Certolizumab pegol para inducir la remisión de la enfermedad de Crohn
Resumen
Antecedentes
La enfermedad de Crohn (EC) es una enfermedad inflamatoria crónica del tubo digestivo, y la modulación de la respuesta inmunitaria es la principal estrategia de tratamiento para inducir la remisión de la EC activa. El certolizumab pegol (CZP) es un inhibidor del factor de necrosis tumoral alfa (TNF‐α) que regula el deterioro de la respuesta inmunitaria.
Objetivos
Los objetivos primarios fueron evaluar la eficacia y la seguridad del CZP para la inducción de la remisión de la EC.
Métodos de búsqueda
Se realizaron búsquedas en MEDLINE, Embase, CENTRAL, el registro especializado del Grupo Cochrane de EII, los registros de ensayos y otras fuentes desde su inicio hasta el 28 de enero de 2019. Además, se estableció contacto con la compañía farmacéutica que fabrica CZP.
Criterios de selección
Se incluyeron ensayos controlados aleatorios que comparaban CZP con placebo o ningún tratamiento en pacientes con EC activa.
Obtención y análisis de los datos
Se utilizaron los procedimientos metodológicos Cochrane estándar. Los resultados principales seleccionados para el análisis GRADE fueron la remisión clínica en la semana ocho (Crohn’s Disease Activity Index [CDAI] ≤ 150), la respuesta clínica en la semana ocho (reducción en el CDAI ≥ 100 o remisión clínica) y los eventos adversos graves. Para los análisis estadísticos se aplicó el método de efectos aleatorios de Mantel‐Haenszel. Para los resultados dicotómicos, se calculó el cociente de riesgos (CR) y el intervalo de confianza (IC) del 95% correspondiente.
Resultados principales
Cuatro estudios con 1 485 participantes con EC moderada a grave cumplieron con los criterios de inclusión y se utilizaron en los metanálisis. Todos los estudios incluyeron a pacientes con EC activa con un CDAI que oscilaba entre 220 y 450. La mayoría de los pacientes eran adultos mayores de 18 años de edad. Un estudio se identificó como de alto riesgo de sesgo debido a la administración de un placebo no idéntico, mientras que los otros estudios se consideraron en riesgo bajo de sesgo.
El CZP (100 mg a 400 mg cada 2 a 4 semanas) demostró ser superior al placebo para lograr la remisión clínica en la semana ocho (CR 1,36; IC del 95%: 1,11 a 1,66; evidencia de certeza moderada). Los números de participantes en bruto que lograron la remisión clínica en la semana ocho fueron 26,9% (225/835) y 19,8% (129/650) en los grupos de CZP y de placebo, respectivamente.
Se demostró que el CZP fue superior al placebo para lograr una respuesta clínica en la semana ocho (CR 1,29; IC del 95%: 1,09 a 1,53; evidencia de certeza moderada). En números brutos, la respuesta clínica a la semana ocho se logró en el 40,2% (336/835) y el 30,9% (201/650) de los participantes en los grupos de CZP y de placebo, respectivamente.
En números brutos, se observaron eventos adversos graves en el 8,7% (73/835) y el 6,2% (40/650) de los participantes de los grupos de CZP y de placebo, respectivamente (CR 1,35; IC del 95%: 0,93 a 1,97; evidencia de certeza moderada). Los eventos adversos graves incluyeron el empeoramiento de la enfermedad de Crohn, las infecciones y las neoplasias malignas.
Conclusiones de los autores
La evidencia de certeza moderada indica que el CZP es efectivo para la inducción de la remisión clínica y la respuesta clínica en pacientes con EC activa. No está claro si difiere el riesgo de eventos adversos graves entre el CZP y el placebo, debido a que el IC del 95% incluye la posibilidad de una pequeña disminución o duplicación de los eventos. Se necesitan estudios futuros para evaluar la eficacia y la seguridad a largo plazo del CZP en pacientes con EC.
PICOs
Resumen en términos sencillos
Certolizumab pegol para el tratamiento de la enfermedad de Crohn activa
Pregunta de la revisión
Se examinó la evidencia sobre los efectos beneficiosos y perjudiciales del certolizumab pegol en pacientes con enfermedad de Crohn activa.
Antecedentes
La enfermedad de Crohn es una enfermedad inflamatoria crónica que afecta principalmente el tubo digestivo, como el intestino delgado y el intestino grueso. Los síntomas frecuentes de la enfermedad de Crohn son la diarrea crónica, el dolor abdominal y la pérdida de peso. Cuando los pacientes con enfermedad de Crohn presentan síntomas, se considera que la enfermedad está "activa". Durante la "remisión", los pacientes no presentan síntomas.
El certolizumab pegol es un fármaco biológico utilizado para modificar la respuesta inmunitaria excesiva que causa la inflamación crónica en la enfermedad de Crohn. Por lo general el certolizumab pegol se inyecta debajo de la piel cada dos a cuatro semanas.
Características de los estudios
Se realizaron búsquedas en la literatura hasta el 28 de enero de 2019. Cuatro estudios con 1 485 pacientes compararon certolizumab pegol con placebo (un fármaco simulado). Todos los estudios incluyeron a pacientes con enfermedad de Crohn activa. La mayoría de los pacientes eran adultos mayores de 18 años de edad, excepto seis pacientes de 16 ó 17 años de edad. Todos los estudios fueron financiados por el fabricante del fármaco.
Resultados clave
En un análisis combinado de los cuatro estudios, los pacientes con enfermedad de Crohn activa que recibieron certolizumab pegol en una dosis que osciló entre 100 mg y 400 mg cada dos a cuatro semanas, respondieron al tratamiento y lograron la remisión a las ocho semanas con más frecuencia que los pacientes que recibieron placebo. No se observaron diferencias considerables en la tasa de efectos secundarios graves entre el certolizumab pegol y el placebo. Los efectos secundarios graves incluyeron el empeoramiento de la enfermedad de Crohn, las infecciones y las neoplasias malignas (es decir, cáncer).
Calidad de la evidencia
La evidencia de certeza moderada indica que el certolizumab pegol es beneficioso para lograr la remisión en pacientes con enfermedad de Crohn de moderada a grave. Debido al número escaso de efectos secundarios graves, la certeza de la evidencia sobre los efectos perjudiciales del certolizumab pegol fue moderada.
Conclusions
La evidencia de certeza moderada indica que el certolizumab pegol es efectivo para la inducción de la remisión clínica y la respuesta clínica en pacientes con enfermedad de Crohn activa. No está claro si el riesgo de efectos secundarios graves difiere entre el certolizumab pegol y el placebo. Se necesitan estudios futuros para evaluar los efectos beneficiosos y perjudiciales a largo plazo del certolizumab pegol en pacientes con enfermedad de Crohn.
Conclusiones de los autores
Summary of findings
| Certolizumab pegol compared to placebo for induction of remission in Crohn's disease | ||||||
| Patient or population: Patients with active Crohn's disease Settings: Outpatient Intervention: Certolizumab pegol Comparison: Placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Certolizummab pegol | |||||
| Clinical remission Follow‐up: 8 weeks | 198 per 1000 | 270 per 1000 (220 to 329) | RR 1.36 (1.11 to 1.66) | 1485 | ⊕⊕⊕⊝ | Certolizumab pegol was shown to be superior to placebo regarding clinical remission at week 8 Clinical remission was defined as a CDAI < 150 |
| Clinical response Follow‐up: 8 weeks | 309 per 1000 | 399 per 1000 (337 to 473) | RR 1.29 (1.09 to 1.53) | 1485 (4 studies) | ⊕⊕⊕⊝ | Clinical response was defined as CDAI reduction ≥ 100 from baseline |
| Serious adverse events Follow‐up: 8 weeks | 62 per 1000 | 83 per 1000 (57 to 121) | RR 1.35 (0.93 to 1.97) | 1485 (4 studies) | ⊕⊕⊕⊝ | Reported serious adverse events included worsening Crohn's disease, infections, and malignancy |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Downgraded one level due to high risk of bias in one study in the pooled analysis 2 Downgraded one level due to imprecision (113 events) | ||||||
Antecedentes
Descripción de la afección
La enfermedad de Crohn (EC) es una enfermedad inflamatoria crónica que afecta principalmente el tubo digestivo. La EC es más común en América del Norte y Europa que en otras regiones. No obstante, tanto la incidencia como la prevalencia de la EC están aumentando en todo el mundo. Se informó que la incidencia anual más alta de EC fue de 5,0; 12,7 y 20,2 por cada 100 000 personas/año en Asia y el Oriente Medio, Europa y América del Norte, respectivamente. Además, se informó que la prevalencia más alta de EC fue de 67,9; 322 y 318,5 por cada 100 000 personas en Asia y el Oriente Medio, Europa y América del Norte, respectivamente (Molodecky 2012).
Los síntomas comunes de la EC incluyen diarrea crónica, dolor abdominal y pérdida de peso (Torres 2016), y los pacientes habitualmente son diagnosticados con EC entre los 20 y los 30 años (Cosnes 2011). Hasta un tercio de los pacientes con EC presentaban complicaciones tales como enfermedades penetrantes y estenosis en el momento del diagnóstico, y la mitad de dichos pacientes necesitaron cirugía dentro de los 10 años posteriores al diagnóstico (Peyrin‐Biroulet 2010). Después de la cirugía inicial, una cuarta parte de los pacientes requirieron una segunda cirugía en un plazo de cinco años (Frolkis 2014). Además, el riesgo de mortalidad ajustado por edad en pacientes con EC fue 50% mayor que el de la población general (Canavan 2007).
Se desconoce la etiología de la EC, aunque se considera que el deterioro de la respuesta inmunitaria anormal de la mucosa y de la función de barrera desempeñan un papel importante en la patogénesis de la EC. Se ha postulado que la alteración de la microflora intestinal y los factores ambientales, como los alimentos y el tabaquismo, causan disfunción del sistema inmunológico en individuos genéticamente susceptibles (Torres 2016). La clave para el tratamiento de la EC es la regulación de la respuesta inmunitaria deficiente.
Descripción de la intervención
La estrategia de tratamiento actual para inducir la remisión de la EC activa se basa en la modulación de la respuesta inmunitaria. Los tratamientos farmacológicos para la inducción de la remisión de la EC incluyen corticosteroides, inhibidores del factor de necrosis tumoral alfa (TNF‐α), anticuerpos contra la α4β7 integrina y anticuerpos contra la interleucina‐12/23p40. El TNF‐α es una citocina pro‐inflamatoria y desempeña un papel central en la cascada inflamatoria de la EC. La regulación del deterioro de la respuesta inmune con inhibidores del TNF‐α puede ser clave para el tratamiento de la EC (Baumgart 2012; Nielsen 2013; Olesen 2016; Torres 2016).
El certolizumab pegol (CZP) es un inhibidor del TNF‐α. A diferencia de otros inhibidores del TNF‐α como el infliximab (IFX) y el adalimumab (ADA), el CZP es un fragmento Fab de polietileno glicolado de un anticuerpo monoclonal humanizado anti‐TNF‐α con alta afinidad por el TNF‐α. El CZP no tiene porción Fc y tiene un perfil mecanicista diferente al de otros inhibidores del TNF‐ α debido a su estructura única. El fármaco carece de la capacidad de inducir la formación de macrófagos reguladores, citotoxicidad celular dependiente de anticuerpos, citotoxicidad dependiente del complemento y apoptosis mediante señalización inversa. Sin embargo, el CZP puede inhibir los mediadores inflamatorios y aumentar la actividad reguladora de las células T con la misma eficacia que el IFX y el ADA (Gomollon 2017; Nesbitt 2007; Olesen 2016; Shao 2009; Torres 2016).
Los inhibidores del TNF‐α, incluido el CZP, se recomiendan para la EC de actividad moderada a grave (Gomollon 2017; Talley 2011; Terdiman 2013; Torres 2016). El CZP está aprobado para el tratamiento de la EC en Estados Unidos y Suiza (Olesen 2016; Torres 2016). El CZP es un fármaco que se administra por vía subcutánea y puede autoadministrarse. Los posibles eventos adversos graves del CZP son la reacción anafiláctica, el trastorno linfoproliferativo, la reactivación de la tuberculosis y las infecciones oportunistas (Gomollon 2017).
De qué manera podría funcionar la intervención
El CZP inhibe la activación de los receptores del TNF‐α mediante la neutralización tanto de la forma transmembrana del TNF‐α (tmTNF) como de la forma soluble del TNF‐α (sTNF). Actualmente, se considera que la señalización del tmTNF tiene un papel central en la patogénesis de la EC, y el CZP puede unirse tanto al sTNF como al tmTNF. El CZP regula el deterioro de la respuesta inmunitaria a través de los siguientes mecanismos de acción posibles: aumento de la frecuencia y la actividad de las células T reguladoras, supresión de los mediadores inflamatorios en las células inmunitarias, disminución de los mediadores inflamatorios mediante la señalización inversa en las células que expresan el tmTNF, y citotoxicidad no apoptótica y apoptosis mediante el bloqueo de la activación de los receptores del TNF‐α mediada por el tmTNF (Olesen 2016).
Por qué es importante realizar esta revisión
Los metanálisis recientes han mostrado resultados inconsistentes, que pueden deberse a las diferencias en la metodología y en los puntos temporales seleccionados para la evaluación de la remisión clínica (Ford 2011; Kawalec 2013). En una revisión (Ford 2011), no hubo diferencias entre el CZP y el placebo en la proporción de participantes que no lograron la remisión en las semanas 6 a 12 (cociente de riesgos [CR] 0,95; intervalo de confianza [IC] del 95%: 0,90 a 1,01). Otra revisión (Kawalec 2013) encontró un beneficio del CZP sobre el placebo para la inducción de la remisión en la cuarta semana (CR 1,63; IC del 95%: 1,32 a 2,13). También se tiene conocimiento de al menos un ensayo no publicado (NCT00291668). La presente revisión resumirá e integrará de forma adecuada toda la evidencia disponible, incluidos los ensayos controlados aleatorios (ECA) no publicados, para proporcionar la mejor evidencia disponible con objeto de evaluar la eficacia y la seguridad del CZP para la inducción de la remisión de la EC.
Objetivos
Los objetivos primarios fueron evaluar la eficacia y la seguridad del CZP para la inducción de la remisión de la EC.
Métodos
Criterios de inclusión de estudios para esta revisión
Tipos de estudios
Esta revisión incluyó ECA independientemente del estado de publicación. No se aplicaron restricciones de idioma.
Tipos de participantes
Los participantes incluyeron a pacientes (≥ 16 años de edad) con EC activa que se diagnosticó mediante una evaluación clínica, endoscópica, radiográfica e histopatológica estándar. La EC activa se definió como una puntuación del Crohn’s Disease Activity Index (CDAI) superior a 150 o una puntuación del Harvey‐Bradshaw Index (HBI) superior a 4 (Best 1976; Harvey 1980).
Tipos de intervenciones
La administración subcutánea de cualquier dosis de CZP cada dos a cuatro semanas en comparación con placebo o ningún tratamiento. Los comparadores activos como el tratamiento convencional (incluido el ácido 5‐aminosalicílico, los inmunomoduladores o los corticosteroides) no se incluyeron en esta revisión.
Tipos de medida de resultado
Resultados primarios
La medida de resultado primaria fue la proporción de pacientes con EC que lograron en la remisión en la semana ocho después de la administración del CZP. Se seleccionó la semana ocho debido a que dicha semana es el tiempo recomendado para cambiar a la dosificación de mantenimiento de acuerdo con el régimen de dosificación aprobado (Schreiber 2011). Cuando el resultado no se evaluó en la semana ocho, se seleccionó la semana más cercana entre las semanas cuatro y 12 como el punto de evaluación del resultado. Cuando solo se dispuso de fechas de dos puntos temporales igualmente distantes de la semana ocho, como las semanas seis y 10; a pesar de haber consultado a los investigadores originales, se planificó la selección del punto más temprano (semana 6). La remisión se definió como un CDAI ≤ 150 o un HBI ≤ 4. Cuando tanto el CDAI como el HBI se informaron en los estudios primarios, se planificó utilizar el CDAI para la evaluación de resultados. La proporción de participantes en remisión se calculó de acuerdo con el principio de intención de tratar (intention‐to‐treat); el denominador fue el número de pacientes asignados al azar en cada brazo. Se asumió que los participantes con datos faltantes para la medida de resultado primaria eran fracasos del tratamiento.
Resultados secundarios
Los resultados secundarios incluyeron la proporción de participantes con:
-
respuesta clínica en la octava semana;
-
mejoría en la proteína C reactiva (PCR) en la octava semana;
-
calidad de vida relacionada con la salud en la octava semana;
-
mejoría endoscópica en la semana doce;
-
cierre de la fístula en la octava semana;
-
eventos adversos;
-
eventos adversos graves; y
-
retiros debidos a los eventos adversos.
La respuesta clínica se definió como una reducción del CDAI desde el valor inicial mayor o igual a 100 o la remisión (CDAI ≤ 150) o una reducción del HBI desde el valor inicial mayor o igual a 3 o la remisión (HBI ≤ 4) (Vermeire 2010). Para los resultados que no estaban disponibles para la semana ocho, se seleccionó la semana más cercana entre las semanas cuatro y 12. Cuando solo se dispuso de dos puntos de evaluación igualmente distantes de la semana ocho, como las semanas seis y 10, a pesar de consultar con los investigadores originales, se planificó seleccionar el punto más temprano (semana 6). Se siguió este procedimiento para todos los resultados que se pretendía evaluar a las ocho semanas. La evaluación de la mejoría en la proteína C reactiva (PCR) en la octava semana se basó en escalas logarítmicas de los cocientes de la media geométrica de PCR entre el inicio y la octava semana. La evaluación de la calidad de vida relacionada con la salud en la octava semana se basó en el cambio medio en las puntuaciones de la calidad de vida a partir del valor inicial medido con un instrumento validado que incluía el Inflammatory Bowel Disease Questionnaire (IBDQ) (Guyatt 1989) (Guyatt 1989) o el cuestionario Medical Outcomes Study Short‐Form 36 (SF‐36) (Ware 1992). Cuando no se informaron resultados endoscópicos para la semana 12 se planificó seleccionar la semana más cercana entre las semanas ocho y 26. También se planificó evaluar la mejoría endoscópica mediante el cálculo del cambio medio desde el inicio en el Crohn’s Disease Endoscopic Index of Severity (CDEIS), la Simplified Endoscopic Activity Score for Crohn’s Disease (SES‐CD), o la puntuación Rutgeerts (Daperno 2004; Mary 1989; Rutgeerts 1990). Cuando solo se dispuso de dos puntos de evaluación igualmente distantes de la semana 12; como las semanas 10 y 14; a pesar de consultar con los investigadores originales, se planificó seleccionar el punto más temprano (semana 10). Los eventos adversos y los eventos adversos graves se basaron en lo que se informó en los estudios primarios.
Métodos de búsqueda para la identificación de los estudios
Búsquedas electrónicas
We conducted a comprehensive literature search without language restrictions. We searched the following databases to identify relevant RCTs:
-
MEDLINE (inception to date);
-
Embase (inception to date);
-
CENTRAL;
-
The Cochrane IBD Group Specialized Register (inception to date);
-
http://ClinicalTrials.gov (trial registry);
-
https://www.clinicaltrialsregister.eu/ (EU Clinical Trials Register);
-
http://apps.who.int/trialsearch/ (International Clinical Trials Registry Platform); and
-
http://www.ucb.com/our‐science/Our‐clinical‐studies/cimzia‐certolizumab‐pegol (web site of a pharmaceutical company producing CZP).
The search strategies are reported in Appendix 1.
Búsqueda de otros recursos
To identify additional studies, we searched the following resources manually or through personal contacts:
-
Abstracts of Digestive Disease Week, United European Gastroenterology Week, European Crohn’s and Colitis Organization Congress, and Advances in Inflammatory Bowel Diseases (2000 to date);
-
References from published articles; and
-
Pharmaceutical companies and experts involved in the development of CZP.
Obtención y análisis de los datos
Selección de los estudios
Two authors (HY and RS) independently screened titles and abstracts, and selected potential eligible studies based on the above criteria. In addition, these authors independently read the full‐text articles of the potential eligible studies and decided which studies should be included in the review. In cases of insufficient information, we contacted the authors of the primary studies to evaluate eligibility for inclusion. In the event of a disagreement regarding study selection, HY and RS discussed the matter together to reach a consensus. NW acted as the arbitrator when consensus was not reached.
Extracción y manejo de los datos
Two authors (HY and RS) independently extracted data using data extraction forms to record data from the selected studies. Any disagreements were resolved through discussion. NW was the arbitrator when consensus was not reached.
We extracted the following data:
-
Characteristics of the primary studies: publication year, country, study recruitment period, study completion date, study type, and risk of bias items;
-
Participant characteristics: country, total number of participants, number of participants randomized, number of participants analyzed in each group, age, sex, ethnicity, body mass index, disease duration, disease site, smoking status, CDAI score, HBI score, CDEIS, SES‐CD, Rutgeerts score, IBDQ score, SF‐36 score, CRP, fistula, concurrent CD treatment, previous CD treatment, inclusion criteria, and exclusion criteria;
-
Intervention characteristics: dose, delivery route, and administration schedule of CZP;
-
Comparator characteristics: placebo or no treatment control;
-
Outcomes: proportion of participants who achieved clinical remission at week 8, proportion of participants with clinical response at week 8, CDAI score at week 8, HBI score at week 8, CDEIS at week 12, SES‐CD at week 12, Rutgeerts score at week 12, IBDQ score at week 8, SF‐36 score at week 8, CRP at week 8, fistula closure at week 8, any adverse events, adverse events leading to withdrawal, serious adverse events, time of outcome assessment in primary studies, length of follow‐up, number of participants lost to follow‐up, reasons for loss to follow‐up, number of participants who did not complete treatment, reasons for incomplete treatment, and criteria for evaluating outcomes in primary studies.
We contacted the authors of the primary studies and the pharmaceutical company that manufactures CZP if information in the published reports was insufficient.
Evaluación del riesgo de sesgo de los estudios incluidos
Two authors (HY and RS) independently assessed the quality of included studies using the Cochrane risk of bias tool (Higgins 2011). Any disagreements were resolved by consensus with a third author (NW). Primary studies were rated as high, low, or unclear risk of bias. We assessed the following risk of bias items: random sequence generation (selection bias), allocation concealment (selection bias), blinding of participants and personnel (performance bias), blinding of outcome assessment (detection bias), incomplete outcome data (attrition bias), selective reporting (reporting bias), and other potential sources of bias.
We rated random sequence generation as low risk of bias if the method for random sequence generation was described as a random number table, computer‐generated, coin tossing, shuffling cards or envelopes, throwing dice, drawing of lots or minimization. We rated random sequence generation as high risk of bias if the method of generation was not random. Examples included a systematic approach, such as date or record number, or a non‐systematic approach, such as preference and availability. We rated random sequence generation as unclear risk of bias if insufficient information was reported to allow for a judgement.
We rated allocation concealment as low risk of bias if allocation could not be foreseen by participants and investigators. Adequate methods of allocation concealment included centralized allocation such as telephone, web‐based, or pharmacy‐controlled randomization, sequentially numbered drug containers of the same appearance, or sequentially numbered, opaque, sealed envelopes. We rated allocation concealment as high risk of bias if the allocation sequence was likely to be foreseen. Examples included an open random allocation schedule or envelopes without safeguards. We rated allocation concealment as unclear risk of bias if insufficient information was reported to allow for a judgement.
We rated blinding of participants and personnel and blinding of outcome assessors as low risk of bias if proper methods were employed to prevent knowledge of treatment assignment (e.g. double‐blinding with an identical placebo) or if no blinding or incomplete blinding of participants or personnel was unlikely to affect assessment of the outcome (e.g. a serious adverse event resulting in death). We rated blinding of participants and personnel and blinding of outcomes assessors as high risk of bias if blinding was likely to be broken and this could affect outcome assessment, or if there was no blinding or incomplete blinding of participants or personnel or outcome assessors which could affect outcome assessment. We rated blinding of participants and personnel and blinding of outcome assessors as unclear risk of bias if insufficient information was reported to allow for a judgement.
We rated incomplete outcome data as low risk of bias when there were no missing outcome data; when missing outcome data were unlikely to be related to the true outcome; when the number of dropouts and reasons for withdrawal were balanced between treatment groups; when compared to the observed event, the proportion of missing outcomes did not have a clinically relevant impact on the effect estimate for dichotomous outcomes; when the expected effect size among missing outcomes did not have clinically relevant impact on the observed effect size for continuous outcome data; or when missing data were imputed using proper methods. We rated incomplete outcome data as high risk of bias when missing outcome data were likely to be related to the true outcome; when numbers or reasons for missing data were imbalanced across treatment groups; when compared with the observed event, the proportion of missing outcomes had a clinically relevant impact on the effect estimate for dichotomous outcomes; when the expected effect size among missing outcomes had a clinically relevant impact on the observed effect size for continuous outcomes; when an 'as‐treated analysis' was substantially performed; and when missing data were imputed using improper methods (e.g. simple imputation). We rated incomplete outcome data as unclear risk of bias when insufficient information was reported to allow for a judgement.
We rated selective reporting as low risk of bias when the protocols of primary studies were available, and all of the primary and secondary study outcomes related to this systematic review, were reported in a pre‐defined way; and when the study protocols were unavailable, but all of the expected outcomes, related to this systematic review, were reported. We rated selective outcome reporting as high risk of bias when pre‐defined primary outcomes related to this systematic review were not thoroughly reported; when those primary outcomes were measured or analyzed in a way that was different to the protocol; when reported primary outcomes related to this systematic review were different from those in the protocol; when the outcomes were not completely reported; and when key outcomes were not included in the primary studies. We rated selective outcome reporting as unclear risk of bias when insufficient information was reported to allow for a judgement.
We rated studies as low risk of bias for other sources of bias when the study appeared to be free of other potential sources of bias. We rated studies as high risk of bias for other sources of bias when other sources of bias could have an impact on the study outcomes. For example, fraudulent studies or baseline imbalances in demographic factors. We rated studies as unclear risk of bias for other sources of bias when the study reported insufficient details to allow for a judgement. We contacted study authors for additional information to clarify the risk of bias when the study reports did not provide enough detail to allow for a clear judgement.
Medidas del efecto del tratamiento
For dichotomous outcomes, we calculated the risk ratio (RR) and corresponding 95% confidence interval (CI). ITT analyses were conducted for dichotomous outcomes, whereby all dropouts were assumed to be treatment failures. We calculated the mean difference (MD) and 95% CI for continuous outcomes. When different scales were used to measure the same construct, we planned to calculate the standardized mean difference (SMD) and 95% CI.
Cuestiones relativas a la unidad de análisis
We collected outcomes per randomized participant. For cross‐over trials, we planned to use data from the first phase before the cross‐over. Cluster RCTs were not included in this review. If events occurred more than once (e.g. adverse events), we reported on the proportion of participants who experienced at least one event. To avoid double counting of the comparator for multi‐arm studies (multiple dose groups), the number of patients in the comparator group (i.e. placebo or no treatment control) were divided across the number of eligible CZP arms. To deal with multiple observations for the same outcome in primary studies, we precisely defined the outcome assessment points for both primary and secondary outcomes.
Manejo de los datos faltantes
We contacted authors of the primary studies and the pharmaceutical company that manufactures CZP to obtain missing data and the reason for missing data. If it was not possible to obtain the missing data, we reported as such in the results. For dichotomous outcomes, all missing data were treated as treatment failures in the ITT analyses. We conducted sensitivity analyses using available case data to assess the impact on the effect estimate. For continuous outcomes, we did not use any imputation methods, and used the available data only.
Evaluación de la heterogeneidad
Clinical heterogeneity was first assessed with regard to patient characteristics, such as previous treatment and concurrent medication. If the studies were clinically homogenous, statistical heterogeneity was evaluated using the Chi² test and I² statistic. A P value of less than or equal to 0.10 for the Chi² test was considered to show statistically significant heterogeneity. The I² statistic estimates the degree of statistical heterogeneity. We considered an I² value of 25% to indicate low heterogeneity, a value of 50% to indicate moderate heterogeneity and a value of 75% to indicate high heterogeneity. If statistical heterogeneity existed, we planned to perform a visual inspection of the forest plots to identify potential outliers causing the heterogeneity. Moreover, sensitivity and subgroup analyses were planned to explore potential sources of heterogeneity when significant or moderate‐high heterogeneity was identified (Higgins 2003; Higgins 2011).
Evaluación de los sesgos de notificación
We searched for both registered and published trials, and reported on the proportion of registered trials that were unpublished. We contacted the investigators of the unpublished trials and the pharmaceutical company that manufactures CZP to provide data related to outcomes in this systematic review. If we could not obtain these data, we reported as such in this review. When there were 10 or more eligible trials in a pooled analysis, we planned to generate funnel plots to evaluate potential publication bias. Moreover, when we found unclear or high risk of bias for selective reporting, we contacted the study authors and the pharmaceutical company that manufactures CZP to provide unpublished outcome data. If we could not obtain these data, we reported so in the results.
Síntesis de los datos
When included studies were sufficiently similar from the clinical and statistical viewpoints, we conducted meta‐analyses. Data were analyzed using Review Manager 5.3, and a random‐effects model was used for the meta‐analyses. A P value of less than 0.05 was considered to be statistically significant.
On the basis of the characteristics of participants, interventions, and outcomes in primary studies, clinical similarity was determined by consensus between HY and KM. In cases of disagreement between HY and KM, the authors consulted with TK to resolve the disagreement. In cases of high heterogeneity (I² statistic ≥ 75%), meta‐analyses were not planned, and each study was planned to be described in detail.
Análisis de subgrupos e investigación de la heterogeneidad
If sufficient data were available, the following subgroup analyses were planned for the primary outcomes:
-
Disease severity at baseline (150 < CDAI < 220, 220 ≤ CDAI ≤ 450, CDAI > 450);
-
CRP levels at baseline (CRP levels < 10 mg/L, CRP levels ≥ 10 mg/L);
-
Doses of CZP (CZP < 200 mg, 200 mg ≤ CZP < 400 mg, 400 mg ≤ CZP < 600 mg, and CZP ≥ 600 mg); and
-
Previous treatment with other TNF‐α inhibitors (yes, no).
Análisis de sensibilidad
We planned to perform the following sensitivity analyses for primary outcomes:
-
Excluding studies judged to be at high risk of bias for any domain of the risk of bias tool;
-
Excluding studies judged to be at high or unclear risk of bias for any domain of the risk of bias tool;
-
Using available case data instead of ITT analysis for missing dichotomous outcome data;
-
Selecting later outcome assessment points if only dates from two time points equally distant from the defined outcome assessment points were available despite inquiring with the original investigators. For example, if only dates from two time points equally distant from week 8, such as weeks 6 and 10, were available, week 10 was planned to be selected in the sensitivity analysis.
-
Limiting the included studies that administered CZP strictly in accordance with the approved regimen which is subcutaneous administration of 400 mg CZP at weeks 0, 2, and 4, and then every 4 weeks.
Summary of findings tables
We produced 'Summary of findings' tables using the GRADEpro Guideline Development Tool for the following outcomes: clinical remission, clinical response, and serious adverse events.
The Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach was used to evaluate the certainty of the evidence supporting each outcome. Evidence from RCTs starts as high quality, but can be downgraded due to risk of bias, inconsistency across studies, indirectness of evidence, imprecision of effect estimate, and publication bias (Schünemann 2011). If serious limitations were present, we downgraded the evidence level by one. Moreover, very serious limitations can lead to downgrading of the evidence by two levels (Schünemann 2011). HY and RS independently assessed the certainty of evidence for each outcome and the overall quality of the evidence was rated as:
-
High: We are very confident that the true effect lies close to that of the effect estimate;
-
Moderate: We are moderately confident in the effect estimate: the true effect is likely to be close to the effect estimate, but it could be substantially different;
-
Low: Our confidence in the effect estimate is limited: the true effect may be substantially different from the effect estimate; or
-
Very low: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the effect estimate.
In cases of disagreement between HY and RS, we consulted with NW.
Results
Description of studies
Results of the search
The literature search was conducted on 28 January 2019 and 623 studies were identified. After removing duplicates, 423 reports remained for the title and abstract screening. Two authors independently screened the 423 reports, and 26 full‐text reports were assessed for further eligibility. Twelve reports of four studies (1485 participants) were finally included in this systematic review and a total of 14 reports of 12 studies were excluded (see Figure 1).
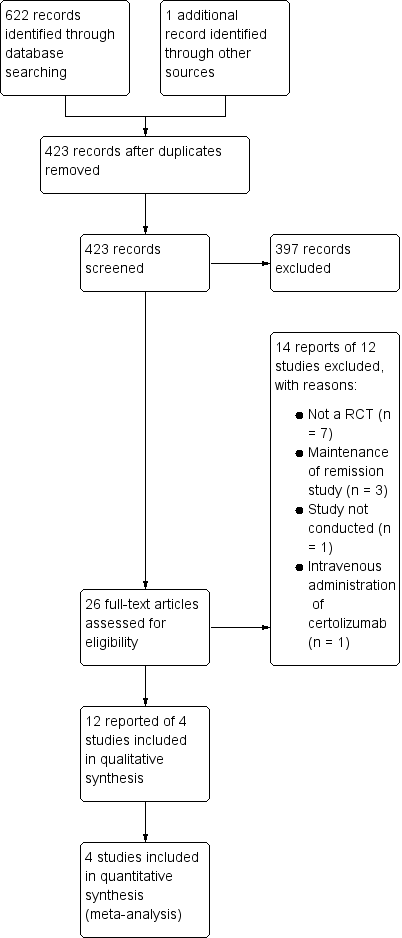
Study flow diagram.
Included studies
The four studies included in this systematic review are summarized in the Characteristics of included studies table. All four studies were randomized, double‐blind, placebo‐controlled multicenter trials sponsored by UCB Inc, the manufacturer of CZP. All the study participants were active CD patients whose CDAI was between 220 and 450. Follow‐up periods ranged between 6 weeks and 28 weeks.
Schreiber 2005 compared three different doses (100 mg, 200 mg, or 400 mg) of CZP with placebo in adult patients (18 to 75 years, N = 292). CZP was administered subcutaneously at weeks zero, four and eight. The primary outcome was clinical response (> 100 points CDAI decrease) or remission (CDAI ≤ 150) at week 12.
Sandborn 2007 compared 400 mg of CZP with placebo in adult patients (≥ 18 years, N = 660). CZP was administered subcutaneously at weeks zero, two and four, and then every four weeks. The primary outcome was clinical response (> 100 points CDAI decrease) at week 6 and at both weeks 6 and 26 in patients with a baseline serum CRP ≥ 10 mg/L. Clinical remission (CDAI ≤ 150) at week 6 was a secondary outcome.
Ogata 2009 compared two different doses (200 mg or 400 mg) of CZP with placebo in patients with CRP ≥ 10 mg/L (16 to 65 years, N = 94). Only two participants were aged 16 or 17 years old in each group. CZP was administered subcutaneously at weeks zero, two and four. The primary outcome was clinical response (> 100 points CDAI decrease) or remission (CDAI ≤ 150) at week 6.
Sandborn 2011 compared 400 mg of CZP with placebo in adult patients without previous treatment with TNF‐α inhibitors (18 to 75 years, N = 439). CZP was administered subcutaneously at weeks zero, two and four. The primary outcome was clinical remission (CDAI ≤ 150) at week 6.
We contacted UCB Inc and obtained the following information: a protocol of Schreiber 2005; a presentation poster from the United European Gastroenterology 2009, detailed information from Ogata 2009, and CRP and IBDQ data from Schreiber 2005, Sandborn 2007, Ogata 2009, and Sandborn 2011. We confirmed via the UCB Inc website (https://www.ucb.com/our‐science/Our‐clinical‐studies/cimzia‐certolizumab‐pegol) that all RCTs conducted by UCB Inc were included in this review.
Excluded studies
The characteristics of excluded studies by full‐text assessment are shown in the Characteristics of excluded studies table. We found no ongoing studies.
Risk of bias in included studies
Figure 2 and Figure 3 report a summary of risk of bias in the included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
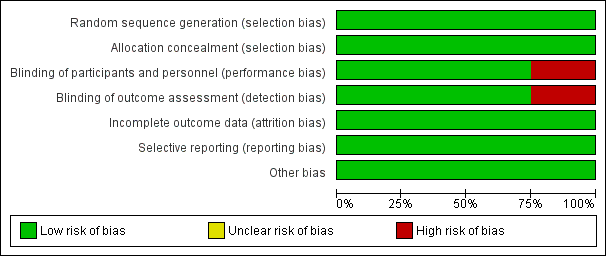
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Centralized randomization schemes with randomization code were used in all studies. Therefore, we considered all studies to be at low risk of selection bias.
Blinding
Schreiber 2005 used a placebo that did not have the same color or viscosity of CZP, and patients may have been able to distinguish between CZP and the placebo. Moreover, outcomes were assessed based on patients' daily diaries. Therefore, we considered this study to have high risk of performance and detection bias. The other three studies used an identical placebo and were considered as low risk of bias regarding both performance and detection bias (Ogata 2009;Sandborn 2007; Sandborn 2011).
Incomplete outcome data
The proportion of loss to follow‐up was less than 2% and was balanced between CZP and placebo in all studies. Therefore, we rated all studies as low risk of attrition bias.
Selective reporting
All of the studies reported our prespecified outcomes properly. Therefore, all studies were rated as having low risk of reporting bias.
Other potential sources of bias
We did not find any other potential sources of bias in all studies. Therefore, we rated all studies as having low risk of other potential sources of bias
Effects of interventions
Main comparison
See Summary of findings table 1: Certolizumab pegol compared to placebo for induction of remission in Crohn’s disease.
Clinical remission at week 8, the only primary outcome in this review, was assessed in four studies involving 1485 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In fact, we could not obtain week eight clinical remission data for two studies involving 533 CD patients (Ogata 2009; Sandborn 2011). As defined in our protocol, we used week six clinical remission data from Ogata 2009 and Sandborn 2011, and these data were combined with week eight data from the other two studies involving 952 CD patients (Sandborn 2007; Schreiber 2005). Heterogeneity was low ( I² = 2%). Clinical remission was achieved in 26.9% (225/835) and 19.8% (129/650) in the CZP and placebo groups, respectively. In the meta‐analysis using the Mantel‐Haenszel random‐effects method, patients treated with CZP (100 mg to 400 mg every 2 to 4 weeks) were significantly more likely to achieve clinical remission at week 8 than those treated with placebo (RR 1.36, 95% CI 1.11 to 1.66; see Figure 4). The certainty of evidence was moderate according to the GRADE system.

Forest plot of comparison: 1 Certolizumab pegol versus placebo, outcome: 1.1 Clinical remission at week 8.
Clinical response at week 8, a secondary outcome in this review, was assessed in four studies involving 1485 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). Clinical response was achieved in 40.2% (336/835) and 30.9% (201/650) in the CZP and placebo groups respectively. In the meta‐analysis using the Mantel‐Haenszel random‐effects method, patients treated with CZP were significantly more likely to achieve clinical response at week 8 than those with treated with placebo (RR 1.29, 95% CI 1.09 to 1.53; see Figure 5). The certainty of evidence was moderate according to the GRADE system.

Forest plot of comparison: 1 Certolizumab pegol versus placebo, outcome: 1.2 Clinical response at week 8.
Serious adverse events, a secondary outcome in this review, were assessed in four studies involving 1485 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). Serious adverse events such as worsening CD, various infections, and malignancy occurred in 8.7% (73/835) and 6.2% (40/650) in CZP and placebo, respectively. In the meta‐analysis using the Mantel‐Haenszel random‐effects method, it is uncertain whether the risk of serious adverse events differs between CZP and placebo (RR 1.35, 95% CI: 0.93 to 1.97; see Figure 6). According to GRADE system, the certainty of evidence was moderate because the number of events was low, and showed imprecision in the results.

Forest plot of comparison: 1 Certolizumab pegol versus placebo, outcome: 1.6 Serious adverse events.
Efficacy outcomes
The log scales of the geometric mean CRP ratio between baseline and week 8 were assessed in four studies involving 1271 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In the meta‐analysis using the inverse variance random‐effects method, patients treated with CZP were significantly more likely to have improvement in CRP at week 8 than those treated with placebo (MD ‐0.37, 95% CI ‐0.49 to ‐0.24).
The mean change in IBDQ score from baseline was assessed in four studies involving 1315 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In the meta‐analysis using the inverse variance random‐effects method, CZP did not appear to make a clear difference in IBDQ at week 8 (MD 2.12, 95% CI ‐1.27 to 5.50).
We did not find any eligible studies assessing endoscopic improvement or fistula closure.
Safety outcomes
Adverse events were assessed in four studies involving 1485 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, CZP did not appear to make any clear difference in the risk of having adverse events (RR 1.03, 95% CI 0.97 to 1.10).
Withdrawals due to adverse events were assessed in four studies involving 1485 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, there was no clear difference between CZP and placebo in the risk of withdrawals due to adverse events (RR 1.01, 95% CI: 0.57 to 1.78).
Subgroup analyses
We conducted three prespecified subgroup analyses to evaluate clinical remission at week 8 based on CZP doses, previous treatment with TNF‐α inhibitors, and CRP levels at baseline. We did not conduct a prespecified subgroup analysis based on disease severity because disease severity was similar among all of the included studies.
Clinical remission at week 8 with CZP 100 mg was assessed in a study involving 99 CD patients (Schreiber 2005). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, it was uncertain whether clinical remission at week 8 differs between CZP 100 mg and placebo (RR 2.48, 95% CI: 0.81 to 7.58). Similarly, it was unclear whether clinical remission at week 8 differs between CZP 200 mg and placebo in 2 studies involving 142 CD patients (RR 1.84, 95% CI: 0.75 to 4.50) (Ogata 2009; Schreiber 2005). However, patients treated with CZP 400 mg were significantly more likely than placebo participants to achieve clinical remission at week 8 (RR 1.30, 95% CI: 1.06 to 1.60, 4 studies; 1244 CD patients) (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). We did not conduct a subgroup analysis of patients with CZP ≥ 600 mg because there were no studies using this dose.
Clinical remission at week 8 among patients with no previous treatment with TNF‐α inhibitors was assessed in a study involving 439 CD patients (Sandborn 2011). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, it was uncertain whether clinical remission at week 8 differs between CZP and placebo patients (RR 1.24, 95% CI: 0.91 to 1.69). We did not conduct a subgroup analysis of CD patients who had previous treatment with TNF‐α inhibitors due to lack of data.
Clinical remission at week 8 among patients with baseline CRP ≥ 10 mg/L was assessed in 4 studies involving 702 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In the meta‐analysis using the Mantel‐Haenszel random‐effects method among patients with baseline CRP ≥ 10 mg/L, patients treated with CZP were significantly more likely to achieve clinical remission at week 8 than those treated with placebo: RR 1.59 (95% CI: 1.17 to 2.16). However, it was uncertain whether clinical remission at week 8 differs between CZP and placebo among patients with baseline CRP < 10 mg/L in three studies involving 762 CD patients (RR 1.15, 95% CI 0.88 to 1.50) (Sandborn 2007; Sandborn 2011; Schreiber 2005).
Sensitivity analyses
We conducted three prespecified sensitivity analyses to evaluate clinical remission at week eight based on excluded studies with high risk of bias, studies using available case data instead of ITT analysis, and studies with the approved regimen of CZP. We did not conduct a sensitivity analysis of selecting later outcome assessment points because we obtained outcomes at prespecified assessment points.
We excluded a study with high risk of bias (Schreiber 2005), and clinical remission at week 8 was assessed in 3 studies involving 1193 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, patients treated with CZP were significantly more likely to achieve clinical remission at week 8 than those treated with placebo (RR 1.29, 95% CI: 1.05 to 1.59). We found no studies with unclear risk of bias in any domain of the risk of bias evaluation.
For the available case data analysis, clinical remission at week 8 was assessed in 4 studies involving 1463 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011; Schreiber 2005). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, patients treated with CZP were significantly more likely to achieve clinical remission at week 8 than those treated with placebo (RR 1.36, 95% CI: 1.11 to 1.67).
Clinical remission at week 8 with the approved regimen of CZP was assessed in 3 studies involving 1147 CD patients (Ogata 2009; Sandborn 2007; Sandborn 2011). In the meta‐analysis using the Mantel‐Haenszel random‐effects method, patients treated with CZP were significantly more likely to achieve clinical remission at week 8 than those treated with placebo (RR 1.28, 95% CI 1.03 to 1.57).
Funnel plots
We did not generate funnel plots to evaluate potential publication bias because there were less than 10 eligible trials for each pooled analysis.
Discusión
Resumen de los resultados principales
Se incluyeron cuatro estudios con 1 485 pacientes en los metanálisis de los resultados principales. Los pacientes tratados con CZP tuvieron significativamente más probabilidades de lograr la remisión clínica (CDAI ≤ 150) en la semana ocho que los tratados con placebo. La eficacia del CZP fue sólida en un análisis de sensibilidad limitado a los estudios con un bajo riesgo de sesgo. De manera similar, los pacientes tratados con CZP presentaron significativamente más probabilidades de lograr una respuesta clínica (reducción en el CDAI ≥ 100 o remisión clínica) en la semana ocho que los tratados con placebo. Estos resultados sugieren un beneficio claro del CZP para los pacientes con EC activa. La mejoría mayor en la PCR en los pacientes que recibieron CZP que en los pacientes que recibieron placebo está de acuerdo con esta evidencia. Con respecto a la seguridad, puede no haber diferencias claras en los eventos adversos y el retiro debido a los eventos adversos. Además, no está claro si el riesgo de eventos adversos graves difiere entre el CZP y el placebo, debido a que el IC del 95% incluye la posibilidad de una pequeña disminución o duplicación de los eventos.
Compleción y aplicabilidad general de las pruebas
Se incluyeron datos publicados y no publicados en esta revisión. Todos los estudios fueron financiados por la compañía farmacéutica y se estableció contacto con la compañía para obtener los datos no informados en los artículos publicados. Hasta donde se conoce, basado en la búsqueda sistemática de la literatura, esta revisión incluyó todos los datos existentes para evaluar la eficacia y la seguridad del CZP para la inducción de la remisión de la EC.
Todos los participantes del estudio tenían EC de moderada a grave (CDAI 220 a 450), y la evidencia no puede aplicarse a los pacientes con EC leve (CDAI < 220) y EC extremadamente grave (CDAI > 450). Además, la mayoría de los pacientes eran adultos (≥ 18 años), y esta revisión no aporta evidencia con respecto a los pacientes con EC pediátrica. Finalmente, en esta revisión no se evaluó la eficacia a largo plazo de más de ocho semanas ni la seguridad de más de 28 semanas. Se justifica una revisión futura para evaluar el mantenimiento de la remisión.
Calidad de la evidencia
De acuerdo con el sistema GRADE, la certeza general de la evidencia fue moderada tanto para la remisión clínica como para la respuesta clínica en la semana ocho. Tres de cada cuatro estudios incluidos fueron clasificados como en bajo riesgo de sesgo para todos los dominios (Ogata 2009; Sandborn 2007; Sandborn 2011), y solo Schreiber 2005 fue considerado en alto riesgo de sesgo con respecto al sesgo de realización y de detección. Se confirmó el efecto beneficioso sólido del CZP en el análisis de sensibilidad, que excluyó a Schreiber 2005. Debido a la escasez de datos, la certeza general de la evidencia fue moderada para los eventos adversos graves. Se optó por no disminuir la calificación de los eventos adversos graves debido al alto riesgo de sesgo (es decir, debido a la inclusión del estudio Schreiber 2005 en el análisis agrupado). Debido a la naturaleza objetiva de este resultado, no se consideró que la posibilidad de ausencia del cegamiento de la asignación al tratamiento tendría un impacto sobre los participantes que experimentaron un evento adverso potencialmente mortal. No se encontraron otras razones para disminuir la certeza de la evidencia con respecto a la remisión clínica, la respuesta clínica y los eventos adversos graves.
Sesgos potenciales en el proceso de revisión
Se realizaron búsquedas sistemáticas en todos los recursos disponibles y se estableció contacto con la compañía farmacéutica que fabrica CZP. Sobre la base de esta búsqueda sistemática, dos autores, de forma independiente, evaluaron los estudios para su inclusión, extrajeron los datos del estudio incluido, evaluaron el riesgo de sesgo para cada estudio y realizaron una evaluación GRADE para los resultados principales. Debido a que los datos se extrajeron de los gráficos de líneas en Schreiber 2005 a causa de la falta de datos detallados, puede haber ligeras diferencias entre estos datos y los datos observados realmente. Con respecto a la seguridad, se incluyeron datos entre la semana cero y la semana 12 en Schreiber 2005 debido a que no fue posible obtener el número total de eventos adversos y eventos adversos graves para el período de seguimiento general de 20 semanas.
Acuerdos y desacuerdos con otros estudios o revisiones
El resultado primario de esta revisión, el efecto significativo del CZP sobre la inducción de la remisión clínica en la semana ocho, no fue consistente con Ford 2011 pero sí con Kawalec 2013. Aunque ambas revisiones sistemáticas (Ford 2011; Kawalec 2013) incluyeron los mismos cuatro estudios (Sandborn 2007; Sandborn 2011; Schreiber 2005; Winter 2004), los puntos temporales utilizados para evaluar la remisión clínica fueron diferentes. El CZP demostró ser efectivo en Kawalec 2013 (en la semana 4), aunque la semana 4 todavía se encuentra en el período de dosificación de inducción (período de administración frecuente). Es importante estar en remisión cuando los pacientes comienzan la dosis de mantenimiento en la semana ocho (Schreiber 2011). Ford 2011 seleccionó puntos temporales más apropiados para evaluar la remisión clínica (en las semanas 6 a 12), aunque no se demostró la eficacia del CZP. En la revisión, se seleccionó la semana ocho debido a que dicha semana es el momento recomendado para cambiar a la dosis de mantenimiento de acuerdo con el régimen aprobado (Schreiber 2011). Además, no se incluyó Winter 2004 basado en el protocolo, debido a que el CZP en dicho estudio se administró por vía intravenosa en lugar de por vía subcutánea. Es importante destacar que se encontró un estudio no publicado (Ogata 2009) a través del contacto con UCB Inc. el cual se incluyó en el metanálisis (Ogata 2009). Por lo tanto, los resultados de esta revisión pueden ser considerados como la revisión más completa con puntos temporales de evaluación adecuados para este tema.
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Forest plot of comparison: 1 Certolizumab pegol versus placebo, outcome: 1.1 Clinical remission at week 8.
Forest plot of comparison: 1 Certolizumab pegol versus placebo, outcome: 1.2 Clinical response at week 8.
Forest plot of comparison: 1 Certolizumab pegol versus placebo, outcome: 1.6 Serious adverse events.

Comparison 1 Certolizumab pegol versus placebo, Outcome 1 Clinical remission at week 8.

Comparison 1 Certolizumab pegol versus placebo, Outcome 2 Clinical response at week 8.

Comparison 1 Certolizumab pegol versus placebo, Outcome 3 CRP at week 8 (log‐scales of geometric mean CRP ratio between baseline and week 8).

Comparison 1 Certolizumab pegol versus placebo, Outcome 4 IBDQ total score at week 8 (mean change from baseline).

Comparison 1 Certolizumab pegol versus placebo, Outcome 5 Adverse events.

Comparison 1 Certolizumab pegol versus placebo, Outcome 6 Serious adverse events.

Comparison 1 Certolizumab pegol versus placebo, Outcome 7 Withdrawals due to adverse events.

Comparison 1 Certolizumab pegol versus placebo, Outcome 8 Clinical remission at week 8 (Subgroup analysis based on CZP doses).

Comparison 1 Certolizumab pegol versus placebo, Outcome 9 Clinical remission at week 8 (Subgroup analysis of no previous treatment with TNF‐α inhibitors).

Comparison 1 Certolizumab pegol versus placebo, Outcome 10 Clinical remission at week 8 (Subgroup analysis of CRP levels at baseline).

Comparison 1 Certolizumab pegol versus placebo, Outcome 11 Clinical remission at week 8 (Sensitivity analysis of excluding studies with high risk of bias).

Comparison 1 Certolizumab pegol versus placebo, Outcome 12 Clinical remission at week 8 (sensitivity analysis of using available case data).

Comparison 1 Certolizumab pegol versus placebo, Outcome 13 Clinical remission at week 8 (Sensitivity analysis of studies with the approval dosing regimen).
| Certolizumab pegol compared to placebo for induction of remission in Crohn's disease | ||||||
| Patient or population: Patients with active Crohn's disease Settings: Outpatient Intervention: Certolizumab pegol Comparison: Placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Certolizummab pegol | |||||
| Clinical remission Follow‐up: 8 weeks | 198 per 1000 | 270 per 1000 (220 to 329) | RR 1.36 (1.11 to 1.66) | 1485 | ⊕⊕⊕⊝ | Certolizumab pegol was shown to be superior to placebo regarding clinical remission at week 8 Clinical remission was defined as a CDAI < 150 |
| Clinical response Follow‐up: 8 weeks | 309 per 1000 | 399 per 1000 (337 to 473) | RR 1.29 (1.09 to 1.53) | 1485 (4 studies) | ⊕⊕⊕⊝ | Clinical response was defined as CDAI reduction ≥ 100 from baseline |
| Serious adverse events Follow‐up: 8 weeks | 62 per 1000 | 83 per 1000 (57 to 121) | RR 1.35 (0.93 to 1.97) | 1485 (4 studies) | ⊕⊕⊕⊝ | Reported serious adverse events included worsening Crohn's disease, infections, and malignancy |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Downgraded one level due to high risk of bias in one study in the pooled analysis 2 Downgraded one level due to imprecision (113 events) | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical remission at week 8 Show forest plot | 4 | 1485 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.11, 1.66] |
| 2 Clinical response at week 8 Show forest plot | 4 | 1485 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [1.09, 1.53] |
| 3 CRP at week 8 (log‐scales of geometric mean CRP ratio between baseline and week 8) Show forest plot | 4 | 1271 | Mean Difference (IV, Random, 95% CI) | ‐0.37 [‐0.49, ‐0.24] |
| 4 IBDQ total score at week 8 (mean change from baseline) Show forest plot | 4 | 1315 | Mean Difference (IV, Random, 95% CI) | 2.12 [‐1.27, 5.50] |
| 5 Adverse events Show forest plot | 4 | 1485 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.97, 1.10] |
| 6 Serious adverse events Show forest plot | 4 | 1485 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [0.93, 1.97] |
| 7 Withdrawals due to adverse events Show forest plot | 4 | 1485 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.57, 1.78] |
| 8 Clinical remission at week 8 (Subgroup analysis based on CZP doses) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Certolizumab pegol 100mg | 1 | 99 | Risk Ratio (M‐H, Random, 95% CI) | 2.48 [0.81, 7.58] |
| 8.2 Certolizumab pegol 200mg | 2 | 142 | Risk Ratio (M‐H, Random, 95% CI) | 1.84 [0.75, 4.50] |
| 8.3 Certolizumab pegol 400mg | 4 | 1244 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [1.06, 1.60] |
| 9 Clinical remission at week 8 (Subgroup analysis of no previous treatment with TNF‐α inhibitors) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Clinical remission at week 8 (Subgroup analysis of CRP levels at baseline) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 CRP ≥ 10 mg/L | 4 | 702 | Risk Ratio (M‐H, Random, 95% CI) | 1.59 [1.17, 2.16] |
| 10.2 CRP < 10 mg/L | 3 | 762 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.88, 1.50] |
| 11 Clinical remission at week 8 (Sensitivity analysis of excluding studies with high risk of bias) Show forest plot | 3 | 1193 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [1.05, 1.59] |
| 12 Clinical remission at week 8 (sensitivity analysis of using available case data) Show forest plot | 4 | 1463 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.11, 1.67] |
| 13 Clinical remission at week 8 (Sensitivity analysis of studies with the approval dosing regimen) Show forest plot | 3 | 1147 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [1.03, 1.57] |