Lamotrigine add‐on therapy for drug‐resistant generalised tonic‐clonic seizures
Abstract
Background
This is an update of the Cochrane Review first published in 2010; it includes one additional study.
Primary generalised tonic‐clonic seizures are a type of generalised seizure. Other types of seizures include: absence, myoclonic, and atonic seizures. Effective control of tonic‐clonic seizures reduces the risk of injury and death, and improves quality of life. While most people achieve seizure control with one antiepileptic drug, around 30% do not, and require a combination of antiepileptic drugs.
Objectives
To assess the effectiveness and tolerability of add‐on lamotrigine for drug‐resistant primary generalised tonic‐clonic seizures.
Search methods
For the latest update, we searched these databases on 19 March 2019: Cochrane Register of Studies (CRS) Web, MEDLINE Ovid, and the WHO International Clinical Trials Registry Platform (ICTRP). The CRS includes records from the Cochrane Epilepsy Group Specialized Register, CENTRAL, Embase, and ClinicalTrials.gov. We imposed no language restrictions. We also contacted GlaxoSmithKline, manufacturers of lamotrigine.
Selection criteria
Randomised controlled parallel or cross‐over trials of add‐on lamotrigine for people of any age with drug‐resistant primary generalised tonic‐clonic seizures.
Data collection and analysis
We followed standard Cochrane methodology; two review authors independently assessed trials for inclusion, evaluated risk of bias, extracted relevant data, and GRADE‐assessed evidence. We investigated these outcomes: (1) 50% or greater reduction in primary generalised tonic‐clonic seizure frequency; (2) seizure freedom; (3) treatment withdrawal; (4) adverse effects; (5) cognitive effects; and (6) quality of life. We used an intention‐to‐treat (ITT) population for all analyses, and presented results as risk ratios (RRs) with 95% confidence intervals (CIs); for adverse effects, we used 99% CIs to compensate for multiple hypothesis testing.
Main results
We included three studies (total 300 participants): two parallel‐group studies and one cross‐over study. We assessed varied risks of bias across studies; most limitations arose from the poor reporting of methodological details. We meta‐analysed data extracted from the two parallel‐group studies, and conducted a narrative synthesis for data from the cross‐over study.
Both parallel‐group studies (270 participants) reported all dichotomous outcomes. Participants taking lamotrigine were almost twice as likely to attain a 50% or greater reduction in primary generalised tonic‐clonic seizure frequency than those taking a placebo (RR 1.88, 95% CI 1.43 to 2.45; low‐certainty evidence). The results between groups were inconclusive for the likelihood of seizure freedom (RR 1.55, 95% CI 0.89 to 2.72; very low‐certainty evidence); treatment withdrawal (RR 1.20, 95% CI 0.72 to 1.99; very low‐certainty evidence); and individual adverse effects: ataxia (RR 3.05, 99% CI 0.05 to 199.36); dizziness (RR 0.91, 99% CI 0.29 to 2.86; very low‐certainty evidence); fatigue (RR 1.02, 99% CI 0.13 to 8.14; very low‐certainty evidence); nausea (RR 1.60, 99% CI 0.48 to 5.32; very low‐certainty evidence); and somnolence (RR 3.73, 99% CI 0.36 to 38.90; low‐certainty evidence).
The cross‐over trial (26 participants) reported that 7/14 participants with generalised tonic‐clonic seizures experienced a 50% or greater reduction in seizure frequency with add‐on lamotrigine compared to placebo. The authors reported four treatment withdrawals, but did not specify during which treatment allocation they occurred. Rash (seven lamotrigine participants; zero placebo participants) and fatigue (five lamotrigine participants; zero placebo participants) were the most frequently reported adverse effects.
None of the included studies measured cognition. One parallel‐group study (N = 153) evaluated quality of life. They reported inconclusive results for the overall quality of life score between groups (P = 0.74).
Authors' conclusions
This review provides insufficient information to inform clinical practice.
Low‐certainty evidence suggests that lamotrigine reduces the rate of generalised tonic‐clonic seizures by 50% or more. Very low‐certainty evidence found inconclusive results between groups for all other outcomes. Therefore, we are uncertain to very uncertain that the results reported are accurate, and suggest that the true effect could be grossly different.
More trials, recruiting larger populations, over longer periods, are necessary to determine lamotrigine's clinical use.
PICO
Plain language summary
Lamotrigine as add‐on therapy for drug‐resistant generalised tonic‐clonic seizures
This is an update of a review first published in 2010.
Background
Epilepsy is a common neurological (brain) condition that is characterised by repeated seizures. Most people can control their seizures with a single antiepileptic medicine, however, about 30% continue to have seizures. These people are said to have drug‐resistant epilepsy. Lamotrigine is an antiepileptic medicine, which can be used as add‐on treatment with other antiepileptic medication to try to manage drug‐resistant epilepsy.
Aim of review
This review studied whether lamotrigine was effective and tolerable when used as add‐on treatment, alongside other antiepileptic medicines, for people with drug‐resistant generalised epilepsy (affecting the entire brain from onset) with tonic clonic‐seizures (seizures where people lose consciousness and jerk quickly and rhythmically).
Results
We found three trials, involving 300 people, that investigated lamotrigine for people with drug‐resistant generalised tonic‐clonic seizures. People who received add‐on lamotrigine were almost twice as likely to have a 50% or greater reduction in the number of generalised tonic‐clonic seizures than people who received add‐on placebo (an inactive, dummy drug). Lamotrigine did not significantly affect: the number of people who were completely free of seizures, the number of people who withdrew from treatment, or the number of people who experienced common adverse effects.
However, we are very uncertain whether these findings are accurate. This is because there were not many people involved in the studies, and we are unclear about the methods some of the studies used. For this reason, we cannot comment on the use of lamotrigine.
More trials, which include more people, and are carried out over longer time periods are needed to properly guide the use of lamotrigine for people with drug‐resistant generalised tonic‐clonic seizures.
The evidence is current to March 2019.
Authors' conclusions
Summary of findings
| Add‐on lamotrigine compared to placebo for drug‐resistant generalised tonic‐clonic seizures | |||||||
|---|---|---|---|---|---|---|---|
| Patient or population: people with drug‐resistant generalised tonic‐clonic seizures Setting: outpatient Intervention: add‐on lamotrigine Comparison: add‐on placebo | |||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | ||
| Risk with placebo | Risk with lamotrigine | ||||||
| 50% or greater reduction in seizure frequency Follow‐up (range): 19 to 24 weeks | Study population | RR 1.88 | 270 | ⊕⊕⊝⊝ | The outcome was reported by a cross‐over study but the data could not be incorporated into the meta‐analysis. In the cross‐over study, 7/17 participants with tonic‐clonic seizures experienced a 50% or greater seizure reduction with add‐on lamotrigine compared to add‐on placebo. Only 1/17 participants had an increased seizure frequency with add‐on lamotrigine compared to placebo. Of importance, seizure frequency was compared between the treatment arms and not to the baseline period. | ||
| 338 per 1000 | 636 per 1000 | ||||||
| Seizure freedom (i.e. total cessation of seizure activity) Follow‐up (range): 19 to 24 weeks | Study population | RR 1.55 | 270 | ⊕⊝⊝⊝ | |||
| 125 per 1000 | 194 per 1000 | ||||||
| Treatment withdrawal Follow‐up (range): 19 to 24 weeks | Study population | RR 1.20 | 270 | ⊕⊝⊝⊝ | The outcome was reported by a cross‐over study but the data could not be incorporated into the meta‐analysis. Four of 26 participants in the cross‐over study withdrew from treatment.Two withdrew from lamotrigine due to rash, one withdrew during their first treatment arm, another was ineligible for inclusion, We do not know whether the participants had tonic‐clonic seizures or which treatment the latter two were receiving at the time of withdrawal.. | ||
| 162 per 1000 | 194 per 1000 | ||||||
| Adverse effects Follow‐up (range): 19 to 24 weeks | Dizziness | Study population | RR 0.91 | 270 | ⊕⊝⊝⊝ | The outcome was reported by a cross‐over study but the data could not be incorporated into the meta‐analysis. 2/26 participants in the cross‐over study experienced dizziness, however, we do not know whether the participants had tonic‐clonic seizures. No participants reported dizziness whilst receiving placebo. | |
| 74 per 1000 | 67 per 1000 | ||||||
| Fatigue | Study population | RR 1.02 | 270 | ⊕⊝⊝⊝ | |||
| 22 per 1000 | 23 per 1000 | ||||||
| Nausea | Study population | RR 1.60 | 270 | ⊕⊝⊝⊝ | |||
| 51 per 1000 | 82 per 1000 | ||||||
| Somnolence | Study population | RR 3.73 | 270 | ⊕⊝⊝⊝ | The outcome was reported by a cross‐over study but the data could not be incorporated into the meta‐analysis. 2/26 participants in the cross‐over study experienced somnolence, however, we do not know whether the participants had tonic‐clonic seizures. No participants reported somnolence whilst receiving placebo. | ||
| 7 per 1000 | 27 per 1000 | ||||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | |||||||
| GRADE Working Group grades of evidence | |||||||
| a Evidence downgraded once for risk of bias. The two studies included in the meta‐analysis failed to clarify whether outcome assessors were blinded. One study also failed to declare the treatment allocation of all randomised participants who withdrew from treatment. Therefore, we awarded unclear risk of bias. | |||||||
Background
This is an update of a Cochrane Review first published in 2010 (Tjia‐Leong 2010); it includes one additional study.
Epilepsy is defined as the occurrence of repeated epileptic seizures. It is a common neurological disorder, with an estimated cumulative incidence of 68 per 100,000 people per year (Fiest 2017), and a worldwide prevalence of four to 10 per 1000 people (WHO 2019). Although the majority of people with epilepsy are able to achieve control of their seizures by using adequate doses of conventional antiepileptic drugs (AEDs), up to 30% of people develop drug‐resistant epilepsy (i.e. drug resistance), and continue to have seizures (Cockerell 1995). The internationally accepted definition of drug‐resistant epilepsy is the failure to respond to adequate trials of two tolerated, appropriately chosen, and compliantly used antiepileptic drug regimens, given with the intention of achieving complete cessation of seizures. The two antiepileptic drugs can be used independently, or in combination (Kwan 2010). For the purposes of this review, we applied this definition when assessing the eligibility of studies for inclusion.
Description of the condition
Around 30% to 40% of people have seizures that are generalised at onset; most of them are thought to have a genetic predisposition, and have primary generalised (idiopathic) epilepsy (Bancaud 1989). Generalised seizures are those in which the first clinical and electroencephalographic changes indicate that large parts of both hemispheres of the brain are involved at the onset of the seizure. There is nearly always impaired consciousness. The common subtypes of generalised seizures are tonic‐clonic seizures (grand mal), absence seizures (petit mal), and myoclonic seizures. Such seizures tend to present in childhood and adolescence; common syndromes include childhood absence, juvenile absence, juvenile myoclonic, and generalised epilepsy with tonic‐clonic seizures on waking. Around 20% to 30% of people with generalised onset seizures fail to achieve seizure control on monotherapy, and often require a combination of drugs to maximise seizure control (Cockerell 1995).
Description of the intervention
In an attempt to achieve seizure control, clinicians often prescribe multiple AEDs. Lamotrigine, the focus of this review, is an antiepileptic drug that can be used as an add‐on for people with drug‐resistant epilepsy. It was initially licensed for people with drug‐resistant focal epilepsy, with few studies examining its effect in people with drug‐resistant generalised seizures. In the UK, it has been licensed for generalised tonic‐clonic seizures as monotherapy and as add‐on therapy. Numerous short‐term, randomised, double‐blinded, placebo‐controlled studies, predominantly of cross‐over designs, have confirmed the efficacy of lamotrigine, when used as add‐on therapy for people with drug‐resistant, focal epilepsy (Ramaratnam 2016).
How the intervention might work
Lamotrigine acts by blocking voltage‐gated sodium channels to stabilise the pre‐synaptic neuronal membrane (Leach 1995). There is some evidence that lamotrigine also selectively inhibits the production of excitatory neurotransmitters, such as glutamate and aspartate (Deng 2013). Its full mechanism of action remains unknown.
Although serum concentrations of lamotrigine are not thought to be influenced by interactions with most drugs, they are affected by interactions with other anti‐epileptic drugs. Specifically, drug levels of lamotrigine are markedly increased by an interaction with valproate, which inhibits glucuronidation, the main metabolic pathway of lamotrigine (Rowland 2006; Weintraub 2005). Levels are decreased in the presence of enzyme‐inducing drugs, such as phenytoin, carbamazepine, and oxcarbazepine (Weintraub 2005). A pharmacodynamic interaction is common when lamotrigine is co‐prescribed with carbamazepine, and the symptoms of neurotoxicity (i.e. headache, nausea, dizziness, diplopia, and ataxia) can be avoided by reducing the dose of carbamazepine (Besag 1998).
Adverse effects of lamotrigine include skin rash, dizziness, nausea, diplopia, and ataxia. Increased toxicity and paradoxic deterioration of seizure control may occur when lamotrigine is used in combination with other AEDs.
Why it is important to do this review
Lamotrigine may be efficacious as an add‐on for generalised tonic‐clonic seizures (Beran 1998). However, compared with focal epilepsies, evidence for the effectiveness of lamotrigine for people with generalised epilepsy syndromes is limited (Marson 2007). This review evaluated whether the currently available literature can adequately determine the efficacy of lamotrigine in people with primary generalised epilepsy. This review does not address the effects of lamotrigine for Lennox‐Gastaut syndrome or for absence seizures, as both have already been discussed elsewhere (Hancock 2013; Brigo 2019).
Objectives
To assess the effectiveness and tolerability of add‐on lamotrigine for drug‐resistant primary generalised tonic‐clonic seizures.
Methods
Criteria for considering studies for this review
Types of studies
We only included trials that met all of the following criteria:
-
randomised and controlled, with an adequate method of concealment of randomisation;
-
double‐blind, single‐blind, or unblinded;
-
placebo‐ or actively‐controlled;
-
parallel‐group or cross‐over design.
Types of participants
Participants of any age, with drug‐resistant, primary generalised epilepsy (i.e. experiencing myoclonic epilepsy, generalised epilepsy with tonic‐clonic seizures on awakening, and other idiopathic seizures). We excluded studies involving participants with absence epilepsy and Lennox Gastaut syndrome.
Types of interventions
-
The active treatment group received treatment with lamotrigine in addition to their conventional antiepileptic drug (AED) treatment.
-
The control group received a matched placebo or an active comparator, such as an alternative AED, in addition to their conventional AED treatment.
Types of outcome measures
Primary outcomes
-
50% or greater reduction in primary generalised tonic‐clonic seizure frequency: the proportion of individuals with a 50% or greater reduction in seizure frequency during the treatment period compared to the pre‐randomizations baseline period. We chose this because it is commonly reported in this type of study, and can be calculated for studies that do not report this outcome, provided that baseline seizure data were recorded.
Secondary outcomes
-
Seizure freedom: the proportion of individuals with a 100% reduction in primary generalised tonic‐clonic seizure frequency (complete cessation seizures) during the treatment period (including maintenance phase) compared to the pre‐randomizations baseline period.
-
Treatment withdrawal. We used the proportion of individuals having treatment withdrawn during the course of a treatment period as a measure of global effectiveness. Treatment is likely to be withdrawn due to adverse effects, lack of efficacy, or a combination. However, in trials of a relatively short duration, adverse effects are likely to be the main reason for treatment withdrawal.
-
Adverse effects:
-
The proportion of individuals experiencing any of the following five adverse effects (we chose these adverse effects because we considered them to be important and common adverse effects of AEDs:
-
ataxia;
-
dizziness;
-
fatigue;
-
nausea;
-
somnolence.
-
-
The proportion of individuals experiencing the five most common adverse effects, if different from those listed above.
-
-
Cognitive effects. At present, there is no consensus as to which instrument should be used to assess the effects of AEDs on cognition. In view of this difficulty, we planned to tabulate results where a specific instrument was used to assess the effects of lamotrigine on cognition, but we did not combine the results in a meta‐analysis.
-
Quality of life. There is no consensus on which instruments should be used to assess quality of life, so we tabulated the results rather than combine them in a meta‐analysis.
Search methods for identification of studies
Electronic searches
We ran searches for the original review in June 2010. We ran subsequent searches in July 2012, October 2013, February 2014, February 2017, and March 2019. For the latest update, we searched the following databases on 19 March 2019:
-
Cochrane Register of Studies Web (CRS Web), using the search strategy shown in Appendix 1;
-
Medline Ovid (1946 to 19 March 2019), using the search strategy shown in Appendix 2;
-
WHO International Clinical Trials Registry Platform (ICTRP), using the search strategy shown in Appendix 3.
The Cochrane Register of Studies includes the Cochrane Epilepsy Group Specialized Register, the Cochrane Central Register of Controlled Trials (CENTRAL), and randomised or quasi‐randomised, controlled trials from Embase, and ClinicalTrials.gov.
Searching other resources
In addition, we contacted the manufacturers of lamotrigine, and checked any cross‐references from identified publications.
Data collection and analysis
Selection of studies
Two review authors (RB and MP) independently assessed the records identified by both the electronic searches and handsearches for their eligibility for this review. First, we (the two review authors), screened the titles and abstracts of all the identified records. At this stage, we eliminated any records that were duplicates, or that did not meet the predefined inclusion criteria. We retrieved the full‐text publications for the remaining, potentially eligible records, and completed a more in‐depth screening. We resolved any disagreements by discussion. If disagreements persisted, the third author (AGM) arbitrated.
Data extraction and management
We extracted the following data from the studies that met our inclusion criteria:
-
Methodological or trial design:
-
method of randomizations and allocation concealment;
-
method of blinding;
-
whether any participants had been excluded from reported analyses;
-
duration of baseline period;
-
duration of treatment period;
-
dose(s) of lamotrigine tested.
-
-
Participant and demographic information:
-
total number of participants allocated to each treatment group;
-
age and sex;
-
seizure types;
-
number and description of background drugs;
-
number of seizures prior to randomisation.
-
-
Treatment data:
-
medication dose per treatment group;
-
route of administration for treatment.
-
-
Follow‐up data:
-
the number of participants in each group achieving 50% or greater reduction in seizures;
-
the number of participants in each group achieving total cessation of seizures;
-
the number of participants in each group who had treatment withdrawn, and reasons for treatment withdrawal;
-
for those excluded; the reason for exclusion, whether any of those excluded completed the treatment phase.
-
Assessment of risk of bias in included studies
Two review authors (RB and MP) independently assessed the risk of bias associated with the included studies, using the Cochrane 'Risk of bias' tool, as outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). The Cochrane 'Risk of bias' tool comprises seven specific domains:
-
random sequence generation;
-
allocation concealment;
-
blinding of participants and personnel;
-
blinding of outcome assessors;
-
incomplete outcome data;
-
selective outcome reporting;
-
other bias.
For each entry, the two review authors made a judgement (low risk of bias, high risk of bias, or unclear risk of bias) and provided support for the decision by an agreed review author comment, or by a quote taken directly from the corresponding trial publication.
When judging overall risk of bias associated with each study, we followed the guidance suggested by Higgins 2017. Specifically, if a study was awarded low risk of bias across all domains, we considered the study to be at low risk of bias overall. If we judged that a study was at unclear risk of bias for one or more domains, then we would award it unclear risk of bias overall. Likewise, if we judged that a study was at high risk of bias for one or more domain, then we would consider the study to be at high risk of overall bias.
Measures of treatment effect
We recorded the number of participants per randomised group who experienced each outcome. We analysed data according to the intention‐to‐treat principle, whereby we assumed that participants who did not complete follow‐up, or who provided insufficient seizure data, were non‐responders. To analyse dichotomous outcomes (50% or greater reduction in seizure frequency, seizure freedom, treatment withdrawal, and adverse effects), we calculated risk ratios (RRs) with 95% confidence intervals (CIs), using the Mantel–Haenszel method. However, for individual adverse effects, we quoted 99% CIs, to make allowance for multiple testing.
Due to the lack of agreement and consistency about how cognitive effects and quality of life should be measured, we tabulated any available results and conducted a narrative synthesis.
Unit of analysis issues
For cross‐over studies, we had planned to extract data from the first treatment period, and incorporate the data into the meta‐analysis, as if the data had been derived from a parallel‐group design study. This would prevent data gathered from the same participant from contributing to both the intervention and control treatment groups, thus avoiding any unit of analysis issues.
Dealing with missing data
Where outcomes of interest were not reported, we contacted the original authors and study sponsors for further information.
Assessment of heterogeneity
We assessed clinical heterogeneity by comparing the characteristics of the participants recruited into the included studies, including factors, such as age and sex. We assessed methodological heterogeneity by comparing the study design of the individual studies, including: method of randomisation, allocation concealment and blinding, and duration of treatment. We assessed statistical heterogeneity using the I² statistic. An I² value < 40% suggested that heterogeneity might not be important, 40% to 60% equated moderate heterogeneity, and 50% to 90% implied substantial heterogeneity.
Assessment of reporting biases
We requested trial protocols to determine the predefined outcomes of the studies. In instances where we were unable to get the trial protocol, we compared the outcomes listed in the methods section to those reported in the results section of the text. We reported any inconsistencies in the 'Risk of bias' table.
Due to the limited number of included studies, we were unable to develop a funnel plot to evaluate reporting bias.
Data synthesis
We analysed all data using Review Manager 5 (Review Manager 2014). Where the I² statistic indicated that heterogeneity was likely unimportant (I² < 40%), we summarised results using a fixed‐effect model. When the I² statistic indicated either moderate or substantial heterogeneity (I² ≥ 40%), or we detected clinical heterogeneity despite insignificant statistical heterogeneity, we used a random‐effects model for the meta‐analysis.
Subgroup analysis and investigation of heterogeneity
Due to the small number of included studies, we were unable to conduct any meaningful subgroup analysis. If it had been possible, we would have undertaken a subgroup analysis according to age groups (children versus adults), seizure type, and baseline severity.
Sensitivity analysis
We had also planned to complete a sensitivity analysis to test the robustness of the meta‐analysis. Specifically, we would have excluded studies from the meta‐analysis that were at high‐risk of bias, to determine the impact on the overall effect size estimated.
Summary of findings and assessment of the certainty of the evidence
We used the GRADE approach to summarise findings, as detailed in the GRADE handbook (Schünemann 2013). We imported data into GRADEpro GDT software from Review Manager 5 software to create a 'Summary of findings' table (GRADEpro GDT; Review Manager 2014). We assessed the certainty of the evidence for the following outcomes in the 'Summary of findings' table: 50% or greater reduction in seizure frequency, seizure freedom, treatment withdrawal, and the adverse effects of dizziness, fatigue, nausea, and somnolence.
We chose to include 50% or greater reduction in seizure frequency and seizure freedom as these were the only two efficacy outcomes included in the review. We also chose to include treatment withdrawal as this is a measure of global effectiveness that encompasses both efficacy and tolerability. Finally, we wanted to include all five adverse effects that are considered to be important and common with AEDs (ataxia, dizziness, fatigue, nausea and somnolence). However, because we were limited by the number of outcomes that can be GRADE‐assessed and included in the summary of findings table, we decided to exclude ataxia as our previous knowledge of the evidence base suggested that ataxia would not be highly reported. We therefore prioritised the GRADE‐assessment of the other four adverse effects.
Results
Description of studies
Results of the search
During the original search, conducted in June 2010, we identified five randomised controlled studies investigating the use of add‐on lamotrigine in participants with drug‐resistant generalised epilepsy (Beran 1998; Biton 2005; Eriksson 1998; Motte 1997; Sander 1990). Of the studies identified during the original searches, only two of the studies fulfilled the protocol criteria for inclusion, one cross‐over and one parallel study (Beran 1998; Biton 2005).
The electronic searches conducted between July 2012 and March 2019, identified a total of 454 records that were potentially eligible for inclusion (Figure 1). We also identified five records through handsearching. We automatically removed 115 records, as they were duplicates. Next, we removed another 240 records, because they were obviously irrelevant. We screened the titles and abstracts of the remaining 104 records, and determined that only eight of the identified records, which related to four individual studies, were potentially eligible for inclusion. We retrieved and screened the full‐text publications of the remaining eight records, five of which we found to be eligible for inclusion. Four related to Biton 2010, and two were additional references for Biton 2005. We excluded two of the full‐texts (Brzakovic 2012; Chung 2009).

Study flow diagram showing the screening results from the searches conducted to March 2019
In total, we assessed that 10 records, relating to three individual studies, were eligible (Beran 1998; Biton 2005; Biton 2010). We excluded five records, relating to five individual studies (Brzakovic 2012; Chung 2009; Eriksson 1998; Motte 1997; Sander 1990).
Included studies
We included three studies in this update (Beran 1998; Biton 2005; Biton 2010). We summarised the relevant information for each study in the 'Characteristics of included studies' tables.
Study design
One study was a cross‐over study (Beran 1998) whilst two studies were parallel‐group studies (Biton 2005; Biton 2010).
The cross‐over study (Beran 1998) featured an eight week baseline period, followed by two eight week treatment periods, separated by a four week washout period. No detailed data were provided for either phase of the cross‐over study.
The parallel‐group studies (Biton 2005; Biton 2010) both used the same study duration for adolescent and adult participants; a two‐week screening period, an eight‐week baseline period, and a 19‐week treatment period (7‐week dose‐escalation plus a 12‐week maintenance period). Biton 2005, however, also included children (aged 2 to 12 years) and for these participants the dose‐escalation was 12‐weeks, leading to an overall treatment period of 24‐weeks, opposed to 19‐weeks for adults. All other phases remained the same in duration.
Participants
Beran 1998 included a small sample of 26 participants, aged between 15 and 50 years (mean 29 years), with drug‐resistant generalised epilepsy. Of the 26 participants, 17 participants had generalised epilepsy with tonic‐clonic seizures (12 with tonic‐clonic and absence seizures, two with tonic‐clonic seizures only, one with tonic‐clonic and myoclonic seizures, two with tonic‐clonic, absence and myoclonic seizures). Only the 17 participants with tonic‐clonic seizures were included in our intention‐to‐treat analysis.
SImilar to Beran 1998, Biton 2010 included both adolescents and adults, but not children. Specifically, Biton 2010 involved 153 randomised participants (76 lamotrigine, 77 placebo), aged 13 years and over. In contrast, Biton 2005 included children, as well as adults and adolescents. Biton 2005 included 121 randomised subjects, aged 2 to 55 years (mean 26 years).
Notably, four of the participants from Biton 2005 did not receive any study medication and it was not clear to which group they had been randomised. As a result, we were only able to include 117 participants (58 lamotrigine, 59 placebo) in our intention‐to‐treat analysis includes.
For both Biton 2005 and Biton 2010, participants had been receiving 1 or 2 established AEDs for at least four weeks prior to screening. In contrast, Beran 1998 included participant who had previously been treated with up to four established antiepileptic drugs (AEDs). For all three studies, participants had to have drug‐resistant seizures despite therapy with a stable AED regimen of at least one or more other AEDs, appropriate for the type of epilepsy when given in adequate doses.
The exclusion criteria for all three studies were similar. They excluded people with: a history of focal seizures, interictal expression of focal seizures revealed by EEG, Lennox‐Gastaut syndrome, confounding organic or psychiatric problems, progressive neurological disease, associated systemic disease, chronic medication that might influence seizure control, recent use of any investigational AED, previous exposure to lamotrigine, and abuse of alcohol, other prescription or non‐prescription drugs. Furthermore, the three studies each excluded women who were pregnant or at risk of pregnancy, and women who were breastfeeding.
Interventions
Participants were not randomised to a single dose in any of the studies but took a range of doses, depending on clinical response, age group, and the concurrent administration of other antiepileptic drugs (AEDs).
Valproate was the most common concomitant AED. Lamotrigine was administered at lower doses when used with an enzyme inhibitor like valproate (75 mg to 250 mg daily). Conversely, higher doses were administered when used with other enzyme inducing AEDs (150 mg to 500 mg daily). Additionally, for Biton 2010, participants who were taking an AED other than valproate and taking multiple enzyme‐inducing AEDs were titrated to 300 mg/day. Conversely, participants under the age of 12 years (N = 23) had a dosing range between 2.25 mg and 7.5 mg/kg/day (Biton 2005).
Excluded studies
We excluded five studies from this review update (Brzakovic 2012; Chung 2009; Eriksson 1998; Motte 1997; Sander 1990). We summarised the reasons for exclusion in the 'Characteristics of excluded studies' tables.
Four of the studies featured ineligible study populations (Chung 2009; Eriksson 1998; Motte 1997; Sander 1990). Two studies included participants with a diagnosis of Lennox‐Gastaut syndrome (Eriksson 1998; Motte 1997); one study included participants with a diagnosis of focal, rather than generalised epilepsy (Chung 2009); the majority of participants in one study had drug‐resistant focal seizures, and although three of the participants did have primary generalised tonic‐clonic seizures, data were not stratified according to seizure subtype or order of treatment (Sander 1990). One study was not a randomised controlled trial (Brzakovic 2012).
Risk of bias in included studies
Summaries of our judgements for each risk of bias domain, across the included studies, are presented in Figure 2 and Figure 3. Reasons supporting our judgements, including quotations taken directly from the text publications and specific review author comments, can be found in the 'Risk of bias' tables.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study
Allocation
One study failed to explicitly state how the randomisation sequence was generated or how allocation was concealed, therefore, we assessed it as an unclear risk of bias (Beran 1998). The other two studies specified that the randomisation schedule was computer‐generated by the study sponsor, GlaxioSmithKline; they used central randomisation and an interactive voice response system to allocate participants to treatment groups, therefore, we assessed them at a low risk of bias.
Blinding
All three studies reported they were double‐blinded. However, one study did not provide a description of the blinding process, and we assessed it as unclear risk of performance and detection bias (Beran 1998). The other two studies specified that matching placebo tablets were used, consequently, we judged them at low risk of performance bias. Since they did not specify whether outcome assessors were blinded, we assessed that the studies were at unclear risk of detection bias.
Incomplete outcome data
All three studies reported losses to follow‐up and treatment withdrawals.
Beran 1998 reported that 4/26 participants withdrew from the trial before completion, but did not specify which treatment group the participants were randomised to at the time of withdrawal, as a result, we judged the study at high risk of attrition bias.
Biton 2005 reported that 16/58 lamotrigine‐randomised participants and 14/59 placebo‐randomised participants withdrew from the trial prior to completion. Data from the 30 participants who withdrew from treatment, but who all received at least one dose of the study drug, were incorporated into the efficacy analysis by using an intention‐to‐treat analysis. An additional 4/117 participants did not receive at least one dose of the study drug, but their treatment group was not specified, so were not included in the meta‐analysis; for this reason, we assessed that the study was at unclear risk of attrition bias.
Biton 2010 reported that 10/76 lamotrigine‐randomised participants and 8/77 placebo‐randomised participants withdrew from the study prior to completion, but they used an intent‐to‐treat analysis. Therefore, we assessed the study had a low risk of attrition bias.
Selective reporting
We were unable to attain a trial protocol for one study, but the outcomes defined in the methods section of the full‐text publication were fully reported in the results section. We obtained the summary of the trial protocol for the remaining two studies, which fully reported all predefined outcomes. Therefore, we assessed all three studies at low risk of reporting bias.
Other potential sources of bias
Beran 1998 experienced difficulties when recording participants' daily seizure frequency. Specifically, 22 of the randomised participants experienced absence seizures. This seizure type can be difficult to accurately record. The authors, therefore, only collected seizure frequency data on days when they were sure that a complete and accurate seizure record had been kept. The mean monthly seizure rate was then calculated based from the number of days were a complete seizure record was available. It is thus possible that the mean monthly seizure frequency calculated could be considerably different from the true monthly seizure frequency. This would be especially problematic if the number of generalised tonic‐clonic seizures correlated with the number of absence seizures, in any way. We thus assessed that the study by Beran 1998 was at unclear risk of other bias.
Effects of interventions
We did not conduct a meta‐analysis in the previous version of this review as we included two studies that were too heterogeneous. Specifically, the two studies varied in: study design (one study was a parallel‐group trial (Biton 2005) whereas the other was a cross‐over trial (Beran 1998)), treatment doses used (200 mg to 400 mg/day versus 75 mg to 150 mg/day), and length of follow‐up (19 weeks to 24 weeks versus 8 weeks).
During this review update, we identified a second parallel group study (Biton 2010) with the same duration of treatment as the previously included parallel‐group study (Biton 2005). We therefore decided to conduct a meta‐analysis of the two parallel‐group studies, whilst continuing to describe the results from the cross‐over study narratively (Beran 1998). Importantly, the narrative synthesis considers data collected from the same participant for both the intervention (lamotrigine) and control (placebo) treatment periods, due to the cross‐over design of the study. Unfortunately. the information regarding only the first treatment period was not available.
The results can be found summarised in the summary of findings Table 1.
Primary outcome
50% or greater reduction in primary generalised tonic‐clonic seizure frequency
All three studies reported this outcome, however, we only used data extracted from the two parallel‐group studies for the meta‐analysis (Biton 2005; Biton 2010). We calculated that participants receiving add‐on lamotrigine were nearly twice as likely to attain a 50% or greater reduction in primary generalised tonic‐clonic seizure frequency, compared to participants who received add‐on placebo (risk ratio (RR) 1.88, 95% confidence interval (CI) 1.43 to 2.45; P < 0.001; 2 studies; 270 participants; Analysis 1.1).
Beran 1998 (N = 17) reported that seven participants with tonic‐clonic seizures experienced a 50% or greater reduction in primary generalised tonic‐clonic seizure frequency with add‐on lamotrigine compared to with add‐on placebo. Importantly, seizure frequency was compared between the two treatment arms to calculate the percentage change rather than to the pre‐randomisation baseline period. Analysis using the non‐parametric Mann Whitney U test revealed a significant reduction in seizure frequency with add‐on lamotrigine compared with add‐on placebo (P = 0.03). Only one participant experienced an increase in seizure frequency when receiving add‐on lamotrigine compared to when receiving add‐on placebo.
Secondary outcomes
1. Seizure freedom
The two parallel‐group studies (Biton 2005; Biton 2010), presented data for the outcome, seizure freedom, however, the cross‐over study by Beran 1998 did not.
We calculated that a greater proportion of participants attained seizure freedom from primary generalised tonic‐clonic seizures when receiving add‐on lamotrigine compared to when receiving add‐on placebo. The meta‐analysis (Analysis 1.2), however, indicated that the results were inconclusive and we were unable to determine whether there was an effect of vigabatrin for seizure freedom (RR 1.55, 95% CI 0.89 to 2.72; P = 0.12; 2 studies; 270 participants).
2. Treatment withdrawal
All three studies measured treatment withdrawal. For the parallel group studies, the results were inconclusive (RR 1.20, 95% CI 0.72 to 1.99; P = 0.48; 2 studies; 270 participants; Analysis 1.3). For participants randomised to placebo, the most common reason for treatment withdrawal was non‐compliance (total 10/22 withdrawals). For participants randomised to lamotrigine groups, the most common reason for treatment withdrawal varied between the two studies. Non‐compliance remained the most common reason for treatment withdrawal in Biton 2010 (7/10 withdrawals); whereas loss to follow‐up and adverse effects were the most common reasons in Biton 2005 (5/16 withdrawals). Other reasons for treatment withdrawal for both treatment groups included withdrawal of consent and lack of efficacy.
Beran 1998 reported that four of the total 26 randomised participants withdrew from treatment. Two participants withdrew from the lamotrigine group due to rash, one participant withdrew during the first treatment phase, and the other participant was later found to be ineligible. There was no information on treatment group for the latter two treatment withdrawals and we do not know the seizure type of the four participants who withdrew.
3. Adverse effects
a. The proportion of individuals experiencing any of the following five listed adverse effects
Both parallel‐group studies reported all five adverse effects. The results were inconclusive for all adverse effects (2 studies, 270 participants; Analysis 1.4):
-
Ataxia – RR 3.05, 99% CI 0.05 to 199.36; P = 0.49;
-
Dizziness – RR 0.91, 99% CI 0.29 to 2.86; P = 0.84;
-
Fatigue – RR 1.02, 99% CI 0.13 to 8.14; P = 0.99;
-
Nausea – RR 1.60, 99% CI 0.48 to 5.32; P = 0.32;
-
Somnolence – RR 3.73, 99% CI 0.36 to 38.90; P = 0.15.
Beran 1998 reported that 3/26 (12%) of the participants experienced ataxia, 2/26 (8%) experienced dizziness, and 2/26 (8%) experienced somnolence, whilst receiving lamotrigine. None of the participants in the placebo group reported any of these symptoms. The report described tiredness rather than fatigue; 1/26 (4%) participants reporting tiredness in the lamotrigine group, 5/26 (19%) reported it in the placebo group. Of significance, the report only listed adverse effects that were reported by more than two participants. Although nausea was not listed amongst the adverse effects, it is possible that two or fewer participants reported it.
b. The proportion of individuals experiencing the five most common adverse effects, if different from those listed above
Both parallel studies reported headache as the most common adverse effect during the studies; there were more reports of headache in the placebo group, but the results were inconclusive (RR 0.75, 99% CI 0.38 to 1.50; P = 0.29; 2 studies, 270 participants; Analysis 1.4).
Biton 2010 reported that vomiting was the second most commonly reported adverse effect, Biton 2005 reported it was the fifth most commonly reported; the results were inconclusive (RR 1.27, 99% CI 0.39 to 4.15; P = 0.60; 2 studies, 270 participants; Analysis 1.4).
Convulsion was the fourth most commonly reported adverse effect in Biton 2005; while more common in the placebo group, the results were inconclusive (RR 0.34, 99% CI 0.05 to 2.13; P = 0.13; 2 studies, 270 participants; Analysis 1.4).
Pyrexia was the fifth most commonly reported adverse effect in Biton 2010; the results were inconclusive (RR 0.87, 99% CI 0.21 to 3.52; P = 0.80; 2 studies, 270 participants; Analysis 1.4).
In addition to the adverse effects listed above, Beran 1998 also reported rash, headache, accidental injury, ataxia, diplopia, and tremor in their top five reported adverse effects. Rash was the most commonly reported adverse effect (lamotrigine = 7/26 (27%); placebo = none); headache and accidental injury were tied for the third most common, with 2/26 reported in each group; ataxia, diplopia, and tremor were tied for the fourth most common (lamotrigine = 3/26 (12%) reported ataxia and diplopia; placebo = none; lamotrigine = 2/26 (8%) reported tremor; placebo = 1/26 (4%)).
4. Cognitive effects and quality of life
None of the three included studies assessed any measures of cognitive effect.
5. Quality of life
Only one study (N = 153 participants) assessed quality of life with the Quality of Life in Epilepsy‐31‐P (QOLIE‐31P) questionnaire. The mean overall quality of life score for both treatment groups increased from baseline to the end of treatment, but the results were inconclusive (P = 0.74; Table 1; Biton 2010).
| Treatment group | Overall score for QOLIE‐31P | Change in overall score for QOLIE‐31P from baseline to week 19 of treatment | |||
|---|---|---|---|---|---|
| Time point | Number of participants analysed | Mean (SD) | |||
| Number of participants analysed | Least squares mean (SE) | ||||
| Placebo | Baseline | 19 | 61.1 (17.1) | 18 | ‐6.5 (4.0) |
| End of study | 20 | 66.4 (14.8) | |||
| Lamotrigine | Baseline | 17 | 54.3 (9.9) | 15 | ‐8.5 (4.4) |
| End of study | 15 | 65.6 (20.4) | |||
SD: standard deviation; SE: standard error; QOLIE‐31P: Quality of Life in Epilepsy Inventory‐31 Patient‐weighted.
Discussion
Summary of main results
We found three short‐term randomised controlled trials that met our inclusion criteria, two of which were parallel‐group studies (Biton 2005; Biton 2010), and one of which was a cross‐over study (Beran 1998). We combined the results from the two parallel‐group studies in a meta‐analysis, and described the results from the cross‐over study narratively.
Low‐certainty evidence suggested that add‐on lamotrigine almost doubled the likelihood that people with generalised tonic‐clonic seizures would attain a 50% or greater reduction in the frequency of their seizures, compared to add‐on placebo. Very low‐certainty evidence suggested inconclusive results for the likelihood of seizure freedom between add‐on lamotrigine and placebo. Very low‐certainty evidence suggested inconclusive results for the likelihood of adverse events, such as treatment withdrawal and individual adverse effects.
Importantly, the low‐ to very low‐certainty evidence for all outcomes means that we are uncertain to very uncertain that the effect sizes that we reported are accurate. It is possible that the true effect could be considerably different to that estimated, both in terms of magnitude and direction. The uncertainty of the results may reflect the very low number of studies and participants that contributed data to the meta‐analysis.
Overall completeness and applicability of evidence
The most notable limitation of this review is the lack of studies that were eligible for inclusion. We included three studies, however, only data from two of the studies were suitable for meta‐analysis. Even after combining the two study populations, the total sample size remained relatively small (270 participants in total). Less frequent events, such as seizure freedom and the incidence of individual adverse effects, require larger sample sizes to detect a significant therapeutic effect (Hajian‐Tillaki 2011). From this viewpoint, it is possible that add‐on lamotrigine might have a therapeutic effect on these outcomes that we have currently been unable to detect.
The limited data also prevented us from performing subgroup analyses. We had originally planned to conduct subgroup analyses according to the age of participants, their seizure type, and their baseline seizure frequency. However, splitting the small sample size further would have rendered the results of any subgroup analysis meaningless, and increased the likelihood of a type II error occurring.
The studies did not report all of the review outcomes; for example, Beran 1998 did not report seizure freedom, and none of the included studies reported any measures of cognitive effect. Only Biton 2010 measured and reported changes in quality of life. This also impacted the completeness and certainty of the evidence.
The design of the included studies also affected the applicability of the evidence. All three included studies were of short duration; hence, longer term outcomes were not assessed. Long‐term outcomes are especially important in chronic conditions, such as epilepsy, where the medications could potentially be taken over a lifetime.
All three studies tried to account for interactions between lamotrigine and enzyme‐inhibiting drugs (valproate) and enzyme‐inducing drugs (e.g. carbamazepine) by altering initial lamotrigine doses to compensate. Of note, the initial target doses were different between the studies. For example, for participants taking add‐on lamotrigine with valproate, but without an enzyme‐inducing drug, Beran 1998 administered a fixed‐dose of 150 mg/d lamotrigine. In contrast, the target dose for participants taking concomitant valproate in Biton 2005 and Biton 2010 was 200 mg/d lamotrigine, the minimum dose was 150 mg/d and the maximum was 250 mg/d. Again, this affected our ability to combine and compare the results of the three studies.
Of further significance, the approach to dosing did not reflect how lamotrigine and other antiepileptic drugs are used in practice. Beran 1998 used a fixed‐dose regimen, which does not reflect everyday practice; in normal clinical practice, individuals would alter their dose, dependent on their tolerability of the drug and their therapeutic response. In contrast, Biton 2005 titrated doses according to plasma levels of lamotrigine, which, again, is not routine clinical practice. Therefore, it is unclear how easily the findings would transfer to clinical practice.
On a more positive note, the two studies that contributed data to the meta‐analysis did include a range of participants; for example, both adults and children were involved in both studies. A mixture of ethnicities were also recruited into the studies, most noticeably, Caucasian, Asian, and Hispanic populations. Due to our inability to complete any subgroup analyses, we were unable to determine whether the findings were generalisable to all participant demographics, or whether a participant's response to lamotrigine was dependent upon certain personal characteristics.
Certainty of the evidence
Data from only two of the included studies contributed to the meta‐analysis. Data from Beran 1998 was excluded from the meta‐analysis. The findings from Beran 1998 are instead described in the comments column of the summary of findings Table 1.
With regard to risk of bias, one of the included trials failed to describe the methodology used for random sequence generation, allocation concealment, and the blinding of participants, study personnel, and outcome assessors (Beran 1998). None of the studies provided details about whether outcome assessors were blinded. There were also issues with the reporting and handling of attrition for two of the included studies (Beran 1998; Biton 2005). Both studies failed to specify the treatment allocation of a subset of participants who withdrew from treatment, and one study did not conduct intention‐to‐treat analyses (Beran 1998). The same study was also at risk of other bias due to the method used to calculate the baseline seizure frequency. As a result, we downgraded the evidence once for risk of bias for all seven outcomes, largely due to the concerns regarding the blinding of outcome assessors and the issue with attrition.
We also downgraded the evidence for imprecision, owing to the insufficient number of events contributing to the data analysis. Specifically, we downgraded the evidence twice for six of the outcomes because fewer than 100 events were reported per outcome. In contrast, we only downgraded the evidence for the primary outcome, 50% or greater reduction in seizure frequency, once, as more events were reported for this outcome (131 events/270 participants for the meta‐analysis).
Overall, we assessed that six of the outcomes were supported by very low‐certainty evidence, and one outcome was supported by low‐certainty evidence. Therefore, we are uncertain to very uncertain that the findings reported are accurate of the true effect of add‐on lamotrigine compared to placebo, and readers should be very cautious about how they interpret and apply the results.
Potential biases in the review process
We conducted the review according to the standard methodological procedures expected by the Cochrane Collaboration. We therefore have no reason to suspect any major potential biases in this review.
Agreements and disagreements with other studies or reviews
The most similar systematic review to the one conducted here was by Bloom 2017. The review by Bloom 2017 investigated the occurrence of adverse effects with lamotrigine monotherapy in randomised controlled trials. The authors identified 122 studies for inclusion, consisting of 18,698 participants. The incidence rate for rash was 8.3%, with 1570 participants affected in total. Although the systematic review by Bloom 2017 focused on the use of lamotrigine as a monotherapy, rather than as an add‐on therapy, the evidence from the review suggests that the incidence of rash should be higher than that which has been reported by our meta‐analysis.
This could be due to the different dosing regimens implemented. For example, Beran 1998 used a fixed‐dose method and did not describe any dose adjustments. In our review, Beran 1998 reported that 27% of participants developed rash whilst receiving add‐on lamotrigine, of which, two cases were serious enough for treatment to be withdrawn. No participants developed rash with add‐on placebo. Biton 2005 and Biton 2010, however, allowed flexibility within their target doses by permitting a range of doses for each target dose. Participants were required to remain within the dose range but were allowed to adjust their dosage, as necessary, dependent on tolerability and efficacy. Biton 2005 reported one mild case of generalised urticaria whilst Biton 2010 reported a higher incidence rate for rash among placebo‐randomised participants compared to lamotrigine‐randomised participants (four versus two participants, respectively). Although Bloom 2017 did not describe the dosing regimes of the included studies, we suggest that this presents one possible explanation for the difference in incidence rates.
Further to this, the systematic review by Bloom 2017 also addressed the occurrence of Stevens‐Johnson syndrome and toxic epidermal necrolysis. Both conditions are classified as being severe adverse skin reactions and are considered under the category of rash. Specifically, Bloom 2017 estimated that Stevens‐Johnson syndrome/ toxic epidermal necrolysis affects one in 2500 participants treated with lamotrigine. Notably, none of the studies, included in our review, reported any occurrence of Stevens‐Johnson syndrome or toxic epidermal necrolysis. This is easily explained by the low number of participants involved across all three studies (300 participants total) and the rarity of the event.
Although neither skin reaction was reported within our review, due to the seriousness and mortality associated with Stevens‐Johnson syndrome/toxic epidermal necrolysis (Kumar 2018), it is important that this eventuality is considered when debating the clinical use of lamotrigine. Multiple studies have demonstrated that the risk of an adverse skin reaction can be minimised by the slow, gradual up‐titration of lamotrigine dosage (Ketter 2005; Lorberg 2009). It is, however, worth noting that people would not be effectively protected against seizures during the titration period.
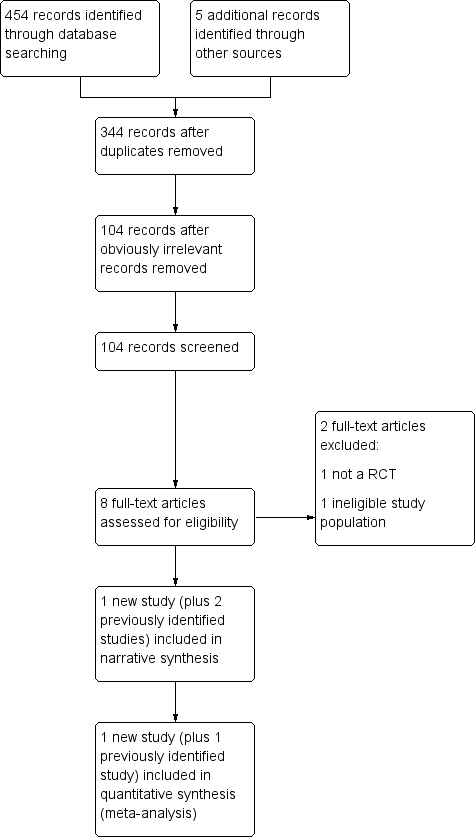
Study flow diagram showing the screening results from the searches conducted to March 2019

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study

Comparison 1: Lamotrigine versus placebo, Outcome 1: 50% or greater reduction in primary generalized tonic‐clonic seizure frequency

Comparison 1: Lamotrigine versus placebo, Outcome 2: Seizure freedom

Comparison 1: Lamotrigine versus placebo, Outcome 3: Treatment withdrawal

Comparison 1: Lamotrigine versus placebo, Outcome 4: Adverse effects
| Add‐on lamotrigine compared to placebo for drug‐resistant generalised tonic‐clonic seizures | |||||||
|---|---|---|---|---|---|---|---|
| Patient or population: people with drug‐resistant generalised tonic‐clonic seizures Setting: outpatient Intervention: add‐on lamotrigine Comparison: add‐on placebo | |||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Certainty of the evidence | Comments | ||
| Risk with placebo | Risk with lamotrigine | ||||||
| 50% or greater reduction in seizure frequency Follow‐up (range): 19 to 24 weeks | Study population | RR 1.88 | 270 | ⊕⊕⊝⊝ | The outcome was reported by a cross‐over study but the data could not be incorporated into the meta‐analysis. In the cross‐over study, 7/17 participants with tonic‐clonic seizures experienced a 50% or greater seizure reduction with add‐on lamotrigine compared to add‐on placebo. Only 1/17 participants had an increased seizure frequency with add‐on lamotrigine compared to placebo. Of importance, seizure frequency was compared between the treatment arms and not to the baseline period. | ||
| 338 per 1000 | 636 per 1000 | ||||||
| Seizure freedom (i.e. total cessation of seizure activity) Follow‐up (range): 19 to 24 weeks | Study population | RR 1.55 | 270 | ⊕⊝⊝⊝ | |||
| 125 per 1000 | 194 per 1000 | ||||||
| Treatment withdrawal Follow‐up (range): 19 to 24 weeks | Study population | RR 1.20 | 270 | ⊕⊝⊝⊝ | The outcome was reported by a cross‐over study but the data could not be incorporated into the meta‐analysis. Four of 26 participants in the cross‐over study withdrew from treatment.Two withdrew from lamotrigine due to rash, one withdrew during their first treatment arm, another was ineligible for inclusion, We do not know whether the participants had tonic‐clonic seizures or which treatment the latter two were receiving at the time of withdrawal.. | ||
| 162 per 1000 | 194 per 1000 | ||||||
| Adverse effects Follow‐up (range): 19 to 24 weeks | Dizziness | Study population | RR 0.91 | 270 | ⊕⊝⊝⊝ | The outcome was reported by a cross‐over study but the data could not be incorporated into the meta‐analysis. 2/26 participants in the cross‐over study experienced dizziness, however, we do not know whether the participants had tonic‐clonic seizures. No participants reported dizziness whilst receiving placebo. | |
| 74 per 1000 | 67 per 1000 | ||||||
| Fatigue | Study population | RR 1.02 | 270 | ⊕⊝⊝⊝ | |||
| 22 per 1000 | 23 per 1000 | ||||||
| Nausea | Study population | RR 1.60 | 270 | ⊕⊝⊝⊝ | |||
| 51 per 1000 | 82 per 1000 | ||||||
| Somnolence | Study population | RR 3.73 | 270 | ⊕⊝⊝⊝ | The outcome was reported by a cross‐over study but the data could not be incorporated into the meta‐analysis. 2/26 participants in the cross‐over study experienced somnolence, however, we do not know whether the participants had tonic‐clonic seizures. No participants reported somnolence whilst receiving placebo. | ||
| 7 per 1000 | 27 per 1000 | ||||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | |||||||
| GRADE Working Group grades of evidence | |||||||
| a Evidence downgraded once for risk of bias. The two studies included in the meta‐analysis failed to clarify whether outcome assessors were blinded. One study also failed to declare the treatment allocation of all randomised participants who withdrew from treatment. Therefore, we awarded unclear risk of bias. | |||||||
| Treatment group | Overall score for QOLIE‐31P | Change in overall score for QOLIE‐31P from baseline to week 19 of treatment | |||
|---|---|---|---|---|---|
| Time point | Number of participants analysed | Mean (SD) | |||
| Number of participants analysed | Least squares mean (SE) | ||||
| Placebo | Baseline | 19 | 61.1 (17.1) | 18 | ‐6.5 (4.0) |
| End of study | 20 | 66.4 (14.8) | |||
| Lamotrigine | Baseline | 17 | 54.3 (9.9) | 15 | ‐8.5 (4.4) |
| End of study | 15 | 65.6 (20.4) | |||
| SD: standard deviation; SE: standard error; QOLIE‐31P: Quality of Life in Epilepsy Inventory‐31 Patient‐weighted. | |||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1.1 50% or greater reduction in primary generalized tonic‐clonic seizure frequency Show forest plot | 2 | 270 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.88 [1.43, 2.45] |
| 1.2 Seizure freedom Show forest plot | 2 | 270 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.89, 2.72] |
| 1.3 Treatment withdrawal Show forest plot | 2 | 270 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.72, 1.99] |
| 1.4 Adverse effects Show forest plot | 2 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 1.4.1 Ataxia | 2 | 270 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.05 [0.05, 199.36] |
| 1.4.2 Dizziness | 2 | 270 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.91 [0.29, 2.86] |
| 1.4.3 Fatigue | 2 | 270 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.02 [0.13, 8.14] |
| 1.4.4 Nausea | 2 | 270 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.60 [0.48, 5.32] |
| 1.4.5 Somnolence | 2 | 270 | Risk Ratio (M‐H, Fixed, 99% CI) | 3.73 [0.36, 38.90] |
| 1.4.6 Headache | 2 | 270 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.75 [0.38, 1.50] |
| 1.4.7 Vomiting | 2 | 270 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.27 [0.39, 4.15] |
| 1.4.8 Convulsion | 2 | 270 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.34 [0.05, 2.13] |
| 1.4.9 Pyrexia | 2 | 270 | Risk Ratio (M‐H, Fixed, 99% CI) | 0.87 [0.21, 3.52] |