Rysperydon w porównaniu z innymi lekami przeciwpsychotycznymi u osób z ciężką chorobą psychiczną jednocześnie nadużywających substancji psychoaktywnych
Abstract
Background
Up to 75% of people with serious mental illness (SMI) such as schizophrenia and bipolar disorder have co‐occurring substance use disorders (dual diagnosis). Dual diagnosis can have an adverse effect on treatment and prognosis of SMI.
Objectives
To evaluate the effects of risperidone compared to treatment with other antipsychotics (first‐generation and other second‐generation antipsychotics) used in people with serious mental illness and co‐occurring substance misuse.
Search methods
On 6 January 2016 and 9 October 2017, we searched the Cochrane Schizophrenia Group’s Study‐Based Register of Trials (including trial registers).
Selection criteria
We selected randomised trials of risperidone versus any other antipsychotic in people with SMI and substance abuse (dual diagnosis). We included trials meeting our inclusion criteria and reporting useable data. We excluded trials that either did not meet our inclusion criteria or met our inclusion criteria but did not report any useable data.
Data collection and analysis
We independently inspected citations and selected studies. For included studies, we independently extracted data and appraised study quality. For binary outcomes we calculated the risk ratios (RRs) and their 95% confidence intervals. For continuous outcomes we calculated the mean differences (MDs) and their 95% confidence intervals. We pooled data using random‐effects meta‐analyses and assessed the quality of evidence, creating a 'Summary of findings' table using the GRADE approach.
Main results
We identified eight randomised trials containing a total of 1073 participants with SMI and co‐occurring substance misuse. Seven of these contributed useable data to the review. There was heterogeneity in trial design and measurement. Risperidone was compared to clozapine, olanzapine, perphenazine, quetiapine and ziprasidone. Few trials compared risperidone with first‐generation agents. Few trials examined participants with a dual diagnosis from the outset and most trials only contained separate analyses of subgroups with a dual diagnosis or were secondary data analyses of subgroups of people with a dual diagnosis from existing larger trials.
For risperidone versus clozapine we found no clear differences between these two antipsychotics in the reduction of positive psychotic symptoms (1 randomised controlled trial (RCT), n = 36, mean difference (MD) 0.90, 95% CI −2.21 to 4.01, very low quality evidence), or reduction in cannabis use (1 RCT, n = 14, risk ratio (RR) 1.00, 95% CI 0.30 to 3.35, very low quality evidence), improvement in subjective well‐being (1 RCT, n = 36, MD −6.00, 95% CI −14.82 to 2.82, very low quality evidence), numbers discontinuing medication (1 RCT, n = 36, RR 4.05, 95% CI 0.21 to 78.76, very low quality evidence), extrapyramidal side‐effects (2 RCTs, n = 50, RR 2.71, 95% CI 0.30 to 24.08; I² = 0%, very low quality evidence), or leaving the study early (2 RCTs, n = 45, RR 0.49, 95% CI 0.10 to 2.51; I² = 34%, very low quality evidence). Clozapine was associated with lower levels of craving for cannabis (1 RCT, n = 28, MD 7.00, 95% CI 2.37 to 11.63, very low quality evidence).
For risperidone versus olanzapine we found no clear differences in the reduction of positive psychotic symptoms (1 RCT, n = 37, MD −1.50, 95% CI −3.82 to 0.82, very low quality evidence), reduction in cannabis use (1 RCT, n = 41, MD 0.40, 95% CI −4.72 to 5.52, very low quality evidence), craving for cannabis (1 RCT, n = 41, MD 5.00, 95% CI −4.86 to 14.86, very low quality evidence), parkinsonism (1 RCT, n = 16, MD −0.08, 95% CI −1.21 to 1.05, very low quality evidence), or leaving the study early (2 RCT, n = 77, RR 0.68, 95% CI 0.34 to 1.35; I² = 0%, very low quality evidence).
For risperidone versus perphenazine, we found no clear differences in the number of participants leaving the study early (1 RCT, n = 281, RR 1.05, 95% CI 0.92 to 1.20, low‐quality evidence).
For risperidone versus quetiapine, we found no clear differences in the number of participants leaving the study early (1 RCT, n = 294, RR 0.96, 95% CI 0.86 to 1.07, low‐quality evidence).
For risperidone versus ziprasidone, we found no clear differences in the number of participants leaving the study early (1 RCT, n = 240, RR 0.96, 95% CI 0.85 to 1.10, low‐quality evidence).
For many comparisons, important outcomes were missing; and no data were reported in any study for metabolic disturbances, global impression of illness severity, quality of life or mortality.
Authors' conclusions
There is not sufficient good‐quality evidence available to determine the effects of risperidone compared with other antipsychotics in people with a dual diagnosis. Few trials compared risperidone with first‐generation agents, leading to limited applicability to settings where access to second‐generation agents is limited, such as in low‐ and middle‐income countries. Moreover, heterogeneity in trial design and measurement of outcomes precluded the use of many trials in our analyses. Future trials in this area need to be sufficiently powered but also need to conform to consistent methods in study population selection, use of measurement scales, definition of outcomes, and measures to counter risk of bias. Investigators should adhere to CONSORT guidelines in the reporting of results.
PICO
Streszczenie prostym językiem
Rysperydon w porównaniu z innymi lekami przeciwpsychotycznymi u osób z "podwójną diagnozą" obejmującą zaburzenie psychiczne oraz uzależnienie od alkoholu lub narkotyków
Co to jest podwójna diagnoza?
"Podwójna diagnoza" jest terminem używanym do opisania współwystępowania problemu ze zdrowiem psychicznym oraz uzależnienia od narkotyków lub alkoholu. Aż u 75% osób z ciężką chorobą psychiczną (ang. serious mental illness, SM; przyp. tłum.I) rozpoznaje się podwójną diagnozę. Sugerowano, że jednym z powodów dużego spożycia substancji psychoaktywnych u osób z SMI jest tzw. "samoleczenie", wśród pacjentów przyjmujących inne leki w celu przeciwdziałania niepokojącym objawom. U osób z podwójną diagnozą odnotowywano więcej powikłań w trakcie leczenia, takich jak: większy odsetek nawrotów choroby i ponownej hospitalizacji, częstsze korzystanie z usług prawnych i śledczych, większe nasilenie objawów psychotycznych, większą liczbę zachowań ryzykownych oraz skutków ubocznych leków przeciwpsychotycznych i mniejszy odsetek osób, które stosowały się do zaleceń lekarskich. Leki przeciwpsychotyczne stanowią główną metodę leczenia SMI. Sugerowano, że stosowanie leków przeciwpsychotycznych drugiej generacji (ang. second‐generation antipsychotics, SGAs), takich jak rysperydon, może być skuteczniejsze niż leki starsze, pierwszej generacji (ang. first‐generation antipsychotics, FGAs) pod względem poprawy w zakresie negatywnych stanów uczuciowych, zmniejszenia pragnienia przyjmowania substancji uzależniającej, samopoczucia oraz mniejszej częstości występowania skutków ubocznych, a co się z tym wiąże zwiększenia odsetka osób, które stosują się do zaleceń lekarskich. Taka poprawa w zakresie objawów, może prowadzić do zmniejszenia częstości przypadków samoleczenia za pomocą alkoholu i narkotyków oraz do ogólnej poprawy stanu zdrowia psychicznego. Jednak nadal pozostaje niejasne, w jakim stopniu rysperydon, jeden z pierwszych atypowych leków przeciwpsychotycznych, jest skuteczniejszy od innych leków przeciwpsychotycznych u osób cierpiących na podwójną diagnozę.
Kogo może zainteresować ten przegląd?
Specjalistów zajmujących się zdrowiem psychicznym, którzy leczą osoby z SMI i podwójną diagnozą oraz przepisują leki przeciwpsychotyczne w obu tych schorzeniach. Osoby, które korzystają z pomocy w zakresie zdrowia psychicznego oraz ich rodziny, które uczestniczą w ich leczeniu oraz opiece.
Na jakie pytanie odpowiada przegląd?
Jak skuteczny oraz bezpieczny jest rysperydon w porównaniu z innymi lekami przeciwpsychotycznymi w leczeniu osób z podwójną diagnozą?
Jakie badania zostały włączone do przeglądu?
Wyszukiwanie istotnych badań z randomizacją przeprowadziliśmy w styczniu 2016 r. oraz w październiku 2017 r. Odnaleźliśmy 8 badań z randomizacją obejmujących 1073 uczestników z rozpoznaniem podwójnej diagnozy. Większość uczestników badań stanowiły osoby dorosłe powyżej 18 r.ż. (4 uczestników miało 17 lat). Rysperydon porównywano z klozapiną, olanzapiną, perfenazyną, kwetiapiną i zyprazydonem.
Jakich danych naukowych dostarcza nam ten przegląd?
Stwierdziliśmy, że nie ma wyraźnie lepszych efektów leczenia rysperydonem w porównaniu z innymi lekami. Dane dotyczące skutków ubocznych były bardzo ograniczone; ponownie, nie stwierdziliśmy żadnych rzeczywistych różnic między stosowaniem rysperydonu i innych leków przeciwpsychotycznych. Ogólnie, jakość dostępnych danych naukowych oceniliśmy jako niską lub bardzo niską, a na chwilę obecną nie ma wystarczających danych naukowych, aby wykazać, że rysperydon jest bardziej lub mniej skutecznym lekiem w porównaniu z innymi lekami przeciwpsychotycznymi w leczeniu osób z ciężką chorobą psychiczną jednocześnie nadużywających substancji psychoaktywnych.
Jaki powinien być następny krok?
Konieczne jest przeprowadzenie dalszych, dobrej jakości badań. Przyszłe badania powinny obejmować grupy wystarczająco liczne, aby umożliwić wykrycie istotnych klinicznie różnic w zakresie ocenianych wyników zdrowotnych.
Authors' conclusions
Summary of findings
| RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: for people with serious mental illness and co‐occurring substance misuse | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with Clozapine | Risk with Risperidone | |||||
| Mental state: positive symptoms ‒average endpoint score (PANSS positive subscale, lower = better) | The mean positive symptoms (PANSS positive subscale, lower = better) in the intervention group was 0.9 higher (2.21 lower to 4.01 higher) | ‐ | 36 | ⊕⊝⊝⊝ | No trial reported "improvement in symptoms of severe mental illness" ‒ this continuous measure is the nearest proxy for this. | |
| Substance use: improvement ‒ (at least 20% reduction in use, TLFB scale) | Study population | RR 1.00 | 14 | ⊕⊝⊝⊝ | ||
| 429 per 1000 | 429 per 1000 | |||||
| Moderate | ||||||
| 429 per 1000 | 429 per 1000 | |||||
| Subjective well‐being: Subjective well‐being under neuroleptics scale ‒ average endpoint scores (SWN scale, higher = better) | The mean subjective well‐being under neuroleptics scale score (SWN scale, higher = better) in the intervention group was 6 lower (14.82 lower to 2.82 higher) | ‐ | 36 | ⊕⊝⊝⊝ | ||
| Craving for substances: Marijuana Craving Questionnaire ‒ average endpoint scores (MCQ, lower = better) | The mean craving for substances score on the Marijuana Craving Questionairre (MCQ, lower = better) in the intervention group was 7 higher (2.37 higher to 11.63 higher) | ‐ | 28 | ⊕⊝⊝⊝ | ||
| Adherence to antipsychotic medication: discontinued medication | Study population | RR 4.05 | 36 | ⊕⊝⊝⊝ | ||
| 0 per 1000 | 0 per 1,000 | |||||
| Moderate | ||||||
| 0 per 1,000 | 0 per 1,000 | |||||
| Adverse effects. 1. Movement disorders ‐ any extrapyramidal | Study population | RR 2.71 | 50 | ⊕⊝⊝⊝ | Many adverse effects reported ‒ none designated 'clinically important' (extrapyramidal used as proxy). | |
| 0 per 1000 | 0 per 1000 | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 | |||||
| Leaving the study early ‒ any reason | Study population | RR 0.49 | 45 | ⊕⊝⊝⊝ | ||
| 318 per 1000 | 156 per 1000 | |||||
| Moderate | ||||||
| 386 per 1000 | 189 per 1000 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 High risk of performance bias and detection bias 2 Sample size is very small, optimal information size (OIS) not met to detect 25% difference 3 Performance bias, attrition bias, selective outcome reporting 4 Sample size is very small (n = 14) 5 High risk of performance bias, detection bias, attrition bias and selective outcomes reporting 6 Total sample size is very small (n<300), total event rate is very low and optimum information size (OIS) is not met | ||||||
| RISPERIDONE versus OLANZAPINE‐ all data short term (up to 6 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: people with serious mental illness and co‐occurring substance misuse | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with Olanzapine | Risk with Risperidone | |||||
| Mental state: 2. Specific‐ Positive symptoms, total score‐ average endpoint scores (SADS‐C‐PD scale, lower = better) | The mean positive symptoms total score at endpoint (SADS‐C‐PD scale, lower = better) in the intervention group was 1.5 lower (3.82 lower to 0.82 higher) | ‐ | 37 | ⊕⊝⊝⊝ | ||
| Substance use: 1. Reduction of cannabis use‐change data (number of joints smoked/week) | The reduction of cannabis joints smoked (number of joints smoked/week‐short term data, up to 6 months) in the intervention group was 0.4 higher (4.72 lower to 5.52 higher) | ‐ | 41 | ⊕⊝⊝⊝ | ||
| Subjective well‐being | ‐ | ‐ | ‐ | No trial reported on this important outcome for participants with a co‐occurring substance use disorder | ||
| Craving for substances: 2. Drug Desires Questionnaire‐ average endpoint scores (DDQ, lower = better) | The mean endpoint. Drug Desires Questionnaire‐ endpoint scores (DDQ, lower = better), short term, up to 6 months‐in the intervention group was5 higher (4.86 lower to 14.86 higher) | ‐ | 41 | ⊕⊝⊝⊝ | ||
| Adherence to antipsychotic medication: number of missed doses, average endpoint data, short term (up to 6 months) | ‐ | ‐ | ‐ | ‐ | no useable data available for this outcome | |
| Adverse effects: Parkinsonism ‐ average endpoint score (SAS, high = worse) | The mean adverse effects: ‐ Parkinsonism‐ average endpoint score (SAS, high = worse)‐ short‐term‐ up to 6 months in the intervention group was 0.08 lower (1.21 lower to 1.05 higher) | ‐ | 16 | ⊕⊝⊝⊝ | ||
| Leaving study early: any reason | Study population | RR 0.68 | 77 | ⊕⊝⊝⊝ | ||
| 357 per 1000 | 243 per 1000 | |||||
| Moderate | ||||||
| 411 per 1000 | 279 per 1000 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 High risk for performance bias, allocation concealment, unknown risk for attrition and slective reporting 2 Very low sample size, optimal information size (OIS) not met 3 High risk of attrition bias, study sponsored by pharmaceutical industry 4 Very low sample size, optimal information criterion not met, CI crosses both appreciable harm and benefit 5 High attrition risk, high other risk of funding by pharmaceutical industry, all other risk items unclear risk of bias 6 High risk of performance, attrition and funding bias. Several domains with unclear risk of bias | ||||||
| RISPERIDONE versus PERPHENAZINE‐long term data (>12 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: people with severe mental illness and co‐occurring substance misuse | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with PERPHENAZINE | Risk with RISPERIDONE | |||||
| Leaving the study early: any reason | Study population | RR 1.05 | 281 | ⊕⊕⊝⊝ | ||
| 750 per 1000 | 788 per 1000 | |||||
| Moderate | ||||||
| 750 per 1000 | 788 per 1000 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 High risk of attrition bias, but this does not affect this particular outcomes 2 Optimal information size criterion is met but the estimate includes no effect with both appreciable harm and benefit | ||||||
| RISPERIDONE versus QUETIAPINE‐ short and long term data (up to 6months and > 12 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: people with severe mental illness and co‐occurring substance misuse | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with QUETIAPINE | Risk with RISPERIDONE | |||||
| Leaving the study early: 1. any reason, long term (>12 months) | Study population | RR 0.96 | 294 | ⊕⊕⊝⊝ | ||
| 825 per 1000 | 792 per 1000 | |||||
| Moderate | ||||||
| 825 per 1000 | 792 per 1000 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Risk of bias unclear across all groups and with high risk of funding bias 2 Sample size meets optimal information threshold/ required sample size to detect 25% difference from control group in in PANSS score; at alpha of 0.05 and power of 80%. 3 Outcome not affected by risk of attrition bias 4 Optimal information criterion not met, estimate includes both appreciable harm and benefit | ||||||
| RISPERIDONE versus ZIPRASIDONE‐ all data long term data (>12 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: people with severe mental illness and co‐occurring substance misuse | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with ZIPRASIDONE | Risk with RISPERIDONE | |||||
| Leaving the study early: any reason | Study population | RR 0.96 | 240 | ⊕⊕⊝⊝ | ||
| 819 per 1000 | 787 per 1000 | |||||
| Moderate | ||||||
| 819 per 1000 | 787 per 1000 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Risk of attrition bias high but this does not affect this outcome 2 Optimal information size criterion met but estimate includes both appreciable harm and benefit. Total sample size small | ||||||
Background
Description of the condition
Serious mental illness is characterised by severe and persisting psychiatric disorder associated with significant functional impairment, and includes disorders such as schizophrenia and bipolar disorder, but is not limited to these conditions (Ruggeri 2000). The prevalence of serious mental illness varies according to survey methods and has been reported to range from 0.4% to 7.7% between different countries (Demyttenaere 2004).
Comorbid substance use frequently occurs in people with severe mental illness such as schizophrenia and major affective disorders. The terms 'dual diagnosis' and more recently 'co‐occurring disorders' have been used to describe persons who have a diagnosis of both a severe mental illness and a drug or alcohol use disorder (Buckley 2006). Depending on the sample, population and assessment method, prevalence rates of drug and alcohol use disorders in persons with serious mental illness have been reported to vary from 25% to 74% in developed countries (Barnett 2007; Fioritti 1997; Fowler 1998; Jablensky 2000; Kessler 2004; Lambert 2005; Menezes 1996; Modestin 1997; Regier 1990; Soyka 1993; van Mastrigt 2004). The prevalence of co‐occurring substance use disorders in persons with severe mental illness from low‐ and middle‐income countries has been found to be in a similar range, with some studies reporting prevalence rates of 51% and up to 68% (Hauli 2011; Weich 2009). Patients participating in studies investigating the use of atypical antipsychotics such as risperidone in the treatment of dual diagnosis have a variety of substance use disorder diagnoses including cocaine, cannabis, opioid and alcohol use disorders; and most have a mental illness diagnosis of schizophrenia or schizoaffective disorder (Kelly 2012; Stuyt 2006). Comorbidity adversely affects the treatment and prognosis of both drug use disorders and mental illness. In comparison to people without substance use disorders, persons with a dual diagnosis have been reported to have higher levels of positive psychotic symptoms (Katz 2010), shorter time to relapse, higher rates of readmission to hospital, lower remission rates (Lambert 2005; Swofford 1996; Wade 2006), higher levels of medication non‐compliance (Ascher‐Svanum 2006; Lacro 2002; Owen 1996), and worse clinical outcomes (Lambert 2005). In addition, dual diagnosis patients with psychotic or manic symptoms treated with antipsychotics are more susceptible to extrapyramidal side‐effects such as akathisia (Salyers 2001).
Description of the intervention
Risperidone is an antipsychotic medication described as a serotonergic and dopaminergic antagonist (SDA) (Horacek 2006, Figure 1). It was the first medication in the second‐generation class of antipsychotics (SGAs), also described as 'atypical' antipsychotics, to be synthesised (Moller 2005). Risperidone is an antagonist on dopamine type 2 (D₂) receptors, and has an even higher affinity for serotonin type 2A receptors, where it also acts as an antagonist (Janssen 1988). These properties result in a relatively low propensity to induce extrapyramidal side‐effects when compared with first‐generation antipsychotics (FGAs) or 'typical' antipsychotics such as haloperidol (Hunter 2003). Important non‐neurological side‐effects include a higher risk of weight gain compared with certain typical and other atypical antipsychotics (Newcomer 2007).

Risperidone
Risperidone is available in an oral as well as a long‐acting injectable formulation (Fleischhacker 2003; Moller 2007). There are no specific recommendations for any particular type of antipsychotic medication to be used preferentially in the dual diagnosis population, and it has been recommended that the same guidance for persons with only a single severe mental illness diagnosis be used in persons with a dual diagnosis (NICE 2011). Accordingly, some treatment guidelines recommend that second‐generation — or 'atypical' — antipsychotics such as risperidone be considered as potential first‐line treatment for persons with a first episode of psychosis and schizophrenia, following discussion with patients and their caregivers (Hasan 2012; Hasan 2013; Lehman 2004; NICE 2009).
How the intervention might work
A number of findings from pre‐clinical and clinical studies have suggested various mechanisms and effects through which second‐generation antipsychotics may result in improved outcomes in the treatment of dual diagnosis.
-
Preclinical studies have shown that risperidone blocks cocaine‐induced dopamine and serotonin release in the nucleus accumbens in the rat brain, and reduces locomotion and stereotypical behaviour in rats (Broderick 2003; Tsibulsky 1998). In turn, treatment of humans with atypical antipsychotics such as risperidone, which act antagonistically on both serotonergic and dopaminergic receptors, may translate to a reduction of conditioned responses such as cue‐elicited craving mediated via dopamine release. This may consequently lead to lower levels of drug use (Drevets 2001; Smelson 1997; Volkow 2006). In addition, research in rats has demonstrated increased sensitivity to cocaine‐induced hyper‐locomotion and increased conditioned place preference following withdrawal from haloperidol, but not from the atypical agent ziprasidone. This serves as a model of dopamine supersensitivity and induced craving which is potentially analogous to situations of non‐compliance with antipsychotic medication in humans (Fukushiro 2007; Fukushiro 2008). Treatment non‐compliance is a phenomenon that can occur in up to three‐quarters of people treated with antipsychotics (Lieberman 2005).
-
Despite the acute reinforcing effects of drug use via increased dopamine release during the early phases of addiction, neuro‐imaging studies in persons with drug dependence have demonstrated lower concentrations of dopaminergic receptors following chronic use (Volkow 1997; Volkow 2007). Lower dopamine receptor concentration may be due to persistently increased dopamine release in the nucleus accumbens induced by drugs of abuse (Kuhar 1996). In turn, increased craving has been associated with lower dopamine receptor levels during protracted withdrawal (Volkow 2011). Consequently, in comparison to drugs with a lower affinity for D₂ receptors such as risperidone, drugs with a high affinity for dopamine receptors, such as the first‐generation antipsychotics, may exacerbate or prolong hypodopaminergic states leading to increased drug craving (Siris 1990).
-
Craving has been shown to be an important predictor of relapse into substance use (Sinha 2006; Sinha 2011). Uncontrolled trials have demonstrated a decrease in craving and drug use in persons treated with risperidone (Smelson 2002). Consequently, the use of risperidone may result in lower scores on craving measures (Rosenberg 2009), and consequently decreased drug use.
-
Results from uncontrolled clinical trials have shown lower levels of psychotic symptoms and depression in persons treated with risperidone when compared to first‐generation antipsychotics (Smelson 1997; Smelson 2002). Risperidone also acts on the serotonergic system by means of 5HT2A receptor antagonism, which may in turn mediate its antidepressant effect (McIntyre 2007; Sajatovic 2002; Yatham 2005). This may affect the 5HT1A system resulting in fewer depressive symptoms and improved cognitive symptoms in persons with serious mental illness such as schizophrenia (Kuroki 2008; Sajatovic 2002). Altogether, these effects may translate into improved ratings on measures of psychosis, mood and quality of life in persons treated with risperidone (Smelson 2002).
-
Clinical studies have demonstrated comparatively lower rates of extrapyramidal side‐effects in participants treated with risperidone (Hunter 2003). As people with dual diagnosis have been shown to be more sensitive to the neurological side‐effects of neuroleptic drugs (Potvin 2009; Salyers 2001), the low propensity of risperidone to induce extrapyramidal side‐effects offers a potentially favourable side‐effect profile. This may result in improved adherence (Perkins 2002; Perkins 2006); and consequently lower rates of symptom re‐emergence and of relapse into illness (Sun 2007).
-
It has been postulated that one of the properties that differentiate atypical antipsychotics from typical antipsychotics may be their lower affinity for D₂ receptors and higher affinity for 5HT2A receptors (Seeman 2002). However, it remains uncertain to what extent potent 5HT2A receptor‐antagonism is responsible for the 'atypical features' of SGAs, given alternative theories of D₂ receptor binding profiles and dissociation rates conferring "atypicality" (Kapur 1995; Kapur 1996; Kapur 2001; Seeman 2002; Tort 2006). In addition, higher levels of D₂ receptor occupancy in the context of neuroleptic treatment has also been associated with lower levels of subjective well‐being (de Haan 2000). Second‐generation agents such as risperidone that have lower D₂ receptor occupancy rates (and higher dissociation rates), therefore have a more favourable profile in that they have been demonstrated to produce higher levels of subjective well‐being, and are associated with less negative affective states and anhedonia compared to first‐generation, typical medications (Smelson 2002; Vothknecht 2011). Therefore, the degree of D₂ binding and D₂ receptor dissociation of antipsychotics may have a differential impact on measures used to assess subjective well‐being. However, within the class of serotonin dopamine antagonists various agents differ in their degree of D₂ receptor affinity and dissociation, with clozapine and olanzapine having the lowest affinities (and highest dissociation rates) and risperidone having higher D₂ affinity. Therefore uncertainty remains as to how medications such as risperidone, that differ only marginally from FGAs with regards to D₂ receptor‐binding profiles (Seeman 2002), offer additional benefit over typical agents.
Figure 2 contains a diagram of a model within a Baxter 2010 logic framework, outlining the relationship between antipsychotic treatment with risperidone, potential effect modifiers and the final target clinical outcomes in a complex causal pathway.
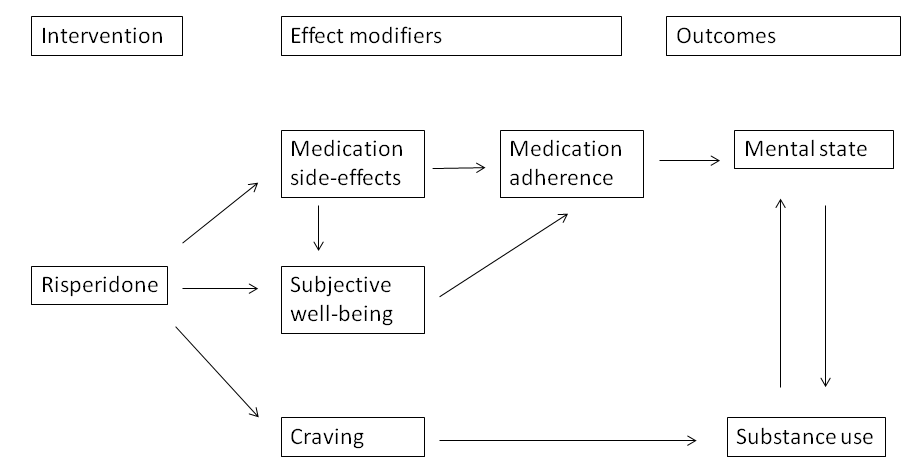
Logic framework model with potential causal pathways: risperidone treatment in persons with dual diagnosis.
Why it is important to do this review
Despite preclinical animal studies and observational research in humans, there remains a paucity of randomised trials on the efficacy of risperidone compared with other antipsychotics in the treatment of people with a dual diagnosis. In fact, most randomised controlled trials that investigate the efficacy of risperidone in the treatment of serious mental disorders such as schizophrenia have excluded people with comorbid drug or alcohol use disorders (Hunter 2003). As comorbid drug and alcohol use disorders occur commonly in people with severe mental illness, it is important to evaluate the impact of risperidone on drug use and symptom control, as well as on other functional outcomes in people with dual diagnosis. Moreover other atypical medications such as olanzapine and clozapine, that have higher D₂ receptor dissociation rates and greater activity on D₁ receptors (Seeman 2002; Tort 2006), have been postulated to reduce drug use to a greater extent compared to risperidone (Akerele 2007). This review aims to assess whether risperidone offers any additional advantage over FGAs or other SGAs in the treatment of people with serious mental illness and co‐occurring substance misuse.
Objectives
To evaluate the effects of risperidone compared to treatment with other antipsychotics (first‐generation and other second‐generation antipsychotics) used in people with serious mental illness and co‐occurring substance misuse.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs), in which risperidone was the antipsychotic treatment that was randomised. If a trial was described as 'double blind' but randomisation was implied, we included such trials after confirming randomisation with the study investigators. We excluded quasi‐randomised studies, such as those allocating by alternate days of the week.
Types of participants
Adults, aged 17 to 65 years, with a severe mental illness and a co‐occurring substance use disorder, where severe mental illness is defined to include the following disorders as diagnosed according to the method specified in each individual trial.
-
Schizophrenia, schizoaffective disorder, schizophreniform disorder.
-
Major affective disorders such as bipolar disorder and major depressive disorder with psychotic features.
Severe mental illness is defined to exclude the following disorders.
-
Organic mental disorders.
-
Moderate to severe intellectual disability.
-
Drug‐induced mental disorders.
-
Personality disorders.
-
Non‐severe mental disorders such as anxiety disorders, mild depression, and somatoform disorders.
-
Factitious disorders or malingering.
Co‐occurring substance use disorder is diagnosed by any means or scale and includes:
-
alcohol or drug abuse or dependence.
Co‐occurring substance use disorder is defined to exclude:
-
nicotine dependence, volatile solvent abuse or dependence, and caffeine use disorders, where the trial has been designed to primarily measure the impact of risperidone on these substance use disorders and related outcomes;
-
studies in which drug use was not classified as either abuse or dependence.
We included RCTs that compared treatment with risperidone with another antipsychotic in people with a dual diagnosis of severe mental disorder and a co‐occurring substance use disorder.
We placed no restrictions on co‐morbid conditions such as anxiety and depressive disorders.
As indicated in the description of the condition, the phenomenon of dual diagnosis is prevalent across various settings as reflected in data from a variety of studies. Therefore we included all settings, embracing those from both the developed and the developing world, as well as where dual diagnosis treatments were delivered in various models of care (such as integrated or parallel service models).
We included studies with participants in any clinical state or stage of illness.
Types of interventions
1. Experimental interventions
Treatment with risperidone in any formulation and any dose.
2. Comparator interventions
Any other antipsychotic, divided into first‐generation ('typical') and second‐generation ('atypical').
In addition to antipsychotic treatment, we included studies in which co‐administration with a mood stabiliser (lithium, valproate, lamotrigine, carbamazepine, topiramate) or antidepressant (serotonin re‐uptake inhibitors, tricyclic, tetracyclic or new generation anti‐depressants) occurred in both treatment arms. Furthermore, studies comparing risperidone with another antipsychotic, where both treatment arms receive the same additional psychosocial intervention, were also included. Studies where only one treatment arm received an additional psychopharmacological or psychosocial intervention were excluded. Where imbalances exist between the treatment arms in terms of the dose, timing or duration of the additional treatments, we intended to note and discuss the impact of such imbalances in the section on assessment of risk of bias (Table 1).
| Sequence generation |
|
| Allocation concealment |
|
| Blinding of participants and personnel |
|
| Blinding of outcome assessment |
|
| Incomplete outcome data |
|
| Selective reporting |
|
| Other forms of bias |
|
Types of outcome measures
We categorised outcomes into short‐term, medium‐term and long‐term outcomes. Short‐term outcomes included outcomes measured in the first six months, medium‐term outcomes include outcomes from six months to one year, and long‐term outcomes include outcomes beyond one year.
Primary outcomes
1. Mental state
1.1 General
1.1.1 Clinically important change in general mental state ‒ as defined by trial authors.
1.2 Specific symptoms
1.2.1 Clinically important change in positive symptoms ‒ as defined by trial authors.
1.2.2 Clinically important change in negative symptoms ‒ as defined by trial authors.
1.2.3 Clinically important change in anxiety symptoms ‒ as defined by trial authors.
2. Substance use (determined according to how it was assessed, i.e. whether by means of self‐report, or determined biochemically, either by positive urine or blood tests, or gas chromatography mass spectroscopy on hair follicles)
2.1 A reduction in substance use (drug or alcohol).
2.2 The presence or absence of drug or alcohol use at the end of the study follow‐up period.
3. Adverse effects *
3.1 Clinically important adverse effect ‒ as defined by trial authors.
Secondary outcomes
1. Mental state
1.1 General
1.1.1 Any change in general mental state ‒ as defined by trial authors.
1.1.2 Average change/endpoint scores mental state scale.
1.1.3 Change in co‐morbid psychopathology.
1.2 Specific symptoms
1.2.1 Any change in positive symptoms ‒ as defined by trial authors.
1.2.2 Average change/endpoint scores positive mental state scale.
1.2.3 Any change in negative symptoms ‒ as defined by trial authors.
1.2.4 Average change/endpoint scores negative mental state scale.
1.2.5 Average change/endpoint scores anxiety scale.
2. Substance use (determined according to how it was assessed, i.e. whether by means of self‐report, or determined biochemically, either by positive urine or blood tests, or gas chromatography mass spectroscopy on hair follicles)
2.1 Time to relapse into drug or alcohol use.
2.2 Frequency of use over the study period.
3. Subjective well‐being as measured by validated rating scales
3.1 Average endpoint/change scores on subjective well‐being scales.
4. Craving for substances
4.1 Average endpoint/change scores on craving scales.
5. Adherence to antipsychotic medication
5.1 Average endpoint/change scores on medication adherence rating scales.
5.2 An improvement in adherence to antipsychotics ‒ as defined by trial authors.
6. Adverse effects
6.1 Any general adverse effects.
6.2 Specific adverse effects.
6.2.1 Allergic reactions.
6.2.2 Blood dyscrasia such as agranulocytosis.
6.2.3 Central nervous system (ataxia, nystagmus, drowsiness, fits, diplopia, tremor).
6.2.4 Metabolic adverse events (weight changes, serum glucose measures, serum triglyceride measures, serum high density lipoprotein (HDL) measures).
6.2.5 Endocrinological dysfunction (hyperprolactinaemia, disturbance of reproductive organ and sexual performance functioning).
6.2.6 Gastrointestinal (nausea, vomiting, diarrhoea).
6.2.7 Kidney dysfunction.
6.2.8 Movement disorders (extrapyramidal side‐effects, including neuroleptic malignant syndrome and tardive dyskinesia).
7. Leaving the study early
7.1 A reduction in numbers leaving study early: any reason.
7.2 A reduction in the proportion of participants lost to follow‐up at the study end‐point.
7.3 An increase in the proportion of participants attending the first follow‐up visit.
7.4 An increase in time to attrition.
7.5 An increase in the mean number of clinic visits.
8. Mortality
8.1 Due to natural causes.
8.2 Due to drug overdose.
8.3 Due to suicide.
8.4 Due to any other unnatural cause.
9. Quality of life
9.1 General quality of life
9.1.1 Clinically important change in general quality of life ‒ as defined by trial authors.
9.1.2 Any change in quality of life ‒ as defined by trial authors.
9.1.3 Average change/endpoint scores quality of life scale.
9.2 Physical health
9.2.1 Clinically important change in physical health ‒ as defined by trial authors.
9.2.2 Any change in physical health ‒ as defined by trial authors.
9.2.3 Average change/endpoint scores physical health. scale
10. 'Summary of findings' table
We used the GRADE approach to interpret findings (Guyatt 2011; Schünemann 2011); and used GRADEpro to export data from our review to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
-
Mental state: General — clinically important change ‒ at study endpoint.
-
Substance use: a reduction in substance use ‒ at study endpoint.
-
Subjective well‐being ‒ improvement in measures of subjective well‐being.
-
Craving for substances ‒ improvement in measures of substance craving.
-
Adherence to antipsychotic medication ‒ improvement in measures of medication adherence.
-
Adverse effects ‒ clinically important adverse effect.
-
Leaving the study early ‒ a reduction in the proportion of participants leaving the study early: any reason.
Search methods for identification of studies
Electronic searches
1. Cochrane Schizophrenia Group’s Study‐Based Register of Trials
On 6 January 2016 and 9 October 2017, the Information Specialist searched the register using the following search strategy:
*Risperidone* in Intervention AND *Substance Abuse* in Healthcare Condition Fields of STUDY
In such study‐based registers, searching the major concept retrieves all the synonyms and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics (Shokraneh 2017).
This register is compiled by systematic searches of major resources (AMED, BIOSIS, CINAHL, ClinicalTrials.Gov, Embase, MEDLINE, PsycINFO, PubMed, WHO ICTRP) and their monthly updates; ProQuest Dissertations and Theses A&I and its quarterly update; Chinese databases (CBM, CNKI, and Wanfang) and their annual updates; handsearches; grey literature; and conference proceedings (see Group’s Module). There are no language, date, document type, or publication status limitations for inclusion of records into the register.
2. Cochrane Common Mental Disorders Group's Trials Register
On 8 January 2016 and 10 October 2017, the Information Specialist of Cochrane's Common Mental Disorders Group searched the trials register of their group using the following search strategy:
Search 1:
#1. (*Risp* AND (*Substance* OR *Cannabis* OR *Amphetamine* OR *Alcohol* OR *Cocaine* OR *Opioid* OR *Drug Dependence* OR *Addict*)) [in Register]
Search 2:
#2. (*Risp*)
#3. (“substance use disorder*” or SUD or SUDs)
#4. “drug abuse”
#5. (abuser* or abusing or addict* or depend* or habit* or misuse or user*)
#6. (abuse not (child* or sex*))
#7. (adinazolam or aerosol* or alcohol* or alprazolam or amphetamin* or anthramycin or anxiolytic* or ativan or barbituat* or bentazepam or benzodiazepin* or bromazepan or brotizolam or buprenorphin* or camazepam or cannabi* or chlordiazepoxid* or cinolazepam or clobazam or clonazepam or clorazepam or clotiazepam or cloxazolam or cocaine* or codeine or crack or crystal or cyprazepam or depressant* or diacetylmorphin* or diazepam* or doxefazepam or ecstasy or estazolam or etizolam or fentanyl or flunitrazepam or flurazepam or flutazoram or flutoprazepam or fosazepam or gases or GHB or girisopam or halazepam or hallucinogen* or haloxazepam or heroin* or hydromorphone or hydroquinone or hypnotic* or inhalant* or ketamin* or ketazolam or librium or loflazepate or loprazolam or lorazepam or lormetazepam or LSD or marihuana* or marijuana* or MDMA or meclonazepam or medazepam or meperidine or mephedrone or mescalin* or metaclazepam or methadone or methamphetamin* or methaqualone or mexazolam or midazepam or midazolam or morphine* or narcotic* or nerisopam or nimetazepam or nitrazepam or nitrites or "nitrous oxide" or "n‐methyl‐3,4‐methylenedioxyamphetamine" or nordazepam or opiate* or opiod* or opium or oxazepam or oxazolam or oxazypam or oxycodone or oxzepam or painkiller* or "pain killer*" or PCP or pethidin* or phencyclidin* or pinasepam or prazepam or propazepam or propoxyphene or psilocybin or psychedelic* or psychoactive* or psychostimulant* or quinazolinone or ripazepam or ritalin or sedative* or serazepin* or solvent* or steroid* or stimulant* or substance* or temazepam or tetrazepam or tofisopam or tramadol or triazolam or triflubazam or valium or vicodin)
#8. (drug* and (recreational or street))
#9. #2 and (#3 or #4 or #5 or #6 or #7 or #8)
#10. #9 not #1 [in Register]
For previous searches, please see Appendix 1.
Searching other resources
1. Reference searching
We inspected reference lists of all included studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials. In addition we contacted pharmaceutical companies regarding unpublished trials.
Data collection and analysis
Selection of studies
HT and TW independently inspected citations from the searches and identified relevant abstracts. NS independently re‐inspected included abstracts to ensure reliability. HT and TW obtained and inspected full reports of the abstracts meeting the review criteria. Where disputes arose, NS re‐inspected reports in order to ensure reliable selection. Where it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study for clarification.
Data extraction and management
1. Extraction
Review authors HT and TW extracted data from all included studies. Again, any disagreements were discussed; decisions documented; and, if necessary, authors of studies were contacted for clarification. NS helped to clarify issues with remaining problems and we documented these final decisions. If data had been presented only in graphs we would have used data in analyses only when both reviewers derived similar results from extraction. One study reported data in graphs only, but we were unable to extract as data were reported for subgroups and the total number of participants at study endpoint for each subgroup was not known, precluding a calculation of standard deviations (SDs) (Akerele 2007). In case of multi‐centre studies we attempted to get information from authors so as to extract data from each component centre separately. In one multi‐centre study, authors were unable to provide this level of data and the data across all participating centres were reported.
2. Management
2.1 Forms
We extracted data onto a standardised form and piloted it on one study prior to use.
2.2 Scale‐derived data
We included continuous data from rating scales only if:
-
the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and
-
the measuring instrument had not been written or modified by one of the trial investigators for that particular trial.
Ideally the measuring instrument should either be i) a self‐report or ii) completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly and recorded if this was the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand calculation of change needs two assessments (baseline and endpoint) which can be difficult to measure in unstable conditions such as schizophrenia. We decided to primarily use endpoint data, and only used change data if the former were not available. We decided that in cases where change data had been used, endpoint and change data would be combined in the analysis. If this had occurred we would have used mean differences (MD) rather than standardised mean differences (Higgins 2011). As pooling of continuous data in a meta‐analysis was not possible for this review we did not use this method.
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion.
-
Standard deviations and means are reported in the paper or obtainable from the authors.
-
When a scale starts from the finite number zero, the standard deviation, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution (Altman 1996).
-
If a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS) which can have values from 30 to 210) we modified the calculation described above to take the scale starting point into account. In these cases skew is present if 2 SD > (S − Smin), where S is the mean score and Smin is the minimum score.
Endpoint scores on scales often have a finite start and end point, and the above rules can be applied. Had we found skewed data from studies of fewer than 200 participants we would have entered such data in additional tables rather than into an analysis. Skewed data pose less of a problem when looking at means if the sample size is large, and had we found such studies we would have entered them into syntheses.
When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. We included change data in statistical analyses regardless of the size of the study.
2.5 Common measure
To facilitate comparisons between trials, we converted variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary data
Where possible, we converted outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS, Kay 1986, this could be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors. As the definition of a clinically significant treatment response may differ across various patient populations, we accepted a 50% symptom reduction as an adequate response in acute, non‐refractory schizophrenia and a 25% reduction in chronic, refractory patients (Leucht 2009), and used these different definitions depending on the population studied. Where possible we also attempted to analyse continuous data.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicates a favourable outcome for risperidone. Where keeping to this makes it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'Not unimproved') we reported data where the left of the line indicates an unfavourable outcome. This was noted in the relevant graphs.
Assessment of risk of bias in included studies
Review authors HT and TW worked independently to assess risk of bias by using criteria described in theCochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article, and includes assessment of sequence generation, allocation concealment, blinding, incomplete outcome data, selective reporting and other forms of bias. Measures to prevent risk of bias were assessed as either 'high', 'low' or 'unclear', according to the definitions of these ratings in the Cochrane Handbook for Systematic Reviews of Interventions (Table 1).
If the raters disagreed, they came to a consensus over the final rating by involving another review author (NS). Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain further information.
We noted the level of risk of bias for each included study in both the text of the review and within 'Risk of bias' tables and incorporated them in the judgement of overall quality across studies in the 'Summary of findings' tables.
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive than odds ratios (Boissel 1999); and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000).
2. Continuous data
For continuous outcomes we estimated the mean difference (MD) and the standard deviation (SD) between groups. However, if scales of very considerable similarity were used, we presumed there was a small difference in measurement, and we calculated the standardised mean difference (SMD) and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster randomised control trials
Analysis of multi‐level data may pose problems and failure to account for clustering in data may result in unit of analysis errors (Divine 1992), whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Where clustering was not accounted for in primary studies, we would have presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999).
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC (Design effect = 1 + (m − 1) * ICC) (Donner 2002). If the ICC is not reported it will be assumed to be 0.1 (Ukoumunne 1999).
If we had found cluster randomised trials in this review and had been able to analyse them appropriately, taking into account ICCs and relevant data documented in the report, we could then have included them in a synthesis with other studies using the generic inverse variance technique. However, we found no cluster randomised trials.
2. Repeated measurements
Multi‐level data may also arise where multiple measurements are conducted on one participant, i.e. counts of the number of urine tests over a period of time per participant that screen positive for drug use. In such cases, unit of analysis issues may also occur. Outcomes could also be reported as the percentage of participants per treatment group with positive urine drug tests, which represents another form of count data that is not compatible with Cochrane Review Manager 5 (RevMan 5) software, that analyses data derived at an individual participant level. In such cases, we contacted the authors to provide actual count data on the number of individual urine tests that screened positive or negative per participant over the study period. In accordance with methods previously described by Mattick 2014, we asked authors to derive a mean and SD of positive urine tests per treatment group over the study period. We would have then analysed differences between treatment groups as continuous data. In cases where the denominator differed for individual participants due to missed study visits and missing data for urine testing, we would have omitted studies where missing data occurred in more than 50% visits per individual participant and for more than 50% of overall participants in either treatment arm. In studies where no data on number of urine tests were obtained, we would have dealt with missing outcome data by the methods described under Dealing with missing data for continuous outcomes. We found one such study where urine tests were measured over time, generating longitudinal data (Akerele 2007). No information was provided by study authors despite attempts to contact them. In a second study authors did report means and standard deviations (van Nimwegen 2008).
3. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, if we obtained data from cross‐over trials we would have only used data from the first phase of such studies. However, we included no cross‐over studies in this review.
4. Studies with multiple treatment groups
When a study involved more than two treatment arms, if relevant we presented the additional treatment arms in comparisons. If data were binary these would have simply been added and combined within a two‐by‐two table, in order to avoid double counting a common comparison group in the same meta‐analysis. If data had been continuous we would have combined data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook of Systematic Reviews of Interventions (Higgins 2011). If the additional treatment arms were not relevant, these data would not have been reproduced.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss to follow‐up, data must lose credibility (Xia 2009). We chose that, for any particular outcome, if more than 50% of data were unaccounted for, we did not reproduce these data or use them within analyses (except for the outcome 'leaving the study early'). If, however, more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we marked such data with (*) to indicate that such a result may well be prone to bias.
2. Binary
In cases where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on a 'once randomised always analyse' basis (an intention‐to‐treat analysis). Those leaving the study early were all assumed to have had the same rates of negative outcome as those who completed, with the exception of the outcomes of death and adverse effects. For these outcomes, the rate of those who stayed in the study — in that particular arm of the trial — was used for those who did not. However, due to the small study sample sizes we were not able to conduct such a sensitivity analysis in order to test how prone the primary outcomes are to change when 'completer' data only are compared to the intention‐to‐treat analysis using the above assumptions.
3. Continuous
3.1 Attrition
Where attrition for a continuous outcome is between 0% and 50%, and completer‐only data were reported, we reproduced these.
3.2 Standard deviations
If standard deviations (SDs) were not reported, we first tried to obtain the missing values from the study authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error and CIs available for group means, and either P value or t value available for differences in mean, we calculated them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). When only the standard error (SE) is reported, SDs are calculated by the formula SD = SE * √(n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) present detailed formula for estimating SDs from P values, t or F values, CIs, ranges or other statistics. If these formulae did not apply, we would have calculated the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome, and thus to lose information. We nevertheless would have examined the validity of the imputations in a sensitivity analysis excluding imputed values. However, as our search only yielded two studies with two different comparisons, we were unable to use this method of imputation.
3.3 Last observation carried forward
We anticipated that in some studies the method of last observation carried forward (LOCF) would have been be employed in the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, when LOCF data had been used in the trial and if less than 50% of the data had been assumed, we would have reproduced these data and indicated that they were the product of LOCF assumptions.
Assessment of heterogeneity
1. Clinical heterogeneity
We inspected all studies to determine whether the studies were similar enough in terms of participant profile and intervention comparisons to combine them, and discussed such differences if any were found.
2. Methodological heterogeneity
We simply inspected all studies for clearly outlying methods and discussed any outlying methods in full.
3. Statistical heterogeneity
3.1 Visual inspection
Where possible we visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I² statistic
We investigated heterogeneity between studies by considering the I² statistic alongside the Chi² test P value. The I² statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I² depends on i) magnitude and direction of effects and ii) strength of evidence for heterogeneity (e.g. P value from Chi² test, or a CI for I²). I² values between 0% to 40% were interpreted as possibly unimportant, 30% to 60% as possibly significant, 50% to 90% as possibly substantial, and 75% to 100% as possibly considerable (Deeks 2011). Had substantial levels of heterogeneity been found in the primary outcome, we would have explored reasons for heterogeneity (see Subgroup analysis and investigation of heterogeneity). As no study data was combined in a meta‐analysis for any primary outcomes, we did not conduct any exploration of heterogeneity.
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in section 10.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We tried to locate protocols of included RCTs by searching clinical trials registries and contacting authors. If the trial protocol was available, outcomes in the protocol and in the published report were compared. If the protocol was not available, outcomes listed in the Methods section of the trial report were compared with actual reported results.
2. Funnel plot
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are again described in section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. However, we were not able to generate funnel plots as there were no outcomes including more than two studies. Should more studies become available in future, and funnel plots become possible, we will seek statistical advice in their interpretation.
Data synthesis
As we anticipated clinical and methodological heterogeneity we used random‐effects models for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
We planned to conduct subgroup analyses based on a number of factors (substance abuse versus dependence, the type and formulation of risperidone, and the presence of additional pharmacological or psychosocial treatment in some studies), but due to the small number of studies included were unable to carry out such subgroup analyses.
Sensitivity analysis
We would have applied the following sensitivity analyses in our review to primary outcome data. However, due to the small number of studies, the low sample size within included studies and single studies within each comparison, none of these sensitivity analyses were possible.
1. Risk of bias
We would have analysed the effects of excluding trials that were judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available), allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcome. In case the exclusion of trials at high risk of bias would have not substantially altered the direction of effect or the precision of the effect estimates, then we would have included data from these trials in the analysis.
2. Assumptions for lost binary data
Where we would have had to make assumptions regarding people lost to follow‐up (see Dealing with missing data), we would have compared the findings of the primary outcomes when we used our assumption with completer data only. If there had been substantial difference, we would have reported results and discussed them but would have continued to employ our assumption.
3. Assumptions for lost continuous data
If we had to make assumptions, such those used in imputation methods, regarding missing SD data (see Dealing with missing data), we intended to compare the findings on primary outcomes when we applied these assumptions with completer data only. We intended to conduct a sensitivity analysis to test how prone results were to change when 'completer' data only were compared to the imputed data using different assumptions (LOCF, imputation from other studies). If there had been a substantial difference, we would have reported results and discussed them but would have continued to employ our assumption.
4. Imputed values
We would have also undertaken a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster randomised trials.
If substantial differences had been noted in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we would not have pooled data from the excluded trials with the other trials contributing to the outcome, but would have presented them separately.
5. Fixed effect and random effects
We expected substantial clinical and methodological heterogeneity in the studies included in the review. We therefore used a random‐effects model to combine data in a meta‐analysis. We intended to carefully inspect the results of our meta‐analysis and if it turned out that smaller studies received a higher weighting, we did intend to conduct a sensitivity analysis using a fixed‐effect model. If a fixed‐effect meta‐analysis did not show a similar beneficial effect compared to the random‐effects analysis, we would have carefully considered whether the conclusions of the random‐effects model were justified in light of the methodological rigour and risk of bias assessments of the larger compared to the smaller studies. If it had turned out that larger studies were indeed more rigorous, we would have restricted our report to the results of the meta‐analysis of the larger studies.
Results
Description of studies
For description of studies please see Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
We identified 197 records from the searches of the Cochrane Schizophrenia Group’s Study‐Based Register of Trials (including trial registers) and the Cochrane Common Mental Disorders' database (Figure 3). In addition we obtained four records from other sources (two through searches of a clinical trials register, one from communication with an author and one from search of reference lists). After removal of 145 abstracts that did not meet eligibility criteria, a total of 56 full text records containing 37 studies that appeared to meet eligibility criteria were inspected in detail. After excluding 23 studies which did not meet our inclusion criteria (see Characteristics of excluded studies) and a further six studies either awaiting classification or ongoing (Characteristics of studies awaiting classification; Characteristics of ongoing studies), we were left with eight studies meeting our inclusion criteria (see Characteristics of included studies).
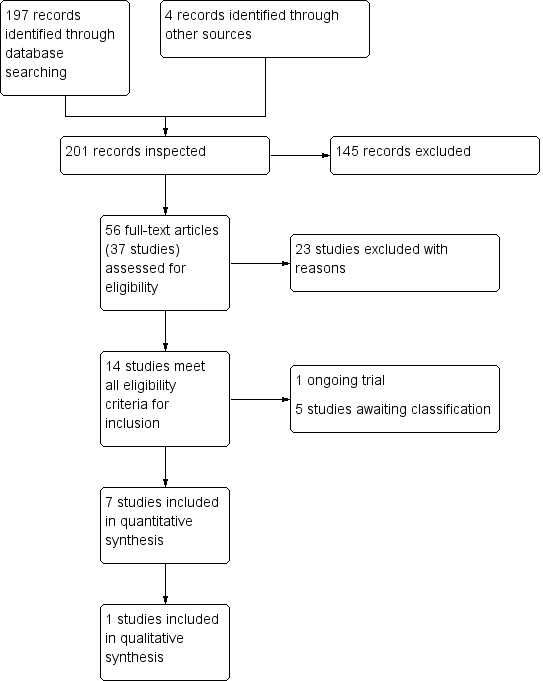
PRISMA flow diagram of study selection from 2016 and 2017 searches
Included studies
A total of eight studies met criteria for inclusion (Akerele 2007; Brunette 2011; Machielsen 2014; Noordsy 2010; Sevy 2011; Smelson 2006; Swartz 2008; van Nimwegen 2008); however, Brunette 2011 did not provide any useable data for analyses.
1. Design and duration
Four of the included studies were parallel‐group superiority trials that randomised participants to risperidone and a comparator drug. These studies all had pre‐specified hypotheses about the comparator drugs. In the study by Akerele 2007 the authors hypothesised that olanzapine would be superior to risperidone in reducing cocaine and cannabis use in people with schizophrenia in a 14‐week trial. In Noordsy 2010 the authors hypothesised that clozapine would be superior to risperidone in reducing substance use and psychiatric symptoms over 24 weeks of study. In a 4‐week trial, Machielsen 2014 hypothesised that clozapine would be superior to risperidone in reducing brain activation in regions associated with attentional bias on fMRI, reduce craving and increase subjective well‐being in people with first episode schizophrenia with comorbid cannabis use disorders. In a 12‐week trial Brunette 2011 randomised participants to either continue their treatment with their treatment as usual (TAU) or to switch participants to clozapine, with the hypothesis that clozapine would reduce cannabis use. In this study five participants in the TAU group were taking risperidone.
Three trials were post‐hoc, secondary data analyses of existing larger parent randomised trials (Sevy 2011; Smelson 2006; Swartz 2008). In Sevy 2011 the authors studied a group of 49 participants from a larger randomised trial of 120 participants (Robinson 2006) over 16 weeks who were randomised to risperidone and olanzapine. Smelson 2006 analysed data on 236 out of 632 substance users from a parent study (Tunis 2006) randomised to risperidone, olanzapine and conventional agents (including perphenazine, loxapine, haloperidol, fluphenazine, thiothixene) over a 12‐month study period. Swartz 2008 studied 643 out of 1432 participants from the CATIE study (Stroup 2003) randomised to risperidone, olanzapine, quetiapine, perphenazine, and ziprasidone, continued over 18 months of study.
One study was a randomised parallel‐group superiority trial comparing risperidone to a comparator drug and reported outcomes on a subgroup of participants with co‐occurring substance use disorders (van Nimwegen 2008).
2. Participants
Participants in all of the included studies had a diagnosis of either schizophreniform disorder, schizophrenia or schizoaffective disorder and co‐occurring substance misuse. In Noordsy 2010 and Sevy 2011, participants were experiencing their first episode of psychosis (schizophrenia or schizoaffective disorder). In Swartz 2008, participants were multi‐episode.
Five studies exclusively randomised participants with a co‐occurring cannabis use disorder (Brunette 2011; Machielsen 2014; Noordsy 2010; Sevy 2011; van Nimwegen 2008). The other three studies included participants with other substance use disorders such as alcohol, amphetamine, cocaine and opioid use disorders (Akerele 2007; Smelson 2006; Swartz 2008).
In Akerele 2007, Smelson 2006 and Swartz 2008, participants were adults of mixed ethnicity and in Noordsy 2010 all participants were Caucasian (understood to be white participants). Brunette 2011 had 83.9% Caucasian participants; and in Machielsen 2014, van Nimwegen 2008 and Sevy 2011 ethnicity was not stated. Seven of the studies included adults over the age of 18 years. In the study by Noordsy 2010 four participants (28% of the total sample) were reported to be 17 years; the age range for this study was 17 to 45 years. Both males and females were included in studies but male participants predominated and all participants were male in the Machielsen 2014 study.
3. Settings
Studies were conducted either in the USA or the Netherlands. Most studies included outpatients from single or a small number of sites (1 to 4), and a few recruited from inpatient sites. One study was a large, multi‐centre trial conducted over 57 sites (Swartz 2008).
4. Study size
A total of 2466 people were randomised after giving informed consent to participate in the trials. Of these 1073 had a dual diagnosis. The number of participants with SMI and co‐occurring substance misuse varied from study to study: six studies randomised fewer than 50 dual diagnosis participants (Akerele 2007; Brunette 2011; Machielsen 2014; Noordsy 2010; Sevy 2011; van Nimwegen 2008); one study randomised between 100 and 300 (Smelson 2006); and Swartz 2008 randomised 643 dual diagnosis participants.
5. Interventions
Risperidone (dose range: 1 mg to 9 mg) was compared to olanzapine (dose range: 2.5 mg to 30 mg) in five different studies (Akerele 2007; Sevy 2011; Smelson 2006; Swartz 2008; van Nimwegen 2008). Three studies compared risperidone (dose range: 3.5 mg to 5 mg daily) to clozapine (dose range: 12.5 mg to 400 mg daily) (Brunette 2011; Machielsen 2014; Noordsy 2010). One study compared risperidone (dose range: 1.5 mg to 6 mg) to quetiapine (dose range: 200 mg to 800 mg) (Swartz 2008). Two studies compared risperidone (dose range: 1 mg to 6 mg daily) with first‐generation antipsychotics (perphenazine, loxapine, haloperidol, fluphenazine, thiothixene, various dosages) (Smelson 2006; Swartz 2008). One study compared risperidone (dose range: 1.5 mg to 6 mg) to ziprasidone (dose range: 40 mg to 160 mg daily) (Swartz 2008).
Concomitant psychosocial interventions were delivered in a number of studies. In the trial by Akerele 2007 all participants received weekly psychotherapy over the study period and were asked to nominate a "significant other" to assist with attendance and follow‐up. In Brunette 2011 all participants received weekly individual substance abuse and mental health counselling and attended weekly Alcoholics Anonymous meetings. In Machielsen 2014 it is reported that participants had "supportive treatment as usual". In the trial by Noordsy 2010 participants also received a "Lifestyle Intervention" to help prevent metabolic side‐effects and assist with recovery. In the trial by Sevy 2011 all participants received psychoeducation about schizophrenia, were seen on a regular basis by allocated social workers and also had access to the ancillary treatment service available from two large departments of psychiatry. In Smelson 2006 it is unclear what psychosocial interventions participants received. In Swartz 2008 the investigators did not account for substance abuse treatments received, but they noted that very few were actively engaged in such treatments.
6. Sources of funding
The study by Akerele 2007 received funding from the National Institute on Drug Abuse (NIDA), the National Alliance for Research on Schizophrenia and Depression (NARSAD) and Eli Lilly, the pharmaceutical company. The study by Brunette 2011 was sponsored by Novartis and Janssen; and Machielsen 2014 by the Dutch health research council. Sponsors and collaborators for the study by Noordsy 2010 included the National Institute of Mental Health and the Dartmouth‐Hitchcock Medical Centre; no funding was received from the pharmaceutical industry and the investigators were not employed by the sponsors. In Sevy 2011 several authors declared ties with the pharmaceutical industry but it is stated that the study was sponsored by the NIMH. Funding for Smelson 2006 and van Nimwegen 2008 was received from Eli Lilly; and in Swartz 2008, study medications were provided by several pharmaceutical companies.
7. Outcomes
Scales reported to have been used in the included studies are summarised in Table 2. A description of the scales for which results have been reported is included below.
| Diagnostic tools | Abbreviation | Source of scale/ instrument | Study using instrument | Results reported or usable data for re‐analysis/ quantitative synthesis or qualitative results/data only |
| Structured Clinical Interview for DSM Disorders | SCID‐I | Not an outcome measure | ||
| Mental state scales | ||||
| Brief Psychiatric Rating Scale | BPRS | No results or usable data reported or obtained | ||
| Clinical Global Impression scale | CGI | No results or usable data reported or obtained | ||
| Hamilton Depression Rating Scale | HAM‐D | Results reported; usable data for quantitative synthesis | ||
| Positive and Negative Syndrome Scale | PANSS | Results reported; usable data for quantitative synthesis | ||
| Schedule for Affective Disorders and Schizophrenia ‒ Change Version with Psychosis and Disorganization items | SADS‐C‐PD | Results reported; usable data for quantitative synthesis | ||
| Schedule for the Assessment of Negative Symptoms | SANS | No results or usable data reported or obtained | ||
| Substance use scales | ||||
| Addiction Severity Index | ASI | No results or usable data reported or obtained | ||
| Composite International Diagnostic Interview | CIDI | Not an outcome measure | ||
| Substance Use Questionnaire | SUQ | Sevy 2011, Locally derived instrument/ non‐validated | Results reported, used together with urine testing; usable data for quantitative synthesis | |
| Time‐Line Follow‐Back | TLFB | Results reported in dichotomised format for quantitative synthesis | ||
| Quantitative Substance Use Inventory | Locally derived instrument/ non‐validated | Non‐validated scale | ||
| Subjective‐Wellbeing Scales | ||||
| Subjective Well‐being Under Neuroleptics Scale | SWN | Results reported; usable data for quantitative synthesis | ||
| Craving for substances measures | ||||
| Cocaine Craving Report | No usable data for quantitative synthesis | |||
| Desires for Drug Questionnaire | DDQ | Results reported; usable data for quantitative synthesis | ||
| Marijuana Craving Report | No usable data for quantitative synthesis | |||
| Marijuana Craving Questionnaire | MCQ | Results reported; usable data for quantitative synthesis | ||
| Obsessive Compulsive Drug Use Scale | OCDUS | Results reported; usable data for quantitative synthesis | ||
| Adverse effect scales | ||||
| Abnormal Involuntary Movement Scale | AIMS | No results or usable data reported or obtained | ||
| Barnes Akathisia Rating Scale | BARS | No results or usable data reported or obtained | ||
| Simpson Angus Scale | SAS | Results reported; usable data for quantitative synthesis | ||
| Other measures (categorical or time to event) | ||||
| Urine assay for cannabis and cocaine use (proportion of treatment group positive per week) | No usable data for quantitative synthesis | |||
| Number of participants with improvement in substance use (categorised as improved or not‐improved versus unchanged) | Results reported; usable data for quantitative synthesis (dichotomised) | |||
| Days of self‐reported drug use in past week | No usable data for quantitative synthesis | |||
| Weeks in treatment | Results reported; usable data for quantitative synthesis | |||
| Number of participants not completing the study | Akerele 2007; Machielsen 2014; Noordsy 2010; Sevy 2011; Swartz 2008 | Results reported, usable data for quantitative synthesis | ||
| Compliance with medication (missed doses) | No usable data for quantitative synthesis | |||
7.1 Mental state scales
a. Hamilton Depression Rating Scale (HAM‐D)
This scale has a 17‐item and 21‐item version. The severity of depression is rated for the past week on separate items with 2‐ to 5‐point severity scales. A total of eight items are scored on a 5‐point scale ranging from 0 "absent" to 4 "severe" and 9‐items on a 2‐point scale. Scoring ranges from 0 to 50, with scores on the first 17 items contributing to the final score. A further four items (items 18 to 21) provide additional information on the characteristics of the depressive symptoms (Hamilton 1960). This scale was used by Akerele 2007.
b. Positive and Negative Syndrome Scale (PANSS)
This scale was used in Akerele 2007, Machielsen 2014 and Swartz 2008 and measures positive psychotic symptoms (delusions, thought disorganization, hallucinations, excitement, grandiosity, hostility, persecutory ideation) and negative symptoms (affective blunting, poor rapport, social and emotional withdrawal, stereotypical thinking, poverty of speech, difficulty in abstract thinking) and a range of general psychopathology symptoms. The scale includes seven items measuring positive psychotic symptoms, seven items measuring negative symptoms and 14 items measuring general psychopathology on a 1 to 7 point scale ranging from "absent" to "extreme". It has a range of 30 to 210 (Kay 1986; Kay 1987).
c. Schedule for Affective Disorders and Schizophrenia—Change Version with psychosis and disorganization items (SADS‐C+PD) (Endicott 1978)
This instrument was used to measure positive psychotic symptoms (delusions, hallucinations, thought disorder, and bizarre behaviour) and negative symptoms (affective flattening/blunting, alogia, avolition‐apathy, asociality‐anhedonia) in the trial by Sevy 2011.
7.2 Substance use scales
a. Timeline Follow‐back (TLFB)
This scale is a calendar‐based method that assesses the frequency of drug use over a period of time (usually the past week) (Sobell 1992). This scale can either be self‐administered by participants or clinician‐completed. In the Noordsy 2010 study this scale was used together with other sources of information such as urine tests and reports from collateral sources of information to determine cannabis use. At the end of the study graphs were plotted showing days of cannabis use per week as rated by the TLFB method and were then rated as "Improved", "Unchanged", or "Worse" by a pair of expert judges (Noordsy 2010). Raters were instructed to rate the graph "Improved" or "Worsened" if it appeared to be more than 20% better or worse and to rate it "Unchanged" if there was little or no change (less than ˜20%) (See under "Other measures: categorical and time to event data").
b. Substance Use Questionnaire:
This instrument was used in conjunction with urine testing to derive best estimate of substance discontinuation in Sevy 2011.
7.3 Subjective well‐being scales
a. Subjective Well‐being Under Neuroleptics Scale (SWN): This self‐rated scale (used in the study by Machielsen 2014) measures various aspects of self‐perceived symptoms, treatment experience and quality of life and functioning over the past 7 days. The SWN (short‐version) consists of 20 statements (10 positive and 10 negative) assessing five domains (mental functioning, self‐control, emotional regulation, physical functioning, and social integration), with each domain containing four questions. Rating is on a 6‐point Likert‐type scale yielding total scores varying from 20 to 120 (de Haan 2002; Naber 1995).
7.4 Craving for substances scales
a. Cocaine and Marijuana Craving Report (Weddington 1990)
This scale measures craving of cocaine or cannabis on a 100‐point, 10 cm visual analogue scale with instructions to participants to rate their desire for cocaine or cannabis in the past 24 hours from "not at all" to "more than ever". This scale was used in Akerele 2007.
b. Desires for Drug Questionnaire (DDQ) (Franken 2002)
This instrument measures instantaneous, immediate craving on 14 different items phrased in a positive manner and assessing desires to use, control over using and use to ameliorate negative emotions (negative reinforcement).
c. Marijuana Craving Questionnaire (MCQ) (Heishman 2009)
This instrument measures current cannabis craving on four domains (compulsivity, emotionality, expectancy and purposefulness) containing three questions each using a 7‐point Likert type scale.
d. Obsessive‐Compulsive Drug Use Scale (OCDUS) (Dekker 2012; Franken 2002)
This instrument measures drug craving over the past 7 days on 12 items containing a 5‐point Likert type rating.
7.5 Adverse effects scales
a. Simpson‐Angus Scale (SAS)
In one study — Akerele 2007 — the Simpson‐Angus Scale was used to measure the presence of parkinsonism (Simpson 1970).This scale has 10 items and measures bradykinesia, rigidity and tremor in different body regions on a scale of 0 to 4. It has a range of 0 to 28.
7.6 Leaving the study early: Time to leaving
In the study by Akerele 2007 the time to study attrition was measured in weeks.
7.7 Categorical outcomes and time‐to‐event data
a. Substance use: urine assay for cannabis and cocaine use
In the study by Akerele 2007 urine samples were collected at each of the three meetings per week. Each week over the 10 weeks of follow‐up was classified as either positive or negative for drug use for each participant if any one of the three samples tested positive. Urine tests for cannabis were classified as positive if the cannabis concentration was above a cut‐off of 100 ng/ml; and for cocaine, if the sample concentration was above the cut‐off of 300 ng/ml.
b. Substance use: number of participants with improvement in substance use
In the study by Noordsy 2010 days of cannabis use per week as gathered by the Time‐Line Follow‐Back method were plotted on graphs at the end of the study. A pair of expert judges were then instructed to rate cannabis use as "improved" or "worsened" if it appeared to be more than 20% better or worse and to rate it as "unchanged" if there was little or no change (less than 20% change). Results were reported as the number of participants per intervention group that had "improved" cannabis use versus "unchanged" or "worsened". Machielsen 2014 reports on the number of participants who continued or stopped using cannabis during the study.
c. Adherence with medication
In the study by Akerele 2007 the proportion of missed doses of all administered doses was reported.
d. Leaving the study early
For six studies the number of participants who did not complete the study was reported (Akerele 2007; Machielsen 2014; Noordsy 2010; Sevy 2011; Swartz 2008; van Nimwegen 2008).
Excluded studies
We excluded 23 studies from the 37 studies examined for inclusion. Table 3 contains a summary of the various excluded studies. A total of 12 studies were excluded as there was no measure or reporting of substance use comorbidity (Blin 1996; Gaebel 2010; Harvey 2007; Ikuta 2014; Liemburg 2011; Perlis 2006; Rezayat 2014; Sachs 2002; Sajatovic 2002; Smulevich 2005; van Nimwegen 2008a; Yatham 2007). Three studies were excluded as they were records of study protocols for which no unpublished data were provided after authors were contacted (Green 2001; NCT00169026; NCT00498550). Three studies were excluded due to the use of quasi‐randomisation (Rubio 2006a; Rubio 2006b; Zhangyue 2005). One study was excluded as only 8.3% of participants had a severe mental illness (Nejtek 2008). One study was excluded as it compared risperidone in oral versus depot formulation (NCT00130923). One study was excluded as it included only mental disorders which were due to alcohol use (Liu 2008). One study was excluded as it did not contain any data comparing risperidone with other medications but examined the impact of substance use on prognosis (Kerfoot 2011). Another study was excluded as it was an observational study (NCT00063349).
| Study tag | Participants | Comparison | Relevant review | ||
| Primary problem | Co‐morbidity | Note (reasons for exclusion) | |||
| schizophrenia | Excludes participants with alcohol or drug abuse in past year | Excludes comorbidity | risperidone, haloperidol, methotrimeprazine | ‐ | |
| schizophrenia, schizoaffective disorder | Excludes participants with alcohol or drug abuse in past year | Excludes comorbidity | risperidone, olanzapine, conventional antipsychotics vs. risperidone long‐acting injectable (RLAI),or quetiapine | ‐ | |
| schizophrenia | cannabis use disorder | Excluded as this was a study protocol, authors contacted for unpublished data, no response | risperidone vs. clozapine | ‐ | |
| bipolar type I disorder | No measure of substance use | Excludes comorbidity | risperidone, quetiapine | ‐ | |
| schizophrenia, schizophreniform, psychosis not otherwise specified | No measure of substance use | Excludes comorbidity | risperidone, aripirazole | ‐ | |
| schizophrenia | Co‐occurring substance use in subgroup of 44.9% | No comparison of study medication, mainly a prognostic study of the impact of substance use | risperidone, quetiapine, perphenazine, olanzapine, ziprasidone | ‐ | |
| schizophrenia | No measure of substance use | Excludes comorbidity | risperidone, aripiprazole | ‐ | |
| mental disorders due to alcohol use | substance induced mental disorders | Exclusion criterion | risperidone, olanzapine | Suggested review: risperidone versus other antipsychotics for substance induced psychosis | |
| schizophrenia | Co‐occurring cannabis use disorder | Study protocol, authors contacted for unpublished data no response | risperidone, clozapine | ||
| schizophrenia, schizoaffective disorder | Co‐occurring alcohol use disorder (abuse or dependence) | Comparison of same medication (risperidone) in different preparations (oral vs depot) | oral risperidone, depot risperidone | ‐ | |
| schizophrenia, schizoaffective disorder | Co‐occurring alcohol and substance use disorder | Study protocol, study terminated, authors contacted for unpublished data no response | clozapine, conventional antipsychotics, atypical antipsychotics | ‐ | |
| schizophrenia, schizoaffective disorder | Co‐occurring cannabis use disorder | Study protocol, authors contacted no data provided | clozapine, conventional antipsychotics, atypical antipsychotics | ‐ | |
| bipolar I and II disorder, recent manic or mixed episode with or without psychosis | co‐occurring cocaine or methamphetamine use disorder | Only 8.3% of total sample had psychotic features and 15.9% had bipolar type II disorder. | risperidone, quetiapine | Suggested review: Risperidone versus other antipsychotics in bipolar disorder with co‐occurring substance use disorders | |
| bipolar I disorder with mania or mixed states | Excludes participants with recent substance use | Excludes comorbidity. Excludes bipolar disorder with psychotic features. | risperidone or olanzapine | ‐ | |
| bipolar disorder, acute mania | Excludes participants with drug or alcohol use in past 3 months | Excludes comorbidity | aripiprazole, risperidone | ‐ | |
| schizophrenia | Co‐occurring substance use disorders | Quasi‐randomisation | risperidone long‐acting depot, zuclopenthixol long‐acting depot | ‐ | |
| schizophrenia | Co‐occurring substance use disorders | Quasi‐randomisation | risperidone (oral), zuclopenthixol (oral), zuclopenthixol long acting depot | ‐ | |
| bipolar with current manic or mixed episode | Study excludes participants with drug or alcohol in past 1 months | Excludes comorbidity | mood stabiliser augmentation with risperidone, haloperidol or placebo | ‐ | |
| psychotic disorders: schizoaffective disorder, bipolar I disorder, major depressive disorder, delusional disorder, Alzheimer's dementia, schizophreniform disorder, vascular dementia, and substance abuse dementia | No subgroups with substance use reported | Excludes comorbidity. Authors contacted for unpublished data, no response. | risperidone, quetiapine | ‐ | |
| bipolar I disorder | Excludes participants with recent substance use | Excludes comorbidity | haloperidol, risperidone, placebo | ‐ | |
| schizophrenia, schizophreniform, schizoaffective disorder | No measure of substance use | Excludes comorbidity | haloperidol, risperidone, placebo | ‐ | |
| bipolar I and II | Excludes participants with drug or alcohol use in past 3 months. | Excludes comorbidity | continuation of oral risperidone, olanzapine, quetiapine or switch to long‐acting injectable LAI risperidone | ‐ | |
| schizophrenia or schizophrenia‐like illnesses | Excludes participants with alcohol or substance dependence | Quasi‐randomisation.Excludes comorbidity. | aripiprazole versus risperidone | ‐ | |
Studies awaiting classification
One study appeared to meet inclusion criteria but no results were reported hence it could not be assessed (NCT00208143). This study is described as an open (no masking), randomised, parallel, superiority trial, comparing the efficacy of quetiapine with risperidone in adults with a diagnosis of schizophrenia or schizoaffective disorder and co‐occurring cocaine or methamphetamine abuse or dependence. We contacted the investigators for information and results but received no response. Another study appeared to meet eligibility criteria, but was described only as "double blind" (Greenspan 2005). Authors were contacted to confirm randomisation but did not respond. A third study appeared to meet eligibility criteria but the authors responded after being contacted that no data for the subgroup with a dual diagnosis are available (Yatham 2003). It was also not clear how many participants in this study had psychotic bipolar disorders. Two studies contained subgroups of participants with substance use disorders, but no data were provided for the subgroup after authors were contacted (Johnsen 2010; San 2012).
Ongoing studies
We found one study currently recruiting participants (NCT01639872). This study is described as a double‐blind, randomised, parallel assignment, superiority trial, comparing the efficacy of clozapine with risperidone in people with schizophrenia and a cannabis use disorder. The estimated sample size is 132.
Risk of bias in included studies
We assessed risk of bias for the eight included studies. Overall the risk of bias was unclear across most domains assessed, as reporting of study results lacked particulars of how potential bias was handled and how potential bias could have influenced results. Figure 4 and Figure 5 contains a graphical overview of the risk of bias. The details of the studies are included under Characteristics of included studies.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
1. Random generation
The method of random sequence generation was unclear in all but two studies for which a description of random sequence generation was provided and therefore assessed as low risk of bias (Machielsen 2014; Sevy 2011).
2. Allocation concealment
The method of allocation concealment was unclear in all the included studies, except for one study for which allocation concealment was adequately described, with low risk of bias (Machielsen 2014).
Blinding
1. Performance bias
There was high risk of performance bias in five of the eight included studies, as many studies were described as open label and both participants and personnel were aware of the medications they received (Brunette 2011; Noordsy 2010; Machielsen 2014; Sevy 2011; Smelson 2006). For Noordsy 2010, Brunette 2011 and Machielsen 2014 we assessed risk of unmasking of participants and personnel as high, as the monitoring requirements for clozapine (weekly blood tests) differs from that of risperidone and no description was given how blinding was maintained. No description of blinding of personnel or participants was provided for the remainder of the included studies and risk of bias was assessed as unclear (Akerele 2007; Swartz 2008; van Nimwegen 2008).
2. Detection bias
We judged detection bias to be low for two studies where outcome assessors were reported to be blinded and independent of study physicians (Brunette 2011; Sevy 2011). One study was judged as high risk of bias as outcome assessors were not blinded and were aware of allocation (Machielsen 2014). We judged the risk of detection bias as unclear for five of the included studies as no description or an incomplete description of blinding was reported (Akerele 2007; Noordsy 2010; Smelson 2006; Swartz 2008; van Nimwegen 2008).
Incomplete outcome data
For four studies we judged the risk of attrition bias as being high, due to high levels of attrition (Swartz 2008), unbalanced numbers in leaving between treatments (Akerele 2007), differing reasons for attrition between medications (Noordsy 2010), the use of only single imputation methods (Swartz 2008; van Nimwegen 2008), and the absence of data on baseline differences in groups that dropped out (van Nimwegen 2008). For one study (Smelson 2006), for which the primary — and only outcome — was attrition, we judged the risk to be low. For three studies (Brunette 2011; Sevy 2011; Machielsen 2014), risk of attrition bias was judged as unclear as the number of participants that failed to complete the study was either not reported across the randomised groups, or only simple imputation methods were used to account for missing data in analyses, or the impact of attrition on the study specific outcomes was not clear.
Selective reporting
Risk of selective reporting was judged to be low for two studies for which a study protocol was available and where there were no changes between the protocol‐defined outcomes and outcomes in the study report (Brunette 2011; Machielsen 2014). For five studies risk of selective reporting was judged as unclear as there was either no study protocol available (Akerele 2007), or outcomes stated in the protocol were not mentioned in the study report (van Nimwegen 2008), or the study was a secondary analysis of an existing parent randomised trial, making it unclear whether the outcome selection could have been influenced by preliminary results and analysis of the primary study (Sevy 2011; Smelson 2006; Swartz 2008). In the original protocol for the study by Noordsy 2010, the primary outcome was specified as days of substance use as measured by the Time Line Follow Back method. However, changes were made to the protocol prior to the end of the study and substance use outcomes were judged by experts based on graphs derived from data collected by TLFB method and dichotomised into the proportion of participants with an improvement (20% or more reduction in substance use) versus no improvement or unchanged and worsened groups. This process appeared to have occurred following the collection of data and a change to the protocol was added. Risk of selective reporting was judged to be high for Noordsy 2010.
Other potential sources of bias
Four studies were judged as having high risk of other bias due to sponsorship of the study by the pharmaceutical industry (Akerele 2007; Brunette 2011; Smelson 2006; van Nimwegen 2008). In three studies the risk of other bias was judged as unclear as sponsorship was by academic centres and national funding agencies such as the NIMH but other potential conflicts of interests are not reported and therefore remain unclear, or many authors were supported by the pharmaceutical industry even though the study sponsorship was stated as being independent of industry (Noordsy 2010; Sevy 2011; Swartz 2008). One study was judged to be at low risk of bias as sponsorship was through national funding bodies and university departments with no other potential conflicts of interest declared (Machielsen 2014).
Effects of interventions
See: Summary of findings for the main comparison RISPERIDONE versus CLOZAPINE ‐ all data short term for people with severe mental illness and co‐occurring substance misuse; Summary of findings 2 RISPERIDONE versus OLANZAPINE ‒ short‐ and long‐term data for people with severe mental illness and co‐occurring substance misuse; Summary of findings 3 RISPERIDONE versus PERHENAZINE ‒ long‐term data for people with severe mental illness and co‐occurring substance misuse; Summary of findings 4 RISPERIDONE versus QUETIAPINE ‒ short‐ and long‐term data for people with severe mental illness and co‐occurring substance misuse; Summary of findings 5 RISPERIDONE versus ZIPRASIDONE ‒ long‐term data (> 12 months) for people with severe mental illness and co‐occurring substance misuse
See: summary of findings Table for the main comparison; summary of findings Table 2; summary of findings Table 3; summary of findings Table 4; summary of findings Table 5.
Comparison 1: risperidone versus clozapine
Only two studies containing data on 50 participants contributed data to this comparison (Machielsen 2014; Noordsy 2010). See summary of findings Table for the main comparison. All data are short term ‒ up to 6 months.
1.1 Mental state: 1. General: average endpoint scores (PANSS subscale, lower = better)
One trial reported data on these outcomes (Machielsen 2014). There were no clear differences between risperidone and clozapine at study endpoint in general psychopathology symptoms (1 RCT, n = 36, MD 2.70, 95% CI −2.14 to 7.54), Analysis 1.1.
1.2 Mental state: 2. General: any change in general symptoms
Only one study reported results for 'worsening of psychotic symptoms' (non‐systematic assessment) as an adverse effect (Noordsy 2010). There were no clear differences between the risperidone and clozapine groups for this outcome (1 RCT, n = 14, RR 0.14, 95% CI 0.01 to 2.34), Analysis 1.2.
1.3 Mental state: 3. Specific: positive, negative symptoms ‒ average endpoint scores (PANSS subscales, lower = better)
One trial reported data on these outcomes (Machielsen 2014). There were no differences between risperidone and clozapine at study endpoint in positive symptoms (1 RCT, n = 36, MD 0.90, 95% CI −2.21 to 4.01). For negative symptoms participants with clozapine had significantly lower scores (1 RCT, n = 36, MD 4.00, 95% CI 0.79 to 7.21), Analysis 1.3.
1.4 Mental state: 4. Specific: anxiety symptoms
The Noordsy 2010 study reported emergence of anxiety symptoms (non‐systematic assessment). No clear differences were found between the risperidone and clozapine groups (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15), Analysis 1.4.
1.5.1 Substance use: 1. Improvement (at least 20% reduction in use, TLFB scale)
One study provided data with this outcome (Noordsy 2010). No significant differences in this measure were demonstrated between the risperidone and clozapine groups (1 RCT, n = 14, RR 1.00, 95% CI 0.30 to 3.35), Analysis 1.5.
1.5.2 Substance use: 2. Discontinued substance use
One study provided data for this outcome (Machielsen 2014). There were no significant differences between risperidone and clozapine in the number of participants who stopped using cannabis (1 RCT, n = 28, RR 1.13, 95% CI 0.41 to 3.12), Analysis 1.5.
1.6 Subjective well‐being: average endpoint scores (Subjective Well‐being under Neuroleptics scale, SWN scale, higher = better)
One study provided data for this outcome (Machielsen 2014). There were no significant differences in the endpoint scores on the Subjective Well‐being under Neuroleptics scale for risperidone compared to clozapine (1 RCT, n = 36, MD −6.00, 95% CI −14.82 to 2.82), Analysis 1.6.
1.7 Craving for substances
1.7.1 Specific: current craving ‒ average endpoint scores (Marijuana Craving Questionnaire, MCQ, lower = better)
One study included data on this outcome (Machielsen 2014). Participants treated with clozapine had significantly lower scores on the Marijuana Craving Questionnaire (MCQ) compared to participants treated with risperidone (1 RCT, n = 28, MD 7.00, 95% CI 2.37 to 11.63, P = 0.003), Analysis 1.7.
1.7.2 Specific: past week craving ‒ average endpoint scores (Obsessive Compulsive Drug Use Scale, OCDUS, lower = better)
One study included data on this outcome (Machielsen 2014). Participants treated with clozapine had significantly lower craving scores on the Obsessive Compulsive Craving Scale (OCDUS), compared to participants treated with risperidone (1 RCT, n = 28, MD 14.20, 95% CI 4.45 to 23.95, P = 0.004), Analysis 1.7.
1.8 Adherence to antipsychotic medication: discontinued medication
One study included data on this outcome (Machielsen 2014). There were no significant differences between risperidone‐ and clozapine‐treated participants in the number of participants who discontinued antipsychotic treatment (1 RCT, n = 36, RR 4.05, 95% CI 0.21 to 78.76), Analysis 1.8.
1.9 Adverse effects: 1. Movement disorders
1.9.1 Any extrapyramidal side‐effects
Two studies reported data on extrapyramidal side‐effects (Machielsen 2014; Noordsy 2010). There were no significant differences between risperidone‐ and clozapine‐treated participants in terms of the number of participants who experienced any extrapyramidal side‐effects (2 RCTs, n = 50, RR 2.71, 95% CI 0.30 to 24.08; I² = 0%), Analysis 1.9.
1.9.2 Akathisia
One study provided data for this outcome (Noordsy 2010). There were no statistically significant differences in the risperidone compared to the clozapine groups for akathisia (1 RCT, n = 14, RR 2.00, 95% CI 0.23 to 17.34), Analysis 1.9.
1.10 Adverse effects: 2. Non‐movement disorder related side‐effects
One study — (Noordsy 2010) — provided data for non‐movement disorder‐related adverse effects. In all cases no statistically significant differences were found in the proportion of participants with side‐effects in the risperidone compared to the clozapine groups. Cardiovascular side‐effects: palpitations (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15); hypotension (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02). Central nervous system side‐effects: headache (1 RCT, n = 14, RR 0.20, 95% CI 0.01 to 3.54); somnolence (1 RCT, n = 14, RR 0.20, 95% CI 0.03 to 1.30). Dermatological side‐effects: acne (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15). Endocrinological side‐effects: decreased libido (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02). Ear and labyrinthine: ear canal blockage (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15). Gastrointestinal: abdominal pain (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15); elevated liver function tests (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15); hypersalivation (1 RCT, n = 14, RR 0.11, 95% CI 0.01 to 1.74). General adverse effects: fatigue (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02). Injuries: sprain (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02). Metabolic side‐effects: increased appetite (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02); weight gain (1 RCT, n = 14, RR 1.00, 95% CI 0.19 to 5.24). Musculoskeletal: ankle pain (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02); knee and foot pain (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02); muscle twitch (1 RCT, n = 14, RR 3.00, 95% CI 0.14 to 63.15). Renal side‐effects: retention (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02); urgency (1 RCT, n = 14, RR 0.33, 95% CI 0.02 to 7.02) (Analysis 1.10).
1.11 Leaving the study early
1.11.1 Any reason
Two studies reported data on this outcome (Machielsen 2014; Noordsy 2010). There were no statistically significant differences between risperidone and clozapine groups (2 RCT, n = 45, RR 0.49, 95% CI 0.10 to 2.51; I² = 34%), Analysis 1.11.
1.11.2 Due to inefficacy
One study provided data for this outcome (Noordsy 2010). There were no participants who left the study due to inefficacy.
Missing outcomes (risperidone versus clozapine)
There were no studies reporting data on mortality or quality of life.
Comparison 2: risperidone versus olanzapine
A total of five studies contributed data to this comparison (Akerele 2007; Sevy 2011; Smelson 2006; Swartz 2008; van Nimwegen 2008). See summary of findings Table 2.
2.1 Mental state: 1. Specific: Depression ‒ change scores (HAM‐D, higher = better), short term (up to 6 months)
Only Akerele 2007 included data on this outcome. There were no significant differences between the risperidone and olanzapine groups in the reduction of depressive symptoms from baseline to the end of the study (1 RCT, n = 22, MD −0.11, 95% CI −0.78 to 0.56), Analysis 2.1.
2.2 Mental state: 2. Specific: Positive symptoms, total score ‒ average endpoint scores (SADS‐C‐PD scale, lower = better), short term (up to 6 months)
Only one study provided analysable data for this outcome (Sevy 2011). There were no significant differences between the risperidone and olanzapine groups in positive symptoms scores (1 RCT, n = 37, MD −1.50, 95% CI −3.82 to 0.82), Analysis 2.2.
2.3 Mental state: 3. Specific: Positive symptom subscales ‒ average endpoint scores (SADS‐C‐PD scale, lower = better), short term (up to 6 months) ‒ skewed data
In addition to total scores for positive symptoms, one study reported on endpoint data for specific types of positive symptoms, namely delusions, hallucinations and thought disorder (Sevy 2011). Data were skewed and best viewed in an additional table (Analysis 2.3). No significant differences between risperidone and olanzapine were reported.
2.4 Mental state: 4. Specific: Negative symptoms, subscales ‒ average endpoint scores (SANS subscales, lower = better), short term (up to 6 months)
Only one study provided analysable data (Sevy 2011). There were no significant differences between the risperidone and olanzapine groups in terms of different subscales for negative symptoms, i.e. affective flattening (1 RCT, n = 39, MD 0.50, 95% CI −0.17 to 1.17); alogia (1 RCT, n = 39, MD 0.40, 95% CI −0.22 to 1.02); avolition apathy (1 RCT, n = 39, MD −0.10, 95% CI −0.73 to 0.53), asociality‐anhedonia (1 RCT, n = 39, MD −0.10, 95% CI −0.80 to 0.60), Analysis 2.4.
2.5. Substance use: 1. Substance use: Reduction of cannabis use‐ change data (number of joints smoked/week, LOCF data, higher = better), short‐term data (up to 6 months)
One study provided data (van Nimwegen 2008). There were no significant differences between risperidone and olanzapine in the number of joints smoked (1 RCT, n = 41, MD 0.40, 95% CI −4.72 to 5.52), Analysis 2.5.
2.6 Substance use: 2. Discontinued substance use, short term (up to 6 months)
2.6.1 Substance use: stopped using cannabis (Urine testing and Substance Use Questionnaire)
Only one study provided analysable data (Sevy 2011). There were no significant differences between risperidone and olanzapine in the number of participants who discontinued cannabis (1 RCT, n = 37, RR 1.19, 95% CI 0.68 to 2.08), Analysis 2.6.
2.6.2 Substance use: stopped using alcohol (Substance Use Questionnaire)
One study provided analysable data (Sevy 2011). There were no significant differences between risperidone and olanzapine in the number of participants who discontinued alcohol (1 RCT, n = 28, RR 1.31, 95% CI 0.73 to 2.36), Analysis 2.6.
2.7 Craving for substances: 1. Obsessive Compulsive Drug Use Scale ‒ average endpoint score (OCDUS, lower = better), short term (up to 6 months)
One study provided data for this outcome (van Nimwegen 2008). There were no significant differences between the risperidone and olanzapine groups in craving as measured by this scale (1 RCT, n = 41, MD 1.30, 95% CI −3.51 to 6.11), Analysis 2.7.
2.8 Craving for substances: 2. Desires for Drug Questionnaire ‒ average endpoint scores (DDQ, LOCF data, lower = better), short term (up to 6 months)
One study provided data for this outcome (van Nimwegen 2008). There were no significant differences between the risperidone and olanzapine groups in craving as measured by this scale (1 RCT, n = 41, MD 5.00, 95% CI −4.86 to 14.86), Analysis 2.8.
2.9 Adverse effects
2.9.1 Movement disorders: Parkinsonism ‒ average endpoint score (SAS, high = worse), short term (up to 6 months)
One study provided data on this outcome (Akerele 2007). There were no significant differences in the scores on this scale between the risperidone and olanzapine groups (1 RCT, n = 16, MD −0.08, 95% CI −1.21 to 1.05), Analysis 2.9.
2.9.2 Non‐movement disorder related side‐effects: weight gain‐ average endpoint score (BMI, lower = better), short term (up to 6 months)
Only one study provided data on this outcome (Sevy 2011). There were no significant differences in the scores on this scale between the risperidone and olanzapine groups (1 RCT, n = 37, MD −1.00, 95% CI −3.99 to 1.99), Analysis 2.9.
2.10 Leaving the study early: 1. Various reasons
2.10.1 Any reason, short term (up to 6 months)
Two studies provided data for this outcome (Akerele 2007; Sevy 2011). There were no significant differences between risperidone and olanzapine in the number of participants leaving the studies early (2 RCT, n = 77, RR 0.68, 95% CI 0.34 to 1.35; I² = 0%), Analysis 2.10.
2.10.2 Any reason, long‐term data ( > 12 months)
One study provided data for this outcome (Swartz 2008). There were no significant differences between risperidone and olanzapine in the number of participants leaving the study early (1 RCT, n = 299, RR 1.07, 95% CI 0.94 to 1.21), Analysis 2.10.
2.10.3 Readmission, short term (up to 6 months)
Only one study provided data on this outcome (Akerele 2007). In Akerele 2007 participants leaving the study early as a result of readmission to an inpatient unit were similar across the risperidone and olanzapine groups, with no significant differences (1 RCT, n = 28, RR 1.00, 95% CI 0.07 to 14.45), Analysis 2.10.
2.10.4 Intolerable adverse effect, short term (up to 6 months)
One study provided data on this outcome (Akerele 2007). In Akerele 2007 no participants left the study early in either treatment group due to adverse medication effects (Analysis 2.10).
2.10.5 Participant loss of interest, short term (up to 6 months)
One study provided data on this outcome (Akerele 2007). In Akerele 2007 there were no significant differences between risperidone and olanzapine in leaving the study early due to lack of interest, although the risperidone group were less likely to drop out due to lack of interest (1 RCT, n = 28, RR 0.43, 95% CI 0.14 to 1.33), Analysis 2.10.
2.11 Leaving the study early: 2. Weeks in the study ‒ average endpoint data (high = good), short term (up to 6 months)
Only one study provided data on this outcome (Akerele 2007). In Akerele 2007 there were no differences in time remaining in the study treatment between the risperidone and olanzapine groups (1 RCT, n = 28, MD 0.00, 95% CI −3.35 to 3.35), Analysis 2.11.
2.12 Leaving the study early: 3. Weeks in study ‒ average endpoint data (high = good), short term (up to 6 months) ‒ skewed data
2.12.1 Weeks remained in study ‒ average endpoint data (high = good), short term (up to 6 months) ‒ skewed data
One study provided data for this outcome (Smelson 2006). Data was skewed so is best inspected in an additional table (Analysis 2.12).
Comparison 3: risperidone versus perphenazine
3.1 Leaving the study early: all‐cause discontinuation, long term (> 12 months)
One study provided data for this outcome (Swartz 2008). There were no significant differences between risperidone and perphenazine for this outcome (1 RCT, n = 281, RR 1.05, 95% CI 0.92 to 1.20), Analysis 3.1.
Comparison 4: risperidone versus quetiapine
4.1 Leaving the study early: all‐cause discontinuation, long term (> 12 months)
One study provided data for this outcome (Swartz 2008). There was no statistically significant difference between risperidone and quetiapine (1 RCT, n = 294, RR 0.96, 95% CI 0.86 to 1.07), Analysis 4.1.
Comparison 5: risperidone versus ziprasidone
5.1 Leaving the study early: all‐cause discontinuation, long term (> 12 months)
One study provided data for this outcome (Swartz 2008). There were no significant differences between risperidone and ziprasidone (1 RCT, n = 240, RR 0.96, 95% CI 0.85 to 1.10), Analysis 5.1.
6. Missing outcomes (risperidone versus olanzapine)
There were no studies reporting data for general mental state (CGI), anxiety symptoms, subjective well‐being, adverse effects such as metabolic syndrome (other than parkinsonism and weight gain), mortality and quality of life.
Discussion
Summary of main results
Comparison 1: risperidone versus clozapine
Only two studies (Noordsy 2010; Machielsen 2014), containing 50 participants, provided usable data for this comparison (summary of findings Table for the main comparison). A third study, Brunette 2011, met criteria for inclusion but we were unable to extract any usable data from this study. Studies with useable data reported no statistically significant differences between risperidone and clozapine in terms of worsening of general psychotic symptoms, general anxiety symptoms, reduction in substance use, subjective well‐being, medication adherence, movement disorder‐related side‐effects, other side‐effects or leaving the study early (Machielsen 2014; Noordsy 2010). However, in the study by Machielsen 2014, lower negative symptoms were found in the clozapine group as compared to the risperidone‐treated group. In addition, lower immediate and 'past 1 week' craving scores were found for participants treated with clozapine as opposed to risperidone (Machielsen 2014). No results were reported on mortality or quality of life, and side‐effects such as weight gain and metabolic syndrome were inconsistently reported. The sample size in this study was very low (N = 36), limiting the conclusions that can be drawn from the analysis of data (Machielsen 2014). Moreover in the study by Noordsy 2010 participants in the clozapine group reported the emergence or worsening of psychotic symptoms and more participants in the risperidone group reported the emergence of anxiety symptoms, although these differences did not reach statistical significance.
Comparison 2: risperidone versus olanzapine
Five studies — Akerele 2007; Sevy 2011; Smelson 2006; Swartz 2008; van Nimwegen 2008 — comprising 997 participants provided data for this comparison (summary of findings Table 2). Overall the quality of evidence was low to very low. Studies differed in terms of design and outcomes measured, precluding pooling of outcome data. In addition sample size was low for most of the individual studies, with Akerele 2007, Sevy 2011 and van Nimwegen 2008 having sample sizes below N = 50, limiting the conclusions that can be drawn from them. There were no statistically significant differences between risperidone and olanzapine for a reduction in any mental symptoms (depression, positive or negative symptoms). In one study there was some evidence of significantly fewer days of any substance use for the olanzapine group compared to the risperidone group, whereas the risperidone group showed significantly greater reductions in craving for cannabis use (Akerele 2007). In turn, olanzapine‐treated participants had a significantly longer time to all‐cause antipsychotic treatment discontinuation in one study (Smelson 2006). However, there were no significant differences between the risperidone and olanzapine groups in the proportion of participants with urine‐positive weeks for the cannabis and cocaine use subgroups, cocaine craving, parkinsonism, weight gain or leaving the study early. Results for many outcomes were incompletely reported or not suitable for re‐analysis and we were only able to report in a qualitative, narrative format. Of note, in many studies authors did not report on any measures relating to metabolic side‐effects such as weight gain, abnormalities in glucose or lipid metabolism. For subjective well‐being, authors reported only total sample findings and did not report on subgroups who used substances. Furthermore, no results were reported on mortality or quality of life.
Comparison 3: risperidone versus perphenazine
One study (Swartz 2008), with data on 281 participants, reported on leaving the study early, with no differences between risperidone‐ or perphenazine‐treated participants.
Comparison 4: risperidone versus quetiapine
One study (Swartz 2008), with data on 294 participants, provided data for this comparison. There was no significant difference between the risperidone and quetiapine groups in leaving the study early.
Comparison 5: risperidone versus ziprasidone
One study (Swartz 2008), with data on 240 participants, reported on leaving the study early, with no differences between risperidone‐ or ziprasidone‐treated participants.
Overall completeness and applicability of evidence
1. Completeness
The reporting of outcome data was incomplete and generally poor. In addition, some trials often failed to report on outcomes that were mentioned to have been measured in the protocol and Methods sections (Akerele 2007; Noordsy 2010). Primary outcomes of improvement in mental state and substance use were poorly reported, with many studies not reporting on changes in mental state or reporting incompletely (Noordsy 2010; Smelson 2006; van Nimwegen 2008). Substance use was also not reported on in Smelson 2006 and Swartz 2008.
Many studies also failed to report on important outcomes such as weight gain, metabolic changes and endocrinological adverse effects. No outcomes on craving were reported in Noordsy 2010, Sevy 2011, Smelson 2006, and Swartz 2008; and adherence was not reported by Noordsy 2010, Sevy 2011, Swartz 2008 and van Nimwegen 2008. Many studies failed to report on subjective well‐being, mortality or quality of life (Akerele 2007, Noordsy 2010; Sevy 2011; Smelson 2006; Swartz 2008).
2. Applicability of evidence
Six of the included trials — Akerele 2007, Brunette 2011, Noordsy 2010, Sevy 2011, Smelson 2006 and Swartz 2008 — were conducted in the USA and two in the Netherlands (Machielsen 2014; van Nimwegen 2008). We found no trials conducted in developing countries. All trials involved comparisons of risperidone with SGAs or clozapine; and only two trials included comparison with FGAs (Smelson 2006; Swartz 2008), only one with useable data (Swartz 2008). Some SGAs may have lower availability due to cost in middle‐ and low‐income countries, potentially leading to lower applicability of findings to these settings. In most studies males and females were eligible for inclusion, but the study by Akerele 2007 included mostly men and only males were included in Machielsen 2014, potentially limiting applicability to female populations. Moreover, mixed ethnic groups were included in most studies. Noordsy 2010 and Brunette 2011 included only Caucasians (understood to be white participants).
The sample size of many included trials was very low and below N = 50 (Akerele 2007; Brunette 2011; Machielsen 2014; Noordsy 2010; Sevy 2011; van Nimwegen 2008). One trial had sample sizes between 100 and 300 (Smelson 2006); and one study had a sample size of more than 400 (Swartz 2008).
Quality of the evidence
1. General
Overall, of the eight studies included in this review, across most studies reporting of results was poor and did not adhere to CONSORT standards (1996). As a result many outcomes yielded unusable data.
2. Specific
From the eight included studies there were five comparisons of risperidone with other antipsychotics. For most studies sample sizes were small. Risk of bias was unclear in most domains across the included studies, however in some instances there was high risk of bias particularly with regards to performance bias, attrition bias, selective outcome reporting and other forms of bias. Moreover, due to heterogeneity in design and poor outcome reporting, pooling of data in meta‐analyses was not possible for most outcomes leading to imprecise effect estimates and precluding any meaningful analysis of results and yielding low to very low quality evidence for the main outcomes.
Potential biases in the review process
We conducted a wide search, including a grey literature search, and searches of clinical trial registers and screening of recent relevant conference abstracts. Study selection and data extraction was done in duplicate to minimise the potential for selection and information bias. Our search was conducted within the Cochrane Schizophrenia Group’s Trials Register and the Cochrane Depression, Anxiety and Neurosis Controlled Trials Register. We did not include searches of the Drugs and Alcohol group register. This may have lead us to miss some studies examining treatments for substance use disorders that contain some participants with serious mental illness, but this is unlikely.
We also contacted authors to clarify reporting of studies and to provide missing data. Following our extensive search we are not aware of any additional studies in this field, but we are open to review the evidence and would call upon authors to contact us should there be any trial that warrant consideration for inclusion.
Agreements and disagreements with other studies or reviews
There have been a number of systematic reviews evaluating the efficacy of antipsychotics in people with a dual diagnosis. In Baker 2010 and Baker 2012 the authors reached similar conclusions regarding the evidence for risperidone versus other antipsychotics. In addition Wobrock 2009 reached conclusions similar to ours, although the methods of grading evidence differed from our review. In Lazary 2012, authors suggest certain agents such as olanzapine and clozapine may be superior to others (including risperidone) but express reservations on the quality of evidence as they based their findings on a mixture of observational and randomised trial evidence. Furthermore, Lazary 2012 used different methods from our review to assess study quality and strength of evidence. In Machielsen 2009 the authors included two studies that we excluded in our review (containing mixed single and dual diagnosis groups and a mixed comparator medication group) and reached a conclusion that clozapine may be superior to risperidone, although they caution against a firm conclusion because of the paucity of well‐designed trials. McLoughlin 2014 compared the use of different antipsychotics (classified as antipsychotic A versus B) in people with schizophrenia and co‐occurring cannabis disorders and reached similar conclusion to our review, despite different methods and inclusion of different studies.


Logic framework model with potential causal pathways: risperidone treatment in persons with dual diagnosis.

PRISMA flow diagram of study selection from 2016 and 2017 searches

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
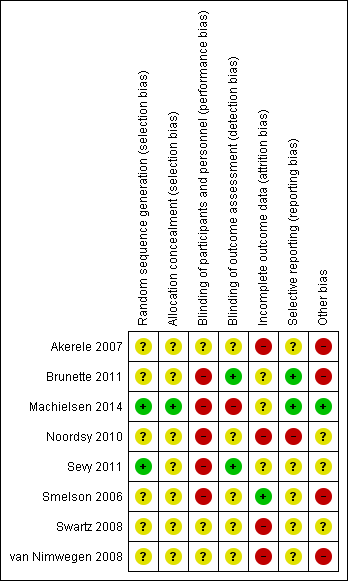
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 1 Mental state: 1. General: average endpoint scores (PANSS subscale, lower=better).

Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 2 Mental state: 2. General: any change in general symptoms:.

Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 3 Mental state: 3. Specific: positive, negative symptoms ‐ average endpoint scores (PANSS subscales, lower = better):.

Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 4 Mental state: 4. Specific: anxiety symptoms.
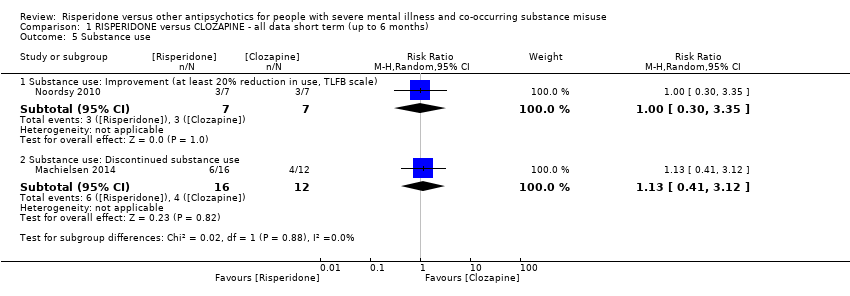
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 5 Substance use.

Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 6 Subjective Well‐being: average endpoint scores (Subjective Well‐being under Neuroleptics scale, SWN scale, higher=better).

Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 7 Craving for substances.
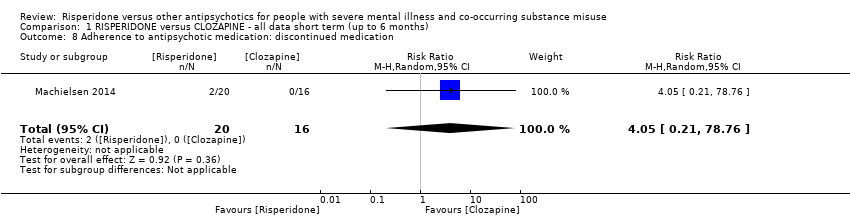
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 8 Adherence to antipsychotic medication: discontinued medication.

Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 9 Adverse effects. 1. Movement disorders.

Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 10 Adverse effects: 2. Non‐movement disorder related side‐effects.
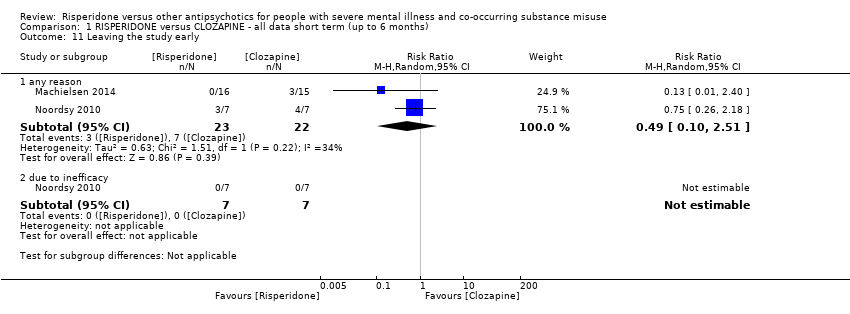
Comparison 1 RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months), Outcome 11 Leaving the study early.
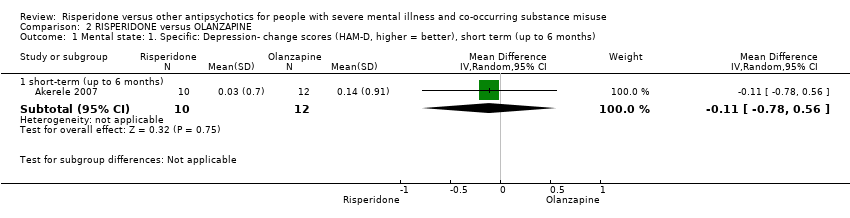
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 1 Mental state: 1. Specific: Depression‐ change scores (HAM‐D, higher = better), short term (up to 6 months).
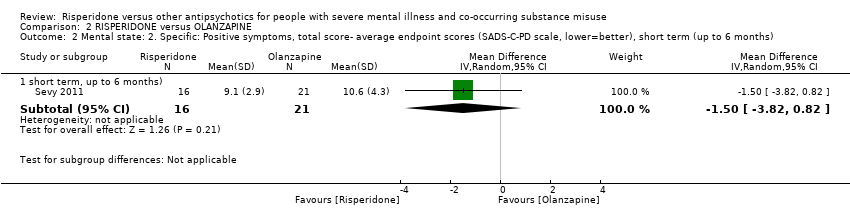
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 2 Mental state: 2. Specific: Positive symptoms, total score‐ average endpoint scores (SADS‐C‐PD scale, lower=better), short term (up to 6 months).
| Study | Intervention | Outcome (symptom subscore) | Mean | SD | N |
| Sevy 2011 | Risperidone | Delusions | 2.6 | 1.7 | 16 |
| Sevy 2011 | Olanzapine | 2.7 | 1.6 | 21 | |
| Sevy 2011 | Risperidone | Hallucinations | 1.8 | 1.2 | 16 |
| Sevy 2011 | Olanzapine | 2 | 1.6 | 21 | |
| Sevy 2011 | Risperidone | Thought disorder | 3.6 | 0.8 | 16 |
| Sevy 2011 | Olanzapine | 4.5 | 2.6 | 21 | |
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 3 Mental state: 3. Specific: Positive symptom subscales‐ average endpoint scores (SADS‐C‐PD subscores, lower=better), short term (up to 6 months)‐ skewed data.

Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 4 Mental state: 4. Specific: Negative symptoms, subscales‐ average endpoint scores (SANS subscales, lower=better), short term (up to 6 months).

Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 5 Substance use: 1. Reduction of cannabis use‐change data (number of joints smoked/week, LOCF data, higher =better)‐ short term data (up to 6 months).
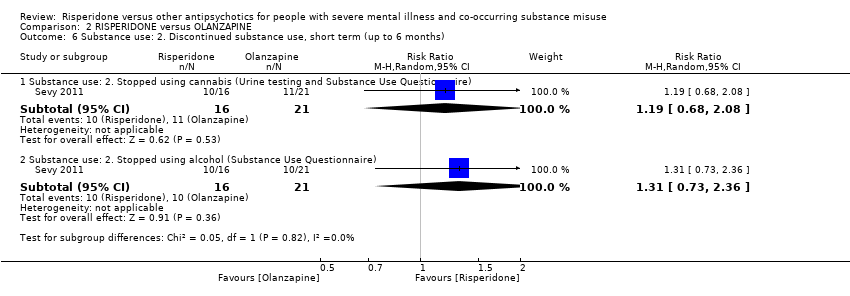
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 6 Substance use: 2. Discontinued substance use, short term (up to 6 months).

Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 7 Craving for substances: 1. Obsessive Compulsive Drug Use Scale‐ average endpoint score (OCDUS, lower=better)‐short term (up to 6 months).

Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 8 Craving for substances: 2. Desires for Drug Questionnaire‐ average endpoint scores (DDQ, LOCF data, lower=better), short term (up to 6 months).

Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 9 Adverse effects.
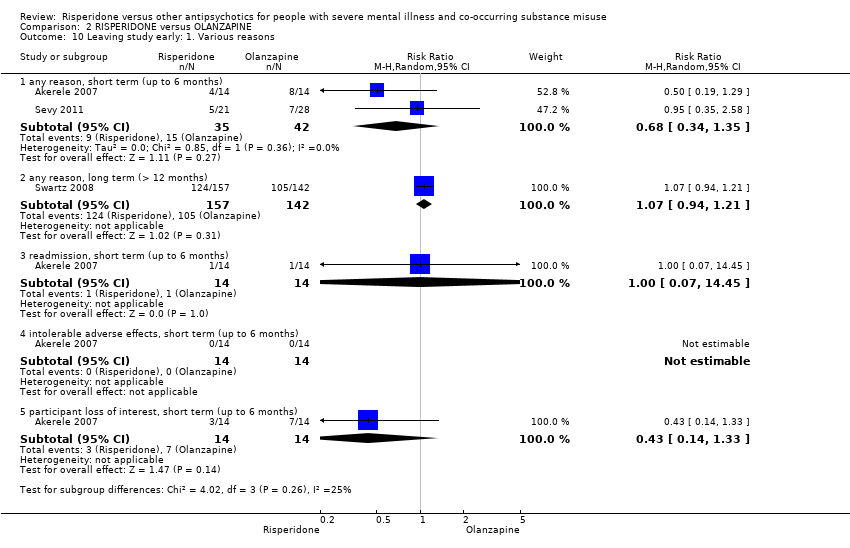
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 10 Leaving study early: 1. Various reasons.

Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 11 Leaving study early: 2. Weeks in the study‐ average endpoint data (high=good), short term (up to 6 months).
| Study | Intervention | Mean (number of weeks) | SD | N |
| Smelson 2006 | Risperidone | 207 | 142.9 | 76 |
| Smelson 2006 | Olanzapine | 267.9 | 127.4 | 85 |
Comparison 2 RISPERIDONE versus OLANZAPINE, Outcome 12 Leaving study early: 3. Weeks in study‐ average endpoint data (high=good), short term (up to 6 months)‐ skewed data.
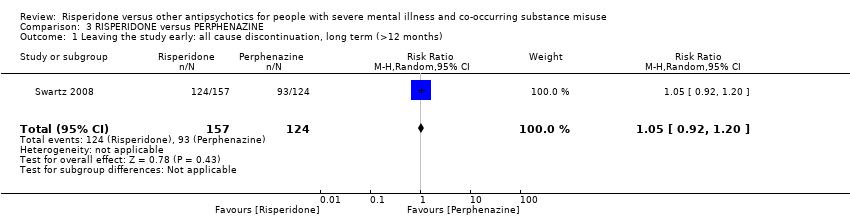
Comparison 3 RISPERIDONE versus PERPHENAZINE, Outcome 1 Leaving the study early: all cause discontinuation, long term (>12 months).

Comparison 4 RISPERIDONE versus QUETIAPINE, Outcome 1 Leaving the study early: all cause discontinuation, long term (>12 months).
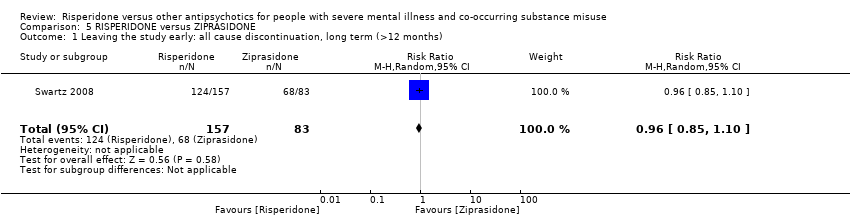
Comparison 5 RISPERIDONE versus ZIPRASIDONE, Outcome 1 Leaving the study early: all cause discontinuation, long term (>12 months).
| Methods | Allocation: centralised sequence generation with table of random numbers or computer‐generated code, stratified by severity of illness, sequence concealed till interventions assigned. |
| Participants | Diagnosis: schizophrenia and co‐occurring ongoing substance misuse (clinical criteria). |
| Interventions | 1. Risperidone: clinically indicated dose. N = 150. 2. Olanzapine: clinically indicated dose. N = 150. |
| Outcomes | Global state: CGI‐I and CGI‐S. Substance use: pragmatic binary/continuous measure. Well‐being: pragmatic binary/continuous measure. Craving: pragmatic binary/continuous measure. Service outcomes: re‐hospitalisation, days in hospital, time attending psychiatric outpatient clinic. Quality of life: important change. Leaving the study early. Other routine data, such as incidents with the police, |
| Notes | * size of study to detect a 10% difference in improvement with 80% certainty. For all outcomes there should be binary cut‐off points of clinically important improvement, defined before the study starts. |
| RISPERIDONE versus CLOZAPINE ‐ all data short term (up to 6 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: for people with serious mental illness and co‐occurring substance misuse | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with Clozapine | Risk with Risperidone | |||||
| Mental state: positive symptoms ‒average endpoint score (PANSS positive subscale, lower = better) | The mean positive symptoms (PANSS positive subscale, lower = better) in the intervention group was 0.9 higher (2.21 lower to 4.01 higher) | ‐ | 36 | ⊕⊝⊝⊝ | No trial reported "improvement in symptoms of severe mental illness" ‒ this continuous measure is the nearest proxy for this. | |
| Substance use: improvement ‒ (at least 20% reduction in use, TLFB scale) | Study population | RR 1.00 | 14 | ⊕⊝⊝⊝ | ||
| 429 per 1000 | 429 per 1000 | |||||
| Moderate | ||||||
| 429 per 1000 | 429 per 1000 | |||||
| Subjective well‐being: Subjective well‐being under neuroleptics scale ‒ average endpoint scores (SWN scale, higher = better) | The mean subjective well‐being under neuroleptics scale score (SWN scale, higher = better) in the intervention group was 6 lower (14.82 lower to 2.82 higher) | ‐ | 36 | ⊕⊝⊝⊝ | ||
| Craving for substances: Marijuana Craving Questionnaire ‒ average endpoint scores (MCQ, lower = better) | The mean craving for substances score on the Marijuana Craving Questionairre (MCQ, lower = better) in the intervention group was 7 higher (2.37 higher to 11.63 higher) | ‐ | 28 | ⊕⊝⊝⊝ | ||
| Adherence to antipsychotic medication: discontinued medication | Study population | RR 4.05 | 36 | ⊕⊝⊝⊝ | ||
| 0 per 1000 | 0 per 1,000 | |||||
| Moderate | ||||||
| 0 per 1,000 | 0 per 1,000 | |||||
| Adverse effects. 1. Movement disorders ‐ any extrapyramidal | Study population | RR 2.71 | 50 | ⊕⊝⊝⊝ | Many adverse effects reported ‒ none designated 'clinically important' (extrapyramidal used as proxy). | |
| 0 per 1000 | 0 per 1000 | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 | |||||
| Leaving the study early ‒ any reason | Study population | RR 0.49 | 45 | ⊕⊝⊝⊝ | ||
| 318 per 1000 | 156 per 1000 | |||||
| Moderate | ||||||
| 386 per 1000 | 189 per 1000 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 High risk of performance bias and detection bias 2 Sample size is very small, optimal information size (OIS) not met to detect 25% difference 3 Performance bias, attrition bias, selective outcome reporting 4 Sample size is very small (n = 14) 5 High risk of performance bias, detection bias, attrition bias and selective outcomes reporting 6 Total sample size is very small (n<300), total event rate is very low and optimum information size (OIS) is not met | ||||||
| RISPERIDONE versus OLANZAPINE‐ all data short term (up to 6 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: people with serious mental illness and co‐occurring substance misuse | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with Olanzapine | Risk with Risperidone | |||||
| Mental state: 2. Specific‐ Positive symptoms, total score‐ average endpoint scores (SADS‐C‐PD scale, lower = better) | The mean positive symptoms total score at endpoint (SADS‐C‐PD scale, lower = better) in the intervention group was 1.5 lower (3.82 lower to 0.82 higher) | ‐ | 37 | ⊕⊝⊝⊝ | ||
| Substance use: 1. Reduction of cannabis use‐change data (number of joints smoked/week) | The reduction of cannabis joints smoked (number of joints smoked/week‐short term data, up to 6 months) in the intervention group was 0.4 higher (4.72 lower to 5.52 higher) | ‐ | 41 | ⊕⊝⊝⊝ | ||
| Subjective well‐being | ‐ | ‐ | ‐ | No trial reported on this important outcome for participants with a co‐occurring substance use disorder | ||
| Craving for substances: 2. Drug Desires Questionnaire‐ average endpoint scores (DDQ, lower = better) | The mean endpoint. Drug Desires Questionnaire‐ endpoint scores (DDQ, lower = better), short term, up to 6 months‐in the intervention group was5 higher (4.86 lower to 14.86 higher) | ‐ | 41 | ⊕⊝⊝⊝ | ||
| Adherence to antipsychotic medication: number of missed doses, average endpoint data, short term (up to 6 months) | ‐ | ‐ | ‐ | ‐ | no useable data available for this outcome | |
| Adverse effects: Parkinsonism ‐ average endpoint score (SAS, high = worse) | The mean adverse effects: ‐ Parkinsonism‐ average endpoint score (SAS, high = worse)‐ short‐term‐ up to 6 months in the intervention group was 0.08 lower (1.21 lower to 1.05 higher) | ‐ | 16 | ⊕⊝⊝⊝ | ||
| Leaving study early: any reason | Study population | RR 0.68 | 77 | ⊕⊝⊝⊝ | ||
| 357 per 1000 | 243 per 1000 | |||||
| Moderate | ||||||
| 411 per 1000 | 279 per 1000 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 High risk for performance bias, allocation concealment, unknown risk for attrition and slective reporting 2 Very low sample size, optimal information size (OIS) not met 3 High risk of attrition bias, study sponsored by pharmaceutical industry 4 Very low sample size, optimal information criterion not met, CI crosses both appreciable harm and benefit 5 High attrition risk, high other risk of funding by pharmaceutical industry, all other risk items unclear risk of bias 6 High risk of performance, attrition and funding bias. Several domains with unclear risk of bias | ||||||
| RISPERIDONE versus PERPHENAZINE‐long term data (>12 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: people with severe mental illness and co‐occurring substance misuse | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with PERPHENAZINE | Risk with RISPERIDONE | |||||
| Leaving the study early: any reason | Study population | RR 1.05 | 281 | ⊕⊕⊝⊝ | ||
| 750 per 1000 | 788 per 1000 | |||||
| Moderate | ||||||
| 750 per 1000 | 788 per 1000 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 High risk of attrition bias, but this does not affect this particular outcomes 2 Optimal information size criterion is met but the estimate includes no effect with both appreciable harm and benefit | ||||||
| RISPERIDONE versus QUETIAPINE‐ short and long term data (up to 6months and > 12 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: people with severe mental illness and co‐occurring substance misuse | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with QUETIAPINE | Risk with RISPERIDONE | |||||
| Leaving the study early: 1. any reason, long term (>12 months) | Study population | RR 0.96 | 294 | ⊕⊕⊝⊝ | ||
| 825 per 1000 | 792 per 1000 | |||||
| Moderate | ||||||
| 825 per 1000 | 792 per 1000 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Risk of bias unclear across all groups and with high risk of funding bias 2 Sample size meets optimal information threshold/ required sample size to detect 25% difference from control group in in PANSS score; at alpha of 0.05 and power of 80%. 3 Outcome not affected by risk of attrition bias 4 Optimal information criterion not met, estimate includes both appreciable harm and benefit | ||||||
| RISPERIDONE versus ZIPRASIDONE‐ all data long term data (>12 months) for people with severe mental illness and co‐occurring substance misuse | ||||||
| Patient or population: people with severe mental illness and co‐occurring substance misuse | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with ZIPRASIDONE | Risk with RISPERIDONE | |||||
| Leaving the study early: any reason | Study population | RR 0.96 | 240 | ⊕⊕⊝⊝ | ||
| 819 per 1000 | 787 per 1000 | |||||
| Moderate | ||||||
| 819 per 1000 | 787 per 1000 | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Risk of attrition bias high but this does not affect this outcome 2 Optimal information size criterion met but estimate includes both appreciable harm and benefit. Total sample size small | ||||||
| Sequence generation |
|
| Allocation concealment |
|
| Blinding of participants and personnel |
|
| Blinding of outcome assessment |
|
| Incomplete outcome data |
|
| Selective reporting |
|
| Other forms of bias |
|
| Diagnostic tools | Abbreviation | Source of scale/ instrument | Study using instrument | Results reported or usable data for re‐analysis/ quantitative synthesis or qualitative results/data only |
| Structured Clinical Interview for DSM Disorders | SCID‐I | Not an outcome measure | ||
| Mental state scales | ||||
| Brief Psychiatric Rating Scale | BPRS | No results or usable data reported or obtained | ||
| Clinical Global Impression scale | CGI | No results or usable data reported or obtained | ||
| Hamilton Depression Rating Scale | HAM‐D | Results reported; usable data for quantitative synthesis | ||
| Positive and Negative Syndrome Scale | PANSS | Results reported; usable data for quantitative synthesis | ||
| Schedule for Affective Disorders and Schizophrenia ‒ Change Version with Psychosis and Disorganization items | SADS‐C‐PD | Results reported; usable data for quantitative synthesis | ||
| Schedule for the Assessment of Negative Symptoms | SANS | No results or usable data reported or obtained | ||
| Substance use scales | ||||
| Addiction Severity Index | ASI | No results or usable data reported or obtained | ||
| Composite International Diagnostic Interview | CIDI | Not an outcome measure | ||
| Substance Use Questionnaire | SUQ | Sevy 2011, Locally derived instrument/ non‐validated | Results reported, used together with urine testing; usable data for quantitative synthesis | |
| Time‐Line Follow‐Back | TLFB | Results reported in dichotomised format for quantitative synthesis | ||
| Quantitative Substance Use Inventory | Locally derived instrument/ non‐validated | Non‐validated scale | ||
| Subjective‐Wellbeing Scales | ||||
| Subjective Well‐being Under Neuroleptics Scale | SWN | Results reported; usable data for quantitative synthesis | ||
| Craving for substances measures | ||||
| Cocaine Craving Report | No usable data for quantitative synthesis | |||
| Desires for Drug Questionnaire | DDQ | Results reported; usable data for quantitative synthesis | ||
| Marijuana Craving Report | No usable data for quantitative synthesis | |||
| Marijuana Craving Questionnaire | MCQ | Results reported; usable data for quantitative synthesis | ||
| Obsessive Compulsive Drug Use Scale | OCDUS | Results reported; usable data for quantitative synthesis | ||
| Adverse effect scales | ||||
| Abnormal Involuntary Movement Scale | AIMS | No results or usable data reported or obtained | ||
| Barnes Akathisia Rating Scale | BARS | No results or usable data reported or obtained | ||
| Simpson Angus Scale | SAS | Results reported; usable data for quantitative synthesis | ||
| Other measures (categorical or time to event) | ||||
| Urine assay for cannabis and cocaine use (proportion of treatment group positive per week) | No usable data for quantitative synthesis | |||
| Number of participants with improvement in substance use (categorised as improved or not‐improved versus unchanged) | Results reported; usable data for quantitative synthesis (dichotomised) | |||
| Days of self‐reported drug use in past week | No usable data for quantitative synthesis | |||
| Weeks in treatment | Results reported; usable data for quantitative synthesis | |||
| Number of participants not completing the study | Akerele 2007; Machielsen 2014; Noordsy 2010; Sevy 2011; Swartz 2008 | Results reported, usable data for quantitative synthesis | ||
| Compliance with medication (missed doses) | No usable data for quantitative synthesis | |||
| Study tag | Participants | Comparison | Relevant review | ||
| Primary problem | Co‐morbidity | Note (reasons for exclusion) | |||
| schizophrenia | Excludes participants with alcohol or drug abuse in past year | Excludes comorbidity | risperidone, haloperidol, methotrimeprazine | ‐ | |
| schizophrenia, schizoaffective disorder | Excludes participants with alcohol or drug abuse in past year | Excludes comorbidity | risperidone, olanzapine, conventional antipsychotics vs. risperidone long‐acting injectable (RLAI),or quetiapine | ‐ | |
| schizophrenia | cannabis use disorder | Excluded as this was a study protocol, authors contacted for unpublished data, no response | risperidone vs. clozapine | ‐ | |
| bipolar type I disorder | No measure of substance use | Excludes comorbidity | risperidone, quetiapine | ‐ | |
| schizophrenia, schizophreniform, psychosis not otherwise specified | No measure of substance use | Excludes comorbidity | risperidone, aripirazole | ‐ | |
| schizophrenia | Co‐occurring substance use in subgroup of 44.9% | No comparison of study medication, mainly a prognostic study of the impact of substance use | risperidone, quetiapine, perphenazine, olanzapine, ziprasidone | ‐ | |
| schizophrenia | No measure of substance use | Excludes comorbidity | risperidone, aripiprazole | ‐ | |
| mental disorders due to alcohol use | substance induced mental disorders | Exclusion criterion | risperidone, olanzapine | Suggested review: risperidone versus other antipsychotics for substance induced psychosis | |
| schizophrenia | Co‐occurring cannabis use disorder | Study protocol, authors contacted for unpublished data no response | risperidone, clozapine | ||
| schizophrenia, schizoaffective disorder | Co‐occurring alcohol use disorder (abuse or dependence) | Comparison of same medication (risperidone) in different preparations (oral vs depot) | oral risperidone, depot risperidone | ‐ | |
| schizophrenia, schizoaffective disorder | Co‐occurring alcohol and substance use disorder | Study protocol, study terminated, authors contacted for unpublished data no response | clozapine, conventional antipsychotics, atypical antipsychotics | ‐ | |
| schizophrenia, schizoaffective disorder | Co‐occurring cannabis use disorder | Study protocol, authors contacted no data provided | clozapine, conventional antipsychotics, atypical antipsychotics | ‐ | |
| bipolar I and II disorder, recent manic or mixed episode with or without psychosis | co‐occurring cocaine or methamphetamine use disorder | Only 8.3% of total sample had psychotic features and 15.9% had bipolar type II disorder. | risperidone, quetiapine | Suggested review: Risperidone versus other antipsychotics in bipolar disorder with co‐occurring substance use disorders | |
| bipolar I disorder with mania or mixed states | Excludes participants with recent substance use | Excludes comorbidity. Excludes bipolar disorder with psychotic features. | risperidone or olanzapine | ‐ | |
| bipolar disorder, acute mania | Excludes participants with drug or alcohol use in past 3 months | Excludes comorbidity | aripiprazole, risperidone | ‐ | |
| schizophrenia | Co‐occurring substance use disorders | Quasi‐randomisation | risperidone long‐acting depot, zuclopenthixol long‐acting depot | ‐ | |
| schizophrenia | Co‐occurring substance use disorders | Quasi‐randomisation | risperidone (oral), zuclopenthixol (oral), zuclopenthixol long acting depot | ‐ | |
| bipolar with current manic or mixed episode | Study excludes participants with drug or alcohol in past 1 months | Excludes comorbidity | mood stabiliser augmentation with risperidone, haloperidol or placebo | ‐ | |
| psychotic disorders: schizoaffective disorder, bipolar I disorder, major depressive disorder, delusional disorder, Alzheimer's dementia, schizophreniform disorder, vascular dementia, and substance abuse dementia | No subgroups with substance use reported | Excludes comorbidity. Authors contacted for unpublished data, no response. | risperidone, quetiapine | ‐ | |
| bipolar I disorder | Excludes participants with recent substance use | Excludes comorbidity | haloperidol, risperidone, placebo | ‐ | |
| schizophrenia, schizophreniform, schizoaffective disorder | No measure of substance use | Excludes comorbidity | haloperidol, risperidone, placebo | ‐ | |
| bipolar I and II | Excludes participants with drug or alcohol use in past 3 months. | Excludes comorbidity | continuation of oral risperidone, olanzapine, quetiapine or switch to long‐acting injectable LAI risperidone | ‐ | |
| schizophrenia or schizophrenia‐like illnesses | Excludes participants with alcohol or substance dependence | Quasi‐randomisation.Excludes comorbidity. | aripiprazole versus risperidone | ‐ | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mental state: 1. General: average endpoint scores (PANSS subscale, lower=better) Show forest plot | 1 | 36 | Mean Difference (IV, Fixed, 95% CI) | 2.70 [‐2.14, 7.54] |
| 2 Mental state: 2. General: any change in general symptoms: Show forest plot | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.34] |
| 3 Mental state: 3. Specific: positive, negative symptoms ‐ average endpoint scores (PANSS subscales, lower = better): Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Mental state: Positive symptoms ‐ average endpoint score (PANSS positive subscale, lower=better) | 1 | 36 | Mean Difference (IV, Random, 95% CI) | 0.90 [‐2.21, 4.01] |
| 3.2 Mental state: Negative symptoms ‐ average endpoint score (PANSS negative subscale, lower=better) | 1 | 36 | Mean Difference (IV, Random, 95% CI) | 4.0 [0.79, 7.21] |
| 4 Mental state: 4. Specific: anxiety symptoms Show forest plot | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 5 Substance use Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Substance use: Improvement (at least 20% reduction in use, TLFB scale) | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.30, 3.35] |
| 5.2 Substance use: Discontinued substance use | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.41, 3.12] |
| 6 Subjective Well‐being: average endpoint scores (Subjective Well‐being under Neuroleptics scale, SWN scale, higher=better) Show forest plot | 1 | 36 | Mean Difference (IV, Random, 95% CI) | ‐6.0 [‐14.82, 2.82] |
| 7 Craving for substances Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 Craving for substances: 1. Specific: current craving‐ average endpoint scores (Marijuana Craving Questionairre, MCQ, lower=better) | 1 | 28 | Mean Difference (IV, Random, 95% CI) | 7.00 [2.37, 11.63] |
| 7.2 Craving for substances: 2. Specific: past week craving‐ average endpoint scores (Obsessive Compulsive Drug Use Scale, OCDUS, lower=better) | 1 | 28 | Mean Difference (IV, Random, 95% CI) | 14.20 [4.45, 23.95] |
| 8 Adherence to antipsychotic medication: discontinued medication Show forest plot | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 4.05 [0.21, 78.76] |
| 9 Adverse effects. 1. Movement disorders Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 any extrapyramidal side‐effects | 2 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 2.71 [0.30, 24.08] |
| 9.2 akathisia | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.23, 17.34] |
| 10 Adverse effects: 2. Non‐movement disorder related side‐effects Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 Cardiovascular: palpitations | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 10.2 Cardiovascular: hypotension | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.3 Central nervous system: headache | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.2 [0.01, 3.54] |
| 10.4 Central Nervous System: somnolence | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.2 [0.03, 1.30] |
| 10.5 Dermatological: acne | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 10.6 Endocrinological: decreased libido | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.7 Ear and labarynthine: ear canal blockage | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 10.8 Gastrointestinal: abdominal pain | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 10.9 Gasstrointesinal: elevated liver function tests | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 10.10 Gastrointestinal: hypersalivation | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.01, 1.74] |
| 10.11 General adverse effects: fatigue | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.12 Injuries: sprain | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.13 Metabolic: increased appetite | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.14 Metabolic: weight gain | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.19, 5.24] |
| 10.15 Musculosceletal: ankle pain | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.16 Musculosceletal: knee and foot pain | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.17 Musculosceletal: muscle twitch | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.14, 63.15] |
| 10.18 Renal: urinary retention | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 10.19 Renal: urinary urgency | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.02, 7.02] |
| 11 Leaving the study early Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 any reason | 2 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.10, 2.51] |
| 11.2 due to inefficacy | 1 | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mental state: 1. Specific: Depression‐ change scores (HAM‐D, higher = better), short term (up to 6 months) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 short‐term (up to 6 months) | 1 | 22 | Mean Difference (IV, Random, 95% CI) | ‐0.11 [‐0.78, 0.56] |
| 2 Mental state: 2. Specific: Positive symptoms, total score‐ average endpoint scores (SADS‐C‐PD scale, lower=better), short term (up to 6 months) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 short term, up to 6 months) | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐1.5 [‐3.82, 0.82] |
| 3 Mental state: 3. Specific: Positive symptom subscales‐ average endpoint scores (SADS‐C‐PD subscores, lower=better), short term (up to 6 months)‐ skewed data Show forest plot | Other data | No numeric data | ||
| 4 Mental state: 4. Specific: Negative symptoms, subscales‐ average endpoint scores (SANS subscales, lower=better), short term (up to 6 months) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Negative symptoms: Affective flattening | 1 | 39 | Mean Difference (IV, Random, 95% CI) | 0.5 [‐0.17, 1.17] |
| 4.2 Negative symptoms: alogia | 1 | 39 | Mean Difference (IV, Random, 95% CI) | 0.40 [‐0.22, 1.02] |
| 4.3 Negative symptoms: avolition‐apathy | 1 | 39 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.73, 0.53] |
| 4.4 Negative symptoms: asociality‐anhedonia | 1 | 39 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.80, 0.60] |
| 5 Substance use: 1. Reduction of cannabis use‐change data (number of joints smoked/week, LOCF data, higher =better)‐ short term data (up to 6 months) Show forest plot | 1 | 41 | Mean Difference (IV, Random, 95% CI) | 0.40 [‐4.72, 5.52] |
| 6 Substance use: 2. Discontinued substance use, short term (up to 6 months) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Substance use: 2. Stopped using cannabis (Urine testing and Substance Use Questionnaire) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.68, 2.08] |
| 6.2 Substance use: 2. Stopped using alcohol (Substance Use Questionnaire) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [0.73, 2.36] |
| 7 Craving for substances: 1. Obsessive Compulsive Drug Use Scale‐ average endpoint score (OCDUS, lower=better)‐short term (up to 6 months) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 short‐term (up to 6 months) | 1 | 41 | Mean Difference (IV, Random, 95% CI) | 1.30 [‐3.51, 6.11] |
| 8 Craving for substances: 2. Desires for Drug Questionnaire‐ average endpoint scores (DDQ, LOCF data, lower=better), short term (up to 6 months) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 Short‐term (up to 6 months) | 1 | 41 | Mean Difference (IV, Random, 95% CI) | 5.0 [‐4.86, 14.86] |
| 9 Adverse effects Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.1 Movement disorders: Parkinsonism‐ average endpoint score (SAS, high = worse)‐ short‐term (up to 6 months) | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐1.21, 1.05] |
| 9.2 Non‐movement disorder related side‐effects: Weight gain‐ average endpoint score (BMI, lower=better)‐ short term (up to 6 months) | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐1.0 [‐3.99, 1.99] |
| 10 Leaving study early: 1. Various reasons Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 any reason, short term (up to 6 months) | 2 | 77 | Risk Ratio (M‐H, Random, 95% CI) | 0.68 [0.34, 1.35] |
| 10.2 any reason, long term (> 12 months) | 1 | 299 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.94, 1.21] |
| 10.3 readmission, short term (up to 6 months) | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 14.45] |
| 10.4 intolerable adverse effects, short term (up to 6 months) | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 10.5 participant loss of interest, short term (up to 6 months) | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.14, 1.33] |
| 11 Leaving study early: 2. Weeks in the study‐ average endpoint data (high=good), short term (up to 6 months) Show forest plot | 1 | 28 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐3.35, 3.35] |
| 12 Leaving study early: 3. Weeks in study‐ average endpoint data (high=good), short term (up to 6 months)‐ skewed data Show forest plot | Other data | No numeric data | ||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Leaving the study early: all cause discontinuation, long term (>12 months) Show forest plot | 1 | 281 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.92, 1.20] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Leaving the study early: all cause discontinuation, long term (>12 months) Show forest plot | 1 | 294 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.86, 1.07] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Leaving the study early: all cause discontinuation, long term (>12 months) Show forest plot | 1 | 240 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.85, 1.10] |