdrawals ‐ adverse events Acute and chronic conditions combined | 49 in 1000 | 11 in 1000 | RR 4.2 (1.5 to 12) NNH 26 (15 to 85) | 7 studies 737 participants | ⊕⊝⊝⊝ | Small numbers of events Acute and chronic conditions combined |
| GRADE Working Group grades of evidence | ||||||
| CI: confidence interval; RR: risk ratio; NNT: number needed to treat; NNTp: number needed to prevent an event happening; NNH: number needed to harm | ||||||
Background
This review is an update of a previously published review on topical rubefacients for acute and chronic pain in adults (Matthews 2009). We made the decision to change the title from "rubefacients" to "salicylate‐containing rubefacients" because all the included studies used salicylates, either alone or in combination with other compounds. We have also specified musculoskeletal pain because topical salicylates are not normally used to treat visceral pain, neuropathic pain, or cancer pain. We felt that the new title better reflected the content of the review.
Rubefacients have been used for many years to treat musculoskeletal pains, but earlier reviews have found little evidence to support their use (Mason 2004; Matthews 2009). There has been confusion about which compounds should be classified as rubefacients. Some, such as salicylates, are related pharmacologically to aspirin and nonsteroidal anti‐inflammatory drugs (NSAIDs), but as topical products their primary action is as skin irritants. Capsaicin applied topically can produce a burning sensation at the application site and has been grouped with rubefacients, although the mechanism of pain relief is different. This review included salicylates, but not capsaicin as this is covered in other reviews (Derry 2012a; Derry 2013).
This review is one of a series on topical analgesics, including topical capsaicin at low and high doses (Derry 2012a; Derry 2013), and topical NSAIDs in acute (Massey 2010), and chronic (Derry 2012b), pain conditions.
Description of the condition
This review looked at the use of salicylate‐containing rubefacients to relieve musculoskeletal pain (pain in muscles, joints, and tendons). We considered acute conditions such as sprains, strains, and bruises (typical of sports injuries) separately from chronic conditions such as osteoarthritis. Acute pain typically lasts for hours, days, or a few weeks, while an injury is healing. Chronic pain lasts beyond the normal time of healing, in situations where healing does not occur (rheumatoid arthritis, for example), or where changes occur in the nervous system that maintain pain. Non‐malignant chronic pain is typically considered to be pain that has been present for at least three, or six, months (Merskey 2002; Turk 2001).
Description of the intervention
The earlier review considered all rubefacients, but searches identified only formulations containing salicylates (Matthews 2009). We have changed the review title to better reflect the content.
Salicylates are derivatives of salicylic acid, and those used in topical preparations are often amine derivatives. They are most often formulated as creams or gels, but sometimes as sprays, which are applied directly onto the affected area two to four times daily. In 2013 there were almost 1.4 million prescriptions for topical salicylates in primary care in England (PCA 2014). Many products that are on sale directly to the public contain salicylates. The quality and cost of these are unknown, but the latter is likely to be substantial.
While topical salicylates are considered relatively safe, particularly in relation to oral NSAIDs, overuse or ingestion can lead to salicylate toxicity, and even death (Davis 2007; O'Malley 2008).
How the intervention might work
Salicylate‐containing rubefacients cause irritation of the skin, and are believed to relieve pain in muscles, joints, and tendons, and other musculoskeletal pains in the extremities, by counter‐irritation (BNF 2008). The term 'counter‐irritant' derives from the fact that they cause a reddening of the skin by dilating the blood vessels of the skin, which gives a soothing feeling of warmth. Irritation of the sensory nerve endings is thought to alter or offset pain in the underlying muscle or joints that are served by the same nerves (Morton 2002).
Salicylates are related pharmacologically to aspirin and NSAIDs, but when used in topical products (often as amine derivatives) their principal action is as skin irritants. By contrast, topical NSAIDs penetrate the skin and underlying tissues where they inhibit cyclo‐oxygenase enzymes responsible for prostaglandin biosynthesis and the development of inflammation.
Why it is important to do this review
The original Cochrane review was published five years ago, and its conclusions were limited by the small number of studies (Matthews 2009). New studies may have been published subsequently. In addition, the standards by which we assess and interpret evidence are now more rigorous (Moore 2010).
Topical salicylates are widely available and are often perceived to be effective and safe. It is important to establish whether new data are available, or whether this will remain an intervention for which there is little evidence.
Objectives
To assess the efficacy and safety of topically applied salicylates in acute and chronic musculoskeletal pain in adults.
Methods
Criteria for considering studies for this review
Types of studies
Randomised, double‐blind studies comparing salicylate‐containing rubefacients with placebo or other active treatment for acute (strains, sprains, and bruises) or chronic (arthritis) musculoskeletal pain, with at least 10 participants per treatment arm. Study duration had to be a minimum of three days for acute conditions and seven days for chronic conditions. We excluded studies published only as short abstracts (usually meeting abstracts) because they do not provide sufficient information to adequately assess the study, and those studying experimentally induced pain because it does not correlate well with clinical pain.
Types of participants
Adult participants (16 years or more) with acute or chronic musculoskeletal pain of at least moderate intensity resulting from any cause.
Types of interventions
Included studies had at least one treatment arm using a topical salicylate preparation, and a comparator arm using placebo or other active treatment, with treatment applied at least once daily.
Types of outcome measures
We sought information on participant characteristics (age, sex, and condition treated) and outcomes at close to 7 days (minimum 3 days, maximum 10 days) for acute conditions, and 14 days (minimum 7 days) for chronic conditions.
Primary outcomes
The primary outcome was 'clinical success', defined as a 50% reduction in pain, measured on a visual analogue scale (VAS) or numerical rating scale (NRS), or an equivalent measure such as a "very good" or "excellent" global assessment of treatment, or "none" or "slight" pain on rest or movement, measured on a categorical scale (or similar wording) (Moore 1998). We used the following hierarchy of outcomes, in order of preference, to extract data for the primary outcome.
-
Participant‐reported reduction in pain of at least 50%.
-
Participant‐reported global assessment of treatment.
-
Pain on movement.
-
Pain on rest, or spontaneous pain.
-
Undefined "improvement".
Only participant‐reported outcomes were used. Physician‐ or investigator‐reported outcomes of efficacy were not used.
Secondary outcomes
-
Numbers of participants with adverse events: local and systemic.
-
Numbers of withdrawals: all cause, lack of efficacy, adverse events.
Outcomes were reported after different durations of treatment, so care was taken to extract data reported as close to specified times as possible, and not less than the minimum. We additionally extracted longer‐duration outcomes where available. We took care to determine whether adverse events were comprehensively reported, and the methods of ascertainment.
Search methods for identification of studies
Electronic searches
We searched the following databases.
-
Cochrane Central Register of Controlled Trials via CRSO (to 22 August 2014).
-
MEDLINE via Ovid (from 1948 to December 2008 for the earlier review, and from 2008 to 22 August 2014 for this update).
-
EMBASE via Ovid (from 1976 to December 2008 for the earlier review, and from 2008 to 22 August 2014 for this update).
-
Oxford Pain Relief Database for the original review (Jadad 1996a).
See Appendix 1 for the CENTRAL search strategy, Appendix 2 for the MEDLINE search strategy, and Appendix 3 for the EMBASE search strategy used for this update of the review.
We did not apply any language restrictions.
Searching other resources
We reviewed the bibliographies of all randomised trials identified and of review articles, and searched clinical trial databases (ClinicalTrials.gov (ClinicalTrials.gov) and the World Health Organization International Clinical Trials Registry Platform (http://apps.who.int/trialsearch/)) to identify additional published or unpublished data.
Manufacturers have previously been asked for details of unpublished studies (Mason 2004), and new manufacturers or UK distributors were sought to ask them about unpublished studies in the earlier review (Matthews 2009). No further attempt was made to contact manufacturers for this update.
Data collection and analysis
Two review authors independently searched for and selected the studies for inclusion, assessed methodological quality, and extracted data. Disagreements were resolved through discussion with a third author.
Selection of studies
We reviewed on screen the titles and abstracts of studies identified by the searches to eliminate those that clearly did not satisfy inclusion criteria and obtained full reports of the remaining studies to determine inclusion in the review. We considered cross‐over studies only if data from the first treatment period were reported separately. We did not include studies in oral, ocular, or buccal diseases.
Data extraction and management
We extracted information on participants, interventions, and outcomes from the original reports using a standard data extraction form. We did not contact study authors for further information.
Assessment of risk of bias in included studies
We used the Oxford Quality Score as the basis for inclusion, limiting inclusion to studies that were randomised and double‐blind as a minimum (Jadad 1996b).
Two authors independently assessed the risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), and adapted from those used by the Cochrane Pregnancy and Childbirth Group, with any disagreements resolved by discussion. We assessed the following for each study.
-
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (any truly random process, for example, random number table; computer random number generator); unclear risk of bias (method used to generate sequence not clearly stated). We excluded studies using a non‐random process (for example, odd or even date of birth; hospital or clinic record number).
-
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions before assignment determines whether intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment. We assessed the methods as: low risk of bias (for example, telephone or central randomisation; consecutively numbered sealed opaque envelopes); unclear risk of bias (method not clearly stated). We excluded studies that did not conceal allocation (for example, open list).
-
Blinding of outcome assessment (checking for possible detection bias). We assessed the methods used to blind study participants and outcome assessors from knowledge of which intervention a participant received. We assessed the methods as: low risk of bias (study stated that it was blinded and described the method used to achieve blinding, for example, identical tablets; matched in appearance and smell); unclear risk of bias (study stated that it was blinded but did not provide an adequate description of how blinding was achieved). We excluded studies that were not double‐blind.
-
Incomplete outcome data (checking for possible attrition bias due to the amount, nature, and handling of incomplete outcome data). We assessed the methods used to deal with incomplete data as: low risk (< 10% of participants did not complete the study or used ‘baseline observation carried forward’ (BOCF) analysis, or both); unclear risk of bias (used 'last observation carried forward' (LOCF) analysis); high risk of bias (used 'completer' analysis).
-
Size of study (checking for possible biases confounded by small size). We assessed studies as being at low risk of bias (≥ 200 participants per treatment arm); unclear risk of bias (50 to 199 participants per treatment arm); high risk of bias (< 50 participants per treatment arm).
Measures of treatment effect
We used risk ratio (or 'relative risk', RR) to establish statistical difference. We used numbers needed to treat (NNT) and pooled percentages as absolute measures of benefit or harm.
We used the following terms to describe adverse outcomes in terms of harm or prevention of harm:
-
when significantly fewer adverse outcomes occurred with salicylate than with control (placebo or active) we used the term the number needed to treat to prevent one event (NNTp);
-
when significantly more adverse outcomes occurred with salicylate compared with control (placebo or active) we used the term the number needed to harm or cause one event (NNH).
Unit of analysis issues
We accepted randomisation by individual patient only.
Dealing with missing data
The most likely source of missing data was expected to be from participants dropping out from the studies. We looked specifically for evidence of LOCF and used a dichotomous responder analysis, where a responder was defined as a participant who experienced the predefined outcome and remained in the study (for example, did not withdraw due to adverse events). LOCF is a potential source of major bias in chronic pain studies (Moore 2012a).
For all outcomes we carried out analyses, as far as possible, on a modified intention‐to‐treat (ITT) basis, including all participants who were randomised and received an intervention. Where sufficient information was reported, we added back missing data in the analyses we undertook.
Assessment of heterogeneity
We assessed heterogeneity of studies visually (L'Abbé 1987). Where data could be pooled, we reported the I2 statistic.
Data synthesis
We undertook meta‐analysis using a fixed‐effect model. A random‐effects model was also used for meta‐analysis if there was significant clinical heterogeneity and it was considered appropriate to combine studies.
We calculated RR estimates with 95% confidence intervals (CI) (Morris 1995). Where appropriate we calculated NNT and NNH, with 95% CIs, using the pooled number of events (Cook 1995). We assumed a statistically significant difference from control when the 95% CI of the RR did not include the number one.
Subgroup analysis and investigation of heterogeneity
We analysed data for acute and chronic conditions separately. The evidence base was known to be small, making analysis of different salicylates impossible, so for each category we combined data for all rubefacients versus placebo for analysis of the primary outcome of clinical success. For secondary outcomes relating to adverse events and withdrawals, data for all rubefacients versus placebo, in acute and chronic conditions, were combined.
Studies comparing rubefacients with an active comparator were also examined.
At least 200 patients were required in any of these different contexts before information was pooled (Moore 1998b).
Sensitivity analysis
We planned sensitivity analyses of the primary outcome only, for:
-
baseline pain intensity (including mild pain versus moderate to severe pain);
-
outcome (undefined "improvement" versus defined outcomes);
-
time of assessment of primary outcome (6 days or less versus 7 days or more for acute conditions, and 13 days or less versus 14 days or more for chronic conditions).
Results
Description of studies
Results of the search
Searches for this update identified 43 potential studies in CENTRAL, 35 in MEDLINE, and 74 in EMBASE. Two studies were read in full, one of which satisfied the inclusion criteria (92 participants) (Zahmatkash 2011). The other did not because its duration was too short (Higashi 2010). No additional studies were identified through the reference lists of included studies or searching clinical trial registries.
Searches for the earlier review identified 28 potentially relevant studies. Twelve were excluded after reading the full publication (Crielaard 1986; Dettoni 1982; He 2006; Heindl 1977; Howell 1955; Jolley 1972; Kantor 1990; Kleinschmidt 1975; Pasila 1980; Shamszad 1986; von Batky 1971; Weisinger 1970) and 16 were included (Algozzine 1982; Camus 1975; Diebschlag 1987; Frahm 1993; Geller 1980; Ginsberg 1987; Golden 1978; Ibanez 1988; Lester 1981; Lobo 2004; Rothhaar 1982; Rutner 1995; Shackel 1997; Stam 2001; von Bach 1979; Wanet 1979).
See Figure 1.
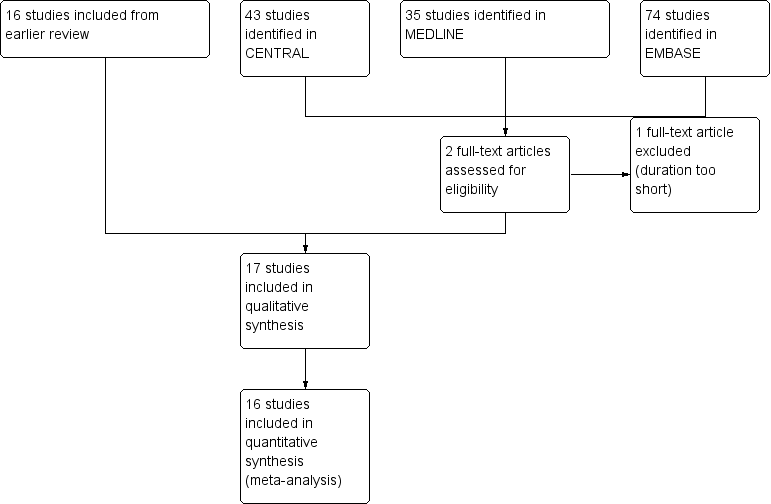
Study flow diagram.
Included studies
Six placebo‐controlled studies of acute injuries were included (Diebschlag 1987; Frahm 1993; Ginsberg 1987; Lester 1981; Rothhaar 1982; Stam 2001), with 560 participants in total, of whom 236 were in two studies that did not have usable information for efficacy and provided data for withdrawals and adverse events only (Diebschlag 1987; Frahm 1993). One acute study with an active comparator was included, involving 137 participants (Ibanez 1988).
Seven placebo‐controlled studies in chronic pain conditions were included, involving 489 participants (including 26 receiving both treatment and placebo in a cross‐over trial) (Algozzine 1982; Camus 1975; Lobo 2004; Rutner 1995; Shackel 1997; von Bach 1979; Wanet 1979). Three studies with active comparators in chronic pain conditions, involving 182 participants, were also included (Geller 1980; Golden 1978; Zahmatkash 2011). Two studies included a minority (20% to 30%) of participants with acute musculoskeletal conditions (Geller 1980; Wanet 1979); we analysed these as chronic studies, subject to a planned sensitivity analysis. Lobo 2004 (52 participants) provided data only for adverse events, and Zahmatkash 2011 (92 participants) provided usable data only for all cause withdrawals.
Acute conditions studied were sprains (Diebschlag 1987; Frahm 1993; Lester 1981), other sports injuries (Ibanez 1988; Rothhaar 1982), or acute lower back pain (Ginsberg 1987; Stam 2001). Chronic conditions included articular musculoskeletal pain (Algozzine 1982; Geller 1980; Golden 1978; Shackel 1997; von Bach 1979; Wanet 1979; Zahmatkash 2011), extra‐articular pain (Camus 1975; Geller 1980; Golden 1978; von Bach 1979), back pain (Geller 1980; Rutner 1995; von Bach 1979; Wanet 1979), and temporomandibular disorders (Lobo 2004).
Our intention was to include only studies of participants with at least moderate pain intensity at baseline. Not all of the studies clearly stated baseline pain intensity and, where stated, the range of pain sometimes included mild pain. We included all levels of pain, with the intention to carry out a sensitivity analysis for this characteristic.
Participants were instructed to apply the study medication directly onto the skin over the painful site, except in one study (Shackel 1997), where the medication was applied distally to the skin of the inner forearm. This site was chosen because the skin is thin and should allow rapid absorption. The aim of this study was to assess the systemic effect of the gel on distant targets, which was fundamentally different from the other studies in the review.
All studies used salicylates as the rubefacient: trolamine salicylate (Algozzine 1982; Golden 1978), diethylamine salicylate (Camus 1975; Geller 1980; Rothhaar 1982; Wanet 1979), salicylic acid (Diebschlag 1987; Frahm 1993; Lester 1981), benzydamine salicylate (Ibanez 1988), methyl salicylate (Lobo 2004), glycol salicylate (Rutner 1995; Stam 2001), copper salicylate (Shackel 1997), ethylene glycol monosalicylate ester (von Bach 1979), a mixture of salicylates (Ginsberg 1987), or unspecified salicylate (Zahmatkash 2011). Formulations varied widely. A variety of additional components were added to the principal ingredient, such as the local anaesthetic myrtecaine (Camus 1975; Wanet 1979), capsicum oleoresin (Ginsberg 1987; Stam 2001), nonivamide (a capsaicinoid) (von Bach 1979), or adrenal extract (Diebschlag 1987; Lester 1981).
The active comparators used were oral aspirin (Golden 1978), the topical NSAIDs etofenamate (Geller 1980) and fepradinol (Ibanez 1988), and a herbal mixture containing cinnamon, ginger, mastic, and sesame oil (Zahmatkash 2011). In some studies participants received additional oral analgesics or physical therapy.
In two studies it was unclear whether the comparator was a placebo or active control, with one study in acute low back pain using a "homeopathic" control (containing appreciable concentrations of herbal ingredients with no known analgesic effects) (Stam 2001), and one study in chronic musculoskeletal conditions using a lower concentration of salicylate, without additional ingredients (von Bach 1979). We analysed these studies as placebo‐controlled trials, but subject to sensitivity analysis.
Of the studies in acute conditions, two were of 7 days duration (Lester 1981; Stam 2001), three between 7 and 14 days (Frahm 1993; Ibanez 1988; Rothhaar 1982), and two of 14 days or more (Diebschlag 1987; Ginsberg 1987). Three studies in chronic conditions lasted for 7 days (Algozzine 1982; Geller 1980; Golden 1978), one for 10 days (Camus 1975), and six for 14 days or more (Lobo 2004; Rutner 1995; Shackel 1997; von Bach 1979; Wanet 1979; Zahmatkash 2011).
Two studies used a cross‐over design (Algozzine 1982; Geller 1980), and the remainder used a parallel‐group design. One of the cross‐over studies did not report outcome data for the first treatment period only (Algozzine 1982).
The dose of rubefacient applied was poorly reported. Even if the application schedule was specified, most studies did not provide details of the volume applied, and some did not provide details of the concentration of the active ingredients. Although outcomes were usually defined, a variety of scales were used to assess efficacy. Adverse events and withdrawals were generally poorly reported with little detail provided.
Details of individual studies are provided in the Characteristics of included studies table.
Excluded studies
Thirteen studies were excluded after reading the full publications (Crielaard 1986; Dettoni 1982; He 2006; Heindl 1977; Higashi 2010; Howell 1955; Jolley 1972; Kantor 1990; Kleinschmidt 1975; Pasila 1980; Shamszad 1986; von Batky 1971; Weisinger 1970). Reasons for exclusion are provided in the Characteristics of excluded studies table.
Risk of bias in included studies
All studies were randomised and double‐blind. Of those in acute conditions, two had a quality score of two (Ibanez 1988; Lester 1981), two of three (Frahm 1993; Ginsberg 1987), two of four (Rothhaar 1982; Stam 2001), and one of five (Diebschlag 1987). One study had a validity score of seven (Ibanez 1988), one of eight (Rothhaar 1982), one of nine (Ginsberg 1987), one of 10 (Frahm 1993), one of 11 (Lester 1981), and two of 12 (Diebschlag 1987; Frahm 1993).
In chronic conditions there were six studies with a quality score of three (Camus 1975; Geller 1980; Lobo 2004; Rutner 1995; Wanet 1979; Zahmatkash 2011), two of four (Algozzine 1982; Golden 1978), and two of five (Shackel 1997; von Bach 1979). Two studies had validity scores of seven (Geller 1980; Lobo 2004), two of nine (Golden 1978; Wanet 1979); three of 10 (Algozzine 1982; Camus 1975; von Bach 1979), one of 11 (Shackel 1997), and one of 12 (Rutner 1995).
Comments on potential biases in individual studies are reported in the Risk of bias section of the Characteristics of included studies table. The findings are displayed in Figure 2 and Figure 3 The greatest risk of bias came from small study size.
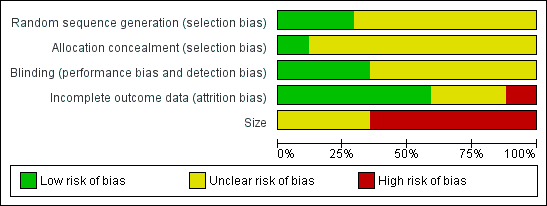
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
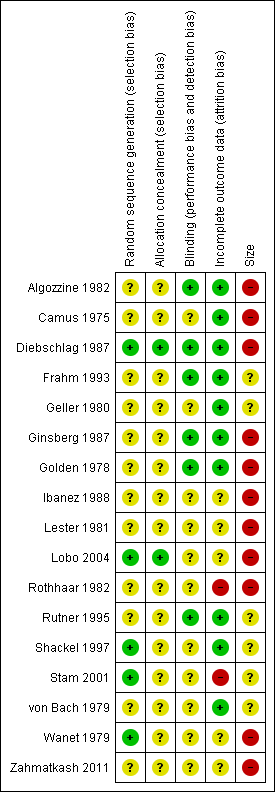
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All studies stated that they were randomised but only five reported the method used to generate the random sequence, and only two adequately described the method used to conceal the allocation sequence.
Blinding
All studies stated that they were double‐blind, but only seven adequately described the method used to conceal the treatment identity from the participants and study personnel.
Incomplete outcome data
Ten studies appeared to account for all participants in a true responder analysis. We judged two studies to be at high risk of bias because of high (> 10%) levels of attrition, poor compliance, and lack of information about any imputation methods used (Rothhaar 1982; Stam 2001). We judged the remaining six studies to be at unclear risk of bias due to a lack of information about withdrawals and participants lost to follow‐up (Ibanez 1988; Lester 1981; Lobo 2004; von Bach 1979; Wanet 1979; Zahmatkash 2011).
Other potential sources of bias
We judged 11 studies to be at high risk of bias because they randomised fewer than 50 participants to each treatment arm (Algozzine 1982; Camus 1975; Diebschlag 1987; Ginsberg 1987; Golden 1978; Ibanez 1988; Lester 1981; Lobo 2004; Rothhaar 1982; Wanet 1979; Zahmatkash 2011), and the remaining six to be at unclear risk because they included between 50 and 90 participants per treatment arm.
Effects of interventions
See: Summary of findings for the main comparison
Summaries of the efficacy outcomes are provided in Appendix 4 and of adverse events and withdrawals in Appendix 5. Because there were a small number of studies with a wide variety of formulations and inadequate reporting of dosage, it was not possible to assess dose‐response relationships. Due to insufficient data it was not possible to perform additional post hoc sensitivity analyses of particular salicylate formulations, most additional active ingredients, and different musculoskeletal conditions.
Number of participants achieving clinical success (at least 50% pain relief or equivalent)
Acute conditions
Four placebo‐controlled studies with 324 participants provided data for efficacy analysis (Ginsberg 1987; Lester 1981; Rothhaar 1982; Stam 2001). The proportion of participants achieving 50% pain relief or equivalent at seven days was 64% (range 25% to 95%; 101/157) for the rubefacient group, and 34% (0% to 59%; 56/167) for the placebo group, giving a RR of 1.9 (95% CI 1.5 to 2.5) and a NNT of 3.2 (2.4 to 4.9) (Figure 4).
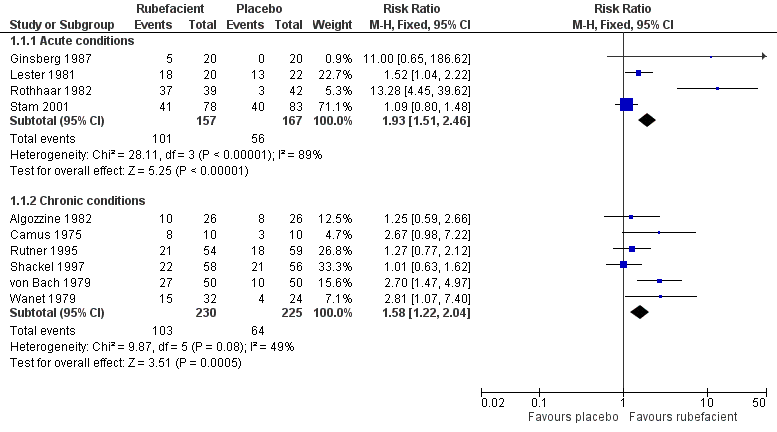
Forest plot of comparison: 1 Rubefacient versus placebo, outcome: 1.1 Clinical success (eg 50% reduction in pain).
Because studies were small, and with high variability between them (I2 = 89%), we also checked this result using the random‐effects model; the RR increased, remaining significant, with wide confidence intervals (RR 2.7 (1.05 to 7.0)). The largest and most recent study showed no difference between topical salicylate and a homeopathic gel, regarded as placebo (Stam 2001).
Only one active‐controlled study was identified (Ibanez 1988). At 12 days, 23/35 participants were reported to be "cured" with salicylate spray, and 85/102 with fepradinol spray. There were insufficient data for statistical analysis.
Sensitivity analyses
The studies differed from one another in a number of factors that might affect the estimate of efficacy. While we planned to carry out sensitivity analyses, for the most part there were too few studies and participants, and too many different factors, to make any such analyses feasible. Instead, we listed the most obvious factors that should be considered when interpreting the results of this analysis.
Baseline pain
Of the four studies contributing data to this outcome, only one clearly stated that the participants had at least moderate baseline pain (Stam 2001). Two stated that the participants had mild to severe pain (Lester 1981; Rothhaar 1982), and the other did not state the level of baseline pain (Ginsberg 1987). Low levels of baseline pain would make the study insensitive to changes in pain intensity associated with the study medication.
Time of assessment
One study had the efficacy outcome measured at < 7 days (Ginsberg 1987), although using additional data from this study measured at 14 days did not appreciably change the result.
Post hoc
The acute study of Stam 2001 used a control treatment containing herbal ingredients that could potentially have represented an active control and underestimated the effect of the rubefacient treatment in acute conditions.
None of the studies used salicylate alone. Lester 1981 included adrenal extracts and mucopolysaccharide; Rothhaar 1982 included escin, an extract of horse chestnut; and Ginsberg 1987 and Stam 2001 included low levels of capsicum oleoresin. The effects of the additional ingredients were unknown.
Chronic conditions
Six placebo‐controlled studies with 455 participants provided data for efficacy analysis (Algozzine 1982; Camus 1975; Rutner 1995; Shackel 1997; von Bach 1979; Wanet 1979). The proportion of participants achieving 50% pain relief or equivalent at 14 days was 45% (range 38% to 80%; 103/230) for the rubefacient group, and 28% (17% to 38%; 64/225) for the placebo group, giving a RR of 1.6 (1.2 to 2.0) and a NNT of 6.2 (4.0 to 13) (Figure 4). The I2 statistic for this analysis was 49%. Using a random‐effects model did not change the result (RR 1.6 (1.1 to 2.4)). The two largest and most recent studies showed no difference from placebo (Rutner 1995; Shackel 1997).
Two active‐controlled studies reported outcomes at seven days. The first found a benefit of rubefacient compared with the topical NSAID etofenamate (50 participants) (Geller 1980), although this topical NSAID has no evidence of efficacy (Massey 2010). The second found no benefit compared with oral aspirin (40 participants; (Analysis 2.1) (Golden 1978). A third active‐controlled study reported outcomes at two, four, and six weeks (92 participants) (Zahmatkash 2011). The mean pain intensity fell in both treatment groups at two weeks, with further, smaller reductions up to six weeks, but there was no difference between the groups. Studies were too small for any of these results to be robust.
Sensitivity analyses
For the most part there were too few studies and participants and too many different factors to make formal sensitivity analyses feasible. We have listed the most obvious factors that should be considered when interpreting the results of this analysis, together with the results of any statistical analysis where it was felt appropriate.
Baseline pain
Of the six studies contributing data to this outcome, only one clearly stated that participants had at least moderate baseline pain (Algozzine 1982). Three studies included participants with mild pain (Camus 1975; Shackel 1997; Wanet 1979), and two did not state the level of baseline pain (Rutner 1995; von Bach 1979). Low levels of baseline pain would make the study insensitive to changes in pain intensity associated with the study medication.
Study outcome
Two studies used undefined improvement as the measure of clinical success (Algozzine 1982; Camus 1975). Outcomes that are easy to achieve can inflate response rates, but excluding these studies did not substantially change the estimated benefit in this data set.
Time of assessment
Two studies had efficacy outcomes measured at < 14 days (Algozzine 1982; Camus 1975), but excluding these did not substantially affect the estimated benefit in this data set.
Post hoc
One study used a cross‐over design and did not report the results for the first treatment period separately (Algozzine 1982). There was no reported assessment for a carry‐over effect.
One study contributing to this analysis included a substantial minority (30%) of participants with acute conditions (Wanet 1979). Acute and chronic conditions may respond differently.
One study (von Bach 1979) used a control treatment containing lower doses of salicylate, which could be considered an active control and lead to underestimation of the beneficial effect of rubefacients.
In Shackel 1997 the rubefacient was applied distant to the site of pain, which could lead to underestimation of any benefit.
Three of the studies contributing to this analysis included additional components, which may have contributed to any observed effect. Two included the local anaesthetic myrtecaine (Camus 1975; Wanet 1979), and one included nonivamide (related to capsaicin) (von Bach 1979). Omitting von Bach 1979 reduced the RR to a barely statistically significant finding (RR 1.4 (1.03 to 1.8)), and omitting all three studies made the result not significant (RR 1.2 (0.84 to 1.6)).
Adverse events
Three studies (Camus 1975; Wanet 1979; Zahmatkash 2011) did not provide any information about adverse events. In the remaining studies, data were collected over periods of 7 to 15 days, except in Shackel 1997 where data were collected over four weeks.
All adverse events
Eleven studies provided data on adverse events with salicylates compared to placebo, six in acute conditions (Diebschlag 1987; Frahm 1993; Ginsberg 1987; Lester 1981; Rothhaar 1982; Stam 2001), and five in chronic conditions (Algozzine 1982; Lobo 2004; Rutner 1995; Shackel 1997; von Bach 1979). Three had no events in either study arm (Algozzine 1982; Diebschlag 1987; Rothhaar 1982). In all studies combined, adverse events were relatively uncommon, with 15% (74/484, range 0% to 83%) of participants in the rubefacient group experiencing an adverse event and 9% (47/500, range 0% to 52%) in the placebo group. The RR with rubefacient compared to placebo was 1.6 (1.2 to 2.0), and the NNH was 17 (9.9 to 58) (Figure 5).
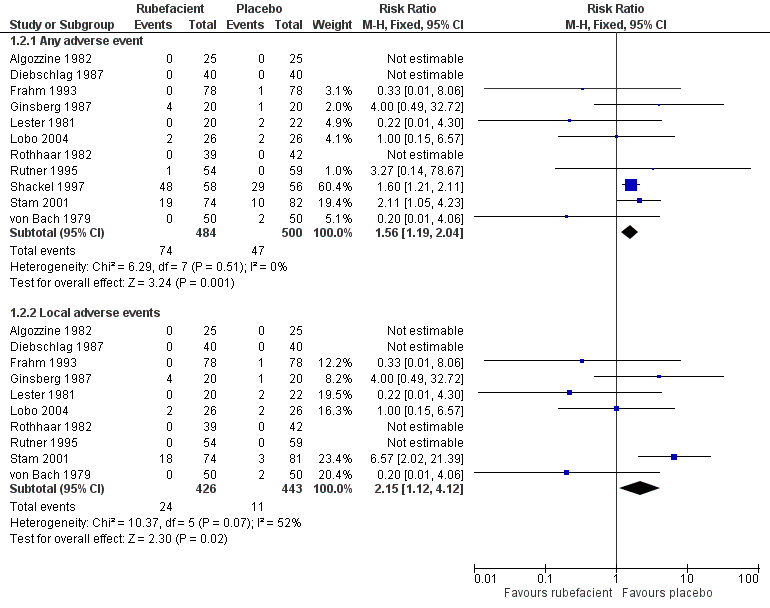
Forest plot of comparison: 1 Rubefacient versus placebo, outcome: 1.4 Adverse events.
In two studies it was not clear that the control was truly a placebo (Stam 2001; von Bach 1979) and this could over estimate the rate of adverse events in the placebo group. Excluding these studies made little difference, with event rates of 15% and 10% for rubefacient and placebo respectively, and RR of 1.5 (1.1 to 2.0). Most of the events were in the single study lasting four weeks (Shackel 1997), which had high rates in both treatment arms, and excluding this study gave event rates of 6% and 4% for rubefacient and placebo respectively, and no significant difference between groups.
Local adverse events
Ten studies provided data on local adverse events, six in acute conditions (Diebschlag 1987; Frahm 1993; Ginsberg 1987; Lester 1981; Rothhaar 1982; Stam 2001), and four in chronic conditions (Algozzine 1982; Lobo 2004; Rutner 1995; von Bach 1979), with four having no events in either study arm (Algozzine 1982; Diebschlag 1987; Rothhaar 1982; Rutner 1995). The local adverse event rates were 6% (24/426, range 0% to 24%) and 2% (11/443, range 0% to 9%) for rubefacient and placebo groups respectively, with a significant RR of 2.2 (1.1 to 4.1) and an NNH of 31 (16 to 300) (Figure 5). The I2 for this analysis was 52%; using a random‐effects model the comparison of the treatment groups was no longer significantly different (RR 1.3 (0.35 to 4.7)).
Excluding Stam 2001 and von Bach 1979 (control not a true placebo) gave an event rate of 2% for both treatment arms.
Post hoc sensitivity analyses
Omitting the three studies that contained the potent irritant capsicum oleoresin or nonivamide (Ginsberg 1987; Stam 2001; von Bach 1979) somewhat reduced the estimated RR for any adverse event (RR 1.4 (1.1 to 1.9)). Local adverse events were reduced to 2/282 and 5/292 in the rubefacient and placebo groups respectively, with too few events for analysis.
The two active‐controlled studies using topical NSAIDs (Geller 1980; Ibanez 1988) found no difference in adverse event rates between the study arms, and the aspirin‐controlled chronic study (Golden 1978) reported high rates of adverse events in the aspirin arm (Analysis 2.2).
Withdrawals
Six studies did not provide information on all cause withdrawals (Camus 1975; Diebschlag 1987; Ginsberg 1987; Ibanez 1988; Lobo 2004; Wanet 1979). In the remaining studies data were collected over periods of 7 to 15 days, except for two studies in which data were collected over four and six weeks (Shackel 1997; Zahmatkash 2011).
Placebo‐controlled studies
Five placebo‐controlled studies had information on withdrawals due to lack of efficacy, two in acute conditions (Frahm 1993; Rothhaar 1982) and three in chronic conditions (Algozzine 1982; Shackel 1997; von Bach 1979), with two having no events in either group (Algozzine 1982; Frahm 1993). The withdrawal rate due to lack of efficacy for all studies combined was 2% (6/250, range 0% to 5%) and 7% (18/251, 0% to 38%) for rubefacient and placebo respectively, giving a RR of 0.36 (0.15 to 0.87) and an NNTp of 21 (12 to 120). (Analysis 1.3)
Seven placebo‐controlled studies provided data on withdrawals due to adverse events, four in acute conditions (Diebschlag 1987; Frahm 1993; Rothhaar 1982; Stam 2001) and three in chronic conditions (Algozzine 1982; Shackel 1997; von Bach 1979), and four of these had no events in either treatment arm (Algozzine 1982; Diebschlag 1987; Frahm 1993; Rothhaar 1982). The withdrawal rate due to adverse events was 5% (18/364, range 0% to 17%) and 1% (4/373, 0% to 4%) for rubefacient and placebo respectively, with a significant relative harm of 4.2 (1.5 to 12) and a NNH of 26 (15 to 85) (Analysis 1.3)
Post hoc sensitivity analyses
All 18 adverse event withdrawals with active treatment were in two studies (Stam 2001 (acute), Shackel 1997 (chronic)). Stam 2001 included the potent irritant capsicum oleoresin in the active treatment, and Shackel 1997 had data collected over four weeks. Although combining all studies gave a significantly greater risk of withdrawal due to adverse events with rubefacients than placebo, the result is not robust, since removing either of these studies resulted in no significant difference between treatment arms.
Active‐controlled studies
The topical NSAID‐controlled study in chronic conditions (Geller 1980) had no withdrawals from either treatment arm, but the aspirin‐controlled study in chronic conditions (Golden 1978) reported one withdrawal due to lack of efficacy in the rubefacient arm, and two due to lack of efficacy and six due to adverse events in the aspirin arm (Analysis 2.3).
Withdrawals and exclusions for reasons other than lack of efficacy and adverse events were uncommon and generally due to protocol violations or loss to follow up.
Discussion
Summary of main results
One new study (92 participants) was identified for this update but it contributed data only for all cause withdrawals and the conclusions of the previous review are unchanged, although the grading and interpretation of the results is now more cautious (summary of findings Table for the main comparison).
Analysis of four studies involving 324 participants with acute musculoskeletal injuries showed a significant benefit of salicylate‐containing rubefacients compared with placebo at 7 days, with an NNT for 50% pain relief of 3.2 (2.4 to 4.9), suggesting a useful therapeutic effect of rubefacients (very low quality evidence). In chronic conditions, six studies involving 455 participant with chronic conditions gave a significant benefit compared with placebo at 14 days, with a NNT for 50% pain relief of 6.2 (4.0 to 13) (very low quality evidence).
In 11 studies (984 participants), in both acute and chronic conditions, rubefacients showed a higher rate of adverse events than placebo with a risk of harm over 1.5‐fold, giving an NNH of 17 over 7 to 14 days (low quality evidence), and a two‐fold risk of local adverse events, giving an NNH of 31 over 7 to 14 days (very low quality evidence). Withdrawals due to adverse events were increased four‐fold in the rubefacient group with a NNH of 26 (very low quality evidence). There were significantly fewer withdrawals due to lack of efficacy with rubefacient than with placebo, giving an NNTp of 21 over 7 to 14 days (very low quality evidence).
There was considerable heterogeneity amongst the trials, particularly for acute conditions, and the results were not robust. They were sensitive to the model used for analysis and the inclusion or exclusion of individual studies.
Overall completeness and applicability of evidence
The number of studies and participants identified for this review was small. Acute conditions that were studied were mainly sprains, strains, and acute low back pain, and are probably representative of the conditions suitable for topical treatment. The timing of enrolment and outcome assessment was generally appropriate for acute, self‐limiting conditions. Chronic conditions studied were not always well described, but appeared to be mainly osteoarthritis, bursitis, and chronic back pain, which again are those potentially suitable for topical treatment. Most studies did not report the duration of the condition at enrolment, so 'chronicity' was taken as reported, and most studies assessed outcomes within two weeks, with only two studies reporting after one month or longer. Studies of longer duration are desirable in chronic conditions.
Not all of the included studies reported outcomes of interest and, when they did, they were not always reported in a form that was clinically useful and that we were able to use in our analyses. This further limited the strength and interpretation of any results.
Quality of the evidence
We identified relatively few studies in either acute or chronic conditions, and all were potentially subject to bias. A major potential source of bias was the size of the studies; there were no studies with over 90 participants in each treatment arm. In addition to random variation, small studies are known to be associated with larger treatment effects (Dechatres 2013; Moore 2012b; Nüesch 2010), and for acute and chronic pain conditions, any statistical significance came from older, smaller studies, while more recent larger studies showed no effect.
Few of the studies adequately described the methods used to generate the random sequence, conceal its allocation, or maintain blinding of the treatments, but this may reflect more on the age of the studies than the conduct; older studies tended not to report such details.
Many of the studies did not report baseline pain intensity or included participants with mild pain, or a range of pain intensities from mild to severe. Measurement of a reduction in pain intensity is difficult when initial pain is mild. Additionally, some studies used poorly defined outcomes, such as 'any improvement'. These factors can make studies insensitive to demonstrating efficacy.
Almost half of the studies did not specifically report the number of participants in each treatment arm who withdrew from the studies, who were excluded from analyses for any reason, or who were lost to follow‐up. Wherever possible we have carried out an ITT analysis, assuming that missing participants were non‐responders, but some uncertainty still remains where studies were not explicit about withdrawals or imputation methods.
Potential biases in the review process
We have combined all acute conditions, and all chronic conditions, for efficacy analyses. Within each category there is heterogeneity in the condition, baseline pain intensity, duration of treatment, outcomes measured, and method of measurement, as well as the treatment applied (salicylate and additional ingredients). While there are too few studies, participants, and events for many sensitivity analyses to be carried out, we have investigated the effect of including individual studies where possible, and highlighted other factors that might influence the results. It is clear that the studies are heterogenous and the results are not robust.
Agreements and disagreements with other studies or reviews
The findings of this update did not change from those of the 2009 review (Matthews 2009), although our interpretation of the results is now more cautious. A systematic review of rubefacients in 2004 found 12 trials that were small and of only moderate quality and validity (Mason 2004). This review concluded that, at best, rubefacients containing salicylates had moderate to poor efficacy in chronic pain and good efficacy in acute pain. These results were judged not robust due to the very limited data. Other reviews have come to similar conclusions about topical rubefacients (Moore 2008).
Guidelines for the treatment of osteoarthritis in England say that rubefacients should not be offered (NICE 2014). In Scotland, guidelines for chronic pain say that "Topical rubefacients should be considered for the treatment of pain in patients with musculoskeletal conditions if other pharmacological therapies have been ineffective" (SIGN 2013).
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Forest plot of comparison: 1 Rubefacient versus placebo, outcome: 1.1 Clinical success (eg 50% reduction in pain).
Forest plot of comparison: 1 Rubefacient versus placebo, outcome: 1.4 Adverse events.

Comparison 1 Rubefacient versus placebo, Outcome 1 Clinical success (eg 50% reduction in pain).

Comparison 1 Rubefacient versus placebo, Outcome 2 Adverse events.

Comparison 1 Rubefacient versus placebo, Outcome 3 Withdrawals.

Comparison 2 Rubefacient versus active control, Outcome 1 Clinical success (eg 50% reduction in pain).

Comparison 2 Rubefacient versus active control, Outcome 2 Adverse events.

Comparison 2 Rubefacient versus active control, Outcome 3 Withdrawals.
| Salicylate‐containing topical rubefacients compared with topical placebo for acute and chronic painful conditions | ||||||
| Patient or population: adults with strains or sprains (acute) or osteoarthritis or low back pain (chronic) Settings: community Intervention: salicylate‐containing topical rubefacient Comparison: topical placebo | ||||||
| Outcomes | Probable outcome with | Probable outcome with | RR NNT, NNTp, or NNH | No of studies, participants | Quality of the evidence | Comments |
| Clinical success (eg 50% reduction in pain) Acute conditions | 640 in 1000 | 335 in 1000 | RR 1.9 (1.5 to 2.5) NNT 3.2 (2.4 to 4.9) | 4 studies 324 participants | ⊕⊝⊝⊝ | Most recent, largest study showed no effect Note NNT cannot be trusted because of low numbers and poor quality studies |
| Clinical success (eg 50% reduction in pain) Chronic conditions | 447 in 1000 | 284 in 1000 | RR 1.6 (1.2 to 2.0) NNT 6.2 (4.0 to 13) | 6 studies 455 participants | ⊕⊝⊝⊝ | Most recent, largest studies showed no effect Note NNT cannot be trusted because of low numbers and poor quality studies |
| Adverse events ‐ any adverse events Acute and chronic conditions combined | 152 in 1000 | 94 in 1000 | RR 1.6 (1.2 to 2.0) NNH 17 (9.9 to 58) | 11 studies 984 participants | ⊕⊕⊝⊝ | Inadequate reporting of adverse events is common Acute and chronic conditions combined |
| Adverse events ‐ local adverse events Acute and chronic conditions combined | 56 in 1000 | 24 in 1000 | RR 2.2 (1.1 to 4.1) NNH 31 (16 to 300) | 10 studies 869 participants | ⊕⊝⊝⊝ | Small numbers of events Acute and chronic conditions combined |
| Withdrawals ‐ lack of efficacy Acute and chronic conditions combined | 24 in 1000 | 72 in 1000 | RR 0.4 (0.2 to 0.9) NNTp 21 (12 to 120) | 5 studies 501 participants | ⊕⊝⊝⊝ | Small numbers of events Acute and chronic conditions combined |
| Withdrawals ‐ adverse events Acute and chronic conditions combined | 49 in 1000 | 11 in 1000 | RR 4.2 (1.5 to 12) NNH 26 (15 to 85) | 7 studies 737 participants | ⊕⊝⊝⊝ | Small numbers of events Acute and chronic conditions combined |
| GRADE Working Group grades of evidence | ||||||
| CI: confidence interval; RR: risk ratio; NNT: number needed to treat; NNTp: number needed to prevent an event happening; NNH: number needed to harm | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical success (eg 50% reduction in pain) Show forest plot | 10 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Acute conditions | 4 | 324 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.93 [1.51, 2.46] |
| 1.2 Chronic conditions | 6 | 455 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [1.22, 2.04] |
| 2 Adverse events Show forest plot | 11 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Any adverse event | 11 | 984 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.56 [1.19, 2.04] |
| 2.2 Local adverse events | 10 | 869 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.15 [1.12, 4.12] |
| 3 Withdrawals Show forest plot | 7 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Lack of efficacy | 5 | 501 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.15, 0.87] |
| 3.2 Adverse events | 7 | 737 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.19 [1.52, 11.56] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical success (eg 50% reduction in pain) Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Acute | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Chronic | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Adverse events Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Any adverse events | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Local adverse events | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawals Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 Lack of efficacy | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Adverse events | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |