Vacuna MVA85A para mejorar la actividad del BCG en la prevención de la tuberculosis
Resumen
Antecedentes
La tuberculosis causa más muertes que otras enfermedades infecciosas a nivel mundial. El Bacillus Calmette‐Guérin (BCG) es la única vacuna disponible, aunque la protección es incompleta y variable. El antígeno que expresa el virus Vaccinia Ankara modificado 85 A (MVA85A) es una vacuna de vectores virales sintetizada para prevenir la tuberculosis.
Objetivos
Evaluar y resumir los efectos de la vacuna MVA85A como refuerzo del BCG en seres humanos.
Métodos de búsqueda
Se hicieron búsquedas en el registro especializado del Grupo Cochrane de Enfermedades Infecciosas (Cochrane Infectious Diseases Group), en el Registro Cochrane Central de Ensayos Controlados (Central Register of Controlled Trials) (CENTRAL), MEDLINE (PubMed); Embase (Ovid); y en otras cuatro bases de datos. Se realizaron búsquedas en el ICTRP de la OMS y en ClinicalTrials.gov. Todas las búsquedas se realizaron hasta el 10 de mayo de 2018.
Criterios de selección
Se evaluaron los ensayos controlados aleatorizados de la vacuna MVA85A administrada con el BCG de forma independiente de la edad o el estado del VIH.
Obtención y análisis de los datos
Dos autores de la revisión, de forma independiente, evaluaron la elegibilidad y el riesgo de sesgo de los ensayos y extrajeron y analizaron los datos. El resultado primario fue la tuberculosis activa. Los resultados dicotómicos se resumieron mediante los cocientes de riesgos (RR) y las diferencias de riesgos (DR), con intervalos de confianza (IC) del 95%. Cuando resultó apropiado, se combinaron los datos en un metanálisis. Cuando no fue apropiado un metanálisis, se resumieron los resultados en forma narrativa.
Resultados principales
La búsqueda identificó seis estudios en relación con cuatro ensayos controlados aleatorizados en fase 2, que incorporaron a 3838 participantes. La financiación provino de organismos gubernamentales, instituciones benéficas y donantes filantrópicos. Cinco estudios incorporaron a lactantes, uno de los cuales incorporó a lactantes nacidos de madres con pruebas positivas para el VIH. Un estudio incluyó a adultos que convivían con el VIH. Todos los ensayos incluyeron a autores de la Universidad de Oxford, encargados del desarrollo en laboratorio de la vacuna. Los participantes recibieron la MVA85A por vía intradérmica después del BCG en algunos estudios, y antes del BCG diferido selectivo en los lactantes expuestos al VIH.
El ensayo más grande en 2797 niños africanos se realizó bien, con bajo riesgo de sesgo para la mayoría de los parámetros. El riesgo de sesgo fue incierto para el informe selectivo porque no hubo variables de definición precisa de casos para la tuberculosis activa publicadas antes del análisis de los ensayos.
La MVA85A añadida a la BCG comparada con la BCG sola probablemente no tiene efecto sobre el riesgo de desarrollar tuberculosis confirmada microbiológicamente (RR 0,97; IC del 95%: 0,58 a 1,62; 3439 participantes, dos ensayos; evidencia de certeza moderada), o el riesgo de iniciar el tratamiento de la tuberculosis (RR 1,10; IC del 95%: 0,92 a 1,33; 3687 participantes, tres ensayos; evidencia de certeza moderada). La MVA85A probablemente no tiene efecto sobre el riesgo de desarrollar tuberculosis latente (RR 1,01; IC del 95%: 0,85 a 1,21; 3831 participantes, cuatro ensayos; evidencia de certeza moderada). La vacunación de las personas con MVA85A además de la BCG no causó efectos adversos graves potencialmente mortales (DR 0,00; IC del 95%: ‐0,00 a 0,00; 3692 participantes, tres ensayos; evidencia de alta certeza). La vacunación con MVA85A probablemente se asocia con un mayor riesgo de efectos adversos locales sobre la piel (3187 participantes, tres ensayos; evidencia de certeza moderada), pero no con efectos adversos sistémicos relacionados con la vacunación (144 participantes, un ensayo; evidencia de certeza baja). Este perfil de seguridad es compatible con los estudios de fase 1 que describieron una reacción local superficial y transitoria en el sitio de inyección y síntomas leves de corta duración, como malestar general y fiebre.
Conclusiones de los autores
La MVA85A administrada en inyección intradérmica además del BCG es segura pero no efectiva para reducir el riesgo de tuberculosis.
PICO
Resumen en términos sencillos
Vacuna MVA85A como refuerzo del BCG para la prevención de la tuberculosis
¿Cuál era el objetivo de esta revisión?
El objetivo de esta revisión Cochrane era evaluar la efectividad y la seguridad del uso de la vacuna MVA85A además del BCG en comparación con la administración del BCG solo para la prevención de la tuberculosis.
Mensajes clave
La MVA85A junto con el BCG no demostró beneficios agregados al BCG en la prevención de la tuberculosis.
¿Qué se estudió en la revisión?
La tuberculosis es una enfermedad infecciosa que se transmite por el aire y afecta a los pulmones y a otros órganos. Puede estar activa (cuando el paciente presenta signos y síntomas o pruebas positivas para la tuberculosis) o latente (cuando el paciente ha inhalado previamente la micobacteria pero no presenta signos ni síntomas de la enfermedad). Actualmente, solo existe una vacuna autorizada para prevenir esta enfermedad, llamada BCG. Sin embargo, existe variabilidad en la capacidad de la vacuna BCG para prevenir la tuberculosis en diferentes ámbitos y grupos de pacientes, lo que lleva a que la enfermedad siga siendo un problema en todo el mundo, a pesar de la vacunación en niños. Se investigó la vacuna MVA85A con la esperanza de que al utilizarla junto con el BCG mejoraría la prevención de la tuberculosis.
¿Cuáles son los principales resultados de esta revisión?
Después de examinar los estudios de investigación publicados hasta el 10 de mayo 2018, se incluyeron seis resultados de estudio de cuatro ensayos controlados aleatorizados (ensayos clínicos en que los individuos son asignados al azar a uno de dos o más grupos de tratamiento), que incorporaron a 3838 niños y adultos. Sobre la base de estos estudios, principalmente con niños y adultos que residían en África, la MVA85A agregada al BCG en comparación con el BCG solo probablemente no tiene ningún efecto sobre el riesgo de tuberculosis activa, definida como tuberculosis confirmada microbiológicamente (evidencia de certeza moderada) ni el riesgo de iniciar el tratamiento de la tuberculosis (evidencia de certeza moderada). La MVA85A no tiene ningún efecto sobre el riesgo de tuberculosis latente (evidencia de certeza moderada). La MVA85A no causa efectos secundarios graves potencialmente mortales (evidencia de certeza alta). Hubo más reacciones locales en la piel de pacientes vacunados con MVA85A; sin embargo, no aumentaron los efectos secundarios generales en los pacientes que recibieron la MVA85A.
¿Cómo de actualizada está esta revisión?
Los autores de la revisión buscaron estudios que se habían publicado hasta mayo 2018.
Authors' conclusions
Summary of findings
| MVA85A compared to placebo for preventing tuberculosis | ||||||
| Patient or population: HIV‐positive and ‐negative adults and children | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | Number of participants | Certainty of the evidence | Comments | |
| Risk with placebo | Risk with MVA85A | |||||
| Active tuberculosis: confirmed by culture or Xpert® MTB/RIF longest reported follow‐up | 17 per 1000 | 16 per 1000 | RR 0.97 | 3439 | ⊕⊕⊕⊝ | Vaccinating people with MVA85A in addition to BCG probably made little or no difference to the risk of developing active tuberculosis. |
| Active tuberculosis: started on tuberculosis treatment | 102 per 1000 | 112 per 1000 | RR 1.10 | 3687 | ⊕⊕⊕⊝ | Vaccinating people with MVA85A in addition to BCG probably made little or no difference to the risk of needing to start tuberculosis treatment. |
| Latent tuberculosis | 114 per 1000 | 115 per 1000 | RR 1.01 | 3831 | ⊕⊕⊕⊝ | Vaccinating people with MVA85A in addition to BCG probably made little or no difference to the risk of developing latent tuberculosis. |
| Serious adverse effects | 1 per 1000 | 1 per 1000 | RD 0.00 | 3692 | ⊕⊕⊕⊕ | Vaccinating people with MVA85A in addition to BCG did not cause life‐threatening serious adverse effects. |
| Adverse effects of any severity (local reactions of the skin) | Vaccination with MVA85A was associated with more reactions at the site of the injection.g | — | 3187 | ⊕⊕⊕⊝ | Vaccinating people with MVA85A in addition to BCG probably increased the risk of having an adverse reaction related to vaccination at the site of the injection. | |
| Adverse effects of any severity (systemic symptoms) | Adverse events reported included malaise, lethargy, fever, and vomiting although differences between groups were not significant at a 95% CI level.g | — | 144 (1 RCT) | ⊕⊕⊝⊝ | Vaccinating people with MVA85A in addition to BCG may not have been associated with an increase in adverse effects related to vaccination. | |
| Adverse events of any severity | 808 per 1000 | 849 per 1000 | RR 1.05 | 3836 | ⊕⊕⊕⊕ | Vaccination with MVA85A alone slightly increased the risk of having an adverse event. |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). BCG: Bacillus Calmette‐Guérin; CI: confidence interval; RCT: randomized controlled trial; RD: risk difference; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aNot downgraded for risk of bias. The largest trial was at unclear risk of bias due to selective reporting; however, the outcomes presented were unlikely to be affected by this (Tameris 2013). | ||||||
Background
Description of the condition
Tuberculosis is an infectious disease caused by Mycobacterium tuberculosis. It was estimated that 10 million people developed tuberculosis in 2017. Tuberculosis now ranks first, followed by HIV, as the leading cause of death from an infectious disease worldwide killing an estimated 1.6 million people in 2017, including 300,000 people living with HIV. Over 95% of these people were living in low‐ and middle‐income countries (WHO 2018).
Tuberculosis can be classed as active when people experience signs or symptoms of tuberculosis or have radiological evidence of it. Tuberculosis can also be classified as latent tuberculosis infection (LTBI) where immunological evidence of previous exposure to M tuberculosis exists without clinical or radiological evidence of the disease (CDC 2000). Of healthy adults with immunological evidence of previous exposure to M tuberculosis, the overall lifetime risk of progressing to active disease if not treated for the infection is 5% to 10% (Harries 2006). Often this happens months or years after the initial infection in response to a weakening of the body's immune system. The probability of developing active disease is higher in HIV‐positive people, people with diabetes, and young children (Baker 2011; Perez‐Velez 2012; Tiemersma 2011). Fifty percent of infants with evidence of LTBI will progress to active disease if untreated (Marais 2004). People with LTBI require early diagnosis and treatment to reduce the pool of active tuberculosis cases. This is particularly important in high‐risk groups, such as those coinfected with HIV (Sharma 2012). Tuberculosis can be treated with long courses of multiple antibiotics, but the rise of HIV and spread of multidrug‐resistant tuberculosis (MDR‐TB) means that tuberculosis is still one of the largest threats to public health worldwide (WHO 2018). Structural determinants such as rapid urbanization of populations and economic inequalities, social determinants such as poverty and poor housing, alongside biological factors such as HIV and drug‐resistant strains of tuberculosis play a vital role in the spread of tuberculosis through vulnerable populations (Daftary 2012).
The Bacillus Calmette‐Guérin (BCG) vaccine is currently the only available vaccine. Epidemiological studies indicate that it has a protective effect against tuberculosis disease in children, particularly against the more severe forms of the disease such as tuberculosis meningitis or miliary tuberculosis (Roy 2014). The effectiveness of BCG differs greatly depending on the site of infection. It has consistent protection against tuberculous meningitis and miliary disease in children but variable protection against pulmonary tuberculosis (Abubakar 2013; Colditz 1995). As a result, despite many areas achieving high coverage of BCG vaccination, the disease remains a problem, and a new tuberculosis vaccine remains an important global research priority (WHO 2018).
Previously it has been impossible to ascertain reliably whether the BCG vaccine protected against active disease or infection with M tuberculosis. This was due to the tuberculin skin test being unable to distinguish between cases of LTBI and people who had been vaccinated with BCG (Roy 2014). Therefore, the development and use of interferon γ release assays (IGRA), which can distinguish between tuberculosis infection and vaccination, has proved useful. This has allowed researchers to establish that BCG vaccination reduces the risk of Mycobacterium infection in some settings (Eisenhut 2009).
Description of the intervention
Many researchers and policy makers emphasize that a new effective vaccine could be a major contribution to tuberculosis control and elimination as a public health problem (de Cassan 2010). There are 12 vaccine candidates in clinical trials: eight in Phase 2 or Phase 3, and four in Phase 1. They include candidates to prevent the development of tuberculosis, and candidates to help improve the outcomes of treatment for tuberculosis disease (WHO 2018).
The modified Vaccinia Ankara virus‐expressing antigen 85A (MVA85A) is a viral vector vaccine based on the modified Vaccinia Ankara (MVA) virus. MVA is an attenuated virus that does not replicate in human tissue and, as such, has been used as a platform to encode multiple antigens and allowing development of multivalent vaccines (Altenburg 2014). In this case, MVA has had pieces of DNA from M tuberculosis inserted into it, so that it expresses the antigen 85A. This antigen complex is an enzyme that is involved in the cell wall biosynthesis of M tuberculosis and constitutes a vital part of the way in which the bacteria forms its outer mycomembrane. This is important for the viability of the mycobacterium and works as an effective barrier to drug therapies by repelling some antibiotics and preventing them from entering the cell (Favrot 2013).
Immunological studies have shown that a prime boost strategy, where MVA85A is used to boost the effects of BCG, is effective in expanding immune responses specific to M tuberculosis (Beveridge 2007). Thus, MVA85A was proposed primarily as a booster to people already vaccinated with BCG (Tameris 2013). Further studies have assessed MVA85A in other regimens including in combination with other viral vector vaccines (Sheehan 2015).
How the intervention might work
MVA85A is the first vaccine since 1968 to be tested in efficacy trials (Tameris 2013). It has been tried with a promise of prolonged antimycobacterial immunity in human UK trials (McShane 2004), and in tuberculosis endemic areas (Hawkridge 2008). The intention is that MVA85A would boost the immune response to tuberculosis above that which is afforded by vaccination with BCG (Roy 2014). MVA85A is administered as a single intradermal dose in people who have already received BCG vaccine (Tameris 2013). Other routes have been studied in animal studies, such as intravenous administration (Romano 2006), and are being considered in humans (Satti 2014).
The researchers who developed the vaccine evaluated its effects in animals and conducted Phase 1 studies in humans. Early literature and reviews by the team noted the vaccine was safe and produced an immune response in several populations (McShane 2004; Rowland 2012).
One independent systematic review of the animal studies, carried out by some members of this Cochrane Review team, raised questions about whether these animal studies provided evidence of efficacy in the various animal models used (Kashangura 2015), when clinical and pathological endpoints were examined in a variety of animal models subjected to challenge studies. This has led to a debate about the reporting of animal studies, in particular the lack of published protocols so that the question being tackled in an animal study is made clear in advance (Cohen 2018). These studies administered BCG, BCG and MVA85A, or no vaccine. Afterwards, animals were exposed to tuberculosis challenge. Clearly progression to clinical trial is not solely based on evidence derived from preclinical efficacy studies, and MVA85A was evaluated in a number of trials in humans before proceeding to an efficacy study (McShane 2018). However, preclinical studies remain an important component of the tuberculosis vaccine development paradigm (Barker 2012; McShane 2014).
The systematic review of animal studies pointed out that there was one study in macaques where more monkeys required euthanasia in the MVA85A plus BCG vaccine group than the BCG control group (Kashangura 2015). This led to considerable controversy as to whether the publication of the results were delayed (Cohen 2018). The findings from this study could be the result of chance; or because the vaccine impaired functional immunity; or the result of a separate adverse effect. The vaccine development team then carried out a relatively large number of safety studies in humans; and, in their words, "none of the 14 trials of MVA85A in over 400 humans (the target species) before the infant efficacy trial showed a safety signal" (McShane 2018). The standard approach for Cochrane Reviews within the Cochrane Infectious Diseases Group is to only summarize efficacy trials. However, as the primary concern of the studies included in this review was safety, we summarized the considerable number of Phase 1 studies that the researchers carried out to exclude severe adverse effects attributable to the vaccine in humans in this 'Background' section of the review. We searched registered clinical trial databases (ClinicalTrials.gov, World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP), Pan African Trials Registry, EU Clinical Trials Register) in June 2017 and summarized the Phase 1 studies identified in Table 1. We found 21 separate studies as registered (prospectively and retrospectively) dating from 2003 with the most recent studies scheduled to complete follow‐up in 2018. In addition, we found an existing narrative review of Phase 1 studies (Rowland 2012), which summarized Phase 1 safety data relating to selected trials including unpublished data and compared this to selected trials in yellow fever and BCG.
| NCT trial number | Route | Dates | Intervention and schedule details | Country | Participants (age) | HIV | Adverse events | Reference |
| ID | 2002–2003 | MVA85A; 1 dose | UK | 14 adults (18–45 years) | –ve | 7 trials (112 participants); combined in 1 report: no serious AE attributable to the vaccine | ||
| ID | 2003–2005 | MVA85A; 1 dose, 2 doses (5 × 107 pfu) | Gambia | 21 adults | NR | No serious AE attributable to the vaccine | ||
| ID | 2003–2005 | MVA85A; 1 dose (5 × 107 pfu) | UK | 21 adults | –ve | No serious AE attributable to the vaccine | McShane 2004; Pathan 2012; Rowland 2012; Tanner 2014; Whelan 2009 | |
| ID | 2003–2005 | MVA85A; 1 dose (5 × 107 pfu) | UK | 10 adults | –ve | No serious AE attributable to the vaccine | ||
| ID | 2005–2007 | MVA85A, (5 × 107 pfu) | UK | 12 adults with latent tuberculosis | –ve | No vaccine‐related serious AEs 7 trials (112 participants; data combined in 1 report) | ||
| ID | 2005–2007 | MVA85A; 1 dose (1 × 108 pfu for 12 participants, and 1 × 107 pfu for 12 participants) | UK | 24 adults | –ve | No serious AE attributable to the vaccine | ||
| ID | 2005–2008 | MVA85A (5 × 107 pfu) | South Africa | 36 adults and adolescents | –ve | No vaccine‐related serious AEs | ||
| ID | 2006–2009 | MVA85A; 1 dose MVA85A (2.5 × 107 pfu, 5 × 107 pfu) Groups
| The Gambia | 214 infants (4 months) | NR | No serious AE judged to be related to the vaccine | ||
| ID | 2006–2010 | MVA85A; 1 dose (5 × 107pfu for 10 participants, and 1 × 108 pfu for 10 participants) | UK | 20 adults | +ve | No serious AE attributable to the vaccine | ||
| ID | 2007–2011 | MVA85A; 1 dose (5 × 107 pfu) 4 groups with background of
| South Africa | 48 adults (18–50 years) | +ve | No vaccine‐related serious AEs | ||
| ID | 2007–2010 | FP85A, MVA85A (5 × 107 pfu) | UK | 31 adults | –ve | No serious AE attributable to the vaccine | ||
| ID | 2007–2010 | MVA85A; 1 dose (1 × 108 pfu), administered as 2 injections (5 × 107 pfu each injection) | UK | 12 adults | –ve | 7 trials (112 participants); data combined in 1 report: no serious AE attributable to the vaccine | Porter (unpublished data: source Rowland 2012) | |
| ID | 2008–2011 | MVA85A; 2 doses (spaced by 6–12 months) (1 × 108 pfu) | Senegal | 24 adults | +ve | No serious AE attributable to the vaccine | ||
| ID IM | 2010–2011 | MVA85A; 1 dose (1 × 108 pfu) | UK | 24 adults | –ve | No serious AE attributable to the vaccine | ||
| ID | 2010–2012 | MVA85A, BCG; 1 dose (1 × 108 pfu) Group A: BCG naive, no MVA85A Group C: BCG vaccinated, no MVA85A Group D: BCG vaccinated, MVA85A. | UK | 49 adults recruited; 48 completed study | –ve | No serious AE attributable to the vaccine | ; Harris 2014b; | |
| Aerosol ID | 2011–2013 | MVA85A; 1 dose: 1 × 108, 1 × 107 pfu | UK | 24 adults | –ve | No vaccine related serious adverse effects. | ||
| ID | 2012–2014 | AERAS‐402 MVA85A; Group A: 2 doses AERAS‐402 then MVA85A Group B: 1 dose AERAS‐402 then MVA85A | UK | 40 adults | –ve | No vaccine related serious AEs | ||
| ID | 2013–2014 | MVA85A IMX313; Group A: low‐dose MVA85A‐IMX313 (1 × 107 pfu) Group B: dose MVA85A‐IMX313 (5 × 107 pfu) Group C: MVA85A (5 × 107 pfu) | UK | 30 BCG vaccinated adults | –ve | No vaccine‐related serious AE | ||
| IM | 2013–2016 | MVA85A, ChAdOx1 85A; Group A: 1 dose ChAdOx1 85A Group B: 1 dose ChAdOx1 85A then MVA85A Group C: 2 doses ChAdOx1 85A then MVA85A (1 × 108 pfu) | UK | 42 adults | –ve | No data reported yet | No publication | |
| Aerosol ID | 2013–2016 | MVA85A; Group 1: aerosol then ID Group 2: ID then aerosol Group 3: ID then ID (5 × 107 pfu) | UK | 37 adults | –ve | No data reported yet | (conference abstract) | |
| Aerosol ID | 2015–2018 | MVA85A; 1 × 107 pfu aerosol inhaled, 5 × 107 aerosol and ID | UK | 15 adults | –ve | No data reported yet |
–ve: negative; +ve: positive; AE: adverse event; ART: antiretroviral therapy; BCG: bacillus Calmette‐Guérin; EPI: Expanded Programme on Immunization; ID: intradermal; IM: intramuscular; MTB: Mycobacterium tuberculosis; NR: not reported; pfu: plaque‐forming unit.
The 21 studies included 712 participants investigated from 2002 with follow‐up expected to be completed by 2018. The studies covered a diverse population in the UK, South Africa, Senegal, and The Gambia with HIV‐positive and HIV‐negative people as well as infants, children, and adults. Intramuscular, intradermal, and aerosolized delivery routes were all investigated. The summary showed most of the adverse effects related to vaccination were mild and were contained locally to the injection site. There were very few serious adverse effects; erythema and mild pain were the most common adverse effects of the vaccine.
Why it is important to do this review
Summarizing the evidence to date will be useful to the public, scientists, and to others interested in innovation in tuberculosis as a case study from laboratory development to field testing. If critical appraisal and systematic review of this vaccine in humans shows no clear effect, this raises questions about any further testing. However, as of November 2017, there were ongoing studies looking at aerosolized delivery of the vaccine (NCT01954563; NCT02532036). In 2017, studies were published that addressed the immunogenicity of the candidate tuberculosis vaccine MVA85A in Schistosomiasis‐infected teenagers (Wajja 2017), and a further efficacy study in HIV‐exposed infants (Nemes 2018).
Objectives
To assess and summarize the effects of the MVA85A vaccine boosting BCG in humans.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials (RCTs) that include measures of clinical efficacy (Phase 2 clinical trials).
Types of participants
Any person regardless of age or HIV status.
Types of interventions
Intervention
MVA85A vaccine regardless of vaccination schedule, dosage, route, or formulation given with BCG.
Control
BCG alone, or Candin® (Candida albicans skin test antigen).
Types of outcome measures
Primary outcomes
-
Active tuberculosis, defined by:
-
clinical signs and symptoms plus confirmation by microscopy, culture, or Xpert® MTB/RIF (an automated nucleic‐acid amplification test);
-
treatment commenced for tuberculosis.
-
Secondary outcomes
-
Latent tuberculosis, diagnosed by IGRA or Mantoux without clinical or radiological evidence of active disease.
Adverse outcomes
-
Adverse effects of any severity, defined as "an adverse event for which the causal relation between the intervention and the event is at least a reasonable possibility" (Loke 2011).
-
Serious adverse effects, defined as an adverse event attributable to the intervention "leading to death, are life threatening, requires inpatient hospitalisation or prolongation of existing hospitalisation, or result in persistent or significant disability or incapacity" (ICH 1994).
-
Adverse events of any severity, defined as "any untoward medical occurrence that may present during treatment with a pharmaceutical product but which does not necessarily have a causal relationship with this treatment" (WHO‐ART 2008).
-
Abnormal haematological tests during the follow‐up period after being vaccinated.
-
Abnormal biochemical tests during the follow‐up period after being vaccinated.
Search methods for identification of studies
We conducted the literature search up to the 10 May 2018 and identified potential studies regardless of language or publication status (published, unpublished, in press, and in progress).
Electronic searches
We searched the following databases using the search terms and strategy described in Appendix 1: the Cochrane Infectious Diseases Group Specialized Register (10 May 2018); the Cochrane Central Register of Controlled Trials (CENTRAL, 2018, Issue 4, published in the Cochrane Library); MEDLINE (PubMed, 1966 to 10 May 2018); Embase (Ovid, 1947 to 10 May 2018); Science Citation Index‐Expanded, Social Sciences Citation index, conference proceedings (Web of Science, 1900 to 10 May 2018); and CINAHL (EBSCOHost (1982 to 10 May 2018). We also searched the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP; www.who.int/ictrp/en/), and ClinicalTrials.gov (clinicaltrials.gov/ct2/home), for trials in progress, up to 10 May 2018, using MVA85A, "modified vaccinia virus Ankara", Ag85A, "Antigen 85A", and tuberculosis OR tuberculosis as search terms.
Searching other resources
We searched the proceedings and abstracts of the following tuberculosis conferences: Union World Conference on Lung Health, European Respiratory Society, and the International Conference of the American Thoracic Society (ATS), from 2012 to 2018. We also handsearched reference lists of relevant papers.
Data collection and analysis
Selection of studies
Two review authors independently screened all abstracts retrieved by the search strategy above using predefined eligibility criteria designed and piloted by the review authors. We excluded clearly irrelevant studies. We searched for multiple publications using studies from the same data set. We retrieved full‐text copies for all trials thought to be potentially relevant. Two review authors (SoJ and SaJ) independently assessed all identified trials for inclusion in the review using the predefined inclusion criteria.
We resolved any disagreements in assessment through discussion. In cases of unresolved differences, a third review author adjudicated. We kept records of the initial results and the changes after discussion. We also kept a list all studies excluded after full‐text assessment in the Characteristics of excluded studies table. We illustrated the study selection process in a PRISMA diagram (Figure 1).

Study flow diagram.
Data extraction and management
We designed and piloted data extraction forms. Two review authors independently performed data extraction. We gathered information from each included trial separately on trial characteristics. These included:
-
study setting, design, study duration, population sample size, and power calculations;
-
baseline characteristics of study population including age, sex, weight, prematurity, HIV, other comorbidity, whether breastfeeding, race, HIV status, antiretroviral therapy (ART), CD4 count, and viral load;
-
intervention and control group vaccine dosages, routes of administration, and times of vaccination;
-
time of outcome measure after administering MVA85A;
-
duration of follow‐up, withdrawals from the study, and reasons for withdrawal.
All outcomes were dichotomous, so we tabulated the numbers of participants who developed tuberculosis or an adverse event (n) with the total sample size number (N) in each comparison group. We documented the different definitions of outcomes in the trials for further consideration and only combined data from endpoints that were similar across studies.
Assessment of risk of bias in included studies
We assessed risk of bias for RCTs using the Cochrane 'Risk of bias' tool (Higgins 2011). Two review authors independently assessed studies for risk of bias. We resolved any disagreement through discussion and, where necessary, through consultation with a third review author.
We assessed sequence generation (if predictable method used) and allocation concealment for selection bias and detection bias by looking at blinding methods. We also considered both the intention of blinding and the success of blinding for each outcome. If there was no description of the procedure, for example how randomization was done, we marked it as unclear.
In addition, we examined the objectivity of outcome measures, use of intention‐to‐treat (ITT) analysis, loss to follow‐up, and selective outcome reporting to assess the risk of bias in included studies. We assessed whether outcome measures were specified a priori and whether the published endpoints matched those specified in study protocols.
We assessed incomplete outcome data in each included trial to determine the proportion of missing results and whether it affected the results in terms of event risk and effect size. We assessed if reasons for missing data were related to adverse events or death from MVA85A and if missing data were balanced in the two experimental groups to have an overall decision on risk associated with incomplete outcome data.
We assessed other dimensions to risk of bias, including conflicts of interest, large differences in baseline characteristics, and early cessation of the trial.
We assessed the included trials for risk of bias of adverse events by examining if monitoring was active or passive; whether participants and outcome assessors were blinded; whether the outcome data reporting was complete; whether all participants were included; and whether data analysis was independent of pharmaceutical companies (Table 2; Bukirwa 2014). We also looked at the times when data were collected in comparison to when they were reported. All this information was included under overall study assessment of blinding, selective outcome reporting, incomplete outcome data, or other biases.
| Criterion | Assessment | Explanation |
| Participant‐reported symptoms | ||
| Was monitoring active or passive? | Active Passive Unclear | We classified monitoring as 'active' when authors reviewed participants at set time points and enquired about symptoms. |
| Was blinding for participants and outcome assessors adequate? | Adequate Inadequate Unclear | We classified blinding as 'adequate' when both participants and outcome assessors were blinded to the intervention group, and the methods of blinding (including use of a placebo) were described. |
| Was outcome data reporting complete or incomplete? | Complete Incomplete | We classified outcome data reporting as 'complete' when data were presented for all the time points where it was collected. |
| Were all participants included in reporting? | Yes No | We reported the percentage of randomized participants included in adverse event reporting. |
| Was the analysis independent of study sponsor? | Yes No Unclear | We classified the analysis of trials sponsored by pharmaceutical companies as independent of the sponsor when it was clearly stated that the sponsor had no input to the trial analysis |
| Laboratory tests | ||
| Number of tests undertaken | — | We extracted the type and number of laboratory tests were taken. |
| Timing of tests: was number and timing of tests adequate? | Adequate Inadequate | We classified the number and timing of tests as 'adequate,' when tests were taken at baseline, plus 2 other time points within the first week after treatment, plus the last day of the study. We classified the number of test taken as 'inadequate,' if either the laboratory controls in the first week or controls at 4 weeks were not performed. |
| Reporting of test results: was reporting of test results complete? | Complete Incomplete | We classified reporting as 'complete' when test results of all time points were reported. For the trials with inadequate number of tests taken, we considered completeness of reporting as inconsequential, and therefore did not record a judgement. |
| Independence of data analysis: was data analysis independent? | Yes No Unclear | We classified the analysis of trials sponsored by pharmaceutical companies as independent of the sponsor when it is clearly stated that the sponsor had no input to the trial analysis. |
Adapted from Bukirwa 2014.
Measures of treatment effect
We analysed all data using Review Manager 5 (Review Manager 2014). We pooled dichotomous data using risk ratios (RR) with their corresponding 95% confidence intervals (CI). When inappropriate due to a small number of events in each group, we presented the pooled data using risk difference (RD) with their 95% CI.
Unit of analysis issues
For included studies that had multiple intervention arms, we included data from these studies by splitting the control group so that participants were only included in the meta‐analysis once.
Dealing with missing data
In our protocol, we anticipated that if the amount of incomplete outcome data was such that the trials were thought to be at a high risk of bias, we may have used imputation and perform sensitivity analyses to investigate the impact of these missing data. However, we identified no studies where missing data affected our ability to measure outcomes. Therefore, we used available‐case analysis, as planned in our protocol.
Assessment of heterogeneity
We assessed extracted data from included trials to find key differences in population groups, study setting, intervention and control groups, dosages and route of vaccine administration, or timing between BCG and boosting. We assessed degree of risk of bias, when and how the outcome was measured, and variation in treatment effects.
We determined the level of heterogeneity by inspecting forest plots for overlapping CIs. We judged a Chi2 P value significance level of 0.1 or less as likely heterogeneity. An I2 statistic value of less than 40% was regarded as not showing any significant heterogeneity.
Assessment of reporting biases
There was an insufficient number of trials included and so we were unable to assess for publication bias using funnel plots or Egger regression.
Data synthesis
We used the fixed‐effect Mantel‐Haenszel model for meta‐analysis where there was little heterogeneity. The intention for meta‐analysis of adverse outcomes was limited to three to five of the most frequent adverse effects and all those that were considered to be serious. However, due to different methods of monitoring adverse effects that in turn lead to different results where meta‐analysis could not be performed, we gave a narrative report.
Subgroup analysis and investigation of heterogeneity
We intended to explore heterogeneity by: subgroup by children and adults; background prevalence of tuberculosis (or tuberculosis incidence in the control group); HIV status; and geographical location. However, there were not enough trials to explore such subgroups when we found high heterogeneity.
We considered random‐effects meta‐analysis if subgroup analysis did not explain the heterogeneity. We applied the I2 statistic according to guidance of: less than 40% as not significant heterogeneity; 30% to 60% representing moderate heterogeneity; 50% to 90% representing substantial heterogeneity; and 75% to 100% considerable heterogeneity (Higgins 2011). We regarded a Chi2 P value significance level of 0.1 or less and an I2 statistic greater than 40% as showing significant heterogeneity, in which case we either considered a random‐effects model or did not perform meta‐analysis.
Sensitivity analysis
We did not perform sensitivity analysis for imputed data, risk of bias, or any other peculiarities between the trials identified during the review process.
Certainty of the evidence
We assessed the certainty of the evidence using the GRADE approach (Schünemann 2013). We constructed a 'Summary of findings' table, which outlines the main review findings alongside the certainty of the evidence.
Results
Description of studies
Results of the search
We identified 153 records, with 152 records remaining after removing duplicates. We excluded 118 records based on title and abstract and assessed the full text of 34 articles. We excluded 28 full‐text articles. Six articles fulfilled the eligibility criteria and were included in the review. See Figure 1 for the flow diagram of inclusion and exclusion of studies in the review.
Included studies
Six studies (3838 participants) that met our inclusion criteria reported findings from four Phase 2 clinical trials (Ndiaye 2015; Nemes 2018; Scriba 2011; Tameris 2013). Andrews 2017 and Bunyasi 2017 presented data based on the Tameris 2013 clinical trial. The six included studies are described in the Characteristics of included studies table.
Setting and time
All took place in South Africa involving rural and urban areas between 2008 and 2015, with one trial that took place at two sites: South Africa and Senegal (Ndiaye 2015).
Source of funding
Aeras sponsored five trials (Andrews 2017; Bunyasi 2017; Ndiaye 2015; Nemes 2018; Tameris 2013). The University of Oxford sponsored one trial (Scriba 2011). The Wellcome Trust funded all the trials. Other funders were Oxford Emergent Tuberculosis Consortium (OETC) for Ndiaye 2015 and Tameris 2013, the European and Developing Countries Clinical Trials Partnership and the Bill and Melinda Gates Foundation for Ndiaye 2015, the UK Medical Research Council for Nemes 2018, and the EuropeAID European Commission for Scriba 2011. Andrews 2017 and Bunyasi 2017 conducted further follow‐up based on the participants enrolled in Tameris 2013, and mentioned that there was no specific additional funding for the analysis performed.
Participants
Five trials included infants (Andrews 2017; Bunyasi 2017; Nemes 2018; Scriba 2011; Tameris 2013). One trial assessed the efficacy and safety of the vaccine in adults with HIV (Ndiaye 2015). Tameris 2013 and Scriba 2011 recruited infants who were HIV‐negative, while Nemes 2018 assessed the vaccine in newborns of HIV‐positive mothers. None of the trials reported other morbidities. In Tameris 2013, 412 (29.4%) participants in the intervention group and 268 (26.4%) participants in the control group were preterm.
Interventions
Intervention
All the infants in the intervention groups received a single dose of intradermal MVA85A. In the trial recruiting adults, the 324 adults allocated in the intervention group received a second dose (booster) of intradermal vaccine six months after the first dose (Ndiaye 2015). The vaccine was given at a dose of 1 × 108 plaque‐forming units (pfu) in Ndiaye 2015, Nemes 2018, and Tameris 2013. Scriba 2011 assessed three different doses of the vaccine by giving a dose of 2.5x107 pfu, 5x107 pfu and 1x 108 pfu to 36 participants in each of the three groups. All the infants in Scriba 2011 and Tameris 2013 received the BCG vaccine in the first four weeks of life, prior to receiving the MVA85A vaccine, as an inclusion criteria. Nemes 2018 gave the MVA85A vaccine to the neonates in the first 96 hours of life, with no prior administration of BCG, and gave BCG at eight weeks of age only to HIV‐negative infants. Ndiaye 2015 did not mention whether the adults they recruited received BCG.
Comparator
Five trials gave Candida skin test antigen (Candin®) as a placebo, using the same route (intradermal) and schedule (one or two doses) as for the intervention group in each of the trial (Andrews 2017; Bunyasi 2017; Ndiaye 2015; Nemes 2018; Tameris 2013). Scriba 2011 gave the infants in the comparator group one dose of pneumococcal 7‐valent conjugate vaccine by the intramuscular route.
Outcomes
Three studies reported different endpoints as measures of tuberculosis disease (Ndiaye 2015; Nemes 2018; Tameris 2013). These are compared in Table 3.
| Study | Endpoint 1 | Endpoint 2 | Endpoint 3 |
| Any of the following criteria.
| "Included all infants who met endpoint 1 criteria; had marginally less stringent criteria to define TB infection and household exposure." Any of the following numerical categories.
| All participants placed on treatment for TB by a health professional with the intent of treating TB regardless of whether they have met the other efficacy endpoints. | |
| Revised endpoint 1 from Tameris 2013 that removed QFT conversion from the diagnostic criteria to avoid bias towards association with QFT status. | Not used | Not used | |
| Not used | Not used | Same definition as for Tameris 2013. | |
| Any of the following numerical categories.
| Any of the following numerical categories:
| Same definition as for Tameris 2013. | |
| Not applicable | Not applicable | Not applicable | |
| Outcomes not specified in the methods section. In results, authors specified that 8 participants were diagnosed as TB: "of whom one was M.tb [Mycobacterium tuberculosis] culture positive and 7 were diagnosed on clinical/ radiographic grounds and TB contact history. Two of the TB cases were QFT positive." | Not used | Not used |
AFB: acid‐fast bacilli; CSF: cerebrospinal fluid; CT: computerized tomography; QFT: quantiFERON; TB: tuberculosis.
aIn Tameris 2013, endpoint 2: criteria in bold indicate where different from endpoint 1.
All the included studies reported data on latent tuberculosis (or tuberculosis infection) to assess either efficacy or safety outcomes. Four trials looked at safety outcomes, including adverse effects of any severity, serious adverse effects, and adverse events of any severity (Ndiaye 2015; Nemes 2018; Scriba 2011; Tameris 2013). Tameris 2013 collected data on biochemical or haematological blood test findings but did not report this element of their primary outcome. Ndiaye 2015 collected data on blood tests but did not report disaggregated findings. Only Scriba 2011 and Nemes 2018 reported on blood test data collected.
Length and method of follow‐up
Scriba 2011 followed up participants for 24 weeks, Nemes 2018 for 52 weeks, Ndiaye 2015 for at least six months after the last participant was enrolled, and Tameris 2013 for up to 39 months. Andrews 2017 was an observational follow‐up study of the participants enrolled in Tameris 2013; authors analysed the data collected at day 336 after the intervention and at the end of the study, which ranged from six to 24 months after day 336. Bunyasi 2017 followed the participants recruited in Tameris 2013 for a median of five years.
Investigators of five studies used diary cards to record adverse events during the seven days following vaccination (Andrews 2017; Bunyasi 2017; Ndiaye 2015; Scriba 2011; Tameris 2013); Nemes 2018 did not mention this. Researchers performed blood investigations at several intervals in all trials, to detect adverse events and to assess immunogenicity. Ndiaye 2015 and Tameris 2013 performed active follow‐up every three months to identify signs, symptoms, or exposure to tuberculosis that merited further investigation, while this was done at irregular but planned intervals in Scriba 2011 and Nemes 2018. The long‐term follow‐up study was based on passive surveillance based on the electronic tuberculosis register database (Bunyasi 2017).
Excluded studies
We excluded 28 studies from the review, with the reasons for exclusion listed in the Characteristics of excluded studies table.
Studies awaiting classification
We did not identify any studies that are awaiting classification.
Ongoing studies
We did not identify any ongoing studies.
Risk of bias in included studies
See Characteristics of included studies table for the assessment of the risk of bias for each included study. See Figure 2 and Figure 3 for the risk of bias summaries.
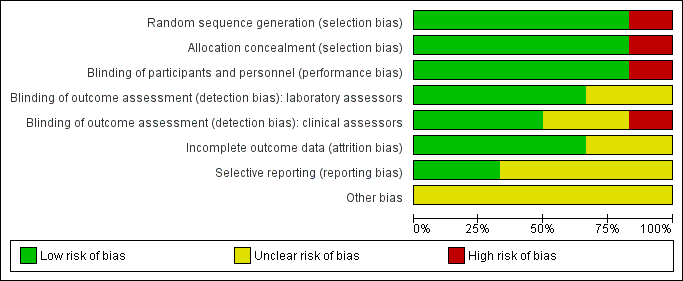
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Five trials were at low risk of selection bias (Andrews 2017; Bunyasi 2017; Ndiaye 2015; Nemes 2018; Tameris 2013). They reported adequate sequence generation and methods of allocation concealment. Scriba 2011 used systematic allocation at a 3:1 ratio allowing predictability of the sequence (high risk of bias).
Blinding
Three studies had adequate blinding of participants, study personnel, laboratory assessors, and clinical assessors and were at low risk for performance and detection bias in all domains (Ndiaye 2015; Nemes 2018; Tameris 2013). Five studies reported blinding of participants and study personnel (Andrews 2017; Bunyasi 2017; Ndiaye 2015; Nemes 2018; Tameris 2013). Scriba 2011, an open‐label trial with different routes of administration for placebo and vaccine, had low risk of detection bias for laboratory assessors as outcomes were objective and high risk of detection bias for subjective assessments by clinicians. Two studies were at unclear risk of detection bias for laboratory assessors and clinicians (Andrews 2017; Bunyasi 2017). Andrews 2017 did not provide any details on blinding, while Bunyasi 2017 reported on post‐trial data and had no information on how data was collected from registers.
Incomplete outcome data
Four trials reported details of all randomized participants (Ndiaye 2015; Nemes 2018; Scriba 2011; Tameris 2013). Only a few participants randomized were not included in the analysis, without resulting in a disbalance between the intervention and control groups. Indeed, three participants were randomized in the control group in Tameris 2013, but not included in the efficacy analysis (two of them were not included either in the safety analysis), while five participants were randomized (four in the intervention group and one in the control group), but not included in the efficacy analysis in Ndiaye 2015. As a result we considered these studies at low risk of attrition bias. There were no details of how many of each group came from the 119 participants excluded from Tameris 2013 for analysis in Bunyasi 2017. Andrews 2017 and Bunyasi 2017 had an unclear risk of attrition bias as these were follow‐up studies from Tameris 2013, and there were unclear discrepancies with those reported previously.
Selective reporting
Nemes 2018 was prospectively registered and appeared free of selective outcome reporting as ascertained from data in trial registers and reports of trials. We also judged Scriba 2011 at low risk of reporting bias, with all the outcomes reported in their methods section presented in the results.
Four studies were at unclear risk of bias due to selective reporting (Andrews 2017; Bunyasi 2017; Ndiaye 2015; Tameris 2013). There were multiple instances where predefined endpoints were poorly defined or were deviated from in the final reported results as laid out in Table 4.
| Study | Protocol | Published findings | Differences between protocol and published findings | ||
| Stated outcomes published prior to commencement of trial that differ to published outcomes | Measurement of outcome as stated a priori | Measurement of outcome as stated in published findings | Reported findings | ||
| No protocol published. | |||||
| No protocol published (extended post‐trial follow‐up of Tameris 2013). | |||||
| Adverse events: blood tests a | "Percentage of participants with adverse events" AEs measured up to day 28 SAEs measured up to 6 months. | "Phlebotomy for routine haematological and biochemical analysis was done at screening, before booster vaccination, and on days 7 and 28 after each vaccination." | "Routine haematological and biochemical test results did not differ between study groups (data not shown)." | Haematological and biochemical blood tests not outlined as a measure of safety in the study protocol. Blood test findings reported unclearly. | |
| Safety | Clinicaltrials.gov – local, regional, and systemic AEs and SAEs which would be reported as cumulative 12‐month incidences. | "Infants followed for safety end points at weeks 1, 4, 6, and 8 after MVA85A/control vaccination and thereafter, at weeks 9, 12, and 16 (corresponding to weeks 1, 4, and 8 following delayed BCG vaccination at 8 weeks of age), and at week 52." | Reported total events for AEs per group after MVA85A and before BCG and for whole follow‐up period. Data including for laboratory AEs were not disaggregated as prespecified. | Data including for laboratory AEs were not disaggregated as prespecified. | |
| Safetya | Local and systemic AEs for the first week. | Diary cards | Local and systemic AEs reported on ≥ 1 day of the first 7 days after MVA85A vaccination. | None | |
| Blood tests (days 7, 28) | Biochemical and haematological tests (days 7, 28) | Reported number and percentages of participants with abnormal results and reported that, "all except one patient that had elevated liver enzymes remained unresolved by day 28." | |||
| Immunology | ESAT‐6/CFP‐10 | Infants converted – suggestive of TB infection but seemed to be reported as safety data not efficacy. | |||
| Safety profile – AEsa | AEs measured up to day 28 SAEs measured throughout follow‐up. | Collected data on solicited and unsolicited local and systemic AEs. Active surveillance for SAEs. | AEs broken down by type of event and reported in supplementary material. Only local events at the injection site were considered to be related to the vaccine. | Causal relationship with AEs other than local injection site reactions was not reported. | |
| Safety profile – blood testsa | Testing up to 28 days postvaccination. | "Peripheral blood for routine haematological and biochemical tests was taken at screening and on day 7 and day 28 after vaccination in an initial safety cohort of at least 330 infants." | Not reported | Primary outcome not reported | |
| Efficacy of MVA85Ab | Using an endpoint derived from epidemiological cohort surveys in BCG vaccinated infants. | Not reported – simply stated clinical endpoints 'developed.' | Composite clinical endpoints 1, 2, 3 (see Table 3) Microbiologically confirmed cases reported in appendix. | The "primary efficacy endpoint" was measured using an endpoint not derived from cohort studies. The endpoint definition differed from all other implied or reported ways of measuring efficacy in the other studies. The point estimate showed clinically significant benefit for endpoint 1 (no benefit seen at the 95% confidence level). This endpoint was reported as the main efficacy finding. All other point estimates show no clinically significant benefit or harm. | |
AE: adverse events; BCG: bacillus Calmette‐Guérin; ESAT‐6/CFP‐10: early secretory antigenic‐6/culture filtrate protein‐10; SAE: severe adverse events; TB: tuberculosis.
aPrimary outcomes as outlined in study protocols.
bSecondary outcomes as outlined in study protocols.
Description of Tameris 2013 published prior to commencement of the trial (NCT00953927) stated that the authors intended to report endpoints of clinical disease based on "observational cohort studies." This was subsequently changed following the publication of the trial in October 2013 to include "clinically‐derived tuberculosis diagnostic criteria." The main trial reports adapting the primary elements proposed in a consensus statement (Graham 2012). There was no record of the change in approach from empirically derived endpoints to endpoints developed by the investigators in the study protocol.
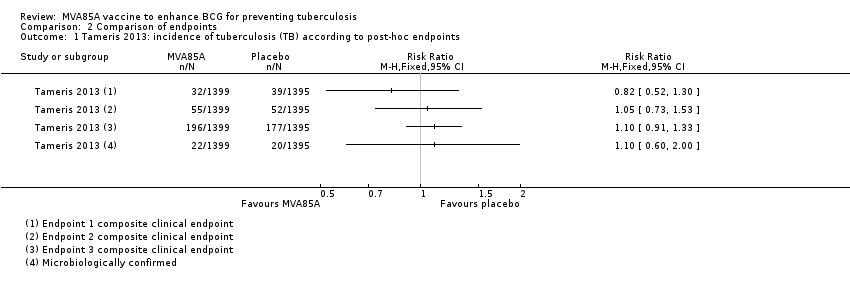
Tameris and colleagues reported on three outcomes with complex definitions (Table 3).
-
Endpoint one, described as "primary efficacy endpoint," comprising nine criteria, which included a binary measure of quantiFERON conversion.
-
Endpoint two, described as "exploratory efficacy endpoint," comprising nine criteria.
-
Endpoint three, described as "exploratory efficacy endpoint," which was defined as "all participants placed on treatment for tuberculosis."
The difference between endpoints one and two, which varied in the direction of the point estimate of the effect, was 5 mm on a tuberculin skin test or household contact with acid‐fast bacilli (AFB) smear‐positive person (Table 3). The process of defining these three endpoints was unexplained, and it is unclear why these specific definitions were used. These endpoint definitions were only used in this trial and not in subsequent studies.
In a subsequent critique, Behr and colleagues noted that the outcomes reported in the trial did not include the simple measure of a positive microbiological endpoint (Behr 2013). The endpoint used in the abstract was endpoint one, which authors have settled as primary efficacy outcome, while endpoints two and three were reported as exploratory outcomes. The complexity of the definitions and the analysis in Behr's paper pointed to the risk of selective reporting. This may not have been intentional, but arose with post‐hoc approaches with different approaches to expressing the results, but could be excluded if outcomes were precisely and clearly defined a priori. The only information publicly available prior to the trial commencing were broad descriptions of the outcome. Hence for selective reporting the classification was unclear.
Andrews 2017 was at unclear risk of reporting bias as this was a nested observational study and there was no prespecified study protocol. Ndiaye 2015 was at unclear risk of reporting bias as the authors commented that there were no differences between biological and haematological tests; however, no data or how these data were analysed to come to this conclusion were reported.
Other potential sources of bias
We considered that the risk of other potential biases was unclear in all included studies. We were concerned as a number of the authors were involved in the private company manufacturing the vaccine or were patent holders for MVA85A. In these circumstances, it would be good practice for this to be declared in the publication. Only one study declared no conflicts in relation to patent holding (Scriba 2011).
Two trials reported a role of funders in design, data analysis, and manuscript writing (Ndiaye 2015; Tameris 2013), and one study had employees of the funder involved in manuscript writing (Andrews 2017). Ndiaye 2015 calculated incident tuberculosis cases from day 28 after vaccination versus from day 0 in Tameris 2013. This was likely to be due to the risk of pre‐existing undiagnosed tuberculosis being inappropriately counted as developing following the intervention. If participants are not followed from the start of the intervention then a period of follow‐up has been excluded, and participants who experienced the outcome soon after intervention will be missing from analyses. We considered this to be of unclear risk of bias as it is unclear if this impacted on outcomes.
Adverse events
For adverse events, we conducted additional assessments on adequacy of safety monitoring and completeness of reporting for participant‐reported outcomes and laboratory tests taken (Table 5). Four trials reported on safety outcomes (Ndiaye 2015; Nemes 2018; Scriba 2011; Tameris 2013). Monitoring of participant‐reported outcomes was active in all trials and blinding was adequate in two trials (Nemes 2018; Tameris 2013). All trials reported specified timing of data collection but only one study reported under some of the days (Scriba 2011). None of the trials completely reported outcomes on prespecified time points including for laboratory results. All trials reported all participants who received intervention per‐protocol. Timing of taking laboratory tests was inadequate in Scriba 2011 and Tameris 2013 as there was no clear indication of tests being taken at the end of the study.
| Study | Participant reported adverse events | Outcome data reporting | Laboratory tests | ||||||||
| Monitoring active or passive | Blinding of participants or outcome assessors | Times data collected | Times data reported | Complete/not complete | Percentage of participants reported on | Analysis independent of study sponsor | Number of tests taken | Timing of tests and adequacy | Complete reporting of test results | Independence of data analysis | |
| Active | Inadequate | 60 min, D 2, 7, 28, 84, and 168 | D 7, 28 | Incomplete | 100% | Unclear | Biochemistry and haematology | Inadequate | Inconsequential | Unclear | |
| Active | Inadequate | D 7, 28, and 84 after boost 3 monthly until end of study | NR | Incomplete | 99.8% | No | Haematology, chemistry, virological markers | Adequate | Incomplete | No | |
| Active | Adequate | Baseline, D 7 and 28, throughout up to D 84 | NR | Incomplete | 99.9% | No | Biochemistry and haematology | Inadequate | Incomplete | No | |
| Active | Adequate | Week 1, 4, 6, 8, 16, and 52 | NR | Incomplete | 85.9% | Unclear | Not specified | Adequate | Incomplete | Unclear | |
D: day; min: minute; NR: not reported.
Effects of interventions
See: Summary of findings for the main comparison MVA85A compared to placebo for preventing tuberculosis
See summary of findings Table for the main comparison.
Active tuberculosis
Studies varies in the way they defined active tuberculosis (see section "description of studies" (Table 3)). Tameris 2013 and Ndiaye 2015 reported hierarchical endpoints including microbiologically confirmed tuberculosis, composite clinical definitions, and participants starting on tuberculosis treatment, with no significant effect consistently seen across endpoints (Analysis 2.1; Analysis 2.2; Table 3; Table 6).
| Active TB | ||||||||||||
| MVA85A | Placebo | MVA85A | Placebo | MVA85A | Placebo | MVA85A | Placebo | MVA85A | Placebo | MVA85A | Placebo | |
| Endpoint 1a | 32/1399 (2.3%) | 39/1395 (2.8%) | 58/2797 (2.1%) with NDD | N/A | N/A | 6/320 (1.9%) | 9/325 (2.8%) | N/A | N/A | 5/123 (4.1%) | 3/125 (2.4%) | |
| Endpoint 2a | 55/1399 (3.9%) | 52/1395 (3.7%) | N/A | N/A | N/A | N/A | 6/320 (1.9%) | 9/325 (2.8%) | N/A | N/A | N/A | N/A |
| Endpoint 3a | 196/1399 (14.0%) | 177/1395 (12.6%) | N/A | N/A | 3.3/100 pyo (95% CI 2.9 to 3.9) | 3.0/100 pyo (95% CI 2.6 to 3.5) | 8/320 (2.5%) | 9/325 (2.8%) | N/A | N/A | N/A | N/A |
CI: confidence interval; N/A: not applicable; NDD: no disaggregated data; pyo: person‐years of observation; TB: tuberculosis.
aSee Table 3 for description of endpoints.
Tameris 2013 reported three endpoints in their main manuscript, with endpoint one described as their primary efficacy endpoint (RR 0.82, 95% CI 0.52 to 1.30, point estimate favouring MVA85A). A fourth endpoint was described in the supplementary material, taking into account the microbiologically confirmed cases of tuberculosis. Other outcomes (endpoint two, endpoint three, and endpoint four of microbiologically confirmed cases) were not statistically different, although their point estimate favoured placebo (endpoint two: RR 1.05, 95% CI 0.73 to 1.53; endpoint 3: RR 1.10, 95% CI 0.91 to 1.33; endpoint four (microbiologically confirmed): RR 1.10, 95% CI 0.60 to 2.00; Analysis 2.1; Figure 4).

Forest plot of comparison: 2 Comparison of endpoints, outcome: 2.1 Tameris 2013: incidence of tuberculosis according to post‐hoc endpoints.
Two studies reported no effect of MVA85A on cases of active tuberculosis confirmed by culture or Xpert® MTB/RIF (RR 0.97, 95% CI 0.58 to 1.62; 3439 participants, two trials) (Analysis 1.1; Figure 5; Ndiaye 2015; Tameris 2013).
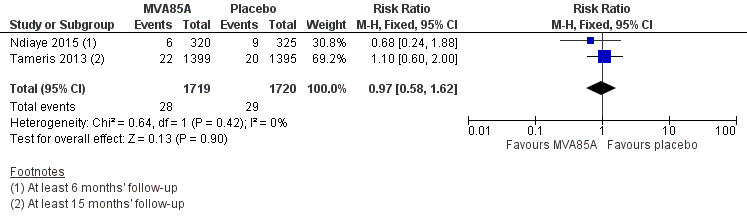
Forest plot of comparison: 1 MVA85A Vs Placebo, outcome: 1.1 Tuberculosis confirmed by culture or Xpert® MTB/RIF longest reported follow‐up.
Three studies (Ndiaye 2015; Nemes 2018; Tameris 2013) reported no effect of MVA85A on cases of active tuberculosis when considering patients started on tuberculosis treatment (RR 1.10, 95% CI 0.92 to 1.33; 3687 participants, 3 trials; Analysis 1.2; Figure 6).
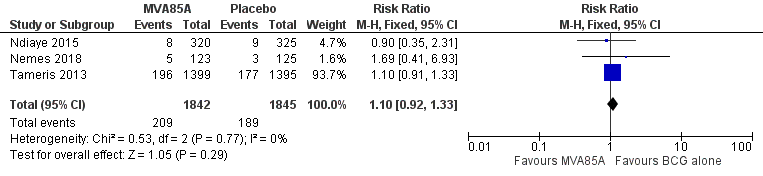
Forest plot of comparison: 1 MVA85A versus placebo, outcome: 1.2 Active tuberculosis: started on tuberculosis treatment.
Nemes 2018 reported active tuberculosis as defined by participants starting tuberculosis treatment. One participant in this trial was diagnosed by culture; however, the authors did not report what intervention this participant received.
Latent tuberculosis
Four studies reported no effect of MVA85A on cases of latent tuberculosis (RR 1.01, 95% CI 0.85 to 1.21; 3831 participants, four trials; Analysis 1.3).
Scriba 2011 was underpowered and not designed to detect measures of efficacy. However, they reported latent tuberculosis, presumably as a measure of safety, as this outcome was poorly defined a priori.
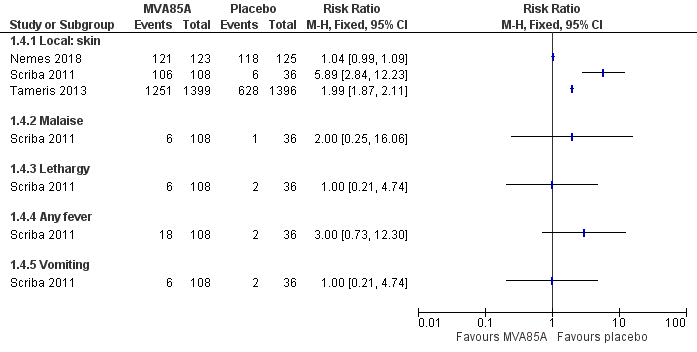
Adverse effects
Four studies reported effects of any severity (Table 7). We presented the effect of the estimates for adverse effects of any severity with disaggregated (Analysis 1.4; Figure 7) and aggregated data (Analysis 1.5) to provide detailed information as provided by the study authors. However, we did not perform meta‐analysis of the estimates due to high heterogeneity. Local reactions of the skin at the injection site was the most common adverse effect associated with the vaccine MVA85A, this was reported in three studies, with the three studies showing direction towards more adverse effects in the intervention group (3187 participants; Nemes 2018; Scriba 2011; Tameris 2013). However, only one study reported systemic symptoms defined as fever, lethargy, malaise, and vomiting (144 participants; Scriba 2011). Therefore, we chose to report adverse effects of any severity disaggregated by local reactions of the skin and systemic symptoms in our summary of findings Table for the main comparison as different amount of information is provided for each group (Scriba 2011).
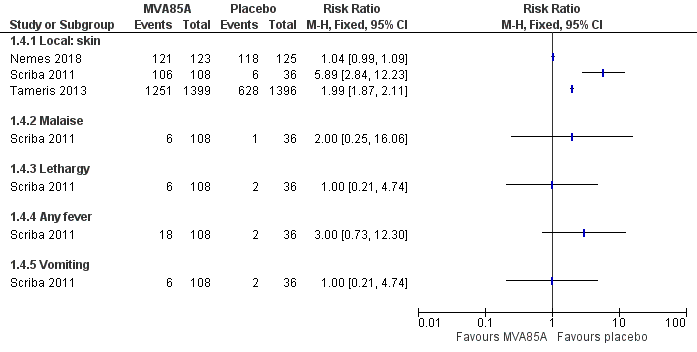
Forest plot of comparison: 1 MVA85A versus placebo, outcome: 1.4 Adverse effects of any severity.
| Study | MVA85A | Placebo | Breakdown | Author conclusions | ||||
| Number of participants with ≥ 1 event caused by the intervention | Total participants | Number of participants with ≥ 1 event caused by the control | Total participants | Detailed AEs | MVA85A | Placebo | ||
| 318 | 324 | 307 | 325 | Solicited AEsa | 288 | 235 | "Solicited adverse events were more common in MVA85A group and most were local injection site reactions." | |
| 105b | 123 | 30b | 125 | Not detailed | N/A | N/A | "Infants in MVA85A arm were more likely to experience an AE than in control arm. Injection site reactions were more frequent in MVA85A recipients and mild." | |
| 106c | 108c | 6 | 36 | Injection sited | 106 | 6 | "Desquamation significantly increased with greater vaccine dose." | |
| Malaise | 6 | 1 | ||||||
| Lethargy | 6 | 2 | ||||||
| Tactile fever | 18 | 0 | ||||||
| Documented fever | 13 | 2 | ||||||
| Vomiting | 6 | 2 | ||||||
| Elevated liver enzyme levels | 13 | 4 | ||||||
| Increased white cell count | 0 | 1 | ||||||
| Local 1251e | 1399 | Local 628e | 1396 | Not detailed | 1251 | 628 | None | |
AE: adverse event; N/A: not applicable.
aIncluded injection reactions, mild influenza‐like symptoms, and regional lymphadenopathy.
bAuthors of the study reported 105 participants with at least one adverse effect in the vaccine group and 30 participants in the placebo group, where causal relationship was defined as definite.
cAggregated between three groups receiving different doses.
dIncluded desquamation (scaling), pain, redness, and swelling.
eAuthors of the study reported local and systemic adverse events. Authors specified in their protocol that, "Solicited adverse events of local injection site reactions will be considered causally related to study vaccine (adverse reaction)." Therefore, we reported such adverse events as adverse effects. Causal relationship with other adverse events was not reported.
Three studies reported no increase in the risk of experiencing a serious adverse effect attributable to MVA85A (3692 participants; Analysis 1.6). Nemes 2018 reported serious adverse events and specified that none of them were related to the investigational product. Therefore, we classified this as no serious adverse effects following the definition of our review.
Adverse events of any severity
Four studies reported a small increase in the risk of experiencing an adverse event of any severity following vaccination with MVA85A (RR 1.05, 95% CI 1.02 to 1.08; 3836 participants; Analysis 1.7; Table 8). Adverse effects related to the vaccine and adverse events not attributed to the vaccine were conflated in the largest trial. No disaggregated data were available.
| Study | Adverse events of any severity | |||
| MVA85A | Placebo | |||
| 1120/1399 (80.1%) | 1059/1396 (75.9%) | |||
| NR | NR | |||
| NR | NR | |||
| 321/324 (99.1%) | 312/325 (96%) | |||
| 2.5 × 107 pfu = 35 μL | 5 × 107 pfu = 70 μL | 1 × 108 pfu = 135 μL | 1/36 | |
| 1/36 | 3/36 | 6/36 | ||
| Mild 122/123 (99.2%) | 121/125 (96.8) | |||
| Moderate 62/123 (50.4%) | 54/125 (3.6%) | |||
| Severe 11/123 (8.9%) | 14/125 (11.2%) | |||
NR: not reported; pfu: plaque‐forming unit.
Abnormal haematological and biochemical tests
Three studies reported abnormal haematological or biochemical laboratory tests. The percentage of those with elevated liver enzymes ranged from 2.8% to 25% in the three different groups reported in Scriba 2011 and there was a dose–response effect of MVA85A. However, none of the doses showed a significant increase at a 95% CI. Ndiaye 2015 reported that routine haematological and biochemical test results did not differ between study groups but disaggregated data were not reported. Nemes 2018 reported no difference between groups in the percentage of people with abnormal biochemical tests (11.4% versus 10.4%), but disaggregated data were not reported. The largest study performed haematological and biochemical tests but did not report data (Tameris 2013). We summarized the report and findings of abnormal haematological and biochemical tests in Table 9, and presented the effect of estimate for abnormal biochemical tests only (Analysis 1.8), as only one study reported disaggregated data for abnormal haematological tests.
| Study | Haematological blood tests | Biochemical blood tests | ||||
| MVA85A | Placebo | MVA85A | Placebo | |||
| NR | NR | NR | NR | |||
| NR | NR | NR | NR | |||
| NR | NR | NR | NR | |||
| NRa | NRa | NRa | NRa | |||
| 0/108b | 1/36b | 2.5 × 107 pfu = 35 μL | 5 × 107 pfu = 70 μL | 1 × 108 pfu = 135 μL | 4/36 (11%) | |
| 1/36 (2.8%) | 3/36 (8.3%) | 9/36 (25%) | ||||
| NR | NR | 14/123 (11.4%) | 13/125 (10.4%) | |||
NR: not reported; pfu: plaque‐forming unit.
aAuthors stated that routine haematological and biochemical test results did not differ between study groups but did not present data.
bOne participant had increased white cell count concurrently with an increase in alanine aminotransferase during an episode of gastroenteritis. Authors did not describe any other case of abnormal haematological test in the rest of the participant, although it was not stated explicitly.
Discussion
Summary of main results
Vaccinating people with MVA85A in addition to BCG:
-
probably makes little or no difference to the risk of developing active tuberculosis (moderate‐certainty evidence);
-
probably makes little or no difference to the risk of needing to start tuberculosis treatment (moderate‐certainty evidence);
-
probably does not have an important effect on the risk of developing latent tuberculosis (moderate‐certainty evidence);
-
does not cause life‐threatening serious adverse effects (high‐certainty evidence);
-
probably increases the risk of having an adverse reaction related to vaccination at the site of the injection (moderate‐certainty evidence);
-
may not be associated with an increase in systemic adverse effects related to vaccination (low‐certainty evidence).
Vaccination with MVA85A alone slightly increases the risk of having an adverse event (high‐certainty evidence).
Overall completeness and applicability of evidence
This review included trials from two countries in Africa. No studies that measured efficacy of the MVA85A vaccine have been carried out elsewhere. The review included studies on HIV‐positive adults, HIV‐negative infants, and infants exposed to HIV. It would be reasonable to generalize the results of these findings to other populations of HIV‐negative infants. The early cessation of the only trial in HIV‐positive adults, resulting in reduced follow‐up from two years to minimum six months and a reduction of study sample size from 1200 to 625, led this study to be underpowered for evaluation of efficacy (Ndiaye 2015). This may have limited the certainty of any inferences made to adults with HIV at high risk of contracting tuberculosis in terms of efficacy of MVA85A in this population. The effect of tuberculosis vaccination would be very similar regardless of geographical variation. Data from this review consistently showed no effect of the vaccine. As such, it is reasonable to generalize these findings to broader populations. For safety outcomes, the Phase 1 studies that we summarized in the Background section and Table 1, included adults, children, and infants from the UK and three African countries. Most of the adverse effects related to vaccination were mild and were contained locally to the injection site. This supports the trial findings summarized in this review.
Certainty of the evidence
Overall the included studies were well‐conducted. For most of our outcomes, there were few events and broad CIs for the pooled estimates of effect which contained clinically appreciable benefit and harm or no effect (see summary of findings Table for the main comparison).
In the largest trial, the main reported endpoint (endpoint one) point estimate was in the direction of benefit of the vaccine on tuberculosis disease (Analysis 2.1; Tameris 2013). Whether this was due to the definition of endpoints or due to statistical heterogeneity was unclear. To minimize the impact of this inconsistency we presented results for cases diagnosed microbiologically and cases defined by being started on treatment. This was felt to reflect the most specific measure of efficacy and a measure of the real‐world situation. As a result of this, the methodological uncertainties surrounding case definition did not reduce our confidence in the effect estimates.
Failure to follow‐up participants from the start of intervention for efficacy measures in Ndiaye 2015 risked biasing outcomes. While it is plausible that participants with undiagnosed active tuberculosis would be inappropriately picked up, it is also plausible that participants who hypothetically could have developed tuberculosis immediately after vaccination would be excluded from analysis. However, the potential impact of this was unclear and as such we did not downgrade due to risk of bias for efficacy outcomes including this study.
In terms of latent tuberculosis, using the online calculator at www.sealedenvelope.com/power/binary‐noninferior/ at a significance level of 5% and with 80% power at a failure rate of 11% and a non‐inferiority limit of 5% a sample size per group of 484 would be sufficient to demonstrate non‐inferiority. Therefore, in terms of risk of developing latent tuberculosis where we had high certainty evidence that MVA85A had no important effect in reducing risk, we are confident that future trials are unlikely to change this result as we had 3831 participants in the analysis versus a minimum number of 484 participants required in each group.
Regarding the safety outcomes, the summary of findings from the Phase I trials for MVA85A performed in adults, adolescents, and infants with 712 participants showed that most of the adverse effects related to vaccination were mild and were contained locally to the injection site, and none of the trials reported a serious adverse event attributable to the vaccine. This supports the certainty of the evidence found in this review.
Potential biases in the review process
We followed standard methods in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). The Cochrane Infectious Disease Group Information Specialist performed a comprehensive literature search with no restriction in language to identify all eligible studies, thus it is unlikely that we missed any large studies. We were unable to formally assess publication bias as fewer than 10 studies met our inclusion criteria.
Agreements and disagreements with other studies or reviews
No previous systematic reviews have been undertaken looking at the effects of MVA85A.
There has been much debate over the contribution of animal studies to the progression of MVA85A vaccine to trial (Cohen 2018; McShane 2018). We systematically assessed Phase 1 and 2 data and we found no difference in tuberculosis incidence in any population, and no increase in the risk of serious adverse effects attributable to the vaccine. There was a small increase in the risk of experiencing any adverse event.
The findings of this review are consistent in that MVA85A is not efficacious for preventing tuberculosis and that there is no evidence that the MVA85A vaccine caused any serious harm to participants in the trials during its investigation.

Study flow diagram.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
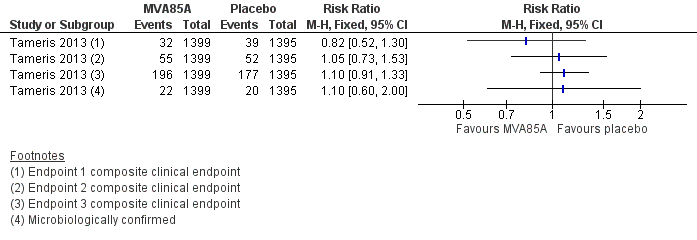
Forest plot of comparison: 2 Comparison of endpoints, outcome: 2.1 Tameris 2013: incidence of tuberculosis according to post‐hoc endpoints.

Forest plot of comparison: 1 MVA85A Vs Placebo, outcome: 1.1 Tuberculosis confirmed by culture or Xpert® MTB/RIF longest reported follow‐up.

Forest plot of comparison: 1 MVA85A versus placebo, outcome: 1.2 Active tuberculosis: started on tuberculosis treatment.
Forest plot of comparison: 1 MVA85A versus placebo, outcome: 1.4 Adverse effects of any severity.
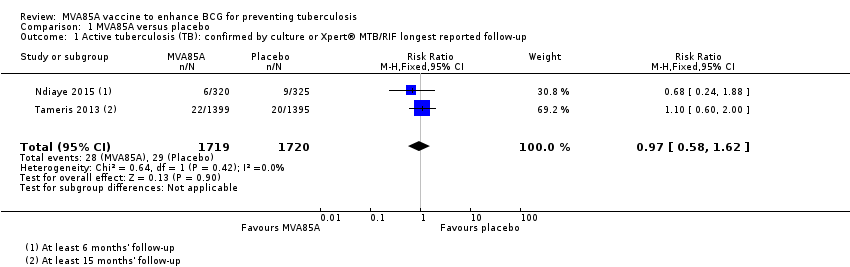
Comparison 1 MVA85A versus placebo, Outcome 1 Active tuberculosis (TB): confirmed by culture or Xpert® MTB/RIF longest reported follow‐up.
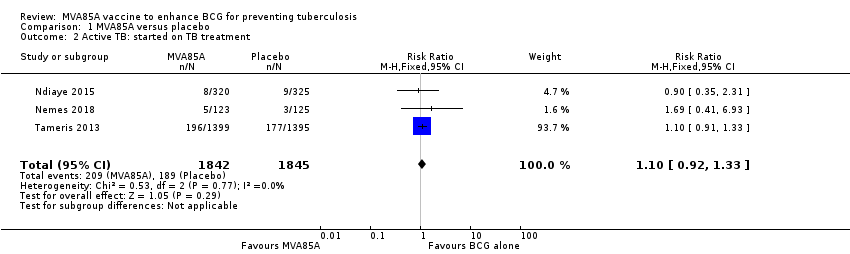
Comparison 1 MVA85A versus placebo, Outcome 2 Active TB: started on TB treatment.

Comparison 1 MVA85A versus placebo, Outcome 3 Latent TB.

Comparison 1 MVA85A versus placebo, Outcome 4 Adverse effects of any severity.
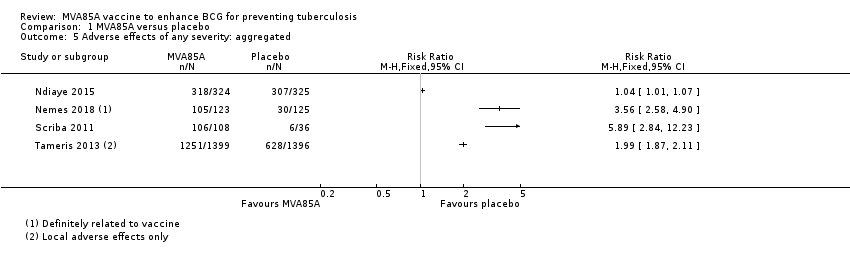
Comparison 1 MVA85A versus placebo, Outcome 5 Adverse effects of any severity: aggregated.

Comparison 1 MVA85A versus placebo, Outcome 6 Serious adverse effects.

Comparison 1 MVA85A versus placebo, Outcome 7 Adverse events of any severity.

Comparison 1 MVA85A versus placebo, Outcome 8 Abnormal biochemical tests.
Comparison 2 Comparison of endpoints, Outcome 1 Tameris 2013: incidence of tuberculosis (TB) according to post‐hoc endpoints.

Comparison 2 Comparison of endpoints, Outcome 2 Ndiaye 2015: incidence of TB according to post hoc defined endpoints.
| MVA85A compared to placebo for preventing tuberculosis | ||||||
| Patient or population: HIV‐positive and ‐negative adults and children | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | Number of participants | Certainty of the evidence | Comments | |
| Risk with placebo | Risk with MVA85A | |||||
| Active tuberculosis: confirmed by culture or Xpert® MTB/RIF longest reported follow‐up | 17 per 1000 | 16 per 1000 | RR 0.97 | 3439 | ⊕⊕⊕⊝ | Vaccinating people with MVA85A in addition to BCG probably made little or no difference to the risk of developing active tuberculosis. |
| Active tuberculosis: started on tuberculosis treatment | 102 per 1000 | 112 per 1000 | RR 1.10 | 3687 | ⊕⊕⊕⊝ | Vaccinating people with MVA85A in addition to BCG probably made little or no difference to the risk of needing to start tuberculosis treatment. |
| Latent tuberculosis | 114 per 1000 | 115 per 1000 | RR 1.01 | 3831 | ⊕⊕⊕⊝ | Vaccinating people with MVA85A in addition to BCG probably made little or no difference to the risk of developing latent tuberculosis. |
| Serious adverse effects | 1 per 1000 | 1 per 1000 | RD 0.00 | 3692 | ⊕⊕⊕⊕ | Vaccinating people with MVA85A in addition to BCG did not cause life‐threatening serious adverse effects. |
| Adverse effects of any severity (local reactions of the skin) | Vaccination with MVA85A was associated with more reactions at the site of the injection.g | — | 3187 | ⊕⊕⊕⊝ | Vaccinating people with MVA85A in addition to BCG probably increased the risk of having an adverse reaction related to vaccination at the site of the injection. | |
| Adverse effects of any severity (systemic symptoms) | Adverse events reported included malaise, lethargy, fever, and vomiting although differences between groups were not significant at a 95% CI level.g | — | 144 (1 RCT) | ⊕⊕⊝⊝ | Vaccinating people with MVA85A in addition to BCG may not have been associated with an increase in adverse effects related to vaccination. | |
| Adverse events of any severity | 808 per 1000 | 849 per 1000 | RR 1.05 | 3836 | ⊕⊕⊕⊕ | Vaccination with MVA85A alone slightly increased the risk of having an adverse event. |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). BCG: Bacillus Calmette‐Guérin; CI: confidence interval; RCT: randomized controlled trial; RD: risk difference; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aNot downgraded for risk of bias. The largest trial was at unclear risk of bias due to selective reporting; however, the outcomes presented were unlikely to be affected by this (Tameris 2013). | ||||||
| NCT trial number | Route | Dates | Intervention and schedule details | Country | Participants (age) | HIV | Adverse events | Reference |
| ID | 2002–2003 | MVA85A; 1 dose | UK | 14 adults (18–45 years) | –ve | 7 trials (112 participants); combined in 1 report: no serious AE attributable to the vaccine | ||
| ID | 2003–2005 | MVA85A; 1 dose, 2 doses (5 × 107 pfu) | Gambia | 21 adults | NR | No serious AE attributable to the vaccine | ||
| ID | 2003–2005 | MVA85A; 1 dose (5 × 107 pfu) | UK | 21 adults | –ve | No serious AE attributable to the vaccine | McShane 2004; Pathan 2012; Rowland 2012; Tanner 2014; Whelan 2009 | |
| ID | 2003–2005 | MVA85A; 1 dose (5 × 107 pfu) | UK | 10 adults | –ve | No serious AE attributable to the vaccine | ||
| ID | 2005–2007 | MVA85A, (5 × 107 pfu) | UK | 12 adults with latent tuberculosis | –ve | No vaccine‐related serious AEs 7 trials (112 participants; data combined in 1 report) | ||
| ID | 2005–2007 | MVA85A; 1 dose (1 × 108 pfu for 12 participants, and 1 × 107 pfu for 12 participants) | UK | 24 adults | –ve | No serious AE attributable to the vaccine | ||
| ID | 2005–2008 | MVA85A (5 × 107 pfu) | South Africa | 36 adults and adolescents | –ve | No vaccine‐related serious AEs | ||
| ID | 2006–2009 | MVA85A; 1 dose MVA85A (2.5 × 107 pfu, 5 × 107 pfu) Groups
| The Gambia | 214 infants (4 months) | NR | No serious AE judged to be related to the vaccine | ||
| ID | 2006–2010 | MVA85A; 1 dose (5 × 107pfu for 10 participants, and 1 × 108 pfu for 10 participants) | UK | 20 adults | +ve | No serious AE attributable to the vaccine | ||
| ID | 2007–2011 | MVA85A; 1 dose (5 × 107 pfu) 4 groups with background of
| South Africa | 48 adults (18–50 years) | +ve | No vaccine‐related serious AEs | ||
| ID | 2007–2010 | FP85A, MVA85A (5 × 107 pfu) | UK | 31 adults | –ve | No serious AE attributable to the vaccine | ||
| ID | 2007–2010 | MVA85A; 1 dose (1 × 108 pfu), administered as 2 injections (5 × 107 pfu each injection) | UK | 12 adults | –ve | 7 trials (112 participants); data combined in 1 report: no serious AE attributable to the vaccine | Porter (unpublished data: source Rowland 2012) | |
| ID | 2008–2011 | MVA85A; 2 doses (spaced by 6–12 months) (1 × 108 pfu) | Senegal | 24 adults | +ve | No serious AE attributable to the vaccine | ||
| ID IM | 2010–2011 | MVA85A; 1 dose (1 × 108 pfu) | UK | 24 adults | –ve | No serious AE attributable to the vaccine | ||
| ID | 2010–2012 | MVA85A, BCG; 1 dose (1 × 108 pfu) Group A: BCG naive, no MVA85A Group C: BCG vaccinated, no MVA85A Group D: BCG vaccinated, MVA85A. | UK | 49 adults recruited; 48 completed study | –ve | No serious AE attributable to the vaccine | ; Harris 2014b; | |
| Aerosol ID | 2011–2013 | MVA85A; 1 dose: 1 × 108, 1 × 107 pfu | UK | 24 adults | –ve | No vaccine related serious adverse effects. | ||
| ID | 2012–2014 | AERAS‐402 MVA85A; Group A: 2 doses AERAS‐402 then MVA85A Group B: 1 dose AERAS‐402 then MVA85A | UK | 40 adults | –ve | No vaccine related serious AEs | ||
| ID | 2013–2014 | MVA85A IMX313; Group A: low‐dose MVA85A‐IMX313 (1 × 107 pfu) Group B: dose MVA85A‐IMX313 (5 × 107 pfu) Group C: MVA85A (5 × 107 pfu) | UK | 30 BCG vaccinated adults | –ve | No vaccine‐related serious AE | ||
| IM | 2013–2016 | MVA85A, ChAdOx1 85A; Group A: 1 dose ChAdOx1 85A Group B: 1 dose ChAdOx1 85A then MVA85A Group C: 2 doses ChAdOx1 85A then MVA85A (1 × 108 pfu) | UK | 42 adults | –ve | No data reported yet | No publication | |
| Aerosol ID | 2013–2016 | MVA85A; Group 1: aerosol then ID Group 2: ID then aerosol Group 3: ID then ID (5 × 107 pfu) | UK | 37 adults | –ve | No data reported yet | (conference abstract) | |
| Aerosol ID | 2015–2018 | MVA85A; 1 × 107 pfu aerosol inhaled, 5 × 107 aerosol and ID | UK | 15 adults | –ve | No data reported yet | ||
| –ve: negative; +ve: positive; AE: adverse event; ART: antiretroviral therapy; BCG: bacillus Calmette‐Guérin; EPI: Expanded Programme on Immunization; ID: intradermal; IM: intramuscular; MTB: Mycobacterium tuberculosis; NR: not reported; pfu: plaque‐forming unit. | ||||||||
| Criterion | Assessment | Explanation |
| Participant‐reported symptoms | ||
| Was monitoring active or passive? | Active Passive Unclear | We classified monitoring as 'active' when authors reviewed participants at set time points and enquired about symptoms. |
| Was blinding for participants and outcome assessors adequate? | Adequate Inadequate Unclear | We classified blinding as 'adequate' when both participants and outcome assessors were blinded to the intervention group, and the methods of blinding (including use of a placebo) were described. |
| Was outcome data reporting complete or incomplete? | Complete Incomplete | We classified outcome data reporting as 'complete' when data were presented for all the time points where it was collected. |
| Were all participants included in reporting? | Yes No | We reported the percentage of randomized participants included in adverse event reporting. |
| Was the analysis independent of study sponsor? | Yes No Unclear | We classified the analysis of trials sponsored by pharmaceutical companies as independent of the sponsor when it was clearly stated that the sponsor had no input to the trial analysis |
| Laboratory tests | ||
| Number of tests undertaken | — | We extracted the type and number of laboratory tests were taken. |
| Timing of tests: was number and timing of tests adequate? | Adequate Inadequate | We classified the number and timing of tests as 'adequate,' when tests were taken at baseline, plus 2 other time points within the first week after treatment, plus the last day of the study. We classified the number of test taken as 'inadequate,' if either the laboratory controls in the first week or controls at 4 weeks were not performed. |
| Reporting of test results: was reporting of test results complete? | Complete Incomplete | We classified reporting as 'complete' when test results of all time points were reported. For the trials with inadequate number of tests taken, we considered completeness of reporting as inconsequential, and therefore did not record a judgement. |
| Independence of data analysis: was data analysis independent? | Yes No Unclear | We classified the analysis of trials sponsored by pharmaceutical companies as independent of the sponsor when it is clearly stated that the sponsor had no input to the trial analysis. |
| Adapted from Bukirwa 2014. | ||
| Study | Endpoint 1 | Endpoint 2 | Endpoint 3 |
| Any of the following criteria.
| "Included all infants who met endpoint 1 criteria; had marginally less stringent criteria to define TB infection and household exposure." Any of the following numerical categories.
| All participants placed on treatment for TB by a health professional with the intent of treating TB regardless of whether they have met the other efficacy endpoints. | |
| Revised endpoint 1 from Tameris 2013 that removed QFT conversion from the diagnostic criteria to avoid bias towards association with QFT status. | Not used | Not used | |
| Not used | Not used | Same definition as for Tameris 2013. | |
| Any of the following numerical categories.
| Any of the following numerical categories:
| Same definition as for Tameris 2013. | |
| Not applicable | Not applicable | Not applicable | |
| Outcomes not specified in the methods section. In results, authors specified that 8 participants were diagnosed as TB: "of whom one was M.tb [Mycobacterium tuberculosis] culture positive and 7 were diagnosed on clinical/ radiographic grounds and TB contact history. Two of the TB cases were QFT positive." | Not used | Not used | |
| AFB: acid‐fast bacilli; CSF: cerebrospinal fluid; CT: computerized tomography; QFT: quantiFERON; TB: tuberculosis. | |||
| Study | Protocol | Published findings | Differences between protocol and published findings | ||
| Stated outcomes published prior to commencement of trial that differ to published outcomes | Measurement of outcome as stated a priori | Measurement of outcome as stated in published findings | Reported findings | ||
| No protocol published. | |||||
| No protocol published (extended post‐trial follow‐up of Tameris 2013). | |||||
| Adverse events: blood tests a | "Percentage of participants with adverse events" AEs measured up to day 28 SAEs measured up to 6 months. | "Phlebotomy for routine haematological and biochemical analysis was done at screening, before booster vaccination, and on days 7 and 28 after each vaccination." | "Routine haematological and biochemical test results did not differ between study groups (data not shown)." | Haematological and biochemical blood tests not outlined as a measure of safety in the study protocol. Blood test findings reported unclearly. | |
| Safety | Clinicaltrials.gov – local, regional, and systemic AEs and SAEs which would be reported as cumulative 12‐month incidences. | "Infants followed for safety end points at weeks 1, 4, 6, and 8 after MVA85A/control vaccination and thereafter, at weeks 9, 12, and 16 (corresponding to weeks 1, 4, and 8 following delayed BCG vaccination at 8 weeks of age), and at week 52." | Reported total events for AEs per group after MVA85A and before BCG and for whole follow‐up period. Data including for laboratory AEs were not disaggregated as prespecified. | Data including for laboratory AEs were not disaggregated as prespecified. | |
| Safetya | Local and systemic AEs for the first week. | Diary cards | Local and systemic AEs reported on ≥ 1 day of the first 7 days after MVA85A vaccination. | None | |
| Blood tests (days 7, 28) | Biochemical and haematological tests (days 7, 28) | Reported number and percentages of participants with abnormal results and reported that, "all except one patient that had elevated liver enzymes remained unresolved by day 28." | |||
| Immunology | ESAT‐6/CFP‐10 | Infants converted – suggestive of TB infection but seemed to be reported as safety data not efficacy. | |||
| Safety profile – AEsa | AEs measured up to day 28 SAEs measured throughout follow‐up. | Collected data on solicited and unsolicited local and systemic AEs. Active surveillance for SAEs. | AEs broken down by type of event and reported in supplementary material. Only local events at the injection site were considered to be related to the vaccine. | Causal relationship with AEs other than local injection site reactions was not reported. | |
| Safety profile – blood testsa | Testing up to 28 days postvaccination. | "Peripheral blood for routine haematological and biochemical tests was taken at screening and on day 7 and day 28 after vaccination in an initial safety cohort of at least 330 infants." | Not reported | Primary outcome not reported | |
| Efficacy of MVA85Ab | Using an endpoint derived from epidemiological cohort surveys in BCG vaccinated infants. | Not reported – simply stated clinical endpoints 'developed.' | Composite clinical endpoints 1, 2, 3 (see Table 3) Microbiologically confirmed cases reported in appendix. | The "primary efficacy endpoint" was measured using an endpoint not derived from cohort studies. The endpoint definition differed from all other implied or reported ways of measuring efficacy in the other studies. The point estimate showed clinically significant benefit for endpoint 1 (no benefit seen at the 95% confidence level). This endpoint was reported as the main efficacy finding. All other point estimates show no clinically significant benefit or harm. | |
| AE: adverse events; BCG: bacillus Calmette‐Guérin; ESAT‐6/CFP‐10: early secretory antigenic‐6/culture filtrate protein‐10; SAE: severe adverse events; TB: tuberculosis. | |||||
| Study | Participant reported adverse events | Outcome data reporting | Laboratory tests | ||||||||
| Monitoring active or passive | Blinding of participants or outcome assessors | Times data collected | Times data reported | Complete/not complete | Percentage of participants reported on | Analysis independent of study sponsor | Number of tests taken | Timing of tests and adequacy | Complete reporting of test results | Independence of data analysis | |
| Active | Inadequate | 60 min, D 2, 7, 28, 84, and 168 | D 7, 28 | Incomplete | 100% | Unclear | Biochemistry and haematology | Inadequate | Inconsequential | Unclear | |
| Active | Inadequate | D 7, 28, and 84 after boost 3 monthly until end of study | NR | Incomplete | 99.8% | No | Haematology, chemistry, virological markers | Adequate | Incomplete | No | |
| Active | Adequate | Baseline, D 7 and 28, throughout up to D 84 | NR | Incomplete | 99.9% | No | Biochemistry and haematology | Inadequate | Incomplete | No | |
| Active | Adequate | Week 1, 4, 6, 8, 16, and 52 | NR | Incomplete | 85.9% | Unclear | Not specified | Adequate | Incomplete | Unclear | |
| D: day; min: minute; NR: not reported. | |||||||||||
| Active TB | ||||||||||||
| MVA85A | Placebo | MVA85A | Placebo | MVA85A | Placebo | MVA85A | Placebo | MVA85A | Placebo | MVA85A | Placebo | |
| Endpoint 1a | 32/1399 (2.3%) | 39/1395 (2.8%) | 58/2797 (2.1%) with NDD | N/A | N/A | 6/320 (1.9%) | 9/325 (2.8%) | N/A | N/A | 5/123 (4.1%) | 3/125 (2.4%) | |
| Endpoint 2a | 55/1399 (3.9%) | 52/1395 (3.7%) | N/A | N/A | N/A | N/A | 6/320 (1.9%) | 9/325 (2.8%) | N/A | N/A | N/A | N/A |
| Endpoint 3a | 196/1399 (14.0%) | 177/1395 (12.6%) | N/A | N/A | 3.3/100 pyo (95% CI 2.9 to 3.9) | 3.0/100 pyo (95% CI 2.6 to 3.5) | 8/320 (2.5%) | 9/325 (2.8%) | N/A | N/A | N/A | N/A |
| CI: confidence interval; N/A: not applicable; NDD: no disaggregated data; pyo: person‐years of observation; TB: tuberculosis. | ||||||||||||
| Study | MVA85A | Placebo | Breakdown | Author conclusions | ||||
| Number of participants with ≥ 1 event caused by the intervention | Total participants | Number of participants with ≥ 1 event caused by the control | Total participants | Detailed AEs | MVA85A | Placebo | ||
| 318 | 324 | 307 | 325 | Solicited AEsa | 288 | 235 | "Solicited adverse events were more common in MVA85A group and most were local injection site reactions." | |
| 105b | 123 | 30b | 125 | Not detailed | N/A | N/A | "Infants in MVA85A arm were more likely to experience an AE than in control arm. Injection site reactions were more frequent in MVA85A recipients and mild." | |
| 106c | 108c | 6 | 36 | Injection sited | 106 | 6 | "Desquamation significantly increased with greater vaccine dose." | |
| Malaise | 6 | 1 | ||||||
| Lethargy | 6 | 2 | ||||||
| Tactile fever | 18 | 0 | ||||||
| Documented fever | 13 | 2 | ||||||
| Vomiting | 6 | 2 | ||||||
| Elevated liver enzyme levels | 13 | 4 | ||||||
| Increased white cell count | 0 | 1 | ||||||
| Local 1251e | 1399 | Local 628e | 1396 | Not detailed | 1251 | 628 | None | |
| AE: adverse event; N/A: not applicable. | ||||||||
| Study | Adverse events of any severity | |||
| MVA85A | Placebo | |||
| 1120/1399 (80.1%) | 1059/1396 (75.9%) | |||
| NR | NR | |||
| NR | NR | |||
| 321/324 (99.1%) | 312/325 (96%) | |||
| 2.5 × 107 pfu = 35 μL | 5 × 107 pfu = 70 μL | 1 × 108 pfu = 135 μL | 1/36 | |
| 1/36 | 3/36 | 6/36 | ||
| Mild 122/123 (99.2%) | 121/125 (96.8) | |||
| Moderate 62/123 (50.4%) | 54/125 (3.6%) | |||
| Severe 11/123 (8.9%) | 14/125 (11.2%) | |||
| NR: not reported; pfu: plaque‐forming unit. | ||||
| Study | Haematological blood tests | Biochemical blood tests | ||||
| MVA85A | Placebo | MVA85A | Placebo | |||
| NR | NR | NR | NR | |||
| NR | NR | NR | NR | |||
| NR | NR | NR | NR | |||
| NRa | NRa | NRa | NRa | |||
| 0/108b | 1/36b | 2.5 × 107 pfu = 35 μL | 5 × 107 pfu = 70 μL | 1 × 108 pfu = 135 μL | 4/36 (11%) | |
| 1/36 (2.8%) | 3/36 (8.3%) | 9/36 (25%) | ||||
| NR | NR | 14/123 (11.4%) | 13/125 (10.4%) | |||
| NR: not reported; pfu: plaque‐forming unit. | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Active tuberculosis (TB): confirmed by culture or Xpert® MTB/RIF longest reported follow‐up Show forest plot | 2 | 3439 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.58, 1.62] |
| 2 Active TB: started on TB treatment Show forest plot | 3 | 3687 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.92, 1.33] |
| 3 Latent TB Show forest plot | 4 | 3831 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.85, 1.21] |
| 4 Adverse effects of any severity Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Local: skin | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Malaise | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Lethargy | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 Any fever | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 Vomiting | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse effects of any severity: aggregated Show forest plot | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6 Serious adverse effects Show forest plot | 3 | 3692 | Risk Difference (M‐H, Fixed, 95% CI) | 0.00 [‐0.00, 0.00] |
| 7 Adverse events of any severity Show forest plot | 4 | 3836 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [1.02, 1.08] |
| 8 Abnormal biochemical tests Show forest plot | 2 | 392 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.60, 1.97] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Tameris 2013: incidence of tuberculosis (TB) according to post‐hoc endpoints Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Ndiaye 2015: incidence of TB according to post hoc defined endpoints Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |