Intervenciones para la queratosis actínica
Referencias
References to studies included in this review
References to studies excluded from this review
References to studies awaiting assessment
References to ongoing studies
Additional references
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | This was a randomised, active‐controlled, double‐blind, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 1% colchicine cream twice daily for 10 days + 10 more days if weak response (N = 8 participants) Control intervention B: 0.5% colchicine cream twice daily for 10 days + 10 more days if weak response (N = 8 participants) | |
| Outcomes | Outcomes of the trial 1) Complete healing (= participant complete clearance) 2) Reduction rate in number of actinic keratoses (= lesion complete response) at 1 month 3) Mean reduction of lesion counts at 1 month 4) Number of participants treated (pooled data) with strong, weak, or no inflammatory reaction 5) Minor adverse events (qualitative) 6) Number of participants with decreased infiltration and disappearance of crust (cosmetic) at 1 month 7) Clinical laboratory tests 8) Relapse at 6 months Efficacy Methods: quantitative assessment by counting visible and palpable actinic keratoses in each test area Time points: at baseline; end of treatment; 1, 2, and 6 months post‐treatment Safety Methods: 1. clinical examination, 2. routine laboratory tests (complete blood cell counts, urinalysis, and fasting chemistry) Time points: 1. each study visit (clinical exam), 2. before and after treatment (laboratory tests) | |
| Funding | The drug was provided by Dr. F Frik Drug Company. | |
| Notes | Thick surface scales were removed by 10% salicylic acid 2 days before treatment. There were severe inflammation reactions in the majority of participants (11/16). In cases of inflammation, a weak antiseptic or antibiotic ointment was applied. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 200): "Patients were randomly assigned to treatment with 0.5% colchicine cream or 1% colchicine cream." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | Both the investigators and the participants were blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | The investigators were blinded. |
| Incomplete outcome data (attrition bias) | Low risk | An intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported even if there was no difference between treatment groups. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, intraindividual study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | A 1‐month run‐in was performed during which participants used a placebo formulation (hydrophilic ointment) twice daily on both right and left forearms. Intervention A: 2‐(Difluoromethyl)‐dl‐ornithine (DFMO) twice daily for 6 months (N = 48 participants) Control intervention B: placebo twice daily for 6 months (N = 48 participants) | |
| Outcomes | Primary outcomes of the trial 1) Mean numbers of lesions at baseline and 6 months (the mean reduction in lesion counts was calculated) 2) Percentage reduction in the number of actinic keratoses 3) Skin concentrations of drug and products due to its mechanism of action at 6 months Secondary outcomes of the trial 1) Tolerance (qualitative) 2) Compliance Efficacy Methods: quantitative assessment by circling and counting of individual lesions on each arm by a dermatologist and photography using a Nikon N5005 camera with a 60‐mm Micor Nikkor lens, SB‐21 Macro Speedlight, and Kodachrome ASA 64 film Time points: at baseline and end of treatment Safety Methods: 1. assessment of clinical toxicity frequency and severity [scale 0 (none) to 3 (severe)] by the study dermatologist, 2. complete blood counts and serum chemistry panels (SMA20s) Time points: 1. before the first application of the placebo ointment, at randomisation, and at each monthly visit (toxicity), 2. run‐in and at the end of the study (laboratory tests) | |
| Funding | This study was supported by USPHS Grant PO1 CA27502. | |
| Notes | There was no evidence of systemic toxicity. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page1282): "Before randomisation, participants were stratified on the basis of gender and numbers of actinic keratoses on the forearms. Participants were then randomly assigned, in a double‐blind fashion, to treatment with hydrophilic DFMO ointment on the right versus the left forearm and placebo hydrophilic ointment on the contralateral forearm twice daily for 6 months." Comment: Stratification was used for randomisation sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) | Low risk | The study was double‐blind. |
| Incomplete outcome data (attrition bias) | Unclear risk | Per‐protocol (PP) analysis was used. Intraindividual study: Intervention ‐ A: 6 dropouts (the reasons were reported) Control ‐ B: 6 dropouts (the reasons were reported) Comment: The associated risk with PP analysis is unclear because the same number of participants were lost in both treatment groups. |
| Selective reporting (reporting bias) | High risk | The percentage reduction in lesion counts was given only for the DFMO‐treated group. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: isotretinoin 0.1% cream twice daily for 24 weeks (N = 50 participants?) Control intervention B: placebo cream twice daily for 24 weeks (N = 50 participants?) | |
| Outcomes | Outcomes of the trial 1) Investigators' evaluation of global therapeutic response (= global improvement indice‐completely cleared) 2) Mean number of actinic keratosis lesions over time by anatomical area 3) Mean reduction of lesion counts by anatomical area at end of treatment 4) Number of participants with severe, moderate, mild, or no local irritation on the face (= skin irritation) 5) Minor adverse events (qualitative) 6) Serious adverse events (including basal and squamous cell carcinoma) 7) Clinical laboratory tests Efficacy Methods: quantitative assessment by lesion counting and photography Time points: 1. at baseline and every 4 weeks (counting), 2. at baseline, week 12, and end of treatment (photography) Definitions for the global evaluation: 1. worsening (increase in lesions in treated area), 2. partial response (between 30% < 100% reduction in the number of lesions), and 3. complete response (total clearing) Safety Methods: 1. local tolerability was scored (absent to severe) by investigator, 2. clinical evaluation and reported adverse events, 3. routine laboratory tests Time points: 1. at each visit (tolerability and adverse events), 2. before and after treatment (laboratory tests) | |
| Funding | ‐ | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 448): "Patients were randomly assigned to treatment with 0.1% isotretinoin or a color‐matched vehicle." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) | Low risk | 2 independent investigators counted lesions. |
| Incomplete outcome data (attrition bias) | High risk | Modified intention‐to‐treat (ITT) analysis was used (i.e. participants with at least 1 postbaseline assessment were included in the analysis, N = 93), but the number of participants lost to follow up was higher than 20%. The numbers used for analysis were unclear. 1 participant in the isotretinoin group was missing in the data for skin irritation. Intervention ‐ A: 11 dropouts (the reasons were reported) Control ‐ B: 10 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Low risk | A poor efficiency was reported. |
| Other bias | High risk | The partial response criteria was very large (30% to 100%). Baseline mean withinparticipant differences in lesion number on the face was significantly different between treatment and control groups (P = 0.04). |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: December 2003 End date: December 2004 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% imiquimod once per day 3 times per week, 4 weeks on, 4 weeks off (repeated if not cleared) (N = 129 participants) Control intervention B: vehicle, once per day 3 times per week, 4 weeks on, 4 weeks off (repeated if not cleared) (N = 130 participants) | |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates at week 8 (1 treatment) and at week 16 (1or 2 treatments) Secondary outcomes of the trial 1) Participant partial (> 75%) clearance rates at week 16 2) Lesion complete response rates at weeks 8 and 16 Other outcomes of the trial 1) Participants experiencing at least 1 adverse event 2) Local skin reactions 3) Minor adverse events 4) Serious adverse events Efficacy Methods: 1. quantitative assessment using lesion counting and mapping with the use of a clear plastic template and photography, 2. histological confirmation using biopsy of a target lesion site Time points: 1. at week 1, week 2, end of treatment (EOT) (week 4 for 1 treatment, week 12 for 2 treatments), and the 4‐week post‐treatment visit (week 8 for 1 treatment, week 16 form 2 treatments), 2. pretreatment and 8‐week post‐treatment visit (week 12 for 1 treatment, week 20 for 2 treatments) (biopsy) Safety Methods: 1. assessment of the presence and intensity of specific local skin reactions by the investigator and rating on a scale [0 (none) to 3 (severe)], 2. safety evaluations including clinical laboratory tests (haematology, blood chemistry, and urinalysis) and urine pregnancy tests for women of child‐bearing potential, 3. physical examination including vital sign measurements, adverse events, and monitoring of concomitant medications Time points: 1. each visit (local skin reactions, adverse events, and medication monitoring), 2. pre‐study and poststudy (safety evaluations), 3. pre‐study, week 4 and week 12 visits (physical exam) | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | Rest periods were allowed in case of local skin reaction or treatment site adverse events. There were significant differences between imiquimod and vehicle groups in term of numbers and intensities of local skin reactions. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 134): "Eligible patients were randomised to either imiquimod 5% cream or vehicle cream in a 1:1 ratio." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blind. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blind and used 2 independent blinded dermatologists for histological evaluation. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used, and all subjects were accounted for. Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 3 dropouts (the reasons were not all reported) |
| Selective reporting (reporting bias) | High risk | The participant complete clearance for the face and scalp was reported for the imiquimod group but not the vehicle group. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, double‐dummy, vehicle‐controlled, parallel‐group study. Start date: September 2006 End date: June 2007 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: once daily 0.025% ingenol mebutate gel (PEP005) for 3 days (N = 50 participants) B: once daily 0.05% ingenol mebutate gel (PEP005) for 2 days (N = 55 participants) C: once daily 0.05% ingenol mebutate gel (PEP005) for 3 days (N = 57 participants) Control intervention D: once daily vehicle for 3 days (N = 60 participants) | |
| Outcomes | Primary outcome of the trial 1) Partial clearance rate (= participant partial clearance) Secondary outcomes of the trial 1) Complete clearance rate (= participant complete clearance for all lesions) 2) Baseline clearance rate (= participant complete clearance for target lesions) 3) Median percentage reduction of target lesions Other outcomes of the trial 1) Application site reactions 2) Local skin reactions overtime (pooled ingenol mebutate data and no data for vehicle) 3) Global severity rating of local reactions 4) Minor adverse events 5) Treatment‐related adverse events 6) Serious adverse events 7) Clinical laboratory tests 8) Cosmetic outcomes: pigmentation and scarring (pooled data) 9) Participants' satisfaction Efficacy Methods: quantitative assessment using counting of clinically‐visible lesions in the selected treatment area (including baseline and new lesions) Time points: at day 57 (end‐of‐study visit) Definitions: 1. partial clearance rate (proportion of participants with > 75% reduction in the number of lesions identified at baseline), 2. complete clearance rate (proportion of participants with no clinically‐visible lesions in the selected treatment area ‐ lesions present at baseline or emergent during the study period), 3. baseline clearance rate (proportion of participants with 100% reduction in the number of lesions identified at baseline), and 4. percentage reduction of the number of lesions (number of lesions present in the treatment area at baseline minus the number of lesions present at the end of the study divided by the number of lesions present at baseline) Safety Methods: 1. assessment of any local skin reactions and global severity rating, and monitoring of adverse events by a qualified dermatologist, 2. clinical laboratory tests Time points: 1. at day 3, follow‐up visits on days 8, 15, 29, and 57 (end‐of‐study visit); 2. at screening visit and on day 8 (laboratory tests) Cosmetic Methods: questionnaire with a 7‐point Likert scale, in which a score of 1 is very negative, 4 is neutral, and 7 is very positive | |
| Funding | This study was supported by Peplin Ltd. | |
| Notes | Participants' satisfaction (P = 0.0005) and overall satisfaction (P < 0.001) were higher in the treatments groups compared with vehicle. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Each centre was allocated an initial block of 4 randomisation numbers and enrolled participants were assigned a participant number in ascending order. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | The centre personnel and the participants were blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | The investigator was blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Modified ITT analysis was used (i.e. at least 1 dose and 1 postbaseline assessment). Intervention ‐ A: 0 dropouts, B: 1 dropout, C: 0 dropouts Control ‐ D: 0 dropouts |
| Selective reporting (reporting bias) | High risk | Data were pooled for safety and cosmetic outcomes. Based on the protocol NCT00375739, safety was supposed to be the primary outcome. However, efficacy data were presented first. |
| Other bias | Unclear risk | ‐ |
| Methods | The was a single‐centre, randomised, double‐blind, placebo‐controlled, intraindividual study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% 5‐fluorouracil twice daily on both arms for 2 to 4 weeks and 0.05% tretinoin nightly on 1 randomised arm for 2 to 4 weeks (N = 20 participants) Control intervention B: 5% 5‐fluorouracil twice daily on both arms for 2 to 4 weeks and placebo nightly on 1 randomised arm for 2 to 4 weeks (N = 20 participants) | |
| Outcomes | Outcomes of the trial 1) Mean number of actinic keratosis lesions at baseline and 3 months (mean reduction of lesion counts was calculated) 2) Relative irritation (skin irritation) Efficacy Methods: quantitative assessment using counting of residual actinic keratoses and biopsy of doubtful lesions Time points: at baseline and week 12 | |
| Funding | The treatments were provided by Ortho Pharmaceutical Corporation and Hoffman‐LaRoche, Inc. | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 550): "Tretinoin 0.05% cream (RETIN‐A Cream, Ortho Pharmaceutical Corp., Raritan, NJ, U.S.A.) and a placebo cream, Eucerin (Beiersdorf Inc., Norwalk, CT, U.S.A.) were supplied to each patient in unmarked jars labelled only with the randomly assigned side to which the medication was to be applied." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) | Low risk | The study was double‐blind. |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear if the only lost participant was included or not in the analysis. Intraindividual study: Intervention ‐ A: 1 dropout (the reasons were reported) Control ‐ B: 1 dropout (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Based on the text, the data should have been presented as percentage of reduction in lesion count, but only the absolute counts at baseline and 3 months were presented. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: January 2002 End date: August 2002 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: imiquimod 5% cream once per day, 3 times per week for 3 weeks on, 4 weeks off (repeat once if 75% of lesions hadn't cleared) Control intervention B: placebo once per day, 3 times per week for 3 weeks on, 4 weeks off (repeat once if 75% of lesions hadn't cleared) | |
| Outcomes | Primary outcome of the trial 1) Participant partial (> 75%) clearance rates at 14 weeks Other outcomes of the trial 1) Mean number of actinic keratosis lesions overtime (graphical representation) 2) Participant complete clearance 3) Local skin reactions Efficacy Methods: quantitative assessment using counting of the number of lesions in the treatment area by the same investigator and photography Time points: at each weekly visit Safety Methods: reporting of local skin reactions [severity on a 0 (none) to 3 (severe) scale] and adverse events Time points: at each weekly visit | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | All adverse effects were gone at follow‐up. An increase in the number of lesions during treatment was observed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 252): "Randomisation codes were prepared using permuted blocks of four and stratifying by study centre." |
| Allocation concealment (selection bias) | Low risk | Quote (page 252): "Randomisation codes were concealed within opaque, sealed envelopes." |
| Blinding of participants and personnel (performance bias) | Low risk | The study was double‐blind, and randomisation codes were not revealed until all final assessments were completed. |
| Blinding of outcome assessment (detection bias) | Low risk | The study was double‐blind, and randomisation codes were not revealed until all final assessments were completed. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol analysis was used. Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 1 dropout (the reasons were reported) 5 participants with protocol violation (4 imiquimod, 1 placebo) were excluded, and 1 with protocol violation was included (imiquimod not cured). |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, intraindividual study. Start date: July 2001 End date: March 2002 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention Control intervention B: placebo‐PDT (N = 17 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: crusts and scales removed by curettage Cream concentration (%): ‐‐ Application of cream: 1 mm thick to lesional field and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: visible non‐coherent light Light source: Waldmann PDT 1200 Wavelength (nm): 600‐730 Energy fluence (J/cm²): 75 inten (mW/cm²): 80 Exposure time: ‐‐ Others: Each participant received 1 g paracetamol orally 1 hour before illumination. Additionally, a fan was used to cool the treated area and to reduce discomfort during illumination. | |
| Outcomes | Outcomes of the trial 1) Complete response rates of the lesional area (= participant complete clearance) at 16 weeks after 2nd treatment 3) Minor adverse events (qualitative) 4) Discomfort on a visual analogue scale (VAS) Efficacy Methods: quantitative assessment by inspection, photography, and palpation of the lesional area Time points: at 1, 4, 8, and 16 weeks after the 2nd treatment Definitions: 1. complete response (complete clinical regression of all lesions within the treated area), 2. partial response (incomplete reduction in size or number of the lesions within the treated area) Safety Methods: reporting of adverse events including the local phototoxicity reactions Time points: before and after illumination, and at 1, 4, 8, and 16 weeks after the 2nd treatment | |
| Funding | ‐ | |
| Notes | There was no quantification of adverse events, but discomfort was reported higher for MAL than placebo. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 197): "Two lesional areas within a patient, measuring a maximum of 4 X 4 cm, were randomised to receive 2 consecutive treatments of topical PDT 1 week apart using either MAL or placebo cream." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | The study was double‐blind, but because discomfort was higher with MAL than placebo cream, the blinding could have been broken. |
| Blinding of outcome assessment (detection bias) | High risk | The study was double‐blind, but because discomfort was higher with MAL than placebo cream, the blinding could have been broken. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Low risk | The same data were reported in the abstract and published paper. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, intraindividual study. Start date: 2003 End date: 2004 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 3% diclofenac/2.5% hyaluronic acid twice daily for 90 days (N = 20 participants, 32 lesions) Control intervention B: 2.5% hyaluronic acid twice daily for 90 days (N = 20 participants, 32 lesions) | |
| Outcomes | Outcomes of the trial 1) Lesion complete response rates 2) Reduction in lesion size 3) Number of participants experiencing irritation (skin irritation) Efficacy Time points: at the end of treatment Definitions: 1. partial response (any reduction in the lesion size compared to baseline), 2. complete response (complete disappearance of the lesion) | |
| Funding | ‐ | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 347): "Sixty‐four lesions of actinic keratosis in 20 patients were evaluated, 32 for active treatment and another 32 lesions with relatively similar characteristics but on the opposite side, as controls. Lesions were randomised to receive either 3% diclofenac in 2.5% hyaluronic acid gel or placebo (the inactive gel vehicle, hyaluronic acid only) 0.5 g twice daily in each 5 cm² treatment area for 90 days." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) | Low risk | The study was double‐blind. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | High risk | The presentation of the data was confusing. The wording in the manuscript for the efficacy analysis created confusion between 'lesion complete response' and 'participant complete clearance', but based on the numbers and the percentages given, the outcome was lesion complete response. |
| Methods | This was a single‐centre, randomised, double‐blind, placebo‐controlled, intraindividual study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 12.5% DL‐α‐tocopherol (vitamin E) on right or left arm twice daily for 6 months (N = 48 participants) Control intervention B: placebo on right or left arm twice daily for 6 months (N = 48 participants) | |
| Outcomes | Primary outcome of the trial 1) Biochemical and immunological outcomes Secondary outcome of the trial 1) Mean reduction of lesion counts Other outcome of the trial 1) Number of reports of symptoms (redness, itchiness, burning, dryness) Efficacy Methods: 1. quantitative assessment using circling of lesions and photographs, 2. shave biopsies by physician Time points: before and at the end of treatment Safety Methods: 1. physical exams, 2. clinical staff inquired about adverse events (severity, date of onset, duration, and date of resolution) Time points: 1. before treatment and at the end of treatment (physical exams), 2. at monthly visits (adverse events) | |
| Funding | This study was supported by NIH grants CA‐27502 and CA‐23074. | |
| Notes | This was a phase IIb study. Vitamin E was well tolerated, i.e. only 14 reports for moderate and severe symptoms, which were similar for treatment and placebo. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A progressive randomisation program was used to make sure that allocation did not vary by gender, age, or actinic keratosis lesions (stratification). |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) | Low risk | The study was double‐blind. |
| Incomplete outcome data (attrition bias) | Unclear risk | Per‐protocol (PP) analysis was used. Intraindividual study: Intervention ‐ A: 6 dropouts (the reasons were reported) Control ‐ B: 6 dropouts (the reasons were reported) Comment: The associated risk with PP analysis is unclear because the same number of participants were lost in both treatment groups. |
| Selective reporting (reporting bias) | Low risk | No statistically different results were reported. |
| Other bias | High risk | Unprecise evaluation: 8 participants at baseline and 6 at the end of treatment had lesions too numerous to count and a number of 78 was used for analysis. |
| Methods | This was a multicentre, randomised, double‐blind, open, placebo‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 88 participants) Control interventions B: placebo ‐PDT: Placebo (N = 23 participants) C: cryotherapy: no prior preparation, variable liquid nitrogen spray unit, 1 to 2 mm rim of frozen tissue beyond marked outline, a single timed freeze‐thaw cycle; mean diameter < 10 mm = mean freeze time of 12 + 13 seconds, 10 to 20 mm = 16 + 15 seconds, > 20 mm = 26 + 11 seconds (N = 89 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: crusts and scales removed by curettage Cream concentration (%): 16 Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): 50 to 250 Exposure time: 10 minutes | |
| Outcomes | Outcomes of the trial 1) Lesion complete response rates at 3 months 2) Participants experiencing at least 1 adverse event 3) Local skin/adverse reactions 4) Minor adverse events (given only for MAL‐PDT) 5) Cosmetic outcomes: overall and individual lesions at 3 months (MAL‐PDT vs cryotherapy) 6) Participant satisfaction Efficacy Methods: quantitative assessment using mapping with acetate sheets, marking of lesions and anatomical landmarks and Polaroid photography Time points: at 3 months after the beginning of treatment Definitions for lesion response: 1. complete response (complete disappearance of the lesion, both visually and by palpation), 2. non‐complete response (incomplete disappearance of the lesion) Safety Methods: adverse events reported by the participant or elicited through open (non‐leading) questioning by the investigator Time points: before, during, and after treatment; at 2 weeks by telephone contact; and at a final examination 3 months after treatment Cosmetic Methods: 1. overall cosmetic outcome of completely cleared participants (by investigator and participant), 2. individual lesion cosmetic outcome for completely cleared lesions (hypopigmentation, hyperpigmentation, scar formation and tissue defect rated as none, slight, or obvious) Time points: at 3 months after the beginning of treatment Definitions for overall outcome: 1. excellent (no scarring, atrophy or induration, and no or slight occurrence of redness or change in pigmentation compared with adjacent skin), 2. good (no scarring, atrophy or induration but moderate redness or change in pigmentation compared with adjacent skin), 3. fair (slight to moderate occurrence of scarring, atrophy, or induration), 4. poor (extensive occurrence of scarring, atrophy, or induration) | |
| Funding | This study was supported by Photocure ASA, Olso, Norway. | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed per participant for each treatment option (first: PDT or cryo, second: MAL or placebo) and stratified by centre. |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes were used to conceal the allocation. |
| Blinding of participants and personnel (performance bias) | High risk | The study was double‐blind for the comparison between MAL‐PDT and placebo‐PDT and open for the comparison between MAL‐PDT and cryotherapy. |
| Blinding of outcome assessment (detection bias) | High risk | The study was double‐blind for the comparison between MAL‐PDT and placebo‐PDT and open for the comparison between MAL‐PDT and cryotherapy. |
| Incomplete outcome data (attrition bias) | High risk | Both intention‐to‐treat (ITT) and per‐protocol (PP) analyses were used, but only values for PP were presented and the authors only mentioned that results were similar for ITT analysis. Intervention ‐ A: 11 dropouts Control ‐ B: 4 dropouts, C: 3 dropouts Thus, there was less lost in cryotherapy (3.4%) than placebo‐PDT (17%) and MAL‐PDT (12.5%). |
| Selective reporting (reporting bias) | High risk | Types of adverse events were not reported separately for PDT and cryotherapy treatments, but more adverse events were reported for PDT than cryotherapy, and more for MAL‐PDT than placebo‐PDT. Risk of bias for more than 2 groups: cosmetic outcomes were not presented for the placebo‐PDT treatment group, and satisfaction was reported only for PDT participants. Similar data were presented in abstract form and published paper. |
| Other bias | High risk | There was a difference in baseline; the placebo PDT group included a greater proportion of men, and slightly more participants with skin type I and fewer with skin type 2. |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: 1994 End date: 1995 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 0.25 g of 3% diclofenac in 2.5% hyaluronic acid gel twice daily for 12 weeks (N = 73 participants) Control intervention B: 2.5% hyaluronic acid gel alone twice daily for 12 weeks (N = 77 participants) | |
| Outcomes | Outcomes of the trial 1) Mean reduction of lesion counts at end of treatment and at 30 days post‐treatment 4) Minor adverse events 5) Serious adverse events 6) Clinical laboratory tests 7) Compliance Efficacy Methods: quantitative assessment using lesion counting by a single doctor in each centre throughout the entire study Time points: at baseline, end of treatment (12 weeks), and at 16 weeks Definitions: 1. complete clearance (proportion of participants with complete resolution of lesions), 2. partial clearance (proportion of participants with a > 50% reduction in lesions) Safety Methods: 1. medical history and physical examination (baseline only), 2. haematology and biochemistry testing, 3. adverse events were recorded in the case report form [type (serious or non‐serious), onset date, severity (mild, moderate or severe), duration, any action taken, assumed relationship to treatment and outcome] Time points: at baseline, week 12 , and week 16 | |
| Funding | This study was supported by Hyal Pharmaceutical Corporation. | |
| Notes | The safety assessment showed no difference between groups. A high compliance was observed for both groups. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 40):"They were randomly allocated to either active treatment (N = 73) or placebo (N = 77)." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) | Low risk | The study was double‐blind. |
| Incomplete outcome data (attrition bias) | Unclear risk | Intention‐to‐treat (ITT) analysis was used and 1 withdrawn participant was accounted for in the wrong treatment group (see below). Intervention ‐ A: 23 dropouts stated but 22 based on the details of the reasons Control ‐ B: 12 dropouts stated but 13 based on the details of the reasons. |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: April 2000 End date: December 2000 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: 5% imiquimod (2 times/week) for 8 weeks (N = 31 participants) B: 5% imiquimod (3 times/week) for 8 weeks (N = 29 participants) C: 5% imiquimod (5 times/week) for 8 weeks (N = 30 participants) D: 5% imiquimod (7 times/week) for 8 weeks (N = 30 participants) Control intervention E: vehicle (2, 3, 5, 7 times/week) for 8 weeks (N = 29 participants, 7 to 8/dosing regimen pooled together) | |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates at week 16 Secondary outcome of the trial 1) Participant partial (> 75%) clearance rates at week 16 Other outcomes of the trial 1) Application site reactions 2) Local skin reactions 3) Treatment‐related adverse events 4) Serious adverse events 5) Clinical laboratory tests 6) Compliance 7) Rest periods Efficacy Methods: 1. quantitative assessment using lesion counts within the target area performed by a qualified dermatologist using a transparent plastic template to track lesions, 2. qualitative assessment using lesion descriptions by investigator, i.e. degree of hyperkeratosis, size and confluence of the lesions, and degree of solar damage between lesions Time points: at baseline, week 4, and end of study (week 16) Definitions: 1. complete clearance rate (proportion of subjects at end of study with no lesions in the treatment area), 2. partial clearance rate (proportion of subjects at their last study visit with at least 75% reduction in lesions in the treatment area) Safety Methods: 1. recording of vital signs and adverse events, 2. assessment Time points: 1. at weeks 1, 2, 3, 4, 6, 8 (end of treatment), 12, and 16 (8 weeks post‐treatment, end of study), 2. at baseline, end of treatment, and end of study (physical exam and laboratory tests) Definitions for grading: 1. mild (subject is aware of the signs and symptoms, but the signs and symptoms are easily tolerated), 2. moderate (signs and symptoms are sufficient to restrict, but not prevent, usual daily activity for the subject), and 3. severe (signs and symptoms are such that the subject is unable to perform usual daily activity) | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | The number of local skin reactions and adverse events increased with dosing frequency. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation schedule was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | The study was double‐blind for intervention versus control but not for the frequency of application. 4 groups of vehicle were used to match the number of application and conceal treatment allocation, but there was not use of the vehicle to have all groups applying cream for 7 days/week, e.g. tubes labelled for each day of the week (with intervention or control) to conceal frequency allocation. |
| Blinding of outcome assessment (detection bias) | Low risk | The study was double‐blind. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 4 dropouts, B: 2 dropouts, C: 9 dropouts, D: 12 dropouts Control ‐ E: 1 dropout The imiquimod 5X/week and 7X/week groups lost 30% and 40% of participants, respectively. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported even if efficacy was low. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: January 2008 End date: July 2008 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: 3.75% imiquimod, once daily for 3 weeks on, 3 weeks off, 3 weeks on (N = 162 participants) B: 2.5% imiquimod, once daily for 3 weeks on, 3 weeks off, 3 weeks on (N = 164 participants) Control intervention C: placebo, once daily for 3 weeks on, 3 weeks off, 3 weeks on (N = 164 participants) | |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates at week 17 Secondary outcomes of the trial 1) Participant partial (> 75%) clearance rates at week 17 2) Median percentage of changes in lesion counts 3) Local skin reactions Other outcomes of the trial 1) Participants experiencing at least 1 adverse event 2) Application site reactions (including irritation) 3) Minor adverse events 4) Treatment‐related adverse events 5) Serious adverse events 6) Clinical laboratory tests 7) Investigator global integrated photodamage (IGIP‐cosmetic outcome) 8) Temporary treatment interruption Efficacy Methods: quantitative assessment using counting of all visible or palpable lesions ‐ baseline or new ‐ in the treatment area by the investigator Time points: baseline; at weeks 1, 2, 3 (end of cycle 1), 6 (beginning of cycle 2), 7, 8, 9 (end of cycle 2), 13, and 17 (end of study) (subjects who discontinued the study prematurely were requested to return for the end‐of‐study visit) Definitions: 1. complete clearance rate (proportion of subjects at the end‐of‐study visit with a count of zero lesions in the treatment area), 2. partial clearance rates (proportion of subjects with 75% or greater reduction in lesion count in the treatment area at the end‐of‐study visit as compared with baseline), and 3. percentage of changes in lesion count (per cent change in lesion number at the end‐of‐study visit as compared with baseline) Safety Methods: 1. measurement of vital signs, 2. recording of adverse events, 3. investigator assessment of local skin reactions (erythema, edema, weeping/exudate, flaking/scaling/dryness, scabbing/crusting, and erosion/ulceration) graded as none, mild, moderate, or severe, 4. hematology, serum chemistry, urinalyses, and urine pregnancy tests (treatment‐emergent adverse events were summarised for each treatment group by preferred term, intensity, and investigator assessment of relationship to study cream. The local skin reactions were summarised by the most intense score for each reaction and by the sum score at each visit and over the course of the study) Time points: 1. baseline, at weeks 1, 2, 3 (end of cycle 1), 6 (beginning of cycle 2), 7, 8, 9 (end of cycle 2), 13, and 17 (end of study), 2. pre‐study visit and end‐of‐study visit (laboratory tests) Cosmetic Methods: qualitative and quantitative assessment (IGIP score) Time points: at end‐of‐study visit Definition: IGIP score (overall assessment of the subject's photodamage change from baseline in the treatment area including an integrated assessment of fine wrinkling, coarse wrinkling, mottled pigmentation, roughness, sallowness, skin laxity, and telangiectasias) [the details on score were not provided, but both numerical results (score with standard deviation) and the number of participants with "significantly or much improved' cosmetic outcome were presented] | |
| Funding | This study was supported by Graceway Pharmaceuticals. | |
| Notes | Data from 2 studies were pooled together. Temporary dosing interruptions could have been instructed by the investigator to manage local skin reactions and adverse events. 96% of subjects were compliant with dosing. A sample size calculation was provided. There was a follow‐up study published (Hanke 2011). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 575): "Eligible subjects were randomised to placebo, imiquimod 2.5%, or imiquimod 3.75% cream in a 1:1:1 treatment allocation." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | The study was double‐blind, but authors mentioned that local effects of imiquimod may have led to investigator and subject bias. |
| Blinding of outcome assessment (detection bias) | High risk | The study was double‐blind, but authors mentioned that local effects of imiquimod may have led to investigator and subject bias. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 10 dropouts, B: 7 dropouts Control ‐ C: 10 dropouts |
| Selective reporting (reporting bias) | Unclear risk | All outcomes from the protocol NCT00603798 were reported, but additional outcomes were also presented in the published paper (e.g. cosmetic). |
| Other bias | Unclear risk | Data for safety were reported differently in the published and the study results section of the protocol NCT00603798 in clinicaltrials.gov. |
| Methods | This was a single‐centre, randomised, active‐controlled, parallel‐group study. Start date: October 1, 2000 End date: October 30, 2002 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 2 passes of carbon dioxide laser resurfacing (N = 8 participants) Control interventions B: 30% trichloroacetic acid peel (N = 10 participants) C: 5% fluorouracil twice daily for 3 weeks (N = 9 participants) D: not randomised control group without treatment (data not presented and not included in our review) (N = 5 participants, 2 participants not included and reasons were given) | |
| Outcomes | Outcomes of the trial 1) Mean number of actinic keratosis lesions at baseline and 3 months (transformed to mean reduction in lesion counts) 2) Mean percentage of reduction of lesion counts at 3 months 3) Incidence of new non‐melanoma skin cancer for 5 years (4 groups) 4) Minor adverse events (qualitative) Efficacy Methods: quantitative assessment using the number and locations of existing lesions charted on a diagram of the head (at each visit, any lesions suggestive of basal or squamous cell carcinoma were biopsied. The VA Palo Alto Health Care System (VAPAHCS) medical and pathologic records were reviewed for each participant through June 30, 2005, to evaluate for any subsequent development of skin cancer in treated areas) Time points: at enrolment and every 3 months for a minimum of 24 months (at the end of the 24‐month study, participants continued routine general dermatology clinic surveillance) Definitions for rates of cancer formation: 1. cancer incidence rates (ratio of the total number of cancers to the total number of participant‐years followed in each group), 2. number of days from baseline/treatment to diagnosis of the first non‐melanoma skin cancer Safety Methods: monitoring for any adverse events Time points: at every 3 months for a minimum of 24 months | |
| Funding | ‐ | |
| Notes | Every 3 months, new or remaining lesions were treated with cryosurgery. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 977): "Patients were prospectively randomised to 1 of 3 treatment arms..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | The study was open because physically different treatments were used. |
| Blinding of outcome assessment (detection bias) | High risk | Blinding of assessor was not stated and physically different treatments were used. |
| Incomplete outcome data (attrition bias) | High risk | The type of analysis was unclear. Intervention ‐ A: 2 dropouts (the reasons were reported) Controls ‐ B: 0 dropouts, C: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blinded, placebo‐controlled, parallel‐group study. Start date: March 2006, End date: December 2007 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 3 to 8 self‐adhesive patches of PD P506A (aminolevulinic acid ‐ ALA)‐photodynamic therapy (PDT) (N = 69 participants) Control intervention B: 3 to 8 self‐adhesive patches of placebo‐PDT (N = 34 participants) Characteristics of PDT intervention: Type of treatment: individual lesions Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: no Cream concentration (%): patches containing 8 mg Application of cream: self‐adhesive patch Incubation with cream: 4 hours Type of light: red light LED Light source: Aktilite CL 128 or Omnilux Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ | |
| Outcomes | Primary outcome of the trial 1) Complete clinical clearance rates on lesion basis (= lesion complete response) at 12 weeks post‐treatment Secondary outcomes of the trial 1) Participant complete clearance rates at 12 weeks post‐treatment 2) Adverse reactions at the treatment site (= application site reactions) during and the day after the treatment 3) Local skin/adverse reactions (presented for ALA‐PDT only) 4) Serious adverse events 5) Treatment‐related adverse events 6) Participant and investigator cosmetic outcomes of cleared lesions 7) Participant satisfaction Efficacy Methods: clinical diagnosis being regarded as usual procedure in dermatological practice Time points: at 12 weeks post‐treatment Definitions: complete clinical clearance of a lesion (no visual evidence of persisting lesion on treated surface; no evidence of adherent scaling plaques on treated skin surface when palpated; lesions no longer perceptible to touch; and slight pink or red foci might be visible at lesion sides) Safety Methods: 1. recording of local reactions by clinical staff, 2. a diary for the documentation of local reactions by participant during the 4 weeks after therapy, 3. blood samples for monitoring hepatic aminotransferases (alanine aminotransferase and aspartate aminotransferase) and ɣ‐glutamyltransferase, 4. documentation of adverse events Time points: 1. during patch application, illumination, and thereafter (local reactions), 2. before and day of treatment (blood tests), 3. each study visit (adverse events) Cosmetic Methods: 1. participants' and investigators' assessment of the cosmetic outcome of cleared lesions ('excellent', 'good', 'fair', or 'poor'), 2. participants' overall satisfaction with the cosmetic outcome ('very satisfied', 'satisfied', 'poorly satisfied', 'not satisfied') Time points: at 12 weeks post‐treatment | |
| Funding | This study was supported by Photonamic GmbH & Co. | |
| Notes | The manuscript included 2 independent phase III studies (AK03 and AK04). This study was AK03. Adverse events were given for individual studies and pooled for ALA‐PDT, but pooled only for placebo‐PDT. Thus, pooled data were used for analysis (under Hauschild 2009a). A follow‐up study was published (Szeimies 2010a). Data for intention‐to‐treat analysis were used for the meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratification was performed by centre. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) | Low risk | The study was double‐blind, and treatment was performed by a second investigator to guarantee an observer‐blinded status. |
| Incomplete outcome data (attrition bias) | Unclear risk | Modified ITT analysis was used (4 participants were not included, and the criteria were not specified but the numbers correspond to the following: ALA‐PDT: 1 missed control visit, 1 curettage before the study, 1 stop of illumination, and placebo‐PDT: 1 consent withdrawn). Intervention ‐ A: 17 dropouts (the reasons were reported) Control ‐ B: 8 dropouts (the reasons were reported) 24% of participants were lost before the end of study, but similar percentages were lost for both treatment arms. |
| Selective reporting (reporting bias) | High risk | Details on investigator cosmetic outcomes and adverse events for placebo group were not given. Outcomes in protocol (NCT00308854) were all presented in published paper. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, open, parallel‐group study. Start date: March 2006 End date: November 2007 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 4 to 8 self‐adhesive patches of PD P506A (aminolevulinic acid ‐ALA)‐ photodynamic therapy (PDT) (N = 148 participants) Control interventions B: 4 to 8 self‐adhesive patches of placebo‐PDT (N = 49 participants) C: cryosurgery: nozzles of size C, 1 cycle and freeze time between 5 and 10 seconds (N = 149 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: no Cream concentration (%): patches containing 8 mg Application of cream: self‐adhesive patch Incubation with cream: 4 hours Type of light: red light LED Light source: Aktilite CL 128 or Omnilux Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ | |
| Outcomes | Primary outcome of the trial 1) Complete clinical clearance rates on lesion basis (= lesion complete response) at 12 weeks post‐treatment Secondary outcomes of the trial 1) Participant complete clearance rates at 12 weeks post‐treatment 2) Adverse reactions at the treatment site (= application site reactions) during and the day after the treatment 3) Local skin/adverse reactions (presented for ALA‐PDT only) 4) Serious adverse events 5) Treatment‐related adverse events 6) Participant and investigator cosmetic outcomes of cleared lesions 7) Participant satisfaction Efficacy Methods: clinical diagnosis being regarded as usual procedure in dermatological practice Time points: at 12 weeks post‐treatment Definitions: complete clinical clearance of a lesion (no visual evidence of persisting lesion on treated surface; no evidence of adherent scaling plaques on treated skin surface when palpated; lesions no longer perceptible to touch; and slight pink or red foci might be visible at lesion sides) Safety Methods: 1. recording of local reactions by clinical staff, 2. a diary for the documentation of local reactions by participant during the 4 weeks post‐treatment, 3. blood samples for monitoring hepatic aminotransferases (alanine aminotransferase and aspartate aminotransferase) and ɣ‐glutamyltransferase, 4. documentation of adverse events Time points: 1. during patch application, illumination, and thereafter for PDT, and during the spraying procedure and thereafter for cryotherapy (local reactions), 2. before and day of treatment (blood tests), 3. each study visit (adverse events) Cosmetic Methods: participants' and investigators' assessment of the cosmetic outcome of cleared lesions ('excellent', 'good', 'fair', or 'poor') Time points: at 12 weeks post‐treatment | |
| Funding | This study was supported by Photonamic GmbH & Co. | |
| Notes | The manuscript included 2 independent phase III studies (AK03 and AK04). This study was AK04. Adverse events were given for individual studies and pooled for ALA‐PDT, but only pooled for placebo‐PDT. Thus, pooled data were used for analysis for ALA‐PDT vs placebo‐PDT (under Hauschild 2009a). A follow‐up study was published (Szeimies 2010a). Data for intention‐to‐treat analysis were used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratification was performed by centre. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | The study was open because the treatments were physically distinct. Similar patches were used for ALA‐PDTand placebo‐PDT, but no concealment was possible for the physically distinct treatments, i.e. PDT versus cryotherapy. |
| Blinding of outcome assessment (detection bias) | High risk | The study was open. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 19 dropouts (the reasons were reported) Controls ‐ B: 6 dropouts (the reasons were reported), C: 23 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Details on investigator cosmetic outcomes and adverse events for placebo group were not given. Outcomes in protocol (NCT00308867) were all presented in published paper. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, assessor‐blinded, active‐controlled, parallel group study. Start date: January 2005 End date: July 2005 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: 1 hour of PD P506A (aminolevulinic acid (ALA) self‐adhesive patch)‐photodynamic therapy (PDT) (N = 38 participants) B: 2 hours of PD P506A‐PDT (N = 34 participants) C: 4 hours of PD P506A‐PDT (N = 34 participants) Control intervention D: 0.5 hour of PD P506A‐PDT (N = 34 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: no Cream concentration (%): patches containing 8 mg Application of cream: self‐adhesive patch Incubation with cream: 0.5 to 4 hours Type of light: red light Light source: Aktilite CL 128 Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ | |
| Outcomes | Primary outcome of the trial 1) Lesion complete response rates at 4 and 8 weeks Secondary outcomes of the trial 1) Participant complete clearance rates at 4 and 8 weeks 2) Local skin reactions presented graphically for 3 periods (during ALA patch application, during illumination, and after illumination) as well as by severity of the reactions 3) Treatment‐related adverse events (minor adverse events) 4) Minor adverse events (pooled) 5) Serious adverse events 6) Clinical laboratory tests 7) New actinic keratoses Efficacy Methods: clinical diagnosis, the usual procedure in dermatological practice Time points: at 4 and 8 weeks after PDT Definitions: complete clinical clearance of a lesion (no visual evidence of persisting lesions on treated surface, no evidence of adherent scaling plaques on treated skin surface when palpated, lesions no longer perceptible to touch, slight pink or red foci might be visible at lesion sides) Safety Methods: 1. inspection of study lesions for tolerability, 2. recording of local reactions and adverse events (local reactions were always assumed to be related to study therapy. For adverse events, the investigator judged the relation to the study therapy) Time points: 1. at 1 day and 1 week after PDT (tolerability), 2. entire study duration (local reactions and adverse events) | |
| Funding | This study was supported by Photoamic. | |
| Notes | Percentages of participants with local skin reactions were given graphically. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 118): "Patients were randomly allocated to treatment." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was no stated. |
| Blinding of participants and personnel (performance bias) | Unclear risk | This was not stated. |
| Blinding of outcome assessment (detection bias) | Low risk | To keep the assessor blinded, 1 investigator performed the evaluation, and another investigator administered the treatments. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis was used. Lost participants were mentioned (9) but not by treatment group and the reasons were not given. |
| Selective reporting (reporting bias) | High risk | Adverse events were not always clearly reported by group, and the number of participants included in the safety analysis was not clear. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single‐centre, randomised, active‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: betulin‐based oleogel applied twice daily (N = 15) B: combination therapy with initial cryotherapy followed by betulin‐based oleogel twice daily (N = 15) Control intervention C: cryotherapy in the form of a spray coat method with liquid nitrogen (20 to 40 seconds) (N = 15) | |
| Outcomes | Outcomes of the trial 1) Complete clearing (= participant complete clearance) rates at 3 months 2) Therapy responders with > 75% of clearing of the lesions (= participant partial clearance) rates at 3 months 3) Histological analysis of biopsies before treatment and at the end of treatment 4) Minor adverse events (qualitative) Efficacy Methods: 1. quantitative assessment using documentation of visual and photographic evaluation in the case report forms, 2. punch biopsies from 4 participants out of the oleogel group, 2 participants out of the cryotherapy group and 2 participants out of the combination therapy group for evaluation of the degree of dysplasia, number of dyskeratoses and thickness of epidermis and stratum corneum Time points: 1. at 1, 2, and 3 months after the beginning of treatment, 2. before treatment and at the end of treatment (biopsy) Definitions: 1. responders [participants with complete (100 %) and with extensive (> 75 %) total clearing of the lesions], and 2. non‐responders (participants with disappearance of < 75 % of the lesions) Safety Methods: assessment of the subjective parameters itching and stinging by a questionnaire and grading as follows: 0 = absent, 1 = mild, 2 = moderate, and 3 = severe | |
| Funding | This study was supported by Birken GmbH. | |
| Notes | This study was a Phase II pilot study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation plan was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | This study was open. This study did not use placebo cream to conceal the allocation to cryotherapy only versus cryotherapy with betulin‐based oleogel. |
| Blinding of outcome assessment (detection bias) | High risk | This study was open. |
| Incomplete outcome data (attrition bias) | Unclear risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 1 dropout (the reason was reported), B: 1 dropout (the reason was reported) Control ‐ C: 1 dropout (the reason was reported) Comment: The effect of PP analysis is difficult to assess because the same number of participants was lost in each treatment group. |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, assessor‐blinded, vehicle‐controlled, intraindividual study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: aminolevulinic acid (ALA)‐photodynamic therapy (PDT) (N = 36 participants) Control intervention B: vehicle‐PDT (N = 36 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 (if complete response not achieved) Interval between treatments: 8 weeks Preparation of lesions: ‐‐ Cream concentration (%): 20% Application of cream: 3 applications onto lesion and a rim of 2 to 4 mm, air dry between applications Incubation with cream: 14 to 18 hours Type of light: blue light Light source: DUSA BLU‐417 Wavelength (nm): 417 Energy fluence (J/cm²): 2, 5, or 10 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ | |
| Outcomes | Outcomes of the trial 1) Clinical response of actinic keratosis lesions including completely cleared (= lesion complete response) rates for individual (at 8 weeks) and all (at 8 and 16 weeks) light doses 2) Number of participants with 0, 1, 2 (all) cleared lesions (= participant complete clearance) for individual (at 8 weeks) and all [at 8 (ALA and placebo) and 16 (ALA only) weeks] light doses 3) Application site reactions during (illumination) and after treatment reported per lesions 4) Clinical laboratory tests 5) Changes in pigmentation (cosmetic) per lesions 6) PpIX fluorescence Efficacy Time points: at baseline; immediately after PDT; at 24 and 72 hours; and at weeks 1, 4, 8, 9 (retreated participants), 12, and 16 Definitions: 1. complete response (completely cleared with no evidence of adherent scale on the surface of the treated skin when palpated), 2. partial response (≥ 50% reduction in lesion size), and 3. no response (< 50% reduction in lesion size) Safety Methods: 1. evaluation of objective changes in erythema, oedema, wheal, vesiculation, ulceration, haemorrhage, and necrosis on a graded scale (0: none; 1: minimal; 2: moderate; 3: severe), 2. subjective assessment of participant discomfort from pain, burning/stinging, and itching was graded (0: none; 1: minimal; 2: moderate; 3: severe), 3. standard hematologic and biochemical laboratory parameters, 4. reporting of adverse events Time points: 1. at baseline; immediately after PDT; at 24 hours; at 72 hours; and at weeks 1, 4, 8, 9 (retreated participants), 12, and 16, 2. at baseline and again at 1 week post‐treatment (laboratory tests) | |
| Funding | This study was supported by DUSA pharmaceuticals, Inc. | |
| Notes | Clinical response was dependant upon dose of light administered (5 or 10 J/cm² were more effective than 2 J/cm²). Visual detection of PpIX confirmed absence of cross contamination of treatment and placebo creams. Grade 1 (30/39 = 77%) lesions had better response than grade 2 (16/31 = 52%). PpIX fluorescence significantly correlated with clinical response (P < 0.001). Only data from 8 week's visit was used, because data from 16 weeks included participants with or without additional treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 97): "Each patient had a minimum of 4 target lesions with 2 lesions being randomised to ALA solution and 2 to vehicle." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | A non‐blinded investigator performed treatments. |
| Blinding of outcome assessment (detection bias) | Low risk | Different investigators were involved for the treatment and the analysis. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intraindividual study: Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 4 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, open (treatment duration), vehicle‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: 0.5% 5‐fluorouracil, once daily for 1 week (N = 47 participants) B: 0.5% 5‐fluorouracil, once daily for 2 weeks (N = 46 participants) C: 0.5% 5‐fluorouracil, once daily for 4 weeks (N = 45 participants) Control intervention D: Vehicle, once daily for 1, 2, or 4 weeks (pooling not clear ‐ see page 336 of the study) (N = 69 participants) | |
| Outcomes | Primary outcomes of the trial 1) Physician Global Assessment of Improvement (PGAI = Global improvement indices) 2) Per cent reduction of lesions (= mean percentage of reduction in lesion counts) 3) Absolute mean reduction in lesion counts Other outcomes of the trial 1) Proportion of participants achieving total clearance (= participant complete clearance) 2) Skin irritation (percentages of participants, severity, overtime) 3) Application and local skin reactions and minor adverse events (pooled data from 2 studies included in Carac product insert, i.e. Jorizzo 2002 and Weiss 2002) 4) Serious adverse events Efficacy Methods: 1. quantitative assessment using lesion counting, 2. qualitative assessment (PGAI mean score, +5 = total clearance and ‐4 = much worse) Time points: at baseline and 4 weeks post‐treatment Safety | |
| Funding | This study was supported by Dermik Laboratories. | |
| Notes | Data from this study were included in the Carac product insert. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 336): "Of the 207 randomised participants,..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | This study was double‐blind (treatment vs placebo) and open (treatment duration). No placebo cream was used to conceal the treatment duration. |
| Blinding of outcome assessment (detection bias) | High risk | This study was double‐blind (treatment vs placebo) and open (treatment duration). No placebo cream was used to conceal the treatment duration. |
| Incomplete outcome data (attrition bias) | High risk | It was unclear which type of analysis was used. Intervention ‐ A: 2 dropouts, B: 1 dropout, C: 1 dropout Control ‐ D: 0 dropouts (the reasons were not reported) |
| Selective reporting (reporting bias) | High risk | Values for absolute reductions in lesion numbers, standard deviations on mean percentages, and PGAI were not given. Details on local skin reactions and adverse events, other than facial irritation, was not reported in the published version of the study. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel group study. Start date: October 2001 End date: February 2002 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: topical 0.5% 5‐fluorouracil, once daily for 7 days. At 4 weeks post‐treatment, residual lesions treated with cryotherapy. (N = 72 participants) Control intervention B: vehicle, once daily for 7 days. At 4 weeks post‐treatment, residual lesions treated with cryotherapy (N = 72 participants) | |
| Outcomes | Primary outcome of the trial 1) Absolute and per cent of mean reduction in lesion counts at 4 weeks (topical only) and 6 months (topical + cryotherapy) Other outcomes of the trial 1) Participant complete clearance rates at 4 weeks and 6 months 2) Application site reactions (also reported in Jorizzo 2006) 3) Eye irritation (= minor adverse events) Efficacy Methods: quantitative assessment using the counting of visible or palpable lesions, or both, by the same evaluator Time points: before initial treatment and at the 4‐week and 6‐month follow‐up visits Safety | |
| Funding | This study was supported by Dermik Laboratories. | |
| Notes | This study (interim analysis) is part of a 3‐cycle study published in Jorizzo 2006, which was also included in this review. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation schedule was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. All study personnel, and participants were blinded to actual treatment assignment. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. Investigators were blinded to actual treatment assignment. |
| Incomplete outcome data (attrition bias) | Unclear risk | Modified Intention‐to‐treat (ITT) analysis was used (i.e. participants at 4 week evaluation based on information, efficacy: 142, safety: 143‐received one treatment). Intervention ‐ A: 2 dropouts (the reasons were reported) Control ‐ B: 7 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 3 topical/cryosurgery cycles: topical 0.5% 5‐fluorouracil, once daily for 7 days. At 4 weeks post‐treatment, residual lesions were treated with cryosurgery (N = 72 participants) Control intervention B: 3 topical/cryosurgery cycles: topical vehicle, once daily for 7 days. At 4 weeks post‐treatment, residual lesions were treated with cryosurgery (N = 70 participants) | |
| Outcomes | Primary outcomes of the trial 1) Mean lesion counts at baseline and different treatment cycles (transformed to mean reduction in lesion counts) 2) Mean percentage of reduction in lesion counts 3) Participant complete clearance rates at 3 topical/cryosurgery cycles (before cryosurgery) 4) New involvement of actinic keratosis lesions Other outcomes of the trial 1) Application site reactions (severe) 2) Treatment‐related adverse events (= minor adverse events) 3) Serious adverse events including basal and squamous cell carcinoma Efficacy Methods: quantitative assessment using the counting of visible or palpable actinic keratoses, or both, by the same evaluator Time points: before initial treatment and at the follow‐up visits Definitions: 1. actinic keratosis reduction (number of lesions present at the 4‐week follow‐up visit for each topical/cryosurgery cycle minus the number of baseline lesions for that cycle), 2. clearance (complete lack of lesions in the treatment area at the 4‐week follow‐up visit for each topical/cryosurgery cycle), 3. new involvement [presence of new lesions in the treatment area at the start of topical/cryosurgery cycle 2 (week 26) or 3 (week 52)] Safety Methods: 1. monitoring the incidence, onset, duration, and severity of adverse events observed, 2. participants reporting the occurrence of any adverse event, and study personnel questioned participants during study visits to monitor safety, 3. all adverse events were categorised by the investigator as serious or not Definitons: serious adverse events [adverse events resulting in death, life threatening; required inpatient hospitalisation or prolongation of existing hospitalisation, resulted in persistent or significant disability or incapacity, those described as important medical events (e.g. diagnosis cancer during the course of treatment)] | |
| Funding | This study was supported by Dermik Laboratories. | |
| Notes | More participants in the vehicle group had cryosurgery. New actinic keratosis lesions were observed over time in both groups, but a lower percentage of participants in the 5‐fluorouracil group than the vehicle group was obtained. The number of participants based on ITT analyses were used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation schedule was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. All study personnel and participants were blinded to the actual treatment assignment. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. Investigators were blinded to the actual treatment assignment. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was stated. Intervention ‐ A: 5 dropouts (the reasons were reported) Control ‐ B: 12 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Standard deviations on percentage of mean reduction in lesion counts were not reported. |
| Other bias | High risk | There was some inconsistency in the numbers in the manuscript as well as with the previous paper, Jorizzo 2004, for efficacy outcomes. |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% imiquimod, once per day, 3 days per week for 4 weeks on, 4 weeks off, 1 or 2 courses (N = 123 participants) Control intervention B: vehicle: once per day, 3 days per week for 4 weeks on, 4 weeks off, 1 or 2 courses (N = 123 participants) | |
| Outcomes | Primary outcome of the trial 1) Recurrence at 1 year Other outcomes of the trial 1) Participant complete and partial (> 75%) clearance rates after course 1 or overall 2) Individual lesion clearance (= lesion complete response) rates after course 1 or overall 3) Minor adverse events (qualitative) 4) Clinical laboratory tests Efficacy Methods: quantitative assessment using lesion counting Time points: at week 8, 16, and 1 year follow‐up (relapse for participants achieving complete clearance) Definitions: 1. complete clearance rate (proportion of participants who cleared all lesions in the treatment area), 2. partial clearance rate (proportion of participants with at least a 75% reduction in baseline lesions) Methods: 1. monitoring for adverse events and local skin reactions, 2. clinical laboratory tests (hematology and chemistry blood tests, and urinalysis), 3. culture of suggested skin infections | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | 1‐year recurrence rate of 39% and 57% were respectively found for imiquimod and vehicle. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 266): "Randomised patients applied imiquimod or vehicle cream (randomised 1:1)..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | High risk | Authors reported that blinded investigators may have been biased toward participants treated with imiquimod identified by treatment site reactions. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 4 dropouts (the reasons were not reported) Control ‐ B: 3 dropouts (the reasons were not reported) |
| Selective reporting (reporting bias) | High risk | Statistically significantly more application site reactions, local skin reactions, and adverse events for imiquimod were reported but not all the numbers supporting it were reported. |
| Other bias | High risk | There was no demographic description. |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel group study. Start date: May 2009 End date: February 2010 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: cryotherapy followed by 3.75% imiquimod, daily for 2 weeks on, 2 weeks off, 2 weeks on (N = 126 participants) B: no cryotherapy followed by 3.75% imiquimod, daily for 2 weeks on, 2 weeks off, 2 weeks on (N = 126 participants) Control interventions C: cryotherapy followed by placebo, daily for 2 weeks on, 2 weeks off, 2 weeks on (N =121 participants) D: no cryotherapy followed by placebo, daily for 2 weeks on, 2 weeks off, 2 weeks on (N =121 participants) Randomisation was performed for imiquimod or placebo treatment, but the method used to select which lesions were treated with or without cryotherapy was not specified. At least 5 lesions were not treated with cryosurgery, and 5 to 14 actinic keratoses were treated with cryosurgery. | |
| Outcomes | Primary outcome of the trial 1) Mean and median per cent changes from baseline for all lesions (= mean percentage of reduction in lesion counts) at week 26 Secondary outcomes of the trial 1) Participant complete clearance rates for all lesions at week 26 2) Local skin reactions (severe) Other outcomes of the trial 1) Application site reactions including irritation 2) Treatment‐related adverse events (= minor adverse events) 3) Minor adverse events 4) Serious adverse events including basal and squamous cell carcinoma 5) Cosmetic outcomes (photodamage) 6) Rest periods 7) Participant satisfaction Efficacy Methods: quantitative assessment using lesion counting and mapping on a facial diagram, including lesions that were initially treated with cryosurgery Time points: 1. at each visit (counting), 2. baseline and end of study at week 26 (mapping) Safety Methods: investigator assessment of local skin reactions (erythema, oedema, weeping/exudate, flaking/scaling/dryness, scabbing/crusting, and erosion/ulceration) graded as none, mild, moderate, or severe and summarised by the most intense score for each reaction, 2. recording of adverse events coded using MedORA® (Medical Dictionary for Regulatory Activities), Version 12.0 (treatment‐emergent AEs were summarised for each treatment group by preferred term, intensity, and investigator assessment of relationship to study cream), 3. serious adverse events and discontinuations due to adverse events Time points: at each clinic visit Cosmetic Methods: assessment of facial photodamage by the investigator using previously published 5‐point scales that rated fine lines, mottled pigmentation, tactile roughness, sallowness, and global photoageing Time points: at baseline (prior to cryosurgery) and at weeks 10, 14, 20, and 26/end of study | |
| Funding | This study was supported by Graceway Pharmaceuticals, LLC. | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated allocation sequence was generated. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | The study was double‐blind for imiquimod versus placebo (subject, caregiver, investigator). |
| Blinding of outcome assessment (detection bias) | Low risk | The study was double‐blind for imiquimod versus placebo (outcomes assessor). |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A & B: 14 dropouts (the reasons were reported) Controls ‐ C & D: 10 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | All outcomes from the protocol were reported, and the data were consistent in conference abstract, published paper, and the study results section of the protocol (NCT00894647) in clinicaltrials.gov. Additonal outcomes were reported in the published report. The data for "no cryotherapy" was not given for all outcomes. Cosmetic outcomes were reported separately for each criteria but were not reported as a global assessment. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, placebo‐controlled, active‐controlled, assessor‐blinded, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: 0.1% adapalene gel, once daily for 4 weeks; twice daily from 4 weeks to 36 weeks (N = 30 participants) B: 0.3% adapalene gel, once daily for 4 weeks; twice daily from 4 weeks to 36 weeks (N = 30 participants) Control intervention C: placebo, once daily for 4 weeks; twice daily from 4 weeks to 36 weeks (N = 30 participants) | |
| Outcomes | Primary outcomes of the trial 1) Mean reduction/changes of lesion counts 2) Morphological changes of target lesions Secondary outcomes of the trial 1) Physician global assessment improvement (PGAI) from worse to clear (= Global Improvement Indices) 2) Histological analysis of biopsies before and after treatment Other outcomes of the trial 1) Tolerability (= local skin reactions) 2) Minor adverse events 3) Serious adverse events 4) Photoaging characteristic improvement (cosmetic outcome) Efficacy Methods: 1. quantitative assessment using total lesion counts, 2. assessment of morphologic changes in target actinic keratoses (induration, scaling, and erythema evaluated on a scale of 0 [none] to 3 [severe]), 3. qualitative assessment of improvement (PGAI) as clear, marked, moderate, slight, no change, or worse, 4. biopsy specimens evaluated in a blinded manner by a board‐certified dermatopathologist at 1 centre (University of Michigan, Ann Arbor, Michigan) Time points: at week 2, 4, 12, 18, 24, 30, and 36 Cosmetic Methods: standardised photographs were taken of 45 participants by a professional photographer at 1 centre (University of Michigan). [The photographs were evaluated retrospectively in a randomised, blinded fashion to assess the effects of the 3 treatments on photoageing characteristics (mottled hyperpigmentation, fine wrinkles, coarse wrinkles, rosy glow‐healthy pink complexion, and global photoageing severity). Each parameter was graded for improvement on a scale of 0 to 6. If there was no difference or worsening between before‐ and after‐treatment photographs, then a score of 0 was given] Time points: at baseline and after 3, 6, and 9 months of treatment | |
| Funding | This study was supported by Galderma Corporation, Texas, US. | |
| Notes | Histology on 36 participants showed no significant difference between treatment groups. There was no follow‐up period. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate randomisation sequence in blocks of 9 using a unique 4‐digit number was generated by a computer program. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Unclear risk | This was not stated. |
| Blinding of outcome assessment (detection bias) | Low risk | Investigators were blinded. |
| Incomplete outcome data (attrition bias) | Unclear risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 2 dropouts (the reasons were not reported), B: 3 dropouts (the reasons were reported) Control ‐ C: 2 dropouts (the reasons were not completely reported) |
| Selective reporting (reporting bias) | High risk | Authors pooled together selected PGAI data, i.e. for clear, marked, moderate (but not slight) improvement to reach statistically significant difference. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, open, active‐controlled, intraindividual study. Start date: January 2005 End date: February 2006 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 121 participants) Control intervention B: cryotherapy: double freeze/thaw (1 to 2 mm frozen rim outside marked outline of lesion), 20 seconds (1 or 2 treatments with a 12‐week interval) (N = 121 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 Interval between treatments: 12 weeks Preparation of lesions: gentle scraping Cream concentration (%): 16 % Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: 3 hours Type of light: red light LED Light source: Aktilite CL128 Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ | |
| Outcomes | Primary outcome of the trial 1) Lesion complete response rates for baseline lesions at week 24 Secondary outcomes of the trial 1) Cosmetic outcome assessed by investigator and participants 2) Participant preference Other outcomes of the trial 1) Mean percentage of reduction in lesion counts 2) Treatment‐related adverse events (= minor adverse events) 3) Participants reporting at least one adverse event 4) Minor adverse events 5) Serious adverse events including squamous cell carcinoma 6) Observation of Bowen's disease and new actinic keratoses Efficacy Methods: quantitative assessment using lesion counting [efficacy evaluations only included lesions present at baseline (i.e. lesions appearing after baseline, if any, were to be reported as an adverse event)] Time points: at baseline and weeks 12 and 24 Definitions for lesion response: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance) Safety Methods: spontaneous reporting of adverse events by the participant or elicited following non‐leading questioning (severity, duration and need for additional therapy) Time points: at each follow‐up visit Definitions for adverse events: 1. phototoxic reaction (any observed erythema, oedema, itching, pain, etc), 2. cryotherapy reaction (any observed blisters, infection, etc) Cosmetic Methods: 1. investigator assessment of cosmetic outcome for all lesions with complete response, 2. participant assessment of cosmetic outcome Time points: at week 24 Definitions for investigator assessment: 1. excellent (only slight occurrence of redness or change in pigmentation), 2. good (moderate redness or change in pigmentation), 3. fair (slight to moderate scarring, atrophy or induration), 4. poor (extensive scarring, atrophy, or induration) Definitions for participant assessment on a 5‐point scale: –2 (cryotherapy a lot better than MALPDT) to 2 (MAL‐PDT a lot better than cryotherapy) | |
| Funding | This study was supported by Galderma. | |
| Notes | A participant questionnaire showed that participants preferred MAL‐PDT over cryotherapy for all questions except for effectiveness of treatment. A sample size calculation was provided. Intention‐to‐treat values were used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 995) : "At the baseline visit, eligible patients received treatment with PDT using MAL... and conventional cryotherapy, randomly allocated to alternate sides of the body." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | There was no blinding. This study was open because physically distinct treatments were compared. |
| Blinding of outcome assessment (detection bias) | High risk | There was no blinding. This study was open because physically distinct treatments were compared. |
| Incomplete outcome data (attrition bias) | High risk | Both per‐protocol (PP) and intention‐to‐treat (ITT) analyses were used. Intraindividual study: Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 4 dropouts (the reasons were reported) Comment: ITT and PP populations were respectively 121 and 106. Thus, 11 participants were not accounted for, and it was not always clear which analysis type was used for the different outcomes reported. |
| Selective reporting (reporting bias) | High risk | The standard deviation associated with the mean percentage of reduction in lesion counts were not provided. |
| Other bias | High risk | Participant's assessment of cosmetic outcomes has negative value if cryotherapy is better and positive value if MAL‐PDT is better. This could influence the participant perception. |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: August 2001 End date: August 2002 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% imiquimod applied 3 times per week for 16 weeks (N = 242 participants) Control intervention B: vehicle cream applied 3 times per week for 16 weeks (N = 250 participants). Applied to entire treatment area at same time of day (before sleeping). Cream to remain in place for approximately 8 hours. Rest periods allowed at discretion of investigator, but did not alter length of 16‐week treatment | |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates for all lesions at 8 weeks post‐treatment Secondary outcome of the trial 1) Participant partial (> 75%) clearance rates for all lesions at 8 weeks post‐treatment Other outcomes of the trial 1) Median percentage reduction of baseline lesions at 8 weeks post‐treatment 2) Participants experiencing at least 1 adverse event 4) Local skin reactions 5) Frequency and severity of adverse events (= minor adverse events) 6) Serious adverse events 7) Rest periods 8) Skin quality (cosmetic outcome) Efficacy Methods: quantitative assessment using lesion counting (baseline and new lesions) Time points: at weeks 4, 8, and 16, and post‐treatment week 8 Definitions: 1. complete clearance rate (proportion of participants at the 8‐week post‐treatment visit with no clinically‐visible lesions in the treatment area), 2. partial clearance rate (proportion of participants at the 8‐week post‐treatment visit with at least a 75% reduction in the number of baseline lesions in the treatment area) Safety Methods: 1. reviewing concomitant medication use, 2. assessing the incidence and severity of adverse events spontaneously reported, 3. assessing of local skin reactions (erythema, edema, erosion/ulceration, scabbing/crusting, weeping/exudation, vesicles, and flaking/scaling/dryness) rated as 0 = none, 1= mild, 2 = moderate, and 3 = severe by investigator (reactions within the treatment area that were not assessed as local skin reactions were reported as adverse events) Time points: at weeks 1, 2, 4, 6, 8, 10, 12, 14, and 16, and post‐treatment weeks 4 and 8 Cosmetic Methods: investigator‐performed (visual, clinical, and tactile examinations) skin quality assessments including skin surface (roughness/dryness/scaliness), hyperpigmentation, hypopigmentation, mottled or irregular pigmentation (hyperpigmentation and hypopigmentation), degree of scarring, and degree of atrophy area rated as 0 = none; 1 = mild; 2 = moderate; and 3 = severe | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | 2 phase III studies were included. Clearance rates increased with intensity of erythema. Increase in lesion counts (new or subclinical lesions) was higher in imiquimod group at any point during the treatment period. Long‐term clinical outcomes were presented in Lee 2005. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate randomisation was achieved by a computer‐generated randomisation schedule. |
| Allocation concealment (selection bias) | Low risk | Participants were assigned the next sequential participant study number and the corresponding study cream. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 15 dropouts (the reasons were reported) Control ‐ B: 15 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Skin quality rating was not reported for placebo. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, open‐label, active‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 3% diclofenac sodium in 2.5% hyaluronic acid, once daily for 12 weeks (N = 24 participants) Control intervention B: 5% imiquimod, 3 times/week for 12 weeks (N = 25 participants) | |
| Outcomes | Outcomes of the trial 1) Investigator (IGII) and participant (PGII) global improvement indices at the end of treatment 2) Lesion severity index 3) Local skin reactions 4) Participants experiencing at least 1 treatment‐related adverse event 5) Clinical laboratory tests Efficacy Methods: 1. quantitative assessment using lesion counting, 2. severity of actinic keratoses at baseline, 3. qualitative assessment (GII) by the investigator and participant Time points: at baseline and monthly up to 1 year follow‐up Definitions for global improvement indices on a 7‐point scale: ‐2 (significantly worse), ‐1 (slightly worse), 0 (no change), 1 (slightly improved), 2 (moderately improved), 3 (significantly improved), and 4 (completely improved) Definitions for baseline severity index: 0 (no lesions visible), 1 (clearly visible lesions), 2 (many visible, small, moderately‐thick lesions, or a few large, thick, rough scaly lesions), 3 (many thick, hypertrophic lesions, which are clearly visible and palpable with well‐defined borders) Safety Methods: 1. assessment of tolerability by investigator (erythema, itching, dry skin, and scaling), 2. clinical examination, 3. reporting of adverse events, 4. routine laboratory tests (complete blood cell counts, urine analysis, and fasting chemistry) Time points: 1. monthly up to 1 year follow‐up, 2. before and after treatment (laboratory tests) | |
| Funding | ‐ | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 159): "Patients were randomly assigned to treatment with DFS or IMI." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | The study was open. The difference in dosing regimen frequency of the 2 topical treatments was not concealed by the use of double‐dummy technique. |
| Blinding of outcome assessment (detection bias) | High risk | The study was open. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropout Control ‐ B: 0 dropout |
| Selective reporting (reporting bias) | High risk | Participant partial (> 75%) clearance was mentioned, but no data were reported. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single‐centre, randomised, active‐controlled, parallel‐group study. Start date: August 2004 End date: February 2005 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 0.25 g of 5% imiquimod cream 3 times per week for 8 hours each over a span of 4 weeks, 1 or 2 treatments with a 4‐week rest period (N = 26 participants) Control interventions B: 5% 5‐fluorouracil cream twice daily for 4 weeks (N= 24 participants) C: cryosurgery using bursts of 20 to 40 seconds, 1 or 2 treatments with a 2‐week rest period (N = 25 participants) | |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates at test of cure and 12 months after the end of treatment Other outcomes of the trial 1) Participant histological clearance rates at test of cure 2) Negative predictive value, i.e. ratio between histological and clinical clearance 3) Serious adverse events 4) Global cosmetic outcome assessments by participant and investigator (presented graphically) 5) Skin quality 6) Recurrence (individual lesion and field) at 12 months after the end of treatment Efficacy Methods: 1. quantitative assessment using precise documentation of location of each lesion on body grid charts with raster and photographs, 2. a 4 mm punch biopsy specimen obtained from 1 of the selected lesions and evaluated independently by 2 expert dermatopathologists Time points: at baseline, test of cure (6, 4, and 8 weeks after last treatment for cryotherapy, 5‐fluorouracil and imiquimod, respectively), and 1 year after the end of treatment (recurrence) Definitions: 1. complete clearance (absence of clinically detectable lesions in treated skin regions), 2. recurrence (for cryotherapy: recurrence of initially cleared lesions determined and expressed as percentage of participants with lesion recurrence in relation to all treated participants; for 5‐fluorouracil and imiquimod: total recurrences within the cleared cancer field determined and expressed as percentage of participants with field recurrence in relation to all treated participants, new lesions considered; missing values counted as recurrence) Cosmetic Methods: global assessment by investigator and participant based on the amount of scarring, atrophy, or indurations and on pigment change within the treatment area by comparison to adjacent, untreated skin Time points: at test of cure Definitions: 1. excellent (treated skin was indistinguishable from normal skin), 2. good (moderate redness or change in pigmentation), 3. fair (moderate redness or change in pigmentation and slight to moderate scarring, atrophy, or induration), 4. poor (extensive scarring, atrophy, or indurations) | |
| Funding | ‐ | |
| Notes | 1 week of rest period for imiquimod and 5‐fluorouracil during treatment in case of acute inflammation was allowed. The data on non‐recurrence at 1 year follow‐up were as follows: 86% (19/22) for imiquimod, 57% (13/23) for 5‐fluorouracil, and 41% (7/17) for cryotherapy. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 35): "Each patient was randomly assigned to one of [the] equally sized treatment groups (cryosurgery, 5‐FU, or IMIQ) by 'on‐call randomisation' provided by a specialised external company." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Low risk | Randomisation involving "on call" randomisation by an external company was used to conceal the allocation. (See previous quote.) |
| Blinding of participants and personnel (performance bias) | High risk | This was not stated, but the study compared physically distinct interventions and topical treatments with different application regimens. |
| Blinding of outcome assessment (detection bias) | High risk | There was no statement about assessor‐blinding. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropouts at test of cure Control ‐ B: 0 dropouts at test of cure, C: 0 dropouts at test of cure |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, active‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 10% masoprocol applied topically twice daily for 4 weeks (N = 27 participants) Control intervention B: 5% 5‐fluorouracil applied topically twice daily for 4 weeks (N = 30 participants) | |
| Outcomes | Outcomes of the trial 1) Investigator global assessment (= global improvement indices) at week 8 3) Number of events for adverse events and their severity 4) Percentage of participants experiencing adverse events represented graphically in function of the event severity (= minor adverse events) 5) Percentage of participants who discontinued treatment 6) Mean max pain score Efficacy Methods: 1. quantitative assessment using lesions counting and rating, 2. qualitative assessment (global assessment) Time points: at baseline; days 7, 14, 21, and 28 (the last day of treatment); at day 42 and day 56; and at 1 year and 2 years (recurrence) Definitions for lesion rating: 1. mild (thin actinic keratoses, visible and palpable), 2. moderate (moderately thick actinic keratoses, easily seen and palpated), 3. severe (thick and florid actinic keratoses with distinct borders) Definitions for Global assessment: 1. cured (clear of palpable lesions, slight residual erythema remaining), 2. marked improvement (majority of lesions absent and scales of remaining lesions barely palpable), 3. moderate improvement (many lesions absent and scales decreased in thickness), 4. slight improvement (some lesions cleared, some decreased in scale, but many lesions remain), 5. no change (slightly worse, more or rougher larger lesions remain), 6. much worse (significantly more lesions or majority of lesions rougher, larger, or both) Safety Methods: 1. recording of all adverse experiences noted by participants and questioning of participants; 2. evaluation by investigator on key adverse reactions based on the appearance of the lesions, i.e. degree of erythema, necrosis, ulceration, and erosion in the lesions, and the degree of erythema and contact dermatitis in tissue surrounding and scored as 0 = none, 1 = mild, 2 = moderate, or 3 = severe; 3. intensity of the pain rated by participant on a 9‐point scale ranging from 0 = no pain to 8 = severe pain Time points: at each visit | |
| Funding | This study was supported by Reed & Carnrick Pharmaceuticals, A Division of Block Drug Company, Inc. | |
| Notes | The numbers for per‐protocol analysis were used for meta‐analyses of mean reduction in lesion counts, and intention‐to‐treat numbers were used for meta‐analysis of global improvement indices. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 162): "Patients were randomised in a double‐blind fashion to twice‐daily treatment with either 10% masoprocol in an emollient cream base or 5% 5‐fluorouracil in a vanishing cream base." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded, and treatments were given with the same application regiment. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Unclear risk | Per‐protocol analysis was used. Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 4 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | High risk | A significantly higher percentage (65.5%) of participants treated with 5‐fluorouracil failed to complete 28 days of treatment than participants treated with masoprocol (16%). |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: September 2001 End date: August 2002 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% imiquimod cream, once per day, twice weekly for 16 weeks or less (N = 215 participants) Control intervention B: vehicle, once per day, twice weekly for 16 weeks or less (N = 221 participants) | |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates (all lesions) at 8 weeks post‐treatment Secondary outcome of the trial 1) Participant partial (> 75% of baseline lesions) clearance rates at 8 weeks post‐treatment Other outcomes of the trial 1) Median per cent reduction in baseline lesions at 8 weeks post‐treatment 2) Clinical laboratory tests 3) Participants experiencing at least 1 adverse event 4) Application site reactions 6) Serious adverse events 7) Skin quality (cosmetic) 8) Increase in the number of lesions during the study Efficacy Methods: quantitative assessment using clinical counting Time points: at baseline; weeks 1, 2, 4, 6, 8, 10, 12, 16 (end of treatment), 20, and 24 Definitions: 1. complete clearance rate (proportion of participants at the 8‐week post‐treatment visit with a count of 0 clinically‐visible lesions in the treatment area), 2. partial clearance rate (proportion of participants at the 8‐week post‐treatment visit with at least a 75% reduction in the number of lesions counted at baseline in the treatment area) Safety Methods: 1. clinical laboratory tests [hematology (haemoglobin, hematocrit, reel blood cell, white blood cell, and platelet counts), serum chemistry (random glucose, blood urea nitrogen, creatinine, total bilirubin, serum glutamic oxaloacetic transaminase, serum glutamic pyruvic transaminase, lactate dehydrogenase, alkaline phosphatase, potassium, sodium, calcium, chloride, total protein, albumin, phosphorous, and cholesterol) and urine analysis for colour/appearance, specific gravity, pH, protein, glucose, and ketones and a microscopic examination]; 2. vital sign measurements and physical examinations; 3. photography; 4. recording of adverse events; 5. assessment of local skin reactions (erythema, oedema, erosion/ulceration, scabbing/crusting, weeping/exudate, vesicles, or flaking/scaling/dryness) rated by a study investigator on a scale of 0 to 3, where 0 = none, 1 = mild, 2 = moderate, and 3 = severe; and 6. recording of concomitant medication use Time points: 1. at each visit, 2. pre‐study visit and end of treatment at week 16 (physical exam and laboratory tests) Definitions for spontaneous participant‐reported adverse events: 1. mild (participant was aware of the signs and symptoms, but the signs and symptoms were easily tolerated), 2. moderate (the signs and symptoms were sufficient to restrict, but not prevent, usual daily activity), and 3. severe (the participant was unable to perform usual daily activity) Methods: assessment of skin quality by visual, clinical, and tactile examinations of the treatment area by investigator [skin surface, hyperpigmentation, hypopigmentation, mottled or irregular pigmentation (both hyperpigmentation and hypopigmentation), degree of scarring, and atrophy on a scale of 0 to 3, where 0 = none, 1 = mild, 2 = moderate, and 3 = severe] Time points: at treatment initiation and 8‐week post‐treatment visit | |
| Funding | This study was supported by 3M Pharmaceuticals, St Paul, Minnesota. | |
| Notes | An increase in lesion counts was observed during treatment. A sample size calculation was provided. Pooled data from 2 phase III studies were presented in Aldara product insert. Long‐term clinical outcomes were presented in Lee 2005. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation schedule was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | High risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 9 dropouts (the reasons were not all reported) Control ‐ B: 11 dropouts (the reasons were not all reported) |
| Selective reporting (reporting bias) | High risk | As a similar study (Szeimies 2004) was also supported by Graceway/3M Pharmaceuticals, not all skin quality outcomes were reported. All outcomes presented in the product insert were reported in the published paper. Another Graceway clinical trial on the arms and hands (NCT00115154) was not published or included in the product insert. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, assessor‐blinded, active‐controlled, intraindividual study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 0.5% 5‐fluorouracil on either side of face, once daily for 4 weeks. Sunscreen/moisturiser was provided when needed (N = 21 participants). Control intervention B: 5% 5‐fluorouracil on either side of face, twice daily for 4 weeks. Sunscreen/moisturiser was provided when needed (N = 21 participants). | |
| Outcomes | Primary outcomes of the trial 1) Absolute and per cent of mean reduction in lesion counts at week 8 2) Total clearance (= participant complete clearance) rates at week 8 Other outcomes of the trial 1) Facial irritation (= skin irritation) 2) Eye irritation (= minor adverse events) 3) Serious adverse events including basal cell carcinoma 4) Participant preference Efficacy Methods: quantitative assessment using counting of palpable or visible (to the unaided eye) lesions by a designated blinded evaluator Time points: during screening period and at 4 weeks post‐treatment Safety Methods: 1. participant‐reported adverse events, 2. photography and evaluation of facial irritation (erythema, edema, dryness, pain, erosion, burning, pruritus, and other signs) on a scale from 0 to 3 (0 = none, 1 = mild, 2 = moderate, 3 = severe) by the same blinded individual and recording of days of onset and resolution (adverse events that occurred on the head were included in the assessment of facial irritation.) Time points: at baseline and weekly throughout the 4‐week treatment (twice weekly for facial irritation) and the post‐treatment periods | |
| Funding | ‐ | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation list was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | Participants and clinic staff were not blinded to the difference in dosing regimens (once versus twice daily). |
| Blinding of outcome assessment (detection bias) | Low risk | This study was evaluator blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | High risk | Only estimates on clearance rates were provided, i.e. exact values were not given and standard deviations for absolute and per cent mean values were not provided. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single‐centre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: September 1994 End date: January 1996 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 3% diclofenac in 2.5% hyaluronic acid gel, applied to single keratosis twice daily for 8 to 24 weeks. Sunscreen was applied after morning application (N = 65 participants). Control intervention B: 2.5% hyaluronic acid gel alone, applied to single keratosis twice daily for 8 to 24 weeks. Sunscreen was applied after morning application (N = 65 participants). | |
| Outcomes | Primary outcome of the trial 1) Response to treatment (including participant complete response) rates at end of treatment Other outcomes of the trial 1) Local adverse reactions 2) Serious adverse events Safety Methods: diary recording of any adverse effects and any change in use of concomitant medications Time points: at 8, 16, and 24 weeks | |
| Funding | This study was supported by Hyal Pharmaceutical Australia Ltd. | |
| Notes | There was no follow‐up period. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A permuted block randomisation design of size 10 was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | The participants, the investigator, and the data managers were kept "blind" as to the treatment until all assessments and data entry had been completed. |
| Blinding of outcome assessment (detection bias) | Low risk | The participants, the investigator, and the data managers were kept "blind" as to the treatment until all assessments and data entry had been completed. |
| Incomplete outcome data (attrition bias) | High risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 31 participants did not complete the 24‐week treatment (the reasons were reported) Control ‐ B: 16 participants did not complete the 24‐week treatment (the reasons were reported), and 29 completed. There was no information given for 20 participants. There were also inconsistency between data presented in a table and the description in the text. |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | High risk | End of treatment varied as participants ceased treatment at varying times. 34% (diclofenac) and 20% (hyaluronic acid) of participants ceased the treatment before 8 weeks. Between 8 and 16 weeks, 11% (diclofenac) and 5% (hyaluronic acid) of participants ceased the treatment. Between 16 and 24 weeks, 55% (diclofenac) and 75% (hyaluronic acid) of participants ceased the treatment. |
| Methods | This was a randomised, double‐blind, active‐controlled, intraindividual study. Start date: January 1988 End date: June 1988 | |
| Participants | Inclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 0.05% (0.5 g) Ro14‐9706 cream (arotinoid methyl sulfone) applied to 1 side of the face twice daily (N = 26 participants) Control intervention B: 0.05% tretinoin cream applied to 1 side of the face twice daily (N = 26 participants) | |
| Outcomes | Outcomes of the trial 1) Participant overall response (= global improvement indices) rates at 16 weeks 2) Mean per cent decrease in the number of lesions (= mean percentage of reduction in lesion counts) at 16 weeks 3) Clinical laboratory tests 4) Tolerability (erythema and scaling) scoring 5) Increase of lesions during treatment 6) Rest periods Efficacy Methods: 1. quantitative assessment using lesion counting by 2 independent investigators, 2. qualitative assessment using photography of the treated areas Time points: 1. at the beginning of the study and weekly intervals (counting), 2. at baseline and after 4, 8, 12, and 16 weeks (photography) Definitions: healed or completely cleared lesion (the site had been replaced by normal, smooth, hypopigmented or hyperpigmented skin, and lesion not palpable) Definitions for overall response: 1. worsening (increase in the number of lesions), 2. no response (no change or less than 50% reduction in the total number of lesions), 3. partial response (reduction greater than 50%, but less than 100%, in the number of lesions), 4. complete response (total clearing of lesions) Safety Methods: 1. assessment of local tolerability (erythema and scaling) on a scale (in case of severe reactions, therapy was interrupted until the inflammation had disappeared), 2. routine laboratory (hematologic and biochemical) tests Time points: 1. weekly, 2. before and after treatment (laboratory tests) Definitions for tolerability scale: 0 (none), 1 (mild, minimal), 2 (moderate, more intense), and 3 (severe, very intense erythema, and scaling with exudation) | |
| Funding | This study was supported by La Roche Ltd. | |
| Notes | Ro 14‐9706 had better tolerability. An initial increase in the number of visible actinic keratoses with tretinoin was observed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 448): "The study was randomised, double‐blind, with each agent applied to opposite sides of the patient's face at a 0.05% concentration, for a period of 16 weeks." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | The assessment was performed by 2 independent investigators. |
| Incomplete outcome data (attrition bias) | Unclear risk | Per‐protocol (PP) analysis was used. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was not reported) Control ‐ B: 1 dropout (the reason was not reported) |
| Selective reporting (reporting bias) | Low risk | Negative (tolerability) data for the sponsored product were reported. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single centre, randomised, double‐blind, active‐controlled, intraindividual study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: aminolevulinic acid (ALA)‐photodynamic therapy (PDT) (N = 16 participants) Control intervention B: methyl aminolevulinate (MAL)‐PDT (N = 16 participants) There was a 2‐week interval between the 2 treatments (right versus left scalp). Characteristics of PDT intervention: Type of treatment: field‐directed treatment Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: hyperkeratotic lesions were treated with white paraffin gel to remove any keratotic debris Cream concentration (%): 20% Application of cream: visible layer Incubation with cream: occlusive dressing over cream for 3 (MAL) or 5 (ALA) hours Type of light: red light Light source: Waldmann PDT lamp MSR 1200 Wavelength (nm): 580‐740 Energy fluence (J/cm²): 50 Intensities (mW/cm²): 50 Exposure time: 16 minutes 40 seconds | |
| Outcomes | Outcomes of the trial 1) Field complete clearance (= participant complete clearance) rates at 1 month post‐treatment 2) Mean number of lesions at baseline and at 1 month post‐treatment 3) Mean reduction in lesion counts at 1 month post‐treatment 4) Minor adverse events (qualitative) 5) Visual analogue score (VAS) for pain 6) Duration of discomfort 7) Participant preference Efficacy Methods: 1. grading of PpIX fluorescence on a scale of 1 to 3 using a Wood's light (1 = light ⁄ pale; 2 = moderate; and 3 = strong), 2. clinical response (clear, improved, or no response, and number of residual palpable lesions) assessed by an investigator not involved in safety assessment Time points: 1. before treatment (fluorescence), 2. at baseline and 1 month post‐treatment Safety Methods: 1. assessment of pain using a VAS (1 to 100 mm) (If treatment had to be discontinued because of pain, the timing of this was recorded), 2. documentation of adverse effects, and 3. assessment of erythema and erosions by 1 investigator Time points: 1. at 3, 6, 12, and 16 minutes during treatment (pain), 2. 4 days after their first and second treatments (erythema and erosion) | |
| Funding | ‐ | |
| Notes | ALA‐PDT was more painful than MAL‐PDT. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 88): "Patients were randomised so that half would receive ALA and half MAL as their first split scalp treatment." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | Quote (page 88): "Both patients and investigators remained blinded until study completion." |
| Blinding of outcome assessment (detection bias) | Low risk | Assessment was performed by a second investigator. |
| Incomplete outcome data (attrition bias) | Unclear risk | Per‐protocol (PP) analysis. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | High risk | Wood's light was used to look at PpIX fluorescence after cream incubation, but the results were not mentioned. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: March 2008 End date: Not available | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 1% nicotinamide, twice daily for 6 months (information from the protocol) (N = 13 participants) Control intervention B: placebo, twice daily for 6 months (information from the protocol) (N = 17 participants) | |
| Outcomes | Primary outcome of the trial 1) Mean percentage of reduction in lesion counts from baseline at 3 and 6 months Secondary outcome of the trial 1) Total count for appearance of new/subclinical lesions at 3 months (protocol) Other outcome of the trial 1) Serious adverse events including basal cell carcinoma and squamous cell carcinoma Efficacy Methods: quantitative assessment using counting of lesions by a single observer and photography Safety Methods: reporting of all adverse events | |
| Funding | This study was supported by Cancer Council NSW, the Dermatology Research Foundation and Epiderm. | |
| Notes | This was a pilot study. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 1138): "Participants were randomised (unstratified, size six randomised block)." |
| Allocation concealment (selection bias) | Low risk | Allocation was concealed within sealed opaque envelopes (protocol). |
| Blinding of participants and personnel (performance bias) | Low risk | Quote (page 1138): "Patients and observers (F.M., M.V.) remained blinded until all patients had completed the study." |
| Blinding of outcome assessment (detection bias) | Low risk | Quote (page 1138): "Patients and observers (F.M., M.V.) remained blinded until all patients had completed the study." |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear if intention‐to‐treat (ITT) or per‐protocol (PP) analysis was used, but ITT was stated in the protocol. Intervention ‐ A: 0 dropouts Control ‐ B: 2 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Appearance of new/subclinical lesions was not reported, but this outcome was included in the protocol ACTRN12607000428460 at anzctr.org.au. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, cross‐over study (2‐part study). The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: etretinate, 75 mg/day (25 mg tablet 3 times daily) for 2 months (N = 25 participants) Control intervention B: placebo. 3 times daily for 2 months (N = 25 participants) Then treatment changes for 2 more months (placebo group gets etretinate, and vice versa) | |
| Outcomes | Outcomes of the trial Part 1 1) Complete remission (= participant complete clearance) rates 2) Partial remission (50% size reduction of 75% of lesions) rates 3) Clinical laboratory tests 1) Complete remission 2) Partial remission rates 3) Clinical laboratory tests 4) Minor adverse events (for the 2 phases) Efficacy Methods: quantitative assessment using direct measurement and photography of the lesions Time points: every month Safety Methods: 1. haematological and biochemical screen, 2. measurement of plasma vitamin A | |
| Funding | ‐ | |
| Notes | 17 participants required dosage reduction due to toxicity. Response was maintained when dosage was reduced. Vitamin A type unwanted effects (dry mouth, skin rash, desquamation, etc) were observed. Only part 1 data has been included in this review. Data for intention‐to‐treat analysis was used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (pages 364 to 365): "Each treatment was given for 2 months and the order of administration was randomised." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 3 dropouts (the reasons were not reported) Control ‐ B: 2 dropouts (the reasons were not reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, open‐label, active‐controlled, intraindividual study. Start date: March 2004 End date: April 2005 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: methyl aminolevulinate (MAL)‐photodynamic (PDT) (N = 119 participants) Control intervention B: cryotherapy: double freeze‐thaw (16 seconds total) using liquid nitrogen spray, lesions with non‐complete response were retreated at 12 weeks 1 (assessment at 12 weeks) or 2 treatments (assessment at 24 weeks) (N = 119 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 Interval between treatments: 12 weeks Preparation of lesions: gentle scraping Cream concentration (%): 16% Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light LED Light source: AktiliteCL 128 Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: 8 to 10 minutes | |
| Outcomes | Primary outcomes of the trial 1) Lesion complete response rates of baseline lesions only at 24 weeks 2) Participant preference Secondary outcomes of the trial 1) Lesion complete response rates of baseline lesions only at 12 weeks 2) Cosmetic outcomes by investigator at 12 and 24 weeks 3) Investigator preference Other outcomes of the trial 1) Mean per cent reduction in lesion counts from baseline at 12 and 24 weeks 2) Skin‐related adverse events 3) Skin discomfort and pain after first or second treatments on a visual analogue scale (VAS) Efficacy Methods: quantitative assessment using lesion counting [efficacy evaluations included only lesions present at baseline (i.e. lesions appearing after baseline, if any, were to be reported as adverse events)] Time points: at baseline, and at weeks 12 and 24 Definitions: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance) Methods: 1. immediate evaluation of skin discomfort by participant after each procedure using a VAS of 0 (no discomfort) to 10 (worst possible skin discomfort), 2. adverse events reported spontaneously by the participant or elicited following non‐leading questioning (severity, duration, and need for additional therapy) (If pain was the only reaction and concomitant treatment was not needed, it was not reported as an adverse event, as it was already recorded as skin discomfort) Time points: at each visit (adverse events) Definitions: 1. phototoxic reaction (any observed erythema, oedema, itching, etc), 2. cryotherapy reaction (any observed blisters, infection, etc) Cosmetic Methods: assessment of the overall cosmetic outcome Time points: at weeks 12 and 24 Definitions: 1. excellent (only slight occurrence of redness or change in pigmentation), 2. good (moderate redness or change in pigmentation), 3. fair (slight to moderate scarring, atrophy, or induration), 4. poor (extensive scarring, atrophy, or induration) | |
| Funding | This study was supported by Galderma France. | |
| Notes | The treatments were comparable in terms of efficacy; however, participants significantly preferred MAL‐PDT over cryotherapy. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1030): "At baseline visit, eligible subjects received treatment with PDT using MAL... and conventional Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated |
| Blinding of participants and personnel (performance bias) | High risk | This study was open because 2 physically distinct treatments were compared. |
| Blinding of outcome assessment (detection bias) | High risk | This study was open because 2 physically distinct treatments were compared. |
| Incomplete outcome data (attrition bias) | Low risk | Per‐protocl (PP) and intention‐to‐treat (ITT) analyses were used. Intraindividual study: Intervention ‐ A: 6 dropouts (the reasons were reported) Control ‐ B: 6 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Standard deviations for the mean percentages of reduction in lesion counts were not provided. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, assessor‐blinded, active‐controlled, intraindividual study. Start date: October 2008 End date: September 2009 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: cryotherapy followed by 5% imiquimod 3 times per week for 4 weeks (N = 27 participants) Control intervention B: cryotherapy (N = 27 participants) | |
| Outcomes | Primary outcome of the trial 1) Mean percentage of reduction in lesion counts at 4 to 8 weeks post‐treatment Secondary outcome of the trial 1) Cosmetic appearance scores by participant and investigator at 4 to 8 weeks post‐treatment Other outcomes of the trial 1) Participant complete clearance rates at 4 to 8 weeks post‐treatment (posthoc analysis) 2) Local skin reactions (severity scores) 3) Minor adverse events (pooled) 4) Serious adverse events Efficacy Methods: quantitative assessment using counting of all (baseline and new) actinic keratoses in each respective treatment area Time points: at baseline and 4 to 8 weeks post‐treatment Definitions: 1. per cent change = [(actinic keratoses count at 4 to 8 weeks post‐treatment)‐(actinic keratoses count at baseline)]/(actinic keratoses count at baseline)]*100%, 2. complete clearance (actinic keratosis count of 0) Safety Methods: 1. mean maximum postbaseline intensity of investigator‐assessed local skin reactions (erythema, edema, weeping/exudate, flaking/scaling/dryness, scabbing/crusting, erosion/ulceration) scored as 0 = none, 1 = mild, 2 = moderate, 3 = severe per treatment area; 2. serious adverse events; 3. adverse events collected by non‐systematic assessment Time points: postbaseline to the end of study Cosmetic Methods: Cosmetic appearance score based on comparison to appearance at baseline by investigator and participant Time points: at 4 to 8 weeks post‐treatment Definitions for 7‐point scale: +3 (treatment area is much better appearing), +2 (treatment area is moderately better appearing), + 1 (treatment area is slightly better appearing), 0 (treatment area appears same), ‐1 (treatment area is slightly worse appearing), ‐2 (treatment area is moderately worse appearing), and ‐3 (treatment area is much worse appearing) | |
| Funding | ‐ | |
| Notes | Local skin reactions were reported as scores, but their values were similar between the imiquimod‐treated and topical untreated sides. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1 of study data document): "Study design: Allocation: Randomised..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | This study was single‐blinded (assessor). |
| Blinding of outcome assessment (detection bias) | Low risk | This study was single‐blinded (assessor). |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis for participant complete clearance and per‐protocol (PP) analysis for other analyses were used. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | High risk | A "favourable" efficacy outcome was analysed posthoc, whereas the prespecified outcome was not favourable. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: June 2008 End date: May 2009 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% imiquimod (Taro), once per day, twice weekly for 16 weeks (N = 183 participants) Control intervention B: vehicle, once per day, twice weekly for 16 weeks (N = 30 participants) | |
| Outcomes | Primary outcome of the trial 1) Number of participants with 100% clearance of lesions (= participant complete clearance) at week 24 Secondary outcome of the trial 1) Participants experiencing at least 1 adverse event Other outcomes of the trial 1) Application site reactions including irritation 2) Minor adverse events 3) Serious adverse events including squamous cell carcinoma Efficacy Methods: quantitative assessment using lesion counting Time points: at baseline and week 24 Definitions: the participant is 100% clear of lesions (all lesions that were identified at baseline are no longer present, and there are no new lesions) Safety Methods: reporting of adverse events and serious adverse events termed from Medical Dictionary for Regulatory Activities (MedDRA) Time points: at each follow‐up visit | |
| Funding | This study was supported by Taro Pharmaceuticals USA. | |
| Notes | This was an equivalence study and was divided into 2 studies for our review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1 of study data document): "Study design: Allocation: Randomised..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded (subject, caregiver, investigator). |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded (outcomes assessor). |
| Incomplete outcome data (attrition bias) | Low risk | Per‐protocol (PP) and modified (no postbaseline assessment but received treatment) intention‐to‐treat ( ITT) analyses were used. Intervention ‐ A: 31 dropouts (the reasons were reported) Control ‐ B: 7 dropouts of the entire group, i.e. 60 participants (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: June 2008 End date: May 2009 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% imiquimod (Aldara), once per day, twice weekly for 16 weeks (N = 179 participants) Control intervention B: vehicle, once per day, twice weekly for 16 weeks (N = 30 participants) | |
| Outcomes | Primary outcome of the trial 1) Number of participants with 100% clearance of lesions (= participant complete clearance) at week 24 Secondary outcome of the trial 1) Participants experiencing at least 1 adverse event Other outcomes of the trial 1) Application site reactions including irritation 2) Minor adverse events 3) Serious adverse events including squamous cell carcinoma Efficacy Methods: quantitative assessment using lesion counting Time points: at baseline and week 24 Definitions: the participant is 100% clear of lesions (all lesions that were identified at baseline are no longer present, and there are no new lesions) Safety Methods: reporting of adverse events and serious adverse events termed from Medical Dictionary for Regulatory Activities (MedDRA) Time points: at each follow‐up visit | |
| Funding | This study was supported by Taro Pharmaceuticals USA. | |
| Notes | This was an equivalence study and was divided into 2 studies for our review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1 of study data document): "Study design: Allocation: Randomised..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded (subject, caregiver, investigator). |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded (outcomes assessor). |
| Incomplete outcome data (attrition bias) | Low risk | Per‐protocol (PP) and modified (no postbaseline assessment but received treatment) Intention‐to‐treat (ITT) analyses were used. Intervention ‐ A: 29 dropouts (the reasons were reported) Control ‐ B: 7 dropouts of the entire group, i.e. 60 participants (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 10% masoprocol cream applied twice daily for 14 to 28 days (N = 131 participants) Control intervention B: placebo applied twice daily for 14 to 28 days (N = 45 participants) | |
| Outcomes | 1) Investigator global assessment (global improvement indices‐cured) at 1 month post‐treatment 2) Mean reduction in lesion counts 3) Median percentage reduction in lesion counts between baseline and 1 month post‐treatment 4) Skin irritation Efficacy Methods: 1. quantitative assessment by counting all actinic keratoses in the test area, 2. qualitative assessment (global assessment) Time points: at baseline and 1 month post‐treatment Definitions: evaluable (participant who completed at least 14 days of therapy Definitions for global assessment: 1. cured (clear of palpable lesions, slight residual Safety Methods: clinical assessment and participant history Time points: at each visit (weekly during treatment) | |
| Funding | This study was supported by Chemex Pharmaceuticals, Denver. | |
| Notes | Inflammatory response was not essential for therapeutic activity of masoprocol. The percentage of reduction in lesion counts did not correlate with baseline lesion severity. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 739): "Patients were randomly assigned to treatment with topical masoprocol or vehicle in a 3:1 ratio, respectively." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis was used, and there was a difference in the percentages of participant lost between treatment (14%) and placebo (9%). Intervention ‐ A: 18 dropouts (the reasons were reported) Control ‐ B: 4 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | High risk | 25 participants stopped treatment between 14 & 28 days due to adverse reactions, but it was stated that their results were comparable to those who completed the full 28 days of treatment. |
| Methods | This was a single‐centre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% imiquimod, applied to 5 lesions, once per day 3 times per week for up to 16 weeks, biopsy of 1 lesion after 2 weeks of treatment (N = 12 participants) Control intervention B: vehicle, applied to 5 lesions, once per day 3 times per week for up to 16 weeks, biopsy of 1 lesion after 2 weeks of treatment (N = 6 participants) | |
| Outcomes | Primary outcome of the trial 1) Immunological outcome Other outcomes of the trial 1) Participant complete clearance rates at the end of treatment 2) Clearance rates 3) Percentage lesion reduction (proportion of baseline lesions cleared at end of treatment = lesion complete response) rates 4) Clinical laboratory tests 5) Application site reactions 6) Local skin reactions 7) Minor adverse events 8) Serious adverse events Efficacy Time points: at the end‐of‐treatment visit Definitions: 1. clearance (clinical resolution of > 50% of the 4 treated, non‐biopsied lesions), 2. complete clearance rate (proportion of participants with 100% clinical clearance of treated lesions) Safety Methods: 1. laboratory tests, 2. recording of local skin reactions and adverse events Time points: pre‐study and end of treatment (laboratory tests) | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | This was a phase I study, mainly on cutaneous immune response (biomarker changes). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation schedule was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Unclear risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: February 2006 End date: January 2007 | |
| Participants | Inclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% imiquimod, once per day, 3 times per week, 4 weeks on, 4 weeks off, 4 weeks on (N = 9 participants) Control intervention B: vehicle, once per day, 3 times per week, 4 weeks on, 4 weeks off, 4 weeks on (N = 3 participants) | |
| Outcomes | Primary outcome of the trial 1) Comparison of evaluation techniques Secondary outcome of the trial 1) Histological clearance (confirmation) Other outcomes of the trial 1) Mean lesion counts at baseline and week 20 (transformed to mean reduction in lesion counts) 2) New/sub‐clinical lesions during the study 3) Minor adverse events (pooled) Efficacy Methods: quantitative assessment using clinical counting Time points: at baseline and weeks 4, 8, 12, and 20 Safety Methods: 1. general physical examination, 2. recording of any adverse events Time points: 1. at the start and end of the study (physical exam), 2. at each visit (adverse events) | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | This was a pilot study. Cross polarised light photography, fluorescence diagnostic, and clinical lesion counting were used for efficacy analysis, but only data obtained with clinical counting were used for analyses because it was the technique used in the other studies. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 641): "Eligible patients were randomised in a 3:1 ratio (active:vehicle) to either imiquimod or vehicle cream." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Low risk | All outcomes in the protocol (NCT00294320) were reported in the published study. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single‐centre, randomised, double‐blind, active‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% 5‐fluorouracil twice daily for 4 to 7 weeks followed by chlorhexidine cream (N = 27 participants) Control intervention B: Er:YAG laser resurfacing with oral prophylactic antibiotics and antivirals, Erbium mode: 7 to 28 J/cm², 10 to 12 pulses per second with 50% CO₂ from 2 to 4 W (N = 28 participants) | |
| Outcomes | Primary outcome of the trial 1) Recurrence rates according to clinical evaluation at 12 months post‐treatment Secondary outcomes of the trial 1) Recurrence rates according to clinical evaluation at 3 and 6 months post‐treatment 2) Recurrence rates according to histological evaluation at 3 months and at time of recurrence Other outcomes of the trial 1) Mean reduction in lesion counts at 3, 6, and 12 months post‐treatment 2) Mean per cent lesions cleared (= mean percentage of reduction in lesion counts) at 6 and 12 months post‐treatment 3) Skin irritation 4) Minor adverse events after treatment, at 3, 6, and 12 months post‐treatment 5) Cosmetic outcome: proportion of participants with improvement of surface with actinic damage 6) Cosmetic outcome: proportion of participants with decrease in photoageing score 7) Cosmetic outcome: number of participants with changes in pigmentation or scarring Efficacy Methods: 1. quantitative assessment using clinical evaluation including the number of lesions and the surface of actinic damage (0% to 25%, 25% to 50%, 50% to 75%, and 75% to 100%) performed by 2 investigators, 2. histopathological evaluation (3 months after treatment and evaluation of recurrence) Time points: at baseline; at 3 days (laser only); at 1 (laser only), 2, and 4 weeks; and at 3, 6, and 12 months post‐treatment Safety Methods: evaluation of adverse effects Time points: at day 3 (laser only), at weeks 1 (laser only), 2 and 4 , and at 3, 6, and 12 months post‐treatment Cosmetic Methods: photoageing score (simplified form of the Glogau score) performed by 2 investigators Time points: at baseline; at day 3 (laser only); at weeks 1 (laser only), 2, and 4; and at 3, 6, and 12 months post‐treatment | |
| Funding | ‐ | |
| Notes | There were significantly less recurrences in the laser group than 5‐fluorouracil group. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer program, Sampsize 2.0, was used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 2 dropouts (the reasons were reported) Control ‐ B: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | High risk | The standard deviations associated with mean values were not provided. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: MAL‐photodynamic therapy (PDT) (N = 42 participants) Control intervention B: placebo‐PDT (N = 38 participants) Characteristics of PDT intervention: Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: crusts and scales removed by curettage and gentle scraping Cream concentration (%): 16% Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: ‐‐ Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): 50 to 200 Exposure time: 8 min | |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates at 3 months after last treatment Other outcomes of the trial 1) Lesion complete response rates at 3 months after last treatment 2) Local adverse events 3) Minor adverse events 4) Serious adverse events 5) Cosmetic outcome with MAL‐PDT in completely cleared participants: physician assessment, participant assessment 6) Participants' satisfaction Efficacy Methods: quantitative assessment using inspection, photography, and palpation of each lesion by the same investigator at each centre Time points: at baseline and at 3 months after the second PDT Definitions: complete response (complete disappearance of the lesion) Methods: 1. recording of local skin reactions, phototoxicity reactions, or both, by study‐centre personnel who were not involved in evaluation of the participant, 2. adverse events reported spontaneously by the participant or elicited after non‐leading questioning (severity, duration, and need for additional therapy) and rated (the clinician assessed the causal relationship of the event to the study treatment as related, uncertain, or not related) Time points: at baseline, during, and immediately after PDT; at week 2; and at 3 months after the second PDT treatment Cosmetic Methods: assessment of overall cosmetic outcome in participants with complete response in all lesions by both the investigator and participant using a 4‐point rating scale Definitions for the 4‐point scale: 1. excellent (no scarring, atrophy, or induration, and no or slight occurrence of redness or change in pigmentation compared with adjacent skin), 2. good (no scarring, atrophy, or induration, but moderate redness or change in pigmentation compared with adjacent skin), 3. fair (slight to moderate occurrence of scarring, atrophy, or induration), 4. poor (extensive occurrence of scarring, atrophy, or induration) | |
| Funding | This study was supported by PhotoCure ASA. | |
| Notes | The response rate was similar for mild and moderate lesions. Most pain and erythema was gone within 24 hours. Data for intention‐to‐treat analysis were used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Prerandomised numbers assigned to participants at screening visit and stratified per centre were used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 3 dropouts (the reasons were reported) Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | High risk | The cosmetic outcomes were reported for MAL‐PDT participants only. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: January 2006 End date: December 2006 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: MAL‐photodynamic therapy (PDT) (N = 49 participants) Control intervention B: placebo‐PDT (N = 47 participants) Characteristics of PDT intervention: Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: crusts and scales removed by curettage and gentle scraping Cream concentration (%): 16.8% Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light LED Light source: Aktilite CL 128 Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: 8 min | |
| Outcomes | Primary outcome of the trial 1) Complete participant response rates (= participant complete clearance) at 3 months post‐treatment Secondary outcomes of the trial 1) Complete lesion response rates (= lesion complete response) at 3 months post‐treatment 2) Application site and local adverse reactions (in general and severe) Other outcomes of the trial 1) Serious adverse events 2) New lesions during the study Efficacy Methods: clinical assessment by inspection, palpation, and characterisation Time points: at baseline and 3 months post‐treatment Definitions: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance of the lesion) (non‐completely responding lesions were treated at the discretion of the investigator) Safety Methods: adverse events reported spontaneously or elicited by non‐leading questioning (severity, localisation, duration, and need for additional treatment) (the clinician assessed the causal relationship of the event to the study treatment as related, uncertain, or not related.) Time points: after lesion preparation before cream application, at the end of the 3‐hour cream application and after illumination during each treatment session, and at 2 weeks and 3 months post‐treatment Definitions for the severity of the adverse events: 1. mild (transient and easily tolerated), 2. moderate (caused the participant's discomfort and interrupted usual activities), 3. severe (caused considerable interference with usual activities and may have been incapacitating or life‐threatening) | |
| Funding | This study was supported by PhotoCure ASA. | |
| Notes | Within the MAL‐PDT group, complete lesion response rates were slightly higher in grade‐1 than grade‐2 lesions (89% vs 80%), and in lesions on the scalp than on the face (93% vs 87%). Larger lesions (diameter > 20 mm) had lower complete response rates than smaller lesions (74% vs 86% to 90%). At 3 months post‐treatment, 31% (15/49) of participants treated with MAL had new lesions compared to 26% (12/47) of participants treated with vehicle. A sample size calculation was provided. This study is study #1 in the Metvixia product insert 2008. The studies included in the Metvixia product insert were changed between 2004 (PhotoCure) and 2008 (Galderma), which correspond to the use of different types of light. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Prerandomised numbers assigned to participants at screening visit and stratified per centre were used for allocation generation. |
| Allocation concealment (selection bias) | Low risk | Randomisation sequence was prepared by sponsor. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was evaluator‐blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropout Control ‐ B: 0 dropout |
| Selective reporting (reporting bias) | Low risk | All outcomes from the protocol (NCT00306800) and protocol mistakes were presented. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single centre, randomised, active‐controlled, intraindividual study. Start date: May 2004 End date: August 2005 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 4 participants) Control intervention B: 5‐fluorouracil twice daily for 3 weeks (N = 4 participants) Characteristics of PDT intervention: Type of treatment: field‐directed treatment Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: scales removed by curettage Cream concentration (%): 16% Application of cream: 1 mm thick onto area Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: Paterson PDT Omnilux Wavelength (nm): 633 + 15 Energy fluence (J/cm²): 75 Intensities (mW/cm²): 80 Exposure time: 8 min | |
| Outcomes | Outcomes of the trial 1) Complete resolution of lesional area (= participant complete clearance) rates at 1, 3, and 6 months 2) Overall reduction in lesional area 3) Local skin reactions (pooled for carcinomas in situ and actinic keratoses) 4) Minor adverse events (pooled for carcinomas in situ and actinic keratoses) 5) Cosmestic outcomes by participant and investigator (pooled for carcinomas in situ and actinic keratoses) 6) Treatment‐associated pain score 7) Participant's preference Efficacy Methods: quantitative assessment using photographic mapping, tracing of the clinical margins of each lesional area onto transparencies and calculating the surface area by overlaying on 1 mm squared graph paper Time points: before treatment, at 1, 3, and 6 months post‐treatment Definitions: 1. complete response (complete clinical resolution of the treated lesion), 2. partial response (at least a 30% reduction in the surface area of the lesion after treatment based upon the European Organisation for Research and Treatment of Cancer (EORTC) guidelines for the evaluation of tumour treatment response), 3. non‐responders (lesions that failed to meet the criteria for partial response) Methods: 1. participants kept a record of pain and erythema using a 4‐point scale (0 = none, 1 = mild, 2 = moderate, and 3 = severe); 2. documentation of other local skin reactions, such as pruritus, erosions, ulceration, crusting, skin infection, and scarring Time points: daily Cosmetic Methods: cosmetic scoring by clinician and participant Time points: at the 6‐month assessment Definitions: 1. poor (extensive scarring, atrophy, or induration), 2. moderate (slight to moderate occurrence of scarring), 3. good (no scarring, atrophy or induration, but moderate redness or pigmentation change compared with adjacent skin), 4. excellent (no scarring, atrophy, or induration, and no or slight occurrence of redness or pigmentation change compared with adjacent skin) | |
| Funding | The light source was provided by Omnilux. | |
| Notes | This study with immunocompromised participants included participants with actinic keratoses (N = 4) or carcinoma in situ (N = 5). Separated data were presented for efficacy but not for local skin reactions, adverse events, and cosmetic outcomes. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 321): "Each patient was randomly assigned to apply topical 5‐FU cream to 1 lesional area twice daily for 3 weeks and to Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | This study was open‐label. |
| Blinding of outcome assessment (detection bias) | High risk | Quote (page 322): "Assessments were not blinded." |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used for the review based on the individual data presented in a table. Intraindividual study: Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, vehicle‐controlled, intraindividual study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% Imiquimod, 3 times per week for 8 weeks or less (clearance achieved) (N = 22 participants) Control intervention B: vehicle, 3 times per week for 8 weeks or less (clearance achieved) (N = 22 participants) | |
| Outcomes | Outcomes of the trial 1) Mean lesion counts (transformed to mean reduction of lesion counts) at baseline and week 16 2) Changes in lesion size 3) Participants experiencing at least 1 adverse event (pooled) 4) Local adverse reactions (pooled) 5) Rest periods Efficacy Methods: quantitative assessment using lesion counting and photography (only participants who completed the 8‐week course of treatment with imiquimod were assessed for changes in lesion size) Time points: at baseline; at weeks 2, 4, 6, 8, and 16 Methods: 1. monitoring of concomitant medications, and 2. recording of the type and severity (mild, moderate, or severe) of local adverse reactions (erythema, itching, scabbing) Time points: at baseline; at weeks 2, 4, 6, and 8, or until total clearance of lesions | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | If needed, a rest period of 3 weeks was allowed and the dosing frequency reduced to 2 times per week. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 554): "Application sites were randomised at the Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Unclear risk | This was not stated. |
| Blinding of outcome assessment (detection bias) | Unclear risk | This was not stated. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis with 23% of lost participants was used. Intraindividual study: Intervention ‐ A: 5 dropouts (the reasons were reported) Control ‐ B: 5 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | The adverse events were not reported separately for the 2 treatments. The standard deviations associated with mean values were not provided. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 88 participants) Control intervention B: placebo‐PDT (N = 23 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: debridement Cream concentration (%): 16.8% Application of cream: onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 2.5 to 4 hours Type of light: red light Light source: CureLight BroadBand Model CureLight 01 Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ | |
| Outcomes | Outcomes of the trial 1) Complete responders (= participant complete clearance) rates at 3 months 2) Participant partial (> 75%) clearance rates at 3 months 3) Lesion complete response rates at 3 months 4) Local adverse reactions 5) Cosmetic outcomes: hyperpigmentation Efficacy Time points: at 3 months after the second treatment session Definitions: 1. cleared lesion (not visible and not palpable), 2. complete responder (participant with all treated lesions cleared) | |
| Funding | This study was supported by Photocure ASA. | |
| Notes | Participants with > 4 lesions had lower success rates than those with < 4 lesions | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 2): "These trials were not identical; however, both were randomised, multicenter, and double‐blinded with patients randomised to Metvixia‐PDT and Vehicle‐PDT study arms that required 2 treatment sessions (7 days apart)." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | High risk | This was not stated, and the information provided did not allow to make any conclusion. There was no information provided on study dropouts. |
| Selective reporting (reporting bias) | High risk | The adverse events were reported for the whole study, i.e. not separated for MAL and vehicle. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 42 participants) Control intervention B: placebo‐PDT (N = 38 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: debridement Cream concentration (%): 16.8% Application of cream: onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 2.5 to 4 hours Type of light: red light Light source: CureLight BroadBand Model CureLight 01 Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ | |
| Outcomes | Outcomes of the trial 1) Complete responders (= participant complete clearance) rates at 3 months 2) Participant partial (> 75%) clearance rates at 3 months 3) Lesion complete response rates at 3 months 4) Local adverse reactions 5) Cosmetic outcomes: hyperpigmentation Efficacy Time points: at 3 months after the second treatment session Definitions: 1. cleared lesion (not visible and not palpable), 2. complete responder (participant with all treated lesions cleared) | |
| Funding | This study was supported by Photocure ASA. | |
| Notes | Lesions that were slightly palpable, i.e. grade 1, had a better success rate than lesions that were visible and palpable, i.e. grade 2. Local skin/adverse reactions and cosmetic outcomes from Photocure ASA Australian and US studies were combined. The studies included in the Metvixia product insert were changed between 2004 (PhotoCure) and 2008 (Galderma), which correspond to the use of different types of light. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 2): "These trials were not identical; however, both were randomised, multicenter, and double‐blinded with patients randomised to Metvixia‐PDT and Vehicle‐PDT study arms that required 2 treatment sessions (7 days apart)." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Unclear risk | This was not stated and the information provided did not allow to make any conclusion. There was no information provided on study dropouts. |
| Selective reporting (reporting bias) | High risk | The adverse events were reported for the whole study, i.e. not separated for MAL and vehicle. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, assessor‐blinded, vehicle‐controlled, parallel group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: aminolevulinic acid (ALA)‐photodynamic therapy (PDT) (N = 181 participants) Control intervention B: placebo‐PDT (N = 62 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 Interval between treatments: 8 weeks Preparation of lesions: ‐‐ Cream concentration (%): 20% Application of cream: ‐‐ Incubation with cream: 14 to 18 hours Type of light: blue light Light source: Blu‐U Wavelength (nm): 417 + 5 Energy fluence (J/cm²): 10 Intensities (mW/cm²): 10 Exposure time: 1000 seconds (16 minutes) | |
| Outcomes | Outcomes of the trial Assessments at 8 weeks (1 treatment) and 12 weeks (1 or 2 treatments): 1) Clearing of individual lesions (= lesion complete response) rates 2) Percentage of participants who experienced 100% clearance of all target lesions (= participant complete clearance) 3) Percentage of participants who experienced 75% or greater clearance of all target lesions (= participant partial clearance) 4) Clinical laboratory tests 5) Application site reactions 6) Local skin reactions by location, i.e. face or scalp 7) Treatment‐related adverse events (= minor adverse events) given for ALA treatment only 8) Minor adverse events in general and by location, i.e. face or scalp 9) Serious adverse events 10) Changes in pigmentation reported per lesion Efficacy Methods: assessments performed by a blinded investigator Time points: at weeks 4, 8, and 12 Safety Methods: 1. laboratory tests, 2. assessment of phototoxic effects such as erythema, edema, stinging or burning, by an unblinded investigator, 3. assessment of adverse events Time points: 1. at baseline and 24 hours after initial light treatment and Cosmetic Methods: assessment of changes in pigmentation by an unblinded investigator Time points: at every visit | |
| Funding | This study was supported by DUSA Pharmaceuticals. | |
| Notes | Pooled results from 2 independent and identical phase III clinical trials also presented in the Levulan kerastick product insert. Recurrence was presented in a follow‐up paper (Fowler 2002). Data for intention‐to‐treat analysis were used for the meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method used for allocation generation is unclear, but it was performed separately for each centre. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | Quote (page 42): "Drug application and activation, light treatment, and all safety evaluations were performed by an unblinded investigator..." |
| Blinding of outcome assessment (detection bias) | High risk | This study was assessor‐blinded for efficacy, but not for safety. Safety and efficacy assessments were performed by different investigators. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 7 dropouts (the reasons were reported) Control ‐ B: 3 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | The Levulan kerastick product insert mentioned 7 clinical trials. Efficacy data from only 2 randomised phase III (Piacquadio 2004) and open studies were presented. Only safety data from the 2 phase III studies were presented. Levulan kerastick reported efficacy outcomes separately for the face and scalp, but Piacquadio 2004 did not. Piacquadio 2004 reported adverse events for ALA‐PDT, whereas Levulan kerastick reported them for both ALA‐PDT and placebo‐PDT. |
| Other bias | High risk | Data between Piacquadio 2004 (PP) and the Levulan kerastick (ITT?) product insert was not always the same. Data from Piacquadio 2004 was used for analyses. |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: 1995 End date: 1996 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: topical 3% diclofenac in 2.5% hyaluronic acid gel, twice daily for 30 days (0.5 g/treatment with 6 hours between applications) (N = 49 participants) B: topical 3% diclofenac in 2.5% hyaluronic acid gel, twice daily for 60 days (0.5 g/treatment with 6 hours between applications) (N = 48 participants) Control interventions C: topical 2.5% hyaluronic acid gel, twice daily for 30 days (0.5 g/treatment with 6 hours between applications) (N = 49 participants) D: topical 2.5% hyaluronic acid gel, twice daily for 60 days (0.5 g/treatment with 6 hours between applications) (N = 49 participants) | |
| Outcomes | Primary outcomes of the trial 1) Investigator global improvement indices (IGII) 2) Participant global improvement indices (PGII) 3) Number of participants with complete target lesion clearance at day 60 (target lesion number score = TLNS) 5) Mean numbers of target lesions at baseline and follow‐up (transformed to mean reduction in lesion counts). 6) Total thickness score (TTS) Other outcomes of the trial 1) Clinical laboratory tests 2) Application site reactions 3) Minor adverse events 4) Serious adverse events (including basal and squamous cell carcinoma) Efficacy Methods: 1. quantitative assessment using outlining and counting of lesions on a 5 cm² transparent grid, 2. TTS determined by palpation and visual assessments, 3. qualitatively assessments of overall improvements by investigator and participant (IGII and PGII), 4. severity of the lesions (mild, moderate, and severe) Time points: at baseline (visit 2, day 1 of treatment) and subsequent visits Definitions for TTS: R (lesion resolved completely), 0 (lesion visible, but not palpable), 1 (lesion visible and palpable), 2 (lesion raised with visible scaling), 3 (lesion hyperkeratotic and > 1 mm in thickness) Safety Methods: 1. participant‐recorded concomitant medications taken and side‐effects experienced, 2. review of adverse events and photography, 3. clinical laboratory analyses (standard haematological, biochemical parameters, assessment of electrolytes and urinalysis), 4. serology (antidiclofenac antibodies) Time points: 1. daily (participant record), 2. at each visit (adverse events review), 3. at screening and end of treatment or onset of reaction (serology) | |
| Funding | This study was supported by Hyal Pharmaceutical Corporation. | |
| Notes | A sample size calculation was provided. Data were reported in the Solaraze gel product insert. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 95): "This randomised, double‐blind, placebo‐controlled, parallel‐group trial was conducted between 1995 and 1996 at 6 Canadian centres." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 3 dropouts, B: 5 dropouts (the reasons were reported) Control ‐ C: 2 dropouts, D: 1 dropout (the reasons were reported) |
| Selective reporting (reporting bias) | Low risk | Statistically significant and non‐significant outcomes were reported. |
| Other bias | High risk | There were different safety data between Rivers 2002 and the Solaraze product insert. |
| Methods | This was a single‐centre, randomised, assessor‐blinded, placebo‐controlled, intraindividual study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: calcipotriol (vitamin D), twice daily for 12 weeks on right/left side (N = 9 participants) Control intervention B: placebo twice daily for 12 weeks on right/left side (N = 9 participants) | |
| Outcomes | Outcomes of the trial 1) Mean numbers of lesions at baseline and week 12 (transformed to mean reduction in lesion counts) 2) Mean diameter of target lesion at baseline and week 12 3) Local skin reactions graded on scale 4) Minor adverse events at week 1 and 12 5) Total score for cosmetic appearance of a target lesion (1 target lesion per treatment side) Efficacy Methods: quantitative assessment using lesion counting and recording of diameters of the target lesions Time points: at baseline and weeks 3, 6, 9, and 12 of therapy Safety Methods: side‐effects, such as erythema, dryness, burning sensation, and pruritus, graded from 0 to 3 (0 = none, 1 = mild, 2 = moderate, and 3 = severe) Time points: at baseline and weeks 3, 6, 9, and 12 of therapy Cosmetic Methods: total scores of the target lesions = the sum of erythema, desquamation, and induration scored between 0 to 3 Time points: at baseline and weeks 3, 6, 9, and 12 of therapy | |
| Funding | ‐ | |
| Notes | Similar number of adverse events reports between to treatment and placebo were reported. Neutrogena sunscreen was used. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate allocation generation was achieved with a random digits table. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Unclear risk | This was not stated. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was assessor‐blinded. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis was used, and there was missing information about participants lost after the baseline evaluation. |
| Selective reporting (reporting bias) | High risk | The severity of local skin reactions was not reported. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, vehicle‐controlled, intraindividual study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: aminolevulinic acid (ALA)‐photodynamic therapy (PDT) followed by imiquimod once per day, twice per week for 16 weeks (N = 25 participants) Control intervention B: ALA‐PDT followed by vehicle once per day, twice per week for 16 weeks (N = 25 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 2 Interval between treatments: 4 weeks Preparation of lesions: microdermabrasion Cream concentration (%): 20% Application of cream: ‐‐ Incubation with cream: 1 hour Type of light: blue light Light source: Blu‐U 4170 Wavelength (nm): (417) Energy fluence (J/cm²): ‐‐ Intensities (mW/cm²): ‐‐ Exposure time: 8 min | |
| Outcomes | Primary outcome of the trial 1) Median and mean per cent reduction of the number of lesions at baseline and month 12 (= mean percentage of reduction in lesion counts) Secondary outcome of the trial 1) Severe local skin reactions (pooled) Other outcomes of the trial 1) Median and mean lesion counts at baseline and month 12 (converted to mean reduction of lesion counts) 2) Participant complete clearance rates 3) Treatment‐related adverse events (= minor adverse events) 4) Rest periods Efficacy Methods: quantitative assessment using lesion counting and mapping by marking their locations on clear acetate templates using permanent marker Time points: at baseline and months 1, 2, 3, 4, 6, and 12 of the study Safety Methods: 1. incidence of severe local skin reactions (erythema, edema, erosion/ulceration, scabbing/crusting, weeping/exudates, vesicles, and flaking/scaling/dryness) associated with imiquimod treatment and compare the incidence to those reported in imiquimod studies in which PDT pretreatment was not utilised, 2. assessment of local skin reactions types on a 4‐point scale (0 = none, 1 = mild, 2 = moderate, 3 = severe) by the investigator Time points: at months 2, 3, 4, and 6 | |
| Funding | This study was supported by 3M Pharmaceuticals and Graceway Pharmaceuticals. | |
| Notes | The data were changed for intention‐to‐treat (ITT) analysis for meta‐analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A previous generated list using a random number generator was used. |
| Allocation concealment (selection bias) | Low risk | Sequential assignment upon participation enrolment was used. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Per‐protocol (PP) analysis was used. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was reported, and the lost was before treatment) Control ‐ B: 1 dropout (the reason was reported, and the lost was before treatment) |
| Selective reporting (reporting bias) | High risk | The number of participants with severe local skin reactions was not given separately for the 2 treatments. The standard deviations associated with mean values were not given. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: March 2005 End date: September 2005 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: 0.0025% ingenol mebutate, once per day at days 1 & 2 or days 1 & 8 (N = 15 participants) B: 0.01% ingenol mebutate, once per day at days 1 & 2 or days 1 & 8 (N = 16 participants) C: 0.05% ingenol mebutate, once per day at days 1 & 2 or days 1 & 8 (N = 15 participants) Control intervention D: vehicle, once per day at days 1 & 2 or days 1 & 8 (N = 12 participants) | |
| Outcomes | Primary outcomes of the trial 1) Application site reactions (pain) 2) Local skin responses (= local skin reactions) 3) Treatment‐related adverse events (= minor adverse events) 4) Serious adverse events 5) Clinical laboratory tests Secondary outcomes of the trial 1) Lesion complete response rates at 85 days (target lesions) 2) Lesion complete and marked clinical clearance rates at 85 days 3) Participant partial (> 80%) clearance rates at 85 days [included in participant partial (> 75%) clearance] 4) Participant histological clearance rates at 85 days Other outcomes of the trial 1) Cosmetic outcomes: changes in pigmentation Efficacy Methods: 1. clinical evaluation of each lesion by the investigator, 2. histological evaluation by a central blinded dermatopathologist based on a repeat biopsy of the lesion biopsied prior to treatment. Time points: at day 85 (end of study) Definitions: 1. complete clearance (no evidence of residual disease), 2. marked clearance (50% to 90% improvement), 3. slight clearance (10% to 50% improvement), 4. unchanged (10%), and 5. worsened (clinically‐observable growth) Safety Methods: 1. vital signs, 2. physical examinations, 3. laboratory tests (haematology, serum chemistry, liver function tests, and urinalysis), 4. recording of local skin reactions (itching, erythema,oedema, erosion/ulceration, scabbing/crusting, weeping/exudates, vesicles, flaking/scaling/dryness) and abnormal skin proliferation (treatment was withheld if a severe local skin reaction occurred prior to the second scheduled dose) Time points: 1. at each visit (vital signs), 2. at screening and final visit (day 85) (physical exam), 3. at screening, last day of treatment, and day 85 (or early exit) (laboratory tests) Cosmetic Methods: recording of hypopigmentation, hyperpigmentation and scarring Time points: at day 85 Definitions for local skin reaction rating: 1. mild (easily tolerated), 2. moderate (associated with discomfort sufficient to interfere with usual activities), and 3. severe (incapacitating with inability to work or perform usual activities). | |
| Funding | This study was supported by Peplin Ltd. | |
| Notes | This was a phase IIa study. Application was done only on predetermined 5 lesions with 2 template diameters. There were 2 application regimens, i.e. days 1 & 2 or days 1 & 8, but no differences were detected and results were pooled together. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 17): "Randomisation was performed by an independent clinical research organisation and identical packaging was used to maintain blinding of both investigator and patients regarding allocation to active or vehicle gel." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Low risk | Randomisation sequence was generated by an independent company (see previous quote in random sequence generation). |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Modified intention‐to‐treat (ITT) analysis was used (participants with no treatment were excluded). Intervention ‐ A: 0 dropouts, B: 1 dropout (the reason was reported), C: 0 dropouts Control ‐ D: 0 dropout |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported based on protocol NCT00107965, as well as the mistakes made in the dose application schedule. |
| Other bias | High risk | There were higher percentages of women and scalp lesions in the vehicle group at baseline. |
| Methods | This was a randomised, active‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: aminolevulinic acid (ALA)‐blue light photodynamic therapy (PDT) (N = 12 participants) B: ALA‐pulsed dye laser (PDL) PDT (N = 12 participants) Control intervention C: 0.5% 5‐fluorouracil once or twice daily for 4 weeks (N = 12 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 2 Interval between treatments: 4 weeks Preparation of lesions: ‐‐ Cream concentration (%): 20% Application of cream: ‐‐ Incubation with cream: 1 hour Type of light: blue light or pulsed dye laser Light source: Blu‐U Photodynamic Therapy Illuminator Wavelength (nm): 595 (PDL) Energy fluence (J/cm²): 7.5 (PDL) Intensities (mW/cm²): ‐‐ Exposure time: 1000 sec (16 min), 10 ms (PDL) | |
| Outcomes | Outcomes of the trial 1) 100% lesions cleared (= participant complete response) at 4 weeks post‐treatment 3) Tolerability, i.e. grading of local skin reactions 4) Photoageing Efficacy Methods: 1. grading of target lesions on a 4‐point scale (from resolved to very thick, markedly keratotic, or both), 2. high magnification digital photography of lesions identified with 3 black ink dots and small adhesive label Time points: at baseline and the end of treatment, 2 weeks and 4 weeks post‐treatment Definitions: therapeutic success (sustained clearance of 75% or more of target lesions) Methods: grading of erythema, edema, crusting/erosions, and stinging/ burning Time points: immediately after PDT treatments Definitions for erythema score: 0 (none), 1 (minimal, scant rare erythema), 2 (mild, easily‐seen erythema up to ⅓ of the treated area), 3 (moderate, easily‐seen erythema involving between ⅓ to ⅔ of the treated area), 4 (severe, easily‐seen erythema involving over ⅔ of the treated area) Definitions for oedema score: 0 (none), 1 (minimal, scant rare oedema), 2 (mild, easily‐seen oedema, minimally palpable, involving up to ⅓ of the treated area), 3 (moderate, easily‐seen oedema and typically palpable involving between ⅓ to ⅔ of the treated area), 4 (severe, easily‐seen oedema, indurated in some areas involving over ⅔ of the treated area) Definitions for crusts and erosions score: 0 (none), 1 (rare, a few 3 mm or smaller areas), 2 (mild, up to 12 lesions 3 mm or less, areas readily seen), 3 (moderate), 4 (severe) Definitions for stinging/burning score: 0 (none), 1 (minimal), 2 (moderate), 3 (severe) Cosmetic Methods: 1. global response, 2. assessment of tactile roughness by lightly palpitating by stroking gently with the index finger and molted hyperpigmentation (including area involved, the colour intensity, and the evenness of pigment distribution) Definitions for global response: 0 (complete response = complete resolution of | |
| Funding | This study was supported by DUSA Laboratories. | |
| Notes | PDT treatments were better tolerated than 5‐fluorouracil. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 630): "Caucasian patients with a minimum of 4 nonhyperkeratotic AK of either the face or scalp were recruited and Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | The blinding was not stated, but 2 physically distinct treatments were compared. |
| Blinding of outcome assessment (detection bias) | High risk | The blinding was not stated, but 2 physically distinct treatments were compared. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A & B: 0 dropouts Control ‐ C: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | High risk | The percentages of participants reporting adverse events were not given except for stinging. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 3% diclofenac in hyaluronic acid for 90 days (N = 53 participants) Control intervention B: hyaluronic acid for 90 days (N = 55 participants) | |
| Outcomes | Outcomes of the trial 1) Complete clearing of lesions (= participant complete clearance) rates at 30 days post‐treatment 2) Application site reactions (for the 3 studies included in insert, i.e. Rivers 2002; Solaraze study 2; Wolf 2001) reported as incidences (i.e. number of events, not number of participants) 3) Minor adverse events (for the 3 studies included in insert, i.e. Rivers 2002; Solaraze study 2; Wolf 2001) reported as incidences (i.e. number of events, not number of participants) | |
| Funding | This study was supported by Nycomed US Inc. | |
| Notes | This study was included in the product package insert as study 2. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation was not stated in the product insert, but the other 2 studies included were randomised. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The blinding was not stated in the product insert, but the other 2 studies were double‐blinded. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The blinding was not stated in the product insert, but the other 2 studies were double‐blinded. |
| Incomplete outcome data (attrition bias) | Unclear risk | The type of analysis was not stated, but the 2 other studies had intention‐to‐treat (ITT) analysis. |
| Selective reporting (reporting bias) | High risk | This was the only study of 3 presented in the Solaraze product insert that was not published and with no significant difference for participant complete clearance. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, active‐controlled, intraindividual study. Start date: September 2007 End date: July 2008 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: aminolevulinic acid (ALA)‐photodynamic therapy (PDT) (N = 30 participants) Control intervention B: 5% imiquimod once per day, 3 times per week for 4 weeks on, 4 weeks off (N = 30 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 15 days Preparation of lesions: crust removed by curettage Cream concentration (%): 20% Application of cream: onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 4 hours Type of light: red light Light source: Waldmann PDT 1200 Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): 75 Exposure time: ‐‐ | |
| Outcomes | Outcomes of the trial 1) Lesion complete response rates at 1 and 6 months 2) Application site reactions 3) Local skin reactions 4) Investigator‐assessed cosmetic outcome 5) Participant's preference Efficacy Methods: quantitative assessment using counting and recording of lesions by the same examiners Time points: at baseline and 1 and 6 months post‐treatment Definitions: 1. clinical lesion response (complete response = complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance of the lesion). Safety Methods: recording of adverse events (severity, duration, and need for additional therapy) Time points: at each visit Methods: assessment by investigators based on the amount of scarring, Time points: at month 6 post‐treatment Definitions: 1. excellent (no erythema, change in pigmentation, scarring, atrophy, or induration), 2. good (slight to moderate erythema or change in pigmentation, but no scarring, atrophy, or induration), 3. fair (slight scarring, atrophy, or induration), 4. poor (moderate to extensive scarring, atrophy, or induration) | |
| Funding | ‐ | |
| Notes | There was a difference in lesion complete response between treatments for grade II lesions, i.e. 57.8% for PDT and 37% for imiquimod, but not for grade I lesions (71% to 72%). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1062): "Eligible patients received PDT treatment and treatment with imiquimod 5% cream randomly allocated to alternate upper extremities." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | The blinding was not stated, but 2 physically distinct treatments were compared. |
| Blinding of outcome assessment (detection bias) | High risk | The blinding was not stated, but 2 physically distinct treatments were compared. |
| Incomplete outcome data (attrition bias) | Unclear risk | Per‐protocol (PP) analysis was used. Intraindividual study: Intervention ‐ A: 2 dropouts (the reasons were reported) Control ‐ B: 2 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported, i.e. significantly different or not. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% imiquimod cream, 3 times (or less based on adverse events) per week for a maximum of 12 weeks (N = 25 participants) Control intervention B: placebo, 3 times (or less based on adverse events) per week for a maximum of 12 weeks (N = 11 participants) | |
| Outcomes | Outcomes of the trial 1) Participant complete clearance rates at 14 weeks. 2) Participant partial clearance rates 3) Local skin reactions (graphical representation) 4) Minor adverse events (graphical representation) 5) Recurrence 6) Compliance 7) Rest periods Efficacy Methods: 1. clinical evaluation, 2. biopsy (histology) assessed by the same dermatopathologist Time points: at baseline and week 14 after treatment initiation Definitions: 1. complete clearance (complete clinical clearance confirmed histologically), 2. partial clearance (the clearance of 1 or more lesions treated with imiquimod) Safety Methods: 1. vital signs recording; 2. photography; 3. assessment and recording of local and systemic adverse or abnormal effects; 4. recording of the incidence and severity of erythema, edema, induration, vesicles, erosion, ulceration, excoriation Time points: at each visit (at weeks 2, 3, 6, 9, and 12) | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | The recurrence rate at 1 year was 10% (2/25) for participants treated with imiquimod. A sample size calculation was based on rate of spontaneous healing of actinic keratoses lesions. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1500): "Each patient was randomly assigned a number that was paired with a box containing 12 sachets." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Low risk | The key to codes were held by the pharmaceutical company. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: January 2008 End date: June 2008 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: 3.75% imiquimod, once daily for 2 weeks on, 2 weeks off, 2 weeks on (N = 160 participants) B: 2.5% imiquimod, once daily for 2 weeks on, 2 weeks off, 2 weeks on (N = 160 participants) Control intervention C: placebo, once daily for 2 weeks on, 2 weeks off, 2 weeks on (N = 159 participants) | |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates at week 14 Secondary outcomes of the trial 1) Participant partial (> 75%) clearance rates at week 14 2) Median percentage of reduction in lesion counts 3) Local skin reactions Other outcomes of the trial 1) Participants experiencing at least 1 adverse event 2) Application site reactions including irritation 3) Treatment‐related adverse events (= minor adverse events) 4) Serious adverse events 5) Clinical laboratory tests 6) Investigator global integrated photodamage (IGIP‐cosmetic outcome) 7) Number of participants with the different cosmetic outcomes 8) Rest periods Efficacy Methods: quantitative assessment by counting all of the visible or palpable lesions (baseline or new) in the treatment area by the investigator Time points: at each visit Definitions: 1. complete clearance rate (proportion of participants at the end‐of‐study visit with a count of 0 lesions in the treatment area), 2. partial clearance rates (proportion of participants with 75% or more reduction in lesion count in the treatment area at the end‐of‐study visit as compared with baseline), 3. per cent change (changes in lesion number at the end‐of‐study visit as compared with baseline) Safety Methods: 1. measurement of vital signs; 2. recording and coding (Medical Dictionary for Regulatory Activities) of adverse events; 3. investigator assessment of local skin reactions (erythema, edema, weeping/exudate, flaking/scaling/dryness, scabbing/crusting, and erosion/ulceration) graded as none, mild, moderate, or severe; 4. laboratory tests (hematology, serum chemistry, and urinalyses) (treatment‐emergent adverse events were summarised for each treatment group by Time points: 1. at each visit, 2. pre‐study visit and end‐of‐study visit (laboratory tests) Cosmetic Methods: an overall assessment (IGIP score) of the participant's photodamage change from baseline in the treatment area (including an integrated assessment of fine wrinkling, coarse wrinkling, mottled pigmentation, roughness, shallowness, skin laxity, and telangiectasias) rated on a 7‐point symmetric scale, ranging from significantly improved = +3 to significantly worse = ‐3 Time points: at end‐of‐study visit | |
| Funding | This study was supported by Graceway Pharmaceuticals LLC. | |
| Notes | Data from 2 studies were pooled together. Temporary dosing interruptions could have been instructed by the investigator to manage local skin reactions and adverse events. A sample size calculation was provided. A follow‐up study was published (Hanke 2011). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 584): "Eligible patients were centrally randomised to placebo, imiquimod 2.5%, or imiquimod 3.75% cream in a 1:1:1 treatment allocation." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Low risk | Centralised randomisation was used. |
| Blinding of participants and personnel (performance bias) | High risk | This study was double‐blinded (subject, caregiver, investigator), but the authors mentioned that adverse events could be an issue for the concealment of the assigned treatment in some participants. |
| Blinding of outcome assessment (detection bias) | High risk | This study was double‐blinded (outcomes assessor), but authors mentioned that adverse events could be an issue for the concealment of the assigned treatment in some participants. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 11 dropouts (the reasons were reported), B: 6 dropouts (the reasons were reported) Control ‐ C: 9 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Low risk | All outcomes from the protocol (NCT00605176) were reported. |
| Other bias | Unclear risk | Data for safety were reported differently in the published study, and the data results linked to the protocol. |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: August 2008 End date: February 2009 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 0.05% ingenol mebutate for 2 days (N = 117 participants) Control intervention B: vehicle for 2 days (N = 118 participants) | |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates at day 57 Secondary outcome of the trial 1) Participant partial (percentage criteria was not specified) clearance rates at day 57 Other outcomes of the trial 1) Median percentage reduction in lesion counts 2) Local skin reactions (qualitative) 3) Treatment‐related adverse events (qualitative) 4) Serious adverse events 5) Pigmentation changes (cosmetic) 6) Compliance Efficacy Time points: on days 3, 8, 15, 29, and 57 Safety Methods: 1. assessment of the incidence rate of adverse events, serious adverse events, and local skin responses; 2. grading of local skin responses Time points: on days 3, 8, 15, 29, and 57 Cosmetic Methods: assessment of pigmentation and scarring Time points: on days 3, 8, 15, 29, and 57 | |
| Funding | This study was supported by Peplin Ltd. | |
| Notes | This study report was a conference abstract and was included in the following study awaiting classification Lebwohl 2012. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page AB2): "A total of 255 patients were randomised to treatment with 0.05'X, ingenol mebutate gel or vehicle." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Unclear risk | The type of analysis was not stated. Only 1 dropout due to adverse events was reported, but the treatment group was not specified. |
| Selective reporting (reporting bias) | Unclear risk | All outcomes in the protocol (NCT00742391) were reported in the published study. Another similar study (NCT00942604) has not been published yet. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, open, active‐controlled, parallel‐group study. Start date: April 1999 End date: November 1999 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 102 participants) Control intervention B: cryotherapy: prior skin preparation, variable liquid nitrogen spray unit, 1 to 2 mm rim of frozen tissue beyond marked outline, 2 freeze‐thaw cycles in the same session; mean freeze time of 24 + 18 seconds, (N = 100 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: once for face and scalp, twice for others (8% of lesions) Interval between treatments: 1 week Preparation of lesions: crusts removed by curettage Cream concentration (%): 16% Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: ‐‐ Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): 70 to 200 Exposure time: 10 min | |
| Outcomes | Outcomes of the trial 1) Lesion complete response rates at 3 months post‐treatment 2) Skin irritation 3) Local adverse reactions 4) Investigator's and participant's cosmetic outcomes in participants with > 75% reduction of total lesions: number of participants (excellent and good pooled together) 5) Participants' satisfaction Efficacy Time points: at 3 months after the initial treatment Definitions: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance of the lesion) Safety Methods: recording of adverse events (including the local phototoxicity due to PDT) Time points: before and after illumination, after 2 weeks by telephone contact, and after a final examination 3 months post‐treatment Cosmetic Methods: assessment and grading of overall cosmetic outcome Time points: at 3 months after the initial treatment Definitions: 1. excellent (only slight occurrence of redness or change in pigmentation), 2. good (moderate redness or change in pigmentation), 3. fair (slight to moderate scarring, atrophy, or induration), and 4. poor (extensive scarring, atrophy, or induration) | |
| Funding | This study was supported by Photocure ASA. | |
| Notes | Higher response rates were obtained with thin lesions. High participant satisfaction was obtained with MAL‐PDT. 43% of participants treated with MAL‐PDT reported local adverse events compared to 26% treated with placebo. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate allocation sequence was generated by stratification with respect to the number of lesions. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | This study was open because 2 physically distinct treatments were compared. |
| Blinding of outcome assessment (detection bias) | High risk | This study was open because 2 physically distinct treatments were compared. |
| Incomplete outcome data (attrition bias) | High risk | There was discrepancy in the text (415) and table (384) for number of lesions in the PDT group. Per‐protocol (PP) analysis was used. Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 5 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Overall cosmetic outcomes were pooled together. Satisfaction was reported for PDT participants only. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel group study. Start date: January 2002 End date: March 2003 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: imiquimod 5% cream, once per day, 3 days per week for 16 weeks or less (N = 147 participants) Control intervention B: vehicle, once per day, 3 days per week for 16 weeks or less (N = 139 participants) | |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates at 8 weeks post‐treatment Secondary outcome of the trial 1) Participant partial (> 75%) clearance rates at 8 weeks post‐treatment Other outcomes of the trial 1) Histological clearance at 8 weeks post‐treatment 2) Clinical laboratory tests 3) Application site reactions (including irritation) 5) Minor adverse events 6) Serious adverse events 7) Skin quality (cosmetic) Efficacy Methods: 1. quantitative assessment using clinical counting and recording the number lesions present in the treatment area, 2. by the histologic result from the biopsy specimen of a predefined lesion biopsy site Time points: 1. at weeks 1, 2, 4, 8, 12, 16 (end of treatment), and 24 (8 weeks post‐treatment); 2. at the 8‐week post‐treatment visit (biopsy) Definitions: 1. complete clearance rate (proportion of participants at the 8‐week post‐treatment visit with no evidence of lesion on the histology result of the post‐treatment target lesion biopsy site and no clinically‐visible lesions in the remainder of the treatment area), 2. partial clearance rate (proportion of participants at the 8‐week post‐treatment visit with at least 75% reduction in the number of lesions counted at baseline in the treatment area) Safety Methods: 1. photography of the treatment area; 2. reviewing adverse events and local skin reactions (erythema, edema, erosion/ulceration, scabbing/crusting, weeping/exudate, vesicles, or flaking/scaling/dryness) rated on a scale of 0 (none) to 3 (severe) and concomitant medication use; 3. clinical laboratory tests (hematology, serum chemistry, urinalyses, and pregnancy test) Time points: 1. at weeks 1, 2, 4, 8, 12, 16 (end of treatment), and 24 (8 weeks post‐treatment), 2. pre‐study and end‐of‐study visits (laboratory tests) Cosmetic Methods: visual, clinical, and tactile examinations of skin quality within the treatment area by investigator [skin surface (roughness/dryness/scaliness), hyperpigmentation, hypopigmentation, mottled or irregular pigmentation (both hyperpigmentation Time points: at the treatment initiation and 8‐week post‐treatment visits | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | This was a phase III study. A high rate of agreement was observed between clinical and histologic lesion clearances. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An adequate method of randomisation was achieved by the use of a computer‐generated randomisation schedule. |
| Allocation concealment (selection bias) | Low risk | Adequate allocation concealment was achieved by sealed, tamper‐proof envelopes containing a number allocated to each participant. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | High risk | Intention‐to‐treat (ITT) analysis was used, but some lost to follow up participants were missing for the description. Intervention ‐ A: 10 dropouts (the reasons were not all reported) Control ‐ B: 18 dropouts (the reasons were not all reported) |
| Selective reporting (reporting bias) | High risk | As a similar study (Lebwohl 2004) was also supported by 3M Pharmaceuticals not all skin quality outcomes were reported. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, active‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: 0.03% resiquimod, once per day, 3 days per week, 4 week on, 8 weeks off, 1or 2 courses (N = 31 participants) B: 0.06% resiquimod, once per day, 3 days per week, 4 week on, 8 weeks off, 1or 2 courses (N = 32 participants) C: 0.1% resiquimod, once per day, 3 days per week, 4 week on, 8 weeks off, 1or 2 courses (N = 34 participants) Control intervention D: 0.01 % resiquimod, once per day, 3 days per week, 4 week on, 8 weeks off, 1or 2 courses (N = 35 participants) The gel application was done using a dosing paper template. | |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates after 1 to 2 treatment courses (week 24) Secondary outcomes of the trial 1) Participant partial (> 75%) clearance rates after 1 to 2 treatment courses 2) Participant complete clearance rates after 1 course only (week 12) . Other outcomes of the trial 1) Application site reactions 2) Severe local skin reactions 3) Treatment‐related adverse events (minor adverse events) 4) Serious adverse events 5) Clinical laboratory tests 6) Compliance Efficacy Methods: quantitative assessment using lesion counting and mapping with a transparent plastic template by a qualified dermatologist Time points: at baseline; weeks 2, 4, 8, and 12 for course 1, and if applicable, at weeks 14, 16, 20, and 24 for course 2 Definitions: 1. overall complete clearance rate (proportion of participants at the end of course 1 (week 12) or course 2 (week 24) with no lesions in the treatment area), 2. partial clearance rate (proportion of participants at their last study visit with at least 75% reduction in the number of lesions in the treatment area) Safety Methods: 1. recording of adverse events, 2. assessment of local skin reactions (erythema, oedema, erosion ⁄ulceration, weeping ⁄exudate, flaking ⁄scaling ⁄dryness, and scabbing ⁄crusting), 3. photographs of treatment area, 4. laboratory tests (haematology, biochemistry, urine analysis, and where applicable, pregnancy tests), 5. vital signs measurements and physical examination, and if appropriate, skin cultures (suspected infection) or skin biopsy (lesion suspicious for malignancy) Time points: at weeks 2, 4, 8, and 12 for course 1, and if applicable, at weeks 14, 16, 20, and 24 for course 2 | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | This was a phase II study. Serious adverse events and local skin reactions were more frequent with higher doses, and there was lowest compliance in the 0.1% group. A sample size calculation was provided. Intention‐to‐treat data were used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Pre‐assigned numbering with 1:1:1:1 randomisation with a block size 4 was used for allocation generation. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Unclear risk | This was not stated. |
| Blinding of outcome assessment (detection bias) | Unclear risk | This was not stated. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) and per‐protocol (P) analyses were used. Intervention ‐ A: 5 dropouts (the reasons were reported), B: 10 dropouts (the reasons were reported), C: 14 dropouts (the reasons were reported) Control ‐ D: 2 dropouts (the reasons were reported) Dropping rates were higher for higher doses, i.e. 6%, 16%, 31%, and 41% for 0.01, 0.03, 0.06, and 0.1% resiquimod groups. |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | High risk | The values for overall partial (> 75%) clearance were lower than overall complete clearance. |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: March 2006 End date: January 2007 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: MAL‐photodynamic therapy (PDT) (N = 57 participants) Control intervention B: placebo‐PDT (N = 58 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: crusts and scales removed by curettage Cream concentration (%): 16% Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light LED Light source: Aktilite CL 128 Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): 56 to 83 Exposure time: 9 min | |
| Outcomes | Primary outcome of the trial 1) Participant complete response (= participant complete clearance) rates at 3 months after last treatment Secondary outcomes of the trial 1) Lesion complete response rates at 3 months post‐treatment 2) Treatment site reactions (= application site reactions) reported by events (i.e. not per participants) 3) Local skin reactions (in general and severe) Other outcomes of the trial 1) Participants experiencing at least one adverse event 2) Treatment‐related adverse events (= minor adverse events) reported by events (i.e. not per participants) 3) Minor adverse events 4) Serious adverse events including squamous cell carcinoma 5) Percentage of participants with new lesions Efficacy Methods: 1. quantitative assessment using inspection, palpation, and characterisation of lesions (Olsen 1991) by the same investigator, who was not involved in the treatment procedure, 2. documentation of any lesions not present at baseline (lesions with a non‐complete response were treated at the discretion of the investigator) Time points: at baseline and 3 months post‐treatment Definitions: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance of the lesion), 3. participant complete response (all participants in whom 100% of lesions had responded completely 3 months post‐treatment) Safety Methods: 1. assessment of tolerability, 2. recording of adverse events (severity, localisation, duration, and need for additional treatment) (the clinician assessed the causal relationship of the event to the study treatment as related, uncertain, or not related) Time points: after lesion preparation before cream application, at the end of the 3‐hour cream application, after illumination during each treatment session, and at 2 weeks and 3 months post‐treatment | |
| Funding | This study was supported by Photocure ASA. | |
| Notes | A sample size calculation was provided. This study was study #2 in the Metvixia product insert 2008. The studies included in the Metvixia product insert were changed between 2004 (PhotoCure) and 2008 (Galderma), which correspond to the use of different types of light. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation scheme was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | A clinician not involved in treatment procedure assessed response. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Low risk | All outcomes were presented based on the protocol (NCT00304239), and protocol mistakes were acknowledged. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: ALA‐photodynamic therapy (PDT) (N = 81 participants) Control intervention B: placebo‐PDT (N = 41 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 Interval between treatments: 12 weeks Preparation of lesions: crusts removed by curettage, roughening, and alcohol wiping Cream concentration (%): BF‐200 gel Application of cream: air dry for 10 min Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: Aktilite CL 128 or PhotoDyn 750 Wavelength (nm): 590‐670 (Aktilite), 595‐1400 (PhotoDyn) Energy fluence (J/cm²): 37 (Aktilite), 170 (PhotoDyn) Intensities (mW/cm²): 50‐70 (Aktilite), 196 (PhotoDyn) Exposure time: 15 minutes (PhotoDyn) | |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance at 12 and 24 weeks Secondary outcome of the trial 1) Lesion complete response at 12 and 24 weeks Other outcomes of the trial 1) Local skin reactions for first and second treatment by light sources and in general 2) Cosmetic outcomes: general Efficacy Methods: 1. quantitative assessment of lesion clearance by visual inspection and by palpation by an investigator not involved in treatment and safety evaluation, 2. histological assessment using biopsy of a lesion defined and marked before the PDT treatment Time points: 1. at baseline; 3, and 12 weeks post‐treatment, 2. end‐of‐study visit (biopsy) Definitions: participant complete clearance (all lesions were considered to be cleared both by the clinical and histological assessment) Safety Methods: 1. recording of adverse effects, 2. documentation of local adverse reactions (pain, itching, burning, erythema, oedema, and induration) at the application site and rated as mild, moderate, and severe by the assessing physician or reporting participants, 3. serious adverse events Time points: 1. at 1 week after PDT (by phone) and 3 weeks, 2. during and after PDT (local adverse reactions) Cosmetic Methods: 1. general cosmetic outcome assessed by the investigator as very good, good, unsatisfactory, and impaired; 2. assessment of skin quality Time points: at 12 weeks post‐treatment | |
| Funding | This study was supported by Biofrontera Bioscience GmbH. | |
| Notes | Pain, itching, and burning were reported separately for 1st and 2nd treatment, anatomical area, and light sources. In general, more symptoms were reported for Aktilite CL128 than PhotoDyn 750. A sample size calculation was provided. Data with intention‐to‐treat analyses were used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A validated SAS programme based on the random number function RANUNI and random blocks for 6 participants was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | Treatment and safety assessment were performed by 1 investigator and efficacy assessment by another. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) and per‐protocol (PP) analyses were used. Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 4 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | All outcomes were reported, but little information was given on adverse events and cosmetic outcomes. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, vehicle‐controlled, parallel‐group study (part 1). Start date: April 2005 End date: December 2006 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Part 1: Intervention A: cryotherapy: 3 to 5 second freeze cycle followed (2 weeks after) by 5% imiquimod cream applied twice weekly for 8 weeks (N = 33 participants) Control intervention B: cryotherapy: 3 to 5 second freeze cycle followed (2 weeks after) by vehicle cream for 8 weeks (N = 32 participants) Part 2: Participants with residual actinic keratosis lesions were offered cryotherapy and open‐label imiquimod twice weekly for 8 weeks. | |
| Outcomes | Primary outcomes of the trial (protocol) 1) Recurrence rate 2) Time to recurrence of lesions Secondary outcomes of the trial (protocol) 1) Time to reach treatment success 2) Proportion of participants completely clear [= participant complete clearance rates for target, subclinical and total lesions at week 22 (i.e. part 1 only)] 3) Participant improvement assessment Other outcomes of the trial 1) Clearance rates of target lesion (= lesion complete response) 2) Skin irritation 3) Treatment‐related adverse events (= minor adverse events) 4) Serious adverse events 5) New actinic keratoses (subclinical) during the study Efficacy Methods: quantitative assessment using lesion mapping on a transparent overlay map Time points: at baseline, at week 22 after cryotherapy (end of part 1) Definitions: 1. target lesions (those within a designated 50 cm² treatment field established at baseline), 2. subclinical lesions (those within the designated treatment field unapparent at baseline), 3. total lesions (the sum of target and subclinical lesions) Safety Methods: monitoring of the frequency and duration of adverse events (local and systemic) Time points: at every study visit | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | An increase in subclinical actinic keratoses was observed within the first 3 weeks of imiquimod treatment with a subsequent progressive reduction there after, but this was not observed with vehicle. This was a 2‐part study: part 1, included in the meta‐analyses, is randomised, double‐blind, and controlled, but part 2 is an optional open study not included in this review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 2): "The initial randomised double‐blind phase in which subjects were allocated to either vehicle or imiquimod 5% cream twice weekly for 8 weeks was followed by an optional open‐label phase..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded for part I. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded for part I. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 2 dropouts (the reasons were reported) Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | High risk | Some outcomes (recurrence rate, time to recurrence, time to reach success, participant improvement assessment) in the protocol (NCT00110682) were not presented in the published study. |
| Other bias | High risk | There were differences between the protocol and the published report. The primary and secondary outcomes in the published report were different than the protocol. The protocol did not mention the second part of the study. |
| Methods | This was a multicentre, randomised, assessor‐blinded, active‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% 5‐fluorouracil twice daily for 2 to 4 weeks (N = 20 participants) Control intervention B: 5% imiquimod twice weekly for 16 weeks (N = 19 participants) | |
| Outcomes | Outcomes of the trial 1) Physician global assessment as scores (presented in a graph) 2) Lesion counts at baseline, during and after treatment (= lesion complete response) 3) Participant complete and partial (> 66%) clearance 4) Mean percentage of reduction in lesion counts 5) Physician's grading of erythema (scores represented in a graph) 6) Local skin reactions (qualitative) 7) Participant's perception of efficacy 8) New/subclinical lesions Efficacy Methods: 1. quantitative assessment using lesion counting, 2. qualitative assessment using physician's global assessment and participant's perception of efficacy Time points: at baseline and weeks 4, 8, 12, 16, and 24 Definitions for physician's global assessment and the participant's perception of efficacy scales: 1 (very effective), 2 (moderately effective), 3 (slightly effective), 4 (not effective at all) Safety Methods: 1. physician's assessment of erythema on a 0 = none to 3 = severe scale, 2. participant perception of discomfort associated with the treatment. Time points: at baseline and weeks 4, 8, 12, 16, and 24 Definitions for participant's perception of discomfort scale: 1 (very painful), 2 (moderately painful), 3 (slightly painful), 4 (not painful at all) | |
| Funding | This study was supported by Valeant Pharmaceuticals International. | |
| Notes | Treatment with 5‐fluorouracil, but not imiquimod, uncovered and treated subclinical lesions. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 145): "Patients were randomly assigned to receive one of the following treatments to be applied in a thin layer to completely cover each affected cosmetic unit:.." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | The 2 distinct dosing schedules were not concealed with a double‐dummy technique. |
| Blinding of outcome assessment (detection bias) | Low risk | The assessor was blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat was used. Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 2 dropouts (the reasons were provided) |
| Selective reporting (reporting bias) | High risk | Values for participant’s perception of efficacy were not presented. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, open, active‐controlled, parallel‐group study. Start date: January 2002 End date: October 2002 | |
| Participants | Inclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) once (N = 105 participants) Control intervention B: MAL‐PDT twice with a 1 week interval (N = 106 participants) There was possible retreatment for single treatment group (not included in meta‐analysis). Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 Interval between treatments: 1 week Preparation of lesions: crusts removed by curettage and gentle scrapping Cream concentration (%): 16 Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: Aktilite CL 16 Wavelength (nm): 590‐670 Energy fluence (J/cm²): 37 Intensities (mW/cm²): 750 to 2050 Exposure time: 8 min | |
| Outcomes | Outcomes of the trial 1) Lesion complete response at 3 months post‐treatment 2) Participant complete response (= participant complete clearance) 3) Participants experiencing at least 1 adverse event 4) Local adverse events 5) Lesion cosmetic outcomes Efficacy Methods: assessment by investigator Time points: at 3 months post‐treatment Definitions: 1. lesion complete response (complete disappearance of the lesion), 2. lesion non‐complete response (incomplete disappearance of the lesion) Safety Methods: recording of adverse events, including local phototoxicity reactions that normally occur after PDT, and rating as mild, moderate, or severe (the clinician assessed the causal relationship of any adverse events to the study treatment as related, uncertain, or not related) Time points: before and after illumination, and at 3 months post‐ treatment Cosmetic Methods: assessment of hypopigmentation, hyperpigmentation, scar formation, and tissue defect by their rating as none, slight, or obvious for each lesion that had responded completely Time points: at 3 months post‐treatment | |
| Funding | This study was supported by PhotoCure ASA. | |
| Notes | A sample size calculation was provided. Data for intention‐to‐treat analysis were used for the meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 425): "The randomisation was performed after the patient was included in the study." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes were used to conceal the allocation. |
| Blinding of participants and personnel (performance bias) | High risk | This study was open. |
| Blinding of outcome assessment (detection bias) | High risk | This study was open. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 0 dropouts Control ‐ B: 6 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Low risk | The results were similar to previously reported data in an abstract. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single ‐centre, randomised, placebo‐controlled, parallel‐group study. Start date: September 1991 End date: March 1992 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: sunscreen SPF 17 (8% 2‐ethyl‐hexyl p‐methoxycinnamate/2% 4‐tert‐butyl‐4‐methoxy‐4‐dibenzoylmethane), as needed daily for 7 months (N = 221 evaluable participants) Control intervention B: placebo, as needed daily for 7 months (N = 210 evaluable participants) | |
| Outcomes | Outcomes of the trial 1) Mean reduction/increase in lesion counts at 7 months (= mean reduction in lesion counts) 2) New lesions 3) Number and per cent of baseline lesion remitting (= lesion complete response) 4) Compliance Efficacy Methods: quantitative assessment using recording of lesions Time points: at baseline and 7 months (end of the trial) Definitions: incident lesions (the number of new lesions appearing during the study) Safety Methods: recording of any untoward reactions to the creams | |
| Funding | This study was supported by grants from the Victorian Health Promotion Foundation, Melbourne; the Skin and Cancer Foundation, Sydney; the Skin and Psoriasis Foundation, Melbourne; the Uoyd Williams Trust, Maryborough; the Sydney Melanoma | |
| Notes | A sex‐based difference in the change in the number of lesions was noted. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate randomisation was achieved by stratification according to sex and self‐rated skin. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This was not stated. Base cream (vehicle) and sunscreen cream had the same consistency by adding 10% mineral oil to the vehicle. Participant blinding concealment was tested by a question at the end of study, and answers were not significant for both treatment arms. |
| Blinding of outcome assessment (detection bias) | Unclear risk | This was not stated. |
| Incomplete outcome data (attrition bias) | High risk | The type of analysis was unclear. The initial number of participants randomised and the number of dropouts (157) were given for the 2 groups together. The reasons for withdrawal were provided in a table, but because 1 participant could have more than 1 reason, it was impossible to determine how many participants withdrew in each treatment group. |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, intraindividual study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: β‐1,3‐D‐glucan, twice daily for 7 days (N = 20 participants) Control intervention B: placebo, twice daily for 7 days (N = 20 participants) | |
| Outcomes | Outcomes of the trial 1) Mean lesion counts (converted to mean reduction of lesion counts) 2) Tolerability (= local skin/adverse reactions) 3) Minor adverse events Efficacy Methods: quantitative assessment using lesion counting by the same investigator Time points: at baseline, and weeks 1, 4, and 8 Safety Methods: 1. grading of erythema and burning/stinging as absent, mild, moderate, or severe, 2. participant‐reported adverse events and concomitant medication use Time points: at baseline and weeks 1, 4, and 8 | |
| Funding | ‐ | |
| Notes | This was a pilot study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 137): "...the respective arms to which each preparation was applied were determined by randomisation. Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Unclear risk | An unclear type of analysis was used. Intraindividual study: Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | High risk | All outcomes were presented even if efficacy was higher for placebo. The standard deviations associated with mean values were not given. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: November 2002 End date: September 2005 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% imiquimod, 3 times per week for 16 weeks (N = 29 participants) Control intervention B: vehicle, 3 times per week for 16 weeks (N = 14 participants) | |
| Outcomes | Primary outcomes of the trial 1) Safety of the graft (rejection) 2) Application site reactions (imiquimod only) 3) Skin irritation (qualitative) 4) Minor adverse events (imiquimod only) 5) Serious adverse events 6) Clinical laboratory tests Secondary outcome of the trial 1) Participant complete or partial (> 75%) clearance Other outcomes of the trial 1) Lesion complete response 2) Skin quality (cosmetic) Efficacy Methods: 1. quantitative assessment using clinical counting of visible lesions in the treatment area, 2. biopsy of a lesion mapped at baseline (week 24) Time points: at weeks 7, 12, and 16 (treatment period) and weeks 19 and 24 (post‐treatment) Safety Methods: 1. monitoring of safety of the graft by an independent and blinded safety committee; monitoring of transplant rejection status, laboratory results, adverse events, local skin reactions, vital signs measurements, and the dosage of immunosuppressive medications, changes in haematology and serum chemistry (specifically: levels of serum creatinine, C‐reactive protein, and proteinuria for renal transplant recipients; levels of gamma glutamyl‐transpeptidase, glutamic‐pyruvic transaminase, glutamic‐oxalacetic transaminase, and bilirubin for liver transplant recipients; GOT and GPT, white cell blood count, serum creatinine, haemoglobin, and signs of heart failure for heart transplant recipients), 2. assessment of local skin reactions (erythema, oedema, erosion/ulceration, scabbing/crusting, weeping/exudate, vesicles, and flaking/scaling/dryness) Time points: at weeks 1, 2, 3, 5, 7, 9, 12, and 16 during the treatment period, and weeks 19 and 24 post‐treatment Cosmetic Methods: assessment of skin quality of the treatment area (skin surface, hyperpigmentation, hypopigmentation, the degree of scarring, and any atrophy) Time points: at the 8‐week post‐treatment visit (week 24) | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | The participants were organ transplant recipients, i.e. immunocompromised participants. There was no graft rejection during the study. There was an increase in the number of lesions in the vehicle group only. Clinical clearance was confirmed histologically. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 3): "Baseline data were collected, and the patients were randomised to study drug." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 6 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Little was reported on skin quality outcomes. A lot of outcomes were reported for the imiquimod‐treated group only. All outcomes from the protocol (NCT00189267) were reported in the published study. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 3% diclofenac in 2.5% hyaluronic acid, twice daily for 16 weeks (N = 24 participants) Control intervention B: vehicle, 2.5% hyaluronic acid, twice daily for 16 weeks (N = 8 participants) | |
| Outcomes | Outcomes of the trial 1) Participant complete clearance at 20 weeks and 24 months (recurrence) 2) Participant partial (> 75%) clearance at 20 weeks and 24 months (recurrence) 3) Average percentage reduction of lesions at 20 weeks (= mean percentage of lesion counts) 4) Safety of the graft (rejection) 5) Minor adverse events (qualitative) 6) Skin irritation (tolerability, presented graphically) 7) Clinical laboratory tests 8) Cosmetic outcomes 9) Skin quality (cosmetic) at 20 weeks 10) 24‐month follow up for development of new lesions and invasive squamous cell carcinoma Efficacy Methods: 1. quantitative assessment using clinical counting of visible lesions supported by a transparent grid, 2. 3 to 4 mm punch biopsy of a target lesion mapped at the initiation visit Time points: at baseline, at each visit (weeks 4, 8, 12, 16) and at the post‐treatment visit (week 20) Definitions: 1. complete clearance rate (proportion of participants at the 4‐week post‐treatment visit who had no evidence of lesions on the histology results of a target biopsy lesion site and no clinically‐visible lesions in the remainder of the treatment area), 2. partial clearance rate (proportion of participants at the 4‐week post‐treatment visit who obtained at least 75% reduction in the number of lesions counted at baseline in the treatment area), 3. clearance rate of individual lesions (percentage reduction of lesions from baseline to the 4‐weeks post‐treatment visit) Safety Methods: monitoring of transplant rejection status, laboratory results, adverse events, local skin reactions, vital signs measurements, and the dosage of immunosuppressive medications; clinical laboratory analyses: serum levels of immunosuppressive medication; levels in serum creatinine, C‐reactive protein, and proteinuria for renal transplant recipients; gamma glutamyltranspeptidase, glutamic‐pyruvic transaminase, glutamicoxalacetic transaminase, and bilirubin for liver transplant recipients; GOT and GPT, white cell blood count, serum creatinine, hemoglobin, and signs of heart failure for heart transplant recipients) Time points: at each visit (weeks 4, 8, 12, 16) and at the post‐treatment visit (week 20) Cosmetic Methods: assessment of skin quality by investigator based on skin surface, hyperpigmentation, hypopigmentation, the degree of scarring, and any atrophy Time points: at the post‐treatment visit (week 20) | |
| Funding | This study was supported by Shire Pharmaceuticals. | |
| Notes | New actinic keratoses developed at an average of 9.3 months, but no invasive squamous cell carcinoma developed within a period of 24 months. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page): "Patients with one of three organ transplant types (kidney, liver, heart) were randomised 3:1 (active:vehicle) in this vehicle‐controlled, double‐blind, parallel group design." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 2 dropouts (the reasons were reported) Control ‐ B: 2 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | There was discrepancy in the number of participants completely cleared between the abstract and published report. The lowest number in the published report was used for meta‐analysis. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, vehicle‐controlled, intraindividual study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 3% diclofenac in 2.5% hyaluronic acid gel twice daily for 4 weeks, 2 weeks off followed by aminolevulinic acid (ALA)‐photodynamic therapy (PDT) (N = 10 participants) Control intervention B: 2.5% hyaluronic acid gel twice daily for 4 weeks, 2 weeks off followed by ALA‐PDT (N = 10 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: no Cream concentration (%): ‐‐ Application of cream: ‐‐ Incubation with cream: occlusive dressing over cream for 4 hours Type of light: red light Light source: Omnilux Wavelength (nm): 633 Energy fluence (J/cm²): 80 Intensities (mW/cm²): ‐‐ Exposure time: 16 minutes (fractions) | |
| Outcomes | Outcomes of the trial 1) Mean reduction of lesion counts in a 5 X 5 cm area 2) Participant and investigator global improvement indices (GIIs) expressed as scores 3) Total thickness scores 4) Pain score 5) Severe local adverse reactions (qualitative) Efficacy Methods: 1. quantitative assessment using lesions counting in a 5 X 5 cm area, 2. assessment of lesion thickness visually and by palpation and scored on a 1 to 4 scale, 3. qualitative assessment of global improvement by the investigator and the participant (an independent dermatologist evaluated the efficacy by using photographs) Time points: at baseline, at 6 weeks and 6 months after PDT, and 8 of 10 participants were examined 12 months post‐treatment. Definitions: 1. total lesion score (number of lesions counted in 5 × 5 cm area), 2. total thickness score (sum of the thickness scores for individual lesions) Definitions for thickness score: 1 (lesion visible, but not palpable), 2 (lesion visible and palpable), 3 (elevated and keratotic lesion), 4 (hyperkeratotic lesion > 1 mm in thickness) Definitions for GIIs: –2 (significantly worse), –1 (slightly worse), 0 (no change), 1 (some improvement), 2 (moderate improvement), 3 (significant improvement), 4 (complete remission) Safety Methods: 1. scoring of pain, 2. participant‐recorded side‐effects in daily diary (number and severity) Time points: 1. during PDT (pain), 2. at each visit (side‐effects) Definitions for pain score: 0 (painless), 1 (mild pain), 2 (moderate pain), 3 (severe pain), 4 (unbearable pain) | |
| Funding | ‐ | |
| Notes | This was a pilot study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 260): "The pharmacist of the hospital randomly assigned the vehicle to 1 hand and the active drug to the other hand on each patient." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Low risk | A third party (the pharmacist) randomly assigned the vehicle to 1 hand. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | An independent dermatologist evaluated the efficacy. |
| Incomplete outcome data (attrition bias) | Low risk | Modified intention‐to‐treat (ITT, exclusion of only 1 participant who withdrew before PDT) was used. Intraindividual study: Intervention ‐ A: 2 dropouts (the reasons were reported) Control ‐ B: 2 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Only mean values were reported i.e. the associated standard deviations were not provided. |
| Other bias | Unclear risk | ‐ |
| Methods | Randomised, double‐blind, active‐controlled, parallel‐group study The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | 2 subgroups: with and without cooling spray Intervention A: methyl aminolevulinate (MAL)‐ visible + water‐filtered infrared A (VIS + wIRA) PDT (N = 40 participants) Control intervention B: MAL‐light‐emitting diode (LED) red light PDT (N = 40 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 1 or 2 Interval between treatments: 3 months Preparation of lesions: gentle removal of scales Cream concentration (%): 16 Application of cream: 1 mm thick to lesion area and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: visible + water‐filtered infrared A (VIS + wIRA) or LED red light Light source: Hydrosun type 505 Broadband with 7‐mm water cuvette and orange filter OG590 (VIS + wIRA), Aktilite CL 128 (red light) Wavelength (nm): 580‐1400 (VIS + wIRA), 630 (red light) Energy fluence (J/cm²): 240 including 60 VIS (VIS + wIRA), 37 (red light) Intensities (mW/cm²): 200 including 50 VIS (VIS + wIRA), 75 (red light) Exposure time: 20 minutes (VIS+ wIRA), 8 minutes (red light) | |
| Outcomes | Outcomes of the trial 1) Participant complete and partial (> 75%) clearance at 3 (1 treatment), 6 (1 or 2 treatments), and 12 (1 or 2 treatments) months 2) Efficacy on a visual assessment scale (VAS) 3) Pain on a VAS (first outcome presented) 4) Local skin reactions 5) Serious adverse events 6) Satisfaction and quality of life on a VAS 7) Number of spray cooling and illumination interruptions Efficacy Methods: 1. documentation of the global aspect of total actinic keratosis area by physicians, 2. rating of efficacy on a VAS [‐50 mm (extreme worsening), 0 mm (unchanged), +50 mm (extreme improvement)], 3. rating of efficacy on a five‐point scale‐rated variable 'percentage of the cleared area in relation to the initial total actinic keratosis area' (100% clearance, > 75% of the total area cleared, > 50% of the total area cleared, > 25% of the total area cleared, no relevant part of the area cleared) Time points: before PDT; at 2 weeks; and at 3, 6, and 12 months after the first PDT Safety Methods: 1. evaluation of the extent of erythema, scaling, crusts, indurations, erosions, ulcerations, and oedema on a VAS [0 (non‐existent) to 100 mm (extremely high)] by physicians, 2. evaluation of the intensity of pain, side‐effects on a VAS [0 (none) to 100 mm (extremely high)] by participants Time points: 1. before PDT; 2 weeks; and 3, 6, and 12 months after the first PDT, 2. 2, 4, 6, 8, 10, 13, 15, 20, 22, and 25 minutes after the start of PDT (pain) Cosmetic Methods: 1. evaluation of the extent of skin atrophy, scar formation, and pigmentation on a visual analogue scale (VAS) [0 (non‐existent) to 100 mm (extremely high)] by physicians, 2. assessment of cosmetic appearance by physicians and participants [VAS: 0 (extremely bad) to 100 mm (extremely good)] Time points: before treatment; at 2 weeks; and at 3, 6, and 12 months after the first PDT | |
| Funding | This study was supported by Erwin Braun Foundation. | |
| Notes | Efficacy was lower in participants receiving cooling spray. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 608): "The patient number was randomly assigned to either group 1 (VIS + wIRA PDT, n = 40 patients) or group 2 (red light PDT, n = 40 patients, Table 1)." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was a double‐blinded. Quote (page 609): "Both patients and investigators remained blinded until study completion." |
| Blinding of outcome assessment (detection bias) | Low risk | The assessor was not involved in treatment. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 1 dropout (the reasons were reported) Control ‐ B: 3 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind (treatment vs placebo),open (treatment duration), vehicle‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Interventions A: 0.5% fluorouracil cream (microsphere) applied once daily to affected areas for 1 week with 4 week follow‐up (N = 38 participants) B: 0.5% fluorouracil cream (microsphere) applied once daily to affected areas for 2 weeks with 4 week follow‐up (N = 41 participants) C: 0.5% fluorouracil cream (microsphere) applied once daily to affected areas for 4 weeks with 4 week follow‐up (N = 40 participants) Control intervention D: vehicle applied once daily to affected areas for 1, 2, or 4 weeks with 4‐week follow‐up (N = 58 participants) | |
| Outcomes | Outcomes of the trial 1) Physician global assessment of improvement (PGAI = global improvement indices) 2) Proportion of participants achieving total clearance (= participant complete clearance) 4) Mean number of lesions at baseline and end of study (transformed to absolute mean reduction in lesion counts) 5) Median number of lesions at baseline and end of study 6) Skin irritation (number of participants, severity, overtime) 7) Serious adverse events Efficacy Methods: 1. quantitative assessment using lesion counting, 2. qualitative assessment using PGAI (+5 = total clearance and ‐4 = much worse) reported as mean score Time points: at baseline and 4 weeks post‐treatment Safety Methods: 1. monitoring of adverse events (onset, duration, severity, and frequency), 2. separate recording for adverse events affecting the facial skin and scalp, 3. monitoring of facial irritation including maximum severity (0 = none, 1 = mild, 2 = moderate, Time points: during treatment: days 1 and 8 (1‐week groups); days 1, 8, and 15 (2‐week groups); or days 1, 8, 15, and 29 (4‐week groups); post‐treatment: weekly visits and a final evaluation 4 weeks after completing or discontinuing treatment | |
| Funding | ‐ | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 23): "Patients were randomised to receive 0.5% fluorouracil cream or vehicle control for 1, 2, or 4 weeks (Figure 1)." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | High risk | This study was double‐blinded (treatment vs placebo) and open (treatment duration). Indeed, placebo cream was not used to conceal allocation to 1, 2 , or 4 weeks. |
| Blinding of outcome assessment (detection bias) | High risk | Different assessment time points were used for 1‐, 2‐, or 4‐week groups. |
| Incomplete outcome data (attrition bias) | Unclear risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 1 dropout (the reason was not reported), B: 1 dropout (the reason was reported), C: 4 dropouts (the reasons were reported) Control ‐ D: 1 dropout (the reason was not reported) |
| Selective reporting (reporting bias) | High risk | The standard deviations associated with the mean number of lesions and percentages were not reported. Adverse events were not reported in this study, but they were reported in a similar study included (Jorizzo 2002). |
| Other bias | High risk | There was slight difference (P = 0.048) in the women ratio [more in 4‐week group (C) and less in placebo group (D)] at baseline. |
| Methods | This was a single‐centre, randomised, assessor‐blind, active‐controlled, intraindividual study. Start date: May 2006 End date: February 2007 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: methyl aminolevulinate (MAL)‐red light photodynamic therapy (PDT) (N = 30 participants) Control intervention B: MAL‐daylight PDT (N = 30 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: crusts and hyperkeratoses removed Cream concentration (%): 16.8 Application of cream: 1 g applied to lesion area Incubation with cream: occlusive dressing over cream for 0.5 hour (daylight) and 3 hours (red light) Type of light: LED red light or daylight Light source: Aktilite CL 128 (red light), sun (daylight) Wavelength (nm): 575‐670 (red light), 290‐670 (daylight) Energy fluence (J/cm²): 37 (red light), 11.7‐65.9 (mean = 43.2, measured with a dosimeter) Intensities (mW/cm²): ‐‐ Exposure time: 2.5 hours (daylight) | |
| Outcomes | Outcomes of the trial 1) Mean reduction in lesion counts at 3 months post‐treatment 2) Local adverse events 3) Participant's pain scores 4) Participant's satisfaction Efficacy Methods: quantitative assessment using counting, grading (Olsen 1991), mapping, and photography of lesions Time points: before treatment and at 3 months post‐treatment Definitions: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance of the lesion) Safety Methods: 1. scoring (0 = no pain to 10 = worst imaginable pain) of the pain in the 2 treated areas by participants, 2. evaluation of adverse events (erythema, crusting or pain) Time points: 1. during daylight exposure, during red LED light illumination, and after treatment (pain), 2. at 1 to 3 days after PDT (adverse events) | |
| Funding | This study was supported by The Eva and Henry Frænkels Memorial Foundation. | |
| Notes | PpIX fluorescence measured before, during, and after treatments showed less fluorescence associated with daylight. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate randomisation sequence generation was achieved by drawing lots. |
| Allocation concealment (selection bias) | Low risk | Opaque sealed envelopes were used to conceal allocation. |
| Blinding of participants and personnel (performance bias) | High risk | The 2 treatments were physically distinct. |
| Blinding of outcome assessment (detection bias) | Low risk | The evaluator was blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Per‐protocol (PP) analysis was used. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | Unclear risk | No outcomes were specified in the protocol (NCT00432224). |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, active‐controlled, intraindividual study. Start date: June 2007 End date: December 2007 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 16% methyl aminolevulinate (MAL)‐daylight photodynamic therapy (PDT) (N = 30 participants) Control intervention B: 8% MAL ‐daylight PDT (N = 30 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: sunscreen SPF20, crusts and hyperkeratoses removed Cream concentration (%):8 or 16 Application of cream: 1 g applied to lesion area Incubation with cream: all day Type of light: daylight Light source: sun (daylight) Wavelength (nm): 290‐670 (daylight) Energy fluence (J/cm²): measured with dosimeter Intensities (mW/cm²): ‐‐ Exposure time: all day | |
| Outcomes | Outcomes of the trial 1) Mean reduction in lesion counts 2) Total lesion count (lesion complete response) 3) Pain scores 4) Erythma scale and per cent by measurements before and after treatment with skin reflectance meter 5) Participant's preference Efficacy Methods: quantitative assessment using counting, grading (Olsen 1991), mapping, and photography of lesions Time points: before treatment and at 3 months post‐treatment Definitions: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance of the lesion) Safety Methods: 1. scoring (0 = no pain to 10 = worst imaginable pain) of the pain in the 2 treated areas by participants in a diary every hour, 2. evaluation of adverse events (erythema, crusting, or pain) visually on a 4‐point scale by a dermatologist, 3. quantitative evaluation of erythema [erythema percentage measured with a skin reflectance meter (Optimize Scientific; Chromo Light Aps,Skodsborg, Denmark)] Time points: 1. during daylight exposure, during red LED light illumination, and after PDT (pain), 2. 1 to 3 days after PDT(adverse events), 3. before curettage and on the day after PDT (erythema evaluation) Definitions for visual evaluation of erythema: 1. 0 (no visible erythema), 2. (+) (just perceptible erythema), 3. + (uniform erythema), 4. ++ (bright red erythema and induration) | |
| Funding | ‐ | |
| Notes | PpIX fluorescence was measured using fluorescence camera, and there was no difference between the 2 cream concentrations. There was a correlation with light exposure/dose and response rate. Pain increased with light exposure. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate randomisation sequence was achieved by drawing lots. |
| Allocation concealment (selection bias) | Low risk | Opaque sealed envelopes were used to conceal allocation. |
| Blinding of participants and personnel (performance bias) | Low risk | Quote (page 1309): "The evaluating dermatologist and participants were blinded to the concentrations of creams." |
| Blinding of outcome assessment (detection bias) | Low risk | Quote (page 1309): "The evaluating dermatologist and participants were blinded to the concentrations of creams." |
| Incomplete outcome data (attrition bias) | Low risk | The type of data analysis was unclear, but only 1 participant was lost to follow up. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | High risk | The number of participants and average time spent outside were different between the abstract and published report. There was a little confusion regarding the type of efficacy outcome reported in the abstract. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, active‐controlled, parallel‐group study. Start date: June 2008 End date: January 2009 | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 2h MAL‐1.5h daylight photodynamic therapy (PDT) (N = 58 participants) Control intervention B: 3h MAL‐2.5h daylight PDT (N = 62 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: sunscreen SPF20, crusts and hyperkeratoses removed Cream concentration (%): 16 Application of cream: thick layer applied to lesion area Incubation with cream: 2 or 3 hours (0.5 hour without light exposure) Type of light: daylight Light source: sun (daylight) Wavelength (nm): 290‐670 (daylight) Energy fluence (J/cm²): 8.6 (1.5 hours), 10.2 (2.5 hours) Intensities (mW/cm²): ‐‐ Exposure time: 131 + 37 min (1.5 hours) or 187 + 52 min (2.5 hours) | |
| Outcomes | Primary outcomes of the trial 1) Mean reduction in lesion counts at 3 months post‐treatment 2) Mean percentage of reduction in lesion counts at 3 months post‐treatment [which correspond to the primary and only outcome "response rate" in the protocol NCT00711178 based on the published report (see efficacy definitions below)] Other outcomes of the trial 1) Pain scores 2) Local adverse reactions: erythema and pustular eruptions (pooled data) 3) Participants' satisfaction 4) New actinic keratosis lesions Efficacy Methods: quantitative assessment using lesion grading and counting using a template Time points: at baseline and 3 months post‐treatment Definitions: 1. lesion response rate (number of completely responding lesions divided by the number of lesions treated within the individual participants), 2. complete response (complete disappearance of the lesion, visually and by palpation ‐ mild erythema might Safety Methods: 1. participant‐recorded pain score (0 = no pain and 10 = worst imaginable pain) in diary, 2. erythema and pustular eruption rating by investigators (other adverse events were recorded if the investigators considered these to be related to treatment) Time points: 1. every half hour during the day of treatment and 4 time‐points the following day (pain), 2. at 2 days after PDT (erythema and pustules) Definitions for erythema rating: 1. none (no redness), 2. mild (visibly pink colour), 3. moderate (red colour), 4. severe (dark red purple) Definitions for pustular eruptions rating: 1. none (no pustules), 2. mild (few pustules), 3. moderate (several pustules), 4. severe (severe pustular eruption with yellow crusting) | |
| Funding | This study was supported by Department of dermatology, Bisebjerg Hospital, Copenhagen. | |
| Notes | The effective daylight dose was measured with electronic wristband dosimeter. The weather conditions were monitored every half hour in diary. An increase in pain and erythema was associated with higher effective light dose. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 2): "The randomisation procedure was performed by a computer‐generated sequence blocked by centre." |
| Allocation concealment (selection bias) | Low risk | Quote (page 2): "Allocations were contained in opaque, sequentially numbered, sealed envelopes and were concealed from assessors throughout the study." |
| Blinding of participants and personnel (performance bias) | High risk | This was not stated, but the participants were exposed to light for different periods of time. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was assessor‐blinded (based on the protocol NCT00711178, clinicaltrials.gov). |
| Incomplete outcome data (attrition bias) | Low risk | Intention‐to‐treat (ITT) analysis was used for primary outcomes, and per‐protocol (PP) analysis was used for secondary outcomes. Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Unclear risk | All primary outcomes were reported based on the protocol NCT00711178. Additional secondary outcomes, which were not included in our review, were also reported. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 3% diclofenac gel in 2.5% hyaluronic acid gel, 0.5 g twice daily for 90 days (N = 58 participants based on safety data) Control intervention B: 2.5% hyaluronic acid gel only, 0.5 g twice daily for 90 days (N = 59 participants based on safety data) | |
| Outcomes | Primary outcomes of the trial 1) Participant Global Improvement Indices = 4 2) Investigator Global Improvement Indices = 4 3) Participants with target lesion number score = 0 at 30 days post‐treatment (= participant complete clearance) Other outcomes of the trial 1) Participants experiencing at least 1 adverse event 2) Application site reactions 3) Minor adverse events 4) Serious adverse events 5) Clinical laboratory tests Efficacy Methods: 1. quantitative assessment using lesion counting, 2. qualitative assessments (IGII and PGII) Time points: at each visit Definitions: 1. target lesion number score (number of lesions identified in the designated treatment blocks at baseline), 2. cumulative lesion number score (number of lesions identified ‐target or new‐in the designated treatment blocks) Definitions for IGII and PGII 7‐point scale: + 2 (significantly worse), +1 (slightly worse), 0 (no change), 1 (slightly improved), 2 (moderately improved), 3 (significantly improved), and 4 (completely improved) Safety Methods: 1. participant‐recorded concomitant medications and adverse events in diary; 2. assessment of adverse events for duration, intensity, and causality by physician; 3. standard laboratory analyses [hematology, biochemistry, urinalysis, and serology (antidiclofenac)] Time points: at screening and end of treatment (laboratory tests) | |
| Funding | This study was supported by Hyal Pharmaceutical Co. | |
| Notes | A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 710): "One week after the Screening Visit, patients were randomised to receive either the active treatment, 3% diclofenac in 2.5% hyaluronan gel (SolarazeTM Bioglan) or placebo, which consisted of the inactive gel vehicle, hyaluronan only." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | Unclear risk | Intention‐to‐treat (ITT) analysis was used, but the initial randomised numbers for each group were not given explicitly. Intervention ‐ A: 14 dropouts (the reasons were reported) Control ‐ B: 8 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single‐centre, randomised, double‐blind, placebo‐controlled, intraindividual study. The start and end dates were not specified. | |
| Participants | Inclusion criteria of the trial
Demographics
| |
| Interventions | Intervention A: 5% imiquimod, once weekly for 24 weeks (N = 20 participants) Control intervention B: placebo, once weekly for 24 weeks (N = 20 participants) | |
| Outcomes | Outcomes of the trial 1) Investigator assessment scale (= global improvement indice) 2) Total lesion number score 3) Local skin reactions (qualitative) 4) Serious adverse events 5) Skin irritation score (graph) Efficacy Methods: 1. qualitative assessment with an investigator assessment scale (7‐point), and 2. quantitative assessment with lesion counting Time points: every 4 weeks during the treatment period and at 4 weeks post‐treatment Definitions for the investigator assessment scale: ‐2 (much worse), ‐1 (slightly worse), 0 (no change), 1 (mild improvement), 2 (moderate improvement), 3 (marked improvement), 4 (cured) Definitions: total lesion number score (total number of lesions present in the target area Methods: 1. monitoring the occurrence of local adverse events and systemic adverse events, 2. rating of skin irritation by participants on a 6‐point scale (there was no objective measure of local side‐effects) Time points: at each visit Definitions for skin irritation scale: 0 (no irritation), 1 (trace irritation), 2 (mild irritation) 3 (moderate irritation), 4 (marked irritation), 5 (severe irritation) | |
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | This was a pilot study. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page): "Enrolled patients were randomised, 1:1, to apply imiquimod to a 20‐cm²area on the right or left side." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) | High risk | Per‐protocol (PP) analysis was used with 25% of subjects lost. Intraindividual study: Intervention ‐ A: 5 dropouts (the reasons were reported) Control ‐ B: 5 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| We assessed actinic damage in general, and the study was not specific to actinic keratoses. Only 60% of participants had clinically‐evaluable actinic keratoses on the forearms. | |
| The study did not randomise all the participants. | |
| This was a conference abstract without numerical values for efficacy. | |
| The study did not meet the outcome requirements of the review. | |
| The efficacy of the intervention was on new actinic keratosis lesions (prevention), not on baseline lesions. | |
| The study was not randomised. Predefined sides were treated with 2 different light sources for PDT, i.e. always the same side for 1 treatment. | |
| The study did not meet the outcome requirements of the review. | |
| The study did not meet the outcome requirements of the review. | |
| The study did not meet the outcome requirements of the review. | |
| The study was not randomised; the participants were allocated based on participant and physician judgement. | |
| The study did not meet the outcome requirements of the review. | |
| The study did not meet the outcome requirements of the review. | |
| The study did not present efficacy results numerically. | |
| The study did not present efficacy results numerically; they were given in graphical form. | |
| The study did not clearly present efficacy measures. The data were presented in graph form with no quantitative numbers. | |
| The study was randomised (90‐ versus 180‐day treatments), but only partial data at 3 months were presented based on the conference abstract. And only data from the 90‐day group were presented in the peer‐reviewed paper, i.e. there was no comparison. | |
| This trial was terminated due to "Orphan Drug Designation for this indication not granted". | |
| The type of interventions were not covered in this review, i.e. injection in participant lesion. | |
| This study was on the prevention of actinic keratosis lesions. | |
| The study did not meet the outcome requirements of the review. | |
| The study did not meet the outcome requirements of the review. | |
| This was a follow‐up study of an included study. | |
| It was not clear if the study was randomised. | |
| The randomisation was based on participant preference, and there was no efficacy comparison between the 2 treatments. | |
| Out of 80 participants, only 4 had actinic keratosis lesions, and mean lesions counts were reported for all subjects. | |
| The randomisation was not mentioned. Little information was given. | |
| The randomisation was not mentioned. | |
| The study did not meet the outcome requirements of the review. | |
| This was a follow‐up study for Hanke 2010; Swanson 2010a. | |
| Actinic keratosis lesions were not distinguished from lentigines for efficacy data. | |
| The criteria for outcomes was not met, i.e. median lesion counts were provided instead of mean lesion counts. | |
| This study did not meet the outcome requirements of the review; it presented results as the mean reduction in lesion area. | |
| There was an inadequate method of randomisation: Every other participant was given a combination treatment on the left side of the face and a monotherapy treatment on the right. All other participants were given the opposite treatment. | |
| This study did not meet the outcome requirements of the review. | |
| This study was on the prevention of actinic keratosis lesions formation, not an intervention to cure. | |
| The trial was terminated because of the low conditional power for a positive study. | |
| This study did not meet the outcome requirements of the review. | |
| This study did not meet the outcome requirements of the review. | |
| 50% of participants were lost to follow up. No statistics were reported. There was too much variability within and between groups as far as following instructions for application. There was no initial counting of lesions. | |
| There was not enough information in this conference abstract of a phase II trial to be able to use the data. | |
| The type of interventions were not covered in this review, i.e. injection in participant lesion, and it was unclear if the study was randomised | |
| There were no numerical results. | |
| This study did not meet the outcome requirements of the review. | |
| This study did not meet the outcome requirements of the review. | |
| No enough numerical information was provided, e.g. number of participants in each of the 8 treatment groups. | |
| This was a long‐term follow‐up to a previous study: Stockfleth 2002 | |
| This was a follow‐up study of Hauschild 2009a; Hauschild 2009b; and Hauschild 2009c. | |
| The study did not present efficacy results numerically; they were given in graph form. | |
| It was unclear if the study was randomised based on this conference abstract. | |
| The randomisation was not mentioned. | |
| This trial was terminated due low accrual. | |
| The study did not present numerical data. | |
| This study was on prevention of new lesions and mixed lesion types, i.e. not only actinic keratoses. | |
| This study does not provide efficacy data on intervention. The primary outcome was mean time to occurrence of first new lesion. | |
| There was not enough information. Results taken from 2 studies and combined, i.e. no direct comparison. |
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | This was a single‐centre, randomised, open, assessor‐blinded, placebo‐ and active‐controlled, parallel‐group study. The start and end dates were not specified. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: 3% diclofenac in 2.5% hyaluronic acid, twice daily for 12 weeks (N = 21 participants) Control interventions B: 5% imiquimod, twice per week for 16 weeks (N = 20 participants) C: placebo (Ultrabase cream; Schering Alman, Istanbul, Turkey), twice daily for 12 weeks (N = 20 participants) |
| Outcomes | Outcomes of the trial 1) Total thickness score (TTS) 2) Patient global improvement index (PGII) 3) Complete clearance (= lesion complete response) 4) Local skin reactions Efficacy Methods: quantitative assessment using a scale and photography of lesions Time points: before treatment and at every 4 weeks up to 24 weeks Definitions: 1. TTS scale, 0 = complete clearance, 1 = lesion visible but not palpable, 2 = lesion visible and palpable, 3 = lesion raised with visible scaling, 4 = lesion hyperkeratotic and > 1 mm in thickness; 2. PGII is a self‐report scale, and participants evaluated themselves according to a 7‐point scale (0 = significantly worse, 1 = slightly worse, 2 = no change, 3 = slightly improved, 4 = moderately improved, 5 = significantly improved, 6 = completely improved) Safety Methods: evaluation of local skin reactions (erythema, oedema, erosion ⁄ ulceration, scabbing ⁄ crusting, weeping⁄ exudates, vesicles, and scaling ⁄ dryness) on a 0 to 3 scale Time points: at every 4 weeks up to 24 weeks Definitions: 0 = none, 1 = mild, 2 = moderate, 3 =severe |
| Notes | ‐ |
| Methods | This was a single‐centre, randomised, open, assessor‐blinded, intraindividual study. Start date: February 2009 End date: May 2010 |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Interventions A: 50 mW/cm² ALA‐red light photodynamic therapy (PDT) (N = 50 participants) B: 75 mW/cm² ALA‐red light PDT (N = 50 participants) Control intervention C: 25 mW/cm² ALA‐red light PDT (N = 50 participants) Characteristics of PDT intervention Type of treatment: individual lesion Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: ‐‐ Cream concentration (%): 20 Application of cream: ‐‐ Incubation with cream: 4 hours with occlusion Type of light: red Light source: Waldmann PDT 1200 (non‐coherent light) Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): 25‐75 Exposure time:‐‐ At the 3‐month follow‐up visit, lesions without a complete response were treated with surgical techniques (cryosurgery or excision). |
| Outcomes | Primary outcomes of the trial 1) Pain on a visual analogue scale (VAS) during illumination Secondary outcomes of the trial 1) Lesion complete response at 3 and 12 months post‐treatment 2) Adverse events (qualitative) Efficacy Methods: 1. quantitative assessment using lesion counting, Time points: at baseline; and at 3, 6, and 12 months post‐treatment Definitions: 1. complete response (CR): complete absence of any clinical sign indicative of actinic keratoses, 2. non‐complete response (non‐CR): remaining clinical signs indicative of actinic keratoses Safety Methods: recording of adverse events Time points: from 5‐ALA application time point until the end‐of‐study |
| Notes | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: January 2010 End date: March 2011 |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: cryotherapy followed by Acnalene 0.1% gel at day 10, twice daily for 3 months Control intervention B: cryotherapy followed by placebo at day 10, twice daily for 3 months |
| Outcomes | Primary outcomes of the trial 1) Changes in the average number of actinic keratosis lesions (= mean reduction in lesion counts) 2) Mean frequency of reduction (= mean percentage of reduction in lesion counts) 3) Clinical outcome based on change in lesion number [including 'recovery rate of over 75%' (= participant partial (> 75%) clearance)] Secondary outcomes of the trial 1) Adverse events 2) Cosmetic outcomes Efficacy Methods: quantitative assessment by lesion counting and photography Time points: at baseline and monthly after the beginning of the topical treatment Definitions: 1. full recovery (greater than or equal to 75% reduction in the number of lesions), 2. relative recovery (30 to 75 per cent reduction in the number of lesions), 3. no response (no reduction or [greater than or equal to] 30% reduction in the number of lesions), 4. worsening (an increase in the number of lesions) Safety Methods: clinical examination and questioning of participants Cosmetic Methods: assessment of changes in pigmentation and scar formation |
| Notes | This study corresponded to the protocol IRCT201010104901N1. |
| Methods | This was a single‐centre, randomised, double‐blind, placebo‐controlled, parallel‐group study. The start and end dates were not specified. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: administration of 500 mg nicotinamide tablet, twice daily for 4 months Control intervention B: placebo tablet made of lactose. The dose, frequency, and duration of treatment are the same as for the nicotinamide group (i.e. 500 mg twice a day for 4 months) |
| Outcomes | Primary outcomes of the trial 1) Reduction in total actinic keratosis count at 4 months compared with baseline count Secondary outcomes of the trial 1) Reduction in site‐specific (i.e. face, arms, scalp) actinic keratosis count at 2 and 4 months compared with baseline count 2) Reduction in skin cancers (posthoc) Efficacy Methods: quantitative assessment by blinded clinical examination by a medically‐qualified observer Time points: at baseline, and 2 and 4 months |
| Notes | ‐ |
| Methods | Randomised, open, active‐controlled, intraindividual study The start and end dates were not specified. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: topical anaesthetic with occlusion (lidocaine 5% and prilocaine 5%) was applied for 2 hours before cryopeeling (on a field) followed by treatment of individual lesions with liquid nitrogen (LN) (N = 16 participants) Control intervention B: topical anaesthetic with occlusion (lidocaine 5% and prilocaine 5%) was applied for 2 hours before cryopeeling (on a field) followed by treatment of individual lesions with dimethyl ether, propane, and isobutane gases in a portable system (PS) (N = 16 participants) Topical Vaseline was used in the postoperative period to moisturise the skin and reduce the discomfort of healing. |
| Outcomes | Outcomes of the trial 1) Percentage of lesions completely healed (= lesion complete response) at 2 months post‐treatment 2) Pain on the visual analogue scale (VAS) from zero (no discomfort) to 10 (worst discomfort possible) 3) Global preference of physician and participant based on a 5‐point scale, which ranged from ‐2 (right much better than left) to +2 (left much better than right) 4) Cosmetic outcomes Efficacy Methods: quantitative assessment using marking of the lesions with acetate sheets and permanent‐ink pen, standardised photographic documentation, and counting Time points: at baseline and days 7, 14, 21, 3, and 60 Definitions: lesions that had healed completely (showed no sign of a previous lesion) Cosmetic Methods: 3 blinded dermatologists evaluated how much the appearance of the skin had improved following a standardised scale based on comparison with pictures taken at baseline Time points: at 60 days Definitions: 0 (no improvement), 1 (a little better), 2 (much better) |
| Notes | ‐ |
| Methods | This was a multicentre, randomised, assessor‐blind, placebo‐ and active‐controlled, parallel‐group study. The start and end dates were not specified. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: BF‐200 (10%) ALA gel‐photodynamic therapy (PDT) (N = 248 participants) Control interventions B: 16% MAL cream ‐PDT (N = 247 participants) C: placebo gel‐PDT (N = 76 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 Interval between treatments: 12 weeks Preparation of lesions: mild curettage, roughening and alcohol wiping Cream concentration (%): ‐‐ Application of cream: ‐‐ Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: Aktilite CL 128, PhotoDyn 750/505, Omnilux PDT, Waldmann PDT 1200L Wavelength (nm): 630 (Aktilite, Omnilux), 580‐1400 (PhotoDyn), 600‐750 (Waldmann) Energy fluence (J/cm²): 37 (Aktilite, Omnilux), 170 (PhotoDyn), 100 (Waldmann) Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance at 12 weeks after the first and last PDT Secondary outcome of the trial 1) Lesion complete response at 12 weeks after the last PDT Other outcomes of the trial 1) Local skin reactions at application sites before and after PDT 2) Adverse events 3) Serious adverse events 3) Pain on a visual analogue scale (VAS: 0 represents no pain, 1 to 3 are interpreted as 'mild', 4 to 7 as 'moderate', and 8 to 10 as 'severe' pain) during PDT 4) Cosmetic outcomes: general Outcomes were stratified for different light sources, mild and moderate lesions, and lesions on the face and scalp. Efficacy Methods: quantitative assessment of lesion clearance by visual inspection and by palpation by an investigator not involved in treatment and safety evaluation Time points: at baseline, and 3 and 12 weeks post‐treatment Definitions: participant complete clearance (all lesions were considered to be cleared both by the clinical assessment) Safety Methods: 1. recording of adverse effects, 2. documentation of local adverse reactions (pain, itching, burning, erythema, oedema, and induration) at the application site and rated as mild, moderate, and severe by the assessing physician or reporting participants, 3. serious adverse events Time points: 1. at 1 week after PDT (by phone) and 3 weeks, 2. during and after PDT (local adverse reactions), 3. throughout the study (serious adverse events) Cosmetic Methods: 1. general cosmetic outcome assessed by the investigator as very good, good, unsatisfactory, and impaired; 2. assessment of skin quality Time points: at 12 weeks post‐treatment |
| Notes | This was a confirmatory phase III study. |
| Methods | This was a multicentre, randomised, assessor‐blind, intraindividual study. The start and end dates were not specified. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: pretreatment with tazarotene gel (0.1 %) twice daily for 1 week + Control intervention B: no pretreatment + ALA‐blue light PDT on the entire treatment area (N = 10 participants) Characteristics of PDT intervention Type of treatment: field‐directed Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: ‐‐ Cream concentration (%): 20 Application of cream: first applied to individual lesions and then to entire treatment area Incubation with cream: 1 hour, with occlusion on the control side Type of light: blue light Light source: BLU‐U Wavelength (nm): ‐‐ Energy fluence (J/cm²): 10 Intensities (mW/cm²): 10 Exposure time: 16 minutes 40 seconds |
| Outcomes | Outcomes of the trial 1) Lesion counts on the target and entire treatment areas after pretreatment and 8 weeks after PDT compared to baseline 2) Median per cent reduction in lesion counts in the target and entire treatment areas after pretreatment and 8 weeks after PDT 3) Participants with different percentage reduction in lesion counts in the target and entire treatment areas (100% = participant complete clearance) after pretreatment (target area only) and 8 weeks after PDT 4) Investigator global assessment (IGA) after pretreatment and 8 weeks after PDT 5) Tolerance (pigmentary changes, erythema, edema, stinging/burning, scaling/dryness, and oozing/vesiculation) at baseline, after pretreatment, after PD, and 8 weeks after PDT 6) Participant satisfaction on 5‐point scale in which 0 = poor and 4 = excellent Efficacy Methods: 1. quantitative assessment by lesion counting, 2. qualitative assessment by investigator (IGA) on a 5‐point scale Time points: at baseline, after pretreatment, and 8 weeks after PDT Definitions: IGA: 0 = clear and 5 = very severe Safety Methods: 1. postinflammatory hyperpigmentation, erythema, scaling & dryness, edema, and oozing/crusting/vesiculation were assessed using a 5‐point ordinal scale (0 = none to 4 = severe), 2. participants rated stinging and burning on a 4‐point scale (0 = none and 3 = severe), 3. adverse events were recorded Time points: at each visit and phone call 24 to 48 hours after PDT |
| Notes | ‐ |
| Methods | This was a randomised, parallel‐group study. The start and end dates were not specified. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Interventions A: 20 J/cm² ALA‐Intense Pulsed Light (IPL) PDT (N = 4 participants) B: 25 J/cm² ALA‐IPL PDT (N = 4 participants) C: 40 J/cm² (2 passes of 20 J/cm²) ALA‐IPL PDT (N = 5 participants) Control intervention E: IPL PDT (N = 3 participants) Characteristics of PDT intervention Type of treatment: ‐‐ Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: ‐‐ Cream concentration (%): 20 Application of cream: twice Incubation with cream: 2 hours Type of light: IPL Light source: ‐‐ Wavelength (nm): ‐‐ Energy fluence (J/cm²): ‐‐ Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
| Outcomes | Outcomes of the trial 1) Global response to treatment (actinic keratoses and photodamage) was determined by a 7‐point scale (= grade) 2) Mean clearance rates for actinic keratoses only (= difference between mean grades before PDT and 8 weeks after PDT) 3) Tolerability (adverse reactions) at 48 hours after PDT 4) Participant discomfort during PDT Efficacy Methods: 1. quantitative assessment of 5 previously identified lesions (non‐hyperkeratotic, < 1 cm in diameter, dry, rough, yellowish, with scales) marked in each participant, numbered from 1 to 5, and documented by the FotoFindermediscope system (photography); 2. qualitative assessment (global response) on a 7‐point scale Time points: at baseline, and 48 hours and 8 weeks after PDT Definitions: 7‐point scale: 0 = complete response, 1 = 90% improvement, 2 = 75% improvement, 3 = 50% improvement, 4 = 10% improvement, 5 = no improvement, and 6 = condition worsened Safety Methods: 1. erythema, edema, crusts, and erosions were graded on a 5‐point scale (0= none to 4= severe), 2. burning or stinging during treatment was graded on a 4‐point scale (0 = none to 3 = severe). Time points: at 48 hours after PDT |
| Notes | The study included participants with actinic keratoses, photodamage, or both. |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group studies (4 studies). Start date: September 2008 End date: October 2009 |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: face or scalp: 0.015% PEP005 gel, once daily for 3 days (N = 277 participants) trunk or extremities: 0.05% PEP005 gel, once daily for 2 days (N = 226 participants) Control intervention B: vehicle gel, once daily for 2 or 3 days (N = 502 participants) |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates at day 57 Secondary outcomes of the trial 1) Participant partial (> 75%) clearance rates at day 57 2) Median percentage changes from baseline in total number of lesions at day 57 and 12 months follow‐up (posthoc) Other outcomes of the trial 1) New actinic keratosis lesions or recurrence at 12 months follow‐up for participants complete cleared in 3 of the studies (posthoc) 2) Local skin reactions on a 5‐point scale (individual scores and time course of the mean composite score) 3) Application‐site adverse reactions 4) Adverse events 5) Serious adverse events 6) Pigmentation changes and scarring (cosmetic) Efficacy Methods: quantitative assessment by a study investigator who examined the selected treatment area in person Time points: at baseline, and day 57 and 12 months follow‐up Safety Methods: 1. assessment of the incidence rate of adverse events, serious adverse events, and local skin responses; 2. grading of local skin responses with photographic guides to ensure uniform reporting for the following: erythema, flaking or scaling, crusting, swelling, vesiculation or pustulation, and erosion or ulceration Time points: on days 3 (trunk or extremities), 4 (face or scalp), 8, 15, 29, and 57 Definitions: composite local‐skin response score (sum of the 6 individual scores that were reported at each study visit for each participant ‐ maximum composite score = 24) Cosmetic Methods: assessment of pigmentation and scarring Time points: on days 3, 8, 15, 29, and 57 |
| Notes | This included 4 studies: NCT00742391 (= Swanson 2010b included in analyses), NCT0091551, NTC00916006, and NCT00942604. |
| Methods | This was a randomised, active‐controlled, parallel‐group study. Start date: January 2009 The end date was not specified. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: MAL‐red light PDT followed 1 month later by 5% imiquimod, 3 times per week for 4 weeks (N = 32 participants) Control interventions B: MAL‐red light PDT (N = 40 participants) C: 5% imiquimod, 3 times per week for 4 weeks (N = 33 participants) Characteristics of PDT intervention Type of treatment: field‐directed Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: curettage of the most hyperkeratotic lesions Cream concentration (%): 16 Application of cream: whole treatment area Incubation with cream: 3 hours with occlusion Type of light: red light Light source: Aktilite CL 1 Wavelength (nm): ‐‐ Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: 8 minutes |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rates (clinical) at 1 month post‐treatment Other outcomes of the trial 1) Participant partial (> 75%) clearance rates (clinical) at 1 month post‐treatment 2) Clinicopathologic response (= clinical complete clearance and histological clearance) at 1 month post‐treatment 3) Local skin reactions for imiquimod treatment at week 4 4) Tolerance (comfort, discomfort, pain, local skin reaction, side‐effects, waiting time, and duration of treatment) evaluated on an analogue scale of 0 (well tolerated) to 10 (very poorly tolerated) after PD, imiquimod treatment, or both 5) Participant satisfaction (benefit, improvement achieved, side‐effects, and tolerance) on an analogue scale of 0 (very dissatisfied) to 10 (very satisfied) at 1 month post‐treatment Efficacy Methods: 1. quantitative assessment by visual examination and palpation, 2. histological evaluation (biopsy) on 2 prespecified lesions identified by photography: lesion 1 before treatment and lesion 2 after treatment Time points: at baseline, and at 1 month post‐treatment Definitions: 1. complete clinical clearance (total absence of actinic keratoses or lesions clinically suspected of being actinic keratoses), 2. clinicopathologic response (complete clinical response and absence of actinic keratoses in the biopsy specimen of lesion 2), 3. absence of actinic keratoses (normalisation of the stratum corneum with no parakeratosis and normal maturation of epidermal keratinocytes with no atypical keratinocytes) Safety Methods: evaluation of the intensity of the local reaction (mild, moderate, or severe) Time points: at week 4 of treatment Definitions: 1. mild local reaction (occasional appearance of limited edema and mild erythema in the treatment area), 2. moderate local reaction (erythema, edema, ulceration, and flaking in at least 50% of the treatment area), 3. severe local reaction (erythema, edema, ulceration, and crusts occupying almost all of the treatment area and even extending beyond the treatment area) |
| Notes | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐ and active‐controlled, parallel‐group study. Start date: 2008 End date: 2009 |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: 0.5% 5‐FU in combination with 10% salicylic acid (SA) solution (LAS41005), once daily for up to 12 weeks (N = 98 participants) Control interventions B: 5‐FU/SA vehicle, once daily for up to 12 weeks (N = 187 participants) C: 3% diclofenac in hyaluronic acid (HA)(LAS106521), twice daily for up to 12 weeks (N = 185 participants) If severe side‐effects occurred, frequency of drug application could be |
| Outcomes | Primary outcome of the trial 1) Histological clearance rate of 1 predefined lesion Secondary outcomes of the trial 1) Lesion counts at baseline, and at week 12 (end of treatment) and 20 (8 weeks post‐treatment) 2) Mean total lesion area at baseline and week 20 (8 weeks post‐treatment) 3) Participant complete clearance (clinical) at week 20 4) Investigator's and participant's global assessment at weeks 6, 12, and 20 5) Treatment‐emergent adverse events (including application site reactions) 6) Tolerance (inflammation and burning) 7) Serious adverse events Efficacy Methods: 1. quantitative assessment by visual inspection of the treatment area and determination of the number and size of lesions, 2. biopsy of 1 representative target lesion performed by 3 mm punch biopsy and a second predefined clinically identical lesion for the biopsy at the post‐treatment visit evaluated by an independent and blinded dermatopathologist. Biopsy sites were selected on the basis of clinical appearance (clinical grade, area, and size). Photodocumentation and grid documentation were performed for the purposes of lesion identification. No skin tattoos were used, and no photodocumentation was performed during interim visits, 3. qualitative assessment by physician and participant ranging from 'very good' to 'none' Time points: at weeks 2, 4, 6, 10, 12 (end of treatment), and 20 (8 weeks Safety Methods: participant‐reported adverse events Time points: at each visit |
| Notes | This corresponded to protocol NCT00987246 and EudraCT No. 2007‐003889‐18. |
| Methods | This was a randomised, assessor‐blinded, active‐controlled, intraindividual study. Start date: December 2008 End date: May 2009 |
| Participants | Inclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: MAL‐2.5h low‐intensity artificial daylight photodynamic therapy (PDT) (N = 20 participants) Control intervention B: MAL‐red light‐emitting diode (LED) PDT (N = 20 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment (100 cm²) Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: scales and hyperkeratoses removed with a curette Cream concentration (%): 16 Application of cream: ‐‐ Incubation with cream: 3 hours (under occlusion for 0.5 hours for daylight and 3 hours for red LED) Type of light: artificial daylight, red light Light source: Xenon H4 light bulbs (daylight) Wavelength (nm): ‐‐ Energy fluence (J/cm²): 37 (red LED) Intensities (mW/cm²): ‐‐ Exposure time: 2.5 hours for artificial daylight |
| Outcomes | Primary outcome of the trial 1) Lesion complete response Other outcomes of the trial 1) Mean reduction in lesion counts at 3 months post‐treatment 2) Participant complete response (= participant complete clearance) at 3 months post‐treatment 3) Pain scores (0 = no pain and 10 = worst imaginable pain) every half hour during 'daylight' exposure and every 1.5 minutes during red LED treatment 4) New actinic keratosis lesions 5) PpIX fluorescence Efficacy Methods: quantitative assessment using lesion grading and counting using a template Time points: at baseline and 3 months post‐treatment Definitions: complete response (complete disappearance of the lesion, visually and by palpation ‐ mild erythema might remain) |
| Notes | ‐ |
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Randomised, double‐blind, placebo‐controlled study to assess efficacy of oral nicotinamide (500 mg daily) in the treatment and prevention of actinic keratoses |
| Methods | This is a randomised, double‐blind, placebo‐controlled, parallel‐group. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: oral nicotinamide 500 mg daily for 4 months Control intervention B: placebo tablets (lactose tablets identical in appearance and size to nicotinamide tablets but without active ingredient) daily for 4 months |
| Outcomes | Primary outcome of the trial 1) Reduction in total actinic keratosis count at 2 and 4 months from baseline |
| Starting date | August 2010 |
| Contact information | A/Prof Diona Damian ([email protected]) Dermatology Australia |
| NCT | ‐ |
| Notes | This study is ongoing and the last update was in August 2010. (April 2012) |
| Trial name or title | Vehicle‐Controlled, Double‐Blind Study to Assess the Safety and Efficacy of Imiquimod 5% Cream for the Treatment of Actinic Keratosis on the Upper Extremities |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: imiquimod 5% cream, once per day, 2 days per week for 16 weeks Control intervention B: vehicle cream, once per day, 2 days per week for 16 weeks |
| Outcomes | Primary outcome of the trial 1) Efficacy of imiquimod 5% cream compared to vehicle cream 1) Safety of treatment with imiquimod 5% cream |
| Starting date | May 2005 |
| Contact information | Graceway Pharmaceuticals, LLC |
| NCT | NCT00115154 |
| Notes | This study has been completed and the last update was in February 2007 (April 2012). |
| Trial name or title | Comparison of the Efficacy and Tolerability of Solaraze for 3 vs. 6 Months in Patients With Mild to Moderate Actinic Keratosis Located at the Face and Head |
| Methods | This is a multicentre, randomised, open, active‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: diclofenac, twice daily for 3 months Control intervention B: diclofenac, twice daily for 6 months |
| Outcomes | Primary outcome of the trial 1) Histologically controlled complete clearance of the actinic keratosis at 6 weeks post‐treatment |
| Starting date | June 2005 |
| Contact information | Claus Garbe, MD Skin Cancer Program, Department of Dermatology, University Hospital Tübingen |
| NCT | NCT00204542 |
| Notes | This study has been completed in December 2010, and the last update was in August 2011 (April 2012). |
| Trial name or title | A Multicentre, Randomised Study of Photodynamic Therapy(PDT) With Metvix® 160 mg/g Cream in Immuno‐compromised Patients With Non‐melanoma Skin Cancer |
| Methods | This was a multicentre, randomised, open, active‐controlled, intraindividual study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: the treatment area on a randomised side (5 x 10 cm²) will be treated at baseline and at visits at 3, 9, and 15 months. At baseline the area will be treated with fractionated Metvix® PDT treatment consisting of 2 treatments 1 week apart and at visits at 3, 9, and 15 months with single Metvix® PDT treatment Control intervention B: in the contralateral control area (5 x 10 cm²), new and recurrent lesions and lesions in non‐complete response will be treated with lesion‐specific treatment at the discretion of the investigator at each study visit |
| Outcomes | Primary outcomes of the trial 1) Occurrence of new lesions (actinic keratosis, basal cell carcinoma, squamous cell carcinoma and warts) at 3, 9, 15, 21, and 27 months after first treatment 1) Number of BCC lesions that show complete response in the treated area with the contralateral control area at 3, 9, 15, 21, and 27 months after first treatment |
| Starting date | July 2003 |
| Contact information | Ann‐Marie Wennberg, MD, PhD (PI) Sahlgrenska University Hospital Gothenburg, Sweden |
| NCT | NCT00472459 |
| Notes | This study has been completed in July 2006, and the last update was in September 2010 (April 2012). |
| Trial name or title | Phase 2a Randomized, Placebo‐Controlled, Double‐Blind Trial of Topical Perillyl Alcohol in Sun Damaged Skin |
| Methods | This is a single‐centre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Disease characteristics
Exclusion criteria of the trial
Demographics
|
| Interventions | Interventions A: perillyl alcohol (POH) cream (0.3%) applied topically to each dorsal forearm twice daily for 3 months in the absence of unacceptable toxicity B: perillyl alcohol (POH) cream (0.76%) applied topically to each dorsal forearm twice daily for 3 months in the absence of unacceptable toxicity Control intervention C: placebo cream applied topically to each dorsal forearm twice daily for 3 months in the absence of unacceptable toxicity |
| Outcomes | Primary outcome of the trial 1) To determine if topical administration of perillyl alcohol (POH) cream can reverse actinic damage as evidenced by normalisation of quantitative skin histopathology scores in skin tissue biopsy samples from participants with moderate to severe sun damage Secondary outcomes of the trial 1) To determine if topically‐administered POH results in significant alterations in surrogate end point biomarkers of epidermal cell proliferation, including optical coherence tomography, p53 expression, c‐Fos expression, and apoptosis (as measured by activated caspase‐3 expression) 2)To determine if topically‐administered POH results in normalisation of nuclear chromatin patterns in skin biopsy tissue from these participants, as determined by karyometric analysis 3) To determine if topical POH can be administered safely to the forearms of these participants |
| Starting date | May 2004 |
| Contact information | Steve Stratton, MD (study chair) University of Arizona |
| NCT | NCT00608634 |
| Notes | The status of this study is unknown, and the last update was in September 2010 (April 2012). |
| Trial name or title | A Randomized Right/Left Clinical Trial to Evaluate the Use of Biafine Cream Versus Standard Care in Subjects With Actinic Keratosis Post Cryotherapy |
| Methods | This is a single‐centre, randomised, assessor‐blinded, active‐controlled, intraindividual study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: cryotherapy followed by Biafine cream Control intervention B: cryotherapy followed by standard care |
| Outcomes | Primary outcome of the trial 1) The change of the target lesions from baseline to end of treatment in the IGA at 4 weeks |
| Starting date | October 2006 |
| Contact information | Steve Feldman, MD, PhD (PI) Wake Forest University |
| NCT | NCT00695578 |
| Notes | This study has been completed in February 2008, and the last update was in February 2009 (April 2012). |
| Trial name or title | A Multicenter, Randomized, Double‐blind, Vehicle‐controlled, Dose‐ranging Study to Evaluate the Safety and Efficacy of 0.005%, 0.01% and 0.015% PEP005 Topical Gel When Used to Treat Actinic Keratoses on the Head (Face or Scalp) |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Interventions A: PEP005 topical gel 0.005%, 2‐day treatment B: PEP005 topical gel 0.01%, 2‐day treatment C: PEP005 topical gel 0.015%, 2‐day treatment D: PEP005 topical gel 0.005%, 3‐day treatment E: PEP005 topical gel 0.01%, 3‐day treatment F: PEP005 topical gel 0.015%, 3‐day treatment Control interventions G: vehicle gel, 2‐day treatment H: vehicle gel, 3‐day treatment |
| Outcomes | Primary outcome of the trial 1) Safety and toleration (incidence of adverse events, serious adverse events, and skin responses) at 57 days 1) Efficacy (clearance of actinic keratosis lesions) at 57 days |
| Starting date | June 2008 |
| Contact information | Peplin |
| NCT | NCT00700063 |
| Notes | This study has been completed in October 2008, and the last update was in July 2010 (April 2012). Incomplete data were published in abstract form in an excluded study (Spencer 2010). |
| Trial name or title | A Randomized Controlled Paired Comparison of Photo‐therapy With a Topical Retinoid Cream Pretreatment Versus PDT Alone for Actinic Keratoses |
| Methods | This is a single‐centre, randomised, double‐blind, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: topical retinoid, for 4 weeks followed by blue‐light photodynamic therapy with photosensitising agent (at week 4) Control intervention B: blue‐light photodynamic therapy with photosensitising agent at week 4 |
| Outcomes | Primary outcomes of the trial 1) Live blinded rater and blinded photo rater analysis of areas at week 0 and week 6 for erythema, edema, crusting, ulceration, palpability, and the need to cease/delay treatment 2) Overall response in reduction of number of AKs at 6 weeks 1) Participants will assess pain, burning, and itching on a scale of 0 to 3 at week 0, during retinoid treatment, during phototherapy, 1 day after, and at week 6 2) Principal investigator will evaluate adverse events at week 6 |
| Starting date | August 2008 |
| Contact information | Murad Alam, MD (PI) Department of Dermatology Northwestern University |
| NCT | NCT00756288 |
| Notes | This study is ongoing and the last update was in April 2011 (April 2012). |
| Trial name or title | Randomized, Multicenter, Double Blind Study to Compare the Efficacy and Tolerability of Oleogel‐S‐10 for 3 Month Versus Placebo Only in Patients With Mild to Moderate Actinic Keratoses Located at the Face and Head Oleogel‐S‐10 in Actinic Keratoses Trial |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Interventions A: oleogel‐S‐10 for 3 months once a day (N = 54 participants) B: oleogel‐S‐10 for 3 months twice a day (N = 54 participants) Control interventions C: placebo (petroleum jelly) for 3 months once a day (N = 27 participants) D: placebo (petroleum jelly) for 3 months twice a day (N = 27 participants) |
| Outcomes | Primary outcome of the trial 1) Objective response of the marker actinic keratosis, defined as histologically complete or partial clearance (partial clearance = down‐grading in Cockerell‐classification) assessed at 18 weeks. The marker actinic keratosis is defined as an initially‐selected lesion within the target area that will be used for final biopsy 1) Histologically‐controlled complete clearance, 2) Histologically‐controlled down staging 75% clearance rate 3) Dose response relationship 4)Time to clinically complete response 5) Tolerability |
| Starting date | October 2008 |
| Contact information | Birken GmbH Principal investigator Claus Garbe, Prof. Dr, Universitätshautklinik Tübingen |
| NCT | NCT00786994 |
| Notes | This study has been completed in November 2010 and the last update was in January 2012 (April 2012). |
| Trial name or title | A Multicenter, Double‐Blind, Vehical‐Controlled Study Comparing Imiquimod Cream, 5% (Apotex Inc.) to Aldara™ Cream, 5%(3M Pharmaceutials, U.S.) and Aldara™ Cream, 5%(3M Pharmaceuticals, Canada) in the Treatments of Actinic Keratosis |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Interventions A: Apotex, 5% imiquimod applied as a thin layer to target area once a day, 2 days each week, 16 weeks B: Aldara US, 5% imiquimod applied as a thin layer to target area once a day, 2 days each week, 16 weeks C: Aldara Canada, 5% imiquimod applied as a thin layer to target area once a day, 2 days each week, 16 weeks Control intervention D: vehicle applied as a thin layer to target area once a day, 2 days each week, 16 weeks |
| Outcomes | Primary outcomes of the trial 1) The primary objectives are to establish the therapeutic equivalence of imiquimod cream 5%, manufactured by Apotex Inc. and 2 Aldara (imiquimod) creams, manufactured by 3M (US & Canada) at 24 weeks 2) Superiority over vehicle in the treatment of AK at 24 weeks 1) The secondary objective is to compare the safety profiles of the 3 creams at 24 weeks |
| Starting date | February 2008 |
| Contact information | William Brooks (study director) Apotex Inc |
| NCT | NCT00859105 |
| Notes | This study has been completed in November 2008, and the last update was in March 2009 (April 2012). |
| Trial name or title | A Multicenter, Double‐Blind, Randomized, Parallel Group, Vehicle‐Controlled Study to Determine the Clinical Equivalence of a Generic Imiquimod Cream, 5% and Aldara™ Cream in Subjects With Actinic Keratosis |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐ and active‐controlled, parallel‐group. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: generic 5% topical cream dispensed in individual 0.25 g sachets applied twice a week for 16 weeks Control interventions B: Aldara 5% topical cream dispensed in individual 0.25 g sachets applied twice a week for 16 weeks C: topical cream vehicle matching generic imiquimod dispensed in individual 0.25 g sachets applied twice a week for 16 weeks |
| Outcomes | Primary outcome of the trial 1) Proportion of participants in each treatment group with complete clearance of actinic keratosis lesions at 8 weeks post‐treatment (week 24, test of cure/TOC) visit 1) The partial clearance rates, defined as the proportion of subjects with at least a 75% reduction in the number of lesions counted at baseline 2) Proportion of participants with complete clearance of lesions at week 16 (end of treatment) and week 24 (TOC) |
| Starting date | May 2008 |
| Contact information | Christine M. Winslow, Ph.D. (study director) Actavis Mid‐Atlantic LLC |
| NCT | NCT00948428 |
| Notes | This study has been completed in April 2009, and last update was in August 2010 (April 2012). |
| Trial name or title | Double‐blind, Randomized, Multi‐centre Phase II Study to Evaluate the Efficacy and Safety of Topically Applied LAS41007 Once Daily and LAS41007 Twice Daily Versus LAS106521 Gel Twice Daily in the Treatment of Actinic Keratosis Grade I to II |
| Methods | This is a multicentre, randomised, double‐blind, active‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Interventions A: LAS41007 once daily B: LAS41007 twice daily Control intervention C: LAS106521 (3% diclofenac in hyaluronic acid) |
| Outcomes | Primary outcomes of the trial 1) Histological clearance of 1 pre‐selected target lesion at day 120 1) Physician's Global Tolerability Assessment (PGT) at day 120 |
| Starting date | August 2009 |
| Contact information | Christoph Willers, MD, MBA Almirall Hermal GmbH |
| NCT | NCT00991861 |
| Notes | This study has been completed in February 2010, and the last update was in July 2010 (April 2012). |
| Trial name or title | An Exploratory, Open‐label Study of Sequential Field‐directed Treatment of Actinic Keratoses of the Face With Imiquimod 3.75% Cream Followed by Photodynamic Therapy |
| Methods | This was a multicentre, randomised, open, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: imiquimod 3.75% cream, up to 2 packets, applied topically daily for 2 2‐week cycles separated by a no‐treatment interval of 2 weeks, followed 4 weeks later by photodynamic therapy with 20% aminolevulinic acid and blue light exposure of the entire face Control intervention B: Imiquimod 3.75% cream, up to 2 packets, applied topically daily for 2 2‐week cycles separated by a no‐treatment interval of 2 weeks, followed by observation |
| Outcomes | Primary outcomes of the trial 1) Actinic keratosis count at week 18 2) The per cent change in actinic keratosis count as compared to the baseline lesion count 1) Complete clearance at week 18 2) The proportion of participants with complete clearance of actinic keratoses in the treatment area (entire face) 4) Change in investigator and participant scores of cosmetic appearance of the treatment area (entire face) |
| Starting date | September 2010 |
| Contact information | Julie Biron [email protected] |
| NCT | NCT01203878 |
| Notes | This study is currently recruiting, and the last update was in July 2011 (April 2012). |
| Trial name or title | An Investigator‐Initiated Study to Assess the Safety and Efficacy of Imiquimod 3.75% Cream When Used After Cryotherapy in the Treatment of Hypertrophic Actinic Keratoses (AK) on Dorsal Hands and Forearms |
| Methods | This is a single‐centre, randomised, assessor‐blinded, intraindividual study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics:
|
| Interventions | Intervention A: cryotherapy + imiquimod 3.75% daily for 2 weeks on, 2 weeks off, 2 weeks on on one arm Control intervention B: cryotherapy alone on the other arm |
| Outcomes | Primary outcomes of the trial 1) Clearance of actinic keratoses assessed at 14 weeks 2) Actinic keratosis lesion count 3) Photography 1) Local skin reactions assessed at 14 weeks |
| Starting date | October 2010 |
| Contact information | Gary S Goldenberg, MD ([email protected]) Giselle Singer, BS ([email protected]) |
| NCT | NCT01229319 |
| Notes | This study is currently recruiting, and the last update was in June 2011 (April 2012). |
| Trial name or title | Conventional Versus Fractional CO2 Laser Assisted Photodynamic Therapy for Basal Call Carcinomas and Actinic Keratoses |
| Methods | This is a single‐centre, randomised, assessor‐blinded, active‐controlled, intraindividual. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: fractional CO₂ laser assisted photodynamic therapy (PDT) pretreatment with fractional CO₂ laser before methyl‐aminolevulinate (MAL)‐red light PDT (37 J/cm²) Control intervention B: conventional photodynamic therapy using methyl‐aminolevulinate (MAL) and red light (37 J/cm²) |
| Outcomes | Primary outcomes of the trial 1) Treatment response at 3 months, clinical evaluation by a blinded physician 1) Pain during treatment, participant score (VAS 0 to 10) 3) Scaring, hyper‐ and hypopigmentation |
| Starting date | October 2010 |
| Contact information | Christina S Haak, MD ([email protected]) Katrine Togsverd‐Bo, MD ([email protected]) |
| NCT | NCT01260987 |
| Notes | This study is currently recruiting, and the last update was June 2011 (April 2012). |
| Trial name or title | Double‐blind, Randomized, Vehicle‐ and Comparator‐controlled, Multi‐center Trial to Evaluate the Efficacy and Safety of LAS41007 in the Treatment of Actinic Keratosis |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐ and active‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: LAS41007, twice daily, once in the morning and once in the evening. Per application, not more than 1.5 g of the immunologically mediated photodermatoses (IMP) should be applied, which is sufficient to cover a total area of 75 cm² (corresponding to 3 single TAs, each with a size of 25 cm² ) in maximum. The IMPs will be applied for 90 days in maximum Control interventions B: LASW1510, twice daily, once in the morning and once in the evening. Per application, not more than 1.5 g of the IMP should be applied, which is sufficient to cover a total area of 75 cm² (corresponding to 3 single TAs, each with a size of 25 cm²) in maximum. The IMPs will be applied for 90 days in maximum C: vehicle, twice daily, once in the morning and once in the evening. Per application, not more than 1.5 g of the IMP should be applied, which is sufficient to cover a total area of 75 cm² (corresponding to 3 single TAs, each with a size of 25 cm²) in maximum. The IMPs will be applied for 90 days in maximum |
| Outcomes | Primary outcomes of the trial 1) Superiority of LAS41007 compared to vehicle at day 1 2) Superiority of LAS41007 compared to LASW1510 assessed by histology to evaluate the histological clearance of one pre‐selected target lesion 4) Superiority of LAS41007 compared toLASW1510 each assessed by histology to evaluate the histological clearance of one pre‐selected target lesion 1) Superiority of LAS41007 compared to vehicle at day 1 2) Improved clinical efficacy of LAS41007 compared to LASW1510 with respect to clinical efficacy at day 1 4) Improved clinical efficacy of LAS41007 compared to LASW1510 with respect to clinical efficacy at day 21 6) Improved clinical efficacy of LAS41007 compared to LASW1510 with respect to clinical efficacy at day 56 8) Improved clinical efficacy of LAS41007 compared to LASW1510 with respect to clinical efficacy at day 90 10) Improved clinical efficacy of LAS41007 compared to LASW1510 with respect to clinical efficacy at day 150 |
| Starting date | November 2010 |
| Contact information | Sven Silberborth, PhD ([email protected]) |
| NCT | NCT01265602 |
| Notes | This study is currently recruiting, and the last update was in December 2010 (April 2012). |
| Trial name or title | Phase 3 Study of Brand Generic and Placebo in Treatment of Actinic Keratosis |
| Methods | This is a randomised, double‐blind, placebo‐ and active‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: generic 0.5% 5‐fluorouracil, once daily (duration of treatment was not specified) Control interventions B: carac 0.5% 5‐fluorouracil, once daily (duration of treatment was not specified) C: placebo, once daily (duration of treatment was not specified) |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance at 6 weeks |
| Starting date | September 2010 |
| Contact information | ‐ |
| NCT | NCT01354717 |
| Notes | This study has been completed in March 2011, and the last update was in JUne 2011 (April 2012). |
| Trial name or title | Prospective Comparator Controlled Randomized Exploratory Study on the Efficacy of LAS 41005 Compared to Cryotherapy in Subjects With Hyperkeratotic Actinic Keratosis |
| Methods | This is a multicentre, randomised, open, active‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: LAS41005 (0.5% 5‐fluorouracil/ 10% salicylic acid) once daily (number of weeks was not specified) Control intervention B: cryotherapy 1 to 2 times during the treatment time |
| Outcomes | Primary outcome of the trial 1) Histological clearance of 1 predefined target lesion at 8 weeks after the end of treatment with LAS41005 or 14 weeks after first cryotherapy 1) Participant complete clearance rates at days 21, 42, and 98 2) Participant partial (% was not specified) clearance rates at days 21, 42, and 98 |
| Starting date | April 2011 |
| Contact information | Rosario Rodríguez Almirall, S.A. |
| NCT | NCT01358851 |
| Notes | This study is ongoing, and the last update was January 2012 (April 2012). |
| Trial name or title | A Double‐blind, Randomized, Placebo‐controlled, 2‐way Crossover Study to Assess the Potential Effect of Topically Applied Imiquimod Cream on Atrial Ectopy in Patients With Actinic Keratosis |
| Methods | This is a single‐centre, randomised, double‐blind, placebo‐controlled, cross‐over study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: 3.75% imiquimod cream, daily for 2 weeks Control intervention B: placebo cream, daily for 2 weeks |
| Outcomes | Primary outcome of the trial 1) Change in 24‐hour supraventricular beat count at day 14 1) Change in 24‐hour supraventricular premature couplet and run counts and atrial fibrillation (per cent time) at day 14 2) Change in 24‐hour mean heart rate at day 14 3) Change in 24‐hour ventricular premature beat count, ventricular premature couplet, and run counts at day 14 |
| Starting date | July 2011 |
| Contact information | Irma Benavides ([email protected]) |
| NCT | NCT01413763 |
| Notes | This study is recruiting, and the last update was in August 2011 (April 2012). |
| Trial name or title | Long‐term Effects of Aldara® 5% Cream and Solaraze® 3% Gel in the Treatment of Actinic Keratoses on the Face or Scalp With Respect to the Risk of Progression to In‐situ and Invasive Squamous Cell Carcinoma |
| Methods | This is a multicentre, randomised, open, active‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: 5% imiquimod, 3 times per week for 4 weeks on, 4 weeks off, once or twice Control intervention B: 3% diclofenac in hyaluronic acid, twice daily for 12 weeks |
| Outcomes | Primary outcome of the trial 1) Long‐term outcome (3 years) with respect to the risk of progression to SCC (in situ, invasive, or both) 1) Recurrence rate at 3 years post‐treatment 2) Time to recurrence at 3 years post‐treatment 3) Need of rescue treatment at 3 years post‐treatment 4) Cosmetic outcome at 3 years post‐treatment |
| Starting date | October 2011 |
| Contact information | Dr Ursula Petzold MEDA Pharma GmbH & Co. KG |
| NCT | NCT01453179 |
| Notes | This study is ongoing, and the last update was in March 2012 (April 2012). |
| Trial name or title | A Phase II Study of Photodynamic Therapy With LEVULAN® Topical Solution + Blue Light Versus LEVULAN® Topical Solution Vehicle + Blue Light for the Treatment of Actinic Keratoses on the Upper Extremities |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: 3 hours 20% ALA‐blue light PDT Control intervention B: 3 hours vehicle‐blue light PDT |
| Outcomes | Primary outcome of the trial 1) Clearance rate for baseline grade I/II lesions at week 12 1) Clearance rate for all baseline lesions at weeks 8 and 12 2) Clearance rate for baseline grade I/II lesions at week 8 3) Per cent change in total actinic keratoses at weeks 8 and 12 4) Participant complete clearance at weeks 8 and 12 5) Participant complete clearance excluding grade III lesions at weeks 8 and 12 6) Participant partial (> 75%) clearance at weeks 8 and 12 7) Participant satisfaction score 8) Changes in pigmentation (hypo and hyper) at 2, 8, and 12 weeks after PDT 9) Local skin reactions during PDT (stinging/burning); 5 minutes after PDT (erythema, edema); and 2, 8, and 12 weeks after PDT (erythema, edema, stinging/burning, scaling/dryness, oozing/vesiculation/crusting) |
| Starting date | November 2011 |
| Contact information | Jim Berg ([email protected]) Dan Piacquadio, MD ([email protected]) |
| NCT | NCT01458587 |
| Notes | This study is recruiting, and the last update was in November 2011 (April 2012). |
| Trial name or title | Evaluation of the Formulation of 5‐aminolevulinic Acid With Dimethylsulfoxide in Photodynamic Therapy for Treatment of Actinic Keratosis |
| Methods | This is a single‐centre, randomised, open, active‐controlled, intraindividual study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: 4 hours 20% ALA‐red light PDT, once or twice with a 3‐month interval Control intervention B: cryotherapy, once or twice with a 3‐month interval |
| Outcomes | Primary outcome of the trial 1) Changes in lesion area at 0, 3, and 6 months 1) Pain on a visual analogue scale (VAS) and a graduated scale at the time 0 and 15 minutes after each intervention 2) Cosmetic outcome evaluated subjectively by the participant and investigator (awful, bad, regular, good, excellent) and objectively by the investigator (presence or absence of 1 or more of these criteria: hypochromia, hyperpigmentation, hyperemia, scar) |
| Starting date | November 2010 |
| Contact information | Catarina Robert, MD René AC Vieira, PHD Fundação Pio XII ‐ Hospital de Câncer de Barretos |
| NCT | NCT01459393 |
| Notes | This study is ongoing, and the last update was in October 2011 (April 2012). |
| Trial name or title | IIntra‐individual Comparison of Efficacy and Safety of Metvix® Natural Daylight Photodynamic Therapy Versus Conventional Metvix® Photodynamic Therapy in Subject With Mild Actinic Keratoses |
| Methods | This is a multicentre, randomised, assessor‐blind, active‐controlled, intraindividual study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A:16% MAL‐daylight photodynamic therapy (PDT), once or twice with a 12‐week interval Control intervention B: 16% MAL‐conventional PDT, once or twice with a 12‐week interval |
| Outcomes | Primary outcome of the trial 1) Per cent change from baseline in total number of treated mild lesions per side at week 12 2) Participant assessment of maximal pain on a scale from 0 (no pain) to 10 (extreme pain) per side at baseline |
| Starting date | March 2012 |
| Contact information | Catherine Bosc ([email protected]) |
| NCT | NCT01475071 |
| Notes | This study is recruiting, and the last update was in March 2012 (April 2012). |
| Trial name or title | A Phase II Study of Photodynamic Therapy With LEVULAN® Topical Solution + Blue Light Versus LEVULAN® Topical Solution Vehicle + Blue Light Using Spot and Broad Area Application and Incubation Times of 1, 2 and 3 Hours for the Treatment of Multiple Actinic Keratoses on the Face or Scalp |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Interventions A: field application 1 hours 20% ALA‐blue light photodynamic therapy (PDT) B: field application 2 hours 20% ALA‐blue light PDT C: field application 3 hours 20% ALA‐blue light PDT D: Individual lesion application 2 hours 20% ALA‐blue light PDT Control intervention E: field/individual application 1 to 3 hours 20% ALA‐blue light PDT |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rate at week 12 1) Participant complete clearance rate at weeks 4, 8, and 24 2) Participant partial (> 75%) clearance rate at weeks 4, 8, 12, and 24 3) Per cent change in lesion number at weeks 4, 8, 12, and 24 4) Participant satisfaction score on a 0 to 3 scale 5) Changes in pigmentation (hyper‐ and hypo‐) at 24 to 48 hours after PDT, and at weeks 2, 4, 8, 12, and 24 6) Local skin reactions during PDT (stinging/burning); at 5 minutes after PDT (erythema, edema); at 24 to 48 hours after PDT; and weeks 2, 4, 8, 12, and 24 (erythema, edema, stinging/burning, scaling/dryness, oozing/vesiculation/crusting) |
| Starting date | December 2011 |
| Contact information | Jim Berg ([email protected]) Dan Piacquadio, MD ([email protected]) |
| NCT | NCT01475955 |
| Notes | This study is recruiting, and the last update was in March 2012 (April 2012). |
| Trial name or title | Prospective, Single‐center, Investigator‐blinded, Randomized, Half‐side, Comparative Study of Photodynamic Therapy vs. CO2 Laser Therapy in Treatment of Actinic Keratoses |
| Methods | This is a single‐centre, randomised, assessor‐blind, active‐controlled, intraindividual study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: CO₂ laser therapy Control intervention B: 4h ALA‐ red light photodynamic therapy (PDT) |
| Outcomes | Primary outcome of the trial 1) Number of actinic keratoses at 3 months post‐treatment 1) Histologic features at 1 month post‐treatment 2) Epidermal thickness in optical coherence tomography at 1 month post‐treatment |
| Starting date | March 2011 |
| Contact information | Dr. Nina Scola, Consultant in Ruhr University of Bochum, Ruhr University of Bochum |
| NCT | NCT01481155 |
| Notes | This study is ongoing, and the last update was in December 2011 (April 2012). |
| Trial name or title | A Randomized, Double‐Blind, Parallel, Vehicle‐Controlled Phase III Trial to Assess the Efficacy and Safety of Topical SR‐T100 Gel in the Treatment of Patients With Actinic Keratosis |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: SR‐T100 gel, once daily with an occlusive dressing for 16 weeks Control intervention B: vehicle gel, once daily with an occlusive dressing for 16 weeks |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance at 8 weeks post‐treatment (week 24) 1) Lesion size reduction at 8 weeks post‐treatment (week 24) 2) Participant partial (> 75%) clearance at 8 weeks post‐treatment (week 24) 3)Tolerance |
| Starting date | October 2011 |
| Contact information | Kou‐Wha Kuo, Ph.D ([email protected]) Tony Chiu, B.S ([email protected]) |
| NCT | NCT01493921 |
| Notes | This study is recruiting, and the last update in March 2012 (April 2012). |
| Trial name or title | A Multicenter, Randomized, Double‐Blind, Vehicle‐Controlled, Parallel Group Comparison Study to Determine the Therapeutic Equivalence of Generic Imiquimod Cream, 3.75% and Zyclara™ (Imiquimod) Cream, 3.75% in Subjects With Actinic Keratoses |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: generic 3.75% imiquimod, once daily for 2 weeks on, 2 weeks off, 2 weeks on Control interventions B: placebo, once daily for 2 weeks on, 2 weeks off, 2 weeks on C: Zyclara 3.75% imiquimod, once daily for 2 weeks on, 2 weeks off, 2 weeks on |
| Outcomes | Primary outcomes of the trial 1) Participant complete clearance rate at 8 weeks post‐treatment (week 14) 2) Compliance 3) Severity and frequency of adverse events 4) Severity and frequency of local skin reactions 1) Participant partial (> 75%) clearance rate at 8 weeks post‐treatment (week 14) 2) Per cent change in the lesion number at 8 weeks post‐treatment (week 14) |
| Starting date | February 2011 |
| Contact information | Daniel Piacquadio, M.D Therapeutics, Inc |
| NCT | NCT01502020 |
| Notes | This study has been completed in November 2011, and the last update was in December 2011 (Apil 2012). |
| Trial name or title | A Double‐blind, Randomized, Vehicle‐controlled, Parallel‐group, Phase II Dose‐ranging Study to Evaluate the Efficacy and Safety of SR‐T100 Gel in Patients With Actinic Keratosis (AK) on the Head (Face and/or Scalp) |
| Methods | This is a randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Interventions A: SR‐T100 with 1.0% of SM in Solanum undatum plant extract for 16 weeks B: SR‐T100 with 2.3% of SM in Solanum undatum plant extract for 16 weeks Control intervention B: placebo for 16 weeks |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance rate at 8 weeks post‐treatment (week 24) 1) Participant partial (> 75%) clearance rate at 8 weeks post‐treatment (week 24) |
| Starting date | ‐ |
| Contact information | Dr Kou‐Wha Kuo ([email protected]) |
| NCT | NCT01516515 |
| Notes | This study is not yet recruiting, and the last update was January 2012 (April 2012). |
| Trial name or title | Combination Therapy With 5‐Fluorouracil and Photodynamic Therapy for the Treatment of Post‐transplant Premalignant Skin Disease |
| Methods | This is a single‐centre, randomised, open, parallel‐group (1 arm with immunocompetent controls and 1 arm with immunosuppressed organ transplant recipients), intraindividual (1 side with topical and PDT and 1 side with only PDT) study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: 5% 5‐fluorouracil once daily for 6 days followed by 3 hours MAL‐red light phototherapy (PDT) in immunocompetent and immunosuppressed participants Control intervention B: 3 hours MAL‐red light PDT in immunocompetent and immunosuppressed participants |
| Outcomes | Primary outcome of the trial 1) Accumulation of PpIX at 3 hours after MAL application 1) Rate of lesion clearance at day 14 and at 3 months 2) Rate of development of new AK at months 3, 6 9, and 12 |
| Starting date | September 2011 |
| Contact information | Margo Riha, BSN, RN ([email protected]) Sara Lohser, MD ([email protected]) |
| NCT | NCT01525329 |
| Notes | This study is recruiting, and the last update was in February 2012 (April 2012). |
| Trial name or title | Topical Imiquimod 5% Cream Therapy Versus Photodynamic Therapy With Methyl‐aminolaevulinate 16% Cream of Actinic Keratoses in Organ Transplant Recipients |
| Methods | This is a single‐centre, randomised, open, active‐controlled, intraindividual study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: 3 hours 16% MAL‐red light photodynamic therapy (PDT) (70 J/cm²), twice with a 2 week interval Control intervention B: 5% imiquimod, 3 times per week for 4 weeks |
| Outcomes | Primary outcome of the trial 1) Lesion complete response rate at 4 weeks post‐treatment 1) Lesion complete response rate at 6 and 12 months post‐treatment 2) Global reduction in the area of specific PpIX fluorescence at 1, 6, and 12 months post‐treatment 3) Global participant's satisfaction on a 10 cm visual analogue scale (VAS, 0 means extremely unsatisfied, 1 to 3 means unsatisfied, 5 to 7 means moderately satisfied, 8 to 10 means highly satisfied) at 3, 6, and 12 months post‐treatment |
| Starting date | April 2012 |
| Contact information | Stanislava Tzaneva, Doz. Dr ([email protected]) Alexandra Geusau, Prof. Dr ([email protected]) |
| NCT | NCT01538901 |
| Notes | This study is not yet recruiting, and the last update was in February 2012 (April 2012). |
| Trial name or title | Clinical Effect of Photodynamic Treatment When Treating Actinic Keratoses With Different Light Doses |
| Methods | This is a single‐centre, randomised, participant‐blinded, active‐controlled, intraindividual study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: 20% ALA‐red light photodynamic therapy (PDT) (70 J/cm²), twice with a 2‐week interval Control intervention B: 20% ALA‐red light PDT (100 J/cm²), twice with a 2‐week interval |
| Outcomes | Primary outcome of the trial 1) Relapse of clinically cleared actinic keratosis, evaluation by two investigators for clinical/histological relapse at 1, 3, 6, 12, and 24 months post‐treatment 1) Pain during treatment, participant scoring on a visual analogue scale (VAS) |
| Starting date | April 2010 |
| Contact information | Evelina Buinauskaite, MD Skaidra Valiukeviciene, Prof. Lithuanian University of Health Sciences, Medical Academy, Department of Skin and Venereal Diseases |
| NCT | NCT01541228 |
| Notes | This study is ongoing, and the last update was in February 2012 (April 2012). |
| Trial name or title | A Sequential Treatment Regimen of Cryotherapy and Picato® for the Treatment of Actinic Keratosis on the Face and Scalp |
| Methods | This is a multicentre, randomised, double‐blinded, placebo‐controlled, parallel‐group study. |
| Participants | Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: cryotherapy followed by 0.015% PEP005 (ingenol mebutate) gel (field) daily for 3 consecutive days Control intervention B: cryotherapy followed by vehicle gel (field) daily for 3 consecutive days |
| Outcomes | Primary outcome of the trial 1) Participant complete clearance at week 11 1) Per cent reduction from baseline in number of lesions at week 11 2) Participant complete clearance for 12 months (recurrence) 4) Participant partial (> 75%) clearance at week 11 through to month 12 |
| Starting date | March 2012 |
| Contact information | Birgitte Vestbjerg (birgitte.vestbjerg@leo‐pharma.com) |
| NCT | NCT01541553 |
| Notes | This study is recruiting, and the last update was in March 2012 (April 2012). |
| Trial name or title | Temperature modulated photodynamic therapy for the treatment of actinic keratoses on the extremities |
| Methods | This is a randomised, blinded, active‐controlled, intraindividual study. |
| Participants | Inclusion criteria of the trial
Demographics
|
| Interventions | Intervention A: ALA + heat followed by blue light photodynamic therapy (PDT) (N= 20 participants) Control interventions B: ALA + no heat followed by blue light PDT (N = 20 participants) Characteristics of PDT intervention Type of treatment: ‐‐ Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: ‐‐ Cream concentration (%): 20 Application of cream: ‐‐ Incubation with cream: 1 hour under occlusion Type of light: blue light Light source: ‐‐ Wavelength (nm): 417 nm Energy fluence (J/cm²): 10 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
| Outcomes | Outcomes of the trial 1) Clearance rates 2) Tolerability during and following treatment on a 4‐point scale Efficacy Methods: quantitative assessment by lesion counting performed by an unblinded investigator and photographs evaluation by a blinded investigator Time points: at baseline, and 2 and 6 months post‐treatment |
| Starting date | ‐ |
| Contact information | ‐ |
| NCT | ‐ |
| Notes | Abstract from 31st Annual Conference of the American Society for Laser Medicine and Surgery, ASLMS 2011 |
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Global Improvement Indices (investigator)‐cleared Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 1.1  Comparison 1 Adapalene gel versus placebo, Outcome 1 Global Improvement Indices (investigator)‐cleared. | ||||
| 2 Mean changes in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 1.2  Comparison 1 Adapalene gel versus placebo, Outcome 2 Mean changes in lesion counts. | ||||
| 2.1 0.1% adapalene gel | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 0.3% adapalene gel | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 1.3  Comparison 1 Adapalene gel versus placebo, Outcome 3 Withdrawal due to adverse events. | ||||
| 4 Minor adverse events excluding skin irritation: dermatitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 1.4  Comparison 1 Adapalene gel versus placebo, Outcome 4 Minor adverse events excluding skin irritation: dermatitis. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Global Improvement Indices (investigator)‐cleared Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 2.1  Comparison 2 0.1% adapalene vs 0.3% adapalene, Outcome 1 Global Improvement Indices (investigator)‐cleared. | ||||
| 2 Mean changes in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 2.2  Comparison 2 0.1% adapalene vs 0.3% adapalene, Outcome 2 Mean changes in lesion counts. | ||||
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 2.3  Comparison 2 0.1% adapalene vs 0.3% adapalene, Outcome 3 Withdrawal due to adverse events. | ||||
| 4 Minor adverse events excluding skin irritation: dermatitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 2.4  Comparison 2 0.1% adapalene vs 0.3% adapalene, Outcome 4 Minor adverse events excluding skin irritation: dermatitis. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 3.1  Comparison 3 Arotinoid Methyl Sulfone (Ro 14‐9706) versus Tretinoin, Outcome 1 Mean percentage of reduction in lesion counts. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean changes in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 4.1  Comparison 4 Calcipotriol (vitamin D) versus placebo, Outcome 1 Mean changes in lesion counts. | ||||
| 2 Cosmetic outcomes: Reduction in total cosmetic appearance score Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 4.2  Comparison 4 Calcipotriol (vitamin D) versus placebo, Outcome 2 Cosmetic outcomes: Reduction in total cosmetic appearance score. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 5.1  Comparison 5 1% colchicine cream versus 0.5% colchicine cream, Outcome 1 Participant complete clearance. | ||||
| 2 Mean reduction in lesion counts‐total Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 5.2  Comparison 5 1% colchicine cream versus 0.5% colchicine cream, Outcome 2 Mean reduction in lesion counts‐total. | ||||
| 3 Mean reduction in lesion counts‐per anatomical locations Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 5.3  Comparison 5 1% colchicine cream versus 0.5% colchicine cream, Outcome 3 Mean reduction in lesion counts‐per anatomical locations. | ||||
| 3.1 Face | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Scalp | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Upper extremities | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Cosmetic outcomes: Number of participants with decreased infiltration and disappearance of crust Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 5.4  Comparison 5 1% colchicine cream versus 0.5% colchicine cream, Outcome 4 Cosmetic outcomes: Number of participants with decreased infiltration and disappearance of crust. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Investigator Global Improvement Indices‐completely improved Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 6.1  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 1 Investigator Global Improvement Indices‐completely improved. | ||||
| 1.1 30 day treatment/30 day follow‐up | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [0.89, 17.89] |
| 1.2 60 day treatment/30 day follow‐up | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 3.06 [1.21, 7.77] |
| 1.3 90 day treatment/30 day follow‐up | 1 | 117 | Risk Ratio (M‐H, Random, 95% CI) | 2.50 [1.37, 4.55] |
| 2 Participant Global Improvement Indices‐completely improved Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 6.2  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 2 Participant Global Improvement Indices‐completely improved. | ||||
| 2.1 30 day treatment/30 day follow‐up | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [0.89, 17.89] |
| 2.2 60 day treatment/30 day follow‐up | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 2.86 [1.12, 7.32] |
| 2.3 90 day treatment/30 day follow‐up | 1 | 117 | Risk Ratio (M‐H, Random, 95% CI) | 2.44 [1.28, 4.64] |
| 3 Participant complete clearance at end of treatment (>56 days) Show forest plot | 2 | 280 | Risk Ratio (M‐H, Random, 95% CI) | 1.95 [1.21, 3.13] |
| Analysis 6.3  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 3 Participant complete clearance at end of treatment (>56 days). | ||||
| 4 Participant complete clearance (target lesions) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 6.4  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 4 Participant complete clearance (target lesions). | ||||
| 4.1 30 day treatment/ 30 day follow‐up | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 3.5 [0.76, 16.01] |
| 4.2 60 day treatment/ 30 day follow‐up | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 3.27 [1.30, 8.21] |
| 4.3 90 day treatment/ 30 day follow‐up | 2 | 267 | Risk Ratio (M‐H, Random, 95% CI) | 2.87 [1.84, 4.48] |
| 5 Participant complete clearance (all lesions) Show forest plot | 3 | 420 | Risk Ratio (M‐H, Random, 95% CI) | 2.46 [1.66, 3.66] |
| Analysis 6.5  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 5 Participant complete clearance (all lesions). | ||||
| 5.1 30 day treatment/ 30 day follow‐up | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 3.5 [0.76, 16.01] |
| 5.2 60 day treatment/ 30 day follow‐up | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 3.83 [1.37, 10.71] |
| 5.3 90 day treatment/30 day follow‐up | 2 | 225 | Risk Ratio (M‐H, Random, 95% CI) | 2.20 [1.40, 3.44] |
| 6 Participant complete clearance for 30 day treatment by locations Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.6  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 6 Participant complete clearance for 30 day treatment by locations. | ||||
| 6.1 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Forehead | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.4 Back of hand | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Participant complete clearance for 60 day treatment by locations Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.7  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 7 Participant complete clearance for 60 day treatment by locations. | ||||
| 7.1 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Forehead | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Arm/forearm | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 Back of hand | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Participant complete clearance for 90 day treatment by locations Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 6.8  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 8 Participant complete clearance for 90 day treatment by locations. | ||||
| 8.1 Scalp | 2 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.25, 6.08] |
| 8.2 Forehead | 2 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [1.03, 2.85] |
| 8.3 Face | 2 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 2.15 [1.05, 4.40] |
| 8.4 Arm/forearm | 2 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.94 [0.26, 14.40] |
| 8.5 Back of hand | 2 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [0.04, 65.87] |
| 9 Participant complete clearance in immunosuppressed participants Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.9  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 9 Participant complete clearance in immunosuppressed participants. | ||||
| 10 Participant partial (>75%) clearance in immunosuppressed participants Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.10  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 10 Participant partial (>75%) clearance in immunosuppressed participants. | ||||
| 11 Mean reduction of lesion counts (30‐90 days ): At the end of study Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 6.11  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 11 Mean reduction of lesion counts (30‐90 days ): At the end of study. | ||||
| 11.1 90 days | 1 | 150 | Mean Difference (IV, Random, 95% CI) | 0.80 [‐1.48, 3.08] |
| 12 Mean reduction of lesion counts (30‐90 days): 30 day follow‐up Show forest plot | 2 | 345 | Mean Difference (IV, Random, 95% CI) | 2.55 [1.56, 3.53] |
| Analysis 6.12  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 12 Mean reduction of lesion counts (30‐90 days): 30 day follow‐up. | ||||
| 12.1 30 days | 1 | 98 | Mean Difference (IV, Random, 95% CI) | 2.00 [0.63, 3.37] |
| 12.2 60 days | 1 | 97 | Mean Difference (IV, Random, 95% CI) | 2.40 [0.73, 4.07] |
| 12.3 90 days | 1 | 150 | Mean Difference (IV, Random, 95% CI) | 3.80 [1.83, 5.77] |
| 13 Withdrawal due to adverse events Show forest plot | 4 | 592 | Risk Ratio (M‐H, Random, 95% CI) | 3.59 [1.92, 6.70] |
| Analysis 6.13  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 13 Withdrawal due to adverse events. | ||||
| 14 Minor adverse event: body as a whole : in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.14  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 14 Minor adverse event: body as a whole : in general. | ||||
| 15 Minor adverse event: body as a whole : "flu" Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.15  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 15 Minor adverse event: body as a whole : "flu". | ||||
| 16 Minor adverse event:: body as a whole : infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.16  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 16 Minor adverse event:: body as a whole : infection. | ||||
| 17 Minor adverse event: cardiovascular: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.17  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 17 Minor adverse event: cardiovascular: in general. | ||||
| 18 Minor adverse event: cardiovascular: sinus bradycardia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.18  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 18 Minor adverse event: cardiovascular: sinus bradycardia. | ||||
| 19 Minor adverse event: dermatological: bursitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.19  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 19 Minor adverse event: dermatological: bursitis. | ||||
| 20 Minor adverse event: dermatological: dry skin Show forest plot | 3 | 462 | Risk Ratio (M‐H, Random, 95% CI) | 2.40 [1.20, 4.78] |
| Analysis 6.20  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 20 Minor adverse event: dermatological: dry skin. | ||||
| 21 Minor adverse event: dermatological: herpes zoster Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.21  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 21 Minor adverse event: dermatological: herpes zoster. | ||||
| 22 Minor adverse event: dermatological: rash vesiculobullous Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.22  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 22 Minor adverse event: dermatological: rash vesiculobullous. | ||||
| 23 Minor adverse event::dermatological: seborrhoea Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.23  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 23 Minor adverse event::dermatological: seborrhoea. | ||||
| 24 Minor adverse event: dermatological: skin exfoliation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.24  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 24 Minor adverse event: dermatological: skin exfoliation. | ||||
| 25 Minor adverse event: dermatological: ulcerated skin Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.25  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 25 Minor adverse event: dermatological: ulcerated skin. | ||||
| 26 Minor adverse event: digestive : in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.26  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 26 Minor adverse event: digestive : in general. | ||||
| 27 Minor adverse event: hemic and lymphatic: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.27  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 27 Minor adverse event: hemic and lymphatic: in general. | ||||
| 28 Minor adverse event: metabolic and nutritional disorders : in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.28  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 28 Minor adverse event: metabolic and nutritional disorders : in general. | ||||
| 29 Minor adverse event: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.29  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 29 Minor adverse event: musculoskeletal and connective tissue: in general. | ||||
| 30 Minor adverse event: musculoskeletal and connective tissue: hypokinesia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.30  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 30 Minor adverse event: musculoskeletal and connective tissue: hypokinesia. | ||||
| 31 Minor adverse event: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.31  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 31 Minor adverse event: nervous system: in general. | ||||
| 32 Minor adverse event: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.32  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 32 Minor adverse event: nervous system: headache. | ||||
| 33 Minor adverse event: nervous system: hyperaesthesia Show forest plot | 2 | 345 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.30, 2.60] |
| Analysis 6.33  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 33 Minor adverse event: nervous system: hyperaesthesia. | ||||
| 34 Minor adverse event: nervous system: paraesthesia Show forest plot | 2 | 345 | Risk Ratio (M‐H, Random, 95% CI) | 2.53 [0.57, 11.20] |
| Analysis 6.34  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 34 Minor adverse event: nervous system: paraesthesia. | ||||
| 35 Minor adverse event: respiratory: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.35  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 35 Minor adverse event: respiratory: in general. | ||||
| 36 Minor adverse event: respiratory: bronchitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.36  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 36 Minor adverse event: respiratory: bronchitis. | ||||
| 37 Minor adverse event: respiratory: pharyngitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.37  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 37 Minor adverse event: respiratory: pharyngitis. | ||||
| 38 Minor adverse event: respiratory: upper respiratory tract infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.38  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 38 Minor adverse event: respiratory: upper respiratory tract infection. | ||||
| 39 Minor adverse event: special senses: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.39  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 39 Minor adverse event: special senses: in general. | ||||
| 40 Minor adverse event: urogenital: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.40  Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 40 Minor adverse event: urogenital: in general. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Investigator Global Improvement Indices‐Complete improvement Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 7.1  Comparison 7 3% diclofenac in 2.5% hyaluronic acid versus 5% imiquimod, Outcome 1 Investigator Global Improvement Indices‐Complete improvement. | ||||
| 2 Participant Global Improvement Indices‐Complete improvement Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 7.2  Comparison 7 3% diclofenac in 2.5% hyaluronic acid versus 5% imiquimod, Outcome 2 Participant Global Improvement Indices‐Complete improvement. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesions counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 8.1  Comparison 8 2‐(Difluoromethyl)‐dl‐ornithine (DFMO) versus placebo, Outcome 1 Mean reduction in lesions counts. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 3 | 522 | Risk Ratio (M‐H, Random, 95% CI) | 8.86 [3.67, 21.40] |
| Analysis 9.1  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 1 Participant complete clearance. | ||||
| 1.1 1 week treatment with 4 week follow‐up | 3 | 267 | Risk Ratio (M‐H, Random, 95% CI) | 8.30 [2.04, 33.76] |
| 1.2 2 week treatment with 4 week follow‐up | 2 | 128 | Risk Ratio (M‐H, Random, 95% CI) | 6.42 [1.27, 32.59] |
| 1.3 4 week treatment with 4 week follow‐up | 2 | 127 | Risk Ratio (M‐H, Random, 95% CI) | 13.07 [2.68, 63.66] |
| 2 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 9.2  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 2 Mean reduction in lesion counts. | ||||
| 3 Mean percentage of reduction in lesion counts Show forest plot | 1 | 142 | Mean Difference (IV, Random, 95% CI) | 33.60 [22.88, 44.32] |
| Analysis 9.3  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 3 Mean percentage of reduction in lesion counts. | ||||
| 4 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.4  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 4 Withdrawal due to adverse events. | ||||
| 5 Skin irritation Show forest plot | 2 | 384 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [1.27, 1.65] |
| Analysis 9.5  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 5 Skin irritation. | ||||
| 6 Minor adverse event excluding skin irritation: body as a whole : in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.6  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 6 Minor adverse event excluding skin irritation: body as a whole : in general. | ||||
| 7 Minor adverse event excluding skin irritation: body as a whole : allergy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.7  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 7 Minor adverse event excluding skin irritation: body as a whole : allergy. | ||||
| 8 Minor adverse event excluding skin irritation: body as a whole : "flu" or common cold Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.8  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 8 Minor adverse event excluding skin irritation: body as a whole : "flu" or common cold. | ||||
| 9 Minor adverse event excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.9  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 9 Minor adverse event excluding skin irritation: musculoskeletal and connective tissue: in general. | ||||
| 10 Minor adverse event excluding skin irritation: musculoskeletal and connective tissue: soreness Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.10  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 10 Minor adverse event excluding skin irritation: musculoskeletal and connective tissue: soreness. | ||||
| 11 Minor adverse event excluding skin irritation:nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.11  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 11 Minor adverse event excluding skin irritation:nervous system: headache. | ||||
| 12 Minor adverse event excluding skin irritation: respiratory: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.12  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 12 Minor adverse event excluding skin irritation: respiratory: in general. | ||||
| 13 Minor adverse event excluding skin irritation: respiratory: sinusitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.13  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 13 Minor adverse event excluding skin irritation: respiratory: sinusitis. | ||||
| 14 Minor adverse event excluding skin irritation: respiratory: upper respiratory tract infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.14  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 14 Minor adverse event excluding skin irritation: respiratory: upper respiratory tract infection. | ||||
| 15 Minor adverse event excluding skin irritation: special senses: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.15  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 15 Minor adverse event excluding skin irritation: special senses: in general. | ||||
| 16 Minor adverse event excluding skin irritation:special senses: eye irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.16  Comparison 9 0.5% 5‐FU versus vehicle, Outcome 16 Minor adverse event excluding skin irritation:special senses: eye irritation. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 10.1  Comparison 10 0.5% 5‐FU at varying application durations, Outcome 1 Participant complete clearance. | ||||
| 1.1 Daily for 1 week versus 4 weeks | 2 | 167 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.19, 0.81] |
| 1.2 Daily for 1 week versus 2 weeks | 2 | 169 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.23, 2.37] |
| 1.3 Daily for 2 weeks versus 4 weeks | 2 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.36, 0.87] |
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 10.2  Comparison 10 0.5% 5‐FU at varying application durations, Outcome 2 Withdrawal due to adverse events. | ||||
| 2.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Skin irritation Show forest plot | 2 | 515 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.91, 1.00] |
| Analysis 10.3  Comparison 10 0.5% 5‐FU at varying application durations, Outcome 3 Skin irritation. | ||||
| 3.1 Daily for 1 week versus 4 weeks | 2 | 170 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.89, 1.03] |
| 3.2 Daily for 1 week versus 2 weeks | 2 | 172 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.86, 1.08] |
| 3.3 Daily for 2 weeks versus 4 weeks | 2 | 173 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.88, 1.02] |
| 4 Minor adverse events excluding skin irritation: body as a whole : in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 10.4  Comparison 10 0.5% 5‐FU at varying application durations, Outcome 4 Minor adverse events excluding skin irritation: body as a whole : in general. | ||||
| 4.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Minor adverse events excluding skin irritation: body as a whole : allergy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 10.5  Comparison 10 0.5% 5‐FU at varying application durations, Outcome 5 Minor adverse events excluding skin irritation: body as a whole : allergy. | ||||
| 5.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Minor adverse events excluding skin irritation: body as a whole : "flu" or common cold Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 10.6  Comparison 10 0.5% 5‐FU at varying application durations, Outcome 6 Minor adverse events excluding skin irritation: body as a whole : "flu" or common cold. | ||||
| 6.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 10.7  Comparison 10 0.5% 5‐FU at varying application durations, Outcome 7 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general. | ||||
| 7.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 10.8  Comparison 10 0.5% 5‐FU at varying application durations, Outcome 8 Minor adverse events excluding skin irritation: nervous system: headache. | ||||
| 8.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: respiratory: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 10.9  Comparison 10 0.5% 5‐FU at varying application durations, Outcome 9 Minor adverse events excluding skin irritation: respiratory: in general. | ||||
| 9.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Minor adverse events excluding skin irritation: respiratory: sinusitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 10.10  Comparison 10 0.5% 5‐FU at varying application durations, Outcome 10 Minor adverse events excluding skin irritation: respiratory: sinusitis. | ||||
| 10.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Minor adverse events excluding skin irritation: special senses: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 10.11  Comparison 10 0.5% 5‐FU at varying application durations, Outcome 11 Minor adverse events excluding skin irritation: special senses: in general. | ||||
| 11.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Minor adverse events excluding skin irritation: special senses: eye irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 10.12  Comparison 10 0.5% 5‐FU at varying application durations, Outcome 12 Minor adverse events excluding skin irritation: special senses: eye irritation. | ||||
| 12.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 11.1  Comparison 11 0.5% 5‐FU versus ALA‐PDT, Outcome 1 Participant complete clearance. | ||||
| 1.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 11.2  Comparison 11 0.5% 5‐FU versus ALA‐PDT, Outcome 2 Withdrawal due to adverse events. | ||||
| 2.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 12.1  Comparison 12 5% 5‐FU with 0.05% tretinoin versus 5% 5‐FU with placebo, Outcome 1 Mean reduction in lesion counts. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 2 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 1.85 [0.41, 8.33] |
| Analysis 13.1  Comparison 13 5% 5‐FU versus 5% imiquimod, Outcome 1 Participant complete clearance. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 14.1  Comparison 14 5% 5‐FU versus cryotherapy, Outcome 1 Participant complete clearance. | ||||
| 1.1 After treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Investigator Global Improvement Indices ‐cleared Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 15.1  Comparison 15 5% 5‐FU versus 10% masoprocol, Outcome 1 Investigator Global Improvement Indices ‐cleared. | ||||
| 2 Mean reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 15.2  Comparison 15 5% 5‐FU versus 10% masoprocol, Outcome 2 Mean reduction of lesion counts. | ||||
| 3 Mean percentage of reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 15.3  Comparison 15 5% 5‐FU versus 10% masoprocol, Outcome 3 Mean percentage of reduction of lesion counts. | ||||
| 4 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 15.4  Comparison 15 5% 5‐FU versus 10% masoprocol, Outcome 4 Withdrawal due to adverse events. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 16.1  Comparison 16 5% 5‐FU versus carbon dioxide laser resurfacing, Outcome 1 Mean percentage of reduction of lesion counts. | ||||
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 16.2  Comparison 16 5% 5‐FU versus carbon dioxide laser resurfacing, Outcome 2 Withdrawal due to adverse events. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 17.1  Comparison 17 5% 5‐FU versus Er:YAG laser resurfacing, Outcome 1 Withdrawal due to adverse events. | ||||
| 2 Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 17.2  Comparison 17 5% 5‐FU versus Er:YAG laser resurfacing, Outcome 2 Skin irritation. | ||||
| 2.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction in lesions Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 18.1  Comparison 18 5% 5‐FU versus Trichloroacetic acid peel, Outcome 1 Mean percentage of reduction in lesions. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance‐number of doses Show forest plot | 11 | 2880 | Risk Ratio (M‐H, Random, 95% CI) | 6.91 [4.25, 11.26] |
| Analysis 19.1  Comparison 19 5% Imiquimod versus placebo, Outcome 1 Participant complete clearance‐number of doses. | ||||
| 1.1 9 or 18 doses (3 times/week for 3 weeks on, 4 weeks off) | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 2.76 [0.39, 19.40] |
| 1.2 12‐16 doses (2 times/week for 8 weeks or 3 times/week for 4 weeks) | 3 | 543 | Risk Ratio (M‐H, Random, 95% CI) | 7.88 [1.09, 56.67] |
| 1.3 12 or 24 doses (3 times/week for 4 weeks on , 4 weeks off, 4 weeks on) | 2 | 505 | Risk Ratio (M‐H, Random, 95% CI) | 8.81 [1.15, 67.32] |
| 1.4 24 doses (3 times/week for 8 weeks) | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.07, 25.08] |
| 1.5 32‐36 doses (2 times/ week for 16 weeks or 3 times/ week for 12 weeks) | 4 | 888 | Risk Ratio (M‐H, Random, 95% CI) | 7.12 [3.06, 16.58] |
| 1.6 40 doses (5 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.03, 17.27] |
| 1.7 48 doses (3 times/ week for 16 weeks) | 3 | 795 | Risk Ratio (M‐H, Random, 95% CI) | 10.90 [3.59, 33.15] |
| 1.8 56 doses (7 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.07, 24.29] |
| 2 Participant complete clearance in immunosuppressed participants Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 19.2  Comparison 19 5% Imiquimod versus placebo, Outcome 2 Participant complete clearance in immunosuppressed participants. | ||||
| 3 Participant partial (>75%) clearance Show forest plot | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 19.3  Comparison 19 5% Imiquimod versus placebo, Outcome 3 Participant partial (>75%) clearance. | ||||
| 3.1 9 or 18 doses (3 times/ week for 3 weeks on, 4 weeks off. 3 weeks on) | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 2.41 [0.91, 6.39] |
| 3.2 12‐16 doses (3 times/week for 4 weeks or 2 times/week for 8 weeks) | 2 | 284 | Risk Ratio (M‐H, Random, 95% CI) | 2.86 [1.53, 5.34] |
| 3.3 12 or 24 doses (3 times/week for 4 weeks on, 4 weeks off) | 2 | 505 | Risk Ratio (M‐H, Random, 95% CI) | 6.23 [0.70, 55.10] |
| 3.4 24 doses (3 times/week for 8 weeks) | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [0.25, 62.85] |
| 3.5 32 doses (2 times/week for 16 weeks) | 1 | 436 | Risk Ratio (M‐H, Random, 95% CI) | 5.02 [3.44, 7.33] |
| 3.6 40 doses (5 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 3.35 [0.21, 53.51] |
| 3.7 48 doses (3 times/ week for 16 weeks) | 2 | 778 | Risk Ratio (M‐H, Random, 95% CI) | 8.46 [2.29, 31.16] |
| 3.8 56 doses (7 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 5.94 [0.39, 90.34] |
| 4 Participant partial (>75%) clearance in immunosuppressed participants Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 19.4  Comparison 19 5% Imiquimod versus placebo, Outcome 4 Participant partial (>75%) clearance in immunosuppressed participants. | ||||
| 5 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 19.5  Comparison 19 5% Imiquimod versus placebo, Outcome 5 Mean reduction in lesion counts. | ||||
| 6 Withdrawal due to adverse events Show forest plot | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 19.6  Comparison 19 5% Imiquimod versus placebo, Outcome 6 Withdrawal due to adverse events. | ||||
| 6.1 12‐16 doses (2 times/week for 8 weeks or 3 times/week for 4 weeks) | 1 | 38 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.03, 16.74] |
| 6.2 12 or 24 doses (3 times/week for 4 weeks on , 4 weeks off, 4 weeks on) | 2 | 505 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [0.31, 8.23] |
| 6.3 24 doses (3 times/week for 8 weeks) | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.07, 25.08] |
| 6.4 32‐36 doses (2 times/ week for 16 weeks or 3 times/ week for 12 weeks) | 3 | 858 | Risk Ratio (M‐H, Random, 95% CI) | 2.29 [0.80, 6.57] |
| 6.5 40 doses (5 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 4.90 [0.32, 75.60] |
| 6.6 48 doses (3 times/ week for 16 weeks) | 2 | 778 | Risk Ratio (M‐H, Random, 95% CI) | 2.69 [1.48, 4.90] |
| 6.7 56 doses (7 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 5.42 [0.35, 82.97] |
| 7 Withdrawal due to adverse events in immunosuppressed participants Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 19.7  Comparison 19 5% Imiquimod versus placebo, Outcome 7 Withdrawal due to adverse events in immunosuppressed participants. | ||||
| 7.1 48 doses (3 times/ week for 16 weeks) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: body as a whole: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 19.8  Comparison 19 5% Imiquimod versus placebo, Outcome 8 Minor adverse events excluding skin irritation: body as a whole: in general. | ||||
| 8.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: body as a whole: "flu" or "cold" Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 19.9  Comparison 19 5% Imiquimod versus placebo, Outcome 9 Minor adverse events excluding skin irritation: body as a whole: "flu" or "cold". | ||||
| 9.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Minor adverse events excluding skin irritation: digestive: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 19.10  Comparison 19 5% Imiquimod versus placebo, Outcome 10 Minor adverse events excluding skin irritation: digestive: in general. | ||||
| 10.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Minor adverse events excluding skin irritation: digestive: nausea Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 19.11  Comparison 19 5% Imiquimod versus placebo, Outcome 11 Minor adverse events excluding skin irritation: digestive: nausea. | ||||
| 11.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 19.12  Comparison 19 5% Imiquimod versus placebo, Outcome 12 Minor adverse events excluding skin irritation: nervous system: in general. | ||||
| 12.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Cosmetic outcome: decrease in roughness/dryness/scaliness of the skin Show forest plot | 2 | 683 | Risk Ratio (M‐H, Random, 95% CI) | 3.23 [1.86, 5.58] |
| Analysis 19.13  Comparison 19 5% Imiquimod versus placebo, Outcome 13 Cosmetic outcome: decrease in roughness/dryness/scaliness of the skin. | ||||
| 13.1 32‐36 doses | 1 | 415 | Risk Ratio (M‐H, Random, 95% CI) | 2.54 [1.91, 3.37] |
| 13.2 48 doses | 1 | 268 | Risk Ratio (M‐H, Random, 95% CI) | 4.43 [2.69, 7.30] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 12 | 3087 | Risk Ratio (M‐H, Random, 95% CI) | 6.73 [5.03, 9.00] |
| Analysis 20.1  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 1 Participant complete clearance. | ||||
| 1.1 5.0% imiquimod | 9 | 1871 | Risk Ratio (M‐H, Random, 95% CI) | 7.70 [4.63, 12.79] |
| 1.2 3.75% imiquimod | 3 | 730 | Risk Ratio (M‐H, Random, 95% CI) | 6.45 [3.87, 10.73] |
| 1.3 2.5% imiquimod | 2 | 486 | Risk Ratio (M‐H, Random, 95% CI) | 4.49 [2.40, 8.39] |
| 2 Participant partial (>75%) clearance Show forest plot | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 20.2  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 2 Participant partial (>75%) clearance. | ||||
| 2.1 5.0% imiquimod | 4 | 1363 | Risk Ratio (M‐H, Random, 95% CI) | 6.71 [3.89, 11.57] |
| 2.2 3.75% imiquimod | 2 | 484 | Risk Ratio (M‐H, Random, 95% CI) | 3.11 [2.08, 4.66] |
| 2.3 2.5% imiquimod | 2 | 485 | Risk Ratio (M‐H, Random, 95% CI) | 2.48 [1.67, 3.68] |
| 3 Mean percentage of reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 20.3  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 3 Mean percentage of reduction in lesion counts. | ||||
| 3.1 3.75% imiquimod | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Minor adverse events excluding skin irritation: body as a whole: 'flu" or "cold" Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 20.4  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: 'flu" or "cold". | ||||
| 4.1 5.0% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Withdrawal due to adverse events Show forest plot | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 20.5  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 5 Withdrawal due to adverse events. | ||||
| 5.1 5.0% imiquimod | 8 | 2290 | Risk Ratio (M‐H, Random, 95% CI) | 2.59 [1.59, 4.23] |
| 5.2 3.75% imiquimod | 2 | 483 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.22, 3.93] |
| 5.3 2.5% imiquimod | 2 | 486 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.09, 2.70] |
| 6 Skin irritation Show forest plot | 5 | 1678 | Risk Ratio (M‐H, Random, 95% CI) | 3.93 [1.56, 9.88] |
| Analysis 20.6  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 6 Skin irritation. | ||||
| 6.1 5.0% imiquimod | 3 | 708 | Risk Ratio (M‐H, Random, 95% CI) | 3.68 [0.86, 15.74] |
| 6.2 3.75% imiquimod | 2 | 484 | Risk Ratio (M‐H, Random, 95% CI) | 4.86 [0.92, 25.83] |
| 6.3 2.5% imiquimod | 2 | 486 | Risk Ratio (M‐H, Random, 95% CI) | 3.45 [0.63, 18.97] |
| 7 Minor adverse events excluding skin irritation: body as a whole: pyrexia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 20.7  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 7 Minor adverse events excluding skin irritation: body as a whole: pyrexia. | ||||
| 7.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: hemic and lymphatic: lymphadenopathy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 20.8  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 8 Minor adverse events excluding skin irritation: hemic and lymphatic: lymphadenopathy. | ||||
| 8.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 20.9  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 9 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia. | ||||
| 9.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Minor adverse events excluding skin irritation: nervous system: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 20.10  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 10 Minor adverse events excluding skin irritation: nervous system: fatigue. | ||||
| 10.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 20.11  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 11 Minor adverse events excluding skin irritation: nervous system: headache. | ||||
| 11.1 5.0% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Minor adverse events excluding skin irritation: respiratory: cough Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 20.12  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 12 Minor adverse events excluding skin irritation: respiratory: cough. | ||||
| 12.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Minor adverse events excluding skin irritation: respiratory: sinusitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 20.13  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 13 Minor adverse events excluding skin irritation: respiratory: sinusitis. | ||||
| 13.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Minor adverse events excluding skin irritation: respiratory: upper respiratory tract infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 20.14  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 14 Minor adverse events excluding skin irritation: respiratory: upper respiratory tract infection. | ||||
| 14.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15 Minor adverse events excluding skin irritation: urogenital: urinary tract infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 20.15  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 15 Minor adverse events excluding skin irritation: urogenital: urinary tract infection. | ||||
| 15.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16 Cosmetic outcome: Participant's significantly or much improved cosmetic outcome assessed by investigator Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 20.16  Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 16 Cosmetic outcome: Participant's significantly or much improved cosmetic outcome assessed by investigator. | ||||
| 16.1 3.75% imiquimod | 2 | 470 | Risk Ratio (M‐H, Random, 95% CI) | 2.71 [2.05, 3.58] |
| 16.2 2.5% imiquimod | 2 | 475 | Risk Ratio (M‐H, Random, 95% CI) | 2.25 [1.62, 3.14] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 21.1  Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 1 Participant complete clearance. | ||||
| 1.1 2 times/week | 4 | 890 | Risk Ratio (M‐H, Random, 95% CI) | 5.36 [2.03, 14.16] |
| 1.2 3 times/week | 6 | 1336 | Risk Ratio (M‐H, Random, 95% CI) | 8.38 [3.79, 18.52] |
| 1.3 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.03, 17.27] |
| 1.4 7 times/week | 4 | 1253 | Risk Ratio (M‐H, Random, 95% CI) | 5.39 [3.65, 7.98] |
| 2 Participant partial (>75%) clearance Show forest plot | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 21.2  Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 2 Participant partial (>75%) clearance. | ||||
| 2.1 2 times/week | 2 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 4.99 [3.43, 7.26] |
| 2.2 3 times/week | 3 | 814 | Risk Ratio (M‐H, Random, 95% CI) | 7.65 [2.51, 23.32] |
| 2.3 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 3.35 [0.21, 53.51] |
| 2.4 7 times/week | 3 | 1006 | Risk Ratio (M‐H, Random, 95% CI) | 2.95 [1.99, 4.37] |
| 3 Withdrawal due to adverse events Show forest plot | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 21.3  Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 3 Withdrawal due to adverse events. | ||||
| 3.1 2 times/week | 4 | 896 | Risk Ratio (M‐H, Random, 95% CI) | 2.04 [0.75, 5.53] |
| 3.2 3 times/week | 5 | 1319 | Risk Ratio (M‐H, Random, 95% CI) | 2.47 [1.42, 4.30] |
| 3.3 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 4.90 [0.32, 75.60] |
| 3.4 7 times/week | 3 | 1006 | Risk Ratio (M‐H, Random, 95% CI) | 1.55 [0.33, 7.18] |
| 4 Minor adverse events excluding skin irritation:body as a whole: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 21.4  Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 4 Minor adverse events excluding skin irritation:body as a whole: in general. | ||||
| 4.1 2 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 3 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 5 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 7 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Minor adverse events excluding skin irritation: body as a whole:"flu" or "cold" Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 21.5  Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 5 Minor adverse events excluding skin irritation: body as a whole:"flu" or "cold". | ||||
| 5.1 2 times/week | 1 | 38 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.03, 16.74] |
| 5.2 3 times/week | 2 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 2.67 [0.36, 19.83] |
| 5.3 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.03, 17.27] |
| 5.4 7 times/week | 2 | 527 | Risk Ratio (M‐H, Random, 95% CI) | 5.20 [0.28, 95.18] |
| 6 Minor adverse events excluding skin irritation: digestive: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 21.6  Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 6 Minor adverse events excluding skin irritation: digestive: in general. | ||||
| 6.1 3 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 5 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 7 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Minor adverse events excluding skin irritation: digestive: nausea Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 21.7  Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 7 Minor adverse events excluding skin irritation: digestive: nausea. | ||||
| 7.1 3 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 5 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 7 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 21.8  Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 8 Minor adverse events excluding skin irritation: nervous system: in general. | ||||
| 8.1 3 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 7 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 21.9  Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 9 Minor adverse events excluding skin irritation: nervous system: headache. | ||||
| 9.1 3 times/week | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | 3.77 [0.23, 63.05] |
| 9.2 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.81 [0.10, 31.53] |
| 9.3 7 times/week | 2 | 527 | Risk Ratio (M‐H, Random, 95% CI) | 4.48 [0.86, 23.31] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 22.1  Comparison 22 5% imiquimod versus 5% 5‐FU, Outcome 1 Participant complete clearance. | ||||
| 2 Cosmetic outcome: Investigator cosmetic outcome "excellent" Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 22.2  Comparison 22 5% imiquimod versus 5% 5‐FU, Outcome 2 Cosmetic outcome: Investigator cosmetic outcome "excellent". | ||||
| 3 Cosmetic outcome: normal skin surface Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 22.3  Comparison 22 5% imiquimod versus 5% 5‐FU, Outcome 3 Cosmetic outcome: normal skin surface. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 23.1  Comparison 23 5% imiquimod versus cryotherapy, Outcome 1 Participant complete clearance. | ||||
| 1.1 5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance of target lesions Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 24.1  Comparison 24 Ingenol mebutate (PEP005) versus placebo, Outcome 1 Participant complete clearance of target lesions. | ||||
| 2 Participant complete clearance of all lesions Show forest plot | 2 | 456 | Risk Ratio (M‐H, Random, 95% CI) | 4.50 [2.61, 7.74] |
| Analysis 24.2  Comparison 24 Ingenol mebutate (PEP005) versus placebo, Outcome 2 Participant complete clearance of all lesions. | ||||
| 3 Participant partial (>75%) clearance of target lesions Show forest plot | 2 | 280 | Risk Ratio (M‐H, Random, 95% CI) | 2.88 [1.81, 4.58] |
| Analysis 24.3  Comparison 24 Ingenol mebutate (PEP005) versus placebo, Outcome 3 Participant partial (>75%) clearance of target lesions. | ||||
| 4 Cosmetic outcomes: changes in pigmentation Show forest plot | 3 | 514 | Risk Ratio (M‐H, Random, 95% CI) | 3.36 [0.63, 17.80] |
| Analysis 24.4  Comparison 24 Ingenol mebutate (PEP005) versus placebo, Outcome 4 Cosmetic outcomes: changes in pigmentation. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance of target lesions Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 25.1  Comparison 25 Ingenol mebutate (PEP005) versus placebo: different concentrations, Outcome 1 Participant complete clearance of target lesions. | ||||
| 1.1 0.025% 3 days | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 0.05% 2‐3 days | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant complete clearance of all lesions Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 25.2  Comparison 25 Ingenol mebutate (PEP005) versus placebo: different concentrations, Outcome 2 Participant complete clearance of all lesions. | ||||
| 2.1 0.025% 3 days | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [1.03, 15.55] |
| 2.2 0.05% 2‐3 days | 2 | 386 | Risk Ratio (M‐H, Random, 95% CI) | 5.14 [2.75, 9.62] |
| 3 Participant partial (>75%) clearance of target lesions Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 25.3  Comparison 25 Ingenol mebutate (PEP005) versus placebo: different concentrations, Outcome 3 Participant partial (>75%) clearance of target lesions. | ||||
| 3.1 0.0025% 2 days | 1 | 19 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.21, 8.41] |
| 3.2 0.01% 2 days | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.06, 4.23] |
| 3.3 0.025% 3 days | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 2.8 [1.13, 6.96] |
| 3.4 0.05% 2‐3 days | 2 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 3.34 [1.84, 6.04] |
| 4 Cosmetic outcomes: changes in pigmentation Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 25.4  Comparison 25 Ingenol mebutate (PEP005) versus placebo: different concentrations, Outcome 4 Cosmetic outcomes: changes in pigmentation. | ||||
| 4.1 0.01% 2 days | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [0.08, 25.88] |
| 4.2 0.05% 2 days | 2 | 253 | Risk Ratio (M‐H, Random, 95% CI) | 4.86 [0.48, 49.39] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance of target lesions Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 26.1  Comparison 26 0.05% Ingenol mebutate (PEP005) versus placebo: number of doses, Outcome 1 Participant complete clearance of target lesions. | ||||
| 1.1 0.05% 2 days | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 0.05% 3 days | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant complete clearance of all lesions Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 26.2  Comparison 26 0.05% Ingenol mebutate (PEP005) versus placebo: number of doses, Outcome 2 Participant complete clearance of all lesions. | ||||
| 2.1 0.05% 2 days | 2 | 319 | Risk Ratio (M‐H, Random, 95% CI) | 4.32 [2.30, 8.11] |
| 2.2 0.05% 3 days | 1 | 87 | Risk Ratio (M‐H, Random, 95% CI) | 4.08 [1.59, 10.47] |
| 3 Participant partial (>75%) clearance of target lesions Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 26.3  Comparison 26 0.05% Ingenol mebutate (PEP005) versus placebo: number of doses, Outcome 3 Participant partial (>75%) clearance of target lesions. | ||||
| 3.1 0.05% 2 days | 2 | 104 | Risk Ratio (M‐H, Random, 95% CI) | 2.65 [1.41, 5.00] |
| 3.2 0.05% 3 days | 1 | 87 | Risk Ratio (M‐H, Random, 95% CI) | 3.23 [1.66, 6.29] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Investigator global improvement indices‐completely cleared Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 27.1  Comparison 27 Isotretinoin versus vehicle, Outcome 1 Investigator global improvement indices‐completely cleared. | ||||
| 1.1 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Upper extremities | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Mean reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 27.2  Comparison 27 Isotretinoin versus vehicle, Outcome 2 Mean reduction of lesion counts. | ||||
| 2.1 Face | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Scalp | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Upper extremities | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 27.3  Comparison 27 Isotretinoin versus vehicle, Outcome 3 Withdrawal due to adverse events. | ||||
| 4 Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 27.4  Comparison 27 Isotretinoin versus vehicle, Outcome 4 Skin irritation. | ||||
| 5 Severe‐Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 27.5  Comparison 27 Isotretinoin versus vehicle, Outcome 5 Severe‐Skin irritation. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Global improvement indices‐cured Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 28.1  Comparison 28 Masoprocol versus placebo, Outcome 1 Global improvement indices‐cured. | ||||
| 2 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 28.2  Comparison 28 Masoprocol versus placebo, Outcome 2 Mean reduction in lesion counts. | ||||
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 28.3  Comparison 28 Masoprocol versus placebo, Outcome 3 Withdrawal due to adverse events. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 29.1  Comparison 29 1% nicotinamide versus placebo, Outcome 1 Mean percentage of reduction in lesion counts. | ||||
| 1.1 At 3 months | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 6 months | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 29.2  Comparison 29 1% nicotinamide versus placebo, Outcome 2 Withdrawal due to adverse events. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.1  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 1 Participant complete clearance. | ||||
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.2  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 2 Participant partial (>75%) clearance. | ||||
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.3  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 3 Withdrawal due to adverse events. | ||||
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.4  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue. | ||||
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.5  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors. | ||||
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.6  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general. | ||||
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.7  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia. | ||||
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.8  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia. | ||||
| 9 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.9  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general. | ||||
| 10 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.10  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache. | ||||
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.11  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy. | ||||
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.12  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders. | ||||
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 30.13  Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.1  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 1 Participant complete clearance. | ||||
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.2  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 2 Participant partial (>75%) clearance. | ||||
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.3  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 3 Withdrawal due to adverse events. | ||||
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.4  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue. | ||||
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.5  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors. | ||||
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.6  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general. | ||||
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.7  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia. | ||||
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.8  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia. | ||||
| 9 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.9  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general. | ||||
| 10 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.10  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache. | ||||
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.11  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy. | ||||
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.12  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders. | ||||
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 31.13  Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.1  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 1 Participant complete clearance. | ||||
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.2  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 2 Participant partial (>75%) clearance. | ||||
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.3  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 3 Withdrawal due to adverse events. | ||||
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.4  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue. | ||||
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.5  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors. | ||||
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.6  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general. | ||||
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.7  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia. | ||||
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.8  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia. | ||||
| 9 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.9  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general. | ||||
| 10 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.10  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache. | ||||
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.11  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy. | ||||
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.12  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders. | ||||
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 32.13  Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.1  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 1 Participant complete clearance. | ||||
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.2  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 2 Participant partial (>75%) clearance. | ||||
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.3  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 3 Withdrawal due to adverse events. | ||||
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.4  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue. | ||||
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.5  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors. | ||||
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.6  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general. | ||||
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.7  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia. | ||||
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.8  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia. | ||||
| 9 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.9  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general. | ||||
| 10 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.10  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache. | ||||
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.11  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy. | ||||
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.12  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders. | ||||
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 33.13  Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.1  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 1 Participant complete clearance. | ||||
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.2  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 2 Participant partial (>75%) clearance. | ||||
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.3  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 3 Withdrawal due to adverse events. | ||||
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.4  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue. | ||||
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.5  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors. | ||||
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.6  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general. | ||||
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.7  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia. | ||||
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.8  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia. | ||||
| 9 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.9  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general. | ||||
| 10 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.10  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache. | ||||
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.11  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy. | ||||
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.12  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders. | ||||
| 13 Minor adverse events excluding skin irritation:skin and subcutaneous disorders: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 34.13  Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 13 Minor adverse events excluding skin irritation:skin and subcutaneous disorders: in general. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.1  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 1 Participant complete clearance. | ||||
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.2  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 2 Participant partial (>75%) clearance. | ||||
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.3  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 3 Withdrawal due to adverse events. | ||||
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.4  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue. | ||||
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.5  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors. | ||||
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.6  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general. | ||||
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.7  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia. | ||||
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.8  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia. | ||||
| 9 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.9  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general. | ||||
| 10 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.10  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache. | ||||
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.11  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy. | ||||
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.12  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders. | ||||
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 35.13  Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean change in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 36.1  Comparison 36 Sunscreen SPF 17 (8% 2‐ethyl‐hexyl p‐methoxycinnamate/2% 4‐tert‐butyl‐4‐methoxy‐4‐dibenzoylmethane) versus placebo, Outcome 1 Mean change in lesion counts. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 37.1  Comparison 37 12.5% DL‐α‐tocopherol (vitamin E) versus placebo, Outcome 1 Mean reduction of lesion counts. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 38.1  Comparison 38 Etretinate versus placebo, Outcome 1 Participant complete clearance. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 39.1  Comparison 39 Carbon dioxide laser resurfacing versus 5% 5‐FU, Outcome 1 Mean percentage of reduction of lesion counts. | ||||
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 39.2  Comparison 39 Carbon dioxide laser resurfacing versus 5% 5‐FU, Outcome 2 Withdrawal due to adverse events. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 40.1  Comparison 40 Carbon dioxide laser resurfacing versus Trichloroacetic acid peel, Outcome 1 Mean percentage of reduction of lesion counts. | ||||
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 40.2  Comparison 40 Carbon dioxide laser resurfacing versus Trichloroacetic acid peel, Outcome 2 Withdrawal due to adverse events. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size | ||||||||||||||||||||
| 1 Mean reduction in lesion counts Show forest plot | Other data | No numeric data | ||||||||||||||||||||||
| Analysis 41.1
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 1 Mean reduction in lesion counts. | ||||||||||||||||||||||||
| 2 Mean percentage of reduction in lesion counts Show forest plot | Other data | No numeric data | ||||||||||||||||||||||
| Analysis 41.2
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 2 Mean percentage of reduction in lesion counts. | ||||||||||||||||||||||||
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |||||||||||||||||||||
| Analysis 41.3  Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 3 Withdrawal due to adverse events. | ||||||||||||||||||||||||
| 4 Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |||||||||||||||||||||
| Analysis 41.4  Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 4 Skin irritation. | ||||||||||||||||||||||||
| 4.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 4.2 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 4.3 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 5 Minor adverse events excluding skin irritation: dermatology: acne Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |||||||||||||||||||||
| Analysis 41.5  Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 5 Minor adverse events excluding skin irritation: dermatology: acne. | ||||||||||||||||||||||||
| 5.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 5.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 5.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 6 Minor adverse events excluding skin irritation: dermatology:crustea Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |||||||||||||||||||||
| Analysis 41.6  Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 6 Minor adverse events excluding skin irritation: dermatology:crustea. | ||||||||||||||||||||||||
| 6.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 6.2 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 6.3 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 6.4 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 7 Minor adverse events excluding skin irritation: dermatology: infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |||||||||||||||||||||
| Analysis 41.7  Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 7 Minor adverse events excluding skin irritation: dermatology: infection. | ||||||||||||||||||||||||
| 7.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 8 Minor adverse events excluding skin irritation: dermatology: milia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |||||||||||||||||||||
| Analysis 41.8  Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 8 Minor adverse events excluding skin irritation: dermatology: milia. | ||||||||||||||||||||||||
| 8.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 8.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 8.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 9 Minor adverse events excluding skin irritation: dermatology:pain Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |||||||||||||||||||||
| Analysis 41.9  Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 9 Minor adverse events excluding skin irritation: dermatology:pain. | ||||||||||||||||||||||||
| 9.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 9.2 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 10 Cosmetic outcomes: changes in pigmentation (hypo) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |||||||||||||||||||||
| Analysis 41.10  Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 10 Cosmetic outcomes: changes in pigmentation (hypo). | ||||||||||||||||||||||||
| 10.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 10.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 10.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 11 Cosmetic outcomes: scarring Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |||||||||||||||||||||
| Analysis 41.11  Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 11 Cosmetic outcomes: scarring. | ||||||||||||||||||||||||
| 11.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 11.2 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 12 Cosmetic outcomes: improvement in photoageing score Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |||||||||||||||||||||
| Analysis 41.12  Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 12 Cosmetic outcomes: improvement in photoageing score. | ||||||||||||||||||||||||
| 12.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 12.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| 12.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |||||||||||||||||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 42.1  Comparison 42 Cryotherapy versus betulin‐based oleogel, Outcome 1 Participant complete clearance. | ||||
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 42.2  Comparison 42 Cryotherapy versus betulin‐based oleogel, Outcome 2 Participant partial (>75%) clearance. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 43.1  Comparison 43 Cryotherapy versus 5% 5‐FU, Outcome 1 Participant complete clearance. | ||||
| 1.1 After treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Cosmetic outcomes: excellent global cosmetic outcome Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 43.2  Comparison 43 Cryotherapy versus 5% 5‐FU, Outcome 2 Cosmetic outcomes: excellent global cosmetic outcome. | ||||
| 3 Cosmetic outcomes: better skin appearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 43.3  Comparison 43 Cryotherapy versus 5% 5‐FU, Outcome 3 Cosmetic outcomes: better skin appearance. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 44.1  Comparison 44 Cryotherapy versus imiquimod, Outcome 1 Participant complete clearance. | ||||
| 1.1 5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Cosmetic outcomes: excellent global cosmetic outcome Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 44.2  Comparison 44 Cryotherapy versus imiquimod, Outcome 2 Cosmetic outcomes: excellent global cosmetic outcome. | ||||
| 3 Cosmetic outcomes: better skin appearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 44.3  Comparison 44 Cryotherapy versus imiquimod, Outcome 3 Cosmetic outcomes: better skin appearance. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size | ||||||||||||||||||||||||
| 1 Mean percentage of reduction in lesion counts Show forest plot | Other data | No numeric data | ||||||||||||||||||||||||||
| Analysis 45.1
Comparison 45 Cryotherapy versus MAL‐red light PDT, Outcome 1 Mean percentage of reduction in lesion counts. | ||||||||||||||||||||||||||||
| 2 Withdrawal due to adverse events Show forest plot | 2 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.16, 7.16] | ||||||||||||||||||||||||
| Analysis 45.2  Comparison 45 Cryotherapy versus MAL‐red light PDT, Outcome 2 Withdrawal due to adverse events. | ||||||||||||||||||||||||||||
| 3 Cosmetic outcomes: excellent or good cosmetic outcomes by investigator Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |||||||||||||||||||||||||
| Analysis 45.3  Comparison 45 Cryotherapy versus MAL‐red light PDT, Outcome 3 Cosmetic outcomes: excellent or good cosmetic outcomes by investigator. | ||||||||||||||||||||||||||||
| 4 Cosmetic outcomes: excellent or good cosmetic outcomes by participant Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |||||||||||||||||||||||||
| Analysis 45.4  Comparison 45 Cryotherapy versus MAL‐red light PDT, Outcome 4 Cosmetic outcomes: excellent or good cosmetic outcomes by participant. | ||||||||||||||||||||||||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 46.1  Comparison 46 Cryotherapy versus ALA‐red light PDT, Outcome 1 Participant complete clearance. | ||||
| 2 Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 46.2  Comparison 46 Cryotherapy versus ALA‐red light PDT, Outcome 2 Skin irritation. | ||||
| 2.1 During treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 One day after treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance [1 treatment] Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 47.1 ![Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 1 Participant complete clearance [1 treatment].](/cdsr/doi/10.1002/14651858.CD004415.pub2/media/CDSR/CD004415/image_n/nCD004415-CMP-047-01.png) Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 1 Participant complete clearance [1 treatment]. | ||||
| 1.1 Blue light | 1 | 243 | Risk Ratio (M‐H, Random, 95% CI) | 6.22 [2.88, 13.43] |
| 1.2 Red light | 3 | 422 | Risk Ratio (M‐H, Random, 95% CI) | 5.94 [3.35, 10.54] |
| 2 Participant complete clearance [1 or 2 treatments] Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 47.2 ![Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 2 Participant complete clearance [1 or 2 treatments].](/cdsr/doi/10.1002/14651858.CD004415.pub2/media/CDSR/CD004415/image_n/nCD004415-CMP-047-02.png) Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 2 Participant complete clearance [1 or 2 treatments]. | ||||
| 2.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Red light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Participant complete clearance [1 or 2 treatments] by anatomical location Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 47.3 ![Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 3 Participant complete clearance [1 or 2 treatments] by anatomical location.](/cdsr/doi/10.1002/14651858.CD004415.pub2/media/CDSR/CD004415/image_n/nCD004415-CMP-047-03.png) Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 3 Participant complete clearance [1 or 2 treatments] by anatomical location. | ||||
| 3.1 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Participant partial (> 75%) clearance [1 treatment] Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 47.4 ![Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 4 Participant partial (> 75%) clearance [1 treatment].](/cdsr/doi/10.1002/14651858.CD004415.pub2/media/CDSR/CD004415/image_n/nCD004415-CMP-047-04.png) Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 4 Participant partial (> 75%) clearance [1 treatment]. | ||||
| 4.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Participant partial (>75%) clearance[1 or 2 treatments] Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 47.5 ![Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 5 Participant partial (>75%) clearance[1 or 2 treatments].](/cdsr/doi/10.1002/14651858.CD004415.pub2/media/CDSR/CD004415/image_n/nCD004415-CMP-047-05.png) Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 5 Participant partial (>75%) clearance[1 or 2 treatments]. | ||||
| 5.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Participant partial (>75%) clearance [1 or 2 treatment] by anatomical location Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 47.6 ![Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 6 Participant partial (>75%) clearance [1 or 2 treatment] by anatomical location.](/cdsr/doi/10.1002/14651858.CD004415.pub2/media/CDSR/CD004415/image_n/nCD004415-CMP-047-06.png) Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 6 Participant partial (>75%) clearance [1 or 2 treatment] by anatomical location. | ||||
| 6.1 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 47.7  Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 7 Skin irritation. | ||||
| 7.1 Red light‐during illumination | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Red light‐after treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: body as a whole: injury Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 47.8  Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 8 Minor adverse events excluding skin irritation: body as a whole: injury. | ||||
| 8.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: cardiovascular: hypertension Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 47.9  Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 9 Minor adverse events excluding skin irritation: cardiovascular: hypertension. | ||||
| 9.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Minor adverse events excluding skin irritation: dermatology: skin discolouration Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 47.10  Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 10 Minor adverse events excluding skin irritation: dermatology: skin discolouration. | ||||
| 10.1 Red light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Minor adverse events excluding skin irritation: dermatology: skin hypertrophy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 47.11  Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 11 Minor adverse events excluding skin irritation: dermatology: skin hypertrophy. | ||||
| 11.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 47.12  Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 12 Minor adverse events excluding skin irritation: nervous system: headache. | ||||
| 12.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Cosmetic outcome: very good or good general cosmetic outcome Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 47.13  Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 13 Cosmetic outcome: very good or good general cosmetic outcome. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 48.1  Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 1 Participant complete clearance. | ||||
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 48.2  Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 2 Participant partial (>75%) clearance. | ||||
| 3 Cosmetic outcome: improvement in global response Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 48.3  Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 3 Cosmetic outcome: improvement in global response. | ||||
| 4 Cosmetic outcome: improvement in tactile roughness Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 48.4  Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 4 Cosmetic outcome: improvement in tactile roughness. | ||||
| 5 Cosmetic outcome: improvement in mottled hyperpigmentation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 48.5  Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 5 Cosmetic outcome: improvement in mottled hyperpigmentation. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance at 4 weeks Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 49.1  Comparison 49 ALA‐red light PDT at different application times, Outcome 1 Participant complete clearance at 4 weeks. | ||||
| 1.1 0.5h versus 1.0h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 0.5h versus 2 h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 1h versus 2h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 1h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 2h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant complete clearance at 8 weeks Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 49.2  Comparison 49 ALA‐red light PDT at different application times, Outcome 2 Participant complete clearance at 8 weeks. | ||||
| 2.1 0.5h versus 1.0h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 0.5h versus 2 h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 1h versus 2h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.5 1h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.6 2h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Minor adverse events excluding skin irritation: metabolic and nutritional disorders: elevated alanine transaminase (ALT) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 49.3  Comparison 49 ALA‐red light PDT at different application times, Outcome 3 Minor adverse events excluding skin irritation: metabolic and nutritional disorders: elevated alanine transaminase (ALT). | ||||
| 3.1 0.5h versus 1.0h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 0.5h versus 2 h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 49.4  Comparison 49 ALA‐red light PDT at different application times, Outcome 4 Minor adverse events excluding skin irritation: nervous system: headache. | ||||
| 4.1 0.5h versus 1h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 0.5h versus 2h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 1h versus 2h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 1h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 2h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Minor adverse events excluding skin irritation: other: epistaxis (nose bleeding) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 49.5  Comparison 49 ALA‐red light PDT at different application times, Outcome 5 Minor adverse events excluding skin irritation: other: epistaxis (nose bleeding). | ||||
| 5.1 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 1h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 2h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 50.1  Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 1 Participant complete clearance. | ||||
| 1.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 50.2  Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 2 Participant partial (>75%) clearance. | ||||
| 2.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 50.3  Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 3 Withdrawal due to adverse events. | ||||
| 3.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Cosmetic outcome: improvement in global response Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 50.4  Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 4 Cosmetic outcome: improvement in global response. | ||||
| 4.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Cosmetic outcome: improvement in tactile roughness Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 50.5  Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 5 Cosmetic outcome: improvement in tactile roughness. | ||||
| 5.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Cosmetic outcome: improvement in mottled hyperpigmentation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 50.6  Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 6 Cosmetic outcome: improvement in mottled hyperpigmentation. | ||||
| 6.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 51.1  Comparison 51 ALA‐red light PDT vs cryotherapy, Outcome 1 Participant complete clearance. | ||||
| 2 Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 51.2  Comparison 51 ALA‐red light PDT vs cryotherapy, Outcome 2 Skin irritation. | ||||
| 2.1 During treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 One day after treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 5 | 482 | Risk Ratio (M‐H, Random, 95% CI) | 4.46 [3.17, 6.28] |
| Analysis 52.1  Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 1 Participant complete clearance. | ||||
| 2 Participant partial (>75%) clearance Show forest plot | 2 | 191 | Risk Ratio (M‐H, Random, 95% CI) | 3.28 [1.73, 6.23] |
| Analysis 52.2  Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 2 Participant partial (>75%) clearance. | ||||
| 3 Withdrawal due to adverse events Show forest plot | 2 | 191 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.23, 17.74] |
| Analysis 52.3  Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 3 Withdrawal due to adverse events. | ||||
| 4 Minor adverse event: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 52.4  Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 4 Minor adverse event: nervous system: headache. | ||||
| 5 Cosmetic outcome: hyperpigmentation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 52.5  Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 5 Cosmetic outcome: hyperpigmentation. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 53.1  Comparison 53 MAL‐red light LED PDT versus MAL‐broad visible + water‐filtered infrared A PDT (1 or 2 treatments), Outcome 1 Participant complete clearance. | ||||
| 1.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 53.2  Comparison 53 MAL‐red light LED PDT versus MAL‐broad visible + water‐filtered infrared A PDT (1 or 2 treatments), Outcome 2 Participant partial (>75%) clearance. | ||||
| 2.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 54.1  Comparison 54 MAL‐red light LED PDT versus MAL‐daylight PDT, Outcome 1 Mean reduction in lesion counts. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 55.1  Comparison 55 2h MAL‐day light PDT versus 3h MAL‐daylight PDT, Outcome 1 Mean reduction in lesion counts. | ||||
| 2 Mean percentage of reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 55.2  Comparison 55 2h MAL‐day light PDT versus 3h MAL‐daylight PDT, Outcome 2 Mean percentage of reduction in lesion counts. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 56.1  Comparison 56 16% MAL‐daylight PDT versus 8% MAL‐daylight PDT, Outcome 1 Mean reduction in lesion counts. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 57.1  Comparison 57 Single MAL‐red light PDT versus multiple MAL‐red light PDT (2 treatments 1 week apart), Outcome 1 Participant complete clearance. | ||||
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 57.2  Comparison 57 Single MAL‐red light PDT versus multiple MAL‐red light PDT (2 treatments 1 week apart), Outcome 2 Withdrawal due to adverse events. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Withdrawal due to adverse events Show forest plot | 2 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.14, 6.36] |
| Analysis 58.1  Comparison 58 MAL‐ red light PDT vs cryotherapy, Outcome 1 Withdrawal due to adverse events. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 59.1  Comparison 59 ALA‐red light PDT versus MAL‐red light PDT, Outcome 1 Mean reduction in lesion counts. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction in lesions Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 60.1  Comparison 60 Trichloroacetic acid peel versus 5% 5‐FU, Outcome 1 Mean percentage of reduction in lesions. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 61.1  Comparison 61 Cryotherapy versus cryotherapy with betulin‐based oleogel, Outcome 1 Participant complete clearance. | ||||
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 61.2  Comparison 61 Cryotherapy versus cryotherapy with betulin‐based oleogel, Outcome 2 Participant partial (>75%) clearance. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance at 6 months Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 62.1  Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 1 Participant complete clearance at 6 months. | ||||
| 1.1 1 cycle (1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 2 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 3 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Mean reduction in lesion counts at 6 months Show forest plot | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| Analysis 62.2  Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 2 Mean reduction in lesion counts at 6 months. | ||||
| 2.1 1 cycle (1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 2 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 3 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Mean percentage of reduction in lesion counts at 6 months Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| Analysis 62.3  Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 3 Mean percentage of reduction in lesion counts at 6 months. | ||||
| 3.1 1 cycle (1 week topical, cryosurgery at 4 weeks, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Minor adverse events excluding skin irritation: body as a whole: allergic reaction Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 62.4  Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 4 Minor adverse events excluding skin irritation: body as a whole: allergic reaction. | ||||
| 5 Minor adverse events excluding skin irritation: dermatology: hyperesthesia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 62.5  Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 5 Minor adverse events excluding skin irritation: dermatology: hyperesthesia. | ||||
| 6 Minor adverse events excluding skin irritation: dermatology: skin discoloration Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 62.6  Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 6 Minor adverse events excluding skin irritation: dermatology: skin discoloration. | ||||
| 7 Minor adverse events excluding skin irritation: dermatology: vesiculobullous rash Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 62.7  Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 7 Minor adverse events excluding skin irritation: dermatology: vesiculobullous rash. | ||||
| 8 Minor adverse events excluding skin irritation: digestive: cheilitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 62.8  Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 8 Minor adverse events excluding skin irritation: digestive: cheilitis. | ||||
| 9 Minor adverse events excluding skin irritation: special senses: conjunctivitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 62.9  Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 9 Minor adverse events excluding skin irritation: special senses: conjunctivitis. | ||||
| 10 Minor adverse events excluding skin irritation: special senses: eye irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 62.10  Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 10 Minor adverse events excluding skin irritation: special senses: eye irritation. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance at 6 months Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 63.1  Comparison 63 (vehicle + cryotherapy) versus (0.5% 5‐FU + cryotherapy), Outcome 1 Participant complete clearance at 6 months. | ||||
| 1.1 1 cycle (1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 2 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 3 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Mean reduction in lesion counts at 6 months Show forest plot | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| Analysis 63.2  Comparison 63 (vehicle + cryotherapy) versus (0.5% 5‐FU + cryotherapy), Outcome 2 Mean reduction in lesion counts at 6 months. | ||||
| 2.1 1 cycle (1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 2 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 3 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Mean percentage of reduction in lesion counts at 6 months Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| Analysis 63.3  Comparison 63 (vehicle + cryotherapy) versus (0.5% 5‐FU + cryotherapy), Outcome 3 Mean percentage of reduction in lesion counts at 6 months. | ||||
| 3.1 1 cycle (1 week topical, cryosurgery at 4 weeks, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance of all lesions Show forest plot | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.05, 0.73] |
| Analysis 64.1  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 1 Participant complete clearance of all lesions. | ||||
| 1.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.11, 1.42] |
| 1.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.04, 0.30] |
| 2 Participant complete clearance of target (cryotherapy treated) lesions Show forest plot | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.36, 1.04] |
| Analysis 64.2  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 2 Participant complete clearance of target (cryotherapy treated) lesions. | ||||
| 2.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.47, 1.60] |
| 2.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.37, 0.68] |
| 3 Participant complete clearance of subclinical lesions Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.3  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 3 Participant complete clearance of subclinical lesions. | ||||
| 3.1 5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Mean percentage of reduction in all lesion counts Show forest plot | 2 | 301 | Mean Difference (IV, Random, 95% CI) | ‐23.69 [‐46.03, ‐1.34] |
| Analysis 64.4  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 4 Mean percentage of reduction in all lesion counts. | ||||
| 4.1 5% imquimod | 1 | 54 | Mean Difference (IV, Random, 95% CI) | ‐11.20 [‐26.53, 4.13] |
| 4.2 3.75% imiquimod | 1 | 247 | Mean Difference (IV, Random, 95% CI) | ‐34.10 [‐41.38, ‐26.82] |
| 5 Mean percentage of reduction in target (cryotherapy treated) lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 64.5  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 5 Mean percentage of reduction in target (cryotherapy treated) lesion counts. | ||||
| 5.1 3.75% imiquimod | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Withdrawal due to adverse events Show forest plot | 2 | 312 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.28, 3.07] |
| Analysis 64.6  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 6 Withdrawal due to adverse events. | ||||
| 6.1 5% imiquimod | 1 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 2.91 [0.12, 68.95] |
| 6.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.21, 2.79] |
| 7 Skin irritation Show forest plot | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.10, 1.54] |
| Analysis 64.7  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 7 Skin irritation. | ||||
| 7.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.20, 2.01] |
| 7.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.02, 1.19] |
| 8 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.8  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 8 Minor adverse events excluding skin irritation: body as a whole: fatigue. | ||||
| 9 Minor adverse events excluding skin irritation: digestive: nausea Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.9  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 9 Minor adverse events excluding skin irritation: digestive: nausea. | ||||
| 10 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.10  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 10 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia. | ||||
| 11 Minor adverse events excluding skin irritation: respiratory: upper respiratory tract infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.11  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 11 Minor adverse events excluding skin irritation: respiratory: upper respiratory tract infection. | ||||
| 12 Minor adverse events excluding skin irritation: respiratory: bronchitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.12  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 12 Minor adverse events excluding skin irritation: respiratory: bronchitis. | ||||
| 13 Minor adverse events excluding skin irritation: respiratory: sinusitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.13  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 13 Minor adverse events excluding skin irritation: respiratory: sinusitis. | ||||
| 14 Minor adverse events excluding skin irritation: special senses: conjunctivitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.14  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 14 Minor adverse events excluding skin irritation: special senses: conjunctivitis. | ||||
| 15 Cosmetic outcomes: Improved global photoageing score Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.15  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 15 Cosmetic outcomes: Improved global photoageing score. | ||||
| 16 Cosmetic outcomes: Improved fine lines Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.16  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 16 Cosmetic outcomes: Improved fine lines. | ||||
| 17 Cosmetic outcomes: Improved tactile roughness Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.17  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 17 Cosmetic outcomes: Improved tactile roughness. | ||||
| 18 Cosmetic outcomes: Improved mottled pigmentation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.18  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 18 Cosmetic outcomes: Improved mottled pigmentation. | ||||
| 19 Cosmetic outcomes: Improved sallowness Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 64.19  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 19 Cosmetic outcomes: Improved sallowness. | ||||
| 20 Cosmetic outcomes: cosmetic appearance score Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 64.20  Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 20 Cosmetic outcomes: cosmetic appearance score. | ||||
| 20.1 Investigator | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 20.2 Participant | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance of all lesions Show forest plot | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 5.04 [1.37, 18.51] |
| Analysis 65.1  Comparison 65 Cryotherapy with imiquimod versus cryotherapy with vehicle, Outcome 1 Participant complete clearance of all lesions. | ||||
| 1.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 2.48 [0.70, 8.76] |
| 1.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 9.12 [3.36, 24.79] |
| 2 Mean percentage of reduction in all lesion counts Show forest plot | 2 | 301 | Mean Difference (IV, Random, 95% CI) | 23.69 [1.34, 46.03] |
| Analysis 65.2  Comparison 65 Cryotherapy with imiquimod versus cryotherapy with vehicle, Outcome 2 Mean percentage of reduction in all lesion counts. | ||||
| 2.1 5% imquimod | 1 | 54 | Mean Difference (IV, Random, 95% CI) | 11.20 [‐4.13, 26.53] |
| 2.2 3.75% imiquimod | 1 | 247 | Mean Difference (IV, Random, 95% CI) | 34.10 [26.82, 41.38] |
| 3 Withdrawal due to adverse events Show forest plot | 2 | 312 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.33, 3.56] |
| Analysis 65.3  Comparison 65 Cryotherapy with imiquimod versus cryotherapy with vehicle, Outcome 3 Withdrawal due to adverse events. | ||||
| 3.1 5% imiquimod | 1 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.13] |
| 3.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.36, 4.73] |
| 4 Skin irritation Show forest plot | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 2.55 [0.65, 10.04] |
| Analysis 65.4  Comparison 65 Cryotherapy with imiquimod versus cryotherapy with vehicle, Outcome 4 Skin irritation. | ||||
| 4.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [0.50, 5.13] |
| 4.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 6.72 [0.84, 53.83] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size | ||||||||||||||||||||
| 1 Global Improvement Indices (‐2 to 4) at 6 months Show forest plot | Other data | No numeric data | ||||||||||||||||||||||
| Analysis 66.1
Comparison 66 (3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT) versus (2.5% hyaluronic acid + ALA‐red light PDT), Outcome 1 Global Improvement Indices (‐2 to 4) at 6 months. | ||||||||||||||||||||||||
| 2 Mean reduction of lesion counts Show forest plot | Other data | No numeric data | ||||||||||||||||||||||
| Analysis 66.2
Comparison 66 (3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT) versus (2.5% hyaluronic acid + ALA‐red light PDT), Outcome 2 Mean reduction of lesion counts. | ||||||||||||||||||||||||

Study flow diagram.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Funnel plot of comparison: 15 Imiquimod versus placebo: different concentrations, outcome: 15.1 Participant complete clearance.

Funnel plot of comparison: 50 MAL‐PDT (red light) versus placebo‐PDT (red light), outcome: 50.1 Participant complete clearance.

Comparison 1 Adapalene gel versus placebo, Outcome 1 Global Improvement Indices (investigator)‐cleared.

Comparison 1 Adapalene gel versus placebo, Outcome 2 Mean changes in lesion counts.

Comparison 1 Adapalene gel versus placebo, Outcome 3 Withdrawal due to adverse events.

Comparison 1 Adapalene gel versus placebo, Outcome 4 Minor adverse events excluding skin irritation: dermatitis.
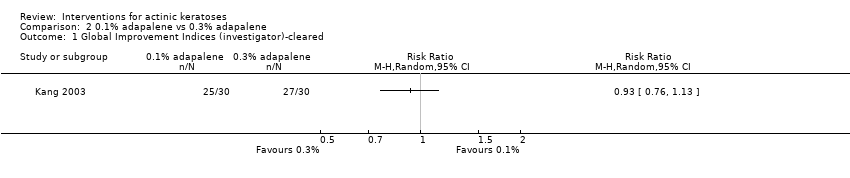
Comparison 2 0.1% adapalene vs 0.3% adapalene, Outcome 1 Global Improvement Indices (investigator)‐cleared.

Comparison 2 0.1% adapalene vs 0.3% adapalene, Outcome 2 Mean changes in lesion counts.

Comparison 2 0.1% adapalene vs 0.3% adapalene, Outcome 3 Withdrawal due to adverse events.

Comparison 2 0.1% adapalene vs 0.3% adapalene, Outcome 4 Minor adverse events excluding skin irritation: dermatitis.
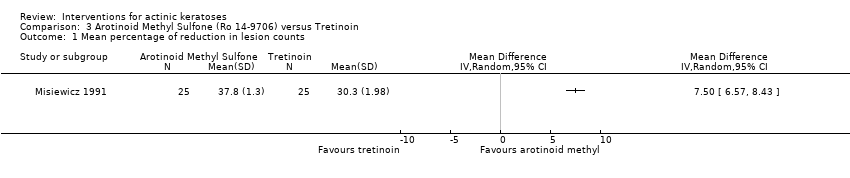
Comparison 3 Arotinoid Methyl Sulfone (Ro 14‐9706) versus Tretinoin, Outcome 1 Mean percentage of reduction in lesion counts.

Comparison 4 Calcipotriol (vitamin D) versus placebo, Outcome 1 Mean changes in lesion counts.
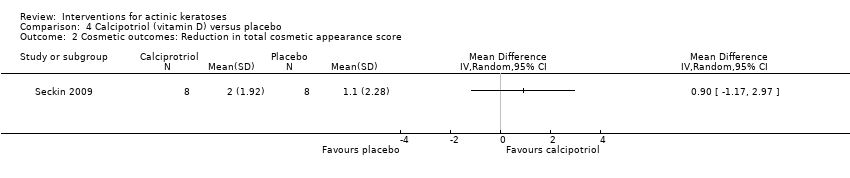
Comparison 4 Calcipotriol (vitamin D) versus placebo, Outcome 2 Cosmetic outcomes: Reduction in total cosmetic appearance score.

Comparison 5 1% colchicine cream versus 0.5% colchicine cream, Outcome 1 Participant complete clearance.

Comparison 5 1% colchicine cream versus 0.5% colchicine cream, Outcome 2 Mean reduction in lesion counts‐total.
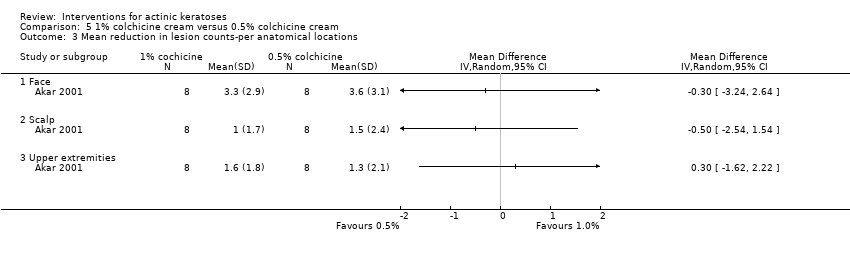
Comparison 5 1% colchicine cream versus 0.5% colchicine cream, Outcome 3 Mean reduction in lesion counts‐per anatomical locations.
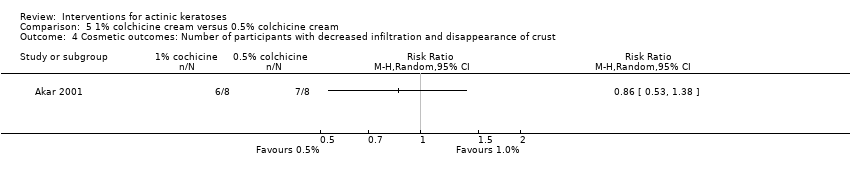
Comparison 5 1% colchicine cream versus 0.5% colchicine cream, Outcome 4 Cosmetic outcomes: Number of participants with decreased infiltration and disappearance of crust.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 1 Investigator Global Improvement Indices‐completely improved.
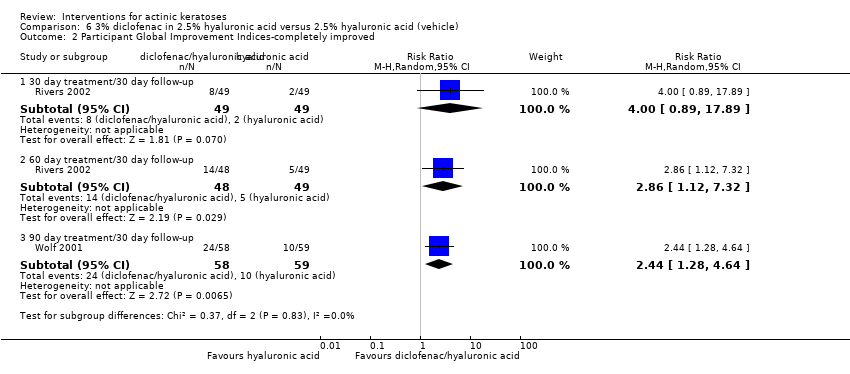
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 2 Participant Global Improvement Indices‐completely improved.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 3 Participant complete clearance at end of treatment (>56 days).

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 4 Participant complete clearance (target lesions).

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 5 Participant complete clearance (all lesions).

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 6 Participant complete clearance for 30 day treatment by locations.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 7 Participant complete clearance for 60 day treatment by locations.
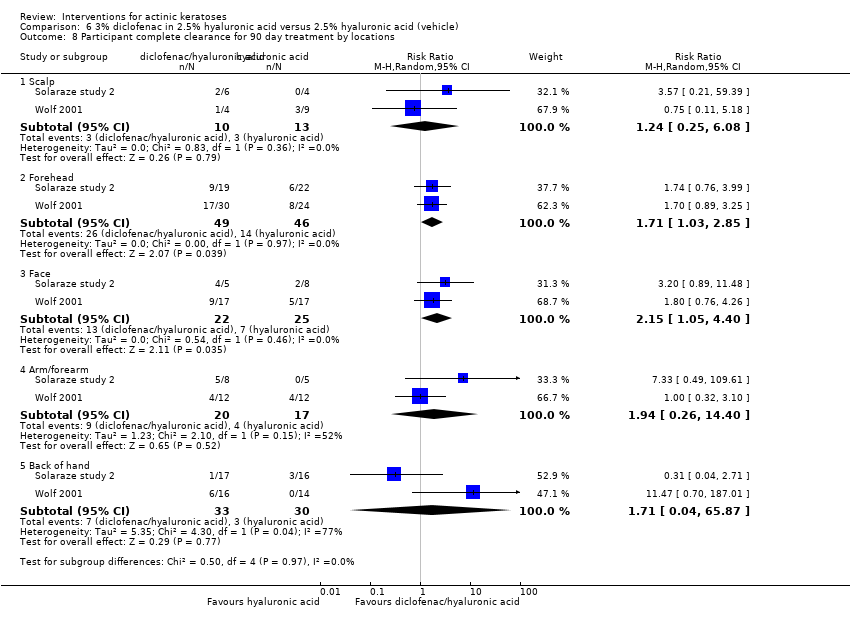
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 8 Participant complete clearance for 90 day treatment by locations.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 9 Participant complete clearance in immunosuppressed participants.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 10 Participant partial (>75%) clearance in immunosuppressed participants.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 11 Mean reduction of lesion counts (30‐90 days ): At the end of study.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 12 Mean reduction of lesion counts (30‐90 days): 30 day follow‐up.
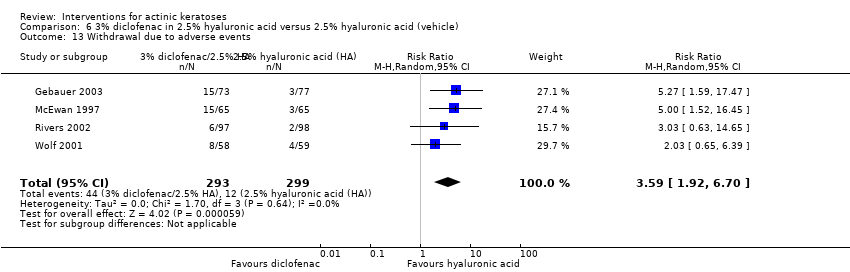
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 13 Withdrawal due to adverse events.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 14 Minor adverse event: body as a whole : in general.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 15 Minor adverse event: body as a whole : "flu".

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 16 Minor adverse event:: body as a whole : infection.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 17 Minor adverse event: cardiovascular: in general.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 18 Minor adverse event: cardiovascular: sinus bradycardia.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 19 Minor adverse event: dermatological: bursitis.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 20 Minor adverse event: dermatological: dry skin.
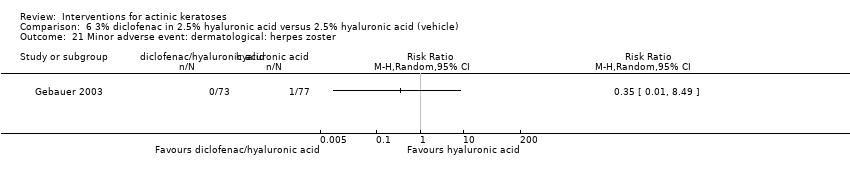
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 21 Minor adverse event: dermatological: herpes zoster.
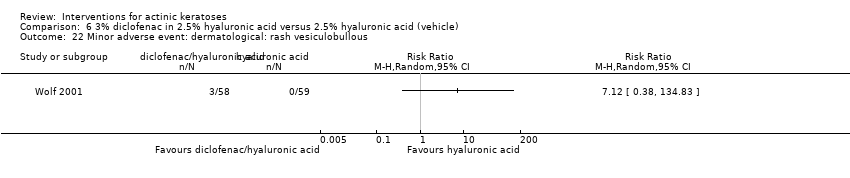
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 22 Minor adverse event: dermatological: rash vesiculobullous.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 23 Minor adverse event::dermatological: seborrhoea.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 24 Minor adverse event: dermatological: skin exfoliation.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 25 Minor adverse event: dermatological: ulcerated skin.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 26 Minor adverse event: digestive : in general.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 27 Minor adverse event: hemic and lymphatic: in general.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 28 Minor adverse event: metabolic and nutritional disorders : in general.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 29 Minor adverse event: musculoskeletal and connective tissue: in general.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 30 Minor adverse event: musculoskeletal and connective tissue: hypokinesia.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 31 Minor adverse event: nervous system: in general.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 32 Minor adverse event: nervous system: headache.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 33 Minor adverse event: nervous system: hyperaesthesia.
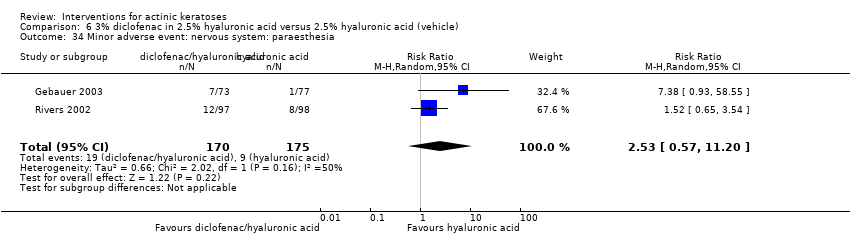
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 34 Minor adverse event: nervous system: paraesthesia.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 35 Minor adverse event: respiratory: in general.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 36 Minor adverse event: respiratory: bronchitis.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 37 Minor adverse event: respiratory: pharyngitis.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 38 Minor adverse event: respiratory: upper respiratory tract infection.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 39 Minor adverse event: special senses: in general.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 40 Minor adverse event: urogenital: in general.
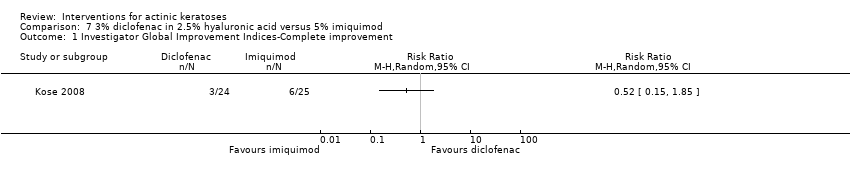
Comparison 7 3% diclofenac in 2.5% hyaluronic acid versus 5% imiquimod, Outcome 1 Investigator Global Improvement Indices‐Complete improvement.

Comparison 7 3% diclofenac in 2.5% hyaluronic acid versus 5% imiquimod, Outcome 2 Participant Global Improvement Indices‐Complete improvement.
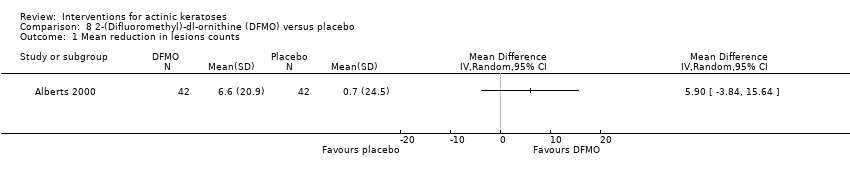
Comparison 8 2‐(Difluoromethyl)‐dl‐ornithine (DFMO) versus placebo, Outcome 1 Mean reduction in lesions counts.
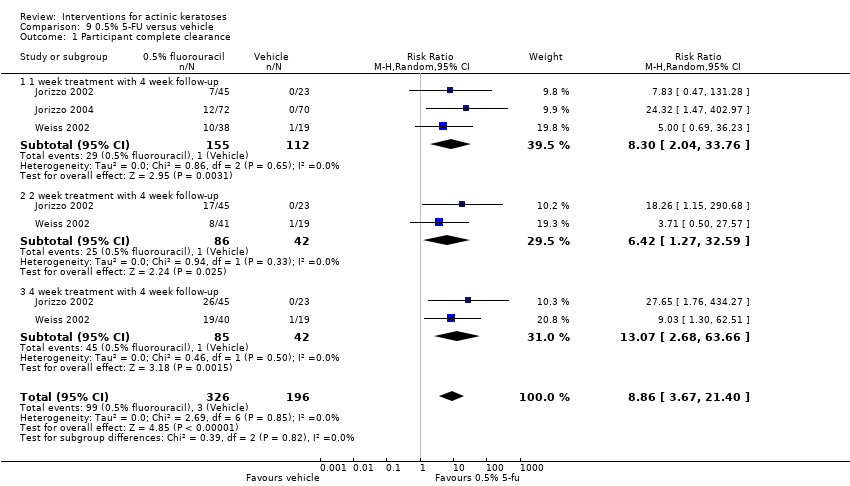
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 1 Participant complete clearance.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 2 Mean reduction in lesion counts.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 3 Mean percentage of reduction in lesion counts.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 4 Withdrawal due to adverse events.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 5 Skin irritation.
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 6 Minor adverse event excluding skin irritation: body as a whole : in general.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 7 Minor adverse event excluding skin irritation: body as a whole : allergy.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 8 Minor adverse event excluding skin irritation: body as a whole : "flu" or common cold.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 9 Minor adverse event excluding skin irritation: musculoskeletal and connective tissue: in general.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 10 Minor adverse event excluding skin irritation: musculoskeletal and connective tissue: soreness.
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 11 Minor adverse event excluding skin irritation:nervous system: headache.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 12 Minor adverse event excluding skin irritation: respiratory: in general.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 13 Minor adverse event excluding skin irritation: respiratory: sinusitis.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 14 Minor adverse event excluding skin irritation: respiratory: upper respiratory tract infection.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 15 Minor adverse event excluding skin irritation: special senses: in general.

Comparison 9 0.5% 5‐FU versus vehicle, Outcome 16 Minor adverse event excluding skin irritation:special senses: eye irritation.
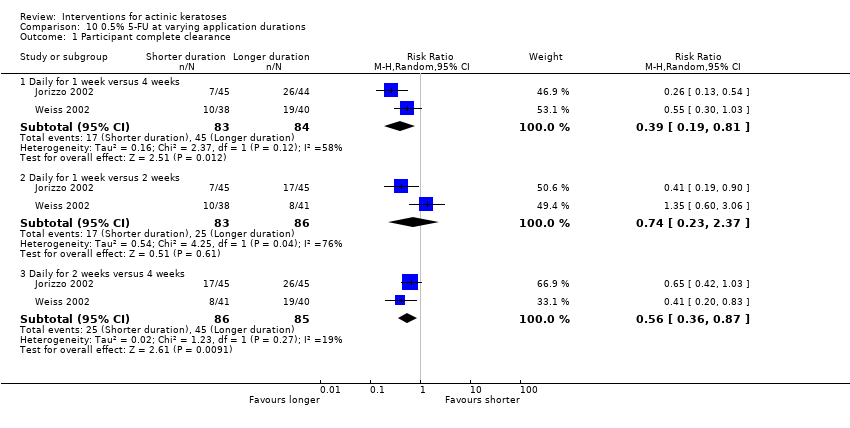
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 1 Participant complete clearance.

Comparison 10 0.5% 5‐FU at varying application durations, Outcome 2 Withdrawal due to adverse events.

Comparison 10 0.5% 5‐FU at varying application durations, Outcome 3 Skin irritation.

Comparison 10 0.5% 5‐FU at varying application durations, Outcome 4 Minor adverse events excluding skin irritation: body as a whole : in general.

Comparison 10 0.5% 5‐FU at varying application durations, Outcome 5 Minor adverse events excluding skin irritation: body as a whole : allergy.

Comparison 10 0.5% 5‐FU at varying application durations, Outcome 6 Minor adverse events excluding skin irritation: body as a whole : "flu" or common cold.

Comparison 10 0.5% 5‐FU at varying application durations, Outcome 7 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 8 Minor adverse events excluding skin irritation: nervous system: headache.

Comparison 10 0.5% 5‐FU at varying application durations, Outcome 9 Minor adverse events excluding skin irritation: respiratory: in general.

Comparison 10 0.5% 5‐FU at varying application durations, Outcome 10 Minor adverse events excluding skin irritation: respiratory: sinusitis.

Comparison 10 0.5% 5‐FU at varying application durations, Outcome 11 Minor adverse events excluding skin irritation: special senses: in general.
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 12 Minor adverse events excluding skin irritation: special senses: eye irritation.

Comparison 11 0.5% 5‐FU versus ALA‐PDT, Outcome 1 Participant complete clearance.
Comparison 11 0.5% 5‐FU versus ALA‐PDT, Outcome 2 Withdrawal due to adverse events.
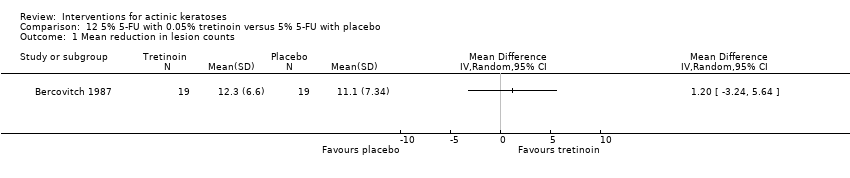
Comparison 12 5% 5‐FU with 0.05% tretinoin versus 5% 5‐FU with placebo, Outcome 1 Mean reduction in lesion counts.

Comparison 13 5% 5‐FU versus 5% imiquimod, Outcome 1 Participant complete clearance.

Comparison 14 5% 5‐FU versus cryotherapy, Outcome 1 Participant complete clearance.

Comparison 15 5% 5‐FU versus 10% masoprocol, Outcome 1 Investigator Global Improvement Indices ‐cleared.

Comparison 15 5% 5‐FU versus 10% masoprocol, Outcome 2 Mean reduction of lesion counts.

Comparison 15 5% 5‐FU versus 10% masoprocol, Outcome 3 Mean percentage of reduction of lesion counts.

Comparison 15 5% 5‐FU versus 10% masoprocol, Outcome 4 Withdrawal due to adverse events.

Comparison 16 5% 5‐FU versus carbon dioxide laser resurfacing, Outcome 1 Mean percentage of reduction of lesion counts.

Comparison 16 5% 5‐FU versus carbon dioxide laser resurfacing, Outcome 2 Withdrawal due to adverse events.

Comparison 17 5% 5‐FU versus Er:YAG laser resurfacing, Outcome 1 Withdrawal due to adverse events.
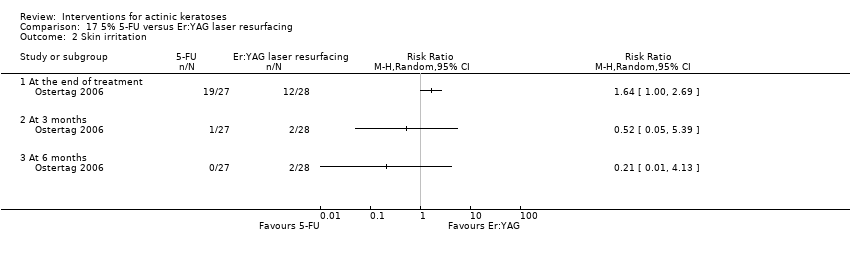
Comparison 17 5% 5‐FU versus Er:YAG laser resurfacing, Outcome 2 Skin irritation.

Comparison 18 5% 5‐FU versus Trichloroacetic acid peel, Outcome 1 Mean percentage of reduction in lesions.

Comparison 19 5% Imiquimod versus placebo, Outcome 1 Participant complete clearance‐number of doses.
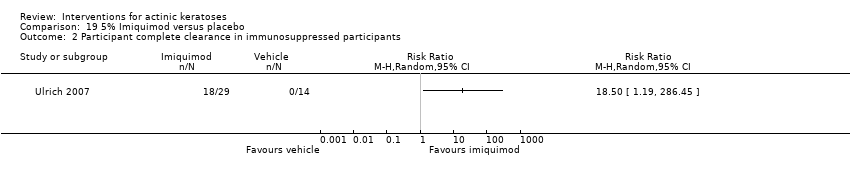
Comparison 19 5% Imiquimod versus placebo, Outcome 2 Participant complete clearance in immunosuppressed participants.
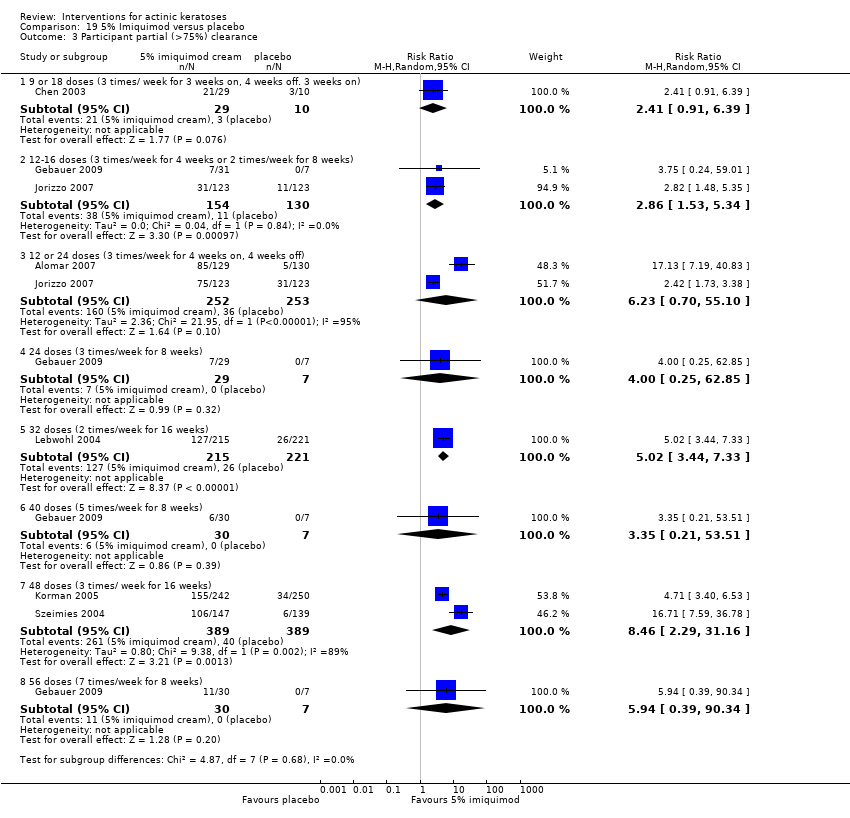
Comparison 19 5% Imiquimod versus placebo, Outcome 3 Participant partial (>75%) clearance.

Comparison 19 5% Imiquimod versus placebo, Outcome 4 Participant partial (>75%) clearance in immunosuppressed participants.

Comparison 19 5% Imiquimod versus placebo, Outcome 5 Mean reduction in lesion counts.
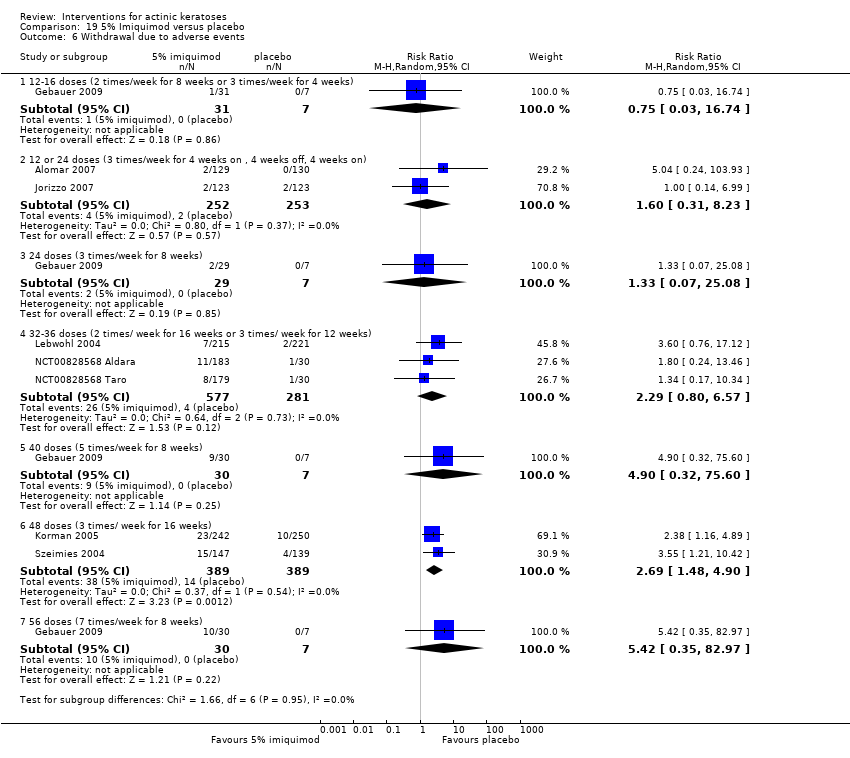
Comparison 19 5% Imiquimod versus placebo, Outcome 6 Withdrawal due to adverse events.
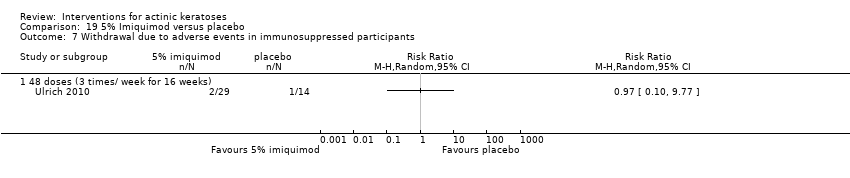
Comparison 19 5% Imiquimod versus placebo, Outcome 7 Withdrawal due to adverse events in immunosuppressed participants.

Comparison 19 5% Imiquimod versus placebo, Outcome 8 Minor adverse events excluding skin irritation: body as a whole: in general.

Comparison 19 5% Imiquimod versus placebo, Outcome 9 Minor adverse events excluding skin irritation: body as a whole: "flu" or "cold".

Comparison 19 5% Imiquimod versus placebo, Outcome 10 Minor adverse events excluding skin irritation: digestive: in general.

Comparison 19 5% Imiquimod versus placebo, Outcome 11 Minor adverse events excluding skin irritation: digestive: nausea.
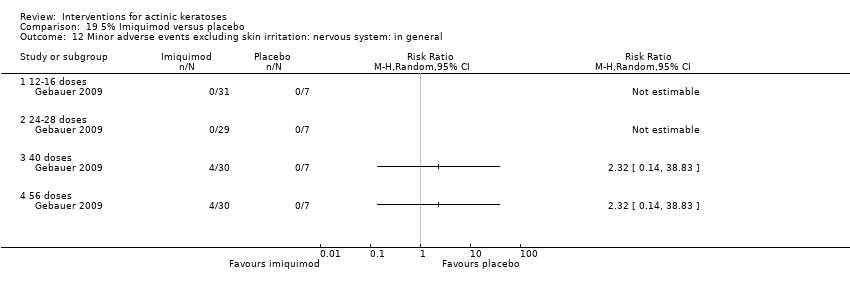
Comparison 19 5% Imiquimod versus placebo, Outcome 12 Minor adverse events excluding skin irritation: nervous system: in general.

Comparison 19 5% Imiquimod versus placebo, Outcome 13 Cosmetic outcome: decrease in roughness/dryness/scaliness of the skin.
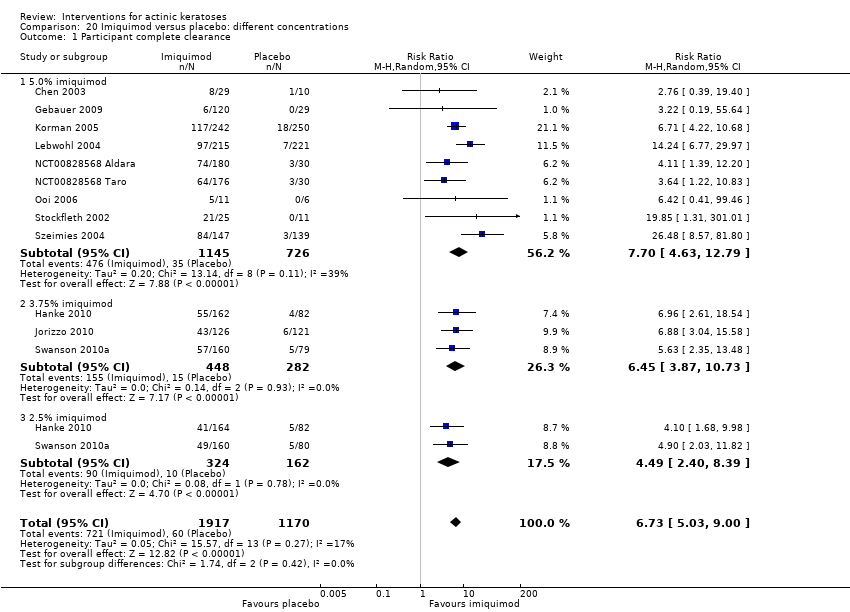
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 1 Participant complete clearance.

Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 2 Participant partial (>75%) clearance.
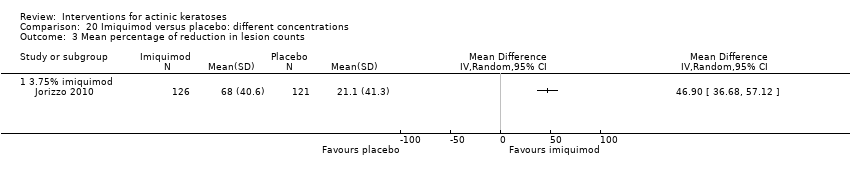
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 3 Mean percentage of reduction in lesion counts.

Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: 'flu" or "cold".

Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 5 Withdrawal due to adverse events.
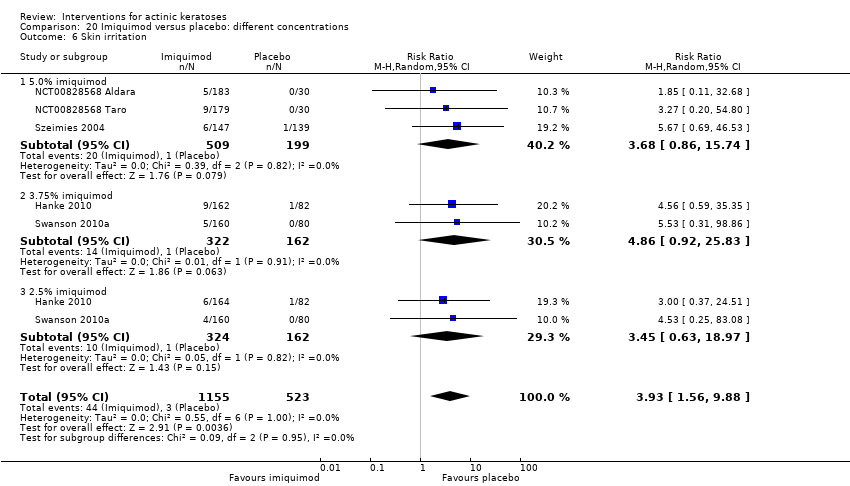
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 6 Skin irritation.
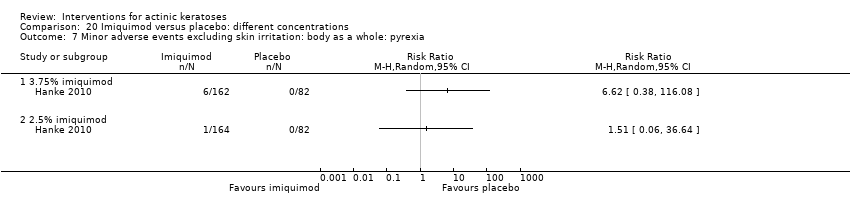
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 7 Minor adverse events excluding skin irritation: body as a whole: pyrexia.

Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 8 Minor adverse events excluding skin irritation: hemic and lymphatic: lymphadenopathy.

Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 9 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.

Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 10 Minor adverse events excluding skin irritation: nervous system: fatigue.

Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 11 Minor adverse events excluding skin irritation: nervous system: headache.

Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 12 Minor adverse events excluding skin irritation: respiratory: cough.

Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 13 Minor adverse events excluding skin irritation: respiratory: sinusitis.
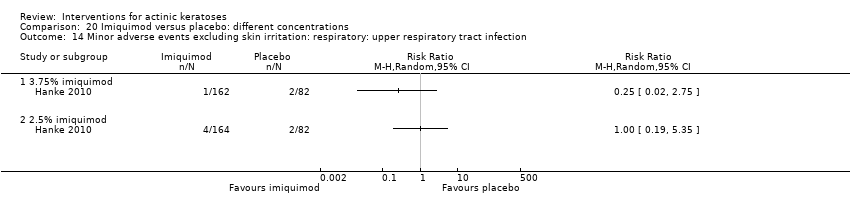
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 14 Minor adverse events excluding skin irritation: respiratory: upper respiratory tract infection.

Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 15 Minor adverse events excluding skin irritation: urogenital: urinary tract infection.
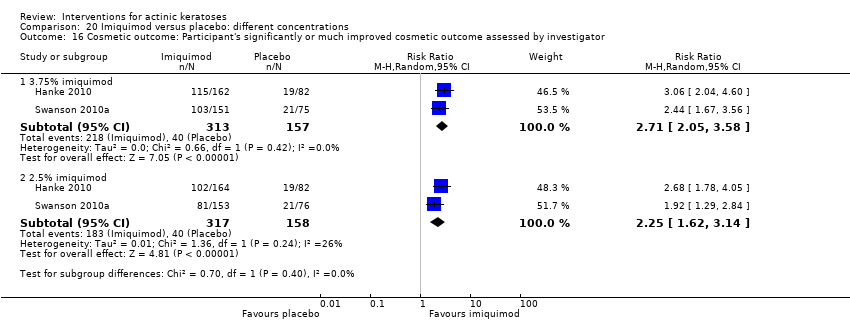
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 16 Cosmetic outcome: Participant's significantly or much improved cosmetic outcome assessed by investigator.

Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 1 Participant complete clearance.

Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 2 Participant partial (>75%) clearance.
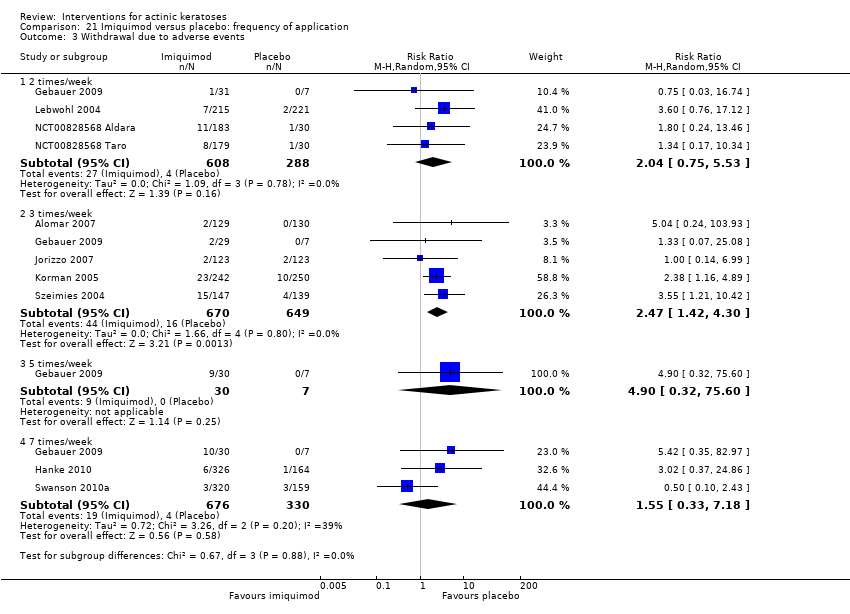
Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 3 Withdrawal due to adverse events.

Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 4 Minor adverse events excluding skin irritation:body as a whole: in general.

Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 5 Minor adverse events excluding skin irritation: body as a whole:"flu" or "cold".

Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 6 Minor adverse events excluding skin irritation: digestive: in general.

Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 7 Minor adverse events excluding skin irritation: digestive: nausea.

Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 8 Minor adverse events excluding skin irritation: nervous system: in general.
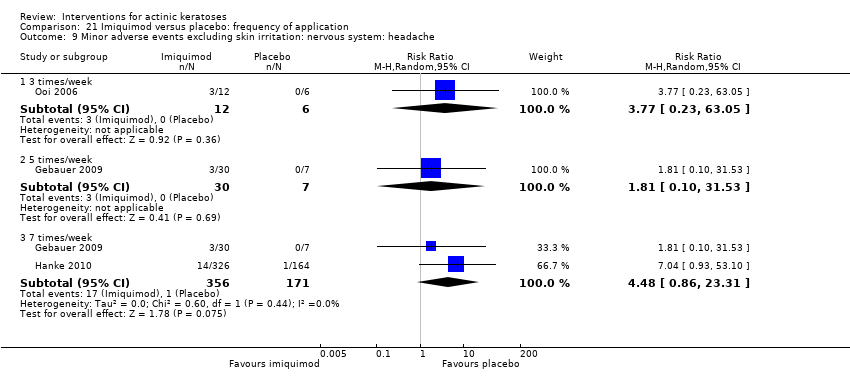
Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 9 Minor adverse events excluding skin irritation: nervous system: headache.

Comparison 22 5% imiquimod versus 5% 5‐FU, Outcome 1 Participant complete clearance.

Comparison 22 5% imiquimod versus 5% 5‐FU, Outcome 2 Cosmetic outcome: Investigator cosmetic outcome "excellent".

Comparison 22 5% imiquimod versus 5% 5‐FU, Outcome 3 Cosmetic outcome: normal skin surface.

Comparison 23 5% imiquimod versus cryotherapy, Outcome 1 Participant complete clearance.

Comparison 24 Ingenol mebutate (PEP005) versus placebo, Outcome 1 Participant complete clearance of target lesions.
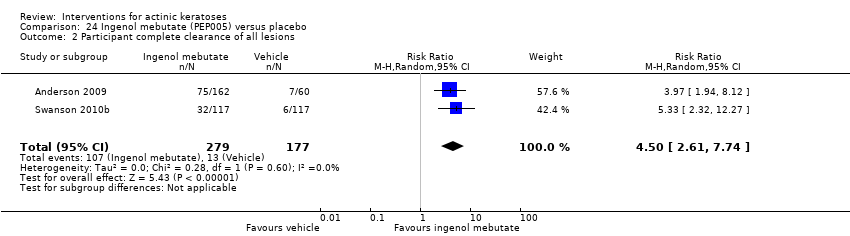
Comparison 24 Ingenol mebutate (PEP005) versus placebo, Outcome 2 Participant complete clearance of all lesions.

Comparison 24 Ingenol mebutate (PEP005) versus placebo, Outcome 3 Participant partial (>75%) clearance of target lesions.

Comparison 24 Ingenol mebutate (PEP005) versus placebo, Outcome 4 Cosmetic outcomes: changes in pigmentation.

Comparison 25 Ingenol mebutate (PEP005) versus placebo: different concentrations, Outcome 1 Participant complete clearance of target lesions.
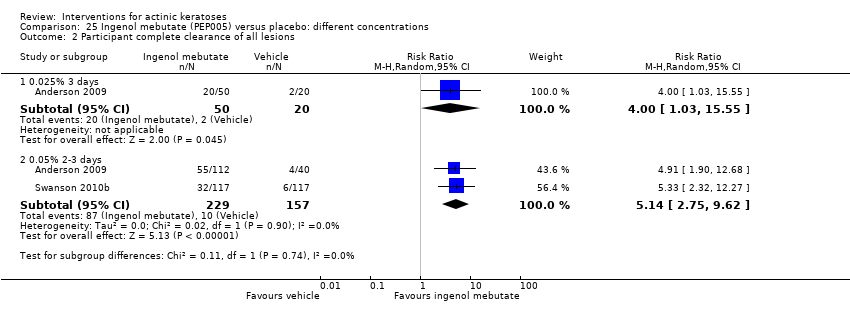
Comparison 25 Ingenol mebutate (PEP005) versus placebo: different concentrations, Outcome 2 Participant complete clearance of all lesions.

Comparison 25 Ingenol mebutate (PEP005) versus placebo: different concentrations, Outcome 3 Participant partial (>75%) clearance of target lesions.

Comparison 25 Ingenol mebutate (PEP005) versus placebo: different concentrations, Outcome 4 Cosmetic outcomes: changes in pigmentation.

Comparison 26 0.05% Ingenol mebutate (PEP005) versus placebo: number of doses, Outcome 1 Participant complete clearance of target lesions.

Comparison 26 0.05% Ingenol mebutate (PEP005) versus placebo: number of doses, Outcome 2 Participant complete clearance of all lesions.
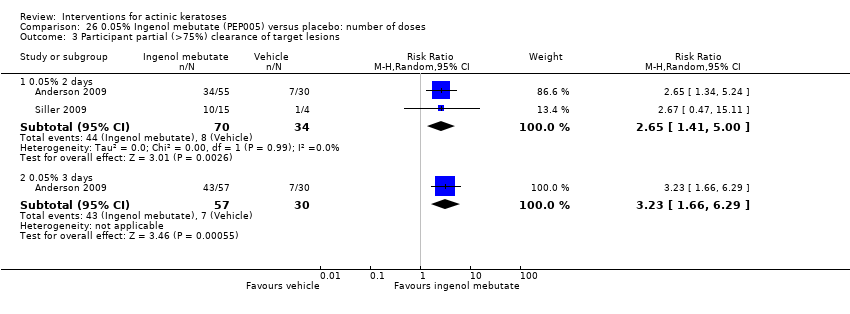
Comparison 26 0.05% Ingenol mebutate (PEP005) versus placebo: number of doses, Outcome 3 Participant partial (>75%) clearance of target lesions.

Comparison 27 Isotretinoin versus vehicle, Outcome 1 Investigator global improvement indices‐completely cleared.
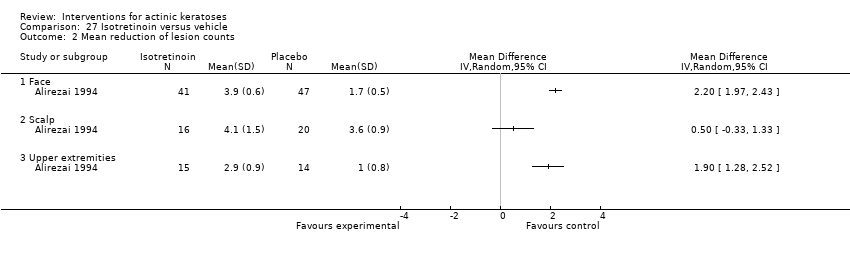
Comparison 27 Isotretinoin versus vehicle, Outcome 2 Mean reduction of lesion counts.
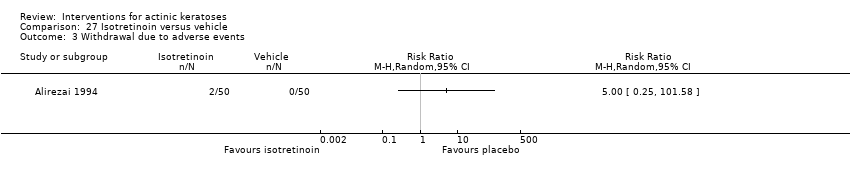
Comparison 27 Isotretinoin versus vehicle, Outcome 3 Withdrawal due to adverse events.

Comparison 27 Isotretinoin versus vehicle, Outcome 4 Skin irritation.

Comparison 27 Isotretinoin versus vehicle, Outcome 5 Severe‐Skin irritation.

Comparison 28 Masoprocol versus placebo, Outcome 1 Global improvement indices‐cured.
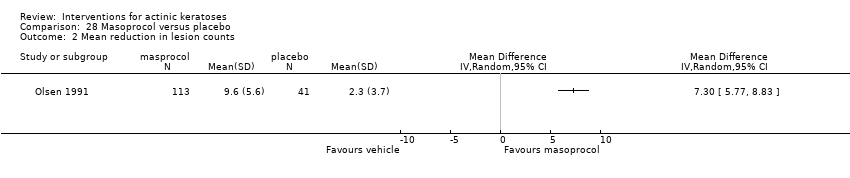
Comparison 28 Masoprocol versus placebo, Outcome 2 Mean reduction in lesion counts.

Comparison 28 Masoprocol versus placebo, Outcome 3 Withdrawal due to adverse events.

Comparison 29 1% nicotinamide versus placebo, Outcome 1 Mean percentage of reduction in lesion counts.

Comparison 29 1% nicotinamide versus placebo, Outcome 2 Withdrawal due to adverse events.

Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 1 Participant complete clearance.

Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 2 Participant partial (>75%) clearance.
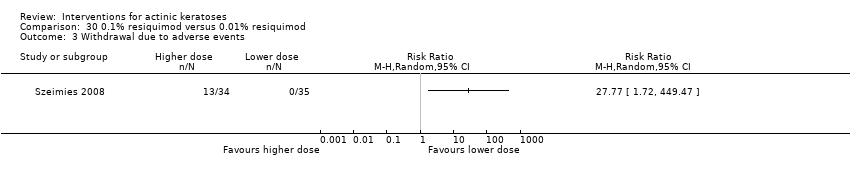
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 3 Withdrawal due to adverse events.

Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue.

Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors.

Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.

Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia.
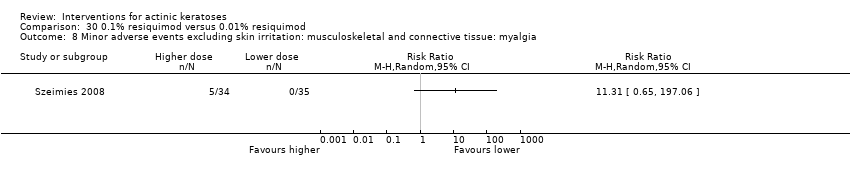
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.

Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general.

Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache.

Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy.

Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders.

Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general.

Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 1 Participant complete clearance.
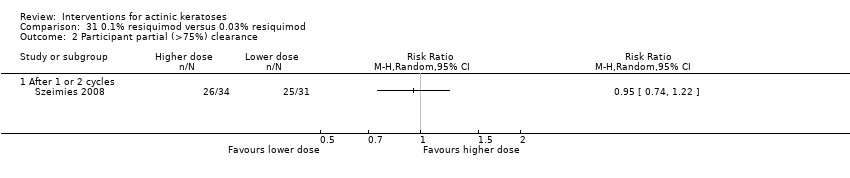
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 2 Participant partial (>75%) clearance.

Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 3 Withdrawal due to adverse events.

Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue.
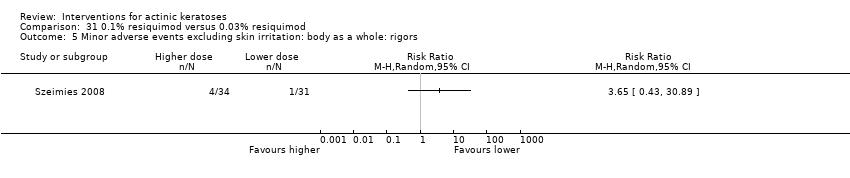
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors.

Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.

Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia.
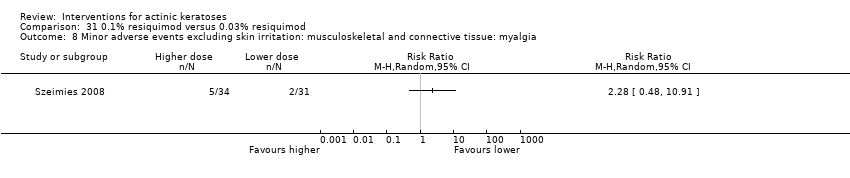
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.

Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general.
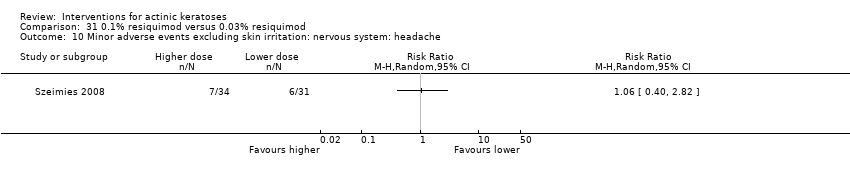
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache.
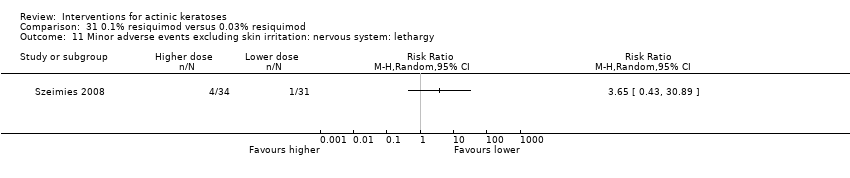
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy.

Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders.

Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general.
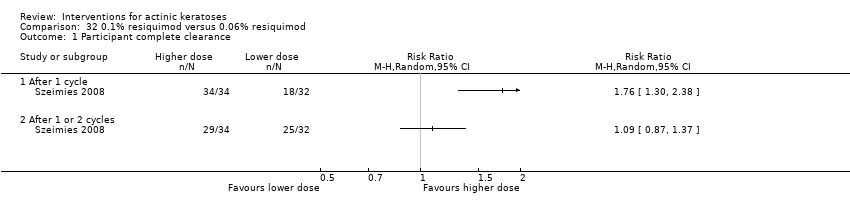
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 1 Participant complete clearance.
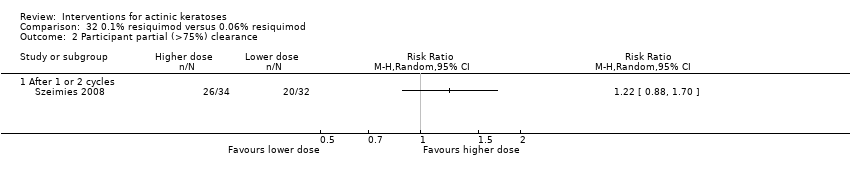
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 2 Participant partial (>75%) clearance.
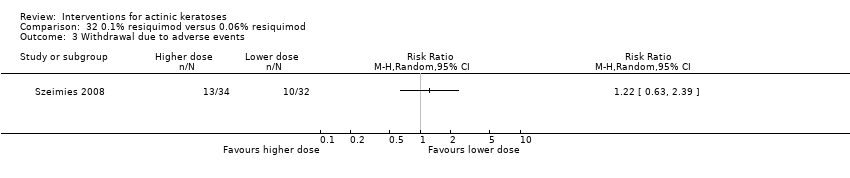
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 3 Withdrawal due to adverse events.
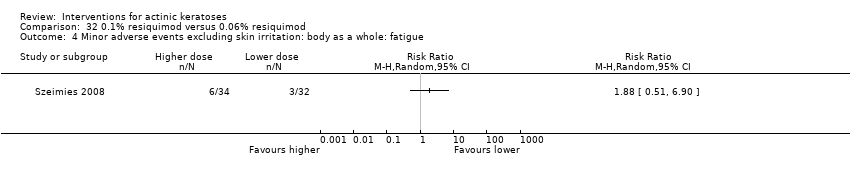
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue.
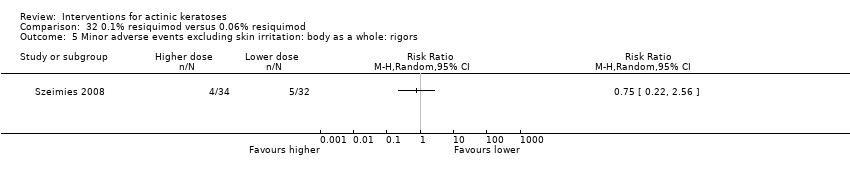
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors.

Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.

Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia.

Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.

Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general.

Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache.

Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy.

Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders.

Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general.

Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 1 Participant complete clearance.
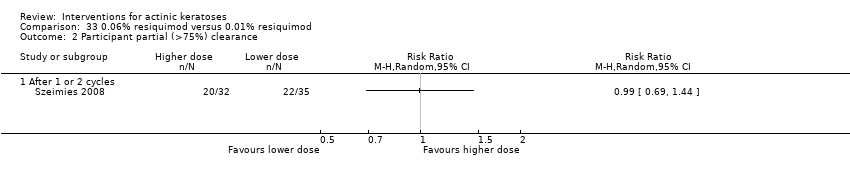
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 2 Participant partial (>75%) clearance.
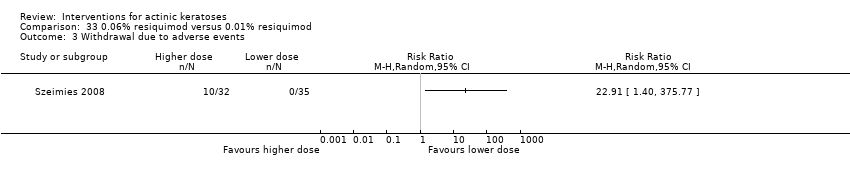
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 3 Withdrawal due to adverse events.
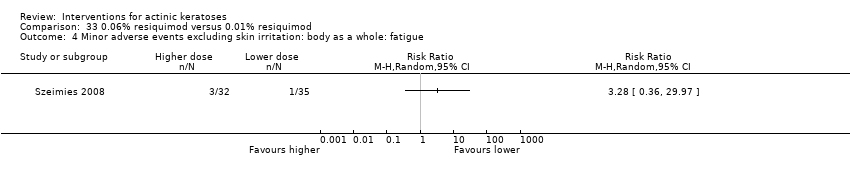
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue.

Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors.

Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.

Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia.

Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.

Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general.
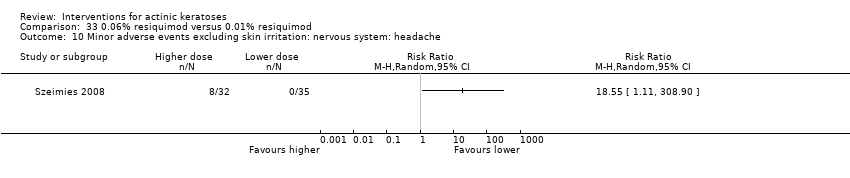
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache.

Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy.

Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders.
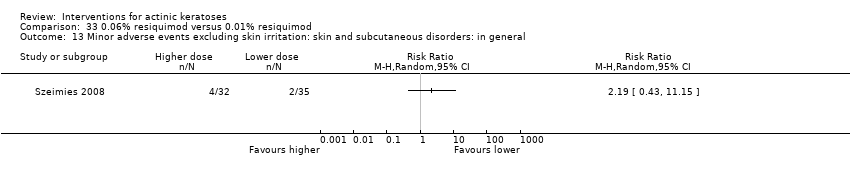
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general.

Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 1 Participant complete clearance.
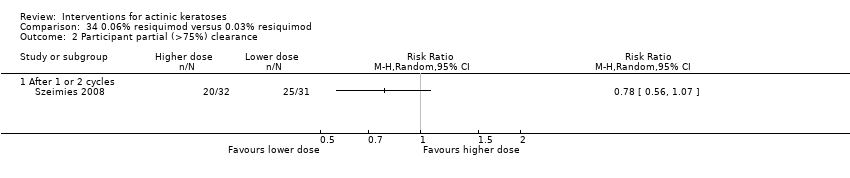
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 2 Participant partial (>75%) clearance.
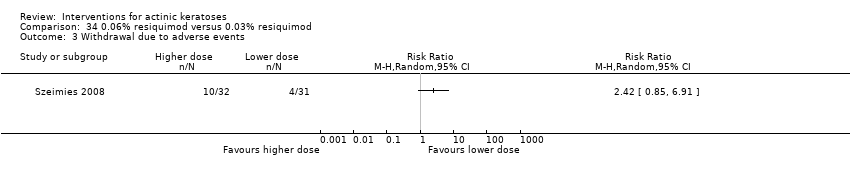
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 3 Withdrawal due to adverse events.

Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue.
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors.

Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.

Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia.

Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general.
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache.

Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy.

Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders.
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 13 Minor adverse events excluding skin irritation:skin and subcutaneous disorders: in general.
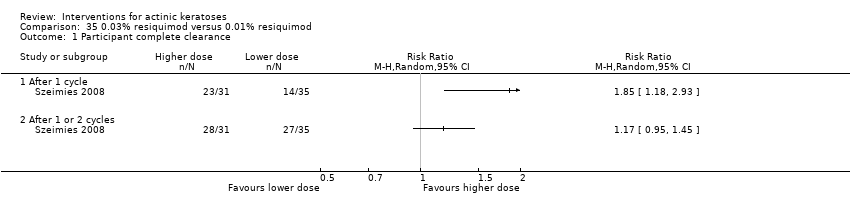
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 1 Participant complete clearance.

Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 2 Participant partial (>75%) clearance.

Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 3 Withdrawal due to adverse events.

Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue.
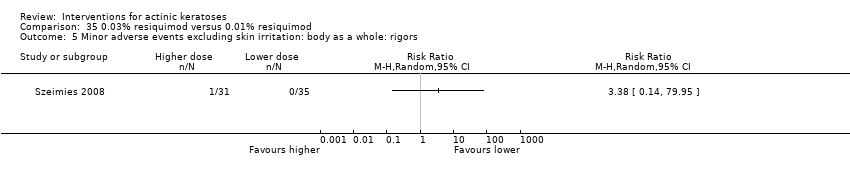
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors.

Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.
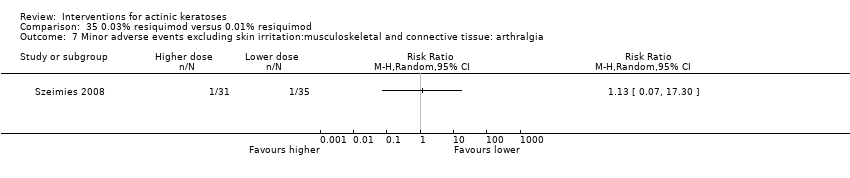
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia.
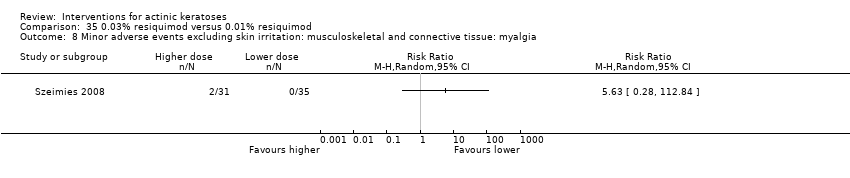
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.
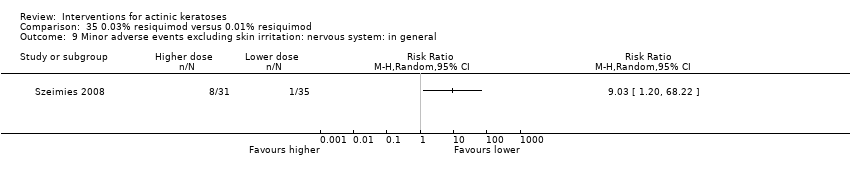
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general.

Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache.

Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy.

Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders.

Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general.

Comparison 36 Sunscreen SPF 17 (8% 2‐ethyl‐hexyl p‐methoxycinnamate/2% 4‐tert‐butyl‐4‐methoxy‐4‐dibenzoylmethane) versus placebo, Outcome 1 Mean change in lesion counts.

Comparison 37 12.5% DL‐α‐tocopherol (vitamin E) versus placebo, Outcome 1 Mean reduction of lesion counts.

Comparison 38 Etretinate versus placebo, Outcome 1 Participant complete clearance.

Comparison 39 Carbon dioxide laser resurfacing versus 5% 5‐FU, Outcome 1 Mean percentage of reduction of lesion counts.

Comparison 39 Carbon dioxide laser resurfacing versus 5% 5‐FU, Outcome 2 Withdrawal due to adverse events.

Comparison 40 Carbon dioxide laser resurfacing versus Trichloroacetic acid peel, Outcome 1 Mean percentage of reduction of lesion counts.
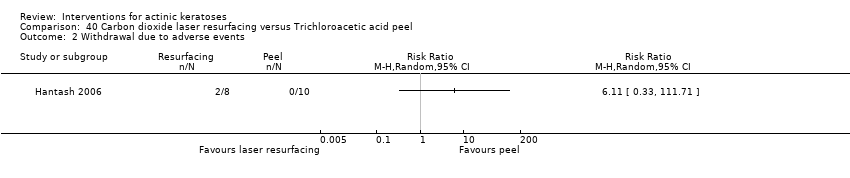
Comparison 40 Carbon dioxide laser resurfacing versus Trichloroacetic acid peel, Outcome 2 Withdrawal due to adverse events.
| Study | Intervention | At 3 months | At 6 months | At 12 months |
| Ostertag 2006 | 5‐fluorouracil | 13.2 | 12.5 | 12.4 |
| Ostertag 2006 | Er:YAG laser resurfacing | 13.8 | 13.9 | 14.2 |
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 1 Mean reduction in lesion counts.
| Study | Assessment | Resurfacing | 5‐FU |
| Ostertag 2006 | At 6 months | 94.4% | 79.2% |
| Ostertag 2006 | At 12 months | 91.1% | 76.6% |
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 2 Mean percentage of reduction in lesion counts.

Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 3 Withdrawal due to adverse events.
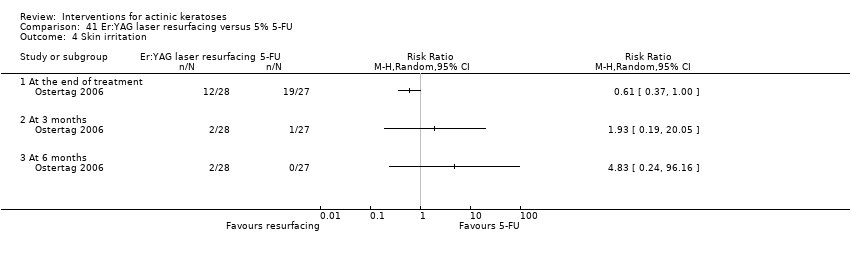
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 4 Skin irritation.

Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 5 Minor adverse events excluding skin irritation: dermatology: acne.

Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 6 Minor adverse events excluding skin irritation: dermatology:crustea.
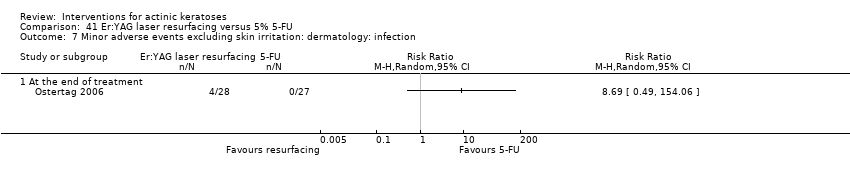
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 7 Minor adverse events excluding skin irritation: dermatology: infection.

Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 8 Minor adverse events excluding skin irritation: dermatology: milia.

Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 9 Minor adverse events excluding skin irritation: dermatology:pain.
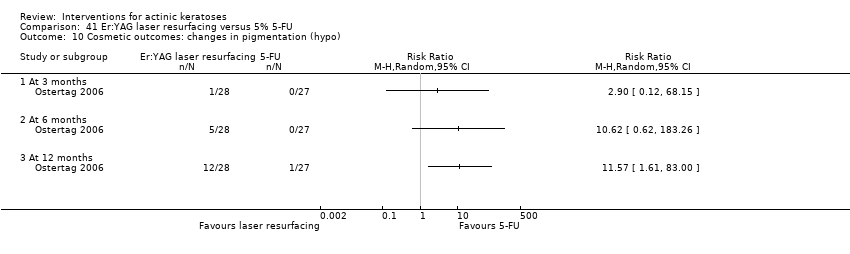
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 10 Cosmetic outcomes: changes in pigmentation (hypo).

Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 11 Cosmetic outcomes: scarring.

Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 12 Cosmetic outcomes: improvement in photoageing score.
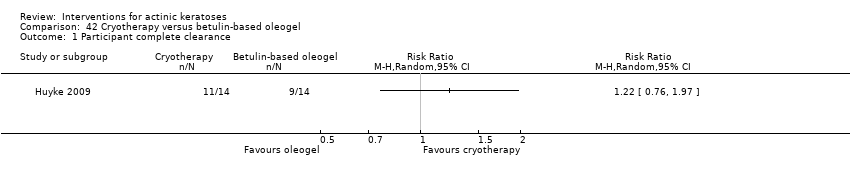
Comparison 42 Cryotherapy versus betulin‐based oleogel, Outcome 1 Participant complete clearance.

Comparison 42 Cryotherapy versus betulin‐based oleogel, Outcome 2 Participant partial (>75%) clearance.

Comparison 43 Cryotherapy versus 5% 5‐FU, Outcome 1 Participant complete clearance.

Comparison 43 Cryotherapy versus 5% 5‐FU, Outcome 2 Cosmetic outcomes: excellent global cosmetic outcome.

Comparison 43 Cryotherapy versus 5% 5‐FU, Outcome 3 Cosmetic outcomes: better skin appearance.

Comparison 44 Cryotherapy versus imiquimod, Outcome 1 Participant complete clearance.

Comparison 44 Cryotherapy versus imiquimod, Outcome 2 Cosmetic outcomes: excellent global cosmetic outcome.
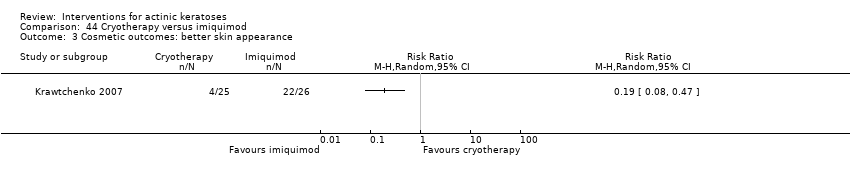
Comparison 44 Cryotherapy versus imiquimod, Outcome 3 Cosmetic outcomes: better skin appearance.
| Study | Assessment at | Cryotherapy | MAL‐PDT |
| Kaufmann 2008 | 12 weeks | N/A | N/A |
| Kaufmann 2008 | 24 weeks | 87% | 75% |
| Morton 2006 | 12 weeks | 74.5% | 84.4% |
| Morton 2006 | 24 weeks | 83.9% | 86.7% |
Comparison 45 Cryotherapy versus MAL‐red light PDT, Outcome 1 Mean percentage of reduction in lesion counts.

Comparison 45 Cryotherapy versus MAL‐red light PDT, Outcome 2 Withdrawal due to adverse events.

Comparison 45 Cryotherapy versus MAL‐red light PDT, Outcome 3 Cosmetic outcomes: excellent or good cosmetic outcomes by investigator.
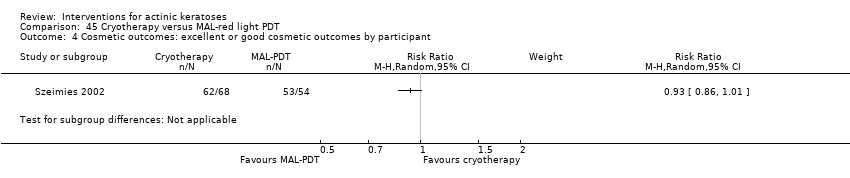
Comparison 45 Cryotherapy versus MAL‐red light PDT, Outcome 4 Cosmetic outcomes: excellent or good cosmetic outcomes by participant.
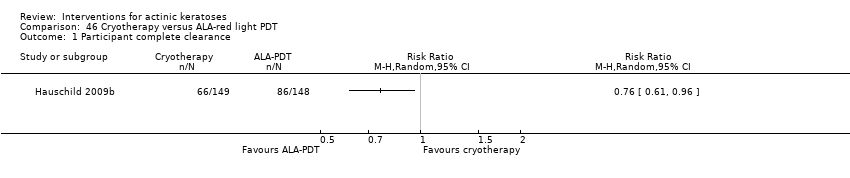
Comparison 46 Cryotherapy versus ALA‐red light PDT, Outcome 1 Participant complete clearance.

Comparison 46 Cryotherapy versus ALA‐red light PDT, Outcome 2 Skin irritation.
![Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 1 Participant complete clearance [1 treatment].](/es/cdsr/doi/10.1002/14651858.CD004415.pub2/media/CDSR/CD004415/image_n/nCD004415-CMP-047-01.png)
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 1 Participant complete clearance [1 treatment].
![Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 2 Participant complete clearance [1 or 2 treatments].](/es/cdsr/doi/10.1002/14651858.CD004415.pub2/media/CDSR/CD004415/image_n/nCD004415-CMP-047-02.png)
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 2 Participant complete clearance [1 or 2 treatments].
![Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 3 Participant complete clearance [1 or 2 treatments] by anatomical location.](/es/cdsr/doi/10.1002/14651858.CD004415.pub2/media/CDSR/CD004415/image_n/nCD004415-CMP-047-03.png)
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 3 Participant complete clearance [1 or 2 treatments] by anatomical location.
![Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 4 Participant partial (> 75%) clearance [1 treatment].](/es/cdsr/doi/10.1002/14651858.CD004415.pub2/media/CDSR/CD004415/image_n/nCD004415-CMP-047-04.png)
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 4 Participant partial (> 75%) clearance [1 treatment].
![Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 5 Participant partial (>75%) clearance[1 or 2 treatments].](/es/cdsr/doi/10.1002/14651858.CD004415.pub2/media/CDSR/CD004415/image_n/nCD004415-CMP-047-05.png)
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 5 Participant partial (>75%) clearance[1 or 2 treatments].
![Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 6 Participant partial (>75%) clearance [1 or 2 treatment] by anatomical location.](/es/cdsr/doi/10.1002/14651858.CD004415.pub2/media/CDSR/CD004415/image_n/nCD004415-CMP-047-06.png)
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 6 Participant partial (>75%) clearance [1 or 2 treatment] by anatomical location.
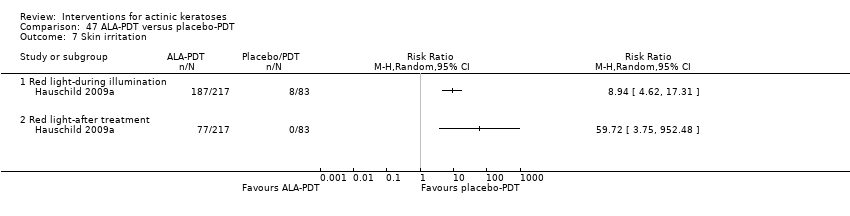
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 7 Skin irritation.
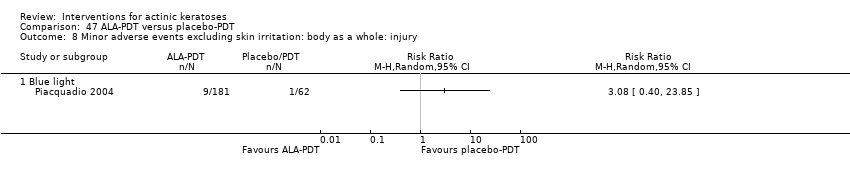
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 8 Minor adverse events excluding skin irritation: body as a whole: injury.
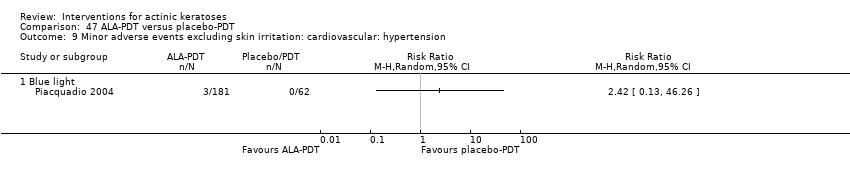
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 9 Minor adverse events excluding skin irritation: cardiovascular: hypertension.

Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 10 Minor adverse events excluding skin irritation: dermatology: skin discolouration.

Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 11 Minor adverse events excluding skin irritation: dermatology: skin hypertrophy.

Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 12 Minor adverse events excluding skin irritation: nervous system: headache.

Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 13 Cosmetic outcome: very good or good general cosmetic outcome.

Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 1 Participant complete clearance.

Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 2 Participant partial (>75%) clearance.
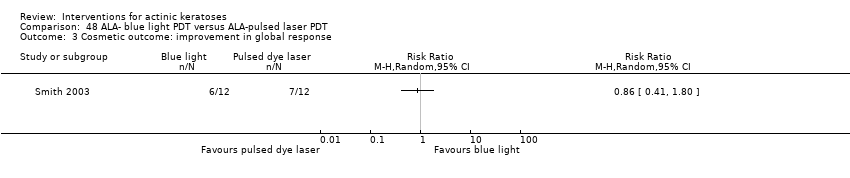
Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 3 Cosmetic outcome: improvement in global response.
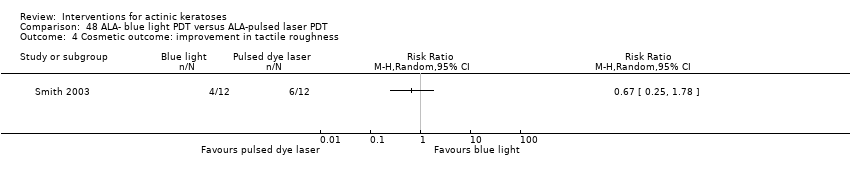
Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 4 Cosmetic outcome: improvement in tactile roughness.

Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 5 Cosmetic outcome: improvement in mottled hyperpigmentation.
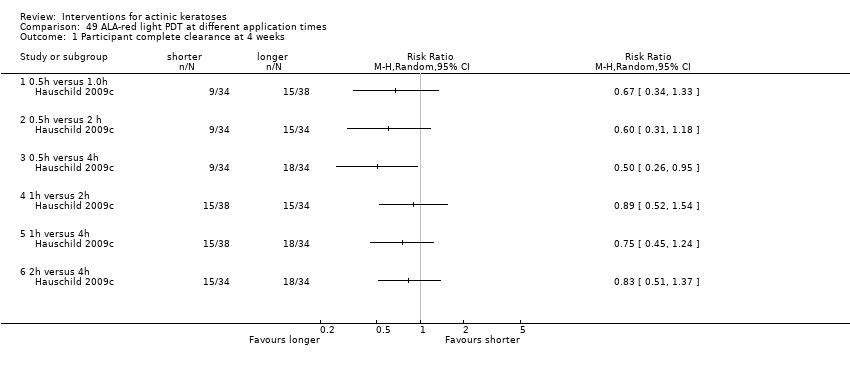
Comparison 49 ALA‐red light PDT at different application times, Outcome 1 Participant complete clearance at 4 weeks.

Comparison 49 ALA‐red light PDT at different application times, Outcome 2 Participant complete clearance at 8 weeks.

Comparison 49 ALA‐red light PDT at different application times, Outcome 3 Minor adverse events excluding skin irritation: metabolic and nutritional disorders: elevated alanine transaminase (ALT).

Comparison 49 ALA‐red light PDT at different application times, Outcome 4 Minor adverse events excluding skin irritation: nervous system: headache.

Comparison 49 ALA‐red light PDT at different application times, Outcome 5 Minor adverse events excluding skin irritation: other: epistaxis (nose bleeding).

Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 1 Participant complete clearance.

Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 2 Participant partial (>75%) clearance.

Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 3 Withdrawal due to adverse events.
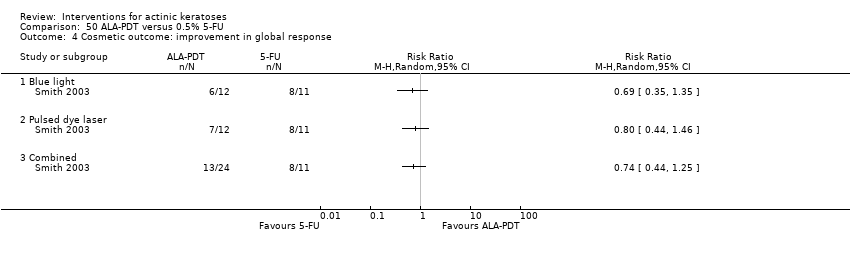
Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 4 Cosmetic outcome: improvement in global response.
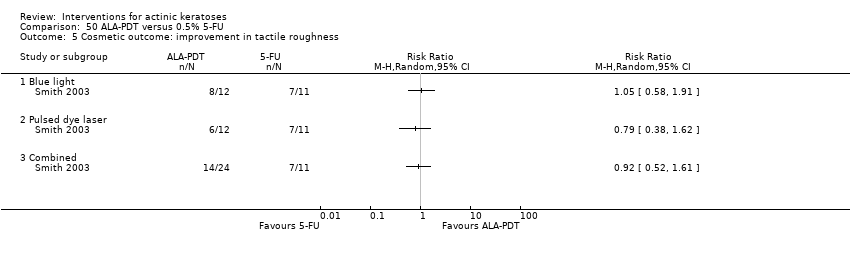
Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 5 Cosmetic outcome: improvement in tactile roughness.
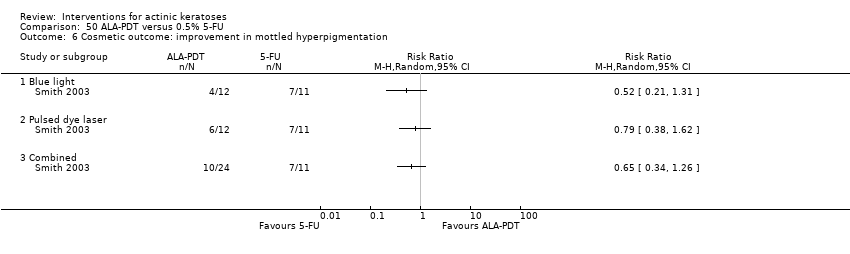
Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 6 Cosmetic outcome: improvement in mottled hyperpigmentation.

Comparison 51 ALA‐red light PDT vs cryotherapy, Outcome 1 Participant complete clearance.

Comparison 51 ALA‐red light PDT vs cryotherapy, Outcome 2 Skin irritation.

Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 1 Participant complete clearance.

Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 2 Participant partial (>75%) clearance.

Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 3 Withdrawal due to adverse events.
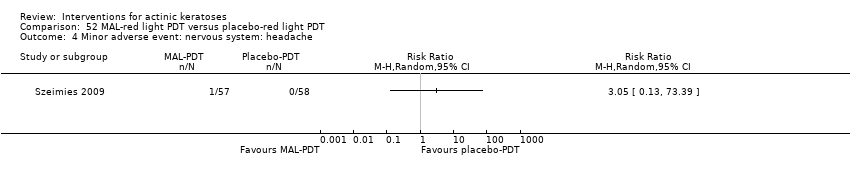
Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 4 Minor adverse event: nervous system: headache.

Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 5 Cosmetic outcome: hyperpigmentation.

Comparison 53 MAL‐red light LED PDT versus MAL‐broad visible + water‐filtered infrared A PDT (1 or 2 treatments), Outcome 1 Participant complete clearance.

Comparison 53 MAL‐red light LED PDT versus MAL‐broad visible + water‐filtered infrared A PDT (1 or 2 treatments), Outcome 2 Participant partial (>75%) clearance.

Comparison 54 MAL‐red light LED PDT versus MAL‐daylight PDT, Outcome 1 Mean reduction in lesion counts.

Comparison 55 2h MAL‐day light PDT versus 3h MAL‐daylight PDT, Outcome 1 Mean reduction in lesion counts.

Comparison 55 2h MAL‐day light PDT versus 3h MAL‐daylight PDT, Outcome 2 Mean percentage of reduction in lesion counts.
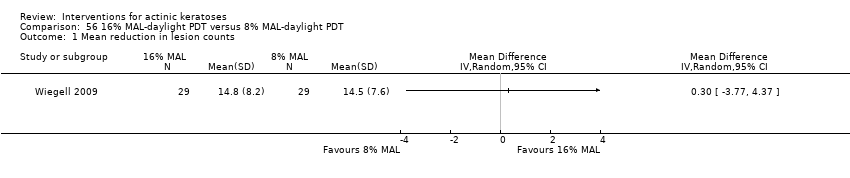
Comparison 56 16% MAL‐daylight PDT versus 8% MAL‐daylight PDT, Outcome 1 Mean reduction in lesion counts.

Comparison 57 Single MAL‐red light PDT versus multiple MAL‐red light PDT (2 treatments 1 week apart), Outcome 1 Participant complete clearance.

Comparison 57 Single MAL‐red light PDT versus multiple MAL‐red light PDT (2 treatments 1 week apart), Outcome 2 Withdrawal due to adverse events.

Comparison 58 MAL‐ red light PDT vs cryotherapy, Outcome 1 Withdrawal due to adverse events.

Comparison 59 ALA‐red light PDT versus MAL‐red light PDT, Outcome 1 Mean reduction in lesion counts.

Comparison 60 Trichloroacetic acid peel versus 5% 5‐FU, Outcome 1 Mean percentage of reduction in lesions.

Comparison 61 Cryotherapy versus cryotherapy with betulin‐based oleogel, Outcome 1 Participant complete clearance.

Comparison 61 Cryotherapy versus cryotherapy with betulin‐based oleogel, Outcome 2 Participant partial (>75%) clearance.
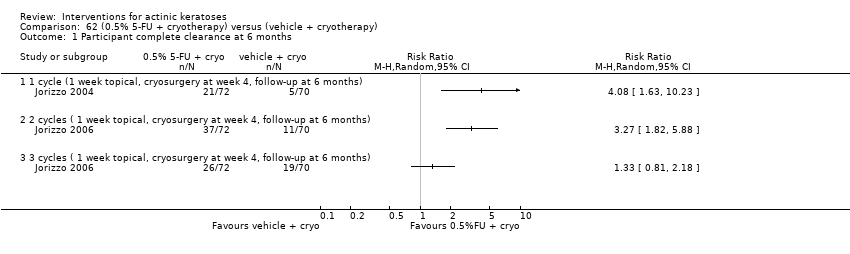
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 1 Participant complete clearance at 6 months.
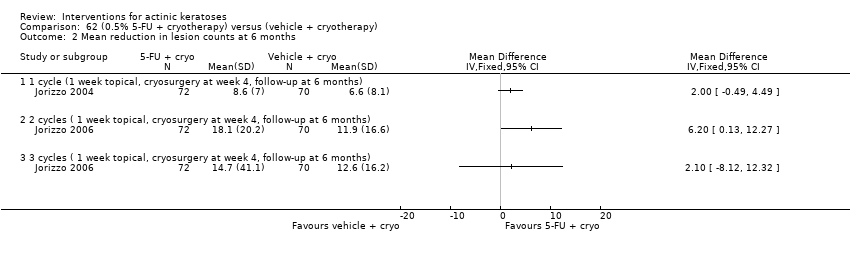
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 2 Mean reduction in lesion counts at 6 months.

Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 3 Mean percentage of reduction in lesion counts at 6 months.

Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 4 Minor adverse events excluding skin irritation: body as a whole: allergic reaction.

Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 5 Minor adverse events excluding skin irritation: dermatology: hyperesthesia.

Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 6 Minor adverse events excluding skin irritation: dermatology: skin discoloration.

Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 7 Minor adverse events excluding skin irritation: dermatology: vesiculobullous rash.

Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 8 Minor adverse events excluding skin irritation: digestive: cheilitis.

Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 9 Minor adverse events excluding skin irritation: special senses: conjunctivitis.
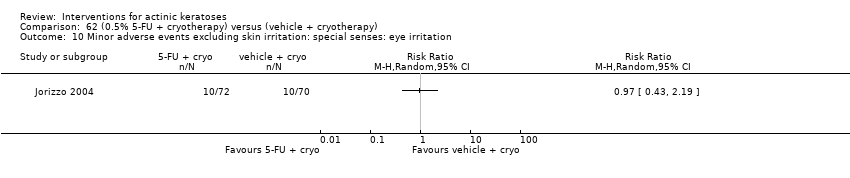
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 10 Minor adverse events excluding skin irritation: special senses: eye irritation.

Comparison 63 (vehicle + cryotherapy) versus (0.5% 5‐FU + cryotherapy), Outcome 1 Participant complete clearance at 6 months.
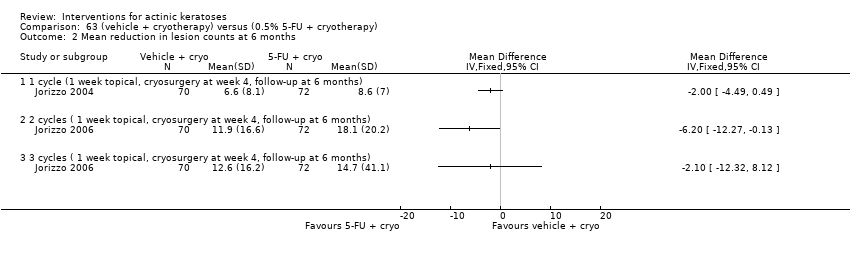
Comparison 63 (vehicle + cryotherapy) versus (0.5% 5‐FU + cryotherapy), Outcome 2 Mean reduction in lesion counts at 6 months.

Comparison 63 (vehicle + cryotherapy) versus (0.5% 5‐FU + cryotherapy), Outcome 3 Mean percentage of reduction in lesion counts at 6 months.

Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 1 Participant complete clearance of all lesions.

Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 2 Participant complete clearance of target (cryotherapy treated) lesions.

Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 3 Participant complete clearance of subclinical lesions.

Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 4 Mean percentage of reduction in all lesion counts.
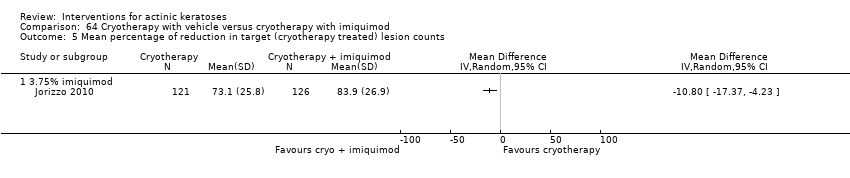
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 5 Mean percentage of reduction in target (cryotherapy treated) lesion counts.
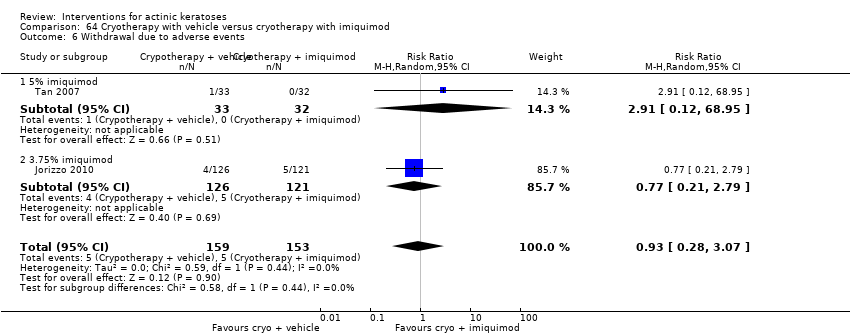
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 6 Withdrawal due to adverse events.
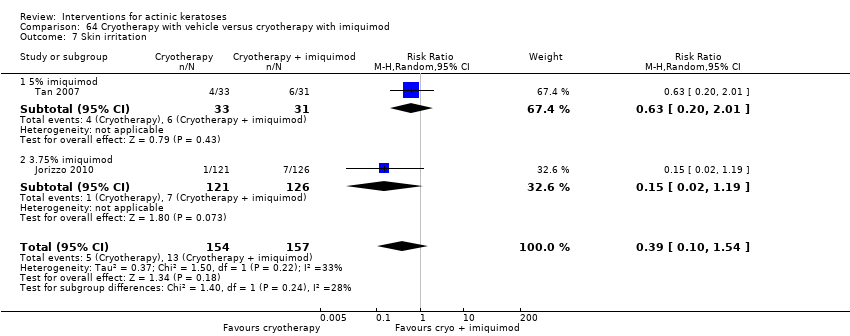
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 7 Skin irritation.
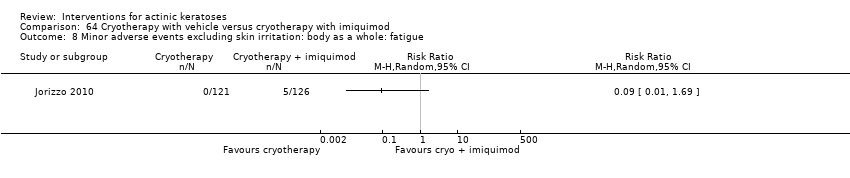
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 8 Minor adverse events excluding skin irritation: body as a whole: fatigue.

Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 9 Minor adverse events excluding skin irritation: digestive: nausea.

Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 10 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.
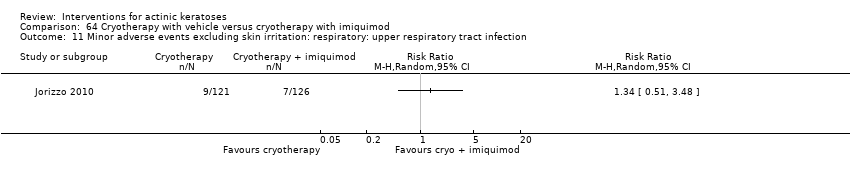
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 11 Minor adverse events excluding skin irritation: respiratory: upper respiratory tract infection.

Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 12 Minor adverse events excluding skin irritation: respiratory: bronchitis.

Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 13 Minor adverse events excluding skin irritation: respiratory: sinusitis.
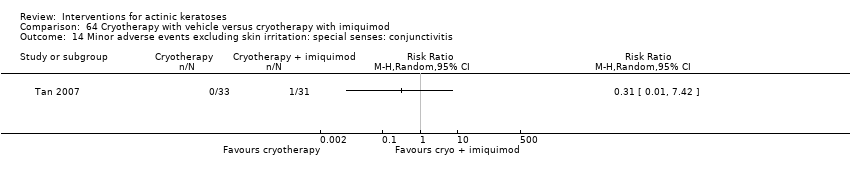
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 14 Minor adverse events excluding skin irritation: special senses: conjunctivitis.

Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 15 Cosmetic outcomes: Improved global photoageing score.

Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 16 Cosmetic outcomes: Improved fine lines.
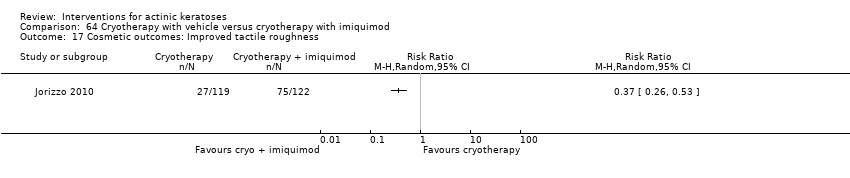
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 17 Cosmetic outcomes: Improved tactile roughness.
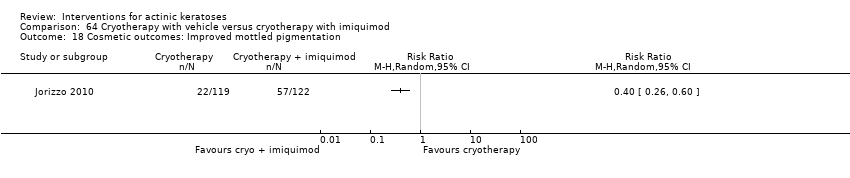
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 18 Cosmetic outcomes: Improved mottled pigmentation.
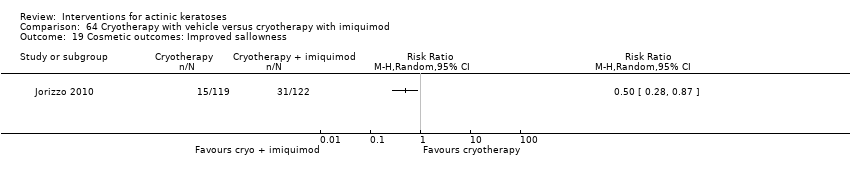
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 19 Cosmetic outcomes: Improved sallowness.

Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 20 Cosmetic outcomes: cosmetic appearance score.
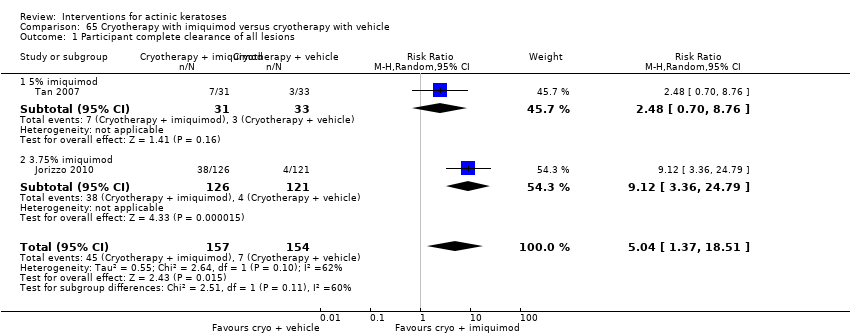
Comparison 65 Cryotherapy with imiquimod versus cryotherapy with vehicle, Outcome 1 Participant complete clearance of all lesions.
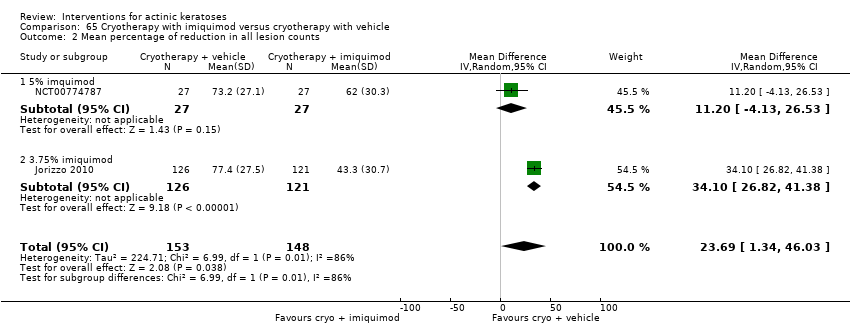
Comparison 65 Cryotherapy with imiquimod versus cryotherapy with vehicle, Outcome 2 Mean percentage of reduction in all lesion counts.

Comparison 65 Cryotherapy with imiquimod versus cryotherapy with vehicle, Outcome 3 Withdrawal due to adverse events.

Comparison 65 Cryotherapy with imiquimod versus cryotherapy with vehicle, Outcome 4 Skin irritation.
| Study | Intervention | Patient | Investigator |
| Van der Geer 2009 | Diclofenac in 2.5% hyaluronic acid + ALA‐PDT | 3.3 | 3.4 |
| Van der Geer 2009 | 2.5% hyaluronic acid + ALA‐PDT | 2.4 | 2.7 |
Comparison 66 (3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT) versus (2.5% hyaluronic acid + ALA‐red light PDT), Outcome 1 Global Improvement Indices (‐2 to 4) at 6 months.
| Study | Intervention | At 6 weeks | At 6 months | At 12 months |
| Van der Geer 2009 | Diclofenac in 2.5% hyaluronic acid + ALA‐PDT | 10.13 | 11.56 | 12.5 |
| Van der Geer 2009 | 2.5% hyaluronic acid + ALA‐PDT | 9.9 | 10.56 | 8.8 |
Comparison 66 (3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT) versus (2.5% hyaluronic acid + ALA‐red light PDT), Outcome 2 Mean reduction of lesion counts.
| Diclofenac in 2.5% hyaluronic acid compared to interventions for actinic keratoses in immunocompetent participants | ||||||
| Intervention/Comparison intervention | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk With comparator | Corresponding risk With intervention | |||||
| Participant complete clearance | ||||||
| 3% diclofenac in 2.5% hyaluronic acid/2.5% hyaluronic acid | Study population | RR 2.46 | 420 | ⊕⊕⊕⊝ | For all lesions, data from 30, 60, and 90 day treatments pooled together, assessment at 30 days after the end of treatment (Analysis 6.5) | |
| 127 per 1000 | 313 per 1000 | |||||
| Moderate | ||||||
| 132 per 1000 | 325 per 1000 | |||||
| 3% diclofenac in 2.5% hyaluronic acid/5% imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT/2.5% hyaluronic acid + ALA‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean reduction in lesion counts | ||||||
| 3% diclofenac in 2.5% hyaluronic acid/2.5% hyaluronic acid | The mean reduction in lesion counts in the control groups was | The mean reduction of lesion counts in the intervention groups was | ‐ | 345 | ⊕⊕⊕⊕ | Data from 30, 60, and 90 day treatments pooled together, assessment 30 days after the end of treatment (Analysis 6.12) |
| 3% diclofenac in 2.5% hyaluronic acid/ 5% imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT/2.5% hyaluronic acid + ALA‐red light PDT | See comment | See comment | Not estimable | 10 | ⊕⊕⊝⊝ | Intraindividual study: at 6 weeks; diclofenac/hyaluronic acid (HA) + ALA‐PDT = 10.13, HA + ALA‐PDT= 9.9, at 6 months; diclofenac/HA + ALA‐PDT = 11.56, HA + ALA‐PDT = 10.56, at 12 months; diclofenac/HA + ALA‐PDT = 12.5, HA + ALA‐PDT = 8.8 |
| Mean percentage of reduction in lesion counts | ||||||
| All comparisons | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Withdrawal due to adverse events | ||||||
| 3% diclofenac in 2.5% hyaluronic acid/2.5% hyaluronic acid | Study population | RR 3.59 | 592 | ⊕⊕⊕⊕ | Additional data from intraindividual study: no participant withdrew because of adverse events (N = 20). GRADE = low. | |
| 40 per 1000 | 144 per 1000 | |||||
| Moderate | ||||||
| 43 per 1000 | 154 per 1000 | |||||
| 3% diclofenac in 2.5% hyaluronic acid/5% imiquimod | 0 per 1000 | 0 per 1000 | Not estimable | 49 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| 3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT/2.5% hyaluronic acid + ALA‐red light PDT | 0 per 1000 | 0 per 1000 | Not estimable | 10 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| Skin irritation | ||||||
| 3% diclofenac in 2.5% hyaluronic acid/2.5% hyaluronic acid | See comment | See comment | Not estimable | 20 | ⊕⊕⊝⊝ | Intraindividual study reported irritation only on the diclofenac treated side of 8 out of 20 participants |
| 3% diclofenac in 2.5% hyaluronic acid/5% imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT/2.5% hyaluronic acid + ALA‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5‐fluorouracil (5‐FU) compared to interventions for actinic keratoses in immunocompetent participants | ||||||
| Intervention/Comparison intervention | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk With comparator | Corresponding risk With intervention | |||||
| Participant complete clearance | ||||||
| 0.5% 5‐FU/Vehicle | Study population | RR 8.86 | 522 | ⊕⊕⊕⊕ | Data from 1, 2, and 4 week treatments were pooled together (Analysis 9.1) | |
| 15 per 1000 | 136 per 1000 | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 | |||||
| 0.5% 5‐FU with cryotherapy/Vehicle with cryotherapy | 71 per 1000 | 291 per 1000 | RR 4.08 | 142 | ⊕⊕⊝⊝ | 1 cycle (Analysis 62.1) |
| 0.5% 5‐FU/ALA‐PDT | 292 per 1000 | 499 per 1000 | RR 1.71 | 48 | ⊕⊝⊝⊝ | Data from blue light and pulsed dye laser were pooled |
| 0.5% 5‐FU/5.0% 5‐FU | See comment | See comment | Not estimable | 21 | ⊕⊝⊝⊝ | Intraindividual study: 0.5% and 5.0% 5‐FU = 9/21 |
| 5% 5‐FU with 0.05% tretinoin /5% 5‐FU with placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU /10% masoprocol | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/5% Imiquimod | Study population | RR 1.85 | 89 | ⊕⊝⊝⊝ | ||
| 600 per 1000 | 1000 per 1000 | |||||
| Moderate | ||||||
| 555 per 1000 | 1000 per 1000 | |||||
| 5% 5‐FU/Carbon dioxide laser resurfacing | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Er:YAG laser resurfacing | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Cryotherapy | 680 per 1000 | 959 per 1000 | RR 1.41 | 49 | ⊕⊕⊝⊝ | Only data after the treatment |
| 5% 5‐FU/Trichloroacetic acid peel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean reduction in lesion counts | ||||||
| 0.5% 5‐FU/Vehicle | The mean reduction in lesion counts in the control groups was | The mean reduction in lesion counts in the intervention groups was | ‐ | 142 | ⊕⊕⊕⊝ | Data from 1, 2, and 4 week treatment were pooled. (Analysis 9.2) Results from another study (N = 177) with no SD: placebo: 2.7 lesions, 5‐FU = 8.8 lesions, GRADE = moderate |
| 0.5% 5‐FU with cryotherapy/Vehicle with cryotherapy | The mean reduction in lesion counts in the control groups was | The mean reduction in lesion counts in the intervention groups was | 142 | ⊕⊕⊕⊝ | 1 cycle (Analysis 62.2) | |
| 0.5% 5‐FU/ALA‐PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5% 5‐FU/5.0% 5‐FU | See comment | See comment | Not estimable | 21 | ⊕⊕⊝⊝ | Intraindividual study: results with no SD: 0.5% 5‐FU = 8.8 lesions, 5.0% 5‐FU = 6.1 lesions |
| 5% 5‐FU with 0.05% tretinoin /5% 5‐FU with placebo | The mean reduction in lesion counts in the control groups was | The mean reduction in lesion counts in the intervention groups was | ‐ | 19 | ⊕⊕⊝⊝ | |
| 5% 5‐FU /10% masoprocol | The mean reduction in lesion counts in the control groups was | The mean reduction in lesion counts in the intervention groups was | ‐ | 49 | ⊕⊕⊝⊝ | |
| 5% 5‐FU/5% Imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Carbon dioxide laser resurfacing | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Er:YAG laser resurfacing | See comment | See comment | Not estimable | 55 | ⊕⊕⊝⊝ | Results with no SD: number of lesions at 3 months:5‐FU = 13.2, resurfacing = 13.8, at 6 months:5‐FU = 12.5, resurfacing = 13.9, at 12 months: 5‐FU = 12.4, resurfacing = 14.2 |
| 5% 5‐FU/Cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Trichloroacetic acid peel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean percentage of reduction in lesion counts | ||||||
| 0.5% 5‐FU/Vehicle | The mean percentage of reduction in lesion counts ranged across control groups from | The mean percentage of reduction in lesion counts in the intervention groups was | ‐ | 142 | ⊕⊕⊕⊝ | Data from 1 week treatment.(Analysis 9.3) Results from two other studies with no SD 1) (N = 207) placebo = 21.6%, 5‐FU = 69.5%, GRADE = low, 2)(N = 177) placebo = 34.4%, 5‐FU = 78.5%, GRADE = moderate |
| 0.5% 5‐FU with cryotherapy/Vehicle with cryotherapy | The mean percentage of reduction in lesion counts in the control groups was | The mean percentage of reduction in lesion counts in the intervention groups was | ‐ | 142 | ⊕⊕⊕⊝ | |
| 0.5% 5‐FU/ALA‐PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5% 5‐FU/5.0% 5‐FU | See comment | See comment | Not estimable | 21 | ⊕⊕⊝⊝ | Intraindividual study: results with no SD: 0.5% 5‐FU = 67% and 5.0% 5‐FU = 47% |
| 5% 5‐FU with 0.05% tretinoin /5% 5‐FU with placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU /10% masoprocol | The mean percentage of reduction in lesion counts in the control groups was | The mean percentage of reduction in lesion counts in the intervention groups was | ‐ | 49 | ⊕⊕⊕⊝ | |
| 5% 5‐FU/5% Imiquimod | See comment | See comment | Not estimable | 39 | ⊕⊕⊝⊝ | Results with no SD: 5% 5‐FU = 94%, 5% imiquimod = 66% |
| 5% 5‐FU/Carbon dioxide laser resurfacing | The mean percentage of reduction in lesion counts in the control groups was | The mean percentage of reduction in lesion counts in the intervention groups was | ‐ | 14 | ⊕⊝⊝⊝ | |
| 5% 5‐FU/Er:YAG laser resurfacing | See comment | See comment | Not estimable | 55 | ⊕⊕⊝⊝ | Results with no SD: at 6 months: 5‐FU = 79.2%, resurfacing 94.5%, at 12 months: 5‐FU = 76.6%, resurfacing = 91.1% |
| 5% 5‐FU/Cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Trichloroacetic acid peel | The mean percentage of reduction in lesion counts in the control groups was | The mean percentage of reduction in lesion counts in the intervention groups was | ‐ | 18 | ⊕⊝⊝⊝ | |
| Withdrawal due to adverse events | ||||||
| 0.5% 5‐FU/Vehicle | 0 per 1000 | N/A (5/119 = 42/1000) | RR 5.41 | 177 | ⊕⊝⊝⊝ | Data from 1, 2, and 4 week treatments were pooled.(Analysis 9.4) Another study reported 24/207 participants withdrew because of adverse events and 12 of them were in 4 week 5‐FU group. GRADE = low |
| 0.5% 5‐FU with cryotherapy/Vehicle with cryotherapy | See comment | See comment | Not estimable | 142 | ⊕⊕⊝⊝ | There were no participant withdrawals in the first part of this three part study (incomplete data were given for the whole study). |
| 0.5% 5‐FU/ALA‐PDT | 0 per 1000 | N/A (1/12 = 83/1000) | RR 5.77 | 36 | ⊕⊕⊝⊝ | Data from blue light and pulsed dye laser were pooled |
| 0.5% 5‐FU/5.0% 5‐FU | See comment | See comment | Not estimable | 21 | ⊕⊕⊝⊝ | Intraindividual study: 16/21 discontinued treatment but did not withdraw: 4 because of 0.5%, 8 because of 5.0% , 4 because of both creams. |
| 5% 5‐FU with 0.05% tretinoin /5% 5‐FU with placebo | See comment | See comment | Not estimable | 19 | ⊕⊕⊝⊝ | Intraindividual study: 1 participant withdrew because of irritation but associated treatment was not specified. |
| 5% 5‐FU /10% masoprocol | 0 per 1000 | N/A (1/30 = 33/1000) | RR 2.71 | 57 | ⊕⊕⊝⊝ | |
| 5% 5‐FU/5% Imiquimod | 0 per 1000 | 0 per 1000 | Not estimable | 89 | ⊕⊕⊝⊝ | There were no participant withdrawals due to adverse events. |
| 5% 5‐FU/Carbon dioxide laser resurfacing | 250 per 1000 | 45 per 1000 | RR 0.18 | 17 | ⊕⊕⊝⊝ | |
| 5% 5‐FU/Er:YAG laser resurfacing | 0 per 1000 | N/A (1/27 = 37/1000) | RR 3.11 | 55 | ⊕⊕⊝⊝ | |
| 5% 5‐FU/Cryotherapy | 0 per 1000 | 0 per 1000 | Not estimable | 49 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| 5% 5‐FU/Trichloroacetic acid peel | 0 per 1000 | 0 per 1000 | Not estimable | 18 | ⊕⊕⊝⊝ | There were no participant withdrawals due to adverse events. |
| Skin irritation | ||||||
| 0.5% 5‐FU/Vehicle | 654 per 1000 | 948 per 1000 | RR 1.45 | 384 | ⊕⊕⊕⊝ | Data from 1, 2, and 4 week treatments were pooled |
| 0.5% 5‐FU with cryotherapy/Vehicle with cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5% 5‐FU/ALA‐PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5% 5‐FU/5.0% 5‐FU | 1000 per 1000 | 1000 per 1000 | ‐ | 21 | ⊕⊕⊕⊝ | Intraindividual study: All participants reported facial irritation in association with both creams |
| 5% 5‐FU with 0.05% tretinoin /5% 5‐FU with placebo | See comment | See comment | Not estimable | 19 | ⊕⊕⊕⊝ | Intraindividual study: 12 had more irritation with tretinoin, 4 had more with placebo, and 3 had equal irritation. |
| 5% 5‐FU /10% masoprocol | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/5% Imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Carbon dioxide laser resurfacing | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Er:YAG laser resurfacing | 429 per 1000 | 703 per 1000 | RR 1.64 | 55 | ⊕⊕⊝⊝ | At the end of treatment |
| 5% 5‐FU/Cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Trichloroacetic acid peel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Photodynamic therapy compared to interventions for actinic keratoses in immunocompetent participants | ||||||
| Intervention/Comparison intervention | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk With comparator | Corresponding risk With intervention | |||||
| Participant complete clearance | ||||||
| ALA‐PDT | ||||||
| 1h ALA‐blue light PDT /1h ALA‐pulsed dye laser PDT (field‐directedtreatments) | 83 per 1000 | 500 per 1000 | RR 6 | 24 | ⊕⊝⊝⊝ | |
| 1h ALA‐blue light PDT /0.5% 5‐FU (field‐directedtreatments) | 500 per 1000 | 500 per 1000 (225 to 1000) | RR 1 | 24 | ⊕⊝⊝⊝ | |
| 14‐18h ALA‐blue light PDT /14‐18h placebo‐blue light PDT (individual lesions) | 97 per 1000 | 602 per 1000 | RR 6.22 | 243 | ⊕⊝⊝⊝ | 1 treatment. (Analysis 47.1) Additional intraindividual study: ALA‐PDT: 16/35, placebo‐PDT = 2/35. GRADE = moderate |
| 1h ALA‐pulsed dye laser PDT /0.5% 5‐FU (field‐directedtreatments) | 500 per 1000 | 85 per 1000 | RR 0.17 | 24 | ⊕⊝⊝⊝ | |
| 0.5h ALA‐red light PDT/1h ALA‐red light PDT (individual lesions) | 474 per 1000 | 237 per 1000 | RR 0.5 | 72 | ⊕⊕⊝⊝ | Data from assessment at 8 weeks after the end of treatment (Analysis 49.2) |
| 0.5h ALA‐red light PDT/2h ALA‐red light PDT (individual lesions) | 471 per 1000 | 235 per 1000 | RR 0.5 | 68 | ⊕⊝⊝⊝ | Data from assessment at 8 weeks after the end of treatment |
| 0.5h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | 735 per 1000 | 235 per 1000 | RR 0.32 | 68 | ⊕⊕⊝⊝ | Data from assessment at 8 weeks after the end of treatment |
| 1h ALA‐red light PDT /2h ALA‐red light PDT (individual lesions) | 471 per 1000 | 475 per 1000 | RR 1.01 | 72 | ⊕⊝⊝⊝ | Data from assessment at 8 weeks after the end of treatment |
| 1h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | 735 per 1000 | 471 per 1000 | RR 0.64 | 72 | ⊕⊕⊝⊝ | Data from assessment at 8 weeks after the end of treatment |
| 2h ALA‐red light PDT/4h ALA‐red light PDT (individual lesions) | 735 per 1000 | 471 per 1000 | RR 0.64 | 68 | ⊕⊕⊝⊝ | Data from assessment at 8 weeks after the end of treatment (Analysis 49.2) |
| 3‐4h ALA‐red light PDT/3 to 4h placebo‐red light PDT (individual lesions) | 89 per 1000 | 527 per 1000 | RR 5.94 | 422 | ⊕⊕⊕⊕ | 1 treatment (Analysis 47.1) |
| 3% diclofenac in 2.5% hyaluronan gel + 4h ALA‐red light PDT /2.5% hyaluronan gel + 4h ALA‐red light PDT (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 4h ALA‐red light PDT/Cryotherapy (individual lesions) | 443 per 1000 | 580 per 1000 | RR 1.31 | 297 | ⊕⊕⊝⊝ | |
| ALA‐red light PDT (individual lesions)/5% imiquimod (field‐directedtreatment) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐blue light PDT + 5% imiquimod / ALA‐blue light PDT + placebo (field‐directedtreatments) | See comment | See comment | Not estimable | 25 | ⊕⊕⊝⊝ | Intraindividual study: ALA‐PDT + 5% imiquimod = 2/25; ALA‐PDT + placebo = 2/25 |
| ALA‐PDT versus MAL‐PDT | ||||||
| 5h ALA‐red light PDT /3h MAL‐red light PDT (field‐directedtreatments) | See comment | See comment | Not estimable | 16 | ⊕⊕⊕⊝ | Intraindividual study: ALA‐PDT = 6/16, MAL‐PDT = 7/16 |
| MAL‐PDT | ||||||
| All day 16% MAL‐daylight PDT /All day 8% MAL‐daylight PDT (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 2h MAL‐daylight PDT /3h MAL‐daylight PDT (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 2.5‐4h MAL‐red light PDT /2.5‐4h placebo‐red light PDT (individual lesions) | 147 per 1000 | 656 per 1000 | RR 4.46 | 482 | ⊕⊕⊕⊝ | |
| 3h MAL‐red light LED PDT /3h MAL‐broad visible + water‐filtered infrared A PDT (individual lesions) | 500 per 1000 | 575 per 1000 | RR 1.15 | 80 | ⊕⊕⊝⊝ | Data from assessment at 12 weeks after the end of treatment.(Analysis 53.1) |
| 3h MAL‐red light LED PDT /3h MAL‐daylight PDT (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Single 3h MAL‐red light PDT /Multiple 3h MAL‐red light PDT [2 treatments 1 week apart] (individual lesions) | 755 per 1000 | 883 per 1000 | RR 1.17 | 211 | ⊕⊕⊝⊝ | |
| 3h MAL‐red light PDT /Cryotherapy (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean reduction in lesion counts | ||||||
| ALA‐PDT | ||||||
| 1h ALA‐blue light PDT /1h ALA‐pulsed dye laser PDT (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐blue light PDT /0.5% 5‐FU (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 14‐18h ALA‐blue light PDT /14‐18h placebo‐blue light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐pulsed dye laser PDT /0.5% 5‐FU (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/1h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/2h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐red light PDT /2h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 2h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3‐4h ALA‐red light PDT /3‐4h placebo‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3% diclofenac in 2.5% hyaluronic acid gel + 4h ALA‐red light PDT /2.5% hyaluronic acid gel + 4h ALA‐red light PDT (field‐directedtreatments) | See comment | See comment | Not estimable | 10 | ⊕⊕⊝⊝ | Intraindividual study: at 6 weeks; diclofenac/hyaluronic acid (HA) + ALA‐PDT = 10.13, HA + ALA‐PDT= 9.9, at 6 months:; diclofenac/HA + ALA‐PDT = 11.56, HA + ALA‐PDT = 10.56, at 12 months; diclofenac/HA + ALA‐PDT = 12.5, HA + ALA‐PDT = 8.8 |
| 4h ALA‐red light PDT /Cryotherapy (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐red light PDT (individual lesions)/5% imiquimod (field‐directedtreatment) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐blue light PDT + 5% imiquimod / ALA‐blue light PDT + placebo (field‐directedtreatments) | See comment | See comment | Not estimable | 25 | ⊕⊕⊝⊝ | Results from intraindividual study without SD: ALA‐PDT + 5% imiquimod = 19.9 lesions; ALA‐PDT + placebo = 16.0 lesions |
| ALA‐PDT versus MAL‐PDT | ||||||
| 5h ALA‐red light PDT /3h MAL‐red light PDT (field‐directedtreatments) | The mean reduction in lesion counts in the control groups was | The mean reduction in lesion counts in the intervention groups was 0.6 higher | ‐ | 15 | ⊕⊕⊝⊝ | |
| MAL‐PDT | ||||||
| All day 16% MAL‐daylight PDT /All day 8% MAL‐daylight PDT (field‐directedtreatments) | The mean reduction in lesion counts in the control groups was | The mean reduction in lesion counts in the intervention groups was 0.3 higher | ‐ | 29 | ⊕⊕⊝⊝ | |
| 2h MAL‐daylight PDT /3h MAL‐daylight PDT (field‐directedtreatments) | The mean reduction in lesion counts in the control groups was | The mean reduction in lesion counts in the intervention groups was 0.1 higher | 120 | ⊕⊕⊝⊝ | ||
| 2.5‐4h MAL‐red light PDT /2.5‐4h placebo‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3h MAL‐red light LED PDT /3h MAL‐broad visible + water‐filtered infrared A PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3h MAL‐red light LED PDT /3h MAL‐daylight PDT (field‐directedtreatments) | The mean reduction in lesion counts in the control groups was | The mean reduction in lesion counts in the intervention groups was 0.4 lower | ‐ | 29 | ⊕⊕⊝⊝ | |
| Single 3h MAL‐red light PDT /Multiple 3h MAL‐red light PDT [2 treatments 1 week apart] (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3h MAL‐red light PDT /Cryotherapy (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean percentage of reduction in lesion count | ||||||
| ALA‐PDT | ||||||
| 1h ALA‐blue light PDT /1h ALA‐pulsed dye laser PDT (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐blue light PDT /0.5% 5‐FU (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 14‐18h ALA‐blue light PDT /14‐18h placebo‐blue light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐pulsed dye laser PDT /0.5% 5‐FU (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/1h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/2h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐red light PDT /2h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 2h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3‐4h ALA‐red light PDT /3‐4h placebo‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3% diclofenac in 2.5% hyaluronic acid gel + 4h ALA‐red light PDT /2.5% hyaluronic acid gel + 4h ALA‐red light PDT (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 4h ALA‐red light PDT /Cryotherapy (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐red light PDT (individual lesions)/5% imiquimod (field‐directedtreatment) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐blue light PDT + 5% imiquimod / ALA‐blue light PDT + placebo (field‐directedtreatments) | See comment | See comment | Not estimable | 25 | ⊕⊕⊝⊝ | Results from intraindividual study without SD: ALA‐PDT + 5% imiquimod = 86.7%; ALA‐PDT + placebo = 73.1% |
| ALA‐PDT versus MAL‐PDT | ||||||
| 5h ALA‐red light PDT /3h MAL‐red light PDT (field‐directedtreatments) | ‐ | See comment | ‐ | ‐ | ‐ | Not reported |
| MAL‐PDT | ||||||
| All day 16% MAL‐daylight PDT /All day 8% MAL‐daylight PDT (field‐directedtreatments) | See comment | See comment | Not estimable | 29 | ⊕⊕⊝⊝ | Data with no SD: 16% MAL‐daylight PDT = 76.9%, 8% MAL‐daylight PDT = 79.5%. |
| 2h MAL‐daylight PDT /3h MAL‐daylight PDT (field‐directedtreatments) | The mean percentage of reduction in lesion counts in the control groups was | The mean percentage of reduction in lesion counts in the intervention groups was 2.6 higher | ‐ | 120 | ⊕⊕⊝⊝ | |
| 2.5‐4h MAL‐red light PDT /2.5‐4h placebo‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3h MAL‐red light LED PDT /3h MAL‐broad visible + water‐filtered infrared A PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3h MAL‐red light LED PDT /3h MAL‐daylight PDT (field‐directedtreatments) | See comment | See comment | Not estimable | 29 | ⊕⊕⊝⊝ | Data with no SD: MAL‐red light LED PDT = 71%, MAL‐daylight PDT = 79%. |
| Single 3h MAL‐red light PDT /Multiple 3h MAL‐red light PDT [2 treatments 1 week apart] (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3h MAL‐red light PDT /Cryotherapy (individual lesions) | See comment | See comment | Not estimable | 240 | ⊕⊝⊝⊝ | Intraindividual studies with no SD: at 12 weeks: MAL‐PDT = 84.4%, cryotherapy = 74.5%, at 24 weeks: MAL‐PDT = 75‐86.7%, cryotherapy = 83.9‐87% |
| Withdrawal due to adverse events | ||||||
| ALA‐PDT | ||||||
| 1h ALA‐blue light PDT /1h ALA‐pulsed dye laser PDT (field‐directedtreatments) | 0 per 1000 | 0 per 1000 | Not estimable | 24 | ⊕⊕⊝⊝ | There were no participant withdrawals due to adverse events. |
| 1h ALA‐blue light PDT /0.5% 5‐FU (field‐directedtreatments) | 83 per 1000 | 28 per 1000 | RR 0.33 | 24 | ⊕⊕⊝⊝ | |
| 14‐18h ALA‐blue light PDT /14‐18h placebo‐blue light PDT (individual lesions) | 0 per 1000 | 0 per 1000 | Not estimable | 271 | ⊕⊕⊝⊝ | There were no participant withdrawals due to adverse events. |
| 1h ALA‐pulsed dye laser PDT /0.5% 5‐FU (field‐directedtreatments) | 83 per 1000 | 28 per 1000 | RR 0.33 | 24 | ⊕⊕⊝⊝ | |
| 0.5h ALA‐red light PDT/1h ALA‐red light PDT (individual lesions) | See comment | See comment | Not estimable | 72 | ⊕⊝⊝⊝ | No details were given for the reasons for withdrawal. |
| 0.5h ALA‐red light PDT/2h ALA‐red light PDT (individual lesions) | See comment | See comment | Not estimable | 68 | ⊕⊝⊝⊝ | No details were given for the reasons for withdrawal. |
| 0.5h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | See comment | See comment | Not estimable | 68 | ⊕⊝⊝⊝ | No details were given for the reasons for withdrawal. |
| 1h ALA‐red light PDT /2h ALA‐red light PDT (individual lesions) | See comment | See comment | Not estimable | 72 | ⊕⊝⊝⊝ | No details were given for the reasons for withdrawal. |
| 1h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | See comment | See comment | Not estimable | 72 | ⊕⊝⊝⊝ | No details were given for the reasons for withdrawal. |
| 2h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | See comment | See comment | Not estimable | 68 | ⊕⊝⊝⊝ | No details were given for the reasons for withdrawal. |
| 3‐4h ALA‐red light PDT /3‐4h placebo‐red light PDT (individual lesions) | 0 per 1000 | 0 per 1000 | Not estimable | 391 | ⊕⊕⊕⊕ | There were no participant withdrawals due to adverse events. |
| 3% diclofenac in 2.5% hyaluronic acid gel + 4h ALA‐red light PDT /2.5% hyaluronic acid gel + 4h ALA‐red light PDT (field‐directedtreatments) | 0 per 1000 | 0 per 1000 | Not estimable | 10 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| 4h ALA‐red light PDT /Cryotherapy (individual lesions) | 0 per 1000 | 0 per 1000 | Not estimable | 255 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| ALA‐red light PDT (individual lesions)/5% imiquimod (field‐directedtreatment) | 0 per 1000 | 0 per 1000 | Not estimable | 30 | ⊕⊕⊝⊝ | There were no participant withdrawals due to adverse events. |
| ALA‐blue light PDT + 5% imiquimod / ALA‐blue light PDT + placebo (field‐directedtreatments) | 0 per 1000 | 0 per 1000 | Not estimable | 25 | ⊕⊕⊝⊝ | There were no participant withdrawals due to adverse events. |
| ALA‐PDT versus MAL‐PDT | ||||||
| 5h ALA‐red light PDT /3h MAL‐red light PDT (field‐directedtreatments) | 0 per 1000 | 0 per 1000 | Not estimable | 15 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| MAL‐PDT | ||||||
| All day 16% MAL‐daylight PDT /All day 8% MAL‐daylight PDT (field‐directedtreatments) | See comment | See comment | Not estimable | 29 | ⊕⊕⊕⊝ | One of 30 participants withdrew because of adverse events unrelated to treatments. |
| 2h MAL‐daylight PDT /3h MAL‐daylight PDT (field‐directedtreatments) | 0 per 1000 | 0 per 1000 | Not estimable | 120 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| 2.5‐4h MAL‐red light PDT /2.5‐4h placebo‐red light PDT (individual lesions) | 0 per 1000 | N/A (3/130 = 23/1000) | RR 2 | 191 | ⊕⊝⊝⊝ | Two additional studies with no participant withdrawals because of adverse events (N = 211). GRADE = low |
| 3h MAL‐red light LED PDT /3h MAL‐broad visible + water‐filtered infrared A PDT (individual lesions) | 0 per 1000 | 0 per 1000 | Not estimable | 78 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| 3h MAL‐red light LED PDT /3h MAL‐daylight PDT (field‐directedtreatments) | 0 per 1000 | 0 per 1000 | Not estimable | 29 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| Single 3h MAL‐red light PDT /Multiple 3h MAL‐red light PDT [2 treatments 1 week apart] (individual lesions) | 9 per 1000 | 3 per 1000 | RR 0.34 | 211 | ⊕⊕⊝⊝ | |
| 3h MAL‐red light PDT /Cryotherapy (individual lesions) | 11 per 1000 | 10 per 1000 | RR 0.94 | 379 | ⊕⊕⊕⊝ | Two additional intraindividual studies: 4 of 119 and 2 of 121 participants withdrew because of adverse events and one of them was related to MAL‐PDT. GRADE = low |
| Skin irritation | ||||||
| ALA‐PDT | ||||||
| 1h ALA‐blue light PDT /1h ALA‐pulsed dye laser PDT (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐blue light PDT /0.5% 5‐FU (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 14‐18h ALA‐blue light PDT /14‐18h placebo‐blue light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐pulsed dye laser PDT /0.5% 5‐FU (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/1h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/2h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐red light PDT/2h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐red light PDT/4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 2h ALA‐red light PDT/4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3 to 4h ALA‐red light PDT /3 to 4h placebo‐red light PDT (individual lesions) | 0 per 1000 | N/A (77/217 = 355/1000) | RR 59.72 | 300 | ⊕⊕⊕⊝ | Data for ALA‐PDT was given separately for two studies but not for placebo. Data from assessment after treatment (Analysis 47.7) |
| 3% diclofenac in 2.5% hyaluronic acid gel + 4h ALA‐red light PDT /2.5% hyaluronic acid gel + 4h ALA‐red light PDT (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 4h ALA‐red light PDT /Cryotherapy (individual lesions) | 101 per 1000 | 371 per 1000 | RR 3.69 | 297 | ⊕⊕⊝⊝ | Assessment one day after the treatment (Analysis 51.2) |
| ALA‐red light PDT (individual lesions)/5% imiquimod (field‐directedtreatment) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐blue light PDT + 5% imiquimod / ALA‐blue light PDT + placebo (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐PDT versus MAL‐PDT | ||||||
| 5h ALA‐red light PDT /3h MAL‐red light PDT (field‐directedtreatments) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| MAL‐PDT | ||||||
| All comparisons | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy compared to interventions for actinic keratoses in immunocompetent participants | ||||||
| Intervention/ Comparison intervention | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| ‐ | With comparator | With intervention | ‐ | ‐ | ‐ | ‐ |
| Participant complete clearance | ||||||
| Cryotherapy /Betulin‐based oleogel | 643 per 1000 | 784 per 1000 | RR 1.22 | 28 | ⊕⊝⊝⊝ | |
| Cryotherapy/cryotherapy with betulin‐based oleogel | 714 per 1000 | 786 per 1000 | RR 1.1 | 28 | ⊕⊝⊝⊝ | |
| Cryotherapy/5% 5‐FU | 958 per 1000 | 680 per 1000 | RR 0.71 | 49 | ⊕⊕⊝⊝ | Assessment after treatment (Analysis 43.1) |
| Vehicle with cryotherapy/0.5% 5‐FU with cryotherapy | 292 per 1000 | 70 per 1000 | RR 0.24 | 142 | ⊕⊕⊝⊝ | 1 cycle (Analysis 63.1) |
| Cryotherapy /Imiquimod | 846 per 1000 | 677 per 1000 | RR 0.8 | 51 | ⊕⊝⊝⊝ | 5% imiquimod (Analysis 44.1) |
| Cryotherapy with vehicle /Cryotherapy with imiquimod | Study population | RR 0.2 | 311 | ⊕⊕⊕⊕ | Pooled data (5% and 3.75% imiquimod)(Analysis 64.1) Results from an additional intraindividual study: cryotherapy + vehicle = 5/27, cryotherapy+imiquimod = 8/27 GRADE = moderate | |
| 287 per 1000 | 57 per 1000 | |||||
| Moderate | ||||||
| 264 per 1000 | 53 per 1000 | |||||
| Cryotherapy /ALA‐red light PDT | 581 per 1000 | 442 per 1000 | RR 0.76 | 297 | ⊕⊕⊝⊝ | |
| Cryotherapy/MAL‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean reduction in lesion counts | ||||||
| Cryotherapy /Betulin‐based oleogel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/cryotherapy with betulin‐based oleogel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/5% 5‐FU | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Vehicle + cryotherapy/0.5% 5‐FU + cryotherapy | The mean reduction in lesion counts in the control groups was | The mean reduction in lesion counts in the intervention groups was | ‐ | 142 | ⊕⊕⊕⊝ | 1 cycle (Analysis 63.2) |
| Cryotherapy /Imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy with vehicle /Cryotherapy with imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy /ALA‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/MAL‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean percentage of reduction in lesion counts | ||||||
| Cryotherapy /Betulin‐based oleogel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/cryotherapy with betulin‐based oleogel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/5% 5‐FU | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Vehicle with cryotherapy/0.5% 5‐FU with cryotherapy | The mean percentage of reduction in lesion counts in the control groups was | The mean percentage of reduction in lesion counts in the intervention groups was | ‐ | 142 | ⊕⊕⊕⊝ | |
| Cryotherapy /Imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy with vehicle /Cryotherapy with imiquimod | See comment | See comment | ‐ | 301 | ⊕⊝⊝⊝ | High heterogeneity (I2=86%) between 3.75% (parallel group, MD ‐34.10, 95% CI ‐41.38 to ‐26.82)) and 5.0% (intraindividual, MD ‐11.20, 95% CI ‐26.53 to 4.13) imiquimod studies. (Analysis 64.4) |
| Cryotherapy /ALA‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/MAL‐red light PDT | See comment | See comment | Not estimable | 240 | ⊕⊝⊝⊝ | Intraindividual studies with no SD: at 12 weeks: cryotherapy = 74.5%, MAL‐PDT= 84.4%, at 24 weeks: cryotherapy = 83.9‐87%, MAL‐PDT = 75‐86.7% |
| Withdrawal due to adverse events | ||||||
| Cryotherapy /Betulin‐based oleogel | 0 per 1000 | 0 per 1000 | Not estimable | 28 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| Cryotherapy/cryotherapy with betulin‐based oleogel | 0 per 1000 | 0 per 1000 | Not estimable | 28 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| Cryotherapy/5% 5‐FU | 0 per 1000 | 0 per 1000 | Not estimable | 49 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| Vehicle with cryotherapy/0.5% 5‐FU with cryotherapy | See comment | See comment | Not estimable | 142 | ⊕⊕⊝⊝ | There were no participant withdrawals due to adverse events in the first part of this three part study (incomplete data were given for the whole study). |
| Cryotherapy /Imiquimod | 0 per 1000 | 0 per 1000 | Not estimable | 51 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| Cryotherapy with vehicle /Cryotherapy with imiquimod | Study population | RR 0.93 | 312 | ⊕⊕⊕⊝ | Pooled data (5% and 3.75% imiquimod) (Analysis 64.6) | |
| 33 per 1000 | 30 per 1000 | |||||
| Moderate | ||||||
| 21 per 1000 | 20 per 1000 | |||||
| Cryotherapy /ALA‐ red light PDT | 0 per 1000 | 0 per 1000 | Not estimable | 297 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| Cryotherapy/MAL‐ red light PDT | 11 per 1000 | 11 per 1000 | RR 1.06 | 379 | ⊕⊕⊕⊝ | Two additional intraindividual studies: 4 of 119 and 2 of 121 participants withdrew because of adverse events and one of them was related to MAL‐PDT. GRADE = low |
| Skin irritation | ||||||
| Cryotherapy /Betulin‐based oleogel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/cryotherapy with betulin‐based oleogel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/5% 5‐FU | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Vehicle with cryotherapy/0.5% 5‐FU with cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy /Imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy with vehicle /Cryotherapy with imiquimod | Study population | RR 0.39 | 311 | ⊕⊕⊕⊝ | Pooled data (5% and 3.75% imiquimod) | |
| 83 per 1000 | 32 per 1000 | |||||
| Moderate | ||||||
| 125 per 1000 | 49 per 1000 | |||||
| Cryotherapy /ALA‐red light PDT | 372 per 1000 | 100 per 1000 | RR 0.27 | 297 | ⊕⊕⊝⊝ | Assessment one day after the treatment (Analysis 46.2) |
| Cryotherapy/MAL‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Imiquimod compared to interventions for actinic keratoses in immunocompetent participants | ||||||
| Intervention/Comparison intervention | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| ‐ | With comparator | With intervention | ‐ | ‐ | ‐ | ‐ |
| Participant complete clearance | ||||||
| 2.5% imiquimod/placebo | 62 per 1000 | 277 per 1000 | RR 4.49 | 486 | ⊕⊕⊕⊕ | |
| 3.75% imiquimod/placebo | Study population | RR 6.45 | 730 | ⊕⊕⊕⊕ | ||
| 53 per 1000 | 343 per 1000 | |||||
| Moderate | ||||||
| 50 per 1000 | 322 per 1000 | |||||
| Cryotherapy + 3.75% imiquimod/Cryotherapy + vehicle | 33 per 1000 | 301 per 1000 | RR 9.12 | 247 | ⊕⊕⊕⊝ | For all lesions |
| 5% imiquimod/placebo | Study population | RR 7.70 | 1871 | ⊕⊕⊕⊕ | ||
| 48 per 1000 | 371 per 1000 | |||||
| Moderate | ||||||
| 32 per 1000 | 246 per 1000 | |||||
| 5% imiquimod/3% diclofenac in 2.5% hyaluronic acid | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod /5% 5‐FU | See comment | See comment | ‐ | 89 | ⊕⊝⊝⊝ | The two studies was associated with high heterogeneity (I²= 93%) and the results could not be pooled together. One study favoured 5‐FU (RR 0.31, 95% CI 0.14 to 0.67] whereas the other did not (RR 0.88, 95% CI 0.73 to 1.06] (Analysis 22.1) |
| 5% imiquimod/Cryotherapy | 680 per 1000 | 843 per 1000 | RR 1.24 | 51 | ⊕⊝⊝⊝ | |
| Cryotherapy + 5% imiquimod/Cryotherapy + vehicle | 91 per 1000 | 225 per 1000 | RR 2.48 | 64 | ⊕⊕⊝⊝ | For all lesions.(Analysis 65.1) Results from an additional intraindividual study: cryotherapy + imiquimod side (8/27 = 30%), cryotherapy alone side (5/27 = 19%), GRADE = low |
| 5% imiquimod/ALA‐PDT | ‐ | Not reported | ||||
| ALA‐PDT + 5% imiquimod/ALA‐PDT + placebo | See comment | See comment | Not estimable | 25 | ⊕⊕⊝⊝ | Intraindividual study: ALA‐PDT + 5% imiquimod = 2/25; ALA‐PDT + placebo = 2/25 |
| Mean reduction in lesion counts | ||||||
| 2.5% imiquimod/placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3.75% imiquimod/placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy + 3.75% imiquimod/Cryotherapy + vehicle | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod/placebo | The mean reduction in lesion counts in the control groups was | The mean reduction in lesion counts in the intervention groups was 2.20 higher | ‐ | 12 | ⊕⊕⊝⊝ | Results from an additional intraindividual study with no SD (N = 21): 5% imiquimod: 3.9 lesions, placebo = 0.5 lesions, GRADE = very low |
| 5% imiquimod/3% diclofenac in 2.5% hyaluronic acid | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod /5% 5‐FU | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod/Cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy + 5% imiquimod/Cryotherapy + vehicle | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod/ALA‐PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐PDT + 5% imiquimod/ALA‐PDT + placebo | See comment | See comment | Not estimable | 25 | ⊕⊕⊝⊝ | Results from intraindividual study without SD: ALA‐PDT + 5% imiquimod= 19.9 lesions; ALA‐PDT + placebo= 16.0 lesions |
| Mean percentage of reduction in lesion counts | ||||||
| 2.5% imiquimod/placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3.75% imiquimod/placebo | The mean percentage of reduction in lesion counts in the control groups was | The mean percentage of reduction in lesion counts in the intervention groups was 46.90 higher | ‐ | 247 | ⊕⊕⊕⊝ | |
| Cryotherapy + 3.75% imiquimod/Cryotherapy + vehicle | The mean percentage of reduction in lesion counts in the control groups was | The mean percentage of reduction in lesion counts in the intervention groups was 34.1 higher | ‐ | 247 | ⊕⊕⊕⊝ | For all lesions (Analysis 65.2) |
| 5% imiquimod/placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod/3% diclofenac in 2.5% hyaluronic acid | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod /5% 5‐FU | See comment | See comment | Not estimable | 39 | ⊕⊕⊝⊝ | Results with no SD: 5% imiquimod = 66%, 5% 5‐FU = 94% |
| 5% imiquimod/Cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy + 5% imiquimod/Cryotherapy + vehicle | The mean percentage of reduction in lesion counts in the control groups was | The mean percentage of reduction in lesion counts in the intervention groups was 11.2 higher | ‐ | 27 | ⊕⊕⊝⊝ | For all lesions.(Analysis 65.2) Results from an additional intraindividual study: cryotherapy‐5% imiquimod = 73.2+27.1%, cryotherapy + vehicle = 62.0+30.3%. GRADE = moderate |
| 5% imiquimod/ALA‐PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐PDT + 5% imiquimod/ALA‐PDT + placebo | See comment | See comment | Not estimable | 25 | ⊕⊕⊝⊝ | Results from intraindividual study without SD: ALA‐PDT + 5% imiquimod = 86.7% ; ALA‐PDT + placebo = 73.1 % |
| Withdrawal due to adverse events | ||||||
| 2.5% imiquimod/placebo | 19 per 1000 | 9 per 1000 | RR 0.5 | 486 | ⊕⊕⊕⊝ | |
| 3.75% imiquimod/placebo | 19 per 1000 | 17 per 1000 | RR 0.92 | 483 | ⊕⊕⊕⊝ | |
| Cryotherapy + 3.75% imiquimod/Cryotherapy + vehicle | 32 per 1000 | 41 per 1000 | RR 1.3 | 247 | ⊕⊕⊝⊝ | |
| 5% imiquimod/placebo | Study population | RR 2.59 | 2290 | ⊕⊕⊕⊝ | (Analysis 20.5) Four small sample size studies with no participant withdrawal are not included in meta‐analysis: pooled data, imiquimod 0/79 and placebo 0/31. Additional two intraindividual studies: no participant withdrew because of adverse events (0/42) GRADE = very low (both studies had more than 20% participant lost). | |
| 21 per 1000 | 56 per 1000 | |||||
| Moderate | ||||||
| 5 per 1000 | 13 per 1000 | |||||
| High | ||||||
| 0 per 1000 | 0 per 1000 | |||||
| 5% imiquimod/3% diclofenac in 2.5% hyaluronic acid | 0 per 1000 | 0 per 1000 | Not estimable | 49 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| 5% imiquimod /5% 5‐FU | 0 per 1000 | 0 per 1000 | Not estimable | 50 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| 5% imiquimod/Cryotherapy | 0 per 1000 | 0 per 1000 | Not estimable | 51 | ⊕⊕⊕⊝ | There were no participant withdrawals due to adverse events. |
| Cryotherapy + 5% imiquimod/Cryotherapy + vehicle | 30 per 1000 | 10 per 1000 | RR 0.34 | 65 | ⊕⊕⊝⊝ | |
| 5% imiquimod/ALA‐PDT | 0 per 1000 | 0 per 1000 | Not estimable | 30 | ⊕⊕⊝⊝ | There were no participant withdrawals due to adverse events. |
| ALA‐PDT + 5% imiquimod/ALA‐PDT + placebo | 0 per 1000 | 0 per 1000 | Not estimable | 25 | ⊕⊕⊝⊝ | There were no participant withdrawals due to adverse events. |
| Skin irritation | ||||||
| 2.5% imiquimod/placebo | 6 per 1000 | 21 per 1000 | RR 3.45 | 486 | ⊕⊕⊕⊝ | |
| 3.75% imiquimod/placebo | 6 per 1000 | 30 per 1000 | RR 4.86 | 484 | ⊕⊕⊕⊝ | |
| Cryotherapy + 3.75% imiquimod/Cryotherapy + vehicle | 8 per 1000 | 56 per 1000 | RR 6.72 | 247 | ⊕⊕⊝⊝ | |
| 5% imiquimod/placebo | 5 per 1000 | 18 per 1000 | RR 3.68 | 708 | ⊕⊕⊕⊝ | Additional intraindividual study: similar mild irritation between the two treatment sides (N = 20) GRADE = very low |
| 5% imiquimod/3% diclofenac in 2.5% hyaluronic acid | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod/5% 5‐FU | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod/Cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy + 5% imiquimod/Cryotherapy + vehicle | 121 per 1000 | 194 per 1000 | RR 1.6 | 64 | ⊕⊕⊝⊝ | |
| 5% imiquimod/ALA‐PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐PDT + 5% imiquimod/ALA‐PDT + placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Global Improvement Indices (investigator)‐cleared Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Mean changes in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2.1 0.1% adapalene gel | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 0.3% adapalene gel | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: dermatitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Global Improvement Indices (investigator)‐cleared Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Mean changes in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: dermatitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean changes in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Cosmetic outcomes: Reduction in total cosmetic appearance score Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Mean reduction in lesion counts‐total Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Mean reduction in lesion counts‐per anatomical locations Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3.1 Face | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Scalp | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Upper extremities | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Cosmetic outcomes: Number of participants with decreased infiltration and disappearance of crust Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Investigator Global Improvement Indices‐completely improved Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 30 day treatment/30 day follow‐up | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [0.89, 17.89] |
| 1.2 60 day treatment/30 day follow‐up | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 3.06 [1.21, 7.77] |
| 1.3 90 day treatment/30 day follow‐up | 1 | 117 | Risk Ratio (M‐H, Random, 95% CI) | 2.50 [1.37, 4.55] |
| 2 Participant Global Improvement Indices‐completely improved Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 30 day treatment/30 day follow‐up | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [0.89, 17.89] |
| 2.2 60 day treatment/30 day follow‐up | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 2.86 [1.12, 7.32] |
| 2.3 90 day treatment/30 day follow‐up | 1 | 117 | Risk Ratio (M‐H, Random, 95% CI) | 2.44 [1.28, 4.64] |
| 3 Participant complete clearance at end of treatment (>56 days) Show forest plot | 2 | 280 | Risk Ratio (M‐H, Random, 95% CI) | 1.95 [1.21, 3.13] |
| 4 Participant complete clearance (target lesions) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 30 day treatment/ 30 day follow‐up | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 3.5 [0.76, 16.01] |
| 4.2 60 day treatment/ 30 day follow‐up | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 3.27 [1.30, 8.21] |
| 4.3 90 day treatment/ 30 day follow‐up | 2 | 267 | Risk Ratio (M‐H, Random, 95% CI) | 2.87 [1.84, 4.48] |
| 5 Participant complete clearance (all lesions) Show forest plot | 3 | 420 | Risk Ratio (M‐H, Random, 95% CI) | 2.46 [1.66, 3.66] |
| 5.1 30 day treatment/ 30 day follow‐up | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 3.5 [0.76, 16.01] |
| 5.2 60 day treatment/ 30 day follow‐up | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 3.83 [1.37, 10.71] |
| 5.3 90 day treatment/30 day follow‐up | 2 | 225 | Risk Ratio (M‐H, Random, 95% CI) | 2.20 [1.40, 3.44] |
| 6 Participant complete clearance for 30 day treatment by locations Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Forehead | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.4 Back of hand | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Participant complete clearance for 60 day treatment by locations Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Forehead | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Arm/forearm | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 Back of hand | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Participant complete clearance for 90 day treatment by locations Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Scalp | 2 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.25, 6.08] |
| 8.2 Forehead | 2 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [1.03, 2.85] |
| 8.3 Face | 2 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 2.15 [1.05, 4.40] |
| 8.4 Arm/forearm | 2 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.94 [0.26, 14.40] |
| 8.5 Back of hand | 2 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [0.04, 65.87] |
| 9 Participant complete clearance in immunosuppressed participants Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Participant partial (>75%) clearance in immunosuppressed participants Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Mean reduction of lesion counts (30‐90 days ): At the end of study Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11.1 90 days | 1 | 150 | Mean Difference (IV, Random, 95% CI) | 0.80 [‐1.48, 3.08] |
| 12 Mean reduction of lesion counts (30‐90 days): 30 day follow‐up Show forest plot | 2 | 345 | Mean Difference (IV, Random, 95% CI) | 2.55 [1.56, 3.53] |
| 12.1 30 days | 1 | 98 | Mean Difference (IV, Random, 95% CI) | 2.00 [0.63, 3.37] |
| 12.2 60 days | 1 | 97 | Mean Difference (IV, Random, 95% CI) | 2.40 [0.73, 4.07] |
| 12.3 90 days | 1 | 150 | Mean Difference (IV, Random, 95% CI) | 3.80 [1.83, 5.77] |
| 13 Withdrawal due to adverse events Show forest plot | 4 | 592 | Risk Ratio (M‐H, Random, 95% CI) | 3.59 [1.92, 6.70] |
| 14 Minor adverse event: body as a whole : in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15 Minor adverse event: body as a whole : "flu" Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16 Minor adverse event:: body as a whole : infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17 Minor adverse event: cardiovascular: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18 Minor adverse event: cardiovascular: sinus bradycardia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 19 Minor adverse event: dermatological: bursitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 20 Minor adverse event: dermatological: dry skin Show forest plot | 3 | 462 | Risk Ratio (M‐H, Random, 95% CI) | 2.40 [1.20, 4.78] |
| 21 Minor adverse event: dermatological: herpes zoster Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22 Minor adverse event: dermatological: rash vesiculobullous Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 23 Minor adverse event::dermatological: seborrhoea Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 24 Minor adverse event: dermatological: skin exfoliation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 25 Minor adverse event: dermatological: ulcerated skin Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 26 Minor adverse event: digestive : in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 27 Minor adverse event: hemic and lymphatic: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 28 Minor adverse event: metabolic and nutritional disorders : in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 29 Minor adverse event: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 30 Minor adverse event: musculoskeletal and connective tissue: hypokinesia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 31 Minor adverse event: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 32 Minor adverse event: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 33 Minor adverse event: nervous system: hyperaesthesia Show forest plot | 2 | 345 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.30, 2.60] |
| 34 Minor adverse event: nervous system: paraesthesia Show forest plot | 2 | 345 | Risk Ratio (M‐H, Random, 95% CI) | 2.53 [0.57, 11.20] |
| 35 Minor adverse event: respiratory: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 36 Minor adverse event: respiratory: bronchitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 37 Minor adverse event: respiratory: pharyngitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 38 Minor adverse event: respiratory: upper respiratory tract infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 39 Minor adverse event: special senses: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 40 Minor adverse event: urogenital: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Investigator Global Improvement Indices‐Complete improvement Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participant Global Improvement Indices‐Complete improvement Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesions counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 3 | 522 | Risk Ratio (M‐H, Random, 95% CI) | 8.86 [3.67, 21.40] |
| 1.1 1 week treatment with 4 week follow‐up | 3 | 267 | Risk Ratio (M‐H, Random, 95% CI) | 8.30 [2.04, 33.76] |
| 1.2 2 week treatment with 4 week follow‐up | 2 | 128 | Risk Ratio (M‐H, Random, 95% CI) | 6.42 [1.27, 32.59] |
| 1.3 4 week treatment with 4 week follow‐up | 2 | 127 | Risk Ratio (M‐H, Random, 95% CI) | 13.07 [2.68, 63.66] |
| 2 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Mean percentage of reduction in lesion counts Show forest plot | 1 | 142 | Mean Difference (IV, Random, 95% CI) | 33.60 [22.88, 44.32] |
| 4 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Skin irritation Show forest plot | 2 | 384 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [1.27, 1.65] |
| 6 Minor adverse event excluding skin irritation: body as a whole : in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse event excluding skin irritation: body as a whole : allergy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse event excluding skin irritation: body as a whole : "flu" or common cold Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse event excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse event excluding skin irritation: musculoskeletal and connective tissue: soreness Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse event excluding skin irritation:nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse event excluding skin irritation: respiratory: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse event excluding skin irritation: respiratory: sinusitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14 Minor adverse event excluding skin irritation: respiratory: upper respiratory tract infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15 Minor adverse event excluding skin irritation: special senses: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16 Minor adverse event excluding skin irritation:special senses: eye irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Daily for 1 week versus 4 weeks | 2 | 167 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.19, 0.81] |
| 1.2 Daily for 1 week versus 2 weeks | 2 | 169 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.23, 2.37] |
| 1.3 Daily for 2 weeks versus 4 weeks | 2 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.36, 0.87] |
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Skin irritation Show forest plot | 2 | 515 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.91, 1.00] |
| 3.1 Daily for 1 week versus 4 weeks | 2 | 170 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.89, 1.03] |
| 3.2 Daily for 1 week versus 2 weeks | 2 | 172 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.86, 1.08] |
| 3.3 Daily for 2 weeks versus 4 weeks | 2 | 173 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.88, 1.02] |
| 4 Minor adverse events excluding skin irritation: body as a whole : in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Minor adverse events excluding skin irritation: body as a whole : allergy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Minor adverse events excluding skin irritation: body as a whole : "flu" or common cold Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: respiratory: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Minor adverse events excluding skin irritation: respiratory: sinusitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Minor adverse events excluding skin irritation: special senses: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Minor adverse events excluding skin irritation: special senses: eye irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 2 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 1.85 [0.41, 8.33] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Investigator Global Improvement Indices ‐cleared Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Mean reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Mean percentage of reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction in lesions Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance‐number of doses Show forest plot | 11 | 2880 | Risk Ratio (M‐H, Random, 95% CI) | 6.91 [4.25, 11.26] |
| 1.1 9 or 18 doses (3 times/week for 3 weeks on, 4 weeks off) | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 2.76 [0.39, 19.40] |
| 1.2 12‐16 doses (2 times/week for 8 weeks or 3 times/week for 4 weeks) | 3 | 543 | Risk Ratio (M‐H, Random, 95% CI) | 7.88 [1.09, 56.67] |
| 1.3 12 or 24 doses (3 times/week for 4 weeks on , 4 weeks off, 4 weeks on) | 2 | 505 | Risk Ratio (M‐H, Random, 95% CI) | 8.81 [1.15, 67.32] |
| 1.4 24 doses (3 times/week for 8 weeks) | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.07, 25.08] |
| 1.5 32‐36 doses (2 times/ week for 16 weeks or 3 times/ week for 12 weeks) | 4 | 888 | Risk Ratio (M‐H, Random, 95% CI) | 7.12 [3.06, 16.58] |
| 1.6 40 doses (5 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.03, 17.27] |
| 1.7 48 doses (3 times/ week for 16 weeks) | 3 | 795 | Risk Ratio (M‐H, Random, 95% CI) | 10.90 [3.59, 33.15] |
| 1.8 56 doses (7 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.07, 24.29] |
| 2 Participant complete clearance in immunosuppressed participants Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Participant partial (>75%) clearance Show forest plot | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 9 or 18 doses (3 times/ week for 3 weeks on, 4 weeks off. 3 weeks on) | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 2.41 [0.91, 6.39] |
| 3.2 12‐16 doses (3 times/week for 4 weeks or 2 times/week for 8 weeks) | 2 | 284 | Risk Ratio (M‐H, Random, 95% CI) | 2.86 [1.53, 5.34] |
| 3.3 12 or 24 doses (3 times/week for 4 weeks on, 4 weeks off) | 2 | 505 | Risk Ratio (M‐H, Random, 95% CI) | 6.23 [0.70, 55.10] |
| 3.4 24 doses (3 times/week for 8 weeks) | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [0.25, 62.85] |
| 3.5 32 doses (2 times/week for 16 weeks) | 1 | 436 | Risk Ratio (M‐H, Random, 95% CI) | 5.02 [3.44, 7.33] |
| 3.6 40 doses (5 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 3.35 [0.21, 53.51] |
| 3.7 48 doses (3 times/ week for 16 weeks) | 2 | 778 | Risk Ratio (M‐H, Random, 95% CI) | 8.46 [2.29, 31.16] |
| 3.8 56 doses (7 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 5.94 [0.39, 90.34] |
| 4 Participant partial (>75%) clearance in immunosuppressed participants Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 6 Withdrawal due to adverse events Show forest plot | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 12‐16 doses (2 times/week for 8 weeks or 3 times/week for 4 weeks) | 1 | 38 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.03, 16.74] |
| 6.2 12 or 24 doses (3 times/week for 4 weeks on , 4 weeks off, 4 weeks on) | 2 | 505 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [0.31, 8.23] |
| 6.3 24 doses (3 times/week for 8 weeks) | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.07, 25.08] |
| 6.4 32‐36 doses (2 times/ week for 16 weeks or 3 times/ week for 12 weeks) | 3 | 858 | Risk Ratio (M‐H, Random, 95% CI) | 2.29 [0.80, 6.57] |
| 6.5 40 doses (5 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 4.90 [0.32, 75.60] |
| 6.6 48 doses (3 times/ week for 16 weeks) | 2 | 778 | Risk Ratio (M‐H, Random, 95% CI) | 2.69 [1.48, 4.90] |
| 6.7 56 doses (7 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 5.42 [0.35, 82.97] |
| 7 Withdrawal due to adverse events in immunosuppressed participants Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 48 doses (3 times/ week for 16 weeks) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: body as a whole: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: body as a whole: "flu" or "cold" Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Minor adverse events excluding skin irritation: digestive: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Minor adverse events excluding skin irritation: digestive: nausea Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Cosmetic outcome: decrease in roughness/dryness/scaliness of the skin Show forest plot | 2 | 683 | Risk Ratio (M‐H, Random, 95% CI) | 3.23 [1.86, 5.58] |
| 13.1 32‐36 doses | 1 | 415 | Risk Ratio (M‐H, Random, 95% CI) | 2.54 [1.91, 3.37] |
| 13.2 48 doses | 1 | 268 | Risk Ratio (M‐H, Random, 95% CI) | 4.43 [2.69, 7.30] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 12 | 3087 | Risk Ratio (M‐H, Random, 95% CI) | 6.73 [5.03, 9.00] |
| 1.1 5.0% imiquimod | 9 | 1871 | Risk Ratio (M‐H, Random, 95% CI) | 7.70 [4.63, 12.79] |
| 1.2 3.75% imiquimod | 3 | 730 | Risk Ratio (M‐H, Random, 95% CI) | 6.45 [3.87, 10.73] |
| 1.3 2.5% imiquimod | 2 | 486 | Risk Ratio (M‐H, Random, 95% CI) | 4.49 [2.40, 8.39] |
| 2 Participant partial (>75%) clearance Show forest plot | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 5.0% imiquimod | 4 | 1363 | Risk Ratio (M‐H, Random, 95% CI) | 6.71 [3.89, 11.57] |
| 2.2 3.75% imiquimod | 2 | 484 | Risk Ratio (M‐H, Random, 95% CI) | 3.11 [2.08, 4.66] |
| 2.3 2.5% imiquimod | 2 | 485 | Risk Ratio (M‐H, Random, 95% CI) | 2.48 [1.67, 3.68] |
| 3 Mean percentage of reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3.1 3.75% imiquimod | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Minor adverse events excluding skin irritation: body as a whole: 'flu" or "cold" Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 5.0% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Withdrawal due to adverse events Show forest plot | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 5.0% imiquimod | 8 | 2290 | Risk Ratio (M‐H, Random, 95% CI) | 2.59 [1.59, 4.23] |
| 5.2 3.75% imiquimod | 2 | 483 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.22, 3.93] |
| 5.3 2.5% imiquimod | 2 | 486 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.09, 2.70] |
| 6 Skin irritation Show forest plot | 5 | 1678 | Risk Ratio (M‐H, Random, 95% CI) | 3.93 [1.56, 9.88] |
| 6.1 5.0% imiquimod | 3 | 708 | Risk Ratio (M‐H, Random, 95% CI) | 3.68 [0.86, 15.74] |
| 6.2 3.75% imiquimod | 2 | 484 | Risk Ratio (M‐H, Random, 95% CI) | 4.86 [0.92, 25.83] |
| 6.3 2.5% imiquimod | 2 | 486 | Risk Ratio (M‐H, Random, 95% CI) | 3.45 [0.63, 18.97] |
| 7 Minor adverse events excluding skin irritation: body as a whole: pyrexia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: hemic and lymphatic: lymphadenopathy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Minor adverse events excluding skin irritation: nervous system: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1 5.0% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Minor adverse events excluding skin irritation: respiratory: cough Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Minor adverse events excluding skin irritation: respiratory: sinusitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Minor adverse events excluding skin irritation: respiratory: upper respiratory tract infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15 Minor adverse events excluding skin irritation: urogenital: urinary tract infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16 Cosmetic outcome: Participant's significantly or much improved cosmetic outcome assessed by investigator Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 3.75% imiquimod | 2 | 470 | Risk Ratio (M‐H, Random, 95% CI) | 2.71 [2.05, 3.58] |
| 16.2 2.5% imiquimod | 2 | 475 | Risk Ratio (M‐H, Random, 95% CI) | 2.25 [1.62, 3.14] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 2 times/week | 4 | 890 | Risk Ratio (M‐H, Random, 95% CI) | 5.36 [2.03, 14.16] |
| 1.2 3 times/week | 6 | 1336 | Risk Ratio (M‐H, Random, 95% CI) | 8.38 [3.79, 18.52] |
| 1.3 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.03, 17.27] |
| 1.4 7 times/week | 4 | 1253 | Risk Ratio (M‐H, Random, 95% CI) | 5.39 [3.65, 7.98] |
| 2 Participant partial (>75%) clearance Show forest plot | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 2 times/week | 2 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 4.99 [3.43, 7.26] |
| 2.2 3 times/week | 3 | 814 | Risk Ratio (M‐H, Random, 95% CI) | 7.65 [2.51, 23.32] |
| 2.3 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 3.35 [0.21, 53.51] |
| 2.4 7 times/week | 3 | 1006 | Risk Ratio (M‐H, Random, 95% CI) | 2.95 [1.99, 4.37] |
| 3 Withdrawal due to adverse events Show forest plot | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 2 times/week | 4 | 896 | Risk Ratio (M‐H, Random, 95% CI) | 2.04 [0.75, 5.53] |
| 3.2 3 times/week | 5 | 1319 | Risk Ratio (M‐H, Random, 95% CI) | 2.47 [1.42, 4.30] |
| 3.3 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 4.90 [0.32, 75.60] |
| 3.4 7 times/week | 3 | 1006 | Risk Ratio (M‐H, Random, 95% CI) | 1.55 [0.33, 7.18] |
| 4 Minor adverse events excluding skin irritation:body as a whole: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 2 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 3 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 5 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 7 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Minor adverse events excluding skin irritation: body as a whole:"flu" or "cold" Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 2 times/week | 1 | 38 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.03, 16.74] |
| 5.2 3 times/week | 2 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 2.67 [0.36, 19.83] |
| 5.3 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.03, 17.27] |
| 5.4 7 times/week | 2 | 527 | Risk Ratio (M‐H, Random, 95% CI) | 5.20 [0.28, 95.18] |
| 6 Minor adverse events excluding skin irritation: digestive: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 3 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 5 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 7 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Minor adverse events excluding skin irritation: digestive: nausea Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 3 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 5 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 7 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 3 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 7 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 3 times/week | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | 3.77 [0.23, 63.05] |
| 9.2 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.81 [0.10, 31.53] |
| 9.3 7 times/week | 2 | 527 | Risk Ratio (M‐H, Random, 95% CI) | 4.48 [0.86, 23.31] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2 Cosmetic outcome: Investigator cosmetic outcome "excellent" Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Cosmetic outcome: normal skin surface Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance of target lesions Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participant complete clearance of all lesions Show forest plot | 2 | 456 | Risk Ratio (M‐H, Random, 95% CI) | 4.50 [2.61, 7.74] |
| 3 Participant partial (>75%) clearance of target lesions Show forest plot | 2 | 280 | Risk Ratio (M‐H, Random, 95% CI) | 2.88 [1.81, 4.58] |
| 4 Cosmetic outcomes: changes in pigmentation Show forest plot | 3 | 514 | Risk Ratio (M‐H, Random, 95% CI) | 3.36 [0.63, 17.80] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance of target lesions Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 0.025% 3 days | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 0.05% 2‐3 days | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant complete clearance of all lesions Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 0.025% 3 days | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [1.03, 15.55] |
| 2.2 0.05% 2‐3 days | 2 | 386 | Risk Ratio (M‐H, Random, 95% CI) | 5.14 [2.75, 9.62] |
| 3 Participant partial (>75%) clearance of target lesions Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 0.0025% 2 days | 1 | 19 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.21, 8.41] |
| 3.2 0.01% 2 days | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.06, 4.23] |
| 3.3 0.025% 3 days | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 2.8 [1.13, 6.96] |
| 3.4 0.05% 2‐3 days | 2 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 3.34 [1.84, 6.04] |
| 4 Cosmetic outcomes: changes in pigmentation Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 0.01% 2 days | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [0.08, 25.88] |
| 4.2 0.05% 2 days | 2 | 253 | Risk Ratio (M‐H, Random, 95% CI) | 4.86 [0.48, 49.39] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance of target lesions Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 0.05% 2 days | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 0.05% 3 days | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant complete clearance of all lesions Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 0.05% 2 days | 2 | 319 | Risk Ratio (M‐H, Random, 95% CI) | 4.32 [2.30, 8.11] |
| 2.2 0.05% 3 days | 1 | 87 | Risk Ratio (M‐H, Random, 95% CI) | 4.08 [1.59, 10.47] |
| 3 Participant partial (>75%) clearance of target lesions Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 0.05% 2 days | 2 | 104 | Risk Ratio (M‐H, Random, 95% CI) | 2.65 [1.41, 5.00] |
| 3.2 0.05% 3 days | 1 | 87 | Risk Ratio (M‐H, Random, 95% CI) | 3.23 [1.66, 6.29] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Investigator global improvement indices‐completely cleared Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Upper extremities | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Mean reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2.1 Face | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Scalp | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Upper extremities | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Severe‐Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Global improvement indices‐cured Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 1.1 At 3 months | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 6 months | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation:skin and subcutaneous disorders: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: nervous system: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean change in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction of lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesion counts Show forest plot | Other data | No numeric data | ||
| 2 Mean percentage of reduction in lesion counts Show forest plot | Other data | No numeric data | ||
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Minor adverse events excluding skin irritation: dermatology: acne Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Minor adverse events excluding skin irritation: dermatology:crustea Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.4 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Minor adverse events excluding skin irritation: dermatology: infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: dermatology: milia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: dermatology:pain Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Cosmetic outcomes: changes in pigmentation (hypo) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Cosmetic outcomes: scarring Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Cosmetic outcomes: improvement in photoageing score Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Cosmetic outcomes: excellent global cosmetic outcome Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Cosmetic outcomes: better skin appearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Cosmetic outcomes: excellent global cosmetic outcome Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Cosmetic outcomes: better skin appearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction in lesion counts Show forest plot | Other data | No numeric data | ||
| 2 Withdrawal due to adverse events Show forest plot | 2 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.16, 7.16] |
| 3 Cosmetic outcomes: excellent or good cosmetic outcomes by investigator Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4 Cosmetic outcomes: excellent or good cosmetic outcomes by participant Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 During treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 One day after treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance [1 treatment] Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Blue light | 1 | 243 | Risk Ratio (M‐H, Random, 95% CI) | 6.22 [2.88, 13.43] |
| 1.2 Red light | 3 | 422 | Risk Ratio (M‐H, Random, 95% CI) | 5.94 [3.35, 10.54] |
| 2 Participant complete clearance [1 or 2 treatments] Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Red light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Participant complete clearance [1 or 2 treatments] by anatomical location Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Participant partial (> 75%) clearance [1 treatment] Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Participant partial (>75%) clearance[1 or 2 treatments] Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Participant partial (>75%) clearance [1 or 2 treatment] by anatomical location Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 Red light‐during illumination | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Red light‐after treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: body as a whole: injury Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: cardiovascular: hypertension Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Minor adverse events excluding skin irritation: dermatology: skin discolouration Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.1 Red light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Minor adverse events excluding skin irritation: dermatology: skin hypertrophy Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Cosmetic outcome: very good or good general cosmetic outcome Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Cosmetic outcome: improvement in global response Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Cosmetic outcome: improvement in tactile roughness Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Cosmetic outcome: improvement in mottled hyperpigmentation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance at 4 weeks Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 0.5h versus 1.0h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 0.5h versus 2 h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 1h versus 2h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 1h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 2h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant complete clearance at 8 weeks Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 0.5h versus 1.0h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 0.5h versus 2 h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 1h versus 2h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.5 1h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.6 2h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Minor adverse events excluding skin irritation: metabolic and nutritional disorders: elevated alanine transaminase (ALT) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 0.5h versus 1.0h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 0.5h versus 2 h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Minor adverse events excluding skin irritation: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 0.5h versus 1h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 0.5h versus 2h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 1h versus 2h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 1h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 2h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Minor adverse events excluding skin irritation: other: epistaxis (nose bleeding) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 1h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 2h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Cosmetic outcome: improvement in global response Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Cosmetic outcome: improvement in tactile roughness Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Cosmetic outcome: improvement in mottled hyperpigmentation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Skin irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 During treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 One day after treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 5 | 482 | Risk Ratio (M‐H, Random, 95% CI) | 4.46 [3.17, 6.28] |
| 2 Participant partial (>75%) clearance Show forest plot | 2 | 191 | Risk Ratio (M‐H, Random, 95% CI) | 3.28 [1.73, 6.23] |
| 3 Withdrawal due to adverse events Show forest plot | 2 | 191 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.23, 17.74] |
| 4 Minor adverse event: nervous system: headache Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Cosmetic outcome: hyperpigmentation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Mean percentage of reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Withdrawal due to adverse events Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Withdrawal due to adverse events Show forest plot | 2 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.14, 6.36] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean reduction in lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean percentage of reduction in lesions Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participant partial (>75%) clearance Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance at 6 months Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 1 cycle (1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 2 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 3 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Mean reduction in lesion counts at 6 months Show forest plot | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 1 cycle (1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 2 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 3 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Mean percentage of reduction in lesion counts at 6 months Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 1 cycle (1 week topical, cryosurgery at 4 weeks, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Minor adverse events excluding skin irritation: body as a whole: allergic reaction Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: dermatology: hyperesthesia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: dermatology: skin discoloration Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation: dermatology: vesiculobullous rash Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: digestive: cheilitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: special senses: conjunctivitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: special senses: eye irritation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance at 6 months Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 1 cycle (1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 2 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 3 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Mean reduction in lesion counts at 6 months Show forest plot | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 1 cycle (1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 2 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 3 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Mean percentage of reduction in lesion counts at 6 months Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 1 cycle (1 week topical, cryosurgery at 4 weeks, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance of all lesions Show forest plot | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.05, 0.73] |
| 1.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.11, 1.42] |
| 1.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.04, 0.30] |
| 2 Participant complete clearance of target (cryotherapy treated) lesions Show forest plot | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.36, 1.04] |
| 2.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.47, 1.60] |
| 2.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.37, 0.68] |
| 3 Participant complete clearance of subclinical lesions Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Mean percentage of reduction in all lesion counts Show forest plot | 2 | 301 | Mean Difference (IV, Random, 95% CI) | ‐23.69 [‐46.03, ‐1.34] |
| 4.1 5% imquimod | 1 | 54 | Mean Difference (IV, Random, 95% CI) | ‐11.20 [‐26.53, 4.13] |
| 4.2 3.75% imiquimod | 1 | 247 | Mean Difference (IV, Random, 95% CI) | ‐34.10 [‐41.38, ‐26.82] |
| 5 Mean percentage of reduction in target (cryotherapy treated) lesion counts Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 5.1 3.75% imiquimod | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Withdrawal due to adverse events Show forest plot | 2 | 312 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.28, 3.07] |
| 6.1 5% imiquimod | 1 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 2.91 [0.12, 68.95] |
| 6.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.21, 2.79] |
| 7 Skin irritation Show forest plot | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.10, 1.54] |
| 7.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.20, 2.01] |
| 7.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.02, 1.19] |
| 8 Minor adverse events excluding skin irritation: body as a whole: fatigue Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: digestive: nausea Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: respiratory: upper respiratory tract infection Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: respiratory: bronchitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation: respiratory: sinusitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14 Minor adverse events excluding skin irritation: special senses: conjunctivitis Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15 Cosmetic outcomes: Improved global photoageing score Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16 Cosmetic outcomes: Improved fine lines Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17 Cosmetic outcomes: Improved tactile roughness Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18 Cosmetic outcomes: Improved mottled pigmentation Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 19 Cosmetic outcomes: Improved sallowness Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 20 Cosmetic outcomes: cosmetic appearance score Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 20.1 Investigator | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 20.2 Participant | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participant complete clearance of all lesions Show forest plot | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 5.04 [1.37, 18.51] |
| 1.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 2.48 [0.70, 8.76] |
| 1.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 9.12 [3.36, 24.79] |
| 2 Mean percentage of reduction in all lesion counts Show forest plot | 2 | 301 | Mean Difference (IV, Random, 95% CI) | 23.69 [1.34, 46.03] |
| 2.1 5% imquimod | 1 | 54 | Mean Difference (IV, Random, 95% CI) | 11.20 [‐4.13, 26.53] |
| 2.2 3.75% imiquimod | 1 | 247 | Mean Difference (IV, Random, 95% CI) | 34.10 [26.82, 41.38] |
| 3 Withdrawal due to adverse events Show forest plot | 2 | 312 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.33, 3.56] |
| 3.1 5% imiquimod | 1 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.13] |
| 3.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.36, 4.73] |
| 4 Skin irritation Show forest plot | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 2.55 [0.65, 10.04] |
| 4.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [0.50, 5.13] |
| 4.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 6.72 [0.84, 53.83] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Global Improvement Indices (‐2 to 4) at 6 months Show forest plot | Other data | No numeric data | ||
| 2 Mean reduction of lesion counts Show forest plot | Other data | No numeric data | ||