Secuenciación de antraciclinas y taxanos en el tratamiento neoadyuvante y adyuvante para el cáncer de mama en estadio inicial
Referencias
Referencias de los estudios incluidos en esta revisión
Referencias de los estudios excluidos de esta revisión
Referencias de los estudios en espera de evaluación
Referencias de los estudios en curso
Referencias adicionales
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Accrual: June 2006 to April 2008 Phase II randomised controlled trial conducted in Japan Adjuvant therapy Multicentre or single centre: not reported Median follow‐up: not reported | |
| Participants | Age: median 52 years, range 29 to 64 years 55% postmenopausal Node‐positive or high‐risk node‐negative women Stage I/IIA/IIB: 100% (83% stage II) Axillary lymph node involvement: 0: 57%; 1–3: 33%; ≥ 4: 10% Hormone receptor‐positive: 55% HER2‐positive: 24% Excluded: prior chemotherapy or hormone therapy as neoadjuvant or adjuvant therapy | |
| Interventions | Arm 1 ('arm B' in the trial publication): docetaxel (100 mg/m²) every 3 weeks for 3 cycles followed by fluorouracil (500 mg/m²), epirubicin (100 mg/m²), cyclophosphamide (500 mg/m²) every 3 weeks for 3 cycles. Arm 2 ('arm A' in the trial publication): fluorouracil (500 mg/m²), epirubicin (100 mg/m²), cyclophosphamide (500 mg/m²) every 3 weeks for 3 cycles followed by docetaxel (100 mg/m²) every 3 weeks for 3 cycles. For both arms: G‐CSF if ANC < 500 μL (full blood count measured on day 8) or febrile neutropenia | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Clinical trial registration record not found Trialists contacted in July 2018 requesting information on mean RDI (mismatch of data in Table 2 and text); as of 16 August 2018, no reply received Funding considerations: no information available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients were "randomised to two arms;" no further details provided; however, baseline characteristics across the 2 arms were balanced. |
| Allocation concealment (selection bias) | Unclear risk | No details provided in trial publication. |
| Blinding of participants and personnel (performance bias) | Low risk | Likely to be an open‐label study. Participants in both groups received the same drugs though in different order. Therefore, it was unlikely that this would lead to a material bias in participants' and physicians' behaviours even when unblinded. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Study used formalised toxicity criteria (NCI CTC version 3) and measured a range of toxicity outcomes where some may have been affected by unblinding (e.g. neuropathy) in borderline cases. Dose‐reduction/delays were based on toxicity assessments. Overall, unblinding may have influenced physicians' assessments on a subset of outcomes assessed. |
| Incomplete outcome data (attrition bias) | Low risk | 1 discontinuation in the intervention arm (due to neutropenia). |
| Selective reporting (reporting bias) | Low risk | Clinical trial registry record not found (trial ran from June 2006 to April 2008). All outcomes reported in the Methods section had the corresponding results in the publication. |
| Other bias | Low risk | None identified |
| Methods | Accrual: 15 September 2007 to 15 December 2011 Open‐label, phase III randomised controlled trial Neoadjuvant therapy Multicentre (36 centres) across the USA and Puerto Rico Follow‐up: yearly for a maximum of 5 years | |
| Participants | Age: 25–39 years: 21% (both arms); 40–49 years: approximately 33% (both arms); 50–59 years: 38.7% (arm 1) and 35.5% (arm 2); 60–69 years: 10.6% (arm 1) and 14.5% (arm 2); > 70 years: approximately 3% (both arms) HER2‐positive disease Stage described as clinical T stage and N stage: generally 7% T1, 55% T2, 29% T3, 9% T4; 36% N0, 51–57% N1, 5% N2, 0.7% to 8% N3 ER‐positive, PR‐positive: 41.5% (arm 1), 35.5% (arm 2); ER‐positive, PR‐negative: 16.2% (arm 1), 22.5% (arm 2); ER‐negative and PR‐negative: 40.8% (arm 1) and 39.1% (arm 2) Excluded: any current breast cancer treatment except hormonal therapy (taken for up to 28 days after diagnosis but had to be stopped before study registration) | |
| Interventions | Arm 1 ('concurrent' in the trial publication): paclitaxel (80 mg/m²) and trastuzumab 4 mg/kg first dose (2 mg/kg after days 1, 8, 15) once a week for 12 weeks followed by fluorouracil (500 mg/m²), epirubicin (75 mg/m²), cyclophosphamide (500 mg/m²) every 3 weeks for 4 cycles and trastuzumab (2 mg/kg after a 4 mg/kg loading dose) once a week for 12 weeks Arm 2 ('sequential' in the trial publication): fluorouracil (500 mg/m²), epirubicin (75 mg/m²), cyclophosphamide (500 mg/m²) every 3 weeks for 4 cycles followed by paclitaxel (80 mg/m²) and trastuzumab (2 mg/kg after a 4 mg/kg loading dose) once a week for 12 weeks | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Clinical trial registration number: NCT00430001 Funding considerations: supported by a National Cancer Institute grant, National Cancer Institute to the Alliance for Clinical Trials in Oncology, and Alliance Statistics and Data Center. Data quality and analysis: completed by Alliance Statistics and Data Center. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to treatment (1:1) with a biased coin minimisation algorithm so that marginal distributions of stratification factors would be similar in each treatment group" (p. 1318). |
| Allocation concealment (selection bias) | Low risk | Central allocation. Quote: "...patients were, unstratified, randomly assigned centrally by the Danish Breast Cancer Cooperative Group Secretariat." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Neither patients nor investigators, except for a cardiac review panel, were masked to treatment assignment." Comment: participants in both groups received the same drugs though in different order. Therefore, it was unlikely that this would lead to a material bias in participants' and physicians' behaviours even when unblinded. |
| Blinding of outcome assessment (detection bias) | Low risk | Outcome collected but not yet reported. Unblinding unlikely to have had an impact on outcome assessment. |
| Blinding of outcome assessment (detection bias) | Low risk | Unblinding unlikely to have had an impact on outcome assessment for this review question where trial participants received both treatment regimens. DFS was determined each year for a maximum of 5 years (data not provided). |
| Blinding of outcome assessment (detection bias) | Unclear risk | Laboratory tests (blood counts) repeated before each cycle. LVEF measured at completion of first 12‐week regimen and second 12‐week regimen. Cardiac review panel reviewed multigated acquisition scan and echocardiography results for all participants each month. Judged at unclear risk of bias because assessors were not blinded to treatment allocation for assessment of other toxicities. Dose reductions/delays were based on toxicities. Knowledge of treatment allocation may have had some influence on physicians' assessments. |
| Blinding of outcome assessment (detection bias) | Low risk | Before each cycle of treatment, assessments for tumour size were completed. Imaging and tumour evaluation was done every 3 months. Mammogram of ipsilateral breast taken at completion of neoadjuvant chemotherapy. pCR was viewed to be an objective outcome and a review by a pathologist on whether pCR had been achieved or not was unlikely to be influenced by unblinding. |
| Incomplete outcome data (attrition bias) | Low risk | 138/140 participants in the comparator group and 142/142 participants in intervention group proceeded to treatment. 4/142 participants in the intervention arm discontinued treatment (2 refused, 1 allergic reaction, 1 disease progression) and 8/138 participants discontinued in the comparator arm (4 refused treatment, 2 based on physicians' discretion, 1 second primary cancer and 1 death unrelated to treatment). All participants who began study treatment were included in the efficacy analysis. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes in Clinical Trials.gov record (NCT00430001) and the Methods section of the trial publication are the same. Most outcomes except OS and DFS (due to immature data), were reported in the results section. Some additional toxicity data were reported in the trial publication that were not included in the trial registry record. |
| Other bias | Low risk | None identified |
| Methods | Accrual: 12 December 2003 to 30 September 2004 Multicentre, phase II randomised controlled trial conducted in France Adjuvant therapy Median follow‐up: not reported | |
| Participants | Age: median 53.9 years, range: 31–69 (arm 1) and 55.4 years, range: 32–72 (arm 2) 56–65% postmenopausal Node‐positive invasive breast adenocarcinoma Majority of women had T1‐2 and N0‐1 (arm 1: 91%; arm 2: 80%) Median number of pathologically involved nodes: 2 (range 1–20) ER‐positive: 68% (arm 1), 71% (arm 2); PR‐positive: 56% (arm 1), 45% (arm 2) HER2‐positive: 17% (arm 1), 6% (arm 2) Excluded: second or inflammatory breast cancer, previous or concomitant anticancer therapy including radiotherapy and hormone therapy | |
| Interventions | 3 arm study Arm 1 ('arm C' in the trial publication): docetaxel (100 mg/m²) every 2 weeks for 4 cycles followed by epirubicin (100 mg/m²), cyclophosphamide (600 mg/m²) every 2 weeks for 4 cycles. Arm 2 ('arm B' in the trial publication): epirubicin (100 mg/m²), cyclophosphamide (600 mg/m²) every 2 weeks for 4 cycles followed by docetaxel (100 mg/m²) every 2 weeks for 4 cycles. Arm 3 ('arm A' in the trial publication – not used in the review): docetaxel (75 mg/m²), epirubicin (75 mg/m²), cyclophosphamide (500 mg/m²) every 3 weeks for 6 cycles. For arms 1 and 2: pegfilgrastim (6 mg) recommended on day 2 after each chemotherapy cycle. Only arms 1 and 2 were used in the review | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Trial not powered to detect differences between treatment arms. Clinical trial registration record not found. Trialists were contacted in July 2018 requesting information on efficacy outcome data; as of 16 August 2018, no reply received. Funding considerations: supported by European Association for Research in Oncology and by grants from Sanofi‐Aventis and Amgen. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were "stratified according to center and number of involved lymph nodes (1‐3, 4‐9, > 9) and randomly assigned to one of three treatment arms." There were no imbalances in baseline characteristics. |
| Allocation concealment (selection bias) | Unclear risk | No details provided in trial publication. |
| Blinding of participants and personnel (performance bias) | Low risk | No information provided on blinding in the trial publication. Participants in both groups would have received the same drugs though in different order. Therefore, it was judged unlikely that this would lead to a material bias in participants' and physicians' behaviours even if unblinded. |
| Blinding of outcome assessment (detection bias) | Low risk | OS data not yet reported. Unlikely that assessment of this outcome would be affected by unblinding. |
| Blinding of outcome assessment (detection bias) | Low risk | Unblinding was unlikely to have an impact on outcome assessment for this review question where trial participants received both treatment regimens. Data for DFS have not been reported (results are not mature, as stated in 2007 publication). |
| Blinding of outcome assessment (detection bias) | Unclear risk | This study used formalised toxicity criteria (NCI CTC version 3) and measured a range of toxicity outcomes where in borderline cases some may be affected by unblinding (e.g. neuropathy). Dose‐reductions/delays were based on toxicity assessments. Overall, unblinding may have influenced the physicians' assessments for a subset of outcomes. |
| Incomplete outcome data (attrition bias) | Low risk | 31/34 (91%) participants in arm 1 and 25/31 (81%) participants in arm 2 completed the study. The reasons for not completing the study were similar across groups. |
| Selective reporting (reporting bias) | High risk | Did not identify clinical trial registry record; trial commenced in 2003 and was completed in 2004. Important efficacy outcome data – OS and DFS – were collected but not reported in trial publication published in 2007 (i.e. immature at time of publication). No data published in last 10 years. Trial authors were contacted in July 2018 and no reply received. |
| Other bias | Low risk | None identified |
| Methods | Accrual: April 2004 to December 2011 Open‐label, phase III randomised controlled trial conducted in Australia Neoadjuvant therapy Multicentre study Follow‐up: not reported | |
| Participants | Age: < 50 years: 56%; ≥ 50 years: 44% T stage: 18% T1, 65% T2, 17% T3 Locally advanced breast cancer Lymph node‐positive: 81% HER2‐positive: 24% Triple‐negative: 24% Luminal A: 20%; luminal B: 41% | |
| Interventions | Arm 1 ('arm B' in the trial publication): docetaxel (100 mg/m²) every 3 weeks for 4 cycles followed by fluorouracil (500 mg/m²), epirubicin (100 mg/m²), cyclophosphamide (500 mg/m²) every 3 weeks for 4 cycles Arm 2 ('arm A' in the trial publication): fluorouracil (500 mg/m²), epirubicin (100 mg/m²), cyclophosphamide (500 mg/m²) every 3 weeks for 4 cycles followed by docetaxel (100 mg/m²) every 3 weeks for 4 cycles | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Clinical trial registration number: ACTRN12605000588695 Funding considerations: ANZCTR record states: primary sponsor – Monash Health; secondary sponsor: Sanofi Aventis; trial publication states support from Victorian Cancer Agency. Trialist was contacted in July 2018 for survival data based on sequencing; as of 16 August 2018, no reply received. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | From trial registry record: random sequence generated through "coin toss with no restriction." |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) | Low risk | Open‐label study. Participants in both groups received the same drugs though in different order. Therefore, it was judged unlikely that this would lead to a material bias in participants' and physicians' behaviours even when unblinded. |
| Blinding of outcome assessment (detection bias) | Low risk | Lack of blinding unlikely to influence this outcome |
| Blinding of outcome assessment (detection bias) | Low risk | Lack of blinding unlikely to have an impact on outcome assessment for this review question where trial participants received both treatment regimens |
| Blinding of outcome assessment (detection bias) | Low risk | Assessments including mammography, ultrasound, etc were repeated before chemotherapy commenced and repeated after 4 cycles of chemotherapy; plus periodic clinical assessments after each cycle of chemotherapy to ensure tumour was not progressing. pCR was viewed to be an objective outcome and a review by a pathologist on whether pCR had been achieved or not was unlikely to be influenced by unblinding. |
| Incomplete outcome data (attrition bias) | Low risk | Although a CONSORT diagram was not provided, it appeared that there were no missing data for the 2 outcomes reported. |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes in ACTRN record included DFS; data for this outcome were not reported in the trial publication. The publication added a new important outcome – pCR – that was not in the trial record but this was considered an important outcome to report in the publication. |
| Other bias | Low risk | None identified |
| Methods | Accrual: June 1999 to October 2002 Single‐centre, phase II randomised controlled trial conducted in US Neoadjuvant study Median follow‐up: not followed up (trial publication specifically states that participants were not followed for recurrence or OS) | |
| Participants | Age: median 50 years, range: 30–64 years (arm 1); and 50 years, range: 36–65 years (arm 2) Inflammatory disease: 17% (arm 1), 23% (arm 2) Median tumour size: 5.5 cm (arm 1), 6.0 cm (arm 2) ER‐positive: 57% (arm 1), 57% (arm 2) HER2‐positive: 23% (arm 1), 17% (arm 2) Excluded: prior breast or chest wall radiation, chemotherapy or hormonal therapy | |
| Interventions | Arm 1: docetaxel (40 mg/m²) weekly for 6 cycles followed by doxorubicin (75 mg/m²) every 2 weeks for 3 cycles with filgrastim on days 2–11 Arm 2: doxorubicin (75 mg/m²) every 2 weeks for 3 cycles with filgrastim on days 2–11 followed by docetaxel (40 mg/m²) weekly for 6 cycles | |
| Outcomes | Trial publication did not appear to define outcomes as primary and secondary. Outcomes collected were:
| |
| Notes | Trial not powered to detect differences between treatment arms. Clinical trial registration record not found. Funding considerations: supported by American Cancer Society, American Society of Clinical Oncology, Breast Cancer Research Foundation, Walter Cancer Institute and unrestricted research grants from Aventis and Amgen. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomly assigned after stratification for tumour size (≤ 5 cm versus > 5 cm) and clinical axillary node status (positive versus negative)". Comment: there did not appear to be imbalances in baseline characteristics between groups. |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) | Low risk | No information provided about blinding in the trial publication. Participants in both groups would have received the same drugs though in different order. Therefore, unlikely that this would lead to a material bias in participants' and physicians' behaviours even if unblinded. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Used formalised toxicity criteria (NCI CTC version 2) and measured a range of toxicity outcomes where in borderline cases some may be affected if unblinded (e.g. neuropathy). Dose‐reductions/delays were based on toxicity assessments. Overall, unblinding may have influenced the physicians' assessments for a subset of outcomes. |
| Blinding of outcome assessment (detection bias) | Low risk | Study pathologist blinded to treatment. |
| Incomplete outcome data (attrition bias) | Low risk | A CONSORT diagram was not provided. There did not appear to be missing data for the outcomes reported. |
| Selective reporting (reporting bias) | Low risk | Did not identify clinical trial registry record (trial started recruitment in June 1999 and completed in October 2002). However, the outcomes outlined in the Methods section were reported in the results section in the publication. Also the Methods section clearly stated that survival data were not collected. |
| Other bias | Low risk | None identified |
| Methods | Accrual: 18 January 2005 to 28 September 2007 Open‐label, phase III randomised controlled trial conducted in the UK Neoadjuvant therapy Multicentre study (57 centres) Median follow‐up: 47 months (interquartile range 37–51) | |
| Participants | Age: < 50 years: 63% (in both arms); ≥ 50 years: 37% (in both arms) 26% (arm 1) and 28% (arm 2) postmenopausal Tumour size > 20 mm Inflammatory or locally advanced disease: 25% Axillary node involvement: 50% ER‐positive: 67% (arm 1), 66% (arm 2); PR‐positive: 49% (arm 1), 53% (arm 2) HER2‐positive: 26% (arm 1), 28% (arm 2) Excluded: prior chemotherapy, radiotherapy or endocrine therapy | |
| Interventions | 4‐arm study with 1 treatment comparison relevant for inclusion in this review (labelled as "sequencing analysis" in trial publication) Arm 2: epirubicin (90 mg/m²) and cyclophosphamide (600 mg/m²) every 3 weeks for 4 cycles followed by paclitaxel (175 mg/m²) with or without gemcitabine (200 mg/m²) every 2 weeks for 4 cycles | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Clinical trial registration record: NCT00070278 Funding considerations: supported by Cancer Research UK, Eli Lilly, Bristol‐Myers Squibb. Trial authors declared that study design, data collection, data analysis, data interpretation, etc did not involve study sponsors. Statistical analysis: Warwick Clinical Trials Unit, Coventry, UK | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Minimisation randomisation. Quote: "…treatment allocations were made by telephoning the Cancer Research UK Trials Unit (Birmingham UK) who used their central computerised minimisation procedure to generate the patient's random allocation." |
| Allocation concealment (selection bias) | Low risk | Central allocation. Quote: "…patients were randomly assigned via a central randomisation procedure to…" |
| Blinding of participants and personnel (performance bias) | Low risk | Open‐label study. Participants in both groups received the same drugs though in different order. Therefore, it was judged to be unlikely that this would lead to a material bias in participants' and physicians' behaviours even when unblinded. |
| Blinding of outcome assessment (detection bias) | Low risk | Lack of blinding unlikely to influence this outcome. |
| Blinding of outcome assessment (detection bias) | Low risk | Unblinding is unlikely to have an impact on outcome assessment for this review question where trial participants received both treatment regimens. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Toxicity was assessed using a range of laboratory tests as well as the CTCAE scale. Dose delays and dose reductions were based on laboratory tests (e.g. ANCs) and severe toxicities. As the study was unblinded, knowledge of treatment allocation may have had some influence on the physicians' assessments. |
| Blinding of outcome assessment (detection bias) | Low risk | Each pathology report was reviewed by 2 people masked to the treatment group – 1 chief investigator and 1 study pathologist. Therefore, judged to have low risk of bias. |
| Incomplete outcome data (attrition bias) | Low risk | All participants randomised to treatment groups, except for 2/416 in the intervention group and 1/415 in the comparator group were included in analysis. 11 (2.6%) participants in the comparator group and 16 (3.85%) in the intervention group were protocol violators (reasons across groups were very similar). These participants were still included in ITT analysis for secondary outcomes. For the primary outcome, there were equal numbers of participants who did not undergo surgery (4 in each group). |
| Selective reporting (reporting bias) | Low risk | Prespecified outcomes in Clinical trials.gov record (NCT00070278) and the Methods section of the trial publication are generally the same. Some important outcomes have been added to the trial publication – i.e. toxicity and treatment adherence data. |
| Other bias | Low risk | None identified |
| Methods | Accrual: October 2004 to June 2006 Multicentre, phase II randomised controlled trial conducted in the USA Adjuvant therapy Median follow‐up: not reported | |
| Participants | Age: median 51 years, range 33–75 years 55% postmenopausal Node‐positive non‐metastatic breast cancer Stage II: 72% Median number of positive lymph nodes: 2 (range 1–35) Hormone receptor‐positive: 66% HER2‐positive: 4% (arm 1); 0% (arm B) Excluded: prior chemotherapy or hormone therapy as neoadjuvant or adjuvant therapy | |
| Interventions | Arm 1 ('arm A' in the trial publication): docetaxel (75 mg/m²) every 2 weeks for 4 cycles followed by doxorubicin (60 mg/m²), cyclophosphamide (600 mg/m²) every 2 weeks for 4 cycles Arm 2 ('arm B' in the trial publication): doxorubicin (60 mg/m²), cyclophosphamide (600 mg/m²) every 2 weeks for 4 cycles followed by docetaxel (75 mg/m²) every 2 weeks for 4 cycles For arm 1 and 2: pegfilgrastim (6 mg) on day 2 after each chemotherapy cycle | |
| Outcomes | Primary outcomes
Secondary outcomes
| |
| Notes | Clinical trial record: NCT00201708 Funding considerations: supported by research grant from Sanofi‐Aventis; data management support by Bridge Site Clinical Research, Columbus | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A 1:1 fixed block randomisation with a block size of 9 was used. After eligibility was confirmed at the coordinating centre randomisation occurred." Comment: baseline characteristics were similar although there were more stage III participants in the comparator arm. |
| Allocation concealment (selection bias) | Low risk | Quote: "at the coordinating centre, randomisation occurred and the group assignment was then sent to the outside institution." |
| Blinding of participants and personnel (performance bias) | Low risk | Open‐label study. Participants in both groups received the same drugs though in different order. Therefore, it was judged to be unlikely that this would lead to a material bias in participants' and physicians' behaviours even when unblinded. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Study used formalised toxicity criteria (NCI CTC version 2) and measured a range of toxicity outcomes, some of which in borderline cases may be affected by unblinding (e.g. neuropathy). Dose‐reductions/delays were based on toxicity assessments. Overall, unblinding may have influenced the physicians' assessments for a subset of outcomes. |
| Blinding of outcome assessment (detection bias) | Low risk | FACT‐B version 4 performed on a small subset of 20 randomly selected participants at 3 time points. Collected for feasibility and not prespecified. Participants were not blinded to treatment allocation. |
| Incomplete outcome data (attrition bias) | Low risk | In the trial publication, there was a mismatch between number of participants in each group in the CONSORT diagram Figure 1 (26 participants in each arm) and Table 1 (28 patients in each arm), with 2 patients unaccounted for. Contacted trial author and received reply confirming that there was an error in the CONSORT diagram and that there were 28 participants in each arm. |
| Selective reporting (reporting bias) | Low risk | Trial record identified (NCT00201708). Outcomes reported (except for 1 outcome that was related to which regimen to take forward for phase III trial). 2 other outcomes were added in the trial publication that were not in trial registration record – RDI and quality of life but these are considered important outcomes to report. |
| Other bias | Low risk | None identified |
| Methods | Accrual: April 1997 to June 2001 Single‐centre, phase II randomised controlled trial conducted in USA Neoadjuvant study Median follow‐up: 24 months (from time of surgery) | |
| Participants | Age: < 50 years: 76%; ≥ 50 years: 24% Stage IIIA: 14 participants, 48%; IIIB: 10 participants, 35%; IV: 5 participants, 17% 55% premenopausal; 31% postmenopausal; 14% perimenopausal Excluded: women with prior hormonal or cytotoxic therapy | |
| Interventions | Arm 1: paclitaxel (250 mg/m²) every 2 weeks for 3 cycles with filgrastim followed by doxorubicin (90 mg/m²) every 2 weeks for 3 cycles with filgrastim; protocol was amended prior to recruitment of final 9 participants: paclitaxel (175 mg/m²) every 2 weeks for 4 cycles with filgrastim followed by doxorubicin (60 mg/m²) every 2 weeks for 4 cycles with filgrastim Arm 2: doxorubicin (90 mg/m²) every 2 weeks for 3 cycles with filgrastim followed by paclitaxel (250 mg/m²) every 2 weeks for 3 cycles with filgrastim; protocol was amended prior to recruitment of final 9 participants: doxorubicin (60 mg/m²) every 2 weeks for 4 cycles with filgrastim followed by paclitaxel (175 mg/m²) every 2 weeks for 4 cycles with filgrastim | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Trial not powered to detect differences between treatment arms Clinical trial registration record not found Funding considerations: cancer research fund of Damon Runyon‐Walter Winchell Foundation, investigator‐initiated grant from Bristol‐Myers Squibb Oncology and Amgen, Footwear Foundation/QVC Presents Shoes on Sale | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was stratified by menopausal status and lesion stage and conducted in blocks of 4 patients as implemented in the RANLST module of the STPLAN software package." |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) | Low risk | No information provided about blinding in the trial publication. Participants in both groups would have received the same drugs though in different order. Therefore, it was unlikely that this would have led to a material bias in participants' and physicians' behaviours even if unblinded. |
| Blinding of outcome assessment (detection bias) | Low risk | If the study was open‐label, a lack of blinding was unlikely to influence this outcome. |
| Blinding of outcome assessment (detection bias) | Low risk | If the study was open‐label, unblinding was unlikely to have an impact on outcome assessment for this review question where trial participants received both treatment regimens. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Collection of toxicity data was not discussed and if study was unblinded, knowledge of treatment allocation may have had some influence on the physicians' assessments. |
| Blinding of outcome assessment (detection bias) | Low risk | Although there was no information regarding blinding of the pathologist who assessed this outcome, pCR was viewed to be an objective outcome. A review by the pathologist on whether pCR had been achieved or not was unlikely to be influenced by unblinding. |
| Incomplete outcome data (attrition bias) | Low risk | A CONSORT diagram was not provided. There do not appear to be missing data for the outcomes reported. |
| Selective reporting (reporting bias) | High risk | Could not identify a clinical trial registry record (trial started recruitment in April 1997 and completed in June 2001). All outcomes reported in the Methods section had the corresponding results in the publication; however, data had not been reported separately by treatment group for OS, DFS or toxicities. |
| Other bias | Low risk | None identified |
| Methods | Accrual: 22 September 2005 to 18 July 2006 Multicentre, phase II randomised controlled trial conducted in Belgium Adjuvant therapy Median follow‐up: not reported | |
| Participants | Age: mean 48.0 years (SD 8.6) (arm 1), mean 48.9 years (SD 9.7) (arm 2) Approximately 50% were > 50 years of age Node‐positive or other features of high risk as per St Gallen criteria Stage I: 20% (arm 1), 26% (arm 2); stage II: 50% (arm 1), 47% (arm 2); stage III: 30% (arm 1), 26% (arm 2) Axillary lymph node involvement: mean 4.8 (SD 7.5) (arm 1), mean 4.9 (SD 9.7) (arm 2) ER‐positive: 70% (arm 1), 74% (arm 2); PR‐positive: 70% (arm 1), 84% (arm 2) HER2‐positive: not reported Excluded: prior systemic anticancer therapy or radiotherapy | |
| Interventions | 4‐arm study with 1 treatment comparison presented as part of Wildiers 2009a (labelled as "conventional" in trial publication) and the other treatment comparison presented in Wildiers 2009b (labelled as "dose‐dense" in trial publication). Arm 1 ('arm B' in the trial publication): docetaxel (100 mg/m²) every 3 weeks for 3 cycles followed by fluorouracil (500 mg/m²), epirubicin (100 mg/m²), cyclophosphamide (500 mg/m²) every 3 weeks for 3 cycles. Arm 2 ('arm A' in the trial publication): fluorouracil (500 mg/m²), epirubicin (100 mg/m²), cyclophosphamide (500 mg/m²) every 3 weeks for 3 cycles followed by docetaxel (100 mg/m²) every 3 weeks for 3 cycles. For both arms: pegfilgrastim allowed for secondary prophylaxis of febrile neutropenia or prolonged grade IV neutropenia | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Clinical trial registration record not found Funding considerations: study and trial publication were supported by an Amgen grant. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomized…(1:1:2:2)." Comment: this process involved stratified randomisation for age (< or > 50 years of age). Baseline characteristics were similar across groups and no major imbalances were noted (e.g. for age, white blood cell count, tumour parameters). |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "…Randomisation of patients was carried out centrally via email or fax…" |
| Blinding of participants and personnel (performance bias) | Low risk | Open‐label study. Participants in both groups received the same drugs though in different order. Therefore, it was unlikely that this would lead to material bias in participants' and physicians' behaviours even when unblinded. |
| Blinding of outcome assessment (detection bias) | Low risk | Study used formalised toxicity criteria (NCI CTC) and the limited number of toxicity outcomes reported (i.e. neutropenia) were based on objective measures from blood tests. Dose‐reductions/delays were based on this toxicity assessment. Given the types of toxicities assessed in this study, unblinding was unlikely to influence the physicians' assessments. |
| Incomplete outcome data (attrition bias) | Low risk | 100% of participants in 'Doc → FEC' (docetaxel → fluorouracil, epirubicin, cyclophosphamide) arm and 95% (19/20) of participants in 'FEC → Doc' (fluorouracil, epirubicin, cyclophosphamide → docetaxel) arm had completed the study. Of the 5 withdrawals, 4 were related to adverse events (perineal abscess, wound problem, mucositis and pharyngitis/gastritis) and 1 due to the physician’s decision. 1 withdrew in the last cycle of chemotherapy. |
| Selective reporting (reporting bias) | Low risk | Could not identify clinical trial registry record (trial started recruitment in September 2005 and completed in July 2006). However, all outcomes reported in the Methods section had the corresponding results in the publication. |
| Other bias | Low risk | None identified |
| Methods | Accrual: 22 September 2005 to 18 July 2006 Multicentre, phase II randomised controlled trial conducted in Belgium Adjuvant therapy Median follow‐up: not reported | |
| Participants | Age: mean 49.2 years (SD 10.0) (arm 1); mean 48.6 years (SD 8.5) (arm 2) 51% (in both arms) were > 50 years of age Node‐positive or other features of high risk as per St Gallen criteria Stage I: 28% (arm 1), 5% (arm 2); stage II: 51% (arm 1), 79% (arm 2); stage III: 21% (arm 1), 15% (arm 2) Axillary lymph node involvement: mean 2.8 (SD 5.3) (arm 1), mean 4.2 (SD 6.9) (arm 2) ER‐positive: 64% (arm 1), 79% (arm 2); PR‐positive: 69% (arm 1), 79% (arm 2) HER2‐positive: not reported Excluded: prior systemic anti‐cancer therapy or radiotherapy | |
| Interventions | 4‐arm study with 1 treatment comparison presented as part of Wildiers 2009a (labelled as "conventional" in trial publication) and the other treatment comparison presented in Wildiers 2009b (labelled as "dose‐dense" in trial publication). Arm 1 ('arm D' in the trial publication): docetaxel (75 mg/m²) every 2 weeks for 4 cycles followed by fluorouracil (375 mg/m²), epirubicin (75 mg/m²), cyclophosphamide (375 mg/m²) every 10 or 11 days for 4 cycles. Arm 2 ('arm C' in the trial publication): fluorouracil (375 mg/m²), epirubicin (75 mg/m²), cyclophosphamide (375 mg/m²) every 10 or 11 days for 4 cycles followed by docetaxel (75 mg/m²) every 2 weeks for 4 cycles. For both arms: pegfilgrastim allowed for secondary prophylaxis of febrile neutropenia or prolonged grade IV neutropenia | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Clinical trial registration record not found Funding considerations: study and trial publication were supported by an Amgen grant. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomized…(1:1:2:2)." Comment: this process involved stratified randomisation for age (< or > 50 years of age). Baseline characteristics were similar across groups and no major imbalances were noted (e.g. for age, white blood cell count, tumour parameters). |
| Allocation concealment (selection bias) | Low risk | Central allocation Quote: "…Randomisation of patients was carried out centrally via email or fax…" |
| Blinding of participants and personnel (performance bias) | Low risk | Open‐label study. Participants in both groups received the same drugs though in different order. Therefore, it was unlikely that this would lead to material bias in participants' and physicians' behaviours even when unblinded. |
| Blinding of outcome assessment (detection bias) | Low risk | Study used formalised toxicity criteria (NCI CTC) and the limited number of toxicity outcomes reported (i.e. neutropenia) were based on objective measures from blood tests. Dose‐reductions/delays were based on this toxicity assessment. Given the types of toxicities assessed in this study, unblinding was unlikely to influence the physicians' assessments. |
| Incomplete outcome data (attrition bias) | Low risk | 92% (36/39) of participants in dose‐dense Doc → FEC (docetaxel → fluorouracil, epirubicin, cyclophosphamide) arm and 97% (38/39) of participants in dose‐dense FEC → Doc (fluorouracil, epirubicin, cyclophosphamide → docetaxel) arm completed the study. 3 withdrawals related to adverse events and 1 due to the physician’s decision. |
| Selective reporting (reporting bias) | Low risk | Could not identify clinical trial registry record (trial started recruitment in September 2005 and completed in July 2006). However, all outcomes reported in the Methods section had the corresponding results in the publication. |
| Other bias | Low risk | None identified |
ANC: absolute neutrophil count; DFS: disease‐free survival; ER: oestrogen receptor; FACT‐B: Functional Assessment of Cancer Therapy – Breast Cancer; G‐CSF: granulocyte‐colony stimulating factor; HER2: human epidermal growth factor receptor 2; ITT: intention to treat; LVEF: left ventricular ejection fraction; NCI CTC: National Cancer Institute Common Toxicity Criteria for Adverse Events; OS: overall survival; pCR: pathological complete response; PR: progesterone receptor; RDI: relative dose intensity; SD: standard deviation.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| Wrong intervention | |
| Wrong study design | |
| Wrong intervention | |
| Wrong intervention | |
| Wrong study design | |
| Wrong participant population | |
| Wrong intervention | |
| Wrong study design | |
| Wrong participant population | |
| Wrong intervention | |
| Wrong intervention | |
| Wrong participant population | |
| Wrong intervention | |
| Wrong intervention | |
| Wrong participant population |
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | Accrual: September 2009 to September 2011 Multicentre, phase II randomised controlled trial conducted in Japan Neoadjuvant study |
| Participants | Median age 54 years (range 33–70 years) Median tumour size 35 mm (range 12–80 mm) 40.8% node‐positive 60.2% ER‐positive 100% HER2‐positive breast cancer |
| Interventions | Arm 1: docetaxel (75 mg/m²), cyclophosphamide (600 mg/m²), trastuzumab (6 mg/kg, loading by 8 mg) for 4 cycles, followed by 5‐fluorouracil (500 mg/m²), epirubicin (100 mg/m²), cyclophosphamide (500 mg/m²) for 4 cycles Arm 3: docetaxel (75 mg/m²), cyclophosphamide (600 mg/m²), trastuzumab (6 mg/kg, loading by 8 mg) for 6 cycles An interim analysis of pCR noted that anthracycline‐containing regimens did not exceed benefit from the current standard regimen and, therefore, study limited allocation only to arm 3. Data for arm 1 and arm 2 only are relevant for this Cochrane review topic |
| Outcomes | Primary outcome
Secondary outcomes
|
| Notes | Clinical trial registry record: UMIN000002365 Last follow‐up date: 1 August 2014 Clinical trials registry record stated that results were partially published at San Antonio Breast Cancer Symposium 2012. Research contact person: Norikazu Masuda, [email protected]; public contact person: Katsumasa Kuroi, [email protected] Authors contacted in May 2018 for data relating to comparison of arm 1 and arm 2 (as data were not reported in San Antonio Breast Cancer Symposium 2012 abstract); no reply. Funding considerations: Japan Breast Cancer Research Group; self‐funded |
| Methods | Accrual: August 2010 to April 2018 Accrual target: 112 participants Phase II randomised controlled trial conducted in Brazil Single centre Neoadjuvant study |
| Participants | Stage IIB to IIIB HER2‐negative breast cancer |
| Interventions | Arm 1: docetaxel (100 mg/m²) intravenously every 3 weeks for 3 cycles followed by 5‐fluorouracil (500 mg/m²), adriamycin (50 mg/m²), cyclophosphamide (500 mg/m²) intravenously every 3 weeks for 3 cycles Arm 2: 5‐fluorouracil (500 mg/m²), adriamycin (50 mg/m²), cyclophosphamide (500 mg/m²) intravenously every 3 weeks for 3 cycles followed by docetaxel (100 mg/m²) every 3 weeks for 3 cycles |
| Outcomes | Primary outcome
Secondary outcome
|
| Notes | Clinical trial registry record: NCT01270373 Sponsor: Instituto Nacional de Cancer Brazil Collaborator: Sanofi |
| Methods | Accrual: February 2000 to March 2005 Multicentre, phase II randomised study conducted in the USA Neoadjuvant study |
| Participants | Age: mean 47.9 years (SD 9.2) (arm 1); mean 50.4 years (SD 8.2) (arm 2) Premenopausal: 59.3% (arm 1), 48.3% (arm 2); postmenopausal: 37.0% (arm 1), 44.8% (arm 2) Clinical T stage: T2 70.4%, T3 29.6% (arm 1); T2 60.0%, T3 40.0% (arm 2) Clinical N stage: N0 55.6%, N1 44.4% (arm 1); N0 46.7%, N1 53.3% (arm 2) ER‐positive: 74.1% (arm 1), 73.3% (arm 2); PR‐positive: 77.8% (arm 1), 60.0% (arm 2) HER2‐positive: 11.1% (arm 1), 30.0% (arm 2) |
| Interventions | Arm 1: paclitaxel (80 mg/m²) every week for 9 cycles followed by doxorubicin (60 mg/m²) every 2 weeks for 4 cycles Arm 2: doxorubicin (60 mg/m²) every 2 weeks for 4 cycles followed by paclitaxel (80 mg/m²) every week for 9 cycles |
| Outcomes |
|
| Notes | Clinical trial registry record: NCT00096291 Trial publications state that overall survival data also collected but not reported. Trialists contacted 27 August 2018 asking for overall survival, disease‐free survival and pCR data if available. Trialists replied that they could make data available but required time. Research contact person: Alphonse Taghian ([email protected]) Funding considerations: supported by Massachusetts Department of Public Health, an Investigator Initiated grant from Bristol‐Myers‐Squibb, Massachusetts General Hospital Cancer Fund, Jane Mailloux Research Fund, NCI Avon Supplement |
ER: oestrogen receptor; HER2: human epidermal growth factor receptor 2; pCR: pathological complete response; PR: progesterone receptor; SD: standard deviation.
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | A randomized study of docetaxel + cyclophosphamide (TC), 5‐fluorouracil + epirubicin + cyclophosphamide (FEC)‐TC and TC‐FEC as preoperative chemotherapy for hormone receptor positive and HER2 negative primary breast cancer JBCRG‐09 |
| Methods | Accrual: 1 September 2009 to undisclosed date Accrual target: 195 Phase II cluster randomised controlled trial conducted in Japan Multicentre Neoadjuvant study Last follow‐up date: 31 May 2017 |
| Participants | Hormone receptor‐positive and HER2‐negative primary breast cancer Primary breast cancer defined as T1c‐3 N0‐1, m0 and tumour size ≤ 7 cm |
| Interventions | Arm 1: docetaxel (75 mg/m²) for 1 hour day 1, cyclophosphamide (600 mg/m²), 15‐30 minutes day 1 for 3 cycles followed 5‐fluorouracil (500 mg/m²) day 1, epirubicin (100 mg/m²) day 1 and cyclophosphamide (500 mg/m²) day 1 for 3 cycles Arm 2: 5‐fluorouracil (500 mg/m²) day 1, epirubicin (100 mg/m²) day 1 and cyclophosphamide (500 mg/m²) day 1 for 3 cycles followed by docetaxel (75 mg/m²) for 1 hour day 1, cyclophosphamide (600 mg/m²), 15‐30 minutes day 1 for 3 cycles Arm 3: docetaxel (75 mg/m²) for 1 hour day 1, cyclophosphamide (600 mg/m²), 15‐30 minutes day 1 for 6 cycles |
| Outcomes | Primary outcome
Secondary outcomes
|
| Starting date | Start date: 1 September 2009 Estimated completion date: 31 May 2017 (last follow‐up date recorded) |
| Contact information | Research contact person: Norikazu Masuda, [email protected]; public contact person: Katsumasa Kuroi, [email protected] |
| Notes | Funding considerations: Japan Breast Cancer Research Group (JBCRG); self‐funded |
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Overall survival Show forest plot | 2 | 947 | Hazard Ratio (Fixed, 95% CI) | 0.80 [0.60, 1.08] |
| Analysis 1.1  Comparison 1 Neoadjuvant, Outcome 1 Overall survival. | ||||
| 2 Disease‐free survival Show forest plot | 1 | 828 | Hazard Ratio (Fixed, 95% CI) | 0.84 [0.65, 1.09] |
| Analysis 1.2  Comparison 1 Neoadjuvant, Outcome 2 Disease‐free survival. | ||||
| 3 Pathological complete response (pCR) includes by hormone or HER2 receptor status Show forest plot | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 1.3  Comparison 1 Neoadjuvant, Outcome 3 Pathological complete response (pCR) includes by hormone or HER2 receptor status. | ||||
| 3.1 No invasive cancer in breast or axilla | 4 | 1280 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.96, 1.38] |
| 3.2 Oestrogen receptor (ER)‐positive | 2 | 708 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.88, 1.58] |
| 3.3 ER‐negative | 2 | 381 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.89, 1.36] |
| 3.4 HER2‐positive | 2 | 467 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.85, 1.33] |
| 3.5 HER2‐negative | 1 | 505 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.92, 2.14] |
| 3.6 HER2‐negative, ER‐negative | 1 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.77, 1.93] |
| 4 Pathological response Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 1.4  Comparison 1 Neoadjuvant, Outcome 4 Pathological response. | ||||
| 4.1 No invasive or in situ carcinoma in breast or axilla | 1 | 119 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.47, 1.84] |
| 4.2 No invasive cancer in breast | 2 | 306 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.79, 1.20] |
| 4.3 No invasive cancer in axillary lymph nodes | 1 | 282 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.66 [1.03, 2.66] |
| 5 Adverse events Show forest plot | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 1.5  Comparison 1 Neoadjuvant, Outcome 5 Adverse events. | ||||
| 5.1 Neutropenia | 1 | 280 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.86, 1.82] |
| 5.2 Neurotoxicity | 2 | 1108 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.55, 1.65] |
| 5.3 Treatment‐related death | 2 | 1108 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.06, 15.93] |
| 6 Treatment adherence: dose reduction Show forest plot | 1 | 280 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.59, 1.11] |
| Analysis 1.6  Comparison 1 Neoadjuvant, Outcome 6 Treatment adherence: dose reduction. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse events Show forest plot | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 2.1  Comparison 2 Adjuvant, Outcome 1 Adverse events. | ||||
| 1.1 Febrile neutropenia | 5 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.24, 2.05] |
| 1.2 Neutropenia | 5 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.40, 0.97] |
| 1.3 Neurotoxicity | 3 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.25, 2.46] |
| 1.4 Treatment‐related death | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Treatment adherence: delay in treatment Show forest plot | 4 | 238 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.52, 1.12] |
| Analysis 2.2  Comparison 2 Adjuvant, Outcome 2 Treatment adherence: delay in treatment. | ||||
| 3 Treatment adherence: dose reduction Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Analysis 2.3  Comparison 2 Adjuvant, Outcome 3 Treatment adherence: dose reduction. | ||||
| 3.1 Taxane (docetaxel) | 3 | 173 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.15, 0.73] |
| 3.2 Anthracycline combination | 2 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.18, 5.44] |
| 4 Treatment adherence: one‐dose reduction Show forest plot | 1 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.14, 2.10] |
| Analysis 2.4  Comparison 2 Adjuvant, Outcome 4 Treatment adherence: one‐dose reduction. | ||||
| 5 Treatment adherence: did not receive planned cycles Show forest plot | 3 | 163 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.15, 1.31] |
| Analysis 2.5  Comparison 2 Adjuvant, Outcome 5 Treatment adherence: did not receive planned cycles. | ||||

Study flow diagram.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
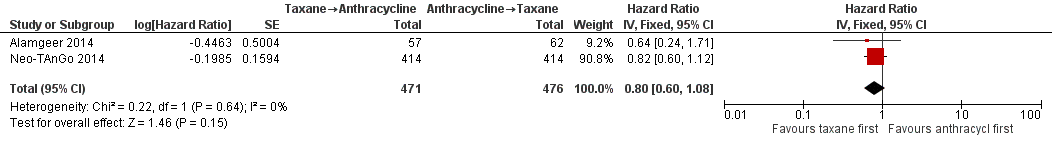
Forest plot of comparison: 1 Neoadjuvant, outcome: 1.1 Overall survival.

Forest plot of comparison: 1 Neoadjuvant, outcome: 1.2 Disease‐free survival.
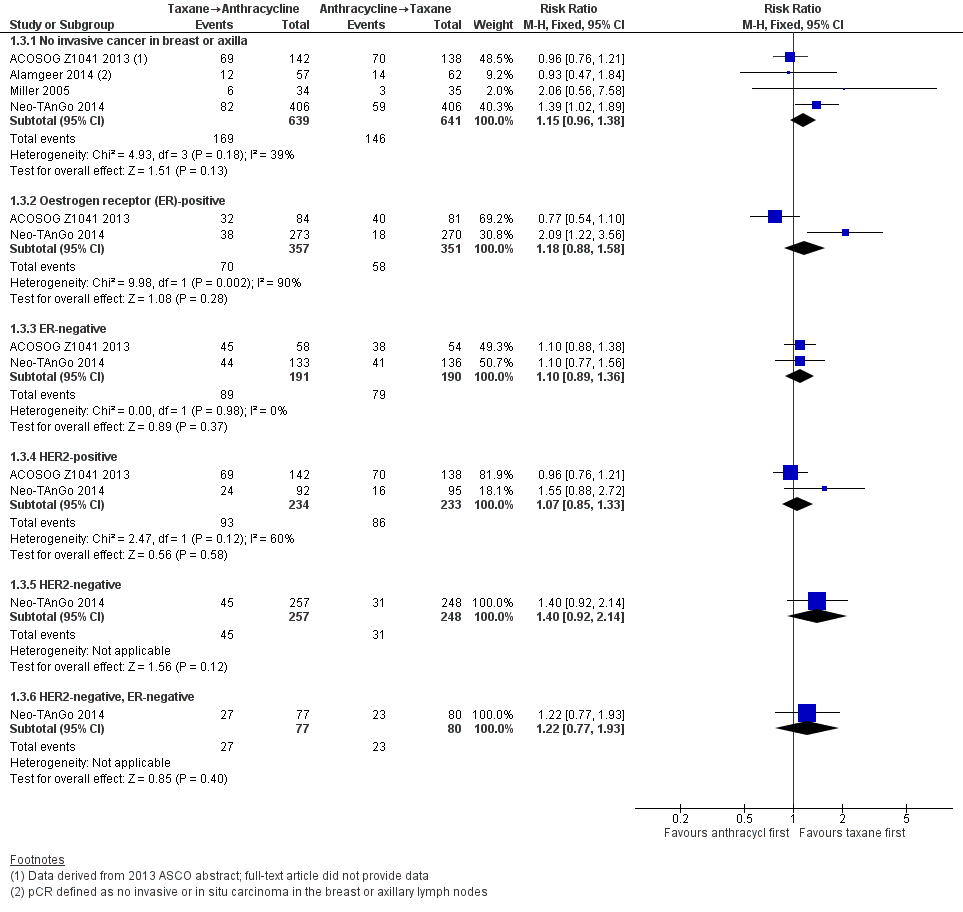
Forest plot of comparison: 1 Neoadjuvant, outcome: 1.3 Pathological complete response (pCR) includes by hormone or HER2 receptor status.
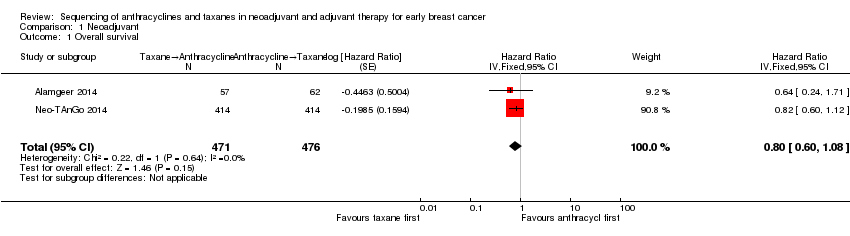
Comparison 1 Neoadjuvant, Outcome 1 Overall survival.
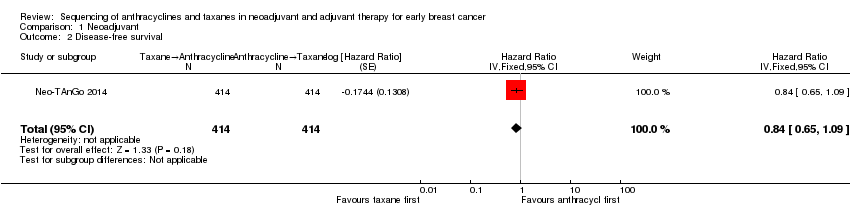
Comparison 1 Neoadjuvant, Outcome 2 Disease‐free survival.
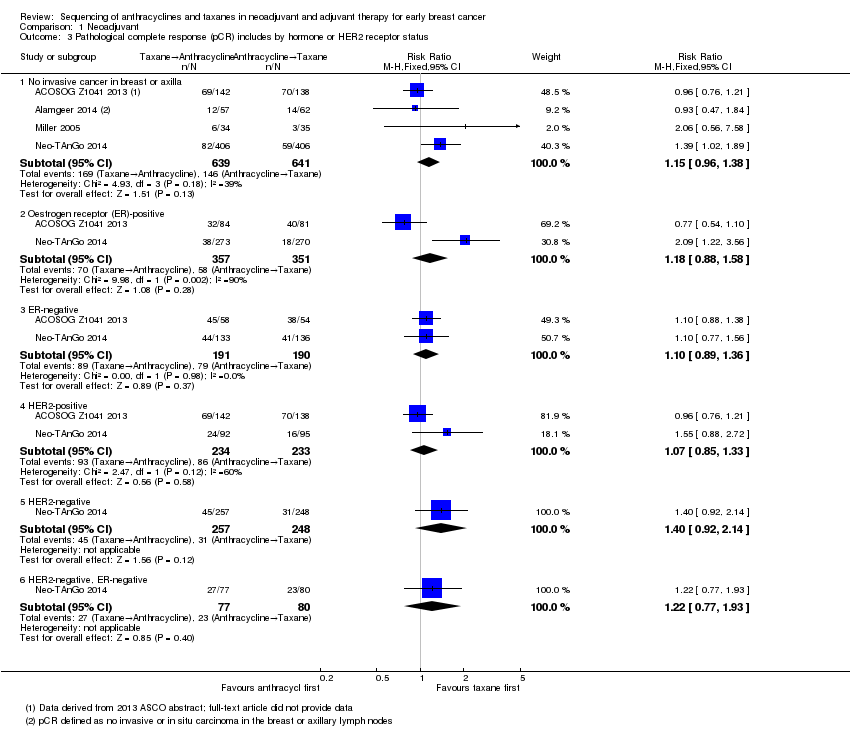
Comparison 1 Neoadjuvant, Outcome 3 Pathological complete response (pCR) includes by hormone or HER2 receptor status.

Comparison 1 Neoadjuvant, Outcome 4 Pathological response.
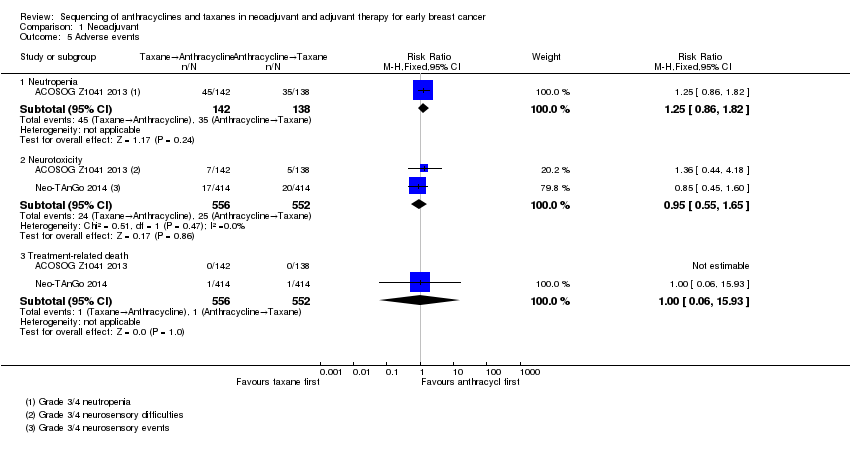
Comparison 1 Neoadjuvant, Outcome 5 Adverse events.

Comparison 1 Neoadjuvant, Outcome 6 Treatment adherence: dose reduction.

Comparison 2 Adjuvant, Outcome 1 Adverse events.

Comparison 2 Adjuvant, Outcome 2 Treatment adherence: delay in treatment.
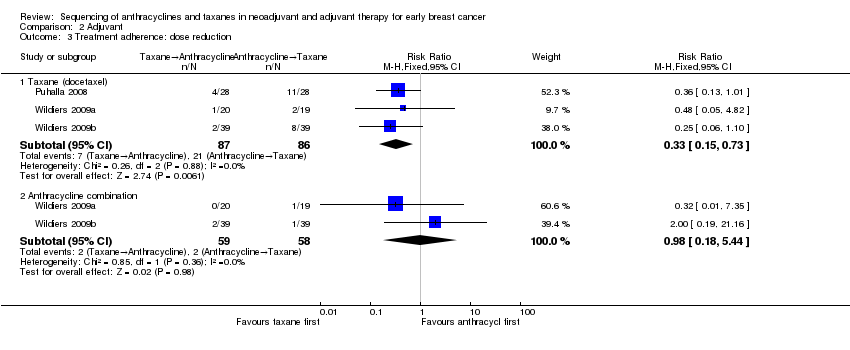
Comparison 2 Adjuvant, Outcome 3 Treatment adherence: dose reduction.

Comparison 2 Adjuvant, Outcome 4 Treatment adherence: one‐dose reduction.
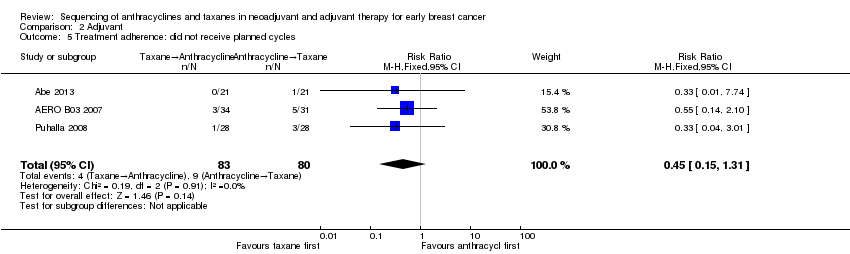
Comparison 2 Adjuvant, Outcome 5 Treatment adherence: did not receive planned cycles.
| Taxane followed by anthracyclines compared to anthracyclines followed by taxane in neoadjuvant therapy for early breast cancer | ||||||
| Patient or population: neoadjuvant therapy for early breast cancer | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with anthracyclines followed by taxane | Risk with taxane followed by anthracyclines | |||||
| Overall survival | 3‐year risk of deatha | HR 0.80 | 947 | ⊕⊕⊕⊝ | — | |
| 702 per 1000 | 620 per 1000 | |||||
| Disease‐free survival | 3‐year risk of recurrencea | HR 0.84 | 828 | ⊕⊕⊕⊝ | — | |
| 616 per 1000 | 552 per 1000 | |||||
| Pathological complete response (no invasive cancer in breast or axilla) (follow‐up: up to 5 years for 2 studies; unreported in 2 studies) | Study population | RR 1.15 | 1280 | ⊕⊕⊕⊕ | — | |
| 228 per 1000 | 262 per 1000 | |||||
| Adverse events: neutropenia (grade 3/4) (follow‐up: up to 6 months based on number of chemotherapy cycles) | Study population | RR 1.25 | 280 | ⊕⊕⊕⊝ | — | |
| 254 per 1000 | 317 per 1000 | |||||
| Adverse events: neurotoxicity (grade 3/4) (follow‐up: up to 5 or 6 months based on number of chemotherapy cycles) | Study population | RR 0.95 | 1108 | ⊕⊕⊝⊝ | — | |
| 45 per 1000 | 43 per 1000 | |||||
| Treatment adherence (defined as dose reduction) (follow‐up: up to 6 months based on number of chemotherapy cycles) | Study population | RR 0.81 | 280 | ⊕⊕⊕⊝ | — | |
| 399 per 1000 | 323 per 1000 | |||||
| Quality of life | — | — | — | — | Not measured | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HR: hazard ratio; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aThe baseline risk in the anthracycline followed by taxane group was based on risk estimates provided in Neo‐TAnGo 2014 (Figure 2D for overall survival; Figure 2B for disease‐free survival). | ||||||
| Taxane followed by anthracyclines compared to anthracyclines followed by taxane in adjuvant therapy for early breast cancer | ||||||
| Patient or population: adjuvant therapy for early breast cancer | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | |
| Risk with anthracyclines followed by taxane | Risk with taxane followed by anthracyclines | |||||
| Overall survival | — | — | — | — | — | Not reported |
| Disease‐free survival | — | — | — | — | — | Not reported |
| Adverse events: neutropenia (grade 3/4) (follow‐up: up to 3.5 or 4.5 months) | Study population | RR 0.62 | 279 | ⊕⊕⊕⊝ | — | |
| 255 per 1000 | 158 per 1000 | |||||
| Adverse events: neurotoxicity (grade 3/4) (follow‐up: up to 4 or 4.5 months) | Study population | RR 0.78 | 162 | ⊕⊕⊝⊝ | — | |
| 63 per 1000 | 49 per 1000 | |||||
| Treatment adherence (defined as dose delay) (follow‐up: up to 3.5 or 4.5 months) | Study population | RR 0.76 | 238 | ⊕⊕⊕⊝ | — | |
| 333 per 1000 | 253 per 1000 | |||||
| Quality of life (follow‐up: up to 4 months) | 1 study reported quality of life data using the FACT‐B version 4 questionnaire (Puhalla 2008). Scores were similar in both groups for a subset of 20 participants who were assessed before, during and after treatment. Numerical or further details were not provided in the trial publication. | — | 20 | ⊕⊕⊕⊝ | — | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aWe did not downgrade the certainty of the evidence. A lack of blinding was judged to be unlikely to influence physician assessment of grade 3/4 neutropenia (blood tests) and there was no heterogeneity detected across studies. | ||||||
| Study | Adherence measure reported | Taxane followed by anthracycline (n/N) | Anthracycline followed by taxane (n/N) |
| Number of participants who received ≥ 90% of planned dose of paclitaxel | 121/142 (85.2%) | 124/138 (89.5%) | |
| Number of participants who received ≥ 90% of planned dose of FEC | 123/142 (86.6%) | 103/138 (74.6%) | |
| Received ≥ 20 weeks of the planned 24 weeks of trastuzumaba or ≥ 10 weeks of planned 12 weeks of trastuzumabb | 127/142 (89.4%)a | 126/138 (91.3%)b | |
| Dose reductions | 46/142 (32.4%) | 55/138 (39.9%) | |
| Discontinued due to intolerance (severe toxic effects) | 9/142 (6.3%) | 9/138 (6.5%) | |
| NR | NR | NR | |
| Dose intensityc | Taxane: 97.% Doxorubicin: 94.2% | Doxorubicin: 95.2% Taxane: 89.2% | |
| Cycle delivered dose intensityd | 83% | 86% | |
| Percentage of cycles with dose reductions | Taxane → epirubicin: 3% | Epirubicin → taxane: 6% | |
| Percentage of cycles with dose delays | Taxane → epirubicin: 15% | Epirubicin → taxane: 15% | |
| NR | NR | NR | |
| FEC: fluorouracil, epirubicin and cyclophosphamide; n: number of participants with delay or dose reduction, etc; N: denominator; NR: not reported. a≥ 20 weeks of the planned 24 weeks of trastuzumab. | |||
| Study | Term used to represent dose intensity | Taxane followed by anthracycline | Anthracycline followed by taxane |
| Mean relative dose intensitya | Docetaxel: 95.2% FEC: 98.9% | Docetaxel: 94.2% FEC: 97.8% | |
| Median relative dose intensity | Docetaxel: 96% (range: 25–104) Epirubicin: 96% (range: 0–102) Cyclophosphamide: 95% (range: 0–102) | Docetaxel: 81% (range: 0–102) Epirubicin: 97% (range: 66–102) Cyclophosphamide: 97% (range: 61–102) | |
| Mean relative dose intensity | Docetaxel: 96% Doxorubicin: 95% | Docetaxel: 82% Doxorubicin: 96% | |
| Mean dose intensityb | Docetaxel: 99% FEC: 97% | Docetaxel: 97% FEC: 97% | |
| Wildiers 2009b (dose‐dense) | Mean dose intensityb | Docetaxel: 111% FEC: 143% | Docetaxel: 108% FEC: 146% |
| FEC: fluorouracil, epirubicin, cyclophosphamide. aData used based on the reporting of findings in the abstract and Table 2 in the trial publication. The docetaxel and FEC percentages were switched in the text in the Results section. Therefore, the consistency of results presented in Table 2 and abstract were considered to be the most accurate reflection of the results. bDefined as the dose intensity achieved in intervention or comparator arm for a participant who received all intended doses with no cycle delay or dose reduction. | |||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Overall survival Show forest plot | 2 | 947 | Hazard Ratio (Fixed, 95% CI) | 0.80 [0.60, 1.08] |
| 2 Disease‐free survival Show forest plot | 1 | 828 | Hazard Ratio (Fixed, 95% CI) | 0.84 [0.65, 1.09] |
| 3 Pathological complete response (pCR) includes by hormone or HER2 receptor status Show forest plot | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 No invasive cancer in breast or axilla | 4 | 1280 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.96, 1.38] |
| 3.2 Oestrogen receptor (ER)‐positive | 2 | 708 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.88, 1.58] |
| 3.3 ER‐negative | 2 | 381 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.89, 1.36] |
| 3.4 HER2‐positive | 2 | 467 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.07 [0.85, 1.33] |
| 3.5 HER2‐negative | 1 | 505 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.40 [0.92, 2.14] |
| 3.6 HER2‐negative, ER‐negative | 1 | 157 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.77, 1.93] |
| 4 Pathological response Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 No invasive or in situ carcinoma in breast or axilla | 1 | 119 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.47, 1.84] |
| 4.2 No invasive cancer in breast | 2 | 306 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.79, 1.20] |
| 4.3 No invasive cancer in axillary lymph nodes | 1 | 282 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.66 [1.03, 2.66] |
| 5 Adverse events Show forest plot | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Neutropenia | 1 | 280 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.86, 1.82] |
| 5.2 Neurotoxicity | 2 | 1108 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.55, 1.65] |
| 5.3 Treatment‐related death | 2 | 1108 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.06, 15.93] |
| 6 Treatment adherence: dose reduction Show forest plot | 1 | 280 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.59, 1.11] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse events Show forest plot | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Febrile neutropenia | 5 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.24, 2.05] |
| 1.2 Neutropenia | 5 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.40, 0.97] |
| 1.3 Neurotoxicity | 3 | 162 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.25, 2.46] |
| 1.4 Treatment‐related death | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Treatment adherence: delay in treatment Show forest plot | 4 | 238 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.52, 1.12] |
| 3 Treatment adherence: dose reduction Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Taxane (docetaxel) | 3 | 173 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.15, 0.73] |
| 3.2 Anthracycline combination | 2 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.98 [0.18, 5.44] |
| 4 Treatment adherence: one‐dose reduction Show forest plot | 1 | 65 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.14, 2.10] |
| 5 Treatment adherence: did not receive planned cycles Show forest plot | 3 | 163 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.45 [0.15, 1.31] |