Vilanterol dan fluticasone furoate untuk rawatan asma
Referencias
References to studies included in this review
References to studies excluded from this review
References to studies awaiting assessment
References to ongoing studies
Additional references
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Randomised double‐blind multi‐centre trial | |
| Participants | Total sample N = 185 participants, 177 completed study FF/VI 100/25 mcg, n = 56 (54 completed study) FF/VI 200/25 mcg, n = 56 (55 completed study) Placebo, n = 58 (55 completed study) Prednisolone, n = 15 (13 completed study) Age FF/VI 100/25 mcg, mean 34.4 (SD 15.63) FF/VI 200/25 mcg, mean 34.0 (SD 13.74) Placebo, mean 36.1 (SD 15.42) Prednisolone, mean 37.5 (SD 14.19) Males FF/VI 100/25 mcg, 25 (45%) FF/VI 200/25 mcg, 33 (59%) Placebo, 31 (53%) Prednisolone, 9 (60%) Baseline FEV1 (% predicted) FF/VI 100/25 mcg, mean 79.9 (SD 12.58) FF/VI 200/25 mcg, mean 77.5 (SD 13.22) Placebo, mean 77.0 (SD 11.88) Prednisolone, mean 78.6 (SD 13.17) Inclusion criteria
Exclusion criteria
| |
| Interventions | Arm 1: FF/VI dose 100/25 mcg inhalation powder once‐daily for 6 weeks' treatment + 1 oral placebo capsule each day on the last 7 days of the study Arm 2: FF/VI 200/25 mcg inhalation powder once‐daily for 6 weeks' treatment + 1 oral placebo capsule each day on the last 7 days of the study Arm 3: placebo inhalation powder once‐daily for 6 weeks' treatment + 1 oral placebo capsule each day on the last 7 days of the study Arm 4: placebo inhalation powder once‐daily for 6 weeks' treatment + 1 oral prednisolone 10 mg capsule each day on the last 7 days of the study | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Data collected from 17 locations in Germany (6), Poland (7) and USA (4) Funded by GlaxoSmithKline Study duration: 6 weeks | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated by the sponsor through a validated computerised system (RandAll, GlaxoSmithKline, Stevenage, UK) |
| Allocation concealment (selection bias) | Low risk | Participants were randomised via the Registration and Medication Ordering System (GlaxoSmithKline) |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind. Placebo inhalers and capsules were identical in appearance to active treatments |
| Blinding of outcome assessment (detection bias) | Low risk | Double‐blind. Placebo inhalers and capsules were identical in appearance to active treatments |
| Incomplete outcome data (attrition bias) | Low risk | Details of the 8 withdrawals included in NCT01086410 report |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
| Methods | Randomised double‐blind parallel‐group trial | |
| Participants | Total sample N = 2020 participants, 1748 completed study FF/VI 100/25, n = 1009 (885 completed study) FF 100, n = 1011 (863 completed study) Age FF/VI 100/25, mean 41.1 (SD 17.10) FF 100, mean 42.3 (SD 16.82) Males FF/VI 100/25, 348 (34%) FF 100, 321 (32%) Baseline FEV1 (% predicted) FF/VI 100/25, mean 24.4 (SD 12.71) FF 100, mean 24.3 (SD 12.10) Inclusion criteria
Exclusion criteria
| |
| Interventions | Arm 1: FF/VI dose 100/25 mcg inhalation powder inhaled orally once‐daily in the evening Arm 2: FF dose 100 mcg inhalation powder inhaled orally once‐daily in the evening | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Data collected from 182 locations in Argentina (9), Australia (6), Germany (28), Japan (14), Mexico (6), Philippines (5), Poland (15), Romania (6), Russian Federation (16), Ukraine (13) and USA (64) Funded by GlaxoSmithKline Study duration: variable (≥ 24 to 78 weeks) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated by the sponsor through a validated computerised system (RandAll, GlaxoSmithKline, Stevenage, UK) |
| Allocation concealment (selection bias) | Low risk | Participants were randomised via the Registration and Medication Ordering System (GlaxoSmithKline) |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | Described as double‐blind |
| Incomplete outcome data (attrition bias) | Low risk | Information on 271 participants failing to complete study included in trial report |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
| Methods | Randomised double‐blind parallel‐group multi‐centre study | |
| Participants | Total sample N = 2019 screened, N = 1039 randomised, 956 completed study FF/VI 100/25, n = 346 (314 completed study) FF 100, n = 347 (296 completed study) FF/VI 200/25, n = 346 (321 completed study) Age FF/VI 100/25, mean 45.9 (SD 16.14) FF 100, mean 44.7 (SD 15.89) FF/VI 200/25, mean 46.6 (SD 14.72) Males FF/VI 100/25, 141 (40.75%) FF 100, 148 (42.65%) FF/VI 200/25, 122 (35.26%) Baseline FEV1 (% predicted) Not reported Inclusion criteria
Exclusion criteria
| |
| Interventions | Randomised 1:1:1 Arm 1: FF/VI dose 100/25 mcg inhalation powder inhaled orally once‐daily in the evening Arm 2: FF 100 mcg inhalation powder inhaled orally once‐daily in the evening Arm 3: FF/VI dose 200/25 mcg inhalation powder inhaled orally once‐daily in the evening | |
| Outcomes | Primary endpoint Weighted mean (WM) serial FEV1 0 to 24 hours post dose at week 12 Secondary endpoints
Adverse events were assessed throughout the study | |
| Notes | Data collected from 137 locations in Argentina (13), Chile (7), Germany (12), Mexico (4), Netherlands (7), Poland (8), Romania (13), Russian Federation (19), Sweden (5), Ukraine (12) and USA (37) Funded by GlaxoSmithKline Study duration: 12 weeks | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised through a validated computerised system (RandAll Version 2.5, GlaxoSmithKline) |
| Allocation concealment (selection bias) | Low risk | The Registration and Medication Ordering System was used to register and randomise participants |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | Described as double‐blind |
| Incomplete outcome data (attrition bias) | Low risk | Information on 108 participants failing to complete study included in NCT01686633 report |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
| Methods | Randomised double‐blind placebo‐controlled parallel‐group multi‐centre trial | |
| Participants | Total sample N = 609 participants, 515 completed study FF/VI 100/25, n = 201 (179 completed study) FF 100, n = 205 (185 completed study) Placebo, n = 203 (151 completed study) Age FF/VI 100/25, mean 40.7 (SD 16.38) FF 100, mean 40.4 (SD 16.78) Placebo, mean 38.1 (SD 16.49) Males FF/VI 100/25, 85 (42%) FF 100, 79 (39%) Placebo, 92 (45%) Baseline FEV1 (% predicted) Not reported Inclusion criteria
Exclusion criteria
| |
| Interventions | Arm 1: FF/VI dose 100/25 mcg inhalation powder inhaled orally once‐daily for 12 weeks Arm 2: FF dose 100 mcg inhalation powder inhaled orally once‐daily for 12 weeks Arm 3: Placebo inhalation powder inhaled orally once‐daily for 12 weeks | |
| Outcomes | Primary outcomes
Secondary outcomes
| |
| Notes | Data collected from 12 sites in Poland (1), Ukraine (10) and USA (1) Funded by GlaxoSmithKline Study duration: 12 weeks | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Details not reported |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | Described as double‐blind |
| Incomplete outcome data (attrition bias) | Low risk | Details of 94 participants withdrawn from the study are included in NCT01165138 report |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
| Methods | Randomised double‐blind double‐dummy parallel‐group multi‐centre trial | |
| Participants | Total sample N = 503 participants, 393 completed study FF/VI 100/25, n = 201 (161 completed study) FF/VI 200/25, n = 202 (161 completed study) FP 500 mcg twice‐daily, n = 100 (71 completed study) Age FF/VI 100/25, mean 39.7 (SD 15.85) FF/VI 200/25, mean 38.5 (SD 15.64) FP 500 mcg twice‐daily, 38.6 (SD 15.97) Males FF/VI 100/25, 71 (35%) FF/VI 200/25, 78 (39%) FP 500 mcg twice‐daily, 38 (38%) Baseline FEV1 (% predicted) FF/VI 100/25, mean 74.2 (SD 13.48) FF/VI 200/25, mean 74.1 (SD 14.13) FP 500 mcg twice‐daily, mean 75.2 (SD 12.46) Inclusion criteria
Exclusion criteria
| |
| Interventions | Arm 1: FF/VI 100/25 mcg once‐daily Arm 2: FF/VI 200/25 mcg once‐daily Arm 3: FP 500 mcg twice‐daily | |
| Outcomes | Primary outcome measures
| |
| Notes | Data collected from 46 sites in Germany (15), Thailand (4), Ukraine (9) and USA (18) Funded by GlaxoSmithKline Study duration: 52 weeks | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated by the sponsor through a validated computerised system (RandAll, GlaxoSmithKline, UK) |
| Allocation concealment (selection bias) | Low risk | Participants were randomised via an automated telephone‐based registration and medication ordering system (Registration and Medication Ordering System (RAMOS)) |
| Blinding of participants and personnel (performance bias) | Low risk | Reported as double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | Reported as double‐blind |
| Incomplete outcome data (attrition bias) | Low risk | Details of 110 participants withdrawn from the study are provided in the ClinicalTrials.gov NCT01018186 report |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
| Methods | Randomised cross‐over trial | |
| Participants | Total sample: 32 adults Age: participants over 20 years of age with severe asthma. Mean age, 62.2 years (SD 13.3) Baseline: % FEV1 mean, 70 (SD 11.9%); ACT mean, 20.3 (SD 2.79) ppb at time of entry to trial suggested relatively poor asthma control status | |
| Interventions | Sequence 1: FF/VI 100/25 once‐daily vs FP/salmeterol 500/50 twice‐daily Sequence 2: FP/salmeterol 500/50 twice‐daily vs FF/VI 100/25 once‐daily Participants randomised to receive 4 weeks of treatment followed by 4‐week washout period, then second 4 weeks of treatment with the remaining intervention | |
| Outcomes | Fractional exhaled nitric oxide (FeNO) measured by NIOX‐MINO Asthma control test Morning PEF Respiratory resistance/reactance measured by Forced Oscillation Technique Mostgraph‐01 | |
| Notes | Reported as conference abstract. Minimal information available Study duration: Each treatment period ran for 4 weeks with a 4‐week washout period between treatment periods | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Details not reported |
| Allocation concealment (selection bias) | Unclear risk | Details not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Details not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Details not reported |
| Incomplete outcome data (attrition bias) | Unclear risk | Details not reported |
| Selective reporting (reporting bias) | Unclear risk | Details not reported |
| Methods | Randomised double‐blind cross‐over trial | |
| Participants | Total sample N = 26 participants Age: mean, 38.1 (SD 11.30) Males: 18 (69%) Baseline FEV1 (% predicted): not reported Inclusion criteria
Exclusion criteria
| |
| Interventions | Sequence 1: placebo, FF/VI 100/25 mcg AM, FF/VI 100/25 mcg PM Sequence 2: placebo, FF/VI 100/25 mcg PM, FF/VI 100/25 mcg AM Sequence 3: FF/VI 100/25 mcg AM, FF/VI 100/25 mcg PM, placebo Sequence 4: FF/VI 100/25 mcg AM, placebo, FF/VI 100/25 mcg PM Sequence 5: FF/VI 100/25 mcg PM, placebo, FF/VI 100/25 mcg AM Sequence 6: FF/VI 100/25 mcg PM, FF/VI 100/25 mcg AM, placebo Participants received all treatments once a day in the evening from a DPI for 14 days. Each 14‐day treatment period was followed by a 14 to 21‐day washout period | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Data collected from 1 site in New Zealand Funded by GlaxoSmithKline Study duration: Each treatment period ran for 14 days, with a 14 to 21‐day washout period between treatment periods | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated by GSK |
| Allocation concealment (selection bias) | Low risk | Investigator or designee received medication assignment information and randomised participants using sequentially numbered containers |
| Blinding of participants and personnel (performance bias) | Unclear risk | Reported as double‐blind |
| Blinding of outcome assessment (detection bias) | Unclear risk | Reported as double‐blind |
| Incomplete outcome data (attrition bias) | Unclear risk | Details of the 2 withdrawals are included in the trial report |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
| Methods | Randomised double‐blind 3‐period cross‐over incomplete block study | |
| Participants | Total sample 706 screened, N = 421 participants, 323 completed study Age Mean, 47.5 (SD 13.84) Males 132 (31%) Baseline FEV1(% predicted) 1.847 L (62.31%) Inclusion criteria
Exclusion criteria
| |
| Interventions | Arm 1: FF 100 mcg once daily for 14 days Arm 2: FF/VI 100/25 mcg once daily for 14 days Arm 3: FF/UMEC 100/variable dose (15.6, 31.25, 62.5, 125, 250 mcg) once daily for 14 days | |
| Outcomes | Primary outcome measure
Secondary outcome measures
| |
| Notes | 32 centres ‐ Argentina (6), Chile (7), Russia (11), Thailand (4) and USA (4) Funded by GlaxoSmithKline Study duration: Each treatment period ran for 14 days with a 12 to 14‐day washout period between treatment periods | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | In a 3‐period cross‐over study, participants were randomised to a sequence of 3 of 7 treatments using SAS‐generated codes in a validated computerised system (RandAll Version 2.5, GlaxoSmithKline) |
| Allocation concealment (selection bias) | Low risk | The Registration and Medication Ordering System was used to register and randomise participants |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blinding in all 3 conditions |
| Blinding of outcome assessment (detection bias) | Low risk | Double‐blinding in all 3 conditions |
| Incomplete outcome data (attrition bias) | Low risk | Information on 98 participants who failed to complete the study included in the trial report |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
| Methods | Randomised double‐blind double‐dummy parallel‐group trial | |
| Participants | Total sample N = 309 participants, 255 completed study FF/VI 200/25, n = 155 (136 completed study) FP 500, n = 154 (119 completed study) Age FF/VI 200/25, mean 46.9 (SD 12.93) FP 500, n = 48.8 (SD 13.41) Males FF/VI 100/25. 59 (38%) FP 500. n = 64 (42%) Baseline FEV1 (% predicted) FF/VI 100/25, mean 67.51 (SD 13.249) FP 500, n = 67.55 (SD 13.432) Inclusion criteria
Exclusion criteria
| |
| Interventions |
| |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Data collected from 24 sites in China (12), Republic of Korea (10) and Philippines (2) Funded by GlaxoSmithKline Study duration: 12 weeks | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated by the sponsor through a validated computerised system (RandAll, GlaxoSmithKline, UK) |
| Allocation concealment (selection bias) | Low risk | Participants were randomised via an automated telephone‐based registration and medication ordering system (Registration and Medication Ordering System (RAMOS)) |
| Blinding of participants and personnel (performance bias) | Low risk | Reported as double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | Reported as double‐blind |
| Incomplete outcome data (attrition bias) | Low risk | Details of attrition bias included in trial report. Three participants (1 FF/VI; 2 FP) reported a total of 5 serious adverse events and were withdrawn from the study |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
| Methods | Randomised double‐blind parallel‐group multi‐centre trial | |
| Participants | Total sample N = 586 participants, 476 completed study FF 200 mcg once‐daily, n = 198 (146 completed study) FF/VI 200/25 mcg once‐daily, n = 197 (169 completed study) FP 500 mcg twice‐daily, n = 195 (161 completed study) Age FF 200 mcg once‐daily, mean 44.6 years (SD 14.33) FF/VI 200/25 mcg once‐daily, mean 46.6 (SD 15.05) FP 500 mcg twice‐daily, mean 47.3 (SD 14.06 ) Males FF 200 mcg once‐daily, 81 (42%) FF/VI 200/25 mcg once‐daily, 81 (42%) FP 500 mcg twice‐daily, 79 (41%) Baseline FEV1 (% predicted) FF 200 mcg once‐daily, mean 66.66 (SD 12.388) FF/VI 200/25 mcg once‐daily, mean 66.59 (SD 12.614) FP 500 mcg twice‐daily, mean 67.57 (SD 12.185) Inclusion criteria
Exclusion criteria
| |
| Interventions | Arm 1: FF 200 mcg once‐daily Arm 2: FF/VI 200/25 mcg once‐daily Arm 3: FP 500 mcg twice‐daily | |
| Outcomes | Primary outcomes
Secondary outcomes
| |
| Notes | Data collected from 71 sites in Germany (10), Japan (12), Poland (8), Romania (7), Russian Federation (11) and USA (23) Funded by GlaxoSmithKline Study duration: 24 weeks | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated by the sponsor through a validated, computerised system (RandAll, GlaxoSmithKline, Stevenage, UK) |
| Allocation concealment (selection bias) | Low risk | Participants were randomised via the Registration and Medication Ordering System (GlaxoSmithKline) |
| Blinding of participants and personnel (performance bias) | Low risk | Trial reported as double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | Trial reported as double‐blind |
| Incomplete outcome data (attrition bias) | Low risk | Information on 110 participants not completing the study is reported at http://clinicaltrials.gov/show/NCT01134042 |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
| Methods | Randomised double‐blind cross‐over trial | |
| Participants | Total sample N = 26 participants, 23 completed study Age: mean 8.1 years (SD 1.97) Males: 15 (58%) Asthma severity, no. (%) Mild (well controlled with GINA step 2 low‐dose ICS), 21 (84%) Moderate (well controlled with GINA step 3 medium‐dose ICS), 4 (16%) Inclusion criteria
Exclusion criteria
| |
| Interventions | Sequence 1: FF 100 mcg/VI 25 mcg in period 1 and FF 100 mcg in period 2 Sequence 2: FF 100 mcg in period 1 and FF 100 mcg/VI 25 mcg in period 2 With a washout period ≥ 7 days | |
| Outcomes | Primary outcomes
Secondary outcomes
| |
| Notes | Data collected from 1 site in California, USA Funded by GlaxoSmithKline Study duration: 2 weeks | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated by the sponsor through a validated computerised system (RandAll, GlaxoSmithKline, Stevenage, UK) |
| Allocation concealment (selection bias) | Low risk | Participants were randomised via the Registration and Medication Ordering System (GlaxoSmithKline) |
| Blinding of participants and personnel (performance bias) | Low risk | Trial reported as double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | Trial reported as double‐blind |
| Incomplete outcome data (attrition bias) | Low risk | Information on the 3 participants not completing the study is provided at http://clinicaltrials.gov/show/NCT01453023 |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
| Methods | Randomised double‐blind cross‐over trial | |
| Participants | Total sample N = 52 participants, 50 completed Age: mean, 35.4 (SD 8.63) Males: 34 years (65%) Pre‐bronchodilator FEV1% predicted: mean, 89.71 (SD 8.848) Inclusion criteria
Exclusion criteria
| |
| Interventions | Sequence 1: placebo, FF 100 mcg, FF/VI 100/25 mcg Sequence 2: placebo, FF/VI 100/25 mcg, FF 100 mcg Sequence 3: FF 100 mcg, FF/VI 100/25 mcg, placebo Sequence 4: FF 100 mcg, placebo, FF/VI 100/25 mcg Sequence 5: FF/VI 100/25 mcg, placebo, FF 100 mcg Sequence 6: FF/VI 100/25 mcg, FF 100 mcg, placebo Following the run‐in period, participants were randomised to 1 of 6 treatment sequences of placebo, FF 100 mcg once‐daily and FF/VI 100/25 mcg once‐daily. The 3 treatment periods were separated by a washout period of ≥ 21 days (from day 28 dose) and maximum of 35 days | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Data collected from 4 sites in Germany (1), New Zealand (1) and UK (2) Funded by GlaxoSmithKline Study duration: 4 weeks | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised by RandAll (GlaxoSmithKline validated internal randomisation software) to 1 of 6 treatment sequences, each comprising 3 treatment periods |
| Allocation concealment (selection bias) | Low risk | Participants were randomised via the Registration and Medication Ordering System (GlaxoSmithKline) |
| Blinding of participants and personnel (performance bias) | Low risk | Trial reported as double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | Trial reported as double‐blind |
| Incomplete outcome data (attrition bias) | Low risk | Details of attrition bias included in trial report. Two participants withdrew: 1 withdrew consent and 1 experienced an SAE |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
| Methods | Randomised double‐blind cross‐over trial | |
| Participants | Total sample N = 27 participants Age: mean, 30.8 years (SD 7.46) Males: 19 (70%) Pre‐bronchodilator FEV1: mean % pred, 92.3 (range 71.3 to 119.8) Inclusion criteria
Exclusion criteria
| |
| Interventions | Sequence 1: VI 25 mcg, placebo, FF 100 mcg, FF/VI 100/25 mcg Sequence 2: FF/VI 100/25 mcg, FF 100 mcg, placebo, VI 25 mcg Sequence 3: placebo, FF/VI 100/25 mcg, VI 25 mcg, FF 100 mcg Sequence 4: FF 100 mcg, VI 25 mcg, FF/VI 100/25 mcg, placebo Participants meeting all inclusion criteria and no exclusion criteria during screening visit, conducted 14 to 42 days before first dose of study medication, entered a 14‐day run‐in period. Participants were then randomised to 4 treatment periods, each lasting 21 days and all separated by a nominal washout period of 21 to 35 days | |
| Outcomes | Primary outcomes
Secondary outcomes
| |
| Notes | Data collected from 4 sites in Australia (1), New Zealand (1) and Sweden (2) Funded by GlaxoSmithKline Study duration: Each treatment period ran for 21 days with a 21 to 35‐day washout period between treatment periods | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation schedule based on a Williams square generated by the sponsor through validated internal software (RandAll, GlaxoSmithKline, London, UK) |
| Allocation concealment (selection bias) | Low risk | Automated telephone‐based interactive voice response system ‐ RAMOS (GlaxoSmithKline, London, UK) ‐ was used by investigators to register participants and obtain randomised treatment assignments in a blinded manner |
| Blinding of participants and personnel (performance bias) | Low risk | Reported as double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | Reported as double‐blind |
| Incomplete outcome data (attrition bias) | Low risk | Details of attrition bias included in trial report. Twenty‐seven participants were randomised, and 26 completed the study. One participant withdrew consent, and 4 protocol deviations were noted during period 1. Data for those participants were excluded from the analysis of relevant study periods |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
| Methods | Randomised double‐blind double‐dummy parallel‐group multi‐centre trial | |
| Participants | Total sample N = 806 participants, 715 completed study FF/VI 100/25 mcg, n = 403 (358 completed study) FP/SAL 250/50 mcg twice‐daily, n = 403 (357 completed study) Age FF/VI 100/25 mcg, mean 43.8 years (SD 15.86) FP/SAL 250/50 mcg twice‐daily, mean 41.9 years (SD 16.90) Males FF/VI 100/25 mcg 159 (44%) FP/SAL 250/50 mcg twice‐daily 158 (44%) Baseline FEV1 (L) FF/VI 100/25 mcg, mean 2.013 (SD 0.653) FP/SAL 250/50 mcg twice‐daily, mean 2.043 (SD 0.6378) Inclusion criteria
Exclusion criteria
| |
| Interventions | Arm 1: FF/VI 100/25 mcg once‐daily Arm 2: FP/SAL 250/50 mcg twice‐daily | |
| Outcomes | Primary outcome
Secondary outcomes
| |
| Notes | Data collected from 63 sites in Argentina (10), Chile (7), Republic of Korea (7), Netherlands (8), Phillipines (6) and US (25). Funded by GlaxoSmithKline Study duration: 24 weeks | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation schedule was generated by the sponsor through a validated computerised system (RandAll, GlaxoSmithKline) |
| Allocation concealment (selection bias) | Low risk | Participants were randomised via the Registration and Medication Ordering System |
| Blinding of participants and personnel (performance bias) | Low risk | Reported as double‐blind 'Neither the patients nor the investigator knew which study medication the patient was receiving' |
| Blinding of outcome assessment (detection bias) | Low risk | Reported as double‐blind 'Neither the patients nor the investigator knew which study medication the patient was receiving' |
| Incomplete outcome data (attrition bias) | Low risk | 89% completed the study. Details of participant withdrawal included in trial report |
| Selective reporting (reporting bias) | Low risk | No apparent indication of reporting bias |
AE: adverse event
ALP: alkaline phosphatase
ALT: alanine aminotransferase
AM: morning
ANC: absolute neutrophil count
AQLQ: asthma quality of life questionnaire
AST: aspartate aminotransferase
AUC: area under the curve
BMI: body mass index
BUN: blood urea nitrogen
CK: creatine kinase
Cmax: maximum serum concentration
CO: carbon monoxide
CO2: carbon dioxide
COPD: chronic obstructive pulmonary disease
DBP: diastolic blood pressure
DPI: dry powder inhaler
EAR: early asthmatic response
ECG: electrocardiogram
EQ‐5D: EuroQuality of Life 5D questionnaire
ETD: ex‐throat dose
FeNO: fractional exhaled nitric oxide
FEV1: forced expiratory volume in one second
FF: fluticasone furoate
FP: fluticasone propionate
GGT: gamma glutamyl transferase
HIV: human immunodeficiency virus
HRT: hormone replacement therapy
ICS: inhaled corticosteroid
IOP: intraocular pressure
LABA: long‐acting beta2‐agonist
LOCS III: Lens Opacities Classification System, Version III
LogMAR: logarithm of the minimum angle of resolution
MCH: mean corpuscular haemoglobin
MCHC: mean corpuscular haemoglobin concentration
MCV: mean corpuscular volume
NHANES: National Health and Nutrition Examination Survey
NIOX‐MINO: first point‐of‐care medical device for measuring fractional exhaled nitric oxide
NSAIDs: non‐steroidal anti‐inflammatory drugs
OCS: oral corticosteroid
OM: evening
PC20: provocative concentration of methacholine estimated to result in a 20% reduction in FEV1
PEF: peak expiratory flow
PEFR: peak expiatory flow rate
PIRF: peak inspiratory flow rate
PK: pharmacokinetics
ppb: parts per billion
QTcB: QT interval using Bazett's correction
QTcF: QT interval using Fridericia's correction
RAMOS: Registration and Medication Ordering System
RBC: red blood cell
SABA: short‐acting beta2‐agonist
SAE: serious adverse event
SAL: salmeterol
SAS: Statistical Analysis System (a software suite developed by SAS Institute)
SBP: systolic blood pressure
SD: standard deviation
TED: total emitted dose
Tlast: time of the last point with quantifiable concentration
Tmax: time to Cmax
ULN: upper limit of normal
UMEC: umeclidinium bromide
VAS: visual analogue scale
VI: vilanterol
WBC: white blood cell count
WM: weighted mean
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| Study focuses on chronic obstructive pulmonary disease participants with community‐acquired pneumonia | |
| Pooled analysis of data from clinical trials | |
| Pooled analysis of data from clinical trials | |
| Comparison includes budesonide/formoterol maintenance and reliever therapy vs fluticasone furoate/vilanterol. Therefore, the comparison is not a direct evaluation of budesonide/formoterol maintenance vs fluticasone furoate/vilanterol | |
| Participants have a diagnosis of asthma/chronic obstructive pulmonary disease overlap syndrome, rather than asthma per se. COPD is included in the review's exclusion criteria | |
| Participants did not have a diagnosis of asthma (healthy participants) | |
| Participants did not have a diagnosis of asthma (healthy participants) | |
| Participants did not have a diagnosis of asthma (healthy participants) | |
| Report of 3 safety, pharmacokinetics and pharmacodynamics studies with healthy participants | |
| VI and FF are not used together in the intervention arm | |
| Inhaled steroid used in the trial was not specifically FF | |
| Dose proportionality study comparing 3 doses of FF/VI without an additional comparison arm in healthy participants | |
| Study was withdrawn before participants were enrolled | |
| Participants did not have a diagnosis of asthma (healthy participants) | |
| Trial focuses on VI and FP, not on VI and FF | |
| Participants did not have a diagnosis of asthma (healthy participants) | |
| Study evaluating exhaled nitric oxide time profile as a biomarker of airway inflammation | |
| Inhaled steroid used in the trial was not specifically FF | |
| Inhaled steroid used in the trial was not specifically FF | |
| Evaluation of DPI among participants with asthma and COPD |
COPD: chronic obstructive pulmonary disease
DPI: dry powder inhaler
FF: fluticasone furoate
FP: fluticasone propionate
VI: vilanterol
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | A randomised, double‐blind, placebo‐controlled, parallel group, multi‐centre study to evaluate the efficacy and safety of FF/VI inhalation powder delivered once‐daily for 12 weeks in the treatment of asthma in adolescent and adult participants of Asian ancestry currently treated with low to mid‐strength ICS or low‐strength combination therapy |
| Methods | Randomised double‐blind placebo‐controlled parallel‐group multi‐centre study |
| Participants | Participants of Asian ancestry with asthma. 12 years of age or older Inclusion criteria
Exclusion criteria
|
| Interventions | FF/VI ICS/LABA combination vs placebo |
| Outcomes | Primary outcome measure
Secondary outcome measures
|
| Starting date | January 2012 |
| Contact information | GlaxoSmithKline Research and Development Limited |
| Notes |
| Trial name or title | A multi‐centee, randomised, double‐blind, dose‐ranging study to evaluate GSK573719 in combination with fluticasone furoate, fluticasone furoate alone and an active control of fluticasone furoate/vilanterol combination in participants with asthma |
| Methods | Randomised double‐blind cross‐over study |
| Participants | Participants with asthma. 18 years of age or older Inclusion criteria
Exclusion criteria
|
| Interventions | FF 100 mcg vs FF/VI 100/25 mcg vs FF/GSK573719 100/15.6 to 250 mcg |
| Outcomes | Primary outcome measures
Secondary outcome measures
|
| Starting date | April 2012 |
| Contact information | GlaxoSmithKline Research and Development Limited; [email protected] |
| Notes |
| Trial name or title | A 12‐month, open label, randomised, effectiveness study to evaluate fluticasone furoate (GW685698)/vilanterol (GW642444) inhalation powder delivered once‐daily via a novel dry powder inhaler compared with usual maintenance therapy in participants with asthma |
| Methods | Randomised parallel‐group study |
| Participants | Participants with asthma. 18 years of age or older Inclusion criteria Participants eligible for enrolment in the study must meet all of the following criteria
Exclusion criteria Participants meeting any of the following criteria must not be enrolled in the study
|
| Interventions | GW685698+GW642444 once‐daily via a novel dry powder inhaler vs existing maintenance therapy (ICS alone or in combination with a LABA) |
| Outcomes | Primary outcome measure
Secondary outcome measures
|
| Starting date | November 2012 |
| Contact information | GlaxoSmithKline Research and Development Limited; [email protected] |
| Notes |
| Trial name or title | A study to assess the bronchodilator effect of a single dose of fluticasone furoate (FF)/vilanterol (VI) 100/25 micrograms (mcg) combination when administered in adult participants with asthma |
| Methods | Randomised double‐blind placebo‐controlled cross‐over study |
| Participants | 32 adult participants with moderately severe asthma Inclusion criteria
Non‐childbearing potential. Females on HRT and whose menopausal status is in doubt will be required to use one of the contraception methods if they wish to continue their HRT during the study. Otherwise, they must discontinue HRT to allow confirmation of post‐menopausal status before study enrolment. Childbearing potential and agrees to use one of the contraception methods for an appropriate period of time (as determined by product label or investigator) before the start of dosing to sufficiently minimise risk of pregnancy at that point. Female participants must agree to use contraception until completion of the follow‐up visit.
Exclusion criteria
|
| Interventions | After screening, participant will be randomised and will be assigned to 1 of 2 treatment sequences (AB or BA, where A is placebo and B is FF/VI 100/25 mcg). Between the 2 treatment periods, a washout period of 7 to 14 days will occur |
| Outcomes | Serial FEV1 measurements will be taken at 15 and 30 minutes, and at 1, 2, 4, 12, 24, 36, 48, 60 and 72 hours post dose. Safety assessments will include vital signs, ECGs, AE monitoring and laboratory safety tests; however, these will not constitute study endpoints. Results of the study will provide supporting information to prescribers on the bronchodilator effect of FF/VI over 72 hours |
| Starting date | Information on http://clinicaltrials.gov/ in October 2013 indicated that study was not yet recruiting. Estimated primary completion date: December 2013 |
| Contact information | GlaxoSmithKline Research and Development Limited; [email protected] |
| Notes |
| Trial name or title | A study to compare the efficacy and safety of fluticasone furoate (FF) 100 mcg once‐daily with fluticasone propionate (FP) 250 mcg twice‐daily and FP 100 mcg twice‐daily in well‐controlled asthmatic Japanese participants |
| Methods | Randomised double‐blind multi‐centre parallel‐group study |
| Participants | Exclusion criteria
Note: Immunotherapy is permitted for treatment of allergies during the study, provided it was initiated ≥ 4 weeks before visit 1 and participants remain in the maintenance phase for the duration of the study; cytochrome P450 3A4 (CYP3A4) inhibitors: participants who have received a potent CYP3A4 inhibitor within 4 weeks of visit 1 (e.g. clarithromycin, atazanavir, indinavir, itraconazole, ketoconazole, nefazodone, nelfinavir; ritonavir; saquinavir; telithromycin, troleandomycin, voriconazole, mibefradil, cyclosporine)
Other exclusion criteria at visit 2 and visit 5
|
| Interventions |
|
| Outcomes | Primary outcome measures
Secondary outcome measures
|
| Starting date | March 2014 |
| Contact information | GlaxoSmithKline Research and Development Limited; [email protected] |
| Notes |
| Trial name or title | An efficacy and safety study of fluticasone furoate/vilanterol 100/25 microgram (mcg) inhalation powder, fluticasone propionate/salmeterol 250/50 mcg inhalation powder and fluticasone propionate 250 mcg inhalation powder in adults and adolescents with persistent asthma |
| Methods | Randomised double‐blind double‐dummy parallel‐group multi‐centre non‐inferiority study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes | Primary outcome measures
Secondary outcome measures
|
| Starting date | February 2015 |
| Contact information | GlaxoSmithKline Research and Development Limited; [email protected] |
| Notes |
| Trial name or title | An efficacy and safety study of fluticasone furoate/vilanterol 100/25 microgram (mcg) inhalation powder, FP/SAL 250/50 mcg inhalation powder and fluticasone propionate 250 mcg inhalation powder in adults and adolescents with persistent asthma |
| Methods | This study is a randomised double‐blind double‐dummy parallel‐group multi‐centre non‐inferiority study |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | The study will enrol adult and adolescent asthmatic participants who are currently receiving mid‐dose inhaled corticosteroids (ICSs) plus a long‐acting beta2‐agonist (LABA) (equivalent to FP/SAL 250/50 microgram (mcg) twice‐daily (BD)), via a fixed‐dose combination product or through separate inhalers. The study consists of a LABA washout period of 5 days and a run‐in period of 4 weeks, followed by a treatment period of 24 weeks and a follow‐up contact period of 1 week. The total duration of the study is 30 weeks. Approximately 1461 participants will be randomised to 1 of the following 3 treatments (487 per treatment): FF/VI 100/25 mcg once‐daily (OD) in the evening (PM) via ELLIPTA inhaler plus placebo BD via ACCUHALER/DISKUS; FP/SAL 250/50 mcg BD via ACCUHALER/DISKUS inhaler plus placebo OD (PM) via ELLIPTA inhaler; FP 250 mcg BD via ACCUHALER/DISKUS inhaler plus placebo OD (PM) via ELLIPTA inhaler. In addition, all participants will be supplied with albuterol/salbutamol inhalation aerosol for use as needed to treat acute asthma symptoms |
| Outcomes | Primary outcome measures
Secondary outcome measures
|
| Starting date | March 2015 |
| Contact information | US GSK Clinical Trials Call Center |
| Notes |
| Trial name or title | A study to compare the efficacy of fluticasone furoate/vilanterol inhalation powder with usual inhaled corticosteroids (ICS)/long‐acting beta‐agonists (LABA) in persistent asthma |
| Methods | Multi‐centre open‐label randomised parallel‐group study |
| Participants | Inclusion criteria
Female participant is eligible to participate if she is not pregnant (as confirmed by a negative urine human chorionic gonadotrophin (hCG) test), is not lactating and at least one of the following conditions applies:
GSK Modified List of Highly Effective Methods for Avoiding Pregnancy in FRP This list does not apply to FRP with same sex partners, when this is their preferred and usual lifestyle, or for participants who are and will continue to be abstinent from penile‐vaginal intercourse on a long‐term and persistent basis: contraceptive subdermal implant that meets standard operating procedure (SOP) effectiveness criteria, including a < 1% rate of failure per year, as stated in the product label; intrauterine device or intrauterine system that meets SOP effectiveness criteria, including a < 1% rate of failure per year, as stated in the product label; oral contraceptive, either combined or progestogen alone; injectable progestogen; contraceptive vaginal ring; percutaneous contraceptive patches; male partner sterilisation with documentation of azoospermia before entry of female participant into the study, and this male is the sole partner for that participant; male condom combined with a vaginal spermicide (foam, gel, film, cream or suppository). These allowed methods of contraception are effective only when used consistently, correctly and in accordance with the product label. The investigator is responsible for ensuring that participants understand how to properly use these methods of contraception.
Exclusion criteria:
|
| Interventions | To evaluate the efficacy and safety of FF/VI compared with 2 usual ICS/LABA fixed combinations (FP/SAL or budesonide/formoterol (BUD/F)) in participants with persistent asthma, in "close to real life" settings. FF/VI will be administered once‐daily (QD) via ELLIPTA dry powder inhaler (DPI), and FP/SAL or BUD/F will be administered twice‐daily (BID) via DISKUS and TURBUHALER DPI, respectively. ELLIPTA is a new powder inhaler designed to be easy to use. The total duration of individual participation will be approximately 6 months (24 weeks) |
| Outcomes | Primary outcome measures
Secondary outcome measures
|
| Starting date | July 2015 |
| Contact information | US GSK Clinical Trials Call Center |
| Notes |
| Trial name or title | Crossover study comparing fluticasone furoate (FF)/vilanterol (VI) once‐daily versus fluticasone propionate (FP) twice‐daily in participants with asthma and exercise‐induced bronchoconstriction (EIB) |
| Methods | Multi‐centre randomised double‐blind double‐dummy cross‐over study with two 2‐week treatment periods separated by a 2‐week washout period |
| Participants | Participants with asthma and exercise‐induced bronchoconstriction (EIB) between 12 and 50 years of age Inclusion criteria Participants eligible for enrolment in the study must meet the following criteria
OR
Exclusion criteria Participants are not eligible for enrolment in the study if they meet the following criteria
|
| Interventions | This study is designed to compare fluticasone furoate (FF)/vilanterol (VI) once‐daily vs fluticasone propionate (FP) twice‐daily in participants with asthma and exercise‐induced bronchoconstriction (EIB) |
| Outcomes | Primary outcome measures
Secondary outcome measures
|
| Starting date | March 2016 |
| Contact information | GlaxoSmithKline |
| Notes |
| Trial name or title | A study to evaluate the effect of fluticasone/formoterol breath actuated inhaler (BAI) or Relvar Ellipta DPI on ventilation heterogeneity in asthma |
| Methods | A randomised assessor‐blinded parallel‐group trial |
| Participants | Inclusion criteria for participants on Seretide Accuhaler 250/50 µg at screening
Inclusion criteria for participants on equivalent /higher dose or other ICS‐LABAs or higher dose of Seretide at screening
Exclusion criteria for all participants
Exclusion criteria for participant or participants undergoing OR‐MRI and HD‐CT
|
| Interventions | Comparison between fluticasone/formoterol BAI vs fluticasone/vilanterol DPI (Relvar Ellipta DPI) |
| Outcomes | Primary outcome measures
Secondary outcome measures
|
| Starting date | April 2016 |
| Contact information | Mundipharma Research Limited |
| Notes |
| Trial name or title | A randomised effectiveness study comparing fluticasone furoate (FF, GW685698)/vilanterol (VI, GW642444) with standard treatment in chronic obstructive pulmonary disease (COPD) |
| Methods | This is a phase III multi‐centre randomised open‐label study |
| Participants | Inclusion criteria Participants eligible for enrolment in the study must meet all of the following criteria
Exclusion criteria Participants meeting any of the following criteria must not be enrolled in the study
|
| Interventions | This study is designed to compare the effectiveness and safety of FF/VI. Inhalation powder (100 mcg FF, GW685698)/25 mcg VI, GW642444)) delivered once‐daily via a novel dry powder inhaler (NDPI) compared with existing COPD maintenance therapy over 12 months in participants diagnosed with COPD. Participants who meet eligibility criteria are randomised and will enter a 12‐month treatment period |
| Outcomes | Primary outcome measures
Secondary outcome measures
|
| Starting date | January 2012 |
| Contact information | GlaxoSmithKline |
| Notes |
| Trial name or title | An effectiveness study comparing fluticasone furoate (FF, GW685698)/vilanterol (VI, GW642444) with standard treatment in asthma |
| Methods | Multi‐centre randomised open‐label study |
| Participants | Inclusion criteria Participants eligible for enrolment in the study must meet all of the following criteria
OR Childbearing potential with a negative urine pregnancy test at visit 2, and agrees to one of the highly effective and acceptable contraceptive methods used consistently and correctly (i.e. in accordance with the approved product label and instructions of the physician for the duration of the study ‐ visit 2 to the end of the study) Exclusion criteria Participants meeting any of the following criteria must not be enrolled in the study
|
| Interventions | This study is designed to compare the effectiveness and safety of FF/VI inhalation powder ((100 mcg FF), GW685698)/25 mcg VI, GW642444) or 200 mcg FF, GW685698)/25 mcg VI, GW642444)) delivered once‐daily via a novel dry powder inhaler (NDPI) compared with the existing asthma maintenance therapy over 12 months in participants diagnosed with asthma. Participants who meet the eligibility criteria are randomised and will enter a 12‐month treatment period |
| Outcomes | Primary outcome measures
Secondary outcome measures
|
| Starting date | November 2012 |
| Contact information | US GSK Clinical Trials Call Center |
| Notes |
ACT: Asthma Control Test
AE: adverse event
AM: morning
AQLQ: Asthma Quality of Life Questionnaire
BTS: British Thoracic Society
COPD: chronic obstructive pulmonary disease
DPI: dry powder inhaler
ECG: electrocardiogram
FEV1: forced expiratory volume in one second
FF: fluticasone furoate
FP: fluticasone propionate
FRP: female reproductive potential
FSH: follicle‐stimulating hormone
GINA: Global Initiative for Asthma
GP: general practitioner
HCP: healthcare practitioner
HRT: hormone replacement therapy
ICS: inhaled corticosteroid
LABA: long‐acting beta2‐agonist
LAMA: long‐acting muscarinic agonist
LTRA: leukotriene receptor antagonist
MAO: monoamine oxidase
NHANES: National Health and Nutrition Examination Survey
OCS: oral corticosteroid
PEF: peak expiratory flow
PM: afternoon
SABA: short‐acting beta2‐agonist
SAL: salmeterol
SIGN: Scottish Intercollegiate Guidelines Network
SOP: standard operating procedure
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Change in quality of life (measured by AQLQ at 12 wk) Show forest plot | 1 | 329 | Mean Difference (Fixed, 95% CI) | 0.3 [0.14, 0.46] |
| Analysis 1.1  Comparison 1 FF/VI 100/25 versus placebo, Outcome 1 Change in quality of life (measured by AQLQ at 12 wk). | ||||
| 2 Exacerbations Show forest plot | 2 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Analysis 1.2  Comparison 1 FF/VI 100/25 versus placebo, Outcome 2 Exacerbations. | ||||
| 3 Serious adverse events Show forest plot | 5 | 721 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Analysis 1.3  Comparison 1 FF/VI 100/25 versus placebo, Outcome 3 Serious adverse events. | ||||
| 4 FEV1 Litres Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.17 [0.09, 0.26] | |
| Analysis 1.4  Comparison 1 FF/VI 100/25 versus placebo, Outcome 4 FEV1 Litres. | ||||
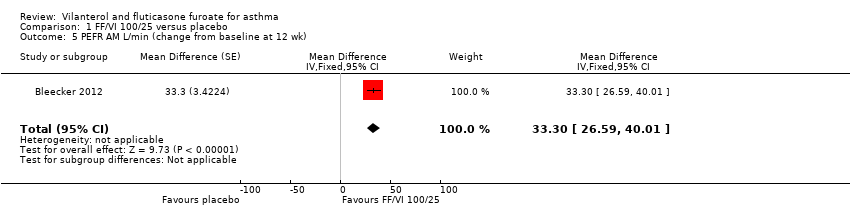
| 5 PEFR AM L/min (change from baseline at 12 wk) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 33.3 [26.59, 40.01] | |
| Analysis 1.5 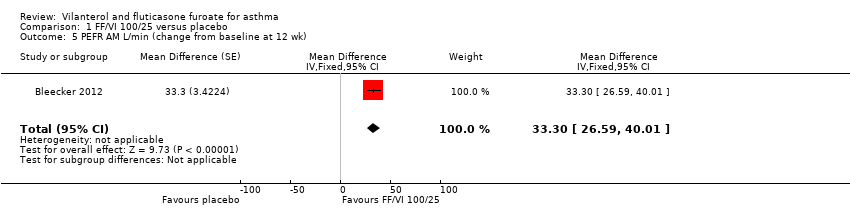 Comparison 1 FF/VI 100/25 versus placebo, Outcome 5 PEFR AM L/min (change from baseline at 12 wk). | ||||
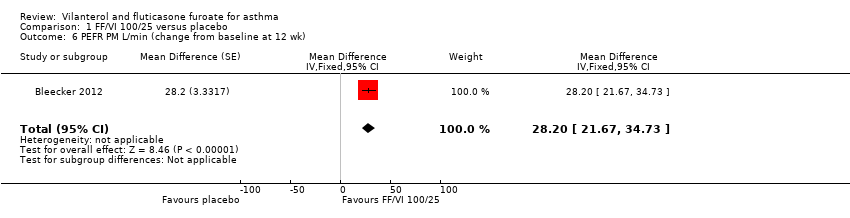
| 6 PEFR PM L/min (change from baseline at 12 wk) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 28.2 [21.67, 34.73] | |
| Analysis 1.6 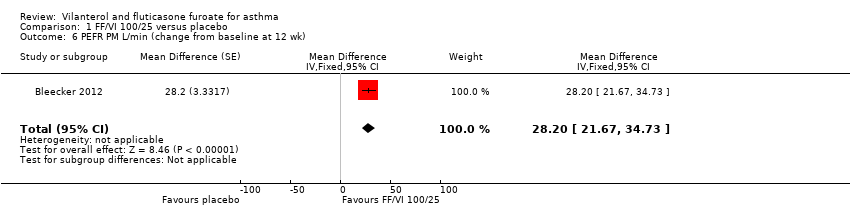 Comparison 1 FF/VI 100/25 versus placebo, Outcome 6 PEFR PM L/min (change from baseline at 12 wk). | ||||
| 7 Change in asthma symptoms (measured by ACT) Show forest plot | 1 | 339 | Mean Difference (Fixed, 95% CI) | 1.9 [1.22, 2.58] |
| Analysis 1.7  Comparison 1 FF/VI 100/25 versus placebo, Outcome 7 Change in asthma symptoms (measured by ACT). | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Change in quality of life (measured by AQLQ at 12 wk) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.15 [‐0.00, 0.30] | |
| Analysis 2.1  Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 1 Change in quality of life (measured by AQLQ at 12 wk). | ||||
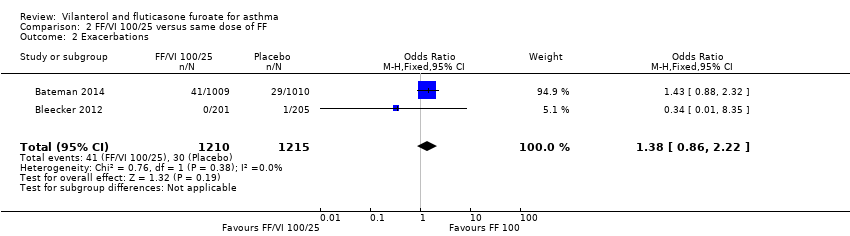
| 2 Exacerbations Show forest plot | 2 | 2425 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.86, 2.22] |
| Analysis 2.2  Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 2 Exacerbations. | ||||
| 3 Serious adverse events Show forest plot | 5 | 1258 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.61 [0.42, 6.17] |
| Analysis 2.3  Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 3 Serious adverse events. | ||||
| 4 Trough FEV1 (L) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.08 [0.02, 0.14] | |
| Analysis 2.4 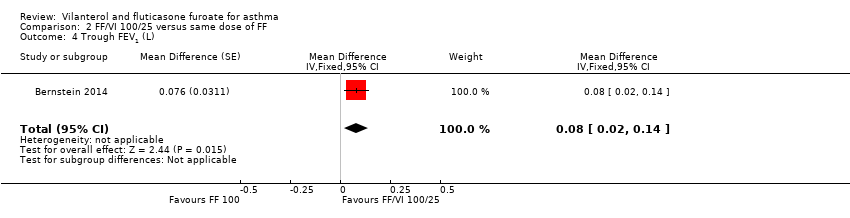 Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 4 Trough FEV1 (L). | ||||
| 5 PEFR AM (change from baseline at 12 wk) Show forest plot | 2 | Mean Difference (Fixed, 95% CI) | 20.29 [15.72, 24.85] | |
| Analysis 2.5  Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 5 PEFR AM (change from baseline at 12 wk). | ||||
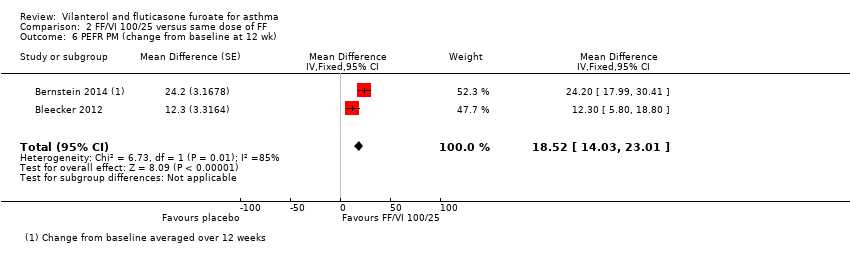
| 6 PEFR PM (change from baseline at 12 wk) Show forest plot | 2 | Mean Difference (Fixed, 95% CI) | 18.52 [14.03, 23.01] | |
| Analysis 2.6  Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 6 PEFR PM (change from baseline at 12 wk). | ||||
| 7 Change in asthma symptoms (measured by ACT) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.6 [‐0.04, 1.24] | |
| Analysis 2.7  Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 7 Change in asthma symptoms (measured by ACT). | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Serious adverse events Show forest plot | 1 | 53 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Analysis 3.1  Comparison 3 FF/VI 100/25 versus same dose VI, Outcome 1 Serious adverse events. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
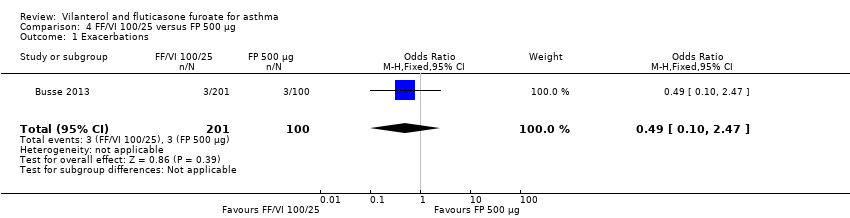
| 1 Exacerbations Show forest plot | 1 | 301 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.10, 2.47] |
| Analysis 4.1  Comparison 4 FF/VI 100/25 versus FP 500 µg, Outcome 1 Exacerbations. | ||||
| 2 Serious adverse events Show forest plot | 1 | 301 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.05, 0.80] |
| Analysis 4.2  Comparison 4 FF/VI 100/25 versus FP 500 µg, Outcome 2 Serious adverse events. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Change in quality of life (measured by AQLQ at 24 wk) Show forest plot | 1 | 677 | Mean Difference (Fixed, 95% CI) | 0.09 [‐0.03, 0.21] |
| Analysis 5.1  Comparison 5 FF/VI 100/25 versus FPS 250/50 bd, Outcome 1 Change in quality of life (measured by AQLQ at 24 wk). | ||||
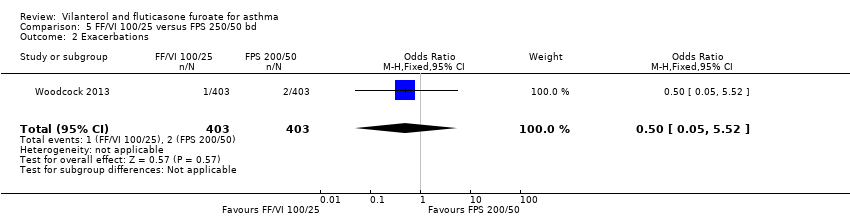
| 2 Exacerbations Show forest plot | 1 | 806 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.50 [0.05, 5.52] |
| Analysis 5.2 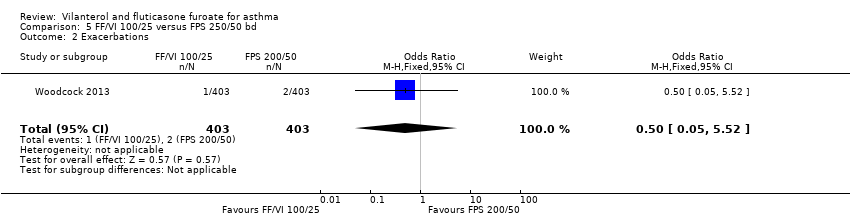 Comparison 5 FF/VI 100/25 versus FPS 250/50 bd, Outcome 2 Exacerbations. | ||||
| 3 Serious adverse events Show forest plot | 1 | 806 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.21, 2.99] |
| Analysis 5.3  Comparison 5 FF/VI 100/25 versus FPS 250/50 bd, Outcome 3 Serious adverse events. | ||||
| 4 FEV1 Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | ‐0.02 [‐0.07, 0.03] | |
| Analysis 5.4  Comparison 5 FF/VI 100/25 versus FPS 250/50 bd, Outcome 4 FEV1. | ||||
| 5 Change in asthma symptoms (measured by ACT) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.24 [‐0.20, 0.68] | |
| Analysis 5.5  Comparison 5 FF/VI 100/25 versus FPS 250/50 bd, Outcome 5 Change in asthma symptoms (measured by ACT). | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Exacerbations Show forest plot | 2 | 515 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.02 [0.50, 8.19] |
| Analysis 6.1  Comparison 6 FF/VI 100/25 µg versus FF/VI 200/25 µg, Outcome 1 Exacerbations. | ||||
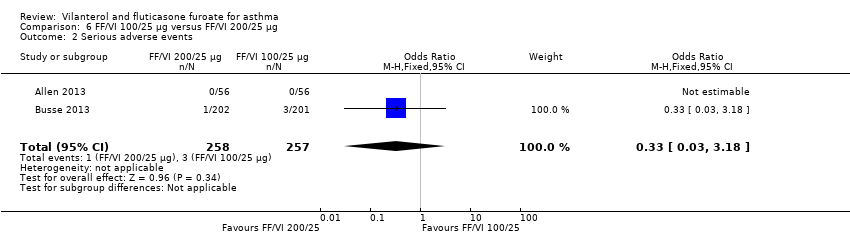
| 2 Serious adverse events Show forest plot | 2 | 515 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.03, 3.18] |
| Analysis 6.2  Comparison 6 FF/VI 100/25 µg versus FF/VI 200/25 µg, Outcome 2 Serious adverse events. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Exacerbations Show forest plot | 1 | 114 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Analysis 7.1  Comparison 7 FF/VI 200/25 versus placebo, Outcome 1 Exacerbations. | ||||
| 2 Serious adverse events Show forest plot | 1 | 114 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Analysis 7.2  Comparison 7 FF/VI 200/25 versus placebo, Outcome 2 Serious adverse events. | ||||
| 3 FEV1 Litres Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.21 [0.13, 0.29] | |
| Analysis 7.3 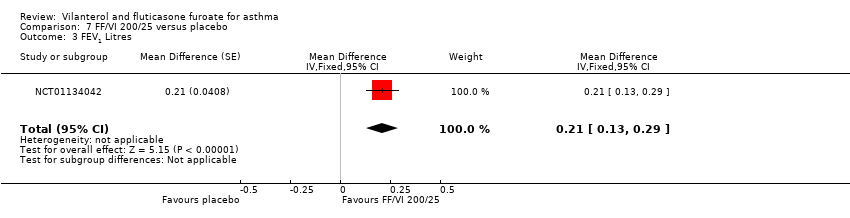 Comparison 7 FF/VI 200/25 versus placebo, Outcome 3 FEV1 Litres. | ||||
| 4 Change in asthma symptoms (measured by ACT) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.9 [0.12, 1.68] | |
| Analysis 7.4  Comparison 7 FF/VI 200/25 versus placebo, Outcome 4 Change in asthma symptoms (measured by ACT). | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
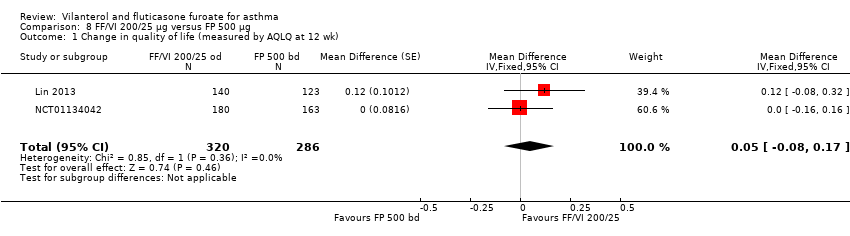
| 1 Change in quality of life (measured by AQLQ at 12 wk) Show forest plot | 2 | 606 | Mean Difference (Fixed, 95% CI) | 0.05 [‐0.08, 0.17] |
| Analysis 8.1  Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 1 Change in quality of life (measured by AQLQ at 12 wk). | ||||
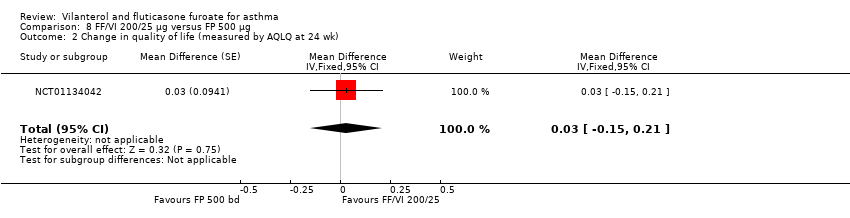
| 2 Change in quality of life (measured by AQLQ at 24 wk) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.03 [‐0.15, 0.21] | |
| Analysis 8.2 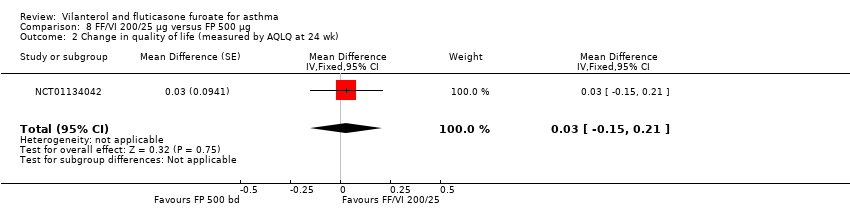 Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 2 Change in quality of life (measured by AQLQ at 24 wk). | ||||
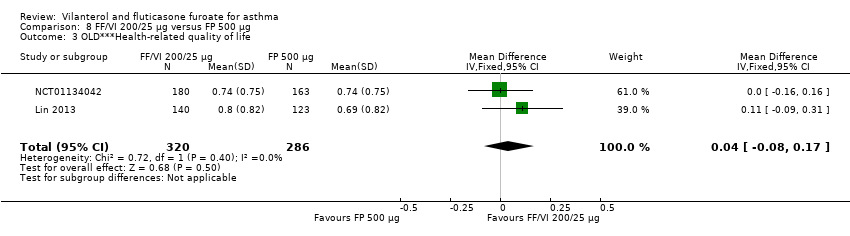
| 3 OLD***Health‐related quality of life Show forest plot | 2 | 606 | Mean Difference (IV, Fixed, 95% CI) | 0.04 [‐0.08, 0.17] |
| Analysis 8.3  Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 3 OLD***Health‐related quality of life. | ||||
| 4 Exacerbations Show forest plot | 2 | 611 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.22, 2.20] |
| Analysis 8.4  Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 4 Exacerbations. | ||||
| 5 Serious adverse events Show forest plot | 3 | 1003 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.25, 1.49] |
| Analysis 8.5  Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 5 Serious adverse events. | ||||
| 6 PEFR Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 28.6 [20.23, 36.97] | |
| Analysis 8.6 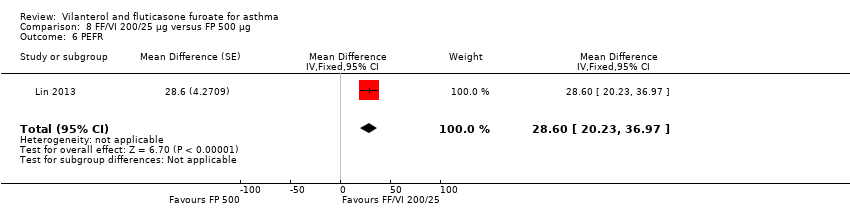 Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 6 PEFR. | ||||
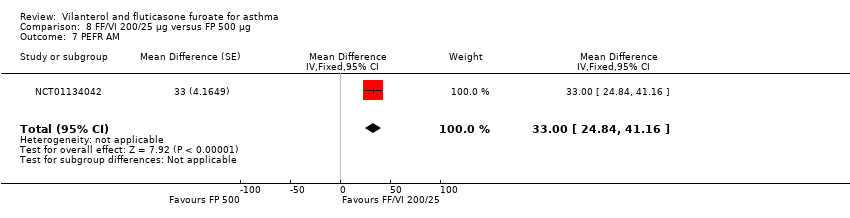
| 7 PEFR AM Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 33.0 [24.84, 41.16] | |
| Analysis 8.7 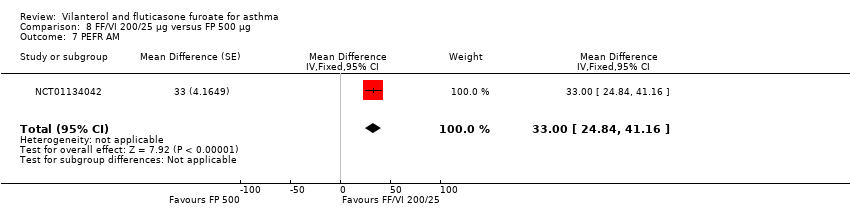 Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 7 PEFR AM. | ||||
| 8 PEFR PM Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 26.2 [18.04, 34.36] | |
| Analysis 8.8  Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 8 PEFR PM. | ||||
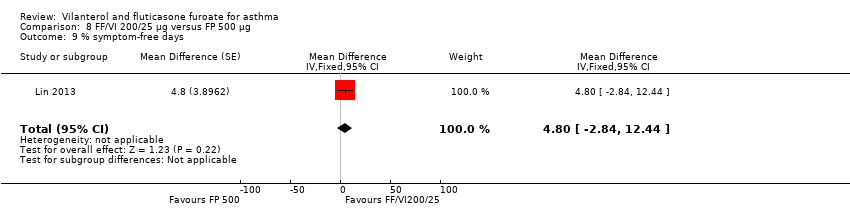
| 9 % symptom‐free days Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 4.8 [‐2.84, 12.44] | |
| Analysis 8.9  Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 9 % symptom‐free days. | ||||
| 10 Change in asthma symptoms (measured by ACT) Show forest plot | 1 | 332 | Mean Difference (Fixed, 95% CI) | 0.8 [0.01, 1.59] |
| Analysis 8.10 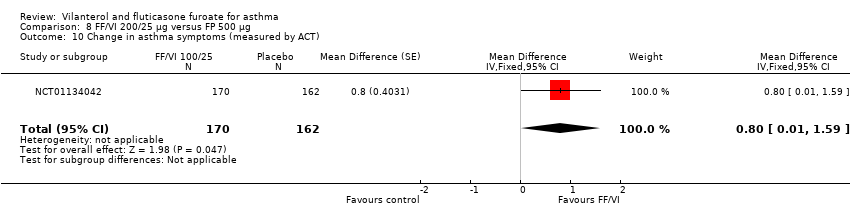 Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 10 Change in asthma symptoms (measured by ACT). | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Change in quality of life (measured by AQLQ at 12 wk) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.08 [‐0.08, 0.24] | |
| Analysis 9.1  Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 1 Change in quality of life (measured by AQLQ at 12 wk). | ||||
| 2 Change in quality of life (measured by AQLQ at 24 wk) Show forest plot | 1 | 307 | Mean Difference (Fixed, 95% CI) | 0.05 [‐0.14, 0.24] |
| Analysis 9.2  Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 2 Change in quality of life (measured by AQLQ at 24 wk). | ||||
| 3 Serious adverse events Show forest plot | 1 | 391 | Odds Ratio (M‐H, Fixed, 95% CI) | 6.06 [0.72, 50.84] |
| Analysis 9.3  Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 3 Serious adverse events. | ||||
| 4 FEV1 Litres Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.19 [0.10, 0.28] | |
| Analysis 9.4  Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 4 FEV1 Litres. | ||||
| 5 PEFR AM Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 33.6 [25.41, 41.79] | |
| Analysis 9.5  Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 5 PEFR AM. | ||||
| 6 PEFR PM Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 30.7 [22.51, 38.89] | |
| Analysis 9.6  Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 6 PEFR PM. | ||||
| 7 Change in asthma symptoms (measured by ACT) Show forest plot | 1 | 317 | Mean Difference (Fixed, 95% CI) | 0.3 [‐0.50, 1.10] |
| Analysis 9.7 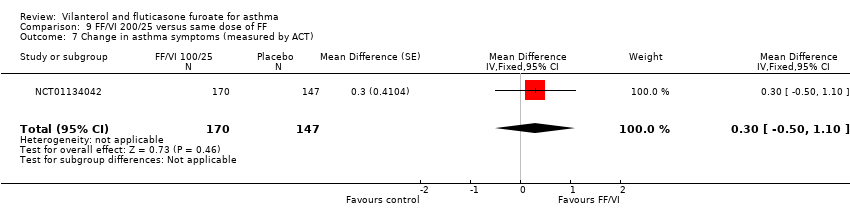 Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 7 Change in asthma symptoms (measured by ACT). | ||||

Study flow diagram.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
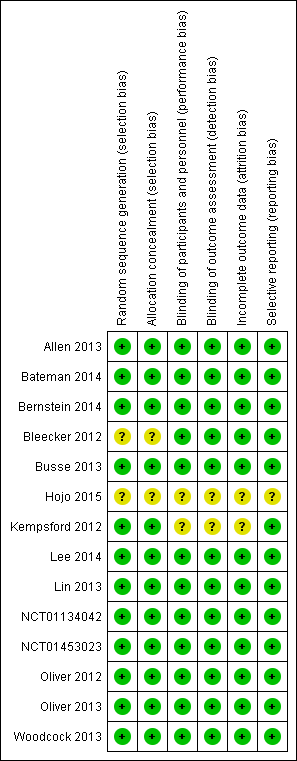
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Comparison 1 FF/VI 100/25 versus placebo, Outcome 1 Change in quality of life (measured by AQLQ at 12 wk).

Comparison 1 FF/VI 100/25 versus placebo, Outcome 2 Exacerbations.

Comparison 1 FF/VI 100/25 versus placebo, Outcome 3 Serious adverse events.
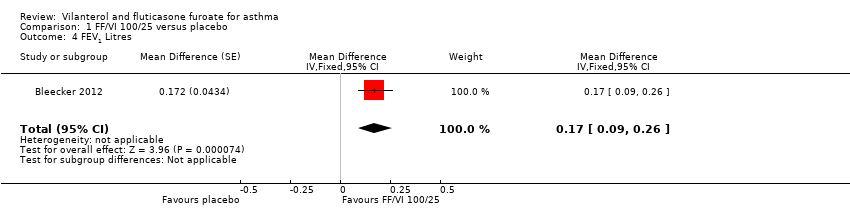
Comparison 1 FF/VI 100/25 versus placebo, Outcome 4 FEV1 Litres.
Comparison 1 FF/VI 100/25 versus placebo, Outcome 5 PEFR AM L/min (change from baseline at 12 wk).
Comparison 1 FF/VI 100/25 versus placebo, Outcome 6 PEFR PM L/min (change from baseline at 12 wk).

Comparison 1 FF/VI 100/25 versus placebo, Outcome 7 Change in asthma symptoms (measured by ACT).

Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 1 Change in quality of life (measured by AQLQ at 12 wk).
Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 2 Exacerbations.

Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 3 Serious adverse events.

Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 4 Trough FEV1 (L).

Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 5 PEFR AM (change from baseline at 12 wk).
Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 6 PEFR PM (change from baseline at 12 wk).

Comparison 2 FF/VI 100/25 versus same dose of FF, Outcome 7 Change in asthma symptoms (measured by ACT).

Comparison 3 FF/VI 100/25 versus same dose VI, Outcome 1 Serious adverse events.
Comparison 4 FF/VI 100/25 versus FP 500 µg, Outcome 1 Exacerbations.

Comparison 4 FF/VI 100/25 versus FP 500 µg, Outcome 2 Serious adverse events.

Comparison 5 FF/VI 100/25 versus FPS 250/50 bd, Outcome 1 Change in quality of life (measured by AQLQ at 24 wk).
Comparison 5 FF/VI 100/25 versus FPS 250/50 bd, Outcome 2 Exacerbations.

Comparison 5 FF/VI 100/25 versus FPS 250/50 bd, Outcome 3 Serious adverse events.

Comparison 5 FF/VI 100/25 versus FPS 250/50 bd, Outcome 4 FEV1.

Comparison 5 FF/VI 100/25 versus FPS 250/50 bd, Outcome 5 Change in asthma symptoms (measured by ACT).

Comparison 6 FF/VI 100/25 µg versus FF/VI 200/25 µg, Outcome 1 Exacerbations.
Comparison 6 FF/VI 100/25 µg versus FF/VI 200/25 µg, Outcome 2 Serious adverse events.

Comparison 7 FF/VI 200/25 versus placebo, Outcome 1 Exacerbations.

Comparison 7 FF/VI 200/25 versus placebo, Outcome 2 Serious adverse events.

Comparison 7 FF/VI 200/25 versus placebo, Outcome 3 FEV1 Litres.

Comparison 7 FF/VI 200/25 versus placebo, Outcome 4 Change in asthma symptoms (measured by ACT).
Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 1 Change in quality of life (measured by AQLQ at 12 wk).
Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 2 Change in quality of life (measured by AQLQ at 24 wk).
Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 3 OLD***Health‐related quality of life.

Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 4 Exacerbations.

Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 5 Serious adverse events.

Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 6 PEFR.
Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 7 PEFR AM.

Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 8 PEFR PM.
Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 9 % symptom‐free days.

Comparison 8 FF/VI 200/25 µg versus FP 500 µg, Outcome 10 Change in asthma symptoms (measured by ACT).

Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 1 Change in quality of life (measured by AQLQ at 12 wk).

Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 2 Change in quality of life (measured by AQLQ at 24 wk).

Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 3 Serious adverse events.
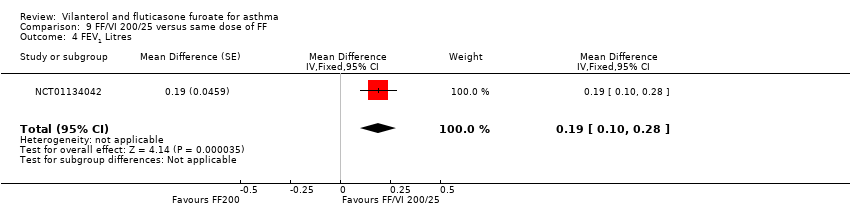
Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 4 FEV1 Litres.

Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 5 PEFR AM.
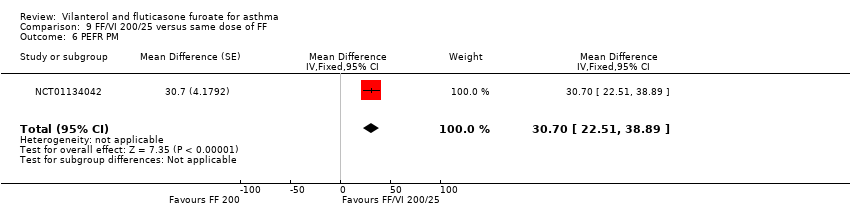
Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 6 PEFR PM.

Comparison 9 FF/VI 200/25 versus same dose of FF, Outcome 7 Change in asthma symptoms (measured by ACT).
| VI and FF compared with placebo for asthma | |||||
| Patient or population: people with asthma Settings: community Intervention: VI and FF Comparison: placebo | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | |
| Assumed risk | Corresponding risk | ||||
| Placebo | VI and FF | ||||
| Health‐related quality of life | 0.61 | 0.91 (SE 0.055), n = 180 | MD 0.30, 95% CI 0.14 to 0.46 | Bleecker 2012 (N = 609 participants, 515 completed study) compared VI/FF 100/25 mcg vs placebo in respect of health‐related quality of life and indicated a significant advantage for VI/FF 100/25 mcg | Moderatea |
| Asthma exacerbation | Not estimable | Only 2 studies (Allen 2013 and Kempsford 2012a) compared VI/FF 100/25 mcg vs placebo in respect of exacerbations; both studies reported no exacerbations in either treatment arm | Very lowb | ||
| Serious adverse events | Not estimable | Five trials (Allen 2013; Bleecker 2012; Kempsford 2012a; Oliver 2012; Oliver 2013) made this same comparison in relation to serious adverse events; all 5 reported no serious adverse events in VI/FF100/25 mcg or placebo arms | Very lowb | ||
| FEV1 | 0.196 L n = 193 | 0.368 L (SE 0.0304), n = 200 | MD 0.17 L, 95% CI 0.09 to 0.26 | Significant difference in favour of VI/FF 100/25 mcg vs placebo with respect to mean change in trough FEV1 (pre‐bronchodilator and pre‐dose) from baseline to week 12 in 1 trial (Bleecker 2012) (N = 609 participants, 515 completed study) (MD 0.17 L, 95% CI 0.09 to 0.26), and a similar effect was found in a small cross‐over trial (Kempsford 2012a) over a 2‐week period in the morning (MD 0.377 L, 90% CI 0.293 to 0.462) and in the evening (MD 0.422 L, 90% CI 0.337 to 0.507) | Moderatec |
| Peak expiratory flow | ‐0.4 L/min (SE 2.42), | 32.9 L/min (SE 2.42), n = 201 | MD 33.30 L/min, 95% CI 26.59 to 40.01 | Bleecker 2012 (N = 609 participants, 515 completed study) compared VI/FF 100/25 mcg vs placebo as mean change from baseline in daily morning (AM) PEF averaged over 12‐week treatment period; researchers noted a significant difference in favour of VI/FF 100/25 mcg (MD 33.30 L/min, 95% CI 26.59 to 40.01). The same trial showed a similar advantage in favour of VI/FF 100/25 mcg vs placebo in the evening over this period (28.20 L/min, 95% CI 21.67 to 34.73). A small cross‐over trial (Kempsford 2012a) produced a similar effect in favour of VI/FF 100/25 mcg vs placebo over a 2‐week period in the morning (MD 44.0 L/min, 90% CI 31.2 to 56.9) and in the evening (MD 69.0 L/min, 90% CI 55.9 to 82.1) | Moderatec |
| Asthma symptoms | 14.6 (SE 2.15), n = 202 | 32.5 (SE 2.14), n = 201 | MD 17.90, 95% CI 11.95 to 23.85 | Only 1 trial (Bleecker 2012) (N = 609 participants, 515 completed study) made VI/FF vs placebo comparison with respect to asthma symptoms, indicating a clear advantage for VI/FF 100/25 mcg | Moderatea |
| Adverse events | Not estimable | Several trials reported a range of adverse events for which overall aggregation was not possible. These are tabulated in Table 8 | Moderated | ||
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) | |||||
| GRADE Working Group grades of evidence | |||||
| aPoint deducted to reflect that these data were derived from only one trial bInvestigators reported no events in either arm of these trials cPoint deducted to reflect that data contributing to the main result (MD 0.17 L, 95% CI 0.09 to 0.26) were obtained from only one trial dPoint deducted, as we were unable to combine data on this outcome; results are presented in a separate table | |||||
| Study | FF/VI 100/25 mcg | FF 100 mcg | FF 200 mcg | FF/VI 200/25 mcg | VI 25 mcg | FP/SAL 250/50 mcg twice‐daily | FP 500 mcg | Prednisolone 10 mg | Placebo |
| 6 weeks' duration. On‐treatment AEs | 23/56 (41.00%) | ‐ | ‐ | 21/56 (38.00%) | ‐ | ‐ | ‐ | 5/15 (33.00%) | 16/58 (28.00%) |
| ≥24 to 78 weeks' duration. On‐treatment AEs | 636/1009 (63.00%) | 652/1010 (65.00%) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 12 weeks' duration | 54/346 (15.61%) | 67/347 (19.31%) | ‐ | 52/346 (15.03%) | ‐‐ | ‐ | ‐ | ‐ | |
| 12 weeks' duration | 29/201 (14.43%) | 20/205 (9.76%) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 22/203 (10.84%) |
| 52 weeks' duration. On‐treatment AEs | 139/201 (69.15%) | ‐ | ‐ | 134/202 (66.34%) | ‐ | ‐ | 73/100 (73.00%) | ‐ | ‐ |
| Cross‐over trial. 3 of 7 treatments (2 weeks) separated by 12 to 14‐day washout periods | 43/172 (25%) | 25/187 (13%) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 12 weeks' duration. Any AE | ‐ | ‐ | ‐ | 40/155 (26.00%) | ‐ | ‐ | 41/154 (27.00%) | ‐ | ‐ |
| Cross‐over trial. Each period lasted 14 days with a 14 to 21‐day washout period between periods | 11/24 (45.83%) AM 12/25 (48.00%) PM | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 8/23 (34.78%) |
| 24 weeks' duration | ‐ | ‐ | 66/194 (34.02%) | 62/197 (31.47%) | ‐ | ‐ | 73/195 (37.44%) | ‐ | ‐ |
| Cross‐over trial. 11 weeks (for a single period) | 4/25 (16.00%) | 1/25 (4.00%) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Cross‐over trial. 28 days for each period | 11/51 (21.57%) | 18/51 (35.29%) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 15/51 (29.41%) |
| Cross‐over trial. 21 days | 20/27 (74.07%) | 19/27 (70.37%) | ‐ | ‐ | 22/26 (84.62%) | ‐ | ‐ | ‐ | 19/27 (70.37%) |
| 24 weeks' duration | 110/403 (27.30%) | ‐ | ‐ | ‐ | ‐ | 106/403 (26.30%) | ‐ | ‐ | ‐ |
| Fractions shown in the table indicate the proportions of people who suffered one or more adverse events of any cause in each treatment arm AE: adverse event; F: fluticasone furoate; FP: fluticasone propionate; SAL: salmeterol; VI: vilanterol | |||||||||
| Study | Duration (weeks) | Severity at baseline | Inclusion criteria | Adverse events |
| 6 | Reversibility > 12%
FEV1 > 50% of predicted | Adults Comply with treatment
Clinical diagnosis of asthma for ≥ 12 weeks | Cortisol urinary excretion, serum AUC and trough | |
| 24 to 78 | Reversibility > 12%
FEV1 > 50% to 90% of predicted | Adults Using ICS
History of ≥ 1 exacerbation requiring hospitalisation or steroids in the past year | None | |
| 12 | Reversibility > 12% FEV1 50% to 80% of predicted | ICS for > 12 weeks before study
> 12 years of age | Yes, not clear | |
| 12 | Pre‐bronchodilator FEV1 40% to 90% of predicted normal Reversibility FEV1 ≥ 12% | ICS for 12 weeks before study
> 12 years of age | Details not stated | |
| 52 | Pre‐bronchodilator FEV1 40% to 90% of predicted normal Reversibility FEV1 ≥ 12%
| Adults Clinical diagnosis of asthma
ICS at high dose | Details not stated | |
| 4 | ACT suggesting poor control and FEV1 mean 70% (SD 11%) | Asthma ≥ 20 years of age | No, conference abstract only | |
| 4 | Pre‐bronchodilator FEV1 40% to 80% of predicted Demonstrated reversibility by ≥ 12% | Need for regular controller therapy for minimum of 8 weeks Stable dose of ICS for ≥ 4 weeks ≥ 18 years of age Diagnosis of asthma for ≥ 6 months
| No | |
| 12 | Reversibility of disease: demonstrated ≥ 12% and FEV1 40% to 90%
| ICS, with or without LABA, for ≥ 12 weeks Clinical diagnosis of asthma for 12 weeks
Adults | No | |
| 6 to 8 | Pre‐bronchodilator FEV1 ≥ 60% of predicted.
| 18 and 70 years of age inclusive Using an ICS, with or without a SABA, for ≥ 12 weeks before screening Participants who are current non‐smokers, who have not used inhaled tobacco products in the 12‐month period preceding screening visit Body weight ≥ 50 kg and BMI within the range 19.0 to 29.9 kg/m2 | Yes, details not stated | |
| 24 | Pre‐bronchodilator FEV1 40% to 90% of predicted Reversibility FEV1 ≥ 12%
| Current asthma therapy that includes an ICS for ≥ 12 weeks before first visit Adults | Cortisol, ECG, mouth swabs, various blood parameters | |
| 14 | Mild to moderate (GINA) | Stable asthma therapy (FP, total daily dose ≤ 400 mcg or equivalent) and SABA inhaler for ≥ 4 weeks before screening 5 to 12 years of age Clinical diagnosis of asthma 6 months before Controlled asthma (Childhood ACT > 19) | Not stated | |
| 8 | Pre‐bronchodilator FEV1 > 70% of predicted at screening
Methacholine challenge PC20 < 8 mg/mL at screening
| Adults Stable asthma therapy (FP, total daily dose ≤ 400 mcg or equivalent) and SABA inhaler for ≥ 4 weeks before screening BMI within the range 18.5 to 35.0 kg/m2
| Not stated | |
| 3 with 3 weeks' washout | Pre‐bronchodilator FEV1 > 70% of predicted at screening
Methacholine challenge PC20 < 8 mg/mL at screening
| Stable asthma therapy (FP, total daily dose ≤ 400 mcg or equivalent) and SABA inhaler for ≥ 4 weeks before screening BMI within the range 18.5 to 35.0 kg/m2 Adults | Not stated | |
| 24 | Reversibility ≥ 12% and 200 mL within 10 to 40 minutes following 2 to 4 inhalations of albuterol FEV1 40% to 85% predicted normal | Currently using ICS therapy Clinical diagnosis of asthma Adults | Not stated | |
| ACT: Asthma Control Test AUC: area under the curve BMI: body mass index ECG: electrocardiogram FEV1: forced expiratory volume in one second FP: fluticasone propionate GINA: Global Initiative for Asthma ICS: inhaled corticosteroid LABA: long‐acting beta2‐agonist PC20: provocative concentration of methacholine estimated to result in a 20% reduction in FEV1 SABA: short‐acting beta2‐agonist | ||||
| Study score (change from baseline) | FF/VI 100/25 mcg Mean (SE), N | FF 100 mcg Mean (SE), N | FF 200 mcg Mean (SE), N | FF/VI 200/25 mcg Mean (SE), N | FP/SAL 250/50 mcg twice‐daily | FP 500 mcg | Placebo | MD (95% CI) |
| AQLQ change from baseline at 12 weeks | 0.91 (0.055), n = 180 | 0.76 (0.055), n = 184 | ‐ | ‐ | ‐ | ‐ | 0.61 (0.061), n = 149 | 0.15 (0.00 to 0.30), 0.30 (0.14 to 0.46), 0.15 (‐0.01 to 0.31) |
| AQLQ change from baseline at 12 weeks | ‐ | ‐ | ‐ | 0.80 (0.069), n = 140 | ‐ | 0.69 (0.074), n = 123 | ‐ | 0.12 (‐0.08 to 0.32) |
| AQLQ change from baseline at 12 weeks | ‐ | ‐ | 0.66 (0.061), n = 154 | 0.74 (0.056), n = 180 | ‐ | 0.74 (0.059), n = 163 | ‐ | ‐0.08 (‐0.24 to 0.08), ‐0.08 (‐0.25 to 0.09), 0.00 (‐0.16 to 0.16) |
| AQLQ change from baseline at 24 weeks | ‐ | ‐ | 0.88 (0.071), n = 140 | 0.93 (0.065), n = 167 | ‐ | 0.90 (0.068), n = 156 | ‐ | ‐0.05 (‐0.24 to 0.14), ‐0.02 (‐0.21 to 0.17), 0.03 (‐0.15 to 0.21) |
| AQLQ change from baseline at 168 days | 0.46 (0.043), n = 342 | ‐ | ‐ | ‐ | 0.37 (0.043), n = 335 | ‐ | ‐ | 0.09 (‐0.03 to 0.21) |
| EQ‐5D change from baseline at 168 days | 5.5 (0.60), n = 343 | ‐ | ‐ | ‐ | 4.1 (0.60), n = 349 | ‐ | ‐ | 1.4 (‐0.3 to 3.0) |
| AQLQ: asthma quality of life questionnaire; CI: confidence interval; EQ‐5D: EuroQuality of Life‐5D questionnaire; FF: fluticasone furoate; FP: fluticasone propionate; MD: mean difference; SAL: salmeterol; SE: standard error; VI: vilanterol | ||||||||
| Study | FF/VI 100/25 mcg | FF 100 mcg | FF 200 mcg | FF/VI 200/25 mcg | FP/SAL 250/50 mcg twice‐daily | FP 500 mcg | Prednisolone 10 mg | Placebo |
| 6 weeks' duration | 0/56 (0.00%) | ‐ | ‐ | 0/56 (0.00%) | ‐ | ‐ | 0/15 (0.00%) | 0/58 (0.00%) |
| ≥ 24 to 78 weeks' duration Time to first severe exacerbation (HR 0.80, 95% CI 0.64 to 0.99). Annualised rate of severe exacerbation 25% reduction (95% CI 5% to 40%) | 154/1009 (15.26%) | 186/1010 (18.42%) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 52 weeks' duration | 3/201 (1.49%) | ‐ | ‐ | 6/202 (2.97%) | ‐ | 3/100 (3.00%) | ‐ | ‐ |
| 12 weeks' duration | ‐ | ‐ | ‐ | 1/155 (0.65%) | ‐ | 3/154 (1.95%) | ‐ | ‐ |
| Cross‐over trial. Each period lasted 14 days with a 14 to 21‐day washout period between periods | 0/24 (0.00%) AM 0/25 (0.00%) PM | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/23 (0.00%) |
| 24 weeks' duration | 1/403 (0.25%) | ‐ | ‐ | ‐ | 2/403 (0.50%) | ‐ | ‐ | ‐ |
| aOne participant in the FF/VI 100/25 mcg group experienced a severe asthma exacerbation concurrent with sinusitis and was withdrawn owing to lack of efficacy. The participant did not require hospitalisation, and the event, which was not classified as an AE, resolved following treatment with prednisone bThe incidence of asthma exacerbations was low, and no difference was noted between groups (3% vs 2% on FP/SAL vs FF/VI, respectively (on‐treatment events)). Eight (2%) participants in the FF/VI group and seven (2%) in the FP/SAL group withdrew because of exacerbation. One patient in the FF/VI group and two in the FP/SAL group were hospitalised because of exacerbation AM: morning; CI: confidence interval; FF: fluticasone furoate; FP: fluticasone propionate; HR: hazard ratio; PM: afternoon; SAL: salmeterol; VI: vilanterol | ||||||||
| Study | FF/VI 100/25 mcg | FF 100 mcg | FF 200 mcg | FF/VI 200/25 mcg | VI 25 mcg | FP/SAL 250/50 mcg twice‐daily | FP 500 mcg | Prednisolone 10 mg | Placebo |
| 6 weeks' duration. Post‐treatment period SAEs | 0/56 (0.00%) | ‐ | ‐ | 0/56 (0.00%) | ‐ | ‐ | ‐ | 0/15 (0.00%) | 0/58 (0.00%) |
| ≥ 24 to 78 weeks' duration. On‐treatment SAEs | 41/1009 (4.06%) | 29/1010 (2.87%) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 12 weeks' duration | 4/346 (1.16%) | 3/347 (0.86%) | ‐ | 1/346 (0.29%) | ‐ | ‐ | ‐ | ‐ | ‐ |
| 12 weeks' duration | 0/201 (0.00%) | 1/205 (0.49%) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/203 (0.00%) |
| 52 weeks' duration. On‐treatment SAEs | 3/201 (1.49%) | ‐ | ‐ | 1/202 (0.50%) | ‐ | ‐ | 7/100 (7.00%) | ‐ | ‐ |
| Cross‐over trial. Three of 7 treatments (2 weeks) separated by 12 to 14‐day washout periods | 1/172 (0.006%) | 0/187 (0%) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 12 weeks' duration | ‐ | ‐ | ‐ | 1/155 (0.65%) | ‐ | ‐ | 2/154 (1.30%) | ‐ | ‐ |
| Cross‐over trial. Each period lasted 14 days with a 14 to 21‐day washout period | 0/24 (0.00%) AM. 0/25 (0.00%) PM. | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/23 (0.00%) |
| 24 weeks' duration | ‐ | ‐ | 1/194 (0.52%) | 6/197 (3.05%) | ‐ | ‐ | 2/195 (1.03%) | ‐ | ‐ |
| Cross‐over trial. 11 weeks (for a single period) | 0/25 (0.00%) | 0/25 (0.00%) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Cross‐over trial. 28 days for each period | 0/51 (0.00%) | 0/51 (0.00%) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 0/51 (0.00%) |
| Cross‐over trial. 21 days | 0/27 (0.00%) | 0/27 (0.00%) | ‐ | ‐ | 0/26 (0.00%) | ‐ | ‐ | ‐ | 0/27 (0.00%) |
| 24 weeks' duration | 4/403 (0.99%) | ‐ | ‐ | ‐ | ‐ | 5/403 (1.24%) | ‐ | ‐ | ‐ |
| aThe main paper reports that 1 of the 52 withdrew during the study owing to an SAE, which occurred 4 days after the last dose in the FF 100 treatment period. This participant was provisionally diagnosed with moderate (grade 2) Still’s disease. Six weeks later, the participant was hospitalised. A diagnosis of histiocytic necrotising lymphadenitis (Kikuchi’s disease) was made on the basis of histology of an excised lymph node. Tapered prednisolone treatment, initiated at 60 mg per day, has been successful FF: fluticasone furoate; FP: fluticasone propionate; SAE: serious adverse event; SAL: salmeterol; VI: vilanterol | |||||||||
| Study measure time point/ duration | FF/VI 100/25 mcg Mean (SE), N, of MD (95% CI) | FF 100 mcg Mean (SE), N | FF 200 mcg Mean (SE), N | FF/VI 200/25 mcg Mean (SE), N | VI 25 mcg Mean (SE), N | FP/SAL250/50 mcg twice‐daily Mean (SE), N | FP 500 mcg Mean (SE), N | Placebo Mean (SE), N | MD (95% CI, unless otherwise stated) |
| Trough FEV1 At 0 to 12 weeks Change in baseline trough FEV1 from baseline to week 12 | 0.441 L (0.022) | 0.365 L (0.022) | ‐ | 0.457 L (0.022) | ‐ | ‐ | ‐‐ | ‐ | ‐ |
| Trough FEV1 At 0 to 12 weeks Mean change in trough FEV1 (pre‐bronchodilator and pre‐dose) from baseline to week 12 | 0.368 L (0.0304), n = 200 | 0.332 L (0.0302), n = 203 | ‐ | ‐ | ‐ | ‐ | ‐ | 0.196 L (0.0310), n = 193 | 0.04 L (‐0.05 to 0.12) 0.17 L (0.09 to 0.26) 0.14 L (0.05 to 0.22) |
| Trough FEV1 combining all treatment periods At 0 to 2 weeks 3 of 7 treatments (2 weeks) separated by 12 to 14‐day washout periods | 0.200 L, n = 158 | 0.087 L, n = 158 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| 12 weeks' duration | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | Adjusted treatment difference 0.108 L (0.040 to 0.176) |
| Weighted mean FEV1 over the day At day 14 Weighted mean FEV1, over 0 to 24 hours post dose at day 14 Cross‐over trial. Each period lasted 14 days with a 14 to 21‐day washout period | AM dose: 3.188 L PM dose: 3.233 L | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 2.811 L n = 20 | AM vs placebo 0.377 L (90% CI 0.293 to 0.462) PM vs placebo 0.422 L (90% CI 0.337 to 0.507) AM vs PM ‐0.44 L (90% CI ‐0.125 to 0.36) |
| Day 14 pre‐treatment (trough) AM FEV1 At day 14 | AM dose: 3.191 L PM dose: 3.285 L n = 25 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 2.788 L n = 22 | AM vs placebo 0.403 L (90% CI 0.272 to 0.533) PM vs placebo 0.496 L (90% CI 0.369 to 0.624) AM vs PM ‐0.094 L (90% CI ‐0.221 to 0.034) |
| Day 14 pre‐treatment (trough) PM FEV1 At day 14 | AM dose: 3.153 L PM dose: 3.188 L | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | 2.879 L n = 23 | AM vs placebo 0.275 L (90% CI 0.169 to 0.380) PM vs placebo 0.309 L (90% CI 0.205 to 0.413) AM vs PM ‐0.034 (90% CI ‐0.138 to 0.070) |
| Change in baseline trough FEV1 At 24 weeks Change from baseline in clinic visit trough (pre‐bronchodilator and pre‐dose) FEV1 at end of 24‐week treatment period | ‐ | ‐ | 0.201 L (0.0303), n = 186 | 0.394 L (0.0302), n = 187 | ‐ | ‐ | 0.183 L (0.0300), n = 190 | ‐ | ‐0.19 L (‐0.28 to ‐0.11) 0.02 L (‐0.06 to 0.10) 0.21 L (0.13 to 0.29) |
| Change from baseline in weighted mean serial FEV1 over 24 hours At 24 weeks Change from baseline in weighted mean serial FEV1 over 0 to 24 hours post dose at week 24 | ‐ | ‐ | 0.328 L (0.0493), n = 83 | 0.464 L (0.0470), n = 89 | ‐ | ‐ | 0.258 L (0.0483), n = 86 | ‐ | ‐0.14 L (‐0.27 to ‐0.00) 0.07 L (‐0.07 to 0.21) 0.21 L (0.07, 0.34) |
| 23 hours post challenge At day 29 Cross‐over trial ‐ 28 days for each period Weighted mean change from baseline in FEV1 between 0 and 2 hours following 22 to 23‐hour post‐treatment allergen challenge at day 29 of each treatment period | ‐0.227 L (0.0550), n = 46 | ‐0.210 L (0.0549), n = 49 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐0.372 L (0.0557), n = 45 | FF vs placebo 0.162 L (0.087 to 0.237) FF/VI vs placebo 0.145 L (0.069 to 0.222) FF/VI vs FF ‐0.017 L (‐0.091 to 0.057) |
| Decrease from baseline 23 hours post challenge At day 29 Maximum % decrease from baseline FEV1 between 0 and 2 hours following 22 to 23‐hour post‐treatment allergen challenge at day 29 of each treatment period (time frame: baseline and at day 29 of each treatment period (up to study day 197)) | ‐13.206% (2.0491), n = 46 | ‐14.040% (2.0435), n = 49 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐24.991% (2.0736), n = 45 | FF vs placebo 10.951% (8.053 to 13.848) (8.849 to 14.721) |
| Change from baseline FEV1 23 hours post challenge Minimum FEV1 absolute change from baseline between 0 and 2 hours following 22 to 23‐hour post‐treatment allergen challenge at day 29 of each treatment period | ‐0.478 L (0.0767), n = 46 | ‐0.479 L (0.0765), n = 49 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐0.809 L (0.0775), n = 45 | FF vs placebo 0.330 L (0.232 to 0.429) |
| Change from baseline 4 to 10 hours post challenge At day 21 Cross‐over trial ‐ 21 days LAR: absolute change from baseline in minimum FEV1 between 4 and 10 hours following 1‐hour post‐treatment allergen challenge at day 21 of each treatment period | ‐0.216 L n = 26 | ‐0.188 L n = 27 | ‐ | ‐ | ‐0.536 L n = 22 | ‐ | ‐ | ‐0.731 L n = 20 | ‐ |
| Change from baseline 4 to 10 hours post challenge At day 21 LAR: absolute change from baseline in weighted mean FEV1 between 4 and 10 hours following 1‐hour post‐treatment allergen challenge at day 21 of each treatment period | 0.018 L | 0.018 L n = 27 | ‐ | ‐ | ‐0.298 L n = 22 | ‐ | ‐ | ‐0.466 L n = 20 | ‐ |
| Change from baseline trough FEV1 At day 168 24 weeks' duration | 0.281 L (0.0191), n = 397 | ‐ | ‐ | ‐ | ‐ | 0.300 L (0.0193), n = 389 | ‐ | ‐ | ‐0.019 L (‐0.073 to 0.034) |
| AM: morning; CI: confidence interval; FEV1: forced expiratory volume in one second; FF: fluticasone furoate; FP: fluticasone propionate; h: hour; LAR: late asthmatic response; MD: mean difference; PM: afternoon; SAL: salmeterol; SE: standard error; VI: vilanterol | |||||||||
| Study | Duration (weeks) | Measure of PEF | FF/VI 100/25 mcg Mean (SD, unless otherwise stated), N | FF 100 mcg Mean (SD, unless otherwise stated), N | FF 200 mcg Mean (SE), N | FF/VI 200/25 mcg Mean (SE ), N | FP 500 mcg Mean (SE), N | Placebo Mean (SE, unless otherwise stated), N | MD (95% CI, unless otherwise stated) |
| 12 | Change from baseline, AM Change from baseline in AM PEF Averaged over 12‐week treatment period | 44.3 L/min (2.25) | 19.1 L/min (2.25) | ‐ | 47.7 L/min (2.25) | ‐ | 25.20 L/min (18.96 to 31.44), 100/25 vs 100 FF | ||
| 12 | Change from baseline, PM Change from baseline in AM PEF Averaged over 12‐week treatment period | 39.7 L/min (2.24) | 15.5 L/min (2.24) | ‐ | 41.7 L/min (2.24) | ‐ | 24.20 L/min (17.99 to 30.41), 100/25 vs 100 FF | ||
| 12 | Change from baseline, PM Mean change from baseline in daily PM PEF averaged over 12‐week treatment period | 26.4 L/min (SE 2.35), n = 201 | 14.1 L/min (SE 2.34), n = 204 | ‐ | ‐ | ‐ | ‐1.8 L/min (2.36), n = 202 | 12.30 L/min (5.80 to 18.80), 28.20 L/min (21.67 to 34.73), 15.90 L/min (9.39 to 22.41) | |
| 4 | Change from baseline, AM Only 1 (FF/VI) condition reported. Trial reported as conference abstract with limited information | ||||||||
| Baseline to day 15 | Least squares mean change calculated from baseline to day 15 Least squares mean change in last 7 days, mean PEF | 24.1 (2.46) AM 21.4 (2.58) PM n = 172 | ‐2.9 (2.44) AM ‐5.2 (2.51) PM n = 187 | ‐ | ‐ | ‐ | ‐ | ‐ | |
| 12 | 12 weeks' duration. | ‐ | ‐ | ‐ | 39.1 L/min (3.01), n = 155 | 10.5 L/min (3.03), n = 154 | ‐ | Adjusted treatment difference 28.5 L/min (20.1 to 36.9) | |
| 12 days | Pre‐treatment Pre‐treatment PEF at days 1 to 12 Cross‐over trial. Each period lasted 14 days with a 14 to 21‐day washout period | AM dose: 510.4 L/min PM dose: 535.3 L/min | ‐ | ‐ | ‐ | ‐ | 466.3 L/min | AM vs placebo 44.0 L/min (90% CI 31.2 to 56.9) PM vs placebo 69.0 L/min (90% CI 55.9 to 82.1) AM vs PM ‐25.0 L/min (90% CI ‐37.9 to ‐12.0) | |
| 12 days | Pre‐treatment Pre‐treatment PEF (PM) at days 1 to 12 | AM dose: 517.6 L/min PM dose: 521.4 L/min | ‐ | ‐ | ‐ | ‐ | 453.2 L/min | AM vs placebo 64.4 L/min (90% CI 52.9 to 76.0) PM vs placebo 68.2 L/min (90% CI 56.5 to 79.8) AM vs PM ‐3.7 L/min (90% CI ‐15.2 to 7.7) | |
| 24 | Change from baseline, AM 4 weeks Mean change from baseline in daily trough (AM) PEF averaged over 24‐week treatment period | ‐ | ‐ | 18.2 L/min (2.97), n = 193 | 51.8L/min (2.94), n = 197 | 18.8L/min (2.95), n = 195 | ‐ | ‐33.60 L/min (‐41.79 to, ‐25.41), ‐0.60 L/min (‐8.80 to 7.60), 33.00 L/min (24.84 to 41.16) | |
| 24 | Change from baseline, PM Mean change from baseline in daily trough (PM) PEF averaged over 24‐week treatment period | ‐ | ‐ | 9.1 L/min (2.98), n = 192 | 39.8 L/min (2.93), n = 197 | 13.6 L/min (2.96), n = 194 | ‐ | ‐30.70 L/min (‐38.89 to ‐22.51), ‐4.50 L/min (‐12.73 to 3.73), 26.20 L/min (18.04 to 34.36) | |
| AM: morning; CI: confidence interval; FF: fluticasone furoate; PEF: peak expiratory flow; PM: evening; SD: standard deviation; SE: standard error; VI: vilanterol | |||||||||
| Study | Measure | FF/VI 100/25 mcg Mean (SE) | FF 100 mcg Mean (SE) | FF 200 mcg Mean (SE) | FF/VI 200/25 mcg Mean (SE) | FP/SAL250/50 mcg twice‐daily Mean (SE) | FP 500 mcg Mean (SE) | Placebo Mean (SE) | MD (95% CI) |
| ≥ 24 to 78 weeks' duration Responder analysis results: ORs for FF/VI vs FF at week 12 (1.49, 95% CI 1.20 to 1.84), week 36 (1.49, 95% CI 1.21 to 1.83) and at endpoint (1.50, 95% CI 1.23 to 1.82) | ACQ7 mean difference and responder analysis | NR | NR | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Change from baseline in percentage of symptom‐free 24‐hour periods during 12‐week treatment | Change from baseline % symptom‐free days | 27.2 (1.74) n = 345 | 19.4 (1.74) n = 346 | ‐ | 29.0 (1.74) n = 346 | ‐ | ‐ | ‐‐ | |
| Change from baseline in % of symptom‐free 24‐hour periods during 12‐week treatment period | Change from baseline % symptom‐free days | 32.5 (2.14), n = 201 | 20.4 (2.13), n = 204 | ‐ | ‐ | ‐ | ‐ | 14.6 (2.15), n = 202 | 12.10 (6.18 to 18.02), 17.90 (11.95 to 23.85), 5.80 (‐0.13 to 11.73) |
| Trial reported as conference abstract with limited information | Change from baseline ACT score | ||||||||
| LS mean change in symptom‐free days during 2‐week treatment period | LS mean change in symptom‐free days (SE) | 7.3 (1.67) n = 172 | 5.8 (1.64) n = 187 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| % of symptom‐free 24‐hour periods, weeks 1 to 12 | % symptom‐free days | ‐ | ‐ | ‐ | 25.4 (2.74), n = 155 | ‐ | 20.6 (2.77), n = 152 | ‐ | 4.9 (‐2.8 to 12.5) |
| Change from baseline in ACT scores at week 12 | Change from baseline ACT score | ‐ | ‐ | 3.9 (0.29), n = 164 | 4.8 (0.27), n = 183 | ‐ | 3.9 (0.28), n = 169 | ‐ | ‐0.90 (‐1.68 to ‐0.12), 0.00 (‐0.79 to 0.79), 0.90 (0.14 to 1.66) |
| Change from baseline in ACT scores at week 24 | Change from baseline ACT score | ‐ | ‐ | 5.2 (0.30), n = 147 | 5.5 (0.28), n = 170 | ‐ | 4.7 (0.29), n = 162 | ‐ | ‐0.30 (‐1.10 to 0.50), 0.50 (‐0.32 to 1.32), 0.80 (0.01 to 1.59) |
| Change from baseline in ACT scores at day 168 and at 24 weeks | Change from baseline ACT score | 2.3 (0.16), n = 354 | ‐ | ‐ | ‐ | 2.0 (0.16), n = 348 | ‐ | ‐ | 0.2 (‐0.2 to 0.7) |
| ACT: asthma control test; CI: confidence interval; FF: fluticasone furoate; FP: fluticasone propionate; LS: least squares; MD: mean difference; NR: not reported; OR: odds ratio; SAL: salmeterol; SE: standard error; VI: vilanterol | |||||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Change in quality of life (measured by AQLQ at 12 wk) Show forest plot | 1 | 329 | Mean Difference (Fixed, 95% CI) | 0.3 [0.14, 0.46] |
| 2 Exacerbations Show forest plot | 2 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Serious adverse events Show forest plot | 5 | 721 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4 FEV1 Litres Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.17 [0.09, 0.26] | |
| 5 PEFR AM L/min (change from baseline at 12 wk) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 33.3 [26.59, 40.01] | |
| 6 PEFR PM L/min (change from baseline at 12 wk) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 28.2 [21.67, 34.73] | |
| 7 Change in asthma symptoms (measured by ACT) Show forest plot | 1 | 339 | Mean Difference (Fixed, 95% CI) | 1.9 [1.22, 2.58] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Change in quality of life (measured by AQLQ at 12 wk) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.15 [‐0.00, 0.30] | |
| 2 Exacerbations Show forest plot | 2 | 2425 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.38 [0.86, 2.22] |
| 3 Serious adverse events Show forest plot | 5 | 1258 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.61 [0.42, 6.17] |
| 4 Trough FEV1 (L) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.08 [0.02, 0.14] | |
| 5 PEFR AM (change from baseline at 12 wk) Show forest plot | 2 | Mean Difference (Fixed, 95% CI) | 20.29 [15.72, 24.85] | |
| 6 PEFR PM (change from baseline at 12 wk) Show forest plot | 2 | Mean Difference (Fixed, 95% CI) | 18.52 [14.03, 23.01] | |
| 7 Change in asthma symptoms (measured by ACT) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.6 [‐0.04, 1.24] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Serious adverse events Show forest plot | 1 | 53 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Exacerbations Show forest plot | 1 | 301 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.10, 2.47] |
| 2 Serious adverse events Show forest plot | 1 | 301 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.05, 0.80] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Change in quality of life (measured by AQLQ at 24 wk) Show forest plot | 1 | 677 | Mean Difference (Fixed, 95% CI) | 0.09 [‐0.03, 0.21] |
| 2 Exacerbations Show forest plot | 1 | 806 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.50 [0.05, 5.52] |
| 3 Serious adverse events Show forest plot | 1 | 806 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.21, 2.99] |
| 4 FEV1 Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | ‐0.02 [‐0.07, 0.03] | |
| 5 Change in asthma symptoms (measured by ACT) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.24 [‐0.20, 0.68] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Exacerbations Show forest plot | 2 | 515 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.02 [0.50, 8.19] |
| 2 Serious adverse events Show forest plot | 2 | 515 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.03, 3.18] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Exacerbations Show forest plot | 1 | 114 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Serious adverse events Show forest plot | 1 | 114 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3 FEV1 Litres Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.21 [0.13, 0.29] | |
| 4 Change in asthma symptoms (measured by ACT) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.9 [0.12, 1.68] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Change in quality of life (measured by AQLQ at 12 wk) Show forest plot | 2 | 606 | Mean Difference (Fixed, 95% CI) | 0.05 [‐0.08, 0.17] |
| 2 Change in quality of life (measured by AQLQ at 24 wk) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.03 [‐0.15, 0.21] | |
| 3 OLD***Health‐related quality of life Show forest plot | 2 | 606 | Mean Difference (IV, Fixed, 95% CI) | 0.04 [‐0.08, 0.17] |
| 4 Exacerbations Show forest plot | 2 | 611 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.22, 2.20] |
| 5 Serious adverse events Show forest plot | 3 | 1003 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.25, 1.49] |
| 6 PEFR Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 28.6 [20.23, 36.97] | |
| 7 PEFR AM Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 33.0 [24.84, 41.16] | |
| 8 PEFR PM Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 26.2 [18.04, 34.36] | |
| 9 % symptom‐free days Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 4.8 [‐2.84, 12.44] | |
| 10 Change in asthma symptoms (measured by ACT) Show forest plot | 1 | 332 | Mean Difference (Fixed, 95% CI) | 0.8 [0.01, 1.59] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Change in quality of life (measured by AQLQ at 12 wk) Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.08 [‐0.08, 0.24] | |
| 2 Change in quality of life (measured by AQLQ at 24 wk) Show forest plot | 1 | 307 | Mean Difference (Fixed, 95% CI) | 0.05 [‐0.14, 0.24] |
| 3 Serious adverse events Show forest plot | 1 | 391 | Odds Ratio (M‐H, Fixed, 95% CI) | 6.06 [0.72, 50.84] |
| 4 FEV1 Litres Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 0.19 [0.10, 0.28] | |
| 5 PEFR AM Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 33.6 [25.41, 41.79] | |
| 6 PEFR PM Show forest plot | 1 | Mean Difference (Fixed, 95% CI) | 30.7 [22.51, 38.89] | |
| 7 Change in asthma symptoms (measured by ACT) Show forest plot | 1 | 317 | Mean Difference (Fixed, 95% CI) | 0.3 [‐0.50, 1.10] |