Pemberhentian berbanding penerusan penggunaan ubat antipsikotik jangka panjang untuk gejala tingkah laku dan psikologi dalam kalangan orang tua dengan demensia
Abstract
Background
Antipsychotic agents are often used to treat neuropsychiatric symptoms (NPS) in people with dementia although there is uncertainty about the effectiveness of their long‐term use for this indication and concern that they may cause harm, including higher mortality. When behavioural strategies have failed and treatment with antipsychotic drugs is instituted, regular attempts to withdraw them have been recommended in guidelines. Physicians, nurses and families of older people with dementia may be reluctant to stop antipsychotics, fearing deterioration of NPS.
This is an update of a Cochrane Review published in 2013.
Objectives
To evaluate whether withdrawal of antipsychotic agents is successful in older people with dementia and NPS in primary care or nursing home settings, to list the different strategies for withdrawal of antipsychotic agents in older participants with dementia and NPS, and to measure the effects of withdrawal of antipsychotic agents on participants' behaviour and assess safety.
Search methods
We searched the Specialized Register of the Cochrane Dementia and Cognitive Improvement Group (ALOIS), the Cochrane Library, MEDLINE, Embase, PsycINFO, CINAHL, LILACS, clinical trials registries and grey literature sources up to 11 January 2018.
Selection criteria
We included all randomised, controlled trials comparing an antipsychotic withdrawal strategy to continuation of antipsychotics in people with dementia who had been treated with an antipsychotic drug for at least three months.
Data collection and analysis
We used standard methodological procedures according to the Cochrane Handbook for Systematic Reviews of Interventions. We rated the quality of evidence for each outcome using the GRADE approach.
Main results
We included 10 studies involving 632 participants. One new trial (19 participants) was added for this update.
One trial was conducted in a community setting, eight in nursing homes and one in both settings. Different types of antipsychotics at varying doses were discontinued in the studies. Both abrupt and gradual withdrawal schedules were used. Reported data were predominantly from studies at low or unclear risk of bias.
We included nine trials with 575 randomised participants that used a proxy outcome for overall success of antipsychotic withdrawal. Pooling data was not possible due to heterogeneity of outcome measures used. Based on assessment of seven studies, discontinuation may make little or no difference to whether or not participants complete the study (low‐quality evidence).
Two trials included only participants with psychosis, agitation or aggression who had responded to antipsychotic treatment. In these two trials, stopping antipsychotics was associated with a higher risk of leaving the study early due to symptomatic relapse or a shorter time to symptomatic relapse.
We found low‐quality evidence that discontinuation may make little or no difference to overall NPS, measured using various scales (7 trials, 519 participants). There was some evidence from subgroup analyses in two trials that discontinuation may reduce agitation for participants with less severe NPS at baseline, but may be associated with a worsening of NPS in participants with more severe NPS at baseline.
None of the studies assessed withdrawal symptoms. Adverse effects of antipsychotics (such as falls) were not systematically assessed. Low‐quality evidence showed that discontinuation may have little or no effect on adverse events (5 trials, 381 participants), quality of life (2 trials, 119 participants), or cognitive function (5 trials, 365 participants).
There were insufficient data to determine whether discontinuation of antipsychotics has any effect on mortality (very low‐quality evidence).
Authors' conclusions
There is low‐quality evidence that antipsychotics may be successfully discontinued in older people with dementia and NPS who have been taking antipsychotics for at least three months, and that discontinuation may have little or no important effect on behavioural and psychological symptoms. This is consistent with the observation that most behavioural complications of dementia are intermittent and often do not persist for longer than three months. Discontinuation may have little or no effect on overall cognitive function. Discontinuation may make no difference to adverse events and quality of life. Based on the trials in this review, we are uncertain whether discontinuation of antipsychotics leads to a decrease in mortality.
People with psychosis, aggression or agitation who responded well to long‐term antipsychotic drug use, or those with more severe NPS at baseline, may benefit behaviourally from continuation of antipsychotics. Discontinuation may reduce agitation for people with mild NPS at baseline. However, these conclusions are based on few studies or small subgroups and further evidence of benefits and harms associated with withdrawal of antipsychotic is required in people with dementia and mild and severe NPS.
The overall conclusions of the review have not changed since 2013 and the number of available trials remains low.
PICO
Ringkasan bahasa mudah
Menghentikan atau meneruskan penggunaan ubat antipsikotik jangka panjang untuk gejala tingkah laku dan psikologi dalam kalangan orang tua dengan demensia
Soalan ulasan
Kami menyiasat kesan menghentikan ubat antipsikotik dalam kalangan orang tua dengan demensia yang telah mengambilnya selama tiga bulan atau lebih.
Latar belakang
Orang yang mengalami demensia mungkin mengalami gejala dan masalah tingkah laku yang membimbangkan dan sukar untuk diurus oleh penjaga. Gejala tersebut (sering digambarkan sebagai gejala neuropsikiatri atau NPS) termasuk kegelisahan, apati, kemurungan, psikosis (halusinasi dan delusi), merayau, mengulang perkataan atau bunyi, menjerit, dan bertindak dengan keresahan atau cara yang agresif, atau kedua‐duanya.
Ubat antipsikotik sering dipreskrib dengan tujuan mengawal gejala dan tingkah laku ini, walaupun panduan terkini menunjukkan ubat‐ubatan ini hanya boleh digunakan untuk jangka masa pendek bagi tingkah laku yang paling mencabar. Ini terutama kerana ubat‐ubatan ini dianggap mempunyai risiko kesan sampingan (termasuk beberapa yang serius), dan kerana banyak masalah tingkah laku bertambah baik tanpa rawatan. Walau bagaimanapun, ramai orang dengan demensia terus mengambil ubat antipsikotik untuk tempoh masa yang lama.
Ulasan ini menyiasat sama ada orang tua dengan demensia dan NPS boleh menghentikan ubat antipsikotik yang mereka ambil selama sekurang‐kurangnya tiga bulan. Ini adalah kemas kini Ulasan Cochrane yang diterbitkan pada 2013.
Kaedah‐kaedah
Kami mencari sehingga 11 Januari 2018 untuk sebarang kajian rawak yang memperuntukkan beberapa orang dengan demensia yang mengambil ubat antipsikotik untuk meneruskan rawatan dan yang lain untuk menghentikan penggunaan ubat antipsikotik. Peserta kajian disusul untuk tempoh masa tertentu untuk melihat apa yang berlaku.
Keputusan
Kami memasukkan 10 kajian dengan jumlah 632 peserta dalam ulasan kami. Kami menambah satu kajian baru dengan 19 peserta untuk kemas kini ini. Kebanyakan peserta tinggal di pusat rawatan. Kajian‐kajian tersebut berbeza dari segi orang yang disertakan, kaedah‐kaedah yang mereka gunakan dan hasilnya yang diukur.
Oleh kerana kajian‐kajian tersebut adalah pelbagai, adalah tidak mungkin untuk menggabungkan semua data secara berangka. Kami mendapati bukti yang berkualiti rendah bahawa orang tua dengan demensia mungkin dapat menghentikan ubat antipsikotik jangka panjang tanpa masalah tingkah laku menjadi semakin buruk. Walau bagaimanapun, dalam sesetengah orang dengan psikosis, agitasi atau keagresifan dan yang telah bertambah baik dengan ketara ketika mereka pertama kali memulakan rawatan antipsikotik, kami mendapati menghentikan ubat‐ubatan mungkin meningkatkan risiko masalah tingkah laku untuk menjadi semakin buruk lagi. Sebaliknya, agitasi menurun selepas menghentikan ubat‐ubatan dalam sesetengah peserta di mana NPS pada permulaan kajian adalah agak ringan.
Kami tidak menemui bukti yang cukup untuk mengetahui sama ada menghentkani ubat antipsikotik mempunyai kesan bermanfaat terhadap kualiti hidup, pemikiran dan ingatan, atau keupayaan menjalankan tugas harian, atau jika risiko kejadian berbahaya ‐ seperti jatuh ‐ dikurangkan. Kami tidak pasti sama ada menghentikan antipsikotik menyebabkan orang hidup lebih lama.
Kualiti bukti
Keseluruhannya, kualiti bukti adalah rendah atau sangat rendah. Ini bermakna kami berkeyakinan terhad atau sedikit dalam keputusan kajian, dan mungkin penyelidikan yang serupa boleh menemukan sesuatu yang berbeza. Sebab‐sebab utama penilaian ini adalah kerana terdapat sedikit kajian‐kajian yang melibatkan segelintir orang, dan berisiko keputusan‐keputusan tidak dilaporkan sepenuhnya. Semua kajian yang dimasukkan bermasalah merekrut peserta yang cukup, menjadikannya lebih sukar bagi mereka untuk mengesan kesan menghentikan antipsikotik.
Kesimpulan
Bukti terhad menunjukkan menghentikan penggunaan ubat antipsikotik jangka panjang dalam kalangan orang tua dengan demensia dan NPS mungkin boleh dilakukan tanpa membuat tingkah laku mereka lebih buruk. Mungkin ada manfaat untuk mereka dengan NPS yang lebih ringan. Mungkin ada orang dengan gejala yang lebih teruk yang mendapat manfaat daripada rawatan yang berterusan, tetapi lebih banyak penyelidikan dalam kalangan orang dengan NPS yang lebih ringan dan lebih parah diperlukan untuk memastikan ini. Kesimpulan keseluruhan tidak berubah sejak versi terakhir ulasan ini dan bilangan kajian yang dimasukkan masih rendah.
Authors' conclusions
Summary of findings
| Discontinuation compared to continuation of antipsychotic medication for behavioural and psychological symptoms in older participants with dementia | ||||||
| Patient or population: older people with dementia who had been taking an antipsychotic drug for at least 3 months | ||||||
| Outcomes | Illustrative comparative risks (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
|---|---|---|---|---|---|---|
| Assumed risk Continuation antipsychotics | Corresponding risk Discontinuation antipsychotics | |||||
| Success of withdrawal from antipsychotics Measured with a variety of outcomes related to failure to complete the study Follow‐up: 1 to 8 months | In 7 studies there was no overall difference in the outcomes reported for success of withdrawal. In two studies of participants with psychosis, aggression or agitation who had responded to antipsychotic treatment, discontinuation accelerated symptomatic relapse without affecting the number of participants experiencing a relapse in one study and was associated with a higher rate of symptomatic relapse in the other study. In one small study a high proportion of the participants in the discontinuation group failed to complete the study. | 575 (9 RCTs) | ⊕⊕⊝⊝ LOWab | Our intended primary outcome, success of withdrawal defined as the ability to complete the study in the allocated study group, i.e. no failure due to worsening of NPS or relapse to antipsychotic drug use, was not reported in any study. We used the difference between groups in the number of non‐completers of the study as a proxy for our primary outcome. However, data could not be pooled due to variability in outcome measures. | ||
| Behavioural and psychological symptoms Assessed with various scales. Follow up: 1 to 8 months | In 2 pooled studies there was no difference in NPI scores between the continuation and discontinuation groups (see Data and analyses and Figure 1). In five non‐pooled studies, there was no difference in the outcomes on scales measuring overall behaviour and psychological symptoms between groups. | 519 (7 RCTs) | ⊕⊕⊝⊝ | Data could only be pooled for 2 studies due to variability in outcome measures. The two pooled studies performed subgroup analyses according to baseline NPI‐score (≤ 14 or > 14). In one study, some participants with milder symptoms at baseline were less agitated at three months in the discontinuation group. In both studies, discontinuation led to worsening of NPS in some participants with more severe baseline NPS. | ||
| Adverse events Assessed with various scales. Follow‐up: 1 to 8 months | In 5 studies, there was no evidence of a difference between groups in adverse events. | 381 (5 RCTs) | ⊕⊕⊝⊝ LOWab | Data could not be pooled due to variability in outcome measures. Adverse events of antipsychotics were not systematically reported. | ||
| Quality of life (QoL) Assessed with DCM or QoL‐AD. Follow‐up: 3 months to 25 weeks | In 2 studies, there was no evidence of an effect on quality of life. | 119 (2 RCTs) | ⊕⊕⊝⊝ | Data could not be pooled due to variability in outcome measures. There was no difference between discontinuation and continuation group in the overall cohort or in subgroups with baseline NPI score above or below the median (14). | ||
| Cognitive function Assessed with various scales. Follow‐up: 1 to 8 months | In 5 studies, there was no evidence of an impact on scales measuring overall cognitive function. In one of these trials, discontinuation improved a measure of verbal fluency. | 365 (5 RCTs) | ⊕⊕⊝⊝ | Data could not be pooled due to variability in outcome measures. | ||
| Use of physical restraint Follow‐up: 1 month | In one study there was no effect on the use of physical restraint. | 36 (1 RCT) | ⊕⊝⊝⊝ | Conclusion made by the authors but not supported by data. | ||
| Mortality Assessed with various scales. Follow‐up: 4 to 12 months | In two studies there was no evidence of an effect on mortality. | 275 (2 RCTs) | ⊕⊝⊝⊝ | Data could not be pooled due to clinical heterogeneity. In a long‐term follow‐up of 36 months after the 12 months randomised discontinuation trial (Devanand 2012), we were uncertain whether discontinuation decreased mortality. | ||
| *The basis for the assumed risk (e.g. the median control group risk across the studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| a Downgraded one level for indirectness. b Downgraded one level for risk of bias. c Downgraded one level for imprecision due to a small number of participants. d Downgraded two level for risk of bias. | ||||||

Forest plot of comparison: 1 Discontinuation versus continuation of long‐term antipsychotic drug use: continuous data, analysis method: mean difference, outcome: 1.1 Behavioural assessment by using Neuropsychiatric Inventory (NPI) measuring neuropsychiatric symptoms (NPS) at 3 months (Ballard 2004 and Ballard DART‐AD) (Analysis 1.1).
Background
Description of the condition
According to the World Health Organization (WHO), the need for healthcare services for older people with dementia will increase significantly over coming years (Ferri 2005). Today, nearly 50 million people worldwide have dementia (Livingston 2017). By 2030, it is estimated that more than 75 million people will be living with dementia, and the number is expected to increase to more than 131 million by 2050, as populations age. (Prince 2016). The risk of dementia rises sharply with age, with an estimated 25% to 30% of people aged 85 years or over having some degree of cognitive decline (WHO 2015).
Although cognitive deficits are the clinical hallmark of dementia, non‐cognitive symptoms are common and can dominate the disease presentation. These symptoms include a wide range of neuropsychiatric symptoms (NPS), such as agitation, aggression, psychosis (hallucinations and delusions), anxiety, apathy, depression, wandering, repetitive vocalisations, shouting and many other symptoms. These NPS have been observed in 60% to 98% of people with dementia, especially in later stages of the disease. The reported prevalence of each type of NPS varies considerably, from 3% to 54% for delusions, 1% to 39% for hallucinations, 8% to 74% for depressed mood, 7% to 69% for anxiety, 17% to 84% for apathy, 48% to 82% for aggression or agitation, and 11% to 44% for physical aggression (Zuidema 2007). Some NPS may be more likely than chance to occur together and different 'clusters' have been described. Petrovic 2007 reports four behavioural syndromes: a cluster with predominantly psychotic symptoms (hallucinations, irritability, agitation and anxiety); a cluster with predominantly mood symptoms (disinhibition, elation and depressive symptoms); a cluster with predominantly psychomotor symptoms (aberrant motor behaviour) and a cluster with predominantly instinctual symptoms (appetite disturbance, sleep disturbance and apathy). Clusters may differ in prevalence, course over time, biological correlates, psychosocial determinants and treatment response. There is probably overlap between clusters. In general, NPS follow a fluctuating course and high placebo response rates have been reported.
NPS can lead to significant carer stress and cause considerable emotional discomfort. They are associated with higher mortality, higher use of physical restraints, increased length of hospitalisation, and often precipitate admission into a nursing home (Gilley 2000). Up to 30% of the costs of caring for people with dementia are directly attributed to the management of NPS (Herrmann 2006).
The treatment of NPS includes non‐pharmacologic and pharmacologic therapies. Non‐pharmacologic therapy is recommended as first‐line treatment of NPS and pharmacologic therapy can be used when non‐pharmacologic therapy fails (NICE 2016).
A wide variety of pharmacological agents are used in the management of neuropsychiatric symptoms but results of individual RCTs on the efficacy and safety of these agents conflict, and most trials investigating the efficacy of drug treatment are only short term (Ballard 2011).
Antipsychotics are often first‐choice drugs for agitation in dementia, however these drugs have low efficacy for managing agitation in dementia. Risperidone has the best evidence for improving agitation and psychotic symptoms, particularly when aggression was the target symptom, but only for 12 weeks. Haloperidol has effects on quelling aggression, although not on other symptoms of agitation. Olanzapine and quetiapine do not improve psychosis, aggression, or agitation, but aripiprazole may improve agitation (Livingston 2017)
Drugs for cognition, such as cholinesterase inhibitors, including donepezil and memantine, have not been shown to be useful for agitation when agitation is the target symptom. Rivastigmine appears to be beneficial in rate of decline of cognitive function and activities of daily living for people with mild to moderate Alzheimers's disease, although the effects were small and of uncertain clinical importance and have poor safety outcomes with increased risk of adverse events (Wang 2015).
Evidence for carbamazepine in managing behavioural and psychological symptoms of dementia, is very limited with an increased risk of adverse effects (NICE 2016)
A Cochrane Review reported that the selective serotonin reuptake inhibitors (SSRIs) sertraline and citalopram were associated with moderate reduction in symptoms of agitation when compared to placebo in two studies (Seitz 2011). Citalopram in higher doses than recommended, may have benefits, especially in individuals with milder Alzheimer's disease and milder agitation, but has some important adverse effects (Porsteinsson 2014). NICE guidelines did not recommend using SSRIs as treatment for NPS (NICE 2016).
The use of benzodiazepines in the treatment of NPS in older people with dementia is not evidence‐based and should be discouraged because of the risk of dependence and falls (CADTH 2010).
A major concern about the use of antipsychotics to treat behavioural symptoms in people with dementia is increased risk of mortality and stroke (Schneider 2005; Schneider 2006). Product side effect and hazard warnings have been issued for atypical antipsychotics (FDA 2005), and for the older typical or first‐generation antipsychotics in the treatment of psychotic symptoms in older people with dementia. In the UK, Banerjee 2009 concluded it was "time for action" in his report to the Minister of State and recommended using antipsychotics only "when they really need it" and that more attention should go to training and non‐pharmacological interventions. The literature review by Banerjee 2009 of antipsychotic treatment in older people with dementia revealed that while improvement in behavioural disturbance was minimal after 6 to 12 weeks of treatment (estimated effect size 0.1 to 0.2), there was an increase in absolute mortality risk of approximately 1%.
Description of the intervention
Withdrawal from antipsychotic agents can be either abrupt (immediate cessation of the active drug) or tapered (gradual withdrawal according to a predefined dosing schedule or following clinical response). In this review, we appraised RCTs investigating interventions aimed at assisting older people with dementia to withdraw from antipsychotics, either by stopping abruptly or by tapering.
How the intervention might work
Withdrawal of antipsychotic agents from older, often frail, people with dementia and NPS might improve cognitive function, quality of life (QoL) of people with dementia and their carers, and decrease mortality and adverse events (e.g. falls and extrapyramidal symptoms). However, drug withdrawal may also cause a recurrence or worsening of the original NPS with a negative impact on QoL, and may cause a temporary withdrawal syndrome.
Why it is important to do this review
Carers looking after people who are agitated and taking drugs that may be suppressing NPS are sometimes understandably reluctant to consider withdrawal of the drug. However, the episodic nature of such symptoms and the harms associated with antipsychotic use, are less well appreciated. Antipsychotic drugs remain in widespread use in this population. An update of our 2013 Cochrane Review (Declercq 2013) of the risks and benefits associated with antipsychotic withdrawal was therefore needed.
Objectives
To evaluate whether withdrawal of antipsychotic agents is feasible in older people with dementia and NPS in primary care or nursing home settings; to list the different strategies for withdrawal of antipsychotic agents in older people with dementia and NPS; and to measure the effects of the withdrawal of antipsychotic agents on peoples’ behaviour and assess safety issues such as mortality, adverse effects or withdrawal symptoms.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials. Withdrawal trials that were not placebo‐controlled were included only if the outcome assessors were blinded to treatment allocation. No language restrictions were applied.
Types of participants
Older participants with dementia living in the community or in nursing homes and taking an antipsychotic drug.
Older participants were defined as 65 years or over without upper age limit.
Dementia was defined as an acquired organic mental disorder with loss of intellectual abilities of sufficient severity to interfere with social or occupational functioning. The dysfunction is multifaceted and involves memory, behaviour, personality, judgment, attention, spatial relations, language, abstract thought, and other executive functions. The intellectual decline is usually progressive, and initially spares the level of consciousness. We accepted studies for inclusion if the reports stated that participants had dementia or any subtype of dementia. If there was any doubt about this diagnosis, first authors of studies were asked to provide further information. All grades of dementia severity were included, regardless of the method of diagnosis. Participants with schizophrenia were excluded if this was reported in the trial.
Nursing homes are defined as institutions in which long‐term care is provided by professional care workers for three or more unrelated, frail, older individuals.
Types of interventions
We included studies in which the intervention was withdrawal of antipsychotic drugs prescribed long‐term for neuropsychiatric symptoms (NPS) in older participants with dementia.
Long‐term antipsychotic drug use is defined as use of at least three months of any antipsychotic agent, either typical (first generation) or atypical (second generation) at a fixed dosage. Although there is no good definition of the subgroup of atypical antipsychotic drugs, we prefer this term to 'new' or 'second generation' antipsychotics. The antipsychotic agents are listed according to the Anatomical Therapeutic Chemical (ATC) classification. Names of drug classes and individual drugs are presented in Table 1 and Table 2, respectively; atypical antipsychotic agents are labelled with an asterisk. Antipsychotic agents should be used in a stable dose, and within the therapeutic range specified in the drug product information insert. Defined daily doses (per os), as mentioned in the ATC classification, are also listed in Table 2. Chlorpromazine is considered to be the reference drug. Baseline dosage regimen is classified as very low, low or high for each antipsychotic agent, according to the dosage table proposed by Ballard 2008 (e.g. for risperidone a dose of 0.5 mg once daily is very low, 0.5 mg twice daily is low and 1 mg twice daily is high; for haloperidol 0.75 mg once daily is very low, 0.75 mg twice daily is low and 1.5 mg twice daily is high; for the referent molecule chlorpromazine 12.5 mg once daily is very low, 12.5 mg twice daily is low and 25 mg twice daily is high).
| Phenothiazines with aliphatic side chain Phenothiazines with piperazine structure Fhenothiazines with piperidine structure Butyrophenone derivatives Indole derivatives Thioxanthene derivatives Diphenylbutylpiperidine derivatives Diazepines, Oxazepines and Thiazepines Benzamides Other antipsychotics |
| Phenothiazines with aliphatic side‐chain N05AA01 Chlorpromazine 0.3 g per os N05AA02 Levomepromazine 0.3 g per os N05AA03 Promazine 0.3 g per os N05AA04 Acepromazine 0.1 g per os N05AA05 Triflupromazine 0.1 g per os N05AA06 Cyamemazine N05AA07 Chlorproethazine |
| Phenothiazines with piperazine structure N05AB01 Dixyrazine 50 mg per os N05AB02 Fluphenazine 10 mg per os N05AB03 Perphenazine 30 mg per os N05AB04 Prochlorperazine 0.1 g per os N05AB05 Thiopropazate 60 mg per os N05AB06 Trifluoperazine 20 mg per os N05AB07 Acetophenazine 50 mg per os N05AB08 Thioproperazine 20 mg per os N05AB09 Butaperazine 10 mg per os N05AB10 Perazine 0.1 g per os N05AB20 Homophenazine |
| Phenothiazines with piperidine structure N05AC01 Periciazine 50 mg per os N05AC02 Thioridazine 0.3 g per os N05AC03 Mesoridazine 0.2 g per os N05AC04 Pipotiazine 10 mg per os |
| Butyrophenone derivatives N05AD01 Haloperidol 8 mg per os N05AD02 Trifluperidol 2 mg per os N05AD03 Melperone* 0.3 g per os N05AD04 Moperon 20 mg per os N05AD05 Pipamperone 0.2 g per os N05AD06 Bromperidol 10 mg per os N05AD07 Benperidol 1.5 mg per os N05AD08 Droperidol N05AD09 Fluanisone |
| N05AE Indole derivatives N05AE01 Oxypertine 0.12 g per os N05AE02 Molindone 50 mg per os N05AE03 Sertindole* 16 mg per os N05AE04 Ziprasidone* 80 mg per os |
| Thioxanthene derivatives N05AF01 Flupentixol 6 mg per os N05AF02 Clopenthixol 0.1 g per os N05AF03 Chlorprothixene 0.3 g per os N05AF04 Tiotixene 30 mg per os N05AF05 Zuclopenthixol 30 mg per os |
| Diphenylbutylpiperidine derivatives N05AG01 Fluspirilene N05AG02 Pimozide 4 mg per os N05AG03 Penfluridol 6 mg per os |
| Diazepines, Oxazepines and Thiazepines N05AH01 Loxapine 0.1 g per os N05AH02 Clozapine* 0.3 g per os N05AH03 Olanzapine* 10 mg per os N05AH04 Quetiapine* 0.4 g per os |
| Benzamides N05AL01 Sulpiride 0.8 g per os N05AL02 Sultopride 1.2 g per os N05AL03 Tiapride 0.4 g per os N05AL04 Remoxipride 0.3 g per os N05AL05 Amisulpride* 0.4 g per os N05AL06 Veralipride N05AL07 Levosulpiride 0.4 g per os |
| Other antipsychotics N05AX07 Prothipendyl 0.24 g per os N05AX08 Risperidone* 5 mg per os N05AX09 Clotiapine 80 mg per os N05AX10 Mosapramine* N05AX11 Zotepine* 0.2 g per os N05AX12 Aripiprazole* 15 mg per os N05AX13 Paliperidone* *atypical antipsychotics |
* Atypical antipsychotic agents.
Types of outcome measures
Primary outcomes
-
Success of withdrawal from antipsychotics over short‐term (four weeks or less) and long‐term (more than four weeks) follow‐up. Success is defined as the ability to complete the study in the allocated study group, i.e. no dropout due to worsening of NPS, or no relapse to antipsychotic drug during the trial.
-
Behavioural and psychological symptoms (especially agitation, aggression and psychotic symptoms) measured with appropriate scales (e.g. Neuropsychiatric Inventory score (NPI), Neuropsychiatric Questionnaire score (NPI‐Q)).
-
Presence or absence of withdrawal symptoms or withdrawal syndrome in the first four weeks.
-
Withdrawal symptoms or withdrawal syndrome include autonomic and behavioural symptoms such as nausea, vomiting, anorexia, rhinorrhoea, diarrhoea, diaphoresis, myalgia, paraesthesia, anxiety, as well as movement disorders, such as withdrawal emergent parkinsonism, withdrawal dyskinesia and covert dyskinesia.
-
Agitation, insomnia and restlessness have also been reported during withdrawal, although it is possible these symptoms occur due to rebound phenomenon. It is impossible to discriminate between these aetiological phenomena.
-
A withdrawal neuroleptic malignant syndrome is a very rare but extremely severe condition that can complicate abrupt antipsychotic discontinuation.
-
-
Adverse events attributable to antipsychotics (e.g. falls, extrapyramidal symptoms, cardiovascular events and diabetes.
Secondary outcomes
-
Cognitive function (general or domain‐specific, e.g. short‐term memory, frontal executive function, language) measured with appropriate scales (e.g. Severe Impairment Battery (SIB) score, Standardised Mini‐Mental State Examination (SMMSE), FAS verbal fluency test, Sheffield Test for Acquired Language Disorder (STALD receptive and STALD expressive skill).
-
Quality of life of participants, carers, family of participants or a combination of these, measured with appropriate scales (e.g. Dementia Care Mapping (DCM) and Quality of life‐Alzheimer Disease (QoL‐AD)).
-
Time, in days, until prescription of any psychotropic or any antipsychotic agent.
-
Use of physical restraint.
-
Mortality.
-
Other secondary outcomes reported in the primary papers (e.g. global functioning, sleep, clinical global impression) measured with appropriate scales.
Search methods for identification of studies
Electronic searches
We searched ALOIS, the Cochrane Dementia and Cognitive Improvement Specialized Register to 10 January 2018. We performed an interim search on 3 March 2017. Searches for the previous version of this review were performed in February 2009, March 2011, June 2011, November 2011, August 2012, and November 2012 (Declercq 2013).
ALOIS is maintained by the Cochrane Dementia and Cognitive Improvement Group's Information Specialists and contains studies in the areas of dementia prevention, dementia treatment and cognitive enhancement in healthy. Studies are identified from:
-
MEDLINE, Embase, CINAHL, PsycINFO and LILACS;
-
trial registers: ISRCTN, UMIN (Japan's Trial Register), the World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) (which covers ClinicalTrials.gov, ISRCTN, the Chinese Clinical Trials Register, the German Clinical Trials Register, the Iranian Registry of Clinical Trials, and the Netherlands National Trials Register, among others);
-
the Cochrane Library's Central Register of Controlled Trials (CENTRAL); and
-
grey literature sources: ISI Web of Knowledge Conference Proceedings, Index to Theses, Australasian Digital Theses.
Aee About ALOIS for all sources searched.
Details of the search strategies used to retrieve reports of trials from healthcare databases, CENTRAL and conference proceedings can be viewed in the 'methods used in reviews' section in editorial information about the Dementia and Cognitive Improvement Group.
Additional searches were performed in many of the sources listed above to cover the timeframe from the last searches performed for ALOIS to ensure that the search for the review was as up‐to‐date and as comprehensive as possible. Search strategies are presented in Appendix 1, Appendix 2 and Appendix 3.
Appendix 4 lists abbreviations used in this review.
Searching other resources
We reviewed reference lists of included and excluded studies to identify any additional studies.
Data collection and analysis
Presentation of results and 'Summary of findings' tables
We included a 'Summary of findings' table, which included seven outcomes, prepared using GRADEpro GDT. We used the GRADE approach to assess evidence quality for all outcomes. Evidence was assessed as high‐, moderate‐, low‐, or very low‐quality, depending on the seriousness of concern about risk of bias, imprecision, inconsistency, indirectness, and publication bias. For each outcome in the 'Summary of findings' table we presented a summary of the available data, the magnitude of the effect size, and the quality of the evidence. We justified all decisions to downgrade the quality of evidence in the footnotes of the 'Summary of findings' table.
Selection of studies
For this update, two review authors (EVL, MP) independently screened study titles and abstracts retrieved from the search for their relevance. We removed obviously irrelevant reports and duplicated reports of the same study. We obtained full‐text versions of potentially relevant reports. We examined these independently to assess compliance with the predefined eligibility criteria. Two review authors independently decided which trials met the inclusion criteria. Differences between authors were resolved by discussion and by consulting other review authors (MVD, TC). We entered all search results into RevMan 5 (Review Manager 2014). We listed excluded studies and reasons for exclusion in the Characteristics of excluded studies tables.
Data extraction and management
Three review authors (TD, MA, EVL) independently extracted data from included studies using a predefined data extraction form. Differences between authors were resolved by discussion and by consulting the review authors (MVD, TC). We extracted the following data:
-
first author, publication year, journal;
-
number, age and gender distribution of the participants included in the trial;
-
withdrawal method (e.g. abruptly versus tapered withdrawal);
-
baseline severity of NPS (e.g. NPI‐score), agitation (e.g. Cohen‐Mansfield Agitation Inventory (CMAI) scale or psychotic symptoms (hallucinations, delusions);
-
baseline severity of dementia as determined by the MMSE score (e.g. mild: 19 to 16; moderate: 15 to 10; severe: 9 to untestable), or other appropriate scales;
-
baseline dose of antipsychotic agent (very low, low, high) and type of antipsychotic agents (typical or atypical); and
-
results (primary and secondary outcomes).
If a paper did not provide sufficient information about either study details or results, we contacted the study authors where possible.
Assessment of risk of bias in included studies
Three review authors (TD, EVL, MVD) independently assessed each included study using the Cochrane’s tool for assessing risk of bias, described in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved disagreements by discussion with co‐authors (MP, TC). We assessed:
-
random sequence generation;
-
allocation concealment;
-
blinding of participants and personnel;
-
blinding of outcome assessors;
-
incomplete outcome data;
-
dropout/selective outcome reporting; and
-
other potential sources of bias.
We judged each potential source of bias as high, low or unclear and provided a quote from the study report together with justification for our judgement in ’Risk of bias' tables. We summarised the risk of bias judgements across different studies for each of the domains listed. We reported the risk of bias using the ’Risk of bias’ tool from the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
In this update, we assessed bias related to blinding of participants and personnel separately from bias related to blinding of outcome assessment (Higgins 2016).
Measures of treatment effect
We entered data into RevMan 5 software for data analysis (Review Manager 2014). For continuous data, we calculated the mean difference (MD) if the same scale was used, or a standardised mean difference (SMD), which is the absolute mean difference divided by the pooled SD, if different scales were used to measure the same construct. We calculated a 95% confidence interval (CI) for each estimate. Dichotomous outcomes were reported as odds ratios (ORs). We pooled data reported as mean differences by using the inverse variance method as described in the Cochrane Handbook for systematic Reviews of Interventions (Higgins 2011).
Unit of analysis issues
Participants in randomised controlled trials (RCTs) was the unit of analysis.
Cross‐over trials were included using the results from paired analyses, which adjust for within‐individual comparisons (Elbourne 2002). The unit of analysis in Cohen‐Mansfield 1999 was paired data for drug versus placebo at the end of intervention period 1 and intervention period 2. Different analysis for assessment of within‐subject variable and between‐subject variable were performed.
Dealing with missing data
We reported where data were missing from published reports. We contacted the original investigators to request missing data. If these data remained unavailable we analysed the available data. We used intention‐to‐treat (ITT) analyses where possible. Any statistical method used by the study authors (e.g. multiple imputation analysis, last observation carried forward) to deal with not‐missing‐at‐random data was reported. If study authors reported outcomes for participants who completed the study, as well as carried forward or otherwise imputed data, we used the latter data for pooling.
Assessment of heterogeneity
We assessed heterogeneity in two ways. First, we explored the presence of heterogeneity at face value by comparing population groups, interventions or outcomes across studies. In the case of clear face value heterogeneity, we reported the outcomes of the studies narratively and did not pool the results. Meta‐analysis was only performed when studies were sufficiently homogeneous in terms of participants, interventions, and outcomes. If there was no obvious clinical heterogeneity we used statistical tests such as the Cochran Chi² (Q) test and the I² statistic to determine the presence and level of statistical heterogeneity for each outcome. An I² value of 50% or higher was considered as significantly heterogeneous (Higgins 2011; Review Manager 2014).
Assessment of reporting biases
To minimise risk of publication bias, a comprehensive search was performed in multiple databases, including searching for unpublished studies. If more than 10 RCTs were identified, we planned to assess the existence of publication bias by constructing a funnel plot (Higgins 2011).
Data synthesis
Trials that did not report comparable outcomes were considered clinically heterogeneous and results were not pooled in meta‐analysis. In this case, we performed critical interpretive synthesis of data from individual studies.
The duration of follow‐up in trials varied considerably. If the range of follow‐up was considered too large to pool results for meta‐analysis, the data were divided into smaller time periods and separate meta‐analyses were conducted for each period. The overall estimate was calculated using a fixed‐effect model in the absence of statistical heterogeneity. In the presence of substantial statistical heterogeneity (I² value of 50% or higher), a random‐effects model was used (Higgins 2011).
Subgroup analysis and investigation of heterogeneity
We conducted only one meta‐analysis including two trials. We were therefore unable to analyse subgroups. We reported the results of subgroup analyses in the included studies.
Sensitivity analysis
We conducted no sensitivity analyses.
Results
Description of studies
See Characteristics of included studies; Characteristics of excluded studies; and Table 3.
| Study IDI | Setting | Duration | Randomised number | Discontinuation group | Continuation group | Discontinuation schedule | Control | Behavioural inclusion criteria | Notes |
|---|---|---|---|---|---|---|---|---|---|
| Residents in long‐term care facilities | 3 months | 100 | 46 | 54 | Abrupt | Typical APa or risperidone | NPIb not higher than 7 | ||
| Residents in long‐term care facilities | 6 months 12 months | 165 | 82 | 83 | Abrupt | Typical and risperidone | NRc | ||
| Residents in nursing homes | 25 weeks | 19 | 9 | 10 | Tapering over 2 week | Risperidone | NRc | Unpublished study | |
| Residents in long‐term care facilities | 1 month | 36 | 22 | 14 | Abrupt + tapering over 2 weeks | Typical APa | Physically aggressive participants identified by nurse supervisors | ||
| Residents in nursing homes | 7 weeks followed by 7 weeks cross‐over | 58 | 29 | 29 | Tapering over 3 weeks | Typical APa + lorazepam | NRc | Cross‐over study | |
| Residents in the community | 6 months (primary analysis) 12 months | 20 | 10 | 10 | Abrupt + tapering over 2 weeks | Haloperidol | Current symptoms of psychosis, agitation or aggression | Participants had a response to haloperidol open treatment for 20 weeks | |
| Residents in the community and nursing homes | 4 months 8 months | 110 | 70 | 40 | Abrupt + tapering over 2 week | Risperidone | NPIb score higher than 4 on psychosis or agitation/aggression subscale | Participants had a response to risperidone open treatment for 16 weeks | |
| Residents in nursing homes | 1 month | 36 | 18 | 18 | Tapering over 1 week | Thioridazine | NRc | ||
| Residents in nursing homes | 1 month | 55 | 27 | 28 | Abrupt | Haloperidol risperidone, olanzapine | All participants regardless individual symptoms | ||
| Residents in nursing homes | 26 weeks | 34 | 17 | 17 | Tapering over 2 weeks | Typical APa | Stable behaviour |
a AP: antipsychotic drug.
b NPI: Neuropsychiatric Inventory.
c NR: not reported.
Results of the search
Searches for this update identified 101 records after a de‐dupliaction and first assessment performed by CDCIG information specialists; an ongoing study identified in the 2013 review was also assessed for inclusion. We removed 13 duplicate records (n = 89 records). We excluded 76 records following assessment of title and abstract (n = 13 full‐text reports). Following assessment, we excluded 12 full‐text articles that did not meet inclusion criteria (see Characteristics of excluded studies). We included one additional randomised controlled trial (RCT) involving 19 participants (Bergh 2011) for this update (Figure 2).
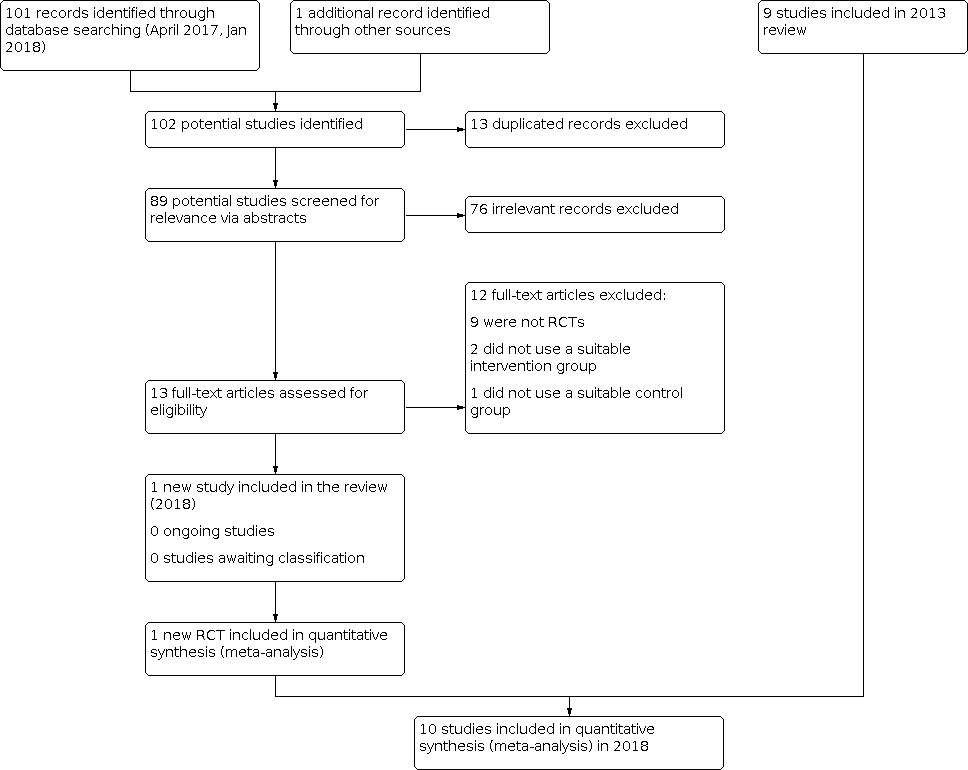
Inclusions of trials of study flow diagram 2018
Included studies
The 2013 review (Declercq 2013) included nine studies (Ballard 2004; Ballard 2008; Bridges‐Parlet 1997; Cohen‐Mansfield 1999; Devanand 2011; Devanand 2012; Findlay 1989; Ruths 2008; van Reekum 2002). One additional study was added for this update (Bergh 2011). The 10 included studies involved a total of 632 participants (Characteristics of included studies; Table 3).
Overview
The included trials were very diverse in terms of study participants (such as the case definition applied and the severity of dementia of the participants), types and dosages of antipsychotics used before withdrawal, exclusion criteria, interventions (i.e. method of discontinuation), outcomes, and times of assessment.
Design
Nine studies were parallel‐group RCTs. One study was a cross‐over RCT (Cohen‐Mansfield 1999).
Sample size
All 10 studies included small numbers of participants. Seven studies included fewer than 100 participants; three studies included between 100 and 200 participants (Ballard 2004; Ballard 2008; Devanand 2012).
Study setting
Eight studies included participants in nursing homes. One pilot study included participants with Alzheimer's disease and psychosis, agitation or aggression who were living in the community (Devanand 2011). One study included participants with Alzheimer's disease and psychosis, agitation or aggression who were living in the community or were residents of assisted‐living facilities or nursing homes (Devanand 2012).
Participants
Clinical characteristics at baseline
See Characteristics of included studies
1. Age status at baseline
Participants' average age was 80 years or over in most studies.
2. Sex status at baseline
Most studies included higher proportions of female participants. Findlay 1989 recruited only female participants.
3. Dementia status at baseline
Different methods were used to diagnose dementia.
-
Ballard 2004 and Ballard 2008 included only participants with Alzheimer's disease who fulfilled the National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer's Disease and Related Disorders Association (NINCDS‐ADRDA) criteria for possible or probable Alzheimer's disease.
-
Bergh 2011 included participants with dementia due to Alzheimer's disease or vascular type or mixed type according to ICD‐10 clinical criteria.
-
Bridges‐Parlet 1997 included residents with diagnoses of dementia or possible or probable dementia.
-
Cohen‐Mansfield 1999 had no explicit diagnostic standard for dementia; the study included nursing home residents aged over 70 years receiving haloperidol, thioridazine, and lorazepam. The study author confirmed by email that the residents participating in the study met the inclusion criteria dementia (Declerck 2009a [pers comm]).
-
Devanand 2011 and Devanand 2012 included participants with diagnoses of dementia using DSM‐IV and probably Alzheimer's Disease by National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (NINCDS‐ADRDA) criteria.
-
Findlay 1989 included participants with Alzheimer's disease classified according to ICD‐9 criteria, assessed by a consultant psychiatrist and based on medical history.
-
Ruths 2008 included participants with diagnoses of dementia according to ICD‐10 clinical criteria.
-
van Reekum 2002 included participants with all forms of dementia based on chart review.
4. Cognitive status at baseline
At baseline, participants in most of the studies were described as having moderate to severe dementia. A variety of methods were used to measure baseline cognitive severity. Several studies had inclusion criteria based on cognitive severity at baseline.
-
Ballard 2008 included participants with a Standardised Mini‐Mental State Examination (SMMSE) score of 6 or a Severe Impairment Battery (SIB) score > 30.
-
In Devanand 2011, study participants had SMMSE scores ranging from 5 to 26.
-
Participants living in the community in the Devanand 2012 study had SMMSE scores of 5 to 26; participants residing in nursing homes had scores between 2 to 26.
-
Cohen‐Mansfield 1999 used the Brief Cognitive Rating Scale (BCRS) at baseline to determine participants' cognitive function without criteria.
-
Findlay 1989 used the Cognitive Assessment Scale (CAS) for measuring cognitive status without criteria.
-
There were no clear cut‐off values reported to indicate the severity of cognitive status severity in seven studies (Ballard 2004; Bergh 2011; Bridges‐Parlet 1997; Cohen‐Mansfield 1999; Findlay 1989; Ruths 2008; van Reekum 2002.
5. Behavioural status at baseline
Several trials applied inclusion criteria based on severity of behavioural problems at baseline. This was not an inclusion criterion for this review.
-
Ballard 2004 included participants with individual scores on the Neuropsychiatric Inventory (NPI) that were not higher than 7 at the time of evaluation.
-
In Bridges‐Parlet 1997, participants were selected by nurse supervisors who identified physically aggressive participants with dementia treated with antipsychotics.
-
In the Devanand 2011 pilot trial, participants needed to have signs of psychosis, agitation or aggression or both to be included in the study. Psychosis was identified using the Columbia University Scale for Psychopathology in Alzheimer's Disease (CUSPAD) and the Brief Psychiatric Rating Scale (BPRS) (psychosis factor of at least 4). Agitation and aggression was measured on the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) Behavioural Rating Scale for Dementia (score > 3 and present for at least 10 days per month, on one or more of the items for agitation, purposeless wandering, verbal aggression or physical aggression).
-
Participants in the Devanand 2012 trial had scores on the NPI of 4 or more at both screening and baseline on the delusions or hallucinations subscale (psychosis score) or the agitation and aggression subscale (agitation/aggression score) (with scores on NPI subscales ranging from 0 to 12).
-
Ruths 2008 included all potential participants regardless of individual neuropsychiatric symptoms (absent = 0, mild = 1, moderate = 2, severe = 3), providing a NPI‐Q sum score ranging from 0 to 36.
-
In van Reekum 2002, participants were included if they had "stable" behaviour.
Four studies (Ballard 2008; Bergh 2011; Cohen‐Mansfield 1999; Findlay 1989) did not use severity of behaviour problems as a criterion for inclusion.
6. Global status at baseline
Several studies specified global functional status at time of inclusion in the study.
-
In Ballard 2004, participants had Clinical Dementia Rating (CDR) Scale severity of stage 1 or greater.
-
In Bergh 2011, inclusion was limited to Dementia Rating 1, 2 or 3 without further specification.
-
van Reekum 2002 used the Clinical Global Impression scale (CGI) without further specifications.
No other studies reported measurements of global functioning at baseline.
Intervention
Antipsychotic treatments to be withdrawn and withdrawal schedules
The included studies used different antipsychotics at different dosages. Antipsychotics used were thioridazine, chlorpromazine, haloperidol, trifluoperazine (classified as 'typical antipsychotics') and risperidone or olanzapine (classified as 'atypical antipsychotics').
Three studies used an abrupt withdrawal schedule (Ballard 2004; Ballard 2008; Ruths 2008).
Two studies (Bridges‐Parlet 1997; Devanand 2012) withdrew most participants abruptly from antipsychotic drugs, but used a tapering schedule when the baseline dose exceeded the equivalent of 50 mg of chlorpromazine. Bridges‐Parlet 1997 did this by halving the baseline antipsychotic dose during week one and discontinuing the antipsychotic drug completely at the beginning of week two. In Devanand 2012, when the baseline dose was 2 mg risperidone or more daily, one‐week tapering was used by means of a sequential double‐blind placebo substitution (e.g. one 2 mg tablet of risperidone was switched to one 1 mg tablet and then to one placebo tablet).
The other studies used a tapering schedule.
-
Most participants in Ballard 2008 were taking risperidone or haloperidol at variable dosages: participants were taking at least 10 mg chlorpromazine equivalents of a typical neuroleptic or at least 0.5 mg daily of risperidone. Dosages were defined as high, low, or very low:
-
very low: risperidone 0.5 mg daily, chlorpromazine 12.5 mg once daily, trifluoperazine 0.5 mg once daily; haloperidol 0.75 mg once daily;
-
low: risperidone 0.5 mg twice daily, chlorpromazine 12.5 mg twice daily, trifluoperazine 0.5 mg twice daily; haloperidol 0.75 mg twice daily; and
-
high: risperidone 1 mg twice daily; chlorpromazine 25 mg twice daily; trifluoperazine 1 mg twice daily; haloperidol 1.5 mg twice daily.
-
-
Most participants in Ballard 2004 took risperidone or thioridazine at variable dosages. Participants used (mean ± SD dose): risperidone 1.3 mg ± 0.7 mg, thioridazine 38.0 mg ± 26.2 mg, haloperidol 0.9 mg ± 0.4 mg, trifluoperazine 3.0 mg ± 1.4 mg or chlorpromazine 20 mg (no SD value for chlorpromazine as there was only one person taking this drug).
-
In Cohen‐Mansfield 1999, participants took haloperidol, thioridazine and lorazepam at variable dosages (mean dosage haloperidol 1.34 mg, thioridazine 27.0 mg and lorazepam 0.94 mg, no SD given). The cross‐over design of this trial led to a three‐week dose‐tapering period followed by seven weeks of placebo. After this placebo period, the placebo group was titrated back to the original dose and groups were switched for the procedure. Participants were withdrawn from antipsychotics (haloperidol and thioridazine), and also from lorazepam, which is a benzodiazepine. Because of the dual drug cross‐over design, it was difficult to interpret the results of this study.
-
In Devanand 2011, participants in a community setting with Alzheimer's disease and symptoms of psychosis, agitation or aggression were included and treated with haloperidol in phase A. In phase B (discontinuation trial) only participants who responded well to haloperidol in phase A were included. Criteria for clinical response were minimum 50% reduction from baseline in the sum score of the three most prominent symptoms of psychosis, agitation or aggression, a sum score of 6 or less on these three items (range 0 to 18), and minimal or greater improvement on the Clinical Global Impression scale (CGI‐C) rated only for symptoms of psychosis, agitation and aggression. Doses of haloperidol used in phase B varied (4 mg daily, 2 mg to 3 mg daily, 0.5 mg to 1 mg daily). According to these different dosages there was a two‐week tapering period (4 mg daily switched to 2 mg daily for one week, 1 mg daily for the next week and then to placebo; participants on 2 mg to 3 mg daily switched to 1 mg daily for two weeks and then to placebo, and participants who received 0.5 mg or 1 mg daily were switched directly to placebo without a tapering period).
-
In Devanand 2012, phase A participants were given flexible‐dose risperidone for 16 weeks: risperidone therapy was initiated at a dose of 0.25 mg to 0.5 mg daily and could be increased to 3 mg daily, depending on the response and side effects. Participants who had a response in phase A entered phase B of the study (discontinuation trial with three regimens: continued risperidone therapy for 32 weeks (group 1), risperidone therapy for 16 weeks followed by placebo for 16 weeks (group 2) or placebo for 32 weeks (group 3)).
-
Findlay 1989 used a half‐dose reduction during the first week and a total placebo substitution over the next week. Original dosages that participants had been receiving were stable dosages between 10 mg and 100 mg thioridazine for at least two months.
-
van Reekum 2002 did not define antipsychotic drug classes and included residents who had been taking typical or atypical antipsychotics for at least six months. In this study, all participants received a standard order for lorazepam (0.5 mg to 1.0 mg) on an as‐needed basis for agitation. The study used a tapering schedule of two weeks in which original medication was halved for the first week and the remaining dose halved during the second week followed by a six‐month study period.
-
In Ruths 2008, participants were taking risperidone 1.0 mg (median; range 0.5 mg to 2.0 mg), olanzapine 5.0 mg (2.5 mg to 5.0 mg), and haloperidol 1.0 mg (0.5 mg to 1.5 mg).
-
In Bergh 2011, all participants were taking risperidone at inclusion. The doses of the continuation group was determined by the participant's dose of antipsychotics prior to recruitment to the study. The study used a tapering schedule over one week for the discontinuation group. Participants received 50% of their original medication dose on day 1, reduced to 25% on day 4 and 12.5% on day 6 and fully discontinued on day 7. The mean dose at inclusion was risperidone 0.92 mg/day.
Outcome measures
Outcome measures were very diverse across included studies and therefore difficult to compare. We could not calculate a standardised mean difference (SMD) for any outcome when different scales were used, because we did not consider the scales to be measuring identical constructs.
Four studies reported outcomes as mean differences with SDs (Ballard 2004; Ballard 2008; Bergh 2011; Ruths 2008). Five studies (Bridges‐Parlet 1997; Cohen‐Mansfield 1999; Devanand 2011; Devanand 2012; van Reekum 2002) reported outcomes as means, but only three also reported SDs (Bridges‐Parlet 1997; Devanand 2011; Devanand 2012). Findlay 1989 reported outcomes as means with a range and number of observations.
Primary outcomes
1. Success of withdrawal from antipsychotics in the short‐term (4 weeks or less) and long‐term (more than 4 weeks)
We defined successful withdrawal as ability to complete the study in the allocated study group (i.e. no withdrawal due to worsening of neuropsychiatric symptoms (NPS), or no relapse to antipsychotic drug use during the trial).
-
Ballard 2004 and Ballard 2008 reported the participant flow in results sections and reasons for withdrawal from the study, for example, withdrawal because of behavioural deterioration. Unfortunately, relapse to antipsychotic drug use was not mentioned.
-
Bridges‐Parlet 1997 reported participants completing the study and relapse to antipsychotic drug use after completion of the trial.
-
Cohen‐Mansfield 1999 reported the participant flow in the results section and reasons why participants discontinued before study completion.
-
Devanand 2011: phase B reported relapse using criteria of 50% worsening of the three target symptoms of psychosis, agitation and aggression, and a severity score ≥ 6 on these three items (range 0 to 18), and minimal or greater worsening on the Clinical Global Impression scale (CGI‐C) (rated for psychosis, agitation and aggression). Time to relapse was also measured in Devanand 2011 phase B.
-
Devanand 2012: phase B reported relapse using criteria of increase in the Neuropsychiatric Inventory (NPI) core score of 30% or more, or a 5‐point increase from the score at the end of phase A, and a score of 6 (much worse) or 7 (very much worse) on the CGI‐C scale. The NPI‐core score is the sum of the subscale scores for agitation‐aggression, hallucinations and delusions. The CGI‐C scale ranged from 1 to 7, with higher scores indicating less improvement for overall psychosis, agitation or aggression.
-
Findlay 1989 did not report withdrawal from the study in the text, but results can be extracted from the table.
-
van Reekum 2002 reported early withdrawals from the study, but did not mention relapse to antipsychotic drugs.
-
Ruths 2008 mentioned relapse of antipsychotic drug use after withdrawal from antipsychotic drugs.
-
Bergh 2011 reported participant flow in the results section and reasons for withdrawal from the study, but relapse to antipsychotic drug use was not mentioned.
2. Behavioural and psychological symptoms (especially agitation, aggression and psychotic symptoms)
Behavioural and psychological symptoms (especially agitation, aggression and psychotic symptoms) were assessed by different scales across included studies:
-
Behavioural and psychological symptoms measured with NPI and NPI‐Q
Two trials using the Neuropsychiatric Inventory (NPI) or Neuropsychiatric Inventory Questionnaire (NPI‐Q) score as a primary outcome performed NPI‐subscore analysis (Ballard 2004; Ballard 2008).
Ruths 2008 assessed agitation as a subscore of the NPI‐Q and Ballard 2004 assessed agitation as subscore of Neuropsychiatric Inventory (NPI) total score.
Only Devanand 2012 reported the effect on the NPI core score, that is, the sum of the NPI‐subscale for agitation and aggression, hallucinations, and delusions.
The NPI covers 12 domains of behavioural and neurovegetative symptoms to assess outcome. Each subscore is rated on a 12‐point scale, assessing severity (0 to 3) and frequency (0 to 4) of a domain, with a theoretical maximum of 144 (i.e. 12 x 12) (range 1 to 144). The NPI‐Q (Neuropsychiatric Inventory Questionnaire) assesses only the severity of each of the same 12 domains (theoretically maximum of 36, range 0 to 36) and can be considered as a shorter version of the NPI.
Bergh 2011 used the primary endpoint changes in the Neuropsychiatric Inventory (NPI‐10). The authors reported NPI‐10, this assesses 10 items out of 12 NPI domains (no sleep/night time behaviour changes and no appetite/eating changes).
-
Behavioural and psychological symptoms measured with other scales
Bergh 2011 used also the primary endpoint changes in Cornell Scale for Depression in Dementia (CSDD) (minimum score 0 and maximum score 38) assesses depressive symptoms of the participants with dementia. A score of 8 points and above is regarded as a sign of a depressive disorder, while a score of 13 and above is regarded as a sign of a severe depressive disorder. The CSDD was divided into two subscales, mood (sadness, anxiety, pessimism, suicidal thoughts, poor self esteem and delusion) and non‐mood (remaining 13 symptoms).
Bridges‐Parlet 1997 used the physically aggressive behaviour scale (PAB) as the main outcome measure. The PAB scale assesses aggressive behaviour identified by type (coded by a barcode system). Five different types of behaviour are identified: hitting, biting, scratching, kicking and pushing. The study authors also assessed verbal aggressiveness, defined as an instance of speaking in an angry tone of voice, swearing or yelling in anger.
Cohen‐Mansfield 1999 used behaviour and agitation measured by different scales. The primary outcome Brief Psychiatric Rating Scale (BPRS) assesses somatic concern, anxiety, emotional withdrawal, conceptual disorganisation, guilt feelings, tension, mannerisms and posturing, grandiosity, depressive mood, hostility, suspiciousness, hallucinatory behaviour, motor retardation, uncooperativeness, unusual thought content, and blunted effect (scale 1 = not present to 7 = extremely severe). Agitation was measured with the Cohen‐Mansfield Agitation Inventory scale (CMAI). This nurse‐rated questionnaire consists of 29 agitated behaviours, each rated on a 7‐point scale of frequency.
van Reekum 2002 used behavioural, cognitive, functional and extrapyramidal signs as outcome measures, but reported the BEHAVE‐AD (Behavioural Pathology in Alzheimer’s disease Rating Scale) measurements only in a figure (no means or SDs reported). Aggression was assessed by the ROAS scale (Retrospective Overt Aggression scale).
3. Presence or absence of withdrawal symptoms in the first four weeks after withdrawal
None of the studies assessed these specific outcomes although it is not easy to distinguish between a withdrawal phenomenon and a relapse of NPS.
4. Adverse events of antipsychotics
Total adverse events likely to be related to antipsychotic use, such as falls, extrapyramidal symptoms, cognitive dysfunction, metabolic changes (including weight gain and diabetes), cardiovascular events and others were not systematically reported in the included studies.
-
Ballard 2008 measured parkinsonism using the Modified Unified Parkinson's Disease Rating Scale (M‐UPDRS).
-
Bridges‐Parlet 1997 gave some attention to observations of tardive dyskinesia but no measurement scales were used. The entire study was based on direct observations by experienced personnel who were blinded to the assigned treatment.
-
Cohen‐Mansfield 1999 reported adverse events as secondary outcomes in a table (without reporting an SD), using the Abnormal Involuntary Movement Scale (AIMS): assessment of neurological and physical side effects associated with psychotropic drug use (9 items: e.g. movement of the face and oral cavity, extremities and trunk, global judgements of abnormal movements). A list of adverse effects (sedation, extrapyramidal reactions, orthostatic hypotension and anticholinergic effects) was provided to the nursing staff, who indicated frequency of occurrence. Nurse managers checked lists of psychomotor adverse effects, including 13 items describing pseudoparkinsonism, akathisia, acute dystonic reaction, and tardive dyskinesia.
-
Devanand 2011 assessed somatic side effects with the Treatment Emergent Symptom Scale (TESS; range from 0 to 26, with higher scores indicating more somatic symptoms), extrapyramidal signs using the Unified Parkinson's Disease Rating Scale (UPDRS) and tardive dyskinesia with the Rockland Tardive Dyskinesia scale. No data were reported for the discontinuation trial.
-
Devanand 2012 assessed extrapyramidal signs using the Simpson‐Angus scale (range from 0 to 40, with higher scores indicating more extrapyramidal signs); tardive dyskinesia using the AIMS (range from 0 to 35, with higher scores indicating more severe symptoms) and general somatic symptoms developing during treatment using the TESS.
-
Findlay 1989 provided additional information on mobility, range of mobility, transferring, response to chest pushing and balance and position sense, vibration sense, reading of a sway for participants standing with eyes open, systolic and diastolic blood pressure, and heart rate. Findlay 1989 reported lying and standing blood pressure and heart rate, the sum of the mobility outcomes, balance while standing, balance on turning head, balance on turning whole body through 360 °.
-
van Reekum 2002 assessed extrapyramidal signs using the Extrapyramidal Symptom Rating Scale (ESRS).
-
Bergh 2011 measured extrapyramidal adverse effects after prescription of antipsychotics using the M‐UPDRS but these results were not reported.
2. Secondary outcomes
1. Cognitive function (e.g. short‐term memory, frontal executive function, language)
-
Ballard 2008 measured cognition using the SMMSE and the SIB, which was the main outcome for this trial. Frontal executive function was assessed by the FAS verbal fluency test, assessing phonemic verbal fluency. Language was assessed by using the Sheffield Test for Acquired Language Disorder (STALD receptive and STALD expressive skill).
-
Cohen‐Mansfield 1999, Devanand 2011 and Devanand 2012 assessed cognitive function using the MMSE. Devanand 2012 also used the Alzheimer's Disease Assessment Scale ‐ cognitive score (ADAS‐cog, range from 0 to 70, with higher scores indicating worse cognition).
-
Findlay 1989 assessed cognitive function with the Cognitive Assessment Scale (CAS) scored by a psychiatrist.
-
van Reekum 2002 assessed cognitive function with the MMSE and the Mattis Dementia Rating Scale (MDRS).
2. Quality of life (QoL) of participants, carers, family of participants, or a combination
-
Ballard 2004 scored QoL using Dementia Care Mapping (DCM) as a measure of participants' well‐being. The method quantifies activity category codes, which are recorded every five minutes over a six hour period of observation during one day.
-
Bergh 2011 assessed changes after 25 weeks on the Quality of Life ‐ Alzheimer disease (QoL‐AD) scale. The QoL‐AD scale evaluates the quality of life of the patient using 13 items which are scored on a 4‐point scale from 'bad' to 'excellent'.
3. Time, in days, until prescription of any psychotropic agent
Time, in days, until repeat prescription of any psychotropic agent with the exception of antipsychotics was not reported systematically. Only Ruths 2008 reported medication changes in a subgroup analysis.
4. Use of physical restraint
Only Bridges‐Parlet 1997 reported use of physical restraint.
5. Mortality
Only Ballard 2008 and Devanand 2012 reported mortality. Mortality data in one of the two papers describing Ballard 2008 were reported at 12, 24 and 36 months follow‐up after randomisation. Devanand 2012 assessed mortality at 16 weeks (4 months) and 32 weeks (8 months).
6. Other secondary outcomes
6.1. Global functioning
-
Ballard 2008 reported global functioning with the BADLS (Bristol Activities of Daily Living Scale) and FAST (Functional Assessment Staging).
-
Cohen‐Mansfield 1999 reported residents' functioning as secondary outcomes by rating levels of activity and positive mood.
-
Devanand 2011 assessed impairment in activities of daily living using the modified Blessed Functional Activity Scale (BFAS).
-
Devanand 2012 assessed physical function with the use of the Physical Self‐Maintenance Scale (PSMS; range from 1 to 30, with higher scores indicating worse functioning).
-
van Reekum 2002 assessed functional outcome with the Blessed Dementia Scale (BDS).
6.2. Sleep
-
Ruths 2008 (and subgroup analysis in Ruths 2004) and Bridges‐Parlet 1997 reported effects on sleep.
-
Cohen‐Mansfield 1999 reported the effect on sleep and activity level (daytime sleep, time to fall asleep and activity level). Daytime sleep was an average of the items “How often does the resident appear drowsy or sleepy during the day?” and “How frequently does the resident actually sleep during the day?" Both items were rated on a frequency scale ranging from 1 (never) to 7 (several times an hour). Time to fall asleep was measured by “On the average, how long did it take the resident to fall asleep at night (from the time he/she went to bed until the time the resident fell asleep)?” and rated on a scale ranging from 1 (falls asleep immediately) to 6 (nearly never sleeps at night). Activity level was an average of 2 items: “How often did the resident participate in social activities?” and “How frequently was the resident involved in activities which is meaningful for his/her level of functioning? and rated on a frequency scale ranging from 1 (never) to 6 (several times a day).
6.3 Clinical global impression
-
Ballard 2008 reported clinical global impression using CGI‐C (Clinical Global Impression‐Change).
-
Cohen‐Mansfield 1999 reported clinical global impressions as secondary outcomes using the CGI‐C scale.
-
Findlay 1989 reported a psychiatric assessment using the Sandoz Clinical Assessment Geriatric Scale (SCAGS)
-
In Devanand 2012, relapse was reported as a predefined deterioration on the NPI and the CGI‐C. The CGI‐C was also measured at different time points, but was not reported in the paper.
Co‐variables
Only Ballard 2008 conducted a post hoc subgroup analysis by type of antipsychotic drug (typical versus atypical).
Time of assessment of outcome measurements
Outcomes were assessed at different times.
-
Ballard 2004 assessed outcomes at three months.
-
Ballard 2008 assessed outcomes in a first paper at 1, 3, 6 and 12 months: only the data assessed at six months were reported. Analysis at 12 months was limited to the two main outcomes: cognitive function and neuropsychiatric features. In a second paper, Ballard assessed the outcome mortality at 12, 24 and 36 months (Ballard 2011). To pool the NPI data we asked Professor Ballard to provide data from the DART‐AD study assessed at three months (Declerck 2009c [pers comm]). These data were extracted from the DART‐AD database by Ly‐Mee Yu from the Oxford Centre for Statistics in Medicine.
-
Bridges‐Parlet 1997 reported outcomes at one, two and four weeks.
-
Cohen‐Mansfield 1999, a cross‐over study, reported that participants were assessed at five time points: one week after start of dosage tapering (week 1), phase one tapering (week 3), phase one end point (week 10), phase two tapering (week 13) and phase two end point (week 20). Results were reported as paired data for time points three and five (comparison of assessments of each phase in the cross‐over study).
-
Devanand 2011 assessed outcomes in phase B at 0, 2, 4, 8, 12, 16, 20 and 24 weeks.
-
Devanand 2012: phase B assessed outcomes at 16 weeks (4 months) and 32 weeks (8 months).
-
Findlay 1989 reported outcomes at two and four weeks.
-
Ruths 2008 assessed outcomes at four weeks (1 month).
-
van Reekum 2002 reported outcomes only in a figure from visit 1 (baseline) to visit 15 (6 months).
-
Bergh 2011 reported outcomes at baseline and after 25 weeks.
Excluded studies
We excluded 12 studies for this update. Of these, eight were commentaries (Devanand 2013; Garner 2015; Gill 2013; Gnjidic 2013; Ling 2013; Lolk 2014; Power 2013; Renard 2014). Two studies did not investigate interventions that were relevant for this review (discontinuation of memantine (Ballard 2015) and discontinuation of antidepressants (Bergh 2012). Patel 2017 was not a randomised controlled trial (presented a post hoc analysis of Devanand 2012). Azermai 2013 did not use a suitable control intervention (it was a pilot study without a control group).
Five studies were excluded in the review 2013 (Other published versions of this review). Horwitz 1995; Westbury 2011; Wessels 2010 were excluded because these were not randomised controlled discontinuation trials. One trial was excluded because it analysed the Findlay 1989 cohort for outcomes that are not relevant to our review (McLennan 1992). Another study was excluded as it seems to be the registration of a not (yet) published (and perhaps still ongoing) trial and further searching did not reveal additional information about this trial (Rule 2003).
Risk of bias in included studies
We assessed risk of bias of included studies according to six specific domains using the Cochrane 'Risk of bias’ assessment tool (Higgins 2011) (Figure 3; Figure 4).

Risk of bias graph for the 10 included studies in the review.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study in the review.
Most studies were assessed at low or unclear risk of bias. Only Ballard 2008 was assessed at low risk of bias for all domains. Bergh 2011 was judged to be at high risk of bias in two domains. Cohen‐Mansfield 1999 and van Reekum 2002 were each assessed at high risk of bias in one domain. The most common unclear risk of bias domains were selection bias, detection bias, attrition bias and reporting bias.
See Characteristics of included studies.
Allocation
Randomisation sequence generation was described and adequate in six trials (Ballard 2008; Bergh 2011; Bridges‐Parlet 1997; Devanand 2012; Ruths 2008; van Reekum 2002) and unclear in four trials (Ballard 2004; Cohen‐Mansfield 1999; Devanand 2011; Findlay 1989).
Allocation concealment was only described in sufficient detail to assess the risk of bias as low in three studies (Ballard 2008; Bergh 2011; Devanand 2012). Risk of allocation concealment bias was unclear in seven studies (Ballard 2004; Bridges‐Parlet 1997; Cohen‐Mansfield 1999; Devanand 2011; Findlay 1989; Ruths 2008; van Reekum 2002).
Blinding
All included studies were double‐blinded. The overall risk of performance bias was low. All studies adequately described methods of blinding participants and personnel. We assessed four studies (Devanand 2011; Findlay 1989; Ruths 2008; van Reekum 2002) at unclear risk of detection bias; information on blinding of outcome assessors was not reported. In these trials, there were several subjective outcomes, so a lack of blinding of outcome assessors could have had an influence.
Incomplete outcome data
Six trials reported the issue of incomplete outcome data, with no unequal numbers across the groups and with adequate reasons provided for dropouts and losses to follow‐up (Ballard 2004; Ballard 2008; Bridges‐Parlet 1997; Devanand 2011; Devanand 2012; van Reekum 2002). In Bergh 2011, attrition bias was judged as high risk due to the high dropout rate with unequal numbers across the groups (7 dropouts of 9 participants in the discontinuation group and no dropouts of 10 participants in the continuation group) and missing data. We considered the risk of incomplete outcome data bias to be unclear in three studies (Cohen‐Mansfield 1999; Findlay 1989; Ruths 2008.
Selective reporting
We judged four studies at low risk of selective reporting bias (Ballard 2004; Ballard 2008; Bridges‐Parlet 1997; Ruths 2008). Findlay 1989 did not describe the primary outcome and was unclear if all outcomes were reported. In van Reekum 2002, some outcomes mentioned in the methods sections of the paper were not reported in the results. In Bergh 2011, an unpublished study, the authors reported that they did not perform an observed case analysis due to the high dropout rate and missing data. In Devanand 2012 the CGI‐C data were not fully reported. Devanand 2011 and Devanand 2012 reported numeric data for several continuous outcomes at the time of randomisation into the discontinuation phase, but only dichotomous data at later time points. In Cohen‐Mansfield 1999, outcome data were not reported separately for each medication discontinued in the trial (i.e. haloperidol, thioridazine or lorazepam).
Other potential sources of bias
It was unclear if participants in the two groups were similar in Ruths 2008. In the Findlay 1989 study there was a baseline imbalance between the placebo group and continuation group in one of the three cognitive/behavioural rating scales used to measure outcomes. It was unclear if this had an impact on the results.
Effects of interventions
We included 10 RCTs with 632 participants. However, although Cohen‐Mansfield 1999 met inclusion criteria, we were unable to use any data from this study. Cohen‐Mansfield 1999 did not report outcome data separately for the different medications discontinued in the trial (which included the benzodiazepine lorazepam as well as the antipsychotics haloperidol and thioridazine). We contacted the study author by for further data, but have not received a response (Declerck 2009b [pers comm]).
For all outcomes, our conclusions were based on studies that reported quantitative data, or on conclusions made by the study authors if data were not provided.
The result and the evidence quality for each outcome for the main comparison (discontinuation compared to continuation of antipsychotic drug use for behavioural and psychological symptoms in older participants with dementia) are described in summary of findings Table 1.
Primary outcomes
1. Success of antipsychotic withdrawal
We defined success of withdrawal as the ability to complete the study, i.e. no dropout due to worsening of neuropsychiatric symptoms (NPS) or no relapse to antipsychotic drugs use during the trial. This was not reported in any of the included studies. Several studies reported the number of participants completing and not completing the study, but did not report the number of participants who failed to complete the study due to worsening NPS or the number of participants who restarted antipsychotics. Therefore, we used the difference between groups in the number of non‐completers of the study as a proxy for our primary outcome. However, we could not pool data because the studies were too heterogeneous clinically and there were considerable discrepancies in the way the success of antipsychotic withdrawal was measured.
Nine studies (575 participants) reported data relevant to this outcome (Ballard 2004; Ballard 2008; Bergh 2011; Bridges‐Parlet 1997; Devanand 2011; Devanand 2012; Findlay 1989; Ruths 2008; van Reekum 2002).
In seven studies (446 participants) discontinuation of the antipsychotic made little or no difference to the ability of participants to complete the study (Ballard 2004; Ballard 2008; Bridges‐Parlet 1997; Devanand 2011; Findlay 1989; Ruths 2008; van Reekum 2002).
In three studies (149 participants) there was some evidence in favour of the continuation group (Devanand 2011; Devanand 2012; Bergh 2011). Although Devanand 2011 reported no difference between groups in the numbers of participants leaving the study group early due to symptomatic relapse (a worsening of psychosis, agitation or aggression), the study also reported that time to a symptomatic relapse was shorter in the discontinuation group than in the continuation group. Devanand 2012 reported a higher rate of participants leaving the study group early due to symptomatic relapse in the discontinuation group compared with the continuation group and that discontinuation led to increased risk of symptomatic relapse (increase in the NPI‐core score at 4 months and 8 months follow‐up). Bergh 2011 reported a very high dropout rate (7 of 9 participants) in the discontinuation group compared to no dropouts (among 10 participants) in the continuation group.
We assessed the overall quality of the evidence for this outcome to be low, downgraded one level for indirectness, as not all included studies directly measured the outcome of our interest, and one level for risk of reporting bias. Several studies did not report the number of participants who failed to complete the study due to worsening NPS or the number of participants who relapsed to antipsychotic use.
In Ballard 2008, we extracted data from the study flow diagram on the same proxy outcome as reported in the pilot study of Ballard 2004 i.e. the number of participants not completing the study (based on intention‐to‐treat (ITT) analysis). In this study, there was a high number of participants not completing the study: 45/82 participants (56%) in the discontinuation group and 43/83 (51%) participants in the continuation group. Three participants in the discontinuation group and four in the continuation group did not complete the study because their behavioural condition deteriorated. No difference between groups was reported. No data were reported for relapse to antipsychotic use.
Ballard 2004 reported the number of non‐completers of the study based on ITT analysis and the proportion of participants developing pronounced behavioural symptoms. About a third (14/46, 30%) participants in the discontinuation group and 14/54 (26%) participants in the continuation group did not complete the study (P = 0.62). Six of 46 participants in the discontinuation and 5/54 participants in the continuation group did not complete the study due to behavioural deterioration (P = 0.55). In this study, there was no difference in completion rates between groups. No data were reported for relapse to antipsychotic use.
In Bergh 2011 there was a high dropout rate and imbalance between the discontinuation and continuation groups: 7/9 participants in the discontinuation group and 0/10 participants in the continuation group failed to complete the study. This could be interpreted as failure of completing the study in the discontinuation group due to worsening NPS. The study authors reported that results were inconclusive.
Bridges‐Parlet 1997 reported the numbers of participants completing the four‐week trial, the number of non‐completers of the study due to increased NPS and also the number of participants relapsing to antipsychotic use. Two of 22 participants (9%) in the discontinuation group and 0/14 participants in the continuation group failed to complete the study. There was no difference in successful completion between groups (Chi² > 0.05). One non‐completer experienced a pronounced increase in behavioural symptoms and was reverted to antipsychotic drug use.
Devanand 2011 reported the total number of participants not completing the study, the rate of leaving the study group early due to symptomatic relapse, and the time to symptomatic relapse. There was a higher rate of participants leaving the study group early due to symptomatic relapse in the discontinuation group (8/10, 80%) compared to the continuation group (4/10, 40%), but the difference in relapse rates between the groups was not statistically significant (Chi² = 3.3, P = 0.07). There was, however, a statistically significantly shorter time to relapse in the discontinuation group: mean 5.8 weeks (SD 6.7) in the discontinuation group compared to mean 8.0 weeks (SD 6.7) in the continuation group (Chi² = 4.1, P = 0.04). The severity of agitation or aggression in the open haloperidol treatment phase of the study did not predict the likelihood of symptomatic relapse in the discontinuation trial.
Devanand 2012 reported the total number of participants not completing the study and the rate of participants leaving the study early due to symptomatic relapse (an increase in the NPI‐core score of 30% or more). During the first 16 weeks of the study (N = 110), 27/40 participants in the discontinuation group and 30/70 participants in the continuation group did not complete the study. The rate of participants leaving the study early due to symptomatic relapse (including relapse, imminent relapse, or mortality) was higher in the discontinuation group (24/40, 60%) than in the continuation group (23/70, 33%) (P = 0.004). The discontinuation group had an increased risk of leaving the study early due to symptomatic relapse compared with the continuation group (hazard ratio (HR) 1.94, 95% CI 1.09 to 3.45, P = 0.02). Crude (unstratified) rates of relapse at four months were 6.5 and 3.0 per 100 patient‐weeks of follow‐up for the discontinuation and continuation groups, respectively. During the subsequent 16 weeks (N = 40), 13/27 participants did not complete the study in the discontinuation group and 3/13 participants did not complete the study in the continuation group. The rate of participants leaving the study early due to symptomatic relapse was higher in the discontinuation group (13/27, 48%) than in the continuation group (2/13, 15%) (HR 4.88, 95% CI 1.08 to 21.98, P = 0.02). At eight months, crude rates of relapse were 4.3 and 1.1 per 100 patient‐weeks of follow‐up for the discontinuation and the continuation groups respectively. The total number of participants completing the eight months discontinuation trial was very low: 10/40 participants in the discontinuation group and 10/32 participants in the continuation group. No difference was reported by the study authors.
In Findlay 1989, all 36 participants (18 in each group) completed the four week study. We therefore assumed that no participants left the study early due to worsening NPS or relapsed to antipsychotic use.
The main outcome measure in Ruths 2008 was successful antipsychotic discontinuation, i.e. still off antipsychotics in the discontinuation group at the end of the one month study. In the discontinuation group 23/27 participants were still off antipsychotics. Four participants did not complete the study in the discontinuation group (4/27) and three participants did not complete the study in the continuation group (3/28) (P = 0.7). There were two non‐completers in the discontinuation group due to behavioural deterioration.
van Reekum 2002 reported that 10/17 participants stopped the allocated treatment early in the discontinuation group and 6/17 stopped early in the continuation group (ITT analysis). The difference in the rate of early stopping between the groups was not significant (RR 1.57, 95% CI 0.76 to 3.26), nor was the difference in number of participants stopping early due to exacerbation of NPS (4/17 in the discontinuation group, 3/17 in the continuation group, P > 0.1.) There were no data on relapse to antipsychotic use.
2. Behavioural and psychological symptoms
Seven studies (519 participants) contributed data for this outcome (Ballard 2004; Ballard 2008; Bergh 2011; Bridges‐Parlet 1997; Devanand 2012; Ruths 2008; van Reekum 2002).
Two studies (265 participants) used the NPI to assess NPS and were considered suitable to pool for meta‐analysis (Ballard 2004; Ballard 2008). There was little or no difference in NPS between groups after three months (negative values favour discontinuation): MD ‐1.49, 95% CI ‐5.39 to 2.40; participants = 194; studies = 2 (Analysis 1.1 and Figure 1). Initially, assessments of the NPI scores in these two Ballard studies were not made at the same time (Ballard 2004 assessed at three months and Ballard 2008 assessed at 1, 3, 6 and 12 months, but data in the publication were only available for six months). With the permission of Clive Ballard and the help of Ly‐Mee Yu we calculated means and mean differences from individual participant data of the DART‐AD trial for the NPI score at three months using SPSS software (Declerck 2009c [pers comm]).
We could not pool data from five studies because they were clinically too heterogeneous (different outcomes, different outcome scales, different time of follow‐up) or reported insufficient data. However, results from these five studies (254 participants) also suggested that discontinuation may make little or no difference to NPS (Bergh 2011; Bridges‐Parlet 1997; Devanand 2012; Ruths 2008; van Reekum 2002).
Overall, we considered the quality of evidence relating to this outcome to be low, downgraded one level for imprecision due to the wide confidence interval and small number of participants and one level for risk of reporting bias (study authors' conclusions were not supported with reported data in four studies).
Subgroup analyses of Ballard 2004 and Ballard 2008 suggested that the effect of antipsychotic discontinuation may differ depending on the severity of NPS at baseline. Ballard 2004 reported that some participants with less severe NPS (NPI score ≤ 14) may benefit from discontinuation of antipsychotics in terms of agitation (a subscore of the NPI). Ballard 2008 and Ballard 2004 suggest that some participants with more severe NPS (total NPI > 14) may benefit from continuing antipsychotic treatment.
Behavioural and psychological symptoms measured with versions of the NPI
Pooled studies
There was no difference between groups in change on the NPI total score after three months in Ballard 2004 or the key psychiatric/behavioural factors of agitation, mood and psychosis. Results were reported for on‐treatment‐analysis only (i.e. all participants who completed the study).
In the subgroup of participants with baseline NPI scores at or below the median (≤ 14) there was a trend that the discontinuation group was less likely to develop pronounced behavioural or psychiatric symptoms (Chi² = 3.6; P = 0.06) although there was no difference in the total NPI score between groups (Mann‐Whitney U test z = 1.7; P = 0.9). A pronounced behavioural problem was defined as a score of 8 or above on an individual item of the NPI (Declerck 2009c [pers comm]). There was a greater reduction of agitation (a subscore of the NPI) in the discontinuation group: 1.0 point (SD 3.1) improvement in the discontinuation group and 1.5 point (SD 2.5) deterioration in the continuation group (Mann‐Whitney U test z = 2.4; P = 0.018).
A subgroup of participants with higher baseline NPI scores (> 14) were more likely to develop pronounced behavioural problems if antipsychotics were discontinued (Chi² = 6.8; P = 0.009). There were no differences in total NPI score (Mann‐Whitney U test z = 0.34; P = 0.73) or agitation (Mann‐Whitney U test z = 0.82; P = 0.38).
In the Ballard The DART-AD Trial 2008 there was no difference between groups in the estimated mean change in NPI scores between baseline and six months. However, there was a significant difference between groups in the estimated mean change in NPI scores between baseline and 12 months. Results are reported for the modified intention‐to‐treat (mITT) analysis (i.e. only participants who had at least one dose of treatment were included in the analysis).
For all participants, there was no clear difference between groups in the estimated mean change in NPI scores between baseline and six months: 4.5 points (SD 17.6) deterioration for the discontinuation group (N = 53) compared to 1.3 points (SD 15.5) deterioration for the continuation group (N = 56); estimated mean difference in NPI change (favouring continue treatment) 2.4, 95% CI ‐8.2 to 3.5, adjusted for baseline value: P = 0.4.
For participants with baseline NPI ≤ 14, the change in NPI over six months was very similar between groups (estimated mean difference in NPI change 0.49, 95% CI ‐5.63 to 6.60).
For participants with baseline NPI ≥ 14, there was no clear difference between groups in the estimated mean change in NPI scores between baseline and 6 months (estimated mean difference in NPI change ‐5.33, 95% CI ‐15.82 to 5.17).
For all participants, continuation leads to small advantages at 12 months for those who continued antipsychotics: 11.4 points (SD 17.7) deterioration in the discontinuation group (N = 31) compared to 1.4 points (SD 22.1) deterioration in the continuation group (N = 28); estimated mean difference in NPI change (favouring continuation group) ‐10.9, 95% CI ‐20.1 to ‐1.7, adjusted for baseline: P = 0.02. Over 12 months there was a large amount of missing data, which could limit the validity of the results.
For participants with baseline NPI ≤ 14, there was no clear difference between groups in the estimated mean change in NPI scores between baseline and 12 months: estimated mean difference in NPI change (favouring continuation of treatment) ‐5.2, 95% CI ‐15.8 to 5.4.
For participants with baseline NPI > 14, continuation lead to advantages at 12 months for those who continued on antipsychotics: estimated mean difference in NPI change (favouring the continuation group) ‐16.9, 95% CI ‐32.5 to ‐1.2. The study authors mentioned that the test for interaction (although underpowered) was not significant (P = 0.2) and therefore concluded there was no evidence of interaction between treatment group and severity of symptoms at baseline (Ballard 2008).
Studies that could not be pooled
Bergh 2011 reported NPI‐10 as a primary outcome, measuring 10 of the 12 NPI items, but without a score for sleep/nighttime behaviour and appetite/eating changes. The mean total score for the NPI‐10 decreased by 3.50 (SD 13.53) in the discontinuation group and decreased by 5.40 (SD 10.78) in the continuation group (P = 0.76).
Devanand 2012 measured and reported the NPI core score for both groups at baseline and at the time of randomisation, and measured but not reported at later time points, although change in the NPI core score was used to define relapse. The study authors reported that the total NPI score at baseline did not predict a relapse during the first 16 weeks of phase B and that the presence of psychosis at baseline or randomisation did not predict a relapse after discontinuation of risperidone.
Ruths 2008 used the NPI‐Q, which measures severity but not frequency of NPS, as a primary outcome. The NPI‐Q scores were reported for all 55 participants at one month follow‐up. Changes from baseline did not differ significantly between groups for total NPI‐Q scores, or the 12 individual symptoms of the NPI, or the agitation subscore. There was no difference between groups in the number of participants whose NPI‐Q scores remained stable or decreased (18/27 in the discontinuation group, 24/28 in the continuation group, P = 0.18). Participants with behavioural deterioration after antipsychotic cessation used higher daily drug doses at baseline (P = 0.042).
van Reekum 2002 used the NPI as an outcome but did not report data in the paper. The study authors reported no conclusions relating to NPI score.
Behavioural and psychological symptoms measured with other scales
We could not pool data from any of the studies using other scales to assess behavioural and psychological symptoms.
Bergh 2011 reported depression measured with the CSDD. They found no evidence of a difference between groups. The mean change from baseline to 25 weeks on the CSDD was a small deterioration of 5.83 points (SD 36.40) in the discontinuation group and a small improvement of 5.30 points (SD 11.25) in the continuation group (P = 0.375).
Bridges‐Parlet 1997 concluded there was no difference between continuation and discontinuation groups in observed instances of physically aggressive behaviour: discontinuation group 1.27 (SD 3.95) versus continuation group 4.50 (SD 8.83), P > 0.05. Verbally aggressive behaviour was reported not to differ significantly between groups, although no data were provided to support this conclusion made by the study authors.
van Reekum 2002 reported no statistically significant difference in BEHAVE‐AD (measuring behaviour) and ROAS (measuring physical aggression towards themselves or others) scores between the discontinuation and continuation groups (P > 0.05). The discontinuation group showed more apathy than the continuation group (P = 0.04). However, not all of the study authors' conclusions were supported by data reported.
3. Presence of withdrawal symptoms or withdrawal syndrome in the first four weeks after withdrawal
No studies reported withdrawal symptoms or withdrawal syndrome in participants who discontinued antipsychotics.
4. Adverse events
Five studies (381 participants) (Ballard 2008; Bridges‐Parlet 1997; Devanand 2012; Findlay 1989; van Reekum 2002) contributed data on adverse events. Devanand 2011 reported adverse events only for the initial phase of open treatment with haloperidol and not for the discontinuation phase. None of the studies systematically reported all adverse and serious adverse events. Studies reported only a selection of adverse events such as parkinsonism, movement disorders, falls, mobility, balance, extrapyramidal symptoms, heart rate and blood pressure. We could not pool data because of the diverse ways adverse events were assessed.
Discontinuation may make little or no difference to adverse events (Devanand 2012; Ballard 2008; Bridges‐Parlet 1997; Findlay 1989; van Reekum 2002). Overall, we considered the quality of evidence for this outcome to be low, downgraded one level for indirectness because only a selection of adverse events was systematically assessed and one level for risk of bias because there was a high dropout in two studies (Ballard 2008; Devanand 2012) and risk of reporting bias (no data were provided to support the authors' conclusions in two studies).
Ballard 2008 measured change in severity of parkinsonism from baseline to six months using the MUPDRS. There was a small 0.4 point (SD 3.2) improvement in the discontinuation group and a small 0.8 point (SD 4.1) deterioration in the continuation group. The study authors reported that this difference was not statistically significant: estimated mean difference (favouring placebo): 1.1, 95% CI 0.4 to 2.6, adjusted for baseline value: P = 0.1. This may be due to the small sample size and high dropout.
Bridges‐Parlet 1997 reported that three participants in the discontinuation group experienced adverse events (2 participants had behaviour deterioration and 1 had tardive dyskinesia). There were no adverse events reported for the continuation group.
In Devanand 2012 all adverse events (extrapyramidal signs, akathisia or restlessness, sedation, insomnia, confusion, agitation‐aggression, falls, nausea or vomiting and other) and serious adverse events (death, cardiovascular event, neurologic event, agitation‐aggression, pulmonary event, fall or fracture and other) were reported in a separate table, and an expanded version of this table was provided in the Supplementary appendix. Only this study assessed falls. A serious adverse event was defined as an adverse event that resulted in any of the following outcomes: death, a life‐threatening condition, hospital admission or prolongation of hospital stay or an unexpected event leading to clinically significant disability or incapacity. There were no differences in rates of serious adverse events, adverse events and death. There was no difference in adverse events measured with the Simpson‐Angus, AIMS, TESS, although comparisons were based on small numbers of participants, especially during the final 16 weeks and on truncated observation period for adverse events in the case of participants who had an early relapse. No data for difference between groups was reported.
Findlay 1989 reported numerical data for mobility, range of mobility, transferring, response to chest pushing and balance and position sense, vibration sense, reading of a sway for the participants standing with eyes open, systolic and diastolic blood pressure and heart rate, lying and standing blood pressure and heart rate, the sum of the mobility outcomes, balance while standing, balance on turning head, balance on turning whole body through 360 °. Only means, ranges and numbers of observations were reported for each of these outcomes. The study authors concluded that discontinuation had no apparent effect on mental function, mobility or balance, and that the drugs had few side effects.
van Reekum 2002 assessed extrapyramidal signs using ESRS, but reported results for this outcome only at baseline and not at the end of the study. The study authors concluded that both groups scored similarly on the assessment measures. The data to support this conclusion were not reported.
Secondary outcomes
1. Cognitive function
We included five studies (365 participants) for this outcome (Ballard 2008; Devanand 2011; Devanand 2012; Findlay 1989; van Reekum 2002). Outcome measures differed across studies and we could not pool data for this outcome.
Discontinuation may make no difference to cognitive function (Ballard 2008; Devanand 2011; Devanand 2012; Findlay 1989; van Reekum 2002). However, one trial found that discontinuation improves measures of verbal fluency (Ballard 2008). Overall, we assessed the quality of the evidence for this outcome to be low, downgraded one level for imprecision (no meta‐analysis and most included studies had few participants) and one level for risk of reporting bias (no data were provided to support the authors' conclusions in 4 studies).
Ballard 2008 reported four outcomes measuring aspects of cognitive function. One outcome, the FAS, measuring verbal fluency, favoured the discontinuation group after six months: the estimated change in FAS totals between baseline and six months was a 0.6 point (SD 6.2) improvement in the discontinuation group and a 3.2 points (SD 6.6) deterioration in the continuation treatment group: estimated mean difference (favouring discontinuation) ‐4.5, 95% CI ‐7.3 to ‐1.7, adjusted for baseline: P = 0.002. For the main outcome, the SIB, used to assess overall cognition, there was no evidence of a difference between groups. The mean change from baseline to six months was a deterioration of 5.7 points (SD 14.2) in the discontinuation group and a deterioration of 6.2 points (SD 16.0) in the continuation group (estimated difference ‐0.4, 95% CI ‐6.4 to 5.5, adjusted for baseline: P = 0.9). For the SMMSE, used to assess overall cognition, there was also no evidence of a difference between groups. The mean change from baseline to six months for the SMMSE was a deterioration of 1.0 point (SD 4.2) in the discontinuation group and a deterioration of 1.8 point (SD 3.6) in the continuation group (estimated MD ‐1, 95% CI ‐2.7 to 0.7, adjusted for baseline: P = 0.2). For the STALD (receptive), used to assess receptive language skills, there was no evidence of a difference between the continuation and discontinuation groups. The mean change in STALD scores was 0.3 points (SD 2.1) deterioration for the discontinuation group and a 0.5 point (SD 1.7) deterioration for the continuation group: estimated MD ‐0.2, 95% CI ‐1.1 to 0.6, adjusted for baseline value: P = 0.6. For the STALD (expressive), used to assess expressive language skills, there was no clear evidence of a difference between the continuation and discontinuation groups. The mean change in receptive language scores between baseline and six months was a 0.2 point (SD 2.5) improvement in the discontinuation group and a 0.6 point (SD 1.8) deterioration in the continuation group: estimated MD ‐1.0, 95% CI ‐2.0 to 0.04, adjusted for baseline: P = 0.06.
In Devanand 2011 the study authors reported that cognition measured by change in MMSE did not differ by treatment group in phase B. No data were provided to support this conclusion. MMSE cognitive scores were measured and reported only at baseline (phase A) and at time of randomisation into phase B, and measured but not reported at other time points or at the end of the study.
In Devanand 2012 the study authors reported that the changes in MMSE and ADAS‐cog score did not differ between the continuation and discontinuation groups. Data were not reported to support these conclusions. Total MMSE and ADAS‐cog scores were measured and reported only at baseline of the open risperidone treatment and at the time of randomisation into the discontinuation trial and measured but not reported at other time points or at the end of the study.
Findlay 1989 concluded there was no difference in cognitive function measured by CAS over a four‐week study period. Outcomes were reported only as means with a range and number of observations. However, the difference between the discontinuation and continuation groups at baseline could have influenced the result.
van Reekum 2002 concluded there was no difference between the continuation and discontinuation groups in cognition, measured by MMSE and MDRS. Data were not reported to support these conclusions.
2. Quality of life of participants, carers, families or a combination
Quality of life was reported in two studies (119 participants) (Ballard 2004; Bergh 2011). We could not pool data because different outcome measures were used.
There may be no difference in quality of life between groups, but we considered the quality of the evidence for this outcome to be low, downgraded one level for imprecision and one level for risk of bias (high risk of attrition bias and reporting bias in Bergh 2011).
There was no clear evidence of a difference between groups in well‐being measured by Dementia Care Mapping in Ballard 2004. There was a small improvement in well‐being (mean ‐0.18 (SD 1.72)) in the discontinuation group and a slight worsening (mean 0.35 (SD 2.41)) in the continuation group (MD ‐0.53, 95% CI ‐1.42 to 0.36).
Bergh 2011 reported there were no statistically significant differences in QoL‐AD between groups. No data were provided to support this conclusion.
3. Time, in days, until repeat prescription for any psychotropic or any antipsychotic agent
Time in days until repeat prescription for any psychotropic or any antipsychotic agent was reported for a subgroup 30 participants in one nursing home. Ruths 2008 reported that standing orders for antidepressants, hypnotic and anxiolytic medications remained unchanged for all participants during the intervention, but did not provide supporting data. We considered this to be very low‐quality evidence, downgraded one level for imprecision (single study with a small number of participants) and two levels for risk of bias (unclear risks of selection, detection bias and reporting bias; no data provided to support conclusion).
4. Use of physical restraint
Use of physical restraint was reported as an outcome in Bridges‐Parlet 1997 (36 participants). The study authors reported no difference in time being restrained between the discontinuation and continuation groups, but provided no supporting data. We considered the quality of this evidence to be very low, downgraded one level for imprecision (single study with a small number of participants) and two levels for risk of bias (no data reported to support conclusion and unclear risk of selection).
5. Mortality
Mortality was reported as an outcome in two studies (275 participants) (Ballard 2008; Devanand 2012). We could not pool data due to heterogeneity of the outcome measures. It was not possible to draw conclusions about the effect of discontinuation on mortality because of the very low‐quality of the evidence, downgraded one level for imprecision (small numbers of participants and events in both studies) and two levels for risk of attrition bias (high dropout in both studies).
Ballard reported mortality data as cumulative probability of survival at 12, 24 and 36 months in a long‐term follow‐up for participants randomised in the 12 month discontinuation trial (Ballard 2008). In a mITT analysis including participants who received at least one dose of treatment, there was no evidence of a difference between groups. The cumulative probability of survival during the first 12 months was 70% (95% CI 58% to 80%) in the continuation group compared with 77% (95% CI 64% to 85%) in the discontinuation group. The small sample size meant a lack of power to detect differences. The difference between groups in cumulative survival rate became more pronounced after the 12 month randomised phase of the trial. The cumulative survival rate was higher in the discontinuation group (71%) compared to the continuation group (46%) after 24 months follow‐up, and also higher in the discontinuation group (59%) compared to the continuation group (30%) after 36 months follow‐up (reported as a significant difference between groups, no further details reported). Due to high dropout and uncertainty about the use of antipsychotics, Ballard 2008 reported that the lower mortality in the discontinuation group should be interpreted with caution. The survival rates were similar in additional analyses that focused on the participants who continued their allocated treatment for at least 12 months.
In Devanand 2012 mortality measured after 16 and 32 weeks did not differ between the continuation and discontinuation groups. Three deaths (1 in the discontinuation group and 2 in the continuation group) occurred during the discontinuation trial. There were small numbers of participants, especially during weeks 16 to 32.
6. Other secondary outcomes
6.1 Global functioning
We included four studies (329 participants) for this outcome (Ballard 2008; Devanand 2011; Devanand 2012; van Reekum 2002). Outcome measures differed among included studies and data could not be pooled for this outcome.
Discontinuation may make no difference to global functioning. Overall, we assessed the quality of the evidence for this outcome to be low, downgraded one level for imprecision (studies with small numbers of participants) and one level for risk of reporting bias in three studies.
In Ballard 2008 global function assessed with the BADLS showed no clear difference between continuation and discontinuation groups: there was an improvement in function of 0.2 points (SD 7.2) in the discontinuation group and an improvement of 1.8 points (SD 8.9) in the continuation group. However there was no difference between groups (estimated MD 1.7, 95% CI ‐1.2 to 4.6, adjusted for baseline: P = 0.2). For the change on FAST, which measures global outcome, there were no differences between the continuation and discontinuation groups in terms of the dementia stage (P = 0.9).
Devanand 2011 reported no evidence of a difference in BFAS scores between the continuation and discontinuation groups in phase B, although the supporting data were not reported.
Devanand 2012 reported no evidence of a difference in physical function between groups on the Physical Self‐Maintenance Scale, although the supporting data were not reported.
van Reekum 2002 concluded that discontinuation of antipsychotics did not lead to differences between groups on the BDS, measuring activities of daily living and motivational behaviour. Supporting data were not provided.
6.2 Sleep
We included two studies (66 participants) for this outcome (Bridges‐Parlet 1997; Ruths 2008). Outcome measures differed and we could not pool data for this outcome.
Discontinuation may result in a large reduction in sleep in a subgroup of one study (Ruths 2008) and may make no or little difference to sleep in
another study (Bridges‐Parlet 1997). Overall, we assessed the quality of evidence for this outcome to be low, downgraded one level for imprecision (only 2 studies with few participants) and one level for risk of bias (conclusions were from subgroup analyses and there was a risk of reporting bias in Ruths 2008).
In a subgroup of the Ruths 2008 study, sleep was measured by actigraphy in 30 participants over four weeks. Abrupt discontinuation of antipsychotics lead to a large reduction in average sleep efficiency from 86% to 75% (i.e. 54 minutes less sleep), in the discontinuation group compared to a reduction from 90% to 87% in the continuation group, with a difference between groups (p=0.029).
In Bridges‐Parlet 1997, the study authors reported no difference in time sleeping between treatment groups. No data were provided to support the conclusion.
6.3 Clinical global impression
We included three studies (311 participants) for this outcome (Ballard 2008; Devanand 2012; Findlay 1989). Outcome measures differed among included studies and we could not pool data for this outcome.
Based on these three studies, we are uncertain whether discontinuation improves clinical global functioning. Overall, we assessed the quality of the evidence for this outcome to be low, downgraded one level for imprecision (small number of participants) and one level for risk of reporting bias (data not provided to support conclusion in either study).
In Ballard 2008, there was no evidence of differences between the continuation and discontinuation groups in clinical global impression rated on the CGI‐C (P = 0.9).
Findlay 1989 reported no difference in global impression using the Sandoz Clinical Assessment Geriatric Scale (SCAGS). Authors' conclusions were not supported with extractable data; results were given only as means, ranges and numbers of observations.
In Devanand 2012, the CGI‐C was also measured at different times, but assessment at the end of the discontinuation trial was not reported in the paper, although this score was a criterion of the predefined threshold score for relapse in the discontinuation trial. No conclusions were made by the study authors.
Covariables
In a post hoc analysis reported in Ballard 2008 (N = 100, follow‐up 3 months) there was no indication of a difference between participants taking typical or atypical antipsychotics. Most participants were taking risperidone or haloperidol; the number of participants taking other drugs was too small for any meaningful comparison.
Discussion
Summary of main results
We included 10 studies that included a total of 632 participants. One new trial was added for this update (19 participants).
Our conclusions for all outcomes were based on studies that reported quantitative data, or on conclusions made by the study authors if data were not provided.
Pooling was only possible for behavioural outcomes assessed by Neuropsychiatric Inventory score (NPI) and not possible for all other outcomes due to the clinical heterogeneity of the studies, and considerable discrepancies in the ways outcomes were measured.
The results and quality of evidence assessment for each outcome in the main comparison (discontinuation compared to continuation of antipsychotic drug use for behavioural and psychological symptoms in older participants with dementia) are described in summary of findings Table 1.
Primary outcomes
Our predefined outcome success of withdrawal was not reported in the included studies. Therefore, we used the difference between groups in the number of non‐completers of the study as a proxy for our primary outcome. Low‐quality evidence in seven studies suggests little or no overall difference in the ability of participants to complete the study. However, in two studies of participants with psychosis, aggression or agitation who had responded to antipsychotic treatment, we found there may be a benefit from continuing antipsychotics. One small study reported that a high proportion of participants in the discontinuation group failed to complete the study.
We found low‐quality evidence in two pooled studies and five non‐pooled studies for the outcome behavioural and psychological symptoms. In the two pooled studies, there was no difference in NPI scores between groups. In five non‐pooled studies, discontinuation may make little or no difference in scales measuring overall behaviour and psychological symptoms between groups. The two pooled studies performed subgroup analyses according to baseline NPI‐score (≤ 14 or > 14). In one study, some participants with milder symptoms at baseline were less agitated at three months in the discontinuation group. Both studies suggest that participants with more severe neuropsychiatric symptoms (NPS) (total NPI above 14) may benefit from continuing antipsychotic treatment.
No studies reported withdrawal symptoms.
Low‐quality evidence from five studies suggested discontinuation may make little or no difference in adverse events between groups.
Secondary outcomes
Low‐quality evidence from five studies suggested discontinuation may make no difference to cognitive function. However, one trial found that discontinuation improved measures of verbal fluency. Low‐quality evidence from two studies indicated there may be no difference in quality of life between discontinuation and continuation group participants.
It remains unclear if discontinuation reduced time to repeat prescription of any psychotropic or antipsychotic agent (1 study) or discontinuation increased use of physical restraint (1 study).
We found low‐quality evidence that discontinuation may make no difference to global functioning (4 studies) and clinical global functioning (3 studies). We found low‐quality evidence for sleep in two non‐pooled studies. In a subgroup of a study, there was a large reduction in sleep efficiency after discontinuation. One study reported no or little difference in sleep after discontinuation.
Based on very low‐quality evidence from two studies, It was not possible to draw conclusions about the effect of discontinuation on mortality.
Overall completeness and applicability of evidence
The main limitation of this review was the lack of consistency regarding study participants (such as the case definition applied and the severity of dementia of the participants), types and dosages of antipsychotics used before withdrawal, exclusion criteria, interventions (i.e. method of withdrawal), outcomes, and times of assessment among the individual studies.
We included all types and grades of dementia severity, regardless of the method of diagnosis. This reflects the current situation in clinical practice where many nursing home residents with dementia are not formally diagnosed. We believe that including all potential participants will make the review findings as widely applicable as possible.
It is possible that the profile of the original symptoms (i.e. a specific cluster of NPS) for which the antipsychotics were prescribed influenced the assessed outcome. Therefore, it would be useful to know why the antipsychotics were prescribed. Devanand 2011 and Devanand 2012 tried to overcome this problem by including only participants with symptoms of psychosis, agitation or aggression who had responded to antipsychotic treatment.
Most of the available evidence only applies to nursing home residents or to people in long‐stay psychogeriatric or geriatric wards (i.e. hospital setting). Only one small pilot study and its larger subsequent trial included participants living in the community (outpatients). Therefore, the results of this review may not be applicable to community settings.
Adverse events, withdrawal symptoms or syndromes, initiation of other psychoactive drug use after withdrawal and baseline antipsychotic dose were not systematically reported. Consequently, the effect of these on clinical outcomes is unknown, which is a major gap in the evidence. It was not possible to draw conclusions about the comparative efficacy of a tapered withdrawal schedule or abrupt withdrawal.
This review assessed the effect of antipsychotic withdrawal only. The results cannot be extrapolated to other drug types which may be prescribed for NPS in dementia and which are also potentially inappropriate or harmful, such as benzodiazepines.
Quality of the evidence
Overall, data on the effect of withdrawal of antipsychotics in older participants with dementia and NPS remain very sparse. We summarised the quality of evidence for comparisons in summary of findings Table 1. Evidence for all outcomes was low‐ or very low‐quality. The reasons for downgrading were imprecision, risk of bias and indirectness.
Limitations in study design or execution
Many included studies had methodological limitations. Only one RCT was assessed at low risk of bias for all domains (Ballard 2008). In almost half of the studies, there was insufficient information on random sequence generation or allocation concealment or both. Participants and personnel were blinded in all studies, but information on blinding of outcome assessment was unclear for four studies, although we thought this was unlikely to seriously alter the results. One study had a high dropout rate with imbalance between groups which could influence the results. Three studies were identified as having a potential risk of reporting bias, although most results reported were negative, suggesting they were not tending to favour the reporting of positive results. We judged three studies to be at high risk of reporting bias, possibly influencing results. Because of these limitations, we downgraded the level of evidence by one or two levels for risk of bias.
Mortality outcomes were measured in two studies from four months to 12 months (Ballard 2008; Devanand 2012). In a long‐term follow‐up of 36 months, after the 12 months randomised discontinuation trial, we were uncertain whether discontinuation led to a decreased mortality (Ballard 2008). These two studies were possibly too short‐term to detect an effect of discontinuation of antipsychotics on mortality rates.
Other limitations of study design were a lack of information about the indications for antipsychotic use and a lack of systematic reporting of adverse events which may be related to antipsychotics use, such as falls, extrapyramidal symptoms, cognitive dysfunction, metabolic changes, cardiovascular events and others. Only one study assessed falls (Devanand 2012).
Inconsistency of results
The two studies that reported our primary outcome NPS and provided data that could be pooled for meta‐analysis, showed consistency of results between the studies. On the whole, the diversity of the studies and their outcome measures precluded meta‐analyses. We therefore summarised evidence in a narrative synthesis, showing good consistency of conclusions across studies.
Indirectness
We defined success of withdrawal as the ability to complete the study (i.e. no withdrawal due to worsening of NPS or no relapse to antipsychotic drugs use during the trial). However none of the studies included this outcome defined in this way. Therefore, we used the number of non‐completers of the study and the difference between groups as a proxy for our primary outcome. We downgraded the quality of evidence for this outcome by one level due to this indirectness.
Most of the included trials assessed a wide range of neuropsychiatric symptoms (anxiety, apathy, depression, delusions, wandering, repetitive vocalisations, shouting, disinhibition, aberrant motor behaviour and appetite behaviour and many other symptoms). Two RCTs (Devanand 2011; Devanand 2012) focused on participants with psychosis, agitation or aggression. This could limit generalisability of the results of these studies, although we did not think this had an impact on the overall conclusions, because this subgroup was small. We discussed this subgroup separately because it may be clinically relevant.
We downgraded quality by one level for indirectness when considering the outcome adverse events because the included studies measured only a selection of potential adverse events.
Imprecision
All included studies had problems including 'frail' older participants (a group with high mortality) and had small sample sizes. Therefore, the statistical power of the studies was low, and very few outcomes showed clinical differences between the groups. Because it was not possible to pool data for most outcomes due to variability in outcome measures, the potential benefit of a meta‐analysis to produce a more precise effect estimate could not be realised.
The effect estimate for the two pooled studies on NPS had a wide 95% confidence interval which includes the null effect of no difference between treatments. This means NPS could get either better or worse after discontinuation.
Publication bias
We conducted a comprehensive search for published and unpublished studies that would have reduced the risk of publication bias. Funnel plots could not be constructed because we included only 10 studies.
Potential biases in the review process
We searched a wide range of databases with no restriction of language. We identified one unpublished study. We may have missed relevant studies; however, we think it is unlikely that we did not capture all available RCT evidence in this review.
Three review authors independently conducted all data selection and extraction; another review author acted as arbiter to minimise the risk of error and bias.
We pooled two studies that used the same NPI scale, although assessed at different times. It was unclear whether this difference in time of assessment had an important impact on the conclusions.
A critical narrative synthesis of the results could introduce bias, although there was consistency in the effects of the intervention across studies.
Overall, data on the effect of withdrawal of antipsychotics in older people with dementia and NPS remain very sparse, and conclusions should be interpreted with caution.
None of the review authors was involved in the included trials or had conflicts of interest in the field of antipsychotics.
Agreements and disagreements with other studies or reviews
A systematic review of 10 studies (Pan 2014) found that discontinuation of antipsychotics had no effect on behavioural and psychological symptoms severity, early study termination and mortality. Pan 2014 did not identify the unpublished study by Bergh 2011.
One cluster RCT (Ballard 2016) in participants with dementia living in nursing homes found advantages on quality of life if antipsychotic review is combined with non‐pharmacological interventions (social interaction or exercise) compared with antipsychotic review without non‐pharmacological interventions. This reinforces the urgency to establish safe and effective pharmacological and non‐pharmacological alternatives to antipsychotics in older people with dementia and NPS.
A literature review by Banerjee 2009 revealed an increase in absolute mortality risk of approximately 1% after antipsychotic treatment in older people with dementia. We found insufficient data to determine whether discontinuation of antipsychotic medication has any effect on mortality.

Forest plot of comparison: 1 Discontinuation versus continuation of long‐term antipsychotic drug use: continuous data, analysis method: mean difference, outcome: 1.1 Behavioural assessment by using Neuropsychiatric Inventory (NPI) measuring neuropsychiatric symptoms (NPS) at 3 months (Ballard 2004 and Ballard DART‐AD) (Analysis 1.1).
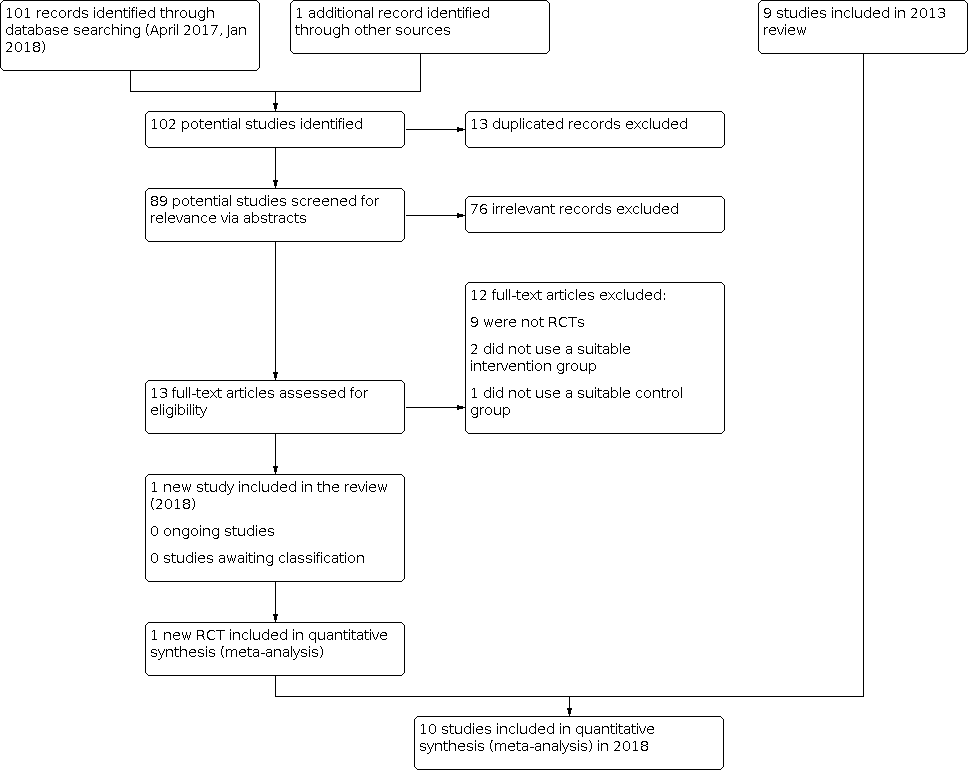
Inclusions of trials of study flow diagram 2018

Risk of bias graph for the 10 included studies in the review.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study in the review.

Comparison 1: Discontinuation versus continuation of long‐term antipsychotic drug use (continuous data, analysis method mean difference), Outcome 1: Behavioural assessment
| Discontinuation compared to continuation of antipsychotic medication for behavioural and psychological symptoms in older participants with dementia | ||||||
| Patient or population: older people with dementia who had been taking an antipsychotic drug for at least 3 months | ||||||
| Outcomes | Illustrative comparative risks (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
|---|---|---|---|---|---|---|
| Assumed risk Continuation antipsychotics | Corresponding risk Discontinuation antipsychotics | |||||
| Success of withdrawal from antipsychotics Measured with a variety of outcomes related to failure to complete the study Follow‐up: 1 to 8 months | In 7 studies there was no overall difference in the outcomes reported for success of withdrawal. In two studies of participants with psychosis, aggression or agitation who had responded to antipsychotic treatment, discontinuation accelerated symptomatic relapse without affecting the number of participants experiencing a relapse in one study and was associated with a higher rate of symptomatic relapse in the other study. In one small study a high proportion of the participants in the discontinuation group failed to complete the study. | 575 (9 RCTs) | ⊕⊕⊝⊝ LOWab | Our intended primary outcome, success of withdrawal defined as the ability to complete the study in the allocated study group, i.e. no failure due to worsening of NPS or relapse to antipsychotic drug use, was not reported in any study. We used the difference between groups in the number of non‐completers of the study as a proxy for our primary outcome. However, data could not be pooled due to variability in outcome measures. | ||
| Behavioural and psychological symptoms Assessed with various scales. Follow up: 1 to 8 months | In 2 pooled studies there was no difference in NPI scores between the continuation and discontinuation groups (see Data and analyses and Figure 1). In five non‐pooled studies, there was no difference in the outcomes on scales measuring overall behaviour and psychological symptoms between groups. | 519 (7 RCTs) | ⊕⊕⊝⊝ | Data could only be pooled for 2 studies due to variability in outcome measures. The two pooled studies performed subgroup analyses according to baseline NPI‐score (≤ 14 or > 14). In one study, some participants with milder symptoms at baseline were less agitated at three months in the discontinuation group. In both studies, discontinuation led to worsening of NPS in some participants with more severe baseline NPS. | ||
| Adverse events Assessed with various scales. Follow‐up: 1 to 8 months | In 5 studies, there was no evidence of a difference between groups in adverse events. | 381 (5 RCTs) | ⊕⊕⊝⊝ LOWab | Data could not be pooled due to variability in outcome measures. Adverse events of antipsychotics were not systematically reported. | ||
| Quality of life (QoL) Assessed with DCM or QoL‐AD. Follow‐up: 3 months to 25 weeks | In 2 studies, there was no evidence of an effect on quality of life. | 119 (2 RCTs) | ⊕⊕⊝⊝ | Data could not be pooled due to variability in outcome measures. There was no difference between discontinuation and continuation group in the overall cohort or in subgroups with baseline NPI score above or below the median (14). | ||
| Cognitive function Assessed with various scales. Follow‐up: 1 to 8 months | In 5 studies, there was no evidence of an impact on scales measuring overall cognitive function. In one of these trials, discontinuation improved a measure of verbal fluency. | 365 (5 RCTs) | ⊕⊕⊝⊝ | Data could not be pooled due to variability in outcome measures. | ||
| Use of physical restraint Follow‐up: 1 month | In one study there was no effect on the use of physical restraint. | 36 (1 RCT) | ⊕⊝⊝⊝ | Conclusion made by the authors but not supported by data. | ||
| Mortality Assessed with various scales. Follow‐up: 4 to 12 months | In two studies there was no evidence of an effect on mortality. | 275 (2 RCTs) | ⊕⊝⊝⊝ | Data could not be pooled due to clinical heterogeneity. In a long‐term follow‐up of 36 months after the 12 months randomised discontinuation trial (Devanand 2012), we were uncertain whether discontinuation decreased mortality. | ||
| *The basis for the assumed risk (e.g. the median control group risk across the studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| a Downgraded one level for indirectness. b Downgraded one level for risk of bias. c Downgraded one level for imprecision due to a small number of participants. d Downgraded two level for risk of bias. | ||||||
| Phenothiazines with aliphatic side chain Phenothiazines with piperazine structure Fhenothiazines with piperidine structure Butyrophenone derivatives Indole derivatives Thioxanthene derivatives Diphenylbutylpiperidine derivatives Diazepines, Oxazepines and Thiazepines Benzamides Other antipsychotics |
| Phenothiazines with aliphatic side‐chain N05AA01 Chlorpromazine 0.3 g per os N05AA02 Levomepromazine 0.3 g per os N05AA03 Promazine 0.3 g per os N05AA04 Acepromazine 0.1 g per os N05AA05 Triflupromazine 0.1 g per os N05AA06 Cyamemazine N05AA07 Chlorproethazine |
| Phenothiazines with piperazine structure N05AB01 Dixyrazine 50 mg per os N05AB02 Fluphenazine 10 mg per os N05AB03 Perphenazine 30 mg per os N05AB04 Prochlorperazine 0.1 g per os N05AB05 Thiopropazate 60 mg per os N05AB06 Trifluoperazine 20 mg per os N05AB07 Acetophenazine 50 mg per os N05AB08 Thioproperazine 20 mg per os N05AB09 Butaperazine 10 mg per os N05AB10 Perazine 0.1 g per os N05AB20 Homophenazine |
| Phenothiazines with piperidine structure N05AC01 Periciazine 50 mg per os N05AC02 Thioridazine 0.3 g per os N05AC03 Mesoridazine 0.2 g per os N05AC04 Pipotiazine 10 mg per os |
| Butyrophenone derivatives N05AD01 Haloperidol 8 mg per os N05AD02 Trifluperidol 2 mg per os N05AD03 Melperone* 0.3 g per os N05AD04 Moperon 20 mg per os N05AD05 Pipamperone 0.2 g per os N05AD06 Bromperidol 10 mg per os N05AD07 Benperidol 1.5 mg per os N05AD08 Droperidol N05AD09 Fluanisone |
| N05AE Indole derivatives N05AE01 Oxypertine 0.12 g per os N05AE02 Molindone 50 mg per os N05AE03 Sertindole* 16 mg per os N05AE04 Ziprasidone* 80 mg per os |
| Thioxanthene derivatives N05AF01 Flupentixol 6 mg per os N05AF02 Clopenthixol 0.1 g per os N05AF03 Chlorprothixene 0.3 g per os N05AF04 Tiotixene 30 mg per os N05AF05 Zuclopenthixol 30 mg per os |
| Diphenylbutylpiperidine derivatives N05AG01 Fluspirilene N05AG02 Pimozide 4 mg per os N05AG03 Penfluridol 6 mg per os |
| Diazepines, Oxazepines and Thiazepines N05AH01 Loxapine 0.1 g per os N05AH02 Clozapine* 0.3 g per os N05AH03 Olanzapine* 10 mg per os N05AH04 Quetiapine* 0.4 g per os |
| Benzamides N05AL01 Sulpiride 0.8 g per os N05AL02 Sultopride 1.2 g per os N05AL03 Tiapride 0.4 g per os N05AL04 Remoxipride 0.3 g per os N05AL05 Amisulpride* 0.4 g per os N05AL06 Veralipride N05AL07 Levosulpiride 0.4 g per os |
| Other antipsychotics N05AX07 Prothipendyl 0.24 g per os N05AX08 Risperidone* 5 mg per os N05AX09 Clotiapine 80 mg per os N05AX10 Mosapramine* N05AX11 Zotepine* 0.2 g per os N05AX12 Aripiprazole* 15 mg per os N05AX13 Paliperidone* *atypical antipsychotics |
| * Atypical antipsychotic agents. |
| Study IDI | Setting | Duration | Randomised number | Discontinuation group | Continuation group | Discontinuation schedule | Control | Behavioural inclusion criteria | Notes |
|---|---|---|---|---|---|---|---|---|---|
| Residents in long‐term care facilities | 3 months | 100 | 46 | 54 | Abrupt | Typical APa or risperidone | NPIb not higher than 7 | ||
| Residents in long‐term care facilities | 6 months 12 months | 165 | 82 | 83 | Abrupt | Typical and risperidone | NRc | ||
| Residents in nursing homes | 25 weeks | 19 | 9 | 10 | Tapering over 2 week | Risperidone | NRc | Unpublished study | |
| Residents in long‐term care facilities | 1 month | 36 | 22 | 14 | Abrupt + tapering over 2 weeks | Typical APa | Physically aggressive participants identified by nurse supervisors | ||
| Residents in nursing homes | 7 weeks followed by 7 weeks cross‐over | 58 | 29 | 29 | Tapering over 3 weeks | Typical APa + lorazepam | NRc | Cross‐over study | |
| Residents in the community | 6 months (primary analysis) 12 months | 20 | 10 | 10 | Abrupt + tapering over 2 weeks | Haloperidol | Current symptoms of psychosis, agitation or aggression | Participants had a response to haloperidol open treatment for 20 weeks | |
| Residents in the community and nursing homes | 4 months 8 months | 110 | 70 | 40 | Abrupt + tapering over 2 week | Risperidone | NPIb score higher than 4 on psychosis or agitation/aggression subscale | Participants had a response to risperidone open treatment for 16 weeks | |
| Residents in nursing homes | 1 month | 36 | 18 | 18 | Tapering over 1 week | Thioridazine | NRc | ||
| Residents in nursing homes | 1 month | 55 | 27 | 28 | Abrupt | Haloperidol risperidone, olanzapine | All participants regardless individual symptoms | ||
| Residents in nursing homes | 26 weeks | 34 | 17 | 17 | Tapering over 2 weeks | Typical APa | Stable behaviour | ||
| a AP: antipsychotic drug. b NPI: Neuropsychiatric Inventory. c NR: not reported. | |||||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1.1 Behavioural assessment Show forest plot | 2 | 194 | Mean Difference (IV, Fixed, 95% CI) | ‐1.49 [‐5.39, 2.40] |