Dosis única de dipirona (metamizol) para el dolor postoperatorio agudo en adultos
Resumen
Antecedentes
La dipirona (metamizol) es un fármaco antiinflamatorio no esteroide utilizado en algunos países para tratar el dolor (postoperatorio, cólico, cáncer y migraña); está prohibido en otros países debido a su asociación con trastornos sanguíneos potencialmente mortales. Esta revisión reemplaza una revisión Cochrane de 2010 que se ha retirado.
Objetivos
Evaluar la eficacia analgésica y los efectos adversos asociados con una dosis única de dipirona para el dolor posoperatorio agudo moderado a intenso con métodos que permitan la comparación con otros analgésicos evaluados en ensayos estandarizados que utilicen métodos y resultados casi idénticos.
Métodos de búsqueda
Se realizaron búsquedas en el Registro Cochrane Central de Ensayos Controlados (Cochrane Central Register of Controlled Trials, CENTRAL), MEDLINE, EMBASE y LILACS hasta el 11 de agosto de 2015; la Oxford Pain Relief Database; dos registros de ensayos clínicos; y las listas de referencias de los artículos.
Criterios de selección
Se incluyeron los ensayos aleatorizados, doble ciego, controlados con placebo de una dosis única de dipirona para el alivio del dolor postoperatorio moderado a intenso en adultos. Se aceptaron las vías orales, rectales, intramusculares e intravenosas de administración.
Obtención y análisis de los datos
Dos autores de la revisión consideraron de forma independiente los estudios para la inclusión en la revisión, evaluaron el riesgo de sesgo y extrajeron los datos. Se utilizó la suma total de alivio del dolor o la diferencia en la intensidad del dolor (TOTPAR o SPID) durante cuatro a seis horas para calcular el número de participantes que lograron al menos un alivio del dolor del 50%. A partir de los resultados obtenidos se calculó el riesgo relativo y el número necesario a tratar para lograr un resultado beneficioso adicional (NNTB), con intervalos de confianza (IC) del 95%, para que un participante presentara al menos un alivio del dolor del 50% durante cuatro a seis horas en comparación con placebo. Se analizó el uso de medicación de rescate y el tiempo transcurrido hasta el uso de la medicación de rescate como medidas adicionales de eficacia. También se buscó información sobre los eventos adversos y los retiros.
Resultados principales
Se incluyeron ocho estudios con 809 participantes que compararon dipirona oral 500 mg (143 participantes), dipirona oral 1000 mg (57 participantes) y dipirona intramuscular 2000 mg (35 participantes) con placebo (236 participantes). Además del placebo, todos los estudios utilizaron controles activos (ibuprofeno, paracetamol, aspirina, flurbiprofeno, ketoprofeno; 338 participantes). Siete estudios utilizaron la vía oral de administración, y un estudio utilizó la vía intramuscular. La media de la edad varió entre 23 y 62 años. Seis estudios incluyeron hombres y mujeres y dos estudios sólo incluyeron mujeres. Todos los estudios fueron pequeños pero tuvieron una calidad moderada a buena.
Más del 70% de los participantes presentaron el resultado primario de al menos un alivio del dolor del 50% durante cuatro a seis horas con dipirona oral 500 mg en comparación con el 30% con placebo (cinco estudios, 288 participantes; NNT 2,4 (IC del 95%: 1,8 a 3,1)) (evidencia de calidad moderada). No hubo datos suficientes para evaluar otras dosis o vías de administración de la dipirona.
Menos participantes necesitaron medicación de rescate en el transcurso de cuatro a seis horas con dipirona 500 mg que con placebo (7% con dipirona versus 34% con placebo; cuatro estudios, 248 participantes) (evidencia de calidad baja).
Los datos sobre los números de participantes que presentaron cualquier evento adverso se informaron de forma inconsistente y no fue posible realizar un análisis. No se informaron eventos adversos graves o retiros debido a eventos adversos (evidencia de calidad muy baja).
Hubo muy pocos datos para comparar la dipirona directamente con otros tratamientos activos.
Conclusiones de los autores
Según información muy limitada, una dosis única de dipirona 500 mg proporciona buen alivio del dolor en cerca del 70% de los pacientes tratados, en comparación con cerca del 30% con placebo. Por cada cinco pacientes que reciben dipirona 500 mg, dos pacientes presentarán este nivel de analgesia durante cuatro a seis horas y no lo habrían logrado con placebo y menos pacientes necesitarían medicación de rescate.
No fue posible comparar la dipirona directamente con otros tratamientos activos, ni evaluar los efectos de diferentes dosis o vías de administración, ni determinar el número de participantes que presentan eventos adversos debido a que no hubo suficientes datos y el informe fue insuficiente.
PICO
Resumen en términos sencillos
Dosis única de dipirona para el tratamiento del dolor postoperatorio agudo
Conclusión
La dipirona (metamizol) a una dosis oral única de 500 mg produce un alivio aceptable del dolor en cerca de siete de diez pacientes con dolor agudo moderado o intenso.
Antecedentes
El dolor agudo dura un tiempo corto y a menudo se percibe poco después de la lesión. La mayoría de los pacientes a los que se les realiza cirugía presenta dolor moderado o intenso después del procedimiento. Los analgésicos se prueban al administrárselos a pacientes que presentan dolor, a menudo después de la extracción de los cordales u otra cirugía menor. Habitualmente este dolor se trata con analgésicos tomados por vía oral. Los resultados pueden aplicarse a otras formas de dolor agudo. Esta es una revisión de una serie de revisiones Cochrane que analizan la eficacia de los analgésicos.
La dipirona es un fármaco popular para el alivio del dolor en algunos países y se utiliza para tratar el dolor postoperatorio, el dolor tipo cólico (dolor repentino en la barriga), el dolor por cáncer y la migraña (cefalea grave). Otros países (Japón, Reino Unido, EE.UU.) han prohibido su uso debido a una asociación con trastornos sanguíneos potencialmente mortales como la agranulocitosis (deficiencia de ciertos glóbulos).
Características de los estudios
Se efectuaron búsquedas en las bases de datos médicas de estudios de la dipirona utilizada para tratar el dolor posterior a la cirugía en adultos y en comparación con placebo (un tratamiento simulado). Los fármacos se podían administrar por vía oral, en una vena, en un músculo, o en el recto. La evidencia está actualizada hasta el 11 de agosto de 2015. Se encontraron ocho estudios con 809 participantes tratados con dipirona, placebo y otros analgésicos. Todos los estudios fueron pequeños, pero tuvieron una calidad moderada a buena.
Resultados clave
Una dosis única de 500 mg de dipirona proporcionó alivio efectivo del dolor (50% o más de reducción del dolor durante cuatro a seis horas) para siete de cada diez (70%) participantes, en comparación con tres de cada diez (30%) con placebo (cinco estudios, 288 participantes en la comparación; evidencia de calidad moderada), y menos participantes necesitan analgésicos adicionales dentro de las cuatro a seis horas (7% con dipirona, 34% con placebo; cuatro estudios, 248 participantes; evidencia de calidad baja).
Hubo muy pocos datos para comparar la dipirona directamente con otros analgésicos.
Hubo muy poca información disponible para establecer cualquier conclusión acerca de otras dosis y formas de administrar la dipirona utilizadas en estos estudios, o acerca del número de pacientes que presentaron efectos secundarios. Los estudios informaron que no hubo efectos secundarios graves ni pacientes que abandonaran los estudios debido a los efectos secundarios, aunque no todos los estudios proporcionaron información sobre estos resultados.
Authors' conclusions
Summary of findings
| Oral dipyrone 500 mg compared with placebo for acute postoperative pain | ||||||
| Patient or population: adults with acute postoperative pain Settings: clinic Intervention: oral dipyrone 500 mg Comparison: placebo | ||||||
| Outcomes | Probable outcome with | Relative effect and NNT or NNTp | Number of studies, participants, events | Quality of the evidence | Comments | |
| intervention | comparator | |||||
| At least 50% of maximum pain relief over 4 to 6 h | 730 in 1000 | 320 in 1000 | RR 2.4 (95% CI 1.8 to 3.1) NNT 2.4 (1.9 to 3.1) | 5 studies, 288 participants, 151 events | Moderate | Small studies, few events |
| Participants remedicating within 4 to 6 h | 70 in 1000 | 340 in 1000 | RR 0.21 (0.11 to 0.40) NNTp 3.6 (2.7 to 5.4) | 4 studies, 248 participants, 51 events | Low | Small studies, very few events |
| Participants with at least one adverse event | Insufficient data for analysis | ‐ | ‐ | ‐ | ‐ | |
| Participants with a serious adverse event | None reported | None reported | ‐ | 5 studies, 288 participants, no events | Very low | Small studies, no events |
| CI: confidence interval; h: hour; RR: risk ratio; NNT: number needed to treat for an additional beneficial outcome; NNTp: number needed to treat to prevent an event. | ||||||
| GRADE Working Group grades of evidence | ||||||
Background
This is one of a series of reviews whose aims are:
-
to increase awareness of the range of analgesics that are available for acute postoperative pain;
-
to present evidence for relative analgesic efficacy through indirect comparisons with placebo in very similar trials performed in a standard manner, with very similar outcomes, and over the same duration.
Such relative analgesic efficacy does not in itself determine choice of drug for any situation or patient but guides policy‐making at the local level. The series covers all analgesics licensed for acute postoperative pain in the UK, and dipyrone (metamizole) because it is commonly used in Spain, Portugal, Bulgaria, Israel, Turkey, India, and Latin‐American countries. Two overviews of efficacy and adverse events have examined the results (Moore 2015a; Moore 2015b). This new review of dipyrone replaces an earlier review, which has been withdrawn and replaced with an up‐to‐date protocol (Derry 2010).
Description of the condition
Acute pain usually occurs as a result of tissue damage, either accidentally due to an injury or as a result of surgery. Acute postoperative pain is a manifestation of inflammation due to tissue injury, nerve injury, or both. The management of postoperative pain and inflammation is a critical component of patient care.
Description of the intervention
Dipyrone is a nonsteroidal anti‐inflammatory drug (NSAID). It was first synthesised in 1920 in Germany and the drug was launched there in 1922. NSAIDs have pain‐relieving, antipyretic, and anti‐inflammatory properties, and have proven efficacy following day surgery and minor surgery. The usual adult dose of dipyrone is 1.0 to 2.5 mg daily when given orally. It is also available for intravenous (IV), intramuscular (IM), or rectal administration. Dipyrone (metamizole) is manufactured by a very large number of different manufacturers under 21 different generic names and hundreds of brand names (http://www.drugs.com/international/metamizole.html).
Dipyrone is a controversial analgesic. It is used most commonly to treat postoperative pain, colic pain, cancer pain, and migraine, and in many countries (eg Russia, Spain, Mexico, and in many parts of South America, Asia, and Africa) it remains a popular non‐opioid first‐line analgesic, either by prescription only, as in Germany and Spain, or non‐prescription (over the counter (OTC)), as in Bulgaria and Mexico. In other countries, it has been banned (eg the USA, the UK, Japan, Canada, and parts of Europe) because of its association with potentially life‐threatening blood disorders such as agranulocytosis (a deficiency of certain blood cells). In countries where it is banned, it may still be available and widely used by immigrant populations (Bonkowsky 2002). It is sold under many different brand names, including Analgin and Novalgin, and is also known in some areas of the USA as 'Mexican aspirin'. In addition to use as a single agent, it is commonly used in combination products.
One systematic review and meta‐analysis of dipyrone use lasting less than two weeks concluded that for short‐term use in the hospital setting, dipyrone appeared to be a safe choice when compared to other widely used analgesics (Kötter 2015). However, the main concern with dipyrone has been an association with agranulocytosis.
There is a wealth of literature on agranulocytosis associated with dipyrone: one large, international study found vastly differing rates of agranulocytosis in the 11 countries from which information was collected (IAAAS 1986). There are a number of published criticisms of this study (Kramer 1988). None of these criticisms mention the importance of size (of the population studied and of the analyses) for detecting true incidence rates for rare events. Size is an important criterion of study validity (Moore 1998). One report from Sweden suggested a rate of 1 case of agranulocytosis in 1439 prescriptions in a small study with high cumulative doses (Hedenmalm 2002). One case‐control study in Berlin identified 10 probable dipyrone‐induced cases (of 63 drug‐related cases) of agranulocytosis between 2000 and 2010, more than for any other drug (Huber 2014). One review of non‐chemotherapy drug‐induced agranulocytosis identified dipyrone in six definite and five probable high quality case reports, with a median time to onset of only two days (Andersohn 2007). One case‐control analysis estimated incidence of agranulocytosis at less than one per million per year; the risk increased with duration of use and disappeared 10 days after the last dose (Ibáñez 2005). One review of the clinical profile of dipyrone concluded that the risk of agranulocytosis with short‐term treatment was low and risks of serious complications were much lower than with aspirin and diclofenac and comparable to those of paracetamol (Nikolova 2013).
While the risk of agranulocytosis remains uncertain (Edwards 2002), and there may be differences between populations in their susceptibility to agranulocytosis (Mérida Rodrigo 2009), dipyrone is one of the 10 drugs most commonly associated with it (Andersohn 2007).
The use of dipyrone has been reported to be associated with other potentially serious adverse events, such as chronic interstitial nephritis and gastrointestinal disturbances (Zukowski 2009), as well as allergic or idiosyncratic reactions such as anaphylaxis, bronchospasm, and toxic epidermal necrolysis (Arellano 1990). One review of hospital admissions for adverse drug reactions in Brazil identified 20 dipyrone‐related admissions over an eight‐month period (Lobo 2013). However, the risk of gastrointestinal bleeding with dipyrone was low in one study of 18 hospitals in Spain and Italy (Laporte 2004). It has been reported that the excess mortality due to agranulocytosis, aplastic anaemia, anaphylaxis, and serious upper gastrointestinal complications is 0.25 per million users for dipyrone (0.2 for paracetamol, 1.85 for aspirin, and 5.92 for diclofenac) (Andrade 1998).
Acute pain trials
Single dose trials in acute pain are commonly short in duration, rarely lasting longer than 12 hours. The numbers of participants are small, allowing no reliable conclusions to be drawn about safety. To show that the analgesic is working, it is necessary to use placebo comparison (McQuay 2005). There are clear ethical considerations in doing this. These ethical considerations are answered by using acute pain situations where the pain is expected to go away, and by providing additional analgesia, commonly called rescue analgesia, if the pain has not diminished after about one hour. This is reasonable because not all participants given an analgesic will necessarily have significant pain relief anyway. Approximately 18% of participants given placebo will have significant pain relief (Moore 2006), while up to 50% may have inadequate analgesia with active medicines. Hence, the use of additional or rescue analgesia is important for all participants in the trials.
Clinical trials measuring the efficacy of analgesics in acute pain have been standardised over many years. Trials have to be randomised and double‐blind. Typically, in the first few hours or days after an operation, people develop pain that is moderate to severe in intensity and will then be given the test analgesic or placebo. Pain is measured using standard pain intensity scales immediately before the intervention and then, using pain intensity and pain relief scales, over the following four to six hours for shorter‐acting drugs, and over 12 to 24 hours for longer‐acting drugs. Pain relief of half the maximum possible or better (at least 50% pain relief) is typically regarded as a clinically useful outcome. For participants given rescue medication, it is usual for no additional pain measurements to be made and for all subsequent measures to be recorded as initial pain intensity or baseline (zero) pain relief (baseline observation carried forward (BOCF)). This process ensures that analgesia from the rescue medication is not wrongly ascribed to the test intervention. In some trials, the last observation is carried forward (LOCF), which gives an inflated response for the test intervention compared to placebo, but the effect of this has been shown to be negligible over four to six hours (Moore 2005). Participants usually remain in the hospital or clinic for at least the first six hours following the intervention, with measurements supervised, although they may then be allowed home to make their own measurements in trials of longer duration.
How the intervention might work
NSAIDs have pain‐relieving, antipyretic, and anti‐inflammatory properties, and are thought to relieve pain by inhibiting cyclo‐oxygenases (COX) (prostaglandin endoperoxide synthases) and thus the production of prostaglandins (Hawkey 1999). Prostaglandins occur throughout body tissues and fluids and act to stimulate pain nerve endings and inhibit the aggregation of blood platelets. Dipyrone's mechanism of action is not entirely clear but it seems to be an inhibitor of COX enzymes and thus inhibits the production of prostaglandins. It may also be associated with the endogenous opioid system (Nikolova 2013). Dipyrone, and some of its active metabolites, may also act by directly blocking ongoing inflammatory hypersensitisation (hyperalgesia).
Inhibition of prostaglandin production may be involved with some of the known problems associated with NSAIDs, including gastrointestinal, cardiovascular, renal, and hypertensive adverse effects (FitzGerald 2001; Hawkey 1999; Hawkey 2002; Patrono 2009). Interestingly, dipyrone has not been linked with gastrointestinal bleeding (Laporte 1991).
Why it is important to do this review
Although use of dipyrone is banned or restricted in many countries, it remains a drug of choice in other countries. It is important that information about its benefits and harms be carefully reviewed and made available to a worldwide audience.
Objectives
To assess the analgesic efficacy and associated adverse events of single dose dipyrone for moderate to severe acute postoperative pain using methods that permit comparison with other analgesics evaluated in standardised trials using almost identical methods and outcomes.
Methods
Criteria for considering studies for this review
Types of studies
We included double‐blind studies of a single dose of dipyrone compared with placebo for the treatment of moderate to severe postoperative pain in adults, with at least 10 participants randomly allocated to each treatment group. We included multiple dose studies, if appropriate data from the first dose were available, and cross‐over studies, provided that data from the first arm were presented separately.
We excluded the following.
-
Review articles, case reports, and clinical observations.
-
Studies of experimental pain.
-
Studies where pain relief was assessed only by clinicians, nurses, or carers (ie not participant‐reported).
-
Studies of less than four hours' duration or studies that did not present data over a four to six hour period post dose.
For postpartum pain, we included studies if the pain investigated was due to episiotomy or Caesarean section, irrespective of the presence of uterine cramps; we excluded studies investigating pain due to uterine cramps alone.
Types of participants
We included studies of adults (aged 15 years or older) with established postoperative pain of moderate to severe intensity following day surgery or inpatient surgery. For studies using a visual analogue scale (VAS), we assumed that pain intensity of greater than 30/100 mm equated to pain of at least moderate intensity (Collins 1997).
Types of interventions
Dipyrone, administered as a single dose, compared with matched placebo, administered postoperatively for pain relief. Where studies also included an active comparator, we extracted data for direct comparison. We included oral, rectal, IV, and IM routes of administration.
Types of outcome measures
We collected the following data where available.
-
Participant characteristics.
-
Dose and route of administration.
-
Participant‐reported pain at baseline (physician, nurse, or carer‐reported pain was not included in the analysis).
-
Participant‐reported pain relief expressed at least hourly over a four to six hour period using validated pain scales (pain intensity and pain relief in the form of VAS or categorical scales, or both).
-
Patient Global Evaluation of treatment (PGE), using a standard categorical scale.
-
Time to use of rescue medication.
-
Number of participants using rescue medication.
-
Number of participants with one or more adverse events.
-
Number of participants with serious adverse events.
-
Number of withdrawals (all causes; adverse events).
Primary outcomes
-
Participants achieving at least 50% pain relief over a four to six hour period.
Secondary outcomes
-
Median (or mean) time to use of rescue medication.
-
Number of participants using rescue medication.
-
Number of participants with: any adverse event; any serious adverse event (as reported in the study); withdrawal due to an adverse event.
-
Other withdrawals: withdrawals for reasons other than lack of efficacy (participants using rescue medication).
Search methods for identification of studies
Two review authors independently searched for studies.
Electronic searches
We searched the following databases.
-
The Cochrane Central Register of Controlled Trials (CENTRAL via CRSO on 11 August 2015).
-
MEDLINE (via Ovid from 1946 to 11 August 2015).
-
EMBASE (via Ovid from 1974 to 11 August 2015).
-
LILACS (via VHL on 11 August 2015).
-
Oxford Pain Relief Database on 6 February 2015 (Jadad 1996a).
See Appendix 1 for the CENTRAL search strategy, Appendix 2 for the MEDLINE search strategy, Appendix 3 for the EMBASE search strategy, and Appendix 4 for the LILACS search strategy. We did not limit the searches by language or date.
Searching other resources
We searched for additional studies in reference lists of the earlier Cochrane review, retrieved articles, and other reviews, and in two clinical trials databases (clinicaltrials.gov and apps.who.int/trialsearch/).
Data collection and analysis
Selection of studies
Two review authors independently assessed the search results and agreed on the studies to be included in the review. We intended to resolve disagreements by consensus or referral to a third review author but this was not necessary.
Data extraction and management
Two review authors extracted data and recorded them on a standard data extraction form. One review author entered data suitable for pooling into Review Manager 5 (RevMan 2014).
Assessment of risk of bias in included studies
We used the Oxford Quality Score as the basis for study inclusion, limiting inclusion to studies that were randomised and double‐blind as a minimum (Jadad 1996b).
We also completed a 'Risk of bias' table, using methods adapted from those described by the Cochrane Pregnancy and Childbirth Group. Two review authors independently assessed risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Chapter 8.5, Higgins 2011), and resolved any disagreements by discussion. We assessed the following for each study.
-
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (ie any truly random process, such as random number table or computer random number generator); unclear risk of bias (when the method used to generate the sequence was not clearly stated). We excluded studies at a high risk of bias that used a non‐random process (eg odd or even date of birth; hospital or clinic record number).
-
Allocation concealment (checking for possible selection bias). We assessed the method used to conceal allocation to interventions prior to assignment as to whether intervention allocation could have been foreseen in advance of or during recruitment, or changed after assignment. We assessed the methods as: low risk of bias (eg telephone or central randomisation; consecutively numbered, sealed, opaque envelopes); unclear risk of bias (when the method was not clearly stated); and high risk of bias (eg open random allocation; unsealed or non‐opaque envelopes; alternation; date of birth).
-
Blinding of outcome assessment (checking for possible detection bias). We assessed the methods used to blind study participants and outcome assessors from knowledge of which intervention a participant received. We considered studies to be at low risk of bias if they stated that they were blinded and described the method used to achieve blinding (eg identical tablets; matched in appearance and smell); or at unclear risk of bias if they stated that they were blinded, but did not provide an adequate description of how this was achieved. We excluded single‐blind and open studies at a high risk of bias.
-
Size (checking for possible biases confounded by small size). Small studies overestimate treatment effects, probably due to methodological weaknesses (Dechartres 2013; Nüesch 2010). We considered studies to be at low risk of bias if they had 200 participants or more; at unclear risk of bias if they had 50 to 200 participants; and at high risk of bias if they had fewer than 50 participants.
Measures of treatment effect
We used risk ratio (RR) to establish statistical difference. We used numbers needed to treat for an additional beneficial outcome (NNT) and pooled percentages as absolute measures of benefit or harm. We reported these with their 95% confidence intervals (CI).
We used the following terms to describe adverse outcomes in terms of harm or prevention of harm.
-
When significantly fewer adverse outcomes occurred with treatment than with control (placebo or active), we used the term the number needed to treat to prevent one additional event (NNTp).
-
When significantly more adverse outcomes occurred with treatment compared with control (placebo or active), we used the term the number needed to treat for an additional harmful outcome or cause one event (NNH).
Unit of analysis issues
We accepted only randomisation of the individual participant.
Dealing with missing data
The only likely issue with missing data in these studies was from imputation using LOCF when a participant requested rescue medication. We have previously shown that this does not affect results for up to six hours after taking study medication (Barden 2004).
Assessment of heterogeneity
We examined heterogeneity visually using L'Abbé plots (L'Abbé 1987), which is a visual method for assessing differences in results of individual studies, and using the I2 statistic.
Assessment of reporting biases
We assessed publication bias using a method designed to detect the amount of unpublished data with a null effect required to make any result clinically irrelevant (NNT of 10 or higher in this condition) (Moore 2008).
Data synthesis
For efficacy analyses, we used the number of participants in each treatment group who were randomised, received medication, and provided at least one post‐baseline assessment. For safety analyses, we used the number of participants randomised to each treatment group who took the study medication. We analysed results for different doses separately.
For each study, we converted the mean total pain relief (TOTPAR), summed pain intensity difference (SPID), VAS TOTPAR, or VAS SPID (see Appendix 5) values for active and placebo groups to %maxTOTPAR or %maxSPID by division into the calculated maximum value (Cooper 1991). We then calculated the proportion of participants in each treatment group who achieved at least 50%maxTOTPAR using verified equations (Moore 1996; Moore 1997a; Moore 1997b). We then converted these proportions into the number of participants achieving at least 50%maxTOTPAR by multiplying by the total number of participants in the treatment group.
We used dichotomous information on the number of participants with an outcome of interest in the active and placebo groups to calculate the RR with 95% CI, using a fixed‐effect model (Morris 1995). We assumed a statistically significant difference from control when the 95% CI of the RR did not include the number '1'. We calculated NNT and NNH with 95% CIs using the pooled number of events by the method of Cook and Sackett (Cook 1995).
We accepted the following pain measures for the calculation of TOTPAR or SPID.
-
Five‐point categorical pain relief (PR) scales with comparable wording to 'none', 'slight', 'moderate', 'good', or 'complete'.
-
Four‐point categorical pain intensity (PI) scales with comparable wording to 'none', 'mild', 'moderate', or 'severe'.
-
VAS for pain relief.
-
VAS for pain intensity.
If none of these measures was available, we used the number of participants reporting 'very good' or 'excellent' on a 5‐point categorical global scale with the wording 'poor', 'fair', 'good', 'very good', or 'excellent' for the number of participants achieving at least 50% pain relief (Collins 2001).
Quality of the evidence
We have used the GRADE (Grades of Recommendation, Assessment, Development and Evaluation) system to assess the quality of the evidence related to the key outcomes listed in Types of outcome measures, as appropriate (Appendix 6; Chapter 12.2, Higgins 2011). Two review authors independently rated the quality of evidence for each outcome.
Summary of findings table
We have included 'Summary of findings' tables as set out in the PaPaS author guide (PaPaS 2012), and recommended in the Cochrane Handbook (Chapter 4.6.6, Higgins 2011). The tables include outcomes of at least 30% and at least 50% pain intensity reduction, PGIC (possibly at least substantial improvement and at least moderate improvement), serious adverse events, withdrawals due to adverse events, and death (a particular serious adverse event).
Subgroup analysis and investigation of heterogeneity
We analysed separately the data for different routes of administration. We planned subgroup analyses to determine the effect of dose and presenting condition (pain model: dental versus other postoperative pain).
Sensitivity analysis
We planned sensitivity analyses for quality score (two versus three or more) and trial size (39 or fewer versus 40 or more per treatment arm).
A minimum of two studies and 200 participants had to be available in any subgroup or sensitivity analysis (Moore 1998), which was restricted to the primary outcome (50% of maximum pain relief over a four to six hour period) and the dose with the greatest amount of data. We planned to determine significant differences between NNT or NNH for different groups in subgroup and sensitivity analyses using the z test (Tramèr 1997).
Results
Description of studies
Results of the search
We identified 315 reports in CENTRAL, 231 in MEDLINE, 442 in EMBASE, and 24 in LILACS. We found no additional studies in the Oxford Pain Relief Database and in clinical trials registries. We excluded 28 studies after obtaining and reading the full report (Figure 1).

Study flow diagram.
Eight studies met the inclusion criteria (Bhounsule 1990; Boraks 1987; De Miguel Rivero 1997; Dos Santos Pereira 1986; Olson 1999; Pinto 1984; Rubinstein 1986; Sakata 1986). One study included one or more participants aged 14 years, but was included, subject to sensitivity analysis, because we believed that the number was small and unlikely to affect results (Pinto 1984).
Included studies
The eight included studies used both placebo and active controls (Bhounsule 1990; Boraks 1987; De Miguel Rivero 1997; Dos Santos Pereira 1986; Olson 1999; Pinto 1984; Rubinstein 1986; Sakata 1986); the active controls were oral ibuprofen 400 mg, paracetamol 500 mg or 1000 mg, aspirin 600/650 mg, flurbiprofen 50 mg, and ketoprofen 25 mg or 50 mg.
In five studies, 143 participants used oral dipyrone 500 mg (Bhounsule 1990; Boraks 1987; Olson 1999; Pinto 1984; Rubinstein 1986), and in two studies, 58 participants used oral dipyrone 1000 mg (Dos Santos Pereira 1986; Sakata 1986). In one study, 35 participants used IM dipyrone 2000 mg (De Miguel Rivero 1997).
The mean age ranged from 23 to 62 years. Six studies included both men and women, and two studies included only women. One study was carried out in participants who had undergone dental surgery (Boraks 1987), three studies following orthopaedic surgery (De Miguel Rivero 1997; Dos Santos Pereira 1986; Sakata 1986), two studies following episiotomy (Bhounsule 1990; Olson 1999), one study following tonsillectomy (Pinto 1984), and one study following urological surgery (Rubinstein 1986).
All studies used a parallel group design. One study used multiple doses of dipyrone but reported outcomes for the first dose separately (De Miguel Rivero 1997). All studies reported single dose efficacy over four to six hours.
Full details of included studies are in the Characteristics of included studies table.
Excluded studies
The main reasons for exclusion of studies were lack of placebo, not randomised controlled trials, dipyrone was given before the end of surgery (before pain was established), time to data collection was too short or too long, no data for single doses, and use of non‐standard pain scales.
Details are in the Characteristics of excluded studies table.
Risk of bias in included studies
Three studies scored the minimum of 2/5 on the Oxford Quality Scale (Bhounsule 1990; Boraks 1987; Sakata 1986), three studies scored 3/5 (Dos Santos Pereira 1986; Olson 1999; Pinto 1984), and two studies scored 4/5 (De Miguel Rivero 1997; Rubinstein 1986).
We completed a 'Risk of bias' table for randomisation, allocation concealment, blinding, and size (Figure 2; Figure 3).
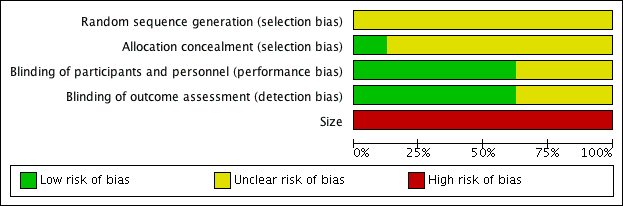
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
All studies were randomised, but none described the method used to generate the random sequence, and only one described the method used to conceal the allocation of the sequence (Olson 1999).
Blinding
All studies were double‐blind, but three did not adequately describe the method used to maintain blinding (Bhounsule 1990; Boraks 1987; Sakata 1986).
Other potential sources of bias
All the studies had fewer than 50 participants in each treatment arm, and we judged them at high risk for size.
Effects of interventions
See: Summary of findings for the main comparison
Eight studies compared dipyrone (500 mg, 1000 mg, or 2000 mg) with placebo over four to six hours (Boraks 1987; Bhounsule 1990; De Miguel Rivero 1997; Dos Santos Pereira 1986; Olson 1999; Pinto 1984; Rubinstein 1986; Sakata 1986). Only the 500 mg dose of oral dipyrone provided sufficient data for statistical analysis. summary of findings Table for the main comparison presents results for this comparison.
Number of participants experiencing at least 50% pain relief
Oral dipyrone 500 mg versus placebo
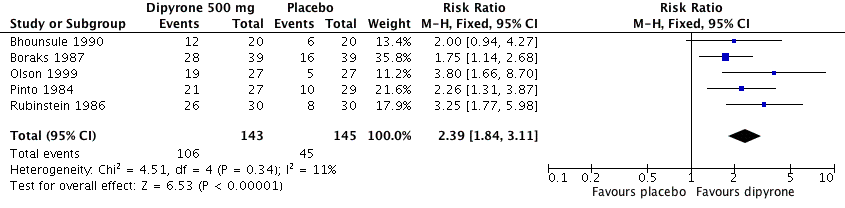
Five studies compared oral dipyrone 500 mg with placebo over four to six hours (Bhounsule 1990; Boraks 1987; Olson 1999; Pinto 1984; Rubinstein 1986). There were 288 participants in the comparison.
-
The proportion of participants experiencing at least 50% pain relief with dipyrone 500 mg was 73% (106/143; range 60% to 87%).
-
The proportion of participants experiencing at least 50% pain relief with placebo was 32% (45/145; range 19% to 41%).
-
The RR of treatment compared with placebo was 2.4 (95% CI 1.8 to 3.1); the NNT for at least 50% pain relief over four to six hours was 2.4 (95% CI 1.9 to 3.1) (Figure 4).

Forest plot of comparison: 1 Oral dipyrone 500 mg versus placebo, outcome: 1.1 Participants with ≥ 50% pain relief over 4 to 6 hours.
Removing Pinto 1984 from the analysis, because it included an unknown number of participants aged 14 years, did not change the result.
Oral dipyrone 1000 mg versus placebo
Two studies (113 participants) compared oral dipyrone 1000 mg with placebo (Dos Santos Pereira 1986; Sakata 1986); 38/57 (67%) of participants treated with dipyrone 1000 mg experienced at least 50% pain relief over four hours compared with 10/56 (18%) participants treated with placebo. There were insufficient data for statistical analysis.
Intramuscular dipyrone 2000 mg versus placebo
One study (70 participants) compared IM dipyrone 2000 mg with placebo (De Miguel Rivero 1997); 26/35 (74%) of participants treated with IM dipyrone 2000 mg experienced at least 50% pain relief over five hours compared with 16/35 (46%) of participants treated with placebo. There were insufficient data for statistical analysis.
Oral dipyrone 500 mg versus active comparators
Three studies (156 participants) compared oral dipyrone 500 mg with paracetamol 500 mg (Pinto 1984; Rubinstein 1986) or 600 mg (Bhounsule 1990); 58/77 (75%) of participants treated with dipyrone 500 mg experienced at least 50% pain relief over four to six hours compared with 53/79 (67%) participants treated with paracetamol 500 mg or 600 mg. There were insufficient data for statistical analysis.
Two studies (120 participants) compared oral dipyrone 500 mg with aspirin 600 mg (Bhounsule 1990) or 650 mg (Boraks 1987); 39/59 (66%) of participants treated with dipyrone 500 mg experienced at least 50% pain relief over four to six hours compared with 30/61 (49%) of participants treated with aspirin 600 mg or 650 mg. There were insufficient data for statistical analysis.
Single studies compared oral dipyrone 500 mg with oral ibuprofen 400 mg (40 participants, Bhounsule 1990), flurbiprofen 50 mg (79 participants, Boraks 1987), or ketoprofen 25 mg or 50 mg (81 participants, Olson 1999). Percentages experiencing at least 50% pain relief ranged from 60% to 72% but were virtually identical for the different arms of these studies. There were insufficient data for statistical analysis.
Oral dipyrone 1000 mg versus active comparators
Two studies (115 participants) compared oral dipyrone 1000 mg with paracetamol 1000 mg (Dos Santos Pereira 1986; Sakata 1986); 38/57 (67%) of participants treated with dipyrone 1000 mg experienced at least 50% pain relief over four hours compared with 38/58 (66%) participants treated with paracetamol 1000 mg. There were insufficient data for statistical analysis.
Intramuscular dipyrone 2000 mg versus active comparators
One study (71 participants) compared IM dipyrone 2000 mg with oral ibuprofen arginine 400 mg (De Miguel Rivero 1997). Despite the differences in dose and route of administration, the numbers experiencing at least 50% pain relief were virtually identical at 26/35 (74%) with dipyrone 2000 mg and 25/36 (69%) with ibuprofen. There were insufficient data for statistical analysis.
Subgroup and sensitivity analyses of the primary outcome
There were insufficient data to carry out subgroup analyses on the primary outcome for route of administration or pain model, or to carry out sensitivity analysis for study quality.
Results for individual studies are available in Summary of efficacy outcomes in individual studies (Appendix 7).
Time to use of rescue medication
One study comparing oral dipyrone 500 mg with placebo following episiotomy reported a mean time to use of rescue medication of more than six hours for dipyrone, more than six hours for ketoprofen 25 mg and 50 mg, and 5.3 hours for placebo (Olson 1999). Another study using IM dipyrone 2000 mg following orthopaedic surgery reported a median time to use of rescue medication of 2.6 hours for dipyrone, 3.5 hours for ibuprofen arginine 400 mg, and 1.8 hours for placebo (De Miguel Rivero 1997).
Number of participants needing rescue medication
The number of participants needing rescue medication during the study period was not reported in three studies (Bhounsule 1990; De Miguel Rivero 1997; Sakata 1986). It is unclear whether there were any withdrawals for this reason in these studies.
Four studies comparing dipyrone 500 mg with placebo provided data on the number of participants using rescue medication before the end of the study (within four to six hours) (Boraks 1987; Olson 1999; Pinto 1984; Rubinstein 1986). There were 248 participants in the comparison.
-
The proportion of participants using rescue medication with dipyrone 500 mg was 7% (8/123; range 0% to 15%).
-
The proportion of participants using rescue medication with placebo was 34% (43/125; range 20% to 51%).
-
The relative benefit of treatment compared with placebo was 0.21 (0.11 to 0.40), and the NNTp was 3.6 (2.7 to 5.4) (Analysis 1.2).
Removing Pinto 1984 from the analysis, because it included an unknown number of participants aged 14 years, did not change the result.
There were insufficient data for analysis of any other dose or route of administration of dipyrone for this outcome. However, Dos Santos Pereira 1986 reported that, for oral dipyrone 1000 mg, 1/28 participants needed rescue medication at four hours, compared to 11/29 participants with placebo. For IV dipyrone 2000 mg following orthopaedic surgery, De Miguel Rivero 1997 reported 3/33 (9%) participants needing rescue medication, compared with 4/34 (12%) participants with placebo, and 5/36 (14%) participants with ibuprofen arginine 400 mg.
Appendix 7 shows the results for individual studies.
Adverse events
One study did not mention adverse events (Sakata 1986), and another study reported only that the study medication was "well tolerated" (Dos Santos Pereira 1986). Reporting of adverse events in the other studies was inconsistent, with some reporting only those that were considered related to the test drug, or that were "clinically relevant". Few events were reported, and we could not carry out an analysis for participants experiencing one or more adverse events. They did not report any serious adverse events.
Appendix 8 shows the details of events in individual studies.
Withdrawals
None of the studies reported any adverse event withdrawals. We have reported withdrawals due to lack of efficacy above under 'Number of participants needing rescue medication'. Five studies did not specifically report on all cause withdrawals (Bhounsule 1990; Boraks 1987; Dos Santos Pereira 1986; Pinto 1984; Sakata 1986).
Discussion
Summary of main results
This review found eight studies using various doses of dipyrone (500 to 2000 mg) administered by different routes (oral or IM) and following different surgical procedures, with comparisons to placebo and a variety of active comparators.
For the primary outcome of at least 50% pain relief over four to six hours, there were sufficient data from placebo‐controlled comparisons to analyse only oral dipyrone 500 mg versus placebo (288 participants). The RR was 2.4 (95% CI 1.8 to 3.1), and the NNT was 2.4 (1.9 to 3.2). For every five people treated, two would experience at least 50% pain relief who would not have done so with placebo (moderate quality data). For the same comparison (248 participants), the RR for needing rescue medication within four to six hours was 0.21 (95% CI 0.11 to 0.40), and the NNTp was 3.6 (2.7 to 5.4) (low quality data). For every seven people treated, two would not need rescue medication who would have done with placebo.
There was very little information on the mean or median time to use of rescue medication, a useful indicator of the duration of analgesia. Reporting of adverse events was inconsistent, with few events reported, and no analysis was possible. The studies reported no serious adverse events or adverse event withdrawals.
Results from studies using different doses and routes of administration were all consistent with a benefit of dipyrone over placebo, but based on very few data.
For active‐controlled comparisons, there were insufficient data for analysis.
Indirect comparisons of NNTs for at least 50% pain relief over four to six hours in reviews of other analgesics using identical methods indicate that dipyrone has similar efficacy to standard ibuprofen 400 mg (NNT 2.5 (2.4 to 2.6)), diclofenac potassium (NNT 2.1 (1.9 to 2.5)), and ketoprofen 12.5 mg (NNT 2.4 (1.9 to 3.1)) (Moore 2015a).
Overall completeness and applicability of evidence
The studies involved participants who had undergone a diverse range of surgical procedures, from episiotomy to total hip replacement. They probably represent the majority of adults likely to be given the drug, although the studies excluded older people, pregnant women, and people with contraindications. Only one study involved dental extractions, which is usually the most common procedure used in these single dose studies.
Quality of the evidence
Overall, the methodological quality of the studies was moderate; all studies had to be randomised and double blind to satisfy inclusion criteria, but some did not fully report on withdrawals, and few gave details of the randomisation and blinding procedures, or of how missing data were handled. Treatment group sizes were small, so that even when several studies contributed data for an outcome, the number of events was small, and confidence in the result must therefore be limited.
All studies enrolled participants with established pain following surgery, with pain levels sufficient to demonstrate reduction, or otherwise, due to treatment. Adverse event data were not well reported, with no information on whether data were collected after use of rescue medication (which may cause its own adverse events). The small size of each treatment arm and small number of studies means that this review is greatly underpowered to address the safety of dipyrone. In particular, rare, but potentially serious, adverse events such as agranulocytosis, are very unlikely to be detected in these studies.
Potential biases in the review process
We identified the included studies from a comprehensive search of published papers, and used standard methods for analysis. We excluded six studies because they used non‐standard measurement scales. We judged that it was unlikely these studies could have given different results that would have changed the findings of this review.
We can estimate the number of participants in studies with zero effect (RR of 1) required in order to change the NNT for at least 50% pain relief to an unacceptably high level (in this case 10) (Moore 2008). Data from over 900 participants in studies comparing oral dipyrone 500 mg with placebo would be required, and it is unlikely that such data exist.
Agreements and disagreements with other studies or reviews
We are not aware of any other systematic reviews or meta‐analyses of dipyrone in acute postoperative pain. One narrative review reported that dipyrone has similar efficacy to other NSAIDs and IV paracetamol, has a notable spasmolytic effect, and is rarely associated with agranulocytosis and other disorders of haematopoiesis (Jage 2008). The results were almost identical to that of the previous version of this review (Derry 2010).

Study flow diagram.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Forest plot of comparison: 1 Oral dipyrone 500 mg versus placebo, outcome: 1.1 Participants with ≥ 50% pain relief over 4 to 6 hours.

Comparison 1 Oral dipyrone 500 mg versus placebo, Outcome 1 Participants with ≥ 50% pain relief over 4 to 6 hours.

Comparison 1 Oral dipyrone 500 mg versus placebo, Outcome 2 Participants using rescue medication over 4 to 6 hours.
| Oral dipyrone 500 mg compared with placebo for acute postoperative pain | ||||||
| Patient or population: adults with acute postoperative pain Settings: clinic Intervention: oral dipyrone 500 mg Comparison: placebo | ||||||
| Outcomes | Probable outcome with | Relative effect and NNT or NNTp | Number of studies, participants, events | Quality of the evidence | Comments | |
| intervention | comparator | |||||
| At least 50% of maximum pain relief over 4 to 6 h | 730 in 1000 | 320 in 1000 | RR 2.4 (95% CI 1.8 to 3.1) NNT 2.4 (1.9 to 3.1) | 5 studies, 288 participants, 151 events | Moderate | Small studies, few events |
| Participants remedicating within 4 to 6 h | 70 in 1000 | 340 in 1000 | RR 0.21 (0.11 to 0.40) NNTp 3.6 (2.7 to 5.4) | 4 studies, 248 participants, 51 events | Low | Small studies, very few events |
| Participants with at least one adverse event | Insufficient data for analysis | ‐ | ‐ | ‐ | ‐ | |
| Participants with a serious adverse event | None reported | None reported | ‐ | 5 studies, 288 participants, no events | Very low | Small studies, no events |
| CI: confidence interval; h: hour; RR: risk ratio; NNT: number needed to treat for an additional beneficial outcome; NNTp: number needed to treat to prevent an event. | ||||||
| GRADE Working Group grades of evidence | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Participants with ≥ 50% pain relief over 4 to 6 hours Show forest plot | 5 | 288 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.39 [1.84, 3.11] |
| 2 Participants using rescue medication over 4 to 6 hours Show forest plot | 4 | 248 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.11, 0.40] |