氯丙嗪比较甲硫平治疗精神分裂症
摘要
研究背景
氯丙嗪(Chlorpromazine)是一种广泛使用且价格低廉的抗精神病药物,被认为是世界范围内精神分裂症的基准治疗方法。甲硫平(Metiapine)是一种二苯并噻氮衍生物,被报告具有强效的抗精神病特性。然而,目前尚无证据表明氯丙嗪与新型抗精神病药甲硫平相比在治疗精神分裂症患者方面的有效性。
研究目的
本系统综述旨在将氯丙嗪比较甲硫平治疗精神分裂症的效果
检索策略
我们检索了Cochrane精神分裂症小组基于研究的试验注册库(Cochrane Schizophrenia Group's Study‐Based Register),于2015年和2016年11月。
纳入排除标准
所有着眼于氯丙嗪比较甲硫平对精神分裂症成年患者疗效的随机对照试验(randomised controlled trials, RCT)。我们纳入了符合选取标准并报告了可用资料的试验。
资料收集与分析
我们独立进行资料提取。对于二分类结局,我们在意向性分析的基础上,计算风险比(risk ratio, RR)及其95%置信区间(confidence interval, CI)。对于连续性资料,我们估计了组间平均差(mean difference, MD)及其95%CI。我们采用随机效应模型进行分析。我们评价了被纳入研究的偏倚风险,并使用GRADE创建了“结果总结”表。
主要结果
我们纳入了三项研究,共涉及随机分组的161名精神分裂症患者。我们预先设定的七个主要结局中只有两个的资料可用。使用临床整体印象(Clinical Global Impression,CGI)测量整体状态的临床重要改善。氯丙嗪组和甲硫平组之间没有明显差异(2项随机对照试验,n=120,RR=1.11,95%CI [0.84,1.47], 极低质量证据 ),8周时出现帕金森症的受试者人数相近(2项随机对照试验,n=70,RR=0.97,95%CI [0.46,2.03], 极低质量证据 )。对于临床上重要的心理状态改善、因复发而再次入院、对治疗的满意度、攻击性或暴力行为或护理成本等其他关键结局,没有可用的资料。
作者结论
几十年来,氯丙嗪一直是精神分裂症的主要治疗药物,但将这种药物与甲硫平进行比较的现有证据未能提供高质量的试验资料。然而,确定甲硫平是否比氯丙嗪有更多疗效或更少疗效的需要似乎缺乏临床相关性,未来似乎不太可能对这种比较进行研究。
PICO
简语概要
氯丙嗪比较甲硫平治疗精神分裂症
本综述的目的是寻找高质量的证据来比较氯丙嗪与美硫平治疗精神分裂症的疗效。
研究背景
精神分裂症是一种世界范围内常见的致残和持久性心理疾病。精神分裂症患者通常会出现妄想和幻觉等阳性症状,以及冷漠(缺乏兴趣)和情绪低落等阴性症状。
抗精神病药物已成功治疗阳性症状,但阴性症状仍然难以治疗并且通常对常规抗精神病药物没有反应。此外,抗精神病药物通常有令人不快的副作用。
氯丙嗪是20世纪50年代推出的一种广泛使用且价格低廉的抗精神病药,被认为是世界范围内精神分裂症的基准治疗方法。初步研究表明,与安慰剂(假治疗)相比,氯丙嗪有助于全面改善并有效预防复发。然而据报告,氯丙嗪的一些副作用,特别是运动障碍的发生率,会很严重或者使人虚弱。甲硫平是一种相对较新的抗精神病药物,据报告可有效治疗精神分裂症的症状,同时引起较少的副作用。然而,目前没有关于与氯丙嗪直接比较,甲硫平有效性的高质量信息。
证据检索
2015年11月,Cochrane精神分裂症组(Cochrane Schizophrenia group)的文献检索信息专员在他们的专业注册库中检索了相关的临床试验。检索确定了四份报告。我们查看了这些报告,发现它们提到了三项试验,将精神分裂症患者随机分配接受氯丙嗪或甲硫平。
主要结果
我们在本综述中纳入了3项研究,共161名受试者。研究表明,氯丙嗪和美硫平在改善整体状态或帕金森症(一系列症状的总称,如震颤(颤抖)、运动迟缓(运动缓慢)、僵硬(僵硬)和姿势不稳(平衡困难) )发病率方面没有真正的区别。没有报告其他主要关注研究领域的资料,如:心理状态、服务使用、治疗满意度、行为或护理成本。
研究结论
我们无法从提供的资料中得出明确的结论。研究数量和每项研究的研究对象人数很少,所有研究都是短期的。因此,我们将报告的证据评为极低质量。然而,甲硫平不是一种开具处方率或使用率很高的抗精神病药物,因此尽管我们的证据不足,但它可能仍然是现有的最佳证据,因为未来不太可能进行比较甲硫平与氯丙嗪的新试验。
Authors' conclusions
Summary of findings
| Chlorpromazine versus metiapine for schizophrenia | ||||||
| Patient or population: people with schizophrenia | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Chlorpromazine | Metiapine | |||||
| Global state: clinically important improvement | 600 per 1000 | 684 per 1000 | RR 1.11 | 120 | ⊕⊝⊝⊝ | 1 trial reported CGI at 4 weeks, and 1 trial reported CGI at 6 weeks' follow‐up, both indicated no significant difference between the 2 drugs. |
| Mental state: clinically important improvement | No studies reported this outcome. | |||||
| Adverse effects. Specific: movement disorders ‐ parkinsonism | 168 per 1000 | 144 per 1000 | RR 0.97 | 70 | ⊕⊝⊝⊝ | ‐ |
| Service use: readmission due to relapse | No studies reported on these outcomes. | |||||
| Satisfaction of participant or care provider with treatment | ||||||
| Behaviour: aggressive or violent behaviour | ||||||
| Cost of care | ||||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Risk of bias: serious (downgraded by 1 level): study had unclear risk of other biases as supported by a pharmaceutical company for drug supply. | ||||||
Background
Description of the condition
Schizophrenia is considered a common disabling and enduring mental problem with a global lifetime prevalence of about 1% (Dickenson 2013; Tardy 2011). According to World Health Organization (WHO) reports, an estimated 24 million people are currently living with schizophrenia worldwide. Low‐ and middle‐income countries account for the majority of those people diagnosed with schizophrenia with few gender disparities (WHO 2001). Schizophrenia appears among the WHO seven top leading causes of disability and loss of years of lives due to disability (Dold 2012).
Schizophrenia is often grouped by two broad categories of positive or negative symptoms. Positive symptoms represent changes in behaviour or thought including delusions, hallucinations, catatonic symptoms such as mannerisms and bizarre posturing (Carpenter 1994; Crow 1980). In contrast, negative symptoms often represent an absence or deficit of normal emotional responses that are typically observed in healthy people. These negative symptoms can include flattening and poverty of speech, social withdrawal, inability to find pleasure in activities normally considered pleasurable and a lack of drive (Elis 2013). Different medications have been used to reduce symptoms of schizophrenia although the negative symptoms remain difficult to treat and usually do not respond to routine antipsychotic treatments.
Description of the intervention
Chlorpromazine, a widely available and inexpensive antipsychotic drug, is considered one benchmark treatment for schizophrenia worldwide. Since its discovery in 1951, with an outstanding impact, chlorpromazine emerged as the second revolution in psychiatry (Grozier 1973). Despite the advent of many new antipsychotic and neuroleptic drugs, chlorpromazine remains one of the first‐line treatments for schizophrenia cases in low‐to‐moderate income regions as well as high‐income regions in the world and is listed among the essential drugs for schizophrenia treatment by the WHO (WHO 2011). India and South East Asia are areas in which chlorpromazine is the most commonly used treatment, rather than other antipsychotic drugs (Adams 2014).
Primary evidence of using chlorpromazine for schizophrenia has indicated that it improved clinical recovery progress, facilitated global improvements and was effective at preventing relapse in comparison with placebo (Adams 2014). While chlorpromazine is considered an inexpensive drug, some of chlorpromazine's adverse effects could be expensive when it comes to human suffering and cost of treatments. Therefore, it might be better to use a safer, costly drug if the latter is equally potent, but has fewer, less severe adverse effects.
Metiapine is among those antipsychotic drugs, introduced in the 1970s, reported to be effective for treating the symptoms of schizophrenia with less severe adverse effects than chlorpromazine. It was originally produced by a US company ‐ Merrell‐Dow. This company was merged into Marion Merrell Dow, then acquired to become Hoechst Marion Roussel and finally subsumed into the Sanofi company. We are unsure of whether this compound is licenced for clinical use anymore.
How the intervention might work
Chlorpromazine, one of the widely used typical antipsychotic drugs, is an aliphatic phenothiazine (2‐chloro‐10‐(3‐dimethylaminopropyl)phenothiazine, Figure 1; Figure 2). Chlorpromazine acts as a blocking agent on different postsynaptic receptors by blocking alpha 1 adrenergic, 5HT2A, D2 and D1 receptors in the brain (Saha 2013). Chlorpromazine is classified as a low‐potency typical antipsychotic drug. Previously it was used for treating people with both acute and chronic psychoses, including schizophrenia and the manic phase of bipolar disorder as well as amphetamine‐induced psychoses. There is evidence that chlorpromazine increases a person's chances of experiencing acute movement disorders (parkinsonism, rigidity, tremor, fits, sleepiness and weakness). In addition, chlorpromazine causes other adverse effects such as eye opacities, jaundice, photosensitivity, dry mouth, constipation, hypotension, urinary retention and blurred vision (Adams 2014). Chlorpromazine is also a sedating medication, prone to cause a variety of movement problems and increased weight. Evidence suggests that chlorpromazine leads to several anxiolytic, antidepressive and antiaggressive properties as well as an attenuation of extrapyramidal adverse effects, but may also cause metabolic syndromes, and movement disorders (Saha 2013).

Chlorpromazine ‐ structure.
Metiapine (2‐methyl‐11‐(4‐methyl‐1‐piperazinyl)dibenzo [b,f][1,4]thiazepine, Figure 2), a dibenzothiazepine derivative, has been reported to have potent neuroleptic and antipsychotic characteristics in humans (Gallant 1970). Early evidence found metiapine's pharmacological properties in animals similar to those of other neuroleptic drugs (Ketteler 1970). Its reported characteristics included reduced spontaneous motor activity, inhibition of social and aggressive activity, and cataleptoid postures. There were unusually wide differences between the doses that altered behavioural patterns and those that produced incapacitation alterations of motor function (Gibson 1973).

Metiapine structure.
Why it is important to do this review
Despite its well‐documented adverse effects, chlorpromazine is likely to remain one of the most globally prescribed treatments for schizophrenia. It is unclear whether chlorpromazine's efficacy and potential performance leads to better results in direct comparison with other antipsychotic drugs such as metiapine. We are also unclear if metiapine is of any value, and, if of value, whether metiapine is a largely forgotten antipsychotic drug.
This review is one of a series of Cochrane Reviews that will build up an over‐review investigating the effects of chlorpromazine (Table 1).
| Review title | Reference |
| Acetophenazine versus chlorpromazine for schizophrenia. | |
| Chlorpromazine dose for people with schizophrenia. | |
| Cessation of medication for people with schizophrenia already stable on chlorpromazine. | |
| Chlorpromazine versus atypical antipsychotic drugs for schizophrenia. | |
| Chlorpromazine versus clotiapine for schizophrenia. | |
| Chlorpromazine versus metiapine for schizophrenia. | This review |
| Chlorpromazine versus penfluridol for schizophrenia. | |
| Chlorpromazine versus piperacetazine for schizophrenia. | |
| Chlorpromazine versus placebo for schizophrenia. | |
| Chlorpromazine for psychosis induced aggression or agitation. | |
| Haloperidol versus chlorpromazine for schizophrenia. |
*
Objectives
To compare the effect of chlorpromazine versus metiapine for the treatment of people with schizophrenia
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials (RCTs). If a trial had been described as 'double blind' but implied randomisation, we would have included such a trial in a sensitivity analysis (see Sensitivity analysis). We excluded quasi‐randomised studies, such as those allocating by alternate days of the week. If people had been given additional treatments within chlorpromazine versus metiapine, we would only have included the data if the adjunct treatment had been evenly distributed between groups and it was only the chlorpromazine versus metiapine that was randomised.
Types of participants
Adults, however defined, with schizophrenia or related disorders, including schizophreniform disorder, schizoaffective disorder and delusional disorder, again, by any means of diagnosis. If we had found a trial with a range of diagnoses, we would have only included if the majority of participants had schizophrenia.
To ensure that information was as relevant to the current care of people with schizophrenia as possible we tried to clearly highlight the current clinical state (acute, early post‐acute, partial remission, remission) and the stage (prodromal, first episode, early illness, persistent), and whether the studies primarily focused on people with particular problems (e.g. negative symptoms, treatment‐resistant illnesses).
Types of interventions
1. Chlorpromazine
Any dose, timing or means of administration.
2. Metiapine
Any dose, timing or means of administration.
Types of outcome measures
Where possible, we divided available outcomes into short term (less than six months), medium term (seven to 12 months) and long term (over one year).
Primary outcomes
1. Global state
1.1. Clinically important improvement in global state as defined by each study.
2. Mental state
2.1. Clinically important improvement in mental state as defined by each study.
3. Adverse events
3.1. Incidence of clinically important movement disorder as defined by each study.
Secondary outcomes
1. Global state
1.1. Mean scores for global state.
1.2. Relapse.
2. Mental state
2.1. General symptoms ‐ prevalence or mean scores.
2.2. Specific symptoms ‐ prevalence or mean scores.
2.2.1. Positive symptoms (delusions, hallucinations, disordered thinking).
2.2.2. Negative symptoms (avolition, poor self‐care, blunted affect).
2.2.3. Mood ‐ depression
3. Adverse effects
3.1. General ‐ prevalence or mean scores.
3.2. Specific ‐ prevalence or mean scores.
3.2.1. Deaths by suicide or natural causes.
3.2.2. Movement disorders (extrapyramidal adverse effects, specifically tardive dyskinesia and neuroleptic malignant syndrome).
3.2.3. Sedation.
3.2.4. Dry mouth.
3.2.5. Others ‐ categorised by system.
4. Leaving the study
5. Behaviour
5.1. General behaviour ‐ prevalence or mean scores.
5.2. Specific behaviour ‐ prevalence or mean scores.
5.2.1. Social functioning.
5.2.2. Employment status during trial (employed/unemployed).
5.2.3. Occurrence of violent incidents (to self, others or property).
6. Service utilisation outcome
6.1. Days in hospital.
6.2. Readmission due to relapse.
7. Quality of life
7.1. Important or mean change in person's quality of life as defined by each study.
8. Satisfaction with care
8.1. Important or mean change in satisfaction of participant or care provider as defined by each study.
9. Cost of care
10. 'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2011) and used GRADE profiler (GRADEpro) to import data from Review Manager 5 (RevMan 2014) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined and the sum of available data on all outcomes we rated as important to patient‐care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
-
Global state: clinically important improvement.
-
Mental state: clinically important improvement.
-
Adverse events: incidence of any adverse event ‐ movement disorders (See Differences between protocol and review).
-
Service use: readmission due to relapse.
-
Satisfaction of participant or care provider with treatment.
-
Behaviour: aggressive or violent behaviour.
-
Cost of care.
Search methods for identification of studies
Electronic searches
1. Cochrane Schizophrenia Group's Study‐Based Register of Trials
The information Specialist searched the Register on 20 November 2015 and 23 November 2016 using the following search strategy:
(*Chlorpromazine* AND *Metiapine*) in Intervention Field of STUDY
In such a study‐based register, searching the major concept retrieves all the synonyms and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics.
This register is compiled by systematic searches of major resources (including MEDLINE, Embase, AMED, BIOSIS, CINAHL, PsycINFO, PubMed and registries of clinical trials) and their monthly updates, handsearches, grey literature and conference proceedings (see Group’s Module). There is no language, date, document type or publication status limitations for inclusion of records into the register.
Searching other resources
1. Reference searching
We inspected references of all included studies for further relevant studies.
2. Personal contact
We did not need to contact the first author of any included study for information regarding unpublished trials.
Data collection and analysis
Selection of studies
One review author (MZ) independently inspected citations from the searches and identified relevant abstracts and the second review author (AB) independently reinspected a random 50% sample to ensure reliability. One review author (MZ) obtained and inspected full reports of the abstracts meeting the review criteria and the second review author (AB) reinspected a random 50% of the full reports to ensure reliable selection. Had it not been possible to resolve disagreement by discussion, we would have attempted to contact authors of studies for clarification.
Data extraction and management
1. Extraction
One review author (MZ) extracted data from all included studies. In addition, to ensure reliability, the second review author (AB) independently extracted data from a random sample of these studies, comprising 50% of the total. We extracted data presented only in graphs and figures whenever possible, but included only if both review authors independently had the same result. If studies had been multicentre, we would have extracted data relevant to each component centre separately.
2. Management
2.1. Forms
We extracted data onto standard, simple forms.
2.2. Scale‐derived data
We included continuous data from rating scales only if:
-
the psychometric properties of the measuring instrument were described in a peer‐reviewed journal (Marshall 2000); and
-
the measuring instrument was not written or modified by one of the trialists for that particular trial;
-
the instrument was a global assessment of an area of functioning and not subscores which are not, in themselves, validated or shown to be reliable. However, there are exceptions, we would have included subscores from mental state scales measuring positive and negative symptoms of schizophrenia (see Differences between protocol and review).
Ideally, the measuring instrument should either be a self‐report or completed by an independent rater or relative (not the therapist). We realised that this was not often reported clearly, therefore, we noted if this was the case or not in 'Description of studies'.
2.3. Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. However, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult‐to‐measure conditions such as schizophrenia. We decided primarily to use endpoint data, and only use change data if endpoint data were not available. Where relevant, we would have combined endpoint and change data in the analysis as we preferred to use mean differences (MD) rather than standardised mean differences (SMD) throughout (Higgins 2011a).
2.4. Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we intended to apply the following standards to relevant data.
We would have entered data from studies of at least 200 participants in the analysis irrespective of the following rules, because skewed data pose less of a problem in large studies. We also entered all useable change data as when continuous data are presented on a scale that included a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not.
For endpoint data from studies with fewer than 200 participants:
-
if a scale started from the finite number zero, we would have subtracted the lowest possible value from the mean, and divide this by the standard deviation (SD). If this value is lower than one, it strongly suggests a skew and such data would not be used. If this ratio is higher than one but below two, there is a suggestion of skew. We would have entered these data and tested whether their inclusion or exclusion had changed the results substantially. Finally, if the ratio is larger than two we would have used these data, because skew is less likely (Altman 1996; Higgins 2011b);
-
if a scale starts from a positive value (such as the Positive and Negative Syndrome Scale (PANSS) (Kay 1986)), which could have values from 30 to 210), the calculation described above is modified to take the scale starting point into account. In these cases, skew is present if 2 SD > (S ‐ Smin), where S is the mean score and 'Smin' is the minimum score.
2.5. Common measure
To facilitate comparison between trials, we intended to convert variables that could be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6. Conversion of continuous to binary
Where possible, we made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there was a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS (Kay 1986), this can be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7. Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for chlorpromazine versus metiapine. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'not un‐improved') we reported data where the left of the line indicated an unfavourable outcome and noted it in the relevant graphs.
Assessment of risk of bias in included studies
Both review authors worked independently to assess risk of bias using criteria described in the Cochrane Handbook for Systemic Reviews of Interventions (Higgins 2011c) to assess trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting. We did not need to contact the authors of the studies to obtain further information.
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and those odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). The number needed to treat for an additional beneficial (NNTB) or harmful NNTH) outcome statistic with its CIs is intuitively attractive to clinicians but is problematic both in its accurate calculation in meta‐analyses and interpretation (Hutton 2009). For binary data presented in the 'Summary of findings' table, where possible, we calculated illustrative comparative risks.
2. Continuous data
For continuous outcomes, we estimated MD between groups. We preferred not to calculate effect size measures (SMD). However, if scales of very considerable similarity had been used, we would have presumed there was a small difference in measurement, and calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Authors often fail to account for intraclass correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
If clustering had not been accounted for in primary studies, we would have presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error and attempt to contact first authors of studies to obtain intraclass correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). If clustering was incorporated into the analysis of primary studies, we would have presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC (design effect = 1 + (m ‐ 1) × ICC) (Donner 2002). If the ICC is not reported in future trials, we will assume it to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed taking into account ICCs and relevant data documented in the report, synthesis with other studies will be possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. This occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a washout phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used data from the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, we would have presented the additional treatment arms in comparisons. If data were binary, we would have simply added these and combined them within the two‐by‐two table. If data were continuous, we would have combined data following the formula in the Cochrane Handbook for Systemic Reviews of Interventions (Higgins 2011a). Where the additional treatment arms were not relevant, we did not use these data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss to follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data or use them within analyses. However, if more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we would have addressed this within the 'Summary of findings' table by downgrading quality. Finally, we also downgraded quality within the 'Summary of findings' table where loss was 25% to 50% in total.
2. Binary
In the case where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis (ITT)); where studies did not use an ITT analysis, we presented completer only data.
3. Continuous
3.1. Attrition
In the case where attrition for a continuous outcome was between 0% and 50%, and data only from people who completed the study to that point were reported, we reproduced these.
3.2. Standard deviations
If SDs were not reported, we would have tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and CIs available for group means, and either a 'P' value or 't' value available for differences in mean, we can calculate them according to the rules described in the Cochrane Handbook for Systemic Reviews of Interventions (Deeks 2011): when only the SE is reported, SDs are calculated by the formula SD = SE × square root (n). The Cochrane Handbook for Systemic Reviews of Interventions presents detailed formulae for estimating SDs from P values, t or F values, CIs, ranges or other statistics (Deeks 2011). If these formulae did not apply, we can calculate the SDs according to a validated imputation method, which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies could introduce error, the alternative is to exclude a given study's outcome and thus to lose information. We did not impute any SDs, if we had we would have examined the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3. Assumptions about participants who left the trials early or were lost to follow‐up
Various methods are available to account for participants who left the trials early or are lost to follow‐up. Some trials just present the results of study completers, others use the method of last observation carried forward (LOCF), while more recently, methods such as multiple imputation or mixed‐effects models for repeated measurements (MMRM) have become more of a standard. While the latter methods seem to be somewhat better than LOCF (Leon 2006), we feel that the high percentage of participants leaving the studies early and differences in the reasons for leaving the studies early between groups are often the core problem in randomised trials of schizophrenia. Therefore, we did not exclude studies based on the statistical approach used. However, we preferred to use the more sophisticated approaches (e.g. we preferred MMRM or multiple‐imputation to LOCF and to only present completer analyses if some type of ITT data were not available at all). Moreover, we addressed this issue in the item "incomplete outcome data" of the 'Risk of bias' tool.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations which we had not predicted would arise. If such situations or participant groups arose, we would have fully discussed them.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. If such methodological outliers arose, we would have fully discussed them.
3. Statistical heterogeneity
3.1. Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2. Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 statistic alongside the Chi2 'P' value. The I2 statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 statistic depends on the magnitude and direction of effects and the strength of evidence for heterogeneity (e.g. a 'P' value from the Chi2 test, or a CI for the I2 statistic). We considered an I2 statistic estimate of 50% or greater accompanied by a statistically significant Chi2 statistic as evidence of substantial levels of heterogeneity (Deeks 2011). Had we found substantial levels of heterogeneity in the primary outcome, we would have explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in the Cochrane Handbook for Systemic Reviews of Interventions (Sterne 2011). We tried to locate protocols of included randomised trials. If the protocol had been available, we would have compared outcomes in the protocol and in the published report. If the protocol was not available, we compared outcomes listed in the methods section of the trial report with actually reported results.
2. Funnel plot
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Chapter 10 of the Cochrane Handbook for Systemic Reviews of Interventions (Sterne 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We would not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar size. In future updates, where funnel plots are possible, we will seek statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies, even if there is no statistically significant heterogeneity. However, there is a disadvantage to the random‐effects model. It puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose to use random‐effects models in data analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
1.1. Primary outcomes
No subgroup analysis was anticipated.
1.2. Clinical state, stage or problem
We proposed to undertake this review and provide an overview of the effects of chlorpromazine versus metiapine for people with schizophrenia in general. In addition, we planned to report data on subgroups of people in the same clinical state, stage and with similar problems if the data were available.
2. Investigation of heterogeneity
We intended to report high inconsistency. First, we would have investigated whether data had been entered correctly. Second, if data were correct, we would have visually inspected the graph and removed outlying studies to see if homogeneity was restored. For this review, we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would present such data. If not, we would not pool these data but discuss them instead. We knew of no supporting research for this 10% cut off but are investigating the use of prediction intervals as an alternative to this unsatisfactory state.
If unanticipated clinical or methodological heterogeneity were obvious, we would have simply stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
We did not include any sensitivity analyses in this review as most outcomes were reported by only one study. If we had, we would have followed the methods below
1. Implication of randomisation
Where included studies implied randomisation, we would have included them in a sensitivity analysis. For the primary outcomes, we would have included these studies and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then we would have used all relevant data from these studies.
2. Assumptions for lost binary data
Where assumptions had to be made regarding people lost to follow‐up (see Dealing with missing data), we would have compared the findings of the primary outcomes when we used our assumption/s and when we used data only from people who completed the study to that point. If there was a substantial difference, we would have reported results and discussed them but continued to employ our assumption.
Where assumptions had to be made regarding missing SDs data (see Dealing with missing data), we would have compared the findings of the primary outcomes when we used our assumption/s and when we used data only from people who completed the study to that point. We would have undertaken a sensitivity analysis to test how prone results were to change when completer‐only data were compared to the imputed data using the above assumption. If there was a substantial difference, we would have reported results and discussed them but continued to employ our assumption.
3. Risk of bias
We would have analysed the effects of excluding trials that were judged at high risk of bias across one or more of the domains of randomisation (see Assessment of risk of bias in included studies). If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then we would have included data from these trials in the analysis.
4. Imputed values
We also would have undertaken a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster randomised trials.
If there were substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we would not have pooled data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
5. Fixed and random effects
We synthesised all data using a random‐effects model; however, we also synthesised data for the primary outcome using a fixed‐effect model to evaluate whether this altered the significance of the results. If results had been different, we would have discussed this issue.
Results
Description of studies
See Characteristics of included studies table.
Results of the search
Details of the search results are illustrated in the PRISMA table (Figure 3).

Study flow diagram.
Initially we identified four records that seemed relevant to this review. After checking for duplicates, we inspected full text of all remaining reports. We then grouped these into 'studies' where two reports referred to the same trial. Of these, after scrutiny, we included four publications reporting three studies and did not exclude any.
Included studies
We included three studies (Kramer 1975; Simpson 1973; Steinbook 1975).
1. Methods
Two included studies were parallel (Kramer 1975; Steinbook 1975) and one was cross‐over (Simpson 1973). All studies were randomised.
2. Length of trials
All studies were short term. Kramer 1975 was four weeks and Steinbook 1975 was six weeks. Simpson 1973 was the longest with a total duration of 28 weeks, 12 weeks up to the point of cross‐over.
3. Participants
All studies stated that their participants were adults (aged 18 to 60 years) with schizophrenia. Only Kramer 1975 described the diagnostic criteria using clinical diagnosis of schizophrenia of recent onset or exacerbation and an elevated combined score of 12 on five selected items of the BPRS. All participants were inpatients, participants in Kramer 1975 were newly admitted.
4. Setting
All studies were single site and conducted in hospital.
5. Study size
Study size was small ranging from 11 (Simpson 1973) to 90 (Kramer 1975) participants per trial. A total of 161 participants were randomised.
6. Intervention
6.1. Chlorpromazine
The doses of chlorpromazine ranged from 50 mg/day to 1200 mg/day with a mean of 427 mg/day (Kramer 1975); 150 mg/day to 900 mg/day with a mean of 627 mg/day (Steinbook 1975); and 100 mg/day to 600 mg/day (mean dose was not provided) (Simpson 1973).
6.2. Metiapine
The doses of metiapine in included studies ranged from 25 mg/day to 600 mg/day (mean dose 219 mg/day) (Kramer 1975); 75 mg/day to 450 mg/day (mean dose 287 mg/day) (Steinbook 1975); and 100 mg/day to 600 mg/day (mean dose was not provided) (Simpson 1973).
6.3. Combination of butabarbital and atropine
Kramer 1975 had a third treatment arm where participants were randomised to receive a combination of butabarbital and atropine. We did not use these data for this review.
7. Outcomes
The studies reported the following outcomes: global state, adverse effects, leaving the study early and service utilisation. None of the included studies reported data for mental state, relapse, quality of life, levels of satisfaction, service use, behaviour or cost of care. Most outcomes reported were dichotomous. Kramer 1975 reported continuous outcomes that could be dichotomised.
7.1. Outcome scales
The following scale provided continuous data for the analysis.
7.1.1. Global state
i. Clinical Global Impression (CGI) (Guy 1976)
The CGI scale enables clinicians to quantify severity of illness and overall clinical improvement. In this 7‐point scoring system, lower scores indicated decreased severity or better recovery (used by Kramer 1975; Steinbook 1975).
ii. Nurse's Observation Scale for Inpatient Evaluation (NOSIE) (Honigfeld 1965)
The NOSIE is an 80‐item scale with items rated on a 5‐point scale from 0 (not present) to 4 (always present). Ratings are based on behaviour over the previous three days. The seven headings are social competence, social interest, personal neatness, cooperation, irritability, manifest psychosis and psychotic depression. The total score ranges from 0 to 320 with high scores indicating a poor outcome.
Excluded studies
We have no excluded studies in this review.
Risk of bias in included studies
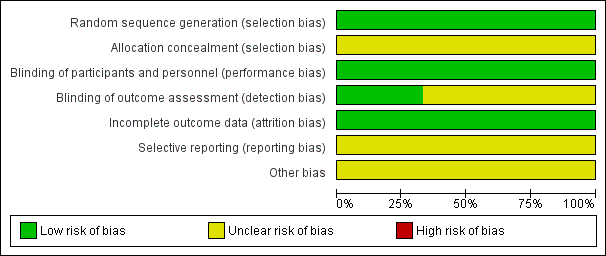
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All studies described the methods used for random sequence generation and rated as low risk. One study reported that participants were randomly assigned in blocks of six (Kramer 1975), and one study reported that participants were randomised using table of random numbers (Simpson 1973). Steinbook 1975 reported participants were randomly allocated to groups and the resulting groups did not differ significantly with respect to age, race, education or number of previous hospitalisations. However, none of the studies described allocation concealment and we rated all three as unclear risk for selection bias.
Blinding
All studies reported as double‐blind studies and rated as low risk for performance bias. However, only one study stated that in addition to blinding of treatment, the physicians or nurses conducting outcome assessment were also blind (Simpson 1973). Kramer 1975 and Steinbook 1975 did not provide this information and we rated these two studies as unclear risk for detection bias.
Incomplete outcome data
All studies were at low risk of bias for incomplete outcome data as all studies addressed missing data issues clearly.
Selective reporting
All studies had unclear risks of bias for selective reporting as, although all expected outcomes were reported in the studies, some means and SDs were not reported.
Other potential sources of bias
Kramer 1975 and Simpson 1973 had an unclear risk of other bias as the treatments used were supplied by the pharmaceutical industry but declared no source of funding. Steinbook 1975 did not report source of funding, again we rated 'other bias' for this study to be unclear.
Effects of interventions
See: Summary of findings for the main comparison Chlorpromazine versus metiapine for schizophrenia
Studies relevant to this review fall into a single comparison. We were able to extract numerical data from three randomised studies.
1. COMPARISON 1: CHLORPROMAZINE versus METIAPINE (all data short term)
1.1 Global state: 1a. Clinically important improvement (CGI, high=poor)
Two trials with a total of 120 participants reported data. We did not find evidence of clear difference between chlorpromazine and metiapine (RR 1.11 95% CI 0.84 to 1.47, Analysis 1.1).
1.2 Global state: 2a. Overall: remaining symptomatic
One trial involving ten participants reported data for numbers of participants remaining symptomatic throughout the trial. We did not find evidence that chlorpromazine was clearly different in its effects compared with metiapine (RD 0.00 95% CI ‐0.31 to 0.31, Analysis 1.2).
1.3 Global state: 3. Overall: mean endpoint score (NOSIE, high=poor)
One trial, with a total of 60 participants reported data. We did not find evidence of a clear difference between chlorpromazine and metiapine (MD 0.20 95% CI ‐3.49 to 3.89, Analysis 1.3).
1.4 Adverse effects: 1. General: "severe reactions"
Three studies involving 140 participants reported data. We did not find evidence of a clear difference between chlorpromazine and metiapine for numbers of participants with "severe reactions" (RR 0.79 95% CI 0.41 to 1.49, Analysis 1.4).
1.6 Adverse effects: 2a. Specific: central nervous system ‐ drowsiness
A single study, with a total of 60 participants reported data. We did not find evidence of a clear difference between chlorpromazine and metiapine (RR 1.00 95% CI 0.15 to 6.64, Analysis 1.5).
1.7 Adverse effects: 2b. Specific: hepatic ‐ abnormal liver function test
Two studies, with a total of 120 participants , reported data. There was not a clear difference between chlorpromazine and metiapine (RR 1.35 95% CI 0.17 to 10.75, Analysis 1.6).
1.8 Adverse effects: 2c. Specific: movement disorders
We did not find evidence of a clear difference between chlorpromazine and metiapine for akathisia (1 RCT, n=60, RR 1.00 95% CI 0.32 to 3.10), dystonic symptoms (1 RCT, n=60, RR 5.00 95% CI 0.25 to 99.95), parkinsonism (2 RCTs, n=70, RR 0.97 95% CI 0.46 to 2.03), rigidity (1 RCT, n=60, RR 0.63 95% CI 0.23 to 1.69), tremor (1 RCT, n=60, RR 0.33 95% CI 0.01 to 7.87) or 'use of anti‐parkinson or other remedial medication' (1 RCT, n=10, RR 1.22 95% CI 0.73 to 2.06, Analysis 1.7).
1.9 Adverse effects: 2d. Specific: others
1.9.1 Autonomic ‐ blurred vision
A single trial, with a total of 60 participants provided data. We did not find evidence of a clear difference between chlorpromazine and metiapine (RR 5.00 95% CI 0.25 to 99.95, Analysis 1.8).
1.9.2 Cardiovascular (heart) problem
Three trials, which included a total of 130 participants reported data. we did not find evidence of a clear difference between chlorpromazine and metiapine (RR 0.52 95% CI 0.09 to 3.08). This subgroup had important levels of heterogeneity (Chi2=2.88; df=1.0; P=0.09; I2=65%, Analysis 1.8).
1.9.3 Ophthalmological ‐ "eye changes"
One trial, with a total of ten participants reported data. No one had eye changes in either group (Analysis 1.8).
1.10 Leaving the study
Two trials, with a total of 70 participants reported data. We did not find evidence of a clear difference between chlorpromazine and metiapine in this comparison (RR 0.33 95% CI 0.01 to 7.87, Analysis 1.9).
1.11 Service utilisation: staying in hospital
A single study, with a total of 60 participants reported data. There was not a clear difference between chlorpromazine and metiapine (RR 1.05 95% CI 0.74 to 1.48, Analysis 1.10).
Discussion
Summary of main results
The summary below indicates the outcomes selected for the summary of findings Table for the main comparison, and highlights other important findings of this review for evidence‐based decision making.
1. Global state
By analysing the results of two trials (total n = 120), there was no indication of a difference between metiapine and the much better established chlorpromazine for improvement of global state. With short‐ and medium‐term data for chlorpromazine versus placebo, around one third of people do improve over and above that improvement that may come even with placebo (Adams 2014). If that held true for metiapine then that would be most important, but results cannot be regarded as conclusive without further trials.
2. Mental state
All data in this review were very limited, but there were no usable data on mental state that could be extracted from the trials. This is not that unusual and certainly not particular to this comparison but the case for retaining metiapine as a useful drug must be damaged by poor reporting or not recording such important outcomes in trials.
3. Adverse effects
Acute movement disorders, parkinsonism, tremor and rigidity are among well‐recognised adverse effects reported by people using chlorpromazine (Adams 2014; Tardy 2014a). As a low‐potency antipsychotic drug, chlorpromazine produces movement disorders, dizziness, sedation and weight gain (Tardy 2014b). The very few studies in this review supported these findings. Our results did not illustrate any clear difference between chlorpromazine and metiapine ‐ although more data are needed to be sure of this.
4. Leaving the study early
There is evidence that using chlorpromazine leads to more people staying in the study ‐ perhaps through a mechanism of decreasing distressing symptoms and increasing the person's concordance with treatment (Adams 2014). In our review, only two studies specifically reported the number of people who left the study early (total n = 70) and there was no clear difference between groups ‐ only one person left. Larger numbers over longer periods of time would have been helpful but, on this low evidence, there was no indication that metiapine is any more unacceptable than chlorpromazine.
5. Service use
One study found no difference between groups for numbers able to be discharged from hospital at the end of the trial. This is a useful and widely understandable outcome. Data were few and of poor quality so confidence in this finding was low.
6. Satisfaction of participant or care provider with treatment/behaviour/cost of care
Because of no data or lack of usable data we can only highlight that we do not have any quantitative data from trials. There are many large gaps in our knowledge to compare metiapine versus chlorpromazine. These gaps could be easily rectified should a trial be designed and conducted.
Overall completeness and applicability of evidence
1. Completeness
Of the three included studies, all reported some data for the primary outcome of global state but this amounted to poor‐quality data on 120 people in the short term. There were no long‐term data. There were no usable data for mental state, a key outcome of this review. Similarly, often only one or two studies reported on secondary outcomes and all trials were small so all data were incomplete or entirely absent. All included studies were characterised by small sample sizes (fewer than 100 participants). Almost 1000 participants are required to be included in psychiatric meta‐analyses for the results to be sound and robust (Trikalinos 2004). This review included 161 participants overall (120 for the primary outcome), so all analyses were underpowered. There were no data on behaviour, quality of life, level of satisfaction with care and cost of care. It seems likely that the data will remain incomplete. New larger well‐designed studies comparing chlorpromazine with metiapine are desirable but probably unlikely. This state of incompleteness probably represents the most compete data set we are likely to get.
2. Applicability
Included studies were from the 1970s. In these studies, participants were diagnosed by less rigorous criteria than tends to be seen in modern trials but this, conversely, could make them more applicable to everyday care rather than less. Clinicians tend not to use very rigid rigorous criteria for diagnosis of schizophrenia in clinical care. Misdiagnoses are made but most people with a clinical diagnosis of schizophrenia probably do really have the illness. These old trials may be more like clinical care than would be seen nowadays. All included studies were conducted in hospitals. Therefore, applicability towards people with schizophrenia in the community must be made by caution. All participants were inpatients, which could have contributed to the very low attrition rate that probably would have been different should the studies have been conducted in the community.
Quality of the evidence
The quality of the current evidence was low to very low based on GRADE (Schünemann 2011). Three studies reported the method of randomisation and only two reported the method of allocation concealment. All studies were reported as double blind, but only one stated that the assessor was blinded to which drug participants were receiving. Studies often reported no usable data on important outcomes as they solely reported statistical measures of probability (P value) or means without any SDs or SEs. Inconsistency, imprecision and publication bias of one included study were also problematic. This led to us grading the evidence as low quality in the 'Summary of findings' table.
Potential biases in the review process
The search was based on Cochrane Schizophrenia Trials Register. There may be some unpublished trials that we were unaware of. At the time this review's trials were published, there was pharmaceutical industry interest in the findings and this could have led to publication or reporting bias.
Agreements and disagreements with other studies or reviews
We are unaware of any other systematic reviews on the efficacy of chlorpromazine versus metiapine for schizophrenia.

Chlorpromazine ‐ structure.

Metiapine structure.

Study flow diagram.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Comparison 1 CHLORPROMAZINE versus METIAPINE (all data short term), Outcome 1 Global state: 1. Clinically important improvement (CGI, high = poor)).

Comparison 1 CHLORPROMAZINE versus METIAPINE (all data short term), Outcome 2 Global state: 2a. Overall: remaining symptomatic.

Comparison 1 CHLORPROMAZINE versus METIAPINE (all data short term), Outcome 3 Global state: 2b. Overall: mean endpoint score (NOSIE, high = poor).

Comparison 1 CHLORPROMAZINE versus METIAPINE (all data short term), Outcome 4 Adverse effects: 1. General: "severe reactions".

Comparison 1 CHLORPROMAZINE versus METIAPINE (all data short term), Outcome 5 Adverse effects: 2a. Specific: central nervous system ‐ drowsiness.

Comparison 1 CHLORPROMAZINE versus METIAPINE (all data short term), Outcome 6 Adverse effects: 2b. Specific: hepatic ‐ abnormal liver function test.

Comparison 1 CHLORPROMAZINE versus METIAPINE (all data short term), Outcome 7 Adverse effects: 2c. Specific: movement disorders.

Comparison 1 CHLORPROMAZINE versus METIAPINE (all data short term), Outcome 8 Adverse effects: 2d. Specific: others.

Comparison 1 CHLORPROMAZINE versus METIAPINE (all data short term), Outcome 9 Leaving the study.

Comparison 1 CHLORPROMAZINE versus METIAPINE (all data short term), Outcome 10 Service utilisation: staying in hospital.
| Methods | Allocation: randomised, with sequence generation and concealment of allocation clearly described. |
| Participants | Diagnosis: people with schizophrenia ‐ however diagnosed.* |
| Interventions | 1. Metiapine: about 200 mg/day. n = 150. |
| Outcomes | Global state ‐ relapse, clinically important change. Mental state ‐ general ‐ clinically important change in mental state, mean change in negative symptoms. Adverse effects ‐ incidence of serious adverse events/effects, clinically important extrapyramidal symptoms. Leaving the study early ‐ for any reason. Cost of care. Service outcomes: admitted, number of admissions, length of hospitalisation, discharge, contacts with psychiatric services. Compliance with drugs. Economic evaluations: cost‐effectiveness, cost‐benefit. |
| Notes | * This could be diagnosed by clinical decision. If funds were permitting all participants could be screened using operational criteria, otherwise a random sample should suffice. ** Size of study with sufficient power to highlight about a 10% difference between groups for primary outcome. |
| n: number of participants. | |
| Chlorpromazine versus metiapine for schizophrenia | ||||||
| Patient or population: people with schizophrenia | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Chlorpromazine | Metiapine | |||||
| Global state: clinically important improvement | 600 per 1000 | 684 per 1000 | RR 1.11 | 120 | ⊕⊝⊝⊝ | 1 trial reported CGI at 4 weeks, and 1 trial reported CGI at 6 weeks' follow‐up, both indicated no significant difference between the 2 drugs. |
| Mental state: clinically important improvement | No studies reported this outcome. | |||||
| Adverse effects. Specific: movement disorders ‐ parkinsonism | 168 per 1000 | 144 per 1000 | RR 0.97 | 70 | ⊕⊝⊝⊝ | ‐ |
| Service use: readmission due to relapse | No studies reported on these outcomes. | |||||
| Satisfaction of participant or care provider with treatment | ||||||
| Behaviour: aggressive or violent behaviour | ||||||
| Cost of care | ||||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Risk of bias: serious (downgraded by 1 level): study had unclear risk of other biases as supported by a pharmaceutical company for drug supply. | ||||||
| Review title | Reference |
| Acetophenazine versus chlorpromazine for schizophrenia. | |
| Chlorpromazine dose for people with schizophrenia. | |
| Cessation of medication for people with schizophrenia already stable on chlorpromazine. | |
| Chlorpromazine versus atypical antipsychotic drugs for schizophrenia. | |
| Chlorpromazine versus clotiapine for schizophrenia. | |
| Chlorpromazine versus metiapine for schizophrenia. | This review |
| Chlorpromazine versus penfluridol for schizophrenia. | |
| Chlorpromazine versus piperacetazine for schizophrenia. | |
| Chlorpromazine versus placebo for schizophrenia. | |
| Chlorpromazine for psychosis induced aggression or agitation. | |
| Haloperidol versus chlorpromazine for schizophrenia. | |
| * | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Global state: 1. Clinically important improvement (CGI, high = poor)) Show forest plot | 2 | 120 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.84, 1.47] |
| 2 Global state: 2a. Overall: remaining symptomatic Show forest plot | 1 | 10 | Risk Difference (M‐H, Random, 95% CI) | 0.0 [‐0.31, 0.31] |
| 3 Global state: 2b. Overall: mean endpoint score (NOSIE, high = poor) Show forest plot | 1 | 60 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐3.49, 3.89] |
| 4 Adverse effects: 1. General: "severe reactions" Show forest plot | 3 | 140 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.41, 1.49] |
| 5 Adverse effects: 2a. Specific: central nervous system ‐ drowsiness Show forest plot | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.64] |
| 6 Adverse effects: 2b. Specific: hepatic ‐ abnormal liver function test Show forest plot | 2 | 120 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [0.17, 10.75] |
| 7 Adverse effects: 2c. Specific: movement disorders Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Akathisia | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.32, 3.10] |
| 7.2 Dystonic symptoms | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 99.95] |
| 7.3 Parkinsonism | 2 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.46, 2.03] |
| 7.4 Rigidity | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.23, 1.69] |
| 7.5 Tremor | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 7.6 Use of anti‐parkinson or other remedial medication | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.73, 2.06] |
| 8 Adverse effects: 2d. Specific: others Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Autonomic ‐ blurred vision | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 99.95] |
| 8.2 Cardiovascular (heart) problem | 3 | 130 | Risk Ratio (M‐H, Random, 95% CI) | 0.52 [0.09, 3.08] |
| 8.3 Ophthalmological ‐ "eye changes" | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 9 Leaving the study Show forest plot | 2 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.87] |
| 10 Service utilisation: staying in hospital Show forest plot | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.74, 1.48] |