Mu‐opioid antagonists for opioid‐induced bowel dysfunction in people with cancer and people receiving palliative care
Abstract
Background
Opioid‐induced bowel dysfunction (OIBD) is characterised by constipation, incomplete evacuation, bloating, and gastric reflux. It is one of the major adverse events of treatment for pain in cancer and in palliative care, resulting in increased morbidity and reduced quality of life.
This is an update of two Cochrane reviews. One was published in 2011, Issue 1 on laxatives and methylnaltrexone for the management of constipation in people receiving palliative care; this was updated in 2015 and excluded methylnaltrexone. The other was published in 2008, Issue 4 on mu‐opioid antagonists (MOA) for OIBD. In this updated review, we only included trials on MOA (including methylnaltrexone) for OIBD in people with cancer and people receiving palliative care.
Objectives
To assess the effectiveness and safety of MOA for OIBD in people with cancer and people receiving palliative care.
Search methods
We searched the Cochrane Central Register of Controlled Trials, MEDLINE, Embase, CINAHL, and Web of Science to August 2017. We also searched clinical trial registries and regulatory websites. We contacted manufacturers of MOA to identify further data.
Selection criteria
We included randomised controlled trials (RCTs) that assessed the effectiveness and safety of MOA for OIBD in people with cancer and people at a palliative stage irrespective of the type of terminal disease they experienced.
Data collection and analysis
Two review authors assessed risk of bias and extracted data. The appropriateness of combining data from the trials depended upon sufficient homogeneity across the trials. Our primary outcomes were laxation, impact on pain relief, and adverse events. Impact on pain relief was a primary outcome because a possible adverse effect of MOAs is a reduction in pain relief from opioids. We assessed the evidence on these outcomes using GRADE.
Main results
We identified four new trials for this update, bringing the total number included in this review to eight. In total, 1022 men and women with cancer irrespective of stage or at a palliative care stage of any disease were randomised across the trials. The MOAs evaluated were oral naldemedine and naloxone (alone or in combination with oxycodone), and subcutaneous methylnaltrexone. The trials compared with MOA with a placebo or with the active intervention administered at different doses or in combination with other drugs. The trial of naldemedine and the two of naloxone in combination with oxycodone were in people with cancer irrespective of disease stage. The trial on naloxone alone was in people with advanced cancer. The four trials on methylnaltrexone were undertaken in palliative care where most participants had cancer. All trials were vulnerable to biases; four were at a high risk as they involved a sample of fewer than 50 participants per arm.
In the trial of naldemedine compared to placebo in 225 participants, there were more spontaneous laxations over the two‐week treatment for the intervention group (risk ratio (RR) 1.93, 95% confidence intervals (CI) 1.36 to 2.74; moderate‐quality evidence). In comparison with higher doses, lower doses resulted in fewer spontaneous laxations (0.1 mg versus 0.2 mg: RR 0.73, 95% CI 0.55 to 0.95; 0.1 mg versus 0.4 mg: RR 0.69, 95% CI 0.53 to 0.89; moderate‐quality evidence). There was moderate‐quality evidence that naldemedine had no effect on opiate withdrawal. There were five serious adverse events. All were in people taking naldemedine (low‐quality evidence). There was an increase in the occurrence of other (non‐serious) adverse events in the naldemedine groups (RR 1.36, 95% CI 1.04 to 1.79, moderate‐quality evidence). The most common adverse event was diarrhoea.
The trials on naloxone taken either on its own, or in combination with oxycodone (an opioid) compared to oxycodone only did not evaluate laxation response over the first two weeks of administration. There was very low‐quality evidence that naloxone alone, and moderate‐quality evidence that oxycodone/naloxone, had no effect on analgesia. There was low‐quality evidence that oxycodone/naloxone did not increase the risk of serious adverse events and moderate‐quality evidence that it did not increase risk of adverse events.
In combined analysis of two trials of 287 participants, we found methylnaltrexone compared to placebo induced more laxations within 24 hours (RR 2.77, 95% CI 1.91 to 4.04. I² = 0%; moderate‐quality evidence). In combined analysis, we found methylnaltrexone induced more laxation responses over two weeks (RR 9.98, 95% CI 4.96 to 20.09. I² = 0%; moderate‐quality evidence). The proportion of participants who had a rescue‐free laxation response within 24 hours of the first dose was 59.1% in the methylnaltrexone arms and 19.1% in the placebo arm. There was moderate‐quality evidence that the rate of opioid withdrawal was not affected. Methylnaltrexone did not increase the likelihood of a serious adverse event; there were fewer in the intervention arm (RR 0.59, 95% CI 0.38 to 0.93; I² = 0%; moderate‐quality evidence). There was no difference in the proportion of participants experiencing an adverse event (RR 1.17, 95% CI 0.94 to 1.45; I² = 74%; low‐quality evidence). Methylnaltrexone increased the likelihood of abdominal pain and flatulence.
Two trials compared differing methylnaltrexone schedules of higher doses with lower doses. For early laxation, there was low‐quality evidence of no clear difference between doses on analgesia and adverse events. Both trials measured laxation response within 24 hours of first dose (trial one: RR 0.82, 95% CI 0.41 to 1.66; trial two: RR 1.07, 95% CI 0.81 to 1.42).
Authors' conclusions
In this update, the conclusions for naldemedine are new. There is moderate‐quality evidence to suggest that, taken orally, naldemedine improves bowel function over two weeks in people with cancer and OIBD but increases the risk of adverse events. The conclusions on naloxone and methylnaltrexone have not changed. The trials on naloxone did not assess laxation at 24 hours or over two weeks. There is moderate‐quality evidence that methylnaltrexone improves bowel function in people receiving palliative care in the short term and over two weeks, and low‐quality evidence that it does not increase adverse events. There is a need for more trials including more evaluation of adverse events. None of the current trials evaluated effects in children.
PICO
Plain language summary
Mu‐opioid antagonists for bowel dysfunction due to opioids in people with cancer and people receiving palliative care
Background
Opioids (morphine‐like drugs) are used to treat severe pain. Unfortunately, they cause side effects. Opioid‐induced bowel dysfunction (OIBD) is a term used to describe constipation, incomplete evacuation of the bowels, bloating, and increased reflux (flowing back) of stomach contents. OIBD may be so severe that a person chooses to limit opioid treatment to improve bowel function. OIBD is common in people with cancer and people receiving palliative care (care given to people with a terminal illness when a cure is no longer possible). Laxatives are often the first‐choice treatment for OIBD. They may not always work. Mu‐opioid antagonists (MOA) are specific medicines for OIBD. Clinical guidelines may recommend them when laxatives fail.
Trial characteristics
The aim of this updated review was to determine what we know about the effectiveness and safety of MOA for the management of OIBD in people with cancer and people receiving palliative care and for whom laxatives have failed. A possible side effect of an MOA is reduced pain relief from opioids; therefore, we looked at its impact on pain relief. We only included randomised controlled trials (RCTs), which are well‐designed clinical trials that provide the most reliable evidence. RCTs needed to evaluate an MOA, such as the medicines naldemedine, methylnaltrexone, and naloxone. The trial comparison groups could be a placebo (a substance with no known active effect), usual care, or another treatment such as a different type of MOA.
Key results
Our search to August 2017 found eight trials involving 1022 adults. The MOAs evaluated in people with cancer were oral naldemedine and naloxone taken in combination with an opioid treatment (for pain). Methylnaltrexone given by injection was evaluated in palliative care where most participants had advanced cancer.
The results were naldemedine or methylnaltrexone compared with placebo. For naloxone, they were either in comparison with a placebo or with opioid treatment only.
We rated the quality of the evidence from studies as very low to moderate. Very low means that we are very uncertain about the results. High means that we are very confident in the results. There were problems with the design of studies, including under‐reporting of trial methods.
Bowel movements within 24 hours and up to two weeks
There was moderate‐quality evidence that naldemedine increased bowel movements up to two weeks. Trials did not measure the effects of naloxone on bowel movements at two weeks. Methylnaltrexone increased bowel movements or laxations (softer stools) within 24 hours and up to two weeks (moderate‐quality evidence).
Pain relief
There was moderate‐quality evidence that naldemedine did not change pain relief. Trials did not measure pain intensity with naldemedine. There was very low‐quality evidence that naloxone taken on its own did not change pain relief. There was moderate‐quality evidence that naloxone taken with an opioid treatment did not change pain relief. There was moderate‐ to low‐quality evidence that methylnaltrexone did not change pain relief.
Risk of serious side effects (hospitalisation, life‐threatening, or fatal) and other side effects
There was low‐quality evidence that naldemedine and naloxone in combination with an opioid treatment did not increase the risk of serious side effects. For naldemedine, there were five serious side effects in the trial, although none were described as relating to the study drug. Methylnaltrexone probably did not increase the risk of serious side effects (moderate‐quality evidence).
There was moderate‐quality evidence that naldemedine increased the risk of side effects. There was moderate‐quality evidence that naloxone taken with oxycodone (an opioid painkiller) did not increase the risk of side effects. There was low‐quality evidence that methylnaltrexone did not increase the overall risk of a side effect. Methylnaltrexone increased the risk of abdominal pain and flatulence.
Conclusion
There was moderate‐quality evidence to suggest that naldemedine improved bowel function over two weeks in adults with cancer and OIBD but increased the risk of side effects; and that methylnaltrexone improved bowel function in people receiving palliative care and low‐quality evidence that methylnaltrexone did not increase side effects. The results of this review need to be interpreted with caution as they were not obtained from high‐quality evidence. There were no studies in children.
Authors' conclusions
Summary of findings
| Naldemedine compared to placebo for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Settings: cancer care Intervention: naldemedine Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Naldemedine | |||||
| Laxation response within 24 hours of dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14a | 375 per 1000 | 724 per 1000 | RR 1.93 (1.36 to 2.74) NNTB 2.88 (2.04 to 4.92) | 225 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: opioid withdrawalc | — | — | 0.1 mg: MD ‐0.13 (‐0.57 to 0.31); 0.2 mg: MD ‐0.40 (‐0.87 to 0.07); 0.4 mg: MD ‐0.02 (‐0.45 to 0.41) | 225 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: pain intensity | — | — | — | — | — | Not reported |
| Serious adverse eventsa | — | — | 5 SAEs occurred, all in naldemedine group. | 225 (1 study) | ⊕⊕⊝⊝ | — |
| Adverse eventsa | 518 per 1000 | 704 per 1000 (539 to 927) | RR 1.36 (1.04 to 1.79) | 225 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; NNTB: number needed to treat for an additional beneficial outcome; RR: risk ratio; SAE: serious adverse events. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by clinician or self‐report and in the case of adverse events using severity grades according to the Common Terminology Criteria for Adverse Events. | ||||||
| Lower‐dose naldemedine compared to higher‐dose naldemedine for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Setting: cancer care Intervention: lower dose naldemedine 0.1 mg daily Comparison: higher dose naldemedine 0.2 mg or 0.4 mg daily | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Higher dose 0.2 mg/0.4 mg daily | Lower dose 0.1 mg daily | |||||
| Laxation response within 24 hours of dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14a | 0.1 mg vs 0.2 mg: 776 per 1000 0.1 mg vs 0.4 mg: 821 per 1000 | 0.1 mg vs 0.2 mg: 564 per 1000 (430 to 739) 0.1 mg vs 0.4 mg: 564 per 1000 (433 to 733) | 0.1 mg vs 0.2 mg: RR 0.73 (0.55 to 0.95) 0.1 mg vs 0.4 mg: RR 0.69 (0.53 to 0.89) | 226 (1 study) 0.1 mg vs 0.2 mg: n = 113 0.1 mg vs 0.4 mg: n = 111 | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: opioid withdrawal | — | — | — | — | — | Not reported |
| Effect on analgesia: pain intensity | — | — | — | — | — | Not reported |
| Serious adverse events | — | — | — | — | — | Not reported |
| Adverse events | — | — | — | — | — | Not reported |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by self‐report. | ||||||
| Naloxone compared with placebo for cancer and people receiving palliative care with opioid‐induced bowel dysfunction | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Settings: cancer care Intervention: naloxone Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Naloxone | |||||
| Laxation response within 24 hours of a dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14 | — | — | — | — | — | Not reported |
| Effect on analgesia: opioid withdrawal | — | — | — | — | — | Not reported |
| Effect on analgesia: pain intensitya | — | — | No statistical difference in pain experienced when taking placebo or naloxone. Full data, including pre‐cross‐over results, were not provided. | 17 (1 study) | ⊕⊝⊝⊝ | — |
| Serious adverse events | — | — | — | — | — | Not reported |
| Adverse events | — | — | — | — | — | Not reported |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured using 4‐point scale (0 = no pain, 3 = severe pain). | ||||||
| Oxycodone/naloxone prolonged release tablets compared with oxycodone prolonged‐released tablets for opioid‐induced bowel dysfunction | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Settings: cancer care Intervention: oxycodone/naloxone prolonged‐release tablets Comparison: oxycodone prolonged‐released tablets | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Oxycodone | Oxycodone/naloxone | |||||
| Laxation response within 24 hours of dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14 | — | — | — | — | — | Not reported |
| Effect on analgesia: opioid withdrawalc | — | — | Intervention group: mean 6.64 (SD 5.97) comparison group: mean 7.29 (SD 4.59) at 7 days | 184 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: pain intensitya | — | — | Intervention group: mean 3.50 (SD 1.88) and comparison group: mean 3.52 (SD 1.80) at 4 weeks | 184 (1 study) | ⊕⊕⊕⊝ Moderateb | Another study, Dupoiron 2017 also found outcome to be similar between trial arms, but did not provide any data. |
| Serious adverse events | 43 per 1000 | 87 per 1000 (27 to 279) | RR 2.00 (95% CI 0.62 to 6.41) | 184 (1 study) | ⊕⊕⊝⊝ Lowb,d | — |
| Adverse events | 754 per 1000 | 815 per 1000 (709 to 935) | RR 1.08 (95% CI 0.94 to 1.24) | 234 (2 studies) | ⊕⊕⊕⊝ Moderateb | — |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; SD: standard deviation. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured using the Brief Pain Inventory‐Short Form. | ||||||
| Methylnaltrexone compared to placebo for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Setting: palliative care Intervention: methylnaltrexone Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Risk with placebo | Risk with methylnaltrexone | |||||
| Laxation response within 24 hours of dosea | 195 per 1000 | 568 per 1000 | RR 2.77 (1.91 to 4.04) | 287 | ⊕⊕⊕⊝ Moderateb | — |
| Laxation response between day 1 and day 14 (specifically within 4 hours after 4 or more of the 7 doses)a | 52 per 1000 | 517 per 1000 | RR 9.98 (4.96 to 20.09) | 305 | ⊕⊕⊕⊝ Moderate b,c | — |
| Effect on analgesia: opioid withdrawald | Study 1: day 1: MD 0.00 (‐0.46 to 0.46); day 14: MD 0.10 (‐0.63 to 0.83) Study 2: median change to day 2 = 0 in both trials arms | 236 (2 studies) | ⊕⊕⊕⊝ Moderateb | ‐ | ||
| Effect on analgesia: pain intensitye | Study 1: at 4 hours (methylnaltrexone 0.15 mg/kg: MD ‐0.76 (‐1.47 to 0.05); methylnaltrexone 0.3 mg/kg: MD ‐0.25 (‐0.91 to 0.41) Study 2: at day 1 and 14 (day 1: MD 0.20 (‐0.62 to 1.02); day 14: MD ‐0.70 (‐1.52 to 0.12) | 287 (2 studies) | ⊕⊕⊝⊝ Lowb,f | Another study, Bull 2015, found similar pain intensity experienced in trial arms, full data not provided. | ||
| Serious adverse events | 238 per 1000 | 142 per 1000 | RR 0.59 (0.38 to 0.93) | 364 | ⊕⊕⊕⊝ Moderateb | — |
| Adverse events | 700 per 1000 | 815 per 1000 | RR 1.17 (CI 0.94 to 1.45) | 518 | ⊕⊕⊝⊝ Lowb,g | Heterogeneity was substantial (74%). It was explained in sensitivity analysis by omitting the trial at a high risk of bias because of small sizes. The effect estimate was reduced. The direction of effect not changed. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by self‐report or clinician report. dMeasured using the modified Himmelsbach Opioid Withdrawal Scale. | ||||||
| Lower dose methylnaltrexone compared to higher dose for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Setting: palliative care Intervention 1: lower‐dose methylnaltrexone (study 1: 3 doses, 1 week, 1 mg; study 2: 1 dose, 0.15 mg/kg) Intervention 2: higher‐dose methylnaltrexone (study 1: 3 doses, 1 week, 5‐12.5 mg; study 2: 1 dose, 0.30 mg/kg) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Higher dose | Lower dose | |||||
| Laxation response within 24 hours of first dosea | Study 1: 609 per 1000 Study 2: 639 per 1000 | Study 1: 499 per 1000 (250 to 100) Study 2: 681 per 1000 (515 to 904) | Study 1: RR 0.82 (0.41 to 1.66) Study 2: RR 1.07 (0.81 to 1.42) | 135 (2 studies) Study 1: n = 33 Study 2: n = 102 | ⊕⊕⊝⊝ Lowb | Unable to combine study data as methylnaltrexone low and higher doses differed per trial |
| Laxation responsea | At 3 days: 706 per 1000 | At 3 days: 332 per 1000 | At 3 days: RR 0.47 (0.18 to 1.25) | 33 participants (1 study) | ⊕⊕⊝⊝ Lowb | Unable to combine study data as methylnaltrexone low and higher doses differed per trial |
| At 5 days: 688 per 1000 | At 5 days: 144 per 1000 | At 3 days: RR 0.21 (0.03 to 1.31) | ||||
| Effect on analgesia: opioid withdrawalc | — | — | MD ‐0.04 (‐0.73 to 0.65) | 102 participants (1 study) | ⊕⊕⊝⊝ Lowb | Another study,Portenoy 2008, also found outcome to be similar between trial arms, but did not provide any data |
| Effect on analgesia: pain intensityd | — | — | MD ‐0.51 (‐1.49 to 0.47) | 102 participants (1 study) | ⊕⊕⊝⊝ Lowb | Another study, Portenoy 2008, also found outcome to be similar between trial arms, but did not provide any data |
| Serious adverse event | — | — | — | — | Not reported | |
| Adverse event | Study 1: 1000 per 1000 Study 2: 800 per 1000 | Study 1: 1000 per 1000 (1000 to 1000) Study 2: 723 per 1000 (580 to 902) | Study 1: RR 1.00 (1.00 to 1.00) Study 2: RR 0.90 (0.73 to 1.13) | 135 (2 studies) Study 1: n = 33 Study 2: n = 102 | ⊕⊕⊝⊝ Lowb | Unable to combine study data as methylnaltrexone low and higher doses differed per trial |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by clinician or self‐report. bDowngraded by two levels for study limitations: one for unclear risk of bias (reporting bias) and one for small sample size (high risk of bias). cMeasured using the modified Himmelsbach Opioid Withdrawal Scale. dMeasured by participant‐rated scale 0‐10. | ||||||
Background
This review is a (partial) update of two previously published reviews in The Cochrane Database of Systematic Reviews.
-
Mu‐opioid antagonists for opioid‐induced bowel dysfunction, 2008, Issue 2 (McNicol 2008).
-
Laxatives or methylnaltrexone for the management of constipation in palliative care patients, 2011, Issue 1 (Candy 2011).
This review update presented the findings on the effectiveness and safety of mu‐opioid antagonists (MOA) for opioid‐induced bowel dysfunction (OIBD) in people with cancer and people receiving palliative care. An updated review on laxatives for the management of constipation in people receiving palliative care has been published (Candy 2015). It is planned that evidence on the effectiveness on MOAs for OIBD in other populations, including, for example, people with chronic non‐malignant pain, will be reviewed in a subsequent Cochrane Review.
Description of the condition
Opioids, such as morphine sulphate, oxycodone, and fentanyl, are potent analgesics. They are recommended by the World Health Organization (WHO) and are in clinical guidelines for the management of moderate‐to‐severe pain from cancer and other populations such as people needing palliative care (WHO 2016). They are widely used, although globally there is wide variation suggesting an under‐utilisation of opioids for pain management in some locations (Manjiani 2014).
However, opioids are associated with adverse events. The most common and disabling of these is bowel dysfunction, which can be severe enough for a person to limit their opioid use (Cook 2008). Opioids, regardless of the method of administration (oral, parenteral, transdermal), interfere with gastrointestinal propulsive motility (Leppert 2010). Opioids increase absorption of fluids from the intestine and decrease epithelial secretion. They delay gastric emptying and decrease peristalsis in the gut.
OIBD has been described as, "A change when initiating opioid therapy from baseline bowel habits that is characterised by any of the following: reduced bowel movement frequency (conventionally less than 3 per week), development or worsening of straining to pass bowel movements, a sense of incomplete rectal evacuation, or harder stool consistency" (Kumar 2012). It may even lead to stool impaction (Camilleri 2014). In addition to constipation, OIBD describes a constellation of symptoms including bloating, abdominal distention, gastric reflux, abdominal cramping, dry mouth, epigastric fullness, nausea, and vomiting (Leppert 2015; Pappagallo 2001). It can cause psychological distress and agitation in terminally ill people. OIBD increases health service use, sometimes necessitates hospitalisation, and it can dramatically reduce an already compromised quality of life. It may lead to people undertreating their pain (Pizzi 2012); however, since the dose that produces constipation may only be 25% of that required for adequate analgesia, dose reduction is not an appropriate option for management of OIBD (Ketwaroo 2013).
Estimated incidence of OIBD in hospice populations and people with advanced disease is high from 65% to 90% (Panchal 2007; Sykes 1998). Although these estimates are relatively old, there is no evidence to suggest that this is no longer the case.
Description of the intervention
The recommended preventive treatment of OIBD in palliative care and advanced disease is the use of a laxative stimulant and a stool softener, in addition to general measures such as increased food, fibre‐rich diet, fluid intake, physical activity, and privacy during defecation (NICE 2012). However, these measures are not always effective; in people taking opioids, it is estimated that over 80% of people remain constipated despite regular use of laxatives (Coyne 2014; Diego 2011).
MOAs, such as methylnaltrexone, naloxone, and naloxegol, are designed specifically to target the pathophysiology of OIBD by 'neutralising' the constipating effect of the opioid. Methylnaltrexone is licensed for the treatment of opioid‐induced constipation in palliative care in more than 50 countries (Bader 2013). In clinical guidelines, where methylnaltrexone is considered, it is described to act as an augmentation to laxatives or as an alternative when laxatives fail (European Association of Palliative Care, Caraceni 2012), and should be used only under advice from a specialist palliative care clinician (Scottish Palliative Care Guidelines 2014). It is important to note that the National Institute for Health and Care Excellence (NICE) is unable to recommend the use in the UK National Health Service (NHS) of methylnaltrexone for treating OIBD in people with advanced illness receiving palliative care because no evidence submission was received from the manufacturer of the technology (NICE 2013).
How the intervention might work
Opioids mediate their gastrointestinal and analgesic effects through the same subclasses of opioid receptors in the human body: mu, kappa, and delta. How each receptor type is involved in OIBD is not fully understood (Neefjes 2014). The peripheral opioid effect on mu‐opioid receptors in the gut wall may play a main role in OIBD (Leppert 2010). Co‐ordination of motility is disrupted by activation of the mu‐opioid receptors that inhibit excitatory and inhibitory neural pathways within the enteric nervous system.
One approach for dissociation of the analgesia effect of opioids is to separate the opioid's central activity from its peripheral activity (Wang 2013). This may be achieved with a peripherally acting opioid receptor antagonist with limited ability to cross the blood‐brain barrier and which therefore does not interfere with analgesia (Brown 1985). Alternatively, this can be achieved by use of a preparation that undergoes extensive 'first‐pass' metabolism by the liver and so does not enter the systemic circulation.
There are several mu‐antagonists in use and others in development. Naloxone is commercially available; it is centrally acting but has a narrow therapeutic effect with certain doses reversing desirable analgesia (Camilleri 2011). It undergoes extensive first‐pass metabolism and in the correct dosage it does not reverse the analgesic effect of opioids. It is administered orally. The development of a prolonged‐release preparation of naloxone to allow as much cover of the small and large intestine as possible when used with oxycodone has led to further studies of the compound (Camilleri 2011). There are several other preparations that do not cross the blood‐brain barrier and these include alvimopan, methylnaltrexone, naloxegol, and naldemedine. Alvimopan has a high affinity for peripheral opioid receptors. It is only recommended for short‐term use, such as postsurgery, because of the possibility of myocardial events (Merck 2015). It is contraindicated in people with advanced disease (Leppert 2015). Methylnaltrexone is less lipid soluble than naloxone and, therefore, less likely to cross the blood‐brain barrier. It is only currently available in subcutaneous formulation. Naloxegol, which is administered orally, has a polyethylene glycol moiety that limits its capacity to cross the blood‐brain barrier (Pritchard 2015). Naldemedine is a derivative of naloxone. There are limited published details on its mechanisms of action, it is currently being evaluated in phase III trials in people with cancer.
Why it is important to do this review
There are reviews of MOAs for OIBD across different populations (e.g. Ford 2013). However, it is important to evaluate their effectiveness specifically in cancer and in palliative care populations (Bader 2012; Clark 2014). This is because of the differences inherent in these groups that may impact, in a likely negative way, on the effect of an MOA. The impact may differ because of the multi‐factorial pathophysiology of constipation in people with cancer and advanced diseases (Leppert 2010). This may include structural abnormalities such as bowel obstruction; pelvic tumours; radiation fibrosis; or metabolic disturbances such as dehydration, hypercalcaemia, and hypokalaemia. It may involve neurological disorders. There may also be general issues increasing the risk and complicating the management of OIBD such as advanced age, depression, drug sedation, chemotherapy, multiple therapies, and a lack of privacy provided as an inpatient for bowel evacuation. As the person's disease progresses, they may have increasing frailty, lower activity, reduced appetite, and eventually multiple organ failure, all of which may impact on bowel function (Bader 2012). Moreover, because of these factors, people with cancer and particularly people at a palliative care stage may have a higher risk than other, less ill populations of experiencing adverse events from an MOA.
Objectives
To assess the effectiveness and safety of MOAs for OIBD in people with cancer and people receiving palliative care.
Methods
Criteria for considering studies for this review
Types of studies
We included double‐blind, randomised controlled trials (RCTs) that investigated the efficacy of MOAs for OIBD. We did not include open‐label extension phases of trials or post‐hoc analyses of trials because they are at an increased risk of bias. We applied no language restrictions. We required full journal publication. In addition to trials presented in full journal publication, we included any online clinical trial results summaries of otherwise unpublished trial data relating to the published trial.
Types of participants
Eligible trials concerned participants of any age or either sex who were:
-
people with cancer or people at a palliative stage irrespective of disease and age, or both;
-
all or the majority (over 95%) of people on a stable opioid regimen and had OIBD that had not resolved from taking laxatives.
We included trials of populations of participants where not all fitted our eligibility criteria so long as at least 50% of the sample were people with cancer or people receiving palliative care or at an advanced stage of their disease or where they provided subgroup analysis in either of these participant groups.
We did not include trials of any populations if the MOAs for bowel dysfunction were for associated postoperative ileus (arrest of intestinal peristalsis). This is because this is not caused primarily by opioids (Marderstein 2008). We also excluded trials of healthy volunteers, participants with constipation because of drug misuse, and participants with constipation arising from bowel obstruction.
Types of interventions
We included trials of interventions evaluating mu‐receptor opioid antagonists that were either peripherally or systemically acting, and administered at any dose and by any route. These included, for example, methylnaltrexone and naloxone.
The comparator intervention of interest was a different MOA, an alternative pharmacological or non‐pharmacological intervention, a placebo, or no treatment.
Types of outcome measures
Primary outcomes
Our primary outcomes of interest were efficacy in regards to laxation response and safety.
-
Efficacy:
-
laxation response in the first 24 hours and between days one and 14 after first dose. Laxation response could have been measured using a validated scale such as the 3‐item Bowel Function Index (BFI) on ease of defecation or person feeling of incomplete bowel emptying;
-
effect on analgesia. This could have been measured as analgesic requirements, opioid withdrawal symptoms, and pain intensity. This was a primary outcome as MOAs may have an adverse effect on pain relief.
-
-
Safety:
-
serious adverse events;
-
number and type of adverse events.
-
Secondary outcomes
-
Number of participants who dropped out due to adverse events.
-
Other measures of laxation response, such as bowel transit time and relief at a time‐point beyond day 14.
-
Relief of other constipation‐associated symptoms, such as abdominal pain and loss of appetite.
-
Use of rescue medication for laxation. This is the need for additional medication because relief from constipation has not occurred within an acceptable time, such as for instance within four hours of administration of the intervention subcutaneous methylnaltrexone. Rescue medication may be in the form of a laxative suppository or an enema.
-
Quality of life, participant satisfaction with bowel movements, and participant preference.
Search methods for identification of studies
Electronic searches
For this update, we searched five databases.
-
CENTRAL (CRSO) 2007 to issue 7 of 12 2017.
-
MEDLINE and MEDLINE in process (Ovid) 2007 to 28 August 2017.
-
Embase (Ovid) 2007 to week 35 2017.
-
CINAHL (EBSCO) 1982 to August 2017.
-
Web of Science (SCI‐Expanded and CPCI‐S) 1945 to 28 August 2017.
The search strategies are listed in Appendix 1.
Searching other resources
We searched three clinical trials registries to March 2016.
-
The metaRegister of controlled trials (mRCT) (www.controlled‐trials.com/mrct).
-
ClinicalTrials. gov (clinicaltrials.gov).
-
The WHO International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/).
We searched two regulatory agency websites, US Food and Drug Administration (FDA) and the European Medicines Agency (EMA), for drug reports.
We searched pharmaceutical companies' websites of known manufacturers of MOAs to identify trial data.
We searched two pharmaceutical company trials registers:
-
AstraZeneca Clinical Trials (www.astrazenecaclinicaltrials.com);
-
GlaxoSmithKline Clinical Trial Register (www.gsk‐clinicalstudyregister.com).
We checked references lists of included trials and any identified systematic reviews. We also undertook a forward citation search of all included trials. We checked conference proceedings of the National Cancer Research Institute (NCRI) Cancer Conference and the European Association of Palliative Care (EAPC). We contacted authors of any identified relevant conference abstracts to ask for full details of their trials.
We wrote to pharmaceutical companies of known manufacturers of MOAs to obtain any trial data not available in peer‐review publications; these were AstraZeneca, Mundipharma GmbH, Progenics, Shionogi, and Valeant. For this purpose, we adapted a letter developed by authors of a previous Cochrane Review; see Appendix 2 for a copy of this letter.
Data collection and analysis
Selection of studies
Two review authors (BC, LJ) independently screened the citations identified in the database searches. Where it was unclear or likely that the studies fulfilled our inclusion criteria, we retrieved the full‐text articles. If disagreements on eligibility had occurred, we would have resolved them by discussion, or if persistent, by a third review author (PS). If necessary for further clarification such as if it was unclear whether the trial identified was completed and whether their findings were available, we sought contact with the author or sponsor.
Data extraction and management
We extracted data (as detailed in Types of outcome measures) from each trial. One review author (BC) extracted the data and another review author checked them (LJ/VV). We resolved disagreements by discussion, or if persistent, we would have involved a third review author (PS).
Assessment of risk of bias in included studies
Two review authors (BC, VV) independently assessed risk of bias for each trial using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions, resolving any disagreements by discussion (Higgins 2011). We completed a 'Risk of bias' table for each included trial. We assessed the following.
-
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (any truly random process: random number table; computer random number generator); and unclear risk of bias (method used to generate sequence not clearly stated). We excluded studies using a non‐random process, which were therefore at high risk of bias (odd or even date of birth; hospital or clinic record number).
-
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions before assignment determines whether the intervention allocation could have been foreseen in advance of, or during, recruitment, or changed after assignment. We assessed the methods as: low risk of bias (telephone or central randomisation; consecutively numbered, sealed, opaque envelopes); and unclear risk of bias if the method was not clearly stated. We excluded trials that did not conceal allocation, which were therefore at high risk of bias (open list).
-
Blinding of participants and personnel (performance bias and detection bias). We assessed the methods used to blind trial participants and personnel (performance bias) and outcome assessors (detection bias) from knowledge after trial assignment of which intervention a participant received. We assessed the methods as: low risk of bias if the trial stated that it was blinded and described the method used to achieve blinding: identical tablets, matched in appearance and smell; and unclear risk of bias if the trial stated that it was blinded but did not provide an adequate description of how blinding was achieved. We judged a trial as high risk if blinding was attempted but it was likely that the blinding could have been broken and that the outcome was likely to be influenced by lack of blinding. We did not include any trial that was reported as not being double blinded.
-
Incomplete outcome data (attrition bias). We assessed whether there was attrition bias due to the amount, nature, or handling of incomplete outcome data. We judged the trial as having low risk of attrition bias if there were no missing outcome data or the reasons for missing data were unlikely to be related to true outcome, or missing data and reasons for it were similar across trial arms, or the missing data had been imputed using appropriate methods. We judged the trial as high risk if the reason for missing outcome data was likely to be related to the outcome, with either imbalance across trial arms in numbers of reasons for missing data and if an inappropriate application of simple imputation was potentially used. We judged the trial as unclear risk if there was insufficient reporting of attrition to permit judgement of low or high risk.
-
Selective outcome reporting (checking if there was a selection of a subset of the original variables recorded on the basis of the results). We assessed selective outcome reporting, if a protocol was available, by comparing outcomes in the protocol and published report. If they were the same we assessed it as low risk in this domain, if they differed, we considered it as high risk. If a protocol was not available, then we compared the outcomes listed in the methods section of an article with the outcomes for which results were reported. If they differed, we considered the trial as high risk. If a protocol was not available and even though the outcomes listed in the methods section and the results section were the same, we considered the trial as having an unclear risk of bias in this domain. Since not all trials have a protocol available, we expected to find a number of trials in this review to be at unclear risk.
-
Sample size (checking for possible biases confounded by small sample size). Small trials have been shown to overestimate treatment effects, probably because the conduct of small trials is more likely to be less rigorous, allowing critical criteria to be compromised (Zhang 2013). We considered trials to be at low risk of bias if they had 200 participants or more per treatment arm, at unclear risk if they had 50 to 199 participants per treatment arm, and at high risk if they had fewer than 50 participants per treatment arm.
We incorporated the results of the 'Risk of bias' assessment into the review through systematic narrative description and commentary about each item.
Measures of treatment effect
We reported trial results organised by type of MOA and comparator evaluated. We measured treatment effects using dichotomous data, an ordinal rating scale, or qualitative evidence. For cross‐over trials, we only generated, as appropriate, a risk ratio (RR) or mean difference (MD) for pre‐cross‐over results.
Dichotomous data
For dichotomous data, we generated RRs and their 95% confidence intervals (CIs). For primary outcomes, we calculated numbers needed to treat (NNT) using the 'treat‐as‐one‐trial' method. To indicate direction of effect, we presented results as either number needed to treat for an additional beneficial outcome (NNTB) or number needed to treat for an additional harmful outcome (NNTH) to indicate direction of effect.
Continuous data
We assessed effects measures for ordinal data as continuous data. We generated the MD for continuous and ordinal data where the data were provided as a mean and standard deviation (SD).
If baseline data were reported preintervention and postintervention, we reported means or proportions for both intervention and control groups and calculated the change from baseline.
If limitations in the trial data prevented reporting a RR or if continuous data, an MD, we reported the results with caution due to lack of transparency of the evidence.
Qualitative evidence
If there had been any qualitative data in the included trials, we planned to extract them in consultation with the Cochrane Qualitative and Implementation Methods Group. Such qualitative data may aim to capture the participant's views on the value of the intervention.
Unit of analysis issues
In our handling of each trial analytic, we considered issues that may have impacted on findings. For these we took guidance from the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). These were:
-
groups of participants randomised together with the same intervention (e.g. cluster‐randomised trials);
-
participants receiving more than one intervention (e.g. cross‐over trials);
-
multiple observations for the same outcomes (such as repeated measures).
Dealing with missing data
Given the nature of this field, there was a significant amount of missing data as a result of trial attrition due to the death of the participant.
We planned to contact trial authors if we had found data to be missing. For trials using continuous outcomes in which SDs were not reported, and no information was available from the authors, we calculated the SDs using the standard error of the mean (SEM).
Assessment of heterogeneity
We assessed statistical heterogeneity using the I² statistic. The I² statistic is a reliable and robust test to quantify heterogeneity, since it does not depend on the number of trials or on the between‐trial variance. I² measures the extent of inconsistency among trials' results, and can be interpreted as the proportion of total variation in trial estimates that is due to heterogeneity rather than sampling error. We considered an I² value of greater than 50% to indicate substantial heterogeneity (Deeks 2006). Where possible, we undertook subanalyses or sensitivity analyses in an attempt to explain heterogeneity.
Assessment of reporting biases
To reduce the risk of reporting bias, we undertook comprehensive database and registry searches, including searches of clinical trial registers and drug regulatory agency websites. We also searched websites of, and wrote to, pharmaceutical companies that are known manufacturers of MOAs to identify trial data.
Owing to an insufficient number of included studies (fewer than 10), as appropriate a test power was not ensured; we did not create funnel plots or conduct Egger's test for funnel plot asymmetry (Egger 1997; Sterne 2011). In applying in combined analysis as appropriate random‐effects estimates of the intervention effect, we decided not to exclude small studies, as this might have led to an inappropriate reduction of studies in a field that is just emerging. Nevertheless, in case of small‐study effects, we cautiously considered sample size when grading and discussing the evidence for each outcome (Roberts 2015). We expect that in updates of the review, when more studies have been published, we will be able to explore reporting biases further by comparing fixed‐effect and random‐effects estimates or L'Abbé plots as a visual method of assessing differences in results of individual studies.
Data synthesis
Where trial data were of sufficient quality and sufficiently similar (in diagnostic criteria, intervention, outcome measure, length of follow‐up, and type of analysis), we combined data in a meta‐analysis to provide a pooled effect estimate. We used a fixed‐effect model in the first instance. If we found no statistical heterogeneity, we used a random‐effects model to check the robustness of the fixed‐effect model. If there was substantial (over 50%) statistical heterogeneity, we reported the random‐effects model only. Where this occurred, we stated we used the random‐effects model.
Subgroup analysis and investigation of heterogeneity
Where heterogeneity was identified in a meta‐analysis, we undertook subgroup and sensitivity analysis to investigate its possible sources. Subgroup analysis explores whether the overall effect varied with different trial populations, and with the nature and content of the interventions. In this update, we planned the following subgroup analysis:
-
studies of participants with advanced disease or in palliative care, as impact of MOAs may differ in such participants than those at an earlier stage of cancer.
Sensitivity analysis
If sufficient trials were available, we sought to perform, in a meta‐analysis, sensitivity analyses to explore the influence of:
-
publication status by excluding unpublished trials;
-
trial quality by excluding trials that had a high risk of bias;
-
use of appropriate measures/validated measures of outcome by excluding trials that did not use appropriate/validated measures.
We presented in a table for ease of comparisons such investigations of heterogeneity.
Quality of evidence
Two review authors (BC, VV) independently rated the quality of the primary outcomes. We used the GRADE system to rank the quality of the evidence using the guidelines provided in Chapter 12.2 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
The GRADE system uses the following criteria for assigning a quality level to a body of evidence (Chapter 12, Higgins 2011).
-
High: randomised trials; or double‐upgraded observational studies.
-
Moderate: downgraded randomised trials; or upgraded observational studies.
-
Low: double‐downgraded randomised trials; or observational studies.
-
Very low: triple‐downgraded randomised trials; or downgraded observational studies; or case series/case reports.
The GRADE approach uses five considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the quality of the body of evidence for each outcome. The GRADE system uses the following criteria for assigning grade of evidence.
-
High: we are very confident that the true effect lies close to that of the estimate of the effect.
-
Moderate: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different.
-
Low: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect.
-
Very low: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect.
Factors that may decrease the quality level of a body of evidence are:
-
limitations in the design and implementation of available studies suggesting high likelihood of bias;
-
indirectness of evidence (indirect population, intervention, control, outcomes);
-
unexplained heterogeneity or inconsistency of results (including problems with subgroup analyses);
-
imprecision of results (wide CIs);
-
high probability of publication bias (0.7854 to 1.1359).
Factors that may increase the quality level of a body of evidence are:
-
large magnitude of effect;
-
all plausible confounding would reduce a demonstrated effect or suggest a spurious effect when results show no effect;
-
dose‐response gradient.
We decreased the grade rating by one (‐1) or two (‐2) (up to a maximum of ‐3 to 'very low') if we identified:
-
serious (‐1) or very serious (‐2) limitation to study quality;
-
important inconsistency (‐1);
-
some (‐1) or major (‐2) uncertainty about directness;
-
imprecise or sparse data (‐1);
-
high probability of reporting bias (‐1).
In certain circumstances, we adjusted the overall rating for a particular outcome as recommended by GRADE guidelines (Guyatt 2013a). For example, we considered whether there were so few data that the results were highly susceptible to the random play of chance, or if a study used last observation carried forward imputation in circumstances where there were substantial differences in adverse event withdrawals, one would have no confidence in the result and would need to downgrade the quality of the evidence by three levels to very low quality (Guyatt 2013b). In other circumstances, we would not downgrade for imprecision if CIs were wide, if the outcome threshold according to how much harm would be acceptable given a benefit or vice versa.
'Summary of findings' table
We included 'Summary of findings' tables to present the main findings in a transparent and simple tabular format. We have summarised the level of overall quality of evidence on all primary outcomes in the 'Summary of findings' tables. This does not include quality evaluations on the individual types of adverse events. This decision was made as we were not judging quality for all types of adverse events using GRADE; we were only judging those adverse events that were most commonly reported. We included key information concerning the quality of evidence, the magnitude of effect of the interventions examined, and the sum of available data on the outcomes laxation response (within 24 hours; between day one and day 14), effect on analgesia (pain intensity, opioid withdrawal), serious adverse events, and number of adverse events.
Results
Description of studies
Results of the search
In this update, we first searched for evidence on 24 September 2014. This search was run without restricting the search terms to only those relating to cancer and palliative care populations. We reran our search to 29 August 2017 updating search terms and restricting to those relating to cancer and palliative care populations. We identified 633 unique citations. See Figure 1 for the flowchart of the screening process.
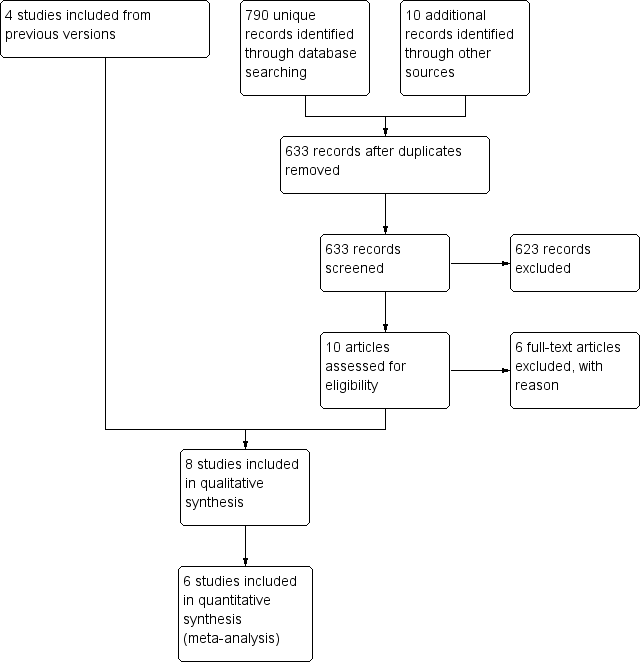
Study flow diagram.
Included studies
We included seven published RCTs of 976 participants (Ahmedzai 2012; Bull 2015; Katakami 2017; Portenoy 2008; Slatkin 2009; Sykes 1996; Thomas 2008). We included one RCT in a general population with a subset analysis in 46 people with cancer (Dupoiron 2017). We include four trials (Portenoy 2008; Slatkin 2009; Sykes 1996; Thomas 2008) that were identified in the earlier Cochrane Reviews (Candy 2011; McNicol 2008) for which this review forms a partial update. For four trials, we also identified regulatory (FDA and the EMA) assessments of the manufactures' Clinical Study Reports (Ahmedzai 2012; Portenoy 2008; Slatkin 2009; Thomas 2008) (these reports are referenced under Ahmedzai 2012 for oxycodone/naloxone, and Slatkin 2009 for methylnaltrexone).
Seven trials were multi‐centre parallel RCTs. The other was a single‐centre cross‐over controlled trial (Sykes 1996). All trials were sponsored by a pharmaceutical company, apart from Sykes 1996. Included trial populations were from North America (Bull 2015; Portenoy 2008; Slatkin 2009; Thomas 2008), Japan (Katakami 2017), South Korea (Katakami 2017) and the UK (Sykes 1996). Two trials involved sites in multiple countries. In one this included sites in Australia, Czech Republic, France, Germany, Hungary, Israel, the Netherlands, Poland, and the UK (Ahmedzai 2012), and in the other France, Germany, Poland, and the UK (Dupoiron 2017).
Three trials evaluated participants with chronic cancer pain who were not described as being at an advanced disease stage (Ahmedzai 2012; Dupoiron 2017; Katakami 2017). Where specified, the healthcare setting was a clinic (Ahmedzai 2012; Dupoiron 2017).
The five other trials evaluated effects in participants with an advanced disease including cancer, and other conditions such as AIDS or circulatory disease. Although in all these trials the majority had a primary diagnosis of cancer. Three of these trials were based in multiple care settings including inpatients and outpatients of a hospice or hospital, and long‐term care facilities (Bull 2015; Slatkin 2009; Thomas 2008). Another was hospice based only (Sykes 1996), and the other did not report the setting (Portenoy 2008).
In all trials, according to inclusion criteria, at baseline all or the majority (over 95%) of participants were on a stable opioid regimen, had OIBD, and were taking laxatives. Six trials specified that the indication for opioids was pain (Ahmedzai 2012; Dupoiron 2017; Katakami 2017; Portenoy 2008; Slatkin 2009; Thomas 2008). The other trials did not state an indication. All studies were on adults. All trials reported laxative use at baseline. For all it was either the need to take regular laxatives was part of the inclusion criteria, or it was stated that all or the majority (90% or greater) used regular laxatives.
In four trials, the intervention of interest was subcutaneous methylnaltrexone (Bull 2015; Portenoy 2008; Slatkin 2009; Thomas 2008). Three trials tested oral naloxone; in one naloxone only (Sykes 1996) and in two oxycodone (an opioid) in combination with naloxone (Ahmedzai 2012; Dupoiron 2017). The other trial evaluated oral naldemedine (Katakami 2017). We identified no trials in cancer or palliative care populations that evaluated naloxegol or another MOA.
Three of the trials involved multiple trial arms (Katakami 2017; Portenoy 2008; Slatkin 2009), the others were two armed. The interventions were either compared with a placebo or with the active intervention administered at different doses or in combination with other drugs. Outcomes on laxation were measured as self‐report or clinician report, for instance on rescue‐free laxation (Bull 2015; Katakami 2017; Portenoy 2008; Slatkin 2009; Thomas 2008), or by using a validated scale such as the BFI (Ahmedzai 2012; Dupoiron 2017), Patient Assessment of Constipation Symptoms (PAC‐SYM), and the Global Clinical Impression of Change (GCIC) (Slatkin 2009). One trial also used small bowel transit time using a lactulose and hydrogen breath test (Sykes 1996). Further details of these trials are shown in the Characteristics of included studies table.
Five trials involved a subsequent open‐label extension phase (Bull 2015; Portenoy 2008; Slatkin 2009; Sykes 1996; Thomas 2008). We did not report results on effectiveness from open‐label extension as the participants were no longer blinded.
Excluded studies
We excluded five trials because they did not include participants with cancer or at the palliative stage of a disease in their sample. These trials are listed in the Characteristics of excluded studies table.
Ongoing studies
We also identified 11 trials known to have been started but results as yet are not published (Dimitroulis 2014; JAPIC‐CTI‐132340; NCT00135577; NCT00331045/00101998; NCT02745353; NCT02839889; NCT01438567; NCT02321397; NCT02574819; Neefjes 2014; Peppin 2013). Four of these trials are evaluating methylnaltrexone (Dimitroulis 2014; NCT02574819; Neefjes 2014; Peppin 2013), one naldemedine (JAPIC‐CTI‐132340), one oxycodone/naloxone (NCT01438567; NCT02321397), two naloxegol (NCT02745353; NCT02839889), and two alvimopan (NCT00135577; NCT00331045/00101998). Further details of these are in the Characteristics of ongoing studies table.
Studies awaiting classification
We are awaiting classification for one trial on naloxegol (Webster 2013). We are unsure until we receive details from the authors or funders whether the trial fulfils our inclusion criteria. See Characteristics of studies awaiting classification for further details.
Risk of bias in included studies
All trials were vulnerable to a number of biases, most commonly reporting bias and small sample sizes. See Figure 2; Figure 3.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
The method of randomisation sequence generation was described adequately in four trials (Ahmedzai 2012; Katakami 2017; Slatkin 2009; Thomas 2008). In the other four trials this was inclear as they did not provide any details. The method of concealment of allocation was described adequately in two trials (Ahmedzai 2012; Slatkin 2009). In the other six trials this was unclear as they did not provide any details.
Blinding
Three trials were at a low risk of performance bias and detection bias (Katakami 2017; Slatkin 2009; Thomas 2008). In the other trials, it was unclear as they provided no details on who was blinded or how blinding was conducted.
Incomplete outcome data
The risk of attrition bias was low in most trials apart from one trial where it was unclear how many had dropped out of the subgroup of people with cancer (Dupoiron 2017).
Selective reporting
The risk of selective reporting was unclear in seven trials as there were no published protocols. One of these trials did not state a primary outcome (Sykes 1996).The eighth study had low risk of bias as it had a protocol (Dupoiron 2017).
Other potential sources of bias
We assessed sample size. Four trials were at a high risk of biased results as they involved fewer than 50 participants in at least one trial arm (Dupoiron 2017; Portenoy 2008; Slatkin 2009; Sykes 1996). In one of these trials, only a subset of their study sample was relevant to this review (Dupoiron 2017). Also in one of these trials while there were fewer than 50 participants in one of their treatments arms, as we combined in our exploration of the impact of MOAs their two treatment groups in comparison with placebo this risk was no longer apparent (Slatkin 2009).
All the other trials were at an unclear risk of bias as they involved treatment arms with between 50 and 199 participants.
Effects of interventions
See: Summary of findings for the main comparison Naldemedine compared to placebo for opioid‐induced bowel dysfunction in cancer and people receiving palliative care; Summary of findings 2 Lower‐dose naldemedine compared to higher‐dose naldemedine for opioid‐induced bowel dysfunction in cancer and people receiving palliative care; Summary of findings 3 Naloxone compared with placebo for cancer and people receiving palliative care with opioid‐induced bowel dysfunction; Summary of findings 4 Oxycodone/naloxone prolonged release tablets compared with oxycodone prolonged‐released tablets for opioid‐induced bowel dysfunction; Summary of findings 5 Methylnaltrexone compared to placebo for opioid‐induced bowel dysfunction in cancer and people receiving palliative care; Summary of findings 6 Lower‐dose methylnaltrexone compared to higher‐dose methylnaltrexone for opioid‐induced bowel dysfunction in cancer and people receiving palliative care
The trials varied in population (participants with any disease at a palliative stage or people with cancer irrespective of disease stage), intervention, and how they reported the outcomes. This limited the number of combined analyses. Subgroup and sensitivity analyses were limited because of the small number of trials included in any combined analysis. We have standardised the results where possible and reported the findings in the trials as fully as possible.
Naldemedine
One trial of 225 participants with cancer evaluated the effectiveness of two weeks of treatment with naldemedine compared with placebo and at different doses in people with cancer irrespective of disease stage (Katakami 2017). Doses of naldemedine were 0.1 mg, 0.2 mg, or 0.4 mg daily. Overall, we rated the quality of the evidence on laxation, effect on analgesia, and adverse events as moderate. See summary of findings Table for the main comparison.
Primary outcomes
Laxation response
The study did not measure laxation response within the first 24 hours following the first drug dose and participant evaluation of improvement in bowel function.
In comparison with placebo, naldemedine increased the number of participants who had spontaneous bowel movements over the two‐week treatment phase (RR 1.93, 95% CI 1.36 to 2.74. NNTB 3, 95% CI 2 to 5). We judged the quality of evidence for laxation response within two weeks to be moderate. We downgraded the quality of evidence by one level for limitations to the study design. This was because of unclear risk of bias (reporting bias).
The proportion of participants who had a rescue‐free laxation response over the two weeks differed by dose of naldemedine, with the higher dose resulting in more laxations (0.1 mg: 56.4%; 0.2 mg: 77.6%; 0.4 mg: 82.1%). All these were clear improvements when compared with placebo, which had a laxation responder rate of 37.5% (0.1 mg: P = 0.0464; 0.2 mg: P = 0.001; and 0.4 mg: P = 0.001). There was a dose difference in laxation response. This was in comparison with higher doses (0.2 mg and 0.4 mg) the dose of 0.1 mg daily resulted in fewer spontaneous bowel movements (0.1 mg versus 0.2 mg: RR 0.73, 95% CI 0.55 to 0.95; 0.1 mg versus 0.4 mg: RR 0.69, 95% CI 0.53 to 0.89). There was no clear difference between the dose of 0.2 mg daily compared to 0.4 mg daily in bowel movements (RR 0.95, 95% CI 0.79 to 1.14). We judged the quality of evidence on dose response to be moderate. We downgraded the quality of evidence by one level for limitations to the study design. This was because of unclear risk of bias (reporting bias).
Effect on analgesia
Naldemedine had no effect on analgesia compared with placebo in that there was no noticeable increase in opiate withdrawal over two weeks (0.1 mg: MD ‐0.13, 95% CI ‐0.57 to 0.31; 0.2 mg: MD ‐0.40, 95% ‐0.87 to 0.07; 0.4 mg: MD ‐ 0.02, 95% CI ‐0.45 to 0.41). The study did not measure the effect on analgesia using pain intensity. We judged the quality of evidence for effect on analgesia (opioid withdrawal) to be moderate. We downgraded the quality of evidence by one level for limitations to the study design. This was because of unclear risk of bias (reporting bias).
Serious adverse events
There were five serious adverse events. All events occurred in the naldemedine group. One participant experienced a gastrointestinal bleed (taking naldemedine 0.1 mg); one participant each experienced pneumonia, anaemia, or asthenia. One participant died due to bile duct cancer. The investigator considered the death unrelated to the study drug. Judgments on whether the other events were related to the study drug were not reported. Four of the serious adverse events occurred in the highest dose (0.4 mg). We judged the quality of evidence for serious adverse events to be low. We downgraded the quality of evidence by two levels, one for limitations to the study design and one for imprecision. This was because of unclear risk of bias (reporting bias) and a limited number of events.
Number and type of adverse events
There was a clear difference in the proportion of participants in the intervention arm compared to participants in the placebo arm who experienced an adverse event (RR 1.36, 95% CI 1.04 to 1.79). We judged the quality of evidence that naldemedine increased the risk of an adverse event to be moderate. We downgraded the quality of evidence by one level for limitations to the study design. This was because of unclear risk of bias (reporting bias).
The most common adverse event was diarrhoea. There was no clear difference in the proportion of participants in the intervention arm compared to participants in the placebo arm who experienced diarrhoea (RR 1.58, 95% CI 0.97 to 2.57). For eight of the participants receiving naldemedine, the diarrhoea was moderate and in another participant, it was severe. Diarrhoea in participants in the placebo group was mild. Other adverse events reported/measured included white blood cell count, abdominal pain, nausea, and vomiting (see Table 1).
| Adverse event | Naldemedine (%) | Placebo (%) |
| Diarrhoea | 67 (39) | 14 (25) |
| Decreased WBC count | 9 (5) | 3 (5) |
| Abdominal pain | 6 (4) | 0 (0) |
| Vomiting | 5 (3) | 0 (0) |
| Bone marrow failure | 3 (2) | 2 (4) |
| Decreased appetite | 6 (4) | 1 (2) |
| Nasopharyngitis | 4 (2) | 1 (2) |
| Nausea | 4 (2) | 4 (7) |
| Rash | 3 (2) | 2 (4) |
| Decreased platelet count | 3 (2) | 0 (0) |
| Decreased total protein | 7 (4) | 1 (2) |
| Glucose in urine | 4 (2) | 1 (2) |
| Abnormal haematology test | 2 (1) | 0 (0) |
| Decreased RBC count | 4 (2) | 0 (0) |
| Hypertension | 2 (1) | 0 (0) |
| Increased blood alkaline phosphatase | 4 (2) | 1 (2) |
| Increased blood lactate dehydrogenase | 2 (1) | 1 (2) |
| Increased blood pressure | 2 (1) | 0 (0) |
| Increased blood urea | 4 (2) | 1 (2) |
| Increased WBC count | 1 (2) | 2 (4) |
| Protein present in urine | 5 (3) | 0 (0) |
| Upper abdominal pain | 3 (2) | 1 (2) |
RBC: red blood cell; WBC: white blood cell.
All comparisons were not statistically significant.
Secondary outcomes
Number of participants who dropped out due to adverse events
There was no clear difference in the proportion of participants who dropped out due to an adverse event between participants taking naldemedine and participants taking placebo (RR 2.68, 95% CI 0.34 to 20.98).
Other measures of laxation response
The trial did not report other measures of laxation response.
Relief of other constipation‐associated symptoms
The trial did not report relief of other constipation‐associated symptoms.
Use of rescue medication for laxation
The trial did not measure use of 'rescue' medication for laxation.
Quality of life, satisfaction with bowel movements, and participant preference
The trial did not report quality of life, satisfaction with bowel movements, and participant preference.
Naloxone
One cross‐over trial evaluated the effectiveness of oral naloxone compared with placebo in 17 participants with advanced cancer (Sykes 1996). The participants received two days on either placebo or naloxone followed (without washout) by another two days on the trial agent that was not received on day one and two. Naloxone was given four‐hourly for a total daily dose of 0.5%, 1%, 2%, 5%, 10%, or 20% of the total daily dose of morphine. Overall, we rated the quality of the evidence on effect on analgesia as very low (laxation and adverse events not reported). See summary of findings Table 3.
Primary outcomes
Laxation response
The trial did not measure laxation response within the first 24 hours or between days one and 14 after first dose.
Effect on analgesia
The authors stated there was no evidence of opioid withdrawal or difference in pain experienced between the comparisons. There was no full data, including pre‐cross‐over results provided. We judged the quality of evidence for effect on analgesia to be very low. We downgraded by three levels. This was because evidence is from one study with small sample size, the study was of cross‐over design with no washout between drug cross‐over, and because of unclear risk of bias (reporting bias).
Serious adverse events
There were no serious adverse events.
Number and type of adverse events
The trial did not provide the overall number of adverse events experienced by the participants.
Secondary outcomes
Number of participants who dropped out due to adverse events
Four participants withdrew from the trial. One participant withdrew because of severe diarrhoea caused by the lactulose taken as part of the test on bowel function, one participant because of general deterioration in health while taking naloxone (although not thought to be a causal relationship), one participant because of diarrhoea experienced while receiving the placebo, and one participant withdrew because of nausea after two doses of naloxone at the 10% level (5 mg in this case).
Other measures of laxation response
On the second day following treatment the trial measured small bowel transit times (SBTTs). This was by lactulose‐hydrogen breath tests to detect the release of hydrogen resulting from breakdown of lactulose by colonic bacteria. They found no clear difference in SBTTs between naloxone and placebo groups. The investigators did not provide full data, including pre‐cross‐over results. They used no other measures of laxation response
Relief of other constipation‐associated symptoms
The trial did not report other constipation‐associated symptoms.
Use of rescue medication for laxation
The trial did not report use of rescue medication for laxation.
Quality of life, participant satisfaction with bowel movements, and participant preference
The trial did not report quality of life, participant satisfaction with bowel movements, and participant preference.
Oxycodone/naloxone prolonged‐release tablets versus oxycodone prolonged‐release tablets
Two trials of 231 participants with cancer evaluated oxycodone/naloxone prolonged‐release tablets (OXN PR) compared with oxycodone prolonged‐release (OXY PR) tablets (Ahmedzai 2012; Dupoiron 2017).
One trial of 185 participants with cancer of any stage evaluated the effectiveness of four weeks of treatment (Ahmedzai 2012). The drug dose in both trials arms was 120 mg daily. In addition to the published trial paper we also reviewed FDA and EMA drug reports; these documents did not add any additional data (for references to these reports see Ahmedzai 2012). The other trial evaluated the effectiveness of five weeks of treatment in a general population. We included the trial's subset data on the 46 people with cancer (Dupoiron 2017). The dose of oxycodone/naloxone was up to daily maximum oxycodone 160 mg/naloxone 80 mg.
Overall, we rated the quality of the evidence on effect on analgesia and adverse events as moderate (effect on laxation not reported) (summary of findings Table 4).
Primary outcomes
Laxation response
The trials did not report laxation response within the first 24 hours and between days one and 14 after first dose.
Effect on analgesia
In one trial using the Brief Pain Inventory‐Short Form at four weeks of treatment, pain scores were similar between the trial arms (OXN PR: mean 3.50, SD 1.88; OXY PR: mean 3.52, SD 1.80) (Ahmedzai 2012). In the other trial, pain scores remained at a low level throughout the study and were comparable between groups (Dupoiron 2017). Neither trial provided full data. We judged the quality of evidence on effect on analgesia in regards to pain intensity as moderate. We downgraded the quality of the evidence by one level because of study limitations (unclear risk of reporting bias).
Serious adverse events
In one trial 18 participants died during the trial, nine in each trial arm (Ahmedzai 2012), in the other trial one person died in the OXN PR arm and three in the OXY PR arm (Dupoiron 2017). None of the deaths were attributed to the trial drugs.
In one trial, there were 12 serious adverse events attributed to the study medication, eight in the OXN PR arm and four in the OXY PR arm (Ahmedzai 2012). There was no clear difference between trial arms in proportion of participants experiencing a serious adverse event (RR 2.00, 95% CI 0.62 to 6.41). The authors did not describe what these events were. In the other trial, there were three serious adverse events in the OXN PR arm and five in the OXY PR arm (Dupoiron 2017). None were attributed to the study medication.
We judged the quality of evidence for serious adverse events to be low. We downgraded the quality of evidence by one level because of study limitations (unclear risk of bias (reporting bias)) and one level for imprecision because of wide CIs.
Number and type of adverse events
In combined analysis of the two trials (234 participants), there was no clear difference between OXN PR in comparison with OXY PR in the proportion of particants experiencing an adverse event (RR 1.08; 95% CO 0.94 to 1.24]. I² = 0%; Analysis 4.1).
In both trials, a common adverse event was gastrointestinal symptoms. In one study, there was no clear difference in the number of such events per trial arm (RR 1.21, 95% CI 0.81 to 1.83) (Ahmedzai 2012); in the other trial, there were two events of gastritis in the OXN PR arm and none in the OXY PR arm (Dupoiron 2017). Also in this trial, there were two events of hypercholesterolaemia and hypertriglyceridaemia in the OXN PR arm and none in the other arm (Dupoiron 2017).
We judged the quality of evidence for number of adverse events to be moderate. We downgraded the quality of evidence by one level because of study limitations (unclear risk of bias (reporting bias).
Secondary outcomes
Number of participants who dropped out due to adverse events
There was no clear difference in the proportion of participants who dropped out of the trial in the OXN PR arm compared to the OXY PR arm (20/92 with OXN PR versus 12/92 OXY PR; RR 1.67, 95% CI 0.87 to 3.21) (Ahmedzai 2012). The other trial did not report the number who dropped out due to adverse events in the subset of people with cancer (Dupoiron 2017).
Other measures of laxation response
One trial measured laxation responses from baseline to four weeks (Ahmedzai 2012). In the assessment using the BFI‐Short Form, the trial reported no significant difference in changes between the trial arms in bowel function (MD ‐0.02, 95% CI ‐0.65 to 0.61). Using the PAC‐SYM on participants' assessment of bowel symptoms there was a difference between the trial arms favouring intervention group in total symptom score (MD ‐5.10, 95% CI ‐8.08 to ‐2.12), and in frequency of symptoms (MD ‐0.56, 95% CI ‐0.94 to ‐0.18). The trial did not report per arm the proportion of participants who had a laxation at each of the time points over the four weeks.
Laxation response was the primary outcome in one trial at five weeks (Dupoiron 2017). At this time point, the researchers found a clinically meaningful and treatment difference in BFI in favour of OXN PR arm (14.0, SD 8.1; P = 0.047; full data not provided).
Neither trial reported other measures of laxation response.
Relief of other constipation‐associated symptoms
One study assessed constipation‐associated symptoms using the PAC‐SYM scale, which primarily focuses on bowel‐related symptoms in addition to those of bloat, stomach cramps, and abdominal pain. However, only the total symptom score was provided (Ahmedzai 2012). Therefore, neither trial reported useful data on relief of other constipation‐associated symptoms.
Use of rescue medication for laxation
The use of laxatives (oral bisacodyl) at the end of follow‐up was not significantly different between the trial arms (MD ‐6.59, 95% CI ‐16.61 to 3.43) (Ahmedzai 2012). The authors reported the need for rescue medication was "generally low"' in both treatment groups; they reported in terms of frequency and dose that the difference in use was not significantly different. At the end of follow‐up in the other trial, the mean daily doses of rescue medication in the OXN PR arm was 0.6 (SD 1.1) and in the OXY PR arm 1.4 (SD 2.3) (Dupoiron 2017).
Quality of life, participant satisfaction with bowel movements, and participant preference
One trial used two measures to assess quality of life; the European QoL EQ‐5D instrument and the European Organization for Research and Treatment of Cancer QoL Questionnaire‐Core 30 (EORTC QLQ‐C30) (Ahmedzai 2012). Using the EQ‐5D they found no clear difference between the trial arms (MD 0.01, 95% CI ‐0.11 to 0.13). They did not provide full results at follow‐up for the EORTC QLQ‐C30 scale. The trial did not assess overall satisfaction and participant preference. The other trial did not measure any of these outcomes (Dupoiron 2017).
Methylnaltrexone
Three trials of 518 participants with advanced disease evaluated the effectiveness of subcutaneous methylnaltrexone compared to placebo (Bull 2015; Slatkin 2009; Thomas 2008). In addition to the three published trial papers, we also reviewed FDA and EMA drug reports, which did not identify any additional data (for references to these see Slatkin 2009).
One trial involved a single dose of either methylnaltrexone 0.15 mg/kg or 0.30 mg/kg (Slatkin 2009). The other two trials administered methylnaltrexone every other day for two weeks. One trial administered methylnaltrexone 0.15 mg/kg of bodyweight (Thomas 2008), and the other trial, with the aim of improving ease of administration, administered methylnaltrexone 8 mg to participants whose bodyweight was between 38 kg and 62 kg, or methylnaltrexone 12 mg if they weighed more than 62 kg (Bull 2015). Overall, we rated the quality of the evidence on laxation, effect on analgesia, and adverse events as moderate. See summary of findings Table 5.
Primary outcomes
Laxation response
In combined analysis of two trials (287 participants), there was a clear difference favouring methylnaltrexone in comparison with placebo in rescue‐free laxation within 24 hours of the first dose (RR 2.77, 95% CI 1.91 to 4.04. I² = 0%; Analysis 1.1) (Slatkin 2009; Thomas 2008). The NNTB was 3 (95% CI 2 to 3). The proportion of participants who had a rescue‐free laxation response with methylnaltrexone within 24 hours of the first dose of treatment was 59.1% and in the placebo arm was 19.5%. We judged the quality of evidence for laxation within 24 hours of the first dose to be moderate. We downgraded by one level for study limitations (unclear risk of bias (reporting bias)).
Two trials measured the laxation response over two weeks (305 participants) (Bull 2015; Thomas 2008). In combined analysis, more participants in the methylnaltrexone group compared to participants in the placebo group had a rescue‐free laxation within four hours of at least four of the maximum seven doses (RR 9.98, 95% CI 4.96 to 20.09. I² = 0%; Analysis 1.2) (Bull 2015; Thomas 2008). The NNTB was 2 (95% CI 2 to 2). The proportion of participants who had a rescue‐free laxation response with methylnaltrexone (within four hours of at least four of the maximum seven doses) was 52.6% and in the placebo arm was 5.3%. We judged the quality of evidence for laxation response over two weeks to be moderate. We downgraded by one level for study limitations (unclear risk of bias (reporting bias)). As the effect size was large, we did not downgrade for imprecision because of wide CIs.
Effect on analgesia
Two trials assessed opioid withdrawal symptoms (Slatkin 2009; Thomas 2008). In one trial, they stated that the median change from baseline to day two in score using the Himmelsbach Opioid Withdrawal Scale in both trials arms was 0 (Slatkin 2009). In the other trial at day one and 14, there was no clear difference between the trial arms (day one: MD 0.00, 95% CI ‐0.46 to 0.46; day 14: MD 0.10, 95% CI ‐0.63 to 0.83). We judged the quality of evidence for effect on analgesia in regards to opioid withdrawal to be moderate. We downgraded the quality of evidence by one level for study limitations because of unclear risk of bias (reporting bias).
In one trial, there was a reduction from baseline to four‐hour follow‐up in pain intensity in the methylnaltrexone 0.15 mg/kg group compared to placebo, but not in the methylnaltrexone 0.3 mg/kg group compared to placebo (0.15 mg/kg: MD ‐0.76, 95% CI ‐1.47 to ‐0.05; 0.3 mg/kg: MD ‐0.25, 95% CI ‐0.91 to 0.41) (Slatkin 2009). In the other two trials, participants had similar pain scores in both trial arms at follow‐up (Bull 2015; Thomas 2008). The dose of methylnaltrexone in the trial by Thomas 2008 was 0.15 mg/kg and by Bull 2015 was either 8 mg or 12 mg dependent on participant's bodyweight. In the trial by Thomas 2008, there was no clear difference in score between the trial arms at day one and day 14 (day one: MD 0.20, 95% CI ‐0.62 to 1.02; day 14: MD ‐0.70, 95% CI ‐1.52 to 0.12). The other trial did not provide full data (Bull 2015). We judged the quality of evidence for effect on analgesia in regards to pain intensity to be low. We downgraded the quality of evidence by two levels, one for study limitations (unclear risk of bias (reporting bias)), and one for inconsistency because of differing estimates of effect.
Serious adverse events
In combined analysis of two trials, with 364 participants, the proportion of participants experiencing a serious adverse event was lower in the methylnaltrexone arm (25/179 in methylnaltrexone arm versus 44/185 in placebo arm; RR 0.59, 95% CI 0.38 to 0.93; I² = 0%; Analysis 2.1) (Bull 2015; Thomas 2008). In Thomas 2008, the type of serious adverse events in the 11 participants who were receiving methylnaltrexone were: aneurysm ruptured, respiratory arrest, exacerbation of dyspnoea, suicidal ideation, aggression, malignant neoplasm progression, concomitant disease progression, myocardial ischaemia, aggravation of coronary artery disease, and aggravation of congestive heart failure. Bull 2015 did not describe the types of serious adverse events. In both trials, the investigators considered all serious adverse events as either not related or unlikely to be related to the trial drug. One trial did not report serious adverse events occurring during the randomised phase (Slatkin 2009), although during the open‐label phase three participants experienced such an event. One participant had flushing, one participant had delirium possibly related to methylnaltrexone, and one participant had severe diarrhoea and subsequent dehydration and cardiovascular collapse considered to be related to methylnaltrexone. We judged the quality of evidence for risk of a serious adverse event to be moderate. We downgraded the quality of evidence by one level for study limitations. This was because of unclear risk of bias (reporting bias).
Number and type of adverse events
In combined analysis of the three trials, with 518 participants, the proportion of participants experiencing adverse events was not clearly different between the trial arms (RR 1.17, 95% CI 0.94 to 1.45; random‐effects model; I² = 74% suggesting substantial heterogeneity between trials; Analysis 3.1) (Bull 2015; Slatkin 2009; Thomas 2008). We considered subgroup and sensitivity analyses to explain the heterogeneity. Sensitivity analysis demonstrated that the primary meta‐analysis was robust. Omitting the trial at high risk of bias because of small sample size (Slatkin 2009) resulted in a smaller estimate of effect. It did not change the overall result or conclusions. See Table 2. We judged the quality of evidence for adverse events to be low. It was downgraded by two levels; one for study limitations (unclear risk of reporting bias) and one because of inconsistency due to substantial statistical heterogeneity between the trials, this was upgraded since sensitivity analysis accounted for heterogeneity.
| Methylnaltrexone vs placeboa | |
| AEs | RR 1.07, 95% CI 0.96 to 1.19 |
| AE of abdominal pain | RR 2.15, 95% CI 1.28 to 3.62 |
| AE of nausea | RR 0.87, 95% CI 0.46 to 1.65 |
| AE of vomiting | RR 0.70, 95% CI 0.33 to 1.47 |
aomitting trial of high risk of bias.
AE: adverse event; CI: confidence intervals; RR: risk ratio.
Two of the trials reported some of the adverse events as severe (Slatkin 2009; Thomas 2008). One reported that 19/102 participants experienced a severe adverse event that was possibly related to the intervention drug; most commonly this event was abdominal pain (Slatkin 2009). The other trial did not describe what the events were and whether they were considered related to methylnaltrexone, but more participants in the placebo group experienced severe adverse events than in the intervention group (5/63 (8%) with methylnaltrexone versus 9/71 (13%) with placebo).
All three trials reported that participants in both trial arms experienced abdominal pain, flatulence, nausea, and vomiting (Bull 2015; Slatkin 2009; Thomas 2008). In combined analysis, participants in the intervention arm compared to participants in the placebo arm were more likely to experience abdominal pain (RR 2.39, 95% CI 1.07 to 5.34; random‐effects model; I² = 65% suggesting substantial heterogeneity between trials). Likewise, in combined analysis participants in the intervention arm compared to participants in the placebo arm were more likely to experience flatulence (RR 2.09, 95% CI 1.07 to 4.08; I² = 0%). In combined analyses, there was no clear difference between the trial arms in the proportion who experienced nausea (RR 0.97, 95% CI 0.89 to 1.06; random‐effects model; I² = 63% suggesting substantial heterogeneity between trials) or vomiting (RR 0.99, 95% CI 0.92 to 1.08; random‐effects model; I² = 67% suggesting substantial heterogeneity between trials). Sensitivity analyses demonstrated that the primary meta‐analyses were robust with regards to risk of experiencing abdominal pain, nausea, and vomiting. Omitting the trial at high risk of bias because of small sample size resulted in a smaller estimate of effect (Slatkin 2009). It did not change the overall result or conclusions. See Table 2.
Two trials also reported that participants experienced diarrhoea (Bull 2015; Thomas 2008), dizziness (Slatkin 2009; Thomas 2008), peripheral oedema (Bull 2015; Thomas 2008), and restlessness (Slatkin 2009; Thomas 2008). In combined analysis, there was no clear difference between trials in the proportion of participants experiencing these adverse events. See Table 3. Two trials also reported on falls (Bull 2015; Thomas 2008), and somnolence (Slatkin 2009; Thomas 2008). For these adverse events in combined analysis, there was also no significant difference between trial arms. See Table 3.
| Adverse event | RR (95% CI) | I²statistic on heterogeneity |
| Abdominal pain | 2.39 (1.07 to 5.34) | 65% |
| Diarrhoea | 1.02 (0.93 to 1.11) | 51% |
| Dizziness | 4.09 (0.99 to 16.83) | 0% |
| Falls | 1.02 (0.89 to 1.16) | 84% |
| Flatulence | 2.09 (1.07 to 4.08) | 0% |
| Nausea | 0.97 (0.89 to 1.06) | 63% |
| Peripheral oedema | 1.01 (0.50 to 2.03) | 0% |
| Restlessness | 0.83 (0.32 to 2.12) | 0% |
| Somnolence | 1.00 (0.93 to 1.08) | 73% |
| Vomiting | 0.99 (0.92 to 1.08) | 67% |
CI: confidence interval; RR: risk ratio.
Only one trial reported other adverse events. See Table 4 for the proportion of participants per trial and per arm that reported these events.
| Adverse event | Methylnaltrexone (%) | Placebo (%) |
| Abdominal distensiona | 1 (2) | 6 (8) |
| Abdominal tendernessa | 1 (2) | 4 (6) |
| Astheniaa | 4 (6) | 4 (6) |
| Anxietyb | 5 (4.9) | 0 (0) |
| Arthralgiab | 3 (2.9) | 1 (1.9) |
| Back painc | 9 (7.8) | 3 (2.9) |
| Confusional statec | 7 (6.0) | 9 (7.9) |
| Dehydrationa | 2 (3) | 4 (6) |
| Fatigueb | 4 (3.9) | 1 (1.9) |
| Hypotensiona | 0 (0) | 4 (6) |
| Increased body temperaturea | 5 (8) | 2 (3) |
| Lethergya | 4 (6) | 4 (6) |
| Malignant‐neoplasm progressiona | 7 (11) | 9 (13) |
| Pain exacerbationb | 8 (8) | 2 (4) |
| Rhinorrhoeab | 6 (5.9) | 1 (1) |
| Sweating increasedb | 8 (7.8) | 4 (7.7) |
| Tachycardiaa | 1 (1) | 4 (6) |
aReported in trial by Thomas 2008.
bReported in trial by Slatkin 2009.
cReported in trial by Bull 2015.
Secondary outcomes
Number of participants who dropped out due to adverse events
Two trials reported that there were dropouts due to adverse events. In combined analysis, there was no significant difference in the proportion of participants who dropped out between the trial arms (RR 1.22, 95% CI 0.54 to 2.76; I² = 0%; Analysis 3.2) (Bull 2015; Thomas 2008). The other trial reported no dropouts due to adverse events.
Other measures of laxation response
In both trials of two‐week treatments there was a shorter time to laxation in the methylnaltrexone group that persisted for each of the seven treatment doses (for all comparisons: P < 0.005 (Bull 2015); P < 0.002 (Thomas 2008)). In combined analysis of all three trials, there was a difference favouring methylnaltrexone in comparison with placebo in rescue‐free laxation within four hours of the first dose (placebo or methylnaltrexone) (RR 3.87, 95% CI 2.83 to 5.28; I² = 0%; Analysis 1.3) (Bull 2015; Slatkin 2009; Thomas 2008). The proportion of participants in total who had a laxation response within four hours of the first (or second) dose of treatment ranged from 61.4% in the methylnaltrexone groups and 16.0% in the placebo groups. The single‐dose trial using the GCIC found that the proportion of participants who reported an improvement in constipation distress at four hours significantly favoured the methylnaltrexone arm (irrespective of dose) compared to placebo (RR 1.84, 95% CI 1.23 to 2.75) (Slatkin 2009). The trial by Bull 2015 measured the mean number of rescue‐free laxation responses within 24 hours after dosing in the first and second week: at both time points participants receiving methylnaltrexone had significantly more laxations than participants receiving placebo (first week: MD 1.90, 95% CI 0.55 to 3.25; second week: MD 1.20, 95% CI 0.15 to 2.25).
Two trials reported clinician ratings of bowel status using the GCIC ratings were assessed in two trials (287 participants) (Slatkin 2009; Thomas 2008). In combined analysis, clinician ratings that bowel status had improved at week one were greater in those in the interventions groups (RR 2.37, 95% CI 1.66 to 3.38; I² = 25%; Analysis 1.7).
All three trials reported that time to first rescue‐free laxation favoured methylnaltrexone (median times in the intervention group versus placebo: 0.8 hours versus 23.6 hours (Bull 2015), 1.1 hours versus > 24 hours (Slatkin 2009), 6.3 hours versus > 48 hours (Thomas 2008)).
Relief of other constipation‐associated symptoms
None of the trials measured the relief of other constipation‐associated symptoms such as bloating, abdominal distention, gastric reflux, abdominal cramping, dry mouth, epigastric fullness, nausea, and vomiting, although some of these symptoms were reported as adverse events.
Use of rescue medication for laxation
Two trials recorded the need for rescue laxatives. In both trials, more participants in the placebo group needed rescue laxatives than participants in the intervention groups.
In one trial, fewer participants used rescue laxatives in the methylnaltrexone group than in the placebo group (31/116 with methylnaltrexone versus 46/114 with placebo; RR 0.66, 95% CI 0.46 to 0.96) (Bull 2015). It was also recorded in the FDA report on the trial by Thomas 2008 that there was an increased use in enemas in both trial arms. This was greater in the placebo group (23.8% with methylnaltrexone versus 35.2% with placebo).
Quality of life, overall satisfaction with bowel movements, and participant preference
None of the trials measured quality of life or participant preference. Two trials (287 participants) assessed participant ratings of bowel status using the GCIC ratings (Slatkin 2009; Thomas 2008). In combined analysis, participant ratings that bowel status had improved at one week were significantly greater in the methylnaltrexone groups (RR 2.32, 95% CI 1.64 to 3.27; I² = 0%; Analysis 1.6). Both trials measured constipation distress at day one (RR 1.87, 95% CI 1.34 to 2.59; I² = 25%).
Methylnaltrexone dose response
Two trials with 135 participants with advanced disease evaluated different dosing regimens of methylnaltrexone (Portenoy 2008; Slatkin 2009). One trial, irrespective of bodyweight, explored fixed doses of methylnaltrexone 1 mg, 5 mg, 12.5 mg, or 20 mg in 33 participants (Portenoy 2008). Because of the limited number of participants in the trials, we provided outcomes for participants taking 1 mg compared to participants of 5 mg or greater. The drug was administered on alternate days over one week. The other trial compared one dose on different dose‐ranging schedules of 0.15 mg/kg for 47 participants with 0.3 mg/kg for 55 participants (Slatkin 2009). We did not consider combining the data because the dosing schedules differed. Overall, we rated the quality of evidence on laxation, effect on analgesia, and adverse events to be low. See summary of findings Table 6.
Primary outcomes
Laxation response
Both trials measured laxation response within 24 hours of the first dose of methylnaltrexone. There was no clear difference between participants taking a higher dose to participants on a lower dose (RR 0.82, 95% CI 0.41 to 1.66 (Portenoy 2008); RR 1.07, 95% CI 0.81 to 1.42 (Slatkin 2009)). We judged the quality of evidence for laxation response within 24 hours as low. We downgraded the quality of evidence by two levels, because of study limitations. This was because of unclear risk of bias (reporting bias) and small sample sizes (high risk of bias).
One trial reported outcomes after dosing at day three and five (Portenoy 2008). Laxation response within 24 hours did not clearly differ in participants receiving 1 mg compared to participants receiving 5 mg or greater on day three or on day five (day three: RR 0.47, 95% CI 0.18 to 1.25; day five: RR 0.21, 95% CI 0.03 to 1.31). The other trial did not measure response beyond one day (Slatkin 2009). We judged the quality of evidence for laxation response at day three and five to be low. We downgraded the quality of evidence by two levels for study limitations. This was because of unclear risk of bias (reporting bias) and results from a single study with a small sample size (high risk of bias).
Effect on analgesia
In both trials, pain intensity did not change from baseline and was similar in both trial arms receiving the different doses of methylnaltrexone (Portenoy 2008; Slatkin 2009). In one trial, the mean change in pain from baseline to four‐hour evaluation was not clearly different (MD ‐0.51, 95% CI ‐1.49 to 0.47) (Slatkin 2009); the other trial did not provide full results. Both trials reported that the use of methylnaltrexone did not result in opioid withdrawal (Portenoy 2008; Slatkin 2009). In one trial, there was a no clear difference in the mean change from baseline to four‐hour evaluation (MD ‐0.04, 95% CI ‐0.73 to 0.65) (Slatkin 2009); the other trial did not provide full results. We judged the quality of evidence for effect on analgesia to be low. We downgraded the quality of evidence by two levels for study limitations. This was because of unclear risk of bias (reporting bias) and small sample sizes (high risk of bias).
Serious adverse events
In one trial, 15 participants experienced a serious adverse event; however, risk of serious adverse events were not reported per trial arm (Portenoy 2008). The events were lymphadenectomy, febrile neutropenia, depressed level of consciousness, suicide attempt, and delirium. All were considered unrelated to study medication. In the other trial, no serious adverse events occurred during the randomised trial phase, although during the open‐label phase three participants experienced such an event of which one had severe diarrhoea and subsequent dehydration and cardiovascular collapse (Slatkin 2009). These were considered to be related to the drug. We did not judge the quality of evidence for serious adverse events as they did not report incidence per trial arm.
Number and type of adverse events
In the fixed‐dose trial, all participants experienced an adverse event (Portenoy 2008). In the other trial, there was no clear difference in dose arms in the occurrence of an adverse event (RR 0.90, 95% CI 0.73 to 1.13) (Slatkin 2009). The most common adverse event in both trials and per trial arm was abdominal pain. We judged the quality of evidence for adverse events to be low. We downgraded the quality of evidence by two levels for study limitations because of unclear risk of bias (reporting bias) and small sample size (high risk of bias).
Secondary outcomes
Number of participants who dropped out due to adverse events
In one trial, one participant discontinued the trial (during the double‐blind treatment phase) because of an adverse event (Portenoy 2008). This was an 84‐year old man who withdrew due to syncope (taking methylnaltrexone 12.5 mg). The event was transient and resolved without sequelae; the investigators assessed that it was related to the medication. This trial also reported that in the open‐label phase, after receiving three doses, a 20‐year old man was withdrawn from the trial due to abdominal cramping that was considered as probably related to the trial medication. In the other trial, none of the participants discontinued because of an adverse event (Slatkin 2009).
Other measures of laxation response
In the single‐dose trial of 154 participants there was no clear difference in the proportion of participants who had a rescue‐free laxation within four hours of receiving methylnaltrexone between participants in the trial arm who received 0.15 mg/kg with participants who received 0.3 mg/kg (RR 1.06, 95% CI 0.77 to 1.46) (Slatkin 2009). In this trial, the proportion of participants who reported an improvement in constipation distress within four hours was similar (30/47 (64.4%) with methylnaltrexone 0.15 mg/kg versus 35/55 (63.5%) with methylnaltrexone 0.3 mg/kg), as was global improvement (27/47 (58.7%) with methylnaltrexone 0.15 mg/kg versus 32/55 (58.8%) with methylnaltrexone 0.3 mg/kg). In the fixed dose trial, 10% (1/10) of participants taking methylnaltrexone 1 mg had a laxation within four hours compared to 43% (3/7) taking methylnaltrexone 5 mg, 60% (6/10) taking methylnaltrexone 12.5 mg, and 33% (2/6) taking methylnaltrexone 20 mg (Portenoy 2008). Laxation response within four hours did not significantly differ in participants receiving 1 mg compared to participants receiving 5 mg or greater (RR 0.21, 95% CI 0.03 to 1.41).
One trial reported outcomes at four hours after dosing at day three and five (Portenoy 2008). Laxation response within four hours did not significantly differ in participants receiving 1 mg compared to participants receiving 5 mg or greater on day three or day five (day three: RR 0.34, 95% CI 0.10 to 1.22; day five: RR 0.09, 95% CI 0.01 to 1.38).
In one trial, the median time to rescue‐free laxation was 1.10 hours in the methylnaltrexone 0.15 mg/kg group and 0.8 hours in the methylnaltrexone 0.3 mg/kg group (Slatkin 2009). In the other trial, for the lowest dose of 1 mg it was more than 48 hours whereas for the higher doses the first laxation was at 1.72 with methylnaltrexone 5 mg, 0.48 with methylnaltrexone 12.5 mg, and 6.75 hours with methylnaltrexone 20 mg (Portenoy 2008).
One of the two‐week trials reported that there was a similar proportion of watery bowel movements in the methylnaltrexone arm compared with the placebo arm (16% (after 28 of 176 drug trial doses) with methylnaltrexone) versus 17% (after 8 out of 48 doses) with placebo) (Thomas 2008). In the single dose trial in participants who had a laxation within four hours of dosing, eight out of the 29 receiving methylnaltrexone had at least one watery laxation compared to none of the seven participants receiving placebo (Slatkin 2009).
Two trials (287 participants) measured self‐report of constipation distress (Slatkin 2009; Thomas 2008). In both trials, distress was reduced more at 24 hours in the methylnaltrexone group than in placebo group (combined analysis: RR 1.87, 95% CI 1.34 to 2.59; I² = 0%; Analysis 1.5).
One trial reported bowel status using the Clinical Global Impression of Change (Slatkin 2009). There was improvement at the end of the double‐blind phase for methylnaltrexone 0.15 mg/kg (58.7%) and methylnaltrexone 0.3 mg/kg (58.8%).
Relief of other constipation‐associated symptoms
None of the trials measured the relief of other constipation‐associated symptoms, although some of these symptoms, such as abdominal pain and nausea, were recorded as adverse events.
Use of rescue medication for laxation
One trial reported that participants in the trial arm receiving the lowest dose (methylnaltrexone 1 mg) required a rescue laxative approximately twice as often as those in the higher dose groups (Portenoy 2008). The other trial did not compare use of rescue medication (Slatkin 2009).
Quality of life, satisfaction with bowel movements, and participant preference
None of the trials assessed the impact of treatment on quality of life or participant preference (Slatkin 2009; Thomas 2008). One trial assessed participants' level of satisfaction with the trial medication using a 7‐point scale (Portenoy 2008). They did not report the level of satisfaction but reported no difference in satisfaction between the trial arms.
Discussion
Summary of main results
This review update sought to establish the effectiveness and safety of MOAs for OIBD in people with cancer and people receiving palliative care. Three of the eight RCTs explored outcomes in cancer populations irrespective of disease stage; one compared oral naldemedine to placebo, two compared oral prolonged released oxycodone/naloxone with oxycodone alone. Oral naloxone only compared to placebo was evaluated in people with advanced cancer. The other four trials compared either subcutaneous methylnaltrexone with placebo or different regimens of methylnaltrexone in palliative care populations in which the majority of participants had advanced cancer. All evaluated the effect of MOAs in populations where all or over 90% were on regular regimens of laxatives. Four trials were at a high risk of bias because of small sample sizes (fewer than 50 participants per trial arm), all were at an unclear risk of selection bias as they under reported allocation concealment or random sequence generation (or both), and seven were at an unclear risk of reporting bias as they did not provide a protocol. Data for all our primary outcomes of interest were only provided for methylnaltrexone compared to placebo. In one trial, only a subset of the sample was eligible for this review. We used the GRADE quality of evidence to assess primary outcomes. Our GRADE judgements differed by trial intervention; overall, it was moderate for naldemedine and methylnaltrexone. For naloxone as an adjunct, it was moderate, and naloxone taken on its own was very low. However, these judgements for naloxone were only for the effect on analgesia and adverse events, not laxation for which there were no data.
For naldemedine, we found moderate‐quality evidence that the drug clearly increased the number of spontaneous laxations over two weeks in people with cancer. There was also a dose response relationship identified with higher doses clearly increasing during this time the number of spontaneous laxations (moderate‐quality evidence). The higher doses were 0.2 mg and 0.4 mg daily compared with the dose of 0.1 mg daily. Evidence that naldemedine had no impact on analgesia in regards to opioid withdrawal symptoms was of moderate quality. There were five serious adverse events that occurred in participants taking naldemedine and none in the placebo group. We judged this as low‐quality evidence. We judged the quality of evidence that there was an increase in adverse events to be moderate. The most common adverse event was diarrhoea.
Evidence on naloxone was limited. Neither the trial of naloxone alone in people with advanced cancer or the two evaluating it in combination with oxycodone in people with cancer evaluated laxation response at 24 hours or over two weeks, and, for some evaluations (e.g. effect on analgesia), the studies did not provide full data. The trial of naloxone alone did not report adverse events.
There was moderate evidence from combined analysis that in people receiving palliative care methylnaltrexone improved laxation, with up to 59.1% of participants having a laxation within 24 hours of the first dose whereas in the placebo group it was up to 19.5% (RR 2.77, 95% CI 1.91 to 4.04; NNTB 3, 95% CI 2 to 3). One trial clearly demonstrated that fewer participants in the methylnaltrexone arm required rescue laxatives compared to placebo. There was also moderate evidence from a combined analysis on laxation response over two weeks, with up to 52.6% of participants having a rescue‐free laxation response (within four hours of at least four of the maximum seven doses), whereas in the placebo group it was up to 5.3% (RR 9.98, 95% CI 4.96 to 20.09; NNTB 2 (95% CI 2 to 3). We judged the evidence of the impact of methylnaltrexone on analgesia in regards to opioid withdrawal symptoms to be moderate and in regards to pain intensity to be low. There was moderate‐quality evidence that the drug did not increase the risk of a serious adverse event. There was low‐quality evidence that methylnaltrexone increased the risk of adverse events in regards to abdominal pain and flatulence. None of the adverse events were severe. However, in one trial, in a subsequent open‐study phase (when both the researchers and participants knew which treatment was being administered), one participant experienced severe diarrhoea and subsequent dehydration and cardiovascular collapse considered related to the drug.
We found low‐quality evidence that there was no impact of different doses of methylnaltrexone on laxation response, on pain analgesia, or adverse events. We were unable to judge the quality on risk of serious adverse events. However, it is important to highlight that during one of the trials, 15 participants experienced a serious adverse event. The investigators did not provide details on what the events were, or whether they were considered to be related to the trial drug. All participants experienced an adverse event in this trial.
Overall completeness and applicability of evidence
We sought trial evidence widely beyond published papers. Where available, we obtained regulatory documents; although these provided few new data.
Our review findings were limited. The trials were few and this limited our combined analyses. In some analyses, there was statistical heterogeneity across the trials. This related to adverse effects of methylnaltrexone in comparison to placebo. Sensitivity analyses demonstrated that the primary meta‐analyses were robust with regards to the risk of overall adverse events, and individual adverse events of abdominal pain, nausea, and vomiting. Omitting the trial at high risk of bias because of small sample size (Slatkin 2009) resulted in all analyses with a smaller estimate of effect but it did not change the direction of effect.
The evidence on naldemedine was only from one trial. We are aware of at least one other trial completed but not published in full; when it becomes available the evidence from this trial may inform better the evidence on this drug.
In the naloxone trials, not all our primary outcomes of interest were the focus of the research. The trial on naloxone only measured one of our three primary outcomes of interest, specifically the effect on analgesia, but not the effect on laxation or adverse events. However, both trials of naloxone in combination with oxycodone did measure laxation at later time points (four and five weeks). Here they found, across the studies, an improvement in bowel function in four out of five assessments.
The body of evidence could be argued as stronger for methylnaltrexone, as it was derived from more studies and with the total number of participants higher than that for trials on other MOAs. It is important to reflect that this is moderate‐quality evidence only, and in one of the four methylnaltrexone trials the assessment of impact of the drug may have been affected as participants in the placebo arm were on higher doses of opioids than those in the methylnaltrexone arm. Our analysis was more limited on methylnaltrexone dose response as we were unable to combine the studies because of different dosing schedules.
Participant outcomes on satisfaction and preference were under evaluated. Only one trial assessed quality of life (Ahmedzai 2012). The trial found no difference in quality of life between participants in the oxycodone/naloxone group and participants in the oxycodone only group. There are also other outcomes that were not measured in any of these trials that earlier research suggests need further exploration. This includes whether MOAs, in particular, methylnaltrexone, increase cancer survival (Janku 2015).
We found no completed trials that fulfilled our inclusion criteria on naloxegol, which in 2014 was approved by the FDA for use in OIBD in people without cancer. However, we are awaiting clarification for one trial from the authors or funders as we are unclear whether it fulfils our inclusion criteria (Webster 2013), and two potentially relevant trials are in progress (NCT02745353; NCT02839889).
Evaluations on the development of new MOAs for OIBD, their effectiveness, and safety is an active research field. We noted 11 trials in populations of people with cancer or people receiving palliative care (or both) that were in progress or were completed but published results were not yet available at the time of publishing this review (Dimitroulis 2014; JAPIC‐CTI‐132340; NCT00135577; NCT00331045/00101998; NCT01438567; NCT02321397; NCT02574819; NCT02745353; NCT02839889; Neefjes 2014; Peppin 2013).
Quality of the evidence
All eight trials were vulnerable to biases. Four were at high risk of bias as they involved a sample of fewer than 50 participants per trial arm. Using the GRADE approach, we assessed the quality of evidence for our primary outcomes of interest.
For both naldemedine and methylnaltrexone, the overall quality of evidence for improving laxation response (in comparison with placebo) was moderate. This means we are moderately confident in the effect estimate and the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. The evidence base to judge quality could be viewed as stronger with methylnaltrexone than naldemedine as it is based on several trials of in total 518 participants, rather than in naldemedine one trial of 225 participants. However, while data from 518 participants in the methylnaltrexone trials was used for adverse event assessments, in comparisons on laxation response data from 287 participants was used.
We judged the quality of evidence that the evaluated MOAs had no impact on the effect of analgesia in regards to naldemedine as moderate (for opioid withdrawal, they did not report pain intensity), in regards to methylnaltrexone low to moderate. The quality of evidence that the MOAs did increase the risk of serious adverse events was low for naldemedine, and moderate for methylnaltrexone. In regards to non‐serious adverse events, there was moderate‐quality evidence that naldemedine increased risk of adverse events and low‐quality evidence that methylnaltrexone did not increase the risk. The overall quality of evidence that there was no difference in our primary outcomes of interest between higher doses compared to lower doses of methylnaltrexone was low. The quality of evidence on naloxone (in comparison with placebo) was very low in regards to no effect on analgesia (adverse events and laxation responses were not reported). The quality of evidence on naloxone in combination with oxycodone was moderate, it had no impact on analgesia or on the risk of adverse events (early laxation responses were not reported).
Potential biases in the review process
We sought trial evidence widely, including five citation databases. We sought unpublished trial data from pharmaceutical and regulatory agencies databases. However, there are limited guidelines in how to seek unpublished data and searching regulatory agency websites is not straightforward.
We limited inclusion to trials that specified that their participants had cancer, or were in palliative care irrespective of disease stage. This is likely to have led to a loss of data, as trials we excluded may have included people with such characteristics, but the trial papers did not provide this 'finer' detail.
We included trials with methodological limitations. In addition, there is a potential problem due to carryover effects in the cross‐over designed trial (Sykes 1996), and our combined analysis was limited by the number of trials available.
Agreements and disagreements with other studies or reviews
This Cochrane systematic review specifically examined the evidence for MOAs for OIBD in cancer and palliative care populations. Our results on methylnaltrexone in palliative care were similar to those in the earlier relevant Cochrane Review that included two of the four trials on methylnaltrexone included in this review (Candy 2011).
There are reviews that have evaluated the effect of MOAs for OIBD in general populations, although no recent (published since 2014) Cochrane Review. One review of a broader population identified 14 trials (Ford 2013). In addition to four of the trials included in this review, they included trials on methadone‐induced constipation and trials involving participants receiving an opioid for chronic non‐malignant pain. In their meta‐analysis of 14 trials of 4101 participants they found the MOAs methylnaltrexone, naloxone, and alvimopan were superior to placebo for the treatment of opioid‐induced constipation. However, the numbers of adverse events were significantly more common. The reviews by Mehta 2016 and Siemens 2015 found positive results on laxation response of methylnaltrexone and other MOAs compared to placebo evaluated in general populations in treating opioid‐induced constipation.

Study flow diagram.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
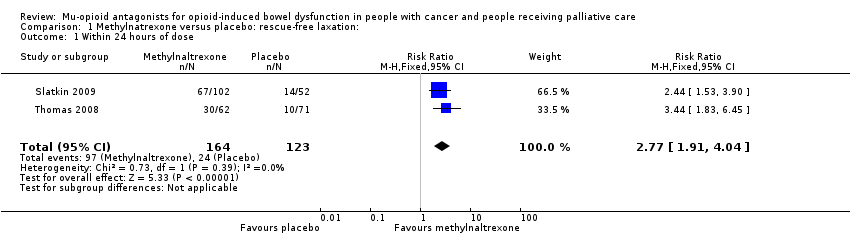
Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 1 Within 24 hours of dose.

Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 2 Within 4 hours after 4 of the 7 doses.
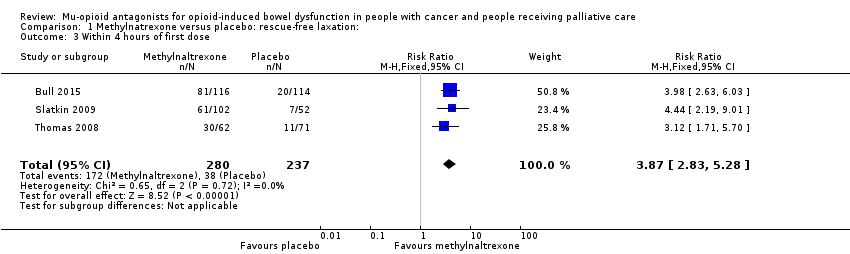
Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 3 Within 4 hours of first dose.

Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 4 Within 4 hours after 1 or 2 doses of the first 4 doses.

Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 5 Improvement in constipation distress at day 1.
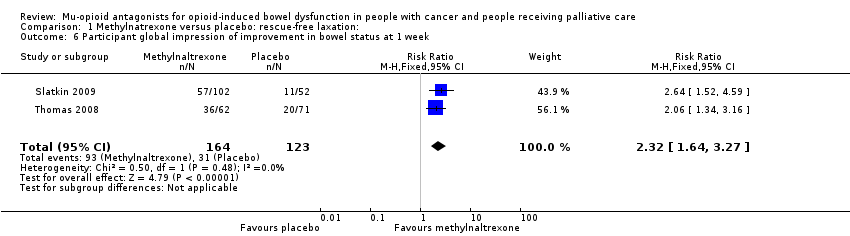
Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 6 Participant global impression of improvement in bowel status at 1 week.
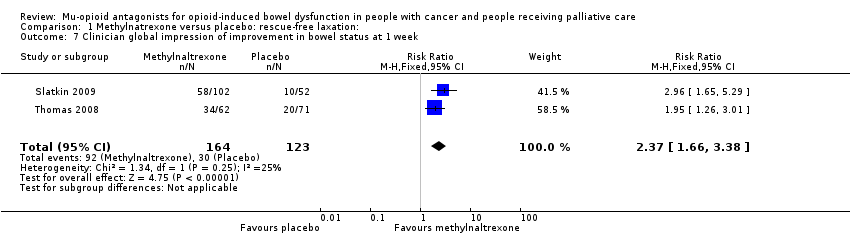
Comparison 1 Methylnatrexone versus placebo: rescue‐free laxation:, Outcome 7 Clinician global impression of improvement in bowel status at 1 week.

Comparison 2 Methylnaltrexone versus placebo: serious adverse event, Outcome 1 Serious adverse event.

Comparison 3 Methylnaltrexone versus placebo: adverse event, Outcome 1 Adverse events.

Comparison 3 Methylnaltrexone versus placebo: adverse event, Outcome 2 Dropouts due to adverse event.

Comparison 4 Oxycodone/naloxone prolonged‐release tablets versus oxycodone prolonged‐release: adverse event, Outcome 1 Adverse events.
| Naldemedine compared to placebo for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Settings: cancer care Intervention: naldemedine Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Naldemedine | |||||
| Laxation response within 24 hours of dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14a | 375 per 1000 | 724 per 1000 | RR 1.93 (1.36 to 2.74) NNTB 2.88 (2.04 to 4.92) | 225 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: opioid withdrawalc | — | — | 0.1 mg: MD ‐0.13 (‐0.57 to 0.31); 0.2 mg: MD ‐0.40 (‐0.87 to 0.07); 0.4 mg: MD ‐0.02 (‐0.45 to 0.41) | 225 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: pain intensity | — | — | — | — | — | Not reported |
| Serious adverse eventsa | — | — | 5 SAEs occurred, all in naldemedine group. | 225 (1 study) | ⊕⊕⊝⊝ | — |
| Adverse eventsa | 518 per 1000 | 704 per 1000 (539 to 927) | RR 1.36 (1.04 to 1.79) | 225 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; NNTB: number needed to treat for an additional beneficial outcome; RR: risk ratio; SAE: serious adverse events. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by clinician or self‐report and in the case of adverse events using severity grades according to the Common Terminology Criteria for Adverse Events. | ||||||
| Lower‐dose naldemedine compared to higher‐dose naldemedine for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Setting: cancer care Intervention: lower dose naldemedine 0.1 mg daily Comparison: higher dose naldemedine 0.2 mg or 0.4 mg daily | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Higher dose 0.2 mg/0.4 mg daily | Lower dose 0.1 mg daily | |||||
| Laxation response within 24 hours of dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14a | 0.1 mg vs 0.2 mg: 776 per 1000 0.1 mg vs 0.4 mg: 821 per 1000 | 0.1 mg vs 0.2 mg: 564 per 1000 (430 to 739) 0.1 mg vs 0.4 mg: 564 per 1000 (433 to 733) | 0.1 mg vs 0.2 mg: RR 0.73 (0.55 to 0.95) 0.1 mg vs 0.4 mg: RR 0.69 (0.53 to 0.89) | 226 (1 study) 0.1 mg vs 0.2 mg: n = 113 0.1 mg vs 0.4 mg: n = 111 | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: opioid withdrawal | — | — | — | — | — | Not reported |
| Effect on analgesia: pain intensity | — | — | — | — | — | Not reported |
| Serious adverse events | — | — | — | — | — | Not reported |
| Adverse events | — | — | — | — | — | Not reported |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by self‐report. | ||||||
| Naloxone compared with placebo for cancer and people receiving palliative care with opioid‐induced bowel dysfunction | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Settings: cancer care Intervention: naloxone Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Naloxone | |||||
| Laxation response within 24 hours of a dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14 | — | — | — | — | — | Not reported |
| Effect on analgesia: opioid withdrawal | — | — | — | — | — | Not reported |
| Effect on analgesia: pain intensitya | — | — | No statistical difference in pain experienced when taking placebo or naloxone. Full data, including pre‐cross‐over results, were not provided. | 17 (1 study) | ⊕⊝⊝⊝ | — |
| Serious adverse events | — | — | — | — | — | Not reported |
| Adverse events | — | — | — | — | — | Not reported |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured using 4‐point scale (0 = no pain, 3 = severe pain). | ||||||
| Oxycodone/naloxone prolonged release tablets compared with oxycodone prolonged‐released tablets for opioid‐induced bowel dysfunction | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Settings: cancer care Intervention: oxycodone/naloxone prolonged‐release tablets Comparison: oxycodone prolonged‐released tablets | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Oxycodone | Oxycodone/naloxone | |||||
| Laxation response within 24 hours of dose | — | — | — | — | — | Not reported |
| Laxation response between day 1 and day 14 | — | — | — | — | — | Not reported |
| Effect on analgesia: opioid withdrawalc | — | — | Intervention group: mean 6.64 (SD 5.97) comparison group: mean 7.29 (SD 4.59) at 7 days | 184 (1 study) | ⊕⊕⊕⊝ Moderateb | — |
| Effect on analgesia: pain intensitya | — | — | Intervention group: mean 3.50 (SD 1.88) and comparison group: mean 3.52 (SD 1.80) at 4 weeks | 184 (1 study) | ⊕⊕⊕⊝ Moderateb | Another study, Dupoiron 2017 also found outcome to be similar between trial arms, but did not provide any data. |
| Serious adverse events | 43 per 1000 | 87 per 1000 (27 to 279) | RR 2.00 (95% CI 0.62 to 6.41) | 184 (1 study) | ⊕⊕⊝⊝ Lowb,d | — |
| Adverse events | 754 per 1000 | 815 per 1000 (709 to 935) | RR 1.08 (95% CI 0.94 to 1.24) | 234 (2 studies) | ⊕⊕⊕⊝ Moderateb | — |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; SD: standard deviation. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured using the Brief Pain Inventory‐Short Form. | ||||||
| Methylnaltrexone compared to placebo for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Setting: palliative care Intervention: methylnaltrexone Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Risk with placebo | Risk with methylnaltrexone | |||||
| Laxation response within 24 hours of dosea | 195 per 1000 | 568 per 1000 | RR 2.77 (1.91 to 4.04) | 287 | ⊕⊕⊕⊝ Moderateb | — |
| Laxation response between day 1 and day 14 (specifically within 4 hours after 4 or more of the 7 doses)a | 52 per 1000 | 517 per 1000 | RR 9.98 (4.96 to 20.09) | 305 | ⊕⊕⊕⊝ Moderate b,c | — |
| Effect on analgesia: opioid withdrawald | Study 1: day 1: MD 0.00 (‐0.46 to 0.46); day 14: MD 0.10 (‐0.63 to 0.83) Study 2: median change to day 2 = 0 in both trials arms | 236 (2 studies) | ⊕⊕⊕⊝ Moderateb | ‐ | ||
| Effect on analgesia: pain intensitye | Study 1: at 4 hours (methylnaltrexone 0.15 mg/kg: MD ‐0.76 (‐1.47 to 0.05); methylnaltrexone 0.3 mg/kg: MD ‐0.25 (‐0.91 to 0.41) Study 2: at day 1 and 14 (day 1: MD 0.20 (‐0.62 to 1.02); day 14: MD ‐0.70 (‐1.52 to 0.12) | 287 (2 studies) | ⊕⊕⊝⊝ Lowb,f | Another study, Bull 2015, found similar pain intensity experienced in trial arms, full data not provided. | ||
| Serious adverse events | 238 per 1000 | 142 per 1000 | RR 0.59 (0.38 to 0.93) | 364 | ⊕⊕⊕⊝ Moderateb | — |
| Adverse events | 700 per 1000 | 815 per 1000 | RR 1.17 (CI 0.94 to 1.45) | 518 | ⊕⊕⊝⊝ Lowb,g | Heterogeneity was substantial (74%). It was explained in sensitivity analysis by omitting the trial at a high risk of bias because of small sizes. The effect estimate was reduced. The direction of effect not changed. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by self‐report or clinician report. dMeasured using the modified Himmelsbach Opioid Withdrawal Scale. | ||||||
| Lower dose methylnaltrexone compared to higher dose for opioid‐induced bowel dysfunction in cancer and people receiving palliative care | ||||||
| Patient or population: people with cancer and people receiving palliative care with opioid‐induced bowel dysfunction Setting: palliative care Intervention 1: lower‐dose methylnaltrexone (study 1: 3 doses, 1 week, 1 mg; study 2: 1 dose, 0.15 mg/kg) Intervention 2: higher‐dose methylnaltrexone (study 1: 3 doses, 1 week, 5‐12.5 mg; study 2: 1 dose, 0.30 mg/kg) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Higher dose | Lower dose | |||||
| Laxation response within 24 hours of first dosea | Study 1: 609 per 1000 Study 2: 639 per 1000 | Study 1: 499 per 1000 (250 to 100) Study 2: 681 per 1000 (515 to 904) | Study 1: RR 0.82 (0.41 to 1.66) Study 2: RR 1.07 (0.81 to 1.42) | 135 (2 studies) Study 1: n = 33 Study 2: n = 102 | ⊕⊕⊝⊝ Lowb | Unable to combine study data as methylnaltrexone low and higher doses differed per trial |
| Laxation responsea | At 3 days: 706 per 1000 | At 3 days: 332 per 1000 | At 3 days: RR 0.47 (0.18 to 1.25) | 33 participants (1 study) | ⊕⊕⊝⊝ Lowb | Unable to combine study data as methylnaltrexone low and higher doses differed per trial |
| At 5 days: 688 per 1000 | At 5 days: 144 per 1000 | At 3 days: RR 0.21 (0.03 to 1.31) | ||||
| Effect on analgesia: opioid withdrawalc | — | — | MD ‐0.04 (‐0.73 to 0.65) | 102 participants (1 study) | ⊕⊕⊝⊝ Lowb | Another study,Portenoy 2008, also found outcome to be similar between trial arms, but did not provide any data |
| Effect on analgesia: pain intensityd | — | — | MD ‐0.51 (‐1.49 to 0.47) | 102 participants (1 study) | ⊕⊕⊝⊝ Lowb | Another study, Portenoy 2008, also found outcome to be similar between trial arms, but did not provide any data |
| Serious adverse event | — | — | — | — | Not reported | |
| Adverse event | Study 1: 1000 per 1000 Study 2: 800 per 1000 | Study 1: 1000 per 1000 (1000 to 1000) Study 2: 723 per 1000 (580 to 902) | Study 1: RR 1.00 (1.00 to 1.00) Study 2: RR 0.90 (0.73 to 1.13) | 135 (2 studies) Study 1: n = 33 Study 2: n = 102 | ⊕⊕⊝⊝ Lowb | Unable to combine study data as methylnaltrexone low and higher doses differed per trial |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MD: mean difference; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence | ||||||
| aMeasured by clinician or self‐report. bDowngraded by two levels for study limitations: one for unclear risk of bias (reporting bias) and one for small sample size (high risk of bias). cMeasured using the modified Himmelsbach Opioid Withdrawal Scale. dMeasured by participant‐rated scale 0‐10. | ||||||
| Adverse event | Naldemedine (%) | Placebo (%) |
| Diarrhoea | 67 (39) | 14 (25) |
| Decreased WBC count | 9 (5) | 3 (5) |
| Abdominal pain | 6 (4) | 0 (0) |
| Vomiting | 5 (3) | 0 (0) |
| Bone marrow failure | 3 (2) | 2 (4) |
| Decreased appetite | 6 (4) | 1 (2) |
| Nasopharyngitis | 4 (2) | 1 (2) |
| Nausea | 4 (2) | 4 (7) |
| Rash | 3 (2) | 2 (4) |
| Decreased platelet count | 3 (2) | 0 (0) |
| Decreased total protein | 7 (4) | 1 (2) |
| Glucose in urine | 4 (2) | 1 (2) |
| Abnormal haematology test | 2 (1) | 0 (0) |
| Decreased RBC count | 4 (2) | 0 (0) |
| Hypertension | 2 (1) | 0 (0) |
| Increased blood alkaline phosphatase | 4 (2) | 1 (2) |
| Increased blood lactate dehydrogenase | 2 (1) | 1 (2) |
| Increased blood pressure | 2 (1) | 0 (0) |
| Increased blood urea | 4 (2) | 1 (2) |
| Increased WBC count | 1 (2) | 2 (4) |
| Protein present in urine | 5 (3) | 0 (0) |
| Upper abdominal pain | 3 (2) | 1 (2) |
| RBC: red blood cell; WBC: white blood cell. All comparisons were not statistically significant. | ||
| Methylnaltrexone vs placeboa | |
| AEs | RR 1.07, 95% CI 0.96 to 1.19 |
| AE of abdominal pain | RR 2.15, 95% CI 1.28 to 3.62 |
| AE of nausea | RR 0.87, 95% CI 0.46 to 1.65 |
| AE of vomiting | RR 0.70, 95% CI 0.33 to 1.47 |
| aomitting trial of high risk of bias. AE: adverse event; CI: confidence intervals; RR: risk ratio. | |
| Adverse event | RR (95% CI) | I²statistic on heterogeneity |
| Abdominal pain | 2.39 (1.07 to 5.34) | 65% |
| Diarrhoea | 1.02 (0.93 to 1.11) | 51% |
| Dizziness | 4.09 (0.99 to 16.83) | 0% |
| Falls | 1.02 (0.89 to 1.16) | 84% |
| Flatulence | 2.09 (1.07 to 4.08) | 0% |
| Nausea | 0.97 (0.89 to 1.06) | 63% |
| Peripheral oedema | 1.01 (0.50 to 2.03) | 0% |
| Restlessness | 0.83 (0.32 to 2.12) | 0% |
| Somnolence | 1.00 (0.93 to 1.08) | 73% |
| Vomiting | 0.99 (0.92 to 1.08) | 67% |
| CI: confidence interval; RR: risk ratio. | ||
| Adverse event | Methylnaltrexone (%) | Placebo (%) |
| Abdominal distensiona | 1 (2) | 6 (8) |
| Abdominal tendernessa | 1 (2) | 4 (6) |
| Astheniaa | 4 (6) | 4 (6) |
| Anxietyb | 5 (4.9) | 0 (0) |
| Arthralgiab | 3 (2.9) | 1 (1.9) |
| Back painc | 9 (7.8) | 3 (2.9) |
| Confusional statec | 7 (6.0) | 9 (7.9) |
| Dehydrationa | 2 (3) | 4 (6) |
| Fatigueb | 4 (3.9) | 1 (1.9) |
| Hypotensiona | 0 (0) | 4 (6) |
| Increased body temperaturea | 5 (8) | 2 (3) |
| Lethergya | 4 (6) | 4 (6) |
| Malignant‐neoplasm progressiona | 7 (11) | 9 (13) |
| Pain exacerbationb | 8 (8) | 2 (4) |
| Rhinorrhoeab | 6 (5.9) | 1 (1) |
| Sweating increasedb | 8 (7.8) | 4 (7.7) |
| Tachycardiaa | 1 (1) | 4 (6) |
| aReported in trial by Thomas 2008. bReported in trial by Slatkin 2009. cReported in trial by Bull 2015. | ||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Within 24 hours of dose Show forest plot | 2 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.77 [1.91, 4.04] |
| 2 Within 4 hours after 4 of the 7 doses Show forest plot | 2 | 305 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.98 [4.96, 20.09] |
| 3 Within 4 hours of first dose Show forest plot | 3 | 517 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.87 [2.83, 5.28] |
| 4 Within 4 hours after 1 or 2 doses of the first 4 doses Show forest plot | 2 | 363 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.89 [4.46, 10.66] |
| 5 Improvement in constipation distress at day 1 Show forest plot | 2 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.87 [1.34, 2.59] |
| 6 Participant global impression of improvement in bowel status at 1 week Show forest plot | 2 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.32 [1.64, 3.27] |
| 7 Clinician global impression of improvement in bowel status at 1 week Show forest plot | 2 | 287 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.37 [1.66, 3.38] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Serious adverse event Show forest plot | 2 | 364 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.38, 0.93] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse events Show forest plot | 3 | 518 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.94, 1.45] |
| 2 Dropouts due to adverse event Show forest plot | 2 | 363 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.54, 2.76] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adverse events Show forest plot | 2 | 234 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.94, 1.24] |