Intervenciones médicas para la rinosinusitis crónica en la fibrosis quística
Resumen
Antecedentes
La rinosinusitis crónica se produce con frecuencia en personas con fibrosis quística. Existen varias intervenciones médicas disponibles para el tratamiento de la rinosinusitis crónica en personas con fibrosis quística; por ejemplo, diferentes concentraciones de irrigaciones nasales de suero fisiológico, corticosteroides tópicos u orales, antibióticos (incluidos los antibióticos nebulizados), dornasa alfa y moduladores del regulador de la conductancia transmembrana de la fibrosis quística (CFTR, por sus siglas en inglés) (como lumacaftor, ivacaftor o tezacaftor). Sin embargo, no se conoce la eficacia de estas intervenciones.
Esta es una actualización de una revisión publicada anteriormente.
Objetivos
El objetivo de esta revisión es comparar los efectos de diferentes intervenciones médicas en personas diagnosticadas con fibrosis quística y rinosinusitis crónica.
Métodos de búsqueda
Se realizaron búsquedas en el registro de ensayos del Grupo Cochrane de Fibrosis quística y trastornos genéticos (Cochrane Cystic Fibrosis and Genetic Disorders Group), compilado a partir de búsquedas de bases de datos electrónicas y búsquedas manuales de revistas y libros de resúmenes de conferencias. Fecha de la última búsqueda en el registro de ensayos: 9 de septiembre de 2021.
También se realizaron búsquedas en bases de datos de ensayos en curso, otras bases de datos médicas y en las listas de referencias de artículos y revisiones pertinentes. Fecha de las últimas búsquedas adicionales: 22 de noviembre de 2021.
Criterios de selección
Ensayos aleatorizados y cuasialeatorizados de diferentes intervenciones médicas comparadas entre sí o con ninguna intervención o placebo.
Obtención y análisis de los datos
Dos autores de la revisión evaluaron de forma independiente los ensayos identificados para su posible inclusión en la revisión. Se planificó realizar la obtención y el análisis de los datos de acuerdo con los métodos Cochrane y evaluar de forma independiente la calidad de la evidencia para cada desenlace mediante las guías de GRADE.
Resultados principales
No se identificaron ensayos que cumplieran con los criterios de inclusión predefinidos. Las búsquedas más recientes identificaron 44 referencias nuevas, aunque ninguna fue elegible para inclusión en la versión actual de esta revisión; 12 estudios se han marcado como excluidos y uno como en curso.
Conclusiones de los autores
No se identificaron ensayos elegibles que evaluaran intervenciones médicas en personas con fibrosis quística y rinosinusitis crónica. Se necesitan ensayos de calidad alta que evalúen la eficacia de las diferentes opciones terapéuticas detalladas anteriormente para el control de la rinosinusitis crónica, la prevención de las exacerbaciones pulmonares y la mejoría de la calidad de vida en personas con fibrosis quística.
PICO
Resumen en términos sencillos
Intervenciones médicas para la inflamación crónica de la nariz y los senos paranasales en la fibrosis quística
Pregunta de la revisión
¿Cuáles son los efectos de las intervenciones médicas para la rinosinusitis crónica en personas con fibrosis quística?
Antecedentes
La rinosinusitis crónica es una infección e inflamación prolongada de la cavidad nasal y de los espacios llenos de aire alrededor de los ojos y la nariz. La fibrosis quística es una afección genética que da lugar a que las secreciones del cuerpo sean espesas y, por lo tanto, a su estancamiento. Cuando lo anterior ocurre dentro y alrededor de la nariz, causa rinosinusitis crónica. La mejoría en la atención de las personas con fibrosis quística ha dado lugar a que vivan más tiempo, lo que aumenta la posibilidad de desarrollar rinosinusitis crónica. Las intervenciones tempranas y efectivas con antibióticos, corticoides, medicamentos que diluyen el moco (p. ej., dornasa alfa) y medicamentos para mejorar el funcionamiento del canal de la membrana celular (moduladores del CFTR) pueden ayudar a mejorar la calidad de vida y prevenir la aparición de enfermedades de las vías respiratorias bajas. Actualmente, no existen guías basadas en ensayos para saber cómo tratar mejor la rinosinusitis crónica en personas con fibrosis quística.
Fecha de la búsqueda
La evidencia está actualizada hasta el: 9 de septiembre de 2021.
Características de los estudios
No se encontraron ensayos en la búsqueda que cumplieran con los criterios de inclusión. En la presente revisión se han incluido 12 ensayos como excluidos y un ensayo como en curso.
Resultados clave
Aunque la rinosinusitis crónica es frecuente en las personas con fibrosis quística, no hay evidencia suficiente disponible sobre su tratamiento. Esta revisión destaca la necesidad de ensayos bien diseñados que evalúen qué tratamiento es mejor para controlar la rinosinusitis crónica, para prevenir las enfermedades de las vías respiratorias bajas y para mejorar la calidad de vida en las personas con fibrosis quística.
Authors' conclusions
Background
Description of the condition
Cystic fibrosis (CF) is a genetic disorder mainly affecting the nasal cavities, paranasal sinuses (air spaces around eyes and nose), lungs and digestive system. It is an autosomal recessively inherited condition which means that children of carrier parents have a 25% chance of having the disease and 50% chance of being a carrier. Carriers transmit the disease to the next generation without suffering from the disease themselves. It is a relatively common debilitating disorder with its incidence differing across different continents. The highest incidence is noted in Europe (ranging between 1 in 2000 to 1 in 3000 live births) and the lowest in Asia (ranging between 1 in 40,000 to 1 in 100,000 live births) (WHO Human Genetics Programme 2004).
The condition is caused by mutations in a gene on chromosome 7 that encodes the CF transmembrane conductance regulator (CFTR) protein (Collins 1992; Drumm 1993). CFTR is an anion (a negatively‐charged molecule, e.g. chloride (Cl‐) and bicarbonate (HCO3‐)) channel usually present on the plasma membrane of epithelial cells lining the airway, pancreas, liver, intestines, sweat ducts, and epididymis (a tube located at the back of the testicles that stores and carries sperm) (Guggino 2004). The normal function of the anion channel promotes anion secretion to the airway surface (or into the extracellular lumen of the intestine and glandular ducts), which impacts the balance of water and electrolyte transport (Johnson 1995; Rowe 2005).
The most common mutation is F508del, although more than 2000 different potentially disease‐causing CFTR gene mutations have been listed in the Cystic Fibrosis Mutation Database (Cystic Fibrosis Mutation Database 2011). Six different classes of mutations have been identified, based on the way the defective CFTR protein is produced. Class I mutations lead to defective protein production, which in turn leads to the premature termination of the mRNA and a complete absence of CFTR protein in the cell. Class II mutations are mutations in protein processing and trafficking and include the most common mutation ‐ F508del. Class III mutations show defective regulation of CFTR channel gating (e.g. G551D). Class IV mutations reduce the duration of channel opening and also the rate of ion flow. Class V mutations have reduced quantities of normal CFTR protein due to abnormalities of mRNA splicing. Finally, class VI mutations have increased channel turnover at the cell surface due to instability, which results in decreased amounts of functional protein. Due to the difference in the mechanisms involved in the disease process, the amount of healthy CFTR produced varies among individuals.
In a normal airway, hair‐like projections from epithelial cells called cilia beat to move the overlying mucus out of the sinuses. The cilia are covered with 'thin' airway surface liquid (ASL) which is made up of a mucus layer overlying a periciliary fluid layer which is in direct contact with the epithelial cells. Normally, oxygen levels are equal throughout the depths of the ASL. In the airways of people with CF, defective Cl‐ ion transport leads to the excessive absorption of sodium and water and consequently the dehydration of the ASL, resulting in a thin periciliary layer (Rowe 2005). Cilia are not able to function correctly in a thin periciliary layer and mucociliary clearance (the transport of mucus by the cilia towards the sinus openings) is impaired. Mucus is still produced and secreted forming a thick layer of static mucus which the cilia are not able to clear adequately and the thick secretions block the sinus openings. Ventilation of the sinuses decreases and hypoxia (low levels of oxygen in tissue) occurs; this hypoxia impairs mucociliary clearance even more by inhibiting Cl‐ transport and further dehydrating ASL (Blount 2011).
Pseudomonas aeruginosa, Staphylococcus aureus, Haemophilus influenzae, Burkholderia cepacia, Achromobacter xylosoxidans and Stenotrophomonas maltophilia are bacteria which commonly cause infection and inflammation (Brook 2016; Ramsey 1992). These bacteria further impair ciliary motion or co‐ordination (or both), exacerbating impaired mucociliary clearance (Wilson 1987). Inflammation increases mucus secretion by mucosa (Mainz 2012), promotes the formation of neutrophil‐predominant nasal polyps (neutrophils are a type of blood cell that increase during infection) (Ryan 2008) and propagates the cycle of infection and inflammation.
Oxygen levels are highest near the surface of this mucus layer and lowest in its depths near the mucosa. P aeruginosa deposited on the surface penetrates into the hypoxic zones of mucus, adapts to the environment and forms microcolonies (Kim 2015). Hypoxic mucus with P aeruginosa microcolonies resists the natural defence mechanism in the lungs and sinuses, resulting in chronic airway infection.
A single type of mucosa (pseudo‐stratified ciliated columnar epithelium) lines the nasal cavity through the sinuses, larynx, and trachea to the distal lungs (Krouse 2007). The pathophysiology of mucostasis (a mass of thick mucus over the mucosa which cannot be pushed away by cilia), infection and inflammation is the same for upper and lower airway. The 'unified airway model' suggests that infection in the upper airway easily spreads to the lower airway and vice versa (Chang 2014; Illing 2014; Tos 1983). Therefore, chronic rhinosinusitis (CRS) can lead to pulmonary exacerbations, a major cause of death in people with CF. Mucostasis with microcolonies of bacteria in the sinuses form a reservoir of infection. It has been shown that in people who have undergone surgery to open up sinuses and help drain the collected mucus, there is reduced pulmonary disease (Chang 2014; Holzmann 2004; Jones 1993; Rosbe 2001).
According to the European Position Paper on rhinosinusitis and nasal polyps, CRS is diagnosed when a person presents having lived for 12 weeks with one of: nasal obstruction or congestion or rhinorrhea, or both; and one of facial pain or pressure or reduction in olfaction (sense of smell), or both; and has either positive endoscopic findings of nasal polyps or mucopurulent discharge (yellow‐green sticky secretions of infected mucus) or mucosal edema or radiological findings of disease on a computer tomography (CT) scan (or both) (Fokkens 2012).
Description of the intervention
Medical intervention is one of the first steps in managing CRS in people with CF and can include oral and topical treatments which are usually given in combination for a long duration. Available treatment options include nasal saline irrigation, oral and topical antibiotics, oral and topical steroids, anti‐inflammatory agents (such as ibuprofen), dornase alfa and CFTR modulators.
Nasal saline irrigation
Nasal saline irrigation involves washing the nasal cavity with saline delivered via squeeze bottles and netti pots. It helps to soften dried mucous crusts and clears them out of the sinonasal cavity. It is usually used in two concentrations: hypertonic (more concentrated than serum, ranging from 3% to 7%); and isotonic (as concentrated as our serum, i.e. 0.9%, such as NasaMist®). Nasal saline irrigation can also include low‐volume treatments like Sterimar® (natural seawater nasal spray).
Corticosteroids
Corticosteroids break the infection‐inflammation cycle, preventing the progression of CRS in people with CF. They are strong immunosuppressants and decrease the inflammatory response to the retained mucus or bacterial microcolonies. Oral corticosteroids are given as tablets, usually in the morning to match the circadian rhythm (bodily rhythm of morning and night). Topical corticosteroids are usually administered as nasal sprays inhaled in the morning. Topical application increases the local delivery of the drug and prevents the potential side effects that could arise by systemic administration.
Antibiotics
Antibiotics are traditionally given as oral tablets or intravenous drips. In CF, nebulized tobramycin and colistin have been used to prevent the formation of bacterial micro‐colonies in the mucus lining the sinuses.
Dornase alfa
Dornase alfa (recombinant human DNase) is a relatively newly developed medicine which is administered topically. It breaks long gene chains present in mucus and decreases its stickiness. It enhances the clearance of mucus from the sinonasal cavities.
CFTR modulators
CFTR modulators (e.g. ivacaftor, lumacaftor and tezacaftor) act on the cause of CF, the defective CFTR protein. They increase the efficacy of these proteins and thereby increase their activity partially. Ivacaftor was approved for individuals with G551D mutations, but has been expanded for use in people with other Class III and residual function mutations (Guigui 2016). Combination treatment of lumacaftor and ivacaftor has been used in children with homozygous F508del‐CFTR with good results (Ratjen 2017). Tezacaftor is a newly approved CFTR modulator widely studied for its efficacy in different CFTR mutations including non‐F508del mutations of CFTR (Sala 2018).
Of these medical interventions, many interventions including saline irrigations, antibiotics and steroids are used empirically and have now become the standard of care.
How the intervention might work
Nasal saline irrigation
Nasal saline irrigation physically removes viscous mucus that impairs the clearance of debris and bacteria. Hypertonic saline could have the additional benefit of creating an osmotic gradient, where water is drawn into the mucus thus decreasing its viscosity. As water transfers from inflamed mucosa to the airway surface, edematous (swollen with fluid) mucosa shrink, thus acting as a decongestant (Kang 2015). Although hypertonic saline increases the osmotic gradient further and is the preferred saline in people with CF, it has been recognized to decrease the function of cilia (ciliostasis) (Boek 1999).
Corticosteroids
Short courses of oral steroids in tapering doses are sometimes given with antibiotics in initial treatment, but their use is controversial. There is a risk of pulmonary exacerbation with the use of oral steroids in people with CF, although long‐term oral steroids have been shown to slow the progression of lung disease, decrease hospitalization rates for respiratory exacerbations and improve quality of life with no effect on sino‐nasal symptoms (Cheng 2015). The short‐term use of oral steroids has not been analyzed.
Topical corticosteroids are commonly prescribed and have the advantage of enhanced local effects without systemic complications. Owing to their anti‐inflammatory properties, topical corticosteroids have been used to decrease the burden associated with nasal polyps (Costantini 1990; Hadfield 2000). As inflammation decreases, edema improves and paranasal sinuses remain open longer, allowing secretions to clear.
Antibiotics
Early aggressive anti‐pseudomonal antibiotic therapy has been advocated to delay the onset of chronic P aeruginosa infection (Doring 2000; Langton Hewer 2017). The duration of antibiotic use for eradication is variable and depends on patient selection criteria, the source of respiratory secretion (upper versus lower airway) and the type of therapy (Gibson 2003). Ratjen reported a 12‐month pathogen‐free period after the use of an inhaled antibiotic (Ratjen 2001).
Anti‐pseudomonal antibiotics are also used as maintenance therapy. These can include inhaled anti‐pseudomonal antibiotics, oral quinolones and macrolides. Advantages of using an inhaled antibiotic, include a greater amount of drug reaching the nose and sinuses directly and limited systemic absorption and toxicity. Inhaled antibiotics that have been studied in people with CF include tobramycin (Ramsey 1999), colistin (Hodson 2002) and aztreonam (Fernandez 1994). Continuous anti‐staphylococcal antibiotics are not used as they propagate the formation of methicillin‐resistant S aureus, increase the colonisation of P aeruginosa and increase the formation of small colony variants (cell wall‐deficient strains that have increased capability to survive defence mechanisms). Use of these antibiotics are appropriate only when they are given intermittently for respiratory symptoms (Gibson 2003).
Disease exacerbations are usually treated by targeting antibiotic therapy to the results of bacterial and antibiotic sensitivities. Antibiotics such as amoxicillin with clavulinic acid, second and third generation cephalosporins (such as cefuroxime axetil, cefprozil, cefixime, cefpodoxime proxetil and loracarbef) are chosen when culture grows H influenzae. Co‐trimoxazole, doxycycline and minocycline are chosen when the culture grows B cepacia. Aminoglycosides are given once daily in order to achieve a high peak blood concentration to enhance anti‐bacterial activity and a low trough blood concentration to minimize toxic effects to the ears and kidneys.
Dornase alfa
The extensive neutrophil degradation observed in mucus from the CF airway results in the accumulation of long‐chain, extracellular DNA. This increases the viscosity of the mucus, contributing to the disease. Dornase alfa targets the highly viscous mucus which lines the mucus membranes (Shak 1990); it cleaves the DNA and decreases the viscosity, helping in its clearance (Lindig 2013).
CFTR modulators
Ivacaftor and lumacaftor are new drugs designed to target the CFTR protein. Ivacaftor is a CFTR potentiator that improves the open probability of the defective Cl‐ channel in those with at least one copy of the mutant G551D‐CFTR allele (Accurso 2010); this mutation is present in 4% to 5% of people with CF (Kaiser 2012). It has also been used successfully in people with other class III mutations such as G178R, S549N, S549R, G551S, G970R, G1244E, S1251N, S1255P, or G1349D (De Boeck 2014). Lumacaftor is a corrector which improves the folding and trafficking of some protein to the cell surface. It is used with ivacaftor as a combination treatment for people with CF aged 12 years or older with two copies of the F508del mutation (Castellani 2008; Liou 2001). The use of these medications improve the function of the CFTR protein. It increases Cl‐ transport and thus decreases the viscosity of the mucus.
Why it is important to do this review
Due to advancements in treating people with CF, many are surviving to an older age and CRS is common, reaching 100% when examined clinically or radiographically (Liang 2014). The unified airway model suggests that the lining of nose and sinuses is in continuity with that of lower respiratory tract. The bacteria that can be found in the upper airway, can also be found in the lower airway. Therefore, if there is an infection in the upper airway, it will easily spread to the lower airway; this can be life‐threatening in people with CF (Tipirneni 2017).
Medical treatment is the initial management option for people with CRS, and several treatment options are currently available. Given the diversity of treatments, it is important to know which combination of intervention and which method of delivery provides the best quality of life (QoL)for people with CF.
This review will help provide high‐quality information regarding medical interventions for CRS in people with CF to researchers, medical professionals, people with CF, parents and the public. This is an updated version of a previously published review (Karanth 2019).
Objectives
The objective of this review is to compare the effects of different medical interventions in people diagnosed with cystic fibrosis (CF) and chronic rhinosinusitis (CRS).
Methods
Criteria for considering studies for this review
Types of studies
We planned to include randomized controlled trials (RCTs), quasi‐RCTs and cluster‐RCTs. We also planned to include RCTs of cross‐over design if data from the first treatment phase were available. Trials were included only if an adequate follow‐up period (i.e. a minimum of three months) was present.
We excluded trials that were randomized by the side of the nose.
Types of participants
Participants diagnosed with CF by sweat test or genetic testing for CFTR mutations and further diagnosed with CRS according to either the International Consensus Statement on Allergy and Rhinology: Rhinosinusitis (ICAR:RS) (Orlandi 2016) or the European Position Paper on Rhinosinusitis and Nasal Polyps (Fokkens 2012).
CRS was defined as persistent sinonasal inflammation lasting at least 12 weeks, with two or more of the following symptoms: nasal blockage or obstruction or congestion; nasal discharge; facial discomfort; decreased sense of smell; and an objective finding by endoscopic or CT imaging showing nasal polyps or mucopurulent discharge or mucosal edema.
Asymptomatic individuals were excluded from this analysis.
Types of interventions
We planned to include both short courses of treatment (up to 28 days) and long courses of treatment (longer than four weeks).
As the Cochrane Review 'Topical nasal steroids for treating nasal polyposis in people with cystic fibrosis' assesses trials addressing the effect of topical nasal steroids on nasal polyposis in CF (Beer 2015), such trials are excluded from this review. We also excluded trials that evaluated the effect of medical interventions on surgical outcomes; the medical interventions that were considered for this review are listed below.
-
Nasal saline irrigation ‐ isotonic (0.9%) saline, hypertonic saline (3% to 7%)
-
Corticosteroids ‐ oral or intravenous (e.g. prednisolone, hydrocortisone); or topical (e.g. beclomethasone, triamcinalone, budesonide, fluticasone, mometasone, betamethasone)‐ considered on people diagnosed with CF and CRS without nasal polyps
-
Antibiotics ‐ anti‐staphylococcal (e.g. dicloxacillin, cephalexin, amoxicillin with clavulinic acid, macrolides (azithromycin, erythromycin, clarithromycin), nafcicillin and vancomycin (for MRSA)), anti‐pseudomonal (e.g. ciprofloxacin, inhaled tobramycin, inhaled colistin, ceftazidime, combination treatment of ticarcillin, piperacillin, imipenem, meropenem, aztreonam, amikacin), for treating H influenzae (e.g. amoxicillin with clavulinic acid, second and third generation cephalosporin (cefuroxime axetil, cefprozil, cefixime, cefpodoxime proxetil, loracarbef) and for treating B cepacia (e.g. co‐trimoxazole, doxycycline, minocycline)
-
Ibuprofen ‐ high dose (more than 2400 mg/day)
-
Dornase alfa
-
CFTR modulators ‐ ivacaftor, lumacaftor or tezacaftor
Methods of delivery for topical application include nebulizers (jet or ultrasonic), soft mist, dry powder inhaler, pressured metered dose inhaler, nasal drops, nasal spray, squeeze bottles and netti pots.
We planned to compare the interventions listed above to no intervention, to placebo or to another medical intervention or class of medical intervention. We also planned to compare the same medical intervention given at a different dose, for a different duration or via a different route. We planned to include comparisons of combinations of the treatments listed above. Concurrent treatments were allowed if they are used in both treatment arms.
Types of outcome measures
If a trial had been found which measured outcomes other than those mentioned below, the trial would have been included if it had met our inclusion criteria and if the outcome had been measured objectively and was relevant to our review. The inclusion of any such additional outcomes would have been noted as a post hoc change in the 'Difference between review and protocol' section.
Primary outcomes
-
QoL measured by questionnaires (e.g. Rhinosinusitis Outcome Measure‐31 (Tipirneni 2017) and Sinonasal Outcome Test‐22 (SNOT‐22) (Tipirneni 2017) for adults; Sinonasal‐5 for children (Tipirneni 2017))
-
Refractory to treatment ‐ defined by persistent symptoms during intervention or recurrence of symptoms soon after stopping the intervention, requiring higher dose or concentration or more potent intervention to obtain any or sustained benefit as measured by Chronic Sinusitis Survey or visual analogue scale
-
Treatment‐related adverse events
-
mild (an adverse event that does not require treatment, does not require the medication to be stopped, e.g. nausea)
-
moderate (an adverse event that needs treatment, does not require the medication to be stopped; or needs the treatment to be stopped for a short duration, e.g. intractable vomiting)
-
severe (an adverse event that is life‐threatening and requires the medication to be stopped permanently, e.g. allergy to an antibiotic)
-
Secondary outcomes
-
Periods of improved symptoms (as measured by number of days of benefit)
-
Change in nasal endoscopic findings
-
nasal polyps
-
mucopurulent discharge (discharge containing both mucus and pus)
-
mucosal edema or change
-
-
Change in Lund‐Mackay scores (scoring based on nose and sinus appearances as seen in CT scans)
-
Pulmonary function tests (as measured by either a change from baseline, post‐treatment values or both)
-
forced expiratory volume in one second (FEV1)
-
functional vital capacity (FVC)
-
-
Change in nutritional status
-
height (cm) (in children)
-
weight (kg) (in children)
-
body mass index (BMI) (in adults)
-
Results were reported at the end of treatment or at one month (or both), up to three months, three to six months, six to 12 months and over 12 months.
Search methods for identification of studies
We searched for all relevant published and unpublished trials without restrictions on language, year or publication status.
Electronic searches
To identify relevant studies, we searched the Cochrane Cystic Fibrosis and Genetic Disorder Group's Cystic Fibrosis Trials Register using the term: sinusitis.
The Cystic Fibrosis Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of the Cochrane Library), weekly searches of MEDLINE, a search of Embase to 1995 and the prospective handsearching of two journals ‐ Pediatric Pulmonology and the Journal of Cystic Fibrosis. Unpublished work was identified by searching the abstract books of three major CF conferences: the International Cystic Fibrosis Conference; the European Cystic Fibrosis Conference and the North American Cystic Fibrosis Conference. For full details of all searching activities for the register, please see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group's website.
Date of latest search: 09 September 2021.
We searched the following databases on 21 November 2021:
-
Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library (www.cochranelibrary.com/);
-
PubMed (www.ncbi.nlm.nih.gov/pubmed);
-
Embase Ovid (1974 to present).
We also searched the following trials registries on 21 November 2021:
-
ClinicalTrials.gov (www.clinicaltrials.gov/);
-
WHO ICTRP (apps.who.int/trialsearch/Default.aspx);
-
EU Clinical Trials Register (www.clinicaltrialsregister.eu/);
See appendices for the full search strategies (Appendix 1; Appendix 2).
Searching other resources
We checked the bibliographies of included trials and any relevant systematic reviews identified for further references to relevant trials. We contacted trial authors if we deemed this to be necessary.
Data collection and analysis
Selection of studies
Two authors (TK and VK) independently reviewed all identified abstracts and reports retrieved from databases and other sources. If the reference appeared relevant to the review topic, they obtained a full text copy. The same two authors assessed and selected any trials which they find to be relevant according to the review's inclusion and exclusion criteria. We resolved disagreements by discussion with a third author (BAW or LK). Any review author who is an investigator on a trial was not active in the selection process for that trial.
Data extraction and management
The review authors created a data extraction sheet based on the 'Checklist of items to consider in data collection or data extraction' as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). Two review authors (TK and VK) planned to independently extract data from the included trials and resolve any disagreements by discussion with a third review author (BAW or LK). If a review author was an author on a trial, that author would not extract data for the trial.
The review authors planned to include key characteristics of the trial, such as trial design, setting, sample size, population, how outcomes were defined and collected in the trial. We also planned to collect baseline information on prognostic factors or effect modifiers, e.g. baseline symptom scores and Lund‐Mackay scores. We planned to note down separately if participants were selected based on bacterial colonization.
For any trials of multiple interventions, the review authors planned to make pair‐wise summaries. If a common intervention group was present, we planned to split the sample size and mark this on the data extraction sheet.
For the outcomes of interest, the review authors planned to extract the findings of the trials on an available‐case‐analysis basis. We planned to include all participants from the point of randomization, irrespective of the treatment they actually received.
The review authors planned to present all the results for each of the broad comparison groups (i.e. nasal saline irrigation, antibiotics, corticosteroids, etc.) together and planned to undertake subgroup analyses to look at the effects of individual agents to investigate any identified heterogeneity in the results (see below).
The review authors planned to extract the following statistics for each trial and each outcome:
-
for dichotomous data ‐ the number of participants experiencing an event and number of participants assessed at that time point;
-
for continuous data ‐ mean values, standard deviations (SDs) and number of participants in each treatment arm, if available, we planned to report absolute values and change from baseline.
We planned to report results at the end of treatment or at one month (or both), up to three months, three to six months, six to 12 months and over 12 months; we would also report the number of days of benefit both at the end of treatment and at one month.
Assessment of risk of bias in included studies
Two review authors (TK and VK) planned to independently assess the risk of bias in each included trial based on the following six components: sequence generation, allocation concealment, blinding or masking, incomplete outcome data, selective outcome reporting, and other biases. For each of these components, we planned to assign a judgment regarding the risk of bias as high, low or unclear (Higgins 2011b). We planned to make these judgments separately for objectively and subjectively ascertained measures for the domains of blinding and incomplete outcome data. The review authors planned to record assessments in the standard risk of bias table for each included trial and planned to use these assessments in making judgments on overall trial quality while preparing a summary of findings table (see below). When methodological details were unclear, the review authors would attempt to contact the trial authors for clarification. The review authors planned to resolve any differences by discussion.
We will include in the review any trials on which the authors themselves are investigators, if these are relevant to the review question, but will note this fact in the section on 'Declarations of interest'. In such cases, the review author who is also an investigator on the trial, will not make any risk of bias judgments for that trial.
Measures of treatment effect
Dichotomous data
For dichotomous data, (refractory to treatment, presence or absence of abnormalities on nasal endoscopy and need for surgery, adverse events), we planned to present results as summary risk ratios (RR) with 95% confidence intervals (CI).
Continuous data
For continuous data (QoL measurements, pulmonary function tests, change in nutritional status and Lund‐Mackay scores), we planned to calculate the mean difference (MD) between groups with 95% CIs if outcomes were measured in the same way in trials. When trials used different assessment scales, the authors planned to calculate the standardized mean difference (SMD) with 95% CIs.
Unit of analysis issues
Cluster‐randomized trials
We planned to include cluster‐RCTs in this review along with individually randomized trials. We planned to use the methods described in the Cochrane Handbook of Systematic Reviews of Interventions to account for any unit of analysis error (Higgins 2011c). We planned to derive an estimate of the intracluster correlation coefficient (ICC) from the trial, from a similarly designed trial or from a trial of a similar population. If planned to use ICCs from other sources in the review, we planned to report this and conduct a sensitivity analysis to recognize the effect of the variation of the ICCs. If planned to identify both cluster‐RCTs and individually randomized trials with little heterogeneity between their methodology, and further if we considered an interaction between the effect of the intervention and choice of randomization unit is unlikely, we planned to pool the relevant information and combine the results. We planned to acknowledge any heterogeneity that may occur in the unit of randomization and planned to conduct a sensitivity analysis to investigate the effects of this.
Cross‐over trials
We planned to include cross‐over trials if we could perform paired analysis on data according to Elbourne (Elbourne 2002). If not, we planned to include the first phase of a cross‐over trial if an adequate follow‐up period (i.e. a minimum of three months) was available.
Trials with multiple treatment groups
The authors planned to include multi‐arm trials in the review if they can make pair‐wise comparisons on the intervention groups and if when investigated alone, the comparison would meet the review's inclusion criteria. The authors would only analyze interventions or groups meeting the inclusion criteria in this review. The authors planned to assess the risk of bias based on the completeness of available data and absence of selective reporting of comparisons of intervention arms. The authors planned to attempt to overcome any unit of analysis errors by combining groups to create a single pair‐wise comparison.
Dealing with missing data
Whenever possible, we tried to contact original investigators to request missing data. We planned to assess missing data to try and establish if they were missing at random or if they had a particular value. Where possible, we planned to extract data to allow an intention‐to‐treat analysis, where all randomized participants were analyzed in the groups to which they were originally assigned (Higgins 2011c). We planned to calculate discrepancies in the numbers randomized and numbers analyzed in each treatment group and report these as the percentage lost to follow‐up. If more than 10% have been lost to follow‐up for any trial, for dichotomous outcomes, we planned to assign the worst outcome and analyze the impact of this assignment using a sensitivity analysis.
If SDs are found to be missing in a small proportion of trials, we planned to impute these from other trials for which full data are available (Higgins 2011c). We planned to carry out sensitivity analyses to assess the impact of changing the assumptions made. If the majority of trials included in this review were missing SDs, we planned not to impute these values. We planned to not make any assumptions regarding loss to follow‐up for continuous data and they will analyze the results for those who completed the trial.
Assessment of heterogeneity
The review authors planned to assess heterogeneity between trials for the outcomes of interest both visually using a forest plot and using the I² statistic as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). For multiple‐intervention trials, we planned to split the sample size for the common intervention group in order to prevent a unit‐of‐analysis error, to perform investigations of heterogeneity.
The interpretation of I² will be as follows:
-
0% to 40%: might not be important;
-
30% to 60%: may represent moderate heterogeneity;
-
50% to 90%: may represent substantial heterogeneity;
-
75% to 100%: considerable heterogeneity (Higgins 2011b).
Assessment of reporting biases
The review authors planned to use funnel plots to assess publication and reporting bias, if the number of trials permitted this. We planned to measure funnel plot asymmetry with a linear regression approach on the logarithm scale of the RR. If the funnel plot was asymmetrical, we planned to explore alternative causes in addition to publication bias.
Data synthesis
The review authors planned to carry out statistical analysis using the Review Manager software (Review Manager 2014). If we found that trials estimate the same underlying treatment effect, we planned to use a fixed‐effect model to combine the data. Alternatively, if there was a high degree of heterogeneity between the trials' populations and methods, or if substantial statistical heterogeneity was detected (I² lies between 60% and 90%), we planned to use a random‐effects model and determine the average treatment effect to decide if the treatment effect is clinically meaningful. The review authors planned to treat the random‐effects model summary as the average range of possible treatment effects and present the results as the average treatment effect with 95% CIs together with the estimates of I².
Subgroup analysis and investigation of heterogeneity
If data had permitted, we planned to carry out subgroup analyses as follows:
-
age (children ‐ aged up to 18 years versus adults ‐ aged more than 18 years);
-
surgical status of participants (pre‐ and post‐surgery);
-
presence of co‐morbidities (e.g. any other chronic disease such as diabetes and hypothyroidism);
-
effects of each category of a type of medical intervention (e.g. fluticasone and mometasone; of inhaled corticosteroids).
Sensitivity analysis
To ensure the conclusions drawn during the review process were robust and representative of reality, if there were sufficient comparable trials and if we identified methodological differences between trials, we planned to conduct sensitivity analyses excluding those trials with clearly inadequate randomisation, concealment of allocation or blinding (high risk of bias) (Deeks 2011).
We also planned to explore the impact of including trials with high levels of missing data in the overall assessment of treatment effect. As stated above, if more than 10% of participants have been lost to follow‐up for any trial and we had assigned the worst outcome for dichotomous outcomes, we planned to analyze the impact of this assignment using a sensitivity analysis. We also planned to assess the effect of any imputed SDs we had used in the analyses.
Furthermore, if we used ICCs from other sources in the review, we planned to report this and conduct a sensitivity analysis to recognize the effect of the variation of the ICCs. We planned to acknowledge heterogeneity that may occur in the randomization unit and we planned to conduct a sensitivity analysis to investigate the effects of the randomization unit.
Summary of findings and assessment of the certainty of the evidence
The authors planned to create a summary of findings table for each comparison presented and use the GRADE approach to interpret the findings. The authors planned to include in each table the following outcomes of this review:
-
QoL questionnaires;
-
refractoriness to treatment;
-
treatment‐related adverse events;
-
periods of improved symptoms;
-
nasal endoscopic findings;
-
Lund‐Mackay scores; and
-
pulmonary function test values.
For each, we planned to state the population, setting, intervention, and comparison. We planned to assess the quality of the body of evidence by considering the overall risk of bias of the included trials, the directness of the evidence, the inconsistency of the results, the precision of the estimates, and the risk of publication bias (Schünemann 2011a; Schünemann 2011b).
Results
Description of studies
Results of the search
For the 2022 update, a search of the Cochrane Cystic Fibrosis and Genetic Disorders (CFGD) Review Group's Cystic Fibrosis Trials Register identified five new potentially eligible references. Authors repeated the additional searches for trials and in identified a total of 44 new references; of these references, four trials (six references) were retrieved and assessed for inclusion in the review, but all were eventually excluded (Di Cicco 2014; IRCT20201009048976N1; Lee 2021; NCT03145051).
When these were added to the previous search results, a total of 21 references to 11 trials have been identified from the CFGD Group's CF Trials Register; all of these trials were assessed as potentially relevant for inclusion, but later excluded. We identified 20 ongoing trials from the ongoing trial registries; we deemed two to be potentially relevant, but on further inspection one was excluded and one other is listed in the review as ongoing (NCT02888730). No additional relevant trials were identified on searching PubMed, Ovid Embase and the Cochrane Library as described in the appendices (Appendix 2).
A 'Preferred Reporting Items for Systematic Reviews and Meta‐Analysis' (PRISMA) flow diagram depicting search results is shown in Figure 1.
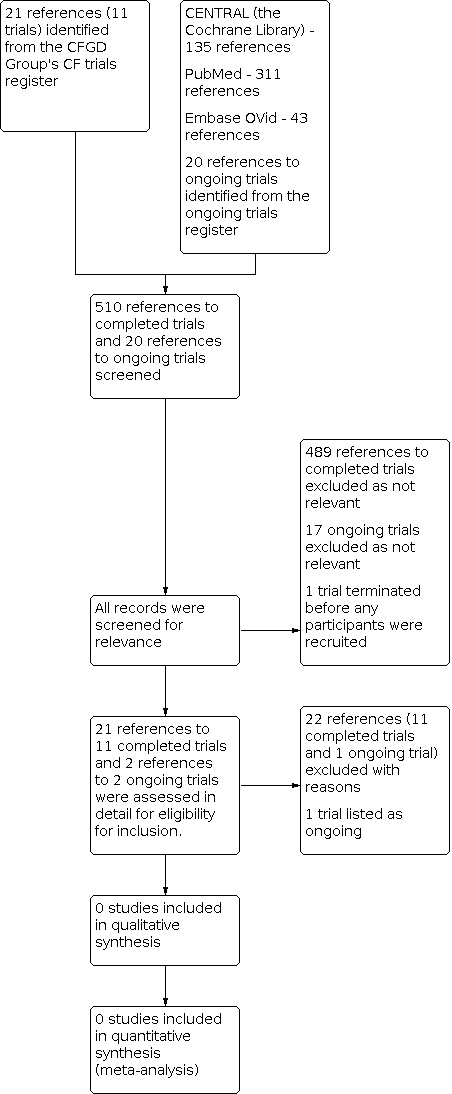
Detail of these trials can be found in the tables (Characteristics of excluded studies; Characteristics of ongoing studies).
Included studies
No trials met the inclusion criteria for this review.
Excluded studies
We excluded 12 trials. The reasons for exclusion were inadequate follow‐up period in six trials (Di Cicco 2014; Mainz 2011; Mainz 2014; Mainz 2016; NCT03145051; NCT03439865), technique of randomisation by the side of the nose in two trials (Wagner 1999; Wagner 2002), usage of an intervention not considered in our protocol in three trials (ACTRN12613000214730; IRCT20201009048976N1; Lee 2021) and assessing the effects of medical intervention on surgical outcomes (NCT00416182).
Ongoing studies
The protocol of the ongoing trial describes a parallel‐assigned triple‐blinded multicentre RCT based in France (NCT02888730). People aged seven years and older with diagnosed CF (by positive sweat test or identification of two CF‐causing mutations) and CRS with bacteria sensitive to tobramycin (as confirmed by an otolaryngologist by endoscopy and culture) will be randomly assigned to receive either tobramycin or 0.9% sodium chloride via nebulizer twice daily for 15 days. Participants will be assessed on day 0, day 15, day 30 and day 90. The trial outcomes include the density of bacteria in sinus ostia, minimum inhibitory concentration of bacteria to antibiotics, symptoms (nasal obstruction, rhinorrhea, mucopurulent secretion, facial pain, dysosmia), endoscopic scores, QoL questionnaires and lung function tests.
Risk of bias in included studies
We did not include any trials and therefore could not assess methodological quality or risk of bias.
Effects of interventions
We did not identify any trials assessing effects of medical intervention for CRS in people with CF.
Discussion
Summary of main results
No trials were identified which met our inclusion criteria for this review.
Although CRS is seen in nearly 100% of people with CF (Liang 2014), adequate trials are not available in order to come to a consensus on the medical management of CRS in people with CF.
Overall completeness and applicability of evidence
We did not identify any trials which met our inclusion criteria. There is a paucity of robust trials that address medical interventions in people diagnosed with CF and CRS. One ongoing trial identified during our searches looks promising and will be assessed fully once completed (NCT02888730). We may have a better consensus on the medical management of CRS in people with CF in future updates of this review.
Quality of the evidence
We did not identify any trials that met our inclusion criteria. Hence, we could not assess the quality of the evidence.
Potential biases in the review process
No bias was encountered. We applied no language restrictions on publication, including during identification of ongoing trials. We used broad search terms in this review which identified a large number of trials. We also conducted searches in other medical databases and online trial registries.
Agreements and disagreements with other studies or reviews
There is a paucity of trials assessing medical interventions for people with CRS and CF.
Three non‐Cochrane systematic reviews have been published on this topic (Liang 2014; Shah 2018; Virgin 2017). All of these reviews evaluate the five completed RCTs that have been listed as excluded in this review. They also have included non‐RCT studies for analysis.
Further investigation of the excluded studies of our Cochrane Review revealed that the five RCTs addressed different aspects of medical management of CRS in people with CF (Mainz 2011; Mainz 2014; Mainz 2016; Wagner 1999; Wagner 2002). In 2011, Mainz conducted a trial to compare the effects of inhaled dornase alfa over normal saline (Mainz 2011). Dornase alfa significantly improved the QoL. However, the trial was conducted on a small sample size of five participants and no difference were found in SNOT‐20 scores, rhinoscopy findings, aerated volume of maxillary sinus as per magnetic resonance imaging and pulmonary function tests. The 2014 publication by Mainz suggested that daily inhalation of dornase alfa produced significant improvement in nasal symptoms, SNOT‐20 scores and pulmonary function tests at the end of 28 days of treatment (Mainz 2014); but similar benefit was not observed with hypertonic saline (6%) (Mainz 2016). Both Wagner trials evaluated the efficacy of adeno‐associated virus vector‐cystic fibrosis trans‐membrane regulator (AAV‐CFTR) in CF maxillary sinus, firstly as a phase I and then as a phase II trial. Both failed to show statistically significant improvement in rate of relapse of sinusitis (Wagner 1999; Wagner 2002).