Cribado neonatal para la galactosemia
Resumen
Antecedentes
La galactosemia clásica se debe a un error congénito autosómico recesivo del metabolismo, causado por una deficiencia de la enzima galactosa‐1‐fosfato uridiltransferasa. Se trata de una enfermedad rara y potencialmente mortal, que aparece por lo general en la primera semana de vida una vez que se inicia la alimentación con leche. En los lactantes afectados, se pueden presentar cualquiera de los siguientes cuadros: cataratas; insuficiencia hepática fulminante; ictericia prolongada; o sepsis por Escherichia coli. Una vez que se presume el diagnóstico, debe interrumpirse de inmediato la leche que contenga galactosa y reemplazarse con leche a base de soja. Aunque la mayoría de los lactantes se recupera, algunos no sobrevivirán. Existen complicaciones a largo plazo de la galactosemia, a pesar del tratamiento, incluidas las dificultades del aprendizaje y la infertilidad femenina. Se ha postulado que la galactosemia podría detectarse en el cribado neonatal y que esto prevendría la disfunción hepática severa inmediata y la sepsis. Esta es una actualización de una revisión publicada anteriormente.
Objetivos
Evaluar si hay evidencia de que el cribado neonatal para la galactosemia previene o reduce la morbimortalidad y mejora los resultados clínicos en los lactantes afectados y la calidad de vida en niños mayores.
Métodos de búsqueda
Se realizaron búsquedas en el registro de ensayos del Grupo Cochrane de Fibrosis Quística y Enfermedades Genéticas (Cochrane Cystic Fibrosis and Genetic Disorders Group) que incluía referencias identificadas de búsquedas en bases de datos electrónicas y búsquedas manuales de revistas relevantes y libros de resúmenes de actas de congresos. También se realizaron búsquedas en registros de ensayos en línea y en las listas de referencias de artículos y revisiones relevantes.
Fecha de la búsqueda más reciente en el registro de ensayos del Grupo Cochrane de Fibrosis Quística: 12 de diciembre de 2019.
Fecha de la búsqueda más reciente de recursos adicionales: 2 de febrero de 2020.
Criterios de selección
Ensayos controlados aleatorizados y estudios clínicos controlados, publicados o no publicados que compararan el uso de cualquier prueba de cribado neonatal para el diagnóstico de lactantes con galactosemia y que presentaran una comparación entre una población cribada y una no cribada.
Obtención y análisis de los datos
No se encontraron estudios sobre cribado neonatal para la galactosemia.
Resultados principales
No se identificaron estudios para ser incluidos en la revisión.
Conclusiones de los autores
No se pudo identificar ningún estudio apto para la inclusión en esta revisión, por lo que no es posible establecer conclusiones basadas en estudios controlados aleatorizados. Sin embargo, se sabe de estudios no controlados que apoyan la eficacia del cribado neonatal para la galactosemia. Existen varias revisiones y análisis económicos de la bibliografía (no ensayos) que indican que el cribado es apropiado.
PICOs
Resumen en términos sencillos
Cribado de neonatos para la galactosemia
Pregunta de la revisión
Se revisó la evidencia del cribado para la galactosemia en neonatos para prevenir o reducir la enfermedad y la mortalidad, mejorar los resultados clínicos en los lactantes afectados y mejorar la calidad de vida en los niños mayores afectados.
Antecedentes
La galactosemia es una enfermedad hereditaria que afecta la capacidad del cuerpo para descomponer la galactosa de la leche. Los neonatos con galactosemia pueden presentar diversos síntomas en las primeras semanas de vida, incluida la inapetencia, cataratas, ictericia, un hígado agrandado con insuficiencia hepática o infección severa. Sin tratamiento, los lactantes con galactosemia se encuentran a menudo en mal estado y tienen una alta probabilidad de morir por la insuficiencia hepática. Lamentablemente, a pesar del tratamiento, las complicaciones a largo plazo para los pacientes con galactosemia incluyen dificultades del aprendizaje y trastornos de fertilidad (en mujeres).
Fecha de la búsqueda
La evidencia está actualizada hasta: 2 de febrero de 2020.
Características de los estudios
No se identificaron estudios para ser incluidos en la revisión.
Resultados clave
No se halló ningún estudio apropiado, aunque se sabe de algunos estudios no controlados que indican que el cribado neonatal para la galactosemia y el tratamiento temprano pueden reducir la mortalidad y la enfermedad. Se necesitan estudios de investigación a largo plazo para aportar evidencia sólida a favor o en contra del cribado.
Calidad de la evidencia
No se ha identificado ningún estudio relevante para su inclusión en esta revisión.
Authors' conclusions
Background
Please see the glossary for an explanation of terms (Appendix 1).
Description of the condition
Galactosaemia is an autosomal recessive disorder of galactose metabolism. It occurs as a consequence of a deficiency of one of three principal enzymes involved in the metabolism of galactose. These enzymes are galactokinase (GALK), galactose‐1‐phosphate uridyltransferase (GALT) and uridine‐diphosphate galactose‐4' epimerase (GALE). The most common deficiency is that of the transferase enzyme, which causes 'classical galactosaemia' (Handerson 2002). Affected infants are born healthy, but experience a rapid, and often, devastating decline following exposure to the galactose found in breast milk or milk formula. Acute symptoms can progress in a matter of days ranging from jaundice, vomiting, and diarrhoea to failure to thrive, hepatomegaly, fulminant liver failure and Escherichia coli (E coli) sepsis. Without treatment, affected infants often die in the neonatal period (Fridovich‐Keil 2008). Females with galactosaemia are at an increased risk of premature ovarian failure and the majority of affected individuals do have some long‐term complications.
The global frequency of galactosaemia is estimated at approximately one in every 62,000 live births; in the USA a recent prevalence of one in 30,000 to one in 60,000 live births has been reported (Pyhtila 2015). Across Europe, the incidence varies greatly (Morel‐Garcia 2014), with a much lower frequency reported in Asian populations (Choi 2014). The Irish Traveller population has a very high incidence of 1 in 480 births (Murphy 1999).
Description of the intervention
Newborn screening for galactosaemia was designed to detect both classical galactosaemia as well as variant forms (e.g. Duarte galactosaemia) as screening largely fulfilled the Wilson Jungner criteria (see glossary) (Pamela 2007). A number of biochemical methods have been used to screen for galactosaemia, the most common being the measurement of galactose and galactose‐1‐phosphate (G‐1‐P) in blood spots (Ohlsson 2011). Galactosaemia is generally screened for in those parts of the world with a high prevalence or an expansive screening programme, or both. In countries where newborn screening is not standard practice, cases can only be detected once the affected individuals present with clinical symptoms.
How the intervention might work
Newborn screening, if performed in the first few days of life, provides an opportunity for a diagnosis either before or just as the infant presents with symptoms. Early diagnosis allows a change to a soya‐based formula and thus reduces the risk of liver failure and its complications and E coli sepsis. In a 10‐year period, mortality was reportedly reduced more than 10‐fold in children with galactosaemia as a result of newborn screening (Padilla 2008). Unfortunately though, newborn screening does not prevent the longer‐term complications of learning disability and ovarian failure as these are due to the endogenous production of galactose. Thus, the importance of newborn screening lies in preventing the initial liver failure and sepsis. Currently, most infants with galactosaemia are hospitalised in neonatal intensive care units, with newborn screening expected to reduce the cost per stay by USD 12,000 per child (Padilla 2008).
Why it is important to do this review
As early as 1978, advocates of newborn screening stated that "Galactosemia screening should be routine for all newborn infants. It is a disorder with definite and severe complications, but one in which the complications can be prevented with simple and inexpensive treatment" (Botlin 2005; Levy 1978). Many rare diseases do not fully fulfil the Wilson Jungner criteria, but galactosaemia is certainly a treatable disorder and early detection can reduce early morbidity and mortality. Robust evidence is required to support the institution of galactosaemia newborn screening programs in those countries where it is not currently undertaken.
Objectives
To determine whether newborn screening for classical galactosaemia prevents or reduces mortality and morbidity and improves clinical outcomes in affected neonates and the quality of life in older children.
Methods
Criteria for considering studies for this review
Types of studies
All randomised controlled trials (RCTs) and quasi‐ randomised controlled trials (quasi‐RCT) where participants are prospectively allocated to either screening via a blood test (e.g. from the heel prick) or no screening. No language restrictions will be placed on studies considered for inclusion in this review, and published or unpublished sources will be considered.
Types of participants
All newborn populations eligible for inclusion in a screening study in the first week of life.
Types of interventions
We will compare the population screened for classical galactosaemia versus the non‐screened population i.e. no intervention. Screening by blood test (heel prick or venous blood specimen) undertaken in the first week of life using any biochemical test such as the Beutler test (also known as the fluorescent spot test), calorimetric test, fluorescent galactose oxidase method, Guthrie's method, etc to measure total galactose or GAL‐1‐P, etc. Other diagnostic methods, such as urine and genetic testing will be excluded.
Types of outcome measures
Primary outcomes
-
Mortality (galactosaemia‐related)
-
Morbidity (in the neonatal period)
-
liver failure
-
sepsis
-
Secondary outcomes
-
Quality of life as measured by a validated scoring system such as SF‐6D questionnaire derived from the SF‐36 form (Ware 1992) (see glossary Appendix 1)
-
Clinical outcomes
-
organ dysfunction or failure, e.g. liver, kidney, eye, ovarian, etc measured by biochemical tests (for liver function, kidney function and for eye), physical examination and sonography (ovarian failure)
-
developmental problems
-
speech difficulties
-
learning difficulties
-
mental retardation assessed by standardised developmental or IQ tests and also Ages and Stages Questionnaire (ASQ) (Squires 1997)
-
-
Reduction in galactose‐1‐phosphate levels
Search methods for identification of studies
We used a combination of electronic and handsearches for this review. We did not restrict the searches by language, year or publication status.
Electronic searches
We searched the Group's Inborn Errors of Metabolism Trials Register to identify relevant studies using the term: galactosaemia.
The Inborn Errors of Metabolism Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated with each new issue of the Cochrane Library), weekly searches of MEDLINE and the prospective handsearching of one journal ‐ Journal of Inherited Metabolic Disease. Unpublished work is identified by searching through the abstract books of the Society for the Study of Inborn Errors of Metabolism conference and the SHS Inborn Error Review Series. For full details of all searching activities for the register, please see the relevant section of the Cystic Fibrosis and Genetic Disorders Group website.
Date of the most recent search: 12 December 2019.
We searched the following databases:
-
Cochrane Central Register of Controlled Trials (CENTRAL; 2020, Issue 2) in the Cochrane Library (searched 02 February 2020);
-
PubMed (www.ncbi.nlm.nih.gov/pubmed; 1946 to 02 February 2020);
We also searched the following trials registries and other resources:
-
US National Institutes of Health Ongoing Trials Register Clinicaltrials.gov (www.clinicaltrials.gov; searched 02 February 2020);
-
World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) (apps.who.int/trialsearch; searched 02 February 2020);
-
Grey Literature Report (www.greylit.org; searched 02 February 2020);
-
System for Information on Grey Literature in Europe (www.opengrey.eu ; searched 02 February 2020);
For details of our search strategies, please see Appendix 2.
Searching other resources
We would have checked the bibliographies of any included studies and any relevant systematic reviews identified for further references to relevant trials had we found any. We hand searched the Journal of Inherited Metabolic Disease and the Galactosemia Foundation Bi‐Annual International Conference, details in Appendix 3.
Data collection and analysis
The review authors planned to follow the recommended strategies for data collection and analysis as documented in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a).
Selection of studies
Two review authors (RL and MNJ) independently scanned the titles and abstracts to determine the studies to be further assessed. They examined all potentially relevant articles, as full text if available. If there had been any differences in opinion, the authors had planned to resolve these by discussion with a third author. If it had not been possible to resolve the disagreement, they would have listed the article as 'Awaiting classification' and contacted the study authors for clarification.
Data extraction and management
To date, we have not identified any studies for inclusion in the review, but if we are able to include any studies in future, we plan to employ the following methods.
Two authors (RL and MNJ) will independently extract data from eligible studies using a standard data extraction form customised for this review. The authors plan to pilot test the data extraction form before using it in the review and will modify it accordingly if needed. They will record information about study and participant characteristics, the intervention and the outcomes. If there are any uncertainties, they will contact the primary investigators of the study in question for clarification. They will check the data for accuracy and consistency, and resolve any disagreements by consensus or by consulting a third author.
The authors will record details of all participants with any type of galactosaemia and their genotypes.
Assessment of risk of bias in included studies
According to the method described in the Cochrane Handbook for Systematic Reviews of Interventions, two authors will independently assess the risk of bias (Higgins 2011b). They will resolve possible disagreements by consensus, or by consulting of a third author. They will assess the following criteria:
-
random sequence generation (selection bias);
-
allocation concealment (selection bias);
-
blinding of participants and personnel (performance bias);
-
blinding of outcome assessment (detection bias);
-
incomplete outcome data (attrition bias);
-
selective reporting (reporting bias); and
-
other bias.
Measures of treatment effect
For dichotomous outcomes (e.g. mortality) the authors will express the measure of effect as a risk ratio (RR) with 95% confidence intervals (CIs); and for continuous outcomes (e.g. quality of life, biochemical tests, growth charts, IQ tests and ASQ), they will express the measure of effect as a mean difference (MD) with 95% CIs. If outcomes are reported using different scales, e.g. quality of life, we will use standardised mean difference (SMD) and corresponding 95% CIs.
Unit of analysis issues
Cluster‐randomised studies and studies of cross‐over design are not appropriate for this review.
Dealing with missing data
The authors plan to obtain any relevant missing data by contacting the primary investigators.
Assessment of heterogeneity
The authors plan to assess any identified heterogeneity between studies using the I² statistic (Higgins 2003). The values of I² lie between 0% and 100%, and a simplified categorisation of heterogeneity that they plan to use is:
-
0% to 40%: might not be important;
-
30% to 60%: may represent moderate heterogeneity;
-
50% to 90%: may represent substantial heterogeneity;
-
75% to 100%: considerable heterogeneity.
Assessment of reporting biases
If there is a sufficient number of included studies in the review (at least 10), the authors will assess reporting bias and small study effects using funnel plots. The authors will identify and report on any selective reporting in the included studies.
Data synthesis
The authors plan to use a fixed‐effect model if they are able to enter data in a meta‐analysis using Cochrane's Review Manager software (Review Manager 2014). If they find statistical heterogeneity between studies (either moderate, substantial or considerable heterogeneity, as defined above), the authors will use a random‐effects model.
Subgroup analysis and investigation of heterogeneity
If the authors identify a high degree of heterogeneity between any included studies, they will carry out subgroup analyses as reported below.
-
Comparison of those screened at different time points (day 1 of life versus day 2 of life versus day 3 etc) since the age at screening is an important factor.
-
Comparison of galactose‐1‐phosphate levels to assess how these affect developmental outcomes (comparing those who maintain higher levels throughout childhood to those who do not).
-
Comparison of those children with a family history of galactosaemia treated with soya formula from birth to those children not treated in this way with regards to clinical outcomes listed above.
Sensitivity analysis
The authors plan to perform sensitivity analyses to investigate the impact of a high risk of bias for generation of randomisation sequence and allocation concealment on the robustness of the results of the included studies. They also plan to perform sensitivity analyses to assess the overall risk of bias by outcome.
Results
Description of studies
Results of the search
A search of the Cystic Fibrosis and Genetic Disorders Group's Cystic Fibrosis Trials Register identified five studies which were potentially eligible for inclusion in the review. However, on closer inspection, all of these studies were excluded (see below for reasons). We did not identify any other studies in our additional searches.( see Figure 1)
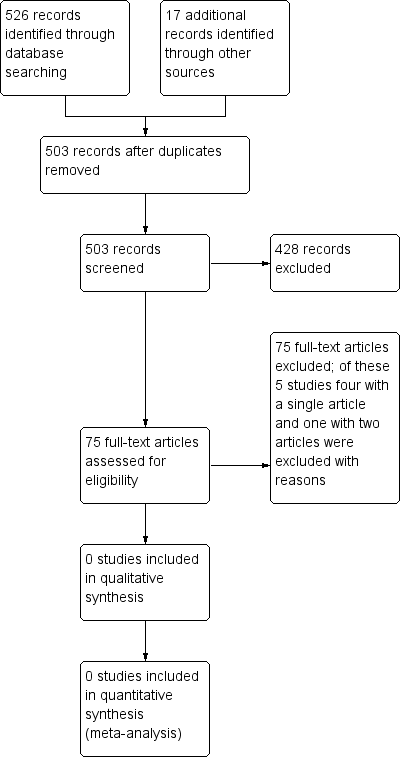
Study flow diagram.
Included studies
No studies were included in the review.
Excluded studies
Five studies were excluded (see Characteristics of excluded studies). One of the excluded studies examined the effectiveness of a particular test for screening and was not a study comparing newborn screening to no screening. The remaining four studies assessed different treatments for galactosaemia and not newborn screening.
Risk of bias in included studies
No studies were found that were eligible for inclusion in the review.
Effects of interventions
No studies were identified for inclusion in the review.
Discussion
Summary of main results
There were no completed or ongoing RCTs that were relevant to this review and there are no results to summarise.
Overall completeness and applicability of evidence
This review did not identify any completed RCTs and therefore there is no evidence that can be assessed.
Quality of the evidence
No RCTs of screening were found for inclusion in this review and there is no evidence that can be assessed.
Potential biases in the review process
We conducted a comprehensive search, searching data sources including multiple databases, and clinical trial registries to ensure that all relevant RCTs would be captured. Two authors independently assessed all of the review processes.
Agreements and disagreements with other studies or reviews
Due to the absence of evidence from RCTs, we were unable to compare our results with other published articles. Our search, only identified a small body of mid‐level evidence derived from observational studies such as cohort, case‐control and cross‐sectional studies, mainly reporting incidence or prevalence, with the bulk of the evidence being low level, derived from retrospective studies, case reports and expert opinion articles.
Currently, there are no adequate comparative studies to determine the effectiveness of neonatal screening for galactosaemia compared to the implementation of other measures designed to prevent severe, acute complications (e.g. protocol alerts, surveillance programmes, opportunistic screening). The only comparative data come from the UK paediatric surveillance programme, which points to similar incidences of severe cases and mortality in the regions which have and have not implemented galactosaemia screening programmes, but does not take into account the characteristics of public healthcare planning in the UK (Honeyman 1993). A recent systematic review concluded that based on the indirect assumptions and descriptive data presented by both the Swedish and German screening programs, one could assume that screening might reduce the risk of mortality and illness in babies if screening results are obtained before the 7th to 8th day of life (Varela Lema 2014). This should be confirmed in properly designed studies.