Umeclidinium bromida berbanding plasebo untuk orang yang mempunyai penyakit paru‐paru obstruktif kronik (COPD)
Abstract
Background
People with chronic obstructive pulmonary disease (COPD) have poor quality of life, reduced survival, and accelerated decline in lung function, especially associated with acute exacerbations, leading to high healthcare costs. Long‐acting bronchodilators are the mainstay of treatment for symptomatic improvement, and umeclidinium is one of the new long‐acting muscarinic antagonists approved for treatment of patients with stable COPD.
Objectives
To assess the efficacy and safety of umeclidinium bromide versus placebo for people with stable COPD.
Search methods
We searched the Cochrane Airways Group Specialised Register (CAGR), ClinicalTrials.gov, the World Health Organization (WHO) trials portal, and the GlaxoSmithKline (GSK) Clinical Study Register, using prespecified terms, as well as the reference lists of all identified studies. Searches are current to April 2017.
Selection criteria
We included randomised controlled trials (RCTs) of parallel design comparing umeclidinium bromide versus placebo in people with COPD, for at least 12 weeks.
Data collection and analysis
We used standard Cochrane methodological procedures. If we noted significant heterogeneity in the meta‐analyses, we subgrouped studies by umeclidinium dose.
Main results
We included four studies of 12 to 52 weeks' duration, involving 3798 participants with COPD. Mean age of participants ranged from 60.1 to 64.6 years; most were males with baseline mean smoking pack‐years of 39.2 to 52.3. They had moderate to severe COPD and baseline mean post‐bronchodilator forced expiratory volume in one second (FEV1) ranging from 44.5% to 55.1% of predicted normal. As all studies were systematically conducted according to prespecified protocols, we assessed risk of selection, performance, detection, attrition, and reporting biases as low.
Compared with those given placebo, participants in the umeclidinium group had a lesser likelihood of developing moderate exacerbations requiring a short course of steroids, antibiotics, or both (odds ratio (OR) 0.61, 95% confidence interval (CI) 0.46 to 0.80; four studies, N = 1922; GRADE: high), but not specifically requiring hospitalisations due to severe exacerbations (OR 0.86, 95% CI 0.25 to 2.92; four studies, N = 1922, GRADE: low). The number needed to treat for an additional beneficial outcome (NNTB) to prevent an acute exacerbation requiring steroids, antibiotics, or both was 18 (95% CI 13 to 37). Quality of life was better in the umeclidinium group (mean difference (MD) ‐4.79, 95% CI ‐8.84 to ‐0.75; three studies, N = 1119), and these participants had a significantly higher chance of achieving a minimal clinically important difference of at least four units in St George's Respiratory Questionnaire (SGRQ) total score compared with those in the placebo group (OR 1.45, 95% CI 1.16 to 1.82; three studies, N = 1397; GRADE: moderate). The NNTB to achieve one person with a clinically meaningful improvement was 11 (95% CI 7 to 29). The likelihood of all‐cause mortality, non‐fatal serious adverse events (OR 1.33; 95% CI 0.89 to 2.00; four studies, N = 1922, GRADE: moderate), and adverse events (OR 1.06, 95% CI 0.85 to 1.31; four studies, N = 1922; GRADE: moderate) did not differ between umeclidinium and placebo groups. The umeclidinium group demonstrated significantly greater improvement in change from baseline in trough FEV1 compared with the placebo group (MD 0.14, 95% CI 0.12 to 0.17; four studies, N = 1381; GRADE: high). Symptomatic improvement was more likely in the umeclidinium group than in the placebo group, as determined by Transitional Dyspnoea Index (TDI) focal score (MD 0.76, 95% CI 0.43 to 1.09; three studies, N = 1193), and the chance of achieving a minimal clinically important difference of at least one unit improvement was significantly higher with umeclidinium than with placebo (OR 1.71, 95% CI 1.37 to 2.15; three studies, N = 1141; GRADE: high). The NNTB to attain one person with clinically important symptomatic improvement was 8 (95% CI 5 to 14). The likelihood of rescue medication usage (change from baseline in the number of puffs per day) was significantly less for the umeclidinium group than for the placebo group (MD ‐0.45, 95% CI ‐0.76 to ‐0.14; four studies, N = 1531).
Authors' conclusions
Umeclidinium reduced acute exacerbations requiring steroids, antibiotics, or both, although no evidence suggests that it decreased the risk of hospital admission due to exacerbations. Moreover, umeclidinium demonstrated significant improvement in quality of life, lung function, and symptoms, along with lesser use of rescue medications. Studies reported no differences in adverse events, non‐fatal serious adverse events, or mortality between umeclidinium and placebo groups; however, larger studies would yield a more precise estimate for these outcomes.
PICO
Ringkasan bahasa mudah
Adakah penyedut yang mengandungi umeclidinium bromida berkesan dan selamat untuk orang yang mempunyai COPD?
Soalan ulasan
Penyelidik mengulas keberkesanan dan keselamatan penyedut umeclidinium berbanding penyedut plasebo pada orang yang mempunyai penyakit paru‐paru obstruktif kronik (COPD).
Latar belakang
Orang dengan COPD sering sesak nafas dan mempunyai kualiti kehidupan yang rendah. Gejala boleh memburukkan lagi semasa serangan, meningkatkan perbelanjaan penjagaan kesihatan dan mengurangkan jangka hayat. Ubat‐ubatan yang meluaskan saluran pernafasan (bronkodilator), yang bertindak selama 12 hingga 24 jam, adalah rawatan yang biasanya diberikan untuk mengurangkan gejala. Umeclidinium adalah rawatan baru seperti ini. Penyelidik ingin mengetahui sama ada umeclidinium lebih baik atau lebih buruk daripada plasebo.
Ciri‐ciri kajian
Penyelidik menyertakan empat kajian yang melibatkan 3798 orang dengan COPD. Kebanyakannya adalah lelaki berusia 60‐an yang sederhana dan perokok berat. Apabila mereka memulakan rawatan, mereka mempunyai gejala COPD yang sederhana hingga teruk. Kajian terdiri dari tiga bulan hingga satu tahun. Kajian telah direka dengan baik dan dibiayai oleh pengeluar ubat. Tiada orang dalam kajian ini atau orang yang melakukan penyelidikan mengenali peserta rawatan yang mana mereka dapati. Orang dalam kajian mengambil sama ada umeclidinium atau plasebo melalui penyedut setiap pagi.
Kesimpulan ulasan ini adalah terkini hingga April 2017.
Keputusan utama
Penyelidik menentukan jumlah orang yang mempunyai serangan sederhana. Serangan sederhana sedang dirawat dengan steroid oral jangka pendek atau antibiotik, atau kedua‐duanya. Orang yang mengambil umeclidinium adalah kurang berkemungkinan daripada mereka yang diberi plasebo untuk mendapatkan serangan sederhana. Lapan belas orang dengan COPD perlu dirawat dengan umeclidinium untuk mengelakkan salah satu daripada serangan‐serangan ini.
Orang yang mengambil umeclidinium mungkin mempunyai kualiti hidup yang lebih baik, dan fungsi paru‐paru mereka lebih baik. Orang yang mengambil umeclidinium kurang sesak nafas dan mengambil sedikit sedutan penyedut pernafasan mereka.
Keputusan menunjukkan sedikit atau tiada perbezaan dengan umeclidinium dalam hasil yang lain, seperti risiko kematian semasa tempoh kajian, kesan sampingan, atau keperluan untuk dimasukkan ke hospital kerana suar‐suar yang teruk.
Kualiti bukti
Penyelidik yakin bahawa penyedut umeclidinium lebih cenderung daripada penyedut "dummy" untuk mengurangkan serangan‐serangan sederhana dan memperbaiki gejala dan fungsi paru‐paru. Walau bagaimanapun, penyelidik kurang pasti mengenai kesan umeclidinium terhadap kualiti hidup, kesan sampingan, dan kesan sampingan yang serius. Penyelidik mempunyai keyakinan yang terhad dari segi kemasukan hospital disebabkan oleh serangan, tetapi ini adalah peristiwa yang jarang berlaku.
Kesimpulan
Pada orang yang mempunyai COPD, penyedut umeclidinium meningkatkan gejala, fungsi paru‐paru, dan kualiti hidup berbanding dengan penyedut "dummy". Mereka juga mengurangkan penggunaan kelegaan ubat‐ubatan yang cepat dan mengurangkan serangan yang memerlukan ubat tambahan. Namun demikian, tiada bukti yang meyakinkan menunjukkan penyedut "dummy" dari segi kemasukan ke hospital, kesan sampingan, kesan sampingan yang serius atau kematian.
Authors' conclusions
Summary of findings
| Umeclidinium bromide vs placebo for stable chronic obstructive pulmonary disease | ||||||
| Patient or population: people with chronic obstructive pulmonary disease (COPD) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Risk with placebo | Risk with umeclidinium bromide | |||||
| Number of participants with exacerbations requiring steroids, antibiotics, or both | 157 per 1000 | 102 per 1000 | OR 0.61 | 1922 | ⊕⊕⊕⊕ | |
| Quality of life: number of participants with ≥ 4 units improvement in SGRQ total score | 342 per 1000 | 429 per 1000 | OR 1.45 | 1397 | ⊕⊕⊕⊝ | Mean quality of life: change from baseline in SGRQ total score was 4.79 lower (8.84 lower to 0.75 lower) in umeclidinium group (1119 participants, 3 RCTs) |
| Non‐fatal serious adverse events | 51 per 1000 | 66 per 1000 | OR 1.33 | 1922 | ⊕⊕⊕⊝ | Larger studies may help refine this estimate |
| Number of participants with hospital admissions due to COPD exacerbation | 20 per 1000 | 18 per 1000 | OR 0.86 | 1922 | ⊕⊕⊝⊝ | Few events, so larger studies may help refine this estimate |
| Number of participants with ≥ 1 unit improvement in TDI focal score | 336 per 1000 | 464 per 1000 | OR 1.71 | 1441 | ⊕⊕⊕⊕ | Mean improvement in TDI focal score change from baseline was 0.76 higher (0.43 higher to 1.09 higher) in umeclidinium group (1193 participants, 3 RCTs) |
| Change from baseline in trough FEV1 (L) | Mean change from baseline in trough FEV1 (L) across control groups ranged from 0.123 to 0.139 | Mean change from baseline in trough FEV1 (L) in the intervention group was 0.14 higher (0.12 higher to 0.17 higher) | ‐ | 1381 | ⊕⊕⊕⊕ | |
| Adverse events (not including serious adverse events) | 239 per 1000 | 250 per 1000 | OR 1.06 | 1922 | ⊕⊕⊕⊝ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on assumed risk in the comparison group and relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| a‐1 for inconsistency: unexplained significant heterogeneity b‐1 for imprecision: the CI includes non‐appreciable benefit and potential harm c‐2 for imprecision: the CI includes both appreciable benefit and harm | ||||||
Background
Description of the condition
Chronic obstructive pulmonary disease (COPD) is a major cause of morbidity and mortality worldwide (GOLD 2017). COPD is characterised by persistent airflow limitation that usually is progressive, is not fully reversible, and is associated with enhanced inflammatory response of the airways and destruction of lung parenchyma (Criner 2015; GOLD 2017). COPD is the third leading cause of death in the United States (USA) and the fourth in Canada, with 12.7 million adults in the USA estimated to have COPD in 2011, and nearly 24 million adults having abnormal lung function (Criner 2015). It is estimated that COPD accounted for more than 3 million deaths worldwide in 2012, corresponding to 6% of all deaths (WHO 2015), and for approximately 30,000 deaths each year in the United Kingdom (UK) (NICE 2010). Owing to continued exposure to tobacco smoke and environmental pollution, COPD is predicted to be the third leading cause of death globally by 2020 (Chapman 2006; GOLD 2017; Raherison 2009). Existing incidence and prevalence data on COPD show wide variation due to differences in survey methods, diagnostic criteria, analytical approaches, under‐reporting, and under‐diagnosis (GOLD 2017; Raluy‐Callado 2015; Rennard 2006; WHO 2015).
Active smoking is the major risk factor for COPD, and more than 1.1 billion smokers worldwide are at risk (Bauer 2013). The prevalence of COPD among heavy smokers is as high as 50% according to current criteria (Mannino 2007). However, COPD is also reported among never‐smokers at a prevalence of 3% to 11% (GOLD 2017). Passive smoking or environmental tobacco smoke; exposure to biomass fuels, chemicals, and occupational elements; and indoor and outdoor air pollution are important risk factors for COPD in non‐smokers (GOLD 2017; Hagstad 2014; Hogg 2009; Rivera 2008; TSANZ 2014; WHO 2015).
A mixture of small airways disease (obstructive bronchiolitis) and parenchymal destruction (emphysema) results in the chronic airflow limitation of COPD (GOLD 2017). Inflammatory cell infiltration of alveoli and lung parenchyma leads to destruction and enlargement of air spaces, thus reducing the elastic pressure that generates expiratory flow (Cosio 2009; GOLD 2017). Onset of symptoms usually occurs when forced expiratory volume at one second (FEV1) has fallen to approximately 50% of the predicted normal value (Sutherland 2004).
Acute exacerbations of COPD are a major cause of morbidity and mortality, which can occur at any stage, but are more frequently seen in patients with severe airflow limitation (Marchetti 2013). The main precipitants of exacerbations are respiratory viral and bacterial infections. Exacerbations are associated with worsened quality of life, increased healthcare costs, accelerated decline in lung function, and reduced survival (Bauer 2013; Marchetti 2013).
Smoking cessation and long‐term oxygen therapy for severe hypoxaemia are the only measures that improve survival in patients with stable COPD (GOLD 2017). In addition to aiding smoking cessation, management of stable COPD is aimed at reducing symptoms and exacerbations, and improving quality of life and exercise capacity (ATS/ERS 2011; GOLD 2017; Sutherland 2004; TSANZ 2014). Long‐acting inhaled bronchodilators ‐ either long‐acting beta2‐agonists (LABAs) or long‐acting muscarinic antagonists (LAMAs) ‐ serve as first‐line maintenance therapy for symptomatic treatment of people with moderate to severe stable COPD (GOLD 2017; NICE 2010).
Description of the intervention
Umeclidinium bromide is a new quinuclidine‐based quaternary ammonium derivative that inhibits cholinergically mediated bronchoconstriction through its potent competitive antagonistic activity at the M3 receptor subtype in airway smooth muscles (Decramer 2013a). It was approved by the US Food and Drug Administration (FDA) on 30 April 2014 for use as long‐term, once‐daily maintenance treatment of airflow obstruction in patients with COPD (FDA 2014). It is marketed as Incruse Ellipta by GlaxoSmithKline in an inhaler, which contains a double‐foil blister strip of powder formulation, with each blister containing 62.5 µg of umeclidinium. The FDA‐approved dosage is 62.5 μg once daily by inhalation via a multi‐dose dry powder inhaler (DPI). The 30‐day cost of Incruse Ellipta is £27.50 (Incruse Ellipta).
According to Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines, a LAMA such as umeclidinium bromide monotherapy is recommended as an alternative in patients with mild, stable COPD (Group A, fewer symptoms/low risk) or as first‐line treatment in Group B patients (more symptoms/low risk). In people with more severe disease, umeclidinium bromide should be used in combination with other treatments as first choice for Group C (fewer symptoms/high risk) and Group D (more symptoms/high risk) patients (GOLD 2017). A fixed‐dose combination of umeclidinium bromide/vilanterol 62.5/25 μg once daily (Anoro Ellipta) has demonstrated greater improvement in FEV1 than is seen with monotherapies (Donohue 2013a; Spyratos 2015) and was approved in December 2013 for long‐term maintenance treatment of patients with moderate to severe stable COPD (FDA 2013). Owing to growing interest in triple therapy with LABA, LAMA, and inhaled corticosteroids, a fixed‐dose combination of umeclidinium/vilanterol/fluticasone furoate 62.5/25/100 µg is currently under clinical development (Cazzola 2014a).
How the intervention might work
Acetylcholine‐mediated airway obstruction plays an important role in the pathogenesis of COPD. Cholinergic parasympathetic nerves contribute to increased tone of airway smooth muscles, and this primary reversible component of airway limitation is sensitive to muscarinic antagonists. Effects of acetylcholine are mediated by five G‐protein‐coupled muscarinic receptors (M1 to M5). However, only M1, M2, and M3 are expressed in human airways; M3 receptors, which are the predominant receptors on airway smooth muscles, submucosal mucous glands, and vascular endothelium, act upon acetylcholine release from the parasympathetic nerves. M2 receptors are located presynaptically and mediate feedback inhibition of acetylcholine release, whereas M1 receptors are expressed in the parasympathetic ganglia and facilitate further neurotransmission (Cazzola 2014b; Manickam 2014; Prakash 2013).
Umeclidinium bromide is a LAMA that inhibits the action of acetylcholine, mainly at M3 muscarinic receptors of airway smooth muscles, causing a decrease in airway resistance. Umeclidinium bromide has fast onset of action (time to maximal plasma concentration 5 to 15 minutes) and slow functional reversibility at M3 receptors, resulting in duration of action longer than 24 hours (Spyratos 2015). Slower dissociation from the M3 receptors (t1/2 = 82 minutes) compared with the M2 subtype (t1/2 = 9 minutes) results in more effective bronchodilator action, with fewer M2‐mediated cardiac side effects (Kelly 2014; Manickam 2014; Salmon 2013).
In placebo‐controlled studies, umeclidinium has shown significant improvement in lung function (forced expiratory volume in one second (FEV1) and forced vital capacity (FVC)) both in healthy volunteers and in patients with moderate to very severe COPD. It has also improved health‐related quality of life and has decreased exacerbations in patients with moderate to severe COPD (Manickam 2014).
Tiotropium bromide, the first LAMA approved for use as once‐daily maintenance therapy in patients with stable COPD, is widely used. It has been shown to be associated with significant improvement in quality of life and reduction in exacerbations, including severe exacerbations leading to hospitalisation. However, it does not decrease hospitalisation for any cause nor mortality (Karner 2012). To date, we are not aware of any trials that have directly compared the licenced dose of umeclidinium monotherapy versus those of other LAMAs, including tiotropium. In a 24‐week phase 3 trial, the fixed‐dose combination of umeclidinium bromide/vilanterol was shown to be associated with improved health‐related quality of life and reduced requirement for use of rescue medication compared with tiotropium (Maleki‐Yazdi 2014a). However, a systematic review on combined umeclidinium/vilanterol reported no significant differences between umeclidinium/vilanterol and tiotropium with respect to dyspnoea, health status, or risk of exacerbation (Rodrigo 2015).
Why it is important to do this review
Several clinical trials have compared umeclidinium bromide versus placebo for treatment of patients with COPD. These trials have demonstrated a long‐lasting bronchodilator effect with reduction in symptoms and improvement in quality of life, as well as a favourable safety profile, in an agent with a once‐daily dose. For patients or for clinicians facing patients, a systematic summary of the effectiveness and safety of umeclidinium would assist in the decision of whether to use this medicine.
Objectives
To assess the efficacy and safety of umeclidinium bromide versus placebo for people with stable COPD.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) of parallel‐group design comparing umeclidinium bromide with placebo; we included studies reported as full text, those published as abstract only, and unpublished data. The minimum duration of trials was 12 weeks. We excluded cross‐over and cluster‐randomised trials.
Types of participants
We included adults over 18 years of age with a diagnosis of COPD according to the criteria of the Global Initiative for Chronic Obstructive Lung Disease (GOLD) (GOLD 2017), the American Thoracic Society (ATS), the European Respiratory Society (ERS) (ATS/ERS 2011), the Thoracic Society of Australia and New Zealand (TSANZ) (TSANZ 2014), the UK National Institute for Health and Clinical Excellence (NICE) (NICE 2010), or the World Health Organization (WHO). Participants had evidence of airway obstruction (post‐bronchodilator FEV1/FVC ratio < 70%) with symptoms of dyspnoea, chronic cough, or sputum production with or without a history of smoking. We included participants with stable COPD who did not have recent exacerbations requiring a short course of oral steroids, antibiotics, or both, and who were taking stable doses of medications for at least four weeks before screening. We excluded participants with the following comorbidities/characteristics: bronchial asthma, bronchiectasis, cystic fibrosis, or other chronic lung diseases.
Types of interventions
We included trials consisting of umeclidinium bromide and placebo arms that compared umeclidinium bromide versus placebo.
We allowed the following co‐interventions, provided they were not part of the randomised treatment: salbutamol or albuterol as rescue medication; oral sustained‐release theophylline, inhaled corticosteroids, or systemic corticosteroids (oral or parenteral) at stable doses; and oxygen therapy given for less than 15 hours per day.
Types of outcome measures
Primary outcomes
-
Exacerbations requiring a short course of an oral steroid or antibiotic, or both
-
Quality of life as measured by a validated scale: St George’s Respiratory Questionnaire (SGRQ) or the Chronic Respiratory Disease Questionnaire (CRQ)
-
Non‐fatal serious adverse events
Secondary outcomes
-
Mortality (all‐cause and respiratory)
-
Hospital admissions due to exacerbations
-
Improvement in symptoms as measured by a validated scale: Transitional Dyspnoea Index (TDI) or EXACT‐Respiratory Symptoms (E‐RS) score
-
Change in lung function
-
Adverse events/side effects
-
Use of rescue medications
Reporting by trial authors of one or more of the outcomes listed here is not an inclusion criterion for this review.
Search methods for identification of studies
Electronic searches
We identified studies from the Cochrane Airways Trials Register, which is maintained by the Information Specialist for the Group. The Cochrane Airways Trials Register contains studies identified from several sources.
-
Monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL), through the Cochrane Register of Studies Online (crso.cochrane.org).
-
Weekly searches of MEDLINE Ovid SP 1946 to date.
-
Weekly searches of Embase Ovid SP 1974 to date.
-
Monthly searches of PsycINFO Ovid SP.
-
Monthly searches of the Cumulative Index to Nursing and Allied Health Literature (CINAHL) EBSCO.
-
Monthly searches of Allied and Complementary Medicine (AMED) EBSCO.
-
Handsearches of the proceedings of major respiratory conferences.
We identified studies contained in the Trials Register through search strategies based on the scope of the Cochrane Airways Review Group. We have provided in Appendix 1 details of these strategies, as well as a list of handsearched conference proceedings. See Appendix 2 for search terms used to identify studies for this review.
We also searched the following trials registries.
-
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (www.clinicaltrials.gov) (Appendix 3).
-
World Health Organization International Clinical Trials Registry Platform (apps.who.int/trialsearch) (Appendix 4).
We searched the Cochrane Airways Trials Register and additional sources from inception to April 2017, with no restriction on language of publication.
Searching other resources
We checked reference lists of all primary studies and review articles for additional references. We also searched relevant manufacturers' websites and the GlaxoSmithKline (GSK) Clinical Study Register (www.gsk‐clinicalstudyregister.com/) for trial information.
We searched for errata or retractions from included studies published in full text on PubMed (www.ncbi.nlm.nih.gov/pubmed) and reported within the review the date this was done.
Data collection and analysis
Selection of studies
Two review authors (SM and AH) independently screened titles and abstracts for inclusion of all potential studies identified as a result of the search and coded them as 'retrieve' (eligible or potentially eligible/unclear) or 'do not retrieve'. We retrieved the full‐text study reports/publications, and two review authors (HN and AH) independently screened these reports, identified studies for inclusion, and identified and recorded reasons for exclusion of ineligible studies. We resolved disagreements through discussion or, if required, through consultation with a third person (SM). We identified and excluded duplicates and collated multiple reports of the same study, so that each study rather than each report is the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses) flow diagram (Moher 2009) and a Characteristics of excluded studies table.
Data extraction and management
We used a data collection form that had been piloted on at least one study in the review to record study characteristics and outcome data. One review author (AH) extracted the following study characteristics from included studies.
-
Methods: study design, total duration of study, details of any 'run‐in' period, number of study centres and locations, study setting, withdrawals, and date of study.
-
Participants: N, mean age, age range, gender, severity of condition, diagnostic criteria, baseline lung function, smoking history, inclusion criteria, and exclusion criteria.
-
Interventions: intervention, comparison, concomitant medications, and excluded medications.
-
Outcomes: primary and secondary outcomes specified and collected, and time points reported.
-
Notes: funding for trial, and notable conflicts of interest of trial authors.
Two review authors (HN and AH) independently extracted outcome data from included studies and noted in the Characteristics of included studies table if outcome data were not reported in a useable way. We resolved disagreements by consensus or by consultation with a third person (SM). One review author (AH) transferred data into the Review Manager (RevMan 2014) file. We double‐checked that data were entered correctly by comparing data presented in the systematic review against the study reports. A second review author (HN) spot‐checked study characteristics against the trial report for accuracy.
Assessment of risk of bias in included studies
Two review authors (AH and SM) independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved disagreements by discussion or by consultation with another review author (HN). We assessed risk of bias according to the following domains.
-
Random sequence generation.
-
Allocation concealment.
-
Blinding of participants and personnel.
-
Blinding of outcome assessment.
-
Incomplete outcome data.
-
Selective outcome reporting.
-
Other bias.
We graded each potential source of bias as high, low, or unclear, and provided a quote from the study report together with a justification for our judgement in the Risk of bias in included studies table. We summarised risk of bias judgements across different studies for each of the domains listed. We considered blinding separately for different key outcomes when necessary (e.g. for unblinded outcome assessment, risk of bias for all‐cause mortality may be very different than for a patient‐reported pain scale). When information on risk of bias was related to unpublished data or correspondence with a trialist, we noted this in the Risk of bias in included studies table.
When considering treatment effects, we took into account the risk of bias for studies that contributed to that outcome.
Assesment of bias in conducting the systematic review
We conducted the review according to the published protocol and reported deviations from it in the Differences between protocol and review section of the systematic review.
Measures of treatment effect
We analysed dichotomous data as odds ratios and continuous data as mean differences or standardised mean differences. We entered data presented as a scale with a consistent direction of effect.
We undertook meta‐analyses only when meaningful (i.e. when treatments, participants, and the underlying clinical question were similar enough for pooling to make sense).
We narratively described skewed data reported as medians and interquartile ranges.
When multiple trial arms were reported in a single trial, we included only the relevant arms. When two comparisons (e.g. drug A vs placebo and drug B vs placebo) were combined in the same meta‐analysis, we halved the control group to avoid double‐counting.
Unit of analysis issues
We analysed the number of participants instead of the number of events for outcomes that may occur more than once, such as exacerbations, hospital admissions, and adverse events. For exacerbation rates, we analysed the data as rate ratios, transformed them into log rate ratios, and combined them across studies by using the generic inverse variance method.
To prevent unit of analysis errors when entering data from studies with multiple intervention arms, for dichotomous data, we divided up both the number of participants and the number of events according to the number of interventions in the study. For continuous data, we divided only the total number of participants and kept means and standard deviations unchanged. If we needed to combine groups, we summed up both sample sizes and numbers of people with events for dichotomous outcomes. For continuous outcomes, we used the formula described in Table 7.7a in Section 7.7.3.8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to combine means and standard deviations.
Dealing with missing data
We contacted investigators or study sponsors to verify key study characteristics and to obtain missing numerical outcome data when possible (e.g. when a study was identified as abstract only). When this was not possible, and when missing data were thought to introduce serious bias, we performed a sensitivity analysis to explore the impact of including such studies in the overall assessment of results.
Assessment of heterogeneity
We used the I2 statistic to measure heterogeneity among the trials in each analysis. If we identified substantial heterogeneity, we reported it and explored possible causes through prespecified subgroup analysis.
Assessment of reporting biases
If we had been able to pool more than 10 trials, we planned to create and examine a funnel plot to explore possible small‐study and publication biases.
Data synthesis
We used a fixed‐effect model and performed a sensitivity analysis with a random‐effects model.
'Summary of findings' table
We created a 'Summary of findings' table using the following outcomes.
-
Exacerbations requiring a short course of an oral steroid or antibiotic, or both.
-
Quality of life.
-
Non‐fatal serious adverse events.
-
Hospital admissions due to exacerbations.
-
Improvement in symptoms.
-
Change in lung function (trough FEV1).
-
Adverse events.
We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the quality of evidence as it relates to studies that contributed data to the meta‐analyses for prespecified outcomes. We used methods and recommendations described in Section 8.5 and Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) and GRADEproGDT software (GRADEproGDT). We justified all decisions to downgrade or upgrade the quality of studies by using footnotes that provided comments to aid readers' understanding of the review, when necessary.
Subgroup analysis and investigation of heterogeneity
If we found significant heterogeneity, we planned the following subgroup analyses.
-
Dose of umeclidinium (e.g. 62.5 µg, 125 µg).
-
Severity of COPD (e.g. GOLD stage 1, 2, 3, 4 or A, B, C, D).
We planned to use the following outcomes in subgroup analyses.
-
Exacerbations requiring a short course of an oral steroid or antibiotic, or both.
-
Quality of life.
-
Non‐fatal serious adverse events.
-
Improvement in symptoms.
We planned to use the formal test for subgroup interactions provided in Review Manager (RevMan 2014).
Sensitivity analysis
We planned to carry out the following sensitivity analyses.
-
Analyses based on both random‐effects and fixed‐effect models.
-
Repeated meta‐analysis after exclusion of trials with high risk of bias or unclear methodological data.
Results
Description of studies
See Characteristics of included studies, Characteristics of excluded studies, and Characteristics of ongoing studies for complete details.
Results of the search
A search of the Cochrane Airways Group Specialised Register of trials (CAGR) initially performed in August 2015 and updated in September 2016 along with a prepublication search in April 2017 yielded a total of 441 records (344 from CAGR, 97 from ClinicalTrials.gov). From the search of other resources, we identified 259 additional records: 110 from the WHO trials portal, 93 from the GSK Clinical Study Register, 36 from ClinicalTrials.gov, and 20 from reference lists. After removing duplicates, we screened 517 records for eligibility and excluded 296 reports. We then studied the remaining 221 references, retrieving full texts when applicable and contacting the manufacturers for any unpublished trials. From the search, we excluded a total of 60 studies reported in 201 references and recorded the details in Characteristics of excluded studies. Four trials with 17 reports met the inclusion criteria for our review, and we identified one ongoing study (NCT02184611) with two references. One reference reported the results of two trials ‐ an excluded study (Maleki‐Yazdi 2014) and an included study (Donohue 2013). A total of 17 references reported two trials; we included these as references for both trials reported ‐ one for included studies and 16 for excluded studies. We also contacted GlaxoSmithKline to ask about additional studies that they had sponsored and received a prompt reply that all GSK‐sponsored clinical studies were made public in the register along with patient‐level data. For details of the search results, please see Figure 1.

Study flow diagram.
Included studies
Study design and duration
All four trials were randomised, double‐blind, parallel‐group, placebo‐controlled phase 3 studies conducted to assess umeclidinium bromide and placebo as the main intervention or as one of several interventions. Duration of studies ranged from 12 weeks (Trivedi 2014) to 52 weeks (Donohue 2014). Two other trials (Celli 2014; Donohue 2013) were of 24 weeks' duration following a run‐in period of 7 to 14 days. All were multi‐centre trials that were conducted in the USA (Celli 2014; Donohue 2013; Donohue 2014; Trivedi 2014), Canada (Donohue 2013), Europe (Celli 2014; Donohue 2013; Donohue 2014; Trivedi 2014), South Africa (Donohue 2013; Donohue 2014), Japan (Celli 2014; Donohue 2013; Trivedi 2014), Thailand (Donohue 2013), and the Philippines (Celli 2014), between 2011 and 2012.
Participants
Studies randomised a total of 3798 participants. Donohue 2013 was the largest trial, with 1536 participants, and Trivedi 2014 included the least number of participants, with 206. Researchers included in the studies adult patients 40 years of age or older, both male and female, current and former smokers with a smoking history of 10 or more pack‐years. They had an established clinical history of COPD according to ATS/ERS criteria, as well as an airflow limitation with post‐bronchodilator FEV1/FVC ratio < 0.70, FEV1 ≤ 70% of predicted normal, and a score ≥ 2 on the modified Medical Research Council dyspnoea scale at screening (Celli 2014; Donohue 2013; Trivedi 2014). Participants in Donohue 2014 had a post‐salbutamol FEV1/FVC ratio < 0.70 and a post‐salbutamol FEV1 ≥ 35% and ≤ 80% of predicted values (as determined by Nutrition Health and Examination Survey III reference equations).
Mean age of participants ranged from 60.1 to 64.6 years, and most were males with baseline mean smoking pack‐years of 39.2 to 52.3. The percentage of current smokers ranged from 47% to 65%, and former smokers from 35% to 53%. The ratio of current to former smokers for umeclidinium and placebo arms was similar in the included studies. Participants had moderate to severe COPD with baseline mean post‐bronchodilator FEV1 ranging from 44.5% to 55.1% of predicted normal. Overall, baseline characteristics of participants were comparable among intervention and control groups.
Interventions
Investigators in all studies administered inhaled umeclidinium bromide once daily via a dry powder inhaler (DPI) in the morning. Celli 2014, Donohue 2014, and Trivedi 2014 studied a dose of 125 µg, and Donohue 2013 and Trivedi 2014 assessed a dose of 62.5 µg. Researchers gave matching placebo once daily in the morning via an identical DPI. Participants were allowed to use the following concomitant medications ‐ inhaled salbutamol (Celli 2014; Donohue 2013; Donohue 2014; Trivedi 2014) and/or ipratropium bromide (Donohue 2014) ‐ as rescue medication along with regular stable doses of inhaled corticosteroids (ICSs) (Celli 2014; Donohue 2013; Trivedi 2014). Investigators discontinued all inhaled long‐acting bronchodilators before screening (Trivedi 2014). Participants who used concomitant ICS during treatment ranged from 22% to 50%, and the proportion was balanced in all included studies across intervention arms, especially umeclidinium and placebo arms. A total of 23% to 55% of participants were taking ICS before the study began. Previous treatment with LABA was seen in 19% to 52% of participants, and 6% to 37% were pretreated with LAMA. Overall, the percentage of participants receiving prior COPD treatment was considered comparable for umeclidinium and placebo arms.
Outcomes
Primary outcomes of the individual studies were not the same as our primary outcomes, as these trials focused mainly on lung function as an efficacy measure. Change from baseline in pre‐dose trough FEV1 at the end of the study was the primary outcome in three trials (Celli 2014; Donohue 2013; Trivedi 2014), and this was a secondary outcome for our review. The numbers of participants with adverse events or serious adverse events studied as primary outcomes in Donohue 2014 were our secondary and primary outcomes, respectively. Health‐related quality of life measured by SGRQ score ‐ one of our primary outcomes ‐ was assessed as a secondary outcome in three trials (Celli 2014; Donohue 2013; Trivedi 2014). Secondary outcomes of our review such as TDI focal score, use of rescue medications, and adverse events were reported also as secondary outcomes in Celli 2014, Donohue 2013, and Trivedi 2014. These trials also studied the numbers of participants with exacerbations, hospital admissions, and death and made results available to the public in published full‐text articles or in GSK clinical study reports.
Funding
GlaxoSmithKline provided funding for all studies.
Excluded studies
We excluded 60 studies discussed in 201 reports, as they did not meet our prespecified criteria (see Characteristics of excluded studies for details).
A total of 16 studies were conducted in healthy adults, 15 lacked umeclidinium monotherapy and placebo as intervention arms, 13 did not study umeclidinium monotherapy, nine assessed umeclidinium without placebo, and five were of cross‐over design. Two other trials that studied umeclidinium versus placebo ‐ Decramer 2013 and Kelleher 2011 ‐ did not meet the minimum study duration of our review, at 28 days and 7 days, respectively.
Risk of bias in included studies
Overall, we found that included studies were of good methodological quality and were at low risk of bias for most domains, as they were pharmaceutical company sponsored and were conducted in accordance with approved protocol procedures and regulations. We provide a detailed assessment in Characteristics of included studies and Figure 2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All studies were GSK sponsored, and randomisation was performed to avoid possible bias. Although one published trial did not provide selection details (Celli 2014), the GSK clinical study report clearly mentioned the procedure used. All trials assigned adults to study treatment in accordance with a central randomisation schedule. GSK generated the randomisation code by using a validated computerised system RandAll, version 2.5. Investigators then randomised adults using RAMOS, an Interactive Voice Response System (IVRS) that was telephone based and was used by the investigator or the designee.
Blinding
Trials were double‐blind with blinding of participant, investigator, and caregivers for all study outcomes. Investigators administered study drugs in a double‐blind fashion by which neither the participant nor the study physician knew which study drug the participant was receiving. The DPIs containing study drug were identical in appearance to placebo DPIs containing only inactive ingredients of lactose and magnesium stearate. Outcome assessors were also blinded with regard to treatment assignments throughout the study period. Independent interviewers and cardiologists, blinded to treatment assignment, were responsible for assessment of dyspnoea scores (Baseline Dyspnoea Index (BDI) and TDI) and for interpretation of electrocardiographic (ECG) and Holter data, respectively.
Incomplete outcome data
All trials reported the number of withdrawals for each study arm along with reasons for withdrawal. In Celli 2014 and Donohue 2014, drop‐out rates were high even though they were relatively similar for umeclidinium and placebo arms. Thus, we assessed the risk of bias for these two studies as unclear, although we scored the other two studies (Donohue 2013; Trivedi 2014) as low risk, as drop‐out was relatively less for umeclidinium and placebo groups for similar reasons.
Researchers performed primary analyses on the intent‐to‐treat (ITT) population, defined as all randomised participants who had received at least one dose of study medication, and applied mixed‐effect model repeated measure (MMRM) for primary analysis of the data, without directly imputing missing data, although the underlying assumption was that data were missing at random, and derived treatment differences were adjusted to take into account missing data. However, for sensitivity analyses, researchers applied multiple imputation methods such as the missing at random (MAR) approach, the copy differences from control (CDC) approach, and the last mean carried forward (LMCF) approach (Celli 2014; Donohue 2013; Trivedi 2014).
Selective reporting
Both full‐text publications and clinical study reports described results of all outcomes prespecified in the protocol for all included studies, without reporting bias. Moreover, GSK had made all trial results public regardless of whether they reflected positively on effects of umeclidinium.
Other potential sources of bias
GSK funded all trials, and study authors disclosed in the published articles any possible conflicts of interest, both financial and non‐financial. We detected no other possible sources of bias.
Effects of interventions
See summary of findings Table for the main comparison. We included data from four trials for meta‐analysis in the comparison of umeclidinium bromide versus placebo (Celli 2014; Donohue 2013; Donohue 2014; Trivedi 2014).
Primary outcomes
Exacerbations requiring a short course of an oral steroid or antibiotic, or both
Analysis of a total of 1922 participants from four trials (Celli 2014; Donohue 2013; Donohue 2014; Trivedi 2014) for the number of people experiencing at least one exacerbation requiring a short course of oral steroids or antibiotics, or both, showed that umeclidinium significantly reduced moderate exacerbations compared with placebo (odds ratio (OR) 0.61, 95% confidence interval (CI) 0.46 to 0.80; high‐quality evidence) (Figure 3; Analysis 1.1). A total of 102 people per 1000 had a moderate COPD exacerbation (95% CI 79 to 130) in the umeclidinium‐treated population over 12 to 52 weeks, along with 157 in 1000 population in the placebo group (summary of findings Table for the main comparison). With umeclidinium, 55 fewer participants (from 27 fewer to 78 fewer) per 1000 experienced at least one moderate COPD exacerbation compared with placebo. Thus, for every 18 people treated with umeclidinium, one additional person was free from a moderate exacerbation requiring a short course of an oral steroid or antibiotic, or both (number needed to treat for an additional beneficial outcome (NNTB) 18, 95% CI 13 to 37).

Forest plot of comparison: 1 Umeclidinium bromide versus placebo, outcome: 1.1 Number of participants with exacerbations requiring steroids, antibiotics, or both.
Quality of life
Three studies (Celli 2014; Donohue 2013; Trivedi 2014) assessed quality of life using SGRQ total score as change from the baseline mean value, as well as percentage of participants who achieved reduction of at least four units, which was the minimal clinically important difference. In the meta‐analysis using a random‐effects model owing to high heterogeneity (I2 = 80%), umeclidinium demonstrated significant improvement in quality of life when compared with placebo, as it decreased SGRQ total score by a mean difference of ‐4.79 units (95% CI ‐8.84 to ‐0.75; three trials, 1119 participants). Subgroup analysis revealed that umeclidinium 62.5 μg was associated with significant change in SGRQ total score (mean difference (MD) ‐4.53, 95% CI ‐6.97 to ‐2.10; two trials, 584 participants), whereas the dose of 125 μg failed to produce a similar significant result (MD ‐5.04, 95% CI ‐15.05 to 4.97; two trials, 535 participants). However, tests for subgroup differences demonstrated no significance (P = 0.92) (Figure 4; Analysis 1.2).

Forest plot of comparison: 1 Umeclidinium bromide versus placebo, outcome: 1.2 Quality of life: change from baseline in SGRQ total score.
The total number of participants attaining the minimal clinically important difference was significantly higher in the umeclidinium group than in the placebo group (OR 1.45, 95% CI 1.16 to 1.82; three trials, 1397 participants). The umeclidinium dose of 62.5 μg demonstrated significant improvement (OR 1.62, 95% CI 1.19 to 2.21; two trials, 732 participants), and the dose of 125 μg showed no similar improvement (OR 1.29, 95% CI 0.93 to 1.79; two trials, 665 participants) with no subgroup significance (P = 0.32) (Analysis 1.3). More participants treated with umeclidinium reported a fall of at least four units in SGRQ total score, which was seen in 429 per 1000 participants in the umeclidinium group compared with 342 per 1000 participants in the placebo group; we rated this as moderate‐quality evidence (summary of findings Table for the main comparison). In absolute terms, SGRQ total score improved in 87 more per 1000 (from 34 more to 144 more) umeclidinium‐treated participants compared with placebo‐treated participants over 12 to 24 weeks. One additional person would experience a clinically meaningful improvement in quality of life for every 11 people treated with umeclidinium (NNTB 11, 95% CI 7 to 29).
Non‐fatal serious adverse events
A pooled analysis of data from four trials (Celli 2014; Donohue 2013; Donohue 2014; Trivedi 2014) revealed no statistically significant differences in non‐fatal serious adverse events between umeclidinium and placebo groups (OR 1.33, 95% CI 0.89 to 2.00; 1922 participants; moderate‐quality evidence) (Analysis 1.4). Among 1000 participants, 66 in the umeclidinium group (95% CI 45 to 96) developed non‐fatal serious adverse events compared with 51 in the placebo group (summary of findings Table for the main comparison).
Secondary outcomes
Mortality
All four trials reported the total number of deaths in the published articles, but Trivedi 2014 recorded no deaths in intervention and placebo arms during the study period of 12 weeks. These investigators noted no significant differences between umeclidinium and placebo in the number of deaths due to all causes (Peto OR 1.68, 95% CI 0.52 to 5.48; four trials, 1922 participants; Analysis 1.5).
Hospital admissions due to exacerbations
Meta‐analysis of data from four trials (Celli 2014; Donohue 2013; Donohue 2014; Trivedi 2014) based on a random‐effects model showed no statistical differences in the numbers of participants reporting one or more severe COPD exacerbations necessitating hospitalisation between umeclidinium and placebo groups (OR 0.86, 95% CI 0.25 to 2.92; four trials, 1922 participants). Moderately high heterogeneity (I2 = 60%) was explained by subgroup analysis using different dosages of umeclidinium, because Donohue 2013 reported more admissions with umeclidinium 62.5 μg than with placebo. However, both 62.5 μg (OR 3.20, 95% CI 0.91 to 11.24; two trials, 801 participants) and 125 μg (OR 0.43, 95% CI 0.18 to 1.03; three trials, 1121 participants) of umeclidinium were not associated with significant reduction in the number of participants requiring hospital admission owing to severe COPD exacerbations compared with participants given placebo (Analysis 1.6). Among a population of 1000 participants, 18 in the umeclidinium group needed hospitalisation (95% CI 5 to 58), whereas 20 in the placebo group required hospital admission (low‐quality evidence; summary of findings Table for the main comparison).
Improvement in symptoms
Three trials (Celli 2014; Donohue 2013; Trivedi 2014) assessed symptomatic improvement by TDI focal score as the change in mean value from baseline to the end of the study, as well as the percentage of participants achieving at least one unit increment in TDI focal score. Umeclidinium significantly increased TDI score, with a mean difference of 0.76 units compared with placebo (95% CI 0.43 to 1.09; three trials, 1193 participants) (Analysis 1.7).
The percentage of participants with a clinically meaningful improvement in TDI focal score was significantly higher with umeclidinium than with placebo (OR 1.71, 95% CI 1.37 to 2.15; three trials, 1441 participants) (Figure 5; Analysis 1.8). In absolute terms, 464 per 1000 participants receiving umeclidinium attained improvement of one or more units in TDI focal score compared with 336 per 1000 participants given placebo, implying that 128 more participants achieved this improvement with umeclidinium than with placebo (74 more to 185 more). For every eight people treated with umeclidinium, one more person will have the minimal clinically important difference of at least one unit increment in TDI focal score (95% CI 5 to 14) (high‐quality evidence; summary of findings Table for the main comparison).

Forest plot of comparison: 1 Umeclidinium bromide versus placebo, outcome: 1.8 Number of participants with ≥ 1 unit improvement in TDI focal score.
Lung function
Four trials (Celli 2014; Donohue 2013; Donohue 2014; Trivedi 2014) studied trough FEV1 and trough FVC as changes in mean value from baseline to the end of the study, and only two trials (Celli 2014; Donohue 2013) reported peak FEV1. Umeclidinium showed significant improvement in trough FEV1 (L) compared with placebo; this served as high‐quality evidence (MD 0.14, 95% CI 0.12 to 0.17; four trials, 1381 participants) (Analysis 1.9; summary of findings Table for the main comparison). Researchers noted similar significant improvement with umeclidinium as with placebo for change from baseline in trough FVC (L) (MD 0.22, 95% CI 0.17 to 0.26; four trials, 1381 participants) (Analysis 1.10) and peak FEV1 (L) (MD 0.17, 95% CI 0.14 to 0.19; two trials, 1035 participants) (Analysis 1.11).
Adverse events
Meta‐analysis of data from all four trials showed no statistical difference between umeclidinium and placebo for the numbers of participants experiencing adverse events, not including serious adverse events (OR 1.06, 95% CI 0.85 to 1.31; 1922 participants) (Analysis 1.12). In absolute terms, 250 of 1000 participants (95% CI 211 to 291) treated with umeclidinium suffered adverse events compared with 239 of 1000 participants given placebo (moderate‐quality evidence; summary of findings Table for the main comparison).
Use of rescue medications
Investigators observed significant reduction in the use of rescue medications (change from baseline in number of puffs per day) with umeclidinium versus placebo (MD ‐0.45, 95% CI ‐0.76 to ‐0.14; four trials, 1531 participants) (Figure 6; Analysis 1.13).

Forest plot of comparison: 1 Umeclidinium bromide versus placebo, outcome: 1.13 Use of rescue medications (change from baseline in number of puffs per day).
Discussion
Summary of main results
For this systematic review, four studies comprising a total of 3798 participants with clinical and spirometrically proven chronic obstructive pulmonary disease (COPD) met our prespecified inclusion criteria. We have summarised the treatment effects of umeclidinium inhalers compared with placebo for clinically relevant outcomes.
In three of the four studies, participants were symptomatic (i.e. short of breath on exertion), indicating that participants on average had moderate to severe COPD. Results showed gender imbalance, with more male than female participants, which reflected key landmark clinical trials on COPD. This imbalance reflects that the prevalence of moderate to severe COPD was higher in males than females (Buist 2007), and that tobacco smoke‐induced COPD was more prevalent in males than in females (Cheng 2015).
Compared with adults taking placebo, those taking umeclidinium were less likely to experience acute exacerbations requiring steroids, antibiotics, or both; this served as high‐quality evidence. The number needed to treat for an additional beneficial outcome (NNTB) to prevent an episode of exacerbation was 18 (95% confidence interval (CI) 13 to 37), representing a reduction in risk from 157 of 1000 taking placebo to 102 of 1000 given umeclidinium. However, no evidence showed that umeclidinium reduced exacerbation‐related hospital admissions. This is a relatively rare event so may represent a type 2 error.
Umeclidinium improved quality of life, as it lowered St George's Respiratory Questionnaire (SGRQ) total score by 4.79 units (from 8.84 to 0.75 units lower), with mean improvement greater than the minimal clinically important difference of four units. Moderate‐quality evidence shows that a significantly higher proportion of participants achieved improvement of at least four units with umeclidinium than with placebo (NNTB 11, 95% CI 7 to 29).
Statistical analysis of quality of life and hospitalisation due to exacerbations revealed substantial heterogeneity, with I2 greater than 50%, for which subgroup analysis of different doses of umeclidinium was performed. This subgroup analysis explained heterogeneity for quality of life, by which the US Food and Drug Administration (FDA)‐approved dose of 62.5 µg produced significant improvement in SGRQ total score with no heterogeneity. However, it failed to demonstrate a significant reduction in hospital admissions, although the heterogeneity was explained by subgroup analysis. One explanation for the lower dose improving quality of life as much as the higher dose is that potential benefits may be offset by other adverse effects. However, a dose effect was not evident in the adverse events analysis.
Symptomatic improvement in breathlessness was seen within the umeclidinium group compared with the placebo group, with a mean difference in Transitional Dyspnoea Index (TDI) focal score from baseline of 0.76 units (from 0.43 to 1.09 units), although this mean increment did not reach the minimal clinically important difference of one unit. The number of participants attaining improvement in TDI focal score of at least one unit was higher in the umeclidinium group, with an increase from 336 of 1000 given placebo to 464 of 1000 taking umeclidinium, and the NNTB was 8 (95% CI 5 to 14). The reduction in use of rescue medication to relieve symptoms was significantly less with umeclidinium than with placebo, as the mean difference between the two groups in the average number of puffs per day was ‐0.45 (95% CI ‐0.76 to‐ 0.14).
The change from baseline in trough forced expiratory volume in one second (FEV1) was significantly greater in umeclidinium‐treated participants, with a mean difference of 0.14 L (from 0.12 to 0.17 L). Similar effects were observed for other spirometric indices such as trough forced vital capacity (FVC) and peak FEV1.
As for safety outcomes, results showed no differences between umeclidinium and placebo groups in terms of adverse events, non‐fatal serious adverse events, and all‐cause mortality. However, the confidence intervals for these outcomes were wide, and this did not allow a confident conclusion about the non‐difference. Three studies (Celli 2014; Donohue 2013; Donohue 2014) reported a total of nine deaths among participants taking umeclidinium that were not considered related to the study drug.
Overall completeness and applicability of evidence
Umeclidinium bromide was approved in April 2014 in the USA and Europe for maintenance treatment of patients with COPD. To date, a considerable number of completed trials have examined umeclidinium as the sole intervention or fixed‐dose combination of umeclidinium and vilanterol. This review synthesises evidence from four studies of high methodological standard, providing a better estimate of true risks and benefits of umeclidinium treatment than is provided by any of the four studies alone. We calculated summary estimates of effects of umeclidinium on clinical outcomes compared with placebo. Some of the data required for the meta‐analysis were not reported in the published full‐text articles; we did not perform an individual participant‐level meta analysis; however, all data that contributed to the outcomes of this review were derived from participant‐level data provided at the GlaxoSmithKline (GSK) website.
Overall evidence reported in this review supports the efficacy of umeclidinium compared with placebo for use in patients with stable COPD. The lung function response to a bronchodilator medication tends to vary with time, with a mean decline in FEV1 of 33 mL per year over the three‐year study period (Vestbo 2011). In our review, we could analyse data for only 52 weeks; longer studies would provide more accurate and reliable evidence for any sustainable improvement in lung function attained with umeclidinium. Furthermore, lack of a significant increase in deaths, non‐fatal serious adverse events, or other adverse events compared with placebo makes umeclidinium a relatively safe medication for use. However, larger studies of longer duration are needed to provide more convincing evidence on safety outcomes. Further evidence on the comparative efficacy and safety of umeclidinium versus other long‐acting muscarinic antagonists (LAMAs) in clinical use, such as tiotropium, which has a strong evidence base for its use, or other new LAMAs, would provide valuable information for appropriate selection of LAMAs as maintenance therapy for people with moderate to severe COPD.
Quality of the evidence
All studies included in this review were industry sponsored and were conducted according to similar strict prespecified protocols; thus they are of good methodological quality. Overall, the quality of evidence ranged from low to high for the outcomes of this review. The estimate of the effect of umeclidinium over placebo for the likelihood of reducing the number of participants with exacerbations requiring a short course of steroids, antibiotics, or both is likely to be accurate, as it is based on high‐quality evidence. Similarly, effects on improvement in lung function (trough FEV1) and symptoms (TDI score) were graded as high quality, and we are confident that the true effect lies close to the effect estimate. We rated evidence on quality of life, adverse events, and non‐fatal serious adverse events as moderate quality because of heterogeneity and imprecision, respectively; we are moderately confident that the effect estimate is likely to be close to the true effect but may be substantially different. However, evidence on hospital admissions due to exacerbations was of low quality owing to imprecision, as the CI includes both appreciable benefit and harm. As these are relatively infrequent events, additional information from future trials might alter our confidence in these results while providing new evidence for clinical practice.
Potential biases in the review process
We believe that incomplete identification of studies for this review is unlikely because we performed a comprehensive search of all possible sources such as the World Health Organization (WHO) portal, the ClinicalTrials.gov website, reference lists, and manufacturers' clinical study registers, in addition to the Cochrane Airways Group systematic electronic search. Two review authors independently selected studies, assessed risk of bias, and extracted data to minimise possible bias in this review. Publication bias was least likely for this review, as GlaxoSmithKline had officially published patient‐level data on its clinical study register website, regardless of positive or negative outcomes of the study medication. Assessment of publication bias through examination of funnel plots was not possible because only four trials were included in this review. Clinical characteristics and baseline lung function of participants were similar for umeclidinium and placebo arms in all included studies.
Concomitant medications, especially inhaled corticosteroids (ICSs), may impact outcomes, but the percentage of participants with ICS use was similar for umeclidinium and placebo arms, making it less likely that ICS treatment had a significant effect on the evidence presented in our review. Similarly, the smoking status of participants was comparable, and the ratio of current to former smokers was relatively balanced. Another potential source of bias is prior use of long‐acting beta2‐agonist (LABA), LAMA, or ICS. However, we noted no obvious differences between the proportions of participants treated with either of these agents in the intervention arms of included studies. Information on previous history of exacerbations ‐ a major predictor of exacerbations ‐ was not available for these studies. However, in light of the comparable characteristics of participants in the intervention arms for other variables, we assume no significant change in the findings of our review. Generally, the number of participants and the minimum study duration of 12 weeks described in this review could be considered adequate for analysis and conclusions, although larger studies of longer duration would yield stronger evidence for outcomes, especially for adverse events, mortality, quality of life, and exacerbations.
Agreements and disagreements with other studies or reviews
We found several published reviews on umeclidinium in our review of the literature. Meta‐analysis of data from four trials showed that use of umeclidinium results in a significant increase in FEV1 compared with placebo, with an adjusted mean difference of 0.14 (95% CI 0.12 to 0.16) for the dose of 62.5 μg (Segreti 2014). This lung function improvement is consistent with the findings of our review. Likewise, another review that conducted a pooled analysis of 10 randomised controlled trials (RCTs) ‐ both parallel and cross‐over studies (Pleasants 2016) ‐ reported a significant increase in trough FEV1 with umeclidinium over placebo (weighted mean difference 0.13, 95% CI 0.11 to 0.14), and the extent was comparable with our finding. Pleasants and colleagues found significant improvement in TDI focal score, with a mean difference in change from baseline of 0.63 (95% CI 0.27 to 0.99), which is similar to the estimate of 0.76 provided in the current review. However, in contrast to our review, Pleasants did not demonstrate statistical or clinical significance for quality of life scores, with a mean difference in SGRQ total score of ‐2.15 (95% CI ‐4.11 to 0.18) compared with the mean difference in the current review of ‐4.79, which exceeded the minimal clinically important difference of four units. Review authors in the Pleasants group analysed exacerbations as time to first exacerbation, whereas we analysed separately the numbers of participants with exacerbations requiring steroids, antibiotics, or both, as well as the number with exacerbations requiring hospital admissions. Umeclidinium prolonged the time to first exacerbation compared with placebo in Pleasants 2016 (mean difference (MD) 0.53, 95% CI 0.40 to 0.70), and in our review, it reduced the number of participants with exacerbations requiring steroids, antibiotics, or both.
Other available evidence comes from Ismaila 2015, which analysed the comparative efficacy of LAMA monotherapies (aclidinium, glycopyrronium, tiotropium, and umeclidinium) versus placebo or each other by performing network meta‐analysis with placebo as the common comparator. Umeclidinium showed a mean change from baseline in trough FEV1 (L) of 0.14 (95% Cl 0.10 to 0.17) at 12 weeks, and 0.12 (95% CI 0.07 to 0.16) at 24 weeks, relative to placebo, both of which were greater than the minimal clinically important difference of 0.1 L. In accordance with our review, network meta‐analysis revealed improvements in TDI score (MD 1.00, 95% CI 0.49 to 1.51) and SGRQ score (MD ‐4.69, 95% CI ‐7.05 to ‐2.31) at 24 weeks. Use of rescue medication was less with umeclidinium than with placebo in both reviews; however, Ismaila 2015 failed to show statistical significance (MD ‐0.30, 95% CI ‐0.81 to 0.21). Review authors concluded that umeclidinium was efficacious relative to placebo, but as noted with other, newer LAMAs, it behaved similarly to tiotropium, which is 'the established class standard'. Our review assessed only the comparative efficacy of umeclidinium and placebo; additional reviews conducted to compare the efficacy of umeclidinium with that of other LAMAs would provide more clinically relevant evidence to guide selection among different LAMAs. Umeclidinium has proved safe, and pooled analysis has revealed no differences in reported adverse events of cough, dry mouth, and exacerbations (Pleasants 2016); these findings are similar to the findings of our review.
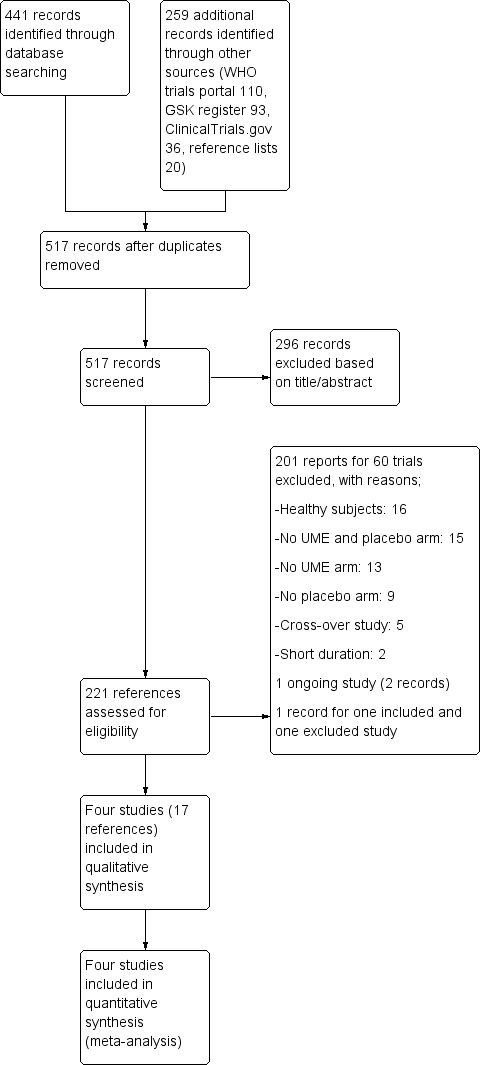
Study flow diagram.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Forest plot of comparison: 1 Umeclidinium bromide versus placebo, outcome: 1.1 Number of participants with exacerbations requiring steroids, antibiotics, or both.
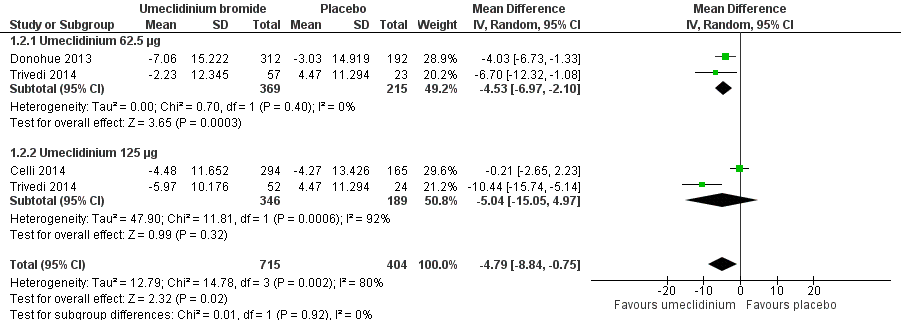
Forest plot of comparison: 1 Umeclidinium bromide versus placebo, outcome: 1.2 Quality of life: change from baseline in SGRQ total score.

Forest plot of comparison: 1 Umeclidinium bromide versus placebo, outcome: 1.8 Number of participants with ≥ 1 unit improvement in TDI focal score.

Forest plot of comparison: 1 Umeclidinium bromide versus placebo, outcome: 1.13 Use of rescue medications (change from baseline in number of puffs per day).
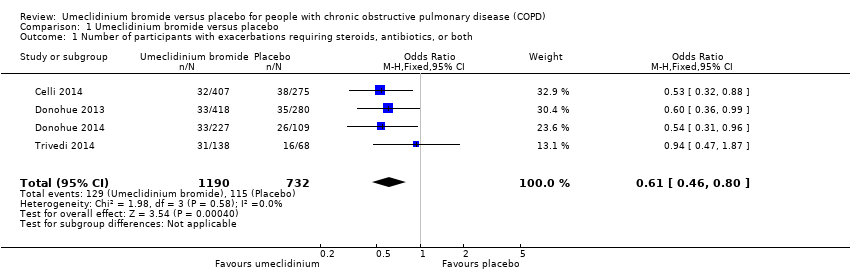
Comparison 1 Umeclidinium bromide versus placebo, Outcome 1 Number of participants with exacerbations requiring steroids, antibiotics, or both.
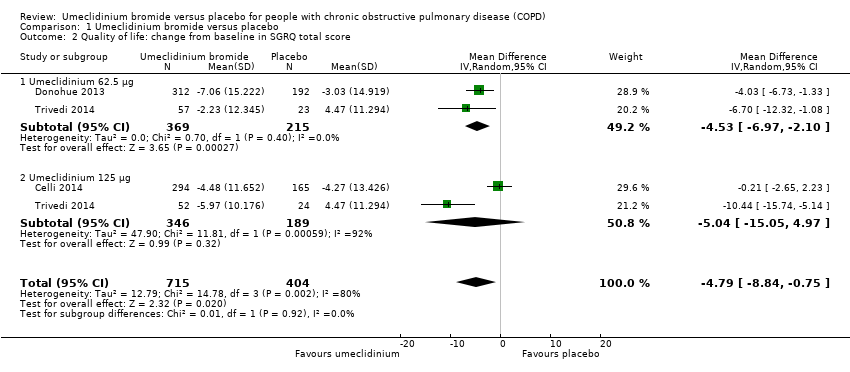
Comparison 1 Umeclidinium bromide versus placebo, Outcome 2 Quality of life: change from baseline in SGRQ total score.
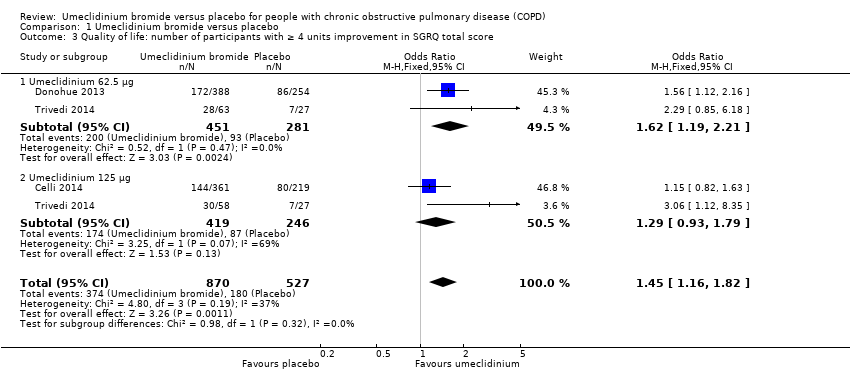
Comparison 1 Umeclidinium bromide versus placebo, Outcome 3 Quality of life: number of participants with ≥ 4 units improvement in SGRQ total score.
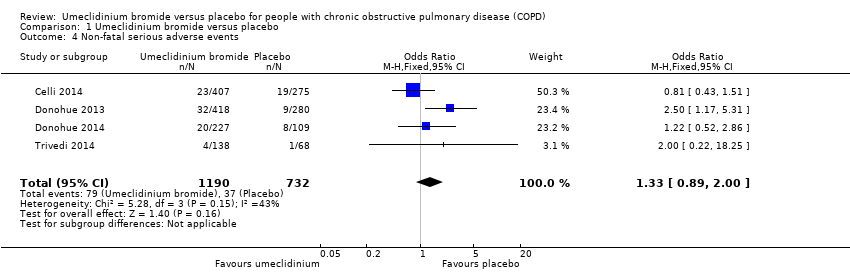
Comparison 1 Umeclidinium bromide versus placebo, Outcome 4 Non‐fatal serious adverse events.

Comparison 1 Umeclidinium bromide versus placebo, Outcome 5 Total number of deaths.

Comparison 1 Umeclidinium bromide versus placebo, Outcome 6 Number of participants with hospital admissions due to COPD exacerbation.

Comparison 1 Umeclidinium bromide versus placebo, Outcome 7 Improvement in symptoms: TDI focal score.

Comparison 1 Umeclidinium bromide versus placebo, Outcome 8 Number of participants with ≥ 1 unit improvement in TDI focal score.

Comparison 1 Umeclidinium bromide versus placebo, Outcome 9 Lung function: change from baseline in trough FEV1 (L).
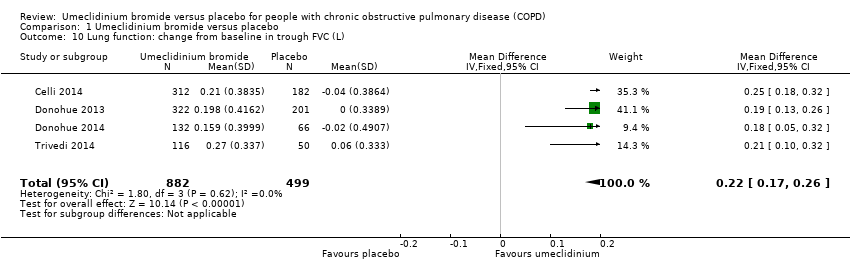
Comparison 1 Umeclidinium bromide versus placebo, Outcome 10 Lung function: change from baseline in trough FVC (L).

Comparison 1 Umeclidinium bromide versus placebo, Outcome 11 Lung function: change from baseline in peak FEV1 (L).

Comparison 1 Umeclidinium bromide versus placebo, Outcome 12 Adverse events (not including serious adverse events).

Comparison 1 Umeclidinium bromide versus placebo, Outcome 13 Use of rescue medications (change from baseline in number of puffs per day).
| Umeclidinium bromide vs placebo for stable chronic obstructive pulmonary disease | ||||||
| Patient or population: people with chronic obstructive pulmonary disease (COPD) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Risk with placebo | Risk with umeclidinium bromide | |||||
| Number of participants with exacerbations requiring steroids, antibiotics, or both | 157 per 1000 | 102 per 1000 | OR 0.61 | 1922 | ⊕⊕⊕⊕ | |
| Quality of life: number of participants with ≥ 4 units improvement in SGRQ total score | 342 per 1000 | 429 per 1000 | OR 1.45 | 1397 | ⊕⊕⊕⊝ | Mean quality of life: change from baseline in SGRQ total score was 4.79 lower (8.84 lower to 0.75 lower) in umeclidinium group (1119 participants, 3 RCTs) |
| Non‐fatal serious adverse events | 51 per 1000 | 66 per 1000 | OR 1.33 | 1922 | ⊕⊕⊕⊝ | Larger studies may help refine this estimate |
| Number of participants with hospital admissions due to COPD exacerbation | 20 per 1000 | 18 per 1000 | OR 0.86 | 1922 | ⊕⊕⊝⊝ | Few events, so larger studies may help refine this estimate |
| Number of participants with ≥ 1 unit improvement in TDI focal score | 336 per 1000 | 464 per 1000 | OR 1.71 | 1441 | ⊕⊕⊕⊕ | Mean improvement in TDI focal score change from baseline was 0.76 higher (0.43 higher to 1.09 higher) in umeclidinium group (1193 participants, 3 RCTs) |
| Change from baseline in trough FEV1 (L) | Mean change from baseline in trough FEV1 (L) across control groups ranged from 0.123 to 0.139 | Mean change from baseline in trough FEV1 (L) in the intervention group was 0.14 higher (0.12 higher to 0.17 higher) | ‐ | 1381 | ⊕⊕⊕⊕ | |
| Adverse events (not including serious adverse events) | 239 per 1000 | 250 per 1000 | OR 1.06 | 1922 | ⊕⊕⊕⊝ | |
| *The risk in the intervention group (and its 95% confidence interval) is based on assumed risk in the comparison group and relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| a‐1 for inconsistency: unexplained significant heterogeneity b‐1 for imprecision: the CI includes non‐appreciable benefit and potential harm c‐2 for imprecision: the CI includes both appreciable benefit and harm | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Number of participants with exacerbations requiring steroids, antibiotics, or both Show forest plot | 4 | 1922 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.46, 0.80] |
| 2 Quality of life: change from baseline in SGRQ total score Show forest plot | 3 | 1119 | Mean Difference (IV, Random, 95% CI) | ‐4.79 [‐8.84, ‐0.75] |
| 2.1 Umeclidinium 62.5 μg | 2 | 584 | Mean Difference (IV, Random, 95% CI) | ‐4.53 [‐6.97, ‐2.10] |
| 2.2 Umeclidinium 125 μg | 2 | 535 | Mean Difference (IV, Random, 95% CI) | ‐5.04 [‐15.05, 4.97] |
| 3 Quality of life: number of participants with ≥ 4 units improvement in SGRQ total score Show forest plot | 3 | 1397 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.45 [1.16, 1.82] |
| 3.1 Umeclidinium 62.5 μg | 2 | 732 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.19, 2.21] |
| 3.2 Umeclidinium 125 μg | 2 | 665 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.93, 1.79] |
| 4 Non‐fatal serious adverse events Show forest plot | 4 | 1922 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.89, 2.00] |
| 5 Total number of deaths Show forest plot | 4 | 1922 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.68 [0.52, 5.48] |
| 6 Number of participants with hospital admissions due to COPD exacerbation Show forest plot | 4 | 1922 | Odds Ratio (M‐H, Random, 95% CI) | 0.86 [0.25, 2.92] |
| 6.1 Umeclidinium 62.5 μg | 2 | 801 | Odds Ratio (M‐H, Random, 95% CI) | 3.20 [0.91, 11.24] |
| 6.2 Umeclidinium 125 μg | 3 | 1121 | Odds Ratio (M‐H, Random, 95% CI) | 0.43 [0.18, 1.03] |
| 7 Improvement in symptoms: TDI focal score Show forest plot | 3 | 1193 | Mean Difference (IV, Fixed, 95% CI) | 0.76 [0.43, 1.09] |
| 8 Number of participants with ≥ 1 unit improvement in TDI focal score Show forest plot | 3 | 1441 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.71 [1.37, 2.15] |
| 9 Lung function: change from baseline in trough FEV1 (L) Show forest plot | 4 | 1381 | Mean Difference (IV, Fixed, 95% CI) | 0.14 [0.12, 0.17] |
| 10 Lung function: change from baseline in trough FVC (L) Show forest plot | 4 | 1381 | Mean Difference (IV, Fixed, 95% CI) | 0.22 [0.17, 0.26] |
| 11 Lung function: change from baseline in peak FEV1 (L) Show forest plot | 2 | 1035 | Mean Difference (IV, Fixed, 95% CI) | 0.17 [0.14, 0.19] |
| 12 Adverse events (not including serious adverse events) Show forest plot | 4 | 1922 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.85, 1.31] |
| 13 Use of rescue medications (change from baseline in number of puffs per day) Show forest plot | 4 | 1531 | Mean Difference (IV, Fixed, 95% CI) | ‐0.45 [‐0.76, ‐0.14] |