Celecoxib para o tratamento da osteoartrose
Referencias
References to studies included in this review
References to studies excluded from this review
References to studies awaiting assessment
References to ongoing studies
Additional references
References to other published versions of this review
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Study design: randomized, double‐blind, placebo‐controlled trial, 2 parallel groups, multicenter study Duration: 6 weeks Study dates: Not indicated Locations: 29 centers in the USA | |
| Participants | Randomized: N = 380 Completed: N = 278 Mean age: 60.3 ± 10 years Female: 60.5% Inclusion: age ≥ 40 years, with diagnosed, active symptomatic osteoarthritis (OA) of the knee in a flare state diagnosed according to the American College of Rheumatology (ACR) criteria, according to the following inclusion criteria: had failed prior treatment with both prescription strength naproxen (at least 750 mg/day for 2 weeks) and ibuprofen (at least 1200 mg/day for 2 weeks) within the past 5 years due to either lack of efficacy and/or tolerability; women of childbearing potential had a negative urine pregnancy test and had to be using an adequate method of contraception; if taking chronic non‐steroidal anti‐inflammatory drug (NSAID) therapy, participants were to complete a wash out period for a minimum of 2 days; participants were to have a functional capacity class of I–III; a willingness to participate for 6 weeks and ability to provide informed consent | |
| Interventions | Celecoxib 200 mg (N = 190) Placebo (N = 190) | |
| Outcomes | Primary efficacy outcomes: change from baseline to week 6 in the Patient’s Assessment of Arthritis Pain using visual analog scale (VAS) measured on a 0 (no pain) to 100 (very severe pain) scale Secondary outcomes: change in Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) from baseline to week 6, change in Patient’s and Physician’s Global Assessment of Pain from baseline to week 6 Harms: number of participants reporting at least one adverse event, number of participants who discontinued due to adverse events, treatment‐related adverse events that led to discontinuation, number of deaths, treatment‐emerging serious adverse events | |
| Funding | Pfizer, Inc | |
| Declaration of interest | All authors were employees of Pfizer, Inc. The authors indicated they had no other conflicts of interest | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomization not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Attrition was 19.5% (37/190) in celecoxib group and 34.2% (65/190) in the placebo group. In the celecoxib group 4.2% of attrition was related to study drug (adverse events, lack of efficacy), while this percent was 13.7% in the placebo group (Table 1, p. 553). Missing values were imputed using last observation carried forward (LOCF) |
| Selective reporting (reporting bias) | Low risk | All primary and secondary outcomes reported in the methods are included in the report of results |
| Other bias | Low risk | From the data provided, no indication of other important risks of bias |
| Methods | Study design: randomized, double‐blind, placebo‐controlled trial, 2 parallel groups, multicentre study Duration: 6 weeks Study dates: Not indicated Locations: 30 centers in the USA | |
| Participants | Randomized: N = 388 Completed: N = 294 Mean age: 58.6 ± 10 years Female: 68% Inclusion: age ≥ 40 years, with diagnosed, active symptomatic osteoarthritis (OA) of the knee in a flare state diagnosed according to the ACR criteria, according to the following inclusion criteria: had failed prior treatment with both prescription strength naproxen (at least 750 mg/day for 2 weeks) and ibuprofen (at least 1200 mg/day for 2 weeks) within the past 5 years due to either lack of efficacy and/or tolerability; women of childbearing potential had a negative urine pregnancy test and had to be using an adequate method of contraception; if taking chronic non‐steroidal anti‐inflammatory drug (NSAID) therapy, participants were to complete a wash‐out period for a minimum of 2 days; participants were to have a functional capacity class of I–III; a willingness to participate for 6 weeks and ability to provide informed consent | |
| Interventions | Celecoxib 200 mg (N = 195) Placebo (N = 193) | |
| Outcomes | Primary efficacy outcomes: change from baseline to week 6 in the Patient’s Assessment of Arthritis Pain (VAS measured on a 0 (no pain) to 100 (very severe pain) scale. Secondary outcomes: change in Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) from baseline to week 6, change in Patient’s and Physician’s Global Assessment of Pain from baseline to week 6 Harms: number of participants reporting at least one adverse event, number of participants who discontinued due to adverse events, treatment‐related adverse events that led to discontinuation, number of deaths, treatment‐emerging serious adverse events | |
| Funding | Pfizer, Inc | |
| Declaration of interest | All authors were employees of Pfizer, Inc. The authors indicated that they had no other conflicts of interest | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomization not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Attrition was 19% (38/195) in celecoxib group and 29% (56/193) in the placebo group. In the celecoxib group 5.6% of attrition was related to study drug (adverse events, lack of efficacy), while this percent was 18.1% in the placebo group (Table 1, p. 553). Missing values were imputed using last observation carried forward (LOCF) |
| Selective reporting (reporting bias) | Low risk | All primary and secondary outcomes reported in the methods are included in the report of results |
| Other bias | Low risk | From the data provided, no indication of other important risks of bias |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 5 parallel groups, multicentre study Duration: 12 weeks Study dates: August 1996 to July 1997 Locations: 71 clinical sites in the USA and Canada | |
| Participants | Randomized N = 1003 Completed N = 569 Mean age: 62 years (range 21 to 89 years) Women: 72% Inclusion: osteoarthritis (OA) of the knee, stage I‐III according to the American College of Rheumatology (ACR) criteria | |
| Interventions | Celecoxib 100 mg/day (50 mg twice daily) (N = 197) Celecoxib 200 mg/day (100 mg twice daily) (N = 201) Celecoxib 400 mg/day (200 mg twice daily) (N = 202) Naproxen 1000 mg/day (500 mg twice daily) (N = 198) Placebo (N = 203) Stable doses of aspirin, 325 mg/d or less, and acetaminophen, up to 2 g/d, taken for no longer than 3 consecutive days – except during the 48‐hour period prior to arthritis assessments – were allowed | |
| Outcomes | Efficacy outcomes: patient's and physician's global assessment of arthritis (1‐5 scale); the patient's assessment of arthritis pain on a visual analog scale (VAS 0‐100 mm); the OA Severity Index (0‐24 scale); Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) Harms outcomes: incidence and type of adverse reactions; the rate of withdrawal because of adverse reactions; laboratory abnormalities | |
| Funding | Supported in part by GD Searle & Co | |
| Declaration of interest | Not reported 7/10 authors were employees of Searle Research and Development | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | All treatment regimens were fully masked with the use of a double‐dummy technique (p. 1096) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | High attrition was observed, as 43% of participants did not complete the study (p. 1097). Attrition per group was reported for withdrawals due to treatment failure: 19.2% in placebo group, 10% in the celecoxib 200 mg/day group and 14% in the naproxen group (Table 2) and for withdrawals due to adverse events as 4% in the placebo group, 8% in the celecoxib group and 4% in the naproxen group (Table 4). Other reasons for attrition were not provided. All efficacy analyses were performed on the intention‐to‐treat (ITT) cohort (all randomized participants who took one dose or more of study medication) and missing values were imputed by carrying forward the last observation for any participant who discontinued the study for any reason (including treatment failure) prior to the full 12 weeks of therapy (p. 1097) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in the methods are also reported in the results section |
| Other bias | Unclear risk | The results may have been influenced by a selection bias in favor of participants tolerant of naproxen, and thus the tolerability comparison in this study may underestimate any actual difference between celecoxib and naproxen in the arthritis population as a whole (p. 1103). Participans were allowed to take stable doses of aspirin (325 mg/d or less) and acetaminophen (up to 2 g/d taken for no longer than 3 consecutive days except during the 48 h period prior to arthritis assessment) (p. 1096). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: two randomized studies, double‐blind, placebo and active controls, 4 parallel groups, multicenter studies Duration: 26 weeks; each study consisting of part one (12 weeks) and part two (14 weeks) Dates of study: March 2004 to February 2005 Locations: 74 centers in the USA | |
| Participants | Randomized N = 599 Completed N = 468 Mean age: 62.3 ± 9.5 years Women: 67.1% Inclusion: Osteoarthritis (OA) of the knee > 6 months, aged ≥ 40 years, American Rheumatology Association (ARA) functional class I, II or III | |
| Interventions | Celecoxib 200 mg (N = 241) Etoricoxib 30 mg (N = 231) First placebo group (N = 64) Second placebo group (N = 63) Participants who successfully completed part one were enrolled directly into part two, an active comparator 14‐week follow‐up. Participants on active treatment in part one remained on the same treatment in part two; participants receiving placebo in part one received either etoricoxib 30 mg or celecoxib 200 mg in part two, based on their initial randomization schedule at enrolment. Only the first part was considered for this systematic review | |
| Outcomes | Efficacy outcomes: Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain sub scale using average of the first five questions of the WOMAC measured by visual analog scale (VAS) 0‐100 mm; WOMAC physical function sub scale (average of questions 8 through 24 of the WOMAC measured by VAS 0‐100 mm); Patient Global Assessment of Disease status (PGADS) measured by VAS 0‐100 mm Harms outcomes: clinical and laboratory assessments and patient reported adverse events (AEs); pre‐defined AEs: discontinuation due to any AE, discontinuation due to oedema, hypertension or gastrointestinal (GI) event or congestive heart failure (CHF) | |
| Funding | The study was funded by Merck & Co., Inc | |
| Declaration of interest | All authors reported potential conflict of interest related to funding from Merck & Co 5/10 authors were employees of Merck & Co | |
| Notes | Two placebo groups in both studies were shown as one group. Corresponding author was contacted, and he responded on May 8, 2013 that he is trying to get the information requested from Merck. The corresponding author did not respond to subsequent multiple queries about the missing information. Therefore, combined data for both placebo group were presented, as in the manuscript Trial registration: ClinicalTrials.gov, http://clinicaltrials.gov/, NCT00092768 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind, double‐dummy study (p. 497) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Attrition was 20% (49/241) in the celecoxib group (33/49 due to lack of efficacy or adverse events), 36% (23/64) in the first placebo group (20/23 due to lack of efficacy or adverse events) and 30% (19/63) in the second placebo group (17/19 due to lack of efficacy or adverse events) The primary efficacy analysis was a modified intention‐to‐treat (mITT) approach on time‐weighted average (TWA) response. All patients with a baseline value and at |
| Selective reporting (reporting bias) | Low risk | All primary and secondary outcomes and harms/adverse events data were included in report of results. Adverse events were categorised and described for each group |
| Other bias | Unclear risk | Acetaminophen 325 mg tablets were available as rescue analgesia at a maximum daily dose of 2600 mg. Patients were requested to use as little acetaminophen as possible, and |
| Methods | Study design: two randomized studies, double‐blind, placebo and active controls, 4 parallel groups, multicenter studies Duration: 26 weeks; each study consisting of part one (12 weeks) and part two (14 weeks) Dates of study: March 2004 to February 2005 Locations: 74 centers in the USA | |
| Participants | Randomized N = 608 Completed N = 474 Mean age: 61.7 ± 9.2 years Women: 65.5% Inclusion: osteoarthritis (OA) of the knee > 6 months, age ≥ 40 years, American Rheumatology Association (ARA) functional class I, II or III | |
| Interventions | Celecoxib 200 mg (N = 247) Etoricoxib 30 mg (N = 244) First placebo group (N = 58) Second placebo group (N = 59) Participants who successfully completed part one were enrolled directly into part two, an active comparator 14‐week follow‐up. Participants on active treatment in part one remained on the same treatment in part two; participants receiving placebo in part one received either etoricoxib 30 mg or celecoxib 200 mg in part two, based on their initial randomization schedule at enrolment. Only the first part was considered for this systematic review | |
| Outcomes | Efficacy outcomes: Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain sub scale using average of the first five questions of the WOMAC measured by visual analog scale (VAS) 0‐100 mm; WOMAC physical function sub scale (average of questions 8 through 24 of the WOMAC measured by VAS 0‐100 mm); Patient Global Assessment of Disease status (PGADS) measured by VAS 0‐100 mm Harms outcomes: clinical and laboratory assessments and patient reported adverse events (AEs); pre‐defined AEs: discontinuation due to any AE, discontinuation due to oedema, hypertension or gastrointestinal (GI) event or chronic heart failure (CHF) | |
| Funding | The study was funded by Merck & Co., Inc | |
| Declaration of interest | All authors reported potential conflict of interest related to funding from Merck & Co 5/10 authors were employees of Merck & Co | |
| Notes | Two placebo groups in both studies were shown as one group. Corresponding author was contacted, and he responded on May 8, 2013 that he is trying to get the information requested from Merck. The corresponding author did not respond to subsequent multiple queries about the missing information. Therefore, combined data for both placebo group were presented, as in the manuscript Trial registration: ClinicalTrials.gov, http://clinicaltrials.gov/, NCT00092791 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind, double‐dummy study (p. 497) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Attrition was 18% (45/247) in the celecoxib group (32/45 due to lack of efficacy or adverse events), 48% (28/58) in the first placebo group (26/28 due to lack of efficacy or adverse events) and 49% (29/59) in the second placebo group (25/29 due to lack of efficacy or adverse events) |
| Selective reporting (reporting bias) | Low risk | All primary and secondary outcomes and harms/adverse events data were included in report of results. Adverse events were categorised and described for each group |
| Other bias | Unclear risk | Acetaminophen 325 mg tablets were available as rescue analgesia at a maximum daily dose of 2600 mg. Patients were requested to use as little acetaminophen as possible, and |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 3 parallel groups, multicenter study Duration: 6 weeks Study dates: February to November 2003 Locations: sites in the USA | |
| Participants | Randomized N = 395 Completed N = 345 Mean age: 60.4 ± 10.3 years Female: 72% Inclusion: osteoarthritis (OA) of the knee ≥ 6 months, class I‐III according to the American Rheumatology Association (ARA) criteria, visual analog scale (VAS) 0‐100 pain walking on flat surface at least 40 mm, which had increased at least 15 mm since discontinuing their original OA medication. Also worsening in Investigator Global Assessment of Disease Status (IGADS) of at least 1 unit on a 5‐point scale. Participants previously taking acetaminophen for OA symptoms were permitted to continue with their medication until 12 hours before the second visit. Acetaminophen participants were required to have a measure of at least 40 mm for Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain walking on a flat surface and a rating of ‘very poor’, ‘poor’ or ‘fair’ on IGADS at both the screening visit and randomization visit while on acetaminophen; no flare of disease was required. Participants who used low‐dose aspirin (81 mg or less daily) for cardioprotective effects were permitted to continue low‐dose aspirin use during the studies. Glucosamine and chondroitin sulfate, if taken for longer than 6 months, were also permitted if taken at the same stable dose for the duration of the studies | |
| Interventions | Celecoxib 200 mg (N = 157) Rofecoxib 12.5 mg (N = 160) Placebo (N = 78) | |
| Outcomes | Efficacy outcomes: Patient global assessment of response to therapy (PGART, 0‐4 scale); WOMAC pain (VAS 0‐100 mm); WOMAC physical function (VAS 0‐100 mm) Harms outcomes: spontaneously reported adverse events (AEs); incidence of serious adverse events (SAEs), drug‐related AEs, AEs which cause discontinuation, edema‐related AEs causing discontinuations, hypertension‐related AEs causing discontinuations, congestive heart failure, and gastro‐intestinal AEs | |
| Funding | Merck & Co. Inc | |
| Declaration of interest | Not reported. 5/8 authors were employees of the Merck & Co. Inc. according to their affiliation data. In September 2004 the drug manufacturer announced the withdrawal of rofecoxib from the market; rofecoxib is no longer available for use | |
| Notes | The manuscript where this study was published reported data for two separate randomized controlled trials (Study 1 and Study 2). Data for adverse events were shown for both studies pooled together. These data were divided proportionally between the two studies and included in the relevant analyses as such | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned by computer‐generated allocation schedule in a 2:2:1 ratio (p. 201) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | All treatment regimens were fully masked with the use of a double‐dummy technique, and both participants and investigators were blinded (p. 201) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | This manuscript reported two separate studies (Study 1 and Study 2) and adverse events were shown as combined for both studies (Table 3) High attrition was observed, with 27% (21/78) in the placebo group (24.4% due to adverse experience or lack of efficacy) and 8.9% (14/157) in the celecoxib group (5.1% due to adverse experience or lack of efficacy), and the main reasons for attrition were lack of efficacy and clinical adverse experience (Table 1). All randomized participants who took at least one dose of study medication, and had at least one on‐treatment assessment, were included in a modified ITT analysis. The last on‐treatment observation was carried forward in the event of missing data for all efficacy analyses (p. 202) |
| Selective reporting (reporting bias) | Low risk | All primary and secondary outcomes and harms/adverse events data were included in report of results. All serious adverse events were adjudicated. Three independent, blinded committees evaluated cardiovascular and gastro‐intestinal AEs. Each of them had three separate subspecialty committees of three members (p. 202) |
| Other bias | Unclear risk | Participants were allowed to take low‐dose aspirin (81 mg or less daily) for cardioprotective effect; glucosamine and chondroitin sulfate (if taken for longer than 6 months, at the same stable dose); acetaminophen (max. dose 2 600 mg/d, as rescue therapy for OA pain if the study medication did not provide adequate pain control, and were instructed to discontinue use 12 hours before study visits) (p. 201). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 3 parallel groups, multicentre study Duration: 6 weeks Study dates: February to November 2003 Locations: sites in the USA | |
| Participants | Randomized N = 413 Completed N = 344 Mean age: 60.8 ± 10.9 years Female: 66% Inclusion: osteoarthritis (OA) of the knee ≥ 6 months, class I‐III according to the American Rheumatology Association (ARA), VAS 0‐100 pain walking on flat surface at least 40 mm, which had increased at least 15 mm since discontinuing their original OA medication. Also worsening in Investigator Global Assessment of Disease Status (IGADS) of at least 1 unit on a 5‐point scale. Participants previously taking acetaminophen for OA symptoms were permitted to continue with their medication until 12 hours before the second visit. Acetaminophen participants were required to have a measure of at least 40 mm for Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain walking on a flat surface and a rating of ‘very poor’, ‘poor’ or ‘fair’ on IGADS at both the screening visit and randomization visit while on acetaminophen; no flare of disease was required. Participants who used low‐dose aspirin (81 mg or less daily) for cardioprotective effects were permitted to continue low‐dose aspirin use during the studies. Glucosamine and chondroitin sulfate, if taken for longer than 6 months, were also permitted if taken at the same stable dose for the duration of the studies. | |
| Interventions | Celecoxib 200 mg (N = 169) Rofecoxib 12.5 mg (N = 159) Placebo (N = 85) | |
| Outcomes | Efficacy outcomes: patient global assessment of response to therapy (PGART, 0‐4 scale); WOMAC pain (VAS 0‐100 mm); WOMAC physical function (VAS 0‐100 mm) Harms outcomes: spontaneously reported adverse events (AEs); incidence of serious adverse events (SAEs), drug‐related AEs, AEs which cause discontinuation, edema‐related AEs causing discontinuations, hypertension‐related AEs causing discontinuations, congestive heart failure, and gastro‐intestinal AEs | |
| Funding | Merck & Co. Inc | |
| Declaration of interest | Not reported. 5/8 authors were employees of the Merck & Co. Inc. according to their affiliation data. In September 2004 the drug manufacturer announced the withdrawal of rofecoxib from the market; rofecoxib is no longer available for use | |
| Notes | The manuscript where this study was published reported data for two separate randomized controlled trials (Study 1 and Study 2). Data for adverse events were shown for both studies pooled together. These data were divided proportionally between the two studies and included in the relevant analyses as such | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned by computer‐generated allocation schedule in a 2:2:1 ratio (p. 201) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | All treatment regimens were fully masked with the use of a double‐dummy technique, and both participants and investigators were blinded (p. 201) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | This manuscript reported two separate studies (Study 1 and Study 2) and adverse events were shown as combined for both studies (Table 3) High attrition was observed, with 30.4% (26/85) in the placebo group (18.8% due to adverse experience and lack of efficacy), while it was 15.4% (26/169) in the celecoxib group (10.1% due to adverse experience and lack of efficacy), and the main reasons for attrition were lack of efficacy and clinical adverse experience (Table 1). All randomized participants who took at least one dose of study medication, and had at least one on‐treatment assessment, were included in a modified intention‐to‐treat (ITT) analysis. The last on‐treatment observation was carried forward in the event of missing data for all efficacy analyses (p. 202) |
| Selective reporting (reporting bias) | Low risk | All primary and secondary outcomes and harms/adverse events data were included in report of results. All serious adverse events were adjudicated. Three independent, blinded committees evaluated cardiovascular and gastro‐intestinal AEs. Each of them had three separate subspecialty committees of three members (p. 202) |
| Other bias | Unclear risk | Participants were allowed to take low‐dose aspirin (81 mg or less daily) for cardioprotective effect; glucosamine and chondroitin sulfate (if taken for longer than 6 months, at the same stable dose); acetaminophen (max. dose 2 600 mg/d, as rescue therapy for OA pain if the study medication did not provide adequate pain control, and were instructed to discontinue use 12 hours before study visits) (p. 201). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: two multicenter, randomized, double‐blind, placebo‐ and active‐controlled, parallel‐group, dose‐ranging studies; study A – a 6‐week, phase 2, non‐flare design study; study B – a 12‐week, phase 3, flare design study Study dates: September 2003 to April 2004 Locations: study A – 50 centers in Europe and Australia; study B – 187 centers in Europe, North America and internationally | |
| Participants | Randomized N = 649 Completed N = 556 Mean age: 63.4 years Female: 68% Inclusion: osteoarthritis (OA) of the knee ≥ 3 months; American College of Rheumatology (ACR) clinical criteria for OA of the knee; recent (≤ 12 months) radiographic evidence of tibiofemoral OA (grade 2 or 3 on the Kellgren & Lawrence scale); and ARA functional class rating I, II or III | |
| Interventions | Celecoxib 200 mg (N = 109) GW406381 10 mg (N = 106) GW406381 20 mg (N = 101) GW406381 35 mg (N = 1108) GW406381 50 mg (N = 109) placebo (N = 107) | |
| Outcomes | Primary efficacy outcomes: Study A – Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) OA index pain sub score change for GW406381 compared with placebo; “pain on walking on a flat surface” (WOMAC pain Q1) for participants receiving GW406381 compared with celecoxib Study B – change from baseline to week 12 in WOMAC pain sub score; WOMAC function sub score; Patient Global Assessment of Change (PGAC) for participants receiving GW406381 compared with placebo Secondary efficacy outcomes (if not specified as primary): the change from baseline in WOMAC pain Q1; WOMAC pain, stiffness, and physical function sub scores at end of treatment; physician’s and patient’s global assessment of arthritis; percentage of participants fulfilling the Outcome Measures in Rheumatology Clinical Trials – Osteoarthritis Research Society International (OMERACT‐OARSI) responder criteria; rate of discontinuation for lack of efficacy; pattern of use of supplemental paracetamol Harms and tolerability outcomes: laboratory testing; electrocardiography; vital signs; assessment of pedal edema; reporting of adverse events and serious adverse events | |
| Funding | GlaxoSmithKline | |
| Declaration of interest | All authors disclosed potential conflict of interest. Of the eight authors, five were current and one was a former employee of the GlaxoSmithKline. One author owned GlaxoSmithKline stock. The eighth author received payment from GlaxoSmithKline for this study | |
| Notes | GW 406381 is an investigational, highly selective COX‐2 inhibitor | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | This was a double‐blind study |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Blinded investigators assessed the knee with greatest pain at screening (“index knee”) throughout the study. For other outcomes, the process of outcome assessment is not explicitly described |
| Incomplete outcome data (attrition bias) | Unclear risk | Total attrition was 14% (93/649), but attrition and reasons for attrition were not reported for each arm separately. The safety population included all participants who received at least one dose of study medication. The efficacy population was a modified intention‐to‐treat (mITT) population that included all participants who received at least one dose of drug and had at least one post‐baseline assessment. Last observation carried forward was used to impute missing data points, and efficacy data are presented for the ITT population using this imputation |
| Selective reporting (reporting bias) | High risk | Data are not provided for the secondary outcomes: physician's and patient's global assessment of arthritis; percentage of participants fulfilling the Outcome Measures in Rheumatology Clinical Trials ‐ Osteoarthritis Research Society International (OMERACT‐OARSI) responder criteria; rate of discontinuation for lack of efficacy; and pattern of use of supplemental paracetamol. Statistical measures of dispersion not reported for Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) physical function sub scale. Adverse events shown only if their incidence was above 5% in any treatment group |
| Other bias | Unclear risk | Participants were allowed to take paracetamol (max. 3 g/d, if pain intensity became unacceptable at any stage of the study, with the exception of the 24‐hour period before a clinic visit) (p. 261). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 7 parallel groups, dose‐ranging phase 3, flare design multicenter study Duration: 12 weeks Study dates: May to December 2005 Locations: 187 centers in Europe, North America and internationally | |
| Participants | Randomized N = 1331 Completed N = 1038 Mean age: 66.5 years Female: 70% Inclusion: osteoarthritis (OA) of the knee ≥ 3 months; American College of Rheumatology (ACR) clinical criteria for OA of the knee; recent (≤12 months) radiographic evidence of tibiofemoral OA (grade 2 or 3 on the Kellgren & Lawrence scale); and American Rheumatology Association (ARA) functional class rating I, II or III | |
| Interventions | Celecoxib 200 mg (N = 185) GW406381 1 mg (N = 188) GW406381 5 mg (N = 187) GW406381 10 mg (N = 188) GW406381 25 mg (N = 189) GW406381 50 mg (N = 186) Placebo (N = 186) | |
| Outcomes | Primary efficacy outcomes: Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain on walking on a flat surface change from baseline to week 6 (Q1) Secondary efficacy outcomes: the change from baseline in WOMAC pain Q1; WOMAC pain, stiffness, and physical function; physician’s and patient’s global assessment of arthritis; percentage of s fulfilling the Outcome Measures for Rheumatology Committee and Osteoarthritis Research Society International Standing Committee for Clinical Trial Response Criteria Initiative (OMERACT‐OARSI) responder criteria; rate of discontinuation for lack of efficacy; pattern of use of supplemental paracetamol Harms and tolerability outcomes: laboratory testing; electrocardiography; vital signs; assessment of pedal edema; reporting of adverse events and serious adverse events | |
| Funding | GlaxoSmithKline | |
| Declaration of interest | All authors disclosed potential conflict of interest. Of the eight authors, five were current and one was former employee of the GlaxoSmithKline. One author owned GlaxoSmithKline stock. The eighth author received payment from GlaxoSmithKline for this study | |
| Notes | GW 406381 is an investigational, highly selective COX‐2 inhibitor | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | This was a double‐blind study |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Blinded investigators assessed the knee with greatest pain at screening (“index knee”) throughout the study. For other outcomes, the process of outcome assessment is not explicitly described |
| Incomplete outcome data (attrition bias) | Unclear risk | Total attrition was high with the total of 22% (293/1131) participants that did not complete the study; attrition and reasons for attrition were not reported for each arm separately. The safety population included all participants who received at least one dose of study medication. The efficacy population was a modified intention‐to‐treat (mITT) population that included all patients who received at least one dose of drug and had at least one post‐baseline assessment. Last observation carried forward was used to impute missing data points, and efficacy data are presented for the ITT population using this imputation |
| Selective reporting (reporting bias) | High risk | Data are not provided for the secondary outcomes: physician's and patient's global assessment of arthritis; percentage of patients fulfilling the Outcome Measures in Rheumatology Clinical Trials ‐ Osteoarthritis Research Society International (OMERACT‐OARSI) responder criteria; rate of discontinuation for lack of efficacy; and pattern of use of supplemental paracetamol. Statistical measures of dispersion not reported for WOMAC physical function sub scale. Adverse events shown only if their incidence was above 5% in any treatment group |
| Other bias | Unclear risk | Patients were allowed to take paracetamol (max. 3 g/d, if pain intensity became unacceptable at any stage of the study, with the exception of the 24‐hour period before a clinic visit) (p. 261). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 5 parallel groups, multicenter study Study acronym: GAIT (Glucosamine/Chondroitin Arthritis Intervention Trial) Duration: 24 weeks Study dates: November 2000 to July 2004 Locations: 16 clinical centers in the USA | |
| Participants | Randomized N = 1583 Completed N = 1258 Mean age: 58.6 ± 10.3 years Female: 64% Inclusion: osteoarthritis (OA) of the knee, age ≥ 40 years, clinical evidence (knee pain for at least six months and on the majority of days during the preceding month) and radiographic evidence (Kellgren and Lawrence grade 2 or 3). Summed pain score of 125 to 400 on the index (more symptomatic) knee according to the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) and class I, II, or III American Rheumatology Association (ARA) criteria | |
| Interventions | Celecoxib 200 mg (N = 318) Glucosamine 1500 mg/day (500 mg three times a day) (N = 317) Chondroitin sulfate 1200 mg/day (400 mg three times a day) (N = 318) Glucosamine 1500 mg plus chondroitin sulfate 1200 mg daily (N = 317) Placebo (N = 313) | |
| Outcomes | Primary efficacy outcome: 20% decrease in the summed score for the WOMAC pain sub scale from baseline to week 24 Secondary efficacy outcomes: Outcome Measures for Rheumatology Committee and Osteoarthritis Research Society International Standing Committee for Clinical Trial Response Criteria Initiative (OMERACT–OARSI) response rate; scores for the stiffness and function sub scales of WOMAC; the patient’s global assessments of disease status and response to therapy using visual analog scale (VAS) 0‐100 mm; the investigator’s global assessment of disease status (VAS 0‐100 mm); the presence or absence of soft‐tissue swelling, effusion, or both in the index knee; Short‐form Health Survey (SF‐36); alternative disability score and pain score on the Health Assessment Questionnaire (HAQ); and acetaminophen use, according to diary entries and tablet counts Harms outcomes: reported adverse events (AEs) and serious adverse events (SAEs); complete blood counts; measurement of serum aspartate aminotransferase, alanine aminotransferase, glucose, creatinine, and partial thromboplastin time; urine analysis | |
| Funding | Supported by a contract from the National Center for Complementary and Alternative Medicine and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NO1AR92236) | |
| Declaration of interest | Authors reported financial ties with companies producing the study drugs, including consulting fees, sitting on company advisory boards, lecture fees, grant support or expert consultants. One author reported receiving royalties from books related to osteoarthritis | |
| Notes | Recruitment began in November 2000, at 13 clinical centers, and 3 centers were added to the study in February 2003 to ensure timely recruitment. Trial registration: ClinicalTrials.gov, http://clinicaltrials.gov/, NCT00032890 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Permuted block randomisation was used with random block sizes stratified according to baseline WOMAC pain stratum and 16 clinical centres. The randomisation code list was developed by the Veterans Affairs Cooperative Studies Program Data Coordinating Centre in Hines, Illinois (p. 796) |
| Allocation concealment (selection bias) | Low risk | The randomisation code list was developed centrally. During data collection, neither the clinical centres nor the coordinating centre at the University of Utah had access to the randomisation codes or statistical summaries of follow‐up data (p. 796) |
| Blinding of participants and personnel (performance bias) | Low risk | Study drug assignment and the investigators were blinded. Study drugs containing glucosamine hydrochloride, chondroitin sulfate, their combination and placebo drugs were manufactured to look the same, celecoxib was over‐encapsulated (for masking) and matching placebo was prepared (p. 797) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Adverse events and serious events were assessed by the blinded investigator at each study visit. For other non‐patient reported outcomes, the process of outcome assessment is not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Attrition of 20.8% (65/313) was reported in the placebo group (10.5% due to adverse event or lack of efficacy) and 16.4% (52/318) in the celecoxib group (5.7% due to adverse event or lack of efficacy) (Figure 1). Analysis of the primary outcome measure was conducted according to the ITT population (p. 797). The last observation carried forward (LOCF) method was used in the analysis of all outcomes among participants who made at least one follow‐up visit but who did not complete follow‐up (p. 798) |
| Selective reporting (reporting bias) | High risk | Certain secondary outcomes, including measure for quality of life, were reported only for baseline, not for the subsequent follow‐up period. Adverse events per each study group were insufficiently described |
| Other bias | Unclear risk | Participants were allowed to take acetaminophen (max. 4 g/d, except during the 24 hours before a clinical evaluation for joint pain) (p. 796). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double blind, placebo and active controls, 6 parallel groups, multicenter Duration: 12 weeks Study dates: May 2008 to April 2009 Locations: 71 centers in the Czech Republic, Germany, Poland and the UK | |
| Participants | Randomized N = 1399 Completed N = 1256 Mean age: 61.2 years Female: 66% Inclusion: age > 45 years with a primary diagnosis of functional class I‐III knee osteoarthritis (OA), meeting American College of Rheumatology (ACR) clinical classification criteria for knee OA, defined by knee pain and 4 or more of the following: (i) age > 50 years; (ii) morning stiffness of < 30 minute duration; (iii) crepitus on active motion; (iv) bony tenderness; (v) bony enlargement and (vi) no palpable warmth of the synovium | |
| Interventions | Celecoxib tablets 200 mg (N = 235) IDEA‐033/ketoprofen 50 mg (N = 233) IDEA‐033/ketoprofen 100 mg (N = 230) 2.2 g TDT 064/vehicle (N = 238) 4.4 g TDT 064/vehicle (N = 235) Oral placebo (N = 228) | |
| Outcomes | Primary efficacy outcomes: Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain sub scale Secondary efficacy outcomes: patient global assessment of response to therapy, WOMAC function sub scale, WOMAC stiffness sub scale, proportion of participants defined as responders and time to onset of clinically meaningful, sustainable pain relief Harms: adverse events, the number of participants requiring therapy with omeprazole and the dose received, vital signs, clinical laboratory assessments | |
| Funding | This work was supported by IDEA AG, Germany | |
| Declaration of interest | 2/5 authors were employees of the sponsor; authors received consultancy fees, investigator grants and honoraria for expert advice from companies producing study drugs | |
| Notes | Trial registration: ClinicalTrials.gov, http://clinicaltrials.gov/, NCT00716547 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants randomized according to a center‐specific assignment list generated by a random permuted block scheme (p. 1304) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Investigators and participants were blinded to whether they were allocated to active or placebo treatment, but not to whether they were allocated topical or oral medication (p. 1304) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Attrition was 11% (26/235) in celecoxib group (7.7% due to adverse events or lack of efficacy) and 17% (39/228) in control group (15% due to adverse events or lack of efficacy) (Figure 1). The primary efficacy analysis was performed on participants who received at least one dose of study medication, the intention‐to‐treat (ITT) population. Missing follow‐up values were replaced using baseline observation carried forward (BOCF) (p. 1305) |
| Selective reporting (reporting bias) | Low risk | All outcomes reported in the methods are also reported in the results section |
| Other bias | Unclear risk | Participants were allowed to take paracetamol (500 mg up to four times per day, although not within 24 h of the next study visit or between B1 i B2). Participants requiring ≥2 g/day of paracetamol or other analgesic medication for longer than 3 consecutive days considered treatment failures and withdrawn from study (p. 1304). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, active controls, 2 parallel groups, multicentre study Duration: 52 weeks Study dates: August 1999 to October 2001 Locations: 124 general practitioner centers in Sweden (45 sites) and Norway (79 sites) | |
| Participants | Randomized N = 925 Completed N = 550 Mean age: 71 ± 7.2 years Female: 69% Inclusion: at least 60 years, clinical diagnosis of osteoarthritis (OA) of the hip or knee, class I‐III according to the American College of Rheumatology (ACR) criteria | |
| Interventions | Celecoxib 200 mg (N = 463) Diclofenac 100 mg/day (50 mg twice daily) (N = 462) | |
| Outcomes | Primary efficacy outcome: incidence of discontinuation of study drug due to AEs Secondary efficacy outcomes: time to discontinuation of study medication; the Patient’s and Physician’s Global Assessment of Arthritis (PaGAA, PhGAA) on a 5‐point scale; the Patient’s Assessment of Arthritis Pain (PAAP) at rest on a visual analog scale (VAS) 0‐100 mm scale; patient’s and physician’s satisfaction assessments on a 10‐point scale; Harms outcomes: incidence and severity of adverse events (AEs); changes in vital signs; haematology and blood chemistry laboratory results; incidence and severity of gastrointestinal (GI) symptoms evaluated with the Severity of Dyspepsia Assessment (SODA); healthcare resource utilization | |
| Funding | Pfizer Inc | |
| Declaration of interest | Authors reported financial ties with companies producing the study drugs, including consulting fees for their work in a steering committee of a clinical trial and clinical research funding | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was done according to a computer‐generated list prepared by Biostat AS unit in Oslo, Norway. Randomisation was stratified by centre with a block size of six (p. 134) |
| Allocation concealment (selection bias) | Low risk | Central randomisation (p. 134) |
| Blinding of participants and personnel (performance bias) | Low risk | To conceal allocation, the double‐blind supplies were identical in physical appearance and were provided to the investigator in bottles marked with randomisation number and visit number (p. 134) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Attrition was high, with 39% (181/463) of participants lost in the placebo group (153/181 due to adverse event or lack of efficacy) and 40% (185/462) in the celecoxib group (152/462 due to adverse event or lack of efficacy). Part of the patients who discontinued drug remained in health economic study (Figure 1). All randomized participants who had taken at least one dose of study medication were included in the modified intention‐to‐treat (mITT) and the safety populations. Unless otherwise indicated, adverse events (AE)‐related discontinuation, efficacy, and health economic analyses were performed on the mITT population. Safety analyses were performed on the safety population (identical to the mITT population). The last observation carried foward (LOCF) approach was used for missing observations (p. 135) |
| Selective reporting (reporting bias) | Unclear risk | All outcomes described in this manuscript were pre‐specified, and all major pre‐specified outcomes are presented in report of results (p. 135). Incidence of adverse events reported only if they occurred in at least 5% of participants |
| Other bias | Unclear risk | Participants were allowed to take paracetamol (for relief of arthritis pain, up to 3 g daily for short periods); aspirin (≤ 75 mg/d, for cardioprotection); occasional use of antacids was also permitted (p. 134). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 5 parallel groups, multicentre study Duration: 12 weeks Study dates: September 2002 to August 2003 Locations: Canada (42 centers), USA (71 centers), Germany (30 centers), Italy (12 centers), and UK (7 centers) | |
| Participants | Randomized N = 1011 Completed N = 555 Mean age: 60 years (range 20 to 80 years) Female: 63% Inclusion: osteoarthritis (OA) of the knee and/or hip, stage I‐III according to the American College of Rheumatology (ACR) criteria, baseline pain intensity of ≥ 40 on a 100‐mm visual analogue scale (VAS) where 0 = no pain, 100 = extreme pain | |
| Interventions | Celecoxib 200 mg (N = 202) Tramadol ER 100 mg (N = 201) Tramadol ER 200 mg (N = 199) Tramadol ER 300 mg (N = 199) Placebo (N = 200) | |
| Outcomes | Efficacy outcomes: patients completed the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC), and they rated arthritis pain intensity in the index and non‐index joints during the past 48 hours using a 100‐mm VAS (0 = no pain, 100 = extreme pain); patient and physician global assessments of disease activity were recorded at each study visit on 100‐mm VAS (0= very good, 100= very poor in response to the question, “how is the patient doing today” and “considering all the ways your arthritis condition affects you, how are you doing today”, respectively); patients assessed sleep with the Chronic Pain Sleep Inventory, which included a 100‐mm visual analogue scale for overall quality of sleep over the past week (0 = very poor, 100 = excellent); patients completed the Short Form‐36 (SF‐36) Health Survey Harms outcomes: reports of adverse events, either spontaneously or in response to non‐directed questioning, and results of physical examinations, vital signs, clinical laboratory tests, and electrocardiograms at study visits; the 49‐item Short Form Addiction Research Center Inventory (ARCI) questionnaire at baseline and week 12; the 16‐item Physical Dependence Questionnaire (PDQ) at baseline, week 12 (or early discontinuation), and week 13 (or 1 week after early discontinuation). | |
| Funding | Biovail Corporation and Ortho‐McNeil Janssen Scientific Affairs, LLC | |
| Declaration of interest | Not reported. According to the author affiliation, 3/6 study authors were employees of Ortho‐McNeil Janssen Scientific Affairs, LLC., one author was an employee of Biopharmatech Consulting Inc and two authors were employees of Biovail Corporation | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | The double‐blind was maintained during the study (p. 218) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Attrition was high. In the placebo group 97/200 (49%) participants discontinued study treatment (Adverse event: 15, Lack of efficacy: 65, Subject choice: 4, Other: 13). In the celecoxib 200 mg group 67/202 (33%) participants discontinued study treatment (Adverse event: 20, Lack of efficacy: 30, Subject choice: 2, Other: 15) (p. 219) |
| Selective reporting (reporting bias) | Low risk | All primary and secondary outcomes were included in report of results. (p. 222). Adverse events were defined and reported (p. 223). |
| Other bias | Unclear risk | The authors write that "No concomitant or rescue analgesic therapy was permitted during the study." However, then they indicate: "Aspirin up to 325 mg/d for cardiovascular prophylaxis was allowed, as was acetaminophen up to 2000 mg/d for no more than 3 consecutive days for reasons other than osteoarthritis or chronic pain if absolutely necessary. The use of acetaminophen was to be avoided during the washout period and within 48 hours before each postscreening study visit" (p. 218). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, active controls, 2 parallel groups, multicenter study Duration: 12 weeks Study dates: August 2003 to February 2005 Locations: 40 centers in the UK | |
| Participants | Randomized N = 249 Completed N = 149 Mean age: 64 ± 8.9 years Female: 46% Inclusion: age ≥ 45 years, osteoarthritis (OA) of the hip class I‐III according to the American College of Rheumatology (ACR) criteria requiring joint replacement surgery in the opinion of the investigator | |
| Interventions | Celecoxib 200 mg (N = 126) Diclofenac 150 mg (50 mg three times a day) (N = 123) | |
| Outcomes | Primary efficacy outcome: change from baseline to week 6 in the patient’s assessment of arthritis pain on walking on a flat surface using visual analog scale (VAS) 0‐100 mm Secondary efficacy outcomes: change from baseline to week 12 in the patient’s assessment of arthritis pain on walking (VAS 0‐100 mm); the Pain Satisfaction Scale score at weeks 6 and 12; the patient’s and physician’s global assessment of arthritis scores (5‐point scale) Harms outcomes: adverse events (AEs) and serious adverse events (SAEs); physical examination findings; sitting blood pressure; pulse | |
| Funding | Pfizer Inc | |
| Declaration of interest | 3/4 authors were employees of Pfizer. The fourth author declared commercial ties with companies producing the study drugs, including undertaking clinical trials and providing expert advice | |
| Notes | The study protocol was amended such that change in VAS at week 6 rather than week 12 became the primary end point. The authors stated that the amendment did not affect the power of the study, but it allowed participation of patients if they had a period of ≥ 8 weeks before any planned surgery The corresponding author Tamas Koncz was contacted twice to clarify pain outcomes. The author did not respond | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Eligible participants were randomized in the order in which they were admitted to the study according to a computer‐generated schedule provided by sponsor (p. 72) |
| Allocation concealment (selection bias) | Low risk | Central randomisation by sponsor (p. 72) |
| Blinding of participants and personnel (performance bias) | Low risk | Study medication was administered in a double‐blind, double‐dummy fashion (p. 72) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Attrition rates were high, with 43% (54/126) participants lost in the celecoxib group (30/54 due to lack of effectiveness and adverse events) and 37% (45/123) in the naproxen group (25/45 due to lack of effectiveness and adverse events (Figure 1, p. 74). The primary effectiveness outcome was determined in the evaluable population. VAS scores at week 6 were also analyzed in the modified intention‐to‐treat (mITT) population, which included all participants who were randomized and received at least one dose of study medication, had a baseline visual analog scale (VAS) score, and had a VAS score at week 6, week 12, or at the withdrawal visit. Although it is indicated that missing data were imputed using last observation carried forward (LOCF) approach (p. 73), this was not done for the outcome measure "arthritis pain on walking" (p. 76) |
| Selective reporting (reporting bias) | High risk | All outcomes specified in the methods were also reported in the results section. The study protocol was amended such that change in VAS at week 6 rather than week 12 became the primary end point. This amendment was reviewed and approved by the institutional review board and the Multicenter Research Ethics Committee. The amendment did not affect the power of the study, but it allowed participation of participants if they had a period of ≥ 8 weeks before any planned surgery (p. 72, 73). Only most frequently reported adverse events were presented, which had incidence above 2% in at least one treatment arm. Data for "withdrawal due to adverse events" from Figure 1 and the same outcome mentioned in text on page 78 do not match. It is indicated that 6 serious adverse events were recorded, but it is unclear in which study group(s) |
| Other bias | Unclear risk | Participants were allowed to take acetaminophen (at a max. daily dose of 4 g as rescue medication for arthritis flare or non‐arthritis pain, but the medication had to be discontinued ≥ 8 hours before arthritis assessments at baseline, week 6, and week 12) and aspirin (for non‐arthritis reasons, at a dose of ≤ 75 mg four times daily) (p. 72). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, active controls, 2 parallel groups, multicentre study Duration: 6 months Study dates: March 2004 to January 2005 Locations: 47 centers in the USA | |
| Participants | Randomized N = 589 Completed N = 391 Mean age: 60.4 ± 10.9 years Female: 66% Inclusion: age ≥ 40 years, osteoarthritis (OA) of the knee class I‐III according to the American College of Rheumatology (ACR) criteria | |
| Interventions | Celecoxib 200 mg (N = 296) Naproxen 1000 mg/day (500 mg twice daily) (N = 293) | |
| Outcomes | Primary efficacy outcome: 20% improvement from baseline to 6 months in Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) total score Secondary efficacy outcomes: changes from baseline to month 6 in total and sub scale WOMAC scores for participants with 20% improvement; the patient’s and physician’s global assessments of arthritis (5‐point scale); the patient’s assessment of arthritis pain using visual analog scale (VAS) 0‐100 mm Harms outcomes: AEs; clinically significant changes in vital signs | |
| Funding | Pfizer Inc | |
| Declaration of interest | All three authors were full‐time employees of Pfizer, Inc | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomized at study entry using a computer‐generated randomisation scheme (p. 1359) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | This was a double‐blind, double‐dummy study (p. 1358) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | AEs were subjectively classified by the blinded investigator as mild, moderate or severe (p. 1360). For other non‐patient‐reported outcomes, the process of outcome assessment is not described |
| Incomplete outcome data (attrition bias) | High risk | Attrition of participants was high with 32% (94/296) of participants lost in the celecoxib group (44/94 due to adverse events) and 35% (104/293) in the naproxen group (64/104 due to adverse events) as shown in Figure 1 (p. 1362). Efficacy analyses were performed on the modified intention‐to‐treat (mITT) population (all who received at least one dose of study drug and had at least one post‐baseline efficacy measurement), and safety analyses were performed on the safety population (all who received at least one dose of study medication) (p. 1360) |
| Selective reporting (reporting bias) | Unclear risk | All outcomes specified in the methods are also reported in the results section. Adverse events reported if their incidence was above 2% in any group (Table 4). All details about statistical analyses were not reported (information about factors used for calculating least squares mean were missing) |
| Other bias | Unclear risk | Participants were allowed to take aspirin (at stable dose of ≤ 325 mg/d for cardiovascular prophylaxis); calcium‐containing antacids (for osteoporosis prevention); paracetamol ( ≤ 2 g/day for ≤ 3 consecutive days, for reasons other than arthritis was permitted if absolutely necessary, but not within 24 h of any visit); topical pain relief (to non‐index joints was allowed except within 48 h of any visit) (pp. 1358‐9). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double blind, placebo and active controls, 3 parallel groups, multicenter Duration: 6 weeks Study dates: 31 October 2001 to 20 November 2002 Locations: 28 centers in the USA | |
| Participants | Randomized N = 322 Completed N = 253 Mean age: 58 ± 8.6 years Female: 79.3% Inclusion: African American participants aged ≥ 45 years, with osteoarthritis (OA) of the knee diagnosed according to American College of Rheumatology (ACR) guidelines, in a flare state, and with a functional capacity classification of I – III | |
| Interventions | Celecoxib 200 mg (N = 127) Naproxen 1000 mg/day (500 mg twice daily) (N = 128) Placebo (N = 67) | |
| Outcomes | Primary efficacy outcomes: Patient’s Assessment of Arthritis Pain measured on a 0‐100 mm visual analog scale (VAS) Secondary efficacy outcomes: Patient’s and Physician’s Global Assessments of Arthritis, Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC), American Pain Society (APS) pain scores, Pain Satisfaction Scale, Patient Health Questionnaire‐9 (PHQ‐9) scores Harms: measurement of upper gastro‐intestinal (UGI) tolerability | |
| Funding | Pfizer, Inc | |
| Declaration of interest | All three authors full‐time employees of Pfizer, Inc. Editorial support was funded by Pfizer | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomization schedule (p. 2255) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | High attrition observed in the study arms, with celecoxib 20% (9/25 due to adverse events of lack of efficacy), naproxen 16% (9/21 due to adverse events or lack of efficacy), placebo 34% (12/23 due to adverse events or lack of efficacy) as shown in Figure 1. Primary efficacy analysis was carried out on evaluable population, while secondary efficacy analyses were performed using modified intention‐to‐treat populations (p. 2356) |
| Selective reporting (reporting bias) | High risk | Data not shown for all secondary outcomes |
| Other bias | Unclear risk | Participants were allowed to take acetaminophen (up to 2 g/day, for ≤ 3 consecutive days, and not within 24 h before any arthritis assessments), aspirin (≤ 325 mg/d, if they had been taking a stable dose for ≥ 30 days before the first dose of study medication) (p. 2255). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double blind, placebo and active controls, 3 parallel groups, multicenter Duration: 6 weeks Study dates: Not indicated Locations: 31 centers in the USA | |
| Participants | Randomized N = 318 Completed N = 236 Mean age: 60.6 ± 10.6 years Female: 65.3% Inclusion: age ≥ 45 years, of self‐reported Hispanic descent with osteoarthritis (OA) of the knee diagnosed according to American College of Rheumatology (ACR) criteria, who were determined to be in a flare state and had a functional capacity classification of I to III | |
| Interventions | Celecoxib 200 mg (N = 127) Naproxen 1000 mg (500 mg twice daily) (N = 129) Placebo (N = 62) | |
| Outcomes | Primary efficacy outcomes: Patient’s Assessment of Arthritis Pain on a 0‐100 mm visual analog scale (VAS) Secondary efficacy outcomes: Patient’s and Physician’s Global Assessments of Arthritis, Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC), American Pain Society (APS) pain scores, Pain Satisfaction Scale and Patient Health Questionnaire‐9 (PHQ‐9) scores Harms: upper gastrointestinal tolerability | |
| Funding | Pfizer, Inc | |
| Declaration of interest | All four authors were full‐time employees of Pfizer, Inc. Editorial support was funded by Pfizer, Inc | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Predetermined computer randomization schedule (p. 228) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Low risk | All assessments were made by individuals who had been blinded (p. 228) |
| Incomplete outcome data (attrition bias) | Unclear risk | Attrition was high with 24% participants lost in the celecoxib group (5/30 due to adverse events or lack of efficacy), 28% in the naproxen group (11/36 due to adverse events or lack of efficacy) and 26% in the placebo group (6/16 due to adverse events or lack of efficacy) as shown in Figure 1. The population evaluable for efficacy was used for the primary efficacy analysis and the modified intent‐to‐treat population was used for secondary efficacy analyses (pp. 228‐9). It is unclear how missing values were handled |
| Selective reporting (reporting bias) | High risk | Data not shown for all secondary outcomes |
| Other bias | Low risk | From the data provided, no indication of other important risks of bias |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 4 parallel groups, multicenter study Duration: 13 weeks Study dates: not reported Locations: Argentina, Chile, Colombia, Peru, Uruguay, Venezuela, Switzerland, Canada, USA | |
| Participants | Randomized N = 1608 Completed N = 1238 Mean age: 61.2 ± 11.3 years Female: 66% Inclusion: age ≥18 years, primary osteoarthritis (OA) of the knee ≥ 3 months according to the American College of Rheumatology (ACR) criteria, visual analog scale (VAS) 0‐100 mm pain in the target knee ≥ 40 mm | |
| Interventions | Celecoxib 200 mg (N = 446) Lumiracoxib 200 mg (N = 465) Lumiracoxib 400 mg (N = 465) Placebo (N = 232) | |
| Outcomes | Primary efficacy outcomes: pain intensity (VAS mm) in the target knee; patient’s global assessment of disease activity (VAS mm); Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain and function Secondary efficacy outcomes: OA pain intensity in the target knee by visit and physician’s and patient’s global assessments of disease activity by visit Harms outcomes: adverse events (AEs) and serious AEs, standard laboratory tests and urinalysis were regularly collected, electrocardiogram measurements were obtained at screening and at week 13 | |
| Funding | Novartis Pharma AG | |
| Declaration of interest | Not reported. According to the author affiliation, 3/6 authors were employees of Novartis | |
| Notes | The corresponding author Roy Fleischmann was contacted twice to clarify pain outcomes. The author did not respond | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Blinding was maintained by means of a double‐dummy technique (p. 43) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Attrition was high, with 22% (99/446) participants lost in the celecoxib group (66/99 due to lack of efficacy or adverse events) and 29% (68/232) in the placebo group (52/68 due to lack of efficacy or adverse events) as shown in Figure 1. Efficacy evaluations were performed using the intention‐to‐treat (ITT) population, with last observation carried forward (LOCF) techniques used for imputing missing results. Some of the efficacy variables were analysed using the ITT modified data set (with no efficacy data being carried forwards) and the per protocol populations at an exploratory level. Tolerability evaluations were performed in all participants randomized to treatment who had been exposed to study medication (p. 44) |
| Selective reporting (reporting bias) | High risk | Quality of life data, measured by SF‐36, not provided. Data for all adverse events that were specified as safety outcomes not provided, only summary of adverse events (Table 4) |
| Other bias | Unclear risk | Participants were allowed to take paracetamol (≤ 2 g/d, as rescue medication during the screening and treatment periods); low‐dose aspirin (≤ 325 mg/d for a CV indication) (p. 43). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 3 parallel groups, multicenter study Duration: 6 weeks Study dates: not reported Locations: 61 centers in the USA and Canada | |
| Participants | Randomized N = 477 Completed N = 383 Mean age: 62.9 ± 10.2 years Female: 67% Inclusion: age ≥ 40 years, osteoarthritis (OA) of the knee class I‐III according to the American College of Rheumatology (ACR) criteria, OA in a flare state at baseline | |
| Interventions | Celecoxib 200 mg (N = 189) Rofecoxib 25 mg (N = 190) Placebo (N = 98) | |
| Outcomes | Primary efficacy outcomes: patient’s assessment of arthritis pain on the visual analog scale (VAS) to measure OA pain and the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) total domain score at week 6 Secondary efficacy outcomes: patient’s and physician’s global assessments patient’s assessment of arthritis pain on the VAS (pain on walking), and WOMAC sub scales for pain, stiffness, and physical function Harms outcomes: adverse events (AEs) and serious AS, laboratory tests | |
| Funding | Pharmacia Corporation | |
| Declaration of interest | One out of four authors was an employee of the Pharmacia Corporation | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Treatment assignment was based on a computer‐generated randomisation schedule prepared prior to the beginning of the study (p. 3103) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Regimens were masked (double‐dummied) so as to be unidentifiable to both participants and study personnel (p. 3103) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | High overall attrition rates were observed, with attrition of 16% (31/189) in the celecoxib group (21/31 due to treatment failure or adverse event) and 35% (34/96) in the placebo group (26/34 due to treatment failure or adverse event) as shown in Figure 1 (p. 3104). Analyses of the primary efficacy measures were based on the evaluable cohort, with supporting analyses based on the intention‐to‐treat (ITT) cohort. The evaluable cohort included all treated participants except those with no post‐baseline data and those with major protocol deviations. The ITT cohort included all randomized participants who received at least one dose of study medication. Analyses of secondary efficacy measures were based on the ITT cohort. Any missing observations were imputed using the last observation carried forward (LOCF) method (p. 3105) |
| Selective reporting (reporting bias) | Low risk | All outcomes specified in the methods are also reported in the results section. Adverse events were compared across treatment groups and included all participants who received at least one dose of study drugs and consisted of laboratory findings and the adverse event (AE) incidences |
| Other bias | Unclear risk | Participants were allowed low‐dose aspirin (≤ 325 mg/d, for cardiovascular prophylaxis); occasional use of acetaminophen (use of acetaminophen had to be discontinued for 48 hours prior to the arthritis assessments) or antacids (p. 3104). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 3 parallel groups, multicenter study Duration: 12 weeks Study dates: April to December 2008 Locations: 157 centers in the USA | |
| Participants | Randomized N = 619 Completed N = 521 Mean age: 61.9 years (range: 49 to 90 years) Female: 64% Inclusion: age ≥ 50 years, osteoarthritis (OA) of the knee class I‐III according to the American College of Rheumatology (ACR) criteria | |
| Interventions | Celecoxib 200 mg (N = 247) Naproxen 1000 mg plus esomeprazole 40 mg magnesium tablets daily (500 mg + 20 mg twice daily) (N = 248) Placebo (N = 124) | |
| Outcomes | Primary efficacy outcomes: Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain, WOMAC function, and PGA‐VAS scores Secondary efficacy outcomes: American Pain Society, the Patient Outcome Questionnaire (APS‐POQ), WOMAC pain (VAS 100 mm); Multi‐Dimensional Health Assessment Questionnaire (MDHAQ), Outcome Measures for Rheumatology Committee and Osteoarthritis Research Society International Standing Committee for Clinical Trial Response Criteria Initiative (OMERACT‐OARSI) Responder Index Harms outcomes: adverse events (AEs), clinical laboratory tests and vital signs and physical examination findings at screening and final visit | |
| Funding | POZEN Inc | |
| Declaration of interest | 4/5 authors were employees of pharmaceutical companies POZEN Inc and AstraZeneca. Authors also reported stock ownership, research grants and consulting fees from pharmaceutical companies producing study drugs | |
| Notes | Trial registration: ClinicalTrials.gov, http://clinicaltrials.gov/, NCT00664560 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization was conducted via the interactive web response system according to unique participant numbers and a randomisation list provided by the sponsor (p. 1244) |
| Allocation concealment (selection bias) | Low risk | Central randomization (p. 1244) |
| Blinding of participants and personnel (performance bias) | Low risk | Participants, investigators, study site staff, those performing study assessments, Pozen staff and analysts remained blinded to treatment identity throughout the study, except in the case of emergency (p. 1244) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Low risk | Investigators and assessors were blinded to treatment identity during the outcome assessment (p. 1244) |
| Incomplete outcome data (attrition bias) | Unclear risk | Attrition rates were 16% (39/243) in the celecoxib group (16/39 due to adverse events) and 15% (19/124) in the placebo group (7/19 due to adverse events) as shown in Figure 1. All efficacy analyses were based on modified intention‐to‐treat (mITT) population (all randomized participants who took at least one dose of study medication and had data available from at least one post‐baseline efficacy assessment). Missing data were imputed using LOCF analysis (p. 1246) |
| Selective reporting (reporting bias) | Unclear risk | Adverse events reported if their incidence was above 2% in any treatment group (Table 3) |
| Other bias | Unclear risk | Participants were allowed to take oral prednisone (≤ 7.5 mg/day), low‐dose aspirin (≤ 325 mg/day), and antiplatelet agents (non‐concomitant with low‐dose aspirin); supplemental use of antacid (≤ 3 g/d, except within 48 hours before efficacy assessments) (p. 1245). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 3 parallel groups, multicenter study Duration: 12 weeks Study dates: April to December 2008 Locations: 157 centers in the USA | |
| Participants | Randomized N = 615 Completed N = 488 Mean age: 61.9 years (range: 50 to 89 years) Female: 64% Inclusion: age ≥50 years, osteoarthritis (OA) of the knee class I‐III according to the American College of Rheumatology (ACR) criteria | |
| Interventions | Celecoxib 200 mg (N = 247) Naproxen 1000 mg plus esomeprazole 40 mg magnesium tablets daily (500 mg + 20 mg twice daily) (N = 244) Placebo (N = 124) | |
| Outcomes | Primary efficacy outcomes: Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain, WOMAC function, and PGA‐VAS scores. Secondary efficacy outcomes: American Pain Society, the Patient Outcome Questionnaire (APS‐POQ), WOMAC pain (VAS 100 mm); Multi‐Dimensional Health Assessment Questionnaire (MDHAQ), Outcome Measures for Rheumatology Committee and Osteoarthritis Research Society International Standing Committee for Clinical Trial Response Criteria Initiative (OMERACT‐OARSI) Responder Index Harms outcomes: adverse events (AEs), clinical laboratory tests and vital signs and physical examination findings at screening and final visit | |
| Funding | POZEN Inc | |
| Declaration of interest | 4/5 authors were employees of pharmaceutical companies POZEN Inc and AstraZeneca. Authors also reported stock ownership, research grants and consulting fees from pharmaceutical companies producing study drugs | |
| Notes | Trial registration: ClinicalTrials.gov, http://clinicaltrials.gov/, NCT00665431 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization was conducted via the interactive web response system according to unique participant numbers and a randomisation list provided by the sponsor (p. 1244) |
| Allocation concealment (selection bias) | Low risk | Central randomization (p. 1244) |
| Blinding of participants and personnel (performance bias) | Low risk | Participants, investigators, study site staff, those performing study assessments, Pozen staff and analysts remained blinded to treatment identity throughout the study, except in the case of emergency (p. 1244) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Low risk | Investigators and assessors were blinded to treatment identity during the outcome assessment (p. 1244) |
| Incomplete outcome data (attrition bias) | Unclear risk | High attrition rates were reported, with 24% (59/247) in the celecoxib group (22/59 due to adverse events) and 21% (26/124) in the placebo group (6/26 due to adverse events) groups as shown in Figure 1. All efficacy analyses were based on modified intention‐to‐treat (mITT) population (all randomized participants who took at least one dose of study medication and had data available from at least one post‐baseline efficacy assessment). Missing data were imputed using last observation carried forward (LOCF) analysis (p. 1246) |
| Selective reporting (reporting bias) | Low risk | All outcomes specified in the methods are also reported in the results section |
| Other bias | Unclear risk | Participants were allowed to take oral prednisone (≤ 7.5 mg/day), low‐dose aspirin (≤ 325 mg/day), and antiplatelet agents (non‐concomitant with low‐dose aspirin); supplemental use of antacid (≤ 3 g/d, except within 48 hours before efficacy assessments) (p. 1245). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 5 parallel groups, multicenter study Duration: 12 weeks Study dates: not reported Locations: 176 sites in the USA and Canada | |
| Participants | Randomized N = 1061 Completed N = 538 Mean age: 62.6 years (range: 30 to 93 years) Female: 66% Inclusion: osteoarthritis (OA) of the hip, American College of Rheumatology (ACR) clinical and radiographic criteria, functional class I‐III, symptomatic OA flare at the baseline visit Taking up to 325 mg/day aspirin for non‐arthritic reasons was allowed | |
| Interventions | Celecoxib 100 mg (N = 216) Celecoxib 200 mg (N = 207) Celecoxib 400 mg (N = 213) Naproxen 1000 mg (N = 207) Placebo (N = 218) | |
| Outcomes | Primary efficacy outcomes: Patient’s and Physician’s Global Assessments, arthritis pain using visual analog scale (VAS), Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain, joint stiffness and physical function Secondary efficacy outcomes: incidence of withdrawal due to lack of arthritis efficacy and time to withdrawal due to lack of arthritis efficacy Harms outcomes: adverse events and changes from baseline in clinical laboratory tests, vital signs, and physical examinations | |
| Funding | Pharmacia Corporation and Pfizer, Inc | |
| Declaration of interest | Not reported According to the author affiliation, 5/7 authors were employees of Pharmacia Corporation | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Random assignment was stratified by site in blocks of 10 (p. 469). Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | This was a double‐blind trial (p. 468) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Attrition rates were high with attrition of 64% (139/218) in the placebo group (129/139 due to treatment failure or adverse events), 46% (96/207) in the celecoxib 200 mg/day group (88/96 due to treatment failure or adverse events) and 42% (89/207) in the naproxen group (80/89 due to treatment failure or adverse events) as shown in Table 2. All efficacy analyses were performed on the intention‐to‐treat (ITT) cohort, which was defined as all participants who were randomly assigned and took one dose or more of study medication. Missing data values were assigned the participant’s most recent observation for that outcome, including those for participants with treatment failure (p. 470) |
| Selective reporting (reporting bias) | Unclear risk | For arthritis pain outcome measured by visual analog scale (VAS) no statistical measure of dispersion provided. Only adverse events affecting 3% or more of any treatment group reported (Table 2) |
| Other bias | Unclear risk | Participants were allowed to take aspirin (up to 325 mg/d); and acetaminophen (up to 2 g/d, for up to 3 consecutive days, when absolutely necessary for non‐arthritic conditions, the use of acetaminophen was prohibited within 48 hours of an assessment of arthritis efficacy) (p. 469). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double blind, placebo and active controls, 4 parallel groups, multicenter study Duration: 13 weeks Study dates: not reported Locations: 116 centers in 15 countries | |
| Participants | Randomized N = 1684 Completed N = 1488 Mean age: 62.4 ± 10.1 years Female: 70% Inclusion: age ≥18 years, osteoarthritis (OA) of the knee, meeting American College of Rheumatology (ACR) criteria and expected to require chronic non‐steroidal anti‐inflammatory drug (NSAID) or other analgesic therapy for ≥ 3 months | |
| Interventions | Celecoxib 200 mg (N = 420) Lumiracoxib 100 mg (N = 420) Lumiracoxib 100 mg with an initial (loading) dose of 200 mg for the first two weeks of the study (N = 420) Placebo (N = 424) | |
| Outcomes | Primary efficacy outcomes: OA pain intensity in the target knee at week 13 (VAS mm), patient’s global assessment of disease activity at week 13 (VAS mm), total score of Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) questionnaire Secondary efficacy outcomes: OA pain intensity in the target knee by visit using visual analog scale (VAS) mm, patient’s and physician’s global assessment of disease activity by visit (VAS mm), WOMAC total and sub scale scores, Short‐Form Health Survey (SF‐36), response to treatment using Outcome Measures for Rheumatology Committee and Osteoarthritis Research Society International Standing Committee for Clinical Trial Response Criteria Initiative (OMERACT‐OARSI) criteria by visit Harms outcomes: reporting of adverse events and serious adverse events, laboratory testing‐liver function parameters; vital signs | |
| Funding | Novartis Pharma AG | |
| Declaration of interest | Not reported. According to the author affiliation, 4/8 authors were employees of Novartis | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomized using a validated automated system (p. 519) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | This was a double‐blind, double‐dummy study (p. 518) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Suspected GI, cardiovascular, cerebrovascular and hepatic events were assessed in a blinded fashion by independent safety committees according to prespecified criteria (p. 519, 520). For other outcomes, the process of outcome assessment is not described |
| Incomplete outcome data (attrition bias) | Low risk | Attrition was 12% (52/420) in the celecoxib group (33/52 due to adverse events or unsatisfactory therapeutic effect) and 15% (64/424) in the placebo group (11%/64 due to adverse events or unsatisfactory therapeutic effect) as shown in Figure 1 (p. 521). All efficacy evaluations were performed on the intention‐to‐treat (ITT) population which included all randomized participants who had been exposed to study medication. All safety evaluations were performed on the safety population which was identical to the ITT population. Last observation carried forward (LOCF) techniques were used to impute missing data (p. 520) |
| Selective reporting (reporting bias) | Low risk | All primary and secondary outcomes specified in the methods are included in the report of results, except for SF‐36, for which authors provided general description of findings, without any numerical results (p. 522) |
| Other bias | Unclear risk | Participants were allowed paracetamol (≤ 2 g/day, as rescue medication throughout the study, including during the screening/washout period); low‐dose aspirin (≤325 mg/d, for prophylaxis against a cardio‐ or cerebrovascular event in participants considered at increased risk); chondroitin sulfate and/or glucosamine sulfate (if established stable dosage and regimen), corticosteroids (topical, ophthalmic, nasal, or inhaled, at the usual labelled doses), H2‐receptor antagonists, proton pump inhibitors, antacids and cytoprotective agents (taken at the usual labelled doses); and physiotherapy as prescribed by the physician (only when ongoing with a stable regimen) (p. p. 518). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double blind, placebo and active controls, 3 parallel groups, multicenter study Duration: 6 weeks Study dates: February and May 2000 Locations: 20 investigational sites in the USA | |
| Participants | Randomized N = 182 Completed N = 142 Mean age: 62.2 ± 10.3 years Female: 71% Inclusion: age ≥ 40 years, osteoarthritis (OA) of the knee according to the American College of Rheumatology (ACR) criteria, functional class I‐III, worsening signs and symptoms | |
| Interventions | Celecoxib 200 mg (N = 63) Rofecoxib 25 mg (N = 59) Placebo (N = 60) | |
| Outcomes | Primary efficacy outcomes: mean changes from baseline in arthritis pain using visual analog scale (VAS), Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC), proportion of participants who improved in the patient’s global assessment Secondary efficacy outcomes: mean changes from baseline in the VAS for pain on walking Harms outcomes: adverse events (AEs) and serious adverse events (SAEs) | |
| Funding | Pharmacia and Pfizer Inc | |
| Declaration of interest | Not reported. According to the author affiliation, 1/5 authors was an employee of Pharmacia Corporation | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned to treatment groups, and separate computer‐generated randomisation schedules were prepared for participants with different baseline characteristics |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | This was a double‐blind, double‐dummy study (p. 152) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Attrition was 22% (14/63) in celecoxib group (9/14 due to treatment failure or adverse events) and 27% (16/60) in the placebo group (13/16 due to treatment failure or adverse event) as shown in Figure 1 (p. 154). All efficacy and safety analyses were conducted using the intention‐to‐treat (ITT) cohort, which included any participant who received at least one dose of study medication (p. 153). ITT cohort was not defined and methods for handling missing values were not described |
| Selective reporting (reporting bias) | Unclear risk | All outcomes specified in the methods are also reported in results section. Arthritis pain on visual analog scale (VAS) reported without statistical measure of dispersion (Table 2). Adverse events reported if they occurred in 5% or more participants in one study group (Table 3) |
| Other bias | Unclear risk | Participants were allowed to take occasional acetaminophen (for non arthritic pain, use was prohibited during the 48 h before the follow‐up visits at Weeks 3 and 6); low‐dose aspirin (≤ 325 mg/d, for cardiovascular prophylaxis); occasional antacid (p. 153). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 3 parallel groups, multicenter study Duration: 6 weeks Study dates: not reported Locations: 54 institutions across the USA | |
| Participants | Randomized N = 600 Completed N = 450 Mean age: 61.7 years (range: 32 to 88 years) Female: 65% Inclusion: osteoarthritis (OA) of the knee according to the American College of Rheumatology (ACR) criteria, symptomatic | |
| Interventions | Celecoxib 200 mg (100 mg twice daily) (N = 199) Diclofenac 150 mg (50 mg three times a day) (N = 199) Placebo (N = 200) | |
| Outcomes | Primary efficacy outcomes: patient’s assessment of arthritis pain using a visual analogue scale (VAS) where 0 mm = no pain, 100 mm = very severe pain, Patient’s Global Assessment of Arthritis and the Physician’ s Global Assessment of Arthritis (both using five grades from 'very good’ to 'very poor’), Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) Secondary efficacy outcomes: incidence of withdrawal due to lack of efficacy of the study medication, time to withdrawal due to lack of efficacy, and the OA Severity Index (range from 1 to 24 with the lower score as better); American Pain Society (APS) pain measure questionnaires Harms outcomes: spontaneously reported adverse events and adverse event related withdrawals. Safety assessments included recording of serious adverse events, evaluating changes in clinical laboratory tests pre‐ and post‐treatment, and monitoring changes in vital signs | |
| Funding | Pharmacia Corporation | |
| Declaration of interest | Not reported. According to the author affiliation, five out of six authors were employees of Pharmacia Corporation | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | This was a double‐blind study (p. 12) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | High attrition disparity reported: 36% (71/200) in placebo group, 21% (42/201) in celecoxib group and 19% (37/199) in diclofenac group (active treatments vs placebo: P < 0.002) (p. 13). It is indicated that the majority of these discontinuations were due to treatment failure or adverse events, but precise number of patients withdrawn for various reasons is not described (p. 13). Analyses were performed on the intention‐to‐treat (ITT) population defined as all s that took at least one dose of study medication (p. 12). Methods for handling missing values were not described. Adverse events reported if their incidence was above 3% of participants in any treatment group (Table 4) |
| Selective reporting (reporting bias) | Low risk | All outcomes specified in the methods are also reported in results section |
| Other bias | Unclear risk | Participants were allowed to take occasional acetaminophen (for non arthritic pain, use was prohibited during the 48 h before the follow‐up visits at Weeks 3 and 6); low‐dose aspirin (≤ 325 mg/d, for cardiovascular prophylaxis); occasional antacid (p. 153). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double blind, placebo and active controls, 3 parallel groups, crossover multicenter clinical study Study acronym: PACES (Patient Preference for Placebo, Acetaminophen or Celecoxib Efficacy Studies) Duration: 14 weeks Study dates: not reported Locations: Finland and different sites in the USA | |
| Participants | Randomized N = 524 Completed N = not reported Mean age: 63.6 years Female: 62% Inclusion: age ≥ 45 years, osteoarthritis (OA) of knee or hip, radiographic Kellgren‐Lawrence grade 2–4, pain intensity on visual analog scale (VAS) 0‐100: 40‐90 mm | |
| Interventions | Participants were randomized to receive a sequence of two of three treatments, each for 6 weeks. Only results of the first sequence were included in the analysis. Celecoxib 200 mg (N = 181) Acetaminophen 1000 mg (four times a day) (N = 171) Placebo (N = 172) | |
| Outcomes | Primary efficacy outcomes: Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain, stiffness and function, Multi‐Dimensional Health Assessment Questionnaire (MDHAQ) using VAS 100 mm, Short‐Form Health Survey (SF‐36) Secondary efficacy outcomes: patient global assessment, also measured on a 100 mm visual analogue scale, SF‐36 pain scores, MDHAQ activities of daily living scale scores, investigator assessment of patient global status, and investigator assessment of patient change in global status Harms: adverse events (AEs) and serious adverse events (SAEs) | |
| Funding | Pfizer Inc | |
| Declaration of interest | Not reported. According to the author affiliation, 4/12 authors were employees of Pfizer Inc | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | These were double‐blind, double‐dummy trials (p. 932) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Completion rates are reported only for the both period of this cross‐over study, but no data are available on attrition in the first period, or reasons for attrition. The last observation carried forward (LOCF) procedure was used to manage missing data. Analyses of total Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) score, pain score, and paired patient preference were conducted on three groups: intention‐to‐treat (ITT) group (all participants who received a single dose of the study drug in period I); protocol adherent (omitting participants with major protocol violations such as noncompliance); all completers (participants with complete data for the relevant period) (p. 933) |
| Selective reporting (reporting bias) | High risk | Data were not shown for the analyses of the patient global scale, Multi‐Dimensional Health Assessment Questionnaire (MDHAQ) activities of daily living scale, investigator assessment of patient global status, investigator assessment of patient change in global status, and Short‐Form Health Survey (SF‐36) pain scores disclosed patterns similar to those of the primary end points. It is indicated that these data are available on request (p. 935). This was crossover trial, but adverse events were reported as combined for both treatment periods (Table 5) |
| Other bias | Unclear risk | Participants were allowed to take propoxyphene (Darvon) 65 mg up to four times per day was given as a rescue analgesic drug; codeine 60 mg or tramadol (Ultram) 100 mg, up to four times per day, were provided as alternatives to fewer than 5% of participants if propoxyphene was poorly tolerated or ineffective. Participants were instructed not to take any rescue drug within 12 hours of any visit (p. 932). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double blind, placebo and active controls, 3 parallel groups, crossover multicenter clinical study Study acronym: PACES (Patient Preference for Placebo, Acetaminophen or Celecoxib Efficacy Studies) Duration: 14 weeks Study dates: not reported Locations: Finland and different sites in USA | |
| Participants | Randomized N = 524 Completed N = not reported Mean age: 63.6 years Female: 62% Inclusion: age ≥ 45 years, osteoarthritis (OA) of knee or hip, radiographic Kellgren‐Lawrence grade 2–4, pain intensity on visual analog scale (VAS) 0‐100: 40‐90 mm | |
| Interventions | Participants were randomized to receive a sequence of 2 of 3 treatments, each for 6 weeks. Only results of the first sequence were included in the analysis. Celecoxib 200 mg (N = 189) Acetaminophen 1000 mg (4 times a day) (N = 185) Placebo (N = 182) | |
| Outcomes | Primary efficacy outcomes: Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain, stiffness and function, Multi‐Dimensional Health Assessment Questionnaire (MDHAQ) using VAS 100 mm, Short‐Form Health Survey (SF‐36) Secondary efficacy outcomes: patient global assessment, also measured on a 100 mm visual analogue scale, SF‐36 pain scores, MDHAQ activities of daily living scale scores, investigator assessment of patient global status, and investigator assessment of patient change in global status Harms: adverse events (AEs) and serious adverse events (SAEs) | |
| Funding | Pfizer Inc | |
| Declaration of interest | Not reported. According to the author affiliation, four out of twelve authors were employees of Pfizer Inc | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | These were double‐blind, double‐dummy trials (p. 932) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Completion rates are reported only for the both period of this cross‐over study, but no data are available on attrition in the first period, or reasons for attrition. he last observation carried forward (LOCF) procedure was used to manage missing data. Analyses of total Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) score, pain score, and paired patient preference were conducted on three groups: intention‐to‐treat (ITT) group (all participants who received a single dose of the study drug in period I); protocol adherent (omitting participants with major protocol violations such as noncompliance); all completers (participants with complete data for the relevant period) (p. 933) |
| Selective reporting (reporting bias) | High risk | Data were not shown for the analyses of the patient global scale, Multi‐Dimensional Health Assessment Questionnaire (MDHAQ) activities of daily living scale, investigator assessment of patient global status, investigator assessment of patient change in global status, and Short‐Form Health Survey (SF‐36) pain scores disclosed patterns similar to those of the primary end points. It is indicated that these data are available on request (p. 935). This was crossover trial, but adverse events were reported as combined for both treatment periods (Table 5) |
| Other bias | Unclear risk | Participants were allowed to take propoxyphene (Darvon) 65 mg up to four times per day was given as a rescue analgesic drug; codeine 60 mg or tramadol (Ultram) 100 mg, up to four times per day, were provided as alternatives to fewer than 5% of participants if propoxyphene was poorly tolerated or ineffective. Participants were instructed not to take any rescue drug within 12 hours of any visit (p. 932). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 4 parallel groups, multicenter study Duration: 6 weeks Study dates: July 2003 and January 2004 Locations: 30 outpatient units in Germany | |
| Participants | Randomized N = 397 Completed N = 324 Mean age: 62.8 ± 9.8 years Women: 60% Inclusion: osteoarthritis (OA) of the knee ≥ 6 months, meeting two of the following three clinical criteria: (1) morning stiffness of 30 minutes duration, crepitus on motion and age > 40 years; (2) rating their pain in the index knee as > 3 on a five‐point Likert scale; and (3) taking oral non‐steroidal anti‐inflammatory drugs (NSAIDs) at least 3 days per week for the past 3 months or for > 25 of the past 30 days. Participants had to meet three osteoarthritis flare criteria: (1) pain in the index knee on walking > 40 mm on a visual analog scale (VAS); (2) increased by > 15 mm compared with pain on p restudy treatment (screening); and patient global assessment (PGA) score for osteoarthritis of 3 to 5 and at least one grade increase from screening | |
| Interventions | Celecoxib 200 mg (100 mg twice daily) (N = 132) Epicutaneous ketoprofen 110 mg in 4.8 g Transfersome (a semi‐solid formulation, IDEA‐033) (N = 138) Both placebo formulations (N = 127) | |
| Outcomes | Primary efficacy outcomes: Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain and physical function, PGA response Secondary efficacy outcomes: changes from baseline to end of study in PGA of osteoarthritis (5‐point classification), WOMAC stiffness sub scale, use of rescue medication, discontinuation due to lack of efficacy, and primary outcome measures after 2 and 4 weeks of treatment Harms outcomes: physical examination, standard haematology and biochemistry tests, adverse events (AEs), plasma concentrations of ketoprofen, scoring of erythema and oedema of the skin area exposed to Transfersome | |
| Funding | IDEA AG and McNeill Consumer & Specialty Pharmaceuticals | |
| Declaration of interest | Not reported. 2/5 authors were employees of the pharmaceutical companies producing study drugs | |
| Notes | Trial registration: ClinicalTrials.gov, http://clinicaltrials.gov/, NCT00317733 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated centralized randomisation list was used (p. 1179) |
| Allocation concealment (selection bias) | Low risk | Randomisation was performed by a centralized telephone procedure (p. 1179) |
| Blinding of participants and personnel (performance bias) | Low risk | Treatment assignments were double‐blind |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Total attrition and attrition was 17% (23/132) in the celecoxib group (21/23 due to adverse events and lack of efficacy) and 19% (25/127) in the placebo group (23/25 due to adverse events and lack of efficacy) as shown in Figure 1 (p. 1180). The primary efficacy analysis was performed on all randomized participants who used at least one dose of study medication in intention to treat (ITT) population. All efficacy outcome measures were also analysed in the per protocol (PP) population, which included participants who did not have significant protocol deviations (p. 1179) |
| Selective reporting (reporting bias) | Low risk | All outcomes specified in the methods are also reported in results section |
| Other bias | Unclear risk | Participants were allowed to take paracetamol (up to 2 g/d, as rescue medication for knee pain for 3 days in any week, apart from the 48 hours preceding a study visit) (p. 1179). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, double‐dummy, placebo and positive‐internal (celecoxib)‐controlled, 3 parallel groups, multicenter study Duration: 13 weeks Study dates: not reported Locations: 162 centers in 5 countries; USA (7 centers), Canada (42 centers), Germany (30 centers), Italy (12 centers), and UK (7 centers) | |
| Participants | Randomized N = 1262 Completed N = 951 Mean age: Lumiracoxib 100 mg once daily group 61.7 years (range 40 to 90 years); celecoxib 200 mg once daily group 61.7 years (range 30 to 86 years); and placebo group 61.4 years (range 41 to 90 years) Female: 61.7 % Inclusion: symptomatic primary osteoarthritis (OA) of the hip according to the American College of Rheumatology (ACR) criteria, who had experienced symptoms for at least 3 months prior screening | |
| Interventions | Lumiracoxib 100 mg (N = 427) Celecoxib 200 mg (N = 419) Placebo (N = 416) | |
| Outcomes | Efficacy outcomes: the patient's global assessment of disease activity measured on a 100‐mm visual analog scale (VAS); the patient's functional status during the previous 48 h using the Western Ontario and McMaster Universities Osteoarthritis Index Likert version 3.1 (WOMAC LK 3.1) sub scale scores (pain, difficulty in performing daily activities (DPDA) as a measure of function, stiffness), and total score; the patient’s overall OA pain intensity in the target hip (measured on a 100‐mm VAS); the physician's global assessment of disease activity (measured on a 100‐mm VAS); actual OA pain intensity in the target joint in the evening during screening and at 12 h post dose for 1 week immediately following the baseline (day 0), visit 3 (day 28), and visit 4 (day 56) using a 0‐100‐mm VAS; and rescue medication use. Harms outcomes: recording adverse events (AEs) and serious adverse events (SAEs); physical examinations, laboratory tests (hematology, blood chemistry, and urinalysis), and vital sign assessments; prespecified cardiovascular (CV)/cerebrovascular, liver, and GI safety events were adjudicated, blinded to treatment, by the appropriate independent CV, liver or gastrointestinal (GI) safety committee | |
| Funding | "Novartis designed the study in collaboration with the independent authors. Both Novartis and the independent authors were involved in the collection, analysis, and interpretation of the date, as well as in the writing of the report and in the decision to submit the paper for publication. Novartis was the sponsor funding the role of the medical editor/writer" (p. 1442) | |
| Declaration of interest | According to the author affiliations, four out of 9 authors were employees of Novartis. For other authors the following was reported in the conflict of interest section: receiving research grants or honoraria and having ownership in relevant pharmaceutical companies (p. 1442) | |
| Notes | Trial registration: ClinicalTrials.gov, http://clinicaltrials.gov/, NCT00154219 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Patients were randomized according to a randomization list, which was computer generated by Novartis Drug Supply Management (Basel, Switzerland) using a validated system (Almedica Drug Label System) (p. 1434) |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Patients, investigator staff, and data analysts remained blind to the identity of the treatment from the time of randomization until database lock. Blinding was maintained by the use of study drugs that were identical in packaging, labelling, schedule of administration, appearance, and odor (p. 1435) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Low risk | Investigator staff and data analysts were blinded (p. 1435) |
| Incomplete outcome data (attrition bias) | High risk | High attrition was reported; 92/419 (22%) treated with celecoxib 200 mg (unsatisfactory therapeutic effect: 32 (7.6%), adverse events (AEs): 22 (5.3%), protocol violation(s): 12 (2.9%), withdrew consent: 14 (3.3%), administrative problems: 6 (1.4%), lost to follow‐up: 3 (0.7%), abnormal lab value(s) or test results: 1 (0.2%), deaths: 2 (0.5%)) and 129/416 (31%) receiving placebo (unsatisfactory therapeutic effect: 80 (19.2%), AEs: 18 (4.3%), protocol violation(s): 11 (2.6%), withdrew consent: 11 (2.6%), administrative problems: 2 (0.5%), lost to follow‐up: 6 (1.4%), abnormal lab value(s) or test results: 1 (0.2%), deaths: 0 (0.0%)) (p. 1437) |
| Selective reporting (reporting bias) | Low risk | All primary outcomes were included in report of results. (pp. 1436, 1438‐9). Secondary outcomes data shown. (pp. 1439‐40). Adverse events were defined and reported. (pp. 1440‐1). |
| Other bias | Unclear risk | The use of rescue medication acetaminophen (paracetamol) was permitted up to a maximum of 3 g/day during the study. Patients who took > 3 g of acetaminophen daily for four or more continuous days during the study were to be discontinued from the trial due to “unsatisfactory therapeutic effect.” No analgesic rescue medications were to be taken in 24 h before a study visit for evaluation (p. 1435). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 4 parallel groups, multicenter study Duration: 13 weeks Study dates: not reported Locations: 38 centers in Canada and 137 centers in the USA | |
| Participants | Randomized N = 1551 Completed N = 1182 Mean age: 60.5 ± 10.8 years Women: 62% Inclusion: age ≥18 years, osteoarthritis (OA) of the knee according to the American College of Rheumatology (ACR) criteria with symptoms requiring NSAID treatment for ≥ 3 months before study entry | |
| Interventions | Celecoxib 200 mg (N = 393) Lumiracoxib 100 mg (4 times a day) (N = 391) Lumiracoxib 100 mg 4 times a day, with a loading dose of 200 mg 4 times daily for the first 2 weeks of the study (N = 385) Placebo (N = 382) | |
| Outcomes | Primary efficacy outcomes: OA pain intensity in the target knee (VAS 100 mm), the patient's global assessment of disease activity using visual analog scale (VAS) 100 mm, Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) total score Secondary efficacy outcomes: OA pain intensity in the target knee, the patient's and physician's global assessments of disease activity, WOMAC pain and physical function, use of rescue medication Harms outcomes: adverse events (AEs) and serious adverse events (SEAs), physical examination, vital signs at each clinical visit, standard laboratory test | |
| Funding | Not reported | |
| Declaration of interest | Not reported. According to the author affiliations, 3/6 authors were employees of Novartis Corporation and lumiracoxib by Novartis was used in the study | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Blinding was maintained through use of the double‐dummy technique (p. 66) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Unblined safety data were reviewed on a quarterly basis by an independent Data Safety Monitoring Board. In addition, independent safety committees reviewed individual cases of suspected events in a blinded fashion (p 67). For other outcomes, the process of outcome assessment not described |
| Incomplete outcome data (attrition bias) | High risk | Attrition was high with 20% (80/393) of participants lost in the celecoxib group (58/80 due to adverse events and unsatisfactory therapeutic effect) and 34% (132/382) in the placebo group (94/132 due to adverse events and unsatisfactory therapeutic effect) as shown in Figure 1 (p. 68). The intention‐to‐treat (ITT) population, defined as all randomized participants who took at least one dose of study medication, was used in all efficacy analyses and was identical to the safety population, which was used in all safety analyses. Last observation carried forward (LOCF) techniques were used to account for missing data (p. 67) |
| Selective reporting (reporting bias) | Unclear risk | All outcomes specified in the methods are also reported in results section. Adverse events reported if their incidence was above 3% in any treatment group (Table 5) |
| Other bias | Unclear risk | Participants were allowed to take low‐dose aspirin (≤ 325 mg/d, for CV prophylaxis in participants at increased cardiovascular risk); acetaminophen (500 mg tablets, as rescue medication throughout the study, including during the screening period); chondroitin sulphate and/or glucosamine sulphate (if the dose and regimen were established and stable, including during the screening period), corticosteroids (topical, ophthalmic, nasal, or inhaled, at the usual labelled doses), histamine‐2‐receptor antagonists, proton pump inhibitors, antacids and cytoprotective agents (taken at the usual labelled doses), and physiotherapy as prescribed by the physician (only if an ongoing, stable regimen) (p. 66). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 4 parallel groups, multicenter study Duration: 6 weeks Study dates: October 1999 and September 2000 Locations: USA | |
| Participants | Randomized N = 1521 Completed N = 1248 Mean age: 61.5 years (range: 39 to 92 years) Women: 68% Inclusion: age ≥ 40 years, osteoarthritis (OA) of the knee or hip ≥ 6 months, class I‐III according to the American College of Rheumatology (ACR) criteria, visual analog scale (VAS) 0‐100 mm pain on walking on flat surface score on Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) at least 40 mm, VAS 0‐100 mm patient assessed pain at night at least 40 mm, an increase of 15 mm from baseline (on therapy) on patient assessed pain on walking on flat surface VAS, and worsening in Investigator Global Assessment of Disease Status (IGADS) of at least one point on five‐point Likert scale compared with the screening visit. | |
| Interventions | Celecoxib 200 mg (N = 456) Rofecoxib 12.5 mg (N = 456) Rofecoxib 25 mg (N = 459) Placebo (N = 150) | |
| Outcomes | Primary efficacy outcomes: WOMAC (VAS 100 mm), Patient Global Assessment of Response to Therapy (PGART) (Likert 0‐4), Investigator’s global assessment of response to therapy (IGART) (Likert 0‐4), Patient Global Assessment of Disease status (PGADS) (VAS 100 mm) Harms outcomes: adverse events (AEs) and serious adverse events (SAEs), physical examination, vital signs (blood pressure and pulse), blood and urine tests | |
| Funding | Merck & Co. Inc | |
| Declaration of interest | 3/6 authors were employees of Merck &Co., Inc. The other authors reported financial ties with pharmaceutical companies, including consultant fees, clinical research support, equity interest, medical advisory bureaus, speaker bureaus, board of directors | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study (p. 1355) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Independent, blinded adjudication committees evaluated cardiovascular and gastrointestinal adverse events (p. 1356). The outcome assessment process was not described for other outcomes |
| Incomplete outcome data (attrition bias) | High risk | Attrition was 18% (80/456) in celecoxib group (68/80 due to lack of efficacy and adverse events) and 38% (57/150) in placebo group (48/57 due to lack of efficacy and adverse events) as shown in Figure 1A (p. 1357). A modified intention‐to‐treat (mITT) analyses method was employed for the analyses of efficacy and safety data. For efficacy analyses, all participants with a baseline value and at least one on‐treatment value were included, with the last observation carried forward for those with missing on‐treatment values (pp. 1356‐7) |
| Selective reporting (reporting bias) | High risk | Data are missing for the outcome of Investigator’s global assessment of response to therapy (IGART) |
| Other bias | Unclear risk | Participants were allowed to take after the first 7 days acetaminophen (max. 2600 mg/d, as rescue therapy for osteoarthritis pain, which was documented and discontinued at least 24 h before each visit); low‐dose aspirin (81 mg or less daily, for cardioprotective effects); glucosamine and chondroitin sulfate (if taken for longer than 6 months, at the same stable dose for the duration of study) (p. 1355). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 3 parallel groups, multicenter study Duration: 6 weeks Study dates: October 1999 and September 2000 Locations: USA | |
| Participants | Randomized N = 1082 Completed N = 897 Mean age: 62.1 years (range: 40 to 91 years) Women: 66% Inclusion: age ≥ 40 years, osteoarthritis (OA) of the knee or hip ≥ 6 months, class I‐III according to the American College of Rheumatology (ACR) criteria, visual analog scale (VAS) 0‐100 mm pain on walking on flat surface score on Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) at least 40 mm, VAS 0‐100 mm patient assessed pain at night at least 40 mm, an increase of 15 mm from baseline (on therapy) on patient assessed pain on walking on flat surface VAS, and worsening in Investigator Global Assessment of Disease Status (IGADS) of at least one point on five‐point Likert scale compared with the screening visit | |
| Interventions | Celecoxib 200 mg (N = 460) Rofecoxib 25 mg (N = 471) Placebo (N = 151) | |
| Outcomes | Primary efficacy outcomes: WOMAC (VAS 100 mm), Patient Global Assessment of Response to Therapy (PGART) (Likert 0‐4), Investigator’s global assessment of response to therapy (IGART) (Likert 0‐4), Patient Global Assessment of Disease status (PGADS) (VAS 100 mm) Harms outcomes: adverse events (AEs) and serious adverse events (SAEs), physical examination, vital signs (blood pressure and pulse), blood and urine tests | |
| Funding | Merck & Co. Inc | |
| Declaration of interest | 3/6 authors were employees of Merck &Co., Inc. The other authors reported financial ties with pharmaceutical companies, including consultant fees, clinical research support, equity interest, medical advisory bureaus, speaker bureaus, board of directors | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study (p. 1355) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Independent, blinded adjudication committees evaluated cardiovascular and gastrointestinal adverse events (p. 1356). For other outcomes, the process of outcome assessment not |
| Incomplete outcome data (attrition bias) | High risk | Attrition was 15% (in celecoxib group (56/68 due to lack of efficacy or adverse events) and 38% (58/151) in placebo group (50/58 due to lack of efficacy and adverse events) as shown in Figure 1B (p. 1357). A modified intention‐to‐treat (mITT) analyses method was employed for the analyses of efficacy and safety data. For efficacy analyses, all participants with a baseline value and at least one on‐treatment value were included, with the last observation carried forward for those with missing on‐treatment values (pp. 1356‐7) |
| Selective reporting (reporting bias) | High risk | Data are missing for the outcome of investigator’s global assessment of response to therapy (IGART) |
| Other bias | Unclear risk | Participants were allowed to take after the first 7 days acetaminophen (max. 2600 mg/d, as rescue therapy for osteoarthritis pain, which was documented and discontinued at least 24 h before each visit); low‐dose aspirin (81 mg or less daily, for cardioprotective effects); glucosamine and chondroitin sulfate (if taken for longer than 6 months, at the same stable dose for the duration of study) (p. 1355). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, active controls, 3 parallel groups, multicenter study Duration: 12 weeks Study dates: May 2001 and April 2002 Locations: 65 centers in 7 countries participated in this trial | |
| Participants | Randomized N = 404 Completed N = 323 Mean age: 63 years Women: 60% Inclusion: osteoarthritis (OA) of the knee or hip according to the American College of Rheumatology (ACR) criteria Ongoing treatment with 81 mg to 325 mg of aspirin daily (if used for at least 30 days before randomization) was permitted if the treatment could be maintained at a stable dose throughout the study | |
| Interventions | Celecoxib 200 mg (N = 136) Rofecoxib 25 mg (N = 138) Naproxen 1000 mg (500 mg twice daily) (N = 130) | |
| Outcomes | Efficacy outcomes: patient’s assessment of arthritis pain (VAS 0‐100 mm), patient’s global assessment of arthritis; Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) scale, WOMAC pain, WOMAC physical function Harms outcomes: AEs | |
| Funding | Pharmacia Corporation and Pfizer Inc | |
| Declaration of interest | 2/9 authors were Pfizer Inc. employees | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | There was a single, computer‐generated randomisation code, and each site was given block sizes of 3 (p. 162) |
| Allocation concealment (selection bias) | Low risk | Single (central) randomisation code |
| Blinding of participants and personnel (performance bias) | Low risk | This was a double‐blind study (p. 162) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | Attrition was 16% (22/136) in the celecoxib group (7/136 due to adverse events) and 21% (27/128) in the naproxen group (11/128 due to adverse events) (p. 163). Detailed reasons for attrition were not reported. For osteoarthritis assessment modified intention‐to‐treat (mITT) analysis was performed (all participants who took at least 1 dose of study medication and had a baseline efficacy assessment) (p. 164) |
| Selective reporting (reporting bias) | High risk | Adverse events not reported |
| Other bias | Unclear risk | Participants were allowed ongoing treatment with estrogen, progesterone, and/or testosterone (if used for at least 2 months before randomization); aspirin (81‐325 mg/d, if used for at least 30 days before randomization) (p. 162). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double blind, placebo and active controls, 4 parallel groups, multicenter study Study dates: not reported Duration: 13 weeks Locations: Canada, France, Belgium, Germany, UK, Hungary, Denmark, Switzerland, Italy, Spain | |
| Participants | Randomized N = 1702 Completed N = 1423 Mean age: 64.3 ± 10.4 years Female: 68% Inclusion: age ≥18 years, osteoarthritis (OA) of the knee according to the American College of Rheumatology (ACR) criteria, pain intensity on visual analog scale (VAS) 0‐100 mm in the affected knee > 40 mm in the past 24 h | |
| Interventions | Celecoxib 200 mg (N = 481) Lumiracoxib 200 mg (N = 487) Lumiracoxib 400 mg (N = 491) Placebo (N = 243) | |
| Outcomes | Primary efficacy outcomes: OA pain intensity in the target knee (VAS 100 mm), patient’s global assessment of disease activity (VAS 100 mm), Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain and total score Secondary efficacy outcomes: OA pain intensity in the target knee, patient’s and physician’s global assessment of disease activity, WOMAC pain and physical function Harms outcomes: adverse events (AEs) and serious adverse events (SEAs), physical examinations, vital signs, and standard laboratory tests, electrocardiogram (ECG) measurements | |
| Funding | Novartis Pharma | |
| Declaration of interest | 3/13 authors were employees of Novartis Pharma. Other potential conflicts not reported | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Blinding was maintained by a double‐dummy technique (p. 1420) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Low risk | Attrition was 17% (80/481) in the celecoxib group (62/80 due to unsatisfactory therapeutic effect and adverse events) and 18% (43/243) in the placebo group (36/43 due to unsatisfactory therapeutic effect and adverse events) as shown in Figure 1 (p. 1420). All safety and efficacy evaluations were performed using the intention‐to‐treat (ITT) population. The safety and ITT populations were identical and included all participants randomized to treatment who had been exposed to the study drug. Conventional last observation carried forward (LOCF) methodology was used when data were missing (p. 1422) |
| Selective reporting (reporting bias) | High risk | All outcomes specified in the methods are also reported in results section. Adverse events reported if their incidence was above 3% in any treatment group (Table 5) |
| Other bias | Unclear risk | Participants were allowed to take paracetamol (≤ 2 g/d, supplied by the investigator, as a rescue drug throughout the trial; however, they were asked to refrain from using the rescue drug from midnight before each clinic visit); low dose aspirin (≤ 325 mg/d, for cardiovascular indication) (p. 1421). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo controls, 3 parallel groups, multicenter study Duration: 6 weeks Study dates: not reported Locations: 50 clinical sites in the USA | |
| Participants | Randomized N = 686 Completed N = 522 Mean age: 62.8 ± 10.9 years Women: 67% Inclusion: osteoarthritis (OA) of the knee, class I‐III according to the American College of Rheumatology (ACR) criteria, in a flare state Participants taking a stable dose of aspirin ≤ 325 mg/day for non‐arthritis conditions were eligible | |
| Interventions | Celecoxib 200 mg (100 mg twice daily) (N = 231) Celecoxib 200 mg (once daily) (N = 223) Placebo (N = 232) | |
| Outcomes | Primary efficacy outcomes: the Patient’s and Physician’s Global Assessments of Arthritis; Lequesne Osteoarthritis Severity Index Secondary efficacy outcomes: Patient’s Assessment of Arthritis Pain using visual analog scale (VAS) 100 mm; Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain, stiffness and physical function Harms outcomes: adverse events (AEs) and serious adverse events (SAEs), physical examination, laboratory tests | |
| Funding | GD Searle & Co. and Pfizer Inc | |
| Declaration of interest | Not reported. According to the author affiliations, 5/8 authors were employees of GD Searle & Co | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study (p. 66) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Total attrition was 37% (86/232) in placebo group and 16% (37/231) and 18% (41/223) in the two celecoxib groups. Attrition due to treatment failure was 24% (56/86) in placebo group, while 8% (18/37) and 9% (21/41) in celecoxib groups. Attrition due to adverse events was 9% (20/86) in placebo group, while 5% (11/37) and 4% (9/41) in celecoxib groups (Figure 1, p. 69). All randomized participants were included in the baseline analyses. All efficacy analyses were based on the ITT cohort, which included all participants who were enrolled and took at least one dose of the study medication. All missing observations were extrapolated by carrying forward the last observation (p. 67) |
| Selective reporting (reporting bias) | High risk | All outcomes specified in the methods are also reported in results section. Adverse events reported if their incidence was above 3% in any treatment group (Table 4) |
| Other bias | Unclear risk | Participants were allowed to take aspirin (≤ 325 mg/d, for conditions other than arthritis, for at least 30 days before the first dose of study medication, at the same dose regimen); acetaminophen (up to 2 g/d, for reasons other than relief of arthritis symptoms and for no more than 3 consecutive days, it must have been avoided within 48 hours before arthritis assessments performed at any visit) (p. 67). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
| Methods | Study design: randomized, double‐blind, placebo controls, 3 parallel groups, multicenter study Duration: 6 weeks Study dates: not reported Locations: 98 clinical sites in the USA | |
| Participants | Randomized N = 718 Completed N = 549 Mean age: 61.5 ± 8.5 years Women: 70% Inclusion: osteoarthritis (OA) of the knee, rheumatology and radiographic criteria, class I‐III according to the American College of Rheumatology (ACR) criteria, in a flare state Participants taking a stable dose of aspirin ≤ 325 mg/day for non‐arthritis conditions were eligible | |
| Interventions | Celecoxib 200 mg (100 mg twice daily) (N = 243) Celecoxib 200 mg (once daily) (N = 231) Placebo (N = 244) | |
| Outcomes | Primary efficacy outcomes: the Patient’s and Physician’s Global Assessments of Arthritis, the Lequesne Osteoarthritis Severity Index Secondary efficacy outcomes: Patient’s Assessment of Arthritis Pain‐Visual Analog Scale using visual analog scale (VAS) 0‐100 mm, Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain and physical function, Patient’s and Physician’s Global Assessment of Arthritis, Lequesne Osteoarthritis Index Harms outcomes: adverse events (AEs) and serious adverse events (SAEs), physical examination, laboratory tests | |
| Funding | Pharmacia Corporation and Pfizer Inc | |
| Declaration of interest | Not reported. According to the author affiliations, 4/5 authors were employees of Pharmacia Corporation | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of random sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study (p. 215) |
| Blinding of outcome assessment (detection bias): Patient‐reported outcomes | Low risk | Participants blinded |
| Blinding of outcome assessment (detection bias): Non‐patient‐reported outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | High risk | Total attrition was 33% (80/244) in placebo group, while 20% (49/243) and 17% (40/231) in celecoxib groups (pp. 217‐8). Reasons for attrition not provided. All randomized participants were included in the baseline analyses. All efficacy analyses were based on intention‐to‐treat (ITT) cohort, which included all participants who were enrolled and took at least one dose of study medication. All missing observations were extrapolated using the last observation carried forward (LOCF) approach. Every randomized participant receiving at least one dose of study drug was included in the safety assessment (p. 217) |
| Selective reporting (reporting bias) | Unclear risk | All outcomes specified in the methods are also reported in results section. Adverse events reported if their incidence was above 3% in any treatment group (Table 4) |
| Other bias | Unclear risk | Participants were allowed to take aspirin (≤ 325 mg/d, for reasons other than arthritis, for ≥ 30 days before the first dose of study medication, were permitted to continue with the same dosing regimen); acetaminophen (up to 2 g/d, for 3 consecutive days, for reasons other than relief of arthritis symptoms, it must not have been taken within 48 hours before OA assessments were performed at any visit) (p. 216). Amount of co‐interventions consumed in each groups was not reported and it is unclear whether this could have affected study results |
Abbreviations, in alphabetical order: ACR ‐ American College of Rheumatology, AE ‐ adverse event, ARA ‐ American Rheumatology Association, ARCI ‐ Addiction Research Center Inventory, APS ‐ American Pain Society, APS‐POQ ‐ American Pain Society, the Patient Outcome Questionnaire, BOCF ‐ baseline observation carried forward, CHF ‐ congestive heart failure, CV ‐ cardiovascular, GI ‐ gastrointestinal, HAQ ‐ Health Assessment Questionnaire, IGADS ‐ Investigator Global Assessment of Disease Status, IGART ‐ Investigator’s global assessment of response to therapy, ITT ‐ intention‐to‐treat analysis, LOCF ‐ last observation carried forward, MDHAQ ‐ Multi‐Dimensional Health Assessment Questionnaire, mITT ‐ modified intention‐to‐treat analysis, NSAID =non‐steroidal anti‐inflammatory drug, OA ‐ osteoarthritis, OMERCT‐OARSI ‐ Outcome Measures for Rheumatology Committee and Osteoarthritis Research Society International Standing Committee for Clinical Trial Response Criteria Initiative, PaGAA ‐ Patient’s Global Assessment of Arthritis, PDQ ‐ Physical Dependence Questionnaire, PGA‐VAS ‐ Patient Global Assessment of osteoarthritis using a visual analog scale, PGAC ‐ Patient Global Assessment of Change, PGADS ‐ Patient Global Assessment of Disease status, PGART ‐ Patient Global Assessment of Response to Therapy, PhGAA ‐ Physician’s Global Assessment of Arthritis, PHQ‐9 ‐ Patient Health Questionnaire‐9, SAE ‐ serious adverse event, SF‐36 ‐ Short Form Health Survey, SODA ‐ Severity of Dyspepsia Assessment, TWA ‐ time‐weighted average, UGI ‐ upper gastrointestinal, VAS ‐ visual analog scale, WOMAC ‐ Western Ontario and McMaster Universities Osteoarthritis Index
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| Included participants with post‐traumatic osteoarthritis | |
| Review article; not a randomized controlled trial | |
| Safety of chronic nonsteroidal anti‐inflammatory drugs and concomitant low‐dose aspirin; pooling results from 5 different studies, not a randomized controlled trial | |
| Systematic review of published and unpublished randomized controlled trials on celecoxib, not a randomized controlled trial | |
| Acetaminophen was the comparator; no comparators of interest | |
| Review article; not a randomized controlled trial | |
| Analysis of 5 tirals; three unpublished, one included in this review (Bensen 1999), one excluded from this review (Simon 1998); not a randomized controlled trial | |
| Fewer than 50 participants in each arm | |
| Duration less than four weeks and fewer than 50 participants in each arm | |
| Secondary pooled analysis of trials; not a randomized controlled trial | |
| Secondary pooled analysis of trials; not a randomized controlled trial | |
| Secondary pooled analysis of trials; not a randomized controlled trial | |
| Not a randomized controlled trial | |
| Comparing efficacy and safety of chondroitin sulfate plus glucosamine hydrochloride versus celecoxib; no comparators of interest | |
| Comparison of a combination of glucosamine hydrochloride and chondroitin sulfate versus celecoxib; no comparators of interest | |
| Combination of celecoxib or naproxen combined with proton pump inhibitors; no intervention/comparator of interest | |
| Duration less than four weeks and fewer than 50 participants in each arm | |
| Pooled analysis of two studies (Lehmann 2005; Sheldon 2005) that are included in this review; not a randomized controlled trial | |
| Comparison of continuous versus intermittent celecoxib only; no comparators of interest | |
| Fewer than 50 participants in each arm; duration less than four weeks (NCT01860833) | |
| Duration less than four weeks | |
| Comparison of a combination chondroitin sulfate and glucosamine versus celecoxib; no comparators of interest | |
| Not a randomized controlled trial | |
| Comparison of combined glucosamine hydrochloride and chondroitin sulfate versus celecoxib; no comparators of interest | |
| Comparison of combined glucosamine hydrochloride and chondroitin sulfate versus celecoxib; no comparators of interest | |
| Practice recommendation based on a systematic review by Deeks 2002; not a randomized controlled trial | |
| Fewer than 50 participants in each arm | |
| Comparison between continuous and intermittent treatment with celecoxib; results for placebo group are not shown | |
| Pooled data from 7 placebo‐controlled, 12‐week studies utilizing etoricoxib 30 or 60 mg, celecoxib 200 mg, naproxen 1000 mg, or ibuprofen 2400 mg; not a randomized controlled trial | |
| Randomized controlled trial that included osteoarthritis patients and compared celecoxib 200 mg, LY2951742, placebo subcutaneous injections and oral placebo. On the ClinicalTrials.gov (NCT02192190) it is indicated that the trial was terminated because interim assessment showed lack of efficacy. Full study results are not available. Two conference abstract with study results have been published (Jin 2016, Smith 2016; available as secondary references for this study in the list of references). They indicate that "LY was generally well tolerated but failed to demonstrate adequate efficacy for the treatment of signs and symptoms of OA in patients with moderate to severe knee pain due to OA.". Jin 2016 abstract indicates that at the time of the interim analysis there were only 36 participants in the celecoxib group. Sponsor: Eli Lilly and Company. Therefore, the study was excluded because there were less than 50 participants per study arm | |
| Patients with osteoarthritis and rheumatoid arthritis were not analyzed separately | |
| Not a randomized controlled trial | |
| Fewer than 50 participants in each arm | |
| Chondroitin sulfate vs. celecoxib; no comparators of interest | |
| Not not a randomized controlled trial | |
| Review article; not a randomized controlled trial | |
| Fewer than 50 participants in each arm | |
| Comparison between continuous and intermittent treatment with celecoxib; no comparators of interest | |
| Randomized controlled trial comparing tanezumab (5 or 10 mg) or placebo every 8 weeks with or without oral naproxen 500 mg twice daily or celecoxib 100 mg twice daily. There were no arms of interest for this systematic review | |
| Randomized controlled trial about gastrointestinal toxicity of celecoxib at 400mg a day. Methods to assess disease severity or progression were not used | |
| Duration less than four weeks | |
| Systematic review | |
| Comparison of different doses and timing of celecoxib treatment; no comparators of interest | |
| Comparison of continuous and intermittent celecoxib use; no comparators of interest (NCT00139776) | |
| Fewer than 50 participants in each arm | |
| Pooled study of three controlled trials, where some groups of participants took naproxen 500 mg twice daily and celecoxib 100 mg twice daily; not a randomized controlled trial | |
| Comparison of celecoxib with rofecoxib and valdecoxib; no comparators of interest | |
| Fewer than 50 participants in each arm | |
| Not a randomized controlled trial | |
| Duration less than four weeks | |
| Not a randomized controlled trial | |
| Randomized controlled trial comparing etoricoxib and celecoxib; no comparators of interest | |
| Compared PG201 (Layla), an ethanol extract, versus celecoxib; no comparators of interest |
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | Randomized, double‐blind, parallel‐group trial Duration: 6 weeks Study dates: not indicated Locations: 31 centers in the US |
| Participants | Asian patients aged ≥ 45 years with defined criteria for knee osteoarthritis (OA) |
| Interventions | Celecoxib 200 mg Naproxen 1000 mg (500 mg twice daily) Placebo |
| Outcomes | Primary outcome: change from baseline to week 6 in the Patient’s Assessment of Arthritis Pain, measured using a standard visual analog scale (VAS) ranging from 0 mm (no pain) to 100 mm (worst pain) Secondary outcomes: change from baseline the Patient’s and Physician’s Global Assessments of Arthritis, Western Ontario and McMaster Universities OA Index (WOMAC), Pain Satisfaction Scale, and the Patient Health Questionnaire (PHQ‐9) (to week 6/early termination), and the American Pain Society (APS) pain score (to day 7). The WOMAC total domain score (range 0–96) was the sum of the pain, stiffness and physical function domain scores. The tolerability of celecoxib versus placebo was also evaluated, by comparing treatment‐emergent treatment‐related adverse events (TEAEs) and measurements of upper gastrointestinal (UGI) tolerability. This evaluation was carried out in the safety population (randomized patients receiving at least one dose of study medication) |
| Notes | Study registration: not indicated Study funding: Pfizer Authors' conflict of interest: all four authors employees of Pfizer The study was found as eligible in the updated search performed on April 11, 2017 and it will be included in the next update of this review |
| Methods | Randomized, double‐blind, parallel‐group trial Duration: 13 weeks Date of first enrolment: October 11, 2005 Global completion date: October 19, 2006 |
| Participants | Male or female outpatients 40 years of age or older who have the highest pain intensity occurring in the target hip joint, relative to other osteoarthritis (OA) joints (including the contralateral hip), with a diagnosis of primary hip osteoarthritis which meets American College of Rheumatology (ACR) criteria and have symptoms present for at least 3 months prior to screening. Diagnosis can be made at screening if symptoms have been present for 3 months by history |
| Interventions | Lumiracoxib 100 mg Celecoxib 200 mg Placebo |
| Outcomes | Primary outcomes: 1. Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain sub‐scale score at 13 weeks, 2. Patient's global assessment of disease activity at 13 weeks (0‐100 mm VAS), 3. WOMAC DPDA (difficulty in performing daily activities; a measure of function) sub‐scale score at 13 weeks Secondary outcomes: none |
| Notes | Study identified at the WHO ICTRP registry. Results available online, but incomplete information available to judge risk of bias (https://www.clinicaltrialsregister.eu/ctr‐search/trial/2005‐002772‐14/results). Query about trials on celecoxib was sent to email address [email protected], with no response. We did not find results of this study published in a peer‐reviewed journal |
| Methods | Randomized, double‐blind, parallel‐group trial Duration: 6 weeks Study dates: February 2003 to January 2004 Locations: 34 centres in Spain, Germany and the UK |
| Participants | Patients diagnosed with osteoarthritis (OA) in a flare state and with a Functional Capacity Classification of I to III were eligible for study participation |
| Interventions | Celecoxib 200 mg Ibuprofen 2400 mg (800 mg three times a day) Placebo |
| Outcomes | Primary outcome: non‐inferiority of celecoxib to ibuprofen in Patient’s Assessment of Arthritis Pain (scored 0–100) Secondary outcomes: Western Ontario and McMaster Universities (WOMAC) Osteoarthritis Index, Pain Satisfaction Scale, and upper gastrointestinal tolerability |
| Notes | Study registration: ClinicalTrials.gov identifier: NCT00630929 Study funding: Pfizer Authors' conflict of interest: all four authors employees of Pfizer The study was found as eligible in the updated search performed on April 11, 2017 and it will be included in the next update of this review |
| Methods | Manuscript reports data from 5 previously unpublished post‐approval randomized studies, placebo and active controls, multicenter studies Duration: 6 weeks Study dates: 2001 to 2003 |
| Participants | Included a total of 1497 participants Inclusion: aged at least 40 years, diagnosed with active and symptomatic osteoarthritis (OA), with a Functional Capacity Classification of I‐III, including people in a flare state |
| Interventions | Celecoxib 200 mg Ibuprofen 2400 mg (800 mg three times a day) or Naproxen 1000 mg (500 mg twice daily) Placebo |
| Outcomes | Patient Health Questionnaire‐9 (PHQ‐9) depression score, pain on visual analog scale (VAS), Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) |
| Notes | Corresponding author was contacted regarding missing information. The corresponding author requested to see a protocol for this systematic review. The protocol was sent, but no further response was received |
| Methods | Study design: 3 randomized studies, double‐blind, placebo and active controls, 4 parallel groups, multicenter studies Duration: 12 weeks Study dates: not indicated Locations: various sites in the USA and Canada |
| Participants | Numbers of participants who were randomized and completed study, their aged and percentage of women, were not provided for any of the 3 separate studies. Total number of participants in all 3 studies was 768 |
| Interventions | Celecoxib 200 mg Celecoxib 400 mg Naproxen 1000 mg Placebo |
| Outcomes | Benefit outcomes: Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) total score and pain, stiffness and physical function sub scales; Short‐Form Health Survey (SF‐36) Harms outcomes: adverse events, rate of withdrawal due to adverse events |
| Notes | The study reports data pooled from 3 randomized controlled trials. The data were not presented separately for each trial separately and methods for each RCT were not described separately, which precludes risk of bias assessment for each study. Although it is indicated that these studies were unpublished at the moment of manuscript publication, it is unclear whether they were published subsequently, since there is no clinical trial registration number. A request for data was submitted to Pfizer in March 2014, the manufacturer and sponsor of the study. The response from Pfizer was received on April 7, 2016, indicating that they will not provide the requested data. This response was received from Daireen Garcia, MPH: "Thank you for your submission entitled “Functional Status and Health‐Related Quality of Life of Elderly Osteoarthritic Patients Treated With Celecoxib.” We regret to inform you that the data sets you requested are not available and we are therefore unable to support your request. Your request falls outside of the scope of Pfizer’s clinical trial data (CTD) policy and while we make every effort to support requests that fall outside of our policy, we cannot guarantee that the data sets from clinical trials pre‐dating 2007 will be available. We are committed to supporting important scientific research proposals such as yours and allowing secure access to Pfizer’s anonymized patient‐level data. We appreciate your interest in Pfizer’s clinical trial data access program and would be pleased to review another submission from you in the future." |
| Methods | Randomized, blinded, trial design not indicated Duration: “at least4 weeks” Study dates: not indicated Locations: not indicated |
| Participants | Patients with severe knee osteoarthritis prior to joint replacement surgery |
| Interventions | celecoxib 2dd200mg celecoxib 2dd200mg stopped 3 days prior to surgery, naproxen 2dd250mg No treatment Total N = 172. Number of participants per group not indicated |
| Outcomes | Western Ontario and McMaster Universities (WOMAC) questionnaires were used to evaluate pain, stiffness and function before and after treatment |
| Notes | Study registration: not indicated Study funding: not indicated This was a conference abstract. We are trying to locate the full text. It is unclear from the abstract how many participants were in each group |
| Methods | Blinded, design not indicated Duration: 182 days Study dates: not indicated Locations: “5 European countries” |
| Participants | Participants with knee OA in accordance with the ACR criteria |
| Interventions | Celecoxib 200 mg Chondroitin Sulfate (CS) 800 mg/day) Placebo Total N = 604. Number of participants per group not indicated |
| Outcomes | pain intensity on visual analog scale (VAS), Lequesne index measuring pain and function, Minimum Clinically Important Improvement (MCII), Patient Acceptable Symptoms State (PASS), safety analyses |
| Notes | Study registration: not indicated Study funding: not indicated This was a conference abstract. It is unclear from the abstract whether the study was randomized and how many participants were in each group. In communication with the lead author (Reginster IY) on April 21, 2017 we found out that the study was “just accepted for publication” in Annals of the Rheumatic Diseases. The study will be analyzed in full text upon publication and screened for potential inclusion |
| Methods | Study design: randomized, double‐blind, placebo and active controls, 3 parallel groups, multicenter study Duration: 12 weeks Study dates: not indicated Locations: Europe, South Africa, Latin America, USA, Canada, Asia, Australia and New Zealand |
| Participants | Number of participants that were randomized and completed study, as well as their age and percent of women, were not provided for each of the separate groups |
| Interventions | Celecoxib 200 mg Celecoxib 400 mg Non‐specific non‐steroidal anti‐inflammatory drug (NSAID) therapy ‐ diclofenac 100 mg or naproxen 1000 mg |
| Outcomes | Benefit outcomes: Patients' assessment of arthritis pain (VAS), Patient's Global Assessment of Arthritis, Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) Index (pain, joint stiffness, physical function) Harms outcomes: adjudication of serious upper gastro‐intestinal events, overall evaluation of investigator‐reported adverse events including cardiovascular events |
| Notes | The study included 13,274 participants from 39 countries. Two randomization schedules were used: one for celecoxib treatment groups and naproxen, and the other for the celecoxib treatment groups and diclofenac. However, data were pooled together and shown as NSAID groups and celecoxib groups. Since data were not presented as randomized, they could not be used. The first author was contacted and indicated that Pfizer, the manufacturer and sponsor of the study, owns the data. A request was submitted to Pfizer to provide summary data for this systematic review. The request for data was denied on October 18, 2013. Details about this communication are presented in Table 1. |
Abbreviations, in alphabetical order: APS ‐ American Pain Society, DPDA ‐ difficulty in performing daily activities, OMERCT‐OARSI ‐ Outcome Measures for Rheumatology Committee and Osteoarthritis Research Society International Standing Committee for Clinical Trial Response Criteria Initiative, INSAID ‐ non‐steroidal anti‐inflammatory drug, OA ‐ osteoarthritis, PHQ‐9 ‐ Patient Health Questionnaire, VAS ‐ visual analog scale, WOMAC ‐ Western Ontario and McMaster Universities OA Index
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Clinical study comparing a new single dose chewable tablet as treatment knee osteoarthritis with moderate to severe pain, compared to placebo and celecoxib |
| Methods | A randomized, multicenter, double‐blind, double dummy and parallel study to evaluate the efficacy of the combination of chondroitin sulfate and glucosamine hydrochloride in a single dose chewable tablet versus placebo, using celecoxib as an active control in patients with knee osteoarthritis with moderate to severe pain Duration: 6 months |
| Participants | Osteoarthritis of the knee diagnosed by the diagnostic criteria of the ACR; pain with moderate to severe score between 301 and 400 in the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) sub scale pain, or well with a score greater than 4 cm visual analogue scale of 10 cm; diagnosis clinical pain and functional limitation in the 6 months prior to the inclusion; male or female patients, aged over 40 years. |
| Interventions | Chondroitin sulfate Celecoxib Placebo |
| Outcomes | Primary outcome: Decrease of 26% in the total score of the questionnaire WOMAC score at 6 months of treatment with initial baseline. Secondary outcomes: 1. Improvement in pain in the WOMAC sub scale. |
| Starting date | Date of first enrolment: 11/10/2012 |
| Contact information | Name: Clinical Operations Department Address: C/ Pamplona 92‐94, 08018, Barcelona, Spain Telephone: 34933005218 Email: [email protected] Affiliation: Recerca Clinica SL |
| Notes | Latest study status checked on: December 14, 2016. The status of the study was: "Recruitment status: Authorised ‐ recruitment may be ongoing or finished" |
| Trial name or title | A Double‐blind, Randomized, Multicenter, Active‐ and Placebo‐Controlled Phase III Study to Evaluate the Efficacy and Safety of CG100649 in Osteoarthritis Patients |
| Methods | Allocation: Randomized Endpoint Classification: Safety/Efficacy Study Intervention Model: Parallel Assignment Masking: Double Blind (Subject, Caregiver, Investigator, Outcomes Assessor) Primary Purpose: Treatment Duration: 6 weeks |
| Participants | Participants with osteoarthritis of the hip or knee |
| Interventions | Drug: CG100649 Drug: Celecoxib Placebo |
| Outcomes | Primary Outcome Measures: Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) Pain Subscale in the index joint |
| Starting date | March 2013 |
| Contact information | Su Jeong Yang, [email protected] Seonggu Ro, PhD, [email protected] |
| Notes | Estimated Study Completion Date: March 2014 URL: https://clinicaltrials.gov/ct2/show/NCT01765296 Latest study status checked on: December 14, 2016. The status of the study was: "The recruitment status of this study is unknown because the information has not been verified recently." The last update of the entry was April 23, 2013. |
| Trial name or title | Evaluation of the Efficacy and Safety of Entelon Tab. 150 mg in Patients With Osteoarthritis of Knee |
| Methods | Allocation: Randomized Endpoint Classification: Efficacy Study Intervention Model: Parallel Assignment Masking: Double Blind (Subject, Investigator) Primary Purpose: Treatment Duration: 12 weeks |
| Participants | Patients with a diagnosis of osteoarthritis (OA) of the knee within 3 months prior to study participation, as determined by the American College of Rheumatology (ACR) clinical and radiographic criteria, Kellgren and Lawrence Scale Grade II˜III, both gender, 35 years ≤ age ≤ 75 years |
| Interventions | Drug: Enteron tab. 150mg (vitis vinifera extract 150mg): twice daily for 12 weeks Drug: Celebrex cap. (celecoxib 200 mg): once daily, for 12 weeks Placebo |
| Outcomes | Primary Outcome Measures: the change of total sum of K‐WOMAC (Korean The Western Ontario and McMaster Universities Arthritis Index) |
| Starting date | July 2012 |
| Contact information | Not provided |
| Notes | Estimated Study Completion Date: June 2014 URL: https://clinicaltrials.gov/ct2/show/NCT01768520 Latest study status checked on: December 14, 2016. The status of the study was: "The recruitment status of this study is unknown. The completion date has passed and the status has not been verified in more than two years." The last update of the entry was January 2013. |
| Trial name or title | A Multicentre, Comparative, randomized, Double‐blind, Double‐dummy Clinical Trial on the Efficacy and Safety of Condrosulf Versus Celebrex and Versus a Placebo in the Treatment of Knee Osteoarthritis |
| Methods | Allocation: Randomized Endpoint Classification: Safety/Efficacy Study Intervention Model: Parallel Assignment Masking: Double Blind (Subject, Caregiver, Investigator, Outcomes Assessor) Primary Purpose: Treatment Duration: 6 months |
| Participants | Outpatients of either sex, aged ≥50 years, Participants affected by primary knee osteoarthritis of the medial or lateral femoro‐tibial compartment, Diagnosis according to the American College of Rheumatology (ACR) criteria, Kellgren & Lawrence grade I‐III, Knee osteoarthritis evolving for more than 6 months |
| Interventions | Drug: Chondroitin sulphate Drug: Celecoxib Placebo |
| Outcomes | Primary Outcome Measures:Lequesne's Index, Pain on visual analog scale (VAS) in mm |
| Starting date | April 2014 |
| Contact information | Jean‐Yves Reginster, Prof. MD PhD, Unité d'Exploration du Métabolisme Osseux, CHU Centre ville, Polycliniques Universitaires L‐Brull, Liège, Belgium |
| Notes | Estimated Study Completion Date: June 2016 URL: https://clinicaltrials.gov/ct2/show/NCT02079727 Latest study status checked on: December 14, 2016. The status of the study was "This study is ongoing, but not recruiting participants." The last update of the entry was February 23, 2016. |
Abbreviations, in alphabetical order: ACR ‐ American College of Rheumatology, AE ‐ adverse events, HAQ ‐ Health Assessment Questionnaire, K‐WOMAC ‐ Korean The Western Ontario and McMaster Universities Arthritis Index, MCII ‐ minimal clinically important improvement, OMERACT‐OARSI ‐ Outcome Measures for Rheumatology Committee and Osteoarthritis Research Society International Standing Committee for Clinical Trial Response Criteria Initiative, PASS ‐ patient acceptable symptom state, PGA ‐ Physician's Global Assessment, SGA ‐ Subject's Global Assessment, VAS ‐ visual analog scale, WOMAC ‐ Western Ontario and McMaster Universities Arthritis Index
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Pain Show forest plot | 4 | 1622 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.32, ‐0.12] |
| Analysis 1.1  Comparison 1 Celecoxib versus placebo, Outcome 1 Pain. | ||||
| 2 Physical function Show forest plot | 4 | 1622 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.27, ‐0.07] |
| Analysis 1.2  Comparison 1 Celecoxib versus placebo, Outcome 2 Physical function. | ||||
| 3 Number withdrawn due to adverse events Show forest plot | 28 | 12965 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.99 [0.85, 1.15] |
| Analysis 1.3  Comparison 1 Celecoxib versus placebo, Outcome 3 Number withdrawn due to adverse events. | ||||
| 4 Number experiencing any serious adverse events Show forest plot | 28 | 13393 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.95 [0.66, 1.36] |
| Analysis 1.4  Comparison 1 Celecoxib versus placebo, Outcome 4 Number experiencing any serious adverse events. | ||||
| 5 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 8 | 3263 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.91 [0.24, 14.90] |
| Analysis 1.5 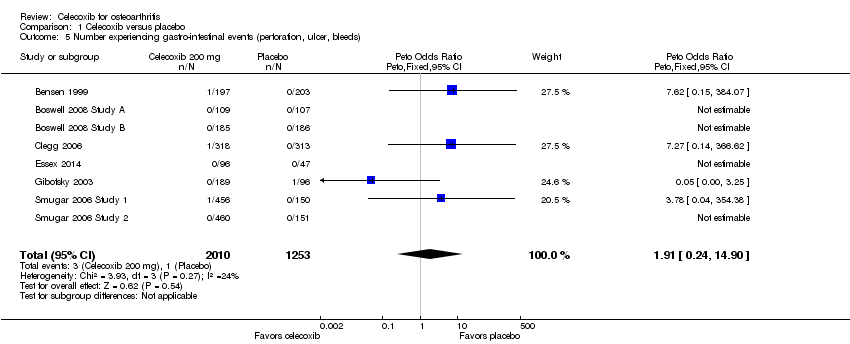 Comparison 1 Celecoxib versus placebo, Outcome 5 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds). | ||||
| 6 Number experiencing cardiovascular events (myocardial infarction, stroke) Show forest plot | 5 | 2947 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.40 [0.73, 15.88] |
| Analysis 1.6  Comparison 1 Celecoxib versus placebo, Outcome 6 Number experiencing cardiovascular events (myocardial infarction, stroke). | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Pain Show forest plot | 2 | 1180 | Mean Difference (IV, Random, 95% CI) | ‐4.52 [‐10.65, 1.61] |
| Analysis 2.1  Comparison 2 Celecoxib versus tNSAIDs, Outcome 1 Pain. | ||||
| 2 Physical function Show forest plot | 1 | 264 | Mean Difference (IV, Random, 95% CI) | ‐4.00 [‐11.40, ‐0.60] |
| Analysis 2.2  Comparison 2 Celecoxib versus tNSAIDs, Outcome 2 Physical function. | ||||
| 3 Number withdrawn due to adverse events Show forest plot | 8 | 3150 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.97 [0.74, 1.27] |
| Analysis 2.3  Comparison 2 Celecoxib versus tNSAIDs, Outcome 3 Number withdrawn due to adverse events. | ||||
| 4 Number experiencing any serious adverse events Show forest plot | 5 | 2404 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.92 [0.66, 1.28] |
| Analysis 2.4  Comparison 2 Celecoxib versus tNSAIDs, Outcome 4 Number experiencing any serious adverse events. | ||||
| 5 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 4 | 1755 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.61 [0.15, 2.43] |
| Analysis 2.5  Comparison 2 Celecoxib versus tNSAIDs, Outcome 5 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds). | ||||
| 6 Number experiencing cardiovascular events (myocardial infarction, stroke) Show forest plot | 1 | 916 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.47 [0.17, 1.25] |
| Analysis 2.6  Comparison 2 Celecoxib versus tNSAIDs, Outcome 6 Number experiencing cardiovascular events (myocardial infarction, stroke). | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 All pain < 24 weeks Show forest plot | 31 | 13069 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.41 [‐0.49, ‐0.32] |
| Analysis 3.1  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 1 All pain < 24 weeks. | ||||
| 2 Pain VAS at 6 weeks Show forest plot | 11 | 3722 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.49 [‐0.64, ‐0.34] |
| Analysis 3.2 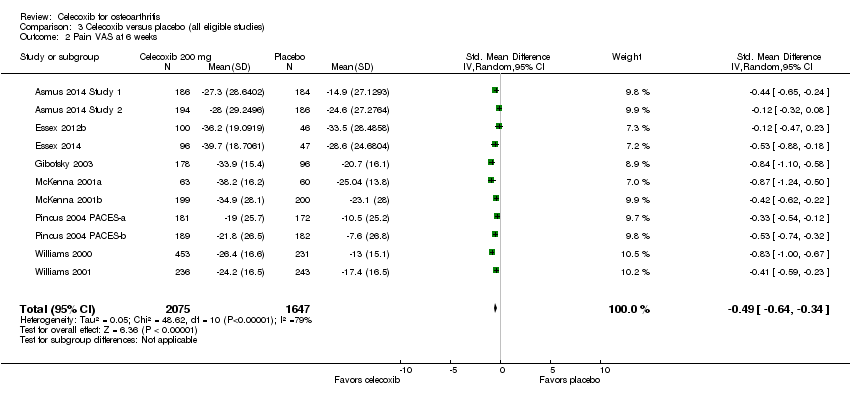 Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 2 Pain VAS at 6 weeks. | ||||
| 3 Pain VAS at 12 weeks Show forest plot | 3 | 1226 | Mean Difference (IV, Random, 95% CI) | ‐11.09 [‐12.68, ‐9.50] |
| Analysis 3.3  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 3 Pain VAS at 12 weeks. | ||||
| 4 Pain VAS at 13 weeks Show forest plot | 5 | 3853 | Mean Difference (IV, Random, 95% CI) | ‐6.35 [‐6.00, ‐4.70] |
| Analysis 3.4 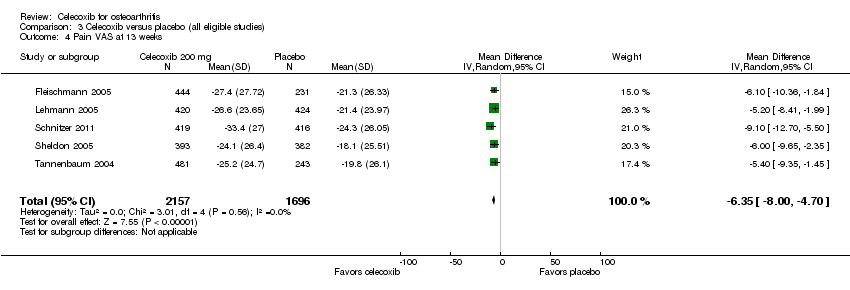 Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 4 Pain VAS at 13 weeks. | ||||
| 5 Pain on walking VAS at 6 weeks Show forest plot | 4 | 1572 | Mean Difference (IV, Random, 95% CI) | ‐12.42 [‐14.83, ‐10.01] |
| Analysis 3.5  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 5 Pain on walking VAS at 6 weeks. | ||||
| 6 Pain on walking WOMAC at 12 weeks Show forest plot | 1 | 402 | Mean Difference (IV, Random, 95% CI) | 7.0 [1.73, 12.27] |
| Analysis 3.6  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 6 Pain on walking WOMAC at 12 weeks. | ||||
| 7 Pain WOMAC at 6 weeks Show forest plot | 12 | 4178 | Mean Difference (IV, Random, 95% CI) | ‐3.28 [‐4.51, ‐2.06] |
| Analysis 3.7 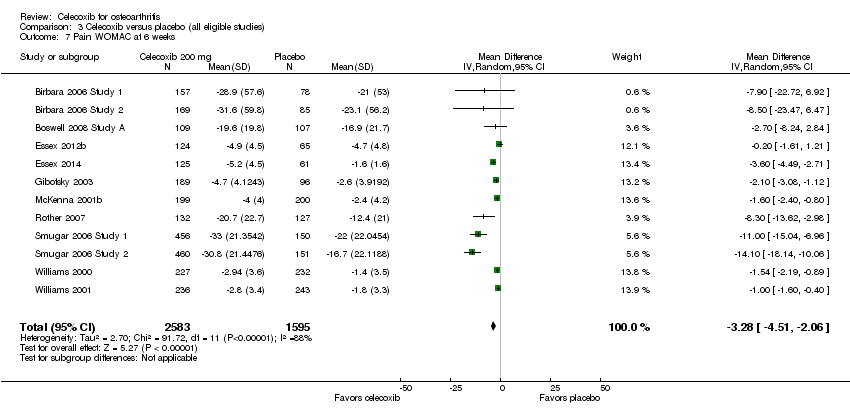 Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 7 Pain WOMAC at 6 weeks. | ||||
| 8 Pain WOMAC at 12 weeks Show forest plot | 9 | 3328 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.32 [‐0.40, ‐0.23] |
| Analysis 3.8  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 8 Pain WOMAC at 12 weeks. | ||||
| 9 Pain WOMAC at 13 weeks Show forest plot | 5 | 3853 | Mean Difference (IV, Random, 95% CI) | ‐1.06 [‐1.31, ‐0.80] |
| Analysis 3.9  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 9 Pain WOMAC at 13 weeks. | ||||
| 10 Pain WOMAC at 24 weeks Show forest plot | 1 | 631 | Mean Difference (IV, Random, 95% CI) | ‐13.10 [‐24.69, ‐1.51] |
| Analysis 3.10  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 10 Pain WOMAC at 24 weeks. | ||||
| 11 All physical function < 24 weeks Show forest plot | 27 | 11940 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.35 [‐0.43, ‐0.27] |
| Analysis 3.11  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 11 All physical function < 24 weeks. | ||||
| 12 Physical function WOMAC at 6 weeks Show forest plot | 12 | 4069 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.41, ‐0.22] |
| Analysis 3.12  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 12 Physical function WOMAC at 6 weeks. | ||||
| 13 Physical function WOMAC at 12 weeks Show forest plot | 9 | 3185 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.46 [‐0.65, ‐0.26] |
| Analysis 3.13  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 13 Physical function WOMAC at 12 weeks. | ||||
| 14 Physical function WOMAC at 13 weeks Show forest plot | 5 | 3853 | Mean Difference (IV, Random, 95% CI) | ‐3.70 [‐4.67, ‐2.74] |
| Analysis 3.14  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 14 Physical function WOMAC at 13 weeks. | ||||
| 15 Physical function WOMAC at 24 weeks Show forest plot | 1 | 631 | Mean Difference (IV, Random, 95% CI) | ‐32.60 [‐81.07, 15.87] |
| Analysis 3.15  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 15 Physical function WOMAC at 24 weeks. | ||||
| 16 Quality of life: SF‐36 physical component scores Show forest plot | 1 | 402 | Mean Difference (IV, Random, 95% CI) | ‐2.2 [‐3.86, ‐0.54] |
| Analysis 3.16  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 16 Quality of life: SF‐36 physical component scores. | ||||
| 17 Quality of life: SF‐36 mental component scores Show forest plot | 1 | 402 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐1.46, 1.86] |
| Analysis 3.17  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 17 Quality of life: SF‐36 mental component scores. | ||||
| 18 Number of responders with at least 50% improvement in WOMAC pain Show forest plot | 4 | 1816 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [1.16, 1.87] |
| Analysis 3.18  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 18 Number of responders with at least 50% improvement in WOMAC pain. | ||||
| 19 Number withdrawn due to adverse events Show forest plot | 28 | 12965 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.99 [0.85, 1.15] |
| Analysis 3.19  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 19 Number withdrawn due to adverse events. | ||||
| 20 Number experiencing any serious adverse events Show forest plot | 28 | 13393 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.95 [0.66, 1.36] |
| Analysis 3.20  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 20 Number experiencing any serious adverse events. | ||||
| 21 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 8 | 3263 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.91 [0.24, 14.90] |
| Analysis 3.21  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 21 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds). | ||||
| 22 Number experiencing cardiovascular events (myocardial infarction, stroke) Show forest plot | 5 | 2947 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.40 [0.73, 15.88] |
| Analysis 3.22  Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 22 Number experiencing cardiovascular events (myocardial infarction, stroke). | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 All pain under 24 weeks Show forest plot | 8 | 2277 | Std. Mean Difference (IV, Random, 95% CI) | 0.00 [‐0.10, 0.10] |
| Analysis 4.1  Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 1 All pain under 24 weeks. | ||||
| 2 All pain over 24 weeks Show forest plot | 1 | 916 | Mean Difference (IV, Random, 95% CI) | ‐2.0 [‐5.32, 1.32] |
| Analysis 4.2  Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 2 All pain over 24 weeks. | ||||
| 3 All physical function under 24 weeks Show forest plot | 7 | 2176 | Std. Mean Difference (IV, Random, 95% CI) | 0.06 [‐0.09, 0.21] |
| Analysis 4.3 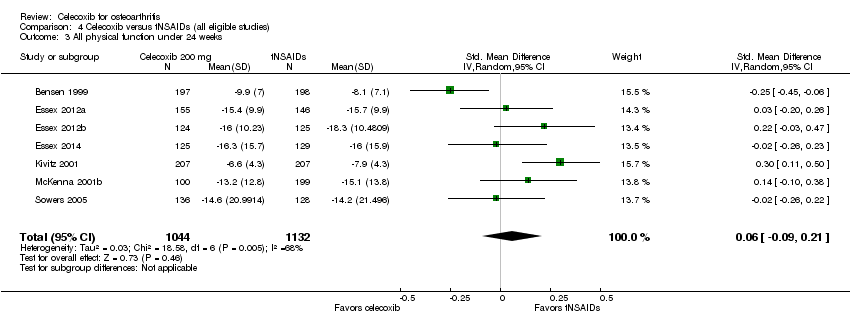 Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 3 All physical function under 24 weeks. | ||||
| 4 Number withdrawn due to adverse events Show forest plot | 9 | 3739 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.85 [0.68, 1.07] |
| Analysis 4.4  Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 4 Number withdrawn due to adverse events. | ||||
| 5 Number experiencing any serious adverse events Show forest plot | 5 | 2404 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.92 [0.66, 1.28] |
| Analysis 4.5  Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 5 Number experiencing any serious adverse events. | ||||
| 6 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 4 | 1755 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.61 [0.15, 2.43] |
| Analysis 4.6  Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 6 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds). | ||||
| 7 Number experiencing cardiovascular events (myocardial infarction, stroke) Show forest plot | 1 | 916 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.47 [0.17, 1.25] |
| Analysis 4.7  Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 7 Number experiencing cardiovascular events (myocardial infarction, stroke). | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 All pain ≤ 24 weeks Show forest plot | 6 | 1781 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.14, 0.05] |
| Analysis 5.1  Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 1 All pain ≤ 24 weeks. | ||||
| 2 Pain VAS at 6 weeks Show forest plot | 2 | 398 | Mean Difference (IV, Random, 95% CI) | 0.55 [‐3.97, 5.07] |
| Analysis 5.2  Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 2 Pain VAS at 6 weeks. | ||||
| 3 Pain VAS at 12 weeks Show forest plot | 3 | 975 | Mean Difference (IV, Random, 95% CI) | ‐0.97 [‐2.69, 0.76] |
| Analysis 5.3  Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 3 Pain VAS at 12 weeks. | ||||
| 4 Pain WOMAC at 6 weeks Show forest plot | 2 | 503 | Mean Difference (IV, Random, 95% CI) | 0.35 [‐0.54, 1.23] |
| Analysis 5.4  Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 4 Pain WOMAC at 6 weeks. | ||||
| 5 Pain WOMAC at 12 weeks Show forest plot | 3 | 1073 | Mean Difference (IV, Random, 95% CI) | ‐0.03 [‐0.65, 0.60] |
| Analysis 5.5  Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 5 Pain WOMAC at 12 weeks. | ||||
| 6 Pain WOMAC at 6 months Show forest plot | 1 | 310 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.46, 0.66] |
| Analysis 5.6  Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 6 Pain WOMAC at 6 months. | ||||
| 7 All physical function ≤ 24 weeks Show forest plot | 6 | 1817 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.18, 0.16] |
| Analysis 5.7  Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 7 All physical function ≤ 24 weeks. | ||||
| 8 Physical function WOMAC at 6 weeks Show forest plot | 2 | 313 | Mean Difference (IV, Random, 95% CI) | ‐2.99 [‐6.43, 0.44] |
| Analysis 5.8  Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 8 Physical function WOMAC at 6 weeks. | ||||
| 9 Physical function WOMAC at 12 weeks Show forest plot | 3 | 1073 | Mean Difference (IV, Random, 95% CI) | ‐1.27 [‐4.32, 1.78] |
| Analysis 5.9  Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 9 Physical function WOMAC at 12 weeks. | ||||
| 10 Physical function WOMAC at 6 months Show forest plot | 1 | 301 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐1.94, 2.54] |
| Analysis 5.10  Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 10 Physical function WOMAC at 6 months. | ||||
| 11 Number withdrawn due to adverse events Show forest plot | 6 | 2173 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.54, 1.23] |
| Analysis 5.11  Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 11 Number withdrawn due to adverse events. | ||||
| 12 Number experiencing any serious adverse events Show forest plot | 2 | 841 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.11 [0.45, 2.75] |
| Analysis 5.12 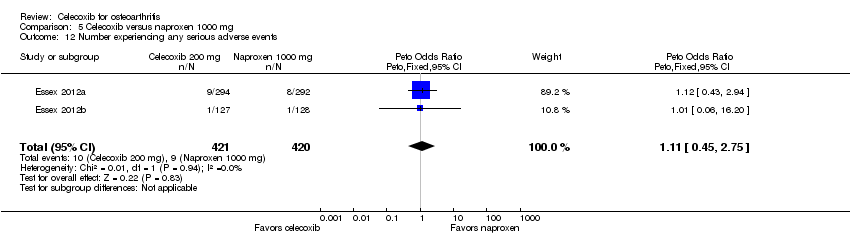 Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 12 Number experiencing any serious adverse events. | ||||
| 13 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 2 | 587 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.37 [0.05, 2.62] |
| Analysis 5.13  Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 13 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds). | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Pain VAS at 1 year Show forest plot | 1 | 916 | Mean Difference (IV, Random, 95% CI) | ‐2.0 [‐5.32, 1.32] |
| Analysis 6.1  Comparison 6 Celecoxib versus diclofenac 100 mg, Outcome 1 Pain VAS at 1 year. | ||||
| 2 Number withdrawn due to adverse events Show forest plot | 1 | 916 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.44 [0.80, 2.61] |
| Analysis 6.2  Comparison 6 Celecoxib versus diclofenac 100 mg, Outcome 2 Number withdrawn due to adverse events. | ||||
| 3 Number experiencing any serious adverse events Show forest plot | 1 | 916 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.62, 1.30] |
| Analysis 6.3  Comparison 6 Celecoxib versus diclofenac 100 mg, Outcome 3 Number experiencing any serious adverse events. | ||||
| 4 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 1 | 916 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.14 [0.01, 2.16] |
| Analysis 6.4  Comparison 6 Celecoxib versus diclofenac 100 mg, Outcome 4 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds). | ||||
| 5 Number experiencing cardiovascular events (myocardial infarction, stroke) Show forest plot | 1 | 916 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.47 [0.17, 1.25] |
| Analysis 6.5  Comparison 6 Celecoxib versus diclofenac 100 mg, Outcome 5 Number experiencing cardiovascular events (myocardial infarction, stroke). | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Pain VAS at 6 weeks Show forest plot | 1 | 398 | Mean Difference (IV, Random, 95% CI) | 1.90 [‐3.68, 7.48] |
| Analysis 7.1  Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 1 Pain VAS at 6 weeks. | ||||
| 2 Pain WOMAC at 6 weeks Show forest plot | 1 | 398 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐0.52, 1.12] |
| Analysis 7.2  Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 2 Pain WOMAC at 6 weeks. | ||||
| 3 Pain on walking at 12 weeks Show forest plot | 1 | 98 | Mean Difference (IV, Random, 95% CI) | 13.0 [3.11, 22.89] |
| Analysis 7.3 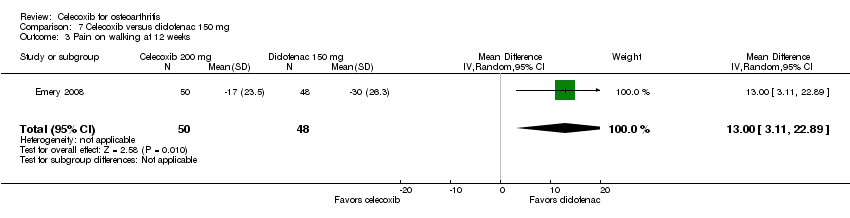 Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 3 Pain on walking at 12 weeks. | ||||
| 4 Physical function WOMAC at 6 weeks Show forest plot | 1 | 398 | Mean Difference (IV, Random, 95% CI) | 1.90 [‐0.72, 4.52] |
| Analysis 7.4  Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 4 Physical function WOMAC at 6 weeks. | ||||
| 5 Number withdrawn due to adverse events Show forest plot | 2 | 650 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.78 [0.46, 1.32] |
| Analysis 7.5  Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 5 Number withdrawn due to adverse events. | ||||
| 6 Number experiencing any serious adverse events Show forest plot | 2 | 647 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.79 [0.21, 2.93] |
| Analysis 7.6  Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 6 Number experiencing any serious adverse events. | ||||
| 7 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 1 | 252 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 7.45 [0.46, 119.74] |
| Analysis 7.7  Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 7 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds). | ||||
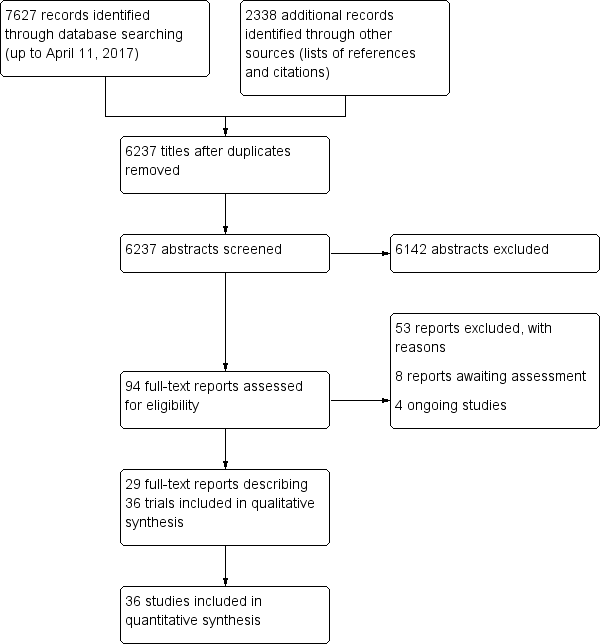
Study flow diagram

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies

Risk of bias summary: review authors' judgements about each risk of bias item for each included study

Funnel plot of comparison: 1 Celecoxib versus placebo, outcome: 1.3 Number withdrawn due to adverse events.

Funnel plot of comparison: 1 Celecoxib versus placebo, outcome: 1.4 Number experiencing any serious adverse events.

Comparison 1 Celecoxib versus placebo, Outcome 1 Pain.

Comparison 1 Celecoxib versus placebo, Outcome 2 Physical function.
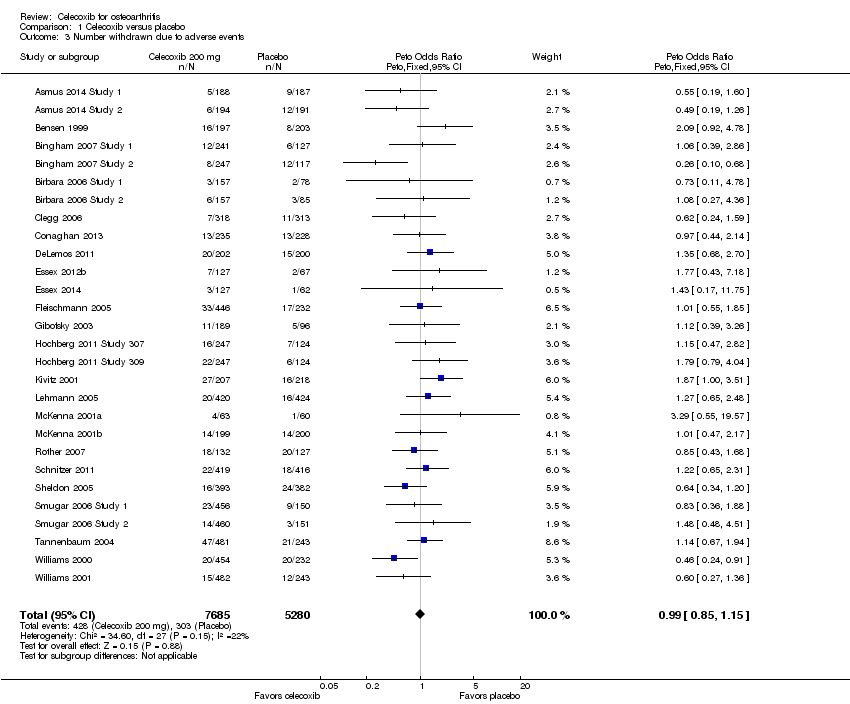
Comparison 1 Celecoxib versus placebo, Outcome 3 Number withdrawn due to adverse events.
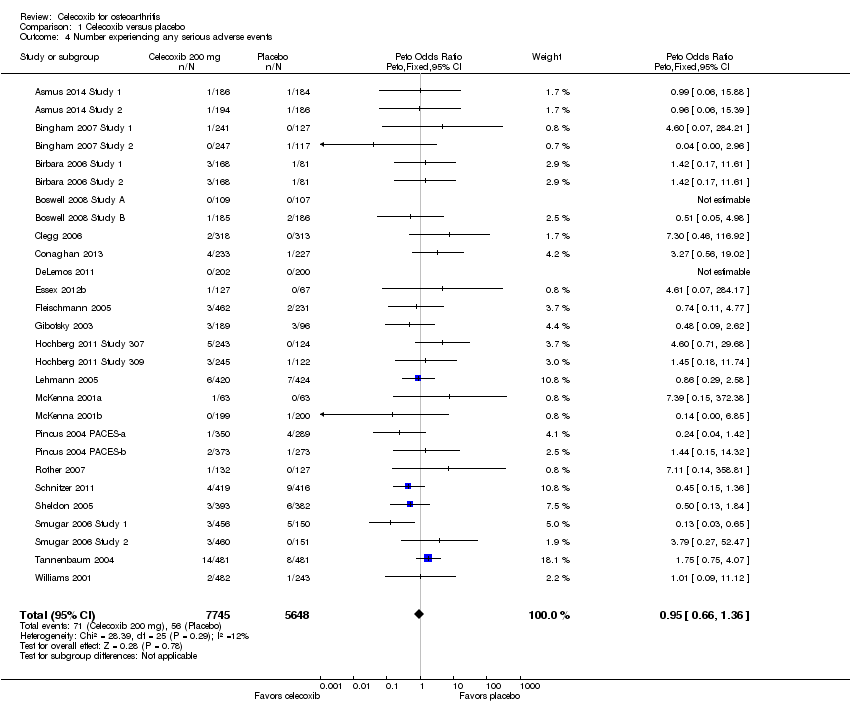
Comparison 1 Celecoxib versus placebo, Outcome 4 Number experiencing any serious adverse events.

Comparison 1 Celecoxib versus placebo, Outcome 5 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds).

Comparison 1 Celecoxib versus placebo, Outcome 6 Number experiencing cardiovascular events (myocardial infarction, stroke).
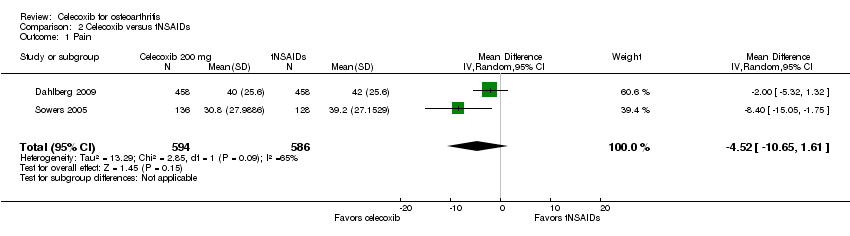
Comparison 2 Celecoxib versus tNSAIDs, Outcome 1 Pain.

Comparison 2 Celecoxib versus tNSAIDs, Outcome 2 Physical function.
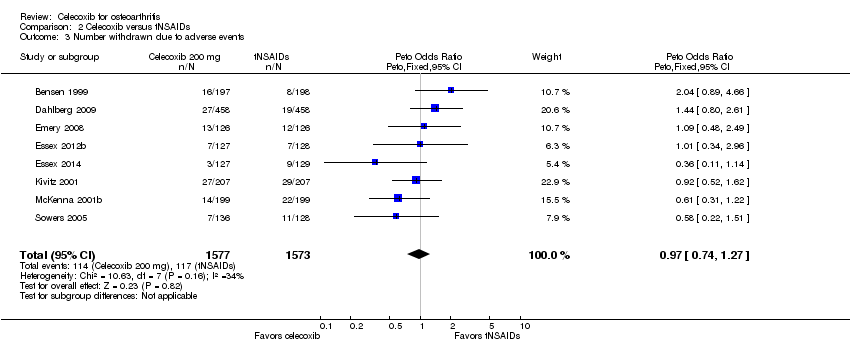
Comparison 2 Celecoxib versus tNSAIDs, Outcome 3 Number withdrawn due to adverse events.

Comparison 2 Celecoxib versus tNSAIDs, Outcome 4 Number experiencing any serious adverse events.

Comparison 2 Celecoxib versus tNSAIDs, Outcome 5 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds).
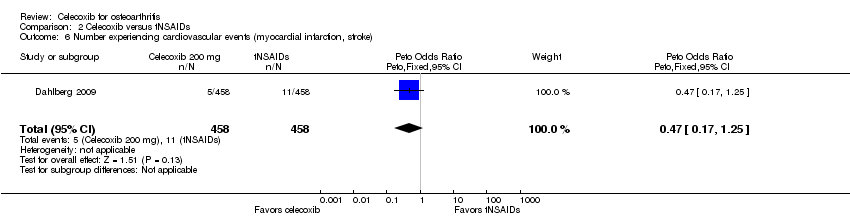
Comparison 2 Celecoxib versus tNSAIDs, Outcome 6 Number experiencing cardiovascular events (myocardial infarction, stroke).

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 1 All pain < 24 weeks.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 2 Pain VAS at 6 weeks.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 3 Pain VAS at 12 weeks.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 4 Pain VAS at 13 weeks.
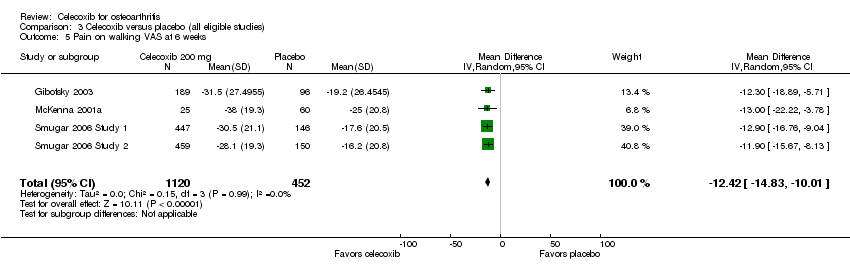
Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 5 Pain on walking VAS at 6 weeks.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 6 Pain on walking WOMAC at 12 weeks.
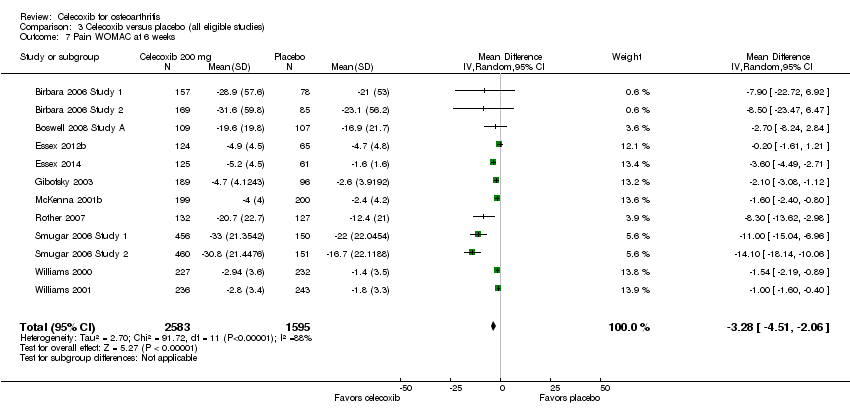
Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 7 Pain WOMAC at 6 weeks.
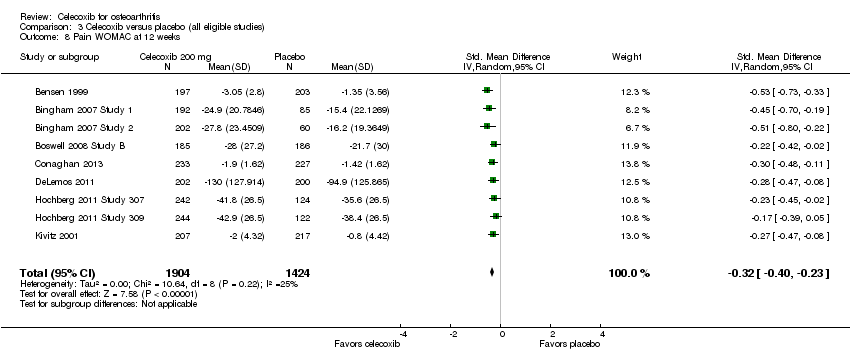
Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 8 Pain WOMAC at 12 weeks.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 9 Pain WOMAC at 13 weeks.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 10 Pain WOMAC at 24 weeks.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 11 All physical function < 24 weeks.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 12 Physical function WOMAC at 6 weeks.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 13 Physical function WOMAC at 12 weeks.
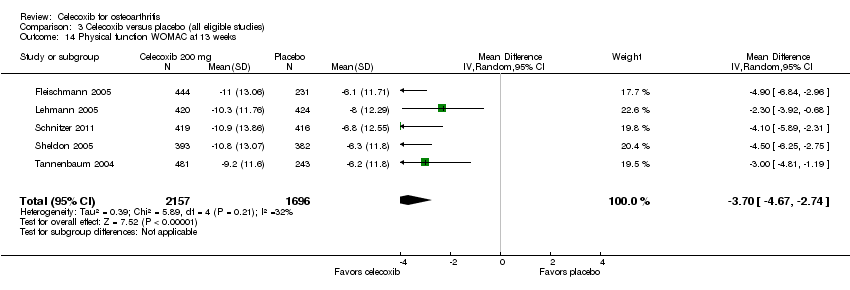
Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 14 Physical function WOMAC at 13 weeks.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 15 Physical function WOMAC at 24 weeks.
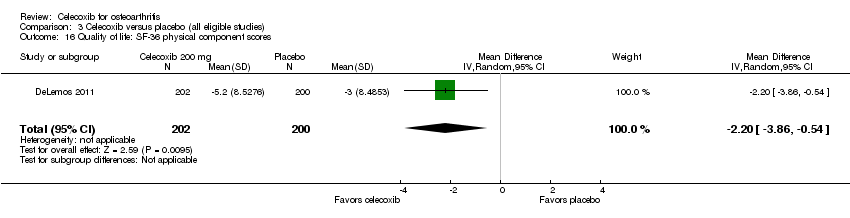
Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 16 Quality of life: SF‐36 physical component scores.
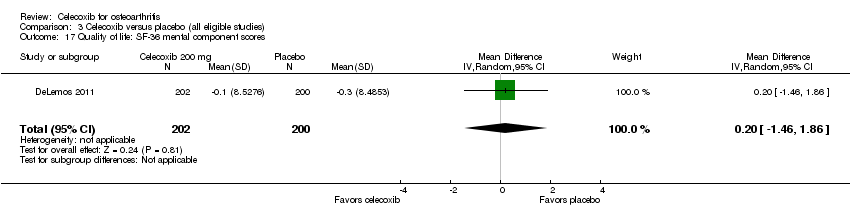
Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 17 Quality of life: SF‐36 mental component scores.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 18 Number of responders with at least 50% improvement in WOMAC pain.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 19 Number withdrawn due to adverse events.

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 20 Number experiencing any serious adverse events.
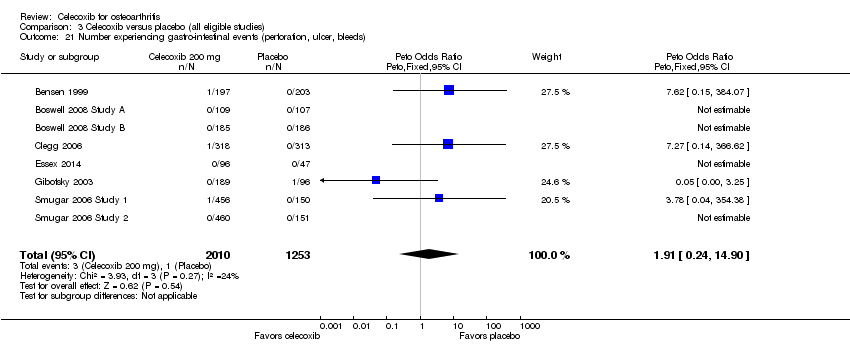
Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 21 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds).

Comparison 3 Celecoxib versus placebo (all eligible studies), Outcome 22 Number experiencing cardiovascular events (myocardial infarction, stroke).
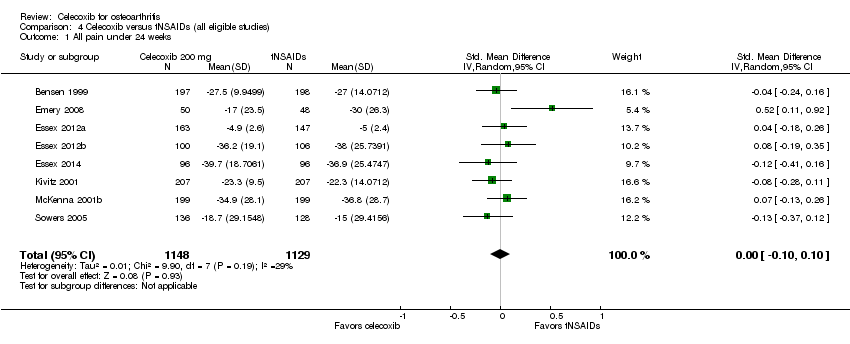
Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 1 All pain under 24 weeks.

Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 2 All pain over 24 weeks.

Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 3 All physical function under 24 weeks.

Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 4 Number withdrawn due to adverse events.

Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 5 Number experiencing any serious adverse events.
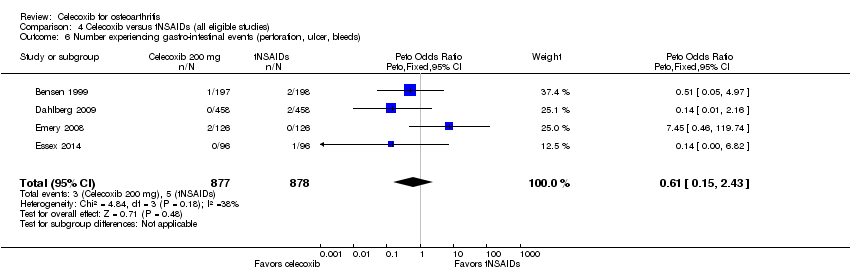
Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 6 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds).

Comparison 4 Celecoxib versus tNSAIDs (all eligible studies), Outcome 7 Number experiencing cardiovascular events (myocardial infarction, stroke).
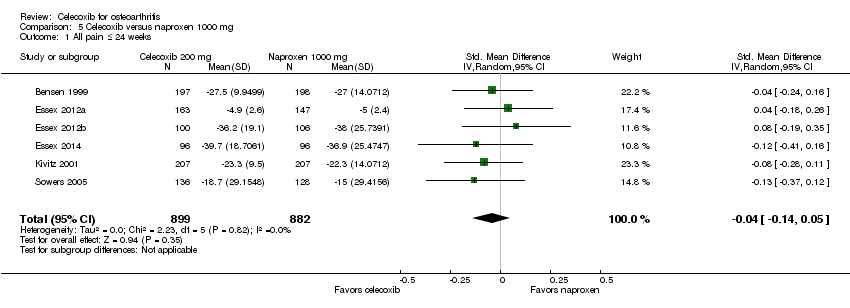
Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 1 All pain ≤ 24 weeks.
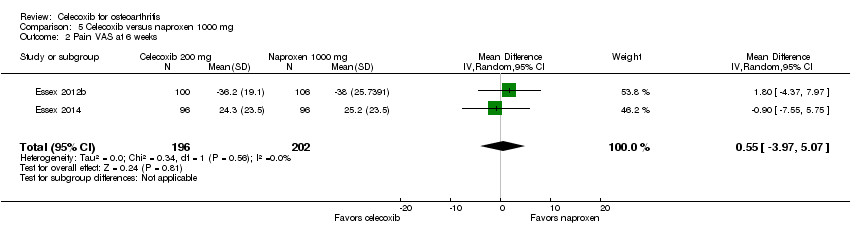
Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 2 Pain VAS at 6 weeks.

Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 3 Pain VAS at 12 weeks.
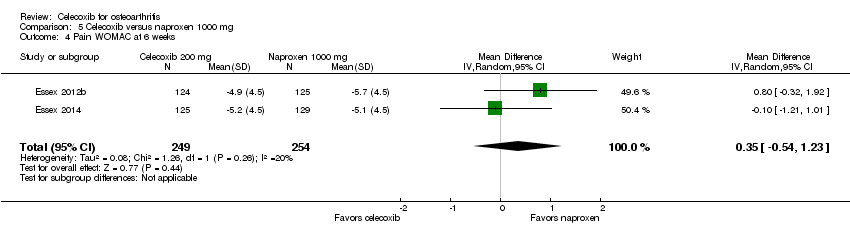
Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 4 Pain WOMAC at 6 weeks.
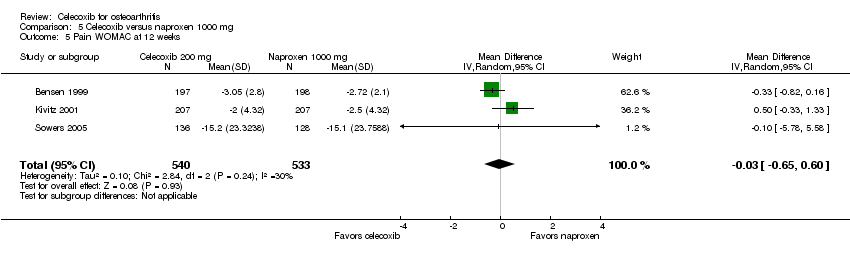
Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 5 Pain WOMAC at 12 weeks.

Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 6 Pain WOMAC at 6 months.

Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 7 All physical function ≤ 24 weeks.
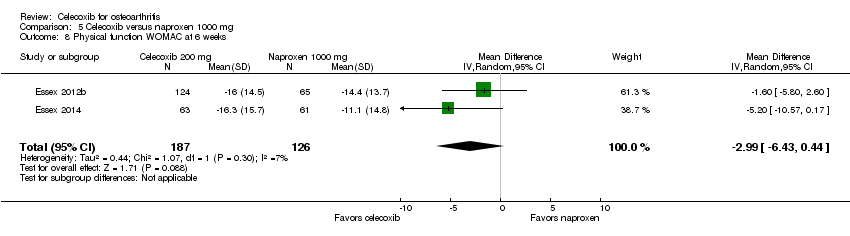
Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 8 Physical function WOMAC at 6 weeks.

Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 9 Physical function WOMAC at 12 weeks.

Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 10 Physical function WOMAC at 6 months.

Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 11 Number withdrawn due to adverse events.
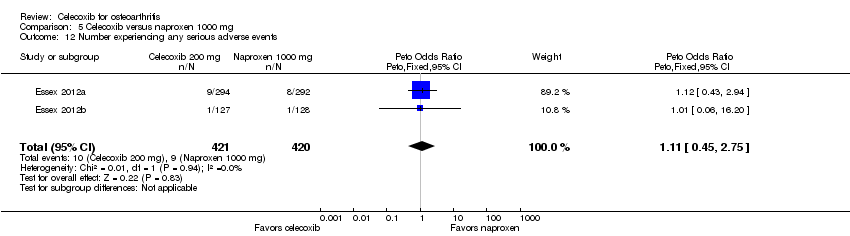
Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 12 Number experiencing any serious adverse events.
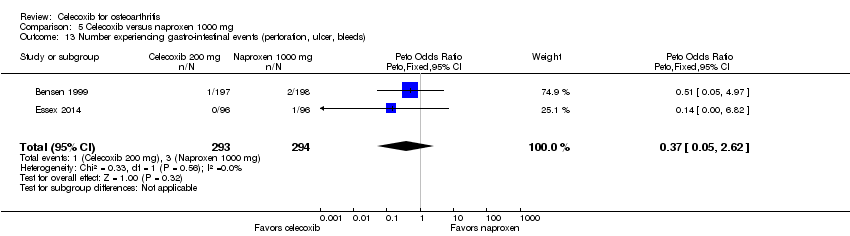
Comparison 5 Celecoxib versus naproxen 1000 mg, Outcome 13 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds).

Comparison 6 Celecoxib versus diclofenac 100 mg, Outcome 1 Pain VAS at 1 year.
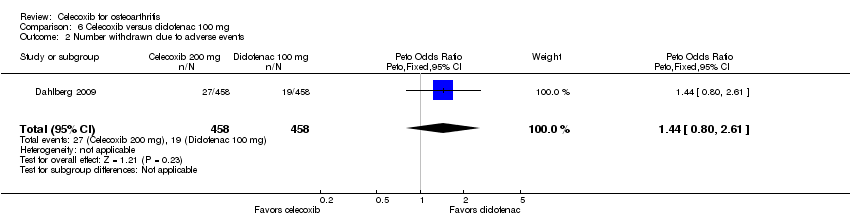
Comparison 6 Celecoxib versus diclofenac 100 mg, Outcome 2 Number withdrawn due to adverse events.

Comparison 6 Celecoxib versus diclofenac 100 mg, Outcome 3 Number experiencing any serious adverse events.

Comparison 6 Celecoxib versus diclofenac 100 mg, Outcome 4 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds).

Comparison 6 Celecoxib versus diclofenac 100 mg, Outcome 5 Number experiencing cardiovascular events (myocardial infarction, stroke).

Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 1 Pain VAS at 6 weeks.

Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 2 Pain WOMAC at 6 weeks.

Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 3 Pain on walking at 12 weeks.

Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 4 Physical function WOMAC at 6 weeks.

Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 5 Number withdrawn due to adverse events.

Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 6 Number experiencing any serious adverse events.
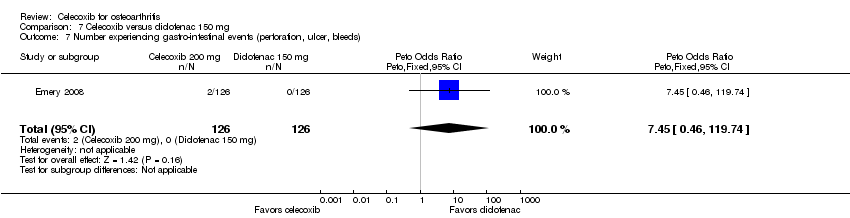
Comparison 7 Celecoxib versus diclofenac 150 mg, Outcome 7 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds).
| Patient or population: osteoarthritis | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with placebo | Risk with celecoxib | |||||
| Pain | The mean pain was 136 | The mean pain in the intervention group was 16 lower (9 lower to 24 lower) 1 | ‐ | 1622 | ⊕⊕⊕⊕ | 3% absolute improvement (95% CI 2% to 5%)1, 12% relative improvement (95% CI 7% to 18% improvement), SMD ‐0.22 (‐0.32 to ‐0.12), NNTB 11 (7 to 18) 2 |
| Physical function | The mean physical function was 540 | The mean physical function in the intervention group was 64 lower (26 lower to 101 lower) 3 | ‐ | 1622 | ⊕⊕⊕⊕ | 4% absolute improvement (95% CI 2% to 6% improvement)3, 12% relative improvement (95% CI 5% to 19% improvement), SMD ‐0.17 (‐0.27 to ‐0.07), NNTB 14 (9 to 34) 2 |
| Quality of life | not estimable | see comment | ‐ | (0 RCTs) | No included studies measured this outcome | |
| Number withdrawn due to adverse events | 57 per 1000 | 55 per 1000 | Peto OR 0.99 | 10996 | ⊕⊕⊕⊝ | 0% absolute change (95% CI 1% less to 1% more), 1% relative change (95% CI 15% less to 15% more) (NNTH = NA) |
| Number experiencing any serious adverse events | 10 per 1000 | 10 per 1000 | Peto OR 0.95 | 10926 | ⊕⊝⊝⊝ | 0% absolute change (95% CI 0% to 0%), 5% relative change (95% CI 34% less to 36% more) (NNTH = NA) |
| Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) | 1 per 1000 | 1 per 1000 | Peto OR 1.91 | 3263 | ⊕⊝⊝⊝ | 0% absolute change (95% CI 0% less to 1% more), 91% relative change (95% CI 76% less to 1390% more) (NNTH = NA) |
| Number experiencing cardiovascular events (myocardial infarction, stroke) | 1 per 1000 | 7 per 1000 | Peto OR 3.40 | 2112 | ⊕⊝⊝⊝ | 0% absolute change (95% CI 0% less to 1% more), 240% relative change (95% CI 27% less to 1488% more) (NNTH = NA) |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| ¹ Calculations based on control group SD for pain of 74.2 (from Clegg 2006); to obtain absolute change, the following formula was used: SMD x SD/500 ² Number needed to treat for an additional beneficial outcome (NNTB) for continuous outcomes calculated using the Wells calculator (from the CMSG Editorial office http://musculoskeletal.cochrane.org/) ³ Calculations based on control group SD for pain of 374.1 (from Clegg 2006); to obtain absolute change, the following formula was used: SMD x SD/500 ⁴ Downgraded one level due to study limitations (all trials had high or unclear risk of at least one type of bias (harms outcomes included all eligible studies)) ⁵ Downgraded two levels for serious imprecision (few events and wide confidence intervals) ⁶ Report Peto OR which can be interpreted as an RR due to the low event rate | ||||||
| Patient or population: osteoarthritis | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with tNSAIDs | Risk with celecoxib | |||||
| Pain | The mean pain was 41 points | The mean pain in the intervention group was 4.52 points lower (10.65 lower to 1.61 higher) | ‐ | 1180 | ⊕⊕⊕⊝ | 5% absolute improvement (95% CI 11% improvement to 2% worse), 11% relative improvement (95% CI 26% improvement to 4% worse), MD ‐4.52 (‐10.65 to 1.61) |
| Physical function | The mean physical function was 37 points | The mean physical function in the intervention group was 6 points lower (0.6 lower to 11 lower) | ‐ | 264 | ⊕⊕⊕⊝ | 6% absolute improvement (95% CI 6% to 11% improvement), 16% relative improvement (95% CI 2% to 30% improvement), MD: ‐6 (‐11.4 to ‐0.6), NNTB 9 (5 to 121) 2 |
| Quality of life | not estimable | see comment | ‐ | (0 study) | no included studies measured this outcome | |
| Number withdrawn due to adverse events | 74 per 1000 | 72 per 1000 | Peto OR 0.97 | 3150 | ⊕⊕⊝⊝ | 0% absolute change (95% CI 3% less to 2% more), 3% relative change (95% CI 26% less to 27% more) (NNTH = NA) |
| Number experiencing any serious adverse events | 68 per 1000 | 63 per 1000 | Peto OR 0.92 | 2404 | ⊕⊝⊝⊝ | 0% absolute change (95% CI 2% less to 1% more), 8% relative change (95% CI 34% less to 28% more) (NNTH = NA) |
| Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) | 6 per 1000 | 4 per 1000 | Peto OR 0.61 | 1755 | ⊕⊝⊝⊝ | 0% absolute change (95% CI 1% less to 0% more), 39% relative change (95% CI 85% less to 143% more) (NNTH = NA) |
| Number experiencing cardiovascular events (myocardial infarction, stroke) follow up: 52 weeks | 24 per 1000 | 11 per 1000 | Peto OR 0.47 | 916 | ⊕⊝⊝⊝ | 1% absolute change (95% CI 3% less to 0% more), 53% relative change (95% CI 83% less to 25% more) (NNTH = NA) |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| ¹ Downgraded one level due to publication bias (missing data from large studies) ² Number needed to treat for an additional beneficial outcome (NNTB) for continuous outcomes calculated using the Wells calculator (from the CMSG editorial office) ³ Downgraded one level for study limitations (all trials had high or unclear risk of at least one type of bias (harms outcomes included all eligible studies)) and one level due to imprecision ⁴ Downgraded two levels for serious imprecision (few events and wide confidence intervals) ⁵ Report Peto OR which can be interpreted as an RR due to the low event rate | ||||||
| March 8, 2013: L Puljak (review author) contacted Prof Gurkirpal Singh to request missing data, because data were not shown as randomized |
| March 8, 2013: Prof Singh responds that the study protocol prespecified data analysis |
| March 8, 2013: L Puljak asked Prof Singh if raw data can be obtained |
| March 8, 2013: Prof Singh responds that data are owned by Pfizer and copies the message to 2 Pfizer employees, Manuela Berger and Gail Cawkwell |
| March 12, 2013: L Puljak writes to Manuela Berger and Gail Cawkwell, repeating the request |
| March 13, 2013: Gail Cawkwell responds, thanks for the interest, directs communication to Dr Peter Park, medical lead for Celebrex, who will “follow up [...] as soon as he returns from a business trip” |
| March 25, 2013: Since there was no response, L Puljak emailed Peter Park |
| March 25, 2013: Automatic reply received, indicating Peter Park is away until March 24 |
| March 25, 2013: Peter Park responds, asks L Puljak to file for "independent data grant application” – Investigator Initiated Research (IIR) request |
| March 29, 2013: L Puljak filed the IIR request in order to obtain summary data (means and standard deviations) for each randomized group separately |
| Shortly afterwards, a gentleman from Croatian Pfizer (Dr Lado Uglesic) called L Puljak on the office phone to ask about the identity of authors, purpose of doing this |
| April 30, 2013: Peter Park responds that a committee will meet in May to decide about the request |
| May 8, 2013: Lado Uglesic from Croatian Pfizer emails L Puljak confirmation of the IIR |
| May 8, 2013: Lado Uglesic asks L Puljak to anonymize her CV submitted with IIR application |
| May 13, 2013: Lado Uglesic confirms that the CV was successfully anonymized |
| October 18, 2013: Lado Uglesic sends email containing the following decision: “Thank you for your submission entitled Celecoxib for osteoarthritis. After careful consideration by the CELEBREX IIR Grant Review Committee, we regret to inform you that we are unable to support it at this time. We receive many promising requests and unfortunately cannot respond favourably to all of them. [...] We appreciate your interest in Pfizer’s IIR program and would be pleased to review another submission from you in the future." |
| Pain instrument | 6 weeks | 12 and 13 weeks | 24 weeks | All follow‐up times combined |
| Pain VAS | SMD = ‐0.49 (95% CI ‐0.56 to ‐0.43), P < 0.001, heterogeneity: 79% (11 studies, 3722 participants) | SMD = ‐0.37 (95% CI ‐0.43 to ‐0.30), P < 0.001, heterogeneity: 93% (6 studies, 3842 participants) | No studies reported pain VAS outcome | SMD = ‐0.43 (95% CI ‐0.48 to ‐0.38), P < 0.001, heterogeneity: 88% (17 studies, 7564 participants) |
| Pain WOMAC | SMD = ‐0.40 (95% CI ‐0.46 to ‐0.33), P < 0.001, heterogeneity: 71% (12 studies, 4718 participants) | SMD = ‐0.28 (95% CI ‐0.33 to ‐0.23), P < 0.001, heterogeneity: 19% (12 studies, 5944 participants) | SMD = ‐0.18 (95% CI ‐0.33 to ‐0.02), P = 0.03, heterogeneity: not applicable (1 study, 631 participants) | SMD = ‐0.32 (95% CI ‐0.36 to ‐0.28), P < 0.001, heterogeneity: 62% (25 studies, 10,753 participants) |
| Both instruments | SMD = ‐0.43 (95% CI ‐0.53 to ‐0.34), P < 0.001, heterogeneity: 76% (17 studies, 7900 participants) | SMD = ‐0.35 (95% CI ‐0.44 to ‐0.25), P < 0.001, heterogeneity: 81% (12 studies, 9786 participants) | SMD = ‐0.18 (95% CI ‐0.33 to ‐0.02), P = 0.03, heterogeneity: not applicable (1 study, 631 participants) | SMD = ‐0.39 (95% CI ‐0.46 to ‐0.32), P < 0.001, heterogeneity: 80% (30 studies, 18,317 participants) |
| Each cell contains the following information: Standardized mean difference (SMD), 95% confidence interval (95% CI), heterogeneity, number of studies, number of participants. Abbreviations: VAS: visual analogue scale; WOMAC: Western Ontario and McMaster Universities Arthritis Index | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Pain Show forest plot | 4 | 1622 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.32, ‐0.12] |
| 2 Physical function Show forest plot | 4 | 1622 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.17 [‐0.27, ‐0.07] |
| 3 Number withdrawn due to adverse events Show forest plot | 28 | 12965 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.99 [0.85, 1.15] |
| 4 Number experiencing any serious adverse events Show forest plot | 28 | 13393 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.95 [0.66, 1.36] |
| 5 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 8 | 3263 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.91 [0.24, 14.90] |
| 6 Number experiencing cardiovascular events (myocardial infarction, stroke) Show forest plot | 5 | 2947 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.40 [0.73, 15.88] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Pain Show forest plot | 2 | 1180 | Mean Difference (IV, Random, 95% CI) | ‐4.52 [‐10.65, 1.61] |
| 2 Physical function Show forest plot | 1 | 264 | Mean Difference (IV, Random, 95% CI) | ‐4.00 [‐11.40, ‐0.60] |
| 3 Number withdrawn due to adverse events Show forest plot | 8 | 3150 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.97 [0.74, 1.27] |
| 4 Number experiencing any serious adverse events Show forest plot | 5 | 2404 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.92 [0.66, 1.28] |
| 5 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 4 | 1755 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.61 [0.15, 2.43] |
| 6 Number experiencing cardiovascular events (myocardial infarction, stroke) Show forest plot | 1 | 916 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.47 [0.17, 1.25] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 All pain < 24 weeks Show forest plot | 31 | 13069 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.41 [‐0.49, ‐0.32] |
| 2 Pain VAS at 6 weeks Show forest plot | 11 | 3722 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.49 [‐0.64, ‐0.34] |
| 3 Pain VAS at 12 weeks Show forest plot | 3 | 1226 | Mean Difference (IV, Random, 95% CI) | ‐11.09 [‐12.68, ‐9.50] |
| 4 Pain VAS at 13 weeks Show forest plot | 5 | 3853 | Mean Difference (IV, Random, 95% CI) | ‐6.35 [‐6.00, ‐4.70] |
| 5 Pain on walking VAS at 6 weeks Show forest plot | 4 | 1572 | Mean Difference (IV, Random, 95% CI) | ‐12.42 [‐14.83, ‐10.01] |
| 6 Pain on walking WOMAC at 12 weeks Show forest plot | 1 | 402 | Mean Difference (IV, Random, 95% CI) | 7.0 [1.73, 12.27] |
| 7 Pain WOMAC at 6 weeks Show forest plot | 12 | 4178 | Mean Difference (IV, Random, 95% CI) | ‐3.28 [‐4.51, ‐2.06] |
| 8 Pain WOMAC at 12 weeks Show forest plot | 9 | 3328 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.32 [‐0.40, ‐0.23] |
| 9 Pain WOMAC at 13 weeks Show forest plot | 5 | 3853 | Mean Difference (IV, Random, 95% CI) | ‐1.06 [‐1.31, ‐0.80] |
| 10 Pain WOMAC at 24 weeks Show forest plot | 1 | 631 | Mean Difference (IV, Random, 95% CI) | ‐13.10 [‐24.69, ‐1.51] |
| 11 All physical function < 24 weeks Show forest plot | 27 | 11940 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.35 [‐0.43, ‐0.27] |
| 12 Physical function WOMAC at 6 weeks Show forest plot | 12 | 4069 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.41, ‐0.22] |
| 13 Physical function WOMAC at 12 weeks Show forest plot | 9 | 3185 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.46 [‐0.65, ‐0.26] |
| 14 Physical function WOMAC at 13 weeks Show forest plot | 5 | 3853 | Mean Difference (IV, Random, 95% CI) | ‐3.70 [‐4.67, ‐2.74] |
| 15 Physical function WOMAC at 24 weeks Show forest plot | 1 | 631 | Mean Difference (IV, Random, 95% CI) | ‐32.60 [‐81.07, 15.87] |
| 16 Quality of life: SF‐36 physical component scores Show forest plot | 1 | 402 | Mean Difference (IV, Random, 95% CI) | ‐2.2 [‐3.86, ‐0.54] |
| 17 Quality of life: SF‐36 mental component scores Show forest plot | 1 | 402 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐1.46, 1.86] |
| 18 Number of responders with at least 50% improvement in WOMAC pain Show forest plot | 4 | 1816 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [1.16, 1.87] |
| 19 Number withdrawn due to adverse events Show forest plot | 28 | 12965 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.99 [0.85, 1.15] |
| 20 Number experiencing any serious adverse events Show forest plot | 28 | 13393 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.95 [0.66, 1.36] |
| 21 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 8 | 3263 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.91 [0.24, 14.90] |
| 22 Number experiencing cardiovascular events (myocardial infarction, stroke) Show forest plot | 5 | 2947 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.40 [0.73, 15.88] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 All pain under 24 weeks Show forest plot | 8 | 2277 | Std. Mean Difference (IV, Random, 95% CI) | 0.00 [‐0.10, 0.10] |
| 2 All pain over 24 weeks Show forest plot | 1 | 916 | Mean Difference (IV, Random, 95% CI) | ‐2.0 [‐5.32, 1.32] |
| 3 All physical function under 24 weeks Show forest plot | 7 | 2176 | Std. Mean Difference (IV, Random, 95% CI) | 0.06 [‐0.09, 0.21] |
| 4 Number withdrawn due to adverse events Show forest plot | 9 | 3739 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.85 [0.68, 1.07] |
| 5 Number experiencing any serious adverse events Show forest plot | 5 | 2404 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.92 [0.66, 1.28] |
| 6 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 4 | 1755 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.61 [0.15, 2.43] |
| 7 Number experiencing cardiovascular events (myocardial infarction, stroke) Show forest plot | 1 | 916 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.47 [0.17, 1.25] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 All pain ≤ 24 weeks Show forest plot | 6 | 1781 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.14, 0.05] |
| 2 Pain VAS at 6 weeks Show forest plot | 2 | 398 | Mean Difference (IV, Random, 95% CI) | 0.55 [‐3.97, 5.07] |
| 3 Pain VAS at 12 weeks Show forest plot | 3 | 975 | Mean Difference (IV, Random, 95% CI) | ‐0.97 [‐2.69, 0.76] |
| 4 Pain WOMAC at 6 weeks Show forest plot | 2 | 503 | Mean Difference (IV, Random, 95% CI) | 0.35 [‐0.54, 1.23] |
| 5 Pain WOMAC at 12 weeks Show forest plot | 3 | 1073 | Mean Difference (IV, Random, 95% CI) | ‐0.03 [‐0.65, 0.60] |
| 6 Pain WOMAC at 6 months Show forest plot | 1 | 310 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.46, 0.66] |
| 7 All physical function ≤ 24 weeks Show forest plot | 6 | 1817 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.18, 0.16] |
| 8 Physical function WOMAC at 6 weeks Show forest plot | 2 | 313 | Mean Difference (IV, Random, 95% CI) | ‐2.99 [‐6.43, 0.44] |
| 9 Physical function WOMAC at 12 weeks Show forest plot | 3 | 1073 | Mean Difference (IV, Random, 95% CI) | ‐1.27 [‐4.32, 1.78] |
| 10 Physical function WOMAC at 6 months Show forest plot | 1 | 301 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐1.94, 2.54] |
| 11 Number withdrawn due to adverse events Show forest plot | 6 | 2173 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.54, 1.23] |
| 12 Number experiencing any serious adverse events Show forest plot | 2 | 841 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.11 [0.45, 2.75] |
| 13 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 2 | 587 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.37 [0.05, 2.62] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Pain VAS at 1 year Show forest plot | 1 | 916 | Mean Difference (IV, Random, 95% CI) | ‐2.0 [‐5.32, 1.32] |
| 2 Number withdrawn due to adverse events Show forest plot | 1 | 916 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.44 [0.80, 2.61] |
| 3 Number experiencing any serious adverse events Show forest plot | 1 | 916 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.62, 1.30] |
| 4 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 1 | 916 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.14 [0.01, 2.16] |
| 5 Number experiencing cardiovascular events (myocardial infarction, stroke) Show forest plot | 1 | 916 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.47 [0.17, 1.25] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Pain VAS at 6 weeks Show forest plot | 1 | 398 | Mean Difference (IV, Random, 95% CI) | 1.90 [‐3.68, 7.48] |
| 2 Pain WOMAC at 6 weeks Show forest plot | 1 | 398 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐0.52, 1.12] |
| 3 Pain on walking at 12 weeks Show forest plot | 1 | 98 | Mean Difference (IV, Random, 95% CI) | 13.0 [3.11, 22.89] |
| 4 Physical function WOMAC at 6 weeks Show forest plot | 1 | 398 | Mean Difference (IV, Random, 95% CI) | 1.90 [‐0.72, 4.52] |
| 5 Number withdrawn due to adverse events Show forest plot | 2 | 650 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.78 [0.46, 1.32] |
| 6 Number experiencing any serious adverse events Show forest plot | 2 | 647 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.79 [0.21, 2.93] |
| 7 Number experiencing gastro‐intestinal events (perforation, ulcer, bleeds) Show forest plot | 1 | 252 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 7.45 [0.46, 119.74] |