Les antiviraux à action directe pour l'hépatite C chronique.
Referencias
References to studies included in this review
References to studies excluded from this review
References to ongoing studies
Additional references
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Randomised phase III clinical trial | |
| Participants | 1088 participants Countries: Europe and USA Inclusion criteria: participants with HCV genotype 1 infection who had not received previous treatment 18‐70 years of age and had HCV genotype 1 infection with evidence of chronic hepatitis, as confirmed by means of a liver biopsy within 1 year before screening for the study; people with compensated liver cirrhosis were eligible. Exclusion criteria: advanced liver disease, co‐infection with HBV or HIV, HCC, other clinical relevant comorbidity. ALT> 5 x the ULN, total bilirubin > 2 mL/dL, albumin < 3.5 g/dL, international normalised ratio > 1.7, platelets < 90 x 109, haemoglobin < 12 g/dL (women) or < 13 g/dL (men). | |
| Interventions | Experimental group 1: telaprevir (orally at a dose of 750 mg every 8 h) and peg‐IFN α‐2a (by subcutaneous injection at a dose of 180 μg per week) and RBV (orally at a dose of 1000 mg per day (in participants who weighed < 75 kg) or 1200 mg per day (in participants who weighed ≥ 75 kg)) for the entire 12 weeks followed by 4 weeks of placebo and peg‐IFN–RBV (T12PR group). Experimental group 2: telaprevir (orally at a dose of 750 mg every 8 h) and peg‐IFN α‐2a (by subcutaneous injection at a dose of 180 μg per week) and RBV (orally at a dose of 1000 mg per day (in participants who weighed < 75 kg) or 1200 mg per day (in participants who weighed ≥ 75 kg) for 8 weeks and placebo with peg‐IFN–RBV for 4 weeks (T8PR group). Control group: placebo with peg‐IFN–RBV for 12 weeks, followed by 36 weeks of peg‐IFN–RBV. Participants in the T12PR and T8PR groups who met the criteria for an extended RVR (defined as undetectable HCV RNA at weeks 4 and 12) received 12 additional weeks of treatment with peg‐IFN–RBV alone, for a total treatment period of 24 weeks. Participants in the T12PR and T8PR groups who had detectable HCV RNA either at week 4 or at week 12 received 36 additional weeks of treatment with peg‐IFN–RBV, for a total treatment period of 48 weeks. The group receiving peg‐IFN α‐2a and RBV alone (PR group) received placebo plus peg‐IFN– RBV for 12 weeks, followed by peg‐IFN–RBV alone for 36 additional weeks. Co‐intervention: peg‐IFN (subcutaneously at 180 µg/week) and RBV orally twice daily dosed according to body weight. | |
| Outcomes | HCV RNA, safety assessment | |
| Notes | We emailed Jacobson and colleagues on 21 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Low risk | < 5% dropped out |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see ADVANCE 2011a2 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Low risk | < 5% dropped out |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 74 participants were randomised Sex: 58 men, 16 women Mean age: 50.2 Inclusion criteria: treatment‐naive adults 18‐65 years of age with chronic HCV genotype 1 infection for > 6 months before study enrolment, with a BMI > 18 and < 35 kg/m2. Chronic HCV infection was defined as 1 of the following: detectable HCV RNA or reactive HCV antibody > 6 months before enrolment; reactive antibody for HCV before screening and a liver biopsy > 6 months before enrolment demonstrating pathology consistent with HCV infection; and reactive HCV antibody or detectable HCV RNA before screening with an HCV risk factor (e.g. unsafe injection practices, blood transfusion before June 1992, receipt of clotting factor before 1987) that had emerged > 6 months before enrolment. In addition, participants had a liver biopsy result with histology consistent with HCV‐induced liver damage and with no evidence of cirrhosis or liver pathology due to any cause other than chronic HCV within the 3‐year period before study enrolment and participants had plasma HCV RNA level > 100.000 IU/mL at screening. Exclusion criteria: participants with METAVIR fibrosis score of 3 or 4 on liver biopsy, a positive test result for hepatitis B surface antigen or anti‐HIV antibodies, a history of major depression within the 2 years before enrolment, or unresolved clinically significant diseases other than HCV were excluded from participation. | |
| Interventions | Experimental group:
Control group: placebo + peg‐IFN/RBV Co‐intervention: peg‐IFN and RBV Participants were treated with ABT‐450/r, ABT‐333, or ABT‐072 monotherapy for 3 days, followed by 81 days (12 weeks minus 3 days of monotherapy) of ABT‐450/r, ABT‐333, or ABT‐072 combined with pegylated IFN/RBV (peg‐IFN/RBV), followed by 36 weeks of peg‐IFN/RBV alone. | |
| Outcomes | Primary outcomes: maximal change from baseline in HCV RNA levels, maximum plasma concentration (Cmax) of ABT‐450, time to maximum plasma concentration (Tmax) of ABT‐450, area under the plasma concentration‐time curve from 0‐24 h (AUC24) post‐dose of ABT‐450, maximum plasma concentration (Cmax) of ritonavir, time to maximum plasma concentration (Tmax) of ritonavir, area under the plasma concentration‐time curve from 0‐24 h (AUC24) post‐dose of ritonavir, maximum plasma concentration (Cmax) of ABT‐072, time to maximum plasma concentration (Tmax) of ABT‐072, area under the plasma concentration‐time curve from 0‐24 h (AUC24) post‐dose of ABT‐072, maximum plasma concentration (Cmax) of ABT‐333, time to maximum plasma concentration (Tmax) of ABT‐333, area under the plasma concentration‐time curve from 0‐12 h (AUC12) post‐dose of abt‐333. Secondary outcomes: percentage of participants with rapid virologic response (RVR) at week 4, percentage of participants with partial early virologic response (EVR) at week 12, Ppercentage of participants with complete early virologic response (cEVR) at week 12. | |
| Notes | We emailed Anderson and colleagues on 20 April 2016 for unpublished data and additional information regarding allocation concealment, random sequence generation, and blinding of outcome but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Investigators and participants were blinded to the study drug treatment regimen, but it was not stated how the blinding was maintained. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% of participants did not complete the study (19%), according to study protocol |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT01074008) |
| Vested‐interest bias | High risk | This study was funded by AbbVie |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Anderson 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Investigators and participants were blinded to the study drug treatment regimen, but it was not stated how the blinding was maintained. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% of participants did not complete the study (19%), according to study protocol |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT01074008) |
| Vested‐interest bias | High risk | This study was funded by AbbVie |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Anderson 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Investigators and participants were blinded to the study drug treatment regimen, but it was not stated how the blinding was maintained. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% of participants did not complete the study (19%), according to study protocol |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT01074008) |
| Vested‐interest bias | High risk | This study was funded by AbbVie |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Anderson 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Investigators and participants were blinded to the study drug treatment regimen, but it was not stated how the blinding was maintained. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% of participants did not complete the study (19%), according to study protocol |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT01074008) |
| Vested‐interest bias | High risk | This study was funded by AbbVie |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Anderson 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Investigators and participants were blinded to the study drug treatment regimen, but it was not stated how the blinding was maintained. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% of participants did not complete the study (19%), according to study protocol |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT01074008) |
| Vested‐interest bias | High risk | This study was funded by AbbVie |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Anderson 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Investigators and participants were blinded to the study drug treatment regimen, but it was not stated how the blinding was maintained. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% of participants did not complete the study (19%), according to study protocol |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT01074008) |
| Vested‐interest bias | High risk | This study was funded by AbbVie |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Anderson 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Investigators and participants were blinded to the study drug treatment regimen, but it was not stated how the blinding was maintained. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% of participants did not complete the study (19%), according to study protocol |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT01074008) |
| Vested‐interest bias | High risk | This study was funded by AbbVie |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Anderson 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Investigators and participants were blinded to the study drug treatment regimen, but it was not stated how the blinding was maintained. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% of participants did not complete the study (19%), according to study protocol |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT01074008) |
| Vested‐interest bias | High risk | This study was funded by AbbVie |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 24 treatment‐naive participants were randomised Inclusion criteria: 18‐65 years male and female, genotype 1, treatment‐naive. Female participants must be surgically sterile or 2 years post‐menopausal and are required to take a pregnancy test. BMI 18‐32 kg/m2, chronically infected with HCV genotype 1. Serum HCV RNA > 5 log 10 IU/mL. No previous treatment with IFNIFN, peg‐IFN, RBV or any investigational HCV antiviral agents. No history or signs of decompensated liver disease. No known history of cirrhosis, no co‐infection with HBV or HIV. No history of any medical condition that may interfere with absorption, distribution or elimination of study drug or with the clinical and laboratory assessments in this study. No history of alcohol abuse, or illicit drug use within 2 years prior to screen or enrolment in a methadone maintenance programme (unless he/she has been enrolled in the programme for at least 3 months with good compliance, stable psychosocial circumstances and no known current risks for recidivism). | |
| Interventions | Experimental group: 50 mg PPI‐461 once a day, 100 mg PPI‐461 once a day, 200 mg PPI‐461 once a day for 3 days Control group: placebo 2 weeks' follow‐up Co‐intervention: none | |
| Outcomes | Primary outcomes: safety and tolerability as measured by clinical AE and laboratory assessments (time frame: up to study day 16, 14 days after the last dose of PPI‐461). Antiviral effects of PPI‐461 measured by HCV RNA levels and pharmacokinetics measured by plasma concentrations of PPI‐461 concentrations. | |
| Notes | This is an unpublished study, only results from 2 abstracts | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial is described as double‐blind |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | No main publication, no drop‐outs |
| Selective reporting (reporting bias) | Unclear risk | Outcomes are only published in the protocol |
| Vested‐interest bias | High risk | Lead sponsor is Presidio Pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Anonymous (PPI‐461) 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial is described as double‐blind |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | No main publication, no drop‐outs |
| Selective reporting (reporting bias) | Unclear risk | Outcomes are only published in the protocol |
| Vested‐interest bias | High risk | Lead sponsor is Presidio Pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Anonymous (PPI‐461) 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial is described as double‐blind |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | No main publication, no drop‐outs |
| Selective reporting (reporting bias) | Unclear risk | Outcomes are only published in the protocol |
| Vested‐interest bias | High risk | Lead sponsor is Presidio Pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised phase IIb clinical trial | |
| Participants | 462 participants Location: Europe and USA Inclusion criteria: participants infected with HCV genotype 1 who had failed to respond to previous peg‐IFN/RBV treatment, adult participants, aged 18‐70 years, chronically infected with HCV genotype 1 and with plasma HCV RNA > 10,000 IU/mL at screening. All participants had to have received at least 1 prior course of peg‐IFN/RBV for 12 consecutive weeks and not discontinued therapy due to tolerability. Exclusion criteria: decompensated liver disease, any other liver disease of non‐HCV aetiology, and infection/co‐infection with nongenotype 1 HCV. | |
| Interventions | Experimental group:
Control group: 48 weeks of simeprevir‐matched placebo plus peg‐IFN/RBV Co‐intervention: peg‐IFN (subcutaneously at 180 µg/week) and RBV orally (1000 mg or 1200 mg/day, depending on body weight). For all participants, the 48‐week treatment period was followed by post‐treatment follow‐up for up to 72 weeks after treatment initiation. | |
| Outcomes | HCV RNA, safety assessment | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial used a computer random‐generation code |
| Allocation concealment (selection bias) | Low risk | The trial used a interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Low risk | < 5% dropped out |
| Selective reporting (reporting bias) | Low risk | All outcomes stated on ClinicalTrials.gov were reported (NCT00980330) |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised phase II clinical trial | |
| Participants | 225 participants Inclusion criteria: HCV treatment‐naïve adults aged 18 years or older with serologic evidence of chronic HCV genotype 1 infection, a serum HCV RNA level 50,000 IU/mL, and an absence of advanced fibrosis or cirrhosis (METAVIR score of F3‐4). Exclusion criteria: participants infected with HCV non‐1 genotypes or co‐infected with HBV or with HIV were excluded, as were participants with liver disease attributable to a cause other than HCV infection, cardiac or renal disease, severe psychiatric disease, uncontrolled seizures, severe retinopathy, immunologically‐mediated disease, poorly controlled diabetes, or who were pregnant or breastfeeding. Participants were also excluded if they had a haemoglobin concentration < 11 g/dL (women), or < 12 g/dL (men); neutrophil count < 1.5 x 109 cells/L; platelet count < 90 x 109 cells/L; serum creatinine concentration > 1.5 times the ULN); or BMI (calculated as kg/m2) < 18 or > 36. The use of agents that could interfere with the metabolism of danoprevir was prohibited. | |
| Interventions | Experimental group:
Control group: placebo with peg‐IFN–RBV for 24 or 48 weeks Co‐intervention: peg‐IFN (subcutaneously at 180 µg/week) and RBV orally twice daily dosed according to body weight | |
| Outcomes | HCV RNA (SVR), safety assessment | |
| Notes | we emailed Marcellin and colleagues on 27 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial used a computer‐generated randomisation code |
| Allocation concealment (selection bias) | Low risk | Interactive voice/web response system |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as "partial‐blind labeling" |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed (NCT00963885) |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Parallel‐group, randomised, placebo‐controlled, double‐blind study (RESPOND‐2)(NCT00708500) | |
| Participants | 403 participants Inclusion criteria: chronic hepatitis C infection genotype 1 HCV RNA ≥ 10,000 IU/mL, demonstrated responsiveness to IFN (minimum duration of therapy 12 weeks); non‐response defined as a decrease in HCV RNA of at least 2 log10 IU/mL by week 12, but with detectable HCV RNA during the therapy period; relapse defined as an undetectable HCV RNA at end of treatment, but without subsequent attainment of SVR. A liver biopsy with histology consistent with chronic hepatitis C, age ≥ 18 years, weight between 40‐125 kg, signed informed consent, acceptable method of contraception for the participant and participant's partner(s) for at least 2 weeks before day 1 and continue until at least 6 months after treatment termination. Exclusion criteria: Hepatitis B infection, HIV infection, other causes of liver disease, decompensated liver disease, uncontrolled diabetes mellitus, a severe psychiatric disorder, active substance abuse, active or suspected malignancy, or a history of malignancy within last 5 years, pregnant or nursing women, severe AE during prior treatment, seizure disorder, cerebrovascular diseases, cardiovascular disease, autoimmune diseases, prior organ transplantation, haemoglobinopathies, coagulopathies, abnormal levels of serum bilirubin, albumin, and creatinine, haemoglobin < 120 g/L (women) and < 130 g/L (men), neutrophil count < 1500/mm3, platelet count < 100,000/mm3. Group 1: 80 participants Age, mean (years): 52.9 Sex: 58 men (72%), 22 women (28%) Race, n(%): white: 62(84), black: 12(15), other: 1(1) Region, n(%): North America: 51(64), European Union: 29(36). Latin America: 0. BMI, mean ± SD (kg/m2): 28.2 ± 4.3 HCV subtype, n(%): 1a: 46(58), 1b: 34(42), missing data: 0 HCV RNA > 800,000 IU/mL, n(%): 65(81) METAVIR fibrosis score, n(%): 0, 1, or 2: 61(76), 3 or 4: 15(19) Cirrhosis, n(%): 10(12) Previous therapy, n(%): peg‐IFN alpha‐2a: 42(53), peg‐IFN alpha‐2b: 38(48) Prior non‐response, n(%): 29(36) Prior relapse, n(%): 51(64) Group 2: 162 participants Age, mean (years): 52.9 Sex: 98 men (60%), 64 women (40%) Race, n(%): white: 142(88), black: 18(11), other: 2(1). Region, n(%): North America: 115(71), European Union: 46(28), Latin America: 1(1). BMI, mean ± SD (kg/m2): 28.8 ± 4.6 HCV subtype, n(%): 1a: 94(58), 1b: 66(41), missing data: 2(1) HCV RNA > 800,000 IU/mL, n(%): 147(91) METAVIR fibrosis score, n(%): 0, 1, or 2: 117(72), 3 or 4: 32(20) Cirrhosis, n(%): 17(10) Previous therapy, n(%): peg‐IFN alpha‐2a: 79(49), peg‐IFN alpha‐2b: 83(51) Prior non‐response, n(%): 57(35) Prior relapse, n(%): 105(65) Group 3: 161 participants Age, mean (yr.): 52.3 Sex: 112 men (70%), 49 women (30%) Race, n(%): white: 135(84), black: 19(12), other: 7(4) Region, n(%): North America: 119(74), European Union: 42(26), Latin America: 0. BMI, mean ± SD (kg/m2): 28.2 ± 4.6 HCV subtype, n(%): 1a: 96(60), 1b: 61(38), missing data, 4(2) HCV RNA > 800,000 IU/mL, n(%): 141(88) METAVIR fibrosis score, n(%): 0, 1, or 2: 119(74), 3 or 4: 31(20) Cirrhosis, n(%): 17(10) Previous therapy, n(%): pegIFN alpha‐2a: 79(49), peg‐IFN alpha‐2b: 83(51) Prior non‐response, n(%): 57(35) Prior relapse, n(%): 105(65) | |
| Interventions | Experimental group: Group 2: oral boceprevir 800 mg thrice‐daily to be taken in with food and with an interval of 7‐9 h, in 4 capsules of 200 mg each, beginning at week 5 for a total of 32 weeks (if HCV RNA undetectable at week 8 and 12, treatment was terminated at week 36; if HCV RNA detectable at week 8 participants received placebo + peg‐IFN + RBV for an additional 12 weeks). Group 3: oral boceprevir 800 mg thrice‐daily to be taken in with food and with an interval of 7‐9 h, in 4 capsules of 200 mg each, beginning at week 5 for a total of 44 weeks Control group: Group 1: boceprevir‐matched placebo beginning at week 5 for a total of 44 weeks. Co‐interventions: Group 1 and 3: peg‐IFN alpha‐2b 1.5 μg/kg body weight subcutaneously once weekly and weight‐based oral RBV at a divided daily dose of 600 to 1400 mg for a total of 48 weeks Group 2: peg‐IFN alpha‐2b 1.5 μg/kg body weight subcutaneously once weekly and weight‐based oral RBV at a divided daily dose of 600 to 1400 mg for 36 weeks (if HCV RNA undetectable at week 8 and 12), and for 48 weeks if (HCV RNA detectable at week 8, but undetectable at week 12). | |
| Outcomes | Primary outcome: achievement of SVR (undetectable HCV RNA at week 24). Secondary outcome: achievement of SVR in randomised participants who received at least 1 dose of experimental study drug or placebo. Proportion of participants with EVR (undetectable HCV RNA at week 2, 4, 8, or 12) who achieved SVR. Proportion of participants with undetectable HCV RNA at week 12. Proportion of participants with undetectable HCV RNA at 72 weeks after randomisation. | |
| Notes | Group 2 received a similar, but not equal co‐intervention as Groups 1 and 3. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence |
| Allocation concealment (selection bias) | Low risk | Allocation of participants through interactive voice‐response system in a 1:2:2 ratio |
| Blinding of participants and personnel (performance bias) | Low risk | A boceprevir‐matched placebo was used. |
| Blinding of outcome assessment (detection bias) | Unclear risk | It was not mentioned if the outcome assessors were blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Treatment discontinuation due to AE was 2% to 12%. Seems no other drop‐outs occurred. |
| Selective reporting (reporting bias) | Low risk | A study protocol was published prior to randomisation (NCT00708500). All pre‐specified outcomes were reported on. |
| Vested‐interest bias | High risk | Trial was sponsored by a pharmaceutical company (Schering‐Plough/Merck). The company was directly involved in trial design and managing, data analysis, and writing of article. |
| Other bias | Low risk | Seems there were no other potential sources of bias. |
| Methods | For characteristics see Bacon 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence |
| Allocation concealment (selection bias) | Low risk | Allocation of participants through interactive voice‐response system in a 1:2:2 ratio |
| Blinding of participants and personnel (performance bias) | Low risk | A boceprevir‐matched placebo was used. |
| Blinding of outcome assessment (detection bias) | Unclear risk | It was not mentioned if the outcome assessors were blinded. |
| Incomplete outcome data (attrition bias) | Low risk | Treatment discontinuation due to AE was 2% to 12%. Seems no other drop‐outs occurred. |
| Selective reporting (reporting bias) | Low risk | A study protocol was published prior to randomisation (NCT00708500). All pre‐specified outcomes were reported on. |
| Vested‐interest bias | High risk | Trial was sponsored by a pharmaceutical company (Schering‐Plough/Merck). The company was directly involved in trial design and managing, data analysis, and writing of article. |
| Other bias | Low risk | Seems there were no other potential sources of bias. |
| Methods | Randomised clinical trial | |
| Participants | 60 adult participants Sex: not described Mean age: not described Inclusion criteria: chronic hepatitis C and with a psychiatric disorder (n = 60, schizophrenia 20/60 (33.3%)), major depression 15/60 (25%), bipolar disorder 20/60 (33.3%), and prior suicidal attempts with depression 5/60 (8.3%). Exclusion criteria: Renal failure with CrCl < 30, sickle cell, thalassaemic syndromes, haemolytic syndrome, co‐infections (HBV, HIV), or CHF NYHA Stage IV. | |
| Interventions | Experimental group: Group 1: simeprevir 150 mg and RBV 1000 mg daily Group 3: simeprevir 150 mg and vitamin D 5000 mg daily. Control group: placebo and RBV 1000 mg daily Co‐intervention: Sofosbuvir 400 mg | |
| Outcomes | Antiviral effect | |
| Notes | Email was sent to Basu and colleagues on 06 June 2016 for additional information on allocation sequence generation and concealment, blinding, incomplete outcome data, protocol, full publication, study sponsor, death, SAE, SVR but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | The trial is described as open‐label |
| Blinding of outcome assessment (detection bias) | High risk | The trial is described as open‐label |
| Incomplete outcome data (attrition bias) | Unclear risk | It is unclear how many participants dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be found |
| Vested‐interest bias | Unclear risk | It was unclear how the trial was funded |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 68 participants Sex: 20 men, 11 women (only reported in Bavisotto trial) Mean age: 43.6 years Country: USA Inclusion criteria: Chronically infected with HCV genotype 1 (genotype‐1) without cirrhosis. 18‐60 years of age and HCV treatment‐naive. | |
| Interventions | Experimental group: ascending doses of GS‐9190 (40, 120, 240, 240‐with food, or 480 mg) orally for 8 days. Control group: placebo orally for 8 days. | |
| Outcomes | Adverse events, GS‐9190 concentration, HCV RNA | |
| Notes | No data could be used. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be found |
| Vested‐interest bias | High risk | Sponsored by Gilead |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 24 participants Sex: 15 men, 9 women Median age: 45.5 years Country: France Inclusion criteria: chronic G4 HCV infection. HCV‐infected treatment‐naive participants aged 18–65 years and in good health (except for chronic G4 HCV infection) were eligible if they had a plasma HCV RNA load of > 10,000 IU/mL, an absolute neutrophil count of ≥ 1500 neutrophils/mm3, and a platelet count of ≥ 100,000 platelets/mm3. Exclusion criteria: contraindications to IFN (peg‐IFN in particular) or RBV treatment; history or evidence of cirrhosis, end‐stage liver disease, or decompensated liver disease (as shown by screening laboratory results); HIV or HBV co‐infection; history of alcohol or illicit drug use; and pregnancy/current breast‐feeding | |
| Interventions | Experimental group 1: oral 750 mg of telaprevir 3 times daily for 2 weeks Experimental group 2: oral 750 mg of telaprevir 3 times daily for 2 weeks + peg‐IFN α‐2a 180 μg once weekly, and RBV 1000–1200 mg/day (weight‐based) Control group: placebo + peg‐IFN α‐2a 180 μg once weekly, and RBV 1000–1200 mg/day (weight‐based) for 2 weeks Co‐intervention: after the 2 weeks of treatment, all participants received peg‐IFN α‐2a 180 μg once weekly, and RBV 1000–1200 mg/day (weight based) (48 weeks for experimental group 1, and 46 weeks for experimental group 2 and control group) | |
| Outcomes | Efficacy assessment, virology assessment, safety and pharmacokinetic assessment | |
| Notes | NCT00580801 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described adequately (computer‐based) |
| Allocation concealment (selection bias) | Unclear risk | Not described adequately (computer‐based) |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was only partially blinded. The participants in the telaprevir group without peg‐IFN and RBV were not blinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was only partially blinded. The participants in the telaprevir group without peg‐IFN and RBV were not blinded |
| Incomplete outcome data (attrition bias) | Unclear risk | < 5% dropped out (1 person) |
| Selective reporting (reporting bias) | Unclear risk | No predefined outcomes were stated in the protocol (NCT00580801) |
| Vested‐interest bias | High risk | The trial was funded by Janssen Pharmaceuticals and Vertex Pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Benhamou 2013a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described adequately (computer‐based) |
| Allocation concealment (selection bias) | Unclear risk | Not described adequately (computer‐based) |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was only partially blinded. The participants in the telaprevir group without peg‐IFN and RBV were not blinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was only partially blinded. The participants in the telaprevir group without peg‐IFN and RBV were not blinded |
| Incomplete outcome data (attrition bias) | Unclear risk | < 5% dropped out (1 person) |
| Selective reporting (reporting bias) | Unclear risk | No predefined outcomes were stated in the protocol (NCT00580801) |
| Vested‐interest bias | High risk | The trial was funded by Janssen Pharmaceuticals and Vertex Pharmaceuticals. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 34 adult participants Inclusion criteria: chronic HCV infection with genotype 1 (1a, 1b or mixed 1a/1b), with an HCV VL ≥ 100,000 IU/ml at screening. For treatment‐naive participants, no prior therapy with IFN, peg‐IFN, or RBV was allowed. For treatment‐experienced participants, virological failure with peg‐IFN/RBV treatment was to be confirmed. treatment‐experienced participants without cirrhosis required histological evidence within 24 months prior to trial enrolment of chronic necroinflammatory activity or the presence of fibrosis; treatment‐experienced participants with compensated cirrhosis required histological evidence of cirrhosis due to HCV infection, without evidence of decompensation. Exclusion criteria: HCV infection of mixed genotype or had been treated previously with at least one dose of any protease. | |
| Interventions | Experimental group: oral BI 201335 NA, 20 mg, 48 mg, 120 mg, or 240 mg once daily. Control group: placebo. Co‐intervention: peg‐IFN/RBV. | |
| Outcomes | Virological response, pharmacokinetics, safety | |
| Notes | Unpublished data only | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being blinded, but it was unclear how this was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being blinded, but it was unclear how this was performed |
| Incomplete outcome data (attrition bias) | Low risk | < 5% dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 10 adult participants Inclusion criteria: with diagnosis of cirrhosis and chronic HCV (genotype 1) infection with a VL greater than 50,000 copies mRNA/ml serum. Country: Germany | |
| Interventions | Experimental group: oral 200 mg twice daily for 2 days. Control group: placebo. | |
| Outcomes | Efficacy assessment, safety assessment | |
| Notes | Unpublished data only | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Low risk | There were no dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 48 adult participants Sex: 35 men, 12 women Mean age: 48.5 years Inclusion criteria: 18‐70 years old with Chronic HCV genotype 1 infection and HCV RNA above 100,000 IU/mL. Participants had no prior treatment or < 4 weeks of total exposure to RBV or peg‐IFN‐based therapy. Participants had to be non‐cirrhotic, documented by liver biopsy obtained within 24 months before randomisation. Exclusion criteria: advanced liver disease, co‐infection with HBC or HIV, hepatocellular carcinoma, other clinical relevant comorbidity. ALT > 5 x the ULN, total bilirubin > 2 mL/dL, albumin < 3.5 g/dL, international normalised ratio > 1.7, platelets < 90x10^9, haemoglobin < 12 g/dL (women) or < 13 g/dL (men). | |
| Interventions | Experimental group 1: oral asunaprevir (200 mg) twice daily for 48 weeks. Experimental group 2: oral asunaprevir (600 mg) twice daily for 48 weeks. Experimental group 3: oral asunaprevir (600 mg) once daily for 48 weeks. Control group: placebo 48 weeks. Co‐intervention: peg‐IFN (subcutaneously at 180 µg/week) and RBV orally twice daily dosed according to body weight. | |
| Outcomes | Proportion of participants with undetectable HCV RNA at week 4 and 12, SAE, AE, mortality, sustained virological response. | |
| Notes | Experimental group 1 vs control. We contacted trial authors on 20 April 2016 for additional information on allocation concealment, specifics of the blinding, what SAE were experienced, and how they dealt with missing data, reached required sample size but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | It was stated that investigators and participants were blinded to treatment assignment throughout the study but it was not stated how the blinding was maintained. |
| Blinding of outcome assessment (detection bias) | High risk | The sponsor was blinded to treatment assignment until the primary end point analysis which was at 12 weeks and we used data at week 24. |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants had missing data and how the trial handled participants with missing data |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the pre published protocol (NCT01030432) were reported |
| Vested‐interest bias | High risk | The study was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Bronowicki 2013a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | It was stated that "Investigators and participants were blinded to treatment assignment throughout the study" but it was not stated how the blinding was maintained. |
| Blinding of outcome assessment (detection bias) | High risk | The sponsor was blinded to treatment assignment until the primary end point analysis which was at 12 weeks and we used data at week 24. |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants had missing data and how the trial handled participants with missing data |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the pre published protocol (NCT01030432) were reported |
| Vested‐interest bias | High risk | The study was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Bronowicki 2013a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | It was stated that investigators and participants were blinded to treatment assignment throughout the study but it was not stated how the blinding was maintained. |
| Blinding of outcome assessment (detection bias) | High risk | The sponsor was blinded to treatment assignment until the primary end point analysis which was at 12 weeks and we used data at week 24. |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants had missing data and how the trial handled participants with missing data |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the pre published protocol (NCT01030432) were reported |
| Vested‐interest bias | High risk | The study was funded by Bristol Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 238 adult participants Sex: 153 men, 85 women Mean age: 47.7 years Inclusion criteria: 18–70 years, with chronic HCV genotype 1 or 4 infection and HCV RNA P100,000 IU/mL. Participants had to have ALT < 5 ULN and no history or evidence of hepatic decompensation. Compensated cirrhotic participants (genotype 1 only) were eligible with a liver biopsy documenting cirrhosis from any period prior to randomisation. For non‐cirrhotic participants, absence of cirrhosis had to be documented by a liver biopsy obtained within 24 months pre‐randomisation. Exclusion criteria: prior exposure to anti‐HCV agents, co‐infection with HBV or HIV, and chronic liver disease other than HCV. | |
| Interventions | Experimental group: 200 mg asunaprevir twice a day for 12 or 24 weeks. Control intervention: placebo twice a day for 12 weeks. Co‐intervention: peg‐IFNa‐2a administered subcutaneously at 180 lg per week, and oral RBV twice a day dosed by body weight (< 75 kg, 1000 mg daily; P75 kg, 1200 mg daily). | |
| Outcomes | SAE, AEs, discontinuations due to AEs, eRVR at week 4 and 12, SVR24, RVR at week 4, complete eRVR, SVR 12, resistant variants associated with virologic failure. | |
| Notes | At week 12, asunaprevir‐treated participants who achieved a protocol‐defined response (HCV RNA < LLOQ at week 4 and undetectable at week 10 were re‐randomised (1:1) to continue triple therapy with asunaprevir plus peg‐IFNα/RBV for a total of 24 weeks (24‐Triple) or to receive placebo plus peg‐IFNα/RBV for an additional 12 weeks (12‐Triple + 12; Fig. 1). Asunaprevir‐treated participants without PDR and those initially assigned to placebo received placebo plus peg‐IFNα/RBV from week 13 to 24. At week 24, PDR‐positive participants who received 24‐Triple or 12‐Triple + 12 stopped treatment and were followed through week 48. PDR‐negative participants and those initially assigned to placebo were switched to open‐label peg‐IFNα/RBV through week 48 and followed through week 72. We report re‐randomisation in Bronowicki 2014a. We contacted the trial authors on 25 February 2016 by email jp.bronowicki@chu‐nancy.fr about allocation concealment, SAE at maximum follow‐up, specific SAE at maximum follow‐up, how the authors accounted for data of missing participants. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | Investigators and participants were blinded to treatment assignment through week 24; the sponsor was blinded through week 12. It was unknown how the blinding was maintained. Additionally, some of the participants had open‐label peg‐IFNα/RBV |
| Blinding of outcome assessment (detection bias) | High risk | Investigators and participants were blinded to treatment assignment through week 24; the sponsor was blinded through week 12. It was unknown how the blinding was maintained. Additionally, some of the participants had open‐label peg‐IFNα/RBV |
| Incomplete outcome data (attrition bias) | Low risk | There were under 5% dropouts |
| Selective reporting (reporting bias) | Low risk | The outcomes stated in the pre published protocol (ClinicalTrials.gov:NCT01030432) were reported on |
| Vested‐interest bias | High risk | Funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 301 participants Sex: 228 men and 73 women Mean age: 47 years Inclusion criteria: documented chronic HCV genotype 1 (genotype1), genotype4, or genotype6 infection with no evidence of genotype2 or genotype3 or non‐typeable genotypes and HCV RNA confirmed by screening lab results prior to randomisation on opiate substitution therapy (methadone, levamethadone, buprenorphine, naloxone, naltrexone) for at least 3 months prior to screening, treatment‐naive to all HCV therapies. HIV‐infected participants enrolled in this study had to meet following criteria: Documented HIV infection, naive to treatment with any antiretroviral therapy or on HIV antiretroviral therapy for at least 8 weeks prior to study entry using a dual nucleoside reverse transcriptase inhibitor backbone of tenofovir or abacavir and either emtricitabine or lamivudine plus raltegravir (or dolutegravir or rilpivirine). Dose modifications or changes in antiretroviral therapy during the 4 weeks prior to study entry (Day 1) were not permitted. Cluster of differentiation 4 (CD4+) T‐cell count > 200 cells/mm^3 if on antiretroviral therapy or > 500 cell/mm^3 if antiretroviral therapy treatment‐naive undetectable plasma HIV‐1 RNA at least 8 weeks prior to screening if on antiretroviral therapy or < 50,000 copies/mL if antiretroviral therapy treatment‐naive. Participants with HIV‐1 infection and on antiretroviral therapy must have at least 1 viable antiretroviral regimen alternative beyond their current regimen in the event of HIV virologic failure or the development of anti‐retroviral drug resistance Women who are of reproductive potential had to agree to avoid becoming pregnant while receiving study drug and for 14 days after the last dose of study drug by complying with 1 of the following: (1) practice abstinence from heterosexual activity OR (2) use (or have her partner use) acceptable contraception during heterosexual activity. Exclusion criteria: evidence of decompensated liver disease. For participants with cirrhosis, participants who are Child‐Pugh Class B or C or who have a Pugh‐Turcotte score > 6 Is co‐infected with HBV. Has cirrhosis and liver imaging within 6 months of Day 1 showing evidence of HCC or is under evaluation for HCC. Currently using or intends to use barbiturates during the treatment period of this study. Is a female and is pregnant or breast‐feeding, or expecting to conceive or donate eggs from Day 1 or anytime during treatment, and 14 days after the last dose of study medication, or longer if dictated by local regulations. Any medical condition requiring or likely to require chronic systemic administration of corticosteroids, Tumor Necrosis Factor‐antagonists, or other immunosuppressant drugs during the course of the trial. Evidence or history of chronic hepatitis not caused by HCV. | |
| Interventions | Experimental group: oral 100 mg of grazoprevir and 50 mg of elbasvir for 12 weeks. Control group: placebo. | |
| Outcomes | Safety assessment, HCV RNA (virological failure). | |
| Notes | Abstract only (still ongoing). Only data for the first 12 weeks could be used, since the control group received the same DAA in the following 12 weeks. (NCT02105688). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | No description of dropouts after 12 weeks for the control group. |
| Selective reporting (reporting bias) | Unclear risk | Only an abstract could be found, and no data on SVR12 and SVR24 were presented. However the trial was still stated as ongoing. (NCT02105688) |
| Vested‐interest bias | High risk | The trial was funded by Merck |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 421 participants Sex: 227 men, 194 women Mean age: 52.6 years Countries: Australia, Czech Republic, France, Germany, Israel, Puerto Rico, South Korea, Sweden, Taiwan, and USA Inclusion criteria: treatment‐naive cirrhotic and non‐cirrhotic adults (aged > 18 years) with HCV RNA levels > 104 IU/mL were eligible. Hepatic fibrosis was staged by biopsy or noninvasive assessment. Exclusion criteria: decompensated liver disease, HCC, HIV or HBV co‐infection, uncontrolled diabetes mellitus (haemoglobin A1c level > 10%), elevated prothrombin time unrelated to anticoagulation, creatinine clearance < 50 mL/min, haemoglobin level < 95 g/L, thrombocytopenia (platelet count < 50 × 109 cells/L), aminotransferase levels more than 10 times the ULN, or hypoalbuminaemia (albumin level < 30 g/L). | |
| Interventions | Experimental group: oral 100 mg of grazoprevir and 50 mg of elbasvir for 12 weeks. Control group: placebo. | |
| Outcomes | HCV RNA, safety assessment. | |
| Notes | Only data for the first 12 weeks could be used, since the control group received the same DAA in the following 12 weeks. We emailed Zeuzem and colleagues on 27 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The trial used a "computer‐generated random allocation schedule" |
| Allocation concealment (selection bias) | Low risk | The trial used a "central interactive voice‐response system" |
| Blinding of participants and personnel (performance bias) | Low risk | The participants and personnel were blinded to treatment assignment for the first 12 weeks (and we used the data from this time point) |
| Blinding of outcome assessment (detection bias) | Low risk | The sponsors performing the analyses were blinded to treatment assignment for the first 12 weeks (and we used the data from this time point) |
| Incomplete outcome data (attrition bias) | Low risk | < 5% dropped out after 12 weeks |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed (NC02105467) |
| Vested‐interest bias | High risk | The trial was funded by Merck‐Sharp which performed the analyses |
| Other bias | Low risk | Trial seems to be free of other potential sources of bias |
| Methods | Randomised clinical trial | |
| Participants | An unknown amount of participants Sex: unknown Mean age: unknown Inclusion criteria: chronic HCV infection (> 6 months) and were treatment‐naive. Participants aged 18‐64 years with ≥ 104 IU/mL HCV RNA levels were enrolled in sequential, ascending dose cohorts of up to 16 participants (12 active, 4 placebo) per cohort. | |
| Interventions | Experimental group: participants received 50, 100, 250, 500, 1000, or 1500 mg oral doses of HCV‐796 or placebo given as monotherapy twice daily. Control group: placebo twice a day. Co‐intervention: none. | |
| Outcomes | Most frequent AE, dose‐limiting toxicities or serious treatment‐emergent AEs, PK parameters, maximal antiviral effects. | |
| Notes | The authors were contacted on VIROPHARMA all bias domains, mortality, SAE, SVR24. mean age, male:female, number of participants, final publication. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | The number of participants with incomplete data was not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | Unclear risk | It was unclear how the trial was funded |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical phase IIb trial | |
| Participants | 395 participants Sex: 262 men, 133 women Mean age: 50.8 years Countries: North and Central America, Australia, North Africa, and Europe. Inclusion criteria: treatment‐naive, aged 18–70 years who had chronic HCV genotype 1 or 4 infection. Compensated cirrhotic, infected with HCV genotype 1, and HCV genotype 4, were each capped at 10% of randomised participants. Cirrhosis was confirmed by biopsy at any time prior to randomisation. For non‐cirrhotic participants, a liver biopsy must have been obtained within 24 months prior to randomisation. Additional inclusion criteria included HCV RNA ≥ 100,000 IU/mL and ALT levels < 5×ULN. Exclusion criteria: history or evidence of hepatic decompensation, prior exposure to any agent with potential anti‐HCV activity, co‐infection with HBV or HIV, or evidence of chronic liver disease other than HCV. | |
| Interventions | Initally the trial was randomised into 3 groups (2 experimental groups, and 1 control group). After week 12, the participants who received a protocol‐defined response, were re‐randomised to placebo or additional 12 weeks of therapy. The participants without a protocol‐defined response were treated with placebo and co‐intervention. Experimental group: oral 20 mg of daclatasvir once a day for 12 weeks (after week 12, the participants with a protocol‐defined response were re‐randomised). Experimental group: oral 60 mg of daclatasvir once a day for 12 weeks (after week 12, the participants with a protocol‐defined response were re‐randomised). Control group: placebo. Co‐intervention: peg‐IFN α‐2a administered subcutaneously at a dose of 180 mg per week and twice a day RBV dosed orally according to body weight (< 75 kg, 1000 mg daily; > 75 kg, 1200 mg daily). After week 24, all participants received standard care (peg‐IFN‐α‐2a and RBV) | |
| Outcomes | Safety assessment, efficacy assessment | |
| Notes | We emailedWe emailed Hezode and colleagues on 21 April 2016 for additional information on sequence generation, missing data, additional data, death but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | The trial used interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | High risk | The participants were only blinded until week 24 |
| Blinding of outcome assessment (detection bias) | High risk | The sponsors, who performed the analyses, were only blinded until week 12 |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see COMMAND‐1 2015a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | The trial used interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | High risk | The participants were only blinded until week 24 |
| Blinding of outcome assessment (detection bias) | High risk | The sponsors, who performed the analyses, were only blinded until week 12 |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised phase III clinical trial | |
| Participants | 188 participants Inclusion criteria: HCV genotype 1 infection, treatment‐naive male and female participants aged 20–70 years with documented chronic genotype 1 HCV infection and plasma HCV RNA P5.0 log10 IU/mL at screening. Exclusion criteria: liver cirrhosis, hepatic failure, any other liver disease of non‐HCV etiology and co‐infection with HIV‐1, HIV‐2, hepatitis B, or non‐genotype 1 HCV. | |
| Interventions | Experimental group: simeprevir 100 mg once a day plus peg‐IFNa‐2a/RBV for 12 weeks followed by response‐guided therapy with peg‐IFNa‐2a/RBV alone for 12 or 36 weeks. Control group: placebo with peg‐IFNa‐2a/RBV for 12 weeks followed by peg‐IFNa‐2a/RBV for 36 weeks. Peg‐IFNα‐2a (Pegasys®, Chugai, Japan) was administered as a subcutaneous injection (180 μg once weekly) and RBV (Copegus®, Chugai) as oral tablets (600‐1000 mg total daily dose, depending on body weight). Co‐intervention: peg‐IFN (subcutaneously at 180 µg/week) and RBV orally twice daily dosed according to body weight. | |
| Outcomes | HCV RNA, safety assessment, ALT/AST elevations. | |
| Notes | We emailedWe emailed Hayashi and colleagues on 21 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed (NCT01292239) |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 34 participants Sex: 21 men, 10 women (analysed only) Mean age: 42.9 years (analysed only) Inclusion criteria: treatment‐naive genotype 1‐infected male or female participants between 18 and 60 years of age with a BMI 633 kg/m2 were recruited. Baseline plasma HCV RNA greater than 100,000 IU/mL, ALT values < 5 times the ULN and a Metavir liver fibrosis stage between 0 and 3 were required. Exclusion criteria: none specified. | |
| Interventions | Experimental group: VCH‐759 doses (400 mg three times a day, 800 mg twice a day and 800 mg three times a day). VCH‐759 was supplied as an oral solution formulation in individual 120 mL clear glass bottles. The oral solution was reconstituted by combining the appropriate VCH‐759 powder‐in‐bottle dose in a 30% polyethylene glycol 400/15% Solutol HS15 aqueous reconstitution vehicle (20 mL for the 400 mg dose and 40 mL for the 800 mg dose). Control group: placebo. Co‐intervention: none. | |
| Outcomes | Absolute change in plasma HCV RNA levels between baseline to nadir, blood samples for evaluation of the plasma HCV RNA viral load, blood samples for NS5B polymerase, the complete PK profile. | |
| Notes | We contacted the trial authors on 26 February 2016 by email [email protected] about random sequence generation, allocation concealment, blinding of participants, personnel and outcome assessment, did the trial account for the missing data, which group the the 2 participants dropped out from and was if there was a prepublished protocol, mortality, SAE, SVR24. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Unclear risk | There were above 5% drop outs and it was unclear how the trial handled participants with missing data |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The authors have declared that this study was funded by ViroChem Pharma Inc. JB, NC, RT, IB, ON, and LP are employees of ViroChem Pharma Inc. The other authors have also declared a relationship with the manufacturers of the drugs involved. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised, active‐controlled phase IIb trial | |
| Participants | Sex: 260 men, 157 women Mean age: 48 years Inclusion criteria: eligible participants were treatment‐naive adults aged > 18 years with chronic HCV genotype 1 or 4 infection and HCV RNA above 50,000 IU/mL. Participants had to have evidence of chronic hepatitis C, documented by liver biopsy obtained within 24 months before randomisation. Exclusion criteria: participants with cirrhosis or incomplete/transition to cirrhosis (Knodell, Metavir, or Batts and Ludwig ≥ 3 or Ishak modified HAI ≥ 4); BMI < 18 or ≥ 36 kg/m2, other forms of liver disease; HIV infection; HCC; severe cardiac disease; severe depression or other psychiatric disease; renal disease; uncontrolled seizure disorders; severe retinopathy; haemoglobin < 12 g/dL for women or < 13 g/dL for men; neutrophil count < 90 cells/nL; serum creatinine > 1.5 times the ULN. | |
| Interventions | Participants were randomised (2:2:2:2:1) to 1 of 5 treatment arms Experimental group 1: ritonavir boosted danoprevir (danoprevir/r) 200/100 mg twice a day for 24 weeks Experimental group 2: ritonavir boosted danoprevir (danoprevir/r) 100/100 mg twice a day for 24 weeks Experimental group 3: ritonavir boosted danoprevir (danoprevir/r) 50/100 mg twice a day for 24 weeks Experimental group 4: ritonavir boosted danoprevir (danoprevir/r) 100/100 mg twice a day for 12 weeks or 24 weeks (participants achieving undetectable HCV RNA from Weeks 2 to 10 (eRVR2) stopped treatment at Week 12) Control group: participants in Arm E with detectable HCV RNA at Week 12 had the option to roll over to treatment with danoprevir/r 200/100 mg twice a day Co‐intervention: peg‐IFN α‐2a (40KD) 180 lg/week and RBV 1000 mg/day (bodyweight < 75 kg) or 1200 mg/day (bodyweight ≥ 75 kg) | |
| Outcomes | Proportion of participants with SVR24, with SAE, AEs, mortality. | |
| Notes | Experimental group 1 vs Control. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | Unblinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unblinded |
| Incomplete outcome data (attrition bias) | Unclear risk | There were above 5% dropouts and it was unclear how the trial handled the missing participants |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were reported on: NCT01220947 |
| Vested‐interest bias | High risk | The trial was funded by F. Hoffmann‐La Roche Ltd |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Dauphine 2015a2 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | Unblinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unblinded |
| Incomplete outcome data (attrition bias) | Unclear risk | There were above 5% dropouts and it was unclear how the trial handled the missing participants |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were reported on: NCT01220947 |
| Vested‐interest bias | High risk | The trial was funded by F. Hoffmann‐La Roche Ltd |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Dauphine 2015a2 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | computer‐generated randomisation |
| Allocation concealment (selection bias) | Unclear risk | not described |
| Blinding of participants and personnel (performance bias) | High risk | unblinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | unblinded |
| Incomplete outcome data (attrition bias) | Unclear risk | There were above 5 % drop outs and it was unclear how the trial handLed the missing participants |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were reported on: NCT01220947 |
| Vested‐interest bias | High risk | the trial was funded by F. Hoffmann‐La Roche Ltd. Support |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Dauphine 2015a2 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | Unblinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unblinded |
| Incomplete outcome data (attrition bias) | Unclear risk | There were above 5 % drop outs and it was unclear how the trial handLed the missing participants |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were reported on: NCT01220947 |
| Vested‐interest bias | High risk | The trial was funded by F. Hoffmann‐La Roche Ltd |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 41 participants Sex: 31 men, 9 women Mean age: 48.8 years Inclusion criteria: 18‐65 years with BMI of 18‐40 kg/m2, HCV genotype 1 (any subtype), and HCV RNA level > 1 105 copies/mL (or equivalent international units). Chronic hepatitis C participants were naive, nonresponders or relapsers to previous IFN‐based treatment. Relapse was defined as undetectable HCV RNA upon completion of a previous IFN‐based treatment, but positive HCV RNA during follow‐up. Nonresponse was defined as positive HCV RNA at the end of a previous IFN‐based treatment or < 2‐log decline in HCV RNA levels at 12 weeks and discontinued treatment. Exclusion criteria: key exclusion criteria included decompensated liver disease, findings consistent with Child‐Pugh class B or C liver cirrhosis, and co‐infection with HIV or HBV. | |
| Interventions | Experimental group: participants received either 800 mg narlaprevir 3 times daily or 400 mg narlaprevir as an oral suspension in combination with for 7 days in the first period and for 14 days in the second period. Control group: placebo. Co‐intervention: 200 mg ritonavir in cohort 3 and 4, a wash‐out period after 1 week of treatment, 1.5 lg/kg/week peg‐IFN‐α‐2b (in period 2) and standard care for 24 weeks after period 2. | |
| Outcomes | Safety assessment, pharmacokinetic assessment, viral assessments. | |
| Notes | Cohort 1 and 3 each included 10 participants naive to HCV treatment; cohorts 2 and 4 each included 10 HCV treatment‐experienced participants. We report here the treatment‐naive participants We contacted the trial authors on 26 February 2016 by email [email protected] about allocation concealment, how the blinding was maintained and who performed the outcome assessment; number of deaths, SAE, which group was the missing participants randomised to. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 participant dropped out |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT01081158) and the outcomes stated in the protocol were reported on |
| Vested‐interest bias | High risk | Sponsored by Schering‐Plough and designed by Schering‐Plough employees and HW Reesink |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias. |
| Methods | For characteristics see De Bruijne 2010a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as double‐blinded and but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 participant dropped out |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT01081158) and the outcomes stated in the protocol are reported on |
| Vested‐interest bias | High risk | Sponsored by Schering‐Plough and designed by Schering‐Plough employees and HW Reesink |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of other bias. |
| Methods | Randomised clinical trial | |
| Participants | 18 participants (only number of experimental group) Country: Moldova Inclusion criteria: healthy, treatment‐naive or experienced HCV genotype 1 participants. | |
| Interventions | Experimental group: oral 400 mg or 600 mg of ACH‐1625 in fasted state for 5 days, or 600 mg of ACH‐1625 once daily following a medium‐fat meal for 5 days. Control group: placebo. | |
| Outcomes | PK, safety, tolerability, effects on viral kinetics. | |
| Notes | It was unclear whether the included participants included healthy participants, or healthy HCV genotype 1 participants. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being placebo blinded, but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being placebo blinded, but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | Several authors were sponsored by Achillion Pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 152 adult participants Sex: 96 men, 55 women (analysed) Mean age: 47.9 years Countries: USA, Australia, Canada, Denmark, France, and Italy. Inclusion criteria: men and women aged 18–70 years, with chronic HCV genotype 2 or 3 infection and no prior exposure to HCV therapeutic agents including DAA, IFN preparations, or RBV. Participants were stratified by HCV genotype (2 or 3) before randomisation. Plasma HCV RNA levels at screening were required to be ≥100,000 IU/mL. Liver disease staging was conducted by liver biopsy within 2 years of screening (biopsies confirming cirrhosis), or by FibroScan (Echosens, Paris, France) within 1 year of screening (14.6 kPa was considered consistent with cirrhosis); participants with compensated cirrhosis were capped at approximately 10% of the study population. Women of childbearing potential and men who were sexually active partners of women of childbearing potential were required to use 2 forms of contraception, including at least 1 barrier method. Exclusion criteria: history or evidence of HCC, decompensated cirrhosis, or chronic liver disease other than hepatitis C; history of cancer within 5 years of enrolment; chronic HBV or HIV infection; presence of any other medical, psychiatric, and/or social reason that would render the patient inappropriate for study participation; gastrointestinal disease or surgical procedure that may impact absorption of the study drug; medical conditions prohibiting use of peg‐α‐2a or RBV, based on their respective product labels; or a history of hypersensitivity to compounds related to NS5A inhibitors. Exclusionary laboratory parameters included ALT level of 5 or more times the ULN; total bilirubin level of ≥ 2 mg/dL; international normalizsed ratio of ≥ 1.7; albumin level of ≤ 3.5 g/dL; haemoglobin level of ≤ 12 g/dL (for women) or ≤ 13 g/dL (for men); absolute neutrophil count of ≤ 1.5 109 cells/L (1.2 109 cells/L for black participants); platelet count of ≤ 90 109 cells/L; creatinine clearance of ≤ 50 mL/min; a fetoprotein level > 100 ng/mL; and corrected QT interval (QTcF) > 450 ms (for men) or > 470 ms (for women). Prohibited concomitant medications included inducers or strong or moderate inhibitors of CYP3A4; P‐glycoprotein substrates with a narrow therapeutic index; strong P‐glycoprotein inhibitors; nonstudy medications with known or potential anti‐ HCV activity; or any prescription or herbal product not prescribed for the treatment of a specific clinical condition. Doses of concomitant medications were required to be stable for 4 weeks or longer before the first dose of study drug. | |
| Interventions | Experimental group: oral 60 mg of daclatasvir for 12 or 16 weeks. Control group: placebo for 24 weeks. Co‐intervention: all participants received antiviral combination therapy with peg‐α‐2a 180 mg weekly, RBV 400 mg twice daily (800 mg/day). | |
| Outcomes | Virological response, safety assessment. | |
| Notes | NCT01257204 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | High risk | The participants were only blinded during treatment period |
| Blinding of outcome assessment (detection bias) | High risk | The study sponsor, who performed the analyses, were only blinded for the first 16 weeks |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | High risk | The trial changed the secondary outcomes |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Dore 2015a2 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | High risk | The participants were only blinded during treatment period |
| Blinding of outcome assessment (detection bias) | High risk | The study sponsor, who performed the analyses, were only blinded for the first 16 weeks |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | High risk | The trial changed the secondary outcomes |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Multicenter, randomised, open‐label, parallel‐group comparison trial (ClinicalTrials.gov number, NCT00996476) | |
| Participants | 93 were randomised to treatment groups, of whom 92 received at least 1 dose of the study drug Mean age: 54 years Sex: 43 men, 49 women Inclusion criteria: eligible participants were treatment‐naive, chronically infected with genotype 1 HCV, aged 20–70 years and had plasma levels of HCV RNA > 5.0 log 10 IU/mL at screening Exclusion criteria:
| |
| Interventions | Eligible participants were randomised to 1 of 5 treatment groups in a 2:1:2:1:1 ratio. | |
| Outcomes | Proportion of participants with undetectable plasma HCV RNA 24 weeks after the end of treatment (SVR24), with SAE, AEs, mortality | |
| Notes | According to predefined virologic stopping rules, participants in the simeprevir groups discontinued simeprevir and continued peg‐IFN α‐2a/RBV if viral breakthrough occurred during the first 24 weeks, and stopped all treatment if the decrease in plasma HCV RNA from baseline to week 12 was < 2 log10 IU/mL, or plasma HCV RNA level at week 24 was > = 1.2 log10 IU/mL. In this review SVR24 rates in the experimental group were analysed only from participants who did not continue treatment after 24 weeks. This is Group 1 vs control. We emailed Hayashi and colleagues on 21 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) | High risk | Unblinded study |
| Blinding of outcome assessment (detection bias) | High risk | Unblinded study |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants dropped out |
| Selective reporting (reporting bias) | Low risk | The outcomes stated in the protocol were reported on |
| Vested‐interest bias | High risk | Janssen Pharmaceutical KK |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see DRAGON 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) | High risk | Unblinded study |
| Blinding of outcome assessment (detection bias) | High risk | Unblinded study |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants dropped out |
| Selective reporting (reporting bias) | Low risk | The outcomes stated in the protocol were reported on |
| Vested‐interest bias | High risk | Janssen Pharmaceutical K.K |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see DRAGON 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) | High risk | Unblinded study |
| Blinding of outcome assessment (detection bias) | High risk | Unblinded study |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants dropped out |
| Selective reporting (reporting bias) | Low risk | The outcomes stated in the protocol were reported on |
| Vested‐interest bias | High risk | Janssen Pharmaceutical KK |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see DRAGON 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) | High risk | Unblinded study |
| Blinding of outcome assessment (detection bias) | High risk | Unblinded study |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants dropped out |
| Selective reporting (reporting bias) | Low risk | The outcomes stated in the protocol were reported on |
| Vested‐interest bias | High risk | Janssen Pharmaceutical KK |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 96 adult men Sex: 96 men Mean age: 44.6 years Country: Germany, Spain, and France Inclusion criteria: chronic HCV genotype 1 with minimal to mild liver fibrosis (Ishak score or Metavir grade < 2, confirmed by liver biopsy within the past 24 months) and HCV RNA viral load > 100.000 IU/mL at screening. No restriction was on the basis of prior IFN treatment experience. Exclusion criteria: laboratory measurements, HIV, HBV, any other additional cause for chronic liver disease, concurrent disease requiring treatment, any use of co‐medication, treatment with IFN and/or RBV within 6 months prior to screening and use of any investigational drug 30 days prior to screening or 5 periods of drug plasma half life. | |
| Interventions | Trial was divided into 8 cohorts and randomised in these cohorts. Control group: placebo. | |
| Outcomes | Antiviral response, pharmacokinetics, safety assessment. | |
| Notes | We emailed Erhardt and colleagues on 20 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The trial was funded by Boehringer‐Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Multicenter, randomised, double‐blind, placebo‐controlled, parallel‐design trial (SAPPHIRE‐I) (NCT01716585) | |
| Participants | 631 participants Location: USA, Europe and Australia Inclusion criteria: age 18‐70 years, chronic hepatitis C infection, genotype 1, HCV RNA level > 10,000 IU/mL, treatment‐naive, no evidence of liver cirrhosis, women had to be post‐menopausal for at least 2 years or surgically sterile or practicing specific forms of birth control. Exclusion criteria: hepatitis B or HIV co‐infection, positive screen for drugs or alcohol, significant sensitivity to any drug, use of contraindicated medications within 2 weeks of dosing, certain predefined abnormal laboratory tests. Group A: 473 participants Sex: 217 men, 256 women Mean age, years (range): 49.4(18.0‐70.0) Race, n(%): white: 428(90.5), black: 26(5.5), other: 19(4.0) Fibrosis score ≥ 2, n(%): 110(23.3), IL28B CC genotype, n(%): 144(30.4), HCV genotype, n(%): 1a: 322(68.1), 1b: 151(31.9) Group B: 158 participants Sex: 73 men, 85 women Mean age, years (range): 51.2(21.0‐70.0) Race, n(%): white: 144(91.1), black: 8(5.1), other: 6(3.8) Fibrosis score ≥ 2, n(%): 42(26.6), IL28B CC genotype, n(%): 50(31.6), HCV genotype, n(%): 1a: 105(66.5), 1b: 53(33.5). | |
| Interventions | Experimental group: ABT‐450 orally at once‐daily dose of 150 mg with ritonavir 100 mg once daily and ombitasvir orally 25 mg once daily for 12 weeks. Dasabuvir orally at a dose of 250 mg twice daily for 12 weeks. Control group: matching placebos for 12 weeks, followed by an open‐label period of 12 weeks' administration of the active treatment. Co‐interventions: weight‐based oral RBV 1000 to 1200 mg in 2 divided doses (1000 mg daily if body weight was < 75 kg, and 1200 mg daily if body weight was ≥ 75 kg). | |
| Outcomes | Primary outcomes: percentage of participants achieving SVR 12 weeks after treatment. Safety of ABT‐450/r/ombitasvir and dasabuvir co‐administered with RBV for 12 weeks. Secondary outcomes: percentage of participants achieving RVR. Percentage of participants achieving end of treatment response. Percentage of participants with ALT normalisation at end of treatment. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated schedules |
| Allocation concealment (selection bias) | Low risk | All participants assigned a unique participant number through the use of interactive response system in order to receive a unique study drug bottle/kit numbers and a unique randomisation number |
| Blinding of participants and personnel (performance bias) | Low risk | Matching placebos were used |
| Blinding of outcome assessment (detection bias) | Low risk | All data were blinded to participants, study personnel, and sponsor. An independent data and safety monitoring committee reviewed safety data. |
| Incomplete outcome data (attrition bias) | Low risk | Number and reasons for withdrawal and discontinuation were clearly stated. |
| Selective reporting (reporting bias) | Low risk | A protocol was published before randomisation and all pre‐specified outcomes were reported on. |
| Vested‐interest bias | High risk | The sponsor (AbbVie) was directly involved in trial design, data analysis, drafting the manuscript and publication. |
| Other bias | Low risk | Seems free of other potential sources of bias. |
| Methods | Randomised clinical trial | |
| Participants | 701 adult participants Sex: 442 men, 298 women (including genotype 5 participants) Mean age: 53.8 years (including genotype 5 participants) Inclusion criteria: chronic infection with HCV genotype 1, 2, 4, or 6, willing and able to provide written informed consent, HCV RNA ≥ 10^4 IU/mL at screening, classification as treatment‐naive or treatment‐experienced. Exclusion criteria: current or prior history of clinically‐significant illness (other than HCV) or any other major medical disorder that may interfere with treatment, assessment, or compliance with the protocol; individuals currently under evaluation for a potentially clinically‐significant illness (other than HCV) are also excluded, screening ECG with clinically significant abnormalities, laboratory results outside of acceptable ranges at screening, prior exposure to sofosbuvir or other nucleotide analogue HCV NS5B inhibitor or any HCV NS5A inhibitor, infection with HBV or HIV. | |
| Interventions | Experimental group: 400 mg of sofosbuvir and 100 mg of velpatasvir administered orally once daily for 12 weeks. Control group: placebo. | |
| Outcomes | SVR12, SAE, AE, viral resistance. | |
| Notes | Participants in the placebo group were eligible for deferred treatment with 12 weeks of sofosbuvir–velpatasvir. Genotype 5 participants were not eligible for randomisation. We contacted the trial authors on health‐related quality of life (HRQoL), allocation sequence generation, if they reported their SVR24 anywhere, at email [email protected]. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | An interactive web response system |
| Blinding of participants and personnel (performance bias) | Low risk | The trial was described as double‐blind (participant, caregiver, investigator, outcomes assessor), and the placebo was described in the protocol |
| Blinding of outcome assessment (detection bias) | Low risk | The trial was described as double‐blind (participant, caregiver, investigator, outcomes assessor), and the placebo was described in the protocol |
| Incomplete outcome data (attrition bias) | Low risk | Only 5 participants had missing data |
| Selective reporting (reporting bias) | High risk | The trial did not report SVR24 as stated as a secondary objective in the protocol (NCT02201940 and supplementary material at NEJM.org) |
| Vested‐interest bias | High risk | "The study was designed and conducted by the sponsor (Gilead Sciences) in collaboration with the principal investigators." |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised, open label, active‐control study | |
| Participants | 527 participants were randomised and 499 participants were treated Sex: 327 men, 172 women Mean age: 48 years Inclusion criteria: eligible participants were treatment‐naive adults aged 18 years or older with chronic hepatitis C genotype 2 or 3 infection and HCV RNA above 10,000 IU/mL. Participants with Childs A cirrhosis were included. Exclusion criteria: BMI < 18 kg/m2; positive HbS‐Ab, positive HbC‐Ag, positive immunoglobulin‐M antibody, positive anti‐HIV antibody, history of other liver disease, current evidence of psychiatric illness, immunologic disorder, haemoglobinopathy, pulmonary disease (including pneumonia or pneumonitis), cardiac disease, seizure disorder or anticonvulsant use, poorly controlled diabetes, cancer, or history of malignancy, clinical signs and symptoms of acute pancreatitis with elevated lipase, clinically significant ECG findings at screening, history of major organ transplantation with an existing functional graft, active substance abuse, history of uncontrolled thyroid disease, haemoglobin < 11 g/dL for women or < 12 g/dL for men; neutrophil count < 1500 cells/nL, serum creatinine > 1.5 times the ULN, ALT or AST ≥ 10 x ULN, albumin ≤ 3.2 g/dL, total bilirubin 1.5 x ULN (except participants with Gilbert's syndrome). | |
| Interventions | Experimental group 1: oral sofosbuvir 400 mg once daily for 12 weeks. Control group: peg‐IFN α‐2a subcutaneous once weekly 180 µg for 24 weeks. Co‐intervention: RBV 1000 mg/day (bodyweight < 75 kg) or 1200 mg/day (bodyweight ≥ 75 kg) for 12 or 24 weeks. | |
| Outcomes | Proportion of participants with undetctable HCV RNA‐level at week 2 and week 4 under treatment, with SVR12, with SAE, AEs, mortality. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Centralised system |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) | High risk | Unblinded |
| Blinding of outcome assessment (detection bias) | High risk | Unblinded |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how the trial handled participants with missing data |
| Selective reporting (reporting bias) | Low risk | All outcomes in the protocol were reported on |
| Vested‐interest bias | Unclear risk | The sponsor (Gilead) collected the data, monitored the conduct of the study, and performed the statistical analysis |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Parallel group, double‐blind, placebo‐controlled, randomised trial | |
| Participants | 201 participants (134 in experimental group, 67 in control group) Sex: 141 men (70%), 60 women (30%) Race: 20 black (10%), 181 non‐black (90%) Cirrhosis, n(%): 32(16%) Location: USA Inclusion criteria: chronic hepatitis C infection genotype 1 HCV RNA ≥ 10,000 IU/mL, demonstrated responsiveness to IFN (minimum duration of therapy 12 weeks); non‐response defined as a decrease in HCV RNA of at least 2 log10 IU/mL by week 12, but with detectable HCV RNA during the therapy period; relapse defined as an undetectable HCV RNA at end of treatment, but without subsequent attainment of SVR. A liver biopsy with histology consistent with chronic hepatitis C, age ≥ 18 years, weight between 40‐125 kg, signed informed consent, acceptable method of contraception for the participant and participant's partner(s) for at least 2 week before day 1 and continue until at least 6 months after treatment termination Exclusion criteria: hepatitis B infection, HIV infection, other causes of liver disease, decompensated liver disease, uncontrolled diabetes mellitus, a severe psychiatric disorder, active substance abuse, active or suspected malignancy, or a history of malignancy within last 5 years, pregnant or nursing women, severe AE during prior treatment, seizure disorder, cerebrovascular diseases, cardiovascular disease, autoimmune diseases, prior organ transplantation, haemoglobinopathies, coagulopathies, abnormal levels of serum bilirubin, albumin, and creatinine, haemoglobin < 120 g/L (women) and < 130 g/L (men), neutrophil count < 1500/mm3, platelet count < 100,000/mm3. | |
| Interventions | Experimental group: oral boceprevir 800 mg thrice daily for 44 weeks, beginning at week 5. Control group: placebo for 44 weeks, beginning at week 5. Co‐interventions: peg‐IFNα‐2a 180 μg subcutaneously once weekly and oral weight‐base RBV 1000 to 1200 mg daily in divided doses for 48 weeks. | |
| Outcomes | Primary outcome: SVR 24 weeks post‐therapy. | |
| Notes | We emailed Flamm and colleagues on 20 April 2016 for additional information (on: random sequence generation; method of allocation concealment; description of blinding procedure; blinding of outcome assessors; potential number and reasons for dropouts; pre‐defined outcomes; sponsorship and its role; type of SAE) but reply not received yet. ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated that trial was randomised, but the method of sequence generation was not described |
| Allocation concealment (selection bias) | Low risk | The trial used an interactive voice‐response system. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Trial defined as double‐blind and placebo was used in the control group. However, method of blinding was not adequately described. |
| Blinding of outcome assessment (detection bias) | Unclear risk | It was not mentioned if the outcome assessors were blinded |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | High risk | The trial authors changed their primary outcomes according to the protocol (NCT00845065) |
| Vested‐interest bias | High risk | The trial was funded by Schering‐Plough |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias. |
| Methods | Randomised clinical trial | |
| Participants | 20 participants. Sex: 12 men, 8 women Mean age: 45.5 years Inclusion criteria: men and women of non‐childbearing potential aged between 18 and 60 years. Participants satisfied the following criteria for inclusion in the study: genotype 1 chronic hepatitis C; had not received any prior therapy for hepatitis C, including approved treatments or participation in studies of investigational treatments; HCV RNA level > 1 × 105 IU/mL ALT concentration < 4.0 times the ULN, no clinically significant deviations from the normal range for haematology or clinical chemistry values; willing to refrain from the concomitant use of herbal dietary supplements or vitamins during the study drug‐dosing period; and willing to initiate standard‐of‐care treatment (peg‐IFNα and RBV) at the conclusion of the study drug‐dosing period. Exclusion criteria: contraindications to peg‐IFNα‐2a or RBV; decompensated liver disease; alcohol‐related cirrhosis or primary biliary cirrhosis; positive screening for hepatitis B surface antigen or HIV co‐infection; donation of blood (500 mL) within 60 days before the first dose of study drug; concurrent antiviral therapy (except for antiviral agents approved for treatment of herpes viruses) within 3 months preceding study entry; regular treatment with nontopical medications or with topical medications with known systemic absorption within 4 weeks before study drug administration (with the exception of oestrogen replacement therapy for women); regular consumption of more than 24 units of alcoholic drinks per week or more than 8 cups of coffee per day; history of drug abuse within 6 months of study entry; history of methadone use within 3 months of study entry; positive urine screen for drugs of abuse; participation in an investigational drug study within 90 days before study drug administration or participation in more than 2 drug studies in the last 12 months (excluding the present study); or participation in a prior clinical study of telaprevir unless it was documented that the participant had been randomised to placebo treatment. Participants were also excluded if they had a history of any illness that, in the opinion of the investigator or the participant’s general practitioner, may have confounded the results of the study or posed an additional risk in administering study drug to the participant. This included but was not limited to a history of relevant drug or food allergies; cardiovascular or central nervous system disease; clinically significant illness; or mental illness that may have affected compliance with study requirements. | |
| Interventions | Experimental group: telaprevir was given as 750 mg oral doses every 8 h. Telaprevir alone every 8 h orally for 14 days (8 participants); or telaprevir every 8 h orally for 14 Control group: placebo every 8 h orally for 14 days and peg‐IFNα‐2a via subcutaneous injection once weekly for 2 weeks (4 participants) Co‐intervention: peg‐IFNα‐2a was given as weekly 180 mg subcutaneous injections After completing study drug dosing, participants were offered the opportunity to begin standard therapy for chronic hepatitis C (180 g/week peg‐IFNα‐2a and 1000 or 1200 mg/day RBV, depending on body weight | |
| Outcomes | Safety assessment, pharmacokinetic assessment, viral assessments | |
| Notes | We contacted trial authors for additional information on allocation concealment, blinding of participants and personal, blinding of outcome assessment, SVR data protocol | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computerised random‐number generator |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The study was placebo‐controlled for telaprevir; peg‐IFNα‐2a treatment was open‐label. Investigators and participants were blinded to HCV RNA |
| Blinding of outcome assessment (detection bias) | Unclear risk | It was unclear how (and if there was any blinding at all) the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol was found |
| Vested‐interest bias | High risk | Supported by Vertex Pharmaceuticals Incorporated |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 50 participants Sex: 40 men, 10 women Mean age: 48 years Inclusion criteria: men and women between 18 and 65 years of age with a history of chronic HCV genotype 1 infection and detectable plasma HCV RNA (> 1 104 IU/mL) at the study screening visit. Additional enrolment criteria included a BMI between 18 and 30, minimum body weight of 45 kg, and a liver biopsy or non‐invasive procedure (liver scan) within the previous 2 years showing no evidence of cirrhosis. In addition, participants in Part A were required to have no history of prior therapy with IFN‐based regimens; participants in Part B were required to have had failed previous IFN‐alfa and RBV‐based therapy as defined above. Exclusion criteria: participants were excluded from the study if they met any of the following criteria: decompensated liver disease; impaired liver function; clinical or histopathologic evidence of cirrhosis; history of non‐hepatitis C chronic liver disease; positive screening for hepatitis B surface antigen or HIV infection; history of active malignancy within the preceding 5 years; history of clinically significant cardiovascular or cerebrovascular disease; treatment with peg‐IFN‐α and RBV (Part A) or treatment with peg‐IFN‐α and RBV within 3 months before screening (Part B); treatment with growth factors within 3 months before screening; history of drug abuse within the previous year; regular consumption of more than 1 glass of alcohol per day for women or 2 glasses of alcohol per day for men; participation in an investigational drug study within 3 months of screening or any prior participation in a study of an experimental HCV therapy; and selected laboratory abnormalities, including serum ALT > 5 times the upper limit of the reference range, creatinine clearance < 30 mL/min, or total bilirubin P26 lmol/L. Pregnant or lactating women, women of childbearing potential, and male partners of pregnant or lactating women were excluded from enrolment. Additionally, anyone who, in the opinion of the investigator, was not a suitable candidate for enrolment or was unlikely to comply with the requirements of the study was also excluded from enrolment. | |
| Interventions | Experimental group: Group 1: danoprevir was administered orally in soft gelatin capsule form in total daily doses of 200, 300, 400 and 600 mg in treatment‐naive participants. Group 2: a single dose level of danoprevir (600 mg daily) was explored in a cohort of non‐responders (NR) Control intervention: placebo | |
| Outcomes | Safety assessments, pharmacokinetics, viral kinetics | |
| Notes | 4 cohorts of 10 participants each were randomised (8:2) to treatment with danoprevir or placebo equivalent. In Part A, treatment‐naive (Cohorts 1–5) were permitted but not required to begin standard of care (SOC) treatment with peg‐IFN‐α/RBV anytime after 24 h following the last dose of the study drug. 3 treatment‐naive participants in the 200 mg every‐12‐h cohort who were mis‐dosed at a single study site were excluded from the efficacy analysis. We sent an email was sent to Forestier and colleagues on 20 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised using an interactive voice‐response system that assigned a participant identification number that corresponded to treatment assignment (danoprevir or placebo) according to the randomisation code. |
| Allocation concealment (selection bias) | Low risk | Participants were randomised using an interactive voice‐response system that assigned a participant identification number that corresponded to treatment assignment (danoprevir or placebo) according to the randomisation code. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as double‐blinded, but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | 3 participants excluded. Clearly stated reason |
| Selective reporting (reporting bias) | Unclear risk | No protocol was found |
| Vested‐interest bias | High risk | The study was sponsored by InterMune, Inc. and Roche |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Forestier 2011a2 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised using an interactive voice‐response system that assigned a participant identification number that corresponded to treatment assignment (danoprevir or placebo) according to the randomisation code. |
| Allocation concealment (selection bias) | Low risk | Participants were randomised using an interactive voice‐response system that assigned a participant identification number that corresponded to treatment assignment (danoprevir or placebo) according to the randomisation code. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as double‐blinded, but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | 3 participants excluded. Clearly stated reason |
| Selective reporting (reporting bias) | Unclear risk | No protocol was found |
| Vested‐interest bias | High risk | The study was sponsored by InterMune, Inc. and Roche |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 59 participants Sex: 46 men, 13 women Mean age: 45.8 years Inclusion criteria: genotype 1 chronic HCV infection with detectable plasma HCV RNA levels (> 1 x 104 IU/mL), no previous treatment for HCV infection, an age of 18–65 years, a BMI (defined as the weight in kilograms divided by the square of the height in meters) of 18–30, and no evidence of cirrhosis during the previous 2 years in a liver biopsy or noninvasive procedure (e.g. elastography). Exclusion criteria: decompensated liver disease; impaired liver function; clinical or histopathologic evidence of cirrhosis; history of non–hepatitis C chronic liver disease; screening positive for hepatitis B surface antigen or HIV infection; history of active malignancy during the preceding 5 years; history of clinically significant cardiovascular or cerebrovascular disease; previous treatment with peg‐IFN‐a and RBV; treatment with growth factors within 3 months before screening; history of drug use within the previous year; regular consumption of > 1 glass of alcohol per day for women or > 2 glasses of alcohol per day for men; participation in an investigational drug study within 3 months before screening or any prior participation in a study of an experimental HCV therapy; and selected laboratory abnormalities, including ALT level .5 times the upper limit of the reference range, creatinine clearance < 30 mL/min, or total bilirubin level >26 mmol/L. Pregnant or lactating women, women of childbearing potential, and male partners of pregnant or lactating women were excluded from enrolment. In addition, anyone who, in the opinion of the investigator, was not a suitable candidate for enrolment or was unlikely to comply with the requirements of the study was also excluded from enrolment. | |
| Interventions | Experimental group: danoprevir was administered orally in soft gelatin capsule form in the following dose regimens: 100 mg 3 times daily, 200 mg 3 times daily, 300 mg 3 times daily, 400 mg twice daily, 600 mg twice daily, and 900 mg twice daily. The 5 lowest dose cohorts consisted of 10 participants randomised (8:2) to receive treatment with danoprevir or placebo equivalent. The highest dose cohort consisted of 9 participants randomised (7:2) to receive treatment with danoprevir or placebo equivalent. 6 dose cohorts (400 mg, 600 mg, and 900 mg twice daily and 100 mg, 200 mg, and 300 mg 3 times daily). Participants also received peg‐IFN a‐2a (180 lg once weekly) and RBV (1000–1200 mg/day) on day 0 and 15. Control group: placebo plus peg‐IFN a‐2a (180 lg once weekly) and RBV (1000–1200 mg/day) Co‐intervention: peg‐IFN‐a 2 a(180 lg once weekly) and RBV (1000–1200 mg/day) | |
| Outcomes | Safety assessments and viral kinetics | |
| Notes | We sent an email to Forestier and colleagues on 20 April 2016 for additional information (missing blinding during assessment of allocation concealment, missing SVR and mortality data ‐ is it investigated) but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomised using an interactive voice‐response system, which assigned a participant identification number corresponding with treatment assignment (danoprevir or placebo), according to the randomisation code. |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | One participant withdrew because of a family emergency after 1 dose of study drug, and 1 participant withdrew because of poor venous access after 4 doses of study drug. A third participant (administered 100 mg 3 times daily) missed 6 danoprevir doses during days 12–14 but was included in efficacy analyses, because 0.90% of danoprevir doses were administered |
| Selective reporting (reporting bias) | Unclear risk | No protocol was found |
| Vested‐interest bias | High risk | This study was supported by InterMune and Roche. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised, multicenter, double‐blind, parallel‐group, placebo‐controlled, phase III clinical trial (PROMISE)(NCT01281839) | |
| Participants | 393 participants (260 in experimental group and 133 in control group) Sex: 258 men, 135 women Mean age: 52 years (range 20‐70 years) Location: Europe, North America, Australia, and New Zealand. Inclusion criteria: age ≥ 18 years. Confirmed chronic genotype 1 HCV infection. Screening plasma HCV RNA levels > 10,000 IU/mL. Treatment‐experienced participants who had relapsed after 24 weeks or more of IFN‐based therapy (undetectable HCV RNA at end of treatment or within 2 months after end of treatment, with documented relapse within 1 year after therapy). A liver biopsy specimen obtained within 3 years of screening showing histology consistent with chronic HCV infection (participants with bridging fibrosis (F3) or cirrhosis (F4) were eligible if they had an ultrasound performed within 6 months before screening (or between the screening and baseline visit) with no findings suspicious for HCC) Exclusion criteria: hepatic decompensation. Non–HCV‐related liver disease. HBV, HIV, or non‐genotype 1 HCV co‐infection. Defined laboratory abnormalities: platelets < 90,000/mm3, white blood cell count < 3000/μL, haemoglobin level < 12 g/dL for women and < 13 g/dL for men, creatinine level > 1.5 mg/dL, ALT and/or AST level > 10 times the upper limit and normal, total serum bilirubin level 1.5 times or more the ULN, and α‐fetoprotein level > 50 ng/mL in participants with cirrhosis. Any other active disease. Pregnant women or planning pregnancy were excluded | |
| Interventions | Experimental group: oral simeprevir 150 mg once daily for 12 weeks Control group: placebo for 12 weeks Co‐interventions: Experimental group: peg‐IFN α‐2a 180 μg subcutaneously once weekly for 24 weeks (if HCV RNA < 25 IU/mL at week 4 and undetectable at week 12) or 48 weeks if not meeting these criteria. Oral weight‐based RBV 1000 to 1200 mg daily for 24 weeks (if HCV RNA < 25 IU/mL at week 4 and undetectable at week 12) or 48 weeks if not meeting these criteria Control group: peg‐IFN α‐2a 180 μg subcutaneously once weekly for 48 weeks. Oral weight‐based RBV 1000 to 1200 mg daily for 48 weeks | |
| Outcomes | Primary outcome: proportion of participants achieving SVR 12 weeks after planned end of treatment (SVR12) Secondary outcomes: comparison of other virologic response rates at other time points. Rate of RVR. Proportion of simeprevir‐treated participants meeting response‐guided treatment criteria to complete treatment at week 24. Incidence of viral breakthrough. Incidence of on‐treatment failure. Incidence of viral relapse. Incidence of AEs. Laboratory abnormalities. Quality‐of‐life measures. | |
| Notes | We sent an email to Forns and colleagues on 20 April 2016 for the following additional information. Reply received on 27 April 2016 with data on baseline number of participants with elevated AST/ALT and randomisation details. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information given |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information given |
| Blinding of participants and personnel (performance bias) | Low risk | Stated that "participants, study personnel, and the sponsor were blinded to the treatment assignments" |
| Blinding of outcome assessment (detection bias) | Low risk | Stated that "participants, study personnel, and the sponsor were blinded to the treatment assignments" |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "The proportion of patients who discontinued simeprevir/placebo intake early was 3.5% and 72.2% in the simeprevir/PR and placebo/PR groups, respectively. The main reason for discontinuation was meeting the week 4 virologic stopping rule for simeprevir or placebo in both arms, with a large proportion of patients in the placebo group (69.9%) stopping placebo at week 4. The proportion of patients who completed PR treatment was 93.5% in the simeprevir/PR group (24 or 48 weeks) and 72.2% in the placebo/PR group (48 weeks)" |
| Selective reporting (reporting bias) | Low risk | A protocol was published before randomisation. Outcomes specified in the protocol are similar, but not completely equal to the ones stated in the article. Not all outcomes stated in the protocol were reported in the article, but results of all outcomes were reported and available on www.ClinicalTrials.gov. |
| Vested‐interest bias | High risk | Trial sponsored by Janssen |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias. |
| Methods | Multicenter randomised clinical trial | |
| Participants | 52 participants Sex: 35 men, 17 women Mean age: 44 years Countries: France, UK, Italy, and Sweden Inclusion criteria: 18–65 years; chronic infection with either genotype 2 or genotype 3 HCV (serum HCV RNA > 10,000 IU/mL); absolute neutrophil count > 1500 mm3 and platelet count > 100,000 mm3; no prior treatment for HCV Exclusion criteria: relevant concomitant medical condition; decompensated liver disease or cirrhosis, or other significant liver disease; HIV or HBV co‐infection; peg‐IFN or RBV contraindication; a history of alcohol or illicit drug use; pregnancy/breast feeding | |
| Interventions | The participants were randomised according to genotype 2 and 3 Experimental group 1: oral 750 mg telaprevir every 8th hour for 2 weeks Experimental group 2: oral 750 mg telaprevir every 8th hour + peg‐IFN‐α‐2a 180 µg once weekly plus RBV 400 mg twice daily for 2 weeks Control group: telaprevir placebo (every 8 h) plus peg‐IFN‐α‐2a 180 µg once weekly plus RBV 400 mg twice daily for 2 weeks Co‐intervention: The peg‐IFN‐α‐2a and RBV were a co‐intervention between control group and experimental group 2 during treatment period, and all participants received peg‐IFN‐α‐2a 180 g once weekly plus RBV 400 mg twice daily for 24 weeks after treatment. | |
| Outcomes | Viral kinetics, efficacy and safety assessment | |
| Notes | We emailed Foster and colleagues on 21 April 2016 for additional information (randomisation, blinding, death, missing data) but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described (central randomisation system) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | The monotherapy group was not blinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% percent dropped out (7 participants) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed |
| Vested‐interest bias | Unclear risk | The trial was funded by Janssen Pharmaceuticals and Vertex Pharmaceuticals) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Foster 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described (central randomisation system) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | The monotherapy group was not blinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% percent dropped out (7 participants) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed |
| Vested‐interest bias | High risk | The trial was funded by Janssen Pharmaceuticals and Vertex Pharmaceuticals) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 558 adult participants Sex: 374 men, 178 women Mean age: 49.5 years Inclusion criteria: chronic hepatitis C genotype 3 who were treatment‐naive or treatment‐experienced, and were required to have liver imaging within 6 months of baseline/Day 1; adults with cirrhosis to exclude HCC , women of childbearing potential (as defined in Appendix 4 must have a negative serum pregnancy test at screening and a negative urine pregnancy test on Baseline/Day 1 prior to randomisation, male participants and female participants of childbearing potential who engage in heterosexual intercourse had to agree to use protocol‐specified method(s) of contraception, lactating women had to agree to discontinue nursing before the study drug was administered, participant had to be of generally good health, with the exception of chronic HCV infection, as determined by the Investigator, participant had to be able to comply with the dosing instructions for study drug administration and able to complete the study schedule of assessments Exclusion criteria: current or prior history of clinically‐significant illness (other than HCV that may interfere with treatment, assessment or compliance with the protocol, screening ECG with clinically significant abnormalities, laboratory results outside of acceptable ranges at screening, pregnant or nursing female or male with pregnant female partner, chronic liver disease of a non‐HCV aetiology (e.g. haemochromatosis, Wilson's disease, alfa‐1 antitrypsin deficiency, cholangitis), infection with HBV or HIV | |
| Interventions | Experimental group: 100 mg of velpatasvir once a day and 400 mg of sofosbuvir once a day for 12 weeks Control group: 400 mg of sofosbuvir plus RBV 1000 or 1200 mg (weight‐based) both for 24 weeks | |
| Outcomes | SVR12, SAE, death, viral resistance | |
| Notes | We could only use data reported at 12 weeks meaning no data were available. We contacted the trial authors for additional information on allocation sequence generation, how many had incomplete outcome data at 12 weeks, SAE, death, health‐related quality of life) at 12 weeks at [email protected] on 21 April 2016 but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | An Interactive Web Response System was used |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as open‐label |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as open‐label |
| Incomplete outcome data (attrition bias) | Unclear risk | There were above 5% dropouts and it was unclear how the trial handled missing participants. It was unclear how many dropouts there were at 12 weeks. |
| Selective reporting (reporting bias) | Unclear risk | SVR 24 was not reported as described in the prepublished protocol NCT02201953 and supplementary material at NEJM.org |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result (Gilead Sciences) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Foster 2015a2 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | An Interactive Web Response System was used |
| Blinding of participants and personnel (performance bias) | High risk | Described as open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Described as open‐label |
| Incomplete outcome data (attrition bias) | Low risk | Only 5 participants dropped out |
| Selective reporting (reporting bias) | High risk | SVR 24 was not reported as described in the prepublished protocol (NCT02220998 and supplementary material at NEJM.org) |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result (Gilead Sciences) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Phase IIb, double‐blind, placebo‐controlled, parallel‐group trial (PILLAR)(NCT00882908) | |
| Participants | 386 participants Sex: 213 men, 173 women Location: 13 countries in North America, Europe, and Asia‐Pacific regions Inclusion criteria: adult participants with chronic hepatitis C, plasma HCV RNA > 100,000 IU/mL, genotype 1, treatment‐naive, eligible to be treated with peg‐IFN‐based regimens according to standard criteria Exclusion criteria: cirrhosis on liver biopsy (required within 24 months of enrolment), HIV or HBV co‐infection, platelet count < 90,000/mm3, haemoglobin< 12 g/dL for women and 13 g/dL for men Group 1: 78 participants Sex: 40 men (51.3%), 38 women (48.7%) Median age: 47 years (range 19‐66) Group 2: 75 participants Sex: 47 men(62,7%), 28 women (37.3%) Median age: 46 years (range 18‐67) Group 3: 77 participants Sex: 43 men (55.8%), 34 women (44.2%) Median age: 47 years (range 18‐69) Group 4: 79 participants Sex: 44 men (55.7%), 35 women (44.3%) Median age: 47 years (range 19‐69) Group 5: 77 participants Sex: 39 men (50.6%), 38 women (49.4%) Median age: 45 years (range 21‐67). | |
| Interventions | Experimental group: Group 1: oral simeprevir 75 mg once daily for 12 weeks, followed by placebo for 12 weeks. Group 2: oral simeprevir 75 mg once daily for 24 weeks. Group 3: oral simeprevir 150 mg once daily for 12 weeks, followed by placebo for 12 weeks. Group 4: oral simeprevir 75 mg once daily for 24 weeks. Control group: Group 5: matched placebo for 24 weeks. Co‐intervention for all groups: peg‐IFN‐α‐2a 180 μg subcutaneously once weekly. Oral RBV 1000‐1200 mg daily. | |
| Outcomes | Primary outcome: proportion of participants with HCV RNA< 25 IU/mL undetectable at week 72 (SVR W72). Secondary outcome: SVR at 12 and 24 weeks after planned end of treatment (SVR12 and SVR24, respectively). Adverse events. Quality‐of‐life measures. Assessment of HCV‐NS3 sequence in participants not achieving SVR. Assessment of simeprevir pharmacokinetics. The influence of interleukin‐28 (IL28)B genotype on efficacy was explored in a subset of participants for whom genomic DNA was available. Influence of IL28B genotype on treatment efficacy. | |
| Notes | We emailed Fried and colleagues on 21 April 2016 for additional information (baseline number of participants with elevated AST/ALT and method of sequence generation but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method of sequence generation was not described |
| Allocation concealment (selection bias) | Low risk | Participants were randomly assigned in equal proportions, using a centralised, interactive voice/web response randomisation system |
| Blinding of participants and personnel (performance bias) | Low risk | Stated that "participants and personnel were blinded to the experimental intervention. A simeprevir‐matched placebo was used." |
| Blinding of outcome assessment (detection bias) | Low risk | Stated as blinded. An external physician monitored individual HCV RNA results and informed investigators regarding protocol‐directed treatment discontinuation |
| Incomplete outcome data (attrition bias) | Low risk | Withdrawals reported with reasons given. Treatment discontinuation rate 7.5% |
| Selective reporting (reporting bias) | Unclear risk | A protocol was published before randomisation began and all outcome results were reported adequately (NCT00882908) |
| Vested‐interest bias | Unclear risk | This study was funded by Janssen Research & Development, LLC. Editorial support was provided by Dr Bethan Hahn, on behalf of Complete Medical Communications, funded by Janssen Research & Development, LLC. |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias |
| Methods | Prospective, double‐blind, multinational, randomised, placebo controlled phase II trial (CDEB025A2210; ClinicalTrials.gov NCT01183169) conducted between 30 August 2010 and 9 May 2013 | |
| Participants | 459 eligible participants Sex: 278 men, 181 women Mean age: 50.6 years Countries: Europe, North America, Asia‐Pacific region Inclusion criteria: 9‐69 years with chronic hepatitis C genotype 1 infection and HCV RNA > = 1000 IU/mL and had failed to respond to or had relapse after prior P/R therapy; all participants had to have a liver biopsy within 3 years or transient elastography within 6 months of enrolment. Participants with compensated cirrhosis were eligible. Exclusion criteria: nongenotype 1 infection, presence or history of hepatic decompensation and haematological abnormalities, and recent treatment with any anti‐HCV drug, concomitant treatment with known substrates or inhibitors of cytochrome P450 3A, P‐gp, OATPs, MRP2 or BSEP was not permitted within 2 weeks of study entry. 459 participants randomised, 77% white, 25% compensated cirrhosis/transition to cirrhosis, 57% prior P/R‐non responders, 79% genotype IL28B 457 treated. | |
| Interventions | Participants were randomised (1:1:1:1) Experimental group 1: alisporivir 600 mg once a day for 48 weeks. Experimental group 2: alisporivir 800 mg once a day for 48 weeks. Experimental group 3: alisporivir 400 mg twice a day for 48 weeks. Control group: placebo for 48 weeks. Co‐intervention: peg‐IFN‐α‐2a 180 lg/week plus RBV 1000 or 1200 mg/day based on body weight for 48 weeks. | |
| Outcomes | eEVR (weeks 12 on treatment), SVR12, SVR24, all‐cause mortality, AEs. | |
| Notes | Following a partial clinical hold imposed by FDA, alisporivir/placebo was discontinued in all participants; at that time, all active participants had received at least 31 weeks of triple therapy out of a total of 48 weeks. Analysis group 1 vs control. In the placebo arm, 57% of participants were switched in a blinded manner to alisporivir plus P/R after Week 16 due to failure to achieve the efficacy criterion (HCV RNA < limit of quantification) at Week 12. We could therefore not use the results from this trial. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) | Low risk | There was only missing data for 2 participants |
| Selective reporting (reporting bias) | High risk | The secondary outcomes were changed from the original secondary outcomes |
| Vested‐interest bias | High risk | The study was funded by Novartis |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Fundamental 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) | Low risk | There was only missing data for 2 participants |
| Selective reporting (reporting bias) | High risk | The secondary outcomes were changed from the original secondary outcomes |
| Vested‐interest bias | High risk | The study was funded by Novartis. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Fundamental 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not reported |
| Incomplete outcome data (attrition bias) | Low risk | There was only missing data for 2 participants |
| Selective reporting (reporting bias) | High risk | The secondary outcomes were changed from the original secondary outcomes |
| Vested‐interest bias | High risk | The study was funded by Novartis |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 25 adult participants Country: New Zealand Inclusion criteria: Non responders for RBV and IFN, infected with genotype 2 or 3. All participants were non‐cirrhotics, and treated with at least 12 weeks of IFN prior to randomisation. | |
| Interventions | Experimental group: 1500 mg R7128 twice daily for 28 days. Control group: placebo twice daily for 28 days. Co‐intervention: 180μg peg‐IFN and 1000‐1200mg RBV. | |
| Outcomes | HCV RNA, SAE, AEs | |
| Notes | We emailed Gane and colleagues on 21 April 2016 for additional information regarding randomisation, blinding, missing data, death, additional data, separate data from Genotype 2 and 3. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | There were missing data from 7 participants (above 5%) |
| Selective reporting (reporting bias) | Unclear risk | A clinicalTrials.gov number was found, but it was unclear which outcome was supposed to be assessed in each part of the trial |
| Vested‐interest bias | High risk | The main author was consulting in pharmaceutical companies |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 71 participants Sex: 54 men, 17 women Mean age: 47.6 years Inclusion criteria: treatment‐naive and treatment‐experienced adults aged 18–65 years, who were chronically infected with HCV genotype 1 but did not have cirrhosis, and who had a minimum HCV RNA of 10⁵ IU/mL. Participants were required to have normal renal and hepatic function and no clinically significant comorbidities. Exclusion criteria: co‐infection with hepatitis B or HIV, concurrent medical or psychiatric disorder (or history of such), history of any neoplastic disease, history of clinically significant cardiovascular or cerebrovascular disease, use of growth factors, or anticipated use or need for significant concomitant medical treatment. | |
| Interventions | Experimental group: Arm B: 500 mg RG7128 twice daily and 100 mg danoprevir every 8 h (treatment‐naive) Arm C1: 500 mg RG7128 twice daily and 200 mg danoprevir every 8 h (treatment‐naive) Arm C2 1000 mg RG7128 twice daily and 100 mg danoprevir every 8 h (treatment‐naive) Arm D: 1000 mg RG7128 twice daily and 200 mg danoprevir every 8 h (treatment‐naive) Arm E: 1000 mg RG7128 twice daily and 600 mg danoprevir twice a day (non‐null responders) Arm F: 1000 mg RG7128 twice daily and 900 mg danoprevir twice a day (null responders) Arm G: 1000 mg RG7128 twice daily and 900 mg danoprevir twice a day (treatment‐naive) Control group: placebo RG7128 and Placebo Danoprevir Co‐intervention: standard of care treatment (180 μg/week peg‐IFN α‐2a, and RBV at 1000 mg/day for participants weighing < 75 kg or 1200 mg/day for those weighing ≥ 75 kg) | |
| Outcomes | Safety, pharmacokinetics, antiviral activity | |
| Notes | We emailed Gane and colleagues on 06 June 2016 for additional information on SVR24 but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The random allocation sequence was computer‐generated |
| Allocation concealment (selection bias) | Low risk | Randomly assigned by interactive voice or web response system to active treatment or placebo |
| Blinding of participants and personnel (performance bias) | High risk | Investigators, personnel at the study centre, and participants were masked to treatment allocation. Study drugs and placebo were identical in colour, size, shape, and taste but "(...) apart from patients in cohort F, who were unmasked after the last assessment was completed" |
| Blinding of outcome assessment (detection bias) | High risk | The pharmacist who prepared the doses, personnel involved in pharmacokinetic sample analyses, statisticians who prepared data summaries, and the clinical pharmacologists who reviewed the data before deciding to initiate dosing in the next cohort were not masked to treatment allocation |
| Incomplete outcome data (attrition bias) | Low risk | There was under 5% dropouts (only 2 dropouts) |
| Selective reporting (reporting bias) | Low risk | The outcomes stated in the protocol were reported on (NCT00801255) |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result (Hoffmann‐La Roche) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 30 adult participants Sex: 21 men, 9 women Mean age: 44.5 years Countries: New Zealand, France, Poland Inclusion criteria: 18‐65 years and with chronic treatment‐naive hepatitis C genotype 1 infection, an HCV RNA level > x105 IU/mL, a BMI between 18 and 35 kg/m2 and without evidence of liver cirrhosis on a liver biopsy or non‐invasive procedure (e.g. Fibroscan) obtained within the preceding 24 months were eligible for the trial. Exclusion criteria: decompensated liver disease; impaired liver function (indicated by a history of ascites, hepatic encephalopathy, HCC or bleeding oesophageal varices); chronic liver disease attributed to a cause other than HCV; or serological evidence of HBV or HIV infection. Increased risk of anaemia; a clinically significant medical condition such as cardiovascular or cerebrovascular disease, chronic pulmonary disease, poorly controlled thyroid function, diabetes mellitus requiring medication, ophthalmic disorders related to diabetes or hypertension, or diseases associated with alterations in immune function; or a history of clinically significant psychiatric disease, a history of excessive alcohol consumption (defined as more than 2 standard drinks per day within the previous 3 months), or a history of drug abuse within the last year, pregnant and lactating women and male partners of pregnant women, any recent use or anticipated need for drugs, herbal preparations or nutrients known to inhibit or induce CYP enzymes, or were substrates of CYP3A or CYP2C9 with a narrow therapeutic index (including oral contraceptives, steroids, antacids, H‐2 blockers or proton‐pump inhibitors). Systemic immunosuppressive drugs, cytotoxic or chemotherapeutic agents, radiation therapy, oral or inhaled corticosteroids, or topical class 1 and 2 steroids. ALT level > 5 times the ULN, creatinine clearance < 50 mL/min, haemoglobin < 120 g/L (if female) or < 130 g/L (if male), an absolute neutrophil count < 1.5 109/L, platelet count < 100 x 109/L, or serum albumin level < 35 g/L | |
| Interventions | The study consisted of 3 cohorts. The randomisation was within each cohort. Experimental group: participants received 100 mg oral danoprevir twice a day, 200 mg oral danoprevir once a day, or 200 mg oral danoprevir twice a day for 15 days. Control group: placebo in same numbers as above. Co‐intervention: both groups received equal amounts of ritonavir (100 mg) pr pill. subcutaneous peg‐IFN α‐2a (40KD) (Pegasys, Roche, Basel, Switzerland) 180 μg once weekly plus oral RBV 1000 mg/day (bodyweight < 75 kg) or 1200 mg/day (bodyweight > 75 kg). After the 15 days, both groups received peg‐IFN α‐2a (40KD) plus RBV for a total of 48 weeks. | |
| Outcomes | Pharmacokinetic parameters (plasma concentration, AUC), HCV RNA level, safety assessment (laboratory test, AEs). | |
| Notes | We emailed Gane and colleagues on 06 June 2016 for additional information on blinding, other outcomes, protocol but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computerised randomisation |
| Allocation concealment (selection bias) | Low risk | Randomisation was managed through a centralised interactive voice and web response system through a 3rd party |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "partially" double‐blinded but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "partially" double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | There were no dropouts |
| Selective reporting (reporting bias) | High risk | The protocol stated "Virological response in prior null‐responders" as a secondary outcome. This outcome was not assessed in any study |
| Vested‐interest bias | High risk | The trial was sponsored by F. Hoffmann‐La Roche Ltd |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 30 adults with chronic hepatitis C infection Sex: 17 men, 13 women Mean age: 45 years Countries: New Zealand and USA | |
| Interventions | Experimental group 1: 12 participants randomised to 50 mg ACH‐3102 (odalasavir) and 400 mg sofosbuvir once a day for 8 weeks Experimental group 2: 6 participants randomised to 50 mg ACH‐3102 (odalasavir) and 400 mg sofosbuvir once a day for 6 weeks Control group 2: 6 participants randomised to observation for 6 weeks. | |
| Outcomes | SVR, SAE. | |
| Notes | Abstract only. After 4 weeks of treatment, group 1 (both experimental and control group) were merged, and received active treatment, therefore data can not be used after week 4. We emailed Gane and colleagues on 21 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | "Observation group" not placebo controlled trial |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | Sponsored by Achillion Pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 16 participants Sex: 14 men, 1 women (analysed) Mean age: 53 years Countries: USA and Puerto Rico Inclusion criteria: treatment‐naive participants men and women 18‐70 years of age with HCV genotype 1 or 4 infection for at least 6 months and HCV RNA ≥ 1,00,000 IU/mL at screening. Eligible participants had no evidence of cirrhosis documented by liver biopsy within 3 years. Fertile men or women were required to use 2 forms of effective contraception between them and their partner during treatment and for 24 weeks afterwards Exclusion criteria: co‐infection with hepatitis B, HIV, clinically significant chronic liver disease, conditions consistent with decompensated liver disease, drug or alcohol abuse, significant ECG findings, history of suicide attempt, major depression or current severe or poorly controlled psychiatric disorder. Abnormal haematological and biochemical parameters that excluded participation were: Neutrophil count (< 1500 cells/mm3 ((or < 1250 cells/mm3 for African American/Black participants)); haemoglobin (< 11 g/dL in women or 12 g/dL in men); creatinine > 1.5 x ULN (ULN); ALT, AST, or alkaline phosphatase > 5 x ULN; total bilirubin > 2.0 x ULN ((except in participants with Gilbert's) syndrome; albumin < 3.0 g/dL and platelet count < 90,000/mm3. Participants were excluded if they received herbal/natural remedies with anti‐HCV activity within 30 days of the baseline visit. The use of systemic antineoplastic or immunomodulatory treatments within 6 months of the baseline visit excluded participation and was not allowed during this study. The use of growth factors was not allowed during this study. In the absence of clinical drug interaction study data, medications that modulate stomach acid and known inhibitors or inducers of the cytochrome P450 3A enzyme and P‐glycoprotein transporter systems were prohibited. | |
| Interventions | Experimental group: oral 60 mg of GSK2336805 for 28 days. Control group: placebo for 28 days. Co‐intervention: peg‐IFN α‐2a (180 μg per week) and RBV (1000–1200 mg daily) from day 2 and for 27 days in total. | |
| Outcomes | Safety assessment, HCV RNA, pharmacokinetics. | |
| Notes | NCT01439373. The trial had 2 parts. Part 1: 1‐day therapy with GSK2336805 versus placebo. Part 2: 27 days of GSK2336805 versus placebo with RBV and peg‐IFN as co‐intervention. We emailed Gardner and colleagues on 21 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out (1 person) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol (NCT01439373) were assessed |
| Vested‐interest bias | High risk | GlaxoSmithKline, LLC |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 37 adult participants (18‐60 years) chronically infected with HCV (genotype 1 (1a or 1b), genotype 2 or genotype 4. | |
| Interventions | Experimental group: oral GSK2878175 10 mg, 30 mg or 60 mg for 2 days. | |
| Outcomes | Safety, pharmacokinetics, HCV RNA. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The participants and personnel were blinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being blinded but it was unclear how the blinding of outcome assessors was performed |
| Incomplete outcome data (attrition bias) | Low risk | < 5% dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The trial was sponsored by Glaxo Smith Kline |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 32 adult treatment‐naive participants with HCV genotype1 Country: USA | |
| Interventions | Experimental group: oral 150 mg, 300 mg, 450 mg of GS‐9256 as a single dose. Control group: placebo. | |
| Outcomes | HCV RNA, pharmacokinetics. | |
| Notes | The trial also had groups with healthy volunteers. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being placebo blinded, but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being placebo blinded, but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | No data |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The trial was funded by Gilead Sciences |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 307 adult participants Sex: 155 men, 152 women Mean age: 54.5 years Countries: Argentina, Australia, Austria, Canada, France, Germany, Ireland, Israel, Italy, Republic of Korea, Netherlands, New Zealand, Poland, Russian Federation, Spain, Taiwan, UK and USA. Inclusion criteria: aged at least 18 years with genotype 1b infection and HCV RNA of 10,000 IU/mL or greater who met inclusion criteria for 1 of 3 cohorts: treatment‐naive, previous non‐responder to peg‐IFNα plus RBV (null or partial response), or ineligible for, intolerant of, or ineligible for and intolerant of peg‐IFN α plus RBV (treatment‐naive and treatment‐experienced). Ineligible or intolerant (or both) participants included those with depression, anaemia or neutropenia, or compensated advanced fibrosis or cirrhosis (F3/F4) with thrombocytopaenia. Anaemia was defined as haemoglobin between 85 g/L and < 120 g/L (women) or < 130 g/L (men), neutropenia as absolute neutrophils between 0.5 x 10⁹ cells per L and < 1.5 x 10⁹ cells per L, and thrombocytopenia as platelets between 50 x 10⁹ cells per L and < 90 x 10⁹ cells per L, at screening or history of these conditions, while receiving peg‐IFN α plus RBV, or both. Exclusion criteria: people with HIV, ascites, oesophageal varices, or other evidence of hepatic decompensation. | |
| Interventions | Experimental group: oral 60 mg once daily of daclatasvir and oral 100 mg twice daily of asunaprevir for 24 weeks. Control group: placebo for 12 weeks. | |
| Outcomes | HCV RNA (SVR), safety assessment. | |
| Notes | Only participants in the treatment‐naive group were randomised. The placebo group entered a new study after 12 weeks, therefore only data for the first 12 weeks could be used. We emailed Manns and colleagues on 27 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random allocation sequence |
| Allocation concealment (selection bias) | Low risk | Interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | Low risk | The participants and personnel were blinded to treatment allocation until week 12, and we used data until week 12 |
| Blinding of outcome assessment (detection bias) | Low risk | The sponsors, who performed the analyses, were blinded until week 12, and we used data until week 12 |
| Incomplete outcome data (attrition bias) | Unclear risk | The amount of drop‐outs until week 12 were not described |
| Selective reporting (reporting bias) | High risk | 2 outcomes were added to the secondary outcomes in the protocol (NCT01581203) |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised, placebo‐controlled, parallel‐group trial | |
| Participants | 107 participants Ethnicity: Korean Race: Asian Country: South Korea, India, Taiwan Inclusion criteria: chronic hepatitis C infection and genotype 1. Previous treatment failure (relapse, non‐responders, and partial responders) | |
| Interventions | Experimental group: boceprevir for 32 weeks, beginning at week 5. Control group: placebo for 44 weeks, beginning at week 5. Co‐interventions: Experimental group: peg‐IFN and RBV for 36 week (participants with detectable HCV RNA at week 8 received additional 12 weeks of treatment, in total 48 weeks). Control group: peg‐IFN and RBV for 48 weeks. | |
| Outcomes | Not specified | |
| Notes | This trial was only available as an abstract of an interim‐analysis. The co‐interventions in both groups (experimental and control) were not completely equal ‐ while all the participants in the control group received Peg‐IFN + RBV for 48 weeks, the experimental group received a response‐guided regimen which implied that some participants received shorter duration of treatment (36 weeks), while others received 48 weeks. The following Information is required: number of participants randomised per group; method of sequence generation; method of allocation concealment; description of blinding; number and reasons for withdrawal; pre‐specified outcomes; sponsorship and its role No contact details of authors | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method of sequence generation was not specified |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided |
| Blinding of participants and personnel (performance bias) | Unclear risk | Use of placebo suggests blinding, but method of blinding was not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Insufficient information provided |
| Incomplete outcome data (attrition bias) | Unclear risk | Insufficient information provided |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information provided. No protocol available |
| Vested‐interest bias | Unclear risk | It was uncertain how the trial was sponsored |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | A phase IIb, randomised, partially double‐blind, placebo‐controlled, parallel‐group trial (PROVE‐2) (NCT00372385) | |
| Participants | 323 participants Sex: 192 men, 131 women Country: France, Germany, the UK, and Austria Inclusion criteria: age between 18 and 65 years. Chronic hepatitis C infection. HCV genotype 1. Detectable plasma HCV RNA levels. treatment‐naive. No histologic evidence of cirrhosis within 2 years before study Day 1. Seronegative for hepatitis B surface antigen and HIV‐1 and 2. Adequate double method of contraception. Negative pregnancy test for women. Exclusion criteria: any medical contraindication to peg‐IFN α‐2a or RBV therapy. Any other cause of significant liver disease in addition to hepatitis C. Diagnosed or suspected HCC. Alcohol/drug abuse or excessive use in the last 12 months. Participation in any investigational drug study within 90 days before drug administration. Group 1: 81 participants: (T12PR24) Sex: 54 men, 27 women Median age: 46 years (range 19‐65) Race: 75 white (93%), 1 black (1%), 3 Asian (4%), 1 Hispanic (1%), 1 other (1%) HCV RNA ≥ 800,000 IU/mL, n(%): 73(90) Fibrosis, n(%): none or minimal: 35(43). Portal: 37(46). Bridging: 9(11). Cirrhosis: 0 HCV genotype, n(%): 1a: 31(38). 1b: 50(62). Intermediate: 0 Group 2: 82 participants (T12PR12) Sex: 49 men, 33 women Median age: 44 years (range 22‐65) Race: 76 white (93%), 2 black (2%), 2 Asian (2%), 1 Hispanic (1%), 1 other (1%) HCV RNA ≥ 800,000 IU/mL, n(%): 67(82) Fibrosis, n(%): none or minimal: 30(37). Portal: 46(56). Bridging: 6(7). Cirrhosis: 0 HCV genotype, n(%): 1a: 37(45). 1b: 45(55). Intermediate: 0 Group 3: 78 participants (T12P12) Sex: 43 men, 55 women Median age: 45 years (range 20‐64) Race: 77 white (99%), 1 black (1%), 0 Asian, 0 Hispanic, 0 other HCV RNA ≥800,000 IU/mL, n(%): 63(81) Fibrosis, n(%): none or minimal: 31(40). Portal: 43(55). Bridging: 3(4). Cirrhosis: 1(1) HCV genotype, n(%): 1a: 40(51). 1b: 38(49). Intermediate: 0 Group 4: 82 participants (PR48) Sex: 46 men, 36 women Median age: 45 years (range 18‐64) Race: 76 white (93%), 2 black (2%), 4 Asian (5%), 0 Hispanic, 0 other HCV RNA ≥800,000 IU/mL, n(%): 68(83) Fibrosis, n(%): none or minimal: 28(34). Portal: 46(56). Bridging: 8(10). Cirrhosis: 0 HCV genotype, n(%): 1a: 35(43). 1b: 45(55). Intermediate: 2(2). | |
| Interventions | Experimental group: 1, 2, and 3: oral telaprevir given as a single dose of 1250 mg on study day 1, followed by a dose of 750 mg every 8 h for 12 weeks. Control group: 4: placebo for 12 weeks. Co‐interventions: 1: peg‐IFN α‐2a 180 μg subcutaneously once weekly plus oral weight‐based RBV 1000 to 1200 mg in 2 divided daily doses for 24 weeks 2: peg‐IFN α‐2a 180 μg subcutaneously once weekly plus oral weight‐based RBV 1000 to 1200 mg in 2 divided daily doses for 12 weeks 3: peg‐IFN α‐2a 180 μg subcutaneously once weekly for 12 weeks 4: peg‐IFN α‐2a 180 μg subcutaneously once weekly plus oral weight‐based RBV 1000 to 1200 mg in 2 divided daily doses for 48 weeks. | |
| Outcomes | Primary outcome: proportion of participants who achieved SVR at 24 weeks after end of treatment (HCV RNA undetectable (< 10 IU/mL) 24 weeks after completion of study treatment) Secondary outcomes: proportion of participants with undetectable HCV RNA at week 12 after end of treatment. Proportion of participants with undetectable HCV RNA at completion of study drug dosing. Number of participants with AEs. Number of participants with viral relapse. Maximum, minimum, and average plasma concentration of telaprevir | |
| Notes | We emailedWe emailed Hezode and colleagues on 21 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided |
| Allocation concealment (selection bias) | Unclear risk | Randomisation was performed through a central telephone‐based system. No other information was provided |
| Blinding of participants and personnel (performance bias) | High risk | Group 3 (T12P12) was not blinded. Other treatment groups were blinded to the interventions |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not enough information provided |
| Incomplete outcome data (attrition bias) | Low risk | Number and reasons for discontinuation were clearly reported on. |
| Selective reporting (reporting bias) | Low risk | Protocol was available and all pre‐specified outcomes were reported on |
| Vested‐interest bias | High risk | The sponsor (Vertex Pharmaceuticals) was directly involved in trial design and protocol development |
| Other bias | Low risk | The trial seems to be free of other potential sources of bias |
| Methods | Randomised clinical trial | |
| Participants | 51 adult participants Sex: 41 men, 10 women Mean age: 47.8 years Countries: Germany, France, and Spain. Inclusion criteria: women or men aged 18 years or older with chronic genotype 1 HCV infection. The line probe assay was used to determine the genotype of the viral infection. A liver biopsy specimen showing changes consistent with chronic HCV infection had to have been performed within the previous 12 months. At screening, the HCV load had to be 50,000 copies/mL serum. Exclusion criteria: women were excluded if they were breast‐feeding or at risk of pregnancy; men had to use an adequate form of contraception if their partner was of childbearing potential. They were not enrolled if there were other or additional reasons for chronic liver disease, including the presence of other hepatitis‐causing viruses and/or a history of alcohol abuse within the previous 12 months and/or evidence of Child’s B or C liver disease at screening. No other antiviral or antimicrobial or investigational therapies were allowed during the study (screening, pretreatment, and treatment phases). Patients were excluded if, at screening, their baseline ALT/AST) plasma levels exceeded the ULN by more than 5‐fold (5 times the ULN) or their total bilirubin or alkaline phosphatase levels were 1.5 times the ULN. Other exclusion criteria included co‐infection with HIV, a platelet count 100,000/mm3, a white blood cell count 2000 cells/mm3, any clinically significant laboratory abnormalities, and a positive test result for illicit or nonprescription drugs. | |
| Interventions | The trial was divided into 3 different cohorts, according to grade of liver disease (Ishak score, Metavir score). Experimental group: 2 days of oral 25 mg, 200 mg or 500 mg of BILN‐2061 in participants with Ishak score 0‐2. Oral 200 mg of BILN 2061 in participants with Ishak score 3‐4. Oral 200 mg of BILN 2061 in participants with Ishak score 5‐6. Control group: placebo. | |
| Outcomes | Virologic efficacy, pharmacokinetics, safety assessment. | |
| Notes | We emailedWe emailed Hinrichsen and colleagues on 21 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Low risk | 0 participants dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained for all 3 stages, and the clinicalTrials.gov information was added after completion |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Phase III, randomised, double‐blind, placebo‐controlled, parallel‐group trial (TIGER)(NCT01725529) | |
| Participants | 457 participants Median age: 48 years (range 18‐68) Sex: 236 men, 221 women Country: China, Korea Ethnicity (%): Chinese (80.3%), Korean (19.7%) HCV genotype (%): 1a (1.1%), 1b (98.9%) Inclusion criteria: treatment‐naive East Asian participants with chronic hepatitis C. A liver biopsy within 3 years prior to the screening visit (or between screening and day of randomisation) with histology consistent with chronic Hepatitis C virus (HCV) infection (presence of contraindications for a liver biopsy in participants who are otherwise deemed eligible for participation does not exclude the patient from participation). Genotype 1 HCV infection (confirmed at screening). Plasma HCV RNA of > 10,000 IU/mL at screening. Age between 18‐70 years. Exclusion criteria: prior treatment with any approved or investigational drug for the treatment of hepatitis C. Co‐infection with HBV or HIV. | |
| Interventions | Experimental group: Group 1: Simeprevir 150 mg orally once daily for 12 weeks. Group 2: Simeprevir 100 mg orally once daily for 12 weeks. Control group: Group 3: matching placebo capsules taken orally with food once‐daily for 48 weeks. Co‐interventions: Group 1 and 2: peg‐IFN α‐2a μg once weekly administered as weekly subcutaneous injections of 0.5 mL for 24 or 48 weeks. RBV 1000 or 1200 mg/day (taken as 100 mg or 200 mg tablets) depending on body weight for 24 or 48 weeks (If body weight is < 75 kg the total daily dose of RBV will be 1000 mg, administered as 400 mg intake with food in the morning and 600 mg intake with food in the evening. If body weight is > or = 75 kg the total daily dose will be 1200 mg, administered as 2 x 600 mg per intake with food, morning and evening). Group 3: peg‐IFN α‐2a μg once weekly administered as weekly subcutaneous injections of 0.5 mL for 48 weeks. RBV 1000 or 1200 mg/day (taken as 100 mg or 200 mg tablets) depending on body weight for 48 weeks (If body weight is < 75 kg the total daily dose of RBV will be 1000 mg, administered as 400 mg intake with food in the morning and 600 mg intake with food in the evening. If body weight is ≥ 75 kg the total daily dose will be 1200 mg, administered as 2 x 600 mg per intake with food, morning and evening). | |
| Outcomes | Primary outcome measures: percentage of participants with SVR 12 weeks after end of study drug treatment (participants considered to have achieved SVR12 if both conditions are met: 1. HCV RNA < 25 IU/mL or undetectable at end of treatment and; 2. HCV RNA is < 25 IU/mL or undetectable at 12 weeks after the planned end of study drug treatment). Secondary outcome measures: percentage of participants with SVR 24 weeks after end of study drug treatment (participants considered to have achieved SVR24 if both conditions are met: 1. HCV RNA < 25 IU/mL or undetectable at end of treatment; 2. HCV RNA < 25 IU/mL or undetectable at 24 weeks after the planned end of study drug treatment). Percentage of participants with SVR at week 72. Percentage of participants with on‐treatment failure (refers to a participant with confirmed detectable HCV RNA at the end of treatment). Percentage of participants with viral breakthrough (defined as a confirmed increase of > 1 log10 IU/mL in HCV RNA level from the lowest level reached, or a confirmed HCV RNA level of > 100 IU/mL in participants whose HCV RNA levels had previously been below the limit of quantification (< 25 IU/mL detectable) or undetectable (< 25 IU/mL undetectable) while on study treatment). Percentage of participants with viral relapse (defined as undetectable HCV RNA at the actual end of treatment and last HCV RNA measurement during follow‐up ≥ 25 IU/mL). Percentage of participants with on‐treatment normalisation of ALT level. | |
| Notes | Abstract. Interim analysis. We emailedWe emailed Hoeben and colleagues on 21 April 2016 for additional information (on method of sequence generation and method of allocation concealment) but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method of sequence generation was not specified |
| Allocation concealment (selection bias) | Unclear risk | The method of allocation concealment was not described |
| Blinding of participants and personnel (performance bias) | Low risk | A simeprevir‐matched placebo was used |
| Blinding of outcome assessment (detection bias) | Low risk | The protocol stated that outcomes assessors were blinded to the intervention |
| Incomplete outcome data (attrition bias) | Low risk | Number and reasons for withdrawal were stated at www.ClinicalTrials.gov (NCT01725529) |
| Selective reporting (reporting bias) | Low risk | A protocol was published before randomisation began and all outcome results were reported adequately |
| Vested‐interest bias | High risk | The trial was sponsored by a pharmaceutical company (Janssen) |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias |
| Methods | For characteristics see Hoeben 2015a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method of sequence generation was not specified |
| Allocation concealment (selection bias) | Unclear risk | The method of allocation concealment was not described |
| Blinding of participants and personnel (performance bias) | Low risk | A simeprevir‐matched placebo was used |
| Blinding of outcome assessment (detection bias) | Low risk | The protocol stated that outcomes assessors were blinded to the intervention |
| Incomplete outcome data (attrition bias) | Low risk | Number and reasons for withdrawal were stated at www.ClinicalTrials.gov (NCT01725529) |
| Selective reporting (reporting bias) | Low risk | A protocol was published before randomisation began and all outcome results were reported adequately |
| Vested‐interest bias | High risk | The trial was sponsored by a pharmaceutical company (Janssen) |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 13 participants Sex: 12 men, 1 woman Mean age: 49 years Countries: Netherlands and USA. Inclusion criteria: chronic hepatitis C participants, both treatment‐naive or treatment‐experienced, aged 18‐65 with a BMI 18‐32. Exclusion criteria: decompensated liver disease, uncontrolled or active major systemic disease and co‐infection with HIV or HBV. Participants with chronic stable haemophilia or on stable methadone substitution treatment. | |
| Interventions | The trial was divided into single and multi ascending cohorts (only cohort 4, 5 and 11, 12 were HCV‐infected participants) Experimental group 1: single ascending dose: 100 mg, 500 mg once daily, or 250 mg twice daily PHX1766 Experimental group 2: multi ascending dose: 400 mg twice daily, 800 mg twice daily PHX1766 Control group: placebo, only in the multi ascending dose | |
| Outcomes | Pharmacokinetics, safety assessment, pharmacodynamics. | |
| Notes | We emailed Hotho and colleagues on 21 April 2016 for additional information but reply not received yet. The trial also included healthy volunteers. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being placebo‐controlled, but method was not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being placebo‐controlled, but method was not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The trial was funded by Phenomix Corporation |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | Treatment‐naive and treatment‐ experienced participants (prior treatment with PR for ≥ 12 weeks had failed) with chronic HCV genotype 1 infection. | |
| Interventions | All participants initially received PR for 4 weeks. Participants randomised to control treatment then received PR for an additional 44 weeks. Treatment‐naive participants randomised to triple therapy received boceprevir (800 mg 3 times daily) plus PR for 24 weeks and then further therapy according to treatment week 8 HCV RNA levels. Treatment‐experienced participants received boceprevir plus PR for 32 wk and then further therapy according to treatment week 8 HCV RNA levels. | |
| Outcomes | SVR defined as undetectable HCV RNA 24 weeks after completing all study therapy. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | < 5% missing data |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Vested‐interest bias | High risk | "Supported by Merck and Co., Inc. Kenilworth, NJ, US."; "Medical writing and editorial assistance were provided by Tim Ibbotson, Ph.D. of ApotheCom, Yardley,PA, United States." |
| Methods | Randomised clinical trial | |
| Participants | 42 adult participants Sex: 20 men, 22 women Mean age: 55 years Country: Japan Exclusion criteria: history of HCC, co‐infection with HBV or HIV, other chronic liver disease, or evidence of hepatic decompensation. Liver cirrhosis, liver biopsy within 24 months, elevated ALT, bilirubin, albumin, decreased haemoglobin, white blood cells, neutrophil count, platelets, creatinine, participants exposed any investigational HCV therapeutic agent 4 weeks prior to dosing. | |
| Interventions | Experimental group: oral 10 mg or 60 mg of daclatasvir once daily. Control group: placebo. Co‐intervention: weight‐based RBV twice daily, once weekly subcutaneous alfa‐2a IFN. Participants receiving protocol‐defined response were treated for 24 weeks. Participants not receiving protocol‐defined response were treated for 48 weeks. | |
| Outcomes | Efficacy assessment, safety assessment, virological response. | |
| Notes | NCT01017575 ‐ only data from the treatment‐naive group could be used, since the non‐responders couldn't be randomised to placebo. We emailed Izumi and colleagues on 21 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | not described |
| Allocation concealment (selection bias) | Low risk | Central randomisation centre |
| Blinding of participants and personnel (performance bias) | High risk | Unblinded after week 24 |
| Blinding of outcome assessment (detection bias) | High risk | Unblinded after week 24 |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 person dropped out |
| Selective reporting (reporting bias) | High risk | The trial changed the primary outcomes |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Izumi 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Central randomisation centre |
| Blinding of participants and personnel (performance bias) | High risk | Unblinded after week 24 |
| Blinding of outcome assessment (detection bias) | High risk | Unblinded after week 24 |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 person dropped out |
| Selective reporting (reporting bias) | High risk | The trial changed the primary outcomes |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 35 adult participants Sex: 18 men, 17 women Mean age: not reported Country: USA and Puerto Rico. Inclusion criteria: 18‐65 years of age treatment‐naive (no prior treatment with IFN‐a +/‐ RBV regimens, discontinued IFN‐a containing regimens after < 2 weeks of therapy due to tolerability issues were considered treatment‐naive, HCV RNA > 100,000 IU/mL at screening, genotype 1, a diagnosis of chronic HCV infection for at least 6 months. Exclusion criteria: evidence of acute or chronic infection with HIV or HBV, exposure within the previous 3 months to an investigational anti‐HCV agent, evidence of severe or decompensated liver disease, participants with liver disease unrelated to HCV infection. | |
| Interventions | Experimental group: oral 200 mg, 300 mg, 500 mg twice daily for 4 weeks. Control group: placebo. Co‐intervention: standard care as per investigator's discretion up to Week 48, then off‐treatment up to Week 72 in open‐label period. Standard of care included peg‐IFN α‐2a 180 μg subcutaneously once weekly starting from day 1 and RBV 1000 mg/day tablet orally in 2 divided doses for participants weighing ≤ 75 kg; 1200 mg/day orally in 2 divided doses for participants weighing > 75 kg. | |
| Outcomes | Plasma HCV, pharmacokinetics, ALT levels, safety assessment. | |
| Notes | NCT00720434 We emailed Jacobson and colleagues on 21 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | Open‐label after week 4 |
| Blinding of outcome assessment (detection bias) | High risk | Open‐label after week 4 |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out (Jacobson 2010, described 2 dropping out) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed |
| Vested‐interest bias | High risk | The trial was funded by Pfizer |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | A phase III, multicenter, randomised, double‐blind, parallel‐group trial (QUEST‐1) (NCT01289782) | |
| Participants | 394 participants Country: Australia, Canada, Germany, Italy, Mexico, New Zealand, Puerto Rico, Romania, Russia, Spain, Ukraine, UK, and USA Inclusion criteria: age ≥ 18 years with chronic hepatitis C infection and HCV genotype 1. Screening HCV RNA level > 10,000 IU/mL, treatment‐naive, an ultrasound performed within 6 months of enrolment showing no signs of HCC in participants with cirrhosis. Exclusion criteria: hepatic decompensation, any non‐HCV‐related liver disease, HIV or HBV co‐infection, non‐genotype 1 HCV infection, significant laboratory abnormalities, any other active disease, male or female participants who had, or were planning to conceive Simeprevir group: 264 participants: Sex: 148 men, 116 women Median age: 48 years (range 39‐54) Race: 227 white (86%), 27 black or African‐American (10%), 5 Asian (2%) HCV genotype 1a: 147 (56%). HCV genotype1b: 117 (44%) Interleukin (IL) 28B genotype CC: 77 (29%). IL28B genotype CT: 150 (57%). IL28B genotype TT: 37(14) HCV RNA > 800,000 IU/mL, n(%): 218(83) Placebo group: 130 participants: Sex: 74 men, 56 women Median age: 48 years (range 36‐54) Race: 122 white (94%), 4 black or African‐American (3%), 3 Asian (2%) HCV genotype, n(%): 1a: 74(57). 1b: 56(43). IL28B genotype, n(%): CC: 37(28). CT: 76(58). TT: 17(13) METAVIR score, n(%): F0‐F1: 50(38). F2: 40(31). F3: 23(18). F4: 17(13). HCV RNA > 800,000 IU/mL, n(%): 96(74) | |
| Interventions | Experimental group: oral simeprevir 150 mg once daily for 12 weeks. Control group: oral placebo 150 mg once daily for 12 weeks. Co‐interventions: Experimental group: peg‐IFN alfa‐2a 180 μg subcutaneously once weekly and oral weight‐based RBV 1000‐1200 mg in 2 divided daily doses for 24‐48 weeks Control group: pegIFN α‐2a 180 μg subcutaneously once weekly and oral weight‐based RBV 1000‐1200 mg in 2 divided daily doses (1000 mg if body weight < 75 kg; 1200 mg if body‐weight ≥ 75 kg) for 48 weeks. | |
| Outcomes | Primary outcome: proportion of participants achieving SVR12 (HCV RNA < 25 IU/mL undetectable at end of treatment and < 25 IU/mL detectable or undetectable 12 weeks after planned end of treatment) Secondary outcomes: comparison of SVR 24 weeks after planned end of treatment. Percentage of participants meeting criteria for response‐guided therapy to complete treatment at week 24. Rapid virological response (HCV RNA < 25 IU/mL undetectable at week 4). On‐treatment failure (detectable HCV RNA at end of treatment). Incidence of viral breakthrough (HCV RNA increase of more than 1 log10 from the lowest level noted or an HCV RNA ≥ 25 IU/mL during follow‐up or at time of SVR assessments after achieving undetectable levels at end of treatment). Incidence of AEs. Incidence of laboratory abnormalities. Patient‐reported symptoms and functioning. Effect of baseline characteristics on treatment response. Assessment of depression severity. Assessment of health status. | |
| Notes | We emailed Jacobson and colleagues on 21 April 2016 for additional information (on blinding of outcomes assessors) but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated schedule prepared by or under the supervision of the sponsor was used |
| Allocation concealment (selection bias) | Low risk | Allocation concealment was performed by "using an interactive voice‐response system (IVRS) which assigned a unique code that dictated the treatment assignment and matching study drug kit for each patient". Randomisation codes were maintained within the IVRS |
| Blinding of participants and personnel (performance bias) | Low risk | Authors stated that "patients, study personnel, and the sponsor were masked to the treatment group assignment", the blinding method was not adequately described. A matched placebo was used |
| Blinding of outcome assessment (detection bias) | Unclear risk | Although RNA levels were monitored by an unmasked independent external person who informed the sponsor of any required changes to treatment, the blinding method for other outcome assessors was not described |
| Incomplete outcome data (attrition bias) | Low risk | Number and reasons for discontinuation were clearly reported on |
| Selective reporting (reporting bias) | Low risk | Protocol is available. All pre‐specified study outcomes were reported on |
| Vested‐interest bias | High risk | The sponsor (Janssen Infectious Diseases‐Diagnostics) was directly involved in trial design, analyses and interpretation of data, writing and reviewing the manuscript |
| Other bias | Low risk | The trial seems free of other potential sources of bias |
| Methods | Phase IIb, randomised, double‐blind, parallel‐group study in treatment‐naive participants with HCV genotype 1 or 4 infection (ClinicalTrials.gov NCT01057667) | |
| Participants | 168 participants were randomised Sex: 118 men, 48 women Mean age: experimental group: 49.7 years/control group: 48.2 years Countries: 25 sites in the USA and Canada. Inclusion criteria: eligible participants were treatment‐niıve adults 18‐70 years of age with chronic hepatitis C of at least 6 months’ duration, a serum HCV RNA titer of at least 50,000 IU/mL (COBAS AmpliPrep/ COBAS TaqMan HCV Test; lower limit of detection ¼ 15 IU/mL), and HCV genotype 1 or 4 infection were eligible for the study. Participants were required to have had a liver biopsy within the previous 24 months (36 months in participants with cirrhosis/bridging fibrosis). Participants with compensated cirrhosis (Child‐Pugh grade A) or transition to cirrhosis were required to have had an abdominal ultrasound, computerised tomography scan, magnetic resonance imaging scan demonstrating the absence of evidence of HCC (within 2 months before randomisation), and a serum alpha‐fetoprotein level < 100 ng/mL. Exclusion criteria: infection with hepatitis A or B viruses or HIV; previous treatment with IFN‐based therapy or any investigational anti‐HCV agent; systemic antiviral therapy within the previous 3 months; history or evidence of medical condition associated with chronic liver disease other than HCV; absolute neutrophil count < 1.5 x 109 cells/L; platelet count < 90 x 109 cells/L; haemoglobin concentration < 12 g/dL in women (< 13 g/dL in men); history of renal disease, serum creatinine > 1.5 times the ULN, an estimated creatinine clearance ≤ 70 mL/min or microproteinuria. | |
| Interventions | Participants were randomised in a 1:1 ratio. 166 participants received at least 1 dose. Experimental group: oral mericitabine (Genentech, San Francisco, CA) 1000 mg twice a day for 24 weeks in participants with eRVR (defined as undetectable HCV RNA from week 4 through 22) or for 48 in participants without eRVR. Control group: placebo twice a day. Co‐intervention: peg‐IFN α‐2a (40 kD) (Pegasys; Roche, Basel,Switzerland) 180 lg subcutaneously once‐weekly and oral RBV (Copegus; Roche) at a dosage of 1000 (body weight: < 75 kg) or 1200 mg/day (body weight: > 75 kg) in 2 divided doses for 24 or 48 weeks. | |
| Outcomes | Proportion of participants with undetectable plasma HCV RNA 24 weeks after the end of treatment (SVR24), with SAE, AEs, mortality. | |
| Notes | We emailed Pockros and colleagues on 06 June 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation list was maintained by the sponsor, and neither study personnel nor investigators had access to the list |
| Allocation concealment (selection bias) | Low risk | Participants were randomised by an interactive voice‐response system. A computer‐generated randomisation list was maintained by the sponsor, and neither study personnel nor investigators had access to the list |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blinding was achieved through the use of matching placebo tablets. Investigators were advised byinteractive voice‐response system at week 24 as to whether a participant was to stop treatment (mericitabine‐treated participants with an eRVR) or continue to week 48 (mericitabine‐treated participants without an eRVR and all placebo‐treated participants). JF: "I guess that all participants were not blinded to maximum‐follow up then? Since it would be obvious that the ones who stopped treatment after 24 weeks, received the study drug?" |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | The trial authors reported that only 55 participants in the experimental group completed 24 weeks of follow‐up. It seems like there are 81 participants in the included analysis of SVR24. The trial authors do not account for how they imputed the participants with missing data |
| Selective reporting (reporting bias) | Low risk | All outcomes in the protocol were reported on |
| Vested‐interest bias | Unclear risk | This research was funded by F. Hoffmann‐La Roche Ltd. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | An open‐label, randomised, multicenter, parallel group, phase II trial (SPRINT‐1) (NCT00423670) | |
| Participants | 520 participants Country: USA, Canada, and Europe Inclusion criteria: chronic hepatitis C infection genotype 1 treatment‐naive, 18‐60 years. Liver biopsy consistent with chronic HCV infection within 5 years of enrolment, haemoglobin ≥ 130 g/L (men), ≥ 120 g/L (women), neutrophil count ≥ 1.5 x 109/L, platelet count ≥ 100 x 109/L. Bilirubin, albumin, and creatinine within normal limits. Exclusion criteria: decompensated liver cirrhosis, HIV infection, previous organ transplantation, other causes of liver disease, pre‐existing psychiatric disease, seizure disorder, cardiovascular disease, haemoglobinopathies, haemophilia, poorly controlled diabetes, autoimmune diseases Group 1: 104 participants Sex: 70 men (67%), 34 women (33%) Mean age ± SD: 48.3 ± 6.9 years Race: 83 white (80%), 2 American Indian or Alaskan (2%), 3 Asian (3%), 16 black (15%), 0 multiracial Weight, mean ± SD (kg): 83.4 ± 16.2 HCV genotype, n(%): 1a: 53(51), 1b: 42(40), 1, no subtype: 9(9) Baseline HCV RNA: log10 of geometric mean: 6.53. > 600,000 IU/mL, n(%): 94(90). Cirrhosis, n(%): 8(8) Group 2: 103 participants Sex: 51 men (50%), 52 women (50%) Mean age ± SD: 47.7 ± 7.4 Race: 85 white (83%), 1 American Indian or Alaskan (1%), 1 Asian (1%), 15 black (15%), 1 multiracial (1%) Weight, mean ± SD (kg): 79.9 ± 14.2 HCV genotype, n(%): 1a: 53(51), 1b: 37(36), 1, no subtype: 13(13) Baseline HCV RNA: log10 of geometric mean: 6.53. > 600,000 IU/mL, n(%): 90(87), cirrhosis, n(%): 7(7) Group 3: 103 participants Sex: 58 men (56%), 45 women (44%) Mean age ± SD: 47.6 ± 8.3 years Race: 85 white (83%), 1 American Indian or Alaskan (1%), 2 Asian (2%), 15 black (15%), 0 multiracial Weight, mean ± SD (kg): 78.4 ± 16.5 HCV genotype, n(%): 1a: 60(58). 1b: 35(34). 1, no subtype: 8(8) Baseline HCV RNA: log10 of geometric mean: 6.53. > 600,000 IU/mL, n(%): 93(90), cirrhosis, n(%): 6(6) Group 4: 107 participants Sex: 63 men (59%), 44 women (41%) Mean age ± SD: 46.4 ± 8.0 years Race: 86 white (80%), 0 American Indian or Alaskan, 2 Asian (2%), 18 black (17%), 1 multiracial (1%) Weight, mean ± SD (kg): 83.4 ± 17.3 HCV genotype, n(%): 1a: 67(63), 1b: 30(28), 1, no subtype: 10(9) Baseline HCV RNA: log10 of geometric mean: 6.64. > 600,000 IU/mL, n(%): 98(92), cirrhosis, n(%): 7(7) Group 5: 103 participants Sex: 63 men (61%), 40 women (39%) Mean age ± SD: 46.7 ± 8.8 Race: 87 white (84%), 0 American Indian or Alaskan, 1 Asian (1%), 14 black (14%), 1 multiracial (1%) Weight, mean ± SD (kg): 80.0 ± 19.4 HCV genotype, n(%): 1a: 55(53), 1b: 36(35), 1, no subtype: 12(12) Baseline HCV RNA: log10 of geometric mean: 6.54. > 600,000 IU/mL, n(%): 94(91), cirrhosis, n(%): 9(9). | |
| Interventions | Experimental group: 2: oral boceprevir 800 mg 3 times per day, starting at week 5 for a total of 24 weeks. 3: oral boceprevir 800 mg 3 times per day, starting at week 5 for a total of 44 weeks. 4: oral boceprevir 800 mg 3 times per day for a total of 28 weeks. 5: oral boceprevir 800 mg 3 times per day for a total of 48 weeks. Control group: 1: no intervention. Co‐interventions: 1‐5: peg‐IFN α‐2b 1.5 μg/kg body weight subcutaneously once weekly ‐ weight‐based oral RBV from 800‐1400 mg daily (if body weight ≤ 65 kg dosage is 800 mg (400 mg twice daily); if body weight is 66‐80 kg dosage is 1000 mg daily (400 mg in the morning and 600 mg in the evening); if body weight is 81‐105 kg dosage is 1200 mg daily (600 mg twice daily); and if body weight is > 105 kg dosage is 1400 mg daily (600 mg in the morning and 800 mg in the evening)). | |
| Outcomes | Primary outcome: SVR, defined as the proportion of participants with undetectable HCV RNA 24 weeks after discontinuation of treatment. Secondary outcomes:
| |
| Notes | 2 additional groups were present in the trial (Groups 6 and 7), which were randomised separately, but did not satisfy inclusion criteria, therefore were not included. We emailed Kwo and colleagues on 26 April 2016 for further explanation on difference between number of SAE stated in published article compared to results published on www.ClinicalTrials.gov but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code |
| Allocation concealment (selection bias) | Low risk | Allocation performed by an external randomisation centre through interactive voice‐response system in 1:1:1:1:1 ratio. Randomisation was stratified according to race (black vs non‐black) and cirrhosis status (cirrhosis vs no cirrhosis). |
| Blinding of participants and personnel (performance bias) | High risk | Trial described as open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Trial described as open‐label |
| Incomplete outcome data (attrition bias) | High risk | Although number and reasons for withdrawal were clearly stated, the proportion of participants who discontinued treatment was high, from 26% to 50%, mostly due to AEs or treatment inefficiency. |
| Selective reporting (reporting bias) | High risk | Although a protocol was available and published before randomisation began, number of SAE were differently stated in the published article compared to data presented on www.ClinicalTrials.gov. Data presented in the latter were somewhat higher. Data reported are from www.ClinicalTrials.gov. |
| Vested‐interest bias | High risk | The sponsor of the study contributed to patient recruitment, trial management, data collection, statistical analyses, and the writing and review of the report. |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias. |
| Methods | For characteristics see Kwo 2010a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code |
| Allocation concealment (selection bias) | Low risk | Allocation performed by an external randomisation centre through interactive voice‐response system in 1:1:1:1:1 ratio. Randomisation was stratified according to race (black vs non‐black) and cirrhosis status (cirrhosis vs no cirrhosis) |
| Blinding of participants and personnel (performance bias) | High risk | Trial described as open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Trial described as open‐label |
| Incomplete outcome data (attrition bias) | High risk | Although number and reasons for withdrawal were clearly stated, the proportion of participants who discontinued treatment was high, from 26% to 50%, mostly due to AEs or treatment inefficiency. |
| Selective reporting (reporting bias) | High risk | Although a protocol was available and published before randomisation began, number of SAE were differently stated in the published article compared to data presented on www.ClinicalTrials.gov. Data presented in the latter were somewhat higher. Data reported are from www.ClinicalTrials.gov |
| Vested‐interest bias | High risk | The sponsor of the study contributed to patient recruitment, trial management, data collection, statistical analyses, and the writing and review of the report |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias |
| Methods | For characteristics see Kwo 2010a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code |
| Allocation concealment (selection bias) | Low risk | Allocation performed by an external randomisation centre through interactive voice‐response system in 1:1:1:1:1 ratio. Randomisation was stratified according to race (black vs non‐black) and cirrhosis status (cirrhosis vs no cirrhosis) |
| Blinding of participants and personnel (performance bias) | High risk | Trial described as open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Trial described as open‐label |
| Incomplete outcome data (attrition bias) | High risk | Although number and reasons for withdrawal were clearly stated, the proportion of participants who discontinued treatment was high, from 26% to 50%, mostly due to AEs or treatment inefficiency |
| Selective reporting (reporting bias) | High risk | Although a protocol was available and published before randomisation began, number of SAE were differently stated in the published article compared to data presented on www.ClinicalTrials.gov. Data presented in the latter were somewhat higher. Data reported are from www.ClinicalTrials.gov |
| Vested‐interest bias | High risk | The sponsor of the study contributed to patient recruitment, trial management, data collection, statistical analyses, and the writing and review of the report |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias |
| Methods | For characteristics see Kwo 2010a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code |
| Allocation concealment (selection bias) | Low risk | Allocation performed by an external randomisation centre through interactive voice‐response system in 1:1:1:1:1 ratio. Randomisation was stratified according to race (black vs. non‐black) and cirrhosis status (cirrhosis vs. no cirrhosis) |
| Blinding of participants and personnel (performance bias) | High risk | Trial described as open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Trial described as open‐label |
| Incomplete outcome data (attrition bias) | High risk | Although number and reasons for withdrawal were clearly stated, the proportion of participants who discontinued treatment was high, from 26% to 50%, mostly due to AEs or treatment inefficiency |
| Selective reporting (reporting bias) | High risk | Although a protocol was available and published before randomisation began, number of SAE were differently stated in the published article compared to data presented on www.ClinicalTrials.gov. Data presented in the latter were somewhat higher. Data reported are from www.ClinicalTrials.gov |
| Vested‐interest bias | High risk | The sponsor of the study contributed to patient recruitment, trial management, data collection, statistical analyses, and the writing and review of the report |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 64 participants Mean age: 50 years Country: USA Inclusion criteria: treatment‐naive adult participants with chronic hepatitis C. | |
| Interventions | Experimental group: oral 200 mg, 400 mg, 800 mg of ACH‐1625 for 28 days. Control group: placebo. Co‐intervention: peg‐IFN‐α 2a/RBV. | |
| Outcomes | Pharmacokinetics, HCV RNA, safety assessment. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as placebo‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as placebo‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | Unclear risk | Not described |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 41 adult participants Sex: 29 men, 12 women Mean age: 48 years Country: USA Inclusion criteria: male or female adults 18‐65 years of age, inclusive; a documented clinical history compatible with chronic hepatitis C, including the presence of HCV RNA in the plasma for least 6 months and a liver biopsy sample within 24 months with histology consistent with chronic HCV infection; HCV genotype 1, plasma HCV RNA > 5 log10 IU/ml, and anti‐HCV antibody positive at screening; and agreement by participants to use a double‐barrier method of birth. Sex: 29 men, 12 women. Exclusion criteria: BMI > 32 kg/m2; pregnancy or breastfeeding; co‐infection with HBV or HIV; history or evidence of decompensated liver disease; history of HCC or findings suggestive of possible HCC; other causes of liver disease; previous antiviral treatment for HCV infection; current abuse of alcohol or illicit drugs or treatment for opioid addiction; use of any known inhibitor and/or inducer of CYP 3A4 or any other investigational drugs within 30 days of dosing; abnormal laboratory values at screening (a hemoglobin level < 12.0 g/dl for males or < 11.0 g/dl for females; an absolute neutrophil count < 1.5 × 109/liter; a platelet count < 130 × 109/liter; an alanine aminotransferase [ALT] or aspartate aminotransferase [AST] level > 2.5 × upper limit of normal [ULN]; an alkaline phosphatase level > 1.25 × ULN; an albumin level < 3.5 g/dl; total bilirubin, amylase, lipase, or international normalized ratio [INR] > ULN; a serum creatinine or blood urea nitrogen value > ULN; creatinine clearance < 80 ml/min as estimated by the Cockcroft‐Gault formula; or any other laboratory abnormality > grade 1, except for asymptomatic cholesterol or triglycerides); or other clinically significant diseases that, in the opinion of the investigator, would jeopardize the safety of the patient or impact the validity of the study results. | |
| Interventions | Experimental group: oral 25 mg, 50 mg, 75 mg, 100 mg of IDX184 for 3 days. Control group: placebo. Co‐intervention: 14 days after treatment the participants were offered extended therapy with peg‐IFN/RBV. | |
| Outcomes | Safety assessment, antiviral activity. | |
| Notes | We emailed Lalezari and colleagues on 26 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Only some outcomes were blinded for outcome assessors |
| Incomplete outcome data (attrition bias) | Unclear risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The trial was funded by Idenix pharmaceuticals Inc |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 81 adult participants Sex: 56 men, 25 women Mean age: 48 years Country: USA Inclusion criteria: male or female participants 18‐65 years old; documented clinical history compatible with chronic hepatitis C, including positive anti‐HCV antibody or presence of HCV RNA in the plasma for at least 6 months and liver biopsy within 24 months with histology consistent with chronic hepatitis C infection; HCV‐genotype 1, plasma HCV RNA> 5 log10 IU/mL; all participants agreed to use double‐barrier birth control (such as condom plus spermicide) from screening through at least 6 months after the last dose of the study drug. Exclusion criteria: pregnancy or breastfeeding; BMI > 35 kg/m2; co‐infection with HBV or HIV; history or evidence of decompensated liver disease; prior clinical or histological evidence of cirrhosis; ALT or AST level > 3 ULN; histology of HCC or findings suggestive of possible HCC; 1 or more additional known primary or secondary causes of liver disease, other than hepatitis C, previous antiviral treatment for HCV; current abuse of alcohol or illicit drugs; current use of any major inhibitor or inducer of cytochrome P450 3A4 or any other investigational drugs within 30 days of dosing, or other clinically significant diseases that, in the opinion of the investigator, would jeopardise the safety of the participants or affect the validity of the study results. | |
| Interventions | Experimental groups: oral rising daily doses of 50, 100, 150 or 200 mg of IDX184 for 2 weeks. Control group: placebo. Co‐intervention: peg‐IFN‐ α 2a and RBV for 2 weeks. All participants received additional 2 weeks of peg‐IFN and RBV. | |
| Outcomes | HCV RNA, Safety, pharmacokinetics. | |
| Notes | We emailed Lalezari and colleagues on 26 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | Despite being a double‐blinded study, there were different doses, syringes plus capsules, different administrations – once vs twice |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | The number of dropouts was unclear |
| Selective reporting (reporting bias) | High risk | Not all outcomes stated in the protocol were assessed (NCT01011166 ) |
| Vested‐interest bias | High risk | The trial was funded by Idenix Pharmaceuticals Inc. |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias |
| Methods | Randomised phase I clinical trial | |
| Participants | 27 participants Sex: 21 men, 6 women Countries: France, Germany, and Switzerland. Inclusion criteria: treatment‐naive participants male or female (with documented hysterectomy or postmenopausal), 18–70 years of age, had chronic hepatitis C infection of genotype‐1, with a HCV viral load > 100,000 IU/mL at screening. Exclusion criteria: cirrhosis was ruled out by biopsy or elastometry (FibroScan; cut‐off used by investigators ranged from 12.5 to 16.0 kPa) performed within 24 months prior to study enrolment. Participants with HBV or HIV co‐infection, concurrent liver disease other than HCV, past treatment with any experimental polymerase inhibitor, or hyperbilirubinaemia (> 1.5 ULN not due to Gilbert’s polymorphism). | |
| Interventions | Experimental group: oral 400 mg, 600 mg, or 800 mg 3 times daily of BI 207127 for 28 days. Control group: placebo. Co‐intervention: peg‐IFN α‐2a was administered subcutaneously at a dose of 180 lg per week, and RBV was given orally at a dose of 1000 mg per day (body weight < 75 kg) or 1200 mg per day (body weight > 75 kg) in 2 divided doses. Participants were advised to use sun protection. After 4 weeks, participants were given the opportunity to continue peg‐IFN α‐2a or 2b and RBV up to week 48 at the investigators' discretion. | |
| Outcomes | Efficacy assessment, safety assessment, drug resistance monitoring, HCV RNA, PK assessment. | |
| Notes | NCT00905632 Only treatment‐naive participants received placebo, and could be used in the analyses. We emailed Larrey and colleagues on 26 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Low risk | No participants in the treatment‐naive group were lost to follow‐up |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol (NCT00905632) were assessed |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 60 participants Sex: 48 men, 12 women Mean age: 50.2 years Inclusion criteria: treatment‐naive or treatment‐experienced participants without cirrhosis or treatment‐experienced participants with compensated cirrhosis female, aged 18‐70 years, with confirmed chronic HCV genotype 1 infection. Deleobuvir had shown activity against HCV genotype 1a and 1b in vitro; therefore, participants with either subgenotype were eligible. All participants had an HCV RNA level > 100,000 IU/mL at screening. The treatment‐experienced group included previous null responders, partial responders, and relapsers. The presence or absence of cirrhosis was confirmed by liver biopsy or transient elastography (Fibroscan 12.5 kPa). Exclusion criteria: hepatitis B or HIV co‐infection, concurrent liver disease other than HCV, past treatment with any experimental polymerase inhibitor, planned or concurrent use of any other approved or investigational pharmacological therapy, or current drug or alcohol abuse. Participants were also excluded if they had hyperbilirubinaemia, abnormal hematologic or laboratory values at screening, or concurrent disease considered clinically significant by the investigator. | |
| Interventions | Experimental group: rising doses of 100 mg, 200 mg, 400 mg, 800 mg, and 1200 mg every 8 h of deleobuvir (BI 207127). Control group: placebo. | |
| Outcomes | N25B variants, safety assessment, pharmacokinetics. | |
| Notes | We emailed Larrey and colleagues on 26 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blinded but method was not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as double‐blinded but method was not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described (it was described that 3 participants dropped out due to AEs) |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 33 participants Sex: 28 men, 5 women Mean age: not described. Inclusion criteria: treatment‐naive and treatment‐experienced noncirrhotic participants,18‐55 years old, with high viral load, genotype 1, chronic HCV infection. | |
| Interventions | Experimental group: oral 125 mg, 600 mg of MK‐7009 once daily for 8 days or 25 mg, 75 mg, 250 mg, or 500 mg of MK‐7009 twice daily for 8 days. Control group: placebo. | |
| Outcomes | HCV RNA, safety assessment. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on random, blinding, missing data, protocol, data, participants' characteristics, funding, number of participants in placebo/exp group but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as placebo‐blinded, but it was not described how blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as placebo‐blinded, but it was not described how blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | Several authors worked for several pharmaceutical companies |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 40 participants Sex: not reported Mean age: not reported Country: USA Inclusion criteria: participants both treatment‐naive and treatment‐experienced with chronic HCV 1 | |
| Interventions | The trial was divided into 4 cohorts, with different experimental intervention. Experimental group: oral 100 mg or 200 mg of VCH‐916 3 times daily for 14 days. Oral 300 or 400 mg of VCH‐916 twice daily for 3 days. Control group: placebo. | |
| Outcomes | Safesty assessment, HCV RNA level, pharmacokinetics | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on random, blinding, missing data, protocol, data, participants characteristics, funding, number of participants in placebo/exp group but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blinded, but it was not described how blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as double‐blinded, but it was not described how blinding was performed |
| Incomplete outcome data (attrition bias) | Low risk | All participants completed the study |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | Unclear risk | Not described |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 54 participants Country: USA Inclusion criteria: Adult treatment‐naive participants in genotype 1 HCV participants. | |
| Interventions | Experimental group: oral 25 mg, 75 mg, or 200 mg of GS‐9256 twice daily, or 300 mg of GS‐9256 once daily for 3 days. Control group: placebo. | |
| Outcomes | Safesty assessment, HCV RNA level, pharmacokinetics. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on random, blinding, missing data, protocol, data, participants characteristics, funding, number of participants in placebo/exp group but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blinded, but it was not described how blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as double‐blinded, but it was not described how blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | Several authors worked for Gilead Sciences |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 63 participants Inclusion criteria: non‐cirrhotic treatment‐naive adult participants with genotype 1 HCV participants. Exclusion criteria: not described. | |
| Interventions | The trial used 3 cohorts Experimental group: oral 100 mg, 200 mg, or 400 mg of PSI‐7977 once daily for 28 days. Control group: placebo. Co‐intervention: epg‐IFN/RBV. | |
| Outcomes | HCV RNA level, pharmacokinetics. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on random, blinding, missing data, protocol, data, participants characteristics, funding, number of participants in placebo/exp group but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blinded, but it was not described how blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as double‐blinded, but it was not described how blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | Main author worked for several pharmaceutical companies |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 63 participants Inclusion criteria: participants received at least 1 dose of the drug and in cohort 200 mg twice a day adults with hepatitis C genotype 1. | |
| Interventions | Experimental group: 200 mg or 400 mg of ANA598 twice a day. Control group: placebo. Co‐intervention: 12 weeks of standard of care treatment. | |
| Outcomes | Safety, antiviral activity, pharmacokinetics. | |
| Notes | Only the cohort with 200 mg is reported here. We emailed Lawitz and colleagues on 26 April 2016 for additional information on sequence generation, blinding, incomplete outcome data, number of deaths, SVR24 but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blind and placebo controlled, but the placebo was not further described. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants were actually randomised to the experimental and control group and therefore, it is unclear how many participants are with missing data |
| Selective reporting (reporting bias) | High risk | The trial did not report on the level of RBV and peg‐IFN in the blood as is stated in the protocol (NCT00978497) |
| Vested‐interest bias | High risk | The trial was supported by a company with an interest in a given result Hoffmann‐La Roche |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of other bias |
| Methods | Randomised clinical trial | |
| Participants | 35 participants Sex: 25 men, 10 women Mean age: 50 years Country: USA Inclusion criteria: treatment‐naive adults diagnosed with hepatitis C genotype 1. Exclusion criteria: not described. | |
| Interventions | The trial used different experimental groups, with different doses of ABT‐450. Experimental group: 50 mg ABT‐450 + 100 mg RBV, 100 mg ABT‐450 + 100 mg RBV, 200 mg ABT‐450 + 100 mg RBV once daily for 3 days. Control group: placebo. Co‐intervention: peg‐IFN α‐2a 180 mg/week + weight‐based RBV 1000–1200 mg/day (standard of care) for 12 weeks. After week 12, participants received standard of care treatment alone for 36 weeks. | |
| Outcomes | Safety assessment, HCV RNA level, pharmacokinetics. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on randomisation, blinding, missing data, protocol, data, funding, IL28b data but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as placebo‐controlled, but it was not described how blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as placebo‐controlled, but it was not described how blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out of the placebo group |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | Several authors worked for Gilead Sciences |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 252 participants Sex: 151 men, 101 women Countries: USA and Europe. Inclusion criteria: non cirrhotic treatment‐naive adult participants with chronic hepatitis C genotype 1. Exclusion criteria: not described. | |
| Interventions | Experimental group 1: oral tegobuvir 40 mg twice daily for 48 weeks. Control group: placebo. Co‐intervention: peg/RBV. | |
| Outcomes | Safety assessment, pharmacokinetics, HCV RNA. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on randomisation, blinding, missing data, protocol, complete trial, data, funding but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as placebo‐controlled, but it was not described how blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as placebo‐controlled, but it was not described how blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% percent dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | Several authors worked for Gilead Sciences |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 72 adult participants Sex: 52 men, 20 women Country: USA Inclusion criteria: 18–65 years of age, with chronic infection with genotype 1a or 1b HCV virus and plasma HCV RNA > 5 log10 IU/mL at screening. Participants were HCV treatment‐naive and had a BMI of 19–35 kg/m2 inclusive, creatinine clearance > 70 mL/min, and a QTcF interval < 450 ms. Exclusion criteria: known cirrhosis, hepatic decompensation, excessive ongoing alcohol intake, Gilbert’s syndrome, evidence of HCC, co‐infection with HIV or HBV, prothrombin time > 1.5 ULN, albumin < 3 g/dL, ALT and AST levels > 5 ULN, total bilirubin > ULN, hemoglobin < 11 g/dL, platelets < 90,000/mm3, or absolute neutrophil count < 1000 cells/mm3 (< 900 cells/mm3 for African Americans). Concomitant prescription or non‐prescription medications were prohibited during the study unless prior approval was received from the medical monitor. The only exception was the use of hormonal contraception; additional double barrier method contraception was mandated for all women of childbearing potential. | |
| Interventions | The trial was divided into 6 different cohorts, and randomised to experimental intervention or placebo. | |
| Outcomes | Safety assessment, pharmacokinetics, HCV RNA, viral sequencing. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on allocation, blinding, protocol, separate data from IL28b but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralised randomisation schedule generated via computer by the sponsor’s Biometrics group |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blinded, but it was not described how blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as double‐blinded, but it was not described how blinding was performed |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 person dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | This trial was supported by Gilead Sciences |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 90 participants Country: USA Inclusion criteria: treatment‐naive adult participants with chronic hepatitis C genotype 1. Exclusion criteria: not described. | |
| Interventions | The trial was divided into 9 cohorts Experimental group: oral 50 mg, 100 mg, or 300 mg of GS‐6620 once daily administered for 5 days. Oral 100 mg, 300 mg, or 900 mg of GS‐6620 once daily administered for 5 days. Oral 450 mg or 900 mg of GS‐6620 twice daily administered for 5 days. Control group: placebo. | |
| Outcomes | Safety assessment, pharmacokinetics, HCV RNA | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on randomisation, blinding, missing data, protocol, data, funding, SAE (non‐treatment related) but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as placebo‐controlled, but it was not described how blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as placebo‐controlled, but it was not described how blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | Several authors worked for Gilead Sciences |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 122 participants Sex: 73 men, 49 women Mean age: 49.4 years Country: USA Inclusion criteria: treatment‐naive participants HCV genotypes 1 had to have an HCV RNA concentration of 50,000 IU/mL or greater. HCV genotypes participants had a liver biopsy within 36 months before enrolment. Inclusion criteria also included the following haematological and biochemical laboratory variables: a neutrophil count of 1.5 × 10⁹/L (or ≥ 1.25 × 10⁹/L for black participants), a haemoglobin concentration of 11 g/dL or higher in women or 12 g/dL or higher in men, a platelet count of greater than 90 × 10⁹/L, total bilirubin within 2 times the ULN (21 μmol/L), and an albumin concentration of 30 g/L or lower. Exclusion criteria: cirrhosis, HBV or HIV, psychiatric illness, pulmonary or cardiac disease, seizure disorder, or other serious comorbid disorders. | |
| Interventions | Experimental group: oral 200 mg, or 400 mg of sofosbuvir once daily for 12 weeks. Control group: placebo. Co‐intervention: 48 weeks of peg‐IFN 180 μg per week subcutaneously; RBV was dosed according to weight (ie, participants < 75 kg received 1000 mg and those > 75 kg received 1200 mg; RBV was given in 2 daily doses. 400 mg in the morning and 600 mg in the evening for participants receiving 1000 mg a day, or 600 mg in the morning and 600 mg in the evening for participants receiving 1200 mg a day). | |
| Outcomes | Virological response, pharmacokinetics, AEs. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code |
| Allocation concealment (selection bias) | Low risk | Interactive online response system |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | High risk | The trial added additional secondary outcomes |
| Vested‐interest bias | High risk | The trial was funded by Gilead Sciences |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Lawitz 2013a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code |
| Allocation concealment (selection bias) | Low risk | Interactive online response system |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | High risk | The trial added additional secondary outcomes |
| Vested‐interest bias | High risk | The trial was funded by Gilead Sciences |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical phase I, multicentre trial | |
| Participants | 44 participants Sex: 32 men, 9 women Median age: 49 years Country: USA Inclusion criteria: 18‐65 years of age and had chronic HCV 1a or 1b and plasma HCV RNA > 5 log10 IU/mL at screening. Participants were HCV treatment‐naive and had a BMI of 19‐35 kg/m2 inclusive, creatinine clearance > 60 mL/min and a QTcF interval < 450 ms. Exclusion criteria: cirrhosis, hepatic decompensation, excessive ongoing alcohol intake, Gilbert's syndrome, evidence of HCC, co‐infection with HIV or HBV, ALT or AST levels > 5 x ULN, total bilirubin > ULN, haemoglobin < 11 g/dL, or absolute neutrophil count 1000 cells/mm2 (750 cells/mm2). Concomitant prescription during the study unless prior approval was received from the medical monitor. Participants using hormonal contraception were required to employ 2 additional barrier methods of contraception. | |
| Interventions | The trial divided into 4 cohorts, and randomised to experimental group or control group Experimental group: oral 60 mg, 200 mg (genotype 1a), 200 mg (genotype 1b), or 400 mg of GS‐9451 once daily for 3 days. Control group: placebo. | |
| Outcomes | Antiviral response, sequence analyses, pharmacokinetics, safety assessment. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on allocation, blinding (placebo pill), protocol but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | It was described that all were blinded, however it was not stated if there were any similarities between the placebo pill and intervention |
| Blinding of outcome assessment (detection bias) | Unclear risk | It was described that all were blinded, however it was not stated if there were any similarities between the placebo pill and intervention |
| Incomplete outcome data (attrition bias) | Unclear risk | There were above 5% dropouts. 3 participants were not included in the efficacy analyses. In addition, 3 participants were withdrawn after enrolment and not included in any analysis due to unknown reasons. It was unclear how the trial handled missing data. |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The trial was funded by Gilead Sciences |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised phase IIb clinical trial | |
| Participants | 211 participants Sex: 131 men, 80 women Mean age: 49.5 years Countries: Australia, Austria, Belgium, Canada, Chile, Czech Republic, France, Germany, Israel, Korea, Lithuania, New Zealand, Poland, South Korea, Sweden, Taiwan, Thailand, UK, and USA Inclusion criteria: treatment‐experienced non‐cirrhotic adults chronic genotype 1 HCV‐infected participants whose previous treatments with P/R had failed, a minimum of 25% of participants prior null responders, men and women 18–65 years of age, and baseline HCV RNA > 4 x 105 IU/mL. Exclusion criteria: non‐HCV‐related chronic hepatitis, HIV co‐infection, evidence of cirrhosis on liver biopsy or approved non‐invasive imaging, or any other condition contraindicated for treatment with P/R | |
| Interventions | 4 different experimental arms Experimental group 1: oral MK‐7009 600 mg twice daily for 24 weeks. Experimental group 3: oral MK‐7009 300 mg twice daily for 48 weeks. Experimental group 4: oral MK‐7009 600 mg twice daily for 48 weeks. Control group: placebo for 48 weeks. Co‐intervention: peg‐IFN 180 μg weekly and RBV 1000–1200 mg/day for 24–48 weeks. | |
| Outcomes | Safety assessment, SVR. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on randomisation, blinding, dealing with missing data, baseline characteristics for IL28B genotype but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | Above 5% dropouts, and it was unclear how the trial handled missing data |
| Selective reporting (reporting bias) | Low risk | All outcomes in the protocol were reported on. NCT00704405 |
| Vested‐interest bias | High risk | The trial was funded by Merck, Sharp & Dohme Corp |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 40 participants Sex: 36 men, 4 women Mean age: 43 years Country: USA Inclusion criteria: treatment‐naive ages between 18 and 65 years, non‐cirrhotic chronic HCV genotype 1 infection and HCV RNA levels of 50,000 IU/mL ages with BMIs ranging from 18‐36 kg/m2 Exclusion criteria: women were to be surgically sterile, postmenopausal for at least 12 months at screening, or taking protocol‐specified contraceptive measures. Positive for anti‐hepatitis A virus immunoglobulin M (IgM) antibodies, hepatitis B surface antigen, anti‐hepatitis B core protein IgM antibodies, or anti‐HIV antibodies. No medication associated with QT interval prolongation was permitted within 30 days prior to dosing or during the study, and any other concurrent medication required approval by the investigator and the sponsor. Participants who had received any systemic antineoplastic or immunomodulatory treatment within 6 months prior to the first dose of study drug or who might have needed such treatments at any time. | |
| Interventions | Participants were randomised in 4 cohorts with different doses of GS‐9851 Experimental group: 3 days of either 50 mg, 100 mg, 200 mg, or 400 mg as oral intake of GS‐9851. Control group: placebo. | |
| Outcomes | Pharmacokinetics, clinical virology assessment, safety and tolerability assessment. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on protocol, randomisation, blinding but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blinded, however it was not stated how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as double‐blinded, however it was not stated how the blinding was performed |
| Incomplete outcome data (attrition bias) | Low risk | All completed the study |
| Selective reporting (reporting bias) | Unclear risk | It was stated that there was a protocol, however the protocol could not be found. |
| Vested‐interest bias | High risk | The trial was funded by Pharmasset, Inc. Severina Moreira and Justin Cook of Niche Science and Technology Ltd. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 40 participants Sex: 33 men, 7 women Country: USA Inclusion criteria: participants aged 18–55 years with a BMI 18.5 to 636 kg/m2 and chronic, compensated, genotype1 HCV infection. All participants had a baseline HCV RNA > 106 IU/mL and no evidence of cirrhosis or bridging fibrosis (according to biopsy within 3 years of screening). Participants also had laboratory values within pre‐specified criteria at study entry. Exclusion criteria: participants previously treated with approved HCV therapy or with a DAA for HCV, or with chronic HBV or HIV infection were excluded. | |
| Interventions | Experimental group: received different doses of vaniprevir orally, for 8 days twice daily (25 mg, 75 mg, 250 mg, 500 mg, 700 mg) or 8 days once daily (125 mg, 600 mg). | |
| Outcomes | Safety, tolerability and efficacy, pharmacokinetics, medication adherence. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on allocation concealment, blinding of outcome assessors, sample size and protocol for trial 1 and 2 but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated centralised randomisation |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Matching placebo delivered in equal amounts |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only on person dropped out |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed |
| Vested‐interest bias | High risk | This study was funded by Merck Sharp & Dohme Corp |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 38 adult participants Country: USA Inclusion criteria: chronic hepatitis C genotype 1 either with cirrhosis, or without cirrhosis | |
| Interventions | Experimental group: oral 100 mg or 400 mg of ACH‐2684 once daily for 3 days. Oral 400 mg of ACH‐2684 twice daily. Control group: placebo. | |
| Outcomes | Safety assessment, pharmacokinetics, HCV RNA. | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on randomisation, blinding, missing data, protocol, data, funding, SAE, participants in each group but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as placebo‐controlled, but it was not described how blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as placebo‐controlled, but it was not described how blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | Several authors worked for Achillion pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 100 participants Sex: 65 men, 35 women Country: USA Inclusion criteria: compensated cirrhotic adults with chronic HCV genotype 1 infection. Exclusion criteria: not described. | |
| Interventions | Experimenatal group: oral 250 mg of GS‐9669 once daily for 8 weeks or oral 500 mg of GS‐9669 once daily for 8 weeks. Control group: RBV. | |
| Outcomes | Adverse events, HCV RNA SVR12 | |
| Notes | We emailed Lawitz and colleagues on 26 April 2016 for additional information on random, blinding, missing data, protocol, data separate from the groups, participants characteristics, funding, IL28b‐databut reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | Several authors worked for several pharmaceutical companies |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 85 adult participants Sex: 68 men, 19 women Mean age: 47 years Countries: USA and Puerto Rico Inclusion criteria: 18–65 years, with treatment‐naive chronic genotype 1–6 HCV infection and HCV RNA levels ≥5 log10 IU/mL at screening. Participants were required to have a BMI of 19–34 kg/m2 inclusive, creatinine clearance > 70 mL/min and QTcF ≤450 ms for men and ≤470 ms for women. Exclusion criteria: co‐infected with HBV or HIV, had prior treatment with a HCV NS5Ainhibitor, evidence of cirrhosis or HCC, history of clinical hepatic decompensation (e.g. ascites, jaundice, encephalopathy or variceal haemorrhage) or any other clinically significant condition other than chronic HCV infection. | |
| Interventions | The trial was divided into 11 dosing cohorts: 5 cohorts of participants with genotype 1a infection; 1 cohort of participants with genotype 1b infection, 1 cohort of participants with genotype 2 infection, 3 cohorts of participants with genotype 3 HCV infection and 1 cohort of participants with genotype 4 HCV infection. Experimental group: oral GS‐5816 (5 mg, 25 mg, 50 mg, 100 mg, 150 mg). | |
| Outcomes | Safety assessment, efficacy analysis, pharmacokinetic analysis. | |
| Notes | ClinicalTrials.gov number: NCT01740791. The trial reported that 87 participants were randomised, however it was also stated that those with genotype 4 (n = 2) were not randomised. Therefore we could not use data from the combined 150 mg group, as the non‐randomised genotype 4 participants were included in this group. We emailed Lawitz and colleagues on 26 April 2016 for additional information on allocation, blinding, how the trial handled missing data but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central computer‐generated randomisation scheme, by the sponsor |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Matching placebo |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | There were more than 5% dropouts and it was unclear how the trial handled missing data. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were reported on |
| Vested‐interest bias | High risk | The study was funded by Gilead Sciences |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical phase Ib trial | |
| Participants | 48 participants Sex: 48 men Country: USA Inclusion criteria: non‐cirrhotic participants aged 18‐60 years (up to 65 years old at the discretion of the investigator) with HCV RNA levels of > 100,000 IU/mL | |
| Interventions | The trial was divided into cohorts, in which randomisation was performed Experimental group: 5 mg, 10 mg, and 50 mg once daily of MK‐8742 for participants infected with genotype 1a or 1b, and 10 mg, 50 mg, and 100 mg once daily of MK‐8742 for participants infected with genotype 3. Control group: placebo. | |
| Outcomes | Activity, pharmacokinetics, safety | |
| Notes | We emailed Liu and colleagues on 26 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | High risk | The trial did not assess safety (NCT01532973) |
| Vested‐interest bias | High risk | The trial was funded by Merck Sharp & Dohme Corp |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 35 participants Sex: 24 men, 11 women Mean age: 47.6 years Inclusion criteria: treatment‐naive male or female participants with chronic hepatitis C aged 18‐55 years, with a BMI of 18‐35 were eligible for a multicenter, double‐blind, randomised, placebo‐controlled study. Participants were required to have HCV genotype 1a or 1b infection, a serum HCV RNA concentration greater than 75,000 IU/mL, a serum ALT concentration under 5 times the ULN, and compensated liver disease. Exclusion criteria: participants with evidence of cirrhosis or decompensated liver disease were excluded, as were participants with a history of or current alcohol abuse, poorly controlled insulin‐dependent diabetes, unstable or poorly controlled asthma, congestive heart failure, unstable cardiopulmonary disease, renal disease, or seizure disorder. Eligible participants in all studies were required to have a negative urine drug screen, serum pregnancy test (if female), and to have a negative hepatitis B surface antigen test and anti‐HIV antibody test. Pregnant and breast feeding female participants were ineligible. Other exclusion criteria included donation of 4500 mL of blood within 30 days (participants with chronic hepatitis C). | |
| Interventions | Experimental group: sequential cohorts of participants were randomly assigned to receive setrobuvir 200 mg, 400 mg, or 800 mg twice a day for 3 days. Control group: received placebo for 3 days. | |
| Outcomes | Safety, kinetics, antiviral activity. | |
| Notes | 5 participants originally enrolled in cohort 2 (400 mg twice a day) were dosed incorrectly. These participants received setrobuvir 200 mg twice a day and were thus included with cohort 1 in the analysis. We emailed Mallalieu and colleagues on 26 April 2016 for additional information random sequence generation + allocation, participants completing the study, blinding but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blinded but there was no further description of the placebo |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It is unclear if any participants dropped out |
| Selective reporting (reporting bias) | Low risk | The trial reports all outcomes stated in the protocol (NCT00782353) |
| Vested‐interest bias | High risk | The trial was sponsored by a company that might have an interest in a given outcome (Hoffmann‐La Roche) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 53 participants were randomised Sex: 27 men, 7 women Mean age: 48.9 years Inclusion criteria: participants with chronic HCV infection of genotype‐1 were recruited to the study, if they were treatment‐naive (no prior therapy with IFN, peg‐IFN, or RBV) or treatment‐experienced (virologic failure during or after treatment with an approved dose of peg‐IFN combined with RBV), had HCV RNA P100,000 IU/mL and were aged 18 years or older. Exclusion criteria: participants with liver cirrhosis, hyperbilirubinaemia (> 1.5x ULN; participants with Gilbert’s disease were accepted), HIV, or HBV co‐infection were excluded. Furthermore, participants who had previously received any treatment with a protease inhibitor and women of child‐bearing potential not agreeing or able to use medically accepted contraception throughout the study were excluded. | |
| Interventions | Experimental group: treatment‐naive participants: BI201335 monotherapy (20 mg, 48 mg, 120 mg, and 240 mg once a day) for 14 days, participants with a HCV RNA decrease P1 log10 from baseline (on Day 10), BI201335 treatment was combined with peg‐IFN α‐2a (180 lg/week) and RBV (1000 mg or 1200 mg/day) from Days 14 to 28. Control group: placebo combined with peg‐IFN α‐2a and RBV. All participants were offered to extend standard of care to Week 48, with an additional 24 weeks of follow‐up. Co‐intervention: peg‐IFN α‐2a (180 lg/week) and RBV (1000 mg or 1200 mg/day). | |
| Outcomes | Primary: virologic response, AEs, SAE, laboratory test abnormalities. Secondary: viral load reduction, change from baseline in viral load, rapid virological response, early virological response, complete early virological response 1+2, end of treatment response and SVR | |
| Notes | We emailed Manns and colleagues on 26 April 2016 for additional information on allocation concealment, random sequence generation, unpublished data, dealing with missing data, SVR data and AE, il28b and blinding in general. Data on SAEs and non‐SAEs distinguishing between treatment‐naive and treatment‐experienced but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as double‐blinded and but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 participant dropped out (The reason for the discontinuation of 1 participant was the diagnosis of an unexpected pregnancy of his partner representing an exclusion criterion for treatment with RBV) |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT00793793) and all outcomes were reported on |
| Vested‐interest bias | High risk | "Michael Manns has received grant support, contributed to clinical trials, and is a member of a speaker bureau and/or consulted for Schering Plough, Roche, Merck, Bristol‐Myers Squibb, Vertex, Tibotec, Astra/Arrows, Novartis, Human Genome Sciences, Boehringer Ingelheim, and Valeant. Peter W. White, Jerry Stern, Gerhard Steinmann, Chan‐Loi Yong, George Kukolj, Joe Scherer and Wulf O. Boecher are employees of Boehringer Ingelheim." |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 95 participants were randomised Sex: 55 men, 39 women Mean age: 46.2 years Inclusion criteria: adult, treatment‐naive participants with chronic, compensated, HCV genotype 1 infection, defined as HCV RNA levels ≥ 4 × 105 IU/mL at screening (i.e. within 75 days preceding the first dose of vaniprevir or placebo), were enrolled. All participants had positive serology for HCV or detectable HCV RNA ≥ 6 months before study initiation. Exclusion criteria: Participants with evidence of cirrhosis by histology, imaging, or physical findings were excluded. | |
| Interventions | Experimental group:
Control group: placebo plus open‐label peg‐IFN α‐2a and RBV 180 μg/week + 1000 mg‐1200 mg/day for 28 days. Co‐intervention: peg‐IFN α‐2a and RBV 180 μg/week + 1000 mg‐1200 mg/day. | |
| Outcomes | Primary: proportion of participants achieving RVR. AEs and participants that discontinued due to AEs. Exploratory: proportion of participants achieving EVR, proportion of participants achieving SVR. | |
| Notes | We emailed Manns and colleagues on 26 April 2016 for additional information on allocation concealment, unpublished data, correlation of il28b genotype data and SVR but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation procedure by an interactive voice‐response system |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The study was described as double‐blinded to investigator and participant |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment |
| Incomplete outcome data (attrition bias) | Unclear risk | 15 participants dropped out |
| Selective reporting (reporting bias) | Unclear risk | A protocol was found (NCT00704184), primary objectives were reported correctly, secondary outcomes changed and new exploratory outcomes were reported in the paper |
| Vested‐interest bias | High risk | This study was funded by Merck Scharp and Dohme Corp. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Manns 2012a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation procedure by an interactive voice‐response system |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The study was described as double‐blinded to investigator and participant |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Unclear risk | 15 participants dropped out |
| Selective reporting (reporting bias) | Unclear risk | A protocol was found (NCT00704184), primary objectives were reported correctly, secondary outcomes changed and new exploratory outcomes were reported in the paper |
| Vested‐interest bias | High risk | This study was funded by Merck Scharp and Dohme Corp. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Manns 2012a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation procedure by an interactive voice‐response system |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The study was described as double‐blinded to investigator and participant |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Unclear risk | 15 participants dropped out |
| Selective reporting (reporting bias) | Unclear risk | A protocol was found (NCT00704184), primary objectives were reported correctly, secondary outcomes changed and new exploratory outcomes were reported in the paper. |
| Vested‐interest bias | High risk | This study was funded by Merck Scharp and Dohme Corp. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Manns 2012a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation procedure by an interactive voice‐response system |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The study was described as double‐blinded to investigator and participant |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Unclear risk | 15 participants dropped out |
| Selective reporting (reporting bias) | Unclear risk | A protocol was found (NCT00704184), primary objectives were reported correctly, secondary outcomes changed and new exploratory outcomes were reported in the paper. |
| Vested‐interest bias | High risk | This study was funded by Merck Scharp and Dohme Corp. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | A phase III, randomised, double‐blind, placebo‐controlled, parallel‐design trial (QUEST‐2)(NCT01290679) | |
| Participants | 391 participants Location: 14 countries in Europe, North America, and South America Inclusion criteria: age ≥ 18 years. Chronic hepatitis C infection. HCV genotype 1. HCV RNA level at screening > 100,000 IU/mL. Treatment‐naive. An ultrasound performed within 6 months of enrolment showing no signs of HCC in participants with cirrhosis. Exclusion criteria: hepatic decompensation. Any non‐HCV‐related liver disease. HIV or HBV co‐infection. Non‐genotype 1 HCV infection. Significant laboratory abnormalities. Any other active disease. Male or female participants who had, or were planning to conceive. Simeprevir group: 257 participants Sex: 140 men, 117 women Median age: 46 years (range 18‐73) Race: 237 white (92%), 16 black or African American (6%), 2 Asian (< 1%), and 2 other (< 1%) HCV genotype 1a: 105 (41%), HCV genotype 1b: 150 (58%), other HCV genotype: 2 (< 1%) IL28B genotype CC: 75 (29%), IL28B genotype CT: 142 (55%), IL28B genotype TT: 40 (16%) METAVIR score F0‐F1: 130 (52%), METAVIR score F2: 65 (26%), METAVIR score F3: 36 (15%), METAVIR score F4: 17 (7%) HCV RNA > 800,000 IU/mL, n(%): 199(77). Placebo group: 134 participants Sex: 77 men, 57 women Median age: 47 years (range 18‐73) Race: 123 white (92%), 10 black or African‐American (10%), 1 Asian (< 1%), and 0 other HCV genotype 1a: 54 (41%), HCV genotype 1b: 77 (58%), other HCV genotype: 2 (2%) IL28B genotype CC: 42 (31%), IL28B genotype CT: 71 (53%), IL28B genotype TT: 21 (16%) METAVIR score, n(%): METAVIR score F0‐F1: 60 (45%), METAVIR score F2: 42 (31%), METAVIR score F3: 17 (13%), METAVIR score F4: 15 (11%) HCV RNA > 800,000 IU/mL, n(%): 98(73). | |
| Interventions | Experimental group: oral simeprevir 150 mg once daily for 12 weeks. Control group: oral placebo 150 mg once daily for 12 weeks. Co‐interventions: Experimental group: peg‐IFN α‐2a 180 μg subcutaneously once weekly or peg‐IFN α‐2b 1.5 μg/kg body weight subcutaneously once weekly and oral weight‐based RBV 1000 mg to 1200 mg in 2 divided daily doses (1000 mg if body weight < 75 kg; 1200 mg if body weight ≥ 75 kg) for 24‐48 weeks Control group: peg‐IFN α‐2a 180 μg subcutaneously once weekly or peg‐IFN α‐2b 1.5 μg/kg body weight subcutaneously once weekly and oral weight‐based RBV 1000 to 1200 mg in 2 divided daily doses (1000 mg if body weight < 75 kg; 1200 mg if body weight ≥75 kg) for 48 weeks. | |
| Outcomes | Primary outcome: proportion of participants achieving SVR12 (HCV RNA < 25 IU/mL undetectable at end of treatment and < 25 IU/mL detectable or undetectable 12 weeks after the planned end of treatment). Secondary outcomes: proportion of participants meeting criteria for response‐guided therapy to complete treatment at week 24. RVA (HCV RNA < 25 IU/mL undetectable at week 4). Activity, safety, and tolerability of simeprevir in the 2 subpopulations of participants who were given peg‐IFN α‐2a or 2b. On‐treatment failure (detectable HCV RNA at end of treatment). Incidence of viral relapse (HCV RNA ≥ 25 IU/mL during follow‐up or at the time of SVR assessments in participants with undetectable levels at end of treatment). Incidence of AEs. Incidence of laboratory abnormalities. Quality‐of‐life measures. SVR at 24 weeks after the planned end of treatment. Assessment of depression severity. Assessment of health status. Assessment of polymorphisms (HCV NS3 protease domain) at baseline and their correlation with efficacy of simeprevir plus peg‐IFN and RBV. | |
| Notes | We emailed Manns and colleagues on 26 April 2016 for additional information blinding of outcome assessors but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A computer‐generated randomisation schedule that was prepared by or under the supervision of the sponsor before the study was used" |
| Allocation concealment (selection bias) | Low risk | Concealment of allocation was obtained by using an interactive web‐based or voice‐response system |
| Blinding of participants and personnel (performance bias) | Low risk | Authors stated that "patients, study personnel, and the sponsor were masked to the treatment group assignment", the blinding method was not adequately described. A matched placebo was used |
| Blinding of outcome assessment (detection bias) | Unclear risk | It was not mentioned if the outcome assessors were blinded |
| Incomplete outcome data (attrition bias) | Low risk | Number and reasons for discontinuation were clearly reported on |
| Selective reporting (reporting bias) | Low risk | Protocol was available. All pre‐specified study outcomes were reported on |
| Vested‐interest bias | Unclear risk | "The sponsor (Janssen Infectious Diseases‐Diagnostics) was directly involved in trial design, data analyses and interpretation, and writing and reviewing the manuscript." |
| Other bias | Low risk | The trial seems to be free of other potential sources of bias |
| Methods | Randomised clinical trial | |
| Participants | 20 participants Inclusion criteria: treatment‐naive for chronic hepatitis C Countries: France, Moldova, Romania, USA | |
| Interventions | Experimental group: oral ALS‐2200 200 mg once daily for 7 days Control group: placebo for 7 days | |
| Outcomes | Safety assessment, HCV RNA | |
| Notes | We emailed Marcellin and colleagues on 27 April 2016 for additional information but reply not received yet. Ongoing study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | Safety assessment was not properly described (NCT01356160) |
| Vested‐interest bias | Unclear risk | Not described |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 351 participants Countries: France, Germany, Poland, and USA Inclusion criteria: treatment‐naive non‐cirrhotic genotype 1 infected HCV participants Exclusion criteria: not described | |
| Interventions | Experimental group: GS‐9451 (200 mg) once daily (those who achieved an extended very rapid virological response (defined as HCV RNA < LLOQ at Weeks 2 and 4 that remained undetectable through week 8) were randomised to stop treatment at either Week 12 or Week 24) Control group: no intervention Co‐intervention: GS‐5885 (30mg once a day) + peg (180 mg/week) + RBV (1000 mg–1200 mg/day) | |
| Outcomes | Adverse events, SVR | |
| Notes | We emailed Marcellin and colleagues on 27 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | Safety assessment was not properly described (NCT01356160) |
| Vested‐interest bias | Unclear risk | Not described |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised, open‐label, parallel‐group trial (ClinicalTrials.gov: NCT01331850) | |
| Participants | 381 participants, randomised: 152 prior partial responders (Cohort A) and 229 prior null responders (cohort B) Sex: 111 men, 40 women (Cohort A) Mean age: 49.4 years Countries: Australia, Austria, Brazil, Canada, France, Germany, Italy, Mexico, Poland, Spain, UK, and USA. Inclusion criteria: non‐cirrhotic adults with HCV genotype 1a or 1b infection, a baseline HCV RNA level P50,000 IU/mL and evidence of prior peg‐IFN α‐2a/RBV treatment failure. The prior course of treatment must have been discontinued > 12 weeks prior to enrolment, must have comprised at least 12 weeks of therapy with approved doses of peg‐IFNα/RBV and participants must have taken a minimum of approximately 80% of the prescribed doses. Prior treatment failure must have been due to either a partial response (> log10 reduction in HCV RNA at week 12, without achieving an undetectable HCV RNA level by the end of treatment), or a null response (< 2 log10 reduction in HCV RNA at week 12). Absence of cirrhosis must have been documented within 24 months of receiving the first dose of study drug either by liver biopsy (Knodell, METAVIR, Batts & Ludwig fibrosis score 63, or Ishak score 64) or, alternatively, by transient elastography (< 14.5 kPa). Participants with a previous liver biopsy were required to have a platelet count > 90 /nL and those with a transient elastography result were required to have a platelet count of 140–400 /nL Exclusion criteria: participants were excluded if they were co‐infected with HBV or HIV, had liver disease attributed to a cause other than HCV infection, had previously received a DAA agent or had a serious concomitant chronic illness. | |
| Interventions | Participants were grouped according to their prior treatment response (A: partial responders; B: null responders) and were randomised (1:1:1) within each cohort to 1 of 3 treatment arms, stratified by HCV genotype 1 subtype and host IL28B genotype. Participants who received at least 1 dose of study medication: 151 prior partial responders (Cohort A) and 228 prior null responders (cohort B). Experimental group A1: oral mericitabine 1000 mg twice a day for 24 weeks. Control group A2: peg‐IFNα‐2a 180 µg once weekly for 24 weeks. Experimental group A3: oral mericitabine 1000 mg twice a day for 24 weeks + peg‐IFN α‐2a 180 µg once weekly for 24 weeks. 24 weeks of peg‐IFNα‐2a/RBV. Co‐intervention: oral danoprevir/r 100/100 mg twice daily (twice a day) for 24 weeks + oral RBV 1000 mg (body weight < 75 kg) or 1200 mg (P75 kg) daily for 24 weeks (group A1,A2,A3,) | |
| Outcomes | Proportion of participants with sustained virological response (SVR24), with SAE, AEs, mortality. | |
| Notes | Due to the parallel design only group A1 and group A3 had an adequate control group (A2), Group B1, B2 and B3 were excluded from the analysis This analysis A1 vs. control. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization was centralised and the computer‐generated randomisation list was maintained |
| Allocation concealment (selection bias) | Low risk | Randomisation list was maintained by Perceptive Informatics (Waltham, MA, USA). "Study sites were informed of participant treatment assignments by an interactive voice/web response system." |
| Blinding of participants and personnel (performance bias) | High risk | Unblinded study |
| Blinding of outcome assessment (detection bias) | High risk | Unblinded study |
| Incomplete outcome data (attrition bias) | Low risk | Only 2 participants had incomplete data. |
| Selective reporting (reporting bias) | High risk | The authors did not report on "Change in danoprevir plasma concentration" as was prespecified in their protocol |
| Vested‐interest bias | High risk | This study was funded by F Hoffmann‐La Roche Ltd |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see MATTERHORN 2015a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was centralised and the computer‐generated randomisation list was maintained |
| Allocation concealment (selection bias) | Unclear risk | Randomisation list was maintained by Perceptive Informatics (Waltham, MA, |
| Blinding of participants and personnel (performance bias) | High risk | Unblinded study |
| Blinding of outcome assessment (detection bias) | High risk | Unblinded study |
| Incomplete outcome data (attrition bias) | Unclear risk | Only 2 participants had incomplete data. |
| Selective reporting (reporting bias) | High risk | The authors did not report on "Change in danoprevir plasma concentration" as was prespecified in their protocol |
| Vested‐interest bias | High risk | This study was funded by F Hoffmann‐La Roche Ltd |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | A phase IIb, randomised, double‐blind, multicenter, parallel‐group trial (PROVE‐1)(NCT00336479) | |
| Participants | 250 participants Sex: 157 men, 93 women Country: USA Inclusion criteria: age between 18 and 65 years. Chronic hepatitis C infection. HCV genotype 1. Treatment‐naive. Seronegative for hepatitis B surface. antigen and antibodies against HIV‐1 and HIV‐2. Absolute neutrophil count ≥ 1500 cells/mm3. Platelet count ≥ 90,000 cells/mm3. Normal haemoglobin level Exclusion criteria: decompensated liver disease. Another cause of clinically significant liver disease. HCC. Histologic evidence of cirrhosis (on liver biopsy, which was required within 2 years before the study). Group 1: 79 participants (T12PR24) Median age: 49 years (range 21‐61) Sex: 54 men, 25 women Race: 60 white (76%), 7 black (9%), 1 Asian (1%), 9 Hispanic (11%), and 2 other (3%) HCV genotype, n(%): 1a: 53(67), 1b: 17(22), intermediate: 9(11) HCV RNA ≥ 800,000 IU/mL, n(%): 66(84) Fibrosis, n(%): none or minimal: 24(30), portal: 41(52), bridging: 14(18) Group 2: 79 participants (T12PR48) Median age: 50 years (range 26‐61) Sex: 48 men, 31 women Race: 60 white (76%), 8 black (10%), 3 Asian (4%), 7 Hispanic (9%), and 1 other (1%) HCV genotype, n(%): 1a: 48(61), 1b: 27(34), intermediate: 4(5) HCV RNA ≥ 800,000 IU/mL, n(%): 68(86) Fibrosis, n(%): none or minimal: 34(43), portal: 31(39), bridging: 14(18) Group 3: 17 participants (T12PR12) Median age: 49 years (range 34‐63) Sex: 12 men, 5 women Race: 13 white (76%), 3 black (18%), 0 Asian, 1 Hispanic (6%), and 0 other HCV genotype, n(%): 1a: 9(53), 1b: 6(35), intermediate: 2(12) HCV RNA ≥800,000 IU/mL, n(%): 15(88) Fibrosis, n(%): none or minimal: 4(24), portal: 9(53), bridging: 4(24) Group 4: 75 participants (PR48) Median age: 49 years (range 24‐59) Sex: 43 men, 32 women Race: 59 white (79%), 9 black (12%), 0 Asian, 6 Hispanic (8%), and 1 other (1%) HCV genotype, n(%): 1a: 50(67), 1b: 20(27), intermediate: 5(7) HCV RNA ≥ 800,000 IU/mL, n(%): 69(92) Fibrosis, n(%): none or minimal: 19(25), portal: 37(49), bridging: 19(25). | |
| Interventions | Experimental group: 1, 2, and 3: oral telaprevir given as a single initial dose of 1250 mg, followed by 750 mg every 8 h for 12 weeks (T12). Control group: 4: Placebo for 12 weeks. Co‐interventions: 1: peg‐IFN α‐2a 180 μg subcutaneously once weekly plus oral weight‐based RBV 1000 mg‐1200 mg daily in 2 divided doses for 24 weeks (PR24). 2 and 4: peg‐IFN α‐2a 180 μg subcutaneously once weekly plus oral weight‐based RBV 1000 mg‐1200 mg daily in 2 divided doses for 48 weeks (PR48). 3: peg‐IFN α‐2a 180 μg subcutaneously once weekly plus oral weight‐based RBV 1000 mg‐1200 mg daily in 2 divided doses for 12 weeks (PR12). | |
| Outcomes | Primary outcome: proportion of participants with undetectable HCV RNA at 24 weeks after completion of study drug dosing (SVR24). Secondary outcomes: proportion of participants with SVR at 12 weeks after completion of study drug dosing. Number of participants with AEs and SAE. Number of participants with viral relapse. Maximum, minimum, and average plasma concentration of telaprevir. | |
| Notes | We emailed McHutchinson and colleagues on 27 April 2016 for additional information on random sequence generation, allocation concealment and SAE but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method of random sequence generation was not described |
| Allocation concealment (selection bias) | Unclear risk | Not enough information was provided |
| Blinding of participants and personnel (performance bias) | Low risk | A telaprevir‐matched placebo given in the same manner was used |
| Blinding of outcome assessment (detection bias) | Low risk | "Data management and interim analyses were performed by the Duke Clinical Research Institute. An independent data‐monitoring committee reviewed the results of all interim analyses" |
| Incomplete outcome data (attrition bias) | High risk | The number of participants who discontinued treatment was clearly stated, but reasons were not mentioned. Up to 36% of participants in a group discontinued study treatment |
| Selective reporting (reporting bias) | Low risk | The protocol was available and all pre‐specified outcomes were reported on |
| Vested‐interest bias | High risk | The sponsor (Vertex Pharmaceuticals) was directly involved in trial design and protocol development |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | A phase II, randomised, partially placebo‐controlled, partially double‐blind, parallel‐group trial (PROVE‐3)(NCT00420784) | |
| Participants | 453 participants Sex: 306 men, 147 women Mean age: 51 years Inclusion criteria: age between 18 and 70 years, chronic hepatitis C infection, HCV genotype 1, previously treated, but without achieving SVR. Seronegative for hepatitis B surface antigen and antibodies against HIV‐1 and HIV‐2, absolute neutrophil count ≥ 1500 cells/mm3, platelet count ≥ 100,000 cell/mm3, normal bilirubin values. Exclusion criteria: decompensated liver disease, HCC, other clinically significant liver disease. Country: Canada, Germany, the Netherlands, Puerto Rico and USA. Group 1: 115 participants (T12PR24) Sex: 78 men, 37 women Median age: 51 years (range 22‐65) Race, n(%): white: 103(90), black: 9(8), Asian: 2(2), other: 1(1) HCV genotype, n(%): 1a: 69(60), 1b: 33(29), unknown: 13(11) HCV RNA ≥ 800,000 IU/mL, n(%): 106(92) Stage of fibrosis or cirrhosis, n(%): none or minimal: 26(23), portal fibrosis: 44(38), bridging fibrosis: 26(23), cirrhosis. 19(17) Group 2: 113 participants (T24PR48) Sex: 80 men, 33 women Median age: 52 years (range 31‐66) Race, n(%): white: 99(88), black: 11(10), Asian: 0, other: 3(3) HCV genotype, n(%): 1a: 61(54), 1b: 42(37), unknown: 10(9) HCV RNA ≥ 800,000 IU/mL, n(%): 104(92) Stage of fibrosis or cirrhosis, n(%): None or minimal: 20(18), portal fibrosis: 40(35), bridging fibrosis: 33(29), cirrhosis. 20(18) Group 3: 111 participants (T24PR24) Sex: 72 men, 39 women Median age: 53 years (range 19‐69) Race, n(%): white: 100(90), black: 10(9), Asian: 1(1), other: 0. HCV genotype, n(%): 1a: 64(58), 1b: 36(32), unknown: 11(10) HCV RNA ≥ 800,000 IU/mL, n(%): 104(94) Stage of fibrosis or cirrhosis, n(%): none or minimal: 17(15), portal fibrosis: 40(36), bridging fibrosis: 32(29), cirrhosis. 22(20) Group 4: 114 participants (PR48) Sex: 76 men, 38 women Median age: 50 years (range 18‐65) Race, n(%): white: 100(88), black: 10(9), Asian: 2(2), other: 2(2) HCV genotype, n(%): 1a: 71(62), 1b: 34(30), unknown: 9(8) HCV RNA ≥ 800,000 IU/mL, n(%): 104(91) Stage of fibrosis or cirrhosis, n(%): none or minimal: 33(29), portal fibrosis: 37(32), bridging fibrosis: 31(27), cirrhosis 13(11). | |
| Interventions | Experimental group: 1: oral telaprevir given in a single initial dose of 1125 mg, followed by 750 mg every 8 h for 12 weeks (T12). 2 and 3: oral telaprevir given in a single initial dose of 1125 mg, followed by 750 mg every 8 h for 24 weeks (T24). Control group: 1: placebo from Week 13 to Week 24. 4: placebo for 24 weeks. Co‐intervention: 1 and 3: peg‐IFN α‐2a 180 μg subcutaneously once weekly plus oral weight‐based RBV 1000 mg to 1200 mg daily in 2 divided doses for 24 weeks (PR24). 2 and 4: peg‐IFN α‐2a 180 μg subcutaneously once weekly plus oral weight‐based RBV 1000 mg to 1200 mg daily in 2 divided doses for 48 weeks (PR48). | |
| Outcomes | Primary outcome: SVR defined as undetectable HCV RNA level 24 weeks after the last dose of study drugs. Secondary outcome measures: proportion of participants with undetectable HCV RNA at completion of study drug dosing. Number of participants with AEs and SAE. Number of participants with viral relapse. Maximum, minimum, and average plasma concentration of telaprevir. | |
| Notes | We emailed McHutchinson and colleagues on 27 April 2016 for additional information on generation of random sequence, allocation concealment, description of blinding but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method of sequence generation was not specified |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information was provided on allocation concealment |
| Blinding of participants and personnel (performance bias) | Unclear risk | The method of blinding was insufficiently described |
| Blinding of outcome assessment (detection bias) | Low risk | "Data management and interim analyses were conducted by the Duke Clinical Research Institute, without revealing the unblinded data" |
| Incomplete outcome data (attrition bias) | Low risk | Number and reasons for discontinuation of treatment were clearly reported. Most participants discontinued treatment due to meeting pre‐specified stopping rules |
| Selective reporting (reporting bias) | Low risk | Protocol was available and all pre‐specified outcomes were reported on |
| Vested‐interest bias | High risk | The sponsor (Vertex Pharmaceuticals) was directly involved in trial design, protocol development, study co‐ordination, drafting and reviewing the manuscript |
| Other bias | Low risk | The trial appeared to be free of other potential sources of bias |
| Methods | Randomised clinical trial | |
| Participants | 40 participants Inclusion criteria: previously untreated adults with chronic hepatitis C genotype 4 infection. Country: Egypt | |
| Interventions | Experimental group: 44 weeks of boceprevir 800 mg 3 times daily. Control group: no intervention. Co‐intervention: peg α‐2b 1.5 lg/kg once per week subcutaneously plus weight‐based dosing RBV 15 mg/kg/day (800 mg‐1400 mg/day) for 48 weeks. | |
| Outcomes | Proportion of participants who achieved early response | |
| Notes | We emailed Mostafa and colleagues on 27 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | Open label study |
| Blinding of outcome assessment (detection bias) | High risk | Open label study |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | The trial is not finished according to ClinicalTrials.gov, therefore not all data might have been collected yet |
| Vested‐interest bias | Low risk | Trial was funded by a non‐profit organisation (Theodor Bilharz Research Institute) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 30 participants Sex: 18 men, 12 women Mean age: 51.7 years Inclusion criteria: adults with chronic hepatitis C, HCV RNA > 10,000 IU/mL at screening, treatment‐naive participants defined as participants who have never received peg‐IFN, RBV, or a DAA agent for the treatment of chronic HCV infection and a liver biopsy within the last 3 years without evidence of cirrhosis. Exclusion criteria: BMI > 36.0, pregnant or nursing (lactating) women, confirmed by a positive human chorionic gonadotropin laboratory test or women contemplating pregnancy, participation in any interventional clinical trial within 35 days prior to first study medication dose administration on Day 1, known HIV‐1 or HIV‐2 infection/serology and/or positive Hepatitis B surface antigen, use of dietary supplements, grapefruit juice, herbal supplements, cytochrome P2C8 substrates, cytochrome P3A4 inducers and inhibitors, P‐glycoprotein inducers and substrates, organic anion transporting polypeptides inhibitors and substrates, and potent inducers of other cytochrome P enzymes within 14 days prior to dosing through 7 days following completion of study meds. Clinically significant laboratory abnormality at screening (specified in protocol), other forms of liver disease, history of severe or uncontrolled psychiatric disease, history of malignancy of any organ system, treated or untreated within the past 5 years, history of major organ transplantation, use of bone marrow colony‐stimulating factor agents within 3 months prior to baseline, history of seizure disorder requiring ongoing medical therapy, history of known coagulopathy including haemophilia, history of haemoglobinopathy, including sickle cell anemia and thalassaemia, history of immunologically‐mediated disease (specified in protocol), history of clinical evidence of significant chronic cardiac disease (specified in protocol), ECG with any clinically significant abnormality, structural or functional cardiac abnormalities (specified in protocol), history of chronic obstructive pulmonary disease, emphysema, or other chronic lung disease, participants currently abusing amphetamines, cocaine or opiates, or with ongoing alcohol abuse in the judgement of the investigator. | |
| Interventions | Experimental group: Arm 1: sovaprevir 200 mg once a day + ACH‐3102 150 mg loading dose on Day 1 followed by 50 mg once a day + RBV weight‐based 1000 mg‐1200 mg once a day for 12 weeks. Arm 2: sovaprevir 400 mg once a day + ACH‐3102 150 mg loading dose on Day 1 followed by 50 mg once a day + RBV weight‐based 1000 mg‐1200 mg once a day for 12 weeks. Control group: placebo for sovaprevir capsule once a day + placebo for ACH‐3102 150 mg loading dose on Day 1 followed by 50 mg capsule once a day + placebo for weight‐based RBV once a day for 12 weeks. | |
| Outcomes | Safety, SVR4 (only experimental group). | |
| Notes | We contacted the trial authors about random sequence generation, allocation, participants completing the study, blinding, number of deaths, SVR24. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blinded but there was no description of the placebo |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as double‐blinded but there was no description of the placebo |
| Incomplete outcome data (attrition bias) | Unclear risk | There were above 5% dropouts (2/30) and it was unclear how the trial dealt with missing data |
| Selective reporting (reporting bias) | High risk | The original secondary outcomes were later removed (NCT01849562) |
| Vested‐interest bias | High risk | The trial was sponsored by a company with a given interest in a result (Achillion Pharmaceuticals) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Phase IIb, randomised, dose‐ranging, parallel‐design trial (PROTON) | |
| Participants | 121 participants Country: not stated Inclusion criteria: chronic hepatitis C, genotype 1, treatment‐naive participants. Exclusion criteria: cirrhosis. | |
| Interventions | Experimental group: Group 1: 95 participants: PSI‐7977 200 or 400 mg daily for 12 weeks. Control group: Group 2: 26 participants: placebo for 12 weeks. Co‐intervention in both groups: peg‐IFN α‐2a for 24‐48 weeks in a response‐guided regimen. RBV for 24‐48 weeks in a response‐guided regimen. | |
| Outcomes | Not clearly stated. | |
| Notes | We contacted the trial authors about whole risk of bias assessment, male:female ratio, SVR results and AEs. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information |
| Blinding of participants and personnel (performance bias) | Unclear risk | Use of placebo suggests blinding, but method not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | There was insufficient information to assess whether missing data were likely to induce bias on the results |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. Not enough information given |
| Vested‐interest bias | Unclear risk | It was uncertain how the trial was sponsored |
| Other bias | Low risk | The trial may or may not have been free of other domains that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 516 adult participants Sex: 311 men, 193 women (analysed only) Mean age: 46.5 years Inclusion criteria: participants aged 18‐65 years with HCV genotype l infection who had never received treatment for chronic hepatitis C were eligible for the trial. Chronic hepatitis C was defined as the presence of anti‐HCV antibodies and an HCV RNA titer ≥ 50,000 IU/mL in serum (COBAS® Ampliprep/COBAS® TaqMan® HCV test; detection limit 15 IU/mL, Roche Diagnostics, Indianapolis, USA) with a liver biopsy obtained within the previous 24 months (36 months in participants with cirrhosis or incomplete/transition to cirrhosis) consistent with chronic hepatitis C. HCV genotype 1 infection was confirmed by a molecular assay (Versant HCV Genotyping 2.0 Assay (LiPA), Bayer Diagnostics And Innogenetics, NY, USA). Participants with advanced fibrosis according to a biopsy obtained within the previous 36 months were required to have compensated liver disease (Child–Pugh grade A), a serum α‐ fetoprotein level < 100 ng/mL, and no evidence of HCC on an ultrasound, computerised tomography, or magnetic resonance imaging scan performed within the previous 2 months. Exclusion criteria: participants were not eligible if they were infected with any HCV genotype other than genotype 1 or had serological evidence of infection with HBV or HIV. Participants were also excluded if they had a BMI < 18 kg/ m2 or ≥ 36 kg/m2, an absolute neutrophil count < 2 × 109 cells/L, a platelet count < 90 × 109 cells/L, a hemoglobin concentration < 120 g/L in women or < 130 g/L in men (or in participants with risk factors for anemia or in whom anemia would be medically problematic), or a serum creatinine level > 1.5 times the ULN. Use of erythropoietin‐stimulating agents or colony‐stimulating factors to elevate haematology parameters to facilitate entry into the study was prohibited. Participants who had previously received any IFN preparation, RBV (or RBV analog), or any investigational HCV protease or polymerase inhibitor were excluded, as were those with a history or evidence of a chronic liver disease other than chronic hepatitis C, a current or past history of chronic disease (including severe psychiatric or pulmonary disease), or a history or evidence of a clinically relevant ophthalmological disorder (e.g. cytomegalovirus infection or macular degeneration). Pregnant or breast‐feeding women and male partners of pregnant women were ineligible for the trial. Female participants of childbearing potential and male participants with partners of childbearing potential were required to use 2 forms of effective contraception during treatment and after the last dose of RBV in accordance with the locally approved label for RBV. | |
| Interventions | Expertimental group:
Control group: placebo. Co‐interventions: Copegus 1000 mg/1200 mg orally daily for 48 weeks. Peg 180 μgsubcutaneously weekly for 24 weeks (groups 1‐3 + control). Copegus 1000 mg/1200 mg orally daily for 48 weeks. Peg 90 μg subcutaneously weekly for 24 weeks (groups 4‐6 + control). | |
| Outcomes | Safety, antiviral activity, SVR12, relapse. | |
| Notes | The planned treatment duration with balapiravir was reduced from 24 to 12 weeks due to safety concerns. We emailed Nelson and colleagues on 06 June 2016 for additional information on incomplete outcome data and SVR but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The computerised randomisation list was generated by the sponsor, maintained in a central repository accessible only to the randomisation list managers, and incorporated in double‐blind labelling of medication containers |
| Allocation concealment (selection bias) | Low risk | The computerised randomisation list was generated by the sponsor, maintained in a central repository accessible only to the randomisation list managers, and incorporated in double‐blind labelling of medication containers |
| Blinding of participants and personnel (performance bias) | High risk | "All patiants were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peg‐IFN α‐2a (40KD) and RBV was permanently discontinued" |
| Blinding of outcome assessment (detection bias) | High risk | "All patients were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peg‐IFN α‐2a (40KD) and RBV was permanently discontinued" |
| Incomplete outcome data (attrition bias) | Unclear risk | There was more than 5% dropouts and it was unclear how the trial accounted for missing data |
| Selective reporting (reporting bias) | High risk | Relapse rate was not reported on despite being stated as an outcome in the protocol (NCT 00517439). |
| Vested‐interest bias | High risk | The trial was supported by a company with an interest in a given result (Hoffmann‐La Roche) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Nelson 2012a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The computerised randomisation list was generated by the sponsor, maintained in a central repository accessible only to the randomisation list managers, and incorporated in double‐blind labelling of medication containers |
| Allocation concealment (selection bias) | Low risk | The computerised randomisation list was generated by the sponsor, maintained in a central repository accessible only to the randomisation list managers, and incorporated in double‐blind labelling of medication containers |
| Blinding of participants and personnel (performance bias) | High risk | All participants were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peg‐IFN alfa‐2a (40KD) and RBV was permanently discontinued |
| Blinding of outcome assessment (detection bias) | High risk | All participants were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peg‐IFN alfa‐2a (40KD) and RBV was permanently discontinued |
| Incomplete outcome data (attrition bias) | Unclear risk | There was more than 5% dropouts and it was unclear how the trial accounted for missing data |
| Selective reporting (reporting bias) | High risk | Relapse rate was not reported on despite being stated as an outcome in the protocol (NCT 00517439). |
| Vested‐interest bias | High risk | The trial was supported by a company with an interest in a given result (Hoffmann‐La Roche) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Nelson 2012a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The computerised randomisation list was generated by the sponsor, maintained in a central repository accessible only to the randomisation list managers, and incorporated in double‐blind labelling of medication containers |
| Allocation concealment (selection bias) | Low risk | The computerised randomisation list was generated by the sponsor, maintained in a central repository accessible only to the randomisation list managers, and incorporated in double‐blind labelling of medication containers |
| Blinding of participants and personnel (performance bias) | High risk | All participants were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peg‐IFN alfa‐2a (40KD) and RBV was permanently discontinued. |
| Blinding of outcome assessment (detection bias) | High risk | All participants were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peg‐IFN alfa‐2a (40KD) and RBV was permanently discontinued |
| Incomplete outcome data (attrition bias) | Unclear risk | There was more than 5% dropouts and it was unclear how the trial accounted for missing data |
| Selective reporting (reporting bias) | High risk | Relapse rate was not reported on despite being stated as an outcome in the protocol (NCT 00517439) |
| Vested‐interest bias | High risk | The trial was supported by a company with an interest in a given result (Hoffmann‐La Roche) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Nelson 2012a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The computerised randomisation list was generated by the sponsor, maintained in a central repository accessible only to the randomisation list managers, and incorporated in double‐blind labelling of medication containers |
| Allocation concealment (selection bias) | Low risk | The computerised randomisation list was generated by the sponsor, maintained in a central repository accessible only to the randomisation list managers, and incorporated in double‐blind labelling of medication containers |
| Blinding of participants and personnel (performance bias) | High risk | All participants were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peg‐IFN alfa‐2a (40KD) and RBV was permanently discontinued |
| Blinding of outcome assessment (detection bias) | High risk | All participants were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peg‐IFN alfa‐2a (40KD) and RBV was permanently discontinued |
| Incomplete outcome data (attrition bias) | Unclear risk | There was more than 5% dropouts and it was unclear how the trial accounted for missing data |
| Selective reporting (reporting bias) | High risk | Relapse rate was not reported on despite being stated as an outcome in the protocol (NCT 00517439). |
| Vested‐interest bias | High risk | The trial was supported by a company with an interest in a given result (Hoffmann‐La Roche) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Nelson 2012a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The computerised randomisation list was generated by the sponsor, maintained in a central repository accessible only to the randomisation list managers, and incorporated in double‐blind labelling of medication containers |
| Allocation concealment (selection bias) | Low risk | The computerized randomizsation list was generated by the sponsor, maintained in a central repository accessible only to the randomizsation list managers, and incorporated in double‐blind labelling of medication containers |
| Blinding of participants and personnel (performance bias) | High risk | All participants were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peg‐IFN alfa‐2a (40KD) and RBV was permanently discontinued. |
| Blinding of outcome assessment (detection bias) | High risk | All participants were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peg‐IFN alfa‐2a (40KD) and RBV was permanently discontinued |
| Incomplete outcome data (attrition bias) | Unclear risk | There was more than 5% dropouts and it was unclear how the trial accounted for missing data |
| Selective reporting (reporting bias) | High risk | Relapse rate was not reported on despite being stated as an outcome in the protocol (NCT 00517439) |
| Vested‐interest bias | High risk | The trial was supported by a company with an interest in a given result (Hoffmann‐La Roche) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Nelson 2012a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The computerised randomisation list was generated by the sponsor, maintained in a central repository accessible only to the randomisation list managers, and incorporated in double‐blind labelling of medication containers |
| Allocation concealment (selection bias) | Low risk | The computerised randomisation list was generated by the sponsor, maintained in a central repository accessible only to the randomisation list managers, and incorporated in double‐blind labelling of medication containers |
| Blinding of participants and personnel (performance bias) | High risk | All participants were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peg‐IFN alfa‐2a (40KD) and RBV was permanently discontinued. |
| Blinding of outcome assessment (detection bias) | High risk | All participants were unblinded to their treatment assignment and, among those who received balapiravir and who had a CD4+ count < 200 cells/mm3, treatment with peg‐IFN alfa‐2a (40KD) and RBV was permanently discontinued. |
| Incomplete outcome data (attrition bias) | Unclear risk | There was more than 5% dropouts and it was unclear how the trial accounted for missing data |
| Selective reporting (reporting bias) | High risk | Relapse rate was not reported on despite being stated as an outcome in the protocol (NCT 00517439). |
| Vested‐interest bias | High risk | The trial was supported by a company with an interest in a given result (Hoffmann‐La Roche) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 323 adult participants Inclusion criteria: chronic HCV infection for at least 6 months prior to baseline (Day 1), liver biopsy results (performed no more than 2 years prior to screening) indicating the absence of cirrhosis, mono‐infection with HCV genotype 1a or 1b, HCV treatment‐naive, BMI between 18 and 36 kg/m2, creatinine clearance >/= 50 mL/min, participant agreed to use highly effective contraception methods if female of childbearing potential or sexually active male, screening laboratory values within defined thresholds for ALT, AST, leukopenia, neutropenia, anaemia, thrombocytopenia, thyroid stimulating hormone, potassium, magnesium. Exclusion criteria: autoimmune disease, decompensated liver disease or cirrhosis, poorly controlled diabetes mellitus, severe psychiatric illness, severe chronic obstructive pulmonary disease, serological evidence of co‐infection with HIV, HBV, or another HCV genotype, suspicion of HCC or other malignancy (with exception of certain skin cancers), history of haemoglobinopathy, known retinal disease. participants who were immunosuppressed, participants with known, current use of amphetamines, cocaine, opiates (i.e. morphine, heroin), methadone, or ongoing alcohol abuse, participants who were on or are expected to be on a potent cytochrome P450 (CYP) 3A4 or Pgp inhibitor, or a QT prolonging medication within 2 weeks of baseline (Day 1) or during the study, participants must have had no history of clinically significant cardiac disease, including a family history of Long QT syndrome, and no relevant ECG abnormalities at screening. | |
| Interventions | Experimental group 1: tegobuvir (20 mg twice a day) + GS‐9256 (150 mg twice a day). Experimental group 2: GS‐9256 (150 mg twice a day). Control group: placebo. Co‐intervention: Peg (180 mg/week) + RBV (1000–1400 mg/day). | |
| Outcomes | Safety, SVR12 (not fully reported so could not be used). | |
| Notes | Participants receiving the 4‐drug therapy who achieved an extended vRVR were randomised to stop treatment at either Week 16 or Week 24. We contacted the trial authors on 06 June 2016 for additional information allocation sequence generation, blinding, dropouts and how this was handled, primary publication, SAE, death, SVR24, number of participants randomised to each group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double blind but the placebo was not further described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants dropped out |
| Selective reporting (reporting bias) | High risk | Not all predefined outcomes in the protocol were reported on (viral resistance, SVR24) |
| Vested‐interest bias | High risk | The trial was supported by a company that might have an interest in a given result (Gilead Sciences) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 18 participants Sex: 10 men, 8 women Mean age: 44 years Inclusion criteria: participants chronically infected with hepatitis C virus genotype 1, treatment‐naive or treatment non‐responders or treatment intolerant; and not co‐infected with HIV or HBV, HCV‐RNA viral load of ≥ 10*5* IU/mL and had a BMI 18‐35 kg/m² Exclusion criteria: any significant acute or chronic medical illness which was not stable or not controlled with medication and not consistent with HCV infection and major surgery within 4 weeks of study drug administration and any gastrointestinal surgery that could impact the absorption of study drug | |
| Interventions | Experimental group:
Control group: placebo | |
| Outcomes | Pharmacokinetics, antiviral activity, safety. | |
| Notes | We contacted trial authors for additional information on allocation sequence generation and concealment, how was blinding maintained, whether HIV participants included. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blinded but the placebo was not described in detail |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% of participants dropped out and it was unclear how the trial handled missing data |
| Selective reporting (reporting bias) | Low risk | All the outcomes stated in the protocol were reported on NCT00546715 |
| Vested‐interest bias | Unclear risk | The trial was funded by a company that might have an interest in a given result (Bristol‐Myers Squibb) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | Sex: 25 men, 5 women Mean age: 44.3 years Inclusion criteria: eligible participants for this study were men and women, ages 18‐60 years inclusive, with a BMI of 18‐35 kg/m2, who were chronically infected (longer than 6 months) with HCV genotype 1, and who were treatment‐naive to IFN and RBV. Additional inclusion criteria were: plasma HCV RNA 100,000 IU/mL; documented FibroTest score of 0.72 and APRI 2, or the absence of cirrhosis based on liver biopsy within 12 months; women of childbearing potential were not to be nursing or pregnant and had to be willing to agree to use double barrier contraception for at least 1 month before dosing, during dosing, and at least 12 weeks after the last dose of study medication. Exclusion criteria: participants with prior documented cirrhosis on liver biopsy; previous exposure to a NS5A replication cofactor inhibitor; co‐infection with HIV; co‐infection with HBV. | |
| Interventions | Experimental group:
Control group: placebo. | |
| Outcomes | Pharmacokinetics, mortality, SAE, antiviral efficacy | |
| Notes | We contacted the trial authors on 06 June 2016 for additional information on blinding of participants, personnel and outcome assessors, SVR24 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Compute‐ generated randomisation scheme |
| Allocation concealment (selection bias) | Low risk | Interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as double‐blinded but the placebo was not described in detail |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 participant did not complete the study until day 28 |
| Selective reporting (reporting bias) | High risk | There were added multiple secondary outcomes to the original protocol after the trial was conducted (NCT00663208) |
| Vested‐interest bias | High risk | The trial was supported by a company that might have an interest in a given result (Bristol‐Myers Squibb) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Nettles 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation scheme |
| Allocation concealment (selection bias) | Low risk | Interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as double‐blinded but the placebo was not described in detail |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 participant did not complete the study until day 28 |
| Selective reporting (reporting bias) | High risk | There were added multiple secondary outcomes to the original protocol after the trial was conducted (NCT00663208) |
| Vested‐interest bias | High risk | The trial was supported by a company that might have an interest in a given result (Bristol‐Myers Squibb) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Nettles 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation scheme |
| Allocation concealment (selection bias) | Low risk | Interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as double‐blinded but the placebo was not described in detail |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 participant did not complete the study until day 28 |
| Selective reporting (reporting bias) | High risk | There were added multiple secondary outcomes to the original protocol after the trial was conducted (NCT00663208) |
| Vested‐interest bias | High risk | The trial was supported by a company that might have an interest in a given result (Bristol‐Myers Squibb) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Nettles 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation scheme |
| Allocation concealment (selection bias) | Low risk | Interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as double‐blinded but the placebo was not described in detail |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 participant did not complete the study until day 28 |
| Selective reporting (reporting bias) | High risk | There were added multiple secondary outcomes to the original protocol after the trial was conducted (NCT00663208) |
| Vested‐interest bias | High risk | The trial was supported by a company that might have an interest in a given result (Bristol‐Myers Squibb) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Nettles 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation scheme |
| Allocation concealment (selection bias) | Low risk | Interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as double‐blinded but the placebo was not described in detail |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 participant did not complete the study until day 28 |
| Selective reporting (reporting bias) | High risk | There were added multiple secondary outcomes to the original protocol after the trial was conducted (NCT00663208) |
| Vested‐interest bias | High risk | The trial was supported by a company that might have an interest in a given result (Bristol‐Myers Squibb) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Nettles 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation scheme |
| Allocation concealment (selection bias) | Low risk | Interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as double‐blinded but the placebo was not described in detail |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 participant did not complete the study until day 28 |
| Selective reporting (reporting bias) | High risk | There were added multiple secondary outcomes to the original protocol after the trial was conducted (NCT00663208) |
| Vested‐interest bias | High risk | The trial was supported by a company that might have an interest in a given result (Bristol‐Myers Squibb) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | Sex: 13 men, 9 women Mean age: 53.9 years Inclusion criteria: treatment‐naive adults aged 20–70 years, with chronic genotype‐1 HCV infection and HCV RNA viral load at screening ≥ 100,000 IU/mL. Exclusion criteria: cirrhosis. | |
| Interventions | Experimental group: 1: faldaprevir 120 mg once a day (treatment‐naive). 2: faldaprevir 240 mg once a day (treatment‐naive). Control group: placebo. Co‐intervention: peg‐IFN α‐2a 180 μg and RBV 600 mg/day (≤ 60 kg), 800 mg/day (> 60 to ≤ 80 kg) or 1000 mg/day (> 80 kg). Both peg‐IFN and RBV were for 44 weeks. | |
| Outcomes | Safety, SVR24. | |
| Notes | We emailed Nishiguchi and colleagues on 24 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial used a "pseudo‐random number generator and supplied seed number" to generate the allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | The trial was only blinded up to week 8 |
| Blinding of outcome assessment (detection bias) | High risk | The trial was only blinded up to week 8 |
| Incomplete outcome data (attrition bias) | Unclear risk | There was above 5% dropouts and it was unclear how the trial accounted for missing data |
| Selective reporting (reporting bias) | High risk | The secondary outcomes were changed after the trial was completed (NCT00947349) |
| Vested‐interest bias | High risk | The trial was supported by a company that might have an interest in a given result (Boehringer Ingelheim) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Nishiguchi 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The trial used a "pseudo‐random number generator and supplied seed number" to generate the allocation sequence |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | The trial was only blinded up to week 8 |
| Blinding of outcome assessment (detection bias) | High risk | The trial was only blinded up to week 8 |
| Incomplete outcome data (attrition bias) | Unclear risk | There was above 5% dropouts and it was unclear how the trial accounted for missing data |
| Selective reporting (reporting bias) | High risk | The secondary outcomes were changed after the trial was completed (NCT00947349) |
| Vested‐interest bias | High risk | The trial was supported by a company that might have an interest in a given result (Boehringer Ingelheim) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Phase IIa, randomised, placebo‐controlled study, parallel‐group design (NCT00561353) | |
| Participants | 77 participants (Cohort 1 and 2) and 39 participants (Cohort 4) Countries: 26 centres in Belgium, France, Germany, the Netherlands, Poland, and the UK Inclusion criteria: eligible participants were aged 18–70 years with documented chronic HCV infection (genotype 1; diagnosis > 6 months prior to screening), a plasma HCV RNA ≥ 10,000 IU/mL (COBAS® TaqMan HCV/HPS assay v2.0 (Roche Molecular Systems, Pleasanton, CA, USA)) and a BMI 18–32 kg/m2. Participants were either treatment‐naive, or were non‐responders or relapsers to prior IFN/RBV or peg‐IFN/RBV therapy who did not discontinue anti‐HCV therapy due to AEs. Participants with compensated cirrhosis (up to Child–Pugh A according to standard criteria) were included. Treatment‐experienced participants were defined as non‐responders or relapsers who had virologically failed prior IFN/RBV or peg‐IFN/RBV therapy. Prior non‐responders were those who had not achieved a 2 log10 IU/ mL decrease in HCV RNA from baseline after 12 weeks of prior IFN‐based therapy. Prior relapsers were those who had detectable HCV RNA during follow‐up after achieving undetectable HCV RNA at the end of previous treatment. Exclusion criteria: other causes of significant liver disease, decompensated cirrhosis, HCC, prolonged Qtc value, platelet count < 90/nl, neutrophile count < 2/nl, bilirubin > 1.5 x ULN, AST or ALT level > 5 x ULN, excessive use of alcohol, positive urinary drug screening, HIV, Hepatitis B, contraindication for treatment with peg‐IFN or RBV. | |
| Interventions | The trial included multiple treatment cohorts. Cohort 1 and 2 included treatment‐naive participants. Participants in Cohort 4 were treatment‐experienced. Cohort 1, Panel A: participants were randomised 3:3:2 Experimental group 1A_1: simeprevir 25 mg once daily for 4 weeks. Experimental group 1A_2: simeprevir 75 mg once daily for 4 weeks. Control group 1A: placebo. Co‐intervention 1A: peg‐IFN α‐2a + RBV in week 2‐4. Cohort 1, Panel B: Participants were randomised 3:3:2 Experimental group 1B_1: simeprevir 25 mg once daily for 4 weeks. Experimental group 1B_2: simeprevir 75 mg once daily for 4 weeks. Control group 1B: placebo. Co‐intervention 1B: peg‐IFN α‐2a + RBV for 4 weeks. Cohort 2, Panel A: participants were randomised 3:1 Experimental group 2A: simeprevir 200 mg once daily for 4 weeks. Control group 2A: placebo. Co‐intervention 2A: peg‐IFN α‐2a + RBV in week 2‐4. Cohort 2, Panel B: participants were randomised 3:1. Experimental group 2B: simeprevir 200 mg once daily for 4 weeks. Control group 2B: placebo. Co‐intervention 2B: peg‐IFN α‐2a + RBV for 4 weeks. Cohort 4: participants randomised 1:1:1:1 Experimental group 4_1: simeprevir 75 mg once daily for 4 weeks. Experimental group 4_1: simeprevir 150 mg once daily for 4 weeks. Experimental group 4_1: simeprevir 250 mg once daily for 4 weeks. Control group 4: placebo. Co‐intervention 4: peg‐IFN α‐2a + RBV for 4 weeks. Participants in all cohorts 1, 2 and 4 could receive P/R up to week 48 following the initial 28‐day TMC435 treatment period. | |
| Outcomes | AE, SAE, change from baseline in HCV RNA level at day 7, percentage of participants with undetectable HCV RNA at week 4. | |
| Notes | A planned cohort 3 should have investigated simeprevir 400 mg once daily, but was cancelled before participant enrolment. This is cohort 125 mg vs control. We emailed Manns and colleagues on 26 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | "Randomisation was achieved using the central interactive web response system, managed by ClinPhone Group Ltd" |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blinded and placebo described as identical |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only 5 participants were not included in the intention‐to‐treat analysis resulting in under 5% with missing data |
| Selective reporting (reporting bias) | Low risk | All outcomes in the protocol were reported on |
| Vested‐interest bias | High risk | This study was sponsored by Tibotec Pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see OPERA 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Randomisation was achieved using the central interactive web response system, managed by ClinPhone Group Ltd |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blinded and placebo described as identical |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only 5 participants were not included in the intention‐to‐treat analysis resulting in under 5% with missing data. |
| Selective reporting (reporting bias) | Low risk | All outcomes in the protocol were reported on |
| Vested‐interest bias | High risk | This study was sponsored by Tibotec Pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see OPERA 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Randomisation was achieved using the central interactive web response system, managed by ClinPhone Group Ltd |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blinded and placebo described as identical |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only 5 participants were not included in the intention‐to‐treat analysis resulting in under 5% with missing data. |
| Selective reporting (reporting bias) | Low risk | All outcomes in the protocol were reported on |
| Vested‐interest bias | High risk | This study was sponsored by Tibotec Pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see OPERA 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Randomisation was achieved using the central interactive web response system, managed by ClinPhone Group Ltd |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blinded and placebo described as identical |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only 5 participants were not included in the intention‐to‐treat analysis resulting in under 5% with missing data. |
| Selective reporting (reporting bias) | Low risk | All outcomes in the protocol were reported on |
| Vested‐interest bias | High risk | This study was sponsored by Tibotec Pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see OPERA 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Randomisation was achieved using the central interactive web response system, managed by ClinPhone Group Ltd |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blinded and placebo described as identical |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only 5 participants were not included in the intention‐to‐treat analysis resulting in under 5% with missing data |
| Selective reporting (reporting bias) | Low risk | All outcomes in the protocol were reported on |
| Vested‐interest bias | High risk | This study was sponsored by Tibotec Pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see OPERA 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | Randomisation was achieved using the central interactive web response system, managed by ClinPhone Group Ltd |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blinded and placebo described as identical |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Only 5 participants were not included in the intention‐to‐treat analysis resulting in under 5% with missing data. |
| Selective reporting (reporting bias) | Low risk | All outcomes in the protocol were reported on |
| Vested‐interest bias | High risk | This study was sponsored by Tibotec Pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | Sex: 18 men, 6 women Mean age: 48 years Inclusion criteria: eligible participants with chronic HCV infection were men or women aged 18‐60 years with a BMI of 18‐35 kg/m2 and chronic infection with HCV genotype 1, either treatment‐naive, treatment nonresponders (including relapsers), or treatment intolerant. Additional inclusion criteria were plasma HCV RNA levels of 100,000 IU/mL, a documented FibroTest score of 0.72 or 0.59, and an AST platelet ratio index of 2 or the absence of cirrhosis based on liver biopsy within 12 months Exclusion criteria: main exclusion criteria included previous exposure to another NS3 protease inhibitor, co‐infection with HIV or HBV, or being women of childbearing potential | |
| Interventions | Experimental group:
Control group: placebo every 12 h | |
| Outcomes | Antiviral activity, safety, pharmacokinetics | |
| Notes | We emailed Pasquinelli and colleagues on 06 June 2016 for additional information on description of the placebo, were outcome assessors blinded, who experienced a SAE, how was missing data handled, SVR24 data.but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation scheme |
| Allocation concealment (selection bias) | Low risk | An interactive voice‐response system was used to assign a unique participant number |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial is described as double‐blinded but the placebo was not described in detail |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | There were no dropouts |
| Selective reporting (reporting bias) | Low risk | The outcomes reported in the protocol are reported (NCT00559247) |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result (Bristol‐Myers Squibb) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Pasquinelli 2012a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation scheme |
| Allocation concealment (selection bias) | Low risk | An interactive voice‐response system was used to assign a unique participant number |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial is described as double‐blinded but the placebo was not described in detail |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | There were above 5% dropouts and it was unclear how the trial handled missing data |
| Selective reporting (reporting bias) | Low risk | The outcomes reported in the protocol are reported (NCT00722358) |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result (Bristol‐Myers Squibb) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 101 participants were randomised to either triple (n = 49) or to double therapy (n = 52) Sex: 63 men, 38 women Mean age: 53 years Inclusion criteria: treatment‐naive, infected with genotype 1 HCV, and had low viral load at baseline (< 600,000 IU/mL). Participants were 18 years of age or older and had a liver biopsy in the past 2 years consistent with chronic hepatitis. Before randomisation, participants had been rapid virologic responders to 4 weeks of peg‐IFN α‐2b. Exclusion criteria: cirrhosis participants. HCV/HIV co‐infection; HCV genotype other than 1; biopsy‐proven or strongly suspected clinical cirrhosis; other causes of liver disease, including co‐infection with hepatitis B; creatinine clearance < 50 mL/min (modification of diet in renal disease equation); platelet count < 80 3 109/L; neutrophil count < 1.5 3 109/L; haemoglobin concentration < 13 g/dL and 12 g/dL in men and women, respectively; coexisting uncontrolled psychiatric or cardiopulmonary disorders; haemoglobinopathy; sarcoidosis; malignant neoplasm; receipt of immunosuppressive or immunomodulatory therapy in the previous 6 months; pregnancy; and men whose partners were pregnant or unwilling to use contraception during the study period. Female participants of childbearing age also agreed to avoid systemic contraception if ultimately randomised into the protease inhibitor‐containing arm. Participants were also excluded if they imbibed significant amounts of alcohol (> 30 g/day), or if they were active substance abusers in the past 6 months. | |
| Interventions | Experimental group: 24 weeks of peg/RBV/BOC (boceprevir 800 mg three times a day) (Group A). Co‐intervention: 20 weeks of peg/RBV only (Group B). | |
| Outcomes | Side effects, viral response. | |
| Notes | We contacted trial authors for additional information on unpublished results, randomisation, blinding of outcome assessment, allocation concealment, SAEs and AEs. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | Open‐label, no blinding |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | 4% in group A and 6% in group B, a total of 10% discontinuations |
| Selective reporting (reporting bias) | Unclear risk | Protocol not found |
| Vested‐interest bias | High risk | Dr. Pearlman consults, advises, and is on the speakers’ bureau for Merck |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 93 participants Sex: 53 men, 29 women (analysed) Mean age: 56.5 (analysed) Country: USA Inclusion criteria: chronic HCV infection. Participants 18 years or older were eligible for enrolment if they had genotype 1a infection and a plasma HCV RNA level greater than 10,000 IU/mL. African American ethnicity was self‐identified by participants at screening. All participants either were previously untreated or had shown a prior null response to peg‐IFN/RBV as defined by < a 2‐log10 decrease at 12 weeks of therapy compared with a baseline value and as verified by laboratory records. Other eligibility criteria included documentation of cirrhosis by means of a liver biopsy (METAVIR stage 4) or a FibroTest (Lab Corp, Burlington, NC) score > 0.75 and an AST:platelet ratio index > 2, with a Child‐Turcotte‐Pugh score of < 7 at screening (class A). Participants needed to have had an ultrasound performed within 6 months before screening, or by the time of the baseline visit, with no findings suspicious for HCC, and to have an international normalised ratio of ≤ 2.3, a total bilirubin level of < 3 mg/dL, a platelet count of ≥ 50,000 per mL3, and a serum albumin level > 2.7 g/dL. There were no upper age or BMI limits. Participants with stable, medicated psychiatric disease and methadone maintenance participants also were eligible. Exclusion criteria: non–genotype 1a, including genotype 1 infection that could not be subtyped; prior treatment with telaprevir or boceprevir; a history of decompensation or history of Child‐Turcotte‐Pugh class B or C; co‐infection with HIV or HBV; a creatinine clearance of < 50 mL/min (modification of diet in renal disease equation); a haemoglobin concentration < 12 g/dL in men and < 11 g/dL in women; co‐existing uncontrolled psychiatric or cardiopulmonary disorders; haemoglobinopathy; sarcoidosis; malignant neoplasm in the past 5 years except localised nonmelanoma skin cancer; receipt of immunosuppressive or immunomodulatory therapy within the previous 6 months; or participants who were either pregnant or planning to be pregnant or were men whose partners were pregnant or unwilling to use contraception during the study period. Participants who had discontinued prior therapy because of an AE were not eligible. | |
| Interventions | Experimental group: oral simeprevir (150 mg) once daily for 12 weeks. Control group: peg‐IFN α‐2b (1.5 μg/kg/wk) (Merck, Whitehouse Station, NJ), oral RBV (1000 mg–1200 mg/day, based on body weight < 75 kg or ≥ 75 kg, respectively) for 12 weeks. Co‐intervention: sofosbuvir (400 mg) once daily for 12 weeks. | |
| Outcomes | Efficacy, quality of life, safety assessment, virological response | |
| Notes | The trial reported it was linked to (NCT021683615) however the NCT number could not be identified on ClinicalTrials.gov. Seperate data from African‐American/white was presented | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | The trial was open‐label |
| Blinding of outcome assessment (detection bias) | High risk | The trial was open‐label |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The trial was funded by Gilead Sciences |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 84 participants Sex: 84 men Inclusion criteria: 18‐65 years old with HCV RNA > 105 IU/L, and genotype‐1 or ‐3 chronic HCV infection without clinical evidence of cirrhosis. | |
| Interventions | Experimental group: doses of 50 mg (genotype‐1) or 100 mg (genotype‐3) to 800 mg MK‐5172) for 7 days. | |
| Outcomes | Plasma HCV RNA, pharmacokinetics. | |
| Notes | NCT00998985 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being placebo‐blinded, but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being placebo‐blinded, but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | Unclear risk | Not described |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 107 adult participants Sex: 67 men, 37 women Mean age: 47.08 years Inclusion criteria: participants were eligible for inclusion if they were aged 18‐65 years and had chronic HCV genotype 1 infection with HCV RNA levels 50,000 IU/mL. Only treatment‐naive participants were enrolled in the study. Other inclusion criteria included chronic liver disease consistent with chronic HCV infection on biopsy, and compensated liver disease (Child‐Turcotte‐Pugh grade A). Women of childbearing potential were required to have a negative blood pregnancy test within the 24‐h period prior to the first dose of study medication. All fertile participants, male and female, were required to use 2 forms of effective contraception during treatment and for 6 months afterward. Exclusion criteria: participants were excluded from the study if they had infection with any HCV genotype other than genotype 1, or an indeterminate or mixed genotype; hepatic cirrhosis (Knodell score of 4, Metavir score of 4, or Ishak modified histological activity index score of 5 or 6) or incomplete/ transition to cirrhosis (Knodell score of 3, Metavir score of 3, or an Ishak modified histological activity index score of 4 with nodules or 3 bridges); a low absolute neutrophil count (1500 cells/mm3); a low platelet count (120,000 cells/mm3); or a low haemoglobin concentration (13 g/dL in women or 14 g/dL in men), HIV, Hepatitis A, Hepatitis B infection. | |
| Interventions | Experimental group:
Control group: placebo + Copegus 1000 mg/1200 mg orally daily. Co‐intervention: Pegasys 180 μg subcutaneously weekly for 4 weeks and 44 weeks of standard of care (peg‐IFN α‐2a (180 μg subcutaneously), RBV (1000 mg orally once a day for those weighing < 75 kg; 1200 mg orally once a day if ≥ 75 kg) for 4 weeks). | |
| Outcomes | Safety, pharmacokinetics, antiviral efficacy. | |
| Notes | We emailed Pockros and colleagues on 06 June 2016 for additional information on allocation sequence generation, allocation concealment, blinding of outcome assessment, how many dropped out but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The participants and care providers were blinded up until week 8. Outcomes were only reported till week 8 and therefore results were blinded. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many dropped out and how the trial dealt with missing data |
| Selective reporting (reporting bias) | Unclear risk | All outcomes stated in the protocol were reported on (NCT00377182) |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result (Hoffmann‐La Roche) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Pockros 2008a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The participants and care providers were blinded up until week 8. Outcomes were only reported till week 8 and therefore results were blinded. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many dropped out and how the trial dealt with missing data |
| Selective reporting (reporting bias) | Unclear risk | All outcomes stated in the protocol were reported on (NCT00377182) |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result (Hoffmann‐La Roche) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Pockros 2008a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The participants and care providers were blinded up until week 8. Outcomes were only reported till week 8 and therefore results were blinded. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many dropped out and how the trial dealt with missing data |
| Selective reporting (reporting bias) | Unclear risk | All outcomes stated in the protocol were reported on (NCT00377182) |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result (Hoffmann‐La Roche) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 244 participants Mean age: 50 years Inclusion criteria: treatment‐naive or prior non‐responders. Exclusion criteria: women who were pregnant or breastfeeding, ALT >/ or = 5 x the ULN, AST >/ or = 5 x the ULN. | |
| Interventions | Experimental group:
Control group: placebo. Co‐intervention: Peg‐Intron subcutaneous injection, weight‐based dosing, weekly and Rebetol capsules, weight‐based dosing, every 12 h daily for 48 weeks. | |
| Outcomes | Primary outcome complete early virologic response. Secondary outcome rapid virological response. | |
| Notes | We contacted trial authors for addition information on whether HIV participants included, allocation sequence generation and concealment, how was blinding maintained, who was blinded, maximum follow‐up, how many participants dropped out, how was missing data handled, SAE, death, SVR24 but reply not received. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial is described as double‐blinded but the placebo was not described in detail |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | The number of dropouts was not described |
| Selective reporting (reporting bias) | Unclear risk | The outcome called upon in the protocol was reported (NCT00367887) |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result (PfizerViroPharma) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 48 participants Sex: 32 men, 16 women Mean age: 51.3 years Countries: USA and France. Inclusion criteria: chronic HCV genotype 1 infection and were treatment‐naive or had < 4 weeks of exposure to RBV or IFN‐based therapy. Participants needed to have an HCV RNA concentration of ≥ 105 IU/mL and be aged 18‐70 years. Exclusion criteria: cirrhosis, by liver biopsy within 24 months of baseline, clinically significant comorbidities, and HIV or hepatitis B co‐infection. | |
| Interventions | Experimental group: oral 3 mg, 10 mg, 60 mg once daily for 48 weeks. Control group: placebo. Co‐intervention: peg‐IFN α‐2a (180 μg per week) and RBV (1000 mg–1200 mg daily). | |
| Outcomes | HCV RNA, safety assessment, virological response. | |
| Notes | We emailed Pol and colleagues on 27 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random allocation sequence |
| Allocation concealment (selection bias) | Low risk | Interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | High risk | The participants and personnel were only blinded until week 12 |
| Blinding of outcome assessment (detection bias) | High risk | The sponsors, who performed the analyses, were only blinded until week 12 |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | High risk | The trial changed outcomes from the protocol |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 239 participants non‐cirrhotic genotype 1 HCV participants Sex: unknown Mean age: unknown Exclusion criteria: none specified. | |
| Interventions | Experimental group: GS‐9451 (200 mg once a day) alone for 16 or 24 weeks (arm 1) or GS‐9451 (200 mg once a day) and tegobuvir (30 mg twice a day) 24 weeks (arm 2). Control group: placebo. Co‐intervention: peg (180 mg/week) + RBV (1000 mg–1200 mg/day) up to 48 weeks based on response to therapy. | |
| Outcomes | Very rapid virological response, rapid virological response, SVR, serious adverse events | |
| Notes | The authors were contacted on 06 June 2016 for additional information on allocation sequence generation, blinding, missing data, SVR24, safety, deaths, full publication | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants had missing data |
| Selective reporting (reporting bias) | High risk | SVR24 was not reported but was stated in the protocol (NCT01271790) |
| Vested‐interest bias | High risk | The trial was sponsored by a company with an interest in a given outcome (Gilead Sciences) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 117 treatment‐naive participants with chronic hepatitis C Exclusion criteria: pregnant, breastfeeding, or co‐infected with HBV and/or HIV. | |
| Interventions | Experimental group: valopicitabine 200 mg once a day. Control group: RBV 1000 mg‐1200 mg daily + valopicitabine placebo once a day. Co‐intervention: peg‐IFNα‐2a 180 μg weekly. | |
| Outcomes | Pharmacokinetics, antiviral activity, SAE (not reported fully, so we could not use the data). | |
| Notes | We contacted the trial authors on 06 June 2016 for additional information on allocation sequence generation and concealment, maximum follow‐up, how many participants dropped out, how was missing data handled, SAE, Death, SVR24, number randomised in each group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | Only described as single blind |
| Blinding of outcome assessment (detection bias) | High risk | Only described as single blind |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many dropped out |
| Selective reporting (reporting bias) | Unclear risk | No outcomes were reported in the protocol (NCT00395421) |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result: Merck Sharp & Dohme Corp. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | A phase III, international, randomised, placebo‐controlled, parallel‐group study (SPRINT‐2)(NCT00705432) | |
| Participants | 1097 participants Country: France, Germany, Italy and USA Inclusion criteria: treatment‐naive participants, age ≥ 18 years, weight of 40‐125 kg, chronic infection with HCV genotype 1, plasma HCV RNA level ≥ 10,000 IU/mL. Exclusion criteria: liver disease of other cause, decompensated cirrhosis, renal insufficiency, HIV or hepatitis B infection, pregnancy, current breastfeeding, active cancer. Group 1: 363 participants Sex: 206 men, 157 women Mean age ± SD: 49 ± 10 years Race, n (%): white: 296 (82), black: 52 (14), Asian: 9 (2), other: 6 (2) Location, n (%): North America: 254 (70), Europe: 99 (27), Latin America: 10 (3) Weight, mean ± SD (kg): 80 ± 16 HCV subtype, n (%): 1a: 227 (63), 1b: 121 (33), missing data: 15 (4) HCV RNA level, n (%): > 400,000 IU/mL: 337 (93), > 800,000 IU/mL: 308 (85) METAVIR fibrosis score, n(%): 0, 1, or 2: 328 (90), 3 or 4: 24 (7), missing data: 11 (3) Group 2: 368 participants Sex: 229 men, 139 women Mean age ± SD: 50 ± 9 years Race, n (%): white: 304 (83), black: 52 (14), Asian: 4 (1), other: 8 (2) Location, n (%): North America: 277 (75), Europe: 79 (21), Latin America: 12 (3) Weight, mean ± SD (kg): 82 ± 17 HCV subtype, n (%): 1a: 234 (64), 1b: 124 (34), missing data: 10 (3) HCV RNA level, n (%): > 400,000 IU/mL: 336 (91), > 800,000 IU/mL: 314 (85) METAVIR fibrosis score, n(%): 0, 1, or 2: 319 (87), 3 or 4: 34 (9), missing data: 15 (4) Group 3: 366 participants Sex: 221 men, 145 women Mean ± SD: 49 ± 9 years Race, n (%): white: 295 (81), black: 55 (15), Asian: 8 (2), other: 8 (2) Location, n (%): North America: 270 (74), Europe: 86 (23), Latin America: 10 (3) Weight, mean ± SD (kg) = 82 ± 17 HCV subtype, n (%): 1a: 237 (65), 1b: 117 (32), missing data: 12 (3) HCV RNA level, n (%): > 400,000 IU/mL: 341 (93), > 800,000 IU/mL: 313 (86) METAVIR fibrosis score, n (%): 0, 1, or 2: 313 (86), 3 or 4: 42 (11), missing data: 11 (3) | |
| Interventions | Experimental group: Group 2: oral boceprevir 800 mg thrice‐daily in 4 capsules of 200 mg each (to be taken with food and at an interval of 7‐9 h between doses) beginning at week 5, for a total of 24 weeks; if HCV RNA levels were undetectable from week 8‐24, treatment was considered complete; if HCV RNA levels were detectable between week 8‐24 (not including week 24), boceprevir was continued for additional 20 weeks (total of 44 weeks). Group 3: oral boceprevir 800 mg thrice‐daily in 4 capsules of 200 mg each (to be taken with food and at an interval of 7‐9 h between doses) beginning at week 5, for a total of 44 weeks. Control group: 1: a matched placebo thrice‐daily beginning at week 5 for 44 weeks. Co‐intervention: All groups: peg‐IFN α‐2b 1.5 μg/kg body weight subcutaneously once weekly and weight‐based oral RBV at a total dose of 600 mg‐1400 mg daily in divided doses for 4 weeks (lead‐in period). Groups 1 and 3: peg‐IFN α‐2b 1.5 μg/kg body weight subcutaneously once weekly and weight‐based oral RBV at a total dose of 600mg‐1400 mg daily in divided doses for additional 44 weeks (total of 48 weeks). Group 2: peg‐IFN α‐2b 1.5 μg/kg body weight subcutaneously once weekly and weight‐based oral RBV at a total dose of 600 mg‐1400 mg daily in divided doses for additional 24 weeks (total of 28 weeks), and those with a detectable HCV RNA level between weeks 8‐24 received the same therapy for an additional 20 weeks (total of 48 weeks). | |
| Outcomes | Primary outcomes: achievement of SVR, defined as undetectable plasma HCV RNA at week 24 (if a participant was missing follow‐up week 24 and had undetectable HCV RNA level at week 12, the participant was considered an SVR). Secondary outcomes: achievement of SVR defined as undetectable HCV RNA at week 24 in non‐black/African American randomised participants who received at least 1 dose of experimental study drug or placebo. The proportion of participants with EVR (e.g. undetectable HCV RNA at weeks 2, 4, 8, or 12) who achieved SVR. The proportion of participants with undetectable HCV RNA at week 12. The proportion of participants with undetectable HCV RNA at 72 weeks after randomisation. | |
| Notes | Co‐intervention in Group 2 was different from Groups 1 and 3. We emailed Poordad and colleagues on 27 April 2016 for additional information about blinding outcome assessors and number of participants experiencing non‐serious AEs but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code |
| Allocation concealment (selection bias) | Low risk | Allocation concealment was done through interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | Low risk | In the trial's protocol it is described that placebo would be matched to boceprevir and would be given in the same manner |
| Blinding of outcome assessment (detection bias) | Unclear risk | It was not mentioned if the outcome assessors were blinded, or the extent of blinding was insufficiently described |
| Incomplete outcome data (attrition bias) | High risk | 49/1099 (4.5%) participants discontinued the peg‐IFN/RBV therapy during the lead‐in period. No specific reasons were given. Due to futility at week 24 another 108, 33, and 36 participants in groups 1, 2, and 3, respectively, discontinued treatment. In total 226/1099 (20,5%) of participants discontinued treatment. No other dropouts were stated |
| Selective reporting (reporting bias) | Low risk | A protocol was published before randomisation began and all outcome results were reported adequately |
| Vested‐interest bias | High risk | The sponsor (Merck) was directly involved in trial's design, managing, analyses, as well as, writing, decision of submission for publication, reviewing and drafting the manuscript |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias |
| Methods | For characteristics see Poordad 2011a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code |
| Allocation concealment (selection bias) | Low risk | Allocation concealment was done through interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | Low risk | In the trial's protocol it is described that placebo would be matched to boceprevir and would be given in the same manner |
| Blinding of outcome assessment (detection bias) | Unclear risk | It was not mentioned if the outcome assessors were blinded, or the extent of blinding was insufficiently described |
| Incomplete outcome data (attrition bias) | High risk | 49/1099 (4.5%) participants discontinued the peg‐IFN+RBV therapy during the lead‐in period. No specific reasons were given. Due to futility at week 24 another 108, 33, and 36 participants in groups 1, 2, and 3, respectively, discontinued treatment. In total 226/1099(20,5%) of participants discontinued treatment. No other drop‐outs were stated |
| Selective reporting (reporting bias) | Low risk | A protocol was published before randomisation began and all outcome results were reported adequately |
| Vested‐interest bias | High risk | The sponsor (Merck) was directly involved in trial's design, managing, analyses, as well as, writing, decision of submission for publication, reviewing and drafting the manuscript |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias |
| Methods | Blinded placebo‐controlled trial (NCT01542788) | |
| Participants | Randomised: 280 underwent randomisation, and 278 began treatment Sex: 151 men, 127 women Countries: 63 sites in the USA, Canada, Australia, and New Zealand from March 2012‐May 2012. Inclusion criteria: eligible participants were cirrhotic or non‐cirrhotic adults with HCV genotype 2 or 3 infection, a baseline HCV RNA level > 10,000 IU/mL unwilling or uneligible or intolerant for IFN‐treatment. Participants had chronic hepatitis C infection (documented by positive anti‐HCV antibody test or positive HCV RNA, or positive HCV genotyping test ≥ 6 months prior to the Baseline/Day 1 visit; or documented by liver biopsy performed prior to the Baseline/Day 1 visit with evidence of chronic HCV). Participants had a BMI > = 18 kg/m2, a screening ECG without clinically significant abnormalities, no evidence of HCC, no Chronic liver disease of a non‐HCV aetiology (e.g. hemochromatosis, Wilson’s disease, α1‐antitrypsin deficiency, and cholangitis) and no co‐infection with HBV or HIV. Participants had no history of significant pulmonary or cardiac disease, or porphyria; no current or prior history of clinical hepatic decompensation (e.g. ascites, jaundice, encephalopathy, or variceal haemorrhage). | |
| Interventions | Randomisation was performed centrally in a 3:1 ratio with stratification according to the presence or absence of cirrhosis. Experimental group: oral sofosbuvir 400 mg once daily + RBV (1000 mg daily in participants with a body weight < 75 kg, and 1200 mg daily in participants with a body weight ≥ 75 kg) for 12 weeks. Control group: placebo. | |
| Outcomes | Proportion of participants with end‐of‐treatment response (week12), SVR12, SAE, AEs, mortality. | |
| Notes | We emailed Jacobson and colleagues on 21 April 2016 for additional information on generation of allocation sequence, how many participants dropped out and how the trial handled missing data but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Low risk | "An Interactive Web Response System (IWRS) will be employed to manage participant randomization and study drug assignment." |
| Blinding of participants and personnel (performance bias) | Low risk | The participant, caregiver, investigator, outcomes assessor were described as being blinded and the placebo was identical in appearance |
| Blinding of outcome assessment (detection bias) | Low risk | The participant, caregiver, investigator, outcomes assessor were described as being blinded and the placebo was identical in appearance |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants dropped out and how the trial handled missing data |
| Selective reporting (reporting bias) | Low risk | The outcomes stated in the protocol were reported on |
| Vested‐interest bias | High risk | The sponsor collected the data, monitored study conduct, and performed the statistical analysis |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 40 adult participants Inclusion criteria: chronic hepatitis C genotype 1 whose alpha‐IFN treatment had failed. Exclusion criteria: non‐cirrhotic. | |
| Interventions | Experimental group:
Control group: placebo | |
| Outcomes | SAE, antiviral activity, safety. | |
| Notes | We contacted the trial authors on allocation sequence generation and concealment, maximum follow‐up, how many participants dropped out, how was missing data handled, death, SVR24, and number randomised in each group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | There was a placebo but it was unclear how well matched the placebo was and who was blinded to it |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants had missing data |
| Selective reporting (reporting bias) | Unclear risk | No prepublished protocol could be found |
| Vested‐interest bias | Unclear risk | It was unclear how the trial was funded |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised phase I clinical trial | |
| Participants | 37 adult participants Sex: 22 men, 12 women (analysed) Mean age: 47 years Countries: Germany, the Netherlands. Inclusion criteria: men or women between the ages of 18 and 65 years, with BMI between 18.5 and 29.0 kg/m2 (men) or 18.5 and 32.5 (women). Entry criteria included an HCV RNA level 1 105 IU/mL as measured using the Roche COBAS TaqMan HCV assay (Roche Molecular Diagnostics, Pleasanton, CA) (confirmed by repeat measure of 2 separate samples taken during the screening period), HCV genotype 1 (any subtype), and an ALT concentration 4 times the ULN. Exclusion criteria: decompensated liver disease, cirrhosis, and positive screening for hepatitis B surface antigen or anti‐HIV 1/2. | |
| Interventions | Experimental group: oral 450 mg or 750 mg of VX‐950 3 times daily, or 1250 mg twice daily for 14 days. Control group: placebo. | |
| Outcomes | Pharmacokinetics, safety assessment, antiviral assessment. | |
| Notes | We emailed Reesink and colleagues on 27 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as double‐blinded with matching placebo, but it was unclear if the participants and investigators were blinded to results (except HCV RNA) |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as double‐blinded with matching placebo, but it was unclear if the participants and investigators were blinded to results (except HCV RNA) |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% did not complete the trial (3 participants were not included in the analyses) |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The trial was funded by Vertex Pharmaceuticals Incorporated |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 10 adult participants Sex: 8 men, 2 women Mean age: 34.5 years Inclusion criteria: women or men aged 18 years or older with chronic genotype 2 or 3 HCV infection. The line probe assay was used to determine the genotype of the viral infection. A liver biopsy specimen showing changes consistent with chronic HCV infection had to have been performed within the previous 12 months. At screening, the HCV load had to be 50,000 copies/mL serum. Exclusion criteria: women were excluded if they were breast‐feeding or at risk of pregnancy; men had to use an adequate form of contraception if their partner was of childbearing potential. They were not enrolled if there were other or additional reasons for chronic liver disease, including the presence of other hepatitis‐causing viruses and/or a history of alcohol abuse within the previous 12 months and/or evidence of Child’s B or C liver disease at screening. No other antiviral or antimicrobial or investigational therapies were allowed during the study (screening, pretreatment, and treatment phases). Participants were excluded if, at screening, their baseline ALT/AST plasma levels exceeded the ULN by more than 5‐fold (5 times the ULN) or their total bilirubin or alkaline phosphatase levels were 1.5 times the ULN. Other exclusion criteria included co‐infection with HIV, a platelet count 100,000/mm3, a white blood cell count 2000 cells/mm3, any clinically significant laboratory abnormalities, and a positive test result for illicit or nonprescription drugs. | |
| Interventions | Experimental group: oral 500 mg of BILN‐2061 for 2 days. Control group: placebo. | |
| Outcomes | Virological efficacy, pharmacokinetics, safety. | |
| Notes | We emailed Reiser and colleagues on 27 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Low risk | 0 participants dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained for all 3 stages, and the ClinicalTrials.gov information was added after completion |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 50 adult participants Inclusion criteria: chronic hepatitis C genotype 1 who were treatment‐naive. Exclusion criteria: none reported. | |
| Interventions | Experimental group:
Control group: placebo. Co‐intervention: 180 μg peg‐IFN α‐2a and 1000 mg‐1200 mg RBV. | |
| Outcomes | Antiviral activity (RVR), SAE, AE. | |
| Notes | We emailed Rodriguez‐Torres and colleagues on 06 June 2016 for additional information on allocation sequence generation and concealment, blinding, incomplete outcome data including which groups the 2 participants who were omitted from the analyses were from, how the trial was funded, prepublished protocol, death, SVR but reply not received. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | A placebo was mentioned but it was unclear who was blinded to the intervention and how well matched the placebo was |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants had missing data |
| Selective reporting (reporting bias) | Unclear risk | No prepublished protocol could be found |
| Vested‐interest bias | Unclear risk | It was unclear how the trial was funded |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 24 participants (first 3 cohorts) Inclusion criteria: participants who were 18‐65 years of age, had laboratory evidence of HCV infection for 6 months, defined by 1. presence of anti‐HCV antibody (genotype 1a and 1b infection), or 2. documented HCV RNA presence by a sensitive and specific assay and 3. histologic evidence of CHC (Fibrosis on a standardised histological grading system), plasma HCV RNA of 100,000 IU/mL, were HIV 1 and HIV2 ab seronegative, BMI ≤ 35 kg/m2 BMI and treatment‐naive. Exclusion criteria: contraindications to peg‐IFN or RBV therapy, have evidence of liver cirrhosis, decompensated liver disease, and Child‐Pugh score > 5, have haemoglobinopathies, unstable cardiac disease, history of organ transplant, active malignant disease or uncontrolled Type I or II diabetes. | |
| Interventions | Experimental group:
Control group: placebo Co‐intervention: peg‐IFN α‐2a plus RBV were offered from day 4 for up to 48 weeks. | |
| Outcomes | Pharmacokinetics, antiviral activity, AEs. | |
| Notes | We emailed Rodriguez‐Torres and colleagues on 06 June 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as double‐blind (participant, caregiver, investigator, outcomes assessor) at ClinicalTrials.gov, but it is not clear how well the placebo was matched |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as double‐blind (participant, caregiver, investigator, outcomes assessor) at ClinicalTrials.gov, but it was not clear how well the placebo was matched |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants had missing data |
| Selective reporting (reporting bias) | Unclear risk | No prepublished protocol could be found. The outcomes stated at ClinicalTrials.gov were submitted after the start of the trial (NCT00911963) |
| Vested‐interest bias | High risk | The trial was funded by companies that might have an interest in a given result (Vertex Pharmaceuticals Incorporated and ViroChem Pharma) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 70 adult participants Inclusion criteria: chronic hepatitis C genotype 1 who were men and women, 18‐65 years of age inclusive (BMI of at least 18kg/m2 not exceeding 36kg/m2), had a diagnosis of chronic HCV by 1 previous PCR result prior to screening, with a positive HCV viral load of at least 100,000 IU/mL at screening measured by quantitative PCR, HCV genotype 1 per central lab testing report, HCV treatment‐naive (defined as no prior treatment with IFN, peg‐IFN, RBV, or any HCV DAA drugs), liver biopsy consistent with chronic HCV infection but non‐cirrhotic as judged by a pathologist (Knodell < 3, Metavir < 2, Ishak < 4, or Batts & Ludwig < 2) within the last 2 years and before Visit 2 (biopsy can be done within screening period), negative urine drug screen for drugs of abuse at screening and Study Day ‐1 (methadone use allowed), women would have a negative serum βHCG pregnancy test at screening & negative urine dipstick pregnancy test upon entry to clinical unit on Study Day ‐1, agreement by both women of childbearing potential and men(who have not been surgically sterilised) to practice an acceptable method of birth control. Surgical sterilisation of either female or male partner must have occurred at least 6 months prior to first dose and women must be post‐menopausal for 2 years to be considered of non‐child‐bearing potential. Acceptable contraceptive methods include 1 of the following: oral and implantable hormonal contraceptives by woman at least 3 months prior to the 1st dose of Study Drug, IUD in place at least 6 months prior to first dose, barrier methods either diaphragm or condom with spermicide. (Abstinence is not an acceptable method of birth control, participants who indicate sexual inactivity must agree to utilise birth control in the event of sexual activity), willing and able to complete all study visits and procedures, and able to communicate with the investigator and other personnel, signed informed consent form executed prior to protocol screening assessments. Exclusion criteria: advanced liver disease, cirrhosis, or with signs of decompensated liver disease such as variceal bleeding, ascites, hepatic encephalopathy, active jaundice (total bilirubin > 2, or other evidence of decompensated liver disease, co‐infection with HBV or HIV (positive test for HBsAg or anti‐HIV Ab), acute cardiac ischaemias, unstable heart disease or clinically symptomatic cardiac abnormalities apparent on ECG & PE, or a QTcB interval at Visit 1 of ≥ to 450 ms by Bazette's correction, or personal or family history of Torsades de pointes, use of the following medications concurrently or within the 30 days prior to screening associated with QT prolongation: macrolides, antiarrhythmic agents, azoles, fluoroquinolones, and tricyclic anti‐depressants (methadone use allowed), use of immunosuppressive or immune‐modulating agents (including corticosteroids and immunosuppressive agents) or presence of an immunologically‐mediated autoimmune disease (other than asthma) or history of organ transplantation (inhaled steroids for asthma and topical steroid for minor skin conditions allowed), use of strong CYP3A4‐inhibiting protease inhibitors (specifically atazanavir, indinavir, nelfinavir, saquinavir, and ritonavir), strong CYP3A4 inhibitors (specifically clarithromycin, itraconazole, ketoconazole, nefazodone, telithromycin), or strong CYP3A4 inducers (specifically rifampin, efavirenz, etravirine, phenobarbital, phenytoin, and carbamazepine); absolute NEUT count of < 1800 cells/mm3 (or < 1500 cells/mm3 for African Americans), or platelet count < 130,000 cells/mm3, or haemoglobin < 11g/dL for women and < 13g/dL for men, a history of abnormal thyroid function not adequately controlled (defined as TSH levels < 0.8 x LLN or > 1.2 x the ULN), serum creatinine concentration > 1.5 times the ULN, or albumin < 3g/dL, presence or history of severe, or uncontrolled, or hospitalisation‐requiring psychiatric disease including severe depression, suicide attempts or any severity of psychosis, any malignancy within the last 5 years other than treated cervical carcinoma in situ or treated basal cell carcinoma with no more than 20% risk of recurrence within 2 years, alcohol abuse (investigator assessment) within the past 2 years or an alcohol use pattern that will interfere with the study conduct, drug abuse (investigator assessment) within the last 6 months with exception of methadone, current lactation or breastfeeding, major surgery within 30 days prior Visit 1, participation in another clinical trial of an investigational drug or device within 6 months prior to visit donation of blood or plasma within 30 days prior to Visit 1. | |
| Interventions | Experimental group:
Control group: Control for arm 1‐3: placebo. Control for arm 4‐6: placebo + RBV. | |
| Outcomes | Adverse events, antiviral activity | |
| Notes | We emailed Rodriguez‐Torres and colleagues on 06 June 2016 for additional information on allocation sequence generation and concealment, how blinding was maintained, if outcome assessors were blinded, how many participants dropped out, SAE, death, SVR, male:female, mean age but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blind but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants dropped out |
| Selective reporting (reporting bias) | Low risk | The outcomes stated in the protocol were reported on (NCT01250366) |
| Vested‐interest bias | High risk | The trial was supported by a company that might have an interest in a given result (Bristol‐Myers Squibb) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 40 adults with chronic hepatitis C genotype 1 who were treatment‐naive. | |
| Interventions | Experimental group:
Control group: placebo | |
| Outcomes | SAE, AE, HCV RNA, HCV mutations. | |
| Notes | We emailed Rodriguez‐Torres and colleagues on 06 June 2016 for additional information on allocation sequence generation and concealment, blinding, incomplete outcome data, how the trial was funded, prepublished protocol, death, SVR but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | A placebo was mentioned but it was unclear who was blinded to the intervention and how well matched the placebo was |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants had missing data |
| Selective reporting (reporting bias) | Unclear risk | No prepublished protocol could be found |
| Vested‐interest bias | Unclear risk | It was unclear how the trial was funded |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 64 participants Sex: 43 men, 20 women Mean age: 45.1 years Inclusion criteria: 64 treatment‐naive participants with chronic HCV genotype 1 infection were enrolled (HCV RNA levels P100,000 IU/mL at screening), 18–65 years of age with a BMI of 18–36 kg/m2. Women of childbearing potential were required to use a protocol‐approved method of contraception. 1 participant in the sofosbuvir 200 mg arm withdrew consent before receiving the first dose of study medication. Exclusion criteria: a liver biopsy within 3 years of dosing was required to exclude cirrhosis. Participants were otherwise in good health, with no significant co‐morbidities. Other key exclusion criteria included positive test for hepatitis B surface antigen, anti‐hepatitis B core protein IgM antibodies and anti‐HIV antibodies. Randomization was stratified by interleukin(IL) 28B status (rs12979860) for CC or CT/TT allele. | |
| Interventions | Participants were randomised in a ratio of active:placebo of 1:1:1:1 Experimental group: participants received 1 of 3 once‐daily doses of sofosbuvir (100 mg, 200 mg, or 400 mg). Control group: placebo plus peg‐IFN α‐2a/RBV for 28 days. Co‐intervention: peg‐IFN α‐2a and RBV were administered according to the package insert for participants with genotype 1 infection. | |
| Outcomes | Primary outcome: AEs. Secondary outcomes: change in circulating HCV RNA at Week 4, percentage of participants with RVR at Week 4, percentage of participants with SVR at 12 and 24 weeks after last dose of peg+RBV following completion of 48 weeks of treatment, pharmacokinetics, percentage of participants who developed resistance to sofosbuvir. | |
| Notes | We emailed Rodriguez‐Torres and colleagues on 27 April 2016 for additional information on blinding during assessment, unpublished data, (mortality data) but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomisation schedule was provided by PharStat, Inc. (NC, USA) |
| Allocation concealment (selection bias) | Low risk | Participants were randomised by a central web‐based system using permutated blocks |
| Blinding of participants and personnel (performance bias) | Low risk | Both investigators and participants were blinded to the treatment assignment |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment |
| Incomplete outcome data (attrition bias) | Low risk | 2 participants dropped out during study |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT01054729) and all outcomes reported on |
| Vested‐interest bias | High risk | This study was funded by Gilead Sciences, Inc. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 74 participants were randomised Sex: 49 men, 25 women Mean age: 54.3 years Inclusion criteria: participants 18‐65 years of age with hepatitis C genotype 1 infection who had had unsuccessful prior treatment with standard P/R therapy and their screening HCV RNA level was 4 x 105 IU/mL or greater. Participants with cirrhosis by liver biopsy or noninvasive assessment (such as Fibroscan ultrasound and other approved methods according to the local standard of care) were enrolled in a separate cohort. The diagnosis of cirrhosis was based on the interpretation provided by the enrolling investigator. Exclusion criteria: complicated cirrhosis (defined per protocol as ascites, bleeding oesophageal varices, hepatic encephalopathy, or other signs or symptoms of decompensated cirrhosis), evidence of HCC, HIV co‐infection, or any condition contraindicating re‐treatment with P/R. Participants also were ineligible if recent laboratory tests showed hyperbilirubinaemia (total, > 2.4 mg/dL; or direct, > 1.0 mg/dL), hypoalbuminaemia (< 3.3 g/dL), anemia (< 13 g/dL for men or < 12 g/dL for women), thrombocytopenia (< 100 ‐103/mL), coagulopathy (international normalised ratio, > 1.2), or renal insufficiency (estimated creatinine clearance < 60 mL/min by the Cockcroft–Gault equation). | |
| Interventions | Experimental group:
Control group: P/R plus placebo for 48 weeks. Co‐intervention: P/R. | |
| Outcomes | Primary: SVR rate, AEs, discontinuations due to AEs. Secondary: cEVR, SVR24 for 300 mg vaniprevir, and SVR24 for 600 mg vaniprevir 24 weeks. | |
| Notes | We emailed Rodriguez‐Torres and colleagues on 27 April 2016 for additional information on allocation concealment, randomisation, blinding of participants and personnel as well as outcome assessment, specification of il28b genotypes and the SVR rates for these. Missing data, number of participants analysed for HCV‐related morbidity, sample size calculation, SAEs, but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as double‐blinded, but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | 4 participants dropped out (5.4%) due to administrative discontinuations |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT00704405) and all outcomes reported on |
| Vested‐interest bias | High risk | This study was sponsored and funded by Merck |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Rodriguez‐Torres 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as double‐blinded, but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | 4 participants dropped out (5.4%) due to administrative discontinuations |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT00704405) and all outcomes reported on |
| Vested‐interest bias | High risk | This study was sponsored and funded by Merck |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Rodriguez‐Torres 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as double‐blinded, but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | 4 participants dropped out (5.4%) due to administrative discontinuations |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT00704405) and all outcomes reported on |
| Vested‐interest bias | High risk | This study was sponsored and funded by Merck |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Rodriguez‐Torres 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as double‐blinded, but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | The studywas described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | 4 participants dropped out (5.4%) due to administrative discontinuations |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT00704405) and all outcomes reported on |
| Vested‐interest bias | High risk | This study was sponsored and funded by Merck |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 288 participants were randomised. Sex: 153 men, 135 women Mean age: 47.8 years Inclusion criteria: treatment‐naive (no prior treatment with IFN ± RBV or investigational anti‐HCV agents). Male and female participants aged ≥ 18 years were eligible for inclusion in the study. All participants were required to be HCV seropositive, infected with a genotype 1 strain, and have plasma HCV RNA levels ≥ 10,000 IU/mL at screening. In addition, a non‐cirrhotic fibrosis classification (i.e. Ishak score ≤ 4 or equivalent) from a liver biopsy obtained within 24 months of screening was required for enrolment. Exclusion criteria: co‐infected with either HIV or hepatitis B, had evidence of severe or decompensated liver disease or liver disease unrelated to HCV infection, or had any pre‐existing medical condition or laboratory abnormality that made them unsuitable for treatment with peg‐IFN/RBV. Additional exclusion criteria included an abnormal ECG suggestive of clinically significant cardiac disease or QTc > 450 ms at screening, and history of solid organ transplant, or active alcohol or substance abuse sufficient to prevent adherence to study medication and/or follow‐up. Lastly, female participants who were pregnant or nursing and male participants whose female partner was pregnant were excluded. | |
| Interventions | Experimental group:
Control group: placebo in combination with peg‐IFN/RBV for 24 weeks peg‐IFN (Pegasys) was administered at a dose of 180 μg subcutaneously once weekly. RBV (Copegus) was administered at 1000 mg twice a day for participants weighing ≤ 75 kg or 1200 mg twice a day for participants weighing > 75 kg. Co‐intervention: peg‐IFN/RBV. | |
| Outcomes | Primary: proportion of participants who achieved SVR. Secondary: the proportion of participants with RVR, complete EVR, end of treatment response (ETR); the proportion of participants with relapsed viraemia; and patterns of AEs and safety measures. | |
| Notes | We emailed Rodriguez‐Torres and colleagues on 27 April 2016 for additional information on randomisation, allocation concealment, blinding of outcome assessment, unpublished data, overview of SAEs and the nature of the SAE but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | All sponsor personnel responsible for the conduct of the trial, with the exception of the sponsor study programmer, remained blinded to the results provided to the data monitoring committee. (Participants and investigators were unblinded to treatment assignment at week 24 to determine eligibility to discontinue therapy) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | 67 participants dropped out |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT00987337) and the outcomes reported on |
| Vested‐interest bias | High risk | This study was sponsored by Pfizer Inc. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Rodriguez‐Torres 2014b1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | All sponsor personnel responsible for the conduct of the trial, with the exception of the sponsor study programmer, remained blinded to the results provided to the data monitoring committee. (Participants and investigators were unblinded to treatment assignment at week 24 to determine eligibility to discontinue therapy) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | 67 participants dropped out |
| Selective reporting (reporting bias) | Low risk | A protocol was found (NCT00987337) and the outcomes reported |
| Vested‐interest bias | High risk | This study was sponsored by Pfizer Inc. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 69 adult participants Sex: 49 men, 20 women Mean age: 50 years Inclusion criteria: chronic genotype 1‐4 HCV infection, for cohorts 1‐9, HCV RNA ≥ 100,000 IU/mL at screening (no HCV RNA restriction for cohort 10), screening laboratory values within defined thresholds and use of 2 effective contraception methods if female of childbearing potential or sexually active male Exclusion criteria: pregnant or nursing woman or man with pregnant female partner, presence of cirrhosis, prior exposure to approved or experimental HCV protease inhibitors, co‐infection with HIV or HBV, current or prior history of clinical hepatic decompensation, chronic use of systemic immunosuppressive agents, history of clinically significant illness or any other medical disorder that may interfere with participant treatment, assessment or compliance with the protocol. | |
| Interventions | Experimental group: 1: GS‐9857 up to 300 mg (genotype 1a) for 3 days. 2: GS‐9857 up to 300 mg (genotype 3) for 3 days. 3: GS‐9857 up to 300 mg (genotype 2) for 3 days. 4‐9: GS‐9857 up to 600 mg (genotype 1a, 1b, 2, 3, or 4) for 3 days. 10: GS‐9857 100 mg on Day 1 and GS‐9857 100 mg plus SOF/GS‐5816 on Days 2 and 3. Control group: placebo. | |
| Outcomes | Safety, antiviral activity. | |
| Notes | We contacted the trial authors about allocation sequence generation and concealment, how blinding was maintained, if outcome assessors were blinded, how many participants dropped out, SAE, death, SVR. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described that the control group received placebo but the similarity of the placebo with the study drug was not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants dropped out |
| Selective reporting (reporting bias) | Unclear risk | No prepublished protocol could be found (NCT02185794 was published after the start of the trial) |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result (Gilead Sciences) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 26 adult participants Inclusion criteria: participants who could be of either sex and any race could be included in this study if they were 18‐60 years of age, were willing to give written informed consent, and were willing to undergo multiple inpatient periods and outpatient visits during the study. Female participants had to be surgically sterile or of non‐childbearing potential, and men had to practice acceptable methods of contraception. Female partners of male enrollees also had to practice acceptable methods of contraception, and all contraception had to have been practiced for 30 days before the dosing period during all dosing periods, and for 30 days after discontinuation of dosing. Participants had to be serum positive for HCV RNA by quantitative polymerase chain reaction assay, with 100,000 IU/mL RNA and be genotype 1a or 1b nonresponders to peg‐IFN‐2b with or without RBV. Nonresponse was defined as achieving < a 2‐log10 decline in HCV RNA levels after at least 12 weeks of dosing with peg‐IFN‐2b at 1.5 g/kg/week. Participants had to have ALT and AST 5 times ULN, ‐fetoprotein values within normal levels, negative screen for drugs with high potential for abuse, normal or clinically acceptable ECG (QTc Exclusion criteria: participants were excluded from the study if they met any of the following criteria: haemophilia or use of anticoagulant therapy; evidence of advanced liver disease (e.g. known cirrhosis, history or presence of ascites, bleeding varices, encephalopathy); presence of organ transplant; known HIV or HBV positivity based on recent tests for anti‐HIV antibodies and hepatitis B surface antigen; or liver disease with a cause other than chronic hepatitis C. The significance of antinuclear antibodies, if present, was to be evaluated by investigators for individual participants to determine whether any interference with the protocol that would warrant exclusion from the study could be expected. | |
| Interventions | Experimental group:
Control group: peg‐IFN‐2b monotherapy administered at 1.5 g/kg once per week. | |
| Outcomes | Antiviral activity, safety, pharmacokinetics. | |
| Notes | We emailed Sarrazin and colleagues on 27 April 2016 for additional information on prepublished protocol, data on SAE, death, SVR24 before the second phase began, allocation concealment but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | The trial was described as an open‐label trial |
| Blinding of outcome assessment (detection bias) | High risk | The trial was described as an open‐label trial |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants completed the first phase of the trial |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The trial was conducted at the Schering‐Plough Research Institute |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 357 participants Inclusion criteria: prior null responders with chronic hepatitis C genotype 1, with no evidence of cirrhosis on liver biopsy, results of physical examination and laboratory tests within specified ranges and abstinence from use of abused substances. Exclusion criteria: women who were pregnant or nursing a child, participants with cirrhosis, co‐infection with Hepatitis B or HIV, and African‐American participants, previous treatment with any HCV polymerase or protease inhibitor, participants who relapsed following response to previous treatment, evidence of advanced liver disease, or liver disease from a cause other than chronic hepatitis C, pre‐existing psychiatric condition. | |
| Interventions | Experimental group: 2: boceprevir 100 mg orally three times a day for 48 weeks. 3: boceprevir 200 mg orally three times a day for 48 weeks. 4: boceprevir 400 mg orally three times a day for 24 weeks 5: boceprevir 400 mg orally three times a day + RBV. 6: boceprevir 400 mg orally three times a day for 48 weeks. 7: boceprevir 800 mg orally three times a day. 8 (added as an amendment): boceprevir 800 mg + RBV. Control group: (arm 1): placebo + a single dose of peg was given first, followed 1 week later by peg + RBV for 12 weeks. If participant was HCV RNA negative, peg + RBV was continued for another 36 weeks. Co‐intervention: peg‐IFN alfa‐2b (1.5 mg/kg/wk). | |
| Outcomes | Pharmacokinetics, antiviral activity, safety. | |
| Notes | Control group crossed over at week 17 if with detectable HCV RNA at week 12. Data needed to be available prior to week 12 before we could report the data. We contacted the trial authors on 06 June 2016 for additional information on allocation sequence generation and concealment, maximum follow‐up, how many participants dropped out, how was missing data handled, was there a prepublished protocol other than ClinicalTrials.gov, SAE, death, SVR24, data at week 12, and how much RBV was given but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as double‐blinded but the placebo was not described in detail |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | There was above 5% dropouts and it was unclear how the trial handled missing data |
| Selective reporting (reporting bias) | Unclear risk | Secondary outcomes were first added after the trial was completed |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result (Merck Sharp and Dohme Corp.) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 39 participants were randomised to treatment Sex: 32 men, 7 women Mean age: 41.5 years Inclusion criteria: male and female participants aged 18–60 years, with a BMI between 18 and 29 kg/m2 were enrolled. All participants were serum positive for HCV RNA by quantitative PCR assay, classified as G2/3, and naive to treatment for HCV infection. They were required to have ALT and AST 65 times ULN, no evidence of HCC (per ultrasound and serum alfa‐fetoprotein levels), and haematologic and biochemical evidence of compensated liver disease. Exclusion criteria: participants with a history of substance abuse within 1 year of study participation, or any clinically significant medical disorder, such as HIV or HBV infection, haemophilia, or evidence of other liver disease not caused by chronic hepatitis C were excluded. | |
| Interventions | Experimental group
Control group: placebo. Co‐intervention: none. | |
| Outcomes | Primary: to evaluate the safety and tolerability of boceprevir. Secondary: pharmacokinetics and changes in HCV RNA viral load. | |
| Notes | We emailed Silva and colleagues on 27 April 2016 for additional information on allocation concealment, unpublished data, SVR data, (AEs and non serious AEs listed) plus published protocols but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code provided by the sponsor (Schering‐Plough Research Institute) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The study was described as double‐blinded, (active drug and matched placebo capsules were used to maintain third‐party blind dispensing) |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 participant dropped out due to AE |
| Selective reporting (reporting bias) | Unclear risk | Protocol not found |
| Vested‐interest bias | High risk | This study was supported by Merck & Co. Inc. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Silva 2013a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random code provided by the sponsor (Schering‐Plough Research Institute) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The study was described as double‐blinded, (active drug and matched placebo capsules were used to maintain third‐party blind dispensing) |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 participant dropped out due to AE |
| Selective reporting (reporting bias) | Unclear risk | Protocol not found |
| Vested‐interest bias | High risk | This study was supported by Merck & Co., Inc. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Silva 2013a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | computer‐generated random code provided by the sponsor (Schering‐Plough Research Institute) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The study was described as double‐blinded, (Active drug and matched placebo cap‐sules were used to maintain third‐party blind dispensing) |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as double‐blinded but it was unclear how the blinding was maintained and who performed the outcome assessment. |
| Incomplete outcome data (attrition bias) | Low risk | Only 1 participant dropped out due to AE |
| Selective reporting (reporting bias) | Unclear risk | Protocol not found |
| Vested‐interest bias | High risk | This study was supported by Merck & Co. Inc. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 24 participants Sex: 18 men, 6 women Mean age: 45.8 years Country: USA Inclusion criteria: men and women aged 18‐60 years with chronic HCV genotype 1 infection, a screening plasma HCV RNA level of at least 100,000 IU/mL, and a BMI between 18 and 35 kg/m2. Participants were noncirrhotic (screening FibroTest score of 0.59 with an aminotransferase/platelet ratio index of 2 or with absence of cirrhosis documented by biopsy within the previous 12 months) and could be either treatment‐naive or have previously received and discontinued alfa IFN, with or without RBV, at least 6 months before enrolment. Exclusion criteria: previous exposure to HCV NS5A or NS5B inhibitors, co‐infected with HIV or HBV or infected with other HCV genotypes. Pregnant or nursing women were also excluded, as were women of childbearing age unwilling to use contraception from 1 month predose through 8 weeks postdose. Men were excluded if unwilling to practice barrier contraception with female partners for at least 12 weeks postdose. | |
| Interventions | The trial was divided into 4 different cohorts comprising Experimental group: oral 100 mg, 300 mg, 600 mg, and 900 mg of BMS‐791325 for 5 days. Control group: placebo. | |
| Outcomes | Safety assessment, HCV RNA assessment, pharmacokinetics | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated scheme |
| Allocation concealment (selection bias) | Low risk | Interactive voice‐response telephone system |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded, but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded, but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Low risk | All participants completed the study |
| Selective reporting (reporting bias) | High risk | The trial added extra primary outcomes in ClinicalTrials.gov (NCT00664625) |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised multicenter phase III clinical trial | |
| Participants | 656 participants Sex: 342 men, 314 women Mean age: 47.6 years Countries: 10 European countries and Japan Inclusion criteria: treatment‐naïve, aged 18–70 years (Europe), or 20–70 years (Japan), with chronic HCV genotype 1 infection diagnosed by positive anti‐HCV antibodies and HCV RNA > 1000 IU/mL at screening plus a positive antibody or HCV RNA test more than 6 months before screening, or a liver biopsy consistent with chronic HCV infection. Participants with compensated liver disease, including cirrhosis, were eligible for inclusion. All participants had a liver biopsy within 3 years or had a FibroScan within 6 months of randomisation to determine fibrosis stage. For participants without a liver biopsy, fibrosis stage was determined by FibroScan results using a cut‐off value of 9.5 kPa to indicate fibrosis stage > F3 (< 9.5 kPa F0–F2; > 9.5 kPa F3–F4), consistent with evaluations of the use of FibroScan in chronic HCV however, there are no reliable cut‐offs in the literature for distinguishing < F3 from > F3. The FibroScan threshold for cirrhosis was > 13 kPa. Exclusion criteria: HCV infection of mixed genotype (1/2, 1/3, and 1/4) diagnosed by genotypic testing at screening, evidence of acute or chronic liver disease due to causes other than chronic HCV infection, HIV co‐infection, HBV infection based on presence of HBs‐Ag, active malignancy, or history of malignancy within the last 5 years prior to screening (with an exception of appropriately treated basal cell carcinoma of the skin or in situ carcinoma of the uterine cervix), active or, history of alcohol or illicit drug abuse other than cannabis within the past 12 months, a condition that is defined as one which in the opinion of investigator may put the patient at risk because of participation in this study, may influence the results of this study, or limit the patient's ability to participate in this study, usage of any investigational drugs within 28 days prior to screening, or planned usage of an investigational drug during the course of this study, received concomitant systemic antiviral, hematopoietic growth factor, or immunomodulatory treatment within 28 days prior to screening. Participants being treated with oral antivirals such as acyclovir, famciclovir or valacyclovir for recurrent herpes simplex infection; or with oseltamivir or zanamivir for influenza A infection, may be screened, received silymarin (milk thistle), glycyrrhizin, or Sho‐saiko‐to (SST) within 28 days prior to screening and throughout the treatment phase, known hypersensitivity to any ingredient of the study drugs, alpha fetoprotein value > 100 ng/mL at screening; if > 20 ng/mL and = 100 ng/mL, participants may be included if there is no evidence of liver cancer in an appropriate imaging study (e.g. ultrasound, CT scan, or MRI) within last 6 months prior to randomisation (Visit 2), decompensated liver disease, or history of decompensated liver disease, as defined by the presence of: hepatic encephalopathy, ascites, or oesophageal variceal bleeding and/or laboratory results of any of the following: international normalized ratio = 1.7; serum albumin = 3.5 g/dL; serum total bilirubin = 2.0 mg/dL (except when the increase is predominately due to unconjugated bilirubin and related to Gilbert's syndrome), pre‐existing psychiatric condition that could interfere with the participant's participation in and completion of the study including but not limited to prior suicidal attempt, schizophrenia, major depression syndrome, severe anxiety, severe personality disorder, a period of disability or impairment due to a psychiatric disease within the past 5 years. | |
| Interventions | Experimental group 1: faldaprevir 120 mg once daily. Those with early treatment success (ETS, HCV RNA < 25 IU/mL target detected() or target not detected() at week 4 and < 25 IU/mL TND at week 8) stopped faldaprevir at week 12 and received placebo plus peg‐IFN and RBV for a further 12 weeks. Participants without ETS received faldaprevir plus peg‐IFN and RBV for 24 weeks. Experimental group 2: faldaprevir 240 mg once daily plus peg‐IFN and RBV for 12 weeks followed by placebo plus peg‐IFN and RBV to week 24, and either stopped treatment (early treatment success) or continued peg‐IFN and RBV to week 48 (no early treatment success). Control group: placebo. Co‐intervention: all participants received peg‐IFNα‐2a administered subcutaneously at 180 lg once weekly. RBV administered orally at a total dose of 1000 or 1200 mg (for bodyweight < 75 kg or P75 kg, respectively) daily in 2 divided doses, except in Japan where the total dose was 600, 800, or 1200 mg (for bodyweight 660 kg, > 60–680 kg, or > 80 kg, respectively) daily in 2 divided doses according to the local label peg‐IFN and RBV for 24 weeks after intervention period. All study medication was stopped in the event of virologic breakthrough at or after week 4 (increase in HCV RNA > 1 log10 from nadir or > 25 IU/mL after an initial decrease to < 25 IU/mL), lack of EVR (decrease in HCV RNA P2 log10 from baseline at week 12), or lack of virologic response (detectable HCV RNA at week 24). | |
| Outcomes | Safety assessment, SVR, AST or ALT normalisation, early treatment success. | |
| Notes | We contacted the trial authors for additional information on sequence generation, blinding, who was blinded for the HCV RNA results, missing data. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | The trial used interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | Low risk | Investigators, sponsor, and participants were blinded to treatment group allocation through the use of matching placebo capsules |
| Blinding of outcome assessment (detection bias) | High risk | HCV RNA results were only blinded up to week 8 |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out (and 34% dropped out of the placebo‐group) and it was unclear if the trial used proper methodology to account for this |
| Selective reporting (reporting bias) | High risk | The trial changed the primary outcomes from the original version |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim Pharmaceuticals, GmbH & Co. KG |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see STARTVerso‐1 2015a2 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | The trial used interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | Low risk | Investigators, sponsor, and participants were blinded to treatment group allocation through the use of matching placebo capsules |
| Blinding of outcome assessment (detection bias) | High risk | HCV RNA results were only blinded up to week 8 |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out (and 34% dropped out of the placebo‐group) and it was unclear if the trial used proper methodology to account for this |
| Selective reporting (reporting bias) | High risk | The trial changed the primary outcomes from the original version |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim Pharmaceuticals, GmbH & Co. KG. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised multicenter phase III clinical trial (STARTverso‐2) | |
| Participants | 658 participants Sex: 389 men, 268 women Mean age: 50.3 Inclusion criteria: treatment‐naive, 18–70 years (Europe), or 20–70 years (Japan), with chronic HCV genotype 1 infection diagnosed by positive anti‐HCV antibodies and HCV RNA > 1000 IU/ml at screening plus a positive antibody or HCV RNA test more than 6 months before screening, or a liver biopsy consistent with chronic HCV infection. Patients with compensated liver disease, including cirrhosis, were eligible for inclusion. All participants had a liver biopsy within 3 years or had a FibroScan within 6 months of randomisation to determine fibrosis stage. For participants without a liver biopsy, fibrosis stage was determined by FibroScan results using a cut‐off value of 9.5 kPa to indicate fibrosis stage > F3 (< 9.5 kPa F0–F2; > 9.5 kPa F3–F4), consistent with evaluations of the use of FibroScan in chronic HCV however, there are no reliable cut‐offs in the literature for distinguishing < F3 from > F3. The FibroScan threshold for cirrhosis was > 13 kPa. Exclusion criteria: mixed genotype HCV; HIV or hepatitis B co‐infection; decompensated liver disease; and contraindications to peg‐IFN or RBV. Asian participants were limited to 20% of the total population. | |
| Interventions | Experimental group 1: faldaprevir (BI 201335) 120 mg once daily (oral), for 24 weeks, with pegylated IFN α‐2a (peg‐IFN/RBV), subcutaneous injection/oral. At week 24, if the participants did not achieve early treatment success they received an additional 24 weeks of peg‐IFN/RBV alone. Experimental group 2: faldaprevir 240 mg once daily. faldaprevir 240 mg once daily (oral), for 12 weeks, with peg‐IFN/RBV (subcutaneous injection/oral). Followed by an additional 12 weeks of placebo plus peg‐IFN/RBV. At week 24, if the participants did not achieve early treatment success they received an additional 24 weeks of peg‐IFN/RBV alone. Control group: placebo (oral) once daily combined with peg‐IFN/RBV (subcutaneous injection) for 24 weeks, followed by an additional 24 weeks of peg‐IFN/RBV (oral) alone. | |
| Outcomes | Safety assessment, SVR, AST or ALT normalisation, early treatment success. | |
| Notes | Email was sent to Asselah and colleagues on 20 April 2016 for additional information on primary publication, randomisation, blinding, all bias, death but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blinded but the placebo was not further described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out and the trial did not report how they dealt with missing data |
| Selective reporting (reporting bias) | High risk | The trial changed the primary outcomes from the original version |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see STARTverso‐2 2014a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blinded but the placebo was not further described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out and the trial did not report how they dealt with missing data |
| Selective reporting (reporting bias) | High risk | The trial changed the primary outcomes from the original version |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 678 participants Sex: 403 men, 274 women Mean age: 53.4 years Inclusion criteria: chronic hepatitis C genotype 1 infection, diagnosed at least 6 months prior to screening, confirmed prior virological failure with an approved dose of peg‐IFN/RBV age 18‐70 years, HCV RNA = 1000 IU/mL at screening. Exclusion criteria: HCV infection of mixed genotype; HBV or HIV co‐infection. Evidence of acute or chronic liver disease due to causes other than chronic HCV infection, decompensated liver disease, or history of decompensated liver disease. Body weight < 40 or > 125 kg, clinical evidence of significant or unstable cardiovascular disease, chronic pulmonary disease, history or evidence of retinopathy or clinically significant ophthalmological disorder. Pre‐existing psychiatric condition that could interfere with the participant's participation in and completion of the study, laboratory parameters disorders (thalassaemia major, sickle cell anaemia or G6PD deficit). Haemoglobin < 12 g/dL for women and < 13 g/dL for men, participants who had been previously treated with at least 1 dose of any antiviral or immunomodulatory drug other than IFN alfa or RBV for acute or chronic HCV infection including and not restricted to protease or polymerase inhibitors. | |
| Interventions | The trial was divided into 3 cohorts according to virological failure (relapse, partial, null response) and randomised to 1 of the following groups: Experimental group 1: participants received faldaprevir 240 mg once daily, in the form of 2 soft gelatin capsules administered orally, combined with peg‐IFN/RBV, administered by injection, for 12 weeks, followed by placebo once daily combined with peg‐IFN/RBV for 12 weeks Experimental group 2: participants received faldaprevir 240mg once daily, in the form of 2 soft gelatin capsules administered orally, combined with peg‐IFN/RBV, administered by injection, for 24 weeks. Control group: received 2 soft gelatin capsules identical to those containing faldaprevir once daily (orally) and peg‐IFN α‐2a/RBV) administered by injection, for 24 weeks. Co‐intervention: At week 24, if the participants did not achieve early treatment success the participants received an additional 24 weeks of peg‐IFN/RBV alone. | |
| Outcomes | SVR, early treatment success, AST, ALT normalisation, safety. | |
| Notes | We emailed Jacobson and colleagues on 26 April 2016 for additional information on primary publication, randomisation, blinding, all bias, death but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being blinded but method was not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being blinded but method was not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out and the trial did not report how they dealt with missing data |
| Selective reporting (reporting bias) | High risk | The trial changed the primary outcomes from the original version (NCT01358864 ) |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see STARTverso‐3 2013a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being blinded but method was not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being blinded but method was not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out and the trial did not report how they dealt with missing data |
| Selective reporting (reporting bias) | High risk | The trial changed the primary outcomes from the original version (NCT01358864 ) |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see ADVANCE 2011a2 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being blinded but method was not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being blinded but method was not described |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out and the trial did not report how they dealt with missing data |
| Selective reporting (reporting bias) | High risk | The trial changed the primary outcomes from the original version (NCT01358864 ) |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 308 participants Sex: 248 men, 60 women Mean age: 46.9 years Inclusion criteria: 18–70 years, had chronic HCV genotype 1 infection (positive anti‐HCV antibody and HCV RNA > 1000 IU/mL at screening, and documented positive anti‐HCV antibody or HCV RNA > 1000 IU/ mL > 6 months prior to screening), and chronic HIV infection (HIV‐1 viral load testing or HIV‐1 western blot at screening and documented for > 6 months prior to screening) with a Karnofsky score greater than 70. HCV treatment‐naive individuals and those with prior relapse after completion of an IFN‐based regimen (detectable HCV/RNA < 24weeks after treatment with undetectable HCV/RNA at end of treatment) were eligible. Individuals naive to highly active antiretroviral therapy (HAART) were required to have a CD4þ cell count at least 500 cells/mL and HIV plasma RNA below 100,000 copies/mL at screening; those stabilised on HAART (HIV‐1 plasma RNA < 40 copies/mL at screening and < 50 copies/mL for > 6 months before randomisation) were required to have been on an acceptable combination of antiretrovirals (as defined in the protocol, Supplemental Table S1, http://links.lww.com/QAD/A638) for at least 6 weeks prior to randomisation and to have a CD4þ cell count at least 200 cells/mL. Individuals prescribed an atazanavir/ritonavir‐containing HAART regimen were required to have total bilirubin 2.5 times or less the ULN at screening. Documentation of a liver biopsy < 3 years or liver elastography < 6 months of randomisation was mandatory. Exclusion criteria: mixed genotype HCV, evidence of non‐HCV‐related liver disease, hepatitis B infection, decompensated liver disease, and hypersensitivity to the study treatments. | |
| Interventions | Experimental group: faldaprevir 240 mg for additional 12 weeks Control group: no intervention Co‐intervention: peg‐IFN and RBV + faldaprevir 240 mg for the first 12 weeks | |
| Outcomes | ALT, AST, SVR, SAE, mortality. | |
| Notes | Only the group with faldaprevir 240 mg 12W and faldaprevir 240 mg 24W could be used for analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Interactive voice‐response system |
| Allocation concealment (selection bias) | Low risk | Interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) | High risk | Open‐label study |
| Incomplete outcome data (attrition bias) | High risk | 12 participants from the experimental group dropped out, while none from the control group dropped out |
| Selective reporting (reporting bias) | Low risk | A protocol were published and the trial reported all outcomes (NCT01399619) |
| Vested‐interest bias | High risk | The trial was funded by Boehringer Ingelheim Pharma GmbH & Co. KG. |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 62 participants Sex: 53 men, 7 women (60 analysed) Mean age: 44.5 years (60 analysed) Countries: France, Germany, Spain and USA. Inclusion criteria: treatment‐naive participants age of 18‐65 years, genotype 1 chronic HCV infection, chronic HIV‐1 infection, no previous HCV treatment, and haemoglobin levels of 120 g/L or greater in women and 130 g/L or greater in men. Participants were required to have stable HIV disease defined as follows: part A (no antiretroviral therapy) participants had CD4 counts of ≥ 0.500 x 10^ cells/L and HIV RNA levels of ≤ 100,000 copies/mL, and part B (antiretroviral therapy for > 12 weeks) participants had CD4 counts of ≥ 0.300 x 10^ cells/L and HIV RNA levels < 50 copies/ mL. For part B, permissible antiretroviral regimens were efavirenz, tenofovir, and emtricitabine, or ritonavir‐boosted atazanavir, tenofovir, and either emtricitabine or lamivudine. Exclusion criteria: hepatic decompensation; other causes of significant liver disease, cancer within 5 years, significant cardiac dysrhythmia, and active AIDS‐related conditions within 6 months. All participants had liver biopsies within 1 year unless previous biopsies indicated cirrhosis; histologic assessment according to the METAVIR scoring system was done by a local pathologist. | |
| Interventions | Experimental group: oral 750 mg of telaprevir 3 times daily for 12 weeks (when the antiretroviral therapy included efavirenz, telaprevir dosage was 1125 3 times daily for 8 weeks). Control group: placebo. Co‐intervention: peg‐IFN 2a (180 μg/wk) and RBV (800 mg/d) for a total of 48 weeks. | |
| Outcomes | Safety assessment, efficacy assessment, SVR, pharmacokinetics. | |
| Notes | NCT00983853 participants were randomised in cohorts according to HIV‐treatment. We emailed Sulkowski and colleagues on 27 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Low risk | The trial used interactive web‐response system |
| Blinding of participants and personnel (performance bias) | High risk | The trial was only blinded for the first 24 weeks |
| Blinding of outcome assessment (detection bias) | High risk | The trial was only blinded for the first 24 weeks |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | High risk | The trial changed the primary outcome. Safety assessments were originally a primary outcome, this was changed |
| Vested‐interest bias | High risk | The trial was funded by Vertex pharmaceuticals |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 99 participants Sex: 68 men, 31 women Mean age: 44 years Countries: Argentina, Belgium, Canada, France and USA. Inclusion criteria: aged 18–65 years who were infected with both HIV and HCV at 30 academic and non‐academic study sites. Eligible participants had to have untreated, chronic HCV genotype 1 infection without hepatic decompensation, plasma HCV RNA of more than 10,000 IU/mL at screening, no infection with other HCV genotypes, and a liver biopsy sample with histological findings consistent for chronic hepatitis C (and no other cause), participants with a history of HIV infection for > 6 months and stable HIV disease, with a CD4 cell count of ≥ 200 cells per μL and HIV‐1 RNA viral load of < 50 copies per mL. Exclusion criteria: HBV surface antigen positive; use of didanosine, zidovudine, efavirenz, or other non‐nucleoside reverse transcriptase inhibitors; a neutrophil count of < 1500 cells per μL; a haemoglobin concentration of < 110 g/L for women and < 120 g/L for men; or a platelet count of < 100,000 platelets per μL. | |
| Interventions | Experimental group: 800 mg of boceprevir (MK‐3034) twice a day for 44 weeks. Control group: placebo. Co‐intervention: peg‐IFN–RBV for 4 weeks prior to intervention period. Additional 44 weeks of Peg‐IFN alfa‐2b 1.5 μg/kg administered once weekly by subcutaneous injection. RBV 600 mg–1400 mg per day (weight‐based) was taken orally twice daily with food. Erythropoietin was permitted if haemoglobin concentrations decreased to < 100 g/L. | |
| Outcomes | Pharmacokinetics, safety assessment, laboratory values. | |
| Notes | After 12 weeks of treatment the control group was allowed to cross‐over to the experimental group, therefore no data could be used. (NCT01482767) We emailed Sulkowski and colleagues on 27 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated sequence |
| Allocation concealment (selection bias) | Low risk | Interactive voice‐response system |
| Blinding of participants and personnel (performance bias) | High risk | All study site personnel (including the investigators), the sponsor, and participants were masked to treatment assignment until final database lock. But it was unclear when final database lock was defined. Additionally control group were allowed to crossover. |
| Blinding of outcome assessment (detection bias) | High risk | All study site personnel (including the investigators), the sponsor, and participants were masked to treatment assignment until final database lock. But it was unclear when final database lock was defined. Additionally control group were allowed to crossover |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed |
| Vested‐interest bias | High risk | The trial was funded by Merck |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | A phase IIb, multicenter, randomised, double‐blind, placebo‐controlled, parallel‐group trial (SILEN‐C1)(NCT00774397) | |
| Participants | 429 participants Sex: 234 men, 195 women Mean age ± SD: 46 ± 10.5 years Country: Argentina, Australia, Austria, Canada, Czech Republic, France, Germany, Republic of Korea, the Netherlands, Portugal, Romania, Spain, Switzerland, UK, and USA. Inclusion criteria: age between 18 and 65 years, chronic hepatitis C infection genotype 1, treatment‐naive, HCV RNA > 100,000 IU/mL. A liver biopsy within 24 months before enrolment providing histologic evidence of any degree of chronic necroinflammatory activity or the presence of fibrosis, but no evidence of cirrhosis, a normal retinal finding on fundoscopy within 6 months before enrolment. Exclusion criteria: HCV of mixed genotype, HBV or HIV co‐infection, decompensated liver disease, hyperbilirubinaemia > 1.5 ULN, concomitant treatment with medications that are substrates of P‐gp, UGT1A1, CYP3A4 or 2C9. Group 1: 71 participants Sex: 41 men, 30 women Mean age ± SD: 46 ± 10.9 years Ethnicity, n(%): Asian: 8(11), black: 4(6), white: 57(80), other: 2(3) HCV genotype, n(%): 1: 1(1), 1a: 32(45), 1b: 38(54). 3a, 4a, 6e, 6q: 0 IL28B genotype, n(%): CC: 11(15), non‐CC: 29(41), missing: 31(44) Group 2: 69 participants Sex: 40 men, 29 women Mean age ± SD: 46 ± 10.9 years Ethnicity, n(%): Asian: 9(13), black: 1(1), white: 58(84), other: 1(1) HCV genotype, n(%): 1: 0, 1a: 19(28), 1b: 50(72). 3a, 4a, 6e, 6q: 0 IL28B genotype, n(%): CC: 8(12), non‐CC: 33(48), missing: 28(41) Group 3: 143 participants Sex: 74 men, 69 women Mean age ± SD: 45 ± 10.2 years Ethnicity, n(%): Asian: 21(15), black: 1(1), white: 119(83), other: 2(1) HCV genotype, n(%): 1: 0, 1a: 67(47), 1b: 74(52). 3a, 4a, 6e, 6q: 2(1) IL28B genotype, n(%): CC: 19(13), non‐CC: 53(37), missing: 71(50) Group 4: 146 participants Sex: 79 men, 67 women Mean age ± SD: 46 ± 10.5 years Ethnicity, n(%): Asian: 17(12), black: 4(3), white: 122(84), other: 3(2) HCV genotype, n(%): 1: 0, 1a: 51(35), 1b: 91(62). 3a, 4a, 6e, 6q: 4(3) IL28B genotype, n(%): CC: 22(15), non‐CC: 48(33), missing: 76(52). | |
| Interventions | Experimental group: 2: faldaprevir 120 mg once daily for 24 weeks, 3: faldaprevir 240 mg once daily for 24 weeks, 4: faldaprevir 240 mg once daily for 24 weeks. Control group: 1: placebo once daily for 24 weeks. Co‐interventions: 2 and 3: peg‐IFN alfa‐2a 180 μg once weekly and oral weight‐based RBV 1000 mg‐1200 mg daily in 2 divided doses for 48 weeks with a 3‐day lead in period given with placebo. 1 and 4: peg‐IFN alfa‐2a 180 μg once weekly and oral weight‐based RBV 1000 mg to 1200 mg daily in 2 divided doses for 48 weeks. | |
| Outcomes | Primary outcome: sustained virological response 24 weeks after end of treatment Secondary outcomes: number of participants with virological rebound (HCV RNA < 1 log10 from nadir, or ≥ 100 IU/mL after previous viral load below the lower limit of detection in 2 consecutive visits at least 2 weeks apart. Number of participants with breakthrough (HCV RNA rebound during treatment). Number of participants with relapse (HCV RNA undetectable at end of treatment, but detectable during the follow‐up period). Number of participants with no response (participants who did not achieve SVR, but did not experience a virological breakthrough or relapse). | |
| Notes | We emailed Sulkowski and colleagues on 27 April 2016 for additional information on random sequence generation, allocation concealment, description of blinding, blinding of outcome assessors but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method of sequence generation was not specified |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided |
| Blinding of participants and personnel (performance bias) | Unclear risk | Authors stated that participants and investigators were blinded to treatment groups until 24 weeks after the end of treatment, but the method of blinding was not sufficiently described |
| Blinding of outcome assessment (detection bias) | Unclear risk | It was not mentioned if outcomes assessors were blinded |
| Incomplete outcome data (attrition bias) | Low risk | Number and reasons for treatment discontinuation and withdrawal were clearly stated. From 23%‐40% in the 3 groups of participants discontinued treatment, mostly due to lack of efficacy |
| Selective reporting (reporting bias) | Low risk | A protocol was published before randomisation began and all outcome results were reported adequately |
| Vested‐interest bias | Unclear risk | The study was sponsored by Boehringer Ingelheim |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 37 adult participants Sex: 22 men, 15 women Mean age: 48.3 years Inclusion criteria: chronic hepatitis C genotype 1, who were treatment‐naive participants, where women had to be either postmenopausal for at least 2 years or surgically sterile and men had to be surgically sterile or practicing specific forms of birth control and had documented FibroTest score in combination with an AST to Platelet Ratio Index, or a liver biopsy within the last 12 months to document absence of cirrhosis. Exclusion criteria: pregnant or breastfeeding woman, use of any medications contraindicated for use with peg‐IFN or RBV 2 weeks prior to study drug administration or 10 half‐lives, whichever was longer, clinically significant cardiac, respiratory (except mild asthma), renal, gastrointestinal, haematologic, neurologic disease, or any uncontrolled medical illness or psychiatric disease or disorder, current or past clinical evidence of cirrhosis or bridging fibrosis, abnormal screening laboratory results. | |
| Interventions | Experimental group:
Control group: placebo Co‐intervention: peg‐IFN α‐2a 180 μg/week + weight‐based RBV 1000 mg‐1200 mg/day for 48 weeks. | |
| Outcomes | ||
| Notes | We emailed Sullivan and colleagues on 27 April 2016 for additional information on allocation sequence generation and concealment, description of placebo, and prepublished protocol but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as double‐blinded (participant, caregiver, investigator, outcomes assessor) but it was unclear how well matched the placebo was |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as double‐blinded (participant, caregiver, investigator, outcomes assessor) but it was unclear how well matched the placebo was |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how the trial handled missing data (many were lost to follow‐up but still 'included' in the analyses) |
| Selective reporting (reporting bias) | High risk | The primary and secondary outcomes were changed after the trial was completed (NCT01314261) |
| Vested‐interest bias | Unclear risk | The trial was funded by a company that might have an interest in a given result (AbbVie) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical phase II trial | |
| Participants | 24 adults with chronic hepatitis C, genotype 1, who were naive to antiviral treatment. Country: Thailand Exclusion criteria: not described. | |
| Interventions | Experimental group: oral 200 mg, 400 mg of BIT225 for 28 days. Control group: placebo. Co‐intervention: IFN alfa 2b and RBV for a total of 48 weeks. | |
| Outcomes | SVR, safety, pharmacokinetics. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being placebo‐blinded, but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being placebo‐blinded, but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | No data |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be obtained |
| Vested‐interest bias | High risk | The trial was funded by Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised phase II clinical trial | |
| Participants | 39 participants Country: USA Inclusion criteria: treatment‐naive adults chronically infected with HCV genotype 1 adult participants. Participants were required to have HCV RNA ≥ 10–5 IU/mL (COBAS TaqMan HCV Test 2.0; Roche Molecular Diagnostics, Pleasanton, California; lower limit of quantitation (LLOQ) 25 IU/mL) at screening, with no evidence of cirrhosis by liver biopsy within 24 months of randomisation. Exclusion criteria: > 4 weeks of prior treatment with IFN or RBV within 6 months prior to randomisation;ALT > 5 x ULN; total bilirubin > 34 μmol/L (> 2 mg/dL) or direct bilirubin > ULN; international normalisation ratio > 1.7; confirmed creatinine clearance < 50 mL/min; or concurrent diagnosis of chronic hepatitis B infection, HIV infection, HCC or other non‐HCV liver disease. | |
| Interventions | Experimental group: oral 75 mg or 150 mg of beclabuvir twice daily for 48 weeks. Control group: placebo. Co‐intervention: once‐weekly subcutaneous peg‐IFN (180 lg) and twice‐daily oral RBV (weight‐based dosing of 1000 mg/day (< 75 kg) or 1200 mg/day (> 75 kg)). | |
| Outcomes | HCV RNA, safety assessment, pharmacokinetics. | |
| Notes | We emailed Tatum and colleagues on 27 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed |
| Vested‐interest bias | High risk | Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | For characteristics see Tatum 2015a1 | |
| Participants | ||
| Interventions | ||
| Outcomes | ||
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Blinding of outcome assessment (detection bias) | Unclear risk | The trial was described as being double‐blinded but it was unclear how the blinding was performed |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were assessed |
| Vested‐interest bias | High risk | Bristol‐Myers Squibb |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 111 participants Sex: 64 men, 47 women Mean age: 46 years Inclusion criteria: adults with chronic hepatitis C genotype 1 with no previous treatment for chronic hepatitis C, 18‐55 years of age, weight between 40 kg and 125 kg, liver biopsy within 2 years of screening with histology consistent with chronic hepatitis C and no evidence of bridging fibrosis or cirrhosis, participant and participant's partner(s) must each agree to use acceptable methods of contraception for at least 2 weeks prior to Day 1 and continue until at least 6 months after last dose of study drugs and participants must be willing to give written informed consent. Exclusion criteria: prior treatment for hepatitis C other than herbal remedies, HIV‐positive or known to be co‐infected with hepatitis B, medically significant gallbladder or hepatobiliary findings on screening ultrasound, use of any known significant inducers or substrates of CYP3A4 2 weeks prior to start of study medications, use of herbal supplements (milk thistle permitted), diabetic and hypertensive participants with clinically significant ocular examination findings, current moderate or severe depression, history of depression associated with any of the following: hospitalisation for depression, electroconvulsive therapy for depression, depression that resulted in a prolonged absence from work and/or significant disruption of daily functions, suicidal or homicidal ideation and/or attempt, history of severe psychiatric disorders, past history or current use of lithium, clinical diagnosis of substance abuse of alcohol, intravenous drugs, inhalational (not including marijuana), psychotropics, narcotics, cocaine use, prescription or over‐the‐counter drugs within 5 years of Day 1, past or current use of opiate agonist substitution therapy, any known pre‐existing medical condition (CNS, cardiac, pulmonary, immune mediated) that could interfere with the participant's participation in and completion of the study, active clinical gout within the last year, haemoglobinopathy or coagulopathy, myelodysplastic syndromes, organ transplants other than cornea and hair, poor venous access that precluded routine peripheral blood sampling or an indwelling venous catheter, participants with a history of gastric surgery (e.g. stapling, banding, bypass) or participants with a history of malabsorption disorders (e.g. celiac sprue disease), evidence of active or suspected malignancy, or a history of malignancy, within the last 5 years (except adequately treated basal cell carcinoma of the skin). Participants under evaluation for malignancy were not eligible, participants who were pregnant or nursing, participants who intended to become pregnant during the study period and male participants with partners who were, or intended to become, pregnant during the study period. | |
| Interventions | Experimental group:
Control group: no intervention. Co‐intervention: peg‐IFN α‐2b (1.5 μg/kg subcutaneously, weekly) and RBV (600 mg‐1400 mg/d based on weight) for 48 weeks. | |
| Outcomes | Antiviral effects, pharmacokinetics, safety. | |
| Notes | Participants from the control group were allowed to cross over to the experimental group after 12 weeks of treatment. We could therefore only use results from the first 12 weeks. We contacted trial authors about allocation sequence generation and concealment, how was missing data accounted for, SAE, number randomised to each group. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | High risk | Described as open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Described as open‐label |
| Incomplete outcome data (attrition bias) | Unclear risk | Above 5% dropouts in the control group and it was unclear how the trial handled missing data |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the protocol were reported on (NCT00797745) |
| Vested‐interest bias | High risk | The trial was funded by a company that might have an interest in a given result (Merck Sharp & Dohme Corp.) |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | Adults with chronic hepatitis C who were naive to treatment | |
| Interventions | Experimental group:
Control group: Control 1: placebo HCV‐796 + peg‐IFN 2b. Control 2: placebo HCV‐796 + peg‐IFN 2a. | |
| Outcomes | Antiviral activity | |
| Notes | We contacted trial authors for additional information on allocation sequence generation and concealment, how was blinding maintained, were outcome assessment blinded, how many dropped out, how many were randomised to each group, SVR, death, SAE, prepublished protocol, how was the trial funded. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as double‐blind but it was unclear how the blinding was maintained |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | It was unclear how many participants dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol could be found |
| Vested‐interest bias | High risk | Multiple authors were employees of Wyers |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Randomised clinical trial | |
| Participants | 64 adult participants Sex: 36 men, 28 women Mean age: 45 years Country: USA Inclusion criteria: male or female participants 18–65 years old inclusive, with a BMI of 18–35 kg/m2; documented clinical history compatible with chronic HCV, including positive anti‐HCV antibody, presence of HCV RNA in the plasma for at least 6 months or liver biopsy within 24 months with histology consistent with chronic HCV infection; HCV genotype 1, 2, 3 or 4; plasma HCV RNA P5 log10 IU/mL; all participants agreed to use double‐barrier birth control (such as a condom plus spermicide) from screening through at least 90 days following the last dose of the study drug. Exclusion criteria: pregnancy or breastfeeding; co‐infection with HBV or HIV; history or evidence of decompensated liver disease; prior clinical or histological evidence of cirrhosis; ALT or AST level > 3.0 ULN; history of HCC or findings suggestive of possible HCC; 1 or more additional known primary or secondary causes of liver disease, other than HCV; previous antiviral treatment for HCV; current abuse of alcohol or illicit drugs; or other clinically significant diseases that, in the opinion of the investigator, would jeopardise the safety of the participant or impact the validity of the study results. | |
| Interventions | Experimental group: oral 25 mg, 50 mg, 100 mg of samatasvir once a day for 3 days, or 50 mg of samatasvir twice a day for 3 days. Control group: placebo. | |
| Outcomes | Safety assessment, pharmacokinetics, antiviral activity, NS5A sequence analysis. | |
| Notes | We emailed Vince and colleagues on 27 April 2016 for additional information but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation sequence |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Randomisation code were "kept blinded to participants and clinical investigators" and matching placebo was used |
| Blinding of outcome assessment (detection bias) | Low risk | Randomisation code were "kept blinded to participants and clinical investigators" and matching placebo was used |
| Incomplete outcome data (attrition bias) | Low risk | There were no dropouts |
| Selective reporting (reporting bias) | High risk | The primary outcomes were changed (NCT01508156) |
| Vested‐interest bias | High risk | Idenix Pharmaceuticals, Inc |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | A phase IIb, randomised, double‐blind, active‐controlled, parallel‐group trial (PROPEL) (NCT00869661) | |
| Participants | 424 participants Sex: 255 men (60.1%), 169 women (39.9%) Location: North America, Europe, and Australia. Inclusion criteria: participants with chronic hepatitis C infection genotype 1 or 4, age 18‐65 years, treatment‐naive, serum HCV RNA level of at least 50,000 IU/mL, liver biopsy consistent with chronic hepatitis C obtained within 24 calendar months before first dose of study drug (36 months for participants with cirrhosis or incomplete/transition to cirrhosis, fibrosis score 3‐4). Participants with fibrosis score 3‐4 were required to have had an abdominal ultrasound, computerised tomography scan, or magnetic resonance imaging scan without evidence of HCC (within 2 months prior to randomisation) and a serum alfa‐fetoprotein < 100 ng/mL. Exclusion criteria: hepatitis A or B co‐infection, HIV co‐infection, history or evidence of other chronic liver disease other than HCV, history or evidence of decompensated liver disease, absolute neutrophil count < 1.5 x 109 cells/L, haemoglobin concentration < 12 g/dL in women and < 13 g/dL in men. Platelet count < 90 x 109 cells/L, history of renal disease, serum creatinine > 1.5 times the ULN, BMI < 18 or ≥ 36 kg/m2. Pregnant or breastfeeding women and male partners of pregnant women, inadequate forms of contraception in women of childbearing age and men with female partners of childbearing age (2 forms of contraception required). Group A: 80 participants Mean age: 47 years (range 18‐62) Race, n(%): white: 70(88), black: 8(10), other: 2(3) HCV genotype, n(%): 1a: 44(55), 1b: 28(35), 4: 8(10) Cirrhosis, n(%): 17(21) Group B: 81 participants Mean age: 47 years (range 23‐62) Race, n(%): white: 69(85), black: 9(11), other: 3(4) HCV genotype, n(%): 1a: 51(63), 1b: 26(32), 4: 4(5) Cirrhosis, n(%): 18(22) Group C: 82 participants Mean age: 47 years (range 21‐65) Race, n(%): white: 70(85), black: 9(11), other: 3(4) HCV genotype, n(%): 1a. 50(61), 1b: 26(32), 4: 6(7) Cirrhosis, n(%): 18(22) Group D: 81 participants Mean age: 48 years (range 23‐60) Race, n(%): white: 71(88), black: 6(7), other: 4(5) HCV genotype, n(%): 1a: 56(69), 1b: 22(27), 4: 3(4) Cirrhosis, n(%): 23(28) Group E: 84 participants Mean age: 48 years (range 22‐65) Race, n(%): white: 75(89), black: 3(4), other: 6(7) HCV genotype, n(%): 1a: 52(62), 1b: 25(30), 4: 7(8) Cirrhosis, n(%): 19(23). | |
| Interventions | Experimental group: Group A: oral mericitabine 500 mg twice daily for 12 weeks. Group B: oral mericitabine 1000 mg twice daily for 8 weeks. Group C: oral mericitabine 1000 mg twice daily for 12 weeks. Group D: oral mericitabine 1000 mg twice daily for 12 weeks. Control group: Group E: matched placebo orally twice daily for 12 weeks. Co‐interventions: Groups A, B, and C: peg‐IFN alfa‐2a 180 μg subcutaneously once weekly for 24 weeks ifeRVR achieved, or for 48 weeks if eRVR not achieved. Weight‐based oral RBV 1000 mg1‐200 mg daily in 2 divided doses for 24 weeks if eRVR achieved, or for 48 weeks if eRVR not achieved (eRVR was defined as undetectable HCV RNA (< 15 IU/mL) by week 4 and maintained through week 22). Groups D and E: peg‐IFN α‐2a 180 μg subcutaneously once weekly for 48 weeks. Weight‐based oral RBV 1000 mg‐1200 mg daily in 2 divided doses for 48 weeks. | |
| Outcomes | Primary outcome: SVR at week 24 after the last dose of study medication. Secondary outcomes: viral responses at clinic visits (HCV RNA was determined at baseline and at weeks 1, 2, 4, 6, 8, 10, 12, 14, 18, 24, 30, 36, 42, 48 of treatment and at weeks 4, 12, and 24 of follow‐up). Proportion of participants with relapse. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was stratified by geographical region and the randomization sequence was generated centrally by the sponsor...The randomization list was not available to personnel at the study centers or to the sponsor’s monitors during the study." |
| Allocation concealment (selection bias) | Low risk | Quote: "...were randomized in enrollment order by central interactive voice‐response system or interactive web response system." |
| Blinding of participants and personnel (performance bias) | Low risk | A mericitabine‐matched placebo was used. Quote: "Patients and investigators remained blinded to individual treatment assignments during 24/48 weeks of study treatment and 24 weeks of study follow‐up." |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "The randomization list was made available to selected individuals from the sponsor at the time of Data Monitoring Committee review of ˜50% of patients in Cohort 2 at week 12, an independent statistician at the sponsor for analysis of ongoing safety data and an independent medical officer to review interim analysis data." |
| Incomplete outcome data (attrition bias) | Low risk | Number and reasons for withdrawal have been clearly stated. |
| Selective reporting (reporting bias) | Low risk | The protocol was published prior to randomisation and all pre‐specified outcomes were reported on. |
| Vested‐interest bias | High risk | Trial funded by Hoffmann‐LaRoche Ltd. |
| Other bias | Low risk | The trial appeared to be free of other bias domains that could put it at risk of bias. |
| Methods | Randomised clinical trial | |
| Participants | 23 adult participants Sex: 20 men, 3 women Mean age: 51.5 years Country: USA Inclusion criteria: chronic HCV (for 6 months) were eligible if they were treatment‐naive and noncirrhotic with HCV RNA levels of > 100,000 IU/mL Exclusion criteria: infected with HIV, HBV. | |
| Interventions | The trial was divided into 5 cohorts Experimental group: oral 1 mg, 10 mg, 30 mg, 60 mg, 120 mg in a single dose of GSK2336805. Control group: placebo. | |
| Outcomes | Safety analysis, pharmacokinetics, metabolite identification, clinical virology assessment. | |
| Notes | We emailed Wilfret and colleagues on 27 April 2016 for additional information but reply not received yet. The study included healthy volunteers. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | It was described that the trial was double‐blinded but it was unclear how the blinding of participants and personnel was performed |
| Blinding of outcome assessment (detection bias) | High risk | Those performing the outcome assessment were not blinded |
| Incomplete outcome data (attrition bias) | Unclear risk | More than 5% dropped out |
| Selective reporting (reporting bias) | High risk | The order of the primary outcomes were changed |
| Vested‐interest bias | High risk | The trial was funded by GlaxoSmithKline |
| Other bias | Low risk | The trial appeared to be free of other components that could put it at risk of bias |
| Methods | Parallel‐group, randomised, placebo‐controlled (SIRIUS) | |
| Participants | 154 participants Sex: 114 men, 40 women Mean age: 56.5 (SD 9.2) years Country: USA Inclusion criteria: treatment‐experienced chronic hepatitis C participants with genotype 1. Compensated cirrhosis. | |
| Interventions | Experimental group: ledipasvir and sofosbuvir for 24 weeks Control group: placebo for 12 weeks, followed by ledipasvir, sofosbuvir, and RBV for 12 weeks. | |
| Outcomes | Not stated. | |
| Notes | Published only as abstract. We emailed Younossi and colleagues on 27 April 2016 for additional information number of participants randomised per group, random sequence generation, method of allocation concealment, description of blinding procedure, blinding of outcome assessors, potential number and reasons for drop‐outs, pre‐defined outcomes, sponsorship and its role, race and ethnicity of participants, full text or at least the figure published in the abstract, and data from quality‐of‐life assessment but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated that trial was randomised, but method of sequence generation was not specified |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | It was unclear if participants and treatment providers were blinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | It was not mentioned if the outcome assessors were blinded |
| Incomplete outcome data (attrition bias) | Unclear risk | Insufficient information provided |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information provided |
| Vested‐interest bias | Unclear risk | It was uncertain how the trial was sponsored |
| Other bias | Low risk | The trial may or may not have been free of other domains that could put it at risk of bias |
| Methods | A randomised, double‐blind, placebo‐controlled, parallel‐group, phase III trial (REALIZE) (NCT00703118) | |
| Participants | 662 participants Location: Europe, South America, and North America Inclusion criteria: age between 18 and 70 years, chronic hepatitis C infection, HCV genotype 1, HCV RNA level ≥ 1000 IU/mL, previously treated, but not achieving SVR, a liver biopsy within 18 months before screening, absolute neutrophil count ≥ 1200 cells/mm3, platelet count ≥ 90,000 cells/mm3, haemoglobin level ≥ 12 g/dL for women, and ≥ 13 g/dL for men Exclusion criteria: decompensated liver disease, other causes of significant liver disease, other severe active diseases Group 1: 266 participants (T12PR48) Sex: 183 men 83 women Mean age: 51 years (range 23‐69) Race, n(%): white: 246(92), black: 11(4), Asian or other: 9(3) HCV genotype, n(%): 1a: 118(44), 1b: 121(45), 1c: 0, unknown: 27(10) HCV RNA ≥ 800,000 IU/mL, n(%): 238(89) Stage of fibrosis or cirrhosis, n(%): no or minimal fibrosis: 51(19), portal fibrosis: 83(31), bridging fibrosis: 60(23), cirrhosis: 72(27). Group 2: 264 participants (lead‐in T12PR48) Sex: 189 men, 75 women Mean age: 51 years (range 24‐70) Race, n(%): white: 252(95), black: 8(3), Asian or other: 4(2) HCV genotype, n(%): 1a: 121(46), 1b: 115(44), 1c: 0, unknown: 28(11) HCV RNA ≥ 800,000 IU/mL, n(%): 234(89) Stage of fibrosis or cirrhosis, n(%): no or minimal fibrosis: 68(26), portal fibrosis: 71(27), bridging fibrosis: 58(22), cirrhosis: 67(25). Control group: 132 participants (PR48) Sex: 88 men, 44 women Mean age: 50 years (range 21‐69) Race, n(%): white: 117(89), black: 11(8), Asian or other: 4(3) HCV genotype, n(%): 1a: 59(45), 1b: 59(45), 1c: 1(1), unknown: 13(10) HCV RNA ≥ 800,000 IU/mL, n(%): 114(86) Stage of fibrosis or cirrhosis, n(%): no or minimal fibrosis: 35(27), portal fibrosis: 38(29), bridging fibrosis: 29(22), cirrhosis: 30(23). | |
| Interventions | Experimental group:
Control group: placebo. Co‐interventions: peg‐IFN α‐2a 180 μg subcutaneously once weekly and oral weight‐based RBV 1000 mg‐1200 mg in 2 divided daily doses for 48 weeks. | |
| Outcomes | Primary outcome: proportion of participants with SVR at week 24 (undetectable HCV RNA 24 weeks after end of treatment). Secondary outcomes: effect of lead‐in treatment. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was performed with the use of a centralized system according to a predefined randomization list, constructed through random permuted blocks..." |
| Allocation concealment (selection bias) | Low risk | Allocation concealment was obtained by use of an interactive voice‐response/web‐response system (IVRS/IWRS). Treatment codes were assigned by the system to the participants, and all codes were kept by IVRS/IWRS and could only be broken by contacting the IVRS/IWRS |
| Blinding of participants and personnel (performance bias) | Low risk | A telaprevir‐matching placebo was used. All participants and study personnel and sponsor were unaware of treatment assignment |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Results of HCV RNA tests up to week 24 were masked and were monitored by an independent reviewer to assess whether participants had met a predefined stopping rule..." |
| Incomplete outcome data (attrition bias) | High risk | Number and reasons for discontinuation were clearly stated. However, the discontinuation rate was very high, from 30%‐38% in the experimental groups, up to 62% in the control group. A majority of participants in the experimental groups discontinued treatment due to AEs, while the main reason for discontinuation in the control group was reaching the virologic stopping rule |
| Selective reporting (reporting bias) | Low risk | The study protocol was available and all pre‐specified outcomes were reported on |
| Vested‐interest bias | High risk | The sponsor (Janssen) was directly involved in trial design and protocol development, as well as editorial assistance in the preparation of the manuscript |
| Other bias | Low risk | Trial seems to be free of other potential sources of bias |
| Methods | A phase III, randomised, placebo‐controlled, double‐blind, parallel‐group trial (SAPPHIRE‐II) (NCT01715415) | |
| Participants | 394 participants Sex: 227 men, 167 women Location: Australia, North America, and Europe Inclusion criteria: age between 18 and 70 years, prior null‐responder, partial responder, or relapser to peg‐IFN/RBV treatment. Chronic hepatitis C HCV genotype 1, no cirrhosis, HCV RNA level > 10,000 IU/mL Exclusion criteria: recent history of drug or alcohol abuse (within 6 months prior to study drug administration), HVB or HIV co‐infection, history of uncontrolled seizures, history of uncontrolled diabetes, active malignancy or history of malignancy, ALT > 5 x ULN, AST > 5 x ULN, calculated creatinine clearance < 60 mL/min, albumin < lower limit of normal (LLN), prothrombin time/international normalised ratio > 1.5, haemoglobin < LLN, platelets < 120,000 cells per mm3, absolute neutrophil count < 1500 cells/μL, indirect bilirubin > 1.5 ULN and direct bilirubin > ULN Group 1: 297 participants Sex: 167 men, 130 women Mean age: 51.7 years (range: 19.0‐71.0) Race, n(%): white: 269(90.6), black: 22(7.4), Asian: 6(2.0) Fibrosis score F2‐F3, n(%): 95(32.0) IL28B genotype CC, n(%): 34(11.4) HCV genotype, n(%): 1a: 173(58.2), 1b: 123(41.4) Group 2: 97 participants Sex: 60 men, 37 women Mean age: 54.9 years (range 30.0‐69.0) Race, n(%): white: 86(88.7), black: 10(10.3), Asian: 0 Fibrosis score F2‐F3, n(%): 32(33.0) IL28B genotype CC, n(%): 7(7.2) HCV genotype, n(%): 1a: 57(58.8), 1b: 40(41.2) | |
| Interventions | Experimental group: Group 1: ABT‐450 orally 150 mg once daily with ritonavir 100 mg once daily and ombitasvir 25 mg once daily for 12 weeks. Dasabuvir orally 250 mg twice daily for 12 weeks Control group: Group 2: matching placebos for 12 weeks, followed by an open‐label period of 12 weeks' administration of the active treatment Co‐intervention: oral weight‐based RBV 1000 mg‐1200 mg in 2 divided daily doses (1000 mg daily if body weight was < 75 kg and 1200 mg daily if body weight was ≥ 75 kg) | |
| Outcomes | Primary outcome: SVR 12 weeks after the end of study treatment. AEs Secondary outcomes: virological failure during treatment. Post‐treatment relapse. Percentage of participants with ALT normalisation at the final treatment visit among participants with ALT > ULN at baseline | |
| Notes | We emailed Zeuzem and colleagues on 27 April 2016 for additional information on SVR for placebo group, normalisation of ALT level after treatment but reply not received yet. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated schedule |
| Allocation concealment (selection bias) | Low risk | Allocation was performed "through IRT (interactive response technology) system in order to receive unique study bottle/kit numbers and a unique randomisation number", which was used only by the sponsor for loading treatment assignments into the database. |
| Blinding of participants and personnel (performance bias) | Low risk | Matching placebos were used identical to study drugs. |
| Blinding of outcome assessment (detection bias) | Low risk | An independent DMC received safety data and provided recommendations. All data were blinded to study all study personnel. |
| Incomplete outcome data (attrition bias) | Low risk | Number and reasons for discontinuation were clearly stated. |
| Selective reporting (reporting bias) | Low risk | Study protocol was published and available before randomisation. All pre‐specified outcomes were reported on. |
| Vested‐interest bias | High risk | The sponsor (AbbVie) was directly involved in study design, data analyses, drafting the manuscript, and submission for publication. |
| Other bias | Low risk | Trial seems to be free of other potential sources of bias. |
AE: adverse events; ALT: alanine aminotransferase;AST: aspartate aminotransferase; BMI: body mass index; DAA: direct‐acting antiviral(s); ECG: electrocardiogram; EVR: early virological response;eRVR: extended rapid virological response; FDA: Food and Drug Administration; h: hour(s); HBV: hepatitis B virus; HCC: hepatocellular carcinoma; HCV: hepatitis C virus; HCV VL (viral load); LLOQ: lower limit of quantification; mRNA: messenger RNA; IFN: interferon; PK: protein kinase; P/R: peg‐interferon/RBV; RBV: ribavirin; RNA: ribonucleic acid; RVR: rapid virologic response; SAE: serious adverse events; SVR: sustained viral response; vs: versus; ULN: upper limit of normal
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| All arms were treated with DAAs | |
| All participants were treated with DAAs | |
| All participants were treated with DAAs | |
| Participants were not treated with DAAs | |
| All participants were treated with DAAs | |
| All participants were treated with DAAs | |
| Not a randomised clinical trial. All participants were treated with DAAs | |
| The trial compared same treatment regimens (simeprevir 150 mg and sofosbuvir 400 mg) with concomitant different dosages of RBV (modified doses vs 1000 mg) and different treatment duration (24 weeks vs 16 weeks) | |
| Short review written as 'Clinical opinion' for RESPOND‐2 and SPRINT‐2 trials | |
| A Markov model simulation | |
| Wrong control (different doses of simeprevir) | |
| All participants were treated with DAAs | |
| All participants were treated with DAAs | |
| Participants were healthy | |
| All participants were treated with DAAs | |
| All participants were treated with DAAs | |
| The trial compared the same treatment in equal or different dosages (telaprevir and VX‐222) combined with or without peg‐IFN and RBV | |
| Pooled analysis from two different trials | |
| Was an analyses of multiple trials. It was not clear which trials the study looked at. | |
| Wrong intervention (trial does not actually compare DAA with placebo/other medical intervention) | |
| A Markov model projection | |
| A Markov model projection | |
| A Markov model projection | |
| Pooled analysis of data from different trials | |
| Parallel‐group design, no control arm | |
| No control arm | |
| Participants were healthy | |
| No control group | |
| No control group | |
| Parallel‐group design, no control arm | |
| Pooled analysis from two different trials | |
| Wrong control | |
| RBV was assessed as active treatment | |
| Wrong control (different doses of DAA) | |
| Wrong control (different doses of DAA) | |
| Wrong control (different doses of DAA) | |
| Wrong control (different doses of DAA) | |
| Wrong control (different doses of DAA) | |
| Wrong control group (control group received another DAA) | |
| Wrong control group (control group received another DAA) | |
| Combined analysis of 3 trials | |
| Compared the same treatment (ledipasvir/sofosbuvir + RBV) of different duration (12 weeks vs 24 weeks) | |
| Not a randomised clinical trial (compared other trials) | |
| Wrong intervention/control (compared RBV vs no RBV) | |
| Not randomised | |
| Not randomised | |
| Not randomised | |
| Single‐group, open label study | |
| No control group | |
| Not randomised | |
| Parallel‐group design, no control arm | |
| Parallel‐group design, no control group | |
| Wrong control (different time points of telaprevir) | |
| The trial compared different treatment durations (12 weeks vs 24 weeks) of the same treatment regimen (ABT‐450/r‐ombitasvir, dasabuvir, and RBV) | |
| Healthy volunteers | |
| Combined analysis of three trials | |
| Wrong control (all groups received DAA) | |
| Retrospective study | |
| Wrong intervention/control (The trial compared ribavirin versus no ribavirin). Same as Sulkowski 2014 (NCT01359644) | |
| Wrong control group (no groups could be used as control) | |
| Trial comparing different dosages of the same DAA | |
| Wrong intervention/control (compared RBV versus no RBV) | |
| Study evaluating 5 arms of participants treated with same drug regimen comparing different dosages, treatment durations, and/or RBV co‐intervention | |
| Evaluated different dosages of the same treatment regimen | |
| The trial was initially designed as a multicenter, phase 3, randomised, placebo‐controlled, double‐blind trial of sofosbuvir + RBV vs placebo + RBV. Based on new published information, the protocol was amended and the study was redefined as a descriptive study in which the groups were unblinded, the placebo group was terminated, and the study assessed sofosbuvir + RBV for 12 weeks vs sofosbuvir + RBV for 24 weeks |
DAA: direct‐acting antivirals; HCV: hepatitis C virus; peg‐IFN: pegylated interferon; RBV: ribavirin; vs: versus
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | D‐Lite |
| Methods | Randomised clinical trial |
| Participants | 165 adults with chronic hepatitis C, genotype 1, HCV RNA > 100,000 IU/mL at screening, seronegative for HIV and Hepatitis B surface antigen, liver biopsy within prior 2 years; subjects with compensated cirrhosis can enrol and will be capped at approximately 10% |
| Interventions | BMS‐790052 or BMS‐650032 |
| Outcomes | |
| Starting date | 4 March 2011 |
| Contact information | |
| Notes | NCT01309932 |
| Trial name or title | A randomised study to evaluate the safety and efficacy of IDX719 in combinations with simeprevir and/or TMC647055/ritonavir with or without ribavirin for 12 weeks in subjects with chronic hepatitis C infection |
| Methods | Randomised clinical trial |
| Participants | Treatment‐naïve, genotype 1b, 4 and 6 hepatitis C virus‐infected participants |
| Interventions | Samatasvir |
| Outcomes | |
| Starting date | 6 May 2013 |
| Contact information | |
| Notes | NCT01852604 |
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Hepatitis C‐related morbidity or all‐cause mortality Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.1  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 1 Hepatitis C‐related morbidity or all‐cause mortality. | ||||
| 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ bias risk Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.2  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ bias risk. | ||||
| 2.1 Trials at high risk of bias | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Trials at low risk of bias | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to type of DAA Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.3 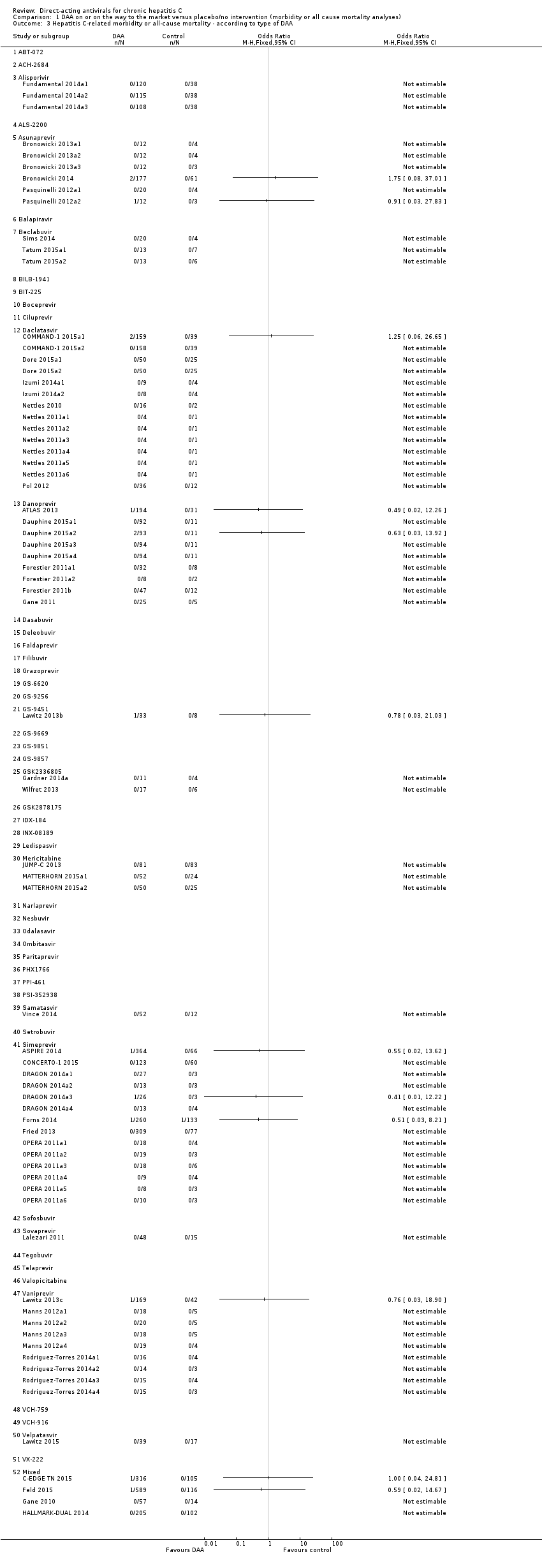 Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 3 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to type of DAA. | ||||
| 3.1 ABT‐072 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 ACH‐2684 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Alisporivir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 ALS‐2200 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 Asunaprevir | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.6 Balapiravir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.7 Beclabuvir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.8 BILB‐1941 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.9 BIT‐225 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.10 Boceprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.11 Ciluprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.12 Daclatasvir | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.13 Danoprevir | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.14 Dasabuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.15 Deleobuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.16 Faldaprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.17 Filibuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.18 Grazoprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.19 GS‐6620 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.20 GS‐9256 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.21 GS‐9451 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.22 GS‐9669 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.23 GS‐9851 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.24 GS‐9857 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.25 GSK2336805 | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.26 GSK2878175 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.27 IDX‐184 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.28 INX‐08189 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.29 Ledispasvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.30 Mericitabine | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.31 Narlaprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.32 Nesbuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.33 Odalasavir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.34 Ombitasvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.35 Paritaprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.36 PHX1766 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.37 PPI‐461 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.38 PSI‐352938 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.39 Samatasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.40 Setrobuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.41 Simeprevir | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.42 Sofosbuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.43 Sovaprevir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.44 Tegobuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.45 Telaprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.46 Valopicitabine | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.47 Vaniprevir | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.48 VCH‐759 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.49 VCH‐916 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.50 Velpatasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.51 VX‐222 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.52 Mixed | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to group of DAA Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.4 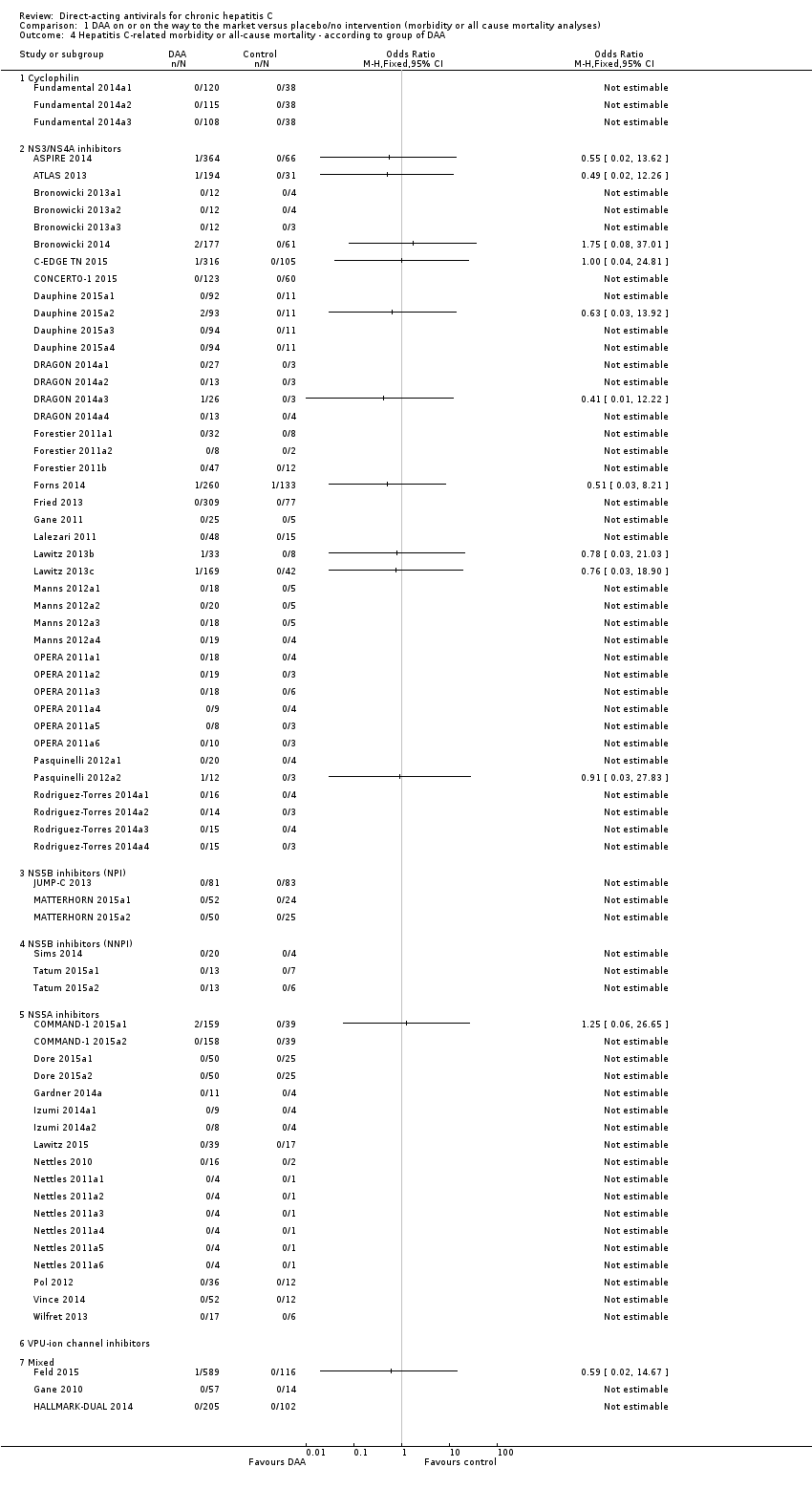 Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 4 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to group of DAA. | ||||
| 4.1 Cyclophilin | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 NS3/NS4A inhibitors | 41 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 NS5B inhibitors (NPI) | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 NS5B inhibitors (NNPI) | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 NS5A inhibitors | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 VPU‐ion channel inhibitors | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.7 Mixed | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to HIV‐infection Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.5  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 5 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to HIV‐infection. | ||||
| 5.1 With HIV‐infection | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Without HIV‐infection | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Mixed (with and without HIV‐infection) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 Unclear | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to comorbidity Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.6  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 6 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to comorbidity. | ||||
| 6.1 With comorbidity | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Without comorbidity | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Unclear | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to viral genotype Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.7  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 7 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to viral genotype. | ||||
| 7.1 Genotype 1 | 57 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Genotype 2 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Genotype 3 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Genotype 4 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 Mixed | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to human genotype (IL28b) Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.8  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 8 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to human genotype (IL28b). | ||||
| 8.1 IL28b (CC) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 IL28B (CT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 IL28B (TT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 IL28B (CT + TT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.5 Mixed | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to Asian‐region Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.9  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 9 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to Asian‐region. | ||||
| 9.1 From Asian region | 8 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 Not from Asian region | 52 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 Mixed | 11 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.4 Unclear | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to specific ethnicities Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.10  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 10 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to specific ethnicities. | ||||
| 10.1 White | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 Black | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 Hispanic | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.4 Mixed | 70 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.5 Unclear | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to reaching planned sample size Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.11  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 11 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to reaching planned sample size. | ||||
| 11.1 Trials reaching planned sample size | 10 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 Trials not reaching planned sample size | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 Unclear | 58 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to prior treatment Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.12  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 12 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to prior treatment. | ||||
| 12.1 Treatment‐naive | 47 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 Treatment‐experienced | 16 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 Mixed | 8 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.4 Unclear | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to interferon Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.13  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 13 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to interferon. | ||||
| 13.1 Trials where both groups received interferon | 52 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.2 Trials where neither group received interferon | 19 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to ribavirin Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.14  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 14 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to ribavirin. | ||||
| 14.1 Trials where both groups received ribavirin | 52 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.2 Trials where neither group received ribavirin | 19 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to chronic kidney disease Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.15  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 15 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to chronic kidney disease. | ||||
| 15.1 With chronic kidney disease | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.2 Without chronic kidney disease | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.3 Unclear | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to cryoglobulinaemia Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.16 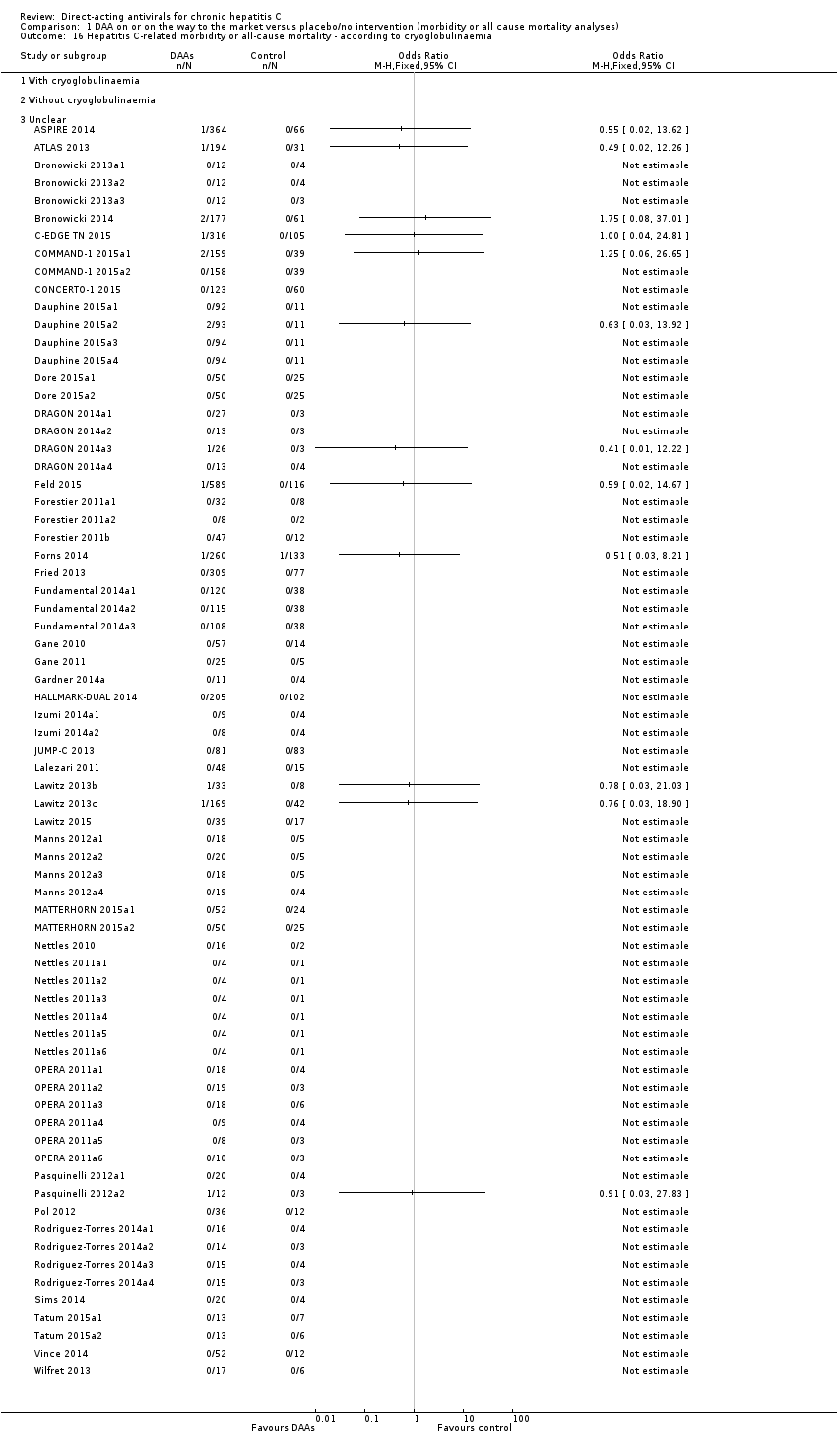 Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 16 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to cryoglobulinaemia. | ||||
| 16.1 With cryoglobulinaemia | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.2 Without cryoglobulinaemia | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.3 Unclear | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to DAA group as co‐intervention Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.17  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 17 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to DAA group as co‐intervention. | ||||
| 17.1 Trials where DAA were used as co‐intervention | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.2 Trials where DAA were not a co‐intervention | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to median dose Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 1.18  Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 18 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to median dose. | ||||
| 18.1 Over or equal to median dose | 41 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18.2 Under median dose | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18.3 Not available | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Serious adverse events Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.1  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 1 Serious adverse events. | ||||
| 2 Serious adverse events ‐ bias risk Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.2  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 2 Serious adverse events ‐ bias risk. | ||||
| 2.1 Trials at high risk of bias | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Trials at low risk of bias | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Serious adverse events ‐ according to type of DAA Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.3  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 3 Serious adverse events ‐ according to type of DAA. | ||||
| 3.1 ABT‐072 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 ACH‐2684 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Alisporivir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 ALS‐2200 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 Asunaprevir | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.6 Balapiravir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.7 Beclabuvir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.8 BILB‐1941 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.9 BIT‐225 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.10 Boceprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.11 Ciluprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.12 Daclatasvir | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.13 Danoprevir | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.14 Dasabuvir | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.15 Deleobuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.16 Faldaprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.17 Filibuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.18 Grazoprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.19 GS‐6620 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.20 GS‐9256 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.21 GS‐9451 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.22 GS‐9669 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.23 GS‐9851 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.24 GS‐9857 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.25 GSK2336805 | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.26 GSK2878175 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.27 IDX‐184 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.28 INX‐08189 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.29 Ledispasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.30 Mericitabine | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.31 Narlaprevir | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.32 Nesbuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.33 Odalasavir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.34 Ombitasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.35 Paritaprevir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.36 PHX1766 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.37 PPI‐461 | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.38 PSI‐352938 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.39 Samatasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.40 Setrobuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.41 Simeprevir | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.42 Sofosbuvir | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.43 Sovaprevir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.44 Tegobuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.45 Telaprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.46 Valopicitabine | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.47 Vaniprevir | 10 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.48 VCH‐759 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.49 VCH‐916 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.50 Velpatasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.51 VX‐222 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.52 Mixed | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Serious adverse events ‐ according to group of DAA Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.4  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 4 Serious adverse events ‐ according to group of DAA. | ||||
| 4.1 Cyclophilin | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 NS3/NS4A inhibitors | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 NS5B inhibitors (NPI) | 8 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 NS5B inhibitors (NNPI) | 5 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 NS5A inhibitors | 25 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 VPU‐ion channel inhibitors | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.7 Mixed | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Serious adverse events ‐ according to HIV‐infection Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.5  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 5 Serious adverse events ‐ according to HIV‐infection. | ||||
| 5.1 With HIV‐infection | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Without HIV‐infection | 94 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Mixed (with and without HIV‐infection) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 Unclear | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Serious adverse events ‐ according to comorbidity Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.6  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 6 Serious adverse events ‐ according to comorbidity. | ||||
| 6.1 With comorbidity | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Without comorbidity | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Unclear | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Serious adverse events ‐ according to viral genotype Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.7  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 7 Serious adverse events ‐ according to viral genotype. | ||||
| 7.1 Genotype 1 | 84 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Genotype 2 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Genotype 3 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Genotype 4 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 Mixed | 17 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Serious adverse events ‐ according to human genotype (IL28b) Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.8  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 8 Serious adverse events ‐ according to human genotype (IL28b). | ||||
| 8.1 IL28b (CC) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 IL28B (CT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 IL28B (TT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 IL28B (CT + TT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.5 Mixed | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Serious adverse events ‐ according to Asian‐region Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.9  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 9 Serious adverse events ‐ according to Asian‐region. | ||||
| 9.1 From Asian region | 10 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 Not from Asian region | 76 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 Mixed | 11 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.4 Unclear | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Serious adverse events ‐ according to specific ethnicities Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.10  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 10 Serious adverse events ‐ according to specific ethnicities. | ||||
| 10.1 White | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 Black | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 Hispanic | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.4 Mixed | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.5 Unclear | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Serious adverse events ‐ according to reaching planned sample size Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.11  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 11 Serious adverse events ‐ according to reaching planned sample size. | ||||
| 11.1 Trials reaching planned sample size | 15 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 Trials not reaching planned sample size | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 Unclear | 83 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Serious adverse events ‐ according to prior treatment Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.12  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 12 Serious adverse events ‐ according to prior treatment. | ||||
| 12.1 Treatment‐naive | 72 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 Treatment‐experienced | 19 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 Mixed | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.4 Unclear | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Serious adverse events ‐ according to interferon Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.13  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 13 Serious adverse events ‐ according to interferon. | ||||
| 13.1 Trials where both groups received interferon | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.2 Trials where neither group received interferon | 29 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.3 Unclear | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Serious adverse events ‐ according to ribavirin Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.14  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 14 Serious adverse events ‐ according to ribavirin. | ||||
| 14.1 Trials where both groups received ribavirin | 73 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.2 Trials where neither group received ribavirin | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.3 Unclear | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15 Serious adverse events ‐ according to chronic kidney disease Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.15  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 15 Serious adverse events ‐ according to chronic kidney disease. | ||||
| 15.1 With chronic kidney disease | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.2 Without chronic kidney disease | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.3 Unclear | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16 Serious adverse events ‐ according to cryoglobulinaemia Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.16  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 16 Serious adverse events ‐ according to cryoglobulinaemia. | ||||
| 16.1 With cryoglobulinaemia | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.2 Without cryoglobulinaemia | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.3 Unclear | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17 Serious adverse events ‐ according to DAA group as co‐intervention Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.17  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 17 Serious adverse events ‐ according to DAA group as co‐intervention. | ||||
| 17.1 Trials where DAA were used as co‐intervention | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.2 Trials where DAA were not a co‐intervention | 99 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18 Serious adverse events ‐ according to median dose Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 2.18  Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 18 Serious adverse events ‐ according to median dose. | ||||
| 18.1 Over or equal to median dose | 58 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18.2 Under median dose | 37 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18.3 Not available | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Without sustained virological response Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| Analysis 3.1  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 1 Without sustained virological response. | ||||
| 2 Without sustained virological response ‐ bias risk Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| Analysis 3.2  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 2 Without sustained virological response ‐ bias risk. | ||||
| 2.1 Trials at high risk of bias | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 2.2 Trials at low risk of bias | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Without sustained virological response ‐ according to type of DAA Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| Analysis 3.3  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 3 Without sustained virological response ‐ according to type of DAA. | ||||
| 3.1 ABT‐072 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 ACH‐2684 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.3 Alisporivir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.4 ALS‐2200 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.5 Asunaprevir | 4 | 285 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.29, 0.85] |
| 3.6 Balapiravir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.7 Beclabuvir | 2 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.43, 1.40] |
| 3.8 BILB‐1941 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.9 BIT‐225 | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 0.27 [0.03, 2.51] |
| 3.10 Boceprevir | 1 | 229 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.29, 0.61] |
| 3.11 Ciluprevir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.12 Daclatasvir | 7 | 619 | Risk Ratio (M‐H, Random, 95% CI) | 0.60 [0.50, 0.73] |
| 3.13 Danoprevir | 5 | 642 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.28, 0.51] |
| 3.14 Dasabuvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.15 Deleobuvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.16 Faldaprevir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.17 Filibuvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.18 Grazoprevir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.19 GS‐6620 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.20 GS‐9256 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.21 GS‐9451 | 1 | 329 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.26, 0.67] |
| 3.22 GS‐9669 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.23 GS‐9851 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.24 GS‐9857 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.25 GSK2336805 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.26 GSK2878175 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.27 IDX‐184 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.28 INX‐08189 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.29 Ledispasvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.30 Mericitabine | 4 | 725 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.49, 1.27] |
| 3.31 Narlaprevir | 2 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.43, 1.09] |
| 3.32 Nesbuvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.33 Odalasavir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.34 Ombitasvir | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.39, 1.07] |
| 3.35 Paritaprevir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.36 PHX1766 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.37 PPI‐461 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.38 PSI‐352938 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.39 Samatasvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.40 Setrobuvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.41 Simeprevir | 19 | 2898 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.33, 0.46] |
| 3.42 Sofosbuvir | 3 | 181 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.20, 0.58] |
| 3.43 Sovaprevir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.44 Tegobuvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.45 Telaprevir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.46 Valopicitabine | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.47 Vaniprevir | 9 | 333 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.25, 0.43] |
| 3.48 VCH‐759 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.49 VCH‐916 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.50 Velpatasvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.51 VX‐222 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.52 Mixed | 2 | 735 | Risk Ratio (M‐H, Random, 95% CI) | 0.06 [0.00, 7.05] |
| 4 Without sustained virological response ‐ according to group of DAA Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| Analysis 3.4  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 4 Without sustained virological response ‐ according to group of DAA. | ||||
| 4.1 Cyclophilin | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 NS3/NS4A inhibitors | 41 | 4756 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.36, 0.46] |
| 4.3 NS5B inhibitors (NPI) | 7 | 906 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.36, 0.90] |
| 4.4 NS5B inhibitors (NNPI) | 2 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.43, 1.40] |
| 4.5 NS5A inhibitors | 9 | 686 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.49, 0.69] |
| 4.6 VPU‐ion channel inhibitors | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 0.27 [0.03, 2.51] |
| 4.7 Mixed | 1 | 705 | Risk Ratio (M‐H, Random, 95% CI) | 0.01 [0.00, 0.02] |
| 5 Without sustained virological response ‐ according to HIV‐infection Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| Analysis 3.5  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 5 Without sustained virological response ‐ according to HIV‐infection. | ||||
| 5.1 With HIV‐infection | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 Without HIV‐infection | 58 | 6726 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 5.3 Mixed (with and without HIV‐infection) | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.4 Unclear | 3 | 389 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.35, 0.72] |
| 6 Without sustained virological response ‐ according to comorbidity Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| Analysis 3.6  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 6 Without sustained virological response ‐ according to comorbidity. | ||||
| 6.1 With comorbidity | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.2 Without comorbidity | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 6.3 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Without sustained virological response ‐ according to viral genotype Show forest plot | 58 | 7098 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.36, 0.51] |
| Analysis 3.7  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 7 Without sustained virological response ‐ according to viral genotype. | ||||
| 7.1 Genotype 1 | 54 | 5984 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.37, 0.50] |
| 7.2 Genotype 2 | 3 | 185 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 3.21] |
| 7.3 Genotype 3 | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.43, 1.43] |
| 7.4 Genotype 4 | 5 | 226 | Risk Ratio (M‐H, Random, 95% CI) | 0.10 [0.02, 0.68] |
| 7.5 Genotype 6 | 1 | 49 | Risk Ratio (M‐H, Random, 95% CI) | 0.01 [0.00, 0.20] |
| 7.6 Mixed | 2 | 574 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.52, 1.62] |
| 8 Without sustained virological response ‐ according to human genotype (IL28b) Show forest plot | 58 | 6745 | Risk Ratio (M‐H, Random, 95% CI) | 0.46 [0.40, 0.54] |
| Analysis 3.8  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 8 Without sustained virological response ‐ according to human genotype (IL28b). | ||||
| 8.1 IL28b (CC) | 25 | 1444 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.29, 0.61] |
| 8.2 IL28B (CT) | 10 | 1304 | Risk Ratio (M‐H, Random, 95% CI) | 0.52 [0.42, 0.66] |
| 8.3 IL28B (TT) | 10 | 359 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.44, 0.67] |
| 8.4 IL28B (CT + TT) | 14 | 1798 | Risk Ratio (M‐H, Random, 95% CI) | 0.37 [0.23, 0.57] |
| 8.5 Unclear | 7 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.33, 0.68] |
| 8.6 Mixed | 26 | 1693 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.40, 0.63] |
| 9 Without sustained virological response ‐ according to Asian‐region Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| Analysis 3.9  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 9 Without sustained virological response ‐ according to Asian‐region. | ||||
| 9.1 From Asian region | 10 | 1128 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.28, 0.42] |
| 9.2 Not from Asian region | 42 | 4910 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.43, 0.60] |
| 9.3 Mixed | 7 | 1010 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.03, 1.17] |
| 9.4 Unclear | 2 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.35, 0.79] |
| 10 Without sustained virological response ‐ according to specific ethnicities Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| Analysis 3.10  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 10 Without sustained virological response ‐ according to specific ethnicities. | ||||
| 10.1 White | 2 | 412 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.15, 0.38] |
| 10.2 Black | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10.3 Hispanic | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10.4 Mixed | 48 | 5384 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.20, 0.27] |
| 10.5 Unclear | 9 | 862 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.20, 0.39] |
| 10.6 Asian | 2 | 457 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.23, 0.63] |
| 11 Without sustained virological response ‐ according to reaching planned sample size Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| Analysis 3.11  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 11 Without sustained virological response ‐ according to reaching planned sample size. | ||||
| 11.1 Trials reaching planned sample size | 13 | 3071 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.18, 0.25] |
| 11.2 Trials not reaching planned sample size | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.3 Unclear | 48 | 4044 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.23, 0.33] |
| 12 Without sustained virological response ‐ according to prior treatment Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| Analysis 3.12  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 12 Without sustained virological response ‐ according to prior treatment. | ||||
| 12.1 Treatment‐naive | 44 | 4777 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.41, 0.56] |
| 12.2 Treatment‐experienced | 13 | 1274 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.36, 0.69] |
| 12.3 Mixed | 4 | 1064 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.02, 0.96] |
| 12.4 Unclear | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 13 Without sustained virological response ‐ according to interferon Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| Analysis 3.13  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 13 Without sustained virological response ‐ according to interferon. | ||||
| 13.1 Trials where both groups received interferon | 57 | 6229 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.41, 0.54] |
| 13.2 Trials where neither group received interferon | 2 | 735 | Risk Ratio (M‐H, Random, 95% CI) | 0.06 [0.00, 7.05] |
| 13.3 Trials where only the experimental group received interferon | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 13.4 Trials where only the control group received interferon | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 13.5 Mixed | 2 | 151 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.15, 2.30] |
| 14 Without sustained virological response ‐ according to ribavirin Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| Analysis 3.14  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 14 Without sustained virological response ‐ according to ribavirin. | ||||
| 14.1 Trials where both groups received ribavirin | 60 | 6410 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.41, 0.55] |
| 14.2 Trials where neither group received ribavirin | 1 | 705 | Risk Ratio (M‐H, Random, 95% CI) | 0.01 [0.00, 0.02] |
| 14.3 Trials where only the experimental group received ribavirin | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 14.4 Trials where only the control group received ribavirin | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 15 Without sustained virological response ‐ according to chronic kidney disease Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| Analysis 3.15  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 15 Without sustained virological response ‐ according to chronic kidney disease. | ||||
| 15.1 With chronic kidney disease | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15.2 Without chronic kidney disease | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15.3 Unclear | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 16 Without sustained virological response ‐ according to cryoglobulinaemia Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| Analysis 3.16 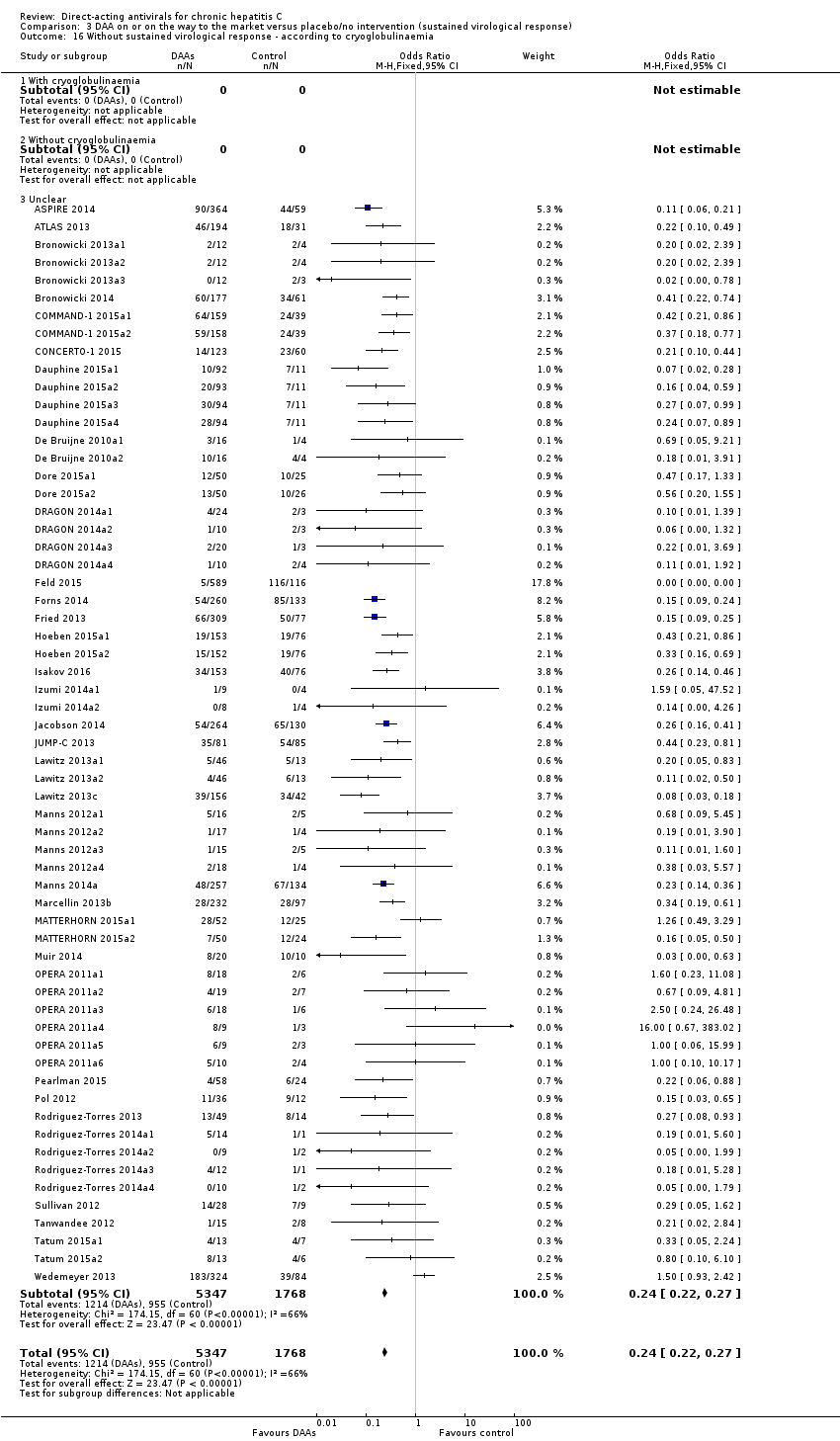 Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 16 Without sustained virological response ‐ according to cryoglobulinaemia. | ||||
| 16.1 With cryoglobulinaemia | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16.2 Without cryoglobulinaemia | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16.3 Unclear | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 17 Without sustained virological response ‐ according to DAA group as co‐intervention Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| Analysis 3.17  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 17 Without sustained virological response ‐ according to DAA group as co‐intervention. | ||||
| 17.1 Trials where DAA were used as co‐intervention | 3 | 480 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.27, 0.66] |
| 17.2 Trials where DAA were not a co‐intervention | 58 | 6635 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.21, 0.26] |
| 18 Without sustained virological response ‐ 'Best‐worst case' scenario Show forest plot | 61 | 7294 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.34, 0.49] |
| Analysis 3.18  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 18 Without sustained virological response ‐ 'Best‐worst case' scenario. | ||||
| 19 Without sustained virological response ‐ 'Worst‐best case' scenario Show forest plot | 61 | 7294 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.43, 0.60] |
| Analysis 3.19  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 19 Without sustained virological response ‐ 'Worst‐best case' scenario. | ||||
| 20 Without sustained virological response ‐ according to median dose Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| Analysis 3.20  Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 20 Without sustained virological response ‐ according to median dose. | ||||
| 20.1 Over or equal to median dose | 34 | 4154 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.32, 0.53] |
| 20.2 Under median dose | 23 | 2086 | Risk Ratio (M‐H, Random, 95% CI) | 0.46 [0.39, 0.55] |
| 20.3 Not available | 4 | 875 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.26, 1.47] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Hepatitis C‐related morbidity or all‐cause mortality Show forest plot | 9 | 781 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.06, 5.19] |
| Analysis 4.1  Comparison 4 Danoprevir versus placebo/no intervention, Outcome 1 Hepatitis C‐related morbidity or all‐cause mortality. | ||||
| 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose Show forest plot | 9 | 781 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.06, 5.19] |
| Analysis 4.2  Comparison 4 Danoprevir versus placebo/no intervention, Outcome 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose. | ||||
| 2.1 Over or equal to median dose | 6 | 606 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.06, 5.19] |
| 2.2 Under median dose | 3 | 175 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.3 Not available | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Serious adverse events Show forest plot | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 4.3  Comparison 4 Danoprevir versus placebo/no intervention, Outcome 3 Serious adverse events. | ||||
| 4 Serious adverse events ‐ according to median dose Show forest plot | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 4.4  Comparison 4 Danoprevir versus placebo/no intervention, Outcome 4 Serious adverse events ‐ according to median dose. | ||||
| 4.1 Over or equal to median dose | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Under median dose | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Not available | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Without sustained virological response Show forest plot | 5 | 642 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.12, 0.32] |
| Analysis 4.5  Comparison 4 Danoprevir versus placebo/no intervention, Outcome 5 Without sustained virological response. | ||||
| 6 Without sustained virological response ‐ according to median dose Show forest plot | 5 | 642 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.12, 0.32] |
| Analysis 4.6  Comparison 4 Danoprevir versus placebo/no intervention, Outcome 6 Without sustained virological response ‐ according to median dose. | ||||
| 6.1 Over or equal to median dose | 4 | 537 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.18 [0.11, 0.32] |
| 6.2 Under median dose | 1 | 105 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.07, 0.99] |
| 6.3 Not available | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Hepatitis C‐related morbidity or all‐cause mortality Show forest plot | 95 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 5.1  Comparison 5 All DAA versus placebo/no intervention/other medical intervention (morbidity or all‐cause mortality analyses), Outcome 1 Hepatitis C‐related morbidity or all‐cause mortality. | ||||
| 1.1 Trials assessing DAAs on or on the way to the market | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Trials assessing DAAs withdrawn from market | 22 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Trials using other medical intervention as control group | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Trials using other medical intervention as experimental group | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ drugs not discontinued | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.1 Trials assessing discontinued drugs | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.2 Trials assessing drugs still used | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Hepatitis C‐related morbidity or all‐cause mortality ‐ bias risk | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.1 Trials with a high risk of bias | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 Trials with a low risk of bias | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to type of DAA | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.1 ABT‐072 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 ACH‐2684 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.3 Alisporivir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.4 ALS‐2200 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.5 Asunaprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.6 Balapiravir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.7 Beclabuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.8 BILB‐1941 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.9 BIT‐225 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.10 Boceprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.11 Ciluprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.12 Daclatasvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.13 Danoprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.14 Dasabuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.15 Deleobuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.16 Faldaprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.17 Filibuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.18 Grazoprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.19 GS‐6620 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.20 GS‐9256 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.21 GS‐9451 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.22 GS‐9669 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.23 GS‐9851 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.24 GS‐9857 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.25 GSK2336805 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.26 GSK2878175 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.27 IDX‐184 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.28 INX‐08189 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.29 Ledispasvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.30 Mericitabine | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.31 Narlaprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.32 Nesbuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.33 Odalasavir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.34 Ombitasvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.35 Paritaprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.36 PHX1766 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.37 PPI‐461 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.38 PSI‐352938 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.39 Samatasvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.40 Setrobuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.41 Simeprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.42 Sofosbuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.43 Sovaprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.44 Tegobuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.45 Telaprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.46 Valopicitabine | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.47 Vaniprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.48 VCH‐759 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.49 VCH‐916 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.50 Velpatasvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.51 VX‐222 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.52 Mixed | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to group of DAA | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.1 Cyclophilin | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 NS3/NS4A inhibitors | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.3 NS5B inhibitors (NPI) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.4 NS5B inhibitors (NNPI) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.5 NS5A inhibitors | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.6 VPU‐ion channel inhibitors | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.7 Mixed | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to HIV‐infection | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.1 With HIV‐infection | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.2 Without HIV‐infection | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.3 Mixed (with and without HIV‐infection) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.4 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to comorbidity | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7.1 With comorbidity | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7.2 Without comorbidity | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7.3 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to viral genotype | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.1 Genotype 1 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.2 Genotype 2 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.3 Genotype 3 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.4 Genotype 4 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.5 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to human genotype (IL28b) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.1 IL28b (CC) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.2 IL28B (CT) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.3 IL28B (TT) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.4 IL28B (CT + TT) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.5 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to Asian‐region | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10.1 From Asian region | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10.2 Not from Asian region | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10.3 Mixed | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10.4 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to specific ethnicities | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.1 White | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.2 Black | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.3 Hispanic | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.4 Mixed | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.5 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to reaching planned sample size | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12.1 Trials reaching planned sample size | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12.2 Trials not reaching planned sample size | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12.3 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to prior treatment | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13.1 Treatment‐naive | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13.2 Treatment‐experienced | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13.3 Mixed | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13.4 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to interferon | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14.1 Trials where both groups received interferon | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14.2 Trials where neither group received interferon | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14.3 Trials where only the experimental group received interferon | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14.4 Trials where only the control group received interferon | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to ribavirin | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15.1 Trials where both groups received ribavirin | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15.2 Trials where neither group received ribavirin | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15.3 Trials where only the experimental group received ribavirin | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15.4 Trials where only the control group received ribavirin | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to chronic kidney disease | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16.1 With chronic kidney disease | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16.2 Without chronic kidney disease | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16.3 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 17 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to cryoglobulinaemia | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 17.1 With cryoglobulinaemia | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 17.2 Without cryoglobulinaemia | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 17.3 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 18 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to DAA group as co‐intervention | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 18.1 Trials where DAA were used as co‐intervention | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 18.2 Trials where DAA were not a co‐intervention | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Serious adverse events Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.1  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 1 Serious adverse events. | ||||
| 1.1 Trials assessing DAAs on or on the way to the market | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Trials assessing DAAs withdrawn from market | 62 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Trials using other medical intervention as control group | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Trials using other medical intervention as experimental group | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Serious adverse events ‐ bias risk Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.2  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 2 Serious adverse events ‐ bias risk. | ||||
| 2.1 Trials with a high risk of bias | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Trials with a low risk of bias | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Serious adverse events ‐ according to type of DAA Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.3  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 3 Serious adverse events ‐ according to type of DAA. | ||||
| 3.1 ABT‐072 | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 ACH‐2684 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Alisporivir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 ALS‐2200 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 Asunaprevir | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.6 Balapiravir | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.7 Beclabuvir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.8 BILB‐1941 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.9 BIT‐225 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.10 Boceprevir | 13 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.11 Ciluprevir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.12 Daclatasvir | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.13 Danoprevir | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.14 Dasabuvir | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.15 Deleobuvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.16 Faldaprevir | 13 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.17 Filibuvir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.18 Grazoprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.19 GS‐6620 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.20 GS‐9256 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.21 GS‐9451 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.22 GS‐9669 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.23 GS‐9851 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.24 GS‐9857 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.25 GSK2336805 | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.26 GSK2878175 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.27 IDX‐184 | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.28 INX‐08189 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.29 Ledispasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.30 Mericitabine | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.31 Narlaprevir | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.32 Nesbuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.33 Odalasavir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.34 Ombitasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.35 Paritaprevir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.36 PHX1766 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.37 PPI‐461 | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.38 PSI‐352938 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.39 Samatasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.40 Setrobuvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.41 Simeprevir | 19 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.42 Sofosbuvir | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.43 Sovaprevir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.44 Tegobuvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.45 Telaprevir | 13 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.46 Valopicitabine | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.47 Vaniprevir | 10 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.48 VCH‐759 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.49 VCH‐916 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.50 Velpatasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.51 VX‐222 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.52 Mixed | 8 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Serious adverse events ‐ according to group of DAA Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.4  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 4 Serious adverse events ‐ according to group of DAA. | ||||
| 4.1 Cyclophilin | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 NS3/NS4A inhibitors | 92 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 NS5B inhibitors (NPI) | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 NS5B inhibitors (NNPI) | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 NS5A inhibitors | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 VPU‐ion channel inhibitors | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.7 Mixed | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Serious adverse events ‐ according to HIV‐infection Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.5  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 5 Serious adverse events ‐ according to HIV‐infection. | ||||
| 5.1 With HIV‐infection | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Without HIV‐infection | 154 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Mixed (with and without HIV‐infection) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 Unclear | 11 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Serious adverse events ‐ according to comorbidity Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.6  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 6 Serious adverse events ‐ according to comorbidity. | ||||
| 6.1 With comorbidity | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Without comorbidity | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Unclear | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Serious adverse events ‐ according to viral genotype Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.7  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 7 Serious adverse events ‐ according to viral genotype. | ||||
| 7.1 Genotype 1 | 138 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Genotype 2 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Genotype 3 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Genotype 4 | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 Mixed | 26 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Serious adverse events ‐ according to human genotype (IL28b) Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.8  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 8 Serious adverse events ‐ according to human genotype (IL28b). | ||||
| 8.1 IL28b (CC) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 IL28B (CT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 IL28B (TT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 IL28B (CT + TT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.5 Unclear | 79 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.6 Mixed IL28b | 88 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Serious adverse events ‐ according to Asian‐region Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.9  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 9 Serious adverse events ‐ according to Asian‐region. | ||||
| 9.1 From Asian region | 12 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 Not from Asian region | 119 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 Mixed | 31 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.4 Unclear | 5 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Serious adverse events ‐ according to specific ethnicities Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.10  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 10 Serious adverse events ‐ according to specific ethnicities. | ||||
| 10.1 White | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 Black | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 Hispanic | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.4 Mixed | 133 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.5 Unclear | 31 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Serious adverse events ‐ according to reaching planned sample size | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 11.1 Trials reaching planned sample size | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 Trials not reaching planned sample size | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 Unclear | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Serious adverse events ‐ according to prior treatment Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.12  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 12 Serious adverse events ‐ according to prior treatment. | ||||
| 12.1 Treatment‐naive | 122 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 Treatment‐experienced | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 Mixed | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.4 Unclear | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Serious adverse events ‐ according to interferon Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.13  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 13 Serious adverse events ‐ according to interferon. | ||||
| 13.1 Trials where both groups received interferon | 126 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.2 Trials where neither group received interferon | 40 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.3 Trials where only the experimental group received interferon | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.4 Trials where only the control group received interferon | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Serious adverse events ‐ according to ribavirin Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.14  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 14 Serious adverse events ‐ according to ribavirin. | ||||
| 14.1 Trials where both groups received ribavirin | 127 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.2 Trials where neither group received ribavirin | 37 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.3 Trials where only the experimental group received ribavirin | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.4 Trials where only the control group received ribavirin | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15 Serious adverse events ‐ according to chronic kidney disease Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.15  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 15 Serious adverse events ‐ according to chronic kidney disease. | ||||
| 15.1 With chronic kidney disease | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.2 Without chronic kidney disease | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.3 Unclear | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16 Serious adverse events ‐ according to cryoglobulinaemia Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.16 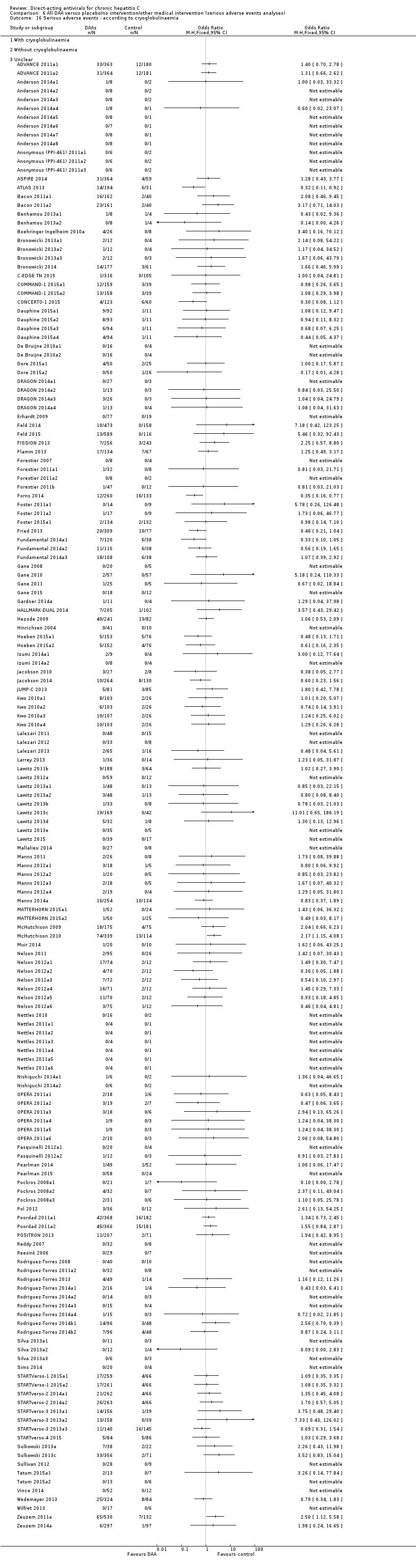 Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 16 Serious adverse events ‐ according to cryoglobulinaemia. | ||||
| 16.1 With cryoglobulinaemia | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.2 Without cryoglobulinaemia | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.3 Unclear | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17 Serious adverse events ‐ according to DAA group as co‐intervention Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 6.17  Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 17 Serious adverse events ‐ according to DAA group as co‐intervention. | ||||
| 17.1 Trials where DAA were used as co‐intervention | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.2 Trials where DAA were not a co‐intervention | 165 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Without sustained virological response Show forest plot | 107 | 17101 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.48, 0.59] |
| Analysis 7.1  Comparison 7 All DAA versus placebo/no intervention/other medical intervention (sustained virological response analyses), Outcome 1 Without sustained virological response. | ||||
| 1.1 Trials assessing DAAs on or on the way to the market | 60 | 6886 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 1.2 Trials assessing DAAs withdrawn from market | 43 | 9075 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.55, 0.69] |
| 1.3 Trials using other medical intervention as control group | 3 | 862 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.36, 1.82] |
| 1.4 Trials using other medical intervention as experimental group | 1 | 278 | Risk Ratio (M‐H, Random, 95% CI) | 0.23 [0.17, 0.29] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 SF‐36 physical score Show forest plot | 1 | 215 | Mean Difference (IV, Fixed, 95% CI) | ‐1.17 [‐3.65, 1.31] |
| Analysis 8.1  Comparison 8 All DAA versus placebo/no intervention/other medical intervention (quality of life scores), Outcome 1 SF‐36 physical score. | ||||
| 2 SF‐36 mental score Show forest plot | 1 | 215 | Mean Difference (IV, Fixed, 95% CI) | 1.36 [‐1.53, 4.25] |
| Analysis 8.2  Comparison 8 All DAA versus placebo/no intervention/other medical intervention (quality of life scores), Outcome 2 SF‐36 mental score. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Hepatitis C‐related morbidity or all‐cause mortality Show forest plot | 14 | 666 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.06, 26.65] |
| Analysis 9.1  Comparison 9 Daclatasvir versus placebo/no intervention, Outcome 1 Hepatitis C‐related morbidity or all‐cause mortality. | ||||
| 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose Show forest plot | 14 | 666 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.06, 26.65] |
| Analysis 9.2  Comparison 9 Daclatasvir versus placebo/no intervention, Outcome 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose. | ||||
| 2.1 Over or equal to median dose | 7 | 374 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.2 Under median dose | 7 | 292 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.06, 26.65] |
| 2.3 Not available | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Serious adverse events Show forest plot | 13 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 9.3  Comparison 9 Daclatasvir versus placebo/no intervention, Outcome 3 Serious adverse events. | ||||
| 4 Serious adverse events ‐ according to median dose Show forest plot | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 9.4  Comparison 9 Daclatasvir versus placebo/no intervention, Outcome 4 Serious adverse events ‐ according to median dose. | ||||
| 4.1 Over or equal to median dose | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Under median dose | 8 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Not available | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Without sustained virological response Show forest plot | 7 | 619 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.27, 0.59] |
| Analysis 9.5  Comparison 9 Daclatasvir versus placebo/no intervention, Outcome 5 Without sustained virological response. | ||||
| 6 Without sustained virological response ‐ according to median dose Show forest plot | 7 | 619 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.27, 0.59] |
| Analysis 9.6  Comparison 9 Daclatasvir versus placebo/no intervention, Outcome 6 Without sustained virological response ‐ according to median dose. | ||||
| 6.1 Over or equal to median dose | 4 | 360 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.26, 0.70] |
| 6.2 Under median dose | 3 | 259 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.19, 0.68] |
| 6.3 Not available | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Hepatitis C‐related morbidity or all‐cause mortality Show forest plot | 14 | 1589 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.08, 2.96] |
| Analysis 10.1  Comparison 10 Simeprevir versus placebo/no intervention, Outcome 1 Hepatitis C‐related morbidity or all‐cause mortality. | ||||
| 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose Show forest plot | 14 | 1589 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.08, 2.96] |
| Analysis 10.2  Comparison 10 Simeprevir versus placebo/no intervention, Outcome 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose. | ||||
| 2.1 Over or equal to median dose | 4 | 441 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.03, 8.21] |
| 2.2 Under median dose | 8 | 705 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.41 [0.01, 12.22] |
| 2.3 Not available | 2 | 443 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.02, 13.62] |
| 3 Serious adverse events Show forest plot | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 10.3  Comparison 10 Simeprevir versus placebo/no intervention, Outcome 3 Serious adverse events. | ||||
| 4 Serious adverse events ‐ according to median dose Show forest plot | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 10.4 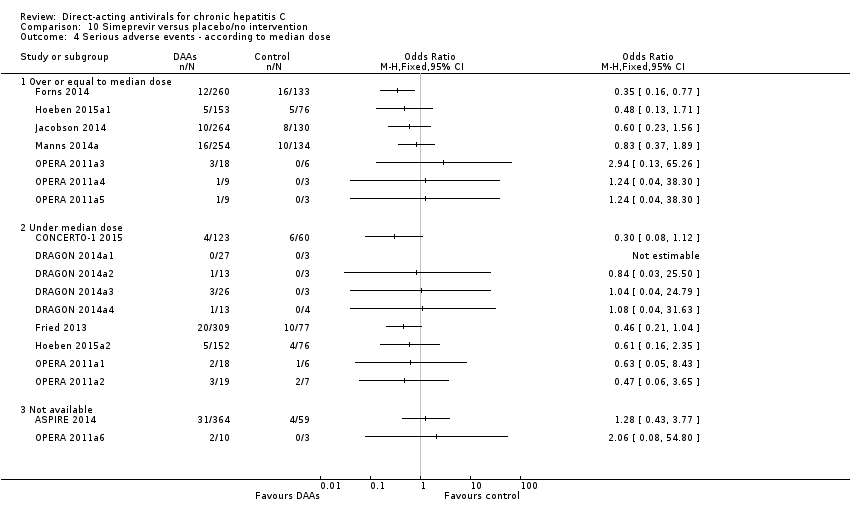 Comparison 10 Simeprevir versus placebo/no intervention, Outcome 4 Serious adverse events ‐ according to median dose. | ||||
| 4.1 Over or equal to median dose | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Under median dose | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Not available | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Without sustained virological response Show forest plot | 19 | 2898 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.19, 0.27] |
| Analysis 10.5  Comparison 10 Simeprevir versus placebo/no intervention, Outcome 5 Without sustained virological response. | ||||
| 6 Without sustained virological response ‐ according to median dose Show forest plot | 19 | 2898 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.19, 0.27] |
| Analysis 10.6  Comparison 10 Simeprevir versus placebo/no intervention, Outcome 6 Without sustained virological response ‐ according to median dose. | ||||
| 6.1 Over or equal to median dose | 9 | 1765 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.25 [0.20, 0.32] |
| 6.2 Under median dose | 8 | 696 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.13, 0.29] |
| 6.3 Not available | 2 | 437 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.13 [0.07, 0.24] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Hepatitis C‐related morbidity or all‐cause mortality Show forest plot | 9 | 379 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.03, 18.90] |
| Analysis 11.1  Comparison 11 Vaniprevir versus placebo/no intervention, Outcome 1 Hepatitis C‐related morbidity or all‐cause mortality. | ||||
| 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose Show forest plot | 9 | 379 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.03, 18.90] |
| Analysis 11.2  Comparison 11 Vaniprevir versus placebo/no intervention, Outcome 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose. | ||||
| 2.1 Over or equal to median dose | 6 | 313 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.03, 18.90] |
| 2.2 Under median dose | 3 | 66 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.3 Not available | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Serious adverse events Show forest plot | 10 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 11.3  Comparison 11 Vaniprevir versus placebo/no intervention, Outcome 3 Serious adverse events. | ||||
| 4 Serious adverse events ‐ according to median dose Show forest plot | 10 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| Analysis 11.4  Comparison 11 Vaniprevir versus placebo/no intervention, Outcome 4 Serious adverse events ‐ according to median dose. | ||||
| 4.1 Over or equal to median dose | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Under median dose | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Not available | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Without sustained virological response Show forest plot | 9 | 333 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.12 [0.06, 0.22] |
| Analysis 11.5  Comparison 11 Vaniprevir versus placebo/no intervention, Outcome 5 Without sustained virological response. | ||||
| 6 Without sustained virological response ‐ according to median dose Show forest plot | 9 | 333 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.12 [0.06, 0.22] |
| Analysis 11.6  Comparison 11 Vaniprevir versus placebo/no intervention, Outcome 6 Without sustained virological response ‐ according to median dose. | ||||
| 6.1 Over or equal to median dose | 6 | 280 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.10 [0.05, 0.20] |
| 6.2 Under median dose | 3 | 53 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.06, 1.04] |
| 6.3 Not available | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Without significant reductions in ALT/AST serum levels Show forest plot | 11 | 2099 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.68, 0.92] |
| Analysis 12.1  Comparison 12 All DAA versus placebo/no intervention, Outcome 1 Without significant reductions in ALT/AST serum levels. | ||||
| 2 Without significant reductions in ALT/AST serum levels ‐ according to DAA status Show forest plot | 11 | 2099 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.68, 0.92] |
| Analysis 12.2  Comparison 12 All DAA versus placebo/no intervention, Outcome 2 Without significant reductions in ALT/AST serum levels ‐ according to DAA status. | ||||
| 2.1 Trials assessing DAAs on or on the way to the market | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2.2 Trials assessing DAAs withdrawn from market | 11 | 2099 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.68, 0.92] |
| 3 Without significant reductions in ALT/AST serum levels ‐ according to type of drug Show forest plot | 11 | 2099 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.68, 0.92] |
| Analysis 12.3  Comparison 12 All DAA versus placebo/no intervention, Outcome 3 Without significant reductions in ALT/AST serum levels ‐ according to type of drug. | ||||
| 3.1 Faldaprevir | 8 | 2019 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.69, 0.96] |
| 3.2 Balaparavir | 3 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.41, 0.92] |


Study flow diagram
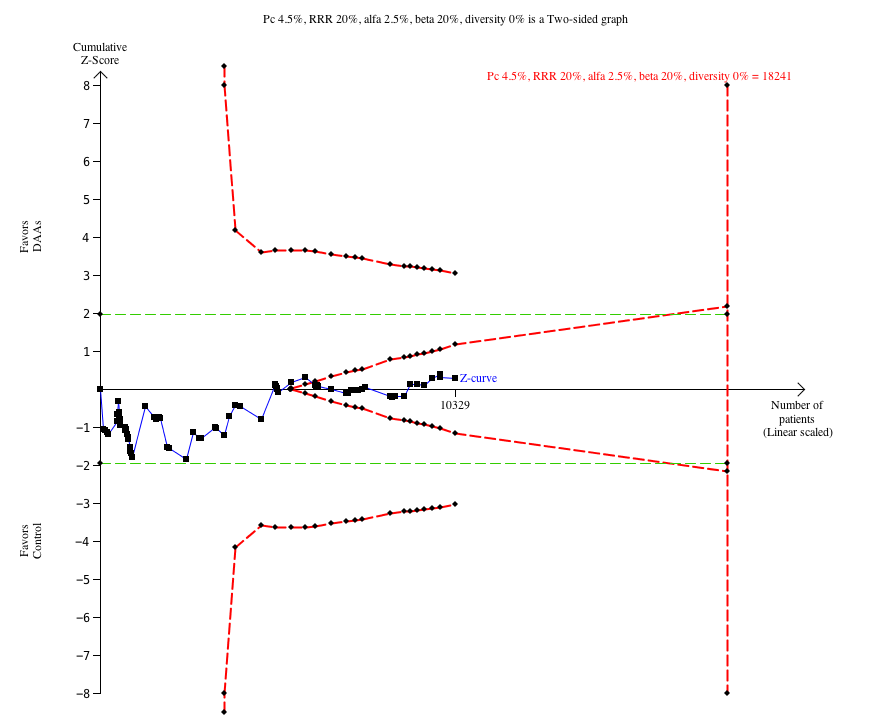
Trial Sequential Analysis of the effects of direct‐acting antivirals on the market or under development versus placebo or no intervention on risk of serious adverse events. The analysis was based on a proportion in the control group (Pc) of 4.5%, a relative risk reduction (RRR) of 20%, and alfa of 2.5%, a beta of 20%, and a diversity of 0%. The cumulative Z‐curve enters the futility area after the randomisation of about 6000 participants.

Trial Sequential Analysis of the effects of simeprevir versus placebo or no intervention on risk of serious adverse events. The analysis was based on a proportion in the control group (Pc) of 8.4%, a relative risk reduction (RRR) of 20%, and alfa of 2.5%, a beta of 20%, and a diversity of 0%. The cumulative Z‐curve crosses the naive type I error level of 5%, but it does not cross the trial monitoring boundary for benefit.

Trial Sequential Analysis of the effects of direct‐acting antivirals on the market or under development versus placebo or no intervention on risk of no sustained virological response. The analysis was based on a proportion in the control group (Pc) of 60.2%, a RRR of 20%, and alfa of 2.5%, a beta of 20%, and a diversity of 83%. After randomisation of about 1000 participants, the cumulative Z‐curve crosses the trial sequential monitoring boundary for benefit.

Trial Sequential Analysis of the effects of withdrawn direct‐acting antivirals versus placebo or no intervention on risk of serious adverse events. The analysis was based on a proportion in the control group (Pc) of 7.5%, a RRR of 20%, and alfa of 2.5%, a beta of 20%, and a diversity of 0%. After randomisation of about 5000 participants, the cumulative Z‐curve crosses the trial sequential monitoring boundary for harm.

Funnel plot of comparison: 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), outcome: 3.1 Without sustained virological response.
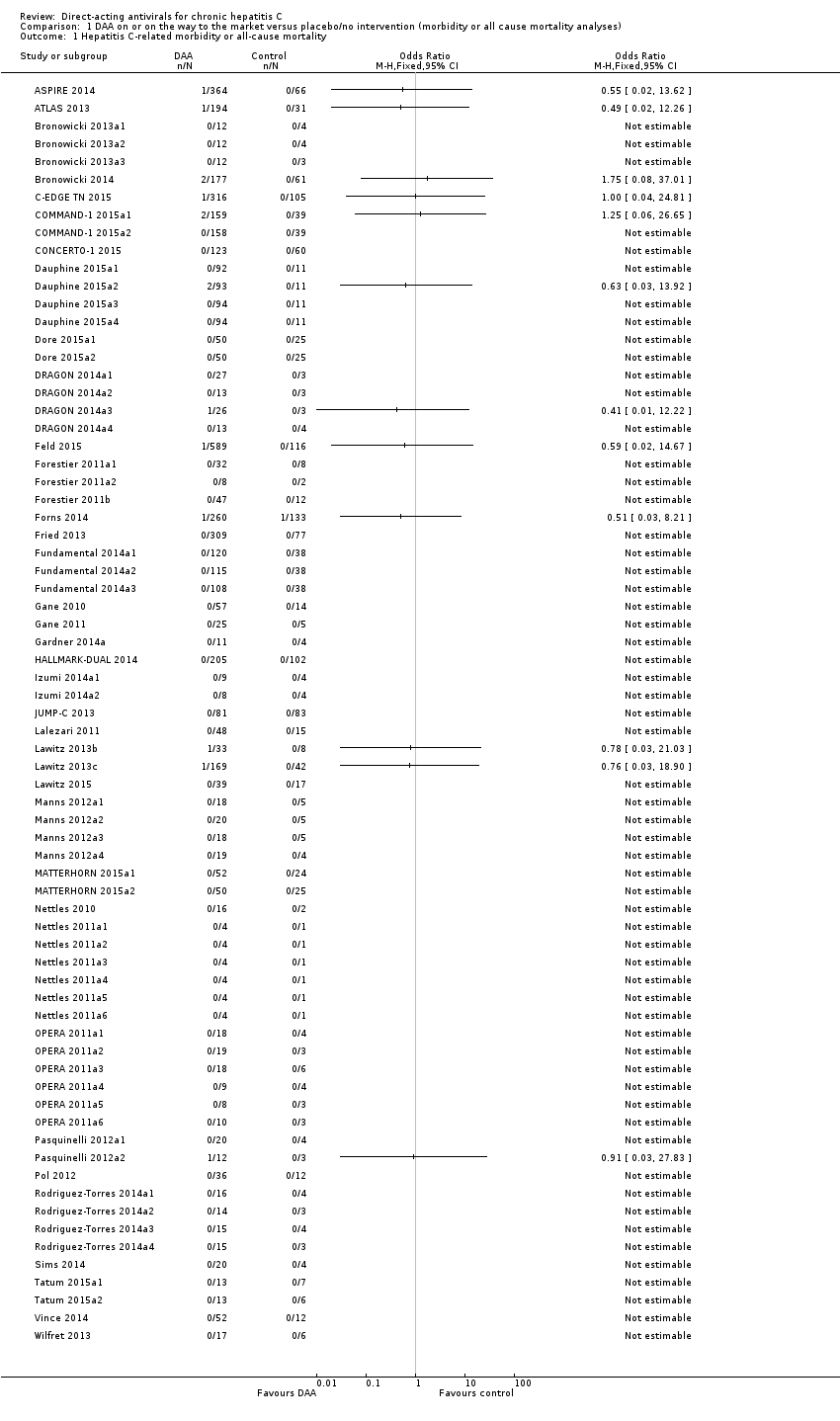
Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 1 Hepatitis C‐related morbidity or all‐cause mortality.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ bias risk.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 3 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to type of DAA.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 4 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to group of DAA.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 5 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to HIV‐infection.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 6 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to comorbidity.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 7 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to viral genotype.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 8 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to human genotype (IL28b).

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 9 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to Asian‐region.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 10 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to specific ethnicities.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 11 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to reaching planned sample size.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 12 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to prior treatment.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 13 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to interferon.
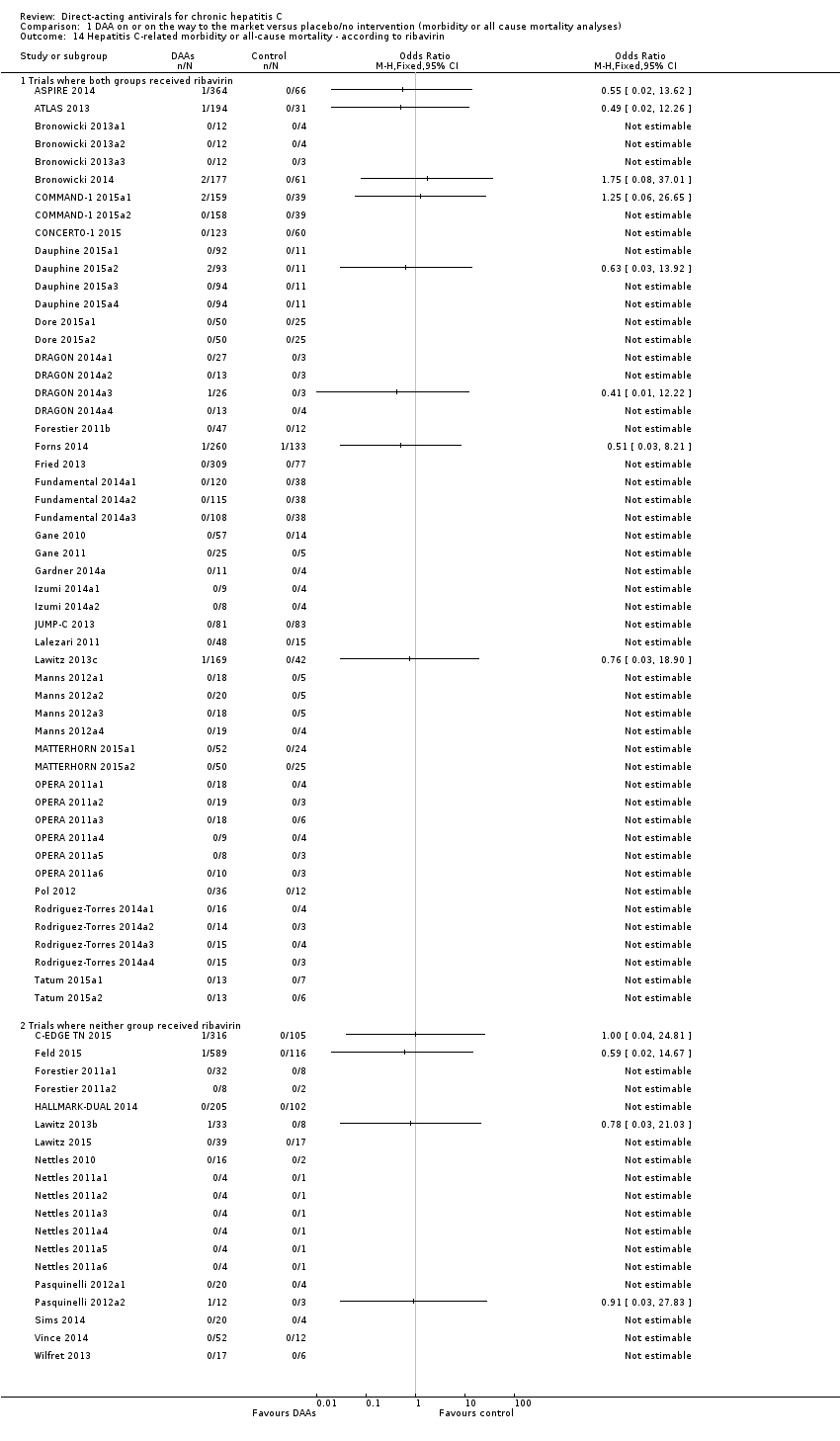
Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 14 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to ribavirin.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 15 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to chronic kidney disease.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 16 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to cryoglobulinaemia.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 17 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to DAA group as co‐intervention.

Comparison 1 DAA on or on the way to the market versus placebo/no intervention (morbidity or all cause mortality analyses), Outcome 18 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to median dose.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 1 Serious adverse events.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 2 Serious adverse events ‐ bias risk.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 3 Serious adverse events ‐ according to type of DAA.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 4 Serious adverse events ‐ according to group of DAA.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 5 Serious adverse events ‐ according to HIV‐infection.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 6 Serious adverse events ‐ according to comorbidity.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 7 Serious adverse events ‐ according to viral genotype.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 8 Serious adverse events ‐ according to human genotype (IL28b).

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 9 Serious adverse events ‐ according to Asian‐region.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 10 Serious adverse events ‐ according to specific ethnicities.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 11 Serious adverse events ‐ according to reaching planned sample size.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 12 Serious adverse events ‐ according to prior treatment.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 13 Serious adverse events ‐ according to interferon.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 14 Serious adverse events ‐ according to ribavirin.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 15 Serious adverse events ‐ according to chronic kidney disease.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 16 Serious adverse events ‐ according to cryoglobulinaemia.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 17 Serious adverse events ‐ according to DAA group as co‐intervention.

Comparison 2 DAA on or on the way to the market versus placebo/no intervention (serious adverse events analyses), Outcome 18 Serious adverse events ‐ according to median dose.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 1 Without sustained virological response.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 2 Without sustained virological response ‐ bias risk.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 3 Without sustained virological response ‐ according to type of DAA.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 4 Without sustained virological response ‐ according to group of DAA.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 5 Without sustained virological response ‐ according to HIV‐infection.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 6 Without sustained virological response ‐ according to comorbidity.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 7 Without sustained virological response ‐ according to viral genotype.
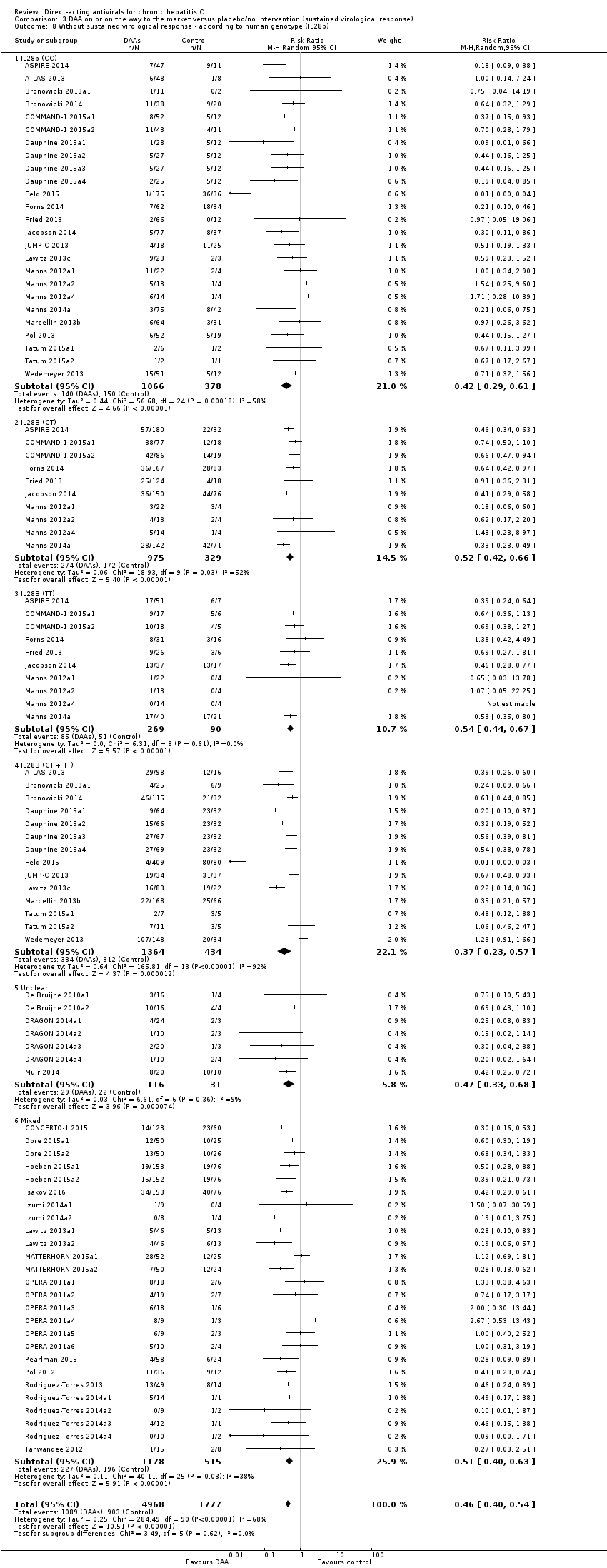
Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 8 Without sustained virological response ‐ according to human genotype (IL28b).

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 9 Without sustained virological response ‐ according to Asian‐region.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 10 Without sustained virological response ‐ according to specific ethnicities.
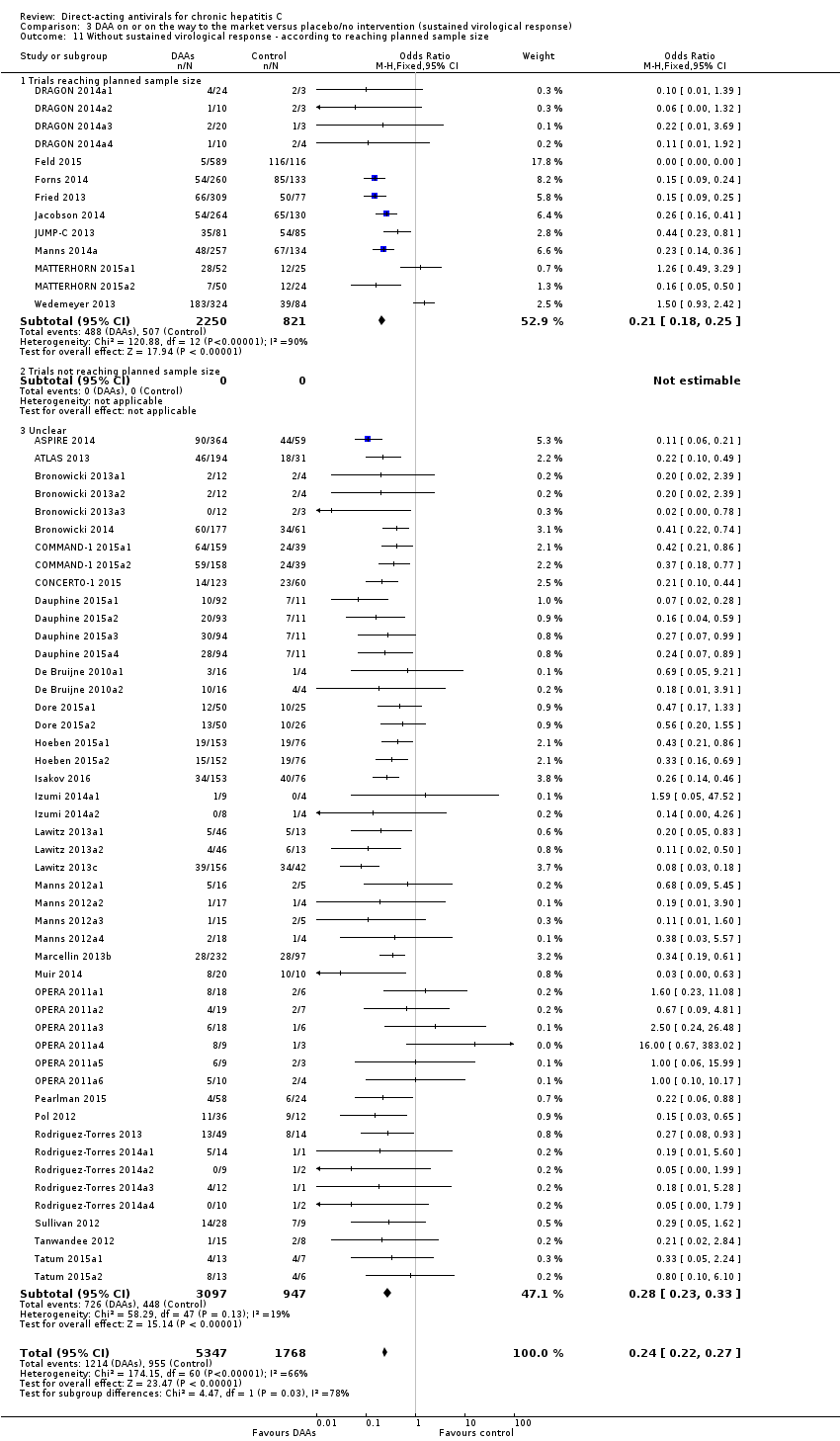
Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 11 Without sustained virological response ‐ according to reaching planned sample size.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 12 Without sustained virological response ‐ according to prior treatment.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 13 Without sustained virological response ‐ according to interferon.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 14 Without sustained virological response ‐ according to ribavirin.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 15 Without sustained virological response ‐ according to chronic kidney disease.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 16 Without sustained virological response ‐ according to cryoglobulinaemia.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 17 Without sustained virological response ‐ according to DAA group as co‐intervention.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 18 Without sustained virological response ‐ 'Best‐worst case' scenario.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 19 Without sustained virological response ‐ 'Worst‐best case' scenario.

Comparison 3 DAA on or on the way to the market versus placebo/no intervention (sustained virological response), Outcome 20 Without sustained virological response ‐ according to median dose.

Comparison 4 Danoprevir versus placebo/no intervention, Outcome 1 Hepatitis C‐related morbidity or all‐cause mortality.

Comparison 4 Danoprevir versus placebo/no intervention, Outcome 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose.

Comparison 4 Danoprevir versus placebo/no intervention, Outcome 3 Serious adverse events.

Comparison 4 Danoprevir versus placebo/no intervention, Outcome 4 Serious adverse events ‐ according to median dose.

Comparison 4 Danoprevir versus placebo/no intervention, Outcome 5 Without sustained virological response.

Comparison 4 Danoprevir versus placebo/no intervention, Outcome 6 Without sustained virological response ‐ according to median dose.

Comparison 5 All DAA versus placebo/no intervention/other medical intervention (morbidity or all‐cause mortality analyses), Outcome 1 Hepatitis C‐related morbidity or all‐cause mortality.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 1 Serious adverse events.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 2 Serious adverse events ‐ bias risk.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 3 Serious adverse events ‐ according to type of DAA.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 4 Serious adverse events ‐ according to group of DAA.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 5 Serious adverse events ‐ according to HIV‐infection.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 6 Serious adverse events ‐ according to comorbidity.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 7 Serious adverse events ‐ according to viral genotype.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 8 Serious adverse events ‐ according to human genotype (IL28b).

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 9 Serious adverse events ‐ according to Asian‐region.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 10 Serious adverse events ‐ according to specific ethnicities.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 12 Serious adverse events ‐ according to prior treatment.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 13 Serious adverse events ‐ according to interferon.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 14 Serious adverse events ‐ according to ribavirin.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 15 Serious adverse events ‐ according to chronic kidney disease.
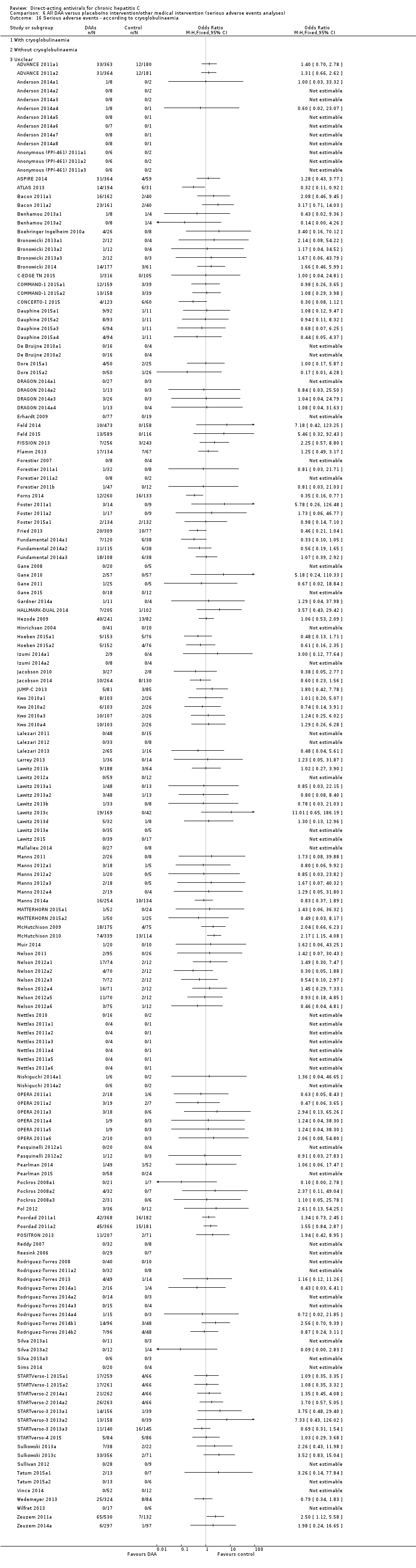
Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 16 Serious adverse events ‐ according to cryoglobulinaemia.

Comparison 6 All DAA versus placebo/no intervention/other medical intervention (serious adverse events analyses), Outcome 17 Serious adverse events ‐ according to DAA group as co‐intervention.

Comparison 7 All DAA versus placebo/no intervention/other medical intervention (sustained virological response analyses), Outcome 1 Without sustained virological response.

Comparison 8 All DAA versus placebo/no intervention/other medical intervention (quality of life scores), Outcome 1 SF‐36 physical score.
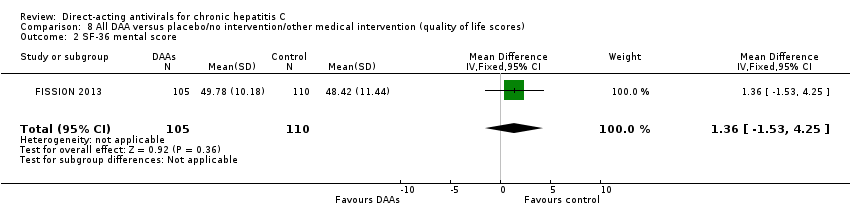
Comparison 8 All DAA versus placebo/no intervention/other medical intervention (quality of life scores), Outcome 2 SF‐36 mental score.
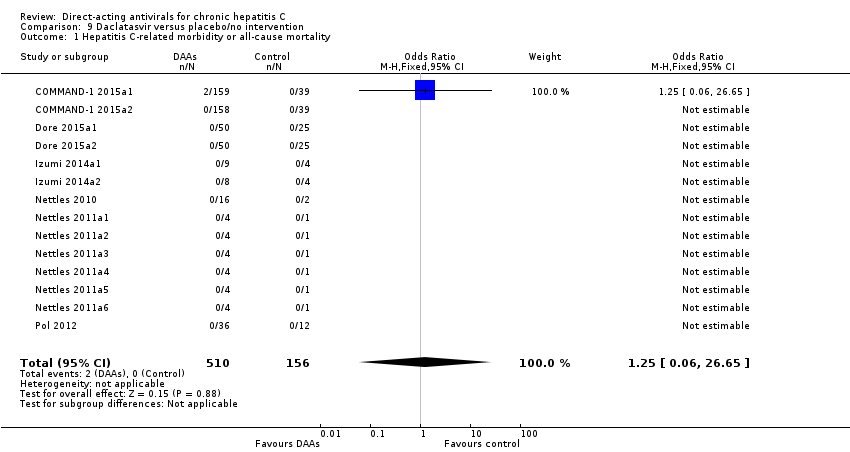
Comparison 9 Daclatasvir versus placebo/no intervention, Outcome 1 Hepatitis C‐related morbidity or all‐cause mortality.

Comparison 9 Daclatasvir versus placebo/no intervention, Outcome 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose.

Comparison 9 Daclatasvir versus placebo/no intervention, Outcome 3 Serious adverse events.

Comparison 9 Daclatasvir versus placebo/no intervention, Outcome 4 Serious adverse events ‐ according to median dose.

Comparison 9 Daclatasvir versus placebo/no intervention, Outcome 5 Without sustained virological response.

Comparison 9 Daclatasvir versus placebo/no intervention, Outcome 6 Without sustained virological response ‐ according to median dose.

Comparison 10 Simeprevir versus placebo/no intervention, Outcome 1 Hepatitis C‐related morbidity or all‐cause mortality.

Comparison 10 Simeprevir versus placebo/no intervention, Outcome 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose.

Comparison 10 Simeprevir versus placebo/no intervention, Outcome 3 Serious adverse events.

Comparison 10 Simeprevir versus placebo/no intervention, Outcome 4 Serious adverse events ‐ according to median dose.
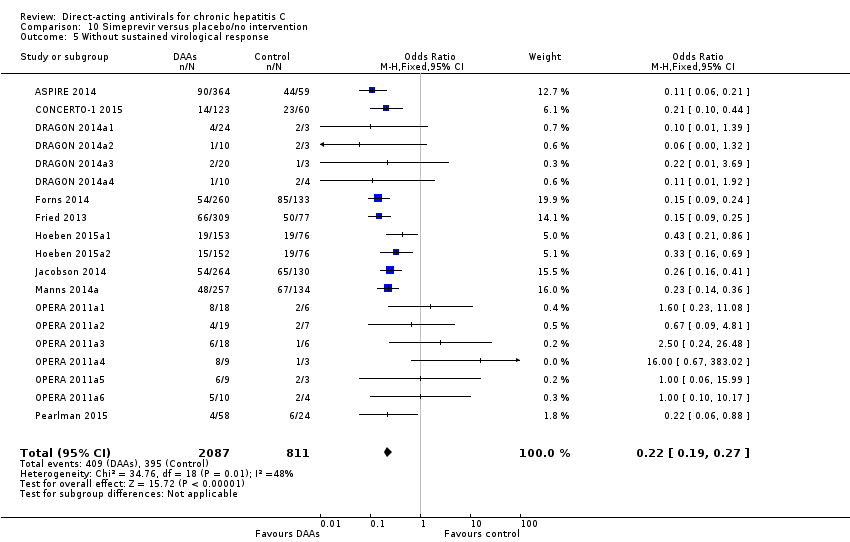
Comparison 10 Simeprevir versus placebo/no intervention, Outcome 5 Without sustained virological response.

Comparison 10 Simeprevir versus placebo/no intervention, Outcome 6 Without sustained virological response ‐ according to median dose.

Comparison 11 Vaniprevir versus placebo/no intervention, Outcome 1 Hepatitis C‐related morbidity or all‐cause mortality.

Comparison 11 Vaniprevir versus placebo/no intervention, Outcome 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose.

Comparison 11 Vaniprevir versus placebo/no intervention, Outcome 3 Serious adverse events.

Comparison 11 Vaniprevir versus placebo/no intervention, Outcome 4 Serious adverse events ‐ according to median dose.

Comparison 11 Vaniprevir versus placebo/no intervention, Outcome 5 Without sustained virological response.

Comparison 11 Vaniprevir versus placebo/no intervention, Outcome 6 Without sustained virological response ‐ according to median dose.

Comparison 12 All DAA versus placebo/no intervention, Outcome 1 Without significant reductions in ALT/AST serum levels.

Comparison 12 All DAA versus placebo/no intervention, Outcome 2 Without significant reductions in ALT/AST serum levels ‐ according to DAA status.
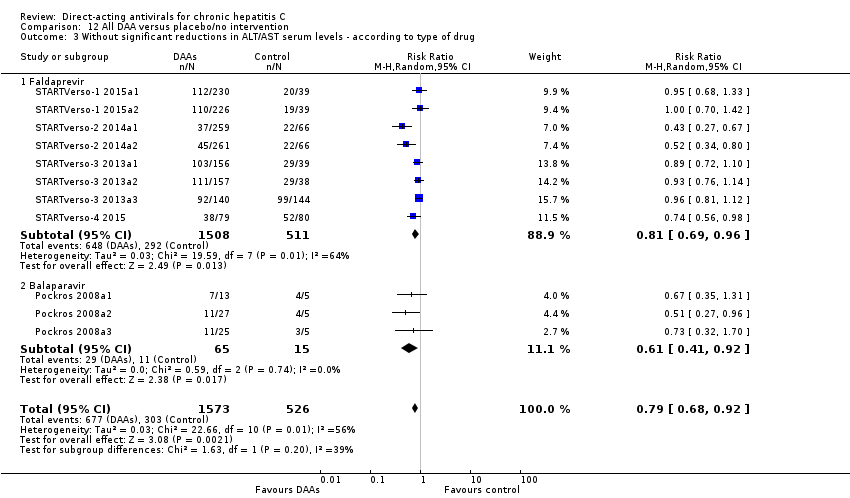
Comparison 12 All DAA versus placebo/no intervention, Outcome 3 Without significant reductions in ALT/AST serum levels ‐ according to type of drug.
| Direct‐acting antivirals versus control | ||||||
| Patient or population: adults with chronic hepatitis C | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (TSA‐adjusted CI) | № of participants | Quality of the evidence | Comments | |
| Risk with placebo or no intervention | Risk with direct‐acting antivirals | |||||
| All‐cause mortality at maximum follow‐up | 2 per 1000 | 7 per 1000 | OR 3.72 (‐) | 2996 | ⊕⊝⊝⊝ | It was not possible to perform Trial Sequential Analysis because of limited data and too few events |
| Proportion of participants with one or more serious adverse event at maximum follow‐up | 56 per 1000 | 52 per 1000 | OR 0.93 (TSA CI 0.71 to 1.33) | 15,817 | ⊕⊝⊝⊝ | Trial Sequential Analysis showed that the boundary for futility was crossed. This leads us to conclude that any possible intervention effect, if any, is less than 20% |
| Proportion of participants with no sustained virological response at maximum follow‐up | 541 per 1000 | 238 per 1000 | RR 0.44 (TSA CI 0.42 to 0.55) | 6886 | ⊕⊕⊝⊝ Low3 | Trial Sequential Analysis showed that the boundary for benefit was crossed. This indicates that DAAs seem to decrease the risk of no sustained virological response by at least 20% if risk of bias and other threats to the validity can be disregarded |
| *The risk in the intervention group (and its 95% confidence interval) is based on the observed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Downgraded two levels because of very serious risk of bias in the included trials (see Figure 1) and two levels due to very serious imprecision (none of the TSA boundaries are crossed, so the information size is too low). | ||||||
| Direct‐acting antivirals withdrawn from the market versus control | ||||||
| Patient or population: adults with chronic hepatitis C | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (TSA‐adjusted CI) | № of participants | Quality of the evidence | Comments | |
| Risk with placebo or no intervention | Risk with direct‐acting antivirals | |||||
| All‐cause mortality at maximum follow‐up | 7 per 1000 | 5 per 1000 | OR 0.64 (‐) | 3045 | ⊕⊝⊝⊝ | It was not possible to perform Trial Sequential Analysis because of limited data and too few events |
| Proportion of participants with one or more serious adverse event at maximum follow‐up | 75 per 1000 | 108 per 1000 | OR 1.45 (TSA 1.16 to 1.82) | 9229 | ⊕⊝⊝⊝ | Trial Sequential Analysis showed that the boundary for harm was crossed. This shows that there is firm evidence that withdrawn DAAs increase the risk of a serious adverse event by at least 20% |
| Proportion of participants with no sustained virological response at maximum follow‐up | 586 per 1000 | 356 per 1000 | RR 0.61 (0.55, 0.69) (TSA CI 0.42 to 0.55) | 9075 | ⊕⊕⊝⊝ Low3 | Trial Sequential Analysis not performed |
| *The risk in the intervention group (and its 95% confidence interval) is based on the observed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DAA: direct‐acting antivirals; OR: odds ratio; RCTs: randomised clinical trials; RR: risk ratio; TSA: Trial Sequential Analysis | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Downgraded two levels because of very serious risk of bias in the included trials (see Figure 1) and two levels due to very serious imprecision (none of the TSA boundaries are crossed so the information size is too low). | ||||||
| Direct‐acting antiviral agents (DAAs) | |||
| NS3/NS4A inhibitors | NS5B inhibitors | NS5A inhibitors | |
| NPI | NNPI | ||
| ACH‐2684 | ALS2200/VX135 | ABT‐072 | ACH‐2928 |
| Asunaprevir | BILB1941 | Beclabuvir | Daclatasvir |
| Boceprevir | GS0938/PSI352938 | BI201127 | Elbasvir |
| Celuprevir | GS6620 | Dasabuvir | GSK2336805 |
| Danoprevir | GS9851(PSI7851) | Deleobuvir | Ledipasvir |
| Faldaprevir | IDX184 | Filibuvir | MK‐8408 |
| Grazoprevir | INX189/BMS986094 | GSK2878175/GSK175 | Odalasvir |
| GS9256 | Mericitabine | IDX375 | Ombitasvir |
| GS9857 | MK‐3682 | MK‐3281 | PPI461 |
| IDX320 | Sofosbuvir | Nesbuvir | Ravidasvir |
| Narlaprevir | VX‐135 | Radalbuvir | Samatasvir |
| Paritaprevir | ‐ | Setrobuvir | Velpatasvir |
| PHX1766 | ‐ | Tegobuvir | ‐ |
| Simperevir | ‐ | TMC‐647055 | ‐ |
| Sovaprevir | ‐ | VCH‐759 | ‐ |
| Telaprevir | ‐ | VCH‐916 | ‐ |
| Vaniprevir | ‐ | VX222 | ‐ |
| Vedroprevir | ‐ | ‐ | ‐ |
| The table presents a list of 58 direct‐acting antiviral agents (DAAs). We have listed the DAAs according to the DAA class they belong to (see Background section). When a DAA has not been assigned a generic or brand name, we have presented it with its experimental compound number prefix. | |||
| Trial | Experimental intervention | Type and number of serious adverse events (experimental group) | Proportion of participants with a serious adverse event (experimental group) | Type and number of serious adverse events (control group) | Proportion of participants with a serious adverse event (control group) |
| Asunaprevir | 1 abdominal pain, 1 lung neoplasm malignant, 1 cytolytic hepatitis, and 2 unspecified events | 5 out of 36 | None reported | 0 out of 11 | |
| Asunaprevir | 2 deaths and 14 unspecified events | 16 out of 177 | 3 unspecified events | 3 out of 61 | |
| Balapiravir | Many events but only a few were specified: 3 deaths, 10 haematological, 10 infection, 8 eye disorders | 49 out of 432 | Many events but not all were specified: 2 infections, 1 death | 9 out of 72 | |
| Beclabuvir | 1 anaemia, 1 constipation, 1 febrile neutropenia, 1 leukopenia | 1 out of 26 | 1 serotonin syndrome | 1 out of 13 | |
| Boceprevir | 5 anaemia, 1 angina pectoris, 1 atrial fibrillation, 1 coronary artery disease, 1 myocardial infarction, 1 myopericarditis, 2 abdominal pain, 1 constipation, 1 diarrhoea, 1 gastritis, 1 irritable bowel syndrome, 1 oesophageal varices haemorrhage, 1 pancreatitis acute, 1 pancreatitis necrotising, 1 peptic ulcer, 1 asthenia, 3 chest pain, 1 oedema peripheral, 1 pyrexia, 1 cholecystitis, 3 appendicitis, 1 bronchopneumonia, 1 catheter site infection, 1 gastroenteritis viral, 1 pneumonia, 1 lower limb fracture, 1 overdose, 1 decreased appetite, 1 dehydration, 1 hyperglycaemia, 1 back pain, 2 intervertebral disc protrusion, 1 pain in extremity, 1 hepatic neoplasm malignant, 1 hepatic encephalopathy, 1 sciatica, 1 syncope, 1 bipolar disorder, 1 completed suicide, 4 depression, 2 homicidal ideation, 5 suicidal ideation, 2 dyspnoea, 1 pleuritic pain, 1 pneumothorax, 1 abdominal hernia repair, 1 deep vein thrombosis, 1 phlebitis | 39 out of 323 | 2 chest pain, 1 cholelithiasis, 1 gastroenteritis | 4 out of 80 | |
| Boceprevir | 1 coronary artery disease, 1 diarrhoea, 1 asthenia, 1 pyrexia, 2 pneumonia, 2 syncope, 1 suicidal ideation, 1 deep vein thrombosis, 1 neutropenia, 1 thrombocytopenia, 1 cardiac failure, 1 upper gastrointestinal haemorrhage , 1 multi‐organ failure, 1 bronchitis, 1 cellulitis, 1 chlamydia infection, 1 influenza, 1 pneumonia staphylococcal, 1 staphylococcal bacteraemia, 1 staphylococcal infection, 1 urosepsis, 1 gun shot wound, 2 hyponatraemia, 1 lethargy, 1 subarachnoid haemorrhage, 1 mental status changes | 18 out of 134 | 1 chest pain, 1 intervertebral disc protrusion, 1 abnormal behaviour, 1 irritability, 1 osteotomy, 1 foreign body, 1 neuralgia, 1 anxiety, 1 renal colic | 7 out of 67 | |
| Boceprevir | 14 neutropenia, 1 intestinal obstruction, 1 osteomyelitis chronic, 1 pneumonia, 1 diabetic ketoacidosis, 1 intervertebral disc protrusion, 1 transient ischaemic attack | 17 out of 159 | 4 neutropenia, 1 general disorders, 1 accidental overdose, 1 prostatitis, 2 hypertension | 9 out of 78 | |
| Boceprevir | 1 anaemia, 1 abdominal pain, 2 asthenia, 2 pyrexia, 2 pneumonia, 1 decreased appetite, 1 dehydration, 2 depression, 2 homicidal ideation, 3 suicidal ideation, 1 dyspnoea, 1 deep vein thrombosis, 3 nausea, 1 vomiting, 3 neutropenia, 1 multi‐organ failure, 2 cellulitis, 2 abdominal pain upper, 1 headache, 1 suicide attempt, 1 accidental overdose, 1 fall, 1 pulmonary embolism, 1 gastroenteritis, 1 erysipelas, 1 panic attack, 1 fatigue, 1 supraventricular tachycardia, 3 pancreatitis, 1 cerebrovascular accident, 1 hypoaesthesia, 1 anxiety, 1 retinal ischaemia, 1 neuropathy peripheral, 1 aggression, 1 scotoma, 1 hypovolaemia, 1 vulval abscess, 1 retinopathy, 1 inguinal hernia, 1 cervix carcinoma, 1 pericarditis, 1 paranoia, 1 neutrophil count decreased, 1 paraesthesia, 1 peritoneal haemorrhage, 1 deafness unilateral, 1 periodontal disease, 1 corneal infection, 1 pneumonia streptococcal, 1 drug toxicity, 1 blood amylase increased, 1 lipase increased, 1 basal cell carcinoma, 1 renal cell carcinoma | 40 out of 527 | 1 suicidal ideation, 1 breast cancer, 1 parathyroid tumour benign, 1 muscle spasms, 1 rib fracture, 1 contusion, 1 inguinal hernia, 1 diplopia, 1 staphylococcal sepsis, 1 animal bite, 1 hand fracture, 1 third nerve paralysis, 1 alcoholism, 1 dependence | 8 out of 104 | |
| Boceprevir | 1 anaemia | 1 out of 49 | 1 anaemia | 1 out of 52 | |
| Boceprevir | 7 anaemia, 1 atrial fibrillation, 1 coronary artery disease, 2 abdominal pain, 1 gastritis, 1 pancreatitis acute, 5 chest pain, 4 pyrexia, 1 cholecystitis, 4 pneumonia, 1 overdose, 1 dehydration, 1 back pain, 1 intervertebral disc protrusion, 5 syncope, 1 completed suicide, 2 depression, 4 suicidal ideation, 1 dyspnoea, 1 nausea, 2 vomiting, 3 neutropenia, 3 thrombocytopenia, 2 bronchitis, 3 cellulitis, 1 staphylococcal infection, 1 hyponatraemia, 1 pancytopenia, 1 breast cancer, 1 malaise, 1 pneumonia pneumococcal, 1 haemoptysis, 1 road traffic accident, 1 suicide attempt, 1 pruritus, 1 rash erythematous, 1 dizziness, 2 pulmonary embolism, 1 haemorrhoids, 4 gastroenteritis, 1 general physical health deterioration, 1 hypertensive crisis, 1 colon cancer, 1 drug abuse, 2 hypokalaemia, 2 chest discomfort, 1 fatigue, 1 perirectal abscess, 1 acute myocardial infarction, 1 gastrointestinal haemorrhage, 1 aplasia pure red cell, 2 leukopenia, 1 atrial flutter, 1 cardiac arrest, 1 hypertrophic cardiomyopathy, 1 tachycardia, 1 deafness, 1 conjunctivitis, 1 optic neuropathy, 1 papilledema, 1 abdominal pain lower, 1 colonic polyp, 1 gastroesophageal reflux disease, 1 hematemesis, 1 haemorrhoidal haemorrhage, 1 Mallory‐weiss syndrome, 1 umbilical hernia, 1 sarcoidosis, 1 abscess, 1 abscess limb, 1 bacteraemia, 1 epiglottitis, 1 infected bites, 1 injection site infection, 1 scrotal abscess, 1 tracheobronchitis, 1 post procedural complication, 1 transfusion reaction, 1 vascular pseudoaneurysm, 1 wound dehiscence, 1 flank pain, 1 groin pain, 1 musculoskeletal chest pain, 1 bladder cancer, 1 pancreatic carcinoma, 1 prostate cancer, 1 carotid artery stenosis, 1 cerebral ischaemia, 1 motor neurone disease, 1 muscle spasticity, 1 affective disorder, 1 alcohol abuse, 1 anxiety, 1 psychiatric decompensation, 1 scrotal pain, 2 cough, 1 pleural fibrosis, 1 alcohol use, 1 laryngeal operation, 1 accelerated hypertension, 1 arterial thrombosis limb, 2 hypotension | 87 out of 734 | 1 anaemia, 1 myocardial infarction, 1 abdominal pain, 2 pyrexia, 1 cholecystitis, 1 appendicitis, 1 pneumonia, 1 hepatic neoplasm malignant, 1 completed suicide, 1 depression, 1 suicidal ideation, 1 pneumothorax, 2 cholelithiasis, 1 nausea, 1 vomiting, 1 cellulitis, 1 breast cancer, 1 colitis, 1 upper respiratory tract infection, 1 suicide attempt, 2 death, 1 accidental overdose, 1 dizziness, 1 loss of consciousness, 1 cholecystitis acute, 1 sinusitis, 2 pancreatitis, 1 leukocytosis, 1 cardiac arrest, 1 cardio‐respiratory arrest, 1 hypothyroidism, 1 cholelithiasis obstructive, 1 atypical mycobacterial infection, 1 diverticulitis, 1 enterocolitis infectious, 1 alcohol poisoning, 1 spinal fracture, 1 white blood cell count decreased, 1 lung adenocarcinoma, 1 prostate cancer, 1 hypoaesthesia, 1 affective disorder, 1 bipolar disorder, 1 drug dependence, 1 intentional self‐injury, 1 personality disorder, 1 glomerulonephritis minimal lesion, 1 renal tubular necrosis, 1 physical assault, 1 cholecystectomy, 1 skin neoplasm excision | 31 out of 363 | |
| Boceprevir | None reported | 0 out of 28 | 1 atrial fibrillation | 1 out of 10 | |
| Boceprevir | 3 anaemia, 2 pneumonia, 1 syncope, 1 depression, 1 deep vein thrombosis, 1 lymphadenopathy, 1 renal failure acute, 2 pulmonary embolism, 1 arthralgia, 1 sinusitis, 1 urinary tract infection, 1 lung infection pseudomonal, 1 pelvic inflammatory disease, 1 pulmonary hypertension, 1 suicide attempt | 11 out of 64 | 2 anaemia, 1 overdose, 1 cholelithiasis, 1 abdominal pain upper, 1 meniscus lesion, 1 pancreatitis, 1 post procedural infection, 1 renal failure, 1 cholecystectomy, 1 vulval abscess, 1 ventricular fibrillation, 1 ligament rupture, 1 lactic acidosis, 1 respiratory failure | 7 out of 34 | |
| Daclatasvir | 1 hepatic neoplasm malignant, 1 rectal ulcer haemorrhage, 1 gastrointestinal inflammation, 1 adhesion, 1 biliary colic, 1 hyperbilirubinaemia, 1 appendiceal abscess, 1 tonsil cancer | 6 out of 196 | 1 abdominal pain upper, 1 epicondylitis, 1 conversion disorder | 3 out of 100 | |
| Daclatasvir | 1 anaemia, 1 abdominal pain, 1 gastritis, 1 chest pain, 2 pneumonia, 1 overdose, 1 syncope, 2 depression, 2 suicidal ideation, 1 dyspnoea, 1 bronchitis, 1 peritonitis, 1 rash generalised, 1 febrile neutropenia, 1 aplastic anaemia, 1 auricular perichondritis, 2 gastric ulcer haemorrhage, 1 death, 1 bile duct stone, 1 clostridium difficile, 1 furuncle, 1 carbuncle, 1 oral herpes, 1 accidental overdose, 2 falls, 1 bursitis, 1 rhabdomyolysis, 1 muscle spasms, 1 costochondritis, 1 dizziness, 1 loss of consciousness, 1 adjustment disorder, 1 hypomania, 1 mental disorder, 1 substance‐induced psychotic disorder, 1 schizophrenia, paranoid type | 25 out of 317 | 2 anaemia, 1 atrial fibrillation, 1 pneumonia, 1 pyelonephritis, 1 haemoglobin decreased, 1 epistaxis, 1 electrocardiogram change, 1 neutrophil count decreased, 1 myalgia, 1 aphasia, 1 paraesthesia | 6 out of 78 | |
| Daclatasvir | 1 pancreatitis acute, 1 back pain | 2 out of 34 | None reported | 0 out of 8 | |
| Daclatasvir | 1 anaemia, 1 chest pain, 2 syncope, 1 bronchitis, 1 epistaxis | 3 out of 36 | None reported | 0 out of 12 | |
| Danoprevir | 28 unspecified SAEs and 2 deaths | 29 out of 373 | 1 unspecified SAE | 1 out of 44 | |
| Danoprevir | 1 benign paroxysmal vertigo | 1 out of 40 | None reported | 0 out of 8 | |
| Danoprevir | 1 gastroenteritis viral | 1 out of 47 | None reported | 0 out of 12 | |
| Danoprevir | 1 altered mood | 1 out of 25 | None reported | 0 out of 5 | |
| Danoprevir | 14 SAEs but not specified, 1 death | 15 out of 194 | 6 SAEs but not specified | 6 out of 31 | |
| Deleobuvir | 1 drug eruption | 1 out of 46 | None reported | 0 out of 14 | |
| Deleobuvir | 1 syncope, 1 rash maculo‐papular, 1 umbilical hernia | 3 out of 49 | None reported | 0 out of 8 | |
| Faldaprevir | 2 anaemia, 1 angina pectoris, 2 diarrhoea, 1 oesophageal varices haemorrhage, 1 cholecystitis, 2 pneumonia, 1 dehydration, 1 back pain, 1 intervertebral disc protrusion, 1 bipolar disorder, 1 depression, 1 suicidal ideation, 1 dyspnoea, 2 nausea, 3 vomiting, 2 neutropenia, 1 thrombocytopenia, 1 cellulitis, 1 mental status changes, 1 pancytopenia, 1 breast cancer, 1 malaise, 2 rash, 2 sepsis, 1 suicide attempt, 1 renal failure acute, 1 rash maculo‐papular, 1 accidental overdose, 1 muscle spasm, 1 tibia fracture, 1 contusion, 1 pulmonary embolism, 2 abortion spontaneous, 1 hypokalaemia, 1 subcutaneous abscess, 1 acute myocardial infarction, 1 pancreatitis, 1 umbilical hernia, 1 diverticulitis, 1 cerebral ischaemia, 1 drug dependence, 1 personality disorder, 1 epidermolysis, 1 ascites, 1 duodenal ulcer haemorrhage, 1 large intestine perforation, 1 hepatic cirrhosis, 2 hepatic failure, 1 hypersensitivity, 1 infective chondritis, 1 vulval abscess, 1 fibula fracture, 1 jaw fracture, 1 ligament sprain, 1 hypocalcaemia, 1 hyponatraemia, 1 hepatocellular carcinoma, 1 papillary thyroid cancer | 47 out of 525 | 1 anaemia, 2 depression, 1 suicidal ideation, 1 bile duct stone, 1 subcutaneous abscess, 1 optic ischaemic neuropathy, 1 laceration, 1 mental status change | 8 out of 132 | |
| Faldaprevir | 3 anaemia, 1 atrial fibrillation, 1 myocardial infarction, 1 asthenia, 1 chest pain, 1 pyrexia, 1 bronchopneumonia, 1 pneumonia, 1 sciatica, 2 vomiting, 1 thrombocytopenia, 1 pancytopenia, 1 headache, 2 rash, 1 drug eruption, 1 dizziness, 1 haemorrhoids, 1 psychotic disorder, 1 urinary tract infection, 1 diabetes mellitus, 1 parapsoriasis, 1 pancreatitis, 1 histiocytosis haematophagic, 1 cerebrovascular accident, 1 muscular weakness, 1 epistaxis, 1 leukopenia, 1 sarcoidosis, 1 hypotension, 1 idiopathic thrombocytopenic purpura, 1 optic ischaemic neuropathy, 1 hypersensitivity, 1 hypoparathyroidism, 1 retinopathy, 1 subdural hematoma, 1 cervix carcinoma, 1 cubital tunnel syndrome, 1 dyspnoea exertional | 34 out of 520 | 1 anaemia, 1 cholecystitis, 1 gun shot wound, 1 rash maculo‐papular, 1 diverticulitis, 1 inguinal hernia, 1 hepatic lesion, 1 polymyositis, 1 blister | 8 out of 132 | |
| Faldaprevir | 5 anaemia, 1 atrial fibrillation, 1 abdominal pain, 5 diarrhoea, 1 pancreatitis acute, 1 asthenia, 1 chest pain, 8 pyrexia, 2 appendicitis, 1 gastroenteritis viral, 2 pneumonia, 1 decreased appetite, 1 dehydration, 1 back pain, 1 hepatic neoplasm malignant, 2 cholelithiasis, 1 biliary colic, 2 hyperbilirubinaemia, 3 nausea, 2 vomiting, 1 thrombocytopenia, 1 cellulitis, 1 bradycardia, 2 presyncope, 2 malaise, 2 headache, 2 sepsis, 1 rash erythematous, 1 rash generalised, 1 fall, 1 multiple injuries, 1 haematochezia, 1 peritonitis bacterial, 1 congestive cardiac failure, 1 gastroenteritis, 1 hypertensive crisis, 1 hypokalaemia, 1 fatigue, 1 pancreatitis, 1 coma, 1 renal colic, 1 leukopenia, 1 cardio‐respiratory arrest, 1 anxiety, 1 psychiatric decompensation, 2 hypotension, 1 viral infection, 2 ascites, 1 hepatic failure, 1 hypoglycaemia, 1 haemolytic anaemia, 1 keratosis follicular, 1 oral lichen planus, 1 peritoneal haemorrhage, 1 salivary gland calculus, 1 hepatorenal failure, 2 jaundice, 1 streptococcal infection, 1 blood lactate dehydrogenase increased, 1 international normalised ratio abnormal, 1 metabolic acidosis, 1 fasciitis, 1 joint instability, 1 musculoskeletal discomfort, 1 haemothorax, 1 venous thrombosis | 54 out of 599 | 1 depression, 1 pleural effusion | 1 out of 78 | |
| Faldaprevir | 1 abdominal pain upper | 1 out of 35 | 1 abdominal pain | 1 out of 8 | |
| Faldaprevir | 1 asthenia, 1 cataract , 1 hypoalbuminaemia, 1 metabolic disorder, 1 ascites | 4 out of 88 | None reported | 0 out of 8 | |
| Faldeprevir | 4 anaemia, 1 angina pectoris, 1 myocardial infarction, 1 diarrhoea, 1 asthenia, 1 chest pain, 1 oedema peripheral, 4 pyrexia, 1 cholecystitis, 1 pneumonia, 2 dehydration, 1 intervertebral disc protrusion, 2 syncope, 1 depression, 1 nausea, 2 vomiting, 1 thrombocytopenia, 1 upper gastrointestinal haemorrhage, 1 influenza, 1 lower respiratory tract infection, 2 photosensitivity reaction, 1 upper respiratory tract infection, 2 headache, 1 rash, 1 road traffic accident, 2 suicide attempt, 3 drug eruption, 2 rash maculo‐papular, 1 rash erythematous, 2 febrile neutropenia, 1 oral herpes, 1 pulmonary embolism, 1 pyelonephritis, 1 cataract, 1 anaemia haemolytic autoimmune, 1 lymphopenia, 1 microvascular angina, 1 prinzmetal angina, 1 anal fistula, 1 haemorrhoids, 1 mouth ulceration, 1 rectal haemorrhage, 1 chest discomfort, 1 fatigue, 1 mucosal inflammation, 1 gallbladder polyp, 1 cryoglobulinaemia, 1 anal abscess, 1 ear infection, 2 H1N1 influenza, 1 infected skin ulcer, 1 lymphangitis, 1 perirectal abscess, 1 pharyngitis, 1 subcutaneous abscess, 1 superinfection bacterial, 1 urinary tract infection, 1 diabetes mellitus, 1 ischaemic stroke, 1 acute psychosis, 1 depressed mood, 1 calculus ureteric, 1 endometrial hyperplasia, 1 dermatitis atopic, 1 eczema, 1 erythema multiforme, 1 lichen planus, 1 palmar‐plantar erythrodysaesthesia syndrome, 1 parapsoriasis, 1 pruritus allergic, 1 rash pruritic, 1 appendicectomy | 61 out of 641 | 1 headache, 1 photophobia, 1 cyst, 1 benign salivary gland neoplasm, 1 migraine | 2 out of 71 | |
| Filibuvir | 1 blood creatinine increased, 1 chronic obstructive pulmonary disease, 1 pulmonary embolism | 3 out of 27 | 1 thyroiditis, 1 gait disturbance | 2 out of 8 | |
| Filibuvir | 1 anaemia, 1 appendicitis, 1 rectal ulcer haemorrhage, 1 craniocerebral injury, 1 vertigo, 1 vestibular disorder, 1 haematochezia, 1 peritonitis bacterial, 1 lymph node tuberculosis, 1 scapula fracture, 1 blood urea nitrogen/creatinine increased, 1 gastric cancer, 1 rectal cancer, 1 abortion spontaneous, 1 cardiac necrosis, 1 pyoderma gangrenosum, 1 depression, 1 breast cancer, 1 chronic obstructive pulmonary disease, 1 lung neoplasm malignant, 1 fall, 1 loss of consciousness, 1 bacterial abscess CNS, 1 actinomyces test positive, 1 pulmonary calcification | 20 out of 192 | 1 neutropenia, 1 sepsis, 1 pulmonary embolism, 1 cerebral haemorrhage, 1 ecchymosis, 1 Appendicitis perforated | 6 out of 96 | |
| GS‐9451 | 1 death, 1 heroin overdose | 1 out of 33 | None reported | 0 out of 8 | |
| GSK2336805 | 1 pneumonia, 1 upper lobe cavitary lesion | 1 out of 11 | None reported | 0 out of 4 | |
| IDX‐184 | 1 pancreatitis, 1 acute cholecystitis | 2 out of 65 | 1 agitation | 1 out of 16 | |
| Mericitabine/danoprevir | 1 multiple drug overdose, 1 ankle fracture | 2 out of 73 | None reported | 0 out of 14 | |
| Mericitabine | 1 nephrolithiasis, 1 porphyria non‐acute | 2 out of 102 | 1 arthritis infective | 1 out of 49 | |
| Mericitabine | 6 SAEs but not specified | 5 out of 81 | 4 SAEs but not specified | 3 out of 85 | |
| Narlaprevir | 1 pyrexia, 1 elevated CRP | 1 out of 32 | None reported | 0 out of 8 | |
| Odalasvir (ACH‐3102) and sovaprevir | 1 non‐cardiac chest pain | 1 out of 20 | None reported | 0 out of 10 | |
| Paritaprevir (ABT‐450)/r–ombitasvir | 1 pneumonia, 1 nausea, 1 vomiting, 1 bradycardia, 1 chronic obstructive pulmonary disease, 1 renal failure acute, | 9 out of 393 | 1 atrial fibrillation | 1 out of 97 | |
| Paritaprevir/ABT‐072/dasabuvir | 1 haemorrhoids, 1 malignant melanoma | 2 out of 63 | None reported | 0 out of 11 | |
| Paritaprevir/ombitasvir | 1 anaemia, 1 abdominal pain, 1 diarrhoea, 1 cholecystitis, 1 appendicitis, 1 overdose, 1 sinus tachycardia, 1 ventricular extrasystoles, 1 nausea, 1 vomiting, 1 chills, 1 non‐cardiac chest pain, 1 lobar pneumonia, 1 postoperative wound infection, 1 lumbar vertebral fracture, 1 non‐small cell lung cancer, 1 encephalopathy, 1 acute respiratory failure, 1 hypoxia, 1 mediastinal mass, 1 aortic stenosis, 1 biliary colic, 1 subcutaneous abscess | 12 out of 630 | None reported | 0 out of 158 | |
| R1626 | 6 SAEs but not specified | 6 out of 84 | 1 SAE but not specified | 1 out of 20 | |
| Simeprevir | 1 abdominal pain, 1 pyrexia, 1 appendicitis, 2 pneumonia, 1 depression, 1 dyspnoea, 1 cholelithiasis, 1 anaemia haemolytic autoimmune, 1 pancytopenia, 1 angina pectoris, 1 bradycardia, 1 myocardial ischaemia, 1 hepatitis, 1 endocarditis, 1 lower respiratory tract infection, 1 septic shock, 1 breast cancer, 1 Guillain‐Barre syndrome, 1 presyncope, 1 confusional state, 1 vaginal haemorrhage, 1 chronic obstructive pulmonary disease, 1 respiratory acidosis, 1 photosensitivity reaction | 14 out of 260 | 1 atrial fibrillation, 1 depression, 1 bronchitis, 1 hypercalcaemia, 1 head injury, 1 bacterial prostatitis, 1 pericarditis, 1 infection, 1 inguinal hernia, 1 neuropathy peripheral, 1 arthritis infective, 1 headache | 11 out of 133 | |
| Simeprevir | 1 cholecystitis, 1 intervertebral disc protrusion, 1 depression, 1 nausea, 1 breast cancer, 1 hyperthyroidism, 1 ocular vasculitis, 1 abdominal pain upper, 1 colitis, 1 small intestinal obstruction, 1 malaise, 1 incision site cellulitis, 1 necrotising fasciitis, 1 perihepatic abscess, 1 pneumonia pneumococcal, 1 upper respiratory tract infection, 1 post‐procedural bile leak, 1 malnutrition, 1 type 1 diabetes mellitus, 1 spinal disorder, 1 parathyroid tumour benign, 1 headache, 1 haemoptysis, 1 cutaneous vasculitis, 1 hypertension | 20 out of 309 | 1 myocardial infarction, 1 myopericarditis, 1 asthenia, 1 appendicitis, 1 vomiting, 1 chronic obstructive pulmonary disease, 1 headache, 1 subcutaneous abscess, 1 vulval abscess, 1 myositis, 1 ovarian neoplasm | 10 out of 77 | |
| Simeprevir | 1 subarachnoid haemorrhage, 1 malaise, 1 cerebral infarction, 1 vulvar erosion, 1 rash, 1 incorrect dose administered | 5 out of 79 | None reported | 0 out of 13 | |
| Simeprevir | 1 depression, 1 non‐cardiac chest pain, 1 angina unstable, 1 nephrolithiasis, 1 ureteric stenosis, 1 colitis ischaemic, 1 incision site infection, 1 craniocerebral injury, 1 foot fracture, 1 meniscus lesion, 1 multiple injuries, 1 rib fracture, 1 tibia fracture, 1 traumatic lung injury, 1 wound, 1 cholesterosis, 1 type 2 diabetes mellitus, 1 shock haemorrhagic | 10 out of 305 | 1 anaemia, 1 decreased appetite, 1 cholelithiasis, 1 contusion, 1 supraventricular tachycardia, 1 ligament sprain, 1 pain, 1 atypical pneumonia, 1 chronic hepatitis C, 1 pulmonary tuberculosis, 1 undifferentiated connective tissue disease, 1 brain neoplasm | 9 out of 152 | |
| Simeprevir | 1 sinus arrest, 1 erysipelas, 1 type 1 diabetes mellitus, 1 psychotic disorder, 1 drug abuse, 1 bronchitis, 1 exostosis, 1 toe deformity, 1 hyperthyroidism, 1 Bowen's disease, 1 neutropenia, 1 thrombocytopenia, 1 breast cancer, 1 sepsis, 1 cupulolithiasis, 1 pneumonia escherichia, 1 panic reaction | 13 out of 88 | 1 pneumonia, 1 sinusitis, 1 panic attack, 1 social stay hospitalisation, 1 pneumonia escherichia | 3 out of 28 | |
| Simeprevir | 2 anaemia, 1 back pain, 1 syncope, 1 hyperthyroidism, 1 death, 1 muscle spasms, 1 colon cancer, 1 anal abscess, 1 urinary tract infection, 1 mixed deafness, 1 hyphaema, 1 visual impairment, 1 enterocutaneous fistula, 1 autoimmune hepatitis, 1 lymphadenitis bacteria, 1 fluid overload, 1 epilepsy, 1 memory impairment, 1 aggression | 16 out of 257 | 1 anaemia, 1 pancreatitis acute, 1 dehydration, 1 vomiting, 1 pancytopenia, 1 loss of consciousness, 1 angina unstable, 1 meniscus lesion, 1 pulmonary embolism, 1 cholecystitis acute, 1 drug abuse, 1 retinal ischaemia, 1 respiratory tract infection viral, 1 viral infection, 1 neuropathy peripheral, 1 thoracic outlet syndrome | 10 out of 134 | |
| Simeprevir | None reported | 0 out of 58 | 1 liver decompensation | 1 out of 24 | |
| Simeprevir | 1 anaemia, 1 abdominal pain, 1 diarrhoea, 1 oedema peripheral, 2 cholecystitis, 1 pneumonia, 1 overdose, 2 | 31 out of 396 | 1 sciatica, 1 nausea, 1 vomiting, 1 lower respiratory tract infection, 1 haemorrhoids, 1 weight decreased, 1 histiocytosis haematophagic, 1 tuberculosis | 4 out of 66 | |
| Sofosbuvir | 1 drug withdrawal syndrome, 1 non‐cardiac chest pain, 1 oedema peripheral, 1 pyrexia, 1 hypersensitivity, 1 abdominal abscess, 1 cellulitis, 2 overdose, 1 injury, 1 road traffic accident, 1 spinal compression fracture, 1 hypoglycaemia, 1 hepatic neoplasm malignant, 1 abnormal behaviour, 1 eczema | 11 out of 207 | 1 pancreatitis, 1 bile duct stone, 1 bronchitis | 2 out of 71 | |
| Sofosbuvir | 1 retinal vein occlusion, 1 depression, 1 suicidal ideation, 1 lymphangitis, 1 acute myocardial infarction | 4 out of 95 | 1 chest pain, 1 electrocardiogram ST segment elevation | 1 out of 26 | |
| Sofosbuvir | 1 anaemia, 1 chest pain, 1 cellulitis, 1 chronic obstructive pulmonary disease, 1 urinary tract infection, 1 allergy to anthropod sting, 1 osteomyelitis chronic , 1 toxicity to various agents | 7 out of 256 | 1 pneumothorax, 1 breast cancer, 1 infection, 1 atrioventricular shock, 1 clavicle fracture, 1 rib fracture | 3 out of 243 | |
| Sofosbuvir | 1 anaemia, 1 depression, 1 peripheral ischaemia, 1 pancreatitis acute | 4 out of 49 | 1 abdominal pain | 1 out of 14 | |
| Sofosbuvir/velpatasvir | 1 bronchitis, 1 cellulitis, 1 influenza, 1 chronic obstructive pulmonary disease, 1 death, 1 gastroenteritis, 1 acute myocardial infarct, 1 ligament sprain, 1 foot abscess, 1 foot necrosis, 1 recurring appendicitis, 1 epileptic seizure, 1 rotator cuff syndrome, 1 lung cancer, 1 mania, 1 palpitations, 1 small bowel obstruction, 1 upper limb fracture, 1 vestibular neuronitis | 15 out of 624 | None reported | 0 out of 116 | |
| Telaprevir | 1 cholelithiasis | 1 out of 16 | None reported | 0 out of 8 | |
| Telaprevir | 5 anaemia, 2 abdominal pain, 1 asthenia, 1 pyrexia, 1 back pain, 3 syncope, 3 depression, 2 dyspnoea, 1 nausea, 1 chills, 1 pancytopenia, 5 rash, 1 lymphadenopathy, 1 hydrocele, 1 retinal haemorrhage, 1 catheter‐related complication, 1 bacterial sepsis, 1 pneumonia, 1 herpes viral, 1 sepsis, 1 road traffic accident, 1 tendon rupture, 1 lung neoplasm malignant, 1 speech disorder, 1 disorientation, 1 emotional distress, 1 suicide attempt, 1 renal failure, 1 acute testicular swelling, 3 pruritis, 2 drug eruption, 2 rash maculo‐papular, 1 rash erythematous, 1 rash generalised, 1 toxic skin eruption, 1 urticaria, 1 splenectomy | 36 out of 241 | 2 anaemia, 1 angina pectoris, 1 syncope, 1 hyperthyroidism, 1 gastroenteritis, 1 haemorrhagic anaemia, 1 alcoholic pancreatitis, 1 paranoia, 1 uterine polyp | 8 out of 82 | |
| Telaprevir | 18 anaemia, 3 pneumonia, 3 syncope, 2 cellulitis, 5 rash, 3 psychiatric disorder, 2 musculoskeletal disorder, 2 cardiac disorder, 2 eye disorder, 3 hepatobiliary disorders, 2 vascular disorder | 64 out of 727 | 4 anaemia, 1 cellulitis, 3 psychiatric disorder, 3 musculoskeletal disorder, 2 cardiac disorder, 4 renal and urinary disorder, 1 eye disorder, 1 vascular disorder | 24 out of 361 | |
| Telaprevir | 2 anaemia, 1 gastroenteritis viral, 1 dehydration, 2 depression, 1 non‐cardiac chest pain, 1 chronic obstructive pulmonary disease, 1 rash, 1 rash generalised, 1 furuncle, 1 colitis ischaemic, 1 acute myocardial infarction, 1 adrenal disorder, 2 scotoma, 1 retinal exudates, 1 retinal infarction, 1 bronchitis bacterial, 1 incisional hernia, 1 lumbar radiculopathy, 1 exfoliative rash | 18 out of 175 | 1 lobar pneumonia, 1 pancytopenia, 1 anxiety, 1 lymphadenitis bacteria, 1 deafness neurosensory | 4 out of 75 | |
| Telaprevir | 6 anaemia, 1 pancreatitis acute, 1 gastroenteritis viral, 1 pneumonia, 1 dehydration, 1 back pain, 1 suicidal ideation, 2 cholelithiasis, 1 postoperative wound infection, 1 upper gastrointestinal haemorrhage, 1 confusional state, 2 small intestinal obstruction, 1 necrotising fasciitis, 1 pneumonia pneumococcal, 1 post‐procedural bile leak, 1 rash, 1 renal failure acute, 1 retinal detachment, 9 gastroenteritis, 1 cholecystitis acute, 1 sinusitis, 1 hypokalaemia, 1 eczema, 2 pancreatitis, 1 dermatitis, 1 diverticulitis, 1 alcohol abuse, 1 hypotension, 1 haemorrhagic anaemia, 1 idiopathic thrombocytopenic purpura, 1 cardiomyopathy, 1 diverticular perforation, 1 gastritis erosive, 1 abscess intestinal, 1 cholecystitis infective, 1 infected insect bite, 1 sepsis syndrome, 1 hypovolaemia, 1 B‐cell unclassifiable lymphoma low grade, 1 migraine with aura, 1 ruptured cerebral aneurysm, 1 neurogenic bladder, 1 lichenoid keratosis, 1 rash macular | 28 out of 339 | 1 anaemia, 1 pneumonia, 2 dehydration, 1 syncope, 1 depression, 1 non‐small cell lung cancer, 1 headache, 2 rash, 1 renal failure acute, 2 gastroenteritis, 1 renal tubular acidosis, 1 decubitus ulcer, 1 hematoma | 9 out of 114 | |
| Telaprevir | 1 myocardial infarction, 1 staphylococcal infection, 1 pyelonephritis acute, 1 haemolytic anaemia, 1 groin infection, 1 cellulitis staphylococcal, 1 staphylococcal abscess, 1 hypokalaemia, 1 hyponatraemia, 1 epididymitis, 1 non‐ cardiac chest pain | 7 out of 38 | 1 anaemia, 1 appendicitis, 1 peritonitis | 2 out of 22 | |
| Telaprevir | 13 anaemia, 1 febrile neutropenia, 2 pancytopenia, 1 thrombocytopenia , 3 acute myocardial infarction, 2 atrial fibrillation, 1 cardiac valve disease, 1 myocardial infarction, 1 supraventricular tachycardia, 1 sudden hearing loss, 1 Basedow's disease, 1 retinal detachment, 1 abdominal pain, 1 anal fissure, 1 caecitis, 1 gastrointestinal haemorrhage, 1 pancreatitis, 2 pancreatitis acute, 1 general physical health deterioration, 1 pyrexia, 1 appendicitis, 2 bronchitis, 1 erysipelas, 1 folliculitis, 1 Helicobacter gastritis, 1 pneumonia, 1 post‐procedural infection, 1 rectal abscess, 2 sepsis, 1 sinusitis, 1 tooth abscess, 2 urinary tract infection, 1 injection site reaction, 1 animal scratch, 1 ankle fracture, 1 femoral neck fracture, 1 multiple drug overdose, 1 blood corticotrophin decreased, 1 weight decreased, 1 anorexia, 1 diabetes mellitus, 1 bronchial carcinoma, 2 gastric cancer, 2 hepatic neoplasm malignant, 1 histiocytosis haematophagic, 1 lung neoplasm malignant, 1 lethargy, 1 subarachnoid haemorrhage, 2 syncope, 1 delirium, 1 depression, 1 insomnia, 1 substance abuse, 1 renal cyst, 1 renal failure, 1 urinary bladder polyp, 1 prostatitis, 1 pulmonary embolism, 1 dermatitis, 1 eczema , 1 erythema multiforme, 1 pruritus 1 pustular psoriasis, 1 rash, 2 toxic skin eruption, 1 orthostatic hypotension, 1 peripheral artery aneurysm | 65 out of 530 | 1 anaemia, 1 atrial fibrillation, 1 abdominal pain, 1 pneumonia, 1 colitis, 1 pyelonephritis, 1 cerebral thrombosis, 1 coma | 7 out of 132 | |
| Vaniprevir | 1 appendicitis, 1 lobar pneumonia, 1 septic shock, 1 confusional state, 1 gastroenteritis, 1 cholecystitis acute, 1 empyema, 1 haemoglobin decreased, 1 myopathy | 8 out of 75 | 1 colon cancer | 1 out of 19 | |
| Vaniprevir | 1 anaemia, 1 pneumonia, 1 syncope, 1 upper gastrointestinal haemorrhage, 1 cellulitis, 1 confusional state, 1 dizziness, 1 nephrolithiasis, 1 malignant melanoma, 3 retinal detachment, 1 joint dislocation, 1 congestive cardiac failure, 2 gastroenteritis, 1 femur fracture, 1 hyperglycaemia, 1 dermatomyositis, 1 retinal vascular thrombosis, 1 general physical health deterioration, 1 anaphylactic reaction, 1 pyelonephritis, 1 pyelonephritis acute, 1 carbon monoxide poisoning, 1 arthralgia, 1 arthritis infective, 1 completed suicide | 22 out of 229 | 1 hypertensive crisis | 1 out of 56 | |
| SAE: serious adverse events. | |||||
| Trial | Experimental intervention | Type and number of participants with a non‐serious adverse events (experimental group) | Proportion of participants with a non‐serious adverse event (experimental group) | Type and number of participants with a non‐serious adverse events (control group) | Proportion of participants with a non‐serious adverse event (control group) |
| Asunaprevir | 18 diarrhoea, 13 nausea, 10 asthenia, 21 fatigue, 14 influenza‐like illness, 7 irritability, 12 decreased appetite, 4 arthralgia, 9 myalgia, 13 headache, 9 depression, 10 insomnia, 7 cough, 10 dyspnoea, 7 alopecia, 11 dry skin, 10 pruritus, 6 rash | 36 out of 36 | 1 diarrhoea, 2 nausea, 4 asthenia, 5 fatigue, 5 influenza‐like illness, 4 irritability, 3 decreased appetite, 4 arthralgia, 1 myalgia, 6 headache, 1 depression, 1 insomnia, 3 cough, 3 dyspnoea, 3 alopecia, 1 dry skin, 2 pruritus, 3 rash | 11 out of 11 | |
| Asunaprevir | 8 anaemia, 63 asthenia, 62 fatigue, 37 influenza‐like illness, 43 decreased appetite, 66 headache, 41 pruritus | 173 out of 177 | 3 anaemia, 1 asthenia, 62 fatigue, 23 influenza‐like illness, 22 decreased appetite, 26 headache, 16 pruritus | 57 out of 61 | |
| Asunaprevir | 1 nausea, 3 headache, 1 flatulence | Not specified out of 20 | None reported | Not specified out of 4 | |
| Asunaprevir | 1 nausea, 3 headache | Not specified out of 12 | 1 nausea, 1 flatulence | Not specified out of 3 | |
| Balapiravir | 120 anaemia, 41 neutropenia, 115 diarrhoea, 162 nausea, 138 chills, 231 fatigue, 117 pyrexia, 100 arthralgia, 156 myalgia, 82 dizziness, 241 headache, 90 depression, 174 insomnia, 86 cough, 88 alopecia, 60 dry skin, 108 pruritus, 90 rash | Not specified out of 432 | 6 anaemia, 3 neutropenia, 16 diarrhoea, 25 nausea, 30 chills, 43 fatigue, 19 pyrexia, 16 arthralgia, 33 myalgia, 15 dizziness, 42 headache, 17 depression, 24 insomnia, 11 cough, 11 alopecia, 16 dry skin, 15 pruritus, 13 rash | Not specified out of 72 | |
| Beclabuvir | 5 anaemia, 5 neutropenia, 5 diarrhoea, 9 nausea, 3 chills, 12 fatigue, 6 influenza‐like illness, 7 irritability, 3 pyrexia, 7 decreased appetite, 4 arthralgia, 5 myalgia, 12 headache, 6 depression, 9 insomnia, 6 cough, 5 pruritus, 2 rash | Not specified out of 26 | 5 anaemia, 1 neutropenia, 1 diarrhoea, 2 nausea, 5 fatigue, 7 influenza‐like illness, 3 irritability, 1 pyrexia, 2 decreased appetite, 3 headache, 3 depression, 3 insomnia, 3 cough, 4 pruritus, 4 rash | Not specified out of 13 | |
| BILB‐1941 | 25 diarrhoea, 7 nausea, 2 vomiting | 30 out of 77 | 2 diarrhoea | 3 out of 19 | |
| BMS‐791325 | 1 diarrhoea, 1 nausea, 1 vomiting, 1 headache, 1 pruritus | 9 out of 20 | 1 nausea, 1 vomiting, 1 headache | 1 out of 4 | |
| Boceprevir | 145 anaemia, 46 neutropenia, 78 diarrhoea, 140 nausea, 47 vomiting, 68 asthenia, 106 chills, 179 fatigue, 79 influenza‐like illness, 67 irritability, 93 pyrexia, 83 decreased appetite, 73 arthralgia, 81 myalgia, 52 dizziness, 142 dysgeusia, 133 headache, 46 depression, 97 insomnia, 70 cough, 69 dyspnoea, 71 alopecia, 72 dry skin, 62 pruritus, 51 rash | 319 out of 323 | 16 anaemia, 8 neutropenia, 13 diarrhoea, 30 nausea, 6 vomiting, 13 asthenia, 24 chills, 40 fatigue, 20 influenza‐like illness, 10 irritability, 20 pyrexia, 13 decreased appetite, 13 arthralgia, 19 myalgia, 8 dizziness, 9 dysgeusia, 39 headache, 12 depression, 19 insomnia, 14 cough, 14 dyspnoea, 13 alopecia, 7 dry skin, 14 pruritus, 5 rash | 77 out of 80 | |
| Boceprevir | 67 anaemia, 41 neutropenia, 33 diarrhoea, 52 nausea, 16 vomiting, 29 asthenia, 14 chills, 67 fatigue, 35 influenza‐like illness, 29 irritability, 18 pyrexia, 27 decreased appetite, 16 arthralgia, 25 myalgia, 17 dizziness, 52 dysgeusia, 37 headache, 22 depression, 32 insomnia, 26 cough, 26 dyspnoea, 22 alopecia, 20 dry skin, 18 pruritus, 31 rash | 133 out of 134 | 22 anaemia, 12 neutropenia, 5 diarrhoea, 18 nausea, 12 asthenia, 8 chills, 36 fatigue, 18 influenza‐like illness, 16 irritability, 8 pyrexia, 12 decreased appetite, 12 arthralgia, 5 myalgia, 10 dizziness, 10 dysgeusia, 21 headache, 6 depression, 20 insomnia, 14 cough, 17 dyspnoea, 5 alopecia, 11 dry skin, 8 pruritus, 5 rash | 67 out of 67 | |
| Boceprevir | 105 anaemia, 103 leukopenia, 141 neutropenia, 16 thrombocytopenia, 22 diarrhoea, 13 dry mouth, 59 nausea, 12 vomiting, 63 asthenia, 24 chills, 40 fatigue, 51 hyperthermia, 356 influenza‐like illness, 9 injection site erythema, 18 irritability, 217 pyrexia, 14 body temperature increased, 33 weight decreased, 10 decreased appetite, 16 arthralgia, 33 myalgia, 9 dizziness, 69 dysgeusia, 89 headache, 2 sleep disorder, 38 cough, 7 dyspnoea, 33 alopecia, 12 dry skin, 20 pruritus, 17 rash | 153 out of 159 | 31 anaemia, 35 leukopenia, 45 neutropenia, 7 thrombocytopenia, 3 diarrhoea, 5 dry mouth, 15 nausea, 2 vomiting, 23 asthenia, 2 chills, 18 fatigue, 12 hyperthermia, 72 influenza‐like illness, 2 injection site erythema, 11 irritability, 124 pyrexia, 2 body temperature increased, 9 weight decreased, 7 decreased appetite, 4 arthralgia, 8 myalgia, 7 dizziness, 5 dysgeusia, 51 headache, 4 sleep disorder, 14 cough, 7 dyspnoea, 16 alopecia, 3 dry skin, 6 pruritus, 2 rash | 71 out of 78 | |
| Boceprevir | 226 anaemia, 96 neutropenia, 109 diarrhoea, 186 nausea, 81 vomiting, 53 asthenia, 130 chills, 259 fatigue, 79 influenza‐like illness, 91 irritability, 129 pyrexia, 49 decreased appetite, 76 arthralgia, 99 myalgia, 70 dizziness, 111 dysgeusia, 190 headache, 91 depression, 146 insomnia, 76 cough, 66 dyspnoea, 131 alopecia, 60 dry skin, 80 pruritus, 27 rash | 413 out of 416 | 35 anaemia, 12 neutropenia, 23 diarrhoea, 45 nausea, 5 vomiting, 14 asthenia, 35 chills, 57 fatigue, 25 influenza‐like illness, 23 irritability, 35 pyrexia, 12 decreased appetite, 21 arthralgia, 17 myalgia, 16 dizziness, 9 dysgeusia, 45 headache, 22 depression, 40 insomnia, 20 cough, 15 dyspnoea, 27 alopecia, 17 dry skin, 16 pruritus, 6 rash | 102 out of 104 | |
| Boceprevir | 361 anaemia, 184 neutropenia, 180 diarrhoea, 334 nausea, 145 vomiting, 125 asthenia, 255 chills, 405 fatigue, 174 influenza‐like illness, 164 irritability, 240 pyrexia, 186 decreased appetite, 141 arthralgia, 170 myalgia, 146 dizziness, 293 dysgeusia, 335 headache, 151 depression, 239 insomnia, 130 cough, 152 dyspnoea, 179 alopecia, 153 dry skin, 181 pruritus, 181 rash | 728 out of 734 | 107 anaemia, 77 neutropenia, 79 diarrhoea, 153 nausea, 57 vomiting, 70 asthenia, 102 chills, 217 fatigue, 93 influenza‐like illness, 86 irritability, 120 pyrexia, 90 decreased appetite, 66 arthralgia, 94 myalgia, 59 dizziness, 64 dysgeusia, 153 headache, 78 depression, 118 insomnia, 76 cough, 59 dyspnoea, 99 alopecia, 66 dry skin, 98 pruritus, 83 rash | 353 out of 363 | |
| Boceprevir | 26 anaemia, 12 neutropenia, 21 diarrhoea, 26 nausea, 18 vomiting, 22 asthenia, 5 chills, 25 fatigue, 16 influenza‐like illness, 10 irritability, 24 pyrexia, 22 decreased appetite, 7 arthralgia, 9 myalgia, 8 dizziness, 18 dysgeusia, 18 headache, 11 depression, 15 insomnia, 9 cough, 5 dyspnoea, 12 alopecia, 8 dry skin, 13 pruritus, 5 rash | 62 out of 64 | 8 anaemia, 2 neutropenia, 6 diarrhoea, 11 nausea, 5 vomiting, 9 asthenia, 5 chills, 12 fatigue, 13 influenza‐like illness, 5 irritability, 7 pyrexia, 6 decreased appetite, 2 arthralgia, 6 myalgia, 2 dizziness, 5 dysgeusia, 6 headache, 4 depression, 9 insomnia, 6 cough, 2 dyspnoea, 6 alopecia, 3 dry skin, 3 pruritus | 33 out of 34 | |
| Daclatasvir | 7 anaemia, 9 neutropenia, 10 diarrhoea, 22 nausea, 4 vomiting, 13 asthenia, 4 chills, 35 fatigue, 19 influenza‐like illness, 17 irritability, 7 pyrexia, 12 decreased appetite, 11 arthralgia, 14 myalgia, 6 dizziness, 5 dysgeusia, 30 headache, 11 depression, 19 insomnia, 8 cough, 12 dyspnoea, 12 alopecia, 13 dry skin, 27 pruritus, 25 rash | 98 out of 100 | 5 anaemia, 8 neutropenia, 3 diarrhoea, 8 nausea, 4 vomiting, 7 asthenia, 19 fatigue, 7 influenza‐like illness, 6 irritability, 2 pyrexia, 8 decreased appetite, 9 arthralgia, 11 myalgia, 6 dizziness, 3 dysgeusia, 9 headache, 9 depression, 17 insomnia, 8 cough, 6 dyspnoea, 5 alopecia, 6 dry skin, 14 pruritus, 12 rash | 48 out of 51 | |
| Daclatasvir | 53 anaemia, 43 neutropenia, 73 diarrhoea, 109 nausea, 34 vomiting, 32 asthenia, 49 chills, 174 fatigue, 94 influenza‐like illness, 72 irritability, 48 pyrexia, 67 decreased appetite, 55 arthralgia, 88 myalgia, 46 dizziness, 25 dysgeusia, 136 headache, 45 depression, 102 insomnia, 54 cough, 58 dyspnoea, 80 alopecia, 88 dry skin, 119 pruritus, 94 rash | 311 out of 317 | 9 anaemia, 9 neutropenia, 14 diarrhoea, 20 nausea, 11 vomiting, 7 asthenia, 16 chills, 46 fatigue, 16 influenza‐like illness, 22 irritability, 15 pyrexia, 17 decreased appetite, 19 arthralgia, 24 myalgia, 9 dizziness, 4 dysgeusia, 36 headache, 10 depression, 30 insomnia, 18 cough, 11 dyspnoea, 13 alopecia, 15 dry skin, 26 pruritus, 25 rash | 76 out of 78 | |
| Daclatasvir | 11 anaemia, 7 neutropenia, 2 diarrhoea, 4 nausea, 6 fatigue, 1 irritability, 11 pyrexia, 7 decreased appetite, 4 arthralgia, 1 myalgia, 1 dizziness, 4 dysgeusia, 3 headache, 7 insomnia, 4 cough, 2 dyspnoea, 8 alopecia, 1 dry skin, 6 pruritus, 7 rash | 34 out of 34 | 5 anaemia, 4 neutropenia, 3 diarrhoea, 2 nausea, 3 vomiting, 4 chills, 4 fatigue, 2 influenza‐like illness, 5 pyrexia, 5 decreased appetite, 2 arthralgia, 2 myalgia, 1 dizziness, 1 dysgeusia, 4 headache, 2 insomnia, 1 cough, 6 alopecia, 1 dry skin, 3 pruritus, 3 rash | 8 out of 8 | |
| Daclatasvir | 14 anaemia, 9 neutropenia, 5 diarrhoea, 13 nausea, 7 vomiting, 9 asthenia, 4 chills, 19 fatigue, 11 influenza‐like illness, 12 irritability, 7 pyrexia, 9 decreased appetite, 2 arthralgia, 8 myalgia, 5 dizziness, 2 dysgeusia, 19 headache, 7 depression, 9 insomnia, 9 cough, 6 dyspnoea, 8 alopecia, 2 dry skin, 12 pruritus, 10 rash | 36 out of 36 | 5 anaemia, 5 neutropenia, 3 diarrhoea, 6 nausea, 1 asthenia, 2 chills, 9 fatigue, 4 influenza‐like illness, 2 irritability, 3 pyrexia, 3 decreased appetite, 3 myalgia, 1 dizziness, 1 dysgeusia, 3 headache, 3 depression, 6 insomnia, 3 cough, 2 dyspnoea, 2 alopecia, 1 dry skin, 3 pruritus, 3 rash | 12 out of 12 | |
| Daclatasvir | 1 diarrhoea, 1 nausea, 4 headache | 7 out of 16 | None reported | 0 out of 2 | |
| Daclatasvir | 2 diarrhoea, 3 fatigue, 1 arthralgia, 1 dizziness, 5 headache, 2 insomnia, 1 dry skin | 16 out of 24 | 1 nausea, 1 vomiting, 2 headache | 4 out of 6 | |
| Danoprevir | 115 diarrhoea, 106 nausea, 77 asthenia, 89 chills, 158 fatigue, 125 pyrexia, 88 decreased appetite, 72 arthralgia, 93 myalgia,158 headache, 102 insomnia, 62 cough, 54 alopecia, 83 pruritus, 78 rash | 364 out of 373 | 5 diarrhoea, 13 nausea, 9 asthenia, 8 chills, 17 fatigue, 15 pyrexia, 6 decreased appetite, 9 arthralgia, 13 myalgia, 24 headache, 16 insomnia, 12 cough, 4 alopecia, 14 pruritus, 6 rash | 42 out of 44 | |
| Danoprevir | 2 diarrhoea, 3 myalgia, 5 headache | 21 out of 40 | 1 diarrhoea, 2 headache | 3 out of 10 | |
| Danoprevir | 6 neutropenia, 5 diarrhoea, 4 nausea, 3 asthenia, 5 chills, 8 fatigue, 4 influenza‐like illness, 2 pyrexia, 2 arthralgia, 17 myalgia, 6 dizziness, 23 headache, 2 depression, 6 insomnia, 3 pruritus | 42 out of 47 | 2 neutropenia, 2 diarrhoea, 1 nausea, 1 chills, 3 fatigue, 1 influenza‐like illness, 1 arthralgia, 5 myalgia, 1 dizziness, 4 headache, 1 depression, 2 insomnia | 12 out of 12 | |
| Danoprevir | 4 diarrhoea, 5 nausea, 5 fatigue, 3 influenza‐like illness, 5 irritability, 5 arthralgia, 5 myalgia, 11 headache, 4 insomnia, 6 rash | 24 out of 25 | 1 diarrhoea, 3 nausea, 1 fatigue, 2 myalgia, 4 headache, 2 insomnia | 5 out of 5 | |
| Danoprevir | 53 anaemia, 70 neutropenia, 56 diarrhoea, 85 nausea, 29 vomiting, 57 chills, 109 fatigue, 38 irritability, 51 pyrexia, 34 decreased appetite, 29 arthralgia, 60 myalgia, 92 headache, 42 depression, 69 insomnia, 31 alopecia, 46 pruritus, 42 rash | Not specified out of 194 | 13 anaemia, 11 neutropenia, 7 diarrhoea, 10 nausea, 4 vomiting, 13 chills, 14 fatigue, 7 irritability, 5 pyrexia, 4 decreased appetite, 9 arthralgia, 11 myalgia, 18 headache, 4 depression, 9 insomnia, 5 alopecia, 6 pruritus, 8 rash | Not specified out of 31 | |
| Deleobuvir | 1 anaemia, 19 diarrhoea, 19 nausea, 11 vomiting, 16 asthenia, 3 chills, 12 fatigue, 14 influenza‐like illness, 7 irritability, 6 pyrexia, 14 decreased appetite, 4 arthralgia, 4 myalgia, 6 dizziness, 4 dysgeusia, 20 headache, 15 insomnia, 6 cough, 3 dyspnoea, 1 alopecia, 6 dry skin, 5 pruritus, 8 rash | 49 out of 49 | 1 nausea, 1 asthenia, 1 chills, 2 fatigue, 1 influenza‐like illness, 2 irritability, 1 decreased appetite, 2 headache, 1 dry skin, 1 pruritus, 2 rash | 7 out of 8 | |
| Faldaprevir | 114 anaemia, 59 neutropenia, 160 diarrhoea, 249 nausea, 110 vomiting, 29 asthenia, 78 chills, 246 fatigue, 39 influenza‐like illness, 67 irritability, 79 pyrexia, 117 decreased appetite, 72 arthralgia, 100 myalgia, 80 dizziness, 31 dysgeusia, 165 headache, 68 depression, 137 insomnia, 89 cough, 53 dyspnoea, 96 alopecia, 67 dry skin, 164 pruritus, 298 rash | 513 out of 525 | 27 anaemia, 14 neutropenia, 23 diarrhoea, 52 nausea, 11 vomiting, 6 asthenia, 25 chills, 70 fatigue, 15 influenza‐like illness, 27 irritability, 20 pyrexia, 26 decreased appetite, 22 arthralgia, 37 myalgia, 25 dizziness, 3 dysgeusia, 45 headache, 11 depression, 38 insomnia, 21 cough, 20 dyspnoea, 22 alopecia, 15 dry skin, 37 pruritus, 41 rash | 130 out of 132 | |
| Faldaprevir | 89 anaemia, 57 neutropenia, 121 diarrhoea, 168 nausea, 79 vomiting, 96 asthenia, 143 fatigue, 92 influenza‐like illness, 37 irritability, 110 pyrexia, 87 decreased appetite, 39 arthralgia, 41 myalgia, 38 dizziness, 23 dysgeusia, 146 headache, 32 depression, 70 insomnia, 58 cough, 36 dyspnoea, 49 alopecia, 80 dry skin, 160 pruritus, 139 rash | 496 out of 520 | 26 anaemia, 18 neutropenia, 17 diarrhoea, 19 nausea, 6 vomiting, 27 asthenia, 35 fatigue, 21 influenza‐like illness, 9 irritability, 32 pyrexia, 22 decreased appetite, 14 arthralgia, 20 myalgia, 13 dizziness, 5 dysgeusia, 40 headache, 8 depression, 22 insomnia, 20 cough, 16 dyspnoea, 15 alopecia, 17 dry skin, 41 pruritus, 25 rash | 120 out of 132 | |
| Faldaprevir | 98 anaemia, 65 neutropenia, 190 diarrhoea, 318 nausea, 171 vomiting, 108 asthenia, 32 chills, 204 fatigue, 107 influenza‐like illness, 49 irritability, 113 pyrexia, 128 decreased appetite, 60 arthralgia, 72 myalgia, 37 dizziness, 39 dysgeusia, 182 headache, 52 depression, 118 insomnia, 99 cough, 47 dyspnoea, 53 alopecia, 108 dry skin, 225 pruritus, 160 rash | 585 out of 599 | 8 anaemia, 12 neutropenia, 10 diarrhoea, 18 nausea, 5 vomiting, 21 asthenia, 5 chills, 16 fatigue, 15 influenza‐like illness, 11 irritability, 14 pyrexia, 10 decreased appetite, 7 arthralgia, 8 myalgia, 6 dizziness, 4 dysgeusia, 22 headache, 10 depression, 13 insomnia, 16 cough, 7 dyspnoea, 4 alopecia, 12 dry skin, 23 pruritus, 16 rash | 74 out of 78 | |
| Faldaprevir | 2 anaemia, 1 neutropenia, 4 diarrhoea, 7 nausea, 10 asthenia, 2 chills, 2 fatigue, 4 influenza‐like illness, 4 irritability, 2 pyrexia, 1 decreased appetite, 7 myalgia, 1 dizziness, 6 headache, 2 depression, 5 insomnia, 2 cough, 1 alopecia, 5 dry skin, 3 pruritis, 1 rash | 32 out of 26 | 1 diarrhoea, 2 nausea, 2 asthenia, 2 headache, 1 depression, 1 insomnia, 1 cough | 5 out of 8 | |
| Faldaprevir | 1 neutropenia, 3 diarrhoea, 3 nausea, 3 vomiting, 2 influenza‐like illness, 8 pyrexia, 2 decreased appetite, 2 arthralgia, 4 dizziness, 1 dysgeusia, 7 headache, 1 depression, 5 insomnia, 1 cough, 3 alopecia, 1 dry skin, 6 pruritus, 6 rash | 33 out of 35 | 2 nausea, 2 vomiting, 1 influenza‐like illness, 1 pyrexia, 1 decreased appetite, 1 headache, 1 insomnia, 1 dyspnoea, 2 dry skin, 3 pruritus, 1 rash | 6 out of 8 | |
| Faldeprevir | 85 anaemia, 49 neutropenia, 188 diarrhoea, 239 nausea, 105 vomiting, 122 asthenia, 54 chills, 194 fatigue, 217 influenza‐like illness, 82 irritability, 86 pyrexia, 133 decreased appetite, 72 arthralgia, 133 myalgia, 48 dizziness, 25 dysgeusia, 243 headache, 68 depression, 107 insomnia, 100 cough, 73 dyspnoea, 106 alopecia, 122 dry skin, 227 pruritus, 163 rash | 620 out of 641 | 12 anaemia, 8 neutropenia, 13 diarrhoea, 14 nausea, 4 vomiting, 15 asthenia, 8 chills, 24 fatigue, 34 influenza‐like illness, 10 irritability, 11 pyrexia, 11 decreased appetite, 5 arthralgia, 12 myalgia, 5 dizziness, 27 headache, 7 depression, 17 insomnia, 9 cough, 11 dyspnoea, 8 alopecia, 10 dry skin, 12 pruritus, 12 rash | 65 out of 71 | |
| Filibuvir | 13 anaemia,4 neutropenia ,6 diarrhoea, 13 nausea, 3 vomiting, 4 chills, 13 fatigue, 3 influenza‐like illness, 2 irritability, 2 pyrexia, 3 decreased appetite, 2 arthralgia, 4 myalgia, 4 dizziness, 14 headache, 5 depression, 11 insomnia, 5 cough, 2 dyspnoea, 2 alopecia, 3 dry skin, 2 pruritus, 3 rash | 27 out of 27 | 3 anaemia, 3 diarrhoea, 4 nausea, 1 vomiting, 1 chills, 5 fatigue, 2 influenza‐like illness, 2 decreased appetite, 2 arthralgia, 2 headache, 3 depression, 2 insomnia, 2 cough, 2 dyspnoea, 1 alopecia, 2 dry skin, 3 pruritus, 1 rash | 8 out of 8 | |
| Filibuvir | 26 anaemia, 26 neutropenia, 24 diarrhoea, 55 nausea, 20 vomiting, 35 asthenia, 25 chills, 73 fatigue, 29 influenza‐like illness, 33 irritability, 28 pyrexia, 36 decreased appetite, 30 arthralgia, 37 myalgia, 20 dizziness, 40 dysgeusia, 61 headache, 32 depression, 55 insomnia, 33 cough, 18 dyspnoea, 34 alopecia, 33 dry skin, 56 pruritus, 34 rash | 174 out of 192 | anaemia, neutropenia, diarrhoea, nausea, vomiting, asthenia, chills, fatigue, influenza‐like illness, irritability, pyrexia, decreased appetite, arthralgia, myalgia, dizziness, dysgeusia, headache, depression, insomnia, cough, dyspnoea, alopecia, dry skin, pruritus, rash. The authors did not report number of adverse events in the control group | 90 out of 96 | |
| Grazoprevir | 9 diarrhoea, 2 nausea, 1 vomiting, 7 fatigue, 1 dysgeusia, 16 headache, 1 insomnia, 2 pruritis | 34 out of 76 | 1 headache | 2 out of 15 | |
| GSK2336805 | 3 anaemia, 3 neutropenia, 4 nausea, 2 vomiting, 2 chills, 5 fatigue, 2 cough | 11 out of 11 | 1 fatigue | 4 out of 4 | |
| IDX‐184 | 1 diarrhoea, 2 fatigue, 1 dizziness, 4 headache | 8 out of 33 | 1 diarrhoea, 1 fatigue, 1 dizziness, 1 headache | 4 out of 8 | |
| IDX‐184 | 6 neutropenia, 7 diarrhoea, 22 nausea, 5 vomiting, 15 chills, 36 fatigue, 10 irritability, 9 pyrexia, 5 decreased appetite, 19 myalgia, 27 headache, 4 depression, 10 insomnia, 6 pruritus | 59 out of 65 | 4 neutropenia, 2 diarrhoea, 3 nausea, 5 chills, 9 fatigue, 4 irritability, 3 decreased appetite, 5 myalgia, 7 headache, 3 depression, 5 insomnia, 2 pruritus | 12 out of 16 | |
| IDX320 | 1 diarrhoea, 1 myalgia, 4 headache | Not specified out of 30 | None reported | 0 out of 8 | |
| Ledispasvir | 2 nausea, 6 headache, 2 rash | 18 out of 59 | 1 nausea | 4 out of 11 | |
| Mericitabine/danoprevir | 7 diarrhoea, 9 nausea, 36 headache, 9 rash | Not specified out of 73 | 1 diarrhoea, 2 nausea, 8 headache, 1 rash | Not specified out of 14 | |
| Mericitabine | 25 diarrhoea, 16 nausea, 6 chills, 47 fatigue, 14 irritability, 16 pyrexia, 12 arthralgia, 19 myalgia, 43 headache, 27 insomnia, 21 cough, 15 pruritus | Not specified out of 102 | 12 diarrhoea, 14 nausea, 12 chills, 25 fatigue, 10 irritability, 14 pyrexia, 11 arthralgia, 18 myalgia, 28 headache, 11 insomnia, 11 cough, 11 pruritus | Not specified out of 49 | |
| Mericitabine | 18 diarrhoea, 33 nausea, 31 chills, 58 fatigue, 21 irritability, 20 pyrexia, 25 decreased appetite, 18 arthralgia, 24 myalgia, 19 dizziness, 42 headache, 31 insomnia, 17 cough, 14 alopecia, 15 pruritus, 17 rash | Not specified out of 81 | 20 diarrhoea, 34 nausea, 33 chills, 58 fatigue, 25 irritability, 27 pyrexia, 22 decreased appetite, 21 arthralgia, 24 myalgia, 20 dizziness, 38 headache, 28 insomnia, 22 cough, 17 alopecia, 28 pruritus, 28 rash | Not specified out of 85 | |
| Narlaprevir | 10 diarrhoea, 8 nausea, 30 influenza‐like illness, 6 dizziness, 11 headache | 32 out of 32 | 1 nausea, 6 influenza‐like illness, 1 dizziness | 7 out of 8 | |
| Narlaprevir | 87 anaemia, 84 diarrhoea, 131 nausea, 54 vomiting, 44 chills, 123 fatigue, 106 influenza‐like illness, 58 irritability, 58 pyrexia, 61 decreased appetite, 60 arthralgia, 39 myalgia, 58 dizziness, 83 headache, 33 depression, 80 insomnia, 28 pruritus, 31 rash | Not specified out of 93 | 6 anaemia, 17 diarrhoea, 50 nausea, 28 vomiting, 17 chills, 56 fatigue, 44 influenza‐like illness, 28 irritability, 17 pyrexia, 44 decreased appetite, 28 arthralgia, 6 dizziness, 39 headache, 22 depression, 39 insomnia, 33 pruritus, 11 rash | Not specified out of 18 | |
| Odalasvir/sovaprevir | 5 anaemia, 2 diarrhoea, 5 fatigue, 2 influenza‐like illness, 2 irritability, 1 decreased appetite, 2 arthralgia, 3 myalgia, 1 dizziness, 1 dysgeusia, 6 headache, 2 insomnia, 4 cough, 1 dyspnoea, 1 pruritus, 1 rash | 20 out of 20 | 2 diarrhoea, 4 fatigue, 1 myalgia, 1 dizziness, 1 headache, 1 cough, pruritus | 10 out of 10 | |
| Ombitasvir | 6 anaemia, 5 neutropenia, 4 diarrhoea, 9 nausea, 7 vomiting, 6 chills, 18 fatigue, 1 influenza‐like illness, 1 irritability, 3 pyrexia, 5 decreased appetite, 2 arthralgia, 2 myalgia, 2 dizziness, 9 headache, 4 depression, 4 insomnia, 3 cough, 2 dyspnoea, 5 dry skin, 3 pruritus, 6 rash | 26 out of 28 | 1 neutropenia, 1 diarrhoea, 3 nausea, 2 vomiting, 6 fatigue, 2 influenza‐like illness, 1 irritability, 1 decreased appetite, 1 myalgia, 2 headache, 1 depression, 2 insomnia, 1 cough, dyspnoea, 1 dry skin, 2 rash | 9 out of 9 | |
| Paritaprevir/ABT‐072/dasabuvir | 12 anaemia, 14 neutropenia, 16 diarrhoea, 15 nausea, 6 vomiting, 4 asthenia, 11 chills, 29 fatigue, 14 influenza‐like illness, 8 irritability, 12 pyrexia, 4 decreased appetite, 9 arthralgia, 14 myalgia, 9 dizziness, 6 dysgeusia, 38 headache, 15 depression, 14 insomnia, 6 cough, 6 dyspnoea, 5 alopecia, 2 dry skin, 8 pruritus, 12 rash | 63 out of 63 | 1 anaemia, 2 neutropenia, 4 diarrhoea, 4 nausea, 1 vomiting, 1 asthenia, 6 fatigue, 1 influenza‐like illness, 1 irritability, 1 pyrexia, 2 decreased appetite, 1 arthralgia, 3 myalgia, 4 dizziness, 2 dysgeusia, 4 headache, 2 insomnia, 2 dyspnoea, 1 alopecia, 2 pruritus, 2 rash | 10 out of 11 | |
| Paritaprevir/ombitasvir | 24 anaemia, 65 diarrhoea, 112 nausea, 23 vomiting, 59 asthenia, 164 fatigue, 26 irritability, 37 decreased appetite, 23 arthralgia, 21 myalgia, 38 dizziness, 156 headache, 67 insomnia, 34 cough, 38 dyspnoea, 27 dry skin, 80 pruritus, 51 rash | 391 out of 473 | 11 diarrhoea, 22 nausea, 6 vomiting, 6 asthenia, 45 fatigue, 4 irritability, 5 decreased appetite, 9 arthralgia, 8 myalgia, 6 dizziness, 42 headache, 12 insomnia, 8 cough, 4 dyspnoea, 2 dry skin, 7 pruritus, 9 rash | 108 out of 158 | |
| Paritaprevir/ombitasvir | 19 anaemia, 47 diarrhoea, 72 nausea, 22 vomiting, 60 asthenia, 115 fatigue, 22 irritability, 24 decreased appetite, 21 arthralgia, 28 myalgia, 30 dizziness, 13 dysgeusia, 126 headache, 52 insomnia, 43 cough, 50 dyspnoea, 27 dry skin, 53 pruritus, 34 rash | 328 out of 394 | 12 diarrhoea, 17 nausea, 11 asthenia, 22 fatigue, 8 irritability, 2 decreased appetite, 7 arthralgia, 10 myalgia, 5 dizziness, 5 dysgeusia, 34 headache, 7 insomnia, 5 cough, 10 dyspnoea, 3 dry skin, 5 pruritus, 6 rash | 74 out of 97 | |
| PHX1766 | 1 nausea, 2 fatigue, 1 dizziness | Not specified | None reported | Not specified | |
| R1626 | 43 neutropenia, 42 diarrhoea, 49 nausea, 26 vomiting, 39 chills, 45 fatigue, 20 irritability, 29 pyrexia, 21 arthralgia, 23 myalgia, 15 dizziness, 47 headache, 27 insomnia, 15 cough, 11 pruritus, 20 rash | Not specified out of 84 | 5 diarrhoea, 10 nausea, 2 vomiting, 9 chills, 10 fatigue, 1 irritability, 8 pyrexia, 5 arthralgia, 11 myalgia, 3 dizziness, 11 headache, 6 insomnia, 4 cough, 6 pruritus, 2 rash | Not specified out of 20 | |
| Samatasvir | 4 nausea, 1 decreased appetite, 6 headache, 1 insomnia | 20 out of 48 | 1 nausea, 1 decreased appetite, 1 headache, 1 insomnia | 6 out of 12 | |
| Simeprevir | 40 anaemia, 37 neutropenia, 36 diarrhoea, 59 nausea, 18 vomiting, 57 asthenia, 17 chills, 87 fatigue, 78 influenza‐like illness, 63 pyrexia, 35 decreased appetite, 26 arthralgia, 39 myalgia, 14 dizziness, 12 dysgeusia, 87 headache, 22 depression, 49 insomnia, 34 cough, 26 dyspnoea, 26 alopecia, 24 dry skin, 16 pruritus, 33 rash | 245 out of 260 | 24 anaemia, 26 neutropenia, 22 diarrhoea, 26 nausea, 9 vomiting, 25 asthenia, 11 chills, 58 fatigue, 27 influenza‐like illness, 30 pyrexia, 24 decreased appetite, 12 arthralgia, 17 myalgia, 6 dizziness, 7 dysgeusia, 48 headache, 10 depression, 33 insomnia, 21 cough, 5 dyspnoea, 17 alopecia, 18 dry skin, 37 pruritus, 19 rash | 123 out of 133 | |
| Simeprevir | 63 anaemia, 75 neutropenia, 47 diarrhoea, 86 nausea, 22 vomiting, 63 asthenia, 25 chills, 107 fatigue, 98 influenza‐like illness, 11 irritability, 64 pyrexia, 17 decreased appetite, 53 arthralgia, 55 myalgia, 29 dizziness, 16 dysgeusia, 142 headache, 32 depression, 69 insomnia, 52 cough, 33 dyspnoea, 53 alopecia, 63 dry skin, 173 pruritus, 65 rash | 302 out of 309 | 16 anaemia, 16 neutropenia, 12 diarrhoea, 21 nausea, 5 vomiting, 16 asthenia, 8 chills, 37 fatigue, 29 influenza‐like illness, 8 irritability, 13 pyrexia, 6 decreased appetite, 11 arthralgia, 17 myalgia, 6 dizziness, 5 dysgeusia, 40 headache, 14 depression, 23 insomnia, 15 cough, 6 dyspnoea, 16 alopecia, 14 dry skin, 35 pruritus, 18 rash | 75 out of 77 | |
| Simeprevir | 24 anaemia, 13 diarrhoea,13 nausea, 6 vomiting, 4 chills, 2 fatigue, 42 pyrexia, 15 decreased appetite, 27 arthralgia, 15 myalgia, 3 dizziness, 6 dysgeusia, 41 headache, 2 depression, 23 insomnia, 8 cough, 25 alopecia, 5 dry skin, 15 pruritus, 47 rash | 79 out of 79 | 5 anaemia, 5 diarrhoea, 2 vomiting, 7 pyrexia, 3 decreased appetite, 2 arthralgia, 2 myalgia, 8 headache, 2 insomnia, 2 cough, 6 alopecia, 6 rash | 13 out of 13 | |
| Simeprevir | 70 anaemia, 8 neutropenia, 20 diarrhoea, 16 nausea, 6 vomiting, 13 fatigue, 75 pyrexia, 28 decreased appetite, 30 arthralgia, 9 myalgia, 4 dizziness, 20 dysgeusia, 54 headache, 27 insomnia, 11 cough, 44 alopecia, 8 dry skin, 35 pruritus, 57 rash | 123 out of 123 | 36 anaemia, 1 neutropenia, 20 diarrhoea, 16 nausea, 6 vomiting, 7 fatigue, 31 pyrexia, 20 decreased appetite, 14 arthralgia, 11 myalgia, 4 dizziness, 8 dysgeusia, 26 headache, 3 depression, 25 insomnia, 8 cough, 28 alopecia, 9 dry skin, 18 pruritus, 37 rash | 60 out of 60 | |
| Simeprevir | 82 anaemia, 59 neutropenia, 14 diarrhoea, 16 nausea, 15 asthenia, 63 fatigue, 39 influenza‐like illness, 67 pyrexia, 28 decreased appetite, 13 arthralgia, 36 myalgia, 39 headache, 17 insomnia, 16 cough, 47 alopecia, 40 pruritus, 57 rash | 298 out of 305 | 53 anaemia, 32 neutropenia, 7 diarrhoea, 10 nausea, 7 asthenia, 36 fatigue, 19 influenza‐like illness, 46 pyrexia, 16 decreased appetite, 4 arthralgia, 22 myalgia, 28 headache, 18 insomnia, 17 cough, 26 alopecia, 17 pruritus, 27 rash | 149 out of 152 | |
| Simeprevir | 44 anaemia, 54 neutropenia, 35 diarrhoea, 65 nausea, 23 vomiting, 25 asthenia, 33 chills, 111 fatigue, 62 influenza‐like illness, 51 pyrexia, 47 decreased appetite, 34 arthralgia, 39 myalgia, 23 dizziness, 16 dysgeusia, 88 headache, 23 depression, 56 insomnia, 25 cough, 23 dyspnoea, 30 alopecia, 33 dry skin, 68 pruritus, 60 rash | 252 out of 264 | 24 anaemia, 15 neutropenia, 19 diarrhoea, 32 nausea, 9 vomiting, 21 asthenia, 18 chills, 53 fatigue, 26 influenza‐like illness, 28 pyrexia, 19 decreased appetite, 21 arthralgia, 18 myalgia, 9 dizziness, 4 dysgeusia, 51 headache, 16 depression, 31 insomnia, 20 cough, 9 dyspnoea, 16 alopecia, 11 dry skin, 20 pruritus, 30 rash | 124 out of 130 | |
| Simeprevir | 16 anaemia, 26 neutropenia, 20 diarrhoea, 31 nausea, 9 vomiting, 26 asthenia, 6 chills, 35 fatigue, 24 influenza‐like illness, 10 irritability, 21 pyrexia, 7 decreased appetite, 20 arthralgia, 14 myalgia, 4 dizziness, 5 dysgeusia, 42 headache, 14 depression, 12 insomnia, 19 cough, 18 dyspnoea, 16 alopecia, 19 dry skin, 18 pruritus, 9 rash | 82 out of 83 | 4 anaemia, 4 neutropenia, 3 diarrhoea, 4 nausea, 3 vomiting, 7 asthenia, 5 chills, 13 fatigue, 5 influenza‐like illness, 4 irritability, 4 pyrexia, 2 decreased appetite, 3 arthralgia, 8 myalgia, 3 dizziness, 3 dysgeusia, 16 headache, 2 depression, 7 insomnia, 10 cough, 4 dyspnoea, 3 alopecia, 5 dry skin, 7 pruritus, 5 rash | 28 out of 28 | |
| Simeprevir | 46 anaemia, 49 neutropenia, 34 diarrhoea, 63 nausea, 17 vomiting, 59 asthenia, 21 chills, 95 fatigue, 66 influenza‐like illness, 80 pyrexia, 46 decreased appetite, 32 arthralgia, 58 myalgia, 21 dizziness, 101 headache, 29 depression, 51 insomnia, 32 cough, 23 dyspnoea, 43 alopecia, 28 dry skin, 65 pruritus, 46 rash | 243 out of 257 | 33 anaemia, 29 neutropenia, 12 diarrhoea, 24 nausea, 7 vomiting, 38 asthenia, 12 chills, 56 fatigue, 35 influenza‐like illness, 53 pyrexia, 21 decreased appetite, 14 arthralgia, 28 myalgia, 9 dizziness, 49 headache, 19 depression, 21 insomnia, 22 cough, 11 dyspnoea, 27 alopecia, 18 dry skin, 34 pruritus, 15 rash | 131 out of 134 | |
| Simeprevir | 1 anaemia, 1 diarrhoea, 6 nausea, 8 fatigue, 1 irritability, 2 myalgia, 7 headache, 3 insomnia, 6 pruritus, 10 rash | 46 out of 58 | 9 anaemia, 5 neutropenia, 2 diarrhoea, 7 nausea, 4 asthenia, 17 fatigue, 6 influenza‐like illness, 3 irritability, 4 myalgia, 8 headache, 6 insomnia, 4 pruritus, 3 rash | 22 out of 24 | |
| Simeprevir | 76 anaemia, 101 neutropenia, 59 diarrhoea, 95 nausea, 21 vomiting, 84 asthenia, 34 chills, 174 fatigue, 116 influenza‐like illness, 53 irritability, 69 pyrexia, 69 decreased appetite, 50 arthralgia, 64 myalgia, 29 dizziness, 22 dysgeusia, 138 headache, 45 depression, 79 insomnia, 76 cough, 49 dyspnoea, 31 alopecia, 72 dry skin, 135 pruritus, 61 rash | 380 out of 396 | 13 anaemia, 11 neutropenia, 13 diarrhoea, 14 nausea, 5 vomiting, 7 asthenia, 6 chills, 174 fatigue, 13 influenza‐like illness, 7 irritability, 9 pyrexia, 9 decreased appetite, 9 arthralgia, 12 myalgia, 6 dizziness, 3 dysgeusia, 24 headache, 6 depression, 9 insomnia, 8 cough, 4 dyspnoea, 5 alopecia, 10 dry skin, 11 pruritus, 9 rash | 63 out of 66 | |
| Sofosbuvir | 19 diarrhoea, 46 nausea, 12 vomiting, 91 fatigue, 19 irritability, 7 decreased appetite, 16 arthralgia, 19 dizziness, 43 headache, 15 depression, 39 insomnia, 11 cough, 19 dyspnoea, 23 pruritus, 18 rash | 184 out of 207 | 4 diarrhoea, 13 nausea, 5 vomiting, 17 fatigue, 1 irritability, 7 decreased appetite, 1 arthralgia, 5 dizziness, 14 headache, 1 depression, 3 insomnia, 2 cough, 1 dyspnoea, 6 pruritus, 6 rash | 55 out of 71 | |
| Sofosbuvir | 19 anaemia, 23 neutropenia, 18 diarrhoea, 38 nausea, 12 vomiting, 2 asthenia, 37 chills, 64 fatigue, 15 irritability, 22 pyrexia, 11 decreased appetite, 10 arthralgia, 17 myalgia, 11 dizziness, 9 dysgeusia, 37 headache, 12 depression, 24 insomnia, 14 cough, 11 dyspnoea, 10 alopecia, 12 dry skin, 13 pruritus, 29 rash | 117 out of 120 | 7 anaemia, 5 neutropenia, 2 diarrhoea, 9 nausea, 2 vomiting, 1 asthenia, 10 chills, 16 fatigue, 5 irritability, 2 pyrexia, 4 decreased appetite, 5 arthralgia, 6 myalgia, 3 dizziness, 15 headache, 3 depression, 9 insomnia, 3 cough, 4 dyspnoea, 2 alopecia, 3 dry skin, 3 pruritus, 4 rash | 26 out of 26 | |
| Sofosbuvir | 20 anaemia, 23 diarrhoea, 46 nausea, 17 vomiting, 7 chills, 92 fatigue, 7 influenza‐like illness, 25 irritability, 6 pyrexia, 17 decreased appetite, 15 arthralgia, 21 myalgia, 27 dizziness, 64 headache, 14 depression, 31 insomnia, 19 cough, 18 dyspnoea, 12 alopecia, 11 dry skin, 19 pruritus, 23 rash | 219 out of 256 | 28 anaemia, 30 neutropenia, 45 diarrhoea, 70 nausea, 23 vomiting, 44 chills, 134 fatigue, 44 influenza‐like illness, 40 irritability, 33 pyrexia, 44 decreased appetite, 35 arthralgia, 40 myalgia, 33 dizziness, 108 headache, 34 depression, 71 insomnia, 21 cough, 20 dyspnoea, 24 alopecia, 23 dry skin, 42 pruritus, 43 rash | 233 out of 243 | |
| Sofosbuvir | 7 anaemia, 17 nausea, 3 vomiting, 22 fatigue, 4 pyrexia, 7 decreased appetite, 12 arthralgia, 7 myalgia, 6 dizziness, 15 headache, 4 depression, 7 insomnia, 9 pruritus | 45 out of 49 | 1 anaemia, 5 nausea, 2 chills, 6 fatigue, 1 pyrexia, 1 decreased appetite, 1 myalgia, 2 dizziness, 2 headache, 2 insomnia,1 pruritus | 13 out of 14 | |
| Sofosbuvir/velpatasvir | 48 diarrhoea, 75 nausea, 41 asthenia, 126 fatigue, 40 arthralgia, 25 myalgia, 182 headache, 50 insomnia, 39 cough | 485 out of 624 | 8 diarrhoea, 13 nausea, 9 asthenia, 23 fatigue, 9 arthralgia, 6 myalgia, 33 headache, 11 insomnia, 4 cough | 89 out of 116 | |
| Telaprevir | 1 diarrhoea, 4 nausea, 1 vomiting, 6 asthenia, 4 fatigue, 10 influenza‐like illness, 3 headache, 2 insomnia, 1 dyspnoea, 1 dry skin, 3 pruritus | 16 out of 16 | 1 anaemia, 1 neutropenia, 3 asthenia, 4 influenza‐like illness, 1 decreased appetite, 1 headache, 1 insomnia, 1 cough, 1 dry skin, 2 pruritus, 2 rash | 8 out of 8 | |
| Telaprevir | 2 diarrhoea, 3 nausea, 1 chills, 5 myalgia, 2 dizziness, 5 headache, 3 dry skin, 3 rash | 14 out of 16 | 1 diarrhoea, 1 nausea, 1 asthenia, 2 chills, 2 myalgia, 1 dizziness, 2 headache, 1 dry skin | 4 out of 4 | |
| Telaprevir | 1 anaemia, 4 diarrhoea, 8 nausea, 5 vomiting, 9 asthenia, 1 chills, 5 fatigue, 11 influenza‐like illness, 1 irritability, 2 pyrexia, 1 arthralgia, 4 myalgia, 3 dizziness, 5 headache, 1 depression, 2 insomnia, 1 cough, 2 dyspnoea, 1 alopecia, 3 dry skin, 11 pruritus, 5 rash | 26 out of 31 | 1 neutropenia, 1 diarrhoea, 1 nausea, 5 asthenia, 3 chills, 2 fatigue, 7 influenza‐like illness, 5 pyrexia, 3 myalgia, 1 dysgeusia, 6 headache, 1 depression, 2 insomnia, 2 cough, 2 dyspnoea, 1 dry skin, 2 pruritus | 16 out of 18 | |
| Telaprevir | 44 anaemia, 11 neutropenia, 66 diarrhoea, 102 nausea, 22 vomiting, 110 asthenia, 12 chills, 70 fatigue, 92 influenza‐like illness, 22 irritability, 44 pyrexia, 30 decreased appetite, 36 arthralgia, 35 myalgia, 12 dizziness, 20 dysgeusia, 105 headache, 51 depression, 62 insomnia, 37 cough, 50 dyspnoea, 29 alopecia, 64 dry skin, 139 pruritus, 71 rash | 240 out of 241 | 14 anaemia, 14 neutropenia, 23 diarrhoea, 33 nausea, 12 vomiting, 26 asthenia, 10 chills, 30 fatigue, 43 influenza‐like illness, 11 irritability, 19 pyrexia, 16 decreased appetite, 14 arthralgia, 17 myalgia, 8 dizziness, 3 dysgeusia, 37 headache, 19 depression, 32 insomnia, 21 cough, 13 dyspnoea, 17 alopecia, 29 dry skin, 29 pruritus, 22 rash | 81 out of 82 | |
| Telaprevir | 276 anaemia, 217 diarrhoea, 302 nausea, 418 fatigue, 203 pyrexia, 304 headache, 233 insomnia, 346 pruritus, 262 rash | 723 out of 727 | 70 anaemia, 80 diarrhoea, 112 nausea, 206 fatigue, 87 pyrexia, 142 headache, 111 insomnia, 131 pruritus, 88 rash | 354 out of 361 | |
| Telaprevir | 58 anaemia, 30 neutropenia, 64 diarrhoea, 93 nausea, 38 vomiting, 29 chills, 127 fatigue, 75 influenza‐like illness, 23 irritability, 33 pyrexia, 22 decreased appetite, 34 arthralgia, 27 myalgia, 41 dizziness, 16 dysgeusia, 80 headache, 34 depression, 68 insomnia, 36 cough, 25 dyspnoea, 21 alopecia, 28 dry skin, 74 pruritus, 62 rash | 175 out of 175 | 20 anaemia, 18 neutropenia, 21 diarrhoea, 22 nausea, 9 vomiting, 14 chills, 57 fatigue, 32 influenza‐like illness, 22 irritability, 22 pyrexia, 9 decreased appetite, 16 arthralgia, 18 myalgia, 14 dizziness, 8 dysgeusia, 45 headache, 13 depression, 29 insomnia, 14 cough, 11 dyspnoea, 8 alopecia, 19 dry skin, 17 pruritus, 20 rash | 75 out of 75 | |
| Telaprevir | 69 anaemia, 31 neutropenia, 115 diarrhoea, 122 nausea, 37 vomiting, 57 chills, 197 fatigue, 93 influenza‐like illness, 63 irritability, 59 pyrexia, 20 decreased appetite, 51 arthralgia, 60 myalgia, 47 dizziness, 130 headache, 43 depression, 83 insomnia, 46 cough, 27 dyspnoea, 54 alopecia, 32 dry skin, 129 pruritus, 126 rash | 329 out of 339 | 9 anaemia, 7 neutropenia, 22 diarrhoea, 39 nausea, 13 vomiting, 15 chills, 64 fatigue, 36 influenza‐like illness, 25 irritability, 14 pyrexia, 12 decreased appetite, 21 arthralgia, 21 myalgia, 18 dizziness, 41 headache, 19 depression, 19 insomnia, 20 cough, 9 dyspnoea, 13 alopecia, 7 dry skin, 17 pruritus, 20 rash | 111 out of 114 | |
| Telaprevir | 35 anaemia, 26 neutropenia, 34 diarrhoea, 44 nausea, 27 vomiting, 22 asthenia, 15 chills, 50 fatigue, 24 influenza‐like illness, 18 irritability, 34 pyrexia, 30 decreased appetite, 9 arthralgia, 20 myalgia, 19 dizziness, 18 dysgeusia, 38 headache, 11 depression, 25 insomnia, 15 cough, 5 dyspnoea, 18 alopecia, 11 dry skin, 30 pruritus, 12 rash | 38 out of 38 | 8 anaemia, 2 neutropenia, 6 diarrhoea, 11 nausea, 5 vomiting, 9 asthenia, 5 chills, 12 fatigue, 13 influenza‐like illness, 5 irritability, 7 pyrexia, 6 decreased appetite, 2 arthralgia, 6 myalgia, 2 dizziness, 5 dysgeusia, 6 headache, 4 depression, 9 insomnia, 6 cough, 2 dyspnoea, 6 alopecia, 3 dry skin, 3 pruritus | 22 out of 22 | |
| Telaprevir | 171 anaemia, 73 neutropenia, 135 diarrhoea, 181 nausea, 68 vomiting, 111 asthenia, 73 chills, 276 fatigue, 179 influenza‐like illness, 74 irritability, 130 pyrexia, 40 decreased appetite, 67 arthralgia, 87 myalgia, 47 dizziness, 65 dysgeusia, 221 headache, 59 depression, 152 insomnia, 128 cough, 82 dyspnoea, 78 alopecia, 97 dry skin, 270 pruritus, 194 rash | 517 out of 530 | 19 anaemia, 14 neutropenia, 18 diarrhoea, 31 nausea, 11 vomiting, 38 asthenia, 73 chills, 53 fatigue, 33 influenza‐like illness, 21 irritability, 36 pyrexia, 9 decreased appetite, 20 arthralgia, 24 myalgia, 7 dizziness, 8 dysgeusia, 49 headache, 19 depression, 34 insomnia, 26 cough, 17 dyspnoea, 17 alopecia, 21 dry skin, 36 pruritus, 25 rash | 126 out of 132 | |
| Vaniprevir | 6 anaemia, 7 neutropenia, 16 diarrhoea, 26 nausea, 16 vomiting, 13 asthenia, 5 chills, 17 fatigue, 17influenza‐like illness, 4 irritability, 8 pyrexia, 13 decreased appetite, 8 arthralgia, 3 myalgia, 5 dizziness, 2 dysgeusia, 26 headache, 8 depression, 15 insomnia, 8 cough, 7 dyspnoea, 6 alopecia, 6 dry skin, 9 pruritus, 10 rash | 71 out of 75 | 3 anaemia, 2 neutropenia, 4 diarrhoea, 6 nausea, 4 asthenia, 2 chills, 7 fatigue, 4 influenza‐like illness, 3 irritability, 2 pyrexia, 2 decreased appetite, 2 arthralgia, 3 myalgia, 7 headache, 3 depression, 2 insomnia, 3 cough, 5 dyspnoea, 3 alopecia, 6 dry skin, 4 pruritus, 4 rash | 19 out of 19 | |
| Vaniprevir | 6 anaemia, 7 neutropenia, 16 diarrhoea, 26 nausea, 16 vomiting, 13 asthenia, 5 chills, 17 fatigue, 17 influenza‐like illness, 4 irritability, 8 pyrexia, 13 decreased appetite, 8 arthralgia, 3 myalgia, 5 dizziness, 2 dysgeusia, 26 headache, 8 depression, 15 insomnia, 8 cough, 7 dyspnoea, 6 alopecia, 6 dry skin, 9 pruritus, 10 rash | 71 out of 75 | 3 anaemia, 2 neutropenia, 4 diarrhoea, 6 nausea, 4 asthenia, 2 chills, 7 fatigue, 4 influenza‐like illness, 3 irritability, 2 pyrexia, 2 decreased appetite, 2 arthralgia, 3 myalgia, 7 headache, 3 depression, 2 insomnia, 3 cough, 5 dyspnoea, 3 alopecia, 6 dry skin, 4 pruritus, 4 rash | 19 out of 19 | |
| Vaniprevir | 43 anaemia, 34 neutropenia, 97 diarrhoea, 110 nausea, 59 vomiting, 50 asthenia, 16 chills, 92 fatigue, 54 influenza‐like illness, 24 irritability, 37 pyrexia, 40 decreased appetite, 37 arthralgia, 38 myalgia, 23 dizziness, 16 dysgeusia, 92 headache, 32 depression, 40 insomnia, 54 cough, 30 dyspnoea, 35 alopecia, 37 dry skin, 75 pruritus, 43 rash | 225 out of 229 | 8 anaemia, 3 neutropenia, 9 diarrhoea, 10 nausea, 3 vomiting, 11 asthenia, 1 chills, 18 fatigue, 12 influenza‐like illness, 8 irritability, 14 pyrexia, 5 decreased appetite, 11 arthralgia, 12 myalgia, 6 dizziness, 3 dysgeusia, 20 headache, 3 depression, 14 insomnia, 14 cough, 8 dyspnoea, 5 alopecia, 10 dry skin, 14 pruritus, 10 rash | 55 out of 56 | |
| Vaniprevir | 43 anaemia, 34 neutropenia, 97 diarrhoea, 110 nausea, 59 vomiting, 50 asthenia, 16 chills, 92 fatigue, 54 influenza‐like illness, 24 irritability, 37 pyrexia, 40 decreased appetite, 37 arthralgia, 38 myalgia, 23 dizziness, 16 dysgeusia, 92 headache, 32 depression, 40 insomnia, 54 cough, 30 dyspnoea, 35 alopecia, 37 dry skin, 75 pruritus, 43 rash | 225 out of 229 | 8 anaemia, 3 neutropenia, 9 diarrhoea, 10 nausea, 3 vomiting, 11 asthenia, 1 chills, 18 fatigue, 12 influenza‐like illness, 8 irritability, 14 pyrexia, 5 decreased appetite, 11 arthralgia, 12 myalgia, 6 dizziness, 3 dysgeusia, 20 headache, 3 depression, 14 insomnia, 14 cough, 8 dyspnoea, 5 alopecia, 10 dry skin, 14 pruritus, 10 rash | 55 out of 56 | |
| VCH‐759 | 18 diarrhoea, 3 nausea, 4 vomiting, 1 chills, 5 fatigue, 8 headache | 20 out of 23 | 5 diarrhoea, 1 nausea, 2 headache | 6 out of 9 | |
| Velpatasvir | 2 diarrhoea, 3 nausea, 4 vomiting, 6 headache, 2 cough | 18 out of 70 | None reported | 3 out of 17 |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Hepatitis C‐related morbidity or all‐cause mortality Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ bias risk Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Trials at high risk of bias | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Trials at low risk of bias | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to type of DAA Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 ABT‐072 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 ACH‐2684 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Alisporivir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 ALS‐2200 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 Asunaprevir | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.6 Balapiravir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.7 Beclabuvir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.8 BILB‐1941 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.9 BIT‐225 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.10 Boceprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.11 Ciluprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.12 Daclatasvir | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.13 Danoprevir | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.14 Dasabuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.15 Deleobuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.16 Faldaprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.17 Filibuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.18 Grazoprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.19 GS‐6620 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.20 GS‐9256 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.21 GS‐9451 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.22 GS‐9669 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.23 GS‐9851 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.24 GS‐9857 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.25 GSK2336805 | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.26 GSK2878175 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.27 IDX‐184 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.28 INX‐08189 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.29 Ledispasvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.30 Mericitabine | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.31 Narlaprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.32 Nesbuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.33 Odalasavir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.34 Ombitasvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.35 Paritaprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.36 PHX1766 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.37 PPI‐461 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.38 PSI‐352938 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.39 Samatasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.40 Setrobuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.41 Simeprevir | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.42 Sofosbuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.43 Sovaprevir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.44 Tegobuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.45 Telaprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.46 Valopicitabine | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.47 Vaniprevir | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.48 VCH‐759 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.49 VCH‐916 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.50 Velpatasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.51 VX‐222 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.52 Mixed | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to group of DAA Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Cyclophilin | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 NS3/NS4A inhibitors | 41 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 NS5B inhibitors (NPI) | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 NS5B inhibitors (NNPI) | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 NS5A inhibitors | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 VPU‐ion channel inhibitors | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.7 Mixed | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to HIV‐infection Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5.1 With HIV‐infection | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Without HIV‐infection | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Mixed (with and without HIV‐infection) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 Unclear | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to comorbidity Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1 With comorbidity | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Without comorbidity | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Unclear | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to viral genotype Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1 Genotype 1 | 57 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Genotype 2 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Genotype 3 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Genotype 4 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 Mixed | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to human genotype (IL28b) Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1 IL28b (CC) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 IL28B (CT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 IL28B (TT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 IL28B (CT + TT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.5 Mixed | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to Asian‐region Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1 From Asian region | 8 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 Not from Asian region | 52 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 Mixed | 11 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.4 Unclear | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to specific ethnicities Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 10.1 White | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 Black | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 Hispanic | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.4 Mixed | 70 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.5 Unclear | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to reaching planned sample size Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 11.1 Trials reaching planned sample size | 10 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 Trials not reaching planned sample size | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 Unclear | 58 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to prior treatment Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.1 Treatment‐naive | 47 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 Treatment‐experienced | 16 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 Mixed | 8 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.4 Unclear | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to interferon Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1 Trials where both groups received interferon | 52 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.2 Trials where neither group received interferon | 19 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to ribavirin Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 14.1 Trials where both groups received ribavirin | 52 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.2 Trials where neither group received ribavirin | 19 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to chronic kidney disease Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 15.1 With chronic kidney disease | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.2 Without chronic kidney disease | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.3 Unclear | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to cryoglobulinaemia Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 16.1 With cryoglobulinaemia | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.2 Without cryoglobulinaemia | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.3 Unclear | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to DAA group as co‐intervention Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 17.1 Trials where DAA were used as co‐intervention | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.2 Trials where DAA were not a co‐intervention | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to median dose Show forest plot | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 18.1 Over or equal to median dose | 41 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18.2 Under median dose | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18.3 Not available | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Serious adverse events Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Serious adverse events ‐ bias risk Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Trials at high risk of bias | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Trials at low risk of bias | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Serious adverse events ‐ according to type of DAA Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 ABT‐072 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 ACH‐2684 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Alisporivir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 ALS‐2200 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 Asunaprevir | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.6 Balapiravir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.7 Beclabuvir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.8 BILB‐1941 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.9 BIT‐225 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.10 Boceprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.11 Ciluprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.12 Daclatasvir | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.13 Danoprevir | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.14 Dasabuvir | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.15 Deleobuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.16 Faldaprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.17 Filibuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.18 Grazoprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.19 GS‐6620 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.20 GS‐9256 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.21 GS‐9451 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.22 GS‐9669 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.23 GS‐9851 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.24 GS‐9857 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.25 GSK2336805 | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.26 GSK2878175 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.27 IDX‐184 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.28 INX‐08189 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.29 Ledispasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.30 Mericitabine | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.31 Narlaprevir | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.32 Nesbuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.33 Odalasavir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.34 Ombitasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.35 Paritaprevir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.36 PHX1766 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.37 PPI‐461 | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.38 PSI‐352938 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.39 Samatasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.40 Setrobuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.41 Simeprevir | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.42 Sofosbuvir | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.43 Sovaprevir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.44 Tegobuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.45 Telaprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.46 Valopicitabine | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.47 Vaniprevir | 10 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.48 VCH‐759 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.49 VCH‐916 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.50 Velpatasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.51 VX‐222 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.52 Mixed | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Serious adverse events ‐ according to group of DAA Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Cyclophilin | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 NS3/NS4A inhibitors | 56 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 NS5B inhibitors (NPI) | 8 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 NS5B inhibitors (NNPI) | 5 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 NS5A inhibitors | 25 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 VPU‐ion channel inhibitors | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.7 Mixed | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Serious adverse events ‐ according to HIV‐infection Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5.1 With HIV‐infection | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Without HIV‐infection | 94 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Mixed (with and without HIV‐infection) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 Unclear | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Serious adverse events ‐ according to comorbidity Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1 With comorbidity | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Without comorbidity | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Unclear | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Serious adverse events ‐ according to viral genotype Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1 Genotype 1 | 84 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Genotype 2 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Genotype 3 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Genotype 4 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 Mixed | 17 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Serious adverse events ‐ according to human genotype (IL28b) Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1 IL28b (CC) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 IL28B (CT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 IL28B (TT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 IL28B (CT + TT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.5 Mixed | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Serious adverse events ‐ according to Asian‐region Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1 From Asian region | 10 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 Not from Asian region | 76 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 Mixed | 11 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.4 Unclear | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Serious adverse events ‐ according to specific ethnicities Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 10.1 White | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 Black | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 Hispanic | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.4 Mixed | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.5 Unclear | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Serious adverse events ‐ according to reaching planned sample size Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 11.1 Trials reaching planned sample size | 15 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 Trials not reaching planned sample size | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 Unclear | 83 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Serious adverse events ‐ according to prior treatment Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.1 Treatment‐naive | 72 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 Treatment‐experienced | 19 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 Mixed | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.4 Unclear | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Serious adverse events ‐ according to interferon Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1 Trials where both groups received interferon | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.2 Trials where neither group received interferon | 29 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.3 Unclear | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Serious adverse events ‐ according to ribavirin Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 14.1 Trials where both groups received ribavirin | 73 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.2 Trials where neither group received ribavirin | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.3 Unclear | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15 Serious adverse events ‐ according to chronic kidney disease Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 15.1 With chronic kidney disease | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.2 Without chronic kidney disease | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.3 Unclear | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16 Serious adverse events ‐ according to cryoglobulinaemia Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 16.1 With cryoglobulinaemia | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.2 Without cryoglobulinaemia | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.3 Unclear | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17 Serious adverse events ‐ according to DAA group as co‐intervention Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 17.1 Trials where DAA were used as co‐intervention | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.2 Trials where DAA were not a co‐intervention | 99 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18 Serious adverse events ‐ according to median dose Show forest plot | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 18.1 Over or equal to median dose | 58 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18.2 Under median dose | 37 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18.3 Not available | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Without sustained virological response Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 2 Without sustained virological response ‐ bias risk Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 2.1 Trials at high risk of bias | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 2.2 Trials at low risk of bias | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Without sustained virological response ‐ according to type of DAA Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 3.1 ABT‐072 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 ACH‐2684 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.3 Alisporivir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.4 ALS‐2200 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.5 Asunaprevir | 4 | 285 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.29, 0.85] |
| 3.6 Balapiravir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.7 Beclabuvir | 2 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.43, 1.40] |
| 3.8 BILB‐1941 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.9 BIT‐225 | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 0.27 [0.03, 2.51] |
| 3.10 Boceprevir | 1 | 229 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.29, 0.61] |
| 3.11 Ciluprevir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.12 Daclatasvir | 7 | 619 | Risk Ratio (M‐H, Random, 95% CI) | 0.60 [0.50, 0.73] |
| 3.13 Danoprevir | 5 | 642 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.28, 0.51] |
| 3.14 Dasabuvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.15 Deleobuvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.16 Faldaprevir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.17 Filibuvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.18 Grazoprevir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.19 GS‐6620 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.20 GS‐9256 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.21 GS‐9451 | 1 | 329 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.26, 0.67] |
| 3.22 GS‐9669 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.23 GS‐9851 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.24 GS‐9857 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.25 GSK2336805 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.26 GSK2878175 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.27 IDX‐184 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.28 INX‐08189 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.29 Ledispasvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.30 Mericitabine | 4 | 725 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.49, 1.27] |
| 3.31 Narlaprevir | 2 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.43, 1.09] |
| 3.32 Nesbuvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.33 Odalasavir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.34 Ombitasvir | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.39, 1.07] |
| 3.35 Paritaprevir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.36 PHX1766 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.37 PPI‐461 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.38 PSI‐352938 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.39 Samatasvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.40 Setrobuvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.41 Simeprevir | 19 | 2898 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.33, 0.46] |
| 3.42 Sofosbuvir | 3 | 181 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.20, 0.58] |
| 3.43 Sovaprevir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.44 Tegobuvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.45 Telaprevir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.46 Valopicitabine | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.47 Vaniprevir | 9 | 333 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.25, 0.43] |
| 3.48 VCH‐759 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.49 VCH‐916 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.50 Velpatasvir | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.51 VX‐222 | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.52 Mixed | 2 | 735 | Risk Ratio (M‐H, Random, 95% CI) | 0.06 [0.00, 7.05] |
| 4 Without sustained virological response ‐ according to group of DAA Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 4.1 Cyclophilin | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 NS3/NS4A inhibitors | 41 | 4756 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.36, 0.46] |
| 4.3 NS5B inhibitors (NPI) | 7 | 906 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.36, 0.90] |
| 4.4 NS5B inhibitors (NNPI) | 2 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.43, 1.40] |
| 4.5 NS5A inhibitors | 9 | 686 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.49, 0.69] |
| 4.6 VPU‐ion channel inhibitors | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 0.27 [0.03, 2.51] |
| 4.7 Mixed | 1 | 705 | Risk Ratio (M‐H, Random, 95% CI) | 0.01 [0.00, 0.02] |
| 5 Without sustained virological response ‐ according to HIV‐infection Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 5.1 With HIV‐infection | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 Without HIV‐infection | 58 | 6726 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 5.3 Mixed (with and without HIV‐infection) | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.4 Unclear | 3 | 389 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.35, 0.72] |
| 6 Without sustained virological response ‐ according to comorbidity Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 6.1 With comorbidity | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.2 Without comorbidity | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 6.3 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Without sustained virological response ‐ according to viral genotype Show forest plot | 58 | 7098 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.36, 0.51] |
| 7.1 Genotype 1 | 54 | 5984 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.37, 0.50] |
| 7.2 Genotype 2 | 3 | 185 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 3.21] |
| 7.3 Genotype 3 | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.43, 1.43] |
| 7.4 Genotype 4 | 5 | 226 | Risk Ratio (M‐H, Random, 95% CI) | 0.10 [0.02, 0.68] |
| 7.5 Genotype 6 | 1 | 49 | Risk Ratio (M‐H, Random, 95% CI) | 0.01 [0.00, 0.20] |
| 7.6 Mixed | 2 | 574 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.52, 1.62] |
| 8 Without sustained virological response ‐ according to human genotype (IL28b) Show forest plot | 58 | 6745 | Risk Ratio (M‐H, Random, 95% CI) | 0.46 [0.40, 0.54] |
| 8.1 IL28b (CC) | 25 | 1444 | Risk Ratio (M‐H, Random, 95% CI) | 0.42 [0.29, 0.61] |
| 8.2 IL28B (CT) | 10 | 1304 | Risk Ratio (M‐H, Random, 95% CI) | 0.52 [0.42, 0.66] |
| 8.3 IL28B (TT) | 10 | 359 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.44, 0.67] |
| 8.4 IL28B (CT + TT) | 14 | 1798 | Risk Ratio (M‐H, Random, 95% CI) | 0.37 [0.23, 0.57] |
| 8.5 Unclear | 7 | 147 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.33, 0.68] |
| 8.6 Mixed | 26 | 1693 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.40, 0.63] |
| 9 Without sustained virological response ‐ according to Asian‐region Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 9.1 From Asian region | 10 | 1128 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.28, 0.42] |
| 9.2 Not from Asian region | 42 | 4910 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.43, 0.60] |
| 9.3 Mixed | 7 | 1010 | Risk Ratio (M‐H, Random, 95% CI) | 0.19 [0.03, 1.17] |
| 9.4 Unclear | 2 | 67 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.35, 0.79] |
| 10 Without sustained virological response ‐ according to specific ethnicities Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 10.1 White | 2 | 412 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.15, 0.38] |
| 10.2 Black | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10.3 Hispanic | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10.4 Mixed | 48 | 5384 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.20, 0.27] |
| 10.5 Unclear | 9 | 862 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.20, 0.39] |
| 10.6 Asian | 2 | 457 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.23, 0.63] |
| 11 Without sustained virological response ‐ according to reaching planned sample size Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 11.1 Trials reaching planned sample size | 13 | 3071 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.18, 0.25] |
| 11.2 Trials not reaching planned sample size | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.3 Unclear | 48 | 4044 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.23, 0.33] |
| 12 Without sustained virological response ‐ according to prior treatment Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 12.1 Treatment‐naive | 44 | 4777 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.41, 0.56] |
| 12.2 Treatment‐experienced | 13 | 1274 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.36, 0.69] |
| 12.3 Mixed | 4 | 1064 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.02, 0.96] |
| 12.4 Unclear | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 13 Without sustained virological response ‐ according to interferon Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 13.1 Trials where both groups received interferon | 57 | 6229 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.41, 0.54] |
| 13.2 Trials where neither group received interferon | 2 | 735 | Risk Ratio (M‐H, Random, 95% CI) | 0.06 [0.00, 7.05] |
| 13.3 Trials where only the experimental group received interferon | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 13.4 Trials where only the control group received interferon | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 13.5 Mixed | 2 | 151 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.15, 2.30] |
| 14 Without sustained virological response ‐ according to ribavirin Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 14.1 Trials where both groups received ribavirin | 60 | 6410 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.41, 0.55] |
| 14.2 Trials where neither group received ribavirin | 1 | 705 | Risk Ratio (M‐H, Random, 95% CI) | 0.01 [0.00, 0.02] |
| 14.3 Trials where only the experimental group received ribavirin | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 14.4 Trials where only the control group received ribavirin | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 15 Without sustained virological response ‐ according to chronic kidney disease Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 15.1 With chronic kidney disease | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15.2 Without chronic kidney disease | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15.3 Unclear | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 16 Without sustained virological response ‐ according to cryoglobulinaemia Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 16.1 With cryoglobulinaemia | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16.2 Without cryoglobulinaemia | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16.3 Unclear | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 17 Without sustained virological response ‐ according to DAA group as co‐intervention Show forest plot | 61 | 7115 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.24 [0.22, 0.27] |
| 17.1 Trials where DAA were used as co‐intervention | 3 | 480 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.42 [0.27, 0.66] |
| 17.2 Trials where DAA were not a co‐intervention | 58 | 6635 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.23 [0.21, 0.26] |
| 18 Without sustained virological response ‐ 'Best‐worst case' scenario Show forest plot | 61 | 7294 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.34, 0.49] |
| 19 Without sustained virological response ‐ 'Worst‐best case' scenario Show forest plot | 61 | 7294 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.43, 0.60] |
| 20 Without sustained virological response ‐ according to median dose Show forest plot | 61 | 7115 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 20.1 Over or equal to median dose | 34 | 4154 | Risk Ratio (M‐H, Random, 95% CI) | 0.41 [0.32, 0.53] |
| 20.2 Under median dose | 23 | 2086 | Risk Ratio (M‐H, Random, 95% CI) | 0.46 [0.39, 0.55] |
| 20.3 Not available | 4 | 875 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.26, 1.47] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Hepatitis C‐related morbidity or all‐cause mortality Show forest plot | 9 | 781 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.06, 5.19] |
| 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose Show forest plot | 9 | 781 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.06, 5.19] |
| 2.1 Over or equal to median dose | 6 | 606 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.06, 5.19] |
| 2.2 Under median dose | 3 | 175 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.3 Not available | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Serious adverse events Show forest plot | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Serious adverse events ‐ according to median dose Show forest plot | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Over or equal to median dose | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Under median dose | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Not available | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Without sustained virological response Show forest plot | 5 | 642 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.12, 0.32] |
| 6 Without sustained virological response ‐ according to median dose Show forest plot | 5 | 642 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.12, 0.32] |
| 6.1 Over or equal to median dose | 4 | 537 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.18 [0.11, 0.32] |
| 6.2 Under median dose | 1 | 105 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.07, 0.99] |
| 6.3 Not available | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Hepatitis C‐related morbidity or all‐cause mortality Show forest plot | 95 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Trials assessing DAAs on or on the way to the market | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Trials assessing DAAs withdrawn from market | 22 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Trials using other medical intervention as control group | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Trials using other medical intervention as experimental group | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ drugs not discontinued | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.1 Trials assessing discontinued drugs | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.2 Trials assessing drugs still used | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Hepatitis C‐related morbidity or all‐cause mortality ‐ bias risk | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.1 Trials with a high risk of bias | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 Trials with a low risk of bias | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to type of DAA | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.1 ABT‐072 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 ACH‐2684 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.3 Alisporivir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.4 ALS‐2200 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.5 Asunaprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.6 Balapiravir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.7 Beclabuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.8 BILB‐1941 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.9 BIT‐225 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.10 Boceprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.11 Ciluprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.12 Daclatasvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.13 Danoprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.14 Dasabuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.15 Deleobuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.16 Faldaprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.17 Filibuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.18 Grazoprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.19 GS‐6620 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.20 GS‐9256 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.21 GS‐9451 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.22 GS‐9669 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.23 GS‐9851 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.24 GS‐9857 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.25 GSK2336805 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.26 GSK2878175 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.27 IDX‐184 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.28 INX‐08189 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.29 Ledispasvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.30 Mericitabine | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.31 Narlaprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.32 Nesbuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.33 Odalasavir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.34 Ombitasvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.35 Paritaprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.36 PHX1766 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.37 PPI‐461 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.38 PSI‐352938 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.39 Samatasvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.40 Setrobuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.41 Simeprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.42 Sofosbuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.43 Sovaprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.44 Tegobuvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.45 Telaprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.46 Valopicitabine | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.47 Vaniprevir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.48 VCH‐759 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.49 VCH‐916 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.50 Velpatasvir | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.51 VX‐222 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.52 Mixed | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to group of DAA | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.1 Cyclophilin | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 NS3/NS4A inhibitors | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.3 NS5B inhibitors (NPI) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.4 NS5B inhibitors (NNPI) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.5 NS5A inhibitors | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.6 VPU‐ion channel inhibitors | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.7 Mixed | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to HIV‐infection | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.1 With HIV‐infection | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.2 Without HIV‐infection | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.3 Mixed (with and without HIV‐infection) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.4 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to comorbidity | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7.1 With comorbidity | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7.2 Without comorbidity | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7.3 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to viral genotype | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.1 Genotype 1 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.2 Genotype 2 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.3 Genotype 3 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.4 Genotype 4 | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.5 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to human genotype (IL28b) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.1 IL28b (CC) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.2 IL28B (CT) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.3 IL28B (TT) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.4 IL28B (CT + TT) | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.5 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to Asian‐region | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10.1 From Asian region | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10.2 Not from Asian region | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10.3 Mixed | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 10.4 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to specific ethnicities | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.1 White | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.2 Black | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.3 Hispanic | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.4 Mixed | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.5 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to reaching planned sample size | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12.1 Trials reaching planned sample size | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12.2 Trials not reaching planned sample size | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12.3 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to prior treatment | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13.1 Treatment‐naive | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13.2 Treatment‐experienced | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13.3 Mixed | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 13.4 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to interferon | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14.1 Trials where both groups received interferon | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14.2 Trials where neither group received interferon | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14.3 Trials where only the experimental group received interferon | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 14.4 Trials where only the control group received interferon | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to ribavirin | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15.1 Trials where both groups received ribavirin | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15.2 Trials where neither group received ribavirin | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15.3 Trials where only the experimental group received ribavirin | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 15.4 Trials where only the control group received ribavirin | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to chronic kidney disease | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16.1 With chronic kidney disease | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16.2 Without chronic kidney disease | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 16.3 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 17 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to cryoglobulinaemia | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 17.1 With cryoglobulinaemia | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 17.2 Without cryoglobulinaemia | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 17.3 Unclear | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 18 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to DAA group as co‐intervention | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 18.1 Trials where DAA were used as co‐intervention | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 18.2 Trials where DAA were not a co‐intervention | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Serious adverse events Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Trials assessing DAAs on or on the way to the market | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Trials assessing DAAs withdrawn from market | 62 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Trials using other medical intervention as control group | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Trials using other medical intervention as experimental group | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Serious adverse events ‐ bias risk Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Trials with a high risk of bias | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Trials with a low risk of bias | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Serious adverse events ‐ according to type of DAA Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1 ABT‐072 | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 ACH‐2684 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Alisporivir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 ALS‐2200 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 Asunaprevir | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.6 Balapiravir | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.7 Beclabuvir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.8 BILB‐1941 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.9 BIT‐225 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.10 Boceprevir | 13 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.11 Ciluprevir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.12 Daclatasvir | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.13 Danoprevir | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.14 Dasabuvir | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.15 Deleobuvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.16 Faldaprevir | 13 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.17 Filibuvir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.18 Grazoprevir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.19 GS‐6620 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.20 GS‐9256 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.21 GS‐9451 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.22 GS‐9669 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.23 GS‐9851 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.24 GS‐9857 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.25 GSK2336805 | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.26 GSK2878175 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.27 IDX‐184 | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.28 INX‐08189 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.29 Ledispasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.30 Mericitabine | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.31 Narlaprevir | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.32 Nesbuvir | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.33 Odalasavir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.34 Ombitasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.35 Paritaprevir | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.36 PHX1766 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.37 PPI‐461 | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.38 PSI‐352938 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.39 Samatasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.40 Setrobuvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.41 Simeprevir | 19 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.42 Sofosbuvir | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.43 Sovaprevir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.44 Tegobuvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.45 Telaprevir | 13 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.46 Valopicitabine | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.47 Vaniprevir | 10 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.48 VCH‐759 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.49 VCH‐916 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.50 Velpatasvir | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.51 VX‐222 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.52 Mixed | 8 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Serious adverse events ‐ according to group of DAA Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Cyclophilin | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 NS3/NS4A inhibitors | 92 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 NS5B inhibitors (NPI) | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 NS5B inhibitors (NNPI) | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 NS5A inhibitors | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 VPU‐ion channel inhibitors | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.7 Mixed | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Serious adverse events ‐ according to HIV‐infection Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5.1 With HIV‐infection | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Without HIV‐infection | 154 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Mixed (with and without HIV‐infection) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 Unclear | 11 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Serious adverse events ‐ according to comorbidity Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1 With comorbidity | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Without comorbidity | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Unclear | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Serious adverse events ‐ according to viral genotype Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1 Genotype 1 | 138 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Genotype 2 | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Genotype 3 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Genotype 4 | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 Mixed | 26 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Serious adverse events ‐ according to human genotype (IL28b) Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1 IL28b (CC) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 IL28B (CT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 IL28B (TT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 IL28B (CT + TT) | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.5 Unclear | 79 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.6 Mixed IL28b | 88 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Serious adverse events ‐ according to Asian‐region Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1 From Asian region | 12 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 Not from Asian region | 119 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 Mixed | 31 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.4 Unclear | 5 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Serious adverse events ‐ according to specific ethnicities Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 10.1 White | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 Black | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 Hispanic | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.4 Mixed | 133 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.5 Unclear | 31 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Serious adverse events ‐ according to reaching planned sample size | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 11.1 Trials reaching planned sample size | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 Trials not reaching planned sample size | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 Unclear | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Serious adverse events ‐ according to prior treatment Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.1 Treatment‐naive | 122 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 Treatment‐experienced | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 Mixed | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.4 Unclear | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Serious adverse events ‐ according to interferon Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1 Trials where both groups received interferon | 126 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.2 Trials where neither group received interferon | 40 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.3 Trials where only the experimental group received interferon | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.4 Trials where only the control group received interferon | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Serious adverse events ‐ according to ribavirin Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 14.1 Trials where both groups received ribavirin | 127 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.2 Trials where neither group received ribavirin | 37 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.3 Trials where only the experimental group received ribavirin | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.4 Trials where only the control group received ribavirin | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15 Serious adverse events ‐ according to chronic kidney disease Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 15.1 With chronic kidney disease | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.2 Without chronic kidney disease | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.3 Unclear | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16 Serious adverse events ‐ according to cryoglobulinaemia Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 16.1 With cryoglobulinaemia | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.2 Without cryoglobulinaemia | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 16.3 Unclear | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17 Serious adverse events ‐ according to DAA group as co‐intervention Show forest plot | 167 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 17.1 Trials where DAA were used as co‐intervention | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.2 Trials where DAA were not a co‐intervention | 165 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Without sustained virological response Show forest plot | 107 | 17101 | Risk Ratio (M‐H, Random, 95% CI) | 0.53 [0.48, 0.59] |
| 1.1 Trials assessing DAAs on or on the way to the market | 60 | 6886 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.37, 0.52] |
| 1.2 Trials assessing DAAs withdrawn from market | 43 | 9075 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.55, 0.69] |
| 1.3 Trials using other medical intervention as control group | 3 | 862 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.36, 1.82] |
| 1.4 Trials using other medical intervention as experimental group | 1 | 278 | Risk Ratio (M‐H, Random, 95% CI) | 0.23 [0.17, 0.29] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 SF‐36 physical score Show forest plot | 1 | 215 | Mean Difference (IV, Fixed, 95% CI) | ‐1.17 [‐3.65, 1.31] |
| 2 SF‐36 mental score Show forest plot | 1 | 215 | Mean Difference (IV, Fixed, 95% CI) | 1.36 [‐1.53, 4.25] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Hepatitis C‐related morbidity or all‐cause mortality Show forest plot | 14 | 666 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.06, 26.65] |
| 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose Show forest plot | 14 | 666 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.06, 26.65] |
| 2.1 Over or equal to median dose | 7 | 374 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.2 Under median dose | 7 | 292 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.25 [0.06, 26.65] |
| 2.3 Not available | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Serious adverse events Show forest plot | 13 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Serious adverse events ‐ according to median dose Show forest plot | 14 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Over or equal to median dose | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Under median dose | 8 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Not available | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Without sustained virological response Show forest plot | 7 | 619 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.27, 0.59] |
| 6 Without sustained virological response ‐ according to median dose Show forest plot | 7 | 619 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.27, 0.59] |
| 6.1 Over or equal to median dose | 4 | 360 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.26, 0.70] |
| 6.2 Under median dose | 3 | 259 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.19, 0.68] |
| 6.3 Not available | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Hepatitis C‐related morbidity or all‐cause mortality Show forest plot | 14 | 1589 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.08, 2.96] |
| 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose Show forest plot | 14 | 1589 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.08, 2.96] |
| 2.1 Over or equal to median dose | 4 | 441 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.03, 8.21] |
| 2.2 Under median dose | 8 | 705 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.41 [0.01, 12.22] |
| 2.3 Not available | 2 | 443 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.02, 13.62] |
| 3 Serious adverse events Show forest plot | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Serious adverse events ‐ according to median dose Show forest plot | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Over or equal to median dose | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Under median dose | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Not available | 2 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Without sustained virological response Show forest plot | 19 | 2898 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.19, 0.27] |
| 6 Without sustained virological response ‐ according to median dose Show forest plot | 19 | 2898 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.19, 0.27] |
| 6.1 Over or equal to median dose | 9 | 1765 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.25 [0.20, 0.32] |
| 6.2 Under median dose | 8 | 696 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.13, 0.29] |
| 6.3 Not available | 2 | 437 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.13 [0.07, 0.24] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Hepatitis C‐related morbidity or all‐cause mortality Show forest plot | 9 | 379 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.03, 18.90] |
| 2 Hepatitis C‐related morbidity or all‐cause mortality ‐ according to dose Show forest plot | 9 | 379 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.03, 18.90] |
| 2.1 Over or equal to median dose | 6 | 313 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.76 [0.03, 18.90] |
| 2.2 Under median dose | 3 | 66 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.3 Not available | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Serious adverse events Show forest plot | 10 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Serious adverse events ‐ according to median dose Show forest plot | 10 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Over or equal to median dose | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Under median dose | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Not available | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Without sustained virological response Show forest plot | 9 | 333 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.12 [0.06, 0.22] |
| 6 Without sustained virological response ‐ according to median dose Show forest plot | 9 | 333 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.12 [0.06, 0.22] |
| 6.1 Over or equal to median dose | 6 | 280 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.10 [0.05, 0.20] |
| 6.2 Under median dose | 3 | 53 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.26 [0.06, 1.04] |
| 6.3 Not available | 0 | 0 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Without significant reductions in ALT/AST serum levels Show forest plot | 11 | 2099 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.68, 0.92] |
| 2 Without significant reductions in ALT/AST serum levels ‐ according to DAA status Show forest plot | 11 | 2099 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.68, 0.92] |
| 2.1 Trials assessing DAAs on or on the way to the market | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2.2 Trials assessing DAAs withdrawn from market | 11 | 2099 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.68, 0.92] |
| 3 Without significant reductions in ALT/AST serum levels ‐ according to type of drug Show forest plot | 11 | 2099 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.68, 0.92] |
| 3.1 Faldaprevir | 8 | 2019 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.69, 0.96] |
| 3.2 Balaparavir | 3 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 0.61 [0.41, 0.92] |