Tratamiento complementario con estiripentol para la epilepsia refractaria focal
Resumen
Antecedentes
Ésta es una versión actualizada de la revisión Cochrane publicada en 2015 (número 10). Las convulsiones no están controladas por los tratamientos actuales en casi el 30% de las personas con epilepsia. El estiripentol es un nuevo fármaco antiepiléptico (FAE) desarrollado en Francia y autorizado por la Agencia Europea de Medicamentos (EMA) en 2007 para el tratamiento del síndrome de Dravet como terapia complementaria con valproato y clobazam, con efectos prometedores.
Objetivos
Evaluar la eficacia y tolerabilidad del estiripentol como tratamiento complementario en pacientes con epilepsia refractaria focal que reciben FAE.
Métodos de búsqueda
Para la última actualización se hicieron búsquedas en las siguientes bases de datos el 21 de agosto de 2017: registro especializado de Cochrane Epilepsia (Cochrane Epilepsy Specialized Register), CENTRAL, MEDLINE, ClinicalTrials.gov, International Clinical Trials Registry Platform (ICTRP) de la OMS. Se estableció contacto con Biocodex (el fabricante del estiripentol) y con expertos en epilepsia para identificar ensayos publicados, no publicados y en curso.
Criterios de selección
Ensayos controlados aleatorizados de tratamientos complementarios con estiripentol en pacientes con epilepsia refractaria focal.
Obtención y análisis de los datos
Los autores de la revisión seleccionaron de forma independiente los ensayos para la inclusión y extrajeron los datos. Los resultados investigados incluyeron una reducción del 50% o más en la frecuencia de las crisis, la ausencia de crisis, los efectos adversos, el retiro del tratamiento y los cambios en la calidad de vida.
Resultados principales
En base a los criterios de selección, no se incluyó ningún estudio nuevo en la presente revisión. Solo se incluyó un estudio en la revisión anterior (32 niños con epilepsia focal). Este estudio adoptó un diseño "ajustado en función de la respuesta" y no encontró evidencia clara de reducción de la frecuencia de crisis (≥ 50% reducción de las crisis) (riesgo relativo [RR] 1,51, intervalo de Confianza (IC) del 95% 0,81 a 2,82, evidencia de calidad baja) ni de ausencia de crisis (RR 1,81, IC del 95%: 0,31 a 4,43, evidencia de calidad baja) al añadir estiripentol en comparación con el placebo. El estiripentol dio lugar a un mayor riesgo de efectos adversos considerados en conjunto (RR 2,65; IC del 95%: 1,08 a 6,47, evidencia de calidad baja) comparado con placebo. Cuando se consideraron los eventos adversos específicos, los intervalos de confianza fueron muy amplios y mostraron la posibilidad de aumentos sustanciales y pequeñas reducciones en los riesgos de efectos adversos neurológicos (RR 2,65; IC del 95%: 0,88 a 8,01, evidencia de baja calidad) o gastrointestinales (RR 11,56; IC del 95%: 0,71 a 189,36, evidencia de baja calidad). Los investigadores no observaron una reducción clara del riesgo de retirada del estudio (RR 0,66; IC del 95%: 0,30 a 1,47; evidencia de baja calidad), que fue alta en ambos grupos (35,0% en el grupo de placebo adicional y 53,3% en el grupo de estiripentol, evidencia de baja calidad). La validez externa de este estudio fue limitada porque sólo los que respondieron al estiripentol (es decir, los pacientes que experimentan una disminución ≥ 50% en la frecuencia de las crisis en comparación con al inicio) se incluyeron en la fase aleatoria, complementaria, controlada por placebo y doble ciego. Además, los efectos de arrastre y retirada probablemente influyeron en los resultados relacionados con la frecuencia de las crisis epilépticas. La información muy limitada derivada del único estudio incluido muestra que los efectos adversos considerados en su conjunto parecían producirse significativamente más a menudo con el estiripentol complementario que con el placebo complementar.
Conclusiones de los autores
No se encontraron estudios nuevos desde que la última versión de esta revisión se publicó. Por lo tanto, no se han realizado cambios en las conclusiones de esta actualización como se presentaron en la revisión inicial. No pueden sacarse conclusiones que apoyen el uso de estiripentol como tratamiento complementario para la epilepsia refractaria focal. Se necesitan ensayos aleatorizados bien realizados adicionales.
PICO
Resumen en términos sencillos
Estiripentol como tratamiento complementario para la epilepsia refractaria focal
Antecedentes
La epilepsia es uno de los trastornos crónicos neurológicos más frecuentes; afecta al 1% de población mundial. Una gran proporción de estas personas (hasta el 30%) sigue teniendo convulsiones a pesar de un tratamiento adecuado con fármacos antiepilépticos (FAE), utilizados de forma individual (como monoterapia) o en combinación (politerapia). A estas personas se las considera que tienen epilepsia refractaria. El estiripentol es un nuevo FAE desarrollado en Francia y autorizado en 2007 por la Agencia Europea de Medicamentos (EMA) para el tratamiento del síndrome de Dravet como terapia complementaria con valproato y clobazam, con efectos prometedores. Esta revisión evaluó la evidencia para el uso de estiripentol como tratamiento complementario para la epilepsia refractaria focal en individuos que reciben FAE.
Resultados
Sobre la base de los criterios de revisión, se incluyó sólo un estudio en la revisión (32 niños con epilepsia focal). Este estudio adoptó un diseño "ajustado en función de la respuesta" y no encontró pruebas claras de reducción de las convulsiones (≥ 50%) ni de libertad de convulsiones con el agregado de estiripentol en comparación con el placebo. El estiripentol dio lugar a un mayor riesgo de efectos adversos considerados en conjunto (riesgo relativo [RR] 2,65; intervalo de confianza [IC] del 95%: 1,08 a 6,47) comparado con placebo. La generalización de los resultados del estudio a una población más amplia está limitada por el hecho de que sólo los que respondieron al stiripentol (es decir, los pacientes que experimentan una disminución en la frecuencia de las crisis de al menos el 50% en comparación con el inicio) se incluyeron en la porción aleatoria, complementaria, controlada por placebo y doble ciego del estudio. Además, el tamaño muy pequeño de la muestra con la correspondiente alta tasa de abandono impide la generalización de los resultados del estudio. Por último, debido al diseño adoptado, los efectos de arrastre y retirada probablemente influyeron en los resultados relacionados con la frecuencia de las crisis epilépticas.
Calidad de la evidencia
Se consideró que los ensayos incluidos estaban en riesgo de sesgo bajo o poco claro. La calidad de la evidencia se calificó mediante el método GRADE.
Actualmente no hay evidencia que apoye el uso de estiripentol como tratamiento complementario para la epilepsia refractaria focal. Se necesitan más ensayos aleatorizados grandes, bien realizados sobre este tema.
La evidencia está actualizada hasta agosto de 2017.
Authors' conclusions
Summary of findings
| Stiripentol compared with placebo for focal refractory epilepsy | ||||||
| Patient or population: people with focal refractory epilepsy Settings: community Intervention: stiripentol Comparison: placebo | ||||||
| Outcomes* | Illustrative comparative risks** (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Stiripentol | |||||
| ≥ 50% seizure reduction | 467 per 1000 | 705 per 1000 | RR 1.51 (0.81 to 2.82) | 32 | ⊕⊕⊖⊖ lowa,b | |
| Seizure freedom | 200 per 1000 | 236 per 1000 | RR 1.18 (0.31 to 4.43) | 32 | ⊕⊕⊖⊖ lowa,b | |
| ≥ 1 adverse effect | 267 per 1000 | 707 per 1000 | RR 2.65 (1.08 to 6.47) | 32 | ⊕⊕⊖⊖ | |
| Neurological adverse effects | 200 per 1000 | 530 per 1000 | RR 2.65 (0.88 to 8.01) | 32 | ⊕⊕⊖⊖ | |
| Gastrointestinal adverse effects | 0 events occurred in the placebo group | 0 events occurred in the stiripentol group | RR 11.56 (0.71 to 189.36) | 32 | ⊕⊕⊖⊖ | |
| Dropouts | 533 per 1000 | 352 per 1000 | RR 0.66 (0.30 to 1.47) | 32 | ⊕⊕⊖⊖ | |
| * Quality of life was not assessed in this study. **The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) and is calculated according to the following formula: corresponding intervention risk, per 1000 = 1000 X ACR X RR. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded once for risk of bias and once for imprecision (small sample size which is made even smaller with dropouts). | ||||||
Background
This is an updated version of the Cochrane Review last published in 2015 (Issue 10) (Brigo 2015).
Description of the condition
Epilepsy is one of the more common chronic neurological disorders; it affects 1% of the population worldwide.
A large proportion of these people (up to 30%) continue to have seizures despite adequate therapy with antiepileptic drugs (AEDs), used singularly or in combination (Cockerell 1995; Granata 2009). These individuals are regarded as having refractory epilepsy. Although there is no universal definition of refractory epilepsy, most definitions refer to continued seizures despite AED treatment, and the definition most often used encompasses continued seizures despite interminable medication changes (French 2006).
Various criteria have been used to define refractory epilepsy. In 2010, an internationally accepted definition of refractory epilepsy was proposed by a Task Force of the International League Against Epilepsy (ILAE) as "failure of adequate trials of two tolerated, appropriately chosen and used AED schedules (whether given as monotherapy or in combination) to achieve sustained seizure freedom" (Kwan 2010). Standard drugs (e.g. carbamazepine, phenytoin, valproate) do not control all patients’ seizures. However, over the past 15 to 20 years, numerous newly available AEDs have offered promise for the treatment of refractory epilepsy.
Seizures may occur within and may rapidly engage bilaterally distributed networks (generalised seizures) or networks limited to one hemisphere and are discretely localised or more widely distributed (focal seizures) (Berg 2010).
In this review, we aimed to investigate the efficacy and tolerability of add‐on stiripentol in people with focal refractory epilepsy.
Description of the intervention
Stiripentol is a new AED that was developed in France and was approved in 2007 by the European Medicines Agency (EMA) for the treatment of Dravet syndrome as adjunctive therapy with valproate and clobazam, with promising effects (Chiron 2007).
The safety profile of stiripentol is good, with most adverse events related to a significant increase in plasma concentrations of valproate and clobazam after the addition of stiripentol (Perez 1999). Adverse events include drowsiness, ataxia, nausea, abdominal pain and loss of appetite with weight loss. Asymptomatic neutropenia is occasionally observed (Chiron 2007).
How the intervention might work
Stiripentol is structurally unrelated to any other marketed AED. A gamma‐aminobutyric acid (GABA)ergic effect of stiripentol, which has been demonstrated in vitro (Quilichini 2006), is probably due to allosteric modulation of the GABA‐A receptor (Fisher 2009). The efficacy of stiripentol could therefore be related to potentiation of GABAergic inhibitory neurotransmission (Quilichini 2006), and enhancement of the action of benzodiazepines (Fisher 2009). In humans, stiripentol also inhibits cytochrome P450 enzymes (CYP) in the liver, resulting in increased plasma concentrations of concomitant AEDs metabolised by CYP (Chiron 2005). In patients affected by severe myoclonic epilepsy in infancy (SMEI), such a pharmacokinetic interaction particularly applies to clobazam (Giraud 2006).
Why it is important to do this review
To date, no studies have systematically reviewed the literature on the role of stiripentol as treatment for focal refractory epilepsy; thus its use in conditions other than SMEI remains to be evaluated.
In this systematic review, we aimed to assess and summarise existing evidence regarding the efficacy and adverse effects of stiripentol as add‐on treatment for people with focal refractory epilepsy.
Objectives
To evaluate the efficacy and tolerability of stiripentol as add‐on treatment for people with focal refractory epilepsy who are taking AEDs.
Methods
Criteria for considering studies for this review
Types of studies
We included studies that met the following criteria.
-
Randomised controlled trials (RCTs)
-
Double‐blind, single‐blind or unblinded trials
We decided to include only the above types of studies, as they are considered to provide the most effective means of evaluating benefits and risks of treatment (Strauss 2005).
We excluded all other study designs, including cohort studies, cross‐over studies, case‐control studies, outcomes research, case studies, case series and expert opinions.
We analysed different treatment groups and controls separately.
We applied no language restrictions.
Types of participants
We considered people with focal epilepsy defined according to ILAE criteria (International League Against Epilepsy 1989). We considered participants regardless of age, sex and ethnicity, including children with disabilities. As no definition of refractory epilepsy has been universally accepted, for the purposes of this review, we included all trials conducted to assess stiripentol in refractory epilepsy, however it was defined, but we noted which definition was used. If possible, on the basis of rough data, we considered individuals to be affected by refractory epilepsy as defined by Kwan 2010. We excluded those affected by SMEI, as another systematic review of ours (Antiepileptic drugs for the treatment of severe myoclonic epilepsy in infancy (Brigo 2013)), specifically assesses the role of stiripentol in such a genetically determined disease.
Types of interventions
-
Active treatment group received stiripentol, in addition to conventional AED treatment
-
Control group received no treatment, and matching add‐on placebo or another AED was used as a comparator.
Types of outcome measures
For each outcome, we performed an intention‐to‐treat primary analysis to include all participants in the treatment group to which they were allocated, irrespective of the treatment they actually received.
Primary outcomes
-
Fifty per cent or greater reduction in seizure frequency: proportion of participants with at least a 50% reduction in seizure frequency at the end of the study compared with the pre‐randomisation baseline period
-
Seizure freedom: proportion of participants achieving total cessation of seizures. We used the most current ILAE‐proposed definition of seizure freedom: no seizures of any type for 12 months, or three times the longest (pre‐intervention) seizure‐free interval, whichever is longest (Kwan 2010).
Secondary outcomes
-
Adverse effects
-
Proportion of participants who experienced at least one adverse effect
-
Proportion of participants who experienced individual adverse effects (to be listed separately)
-
-
Proportion of dropouts or withdrawals due to adverse effects, lack of efficacy or other reasons
-
Improvement in quality of life as assessed by validated and reliable rating scales (e.g. Quality of Life In Epilepsy (QOLIE‐31))
Search methods for identification of studies
Electronic searches
Searches were run for the original review in May 2012. Subsequent searches were run in August 2013 and August 2015. For the latest update, we searched the following databases on 21 August 2017.
-
Cochrane Epilepsy Specialized Register, using the search strategy set out in Appendix 1
-
Cochrane Central Register of Controlled Trials (CENTRAL; 2017, issue 8) via the Cochrane Register of Studies Online (CRSO), using the search strategy set out in Appendix 2
-
MEDLINE (Ovid, 1946‐21 August 2017), using the search strategy set out in Appendix 3
-
ClinicalTrials.gov, using the search strategy: stiripentol OR diacomit | Epilepsy
-
WHO International Clinical Trials Registry Platform (ICTRP) using the search strategy: stiripentol AND epilepsy OR diacomit AND epilepsy
We no longer search Embase, as randomised and quasi‐randomised controlled trials in Embase are now included in CENTRAL.
We imposed no language restrictions.
Searching other resources
We contacted the manufacturers of stiripentol (Biocodex) (contacted by email on 31 May 2012, on 13 August 2015 and on 22 August 2017) and experts in the field (contacted by email on 31 May 2012, on 13 August 2015 and on 22 August 2017) for information about unpublished or ongoing studies. We reviewed the reference lists of retrieved studies to search for additional reports of relevant studies. We also considered conference proceedings of the ILAE.
Data collection and analysis
We did not implement intended methods for assessing heterogeneity, reporting biases, synthesising data and performing subgroup and sensitivity analyses found in the protocol of this systematic review because of the low number of studies (Brigo 2012). In case future review updates identify more than one study, we may conduct data analyses referring to methods reported in the previously published protocol of the present systematic review (Brigo 2012).
Selection of studies
Two review authors (FB and SCI) independently screened titles and abstracts of all publications identified by the searches to assess their eligibility. At this stage, we excluded publications that did not meet inclusion criteria. After screening, we assessed the full‐text articles of potentially eligible citations for inclusion. We reached consensus on selection of trials and on the final list of studies. We discussed and resolved disagreements.
Data extraction and management
Two review authors (FB and SCI) independently extracted the following characteristics of each included trial from the published reports, when possible. We used data extraction forms and resolved disagreements by mutual agreement. We recorded the rawest form of data, when possible. In the case of missing or incomplete data, we contacted the principal investigators of included trials to request the required additional information.
Participant factors
-
Age
-
Sex
-
Epileptic seizure type and epilepsy syndrome
-
Causes of epilepsy
-
Duration of epilepsy
-
Number of seizures or seizure frequency before randomisation
-
Presence of status epilepticus
-
Numbers and types of AEDs previously taken
-
Concomitant AEDs
-
Presence of neurological deficit/signs
-
Neuropsychological status
-
Electroencephalographic (EEG) findings
-
Neuroradiological findings (computed tomography (CT), magnetic resonance imaging (MRI))
Trial design
-
Criteria used to diagnose epilepsy
-
Definition of drug‐resistant or refractory epilepsy
-
Trial design (i.e. RCT, parallel group or cross‐over, single‐blinded or double‐blinded)
-
Inclusion and exclusion criteria
-
Method of randomisation
-
Method of allocation concealment
-
Method of blinding
-
Stratification factors
-
Number of participants allocated to each group
-
Duration of different phases of the trial (baseline, titration, maintenance and optional open‐label extension (if any))
Intervention and control
-
Intervention given to controls
-
Dosage of stiripentol
-
Duration of treatment period
Follow‐up data
-
Duration of follow‐up
-
Reasons for incomplete outcome data
-
Dropout or loss to follow‐up rates
-
Methods of analysis (e.g. intention‐to‐treat, per‐protocol, worst‐case or best‐case scenario)
Primary outcomes
-
Fifty per cent or greater reduction in seizure frequency: proportion of participants with at least 50% reduction in seizure frequency at the end of the study (numerator)/number of participants at pre‐randomisation baseline period (denominator)
-
Seizure freedom: proportion of participants achieving total cessation of seizures (numerator)/number of participants at pre‐randomisation baseline period (denominator)
Secondary outcomes
-
Incidence of adverse effects of any type: numbers of adverse effects (numerator)/total number of participants at pre‐randomisation baseline period (denominator)
Assessment of risk of bias in included studies
Two review authors (FB and NLB) assessed risk of bias of each trial according to approaches described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We assigned risk of bias as yes (low risk of bias), no (high risk of bias) or unclear (uncertain risk of bias).
We evaluated the following characteristics.
-
Random sequence generation (selection bias)
-
Allocation concealment (selection bias)
-
Blinding of participants and personnel (performance bias)
-
Blinding of outcome assessment (detection bias)
-
Incomplete outcome data
-
Selective reporting (reporting bias)
-
Other bias (including outcome reporting bias)
Measures of treatment effect
For dichotomous outcomes, we extracted the number of participants in each arm who experienced the outcome of interest. Data for our chosen outcomes were dichotomous, and our preferred outcome statistic was the risk ratio (RR), calculated with uncertainty in each trial expressed with 95% confidence intervals (CIs).
Dealing with missing data
For each outcome, we performed an intention‐to‐treat primary analysis to include all participants in the treatment group to which they were allocated, irrespective of the treatment they actually received.
Assessment of heterogeneity
As only one study satisfied our inclusion criteria, we did not perform an assessment of heterogeneity.
If we had included more than one study, we would have assessed heterogeneity as follows:
For each outcome, an intention‐to‐treat primary analysis would have been made in order to include all patients in the treatment group to which they were allocated, irrespective of the treatment they actually received. We would have tested heterogeneity of the intervention effects among trials using the standard Chi2 statistic (P value) and the I2 statistic. Homogeneity among trial results would have been evaluated using a standard Chi2 test and the hypothesis of homogeneity would have been rejected if the P value was less than 0.10.
The interpretation of I2 for heterogeneity would have been as follows:
• 0% to 40%, may not be important;
• 30% to 60%, represents moderate heterogeneity;
• 50% to 90%, represents substantial heterogeneity;
• 75% to 100%, represents considerable heterogeneity.
Trial outcomes would have been combined to obtain a summary estimate of effect (and the corresponding confidence interval (CI)) using a fixed‐effect model unless there is a significant heterogeneity (that is I2 > 75%). If there was substantial heterogeneity we would have planned to explore the contributing factors for heterogeneity. If there was substantial heterogeneity that could not readily be explained we would have used a random‐effects model.
We would have assessed possible sources of heterogeneity (for example clinical heterogeneity, methodological heterogeneity or statistical heterogeneity) by using sensitivity analysis as described below.
Assessment of reporting biases
As only one study satisfied our inclusion criteria, we did not carry out an analysis of reporting biases.
If we had included more than one study, we would have assessed reporting bias as follows (Brigo 2012).
We would have used a funnel plot to detect reporting biases when sufficient numbers of studies (10 or more) were available. Possible sources of funnel plot asymmetry can exist, publication bias, language bias, citation bias, poor methodological quality, true heterogeneity etc., and we would have analysed them according to the trials.
Data synthesis
As only one study satisfied our inclusion criteria, we did not perform a meta‐analysis.
We used GRADE (Guyatt 2008) quality assessment criteria in the 'Summary of findings' table, including all outcomes assessed in this review.
If we had included more than one study, we would have synthesized data as follows:
Provided we thought it clinically appropriate, and no important clinical and methodological heterogeneity was found, we would have planned to synthesize the results in a meta‐analysis.
We would have synthesized data on all seizures and also according to seizure type. We would have analysed different treatments and controls separately, including no treatment and placebo together. We would have used Review Manager to combine trial data.
Subgroup analysis and investigation of heterogeneity
As eligible data were limited, we did not perform subgroup analysis.
As per protocol, we planned no subgroup analysis to further investigate heterogeneity (Brigo 2012).
Sensitivity analysis
As eligible data were limited, we did not perform a sensitivity analysis.
If we had included more than one study, we would have performed sensitivity analysis as follows:
In the case of residual unexplained heterogeneity, we would have evaluated the robustness of the results of the meta‐analysis by comparing fixed‐effect and random‐effects model estimates, removing trials with low methodological quality or excluding trials with large effect size. We would have also used the worst‐case and best‐case scenarios whenever possible. If the conclusions we observed remained unchanged (that is if the RR contains 1 and the sensitivity analysis still does or does not contain 1, and the sensitivity analysis still does not), then we would have considered the evidence to be robust.
Results
Description of studies
See Characteristics of included studies and Characteristics of excluded studies.
The only included trial (Chiron 2006), used a 'responder enriched' design, whereby participants responding to stiripentol during a pre‐randomisation baseline phase were randomly assigned to continue stiripentol or to have it withdrawn. This trial therefore compared the effects of continuing versus withdrawing stiripentol. We only included data from the randomised, double‐blind, add‐on, placebo‐controlled portion of the trial in the present review.
Results of the search
The updated search strategy described above yielded 19 results (0 Cochrane Epilepsy Group Specialised Register, 0 CENTRAL, 5 MEDLINE, 9 ClinicalTrials.gov, 5 ICTRP; 0 in reference lists; 0 by handsearching; no ongoing or unpublished trials; total 19 results). After removing five duplicates and two irrelevant items, we reviewed abstracts of the remaining 12 items.
We excluded all 12 of these studies. Thus, since the last version of this review, we have found no new studies.
Searches conducted in the previous versions of this review (Brigo 2014; Brigo 2015), yielded 118 results (10 Cochrane Epilepsy Group Specialised Register, 14 CENTRAL, 31 MEDLINE, 48 EMBASE; one in reference lists; 14 by handsearching; no ongoing or unpublished trials; total 118 results, 94 after removal of 24 duplicates). In the previous versions of this review (Brigo 2014; Brigo 2015), after review of the abstracts, we provisionally selected six studies. We later excluded five studies after reading the full texts, as they did not meet our review criteria. See Characteristics of excluded studies for reasons for exclusion. We identified one study that met our inclusion criteria (Chiron 2006).
See Figure 1.

Study flow diagram. The results shown in this figure refer both to the searches conducted in the present version of the review and in its previous versions (Brigo 2014; Brigo 2015).
Included studies
Chiron 2006
Investigators in Chiron 2006 aimed to study stiripentol as add‐on therapy to carbamazepine for childhood partial epilepsy by adopting a 'responder enriched' design. Participants were 32 children with focal epilepsy. All included participants were defined as "refractory to the usual antiepileptic drugs (including valproate, carbamazepine, benzodiazepines and phenytoin), as well as to vigabatrin". However, presence of refractory epilepsy was not specified among the inclusion criteria. The study included 18 male (seven in the stiripentol group and 11 in the add‐on placebo group) and 14 female (10 in the stiripentol group and four in the add‐on placebo group) participants. Mean age was 8 ± 3 years (mean ± standard deviation) among participants in the stiripentol group and 10.4 ± 3.4 years in the add‐on placebo group.
The first study period consisted of a one‐month baseline with a single‐blind, add‐on placebo. The second period included a four‐month open phase with open, add‐on stiripentol. These first two study periods adopted a non‐randomised before‐after design. At the end of this open phase, responders (defined as participants with at least a 50% decrease in seizure frequency during the open period versus baseline) were randomly assigned to stiripentol or to add‐on placebo for a two‐month, double‐blind period. Then all participants received long‐term open stiripentol.
The following criteria were required for patients to be included in the baseline period: (1) focal seizures; (2) receiving carbamazepine as co‐medication, with a benzodiazepine (clobazam or clonazepam) or vigabatrin, or both, administered in association; and (3) receiving at least 400 mg/day of carbamazepine. Participants had to be responders (i.e. experiencing ≥ 50% decrease in seizure frequency during the third month of the open period versus baseline) in the open phase to be eligible for randomisation. Researchers did not include participants receiving other drugs nor those whose parents were unable to comply regularly with drug delivery and daily seizure diaries.
Investigators did not report conflicts of interest nor study sponsors.
Excluded studies
We excluded three studies, as they were non‐randomised trials (Loiseau 1988; Perez 1999; Rascol 1989). These studies adopted an uncontrolled before‐after design. One study (Chiron 2000 published as a conference proceeding), provided preliminary results (interim analyses) of the study of Chiron 2006, which was published a few years later as an in extenso paper presenting definitive results and was included in the present review. The other excluded study (Loiseau 1990), was a randomised, double‐blind, parallel‐group trial that evaluated the efficacy of stiripentol as add‐on therapy to carbamazepine versus carbamazepine monotherapy in individuals with epilepsy uncontrolled by carbamazepine monotherapy. We excluded this study because it did not clearly specify whether patients with focal epilepsy were included. Moreover, this study was conducted in individuals with epilepsy "uncontrolled by carbamazepine monotherapy". Most available definitions of drug refractory epilepsy require failure of at least two AEDs for such a diagnosis (Berg 2006). As a consequence, we did not consider participants in this study as affected by drug refractory epilepsy, even when we applied the internationally accepted definition of refractory epilepsy: failure of adequate trials of two tolerated, appropriately chosen and used AED schedules (whether given as monotherapy or in combination) to achieve sustained seizure freedom (Kwan 2010).
Risk of bias in included studies
See Characteristics of included studies.
Allocation
Researchers in Chiron 2006 used a computer‐generated list to randomly assign participants, and a pharmacist dosed the tablets, to ensure that investigators were blinded (low risk of selection bias).
Blinding
Study authors described the second part of the trial as double‐blinded (low risk of performance bias). Each participant received tablets of both stiripentol and 'placebo of stiripentol' and tablets of both carbamazepine and 'placebo of carbamazepine', and a pharmacist prepared the individual tablets (low risk of selection bias). However, part of the carbamazepine schedule was administered as 'open carbamazepine', the dose of which could be decreased when necessary.
Incomplete outcome data
Investigators reported the number of dropouts and specified reasons for dropout. Although these reasons were similar among participants in the two groups, and despite the fact that strict escape criteria were specifically required for a 'responder enriched' design, the number of dropouts in both arms (add‐on stiripentol and placebo) was high and far exceeded 20% (53.3 versus 35.3) (high risk of attrition bias).
Selective reporting
Published reports included all expected outcomes (low risk of reporting bias).
Other potential sources of bias
Through its 'responder‐enriched' design, this study conducted a primary efficacy evaluation of an enriched population of participants, as the result of random assignment only of participants who responded to open‐label treatment (high risk of selection bias).
This trial used as a primary endpoint the number of participants who met the escape criteria during the double‐blind period, defined as (1) increased seizure frequency during the double‐blind period compared with the pre‐randomisation period; (2) significantly increased seizure severity during the double‐blind period compared with the open period; and (3) status epilepticus during the double‐blind period. However, this study provided individual participant data only for the randomised, double‐blind portion of the trial, thus allowing us to include this information in the present review.
Length of follow‐up for the randomised, double‐blind study (only two months) was not adequate for evaluation of a change in seizure frequency.
Effects of interventions
Add‐on stiripentol versus add‐on placebo
See summary of findings Table for the main comparison
We found one study (Chiron 2006), that compared add‐on stiripentol with add‐on placebo and recruited 32 participants. As outlined under Description of studies above, this trial used a 'responder enriched' design, whereby participants responding to stiripentol during a pre‐randomisation baseline phase were randomly assigned to continue stiripentol or to have it withdrawn. Therefore, this trial compared the effects of continuing versus withdrawing stiripentol.
Primary outcomes
See Data and analyses.
Fifty per cent or greater reduction in seizure frequency, and seizure freedom
No clear evidence showed a reduction in seizure frequency (≥ 50% seizure reduction) (RR 1.51, 95% CI 0.81 to 2.82, Analysis 1.1) nor occurrence of seizure freedom (RR 1.18, 95% CI 0.31 to 4.43, Analysis 1.2) when add‐on stiripentol was compared with placebo, although a non‐significant trend favouring add‐on stiripentol was reported for both outcomes. In the add‐on placebo group, 4/15 participants experienced worsening of seizure frequency compared with the baseline period.
Secondary outcomes
See Data and analyses.
Adverse effects
Add‐on stiripentol led to greater risk of adverse effects considered as a whole (RR 2.65, 95% CI 1.08 to 6.47, Analysis 1.3) when compared with placebo. When specific adverse events were considered, confidence intervals were very wide and included the possibility of substantial increases and small reductions in risk of neurological ( RR 2.65, 95% CI 0.88 to 8.01, Analysis 1.4) or gastrointestinal adverse effects (RR 11.56, 95% CI 0.71 to 189.36, Analysis 1.5).
Proportion of dropouts or withdrawals due to side effects, lack of efficacy or other reasons
We noted no clear reduction in the risk of study withdrawal (RR 0.66, 95% CI 0.30 to 1.47, Analysis 1.6), which was high in both groups (35.0% in add‐on placebo and 53.3% in stiripentol group). Eight participants in the add‐on placebo group (35.3%) dropped out because of loss of response (seven for an increase in seizure frequency and one for an increase in seizure severity), and four experienced worsening compared with baseline. Six participants in the stiripentol group (53.3%) dropped out (five because of an increase in seizure frequency and one for an increase in seizure severity).
Improvement in quality of life as assessed by validated and reliable rating scales
The included study did not assess this outcome.
Discussion
This review aimed to assess the efficacy and tolerability of stiripentol as add‐on treatment for focal refractory epilepsy.
Since the last version of this review was published, we have found no new studies. Hence we have made no changes to the conclusions of this update as presented in the initial review (Brigo 2014) and in the first updated version (Brigo 2015).
Summary of main results
We included only one study, which we identified in the first version of this review (Chiron 2006). This study adopted a 'responder enriched' design. Although all included participants were "refractory to the usual antiepileptic drugs (including valproate, carbamazepine, benzodiazepines and phenytoin), as well as to vigabatrin as a new drug", the presence of refractory epilepsy was not considered among the inclusion criteria. Furthermore, investigators did not provide a definition of refractory epilepsy.
The only study included in the present review found no clear evidence of seizure reduction (≥ 50%) nor of seizure freedom with add‐on stiripentol compared with placebo. Add‐on stiripentol led to greater risk of adverse effects considered as a whole compared with placebo; however we are uncertain of this effect, because the results are imprecise. No clear difference was found in neurological adverse effects and in gastrointestinal adverse effects between add‐on stiripentol and placebo, although the included study showed a non‐significant trend toward more frequent adverse effects after add‐on stiripentol. The study showed not clear differences in the proportion of dropouts between add‐on stiripentol and add‐on placebo, although with a trend toward increased dropouts among add‐on placebo participants.
Overall completeness and applicability of evidence
Despite an overall 'low risk' of bias, the 'responder enriched' design of the included trial raises several ethical and methodological concerns. This design shifts the focus to a participant subgroup when accumulating data suggest greatest benefit for that subgroup. Only the second portion of this study met the inclusion criteria of the systematic review (randomised, add‐on, placebo‐controlled, double‐blind trial), whereas the first portion of the study adopted a non‐randomised, before‐after design. Inclusion of responders to add‐on stiripentol alone (i.e. those experiencing a ≥ 50% decrease in seizure frequency during the third month of the open period versus baseline) in the second portion of the study may severely reduce the external validity of the results, limiting their generalisation to a more widespread population. Therefore, this study design has resulted in a primary efficacy evaluation of a highly selected 'enriched' population of participants as a result of random assignment only of those who responded to open‐label treatment (high risk of selection bias).
Furthermore, a 'responder enriched' design carries the risk of a carry‐over effect in the add‐on placebo group. A carry‐over effect occurs when the effects of an intervention given during one period persist into a subsequent period, thus interfering with the effects of a different subsequent intervention. Risk of a carry‐over effect in the add‐on placebo group of the included study seems to be high, because in the add‐on placebo group, add‐on stiripentol was withdrawn over three weeks (a long period, especially given that the overall length of the randomised, double‐blind portion of the trial was only two months). Furthermore, investigators included no washout period during the randomised, double‐blind phase, to reduce the carry‐over effect. As a consequence, it is likely that a carry‐over effect may have influenced outcomes related to seizure frequency in the included study, with possible reduction in seizure frequency in the add‐on placebo group. Conversely, a 'responder enriched' design carries the risk of a withdrawal effect secondary to withdrawal of add‐on stiripentol in the add‐on placebo group during the randomised add‐on placebo‐controlled phase of the trial. The withdrawal effect may be responsible for an increase in seizure frequency (which, unlike reduction in seizure frequency, becomes a relevant endpoint within such a study design). This should be carefully taken into account when strict escape criteria are defined, to prevent exposure of participants in the add‐on placebo group to seizures that may become more severe or more prolonged and may even evolve into status epilepticus. Regarding this last aspect, it is noteworthy to consider that in both arms (add‐on stiripentol and add‐on placebo) ‐ not only in the add‐on placebo group ‐ the percentage of dropouts was extremely high as the result of an increase in seizure frequency or severity.
Furthermore, length of follow‐up for the randomised, double‐blind study (only two months) probably was inadequate to permit evaluation of changes in seizure frequency.
Additional research is needed to assess the efficacy and tolerability of add‐on stiripentol for treatment of focal refractory epilepsy. Future studies should be randomised and double‐blinded, should aim to recruit a sufficiently large number of participants and should assess clinically meaningful outcome measures, while adopting an internationally accepted definition of refractory epilepsy (Kwan 2010).
Quality of the evidence
Generalisation of study results to a more widespread population is prevented by the fact that only responders to add‐on stiripentol (i.e. those experiencing a ≥ 50% decrease in seizure frequency versus baseline) were included in the randomised, add‐on, placebo‐controlled, double‐blind portion of the study. Also, the very small sample size with correspondingly high dropout rates prevents generalisation of study results. Finally, because of the adopted design, carry‐over and withdrawal effects probably influenced outcomes related to seizure frequency. Using the GRADE methodology, we rated the quality of evidence as low.
Agreements and disagreements with other studies or reviews
No other studies or reviews on the same topic have been published so far.

Study flow diagram. The results shown in this figure refer both to the searches conducted in the present version of the review and in its previous versions (Brigo 2014; Brigo 2015).

Comparison 1 Add‐on stiripentol versus placebo, Outcome 1 ≥ 50% seizure reduction.

Comparison 1 Add‐on stiripentol versus placebo, Outcome 2 Seizure freedom.

Comparison 1 Add‐on stiripentol versus placebo, Outcome 3 ≥ 1 adverse effect.
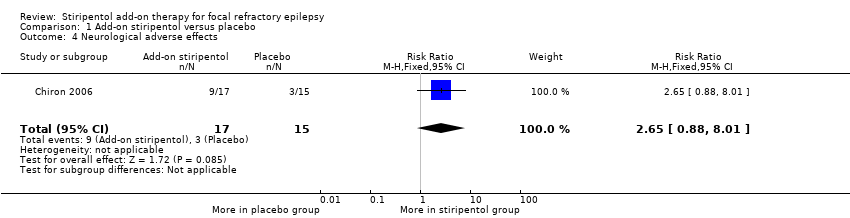
Comparison 1 Add‐on stiripentol versus placebo, Outcome 4 Neurological adverse effects.

Comparison 1 Add‐on stiripentol versus placebo, Outcome 5 Gastrointestinal adverse effects.

Comparison 1 Add‐on stiripentol versus placebo, Outcome 6 Dropouts.
| Stiripentol compared with placebo for focal refractory epilepsy | ||||||
| Patient or population: people with focal refractory epilepsy Settings: community Intervention: stiripentol Comparison: placebo | ||||||
| Outcomes* | Illustrative comparative risks** (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Stiripentol | |||||
| ≥ 50% seizure reduction | 467 per 1000 | 705 per 1000 | RR 1.51 (0.81 to 2.82) | 32 | ⊕⊕⊖⊖ lowa,b | |
| Seizure freedom | 200 per 1000 | 236 per 1000 | RR 1.18 (0.31 to 4.43) | 32 | ⊕⊕⊖⊖ lowa,b | |
| ≥ 1 adverse effect | 267 per 1000 | 707 per 1000 | RR 2.65 (1.08 to 6.47) | 32 | ⊕⊕⊖⊖ | |
| Neurological adverse effects | 200 per 1000 | 530 per 1000 | RR 2.65 (0.88 to 8.01) | 32 | ⊕⊕⊖⊖ | |
| Gastrointestinal adverse effects | 0 events occurred in the placebo group | 0 events occurred in the stiripentol group | RR 11.56 (0.71 to 189.36) | 32 | ⊕⊕⊖⊖ | |
| Dropouts | 533 per 1000 | 352 per 1000 | RR 0.66 (0.30 to 1.47) | 32 | ⊕⊕⊖⊖ | |
| * Quality of life was not assessed in this study. **The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) and is calculated according to the following formula: corresponding intervention risk, per 1000 = 1000 X ACR X RR. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded once for risk of bias and once for imprecision (small sample size which is made even smaller with dropouts). | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 ≥ 50% seizure reduction Show forest plot | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.51 [0.81, 2.82] |
| 2 Seizure freedom Show forest plot | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.31, 4.43] |
| 3 ≥ 1 adverse effect Show forest plot | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.65 [1.08, 6.47] |
| 4 Neurological adverse effects Show forest plot | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.65 [0.88, 8.01] |
| 5 Gastrointestinal adverse effects Show forest plot | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 11.56 [0.71, 189.36] |
| 6 Dropouts Show forest plot | 1 | 32 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.30, 1.47] |