Anticuerpos anti‐IL‐12/23p40 para el mantenimiento de la remisión en la enfermedad de Crohn
Resumen
Antecedentes
El ustekinumab (CNTO 12) y el briakinumab (ABT‐23) son anticuerpos monoclonales dirigidos a la subunidad p40 estándar de las citoquinas interleucina‐12 e interleucina‐23 (IL‐12/23p40), que están involucradas en la patogenia de la enfermedad de Crohn. Una proporción significativa de pacientes con enfermedad de Crohn fracasan con el tratamiento convencional o con productos biológicos (p.ej. infliximab) o presentan eventos adversos significativos. Los anticuerpos anti‐IL‐12/23p40 como el ustekinumab pueden ser una alternativa efectiva para estos pacientes.
Objetivos
Los objetivos de esta revisión fueron evaluar la eficacia y la seguridad de los anticuerpos anti‐IL‐12/23p40 para el mantenimiento de la remisión en la EC.
Métodos de búsqueda
Se realizaron búsquedas en el Registro Especializado de Ensayos Controlados del Grupo Cochrane de EII (Cochrane IBD Group), CENTRAL, MEDLINE, Embase y en los registros de ensayos desde su inicio hasta el 17 de septiembre 2019. Se realizaron búsquedas de estudios adicionales en las referencias y en los resúmenes de congresos.
Criterios de selección
Se consideraron para su inclusión los ensayos controlados aleatorios en que se compararon los anticuerpos monoclonales contra la IL‐12/23p40 con placebo u otro comparador activo en pacientes con EC inactiva.
Obtención y análisis de los datos
Dos autores de la revisión examinaron de forma independiente los estudios para la inclusión, extrajeron los datos y evaluaron la calidad de los estudios mediante la herramienta Cochrane de «Riesgo de sesgo». La medida de resultado primaria fue el fracaso para inducir la remisión clínica, definida como el índice de actividad de enfermedad de Crohn (IAEC) < 150 puntos. Los resultados secundarios incluyeron: el fracaso para mantener la respuesta clínica, los eventos adversos (EA), los eventos adversos graves (EAG) y los retiros debidos a EA. La respuesta clínica se definió como una disminución en la puntuación del IAEC de ≥ 100 puntos de la puntuación inicial. Para cada resultado se calculó el riesgo relativo (RR) y los intervalos de confianza del 95% (IC del 95%). Se analizaron todos los datos sobre la base del análisis de intención de tratar. Se utilizaron los criterios GRADE para evaluar la certeza general de la evidencia que apoyan los resultados.
Resultados principales
Tres ensayos controlados aleatorizados (646 participantes) cumplieron los criterios de inclusión. Dos ensayos evaluaron la eficacia del ustekinumab (542 participantes) y un estudio evaluó la eficacia del briakinumab (104 participantes). Se evaluaron todos los estudios incluidos como de riesgo de sesgo bajo.
Un estudio (N = 145) comparó el ustekinumab subcutáneo (90 mg) administrado a las ocho y 16 semanas en comparación con el placebo. El 58% (42/72) de los participantes del grupo ustekinumab no lograron mantener la remisión clínica a las 22 semanas en comparación con el 73% (53/73) de los participantes del grupo placebo (RR 0,80; IC del 95%: 0,63 a 1,02; evidencia de certeza moderada). Se observó un fracaso en el mantenimiento de la respuesta clínica a las 22 semanas en el 31% (22/72) de los participantes del grupo ustekinumab en comparación con el 58% (42/73) de los participantes en el grupo placebo (RR 0,53; IC del 95%: 0,36 a 0,79; evidencia de certeza moderada). Un estudio (N = 388) comparó el ustekinumab subcutáneo (90 mg) administrado cada ocho semanas o cada 12 semanas con placebo durante 44 semanas. El 49% (126/257) de los participantes del grupo ustekinumab no lograron mantener la remisión clínica a las 44 semanas en comparación con el 64% (84/131) de los participantes del grupo placebo (RR 0,76; IC del 95%: 0,64 a 0,91; evidencia de certeza moderada). El 41% (106/257) de los participantes en el grupo ustekinumab no lograron mantener la respuesta clínica a las 44 semanas en comparación con el 56% (73/131) de los participantes en el grupo placebo (RR 0,74; IC del 95%: 0,60 a 0,91; evidencia de certeza moderada). El 80% (267/335) de los participantes en el grupo ustekinumab tuvieron un AE en comparación con el 84% (173/206) de los participantes en el grupo placebo (RR 0,94; IC del 95%: 0,87 a 1,03; evidencia de certeza alta). Los eventos adversos que se informaron con frecuencia fueron: infecciones, reacciones en el sitio de la inyección, evento de EC, dolor abdominal, náuseas, artralgia y dolor de cabeza. El 11% de los participantes del grupo ustekinumab presentaron un EAG comparado con el 16% (32/206) de los participantes del grupo placebo (RR 0,74; IC del 95%: 0,48 a 1,15; evidencia de certeza moderada). Los EAG incluyeron infecciones graves, neoplasias malignas y carcinoma de células basales. El 7% (5/73) de los participantes del grupo ustekinumab se retiraron del estudio debido a un EA comparado con el 1% (1/72) de los participantes del grupo placebo (RR 4,93; IC del 95%: 0,59 a 41,18; evidencia de certeza baja). El empeoramiento de la EC fue la razón más común para el retiro debido a un EA.
Un estudio comparó el briakinumab intravenoso (200 mg, 400 mg o 700 mg) administrado en las semanas 12, 16 y 20 con placebo. Se observó un fracaso en el mantenimiento de la remisión clínica a las 24 semanas en el 51% (32/63) de los participantes que recibieron briakinumab en comparación con el 61% (22/36) de los participantes que recibieron placebo (RR 0,84; IC del 95%: 0,58 a 1,20; evidencia de certeza baja). Se observó un fracaso en el mantenimiento de la respuesta clínica a las 24 semanas en el 33% (21/63) de los participantes que recibieron briakinumab en comparación con el 53% (19/36) de los participantes que recibieron placebo (RR 0,64; IC del 95%: 0,40 a 1,02; evidencia de certeza baja). El 66% (59/90) de los participantes que recibieron briakinumab presentaron un EA en comparación con el 64% (9/14) de los participantes que recibieron placebo (RR 1,02; IC del 95%: 0,67 a 1,55; evidencia de certeza baja). Los EA comunes fueron: infección de las vías respiratorias superiores, náuseas, dolor abdominal, dolor de cabeza y reacción en el sitio de la inyección. El 2% (2/90) de los participantes que recibieron briakinumab presentaron un EAG en comparación con el 7% (1/14) de los participantes que recibieron placebo (RR 0,31; IC del 95%: 0,03 a 3,21; evidencia de certeza baja). Los EAG incluyeron: obstrucción del intestino delgado, trombosis venosa profunda y dificultad respiratoria. El retiro debido a un EA se observó en el 2% de los participantes que recibieron briakinumab en comparación con el 0% (0/14) de los participantes que recibieron placebo (RR 0,82; IC del 95%: 0,04 a 16,34; evidencia de certeza baja). No se describieron los EA que provocaron el retiro del estudio.
Conclusiones de los autores
La evidencia de certeza moderada indica que es probable que el uso de ustekinumab sea efectivo para el mantenimiento de la remisión clínica y la respuesta en pacientes con EC de moderada a grave en remisión sin un mayor riesgo de eventos adversos (evidencia de certeza alta) o eventos adversos graves (evidencia de certeza moderada) en comparación con el placebo. El efecto del briakinumab sobre el mantenimiento de la remisión clínica y la respuesta en pacientes con enfermedad de Crohn de moderada a grave en remisión fue incierto, ya que la certeza de la evidencia fue baja. El efecto del briakinumab sobre los eventos adversos y los eventos adversos graves también fue incierto debido a que la certeza de la evidencia fue baja. Se requieren estudios adicionales para determinar la eficacia y la seguridad a largo plazo del tratamiento subcutáneo de mantenimiento con ustekinumab en pacientes con enfermedad de Crohn y también para confirmar si debe utilizarse solo o en combinación con otros agentes. Las investigaciones futuras que comparen el ustekinumab con otros fármacos biológicos ayudarán a determinar cuándo es más apropiado el tratamiento con ustekinumab en la EC. En la actualidad, hay un estudio en curso que compara el ustekinumab con el adalimumab. Esta revisión se actualizará cuando los resultados de este estudio estén disponibles. Los fabricantes de briakinumab han interrumpido la producción de este medicamento, por lo que es poco probable que se realicen estudios adicionales.
PICO
Resumen en términos sencillos
Ustekinumab y briakinumab para el tratamiento de la enfermedad de Crohn inactiva
¿Qué es la enfermedad de Crohn?
La enfermedad de Crohn es una enfermedad intestinal inflamatoria de largo plazo (crónica) que puede afectar cualquier parte del aparato digestivo, desde la boca al ano. Los síntomas habituales incluyen dolor abdominal, diarrea y pérdida de peso. Por lo general, la enfermedad de Crohn se controla con tratamientos médicos y quirúrgicos. Cuando un paciente con enfermedad de Crohn presenta síntomas, la enfermedad es «activa». Cuando los síntomas de la enfermedad de Crohn desaparecen, se dice que la enfermedad está en remisión.
¿Qué son el ustekinumab y el briakinumab?
El ustekinumab y el briakinumab son medicamentos biológicos que inhiben el sistema inmunológico y reducen la inflamación asociada con la enfermedad de Crohn. Se pueden inyectar debajo de la piel con una jeringa (subcutánea) o directamente en una vena (intravenosa). Muchos pacientes con enfermedad de Crohn fracasan con el tratamiento convencional con corticosteroides o con productos biológicos (p.ej. infliximab) o presentan efectos secundarios significativos. Un medicamento como el ustekinumab puede ser una alternativa efectiva para estos pacientes.
¿Qué examinó la investigación?
Los investigadores analizaron si el ustekinumab o el briakinumab ayudan a mantener la remisión en pacientes con enfermedad de Crohn y si estos medicamentos tienen efectos perjudiciales (efectos secundarios). Los investigadores buscaron en la literatura médica hasta el 17 de septiembre 2019.
¿Qué encontraron los investigadores?
Los investigadores identificaron tres estudios (646 participantes) que compararon el ustekinumab (2 estudios, 542 participantes) o el briakinumab (1 estudio, 104 participantes) con un placebo (un medicamento falso). El efecto del tratamiento con ustekinumab se examinó en un estudio después de 22 semanas y en otro estudio después de 44 semanas. El estudio de briakinumab examinó el efecto del tratamiento después de 24 semanas. Todos los estudios tuvieron una calidad metodológica elevada.
La evidencia de certeza moderada indica que el ustekinumab es más efectivo que el placebo para mantener la remisión y reducir los síntomas de la enfermedad de Crohn a las 22 y 44 semanas. Las tasas de efectos secundarios (ustekinumab: 80%; placebo: 84%) y de efectos secundarios graves (ustekinumab: 11%; placebo: 16%) fueron inferiores en los participantes con ustekinumab que en los participantes con placebo. La evidencia de certeza alta indica que no hay un mayor riesgo de efectos secundarios con el uso de ustekinumab en comparación con placebo. Los efectos secundarios informados con frecuencia fueron: infecciones, reacciones en el sitio de la inyección, evento de la enfermedad de Crohn (p.ej. empeoramiento de la enfermedad), dolor abdominal, náuseas, artralgia (p.ej. dolor en las articulaciones) y cefalea. La evidencia de certeza moderada indica que no hay un mayor riesgo de efectos secundarios graves con el uso de ustekinumab en comparación con placebo. Los efectos secundarios graves fueron: infecciones graves, neoplasia maligna (es decir, un tumor canceroso) y el carcinoma de células basales (es decir, cáncer de piel). En un estudio, se retiraron más participantes del grupo de ustekinumab (7%) debido a un efecto secundario en comparación con el grupo de placebo (1%). Sin embargo, la certeza de la evidencia fue baja. El efecto secundario más común que llevó al retiro del estudio fue el empeoramiento de la enfermedad de Crohn.
El 51% (32/63) de los participantes que recibieron briakinumab tuvieron recaídas a las 24 semanas en comparación con el 61% (22/36) de los participantes con placebo (evidencia de certeza baja). La proporción de participantes sin reducción de los síntomas de la enfermedad de Crohn a las 24 semanas fue del 33% (21/63) en el grupo que recibió briakinumab en comparación con el 53% (19/36) en el grupo placebo (evidencia de certeza baja). Las tasas de efectos secundarios (briakinumab: 66%; placebo: 64%; evidencia de certeza baja) y el retiro del estudio debido a efectos secundarios (briakinumab: 2%; placebo: 0%; evidencia de certeza baja) fueron comparables en los participantes que recibieron briakinumab y los que recibieron placebo. Los efectos secundarios que se informaron con frecuencia incluyeron: infección del tracto respiratorio superior, náuseas, dolor abdominal, dolor de cabeza y reacción en el sitio de la inyección. La tasa de efectos secundarios graves fue menor en los participantes que recibieron briakinumab (2%) en comparación con los participantes que recibieron placebo (7%) (evidencia de certeza baja). Los efectos secundarios graves incluyeron: obstrucción del intestino delgado, trombosis venosa profunda y dificultad respiratoria.
Conclusiones
La evidencia de certeza moderada indica que es probable que el ustekinumab sea efectivo para el mantenimiento de la remisión clínica y la respuesta en pacientes con enfermedad de Crohn de moderada a grave en remisión sin un mayor riesgo de efectos secundarios (evidencia de certeza alta) o efectos secundarios graves (evidencia de certeza moderada). Se requieren estudios adicionales para determinar los beneficios y los daños a largo plazo del tratamiento de mantenimiento con ustekinumab subcutáneo en pacientes con enfermedad de Crohn y también para definir si se debe utilizar solo o en combinación con otros agentes. Las investigaciones futuras que comparen el ustekinumab con otros fármacos biológicos ayudarán a determinar cuándo es más apropiado el tratamiento con ustekinumab para la enfermedad de Crohn. En la actualidad, hay un estudio en curso que compara el ustekinumab con el adalimumab (otro tipo de medicamento biológico). Los fabricantes de briakinumab han interrumpido la producción de este medicamento, por lo que es poco probable que se realicen estudios adicionales.
Authors' conclusions
Summary of findings
| Ustekinumab compared to placebo for maintenance of remission in Crohn's disease | ||||||
| Patient or population: people with moderate to severe Crohn's disease in remission | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | No. of participants | Certainty of the evidence | Comments | |
| Risk with placebo | Risk with ustekinumab | |||||
| Failure to maintain clinical remission Follow‐up: 22 weeks | 726 per 1000 | 581 per 1000 | RR 0.80 | 145 | ⊕⊕⊕⊝ | Clinical remission was defined as a CDAI < 150 points. |
| Failure to maintain clinical response Follow‐up: 22 weeks | 575 per 1000 | 305 per 1000 | RR 0.53 | 145 | ⊕⊕⊕⊝ | Clinical response was defined as a ≥ 100‐point decrease from the baseline CDAI score. |
| Failure to maintain clinical remission Follow‐up: 44 weeks | 641 per 1000 | 487 per 1000 (410 to 584) | RR 0.76 (0.64 to 0.91) | 388 (1 RCT) | ⊕⊕⊕⊝ | Clinical remission was defined as a CDAI < 150 points. |
| Failure to maintain clinical response Follow‐up: 44 weeks | 557 per 1000 | 412 per 1000 (334 to 507) | RR 0.74 (0.60 to 0.91) | 388 (1 RCT) | ⊕⊕⊕⊝ | Clinical response was defined as a decrease from baseline in the CDAI score of ≥ 100 points or a CDAI score < 150. |
| Adverse events Follow‐up: 44 weeks | 840 per 1000 | 789 per 1000 (731 to 865) | RR 0.94 (0.87 to 1.03) | 541 (2 RCTs) | ⊕⊕⊕⊕ | Commonly reported adverse events included worsening Crohn's disease, abdominal pain, nausea, arthralgia, and headache. |
| Serious adverse events Follow‐up: 44 weeks | 155 per 1000 | 115 per 1000 | RR 0.74 | 541 | ⊕⊕⊕⊝ | Commonly reported serious adverse events included malignant neoplasm, basal cell carcinoma, and injection site reactions. |
| Withdrawal due to adverse events Follow‐up: 44 weeks | 14 per 1000 | 68 per 1000 (8 to 572) | RR 4.93 (0.59 to 41.18) | 145 (1 RCT) | ⊕⊕⊝⊝ | Adverse events leading to withdrawal included worsening Crohn's disease |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Downgraded one level due to sparse data (95 events). | ||||||
| Briakinumab compared to placebo for maintenance of remission in Crohn's disease | ||||||
| Patient or population: people with moderate to severe Crohn's disease in remission | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | No. of participants | Certainty of the evidence | Comments | |
| Risk with placebo | Risk with briakinumab | |||||
| Failure to maintain clinical remission Follow‐up: 24 weeks | 611 per 1000 | 513 per 1000 (354 to 733) | RR 0.84 (0.58 to 1.20) | 99 (1 RCT) | ⊕⊕⊝⊝ | Clinical remission was defined as a CDAI score < 150 points. |
| Failure to maintain clinical response Follow‐up: 24 weeks | 528 per 1000 | 338 per 1000 (211 to 538) | RR 0.64 (0.40 to 1.02) | 99 (1 RCT) | ⊕⊕⊝⊝ | Clinical response was defined as a decrease in CDAI score ≥ 100 points compared with week 0. |
| Adverse events Follow‐up: 24 weeks | 643 per 1000 | 656 per 1000 (431 to 996) | RR 1.02 (0.67 to 1.55) | 104 (1 RCT) | ⊕⊕⊝⊝ | Common adverse events included upper respiratory tract infection, nausea, abdominal pain, and headache. |
| Serious adverse events Follow‐up: 24 weeks | 71 per 1000 | 22 per 1000 (2 to 229) | RR 0.31 (0.03 to 3.21) | 104 (1 RCT) | ⊕⊕⊝⊝ | Serious adverse events included small bowel obstruction, deep vein thrombosis, and respiratory distress. |
| Withdrawals due to adverse events Follow‐up: 24 weeks | 0 per 1000 | 0 per 1000 (0 to 0) | RR 0.82 (0.04 to 16.34) | 104 (1 RCT) | ⊕⊕⊝⊝ | We were unable to calculate absolute effects. 2% of briakinumab participants withdrew due to an adverse event compared to none of the placebo participants. Adverse events leading to withdrawal were not reported. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CDAI: Crohn's Disease Activity Index; CI: confidence interval; RCT: randomized controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Downgraded two levels due to sparse data (54 events) and very wide confidence interval. | ||||||
Background
Description of the condition
Crohn's disease is a chronic, relapsing and remitting, inflammatory condition that results in abdominal pain, diarrhea, and weight loss. Despite highly effective therapies, up to one‐third of people with Crohn's disease develop fistulas or require surgery for disease complications (CCC 2018). Additionally, many of the current therapies are encumbered by unexpected adverse events (Colombel 2004; Colombel 2007; Ford 2011; Raj 2010; Schreiber 2007; Singh 2011; Yang 2002). Given that approximately 40% of patients do not respond to conventional agents such as corticosteroids, immunosuppressants, and biologics (e.g. infliximab), the development of new therapies is a priority for clinical practice (Danese 2011; Hanauer 2002; Hanauer 2006; Targan 1997).
Monoclonal antibodies against tumor necrosis factor alpha (TNF‐α) are commonly used for maintenance of remission in Crohn's disease. However, it is estimated that 25% to 40% of patients receiving TNF‐α antagonists who initially benefit from treatment either lose response or are forced to stop treatment due to intolerable adverse events during the maintenance phase (Danese 2011). Although some patients regain response with an increase in dose, a significant proportion of these individuals experience a relapse of their Crohn's disease due to non‐TNF‐α driven inflammatory pathways (Steenholdt 2016). In such cases continued anti‐TNF‐α treatment would be ineffective and other therapies are needed.
Description of the intervention
Ustekinumab (CNTO 1275) and briakinumab (ABT‐874) are monoclonal antibodies directed against the shared p40 subunit of interleukin‐12 and interleukin‐23 (IL‐12/23p40). Inhibition of these cytokines is beneficial for induction of remission in Crohn's disease (Feagan 2016).
How the intervention might work
Pathogenic immune responses in Crohn’s disease are characterized by dysregulated T‐cell activity stimulated by the release of interleukin‐12 (IL‐12) and IL‐23 by antigen‐presenting cells (Benson 2011a; Duvallet 2011; Peluso 2006; Watanabe 2004). IL‐12 promotes differentiation of naive T‐cells down the Th1 pathway (Xavier 2007), whereas IL‐23 stimulates proliferation of Th17 lymphocytes. Activation of the Th1 pathway culminates in the release of interferon (IFN)‐ɣ and TNF‐α (Benson 2011b; Cingoz 2009; Peluso 2006), two cytokines that have been implicated in the pathogenesis of Crohn's disease. In contrast, Th17 is important in maintaining many chronic inflammatory conditions (Duvallet 2011).
In murine models, inhibition of IL‐12/23p40 results in apoptosis of T cells in the gut mucosa (Fuss 1999), which results in disease improvement (Neurath 1995). Collectively, these data suggest a possible therapeutic role for ustekinumab in the treatment of Crohn’s disease.
Why it is important to do this review
Ustekinumab is widely used for the treatment of psoriasis where it has demonstrated safety and efficacy (Papp 2008). The safety and efficacy of ustekinumab has been established for induction of remission in Crohn's disease (MacDonald 2016). However, the benefits and harms of this therapy for maintenance of remission in Crohn's disease have not been systematically assessed.
Objectives
The objectives of this review were to assess the efficacy and safety of anti‐IL‐12/23p40 antibodies for maintenance of remission in Crohn's disease.
Methods
Criteria for considering studies for this review
Types of studies
We considered randomized controlled trials for inclusion in the review.
Types of participants
We considered participants with quiescent Crohn's disease (as defined by the original study) for inclusion. We applied no age restrictions.
Types of interventions
We considered trials comparing monoclonal antibodies against IL‐12/23p40 to placebo or an active comparator for inclusion.
Types of outcome measures
Primary outcomes
-
Proportion of participants who failed to maintain clinical remission (as defined by the original study).
Secondary outcomes
The proportion of participants:
-
who failed to maintain clinical response (as defined by the original study);
-
who failed to maintain endoscopic response (as defined by the original study);
-
who failed to maintain endoscopic remission (as defined by the original study);
-
who failed to maintain histological response (as defined by the original study);
-
who failed to maintain histological remission (as defined by the original study);
-
who failed to maintain both clinical and endoscopic response (as defined by the original study);
-
who failed to maintain both clinical and endoscopic remission (as defined by the original study);
-
with adverse events;
-
with serious adverse events; and
-
who withdrew from the study due to adverse events.
Search methods for identification of studies
Electronic searches
We searched the following databases for relevant studies:
-
the Cochrane IBD Group Specialized Register (searched 17 September 2019);
-
the Cochrane Central Register of Controlled Trials (CENTRAL) (searched 17 September 2019);
-
MEDLINE (Ovid, 1946 to 17 September 2019); and
-
Embase (Ovid, 1984 to 17 September 2019).
The search strategies are listed in Appendix 1.
Searching other resources
We also searched the references of relevant trials and review articles for additional studies not identified by the search. Furthermore, we searched conference proceedings from major meetings including Digestive Disease Week, the European Crohn's and Colitis Organisation congress, and the United European Gastroenterology Week for the last five years (2013 to 2017) to identify studies published in abstract form only. We contacted leaders in the field and the manufacturers of briakinumab and ustekinumab (Abbott Laboratories, Abbott Park, IL, USA and Centocor, Horsham, PA, USA) to identify any unpublished studies.
Lastly, to identify ongoing trials, we searched trials registers including:
-
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (www.clinicaltrials.gov; searched 17 September 2019);
-
International Federation of Pharmaceutical Manufacturers & Associations (www.ifpma.org; searched 17 September 2019); and
-
ISRCTN registry (www.isrctn.com; searched 17 September 2019).
Data collection and analysis
Selection of studies
Two review authors (SCD and TMN) screened the search results independently for eligible studies based on the described inclusion criteria. Any disagreements were discussed until a consensus was reached or by consulting a third review author (RK) when necessary.
Data extraction and management
Two review authors (SCD and TMN) independently extracted data from the included studies. Any disagreements regarding the extracted data were first discussed and then brought to a third review author (RK) for resolution as required. If the case of missing data, we contacted the study authors for additional information.
Assessment of risk of bias in included studies
Two review authors (SCD and TMN) assessed the methodological quality of the included studies using the Cochrane 'Risk of bias' tool (Higgins 2011). We assessed factors pertaining to the methodological quality of the study including sequence generation, allocation concealment, blinding, incomplete outcome data, selective outcome reporting, and other potential sources of bias. We judged studies to be at low, high, or unclear risk of bias. Any disagreements regarding risk of bias were first discussed and then brought to a third review author (RK) for resolution as required.
We used the GRADE approach to determine the overall certainty of evidence supporting the following outcomes: failure to maintain clinical remission, failure to maintain clinical response, adverse events, serious adverse events, and withdrawal due to adverse events (Schünemann 2011). We considered evidence that was extracted from randomized controlled trials to be of high certainty. The certainty of the evidence could be downgraded due to the following factors:
-
risk of bias;
-
indirect evidence;
-
inconsistency (unexplained heterogeneity);
-
imprecision; and
-
publication bias.
We classified the overall certainty of the evidence as high (we are very confident that the true effect lies close to that of the estimate of the effect); moderate (we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different.); low (our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect); or very low (we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect) (Guyatt 2008).
Measures of treatment effect
We used Review Manager 5 to analyze the data on an intention‐to‐treat basis (Review Manager 2014). We calculated the risk ratio (RR) and corresponding 95% confidence interval (95% CI) for dichotomous outcomes. We planned to calculate the mean difference (MD) and corresponding 95% CI for continuous outcomes.
Unit of analysis issues
For outcomes measured at different time points, we determined the appropriate fixed intervals for the follow‐up for each outcome. We included cross‐over trials if data were available for the first phase of the trial prior to cross‐over. We performed separate comparisons for each type of antibody and for each antibody in the context of other therapies. For reoccurring events (e.g. adverse events), we reported on the proportion of participants who experienced at least one event. If we encountered multiple drug dose groups, we divided the placebo group across the dose groups to avoid a unit of analysis error. We did not encounter any cluster‐randomized trials.
Dealing with missing data
We contacted the authors of the original study if any data were missing. Additionally, participants without treatment outcomes were presumed to be treatment failures. We performed a sensitivity analysis to assess the impact of this assumption on the effect estimate if deemed appropriate.
Assessment of heterogeneity
We assessed heterogeneity using the Chi² test (a P value of 0.10 was considered statistically significant) and the I² statistic. For the I² statistic, 75% indicated high heterogeneity among study data, 50% moderate heterogeneity, and 25% low heterogeneity (Higgins 2003). We conducted sensitivity analysis to explore possible explanations for heterogeneity as appropriate.
Assessment of reporting biases
To assess potential reporting biases, we compared outcomes listed in the study protocol to those outcomes reported in the published manuscript. If we did not have access to the protocol, we compared the outcomes listed in the methods section of the published manuscript with the outcomes reported in the published manuscript. We planned to investigate potential publication bias using funnel plots if a sufficient number of studies (≥ 10) were pooled for meta‐analysis (Egger 1997).
Data synthesis
We combined data from individual trials for meta‐analysis when the interventions, patient groups, and outcomes were similar as deemed by author consensus. We calculated the pooled RR and 95% CI for dichotomous outcomes. Additionally, we planned to calculate the pooled MD and 95% CI for any continuous outcomes. We used a fixed‐effect model to pool data unless there was heterogeneity among the studies (I² of 50% to 75%), in which case we used a random‐effects model. We did not pool data for meta‐analysis if a high degree of heterogeneity (I² ≥ 75%) was found.
Subgroup analysis and investigation of heterogeneity
We performed a subgroup analysis for different drug doses.
Sensitivity analysis
We used a sensitivity analysis to determine the impact of random‐effects and fixed‐effect modelling, risk of bias, type of report (full manuscript, abstract, or unpublished data), and loss to follow‐up on the pooled effect estimate.
Results
Description of studies
Results of the search
The literature search conducted on 17 September 2019 identified 725 records, and an additional two records were identified through other sources (See Figure 1). After removal of duplicates, 479 records remained for screening. Of these, 67 records were selected for full‐text review. Three studies were excluded after reviewing the full text (see Characteristics of excluded studies tables). Sixty‐two reports of 3 studies (646 participants) met the inclusion criteria and were included in the review (Feagan 2016; Panaccione 2015; Sandborn 2012).
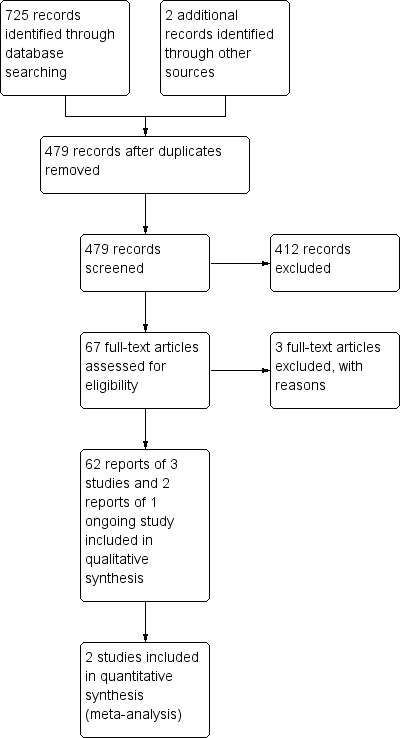
Study flow diagram.
Included studies
This study assessed ustekinumab in two populations (UNITI‐1 and UNITI‐2) with moderate to severe Crohn's disease. Adults (≥ 18 years) who had Crohn's disease for at least three months and a Crohn's Disease Activity Index (CDAI) score of 220 to 450 were enrolled in the induction trials. For the UNITI‐1 study, participants were required to have received one or more TNF‐α antagonists at approved doses and to have met the criteria for primary non‐response or secondary non‐response or to have experienced any unacceptable adverse effects. For the UNITI‐2 study, participants were required to have had treatment failure or unacceptable adverse effects when treated with immunosuppressants or glucocorticosteroids. Participants could have previously received one or more TNF‐α antagonists if they did not experience any unacceptable adverse effects and did not meet the criteria for primary or secondary non‐response to treatment. The primary outcome for both the UNITI‐1 and UNITI‐2 induction trials was clinical response at week 6 (decrease from baseline in CDAI score of at least 100 points or a total CDAI score of less than 150). Secondary endpoints included clinical remission at week 8 (CDAI < 150), clinical response at week 8, and a decrease from baseline in CDAI score of at least 70 points at weeks 3 and 6.
In the IM‐UNITI trial (N = 397) participants who responded to ustekinumab during the induction studies were randomly assigned to receive subcutaneous maintenance injections of 90 mg of ustekinumab (either every 8 weeks or every 12 weeks) or placebo. The primary outcome in the maintenance trial was clinical remission at week 44 (CDAI score < 150). Secondary outcomes included clinical response (decrease in CDAI score of > 100 points from week 0 of induction or clinical remission) at week 44, maintenance of clinical remission among participants in remission at week 0 of the maintenance trial, glucocorticosteroid‐free remission, and remission in participants who met the primary or secondary non‐response criteria or who had unacceptable adverse effects. In IM‐UNITI study, of the 397 randomized patients, 9 patients, who were randomized prior to the study restart, were excluded from the efficacy analysis population. Thus, 388 patients were included in the efficacy analysis population for IM‐UNITI.
This study assessed the effectiveness of briakinumab in adults who had Crohn's disease for at least four months that was confirmed by endoscopy or radiologic evaluation and a CDAI score ranging from 220 to 450. Exclusion criteria included people with colitis other than Crohn's disease, prior exposure to systemic or biologic anti‐IL‐12 therapy, receipt of an investigational TNF‐α antagonist at any time, or receipt of any investigational agent within 30 days of study entry. The study was conducted at 60 sites in Australia, Canada, Europe, and the United States. The study was both an induction and maintenance study that lasted 115 weeks and included 6 phases, starting with screening (< 4 weeks), induction (12 weeks), and maintenance (12 weeks). Participants were randomly assigned 1:1:1:3 to intravenous infusion induction regimens: placebo, 200 mg briakinumab, 400 mg briakinumab, and 700 mg briakinumab administered at 0, 4, and 8 weeks. At week 12, participants in the placebo group and the 400 mg induction group who achieved a clinical response continued into the maintenance phase, receiving the same treatment and dosage. Participants in the 700 mg induction group who achieved a clinical response were re‐randomized 1:1:1 to placebo, 200 mg briakinumab, or 700 mg briakinumab. Participants received placebo or briakinumab at weeks 12, 16, and 20 of the maintenance phase. A total of 104 participants were involved in the maintenance phase.
This study assessed the role of ustekinumab in adults (> 18 years) with a minimum three‐month history of moderate to severe Crohn's disease as defined by a CDAI score of 220 to 450 points. Participants enrolled in CERTIFI were recruited from 153 centers in 12 different countries. All participants were required to meet a specified criteria for a primary non‐response, secondary non‐response, or unacceptable adverse effects after receiving an anti‐TNF‐α at an approved dose. Participants were allowed to receive concomitant medications for the treatment of Crohn's disease, which included oral prednisolone or budesonide, immunomodulators, mesalamine, and antibiotics. Exclusion criteria included undergoing bowel resection within six months of enrollment, short‐bowel syndrome, clinically significant stricture, abscess, active tuberculosis, current infection, or other previous or current infections or cancers. During the induction phase of the study, 526 participants were randomly assigned to receive intravenous ustekinumab in doses of 1, 3, or 6 mg/kg or placebo. The primary outcome during the induction phase was clinical response (≥ 100‐point decrease from the baseline CDAI score) at week 6. Participants with a baseline CDAI score of 248 points or less were considered to have a clinical response if their CDAI score was less than 150. The main secondary outcomes included clinical remission (CDAI < 150) at week 6 and clinical response at week 6. During the maintenance phase of the study (weeks 8 to 36), participants who had a response to ustekinumab during the induction phase (N = 145) and those who did not respond to ustekinumab underwent separate randomizations to receive subcutaneous ustekinumab (90 mg) or placebo at weeks 8 and 16. Clinical remission and clinical response in the maintenance phase were measured at 22 weeks.
Excluded studies
We excluded three studies (Fasanmade 2008; Mannon 2004; Sands 2010). Fasanmade 2008 was a pharmacokinetics study that compared different doses of ustekinumab (4.5 mg/kg to 90 mg) and was not a randomized trial. Mannon 2004 was not a maintenance‐of‐remission study. Sands 2010 assessed a different compound of interest (i.e. a different mechanism of action than briakinumab and ustekinumab).
Risk of bias in included studies
A summary of the 'Risk of bias' assessment is provided in Figure 2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
The three included studies performed central randomization and were rated as at low risk bias. Randomization in Feagan 2016 and Panaccione 2015 was performed centrally using permuted blocks in all trials. Adaptive randomization based on the investigative site and induction dose was performed centrally in Sandborn 2012.
Blinding
We assessed all included studies as at low risk of bias for blinding (Feagan 2016; Panaccione 2015; Sandborn 2012).
Incomplete outcome data
We assessed all included studies as at low risk of bias for incomplete outcome data (Feagan 2016; Panaccione 2015; Sandborn 2012). Dropouts were balanced across treatment groups with similar reasons for withdrawal.
Selective reporting
We assessed all included studies as at low risk of bias for selective reporting, as all expected outcomes were reported (Feagan 2016; Panaccione 2015; Sandborn 2012).
Other potential sources of bias
The included studies appeared to be free from other potential sources of bias and were therefore rated as at low risk of bias for this domain (Feagan 2016; Panaccione 2015; Sandborn 2012).
Effects of interventions
See: Summary of findings for the main comparison Ustekinumab compared to placebo for maintenance of remission in Crohn's disease; Summary of findings 2 Briakinumab compared to placebo for maintenance of remission in Crohn's disease
Ustekinumab versus placebo
Failure to maintain clinical remission at 22 weeks
One placebo‐controlled trial involving 145 participants reported on the proportion of participants who failed to maintain clinical remission (CDAI < 150) at 22 weeks (Sandborn 2012). Fifty‐eight per cent (42/72) of ustekinumab participants failed to maintain clinical remission at week 22 compared to 73% (53/73) of placebo participants (risk ratio (RR) 0.80, 95% confidence interval (CI) 0.63 to 1.02; Analysis 1.1). The GRADE analysis indicated that the overall certainty of the evidence was moderate due to sparse data (95 events; see summary of findings Table for the main comparison).
Failure to maintain clinical response at 22 weeks
One study (N = 145) reported on the proportion of participants who failed to maintain clinical response (≥ 100‐point decrease from the baseline CDAI score) at week 22 (Sandborn 2012). Thirty‐one per cent (22/72) of ustekinumab participants failed to maintain clinical response at week 22 compared to 58% (42/73) of placebo participants (RR 0.53, 95% CI 0.36 to 0.79; Analysis 1.2). The GRADE analysis indicated the overall certainty of the evidence for this outcome was moderate due to sparse data (64 events; see summary of findings Table for the main comparison).
Failure to maintain clinical remission at 44 weeks
Feagan 2016 (N = 388) reported on failure to maintain clinical remission (CDAI < 150) at 44 weeks. Forty‐nine (126/257) per cent of participants in the ustekinumab group failed to maintain clinical remission compared to 64% (84/131) in the placebo group (RR 0.76, 95% CI 0.64 to 0.91; Analysis 1.3). The GRADE analysis indicated that the overall certainty of evidence for this outcome was moderate due to sparse data (210 events; see summary of findings Table for the main comparison).
We performed a subgroup analysis based on dosing schedules of 90 mg every 8 weeks and 90 mg every 12 weeks, which suggested no difference in efficacy between the 8‐week and 12‐week dosing schedules. Among participants who were treated every 8 weeks, 47% (60/128) of ustekinumab participants failed to maintain remission at 44 weeks compared to 65% (42/65) of placebo participants (RR 0.73, 95% CI 0.56 to 0.94; Analysis 1.3). Among participants who were treated every 12 weeks, 51% (66/129) of ustekinumab participants failed to maintain remission at 44 weeks compared to 64% (42/66) of placebo participants (RR 0.80, 95% CI 0.63 to 1.03; Analysis 1.3). The test for subgroup differences showed no difference between the dosing schedule subgroups (test for subgroup differences Chi² = 0.32, df = 1, P = 0.57, I² = 0%).
Failure to maintain clinical response at 44 weeks
Feagan 2016 (N = 388) reported on failure to maintain clinical response (≥ 100‐point decrease from the baseline CDAI score) at 44 weeks. Forty‐one per cent (106/257) of participants in the ustekinumab group failed to maintain clinical response compared to 56% (73/131) of placebo participants (RR 0.74, 95% CI 0.60 to 0.91; Analysis 1.4). The GRADE analysis indicated that the overall certainty of evidence for this outcome was moderate due to sparse data (179 events; see summary of findings Table for the main comparison).
We performed a subgroup analysis based on the dosing schedule of 90 mg every 8 weeks and 90 mg every 12 weeks, which suggested no difference in efficacy between the 8‐week and 12‐week dosing schedules. Among participants who were treated every 8 weeks, 41% (52/128) of ustekinumab participants failed to maintain clinical response at 44 weeks compared to 55% (36/65) of placebo participants (RR 0.73, 95% CI 0.54 to 0.99; Analysis 1.4). Among participants who were treated every 12 weeks, 42% (54/129) of ustekinumab participants failed to maintain clinical response at 44 weeks compared to 56% (37/66) of placebo participants (RR 0.75, 95% CI 0.56 to 1.00; Analysis 1.4). The test for subgroup differences showed no difference between the dosing schedule subgroups (test for subgroup differences Chi² = 0.01, df = 1, P = 0.93, I² = 0%).
Failure to maintain endoscopic response
Sandborn 2012 and Feagan 2016 did not report data on this outcome.
Failure to maintain endoscopic remission
Sandborn 2012 and Feagan 2016 did not report data on this outcome.
Failure to maintain histological response
Sandborn 2012 and Feagan 2016 did not report data on this outcome.
Failure to maintain histological remission
Sandborn 2012 and Feagan 2016 did not report data on this outcome.
Failure to maintain clinical and endoscopic response
Sandborn 2012 and Feagan 2016 did not report data on this outcome.
Failure to maintain clinical and endoscopic remission
Sandborn 2012 and Feagan 2016 did not report data on this outcome.
Adverse events
Two studies provided data on adverse events (Feagan 2016; Sandborn 2012). Eighty per cent (267/335) of ustekinumab participants experienced an adverse event compared to 84% (173/206) of placebo participants (RR 0.94, 95% CI 0.87 to 1.03; I² = 0%; Analysis 1.5). The GRADE analysis indicated the overall certainty of evidence for this outcome was high (see summary of findings Table for the main comparison). Commonly reported adverse events included infections, injection site reactions, Crohn's disease event, abdominal pain, nausea, arthralgia, and headache.
Serious adverse events
Two studies provided data on serious adverse events (Feagan 2016; Sandborn 2012). Eleven per cent (38/335) of ustekinumab participants experienced a serious adverse event compared to 16% (32/206) of placebo participants (RR 0.74, 95% CI 0.48 to 1.15; I² = 0%; Analysis 1.6). The GRADE analysis indicated the overall certainty of evidence for this outcome was moderate due to sparse data (70 events; see summary of findings Table for the main comparison). Serious adverse events included serious infections, malignant neoplasm, and basal cell carcinoma.
Withdrawals due to adverse events
One study provided data for withdrawal due to adverse events (Sandborn 2012). Seven per cent (5/73) of ustekinumab participants withdrew from the study due to an adverse event compared to 1% (1/72) of placebo participants (RR 4.93, 95% CI 0.59 to 41.18; Analysis 1.7). The GRADE analysis indicated the certainty of evidence was low due to very sparse data (6 events; see summary of findings Table for the main comparison). Adverse events leading to study withdrawal included worsening Crohn's disease.
Subgroup analysis
We did not perform a subgroup analysis by dose because participants in both studies received 90 mg subcutaneous ustekinumab as maintenance therapy.
Sensitivity analyses
We did not perform sensitivity analyses as both studies had very similar populations and inclusion criteria (Feagan 2016; Sandborn 2012). Both studies used the same dose and method of administration for treatments, and outcomes were measured using the same criteria. This made fixed‐effect the most appropriate method of analysis. We did not conduct sensitivity analysis for risk of bias as we assessed both studies as at low risk of bias. Both studies were Phase II randomized controlled trials and were reported as full manuscripts, therefore we did not conduct sensitivity analysis for type of report.
Briakinumab versus placebo
Failure to maintain clinical remission at 24 weeks
Panaccione 2015 (99 participants) reported on failure to maintain clinical remission (CDAI < 150) at 24 weeks. Fifty‐one per cent (32/63) of participants in the briakinumab group failed to maintain clinical remission at 24 weeks compared to 61% (22/36) of placebo participants (RR 0.84, 95% CI 0.58 to 1.20; Analysis 2.1). The GRADE analysis indicated the certainty of evidence was low due to sparse data (54 events) and a very wide confidence interval (see summary of findings Table 2).
A subgroup analysis based on dose suggested that there was no difference in remission rates across the 200 mg, 400 mg, or 700 mg dose subgroups. Among participants in the 200 mg group, 57% (12/21) of briakinumab participants failed to maintain remission at 24 weeks compared to 54% (6/11) of placebo participants (RR 1.05, 95% CI 0.54 to 2.02; Analysis 2.1). Among participants in the 400 mg group, 52% (11/21) of briakinumab participants failed to maintain remission at 24 weeks compared to 71% (10/14) of placebo participants (RR 0.73, 95% CI 0.43 to 1.24; Analysis 2.1). Lastly, among participants in the 700 mg group, 43% (9/21) of briakinumab participants failed to maintain remission at 24 weeks compared to 54% (6/11) of placebo participants (RR 0.79, 95% CI 0.38 to 1.63; Analysis 2.1). The test for subgroup differences showed no difference between the dose subgroups (test for subgroup differences Chi² = 0.72, df = 2, P = 0.70, I² = 0%).
Failure to maintain clinical response at 24 weeks
Panaccione 2015 (99 participants) reported on failure to maintain clinical response (≥ 100‐point decrease in CDAI from baseline) at 24 weeks. Thirty‐three per cent (21/63) of briakinumab participants failed to maintain clinical response at 24 weeks compared to 53% (19/36) of placebo participants (RR 0.64, 95% CI 0.40 to 1.02; Analysis 2.2). The GRADE analysis indicated the certainty of evidence was low due to very sparse data (30 events; see summary of findings Table 2).
A subgroup analysis based on dose suggested that there was no difference in response rates across the 200 mg, 400 mg, or 700 mg dose subgroups. Among participants in the 200 mg group, 33% (7/21) of briakinumab participants failed to maintain clinical response at 24 weeks compared with 45% (5/11) of placebo participants (RR 0.73, 95% CI 0.30 to 1.78; Analysis 2.2). Among participants in the 400 mg group, 38% (8/21) of briakinumab participants failed to maintain clinical response at 24 weeks compared with 64% (9/14) of placebo participants (RR 0.59, 95% CI 0.30 to 1.16; Analysis 2.2). Lastly, among participants in the 700 mg group, 29% (6/21) of briakinumab participants failed to maintain clinical response at 24 weeks compared to 45% (5/11) of placebo participants (RR 0.63, 95% CI 0.25 to 1.60; Analysis 2.2). The test for subgroup differences showed no difference between the dose subgroups (test for subgroup differences Chi² = 0.14, df = 2, P = 0.93, I² = 0%).
Failure to maintain endoscopic response
Panaccione 2015 did not report data on this outcome.
Failure to maintain endoscopic remission
Panaccione 2015 did not report data on this outcome.
Failure to maintain histological response
Panaccione 2015 did not report data on this outcome.
Failure to maintain histological remission
Panaccione 2015 did not report data on this outcome.
Failure to maintain clinical and endoscopic response
Panaccione 2015 did not report data on this outcome.
Failure to maintain clinical and endoscopic remission
Panaccione 2015 did not report data on this outcome.
Adverse events
Adverse events were reported in 66% (59/90) of briakinumab participants compared to 64% (9/14) of placebo participants (RR 1.02, 95% CI 0.67 to 1.55; Analysis 2.3) (Panaccione 2015). Commonly reported adverse events included upper respiratory tract infection, nausea, abdominal pain, headache, and injection site reaction. The GRADE analysis indicated the certainty of evidence was low due to sparse data (68 events) and a very wide confidence interval (see summary of findings Table 2).
Serious adverse events
Serious adverse events were reported in 2% (2/90) of briakinumab participants compared to 7% (1/14) of placebo participants (RR 0.31, 95% CI 0.03 to 3.21; Analysis 2.4) (Panaccione 2015). Serious adverse events included small bowel obstruction, deep vein thrombosis, and respiratory distress. The GRADE analysis indicated the certainty of evidence was low due to very sparse data (3 events; see summary of findings Table 2).
Withdrawal due to adverse events
Withdrawals due to adverse events were reported in 2% of briakinumab participants compared to 0% (0/14) of placebo participants (RR 0.82, 95% CI 0.04 to 16.34; Analysis 2.5) (Panaccione 2015). The adverse events leading to withdrawal were not reported in the manuscript. The GRADE analysis indicated the certainty of evidence was low due to very sparse data (2 events; see summary of findings Table 2).
Discussion
Summary of main results
This systematic review included three randomized controlled trials (646 participants) that evaluated the efficacy and safety of ustekinumab or briakinumab as maintenance therapy in people with moderate to severe Crohn's disease. Feagan 2016 (N = 397) assessed maintenance therapy in participants receiving 90 mg of ustekinumab (every 8 or 12 weeks) or placebo. Sandborn 2012 (N = 145) assessed maintenance therapy in participants receiving 90 mg of ustekinumab or placebo at weeks 8 and 16. Panaccione 2015 assessed maintenance therapy (N = 104) in participants receiving 200 mg, 400 mg, or 700 mg of briakinumab or placebo at weeks 12, 16, and 20 of the maintenance phase of the study. The participants in all three studies were responders to ustekinumab or briakinumab induction therapy. Caution is advised in interpreting the results of this review, as the certainty of the evidence was impacted by sparse data.
Moderate‐certainty evidence suggests that ustekinumab is probably effective for the maintenance of clinical remission and response at 22 weeks and 44 weeks in people with moderate to severe Crohn's disease in remission. Subgroup analyses showed no difference in efficacy between 8‐week and 12‐week dosing schedules. High‐certainty evidence suggests there is no increased risk of adverse events with ustekinumab when compared with placebo. Moderate‐certainty evidence suggests there is no increased risk of serious adverse events with ustekinumab compared with placebo. Commonly reported adverse events included infections, injection site reactions, Crohn's disease event, abdominal pain, nausea, arthralgia, and headache. Serious adverse events included serious infections, malignant neoplasm, and basal cell carcinoma.
The effect of briakinumab on maintenance of clinical remission and response in people with moderate to severe Crohn's disease in remission was uncertain as the certainty of the evidence was low. Subgroup analysis showed no difference in efficacy across the 200 mg, 400 mg, and 700 mg dose subgroups. However, the number of participants assessed in each subgroup was small, therefore the ideal dose of briakinumab, should one exist, is unknown. The effect of briakinumab on adverse events and serious adverse events was uncertain as the certainty of the evidence was low. Commonly reported adverse events included upper respiratory tract infection, nausea, abdominal pain, headache, and injection site reaction. Serious adverse events included small bowel obstruction, deep vein thrombosis, and respiratory distress.
Overall completeness and applicability of evidence
The results of this review are applicable to individuals with moderate to severe Crohn's disease in remission. The study participants included people who had failed standard therapy with corticosteroids or immunosuppressants or who had developed unacceptable adverse effects associated with these agents, or people who had previous treatment with TNF‐α antagonists and had lost response or had become intolerant to the drug. However, the overall evidence cannot be considered complete. The three included studies (646 participants) assessed maintenance therapy of moderate to severe Crohn's disease with ustekinumab or briakinumab and reported data on maintenance of clinical remission and clinical response, adverse events, serious adverse events, and withdrawals due to adverse events. We did not pre‐specify quality of life as a secondary outcome. Furthermore, some of our prespecified secondary outcomes, including maintenance of endoscopic response and remission and maintenance of histological response and remission, were not reported in the included studies.
Quality of the evidence
We judged all three included studies as at low risk of bias. For ustekinumab, GRADE analysis indicated the overall certainty of the evidence supporting the primary outcome of failure to maintain clinical remission was moderate. The certainty of the evidence supporting the secondary outcomes of failure to maintain clinical response and serious adverse events was moderate. The certainty of the evidence supporting the outcomes adverse events and withdrawal due to adverse events was high and low, respectively. Outcomes that were downgraded were due to varying degrees of imprecision (sparse data).
For briakinumab, GRADE analysis indicated the overall certainty of the evidence supporting the primary outcome of failure to maintain clinical remission was low. The certainty of the evidence supporting the secondary outcomes failure to maintain clinical response, adverse events, serious adverse events, and withdrawal due to adverse events was low. We downgraded all of the outcomes due to varying degrees of imprecision (sparse data).
Potential biases in the review process
In order to reduce potential bias in the review process we performed a comprehensive literature search to identify all eligible studies. Two review authors independently assessed studies for inclusion, extracted data, and assessed risk of bias. There were some limitations to this review. Due to differing time points for analysis in the two ustekinumab studies, the primary outcome and several secondary outcomes could not be pooled for meta‐analysis. In addition, many of the outcomes assessed in this systematic review were given GRADE ratings of moderate or low certainty due to sparse event data. None of the included studies used endoscopic remission or response as an outcome, despite the increasing use of endoscopic outcomes in Crohn's disease studies. Further research is needed to assess the impact of ustekinumab and briakinumab on these outcomes.
Agreements and disagreements with other studies or reviews
The results of our systematic review with respect to the efficacy and safety of ustekinumab agree with the results of a recently published systematic review, Zaltman 2019, and the results of a recently published network meta‐analysis (Singh 2018). We were unable to identify any systematic reviews dealing with the efficacy and safety of briakinumab for maintenance of remission in people with moderate to severe Crohn's disease in remission.

Study flow diagram.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Comparison 1 Ustekinumab versus placebo, Outcome 1 Failure to maintain clinical remission at 22 weeks.

Comparison 1 Ustekinumab versus placebo, Outcome 2 Failure to maintain clinical response at 22 weeks.

Comparison 1 Ustekinumab versus placebo, Outcome 3 Failure to maintain clinical remission at 44 weeks.
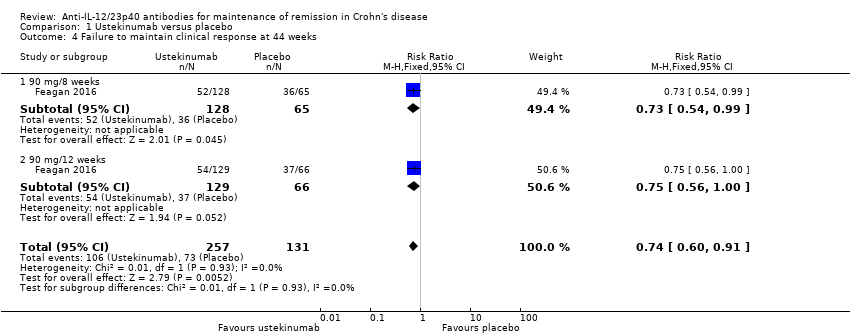
Comparison 1 Ustekinumab versus placebo, Outcome 4 Failure to maintain clinical response at 44 weeks.

Comparison 1 Ustekinumab versus placebo, Outcome 5 Adverse events.
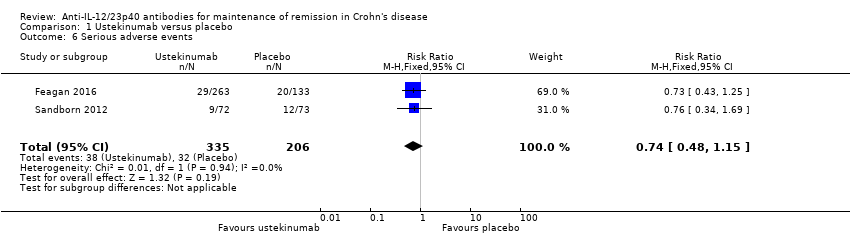
Comparison 1 Ustekinumab versus placebo, Outcome 6 Serious adverse events.

Comparison 1 Ustekinumab versus placebo, Outcome 7 Withdrawal due to adverse events.

Comparison 2 Briakinumab versus placebo, Outcome 1 Failure to maintain clinical remission at 24 weeks.

Comparison 2 Briakinumab versus placebo, Outcome 2 Failure to maintain clinical response at 24 weeks.

Comparison 2 Briakinumab versus placebo, Outcome 3 Adverse events.

Comparison 2 Briakinumab versus placebo, Outcome 4 Serious adverse events.

Comparison 2 Briakinumab versus placebo, Outcome 5 Withdrawal due to adverse events.
| Ustekinumab compared to placebo for maintenance of remission in Crohn's disease | ||||||
| Patient or population: people with moderate to severe Crohn's disease in remission | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | No. of participants | Certainty of the evidence | Comments | |
| Risk with placebo | Risk with ustekinumab | |||||
| Failure to maintain clinical remission Follow‐up: 22 weeks | 726 per 1000 | 581 per 1000 | RR 0.80 | 145 | ⊕⊕⊕⊝ | Clinical remission was defined as a CDAI < 150 points. |
| Failure to maintain clinical response Follow‐up: 22 weeks | 575 per 1000 | 305 per 1000 | RR 0.53 | 145 | ⊕⊕⊕⊝ | Clinical response was defined as a ≥ 100‐point decrease from the baseline CDAI score. |
| Failure to maintain clinical remission Follow‐up: 44 weeks | 641 per 1000 | 487 per 1000 (410 to 584) | RR 0.76 (0.64 to 0.91) | 388 (1 RCT) | ⊕⊕⊕⊝ | Clinical remission was defined as a CDAI < 150 points. |
| Failure to maintain clinical response Follow‐up: 44 weeks | 557 per 1000 | 412 per 1000 (334 to 507) | RR 0.74 (0.60 to 0.91) | 388 (1 RCT) | ⊕⊕⊕⊝ | Clinical response was defined as a decrease from baseline in the CDAI score of ≥ 100 points or a CDAI score < 150. |
| Adverse events Follow‐up: 44 weeks | 840 per 1000 | 789 per 1000 (731 to 865) | RR 0.94 (0.87 to 1.03) | 541 (2 RCTs) | ⊕⊕⊕⊕ | Commonly reported adverse events included worsening Crohn's disease, abdominal pain, nausea, arthralgia, and headache. |
| Serious adverse events Follow‐up: 44 weeks | 155 per 1000 | 115 per 1000 | RR 0.74 | 541 | ⊕⊕⊕⊝ | Commonly reported serious adverse events included malignant neoplasm, basal cell carcinoma, and injection site reactions. |
| Withdrawal due to adverse events Follow‐up: 44 weeks | 14 per 1000 | 68 per 1000 (8 to 572) | RR 4.93 (0.59 to 41.18) | 145 (1 RCT) | ⊕⊕⊝⊝ | Adverse events leading to withdrawal included worsening Crohn's disease |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Downgraded one level due to sparse data (95 events). | ||||||
| Briakinumab compared to placebo for maintenance of remission in Crohn's disease | ||||||
| Patient or population: people with moderate to severe Crohn's disease in remission | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | No. of participants | Certainty of the evidence | Comments | |
| Risk with placebo | Risk with briakinumab | |||||
| Failure to maintain clinical remission Follow‐up: 24 weeks | 611 per 1000 | 513 per 1000 (354 to 733) | RR 0.84 (0.58 to 1.20) | 99 (1 RCT) | ⊕⊕⊝⊝ | Clinical remission was defined as a CDAI score < 150 points. |
| Failure to maintain clinical response Follow‐up: 24 weeks | 528 per 1000 | 338 per 1000 (211 to 538) | RR 0.64 (0.40 to 1.02) | 99 (1 RCT) | ⊕⊕⊝⊝ | Clinical response was defined as a decrease in CDAI score ≥ 100 points compared with week 0. |
| Adverse events Follow‐up: 24 weeks | 643 per 1000 | 656 per 1000 (431 to 996) | RR 1.02 (0.67 to 1.55) | 104 (1 RCT) | ⊕⊕⊝⊝ | Common adverse events included upper respiratory tract infection, nausea, abdominal pain, and headache. |
| Serious adverse events Follow‐up: 24 weeks | 71 per 1000 | 22 per 1000 (2 to 229) | RR 0.31 (0.03 to 3.21) | 104 (1 RCT) | ⊕⊕⊝⊝ | Serious adverse events included small bowel obstruction, deep vein thrombosis, and respiratory distress. |
| Withdrawals due to adverse events Follow‐up: 24 weeks | 0 per 1000 | 0 per 1000 (0 to 0) | RR 0.82 (0.04 to 16.34) | 104 (1 RCT) | ⊕⊕⊝⊝ | We were unable to calculate absolute effects. 2% of briakinumab participants withdrew due to an adverse event compared to none of the placebo participants. Adverse events leading to withdrawal were not reported. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CDAI: Crohn's Disease Activity Index; CI: confidence interval; RCT: randomized controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Downgraded two levels due to sparse data (54 events) and very wide confidence interval. | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Failure to maintain clinical remission at 22 weeks Show forest plot | 1 | 145 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.63, 1.02] |
| 2 Failure to maintain clinical response at 22 weeks Show forest plot | 1 | 145 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.36, 0.79] |
| 3 Failure to maintain clinical remission at 44 weeks Show forest plot | 1 | 388 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.64, 0.91] |
| 3.1 90 mg/8 weeks | 1 | 193 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.56, 0.94] |
| 3.2 90 mg/12 weeks | 1 | 195 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.63, 1.03] |
| 4 Failure to maintain clinical response at 44 weeks Show forest plot | 1 | 388 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.60, 0.91] |
| 4.1 90 mg/8 weeks | 1 | 193 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.54, 0.99] |
| 4.2 90 mg/12 weeks | 1 | 195 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.56, 1.00] |
| 5 Adverse events Show forest plot | 2 | 541 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.87, 1.03] |
| 6 Serious adverse events Show forest plot | 2 | 541 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.48, 1.15] |
| 7 Withdrawal due to adverse events Show forest plot | 1 | 145 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.93 [0.59, 41.18] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Failure to maintain clinical remission at 24 weeks Show forest plot | 1 | 99 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.84 [0.58, 1.20] |
| 1.1 200 mg | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.54, 2.02] |
| 1.2 Continued 400 mg | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.43, 1.24] |
| 1.3 700 mg | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.38, 1.63] |
| 2 Failure to maintain clinical response at 24 weeks Show forest plot | 1 | 99 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.40, 1.02] |
| 2.1 200 mg | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.30, 1.78] |
| 2.2 Continued 400 mg | 1 | 35 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.30, 1.16] |
| 2.3 700 mg | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.25, 1.60] |
| 3 Adverse events Show forest plot | 1 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.67, 1.55] |
| 4 Serious adverse events Show forest plot | 1 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.03, 3.21] |
| 5 Withdrawal due to adverse events Show forest plot | 1 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.04, 16.34] |