Antihistamínicos H1 para la urticaria espontánea crónica
Referencias
Referencias de los estudios incluidos en esta revisión
Referencias de los estudios excluidos de esta revisión
Referencias de los estudios en espera de evaluación
Referencias de los estudios en curso
Referencias adicionales
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Design: randomised double‐blind 2‐arm parallel‐group study of cetirizine hydrochloride vs placebo Duration: 15 days | |
| Participants | Number randomly assigned: 30 participants Sex: 44% male, 56% female Age of participants, years: 21 to 64 Unit of allocation: participant Country and setting: Spain; secondary care, hospital clinic Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (15 days) Length of follow‐up: 15 days | |
| Outcomes | Timing of outcome assessment: baseline and 15 days Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and clinician | |
| Notes | In Spanish with English abstract Investigators concluded that cetirizine was more active than placebo in terms of clinician reports of efficacy; findings were not statistically significantly different | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as random allocation, no details given |
| Allocation concealment (selection bias) | Unclear risk | Not described in published report |
| Blinding of participants and personnel (performance bias) | Unclear risk | Stated to be double‐blind, details of blinding not stated |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) | Unclear risk | 30 randomly assigned: 15 intervention, 15 control. 13/15 completed intervention, 12/15 control. 5 from each group experienced adverse effects, but it is unclear whether they withdrew from the study. Reasons for dropout not stated |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Low risk | None detected. Funder: not stated |
| Methods | Design: randomised open comparative clinical study of loratadine vs levocetirizine Duration: 4 weeks | |
| Participants | Number randomly assigned: 60 (loratadine n = 30; levocetirizine n = 30) Sex: 40% male, 60% female; in loratadine group, 43.3% male; in levocetirizine group, 56.7% female Age of participants, years: 12 to 60 (mean age 33.4 and 34.8 in loratadine and levocetirizine groups, respectively) Unit of allocation: participant Country and setting: India; secondary, outpatient Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: 4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician and participant | |
| Notes | Study authors conclude that this safety and efficacy study proves the superiority of levocetirizine over loratadine for CSU | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Unclear (described as quote from ‘Subjects’ section of report of study as ‘systematic randomisation’) |
| Allocation concealment (selection bias) | High risk | Not stated, but trial described as 'open' |
| Blinding of participants and personnel (performance bias) | High risk | No blinding in this open study |
| Blinding of outcome assessment (detection bias) | High risk | No blinding in this open study |
| Incomplete outcome data (attrition bias) | Low risk | 51/60 completed. Six participants did not report for follow‐up (no reasons given), and 3 participants were non‐compliant with treatment |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Low risk | None detected. Funder: none (drugs free of charge from hospital pharmacy) |
| Methods | Design: randomised double‐blind 2‐arm parallel‐group trial of oxatomide vs clemastine Duration: 6 weeks | |
| Participants | Number randomly assigned: 30 participants (15 in each group) Sex: 43% (13) male, 57% (17) female Age of participants, years: between 15 and 67 Unit of allocation: participant Country and setting: Denmark; setting unclear Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Doses could be increased to 4 capsules daily. Cinnarizine 5 mg every 4 hours could be added if insufficient efficacy in either group Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: 6 weeks (duration of study) | |
| Outcomes | Timing of outcome assessment: 1, 3 and 6 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participants and clinician | |
| Notes | Study investigators concluded that the effect of oxatomide was equal to that of clemastine | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 50): "the 30 patients were randomly assigned to a 6 weeks double‐blind treatment...'' Unclear which method of randomisation was used |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote (page 50): "double‐blind treatment'' Method used not described, no further information available |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote (page 50): "double‐blind treatment'' Method used not described, no further information available |
| Incomplete outcome data (attrition bias) | High risk | 0/30 dropped out |
| Selective reporting (reporting bias) | High risk | No participant numbers given in results, only statistical differences (and percentages for adverse events). No figures given (graph only) Severity of weals, erythema and itching noted but not mentioned in the results. No figures given, graph only. No further information available |
| Other bias | Unclear risk | No clear definition of disease given; washout period not specified; concomitant treatment permitted Funder: not stated |
| Methods | Design: randomised double‐blind 3‐arm parallel‐group multi‐centre study of loratadine vs terfenadine vs placebo Duration: 28 days | |
| Participants | Number of participants randomly assigned: 187 (61 in loratadine group; 64 in terfenadine group; 62 in placebo group) Sex: 46% male, 53% female. Number of male/female: 32/27 loratadine; 36/24 terfenadine; 20/32 placebo Age of participants, years: average 37 Unit of allocation: participant Country and setting: France, Belgium, Germany; setting unclear, included private practice and dermatology clinics Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Loratadine 10 mg (active drug in the morning and placebo in the evening), 60 mg terfenadine twice daily or placebo twice daily for 28 days Duration of intervention: intermediate‐term (28 days) Length of follow‐up: 28 days | |
| Outcomes | Timing of outcome assessment: participants seen at baseline (day 1), then at days 7, 14 and 28 Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator report | |
| Notes | Study investigators concluded that loratadine 10 mg once daily is safe and effective | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ''Qualified patients were randomly assigned...'' Method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated, unclear whether allocation was concealed |
| Blinding of participants and personnel (performance bias) | Unclear risk | Stated to be a double‐blind study, method of blinding not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Stated to be a double‐blind study, method of blinding not described |
| Incomplete outcome data (attrition bias) | High risk | No ITT. 15/187 dropouts due to protocol violation (no details given): 1/61 loratadine; 4/64 terfenadine; 52/62 placebo dropped out |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Low risk | Groups were comparable at baseline Funder: not stated |
| Methods | Design: double‐blind multi‐centre 3‐arm randomised trial of cetirizine vs astemizole vs placebo Duration: 4 weeks | |
| Participants | Number randomly assigned: 187 (62 patients in cetirizine group; 62 in astemizole group; 63 in placebo group) Sex: 27% male, 73% female Age of participants, years: > 12, average 37.7 Unit of allocation: participant Country and setting: USA; university medical centres
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: 1, 2, 3 and 4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and investigator | |
| Notes | Study investigators concluded that cetirizine provides effective relief of symptoms | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 192): "...patients were randomly assigned to receive either 10 mg cetirizine, 10 mg astemizole, or placebo once each night for 4 weeks." No further details given |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Unclear; stated to be ''double‐blind trial'' (page 192) but no details given |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unclear; stated to be ''double‐blind trial'' (page 192) but no details given |
| Incomplete outcome data (attrition bias) | High risk | 51/187 randomly assigned participants dropped out/lost to follow‐up; lost to follow‐up were 51 participants (27.3%). 43 participants were withdrawn before trial completion; 1 failed to take astemizole; 7 were lost to follow‐up Serious adverse events (requiring drug withdrawal): cetirizine: n = 2 (headache n = 1; vasovagal/vomiting/palpitations n = 1); astemizole: n = 1 (dizziness, nausea, lethargy, syncope). Evaluable participants: n = 136 Comment: high loss to follow‐up |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Unclear risk | Severity of urticaria comparable at baseline, but statistical differences between other demographic details (age and race) Funder: Pfizer Labs |
| Methods | Design: randomised double‐blind placebo‐controlled multi‐centred 3‐arm study of cetirizine vs hydroxyzine vs placebo Duration: 4 weeks | |
| Participants | Number randomly assigned: 188 (60 in cetirizine group; 63 in hydroxyzine group; 65 in placebo group) Sex: 32% male, 68% female Age of participants, years: > 12; mean: cetirizine: 36.8; hydroxyzine: 34.5; placebo: 38.8 Unit of allocation: participant Country and setting: USA; allergy practice settings Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: 4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and investigator | |
| Notes | Study investigators concluded that cetirizine 10 mg was equivalent to hydroxyzine 25 mg in symptom control | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1076) "randomised, parallel‐group..." but no further details |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐dummy used, but blinding not fully described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unclear how blinding of outcome assessors was achieved |
| Incomplete outcome data (attrition bias) | Low risk | 9/188 dropouts were recorded for each group as the result of serious adverse events requiring withdrawal of drug (1/60 in cetirizine group; 4/63 in hydroxyzine group; 4/65 in placebo group) Cetirizine: somnolence (n = 1) Hydroxyzine: somnolence (n = 4) Placebo: (n = 4) consisted of somnolence n = 1; sweating, vertigo and vomiting (n = 1); lethargy n = 1; headache n = 1 Dropouts balanced between groups |
| Selective reporting (reporting bias) | High risk | Reporting of results in graph form (means with statistical significance) only |
| Other bias | Unclear risk | No power calculation—may have missed significant differences between groups if underpowered Definition of disease partially defined, with physical urticaria not explicitly excluded Funder: Pfizer Laboratories |
| Methods | Design: multi‐centre double‐blind randomised parallel‐group study comparing desloratadine 5 mg vs placebo Duration: 6 weeks | |
| Participants | Number of participants randomly assigned: 225 (115 in desloratadine group and 110 in placebo group) Sex: not stated Age of participants: not stated Unit of allocation: participant Country and setting: USA; setting unclear Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose duration
Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: 6 weeks | |
| Outcomes | Timing of outcome assessment: twice daily for 6 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: unclear | |
| Notes | Study investigators concluded that desloratadine produced substantial efficacy after just 1 dose, which was maintained throughout study. All measures were statistically significant in favour of desloratadine vs placebo and were sustained at all time points | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Stated to be double‐blind, unclear how this was done |
| Blinding of outcome assessment (detection bias) | Unclear risk | Stated to be double‐blind, unclear how this was done |
| Incomplete outcome data (attrition bias) | Unclear risk | No mention of dropouts/adverse events |
| Selective reporting (reporting bias) | High risk | Pruritus score given for days 1 to 8, then total symptoms score given for days 2 to 8 |
| Other bias | Unclear risk | None detected. Short report (abstract). Funder: not stated |
| Methods | Design: randomised double‐blind multi‐centre 2‐arm trial of mizolastine vs placebo Duration: 28 days | |
| Participants | Number of participants randomly assigned: 56; 28 in each group Sex: 55% male, 45% female Age of participants, years: 18; mean 38 ± 15 Unit of allocation: participant Country and setting: UK; setting research clinics Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration After single‐blind placebo run‐in period of 4 to 10 days:
Duration of intervention: intermediate‐term (28 days) | |
| Outcomes | Timing of outcome assessment: days 7 and 28 Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician and participant | |
| Notes | Study authors concluded that mizolastine controlled symptoms of urticaria | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 321): ''a two‐centre, double‐blind randomised, placebo‐controlled parallel group study... allocated according to the randomisation" |
| Allocation concealment (selection bias) | Unclear risk | No details given about allocation concealment |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote (page 321): "Patients received single blind placebo medication for a variable period of 4‐10 days (initially, then were allocated to one of two treatment groups)" Quote (page 321): "All tablets were identical in appearance, ensuring double blind nature of trial." Unclear how investigators were blinded to treatment |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unclear how investigators were blinded to treatment Comment: participants probably blind, as all tablets were identical |
| Incomplete outcome data (attrition bias) | High risk | 29/56, 51% losses to follow‐up (29/56 with 10/28 in mizolastine arm and 19/28 in placebo arm). A large proportion of participants dropped out; this is unbalanced across trial arms 1 participant in mizolastine group did not take treatment Lack of efficacy in 5 in mizolastine group and in 17 in placebo group Drowsiness in 1 in mizolastine group Loss to follow‐up at day 7 in 2 mizolastine group 1 participant in each group "unco‐operative" 1 in each group discontinued for reasons unrelated to study Analysis in the paper is presented as ITT |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Unclear risk | Funder: Synthelabo Key outcome based on physician VAS estimate of urticaria severity (i.e. totally subjective); no indication of how many participants were cleared on treatment |
| Methods | Design: double‐blind cross‐over 3‐arm randomised controlled trial of cimetidine and chlorpheniramine vs placebo Duration: 2 weeks | |
| Participants | Number of participants randomly assigned: 25 entered study. Numbers in each group not stated Sex: 32% male, 68% female Age of participants, years: 18 to 66 Unit of allocation: cross‐over, without washout (consecutive 2‐week treatments) Country and setting: UK; outpatient clinic Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: short‐term (2 weeks) | |
| Outcomes | Timing of outcome assessment: 2 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician and participant | |
| Notes | Study investigators concluded that chlorpheniramine is effective in controlling symptoms in some patients with urticaria | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not stated. Quote: ''On entry to the trial patients were allocated, on a random double blind basis, to the consecutive 2‐week treatment'' Cross‐over study with no apparent washout |
| Allocation concealment (selection bias) | Unclear risk | No allocation concealment stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Study stated double‐blinding; method unclear, no indication whether identical tablets/capsules given, no details about methods of blinding. Dosages were different for each intervention, so blinding incomplete (intervention group could potentially be guessed by number of tablets) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Study stated double‐blinding; method unclear |
| Incomplete outcome data (attrition bias) | High risk | 6/25 participants recruited (24%) were lost to follow‐up (non‐compliance 5; spontaneous remission 1) and were not included in the analysis. Unclear which group dropouts belonged to. No ITT |
| Selective reporting (reporting bias) | High risk | Results not clearly reported. Results for numbers of participants in each group not stated, only mean scores (no SD) provided |
| Other bias | Unclear risk | Unclear schedule of assignment, unclear whether each phase was given consecutively, as results reported only for intervention and control—not by phase of the study Funder: Smith Kline and French Laboratories Ltd |
| Methods | Design: double‐blind 2‐arm randomised controlled trial of cetirizine vs rupatadine Duration: 6 weeks | |
| Participants | Number of participants randomly assigned: 70 Sex: cetirizine females 64.5%, rupatadine females 61.2% Age of participants, years: cetirizine 41.5 (SD 11.49); rupatadine 43.81 (SD 12.30) Unit of allocation: participant Country and setting: India; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
Previous unresponsiveness to antihistamine: excluded any with previous failure to respond to antihistamine | |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate Length of follow‐up: 6 weeks | |
| Outcomes | Timing of outcome assessment: 6 weeks Study outcomes Primary outcomes of the trial Trial was undertaken to test whether treatment with rupatadine was more successful than treatment with cetirizine in resolving symptoms as follows:
Secondary outcomes of the trial
Adverse events: general clinical follow‐up and monitoring of adverse events, no serious adverse events requiring withdrawal of treatment; cetirizine group: total affected 12, 38.71% (headache n = 2, gastric irritation n = 1, dry mouth n = 1, sedation n = 8). Rupatadine group: total affected 7, 21.21% (headache n = 2, dry mouth n = 1, sedation n = 4) Quality of life measures: none Clinician or participant report: participant and clinician | |
| Notes | Study investigators concluded that rupatadine led to improvement in all outcomes by the end of the trial, and that rupatadine is a particularly attractive therapeutic modality compared with cetirizine for the treatment of CSU | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block randomisation, with block sizes of 4 in equal proportions to ensure a uniform allocation ratio. Randomised treatment allocation sequence was generated by a statistician using a random numbers table |
| Allocation concealment (selection bias) | Low risk | Quote (page 644): "The codes used in this random allocation sequence were retained in a sealed envelope, which was opened only after the completion of the study" |
| Blinding of participants and personnel (performance bias) | Low risk | Both participants and investigators were unaware of the treatment administered. Drugs (21 tablets of cetirizine or rupatadine) were handed over in identical plastic containers to a third person, who was not directly involved in this study. Drugs were presented in identical format in terms of shape, size, texture and packing |
| Blinding of outcome assessment (detection bias) | Low risk | Both participants and investigators were unaware of the treatment administered |
| Incomplete outcome data (attrition bias) | Unclear risk | 64/70 completed. 5 participants were lost to follow‐up at end of first week of the study (3 in cetirizine group and 2 in rupatadine group). One from cetirizine group was dropped from the study and was shifted to another drug because of non‐response. Data for these 6 participants were not integrated into the analysis |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Low risk | None detected. Funder: none |
| Methods | Design: double‐blind 2‐arm randomised controlled trial of fexofenadine vs placebo Duration: 21 days | |
| Participants | Number of participants randomly assigned: 21 (further information obtained from trial investigator); 13 evaluable participants with 6 in fexofenadine group and 7 in placebo group Sex: 38% female Age of participants, years: 38 Unit of allocation: participant Country and setting: Switzerland; hospital allergy clinics Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (21 days) Length of follow‐up: 21 days | |
| Outcomes | Timing of outcome assessment: 21 days Primary outcomes of the trial Assessment scores used were as follows:
Secondary outcomes of the trial
Clinician or participant report: clinician and participant | |
| Notes | Study investigators concluded that fexofenadine had a beneficial effect on urticaria Main report language is German | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Performed by pharmacist who was not involved in the study Comment: Although randomisation sequence was generated offsite, adequacy of sequence generation is unclear, as no further information was provided by trial investigator |
| Allocation concealment (selection bias) | Unclear risk | No details about allocation concealment available Comment: Although only allocation number was visible on sealed medication boxes, allocation concealment up to the point of assignment of the intervention is unclear |
| Blinding of participants and personnel (performance bias) | Low risk | Study drugs and placebo were identical in appearance Comment: Further information from trial investigators stated that identical small and white tablet boxes were sealed with a plastic band (only allocation number could identify the participant) |
| Blinding of outcome assessment (detection bias) | Low risk | Study drugs and placebo were identical in appearance Comment: Further information from trial investigators states that identical small and white tablet boxes were sealed with a plastic band (only allocation number could identify the participant). Outcome assessors were unaware of treatment allocation |
| Incomplete outcome data (attrition bias) | Low risk | 18 commenced active study (Phase II): 3 dropped out from the fexofenadine group (1 lack of efficacy, 1 worsening of condition, 1 moved house and lost contact). No participants dropped out from the placebo group (i.e. 15/18 completed) (further information supplied by trial investigator) |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes were reported, but results were presented in graph format or as percentages; number of once‐daily treated participants in each group is unclear, therefore calculations may be prone to error |
| Other bias | Unclear risk | Funder: Aventis Odd and unclear trial design by which all participants were given active drug as run‐in, then were randomly assigned again to fexofenadine or placebo |
| Methods | Design: randomised parallel‐group 4‐arm study conducted to compare desloratadine vs montelukast vs desloratadine plus montelukast vs placebo Duration: 6 weeks | |
| Participants | Number of participants randomly assigned: 160 with 40 in each group Sex: 31% male, 69% female Age of participants, years: 18 to 69; mean 43.9 (SD 13.4) Unit of allocation: participant Country and setting: Italy; outpatient clinics of university hospitals Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Rescue therapy: loratadine 10 mg allowed (frequency and duration unclear) Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: 8 weeks i.e. follow‐up extended after cessation of treatment. | |
| Outcomes | Timing of outcome assessment: baseline, after 3 weeks of treatment, after 6 weeks, follow‐up at 8 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician and participant | |
| Notes | Study investigators concluded that on average, desloratadine and desloratadine plus montelukast appear to be more effective than placebo or montelukast alone | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Method of randomisation not stated in publication. Further information from trial investigator: "We used the StatsDirect software for the list of randomised patients" |
| Allocation concealment (selection bias) | Unclear risk | No details given about allocation concealment; sealed envelopes were used and were opened after the study had ended. Further details supplied by study investigator |
| Blinding of participants and personnel (performance bias) | Low risk | Quote (page 620): ''The pharmacist of the University Hospital of Verona prepared a specific set with the treatments to be used for the study''; ''The investigators and patients were blinded with respect to the contents of each set'' Comment: unclear whether tablets were of identical appearance but probably adequate |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unclear whether or how outcome assessors were blinded Participants assessed some outcomes and were blinded |
| Incomplete outcome data (attrition bias) | High risk | 62/160 initially randomly assigned participants dropped out, but this is unclear. Study investigator states that dropouts resulted from inefficacy and from requests from participants to discontinue therapy Dropouts from the study were included in the analysis Losses to follow‐up: 68% (27/40)in montelukast plus placebo group; 88% (35/40) in placebo only group. Assumed no dropouts in desloratadine monotherapy or in combined therapy dropped out Comment: high dropout rates unevenly distributed between groups Results presented in graph format or as statistical significance; raw data not given |
| Selective reporting (reporting bias) | Unclear risk | Results obscure; expressed as numbers of participants not given (mean plus 95% CI or graphs) Adverse events: not stated ("low incidence… mild") |
| Other bias | Unclear risk | Funder: Ministero Italiano Universita e Ricerca; no pharmaceutical industry support All data potentially confounded by allowance of rescue medication. Our interpretation suggests that of a possible 1680 patient‐days per group in the active study, Group 1 took rescue medication on average (median) on all but 90.6 days; Group 2 on all but 91 days; Group 3 on all but 45.2 days; Group 4 on all but 54 days |
| Methods | Design: multi‐centre double‐blind 3‐arm placebo‐controlled parallel‐group study of mizolastine vs loratadine vs placebo Duration: 1‐week placebo run‐ in period, then participants received therapy for 4 weeks | |
| Participants | Number of participants randomly assigned: 247 enrolled: 88 to mizolastine; 79 to loratadine; 80 to placebo Sex: 36.8% male, 63.2% female Age of participants, years: 42 ± 15 Unit of allocation: participant Country and setting: France, Spain, Italy; secondary care, hospital clinics Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration After 1‐week placebo run‐in period, participants received 4 weeks:
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: 2 weeks and 4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and clinician | |
| Notes | Study acronym: 'MILOR' Study investigators concluded that mizolastine 10 mg daily is an effective and well‐tolerated agent | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised, but method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | No methods of allocation concealment given in the study report, no further information available |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: ''Patients were randomised to a 4‐week double‐blind treatment...'' but no methods of blinding given |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not described in the study report |
| Incomplete outcome data (attrition bias) | Low risk | 205/247 completed Losses to follow‐up: total 17%; mizolastine: 13/88 (14.8%); loratadine: 10/79 (12.9%); placebo: 19/80 (23.7%), with reasons given Mizolastine: lack of efficacy 3; adverse events 4; non‐compliance 2; "other" 2; loss to follow‐up 2 Loratadine: lack of efficacy 5; adverse events 3; non‐compliance 0; "other" 2; loss to follow‐up 0 Placebo: lack of efficacy 11; non‐compliance 4; "other" 2; loss to follow‐up 2 |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes were reported, but data were given as mean scores plus SD; no participant numbers were given |
| Other bias | Unclear risk | Funder: Synthelabo research |
| Methods | Design: randomised double‐blind placebo‐controlled 4‐arm dose‐ranging study of rupatadine (3 doses) vs placebo Duration: 28 days | |
| Participants | Number of participants randomly assigned: 283 (rupatadine 5 mg n = 68, rupatadine 10 mg n = 73, rupatadine 20 mg n = 67, placebo n = 69) Sex: 28% male, 72% female Age of participants, years: range between 12 and 65; average 38.1 ± 13.0 Unit of allocation: participant Country and setting: France, Romania, Argentina, Hungary; secondary care, hospital clinics and skin research clinics Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
(as a single once‐daily tablet) Duration of intervention: intermediate‐term (28 days) Length of follow‐up: 28 days | |
| Outcomes | Timing of outcome assessment: 14 days and 28 days Primary outcomes of the trial
Secondary outcomes of the trial "All remaining variables," that is:
Clinician or participant report: clinician and participant | |
| Notes | Study investigators concluded that over the 4‐week period, rupatadine 10 mg and 20 mg significantly reduced mean pruritus score compared with placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | No methods of allocation concealment given in study report |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote (page 224): ''This was a phase II dose‐ranging, randomised, double‐blind, placebo‐controlled...'' No details given about method of blinding |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote (page 224): ''This was a phase II dose‐ranging, randomised, double‐blind, placebo‐controlled...'' No details given about method of blinding |
| Incomplete outcome data (attrition bias) | Unclear risk | 244/283 completed study according to protocol 39 participants (14%); 25 (10 given placebo, 9 given 5 mg, 3 given 10 mg, 3 given 20 mg) withdrew because of lack of efficacy: 1 for adverse event, 2 for incorrect treatment allocation, 11 for other or personal reasons However, 6 participants were excluded from analysis without explanation (i.e. 283 randomly assigned); 277 were included in study ITT analysis |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Unclear risk | Funder: Uriach y Compania (Barcelona, Spain); National Scientific Research Program of the Spanish Minister of Science and Technology Unclear clinical meaning of primary outcome (0.5‐point drop in mean pruritus severity score) |
| Methods | Design: double‐blind multi‐centre placebo‐controlled 5‐arm trial to evaluate efficacy and safety of 4 different doses of fexofenadine vs placebo Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 476 from 35 centres Number in each group: placebo 78; 20 mg twice daily 81; 60 mg twice daily 79; 120 mg twice daily 86; 240 mg twice daily 80 Sex: 30% male, 70% female Ethnicity: 90% white, 4% black, 4% "Asian" and 2% multi‐racial Age of participants, years: 12 to 65 Unit of allocation: participants Country and setting: USA; setting research clinics Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration Single‐blind placebo run‐in for 24 hours
Twice a day for 4 weeks Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessments: unclear. Results based on at least 1 postbaseline 12‐hour reflective mean pruritus score (MPS) assessment Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and investigator | |
| Notes | Study investigators concluded that fexofenadine HCl was well tolerated and statistically superior to placebo in treating CSU and in ameliorating interference with sleep and daily activities. They concluded that doses of 60 mg twice daily or greater were most effective | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk | No methods of allocation concealment given in the study report |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: ''...4 week double blind treatment period.'' Methods of blinding not described in the study report |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote: ''...4 week double blind treatment period.'' Methods of blinding not described in the study report |
| Incomplete outcome data (attrition bias) | Unclear risk | 439/476 available for analyses (baseline scores and 1 postbaseline reflective MPS assessment). 103 (21.6%) lost to follow‐up. Issues with losses to follow‐up with participants involved in total discontinuation of treatment accounting for 21.6% Reasons for withdrawal:
Comment: Table 1 in the study report gives a full description of reasons for dropout by group; however as the level of dropout is high at 21.6%, it is unclear whether dropout was a significant source of bias in this study |
| Selective reporting (reporting bias) | High risk | Only participants with baseline and at least 1 postbaseline mean pruritus score were included in analysis |
| Other bias | Unclear risk | Groups comparable at baseline except significant differences in interference with daily activities at baseline Funder: Hoechst Marion Roussel |
| Methods | Design: randomised double‐blind cross‐over 2‐arm study comparing the efficacy of acrivastine vs chlorpheniramine Duration: 24 days | |
| Participants | Number of participants randomly assigned: 20 Sex: 55% male, 45% female Age of participants, years: mean 39.2 (range 18‐27) Unit of allocation: cross‐over (24 days) Country and setting: Australia; setting unclear Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Three times daily for 24 days in this cross‐over study. No wash out period reported, but no participant self‐assessments reported in the first 3 days after cross‐over to eliminate carryover effects from previous therapies Duration of intervention: intermediate‐term (24 days) Length of follow‐up: 24 days | |
| Outcomes | Timing of outcome assessment: after each 24‐day treatment period Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and investigator. Participants self‐assessed daily. Participant reviewed by physician after each 24‐day treatment period—physician recorded opinion on which treatment worked best and suited participant best overall | |
| Notes | Study investigators concluded that both active drugs were effective, with no significant differences noted between them | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Study states that this is a double‐blind study; no methods of blinding given |
| Blinding of outcome assessment (detection bias) | Unclear risk | Method of blinding of outcome assessors not stated |
| Incomplete outcome data (attrition bias) | Unclear risk | 4/20 participants excluded from analysis because of protocol violations (20% lost to follow‐up), with no ITT |
| Selective reporting (reporting bias) | High risk | Incomplete reporting of data (only means given); no comment on adverse events |
| Other bias | Unclear risk | Definition of disease given but CSU defined as > 4 weeks; however, no included participants had urticaria < 2 months Underpowered Funder: Wellcome Research Laboratories |
| Methods | Design: randomised double‐blind 6‐week study of cetirizine vs terfenadine vs placebo in CSU; parallel 3‐arm trial Duration: 6 weeks | |
| Participants | Number of participants randomly assigned: n = 63 took part in the study; however as participants dropped out, they were replaced, so 47 are presented (number given cetirizine n = 17; terfenadine n = 16; placebo n = 14) Sex: cetirizine: 29.41% male, 70.59% female; terfenadine: 18.75% male, 69.23% female; placebo: 69.23% male, 30.77% female Age of participants, years: cetirizine 33.8 ± 13.8; terfenadine 35.88 ± 17.3; placebo 37.8 ± 16.45 Unit of allocation: participant Country and setting: Argentina; outpatient research clinic Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: 6 weeks | |
| Outcomes | Timing of outcome assessment: 3 visits in total, initial at 3 weeks and final at 6 weeks from start of study Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician and participant | |
| Notes | Study report written in Spanish Study investigators concluded that cetirizine is superior to terfenadine in terms of efficacy and tolerability; for symptom control, both active drugs were significantly better than placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quote (page 180): "Randomly divided (in threes) into three equal groups" However, if participants dropped out, they were replaced with new participants (14 did not take the medication, 35 did not return for assessment, 37 did not take the correct medication. All of these participants were replaced). As participants who dropped out were replaced with new participants, it is unclear whether the trial design is truly randomised; it is not clear whether new participants were randomly assigned de novo or were assigned to the group of the most recent dropout. The trial report states: "since the randomisation was performed on groups of 3, it was actually necessary for each loss of a patient [to result in resumed] treatment of three patients" (page 182) (page 186) "9 patients were replaced as three were withdrawn due to protocol violations (lost medication, did not attend tests, did not take correct medication). Therefore 9 new [participants] were recruited, as randomisation was [done] in threes" |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Low risk | Tablets and dosages prepared to be identical (boxes of white round tablets), presented so that each drug or placebo was administered in a uniform way. A scratch‐off label would reveal the drug type in case of emergency |
| Blinding of outcome assessment (detection bias) | Unclear risk | Outcome assessors (clinicians and participants) would not have had indications of treatment group because of uniform packaging, but methods of blinding are not explicitly stated |
| Incomplete outcome data (attrition bias) | High risk | 47/63 completed. Dropped out: 3/17 in cetirizine; 3/16 in terfenadine; 9/14 placebo. 15 left the study because of inefficacy and were not replaced; they were "statistically computable" (page 186). It is unclear how results were computed for participants who dropped out because of inefficacy. Note: This may have introduced bias, as the study was possibly biased towards positive results Other reasons for dropout: adverse events: 2/17 in cetirizine; 2/16 in terfenadine due to adverse events No participant left the study because of intolerable adverse reactions |
| Selective reporting (reporting bias) | High risk | Only results for the cetirizine and terfenadine arms of the study were included in the published report. No results were presented for the placebo arm The researcher was able to withdraw participants on the basis of his opinion |
| Other bias | Unclear risk | Funder: not stated Analyses were not statistically significantly different and placebo results were not presented; therefore conclusions of the study as stated in the study report are unreliable |
| Methods | Design: randomised controlled trial of doxepin 10 mg thrice daily vs pheniramine maleate 22.5 mg thrice daily Duration: 3 weeks | |
| Participants | Number of participants randomly assigned: 56 Sex: 67% female in doxepin group, 60% female in pheniramine group Age of participants, years: 18 to 59 Unit of allocation: participant Duration of urticaria: 8 weeks to 4 years Country and setting: India; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Length of follow‐up: 4 weeks (follow up extended 1 week after cessation of therapy) | |
| Outcomes | Timing of outcome assessment: 3 weeks and 4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: both | |
| Notes | Study investigators concluded that after 3 weeks of therapy, 8 (28.6%) participants in doxepin group and 3 (10.7%) in pheniramine group were symptom free (complete suppression) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants non‐responsive to other antihistamine treatment excluded |
| Allocation concealment (selection bias) | Unclear risk | Unclear allocation concealment |
| Blinding of participants and personnel (performance bias) | Unclear risk | Does not appear to be blinded: "twenty eight subjects were given doxepin 10 mg thrice weekly for 3 weeks"..."Another 28 subjects...were treated with pheniramine maleate 22.5 mg thrice weekly for 3 weeks" |
| Blinding of outcome assessment (detection bias) | Unclear risk | Does not appear to be blinded: "twenty eight subjects were given doxepin 10 mg thrice weekly for 3 weeks"..."Another 28 subjects...were treated with pheniramine maleate 22.5 mg thrice weekly for 3 weeks |
| Incomplete outcome data (attrition bias) | Unclear risk | No dropouts apparent |
| Selective reporting (reporting bias) | Unclear risk | Partial remission/improvement amongst participants is not defined or specific
|
| Other bias | Low risk | None detected. Funder: none |
| Methods | Design: a randomised multi‐centre multi‐country double‐blind parallel‐group placebo‐controlled 3‐arm study of rupatadine at 2 different doses Duration: 6 weeks | |
| Participants | Number of participants randomly assigned: 334 Sex: placebo: male 37.8%, female 62.2%; rupatadine 10 mg: 30% male, 70% female; rupatadine 20 mg: 26.9% male, 73.1% female Age of participants, years: mean (SD): placebo: 35.8 (13.4); rupatadine 10 mg: 40.2 (3.6); rupatadine 20 mg: 37.6 (14.6) Unit of allocation: participant Country and setting: Spain, Romania, Argentina, Poland, Germany, Italy; multi‐centre, research clinics Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Once daily for 6 weeks Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: up to 6 weeks | |
| Outcomes | Timing of outcome assessment: after 2, 4 and 6 weeks of treatment Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and investigator (patient daily diary cards) Adverse events: any adverse events; headache; somnolence; hypertension; "metrorrhagia" | |
| Notes | Investigators conclude that rupatadine 10 mg is a fast long‐acting treatment with a better safety profile than rupatadine 20 mg. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: ''...according to a centralized computer‐generated randomisation code provided by the sponsor of the study'' Further information from the study investigator "The randomisation procedures were carried out at J. Uriach y Compañía, S.A. The Production Quality Management Department of J. Uriach y Compañía, S.A. drew up a randomisation list for the treatments. Afterwards, the Quality Assurance Unit of J. Uriach y Compañía S.A. randomly assigned a treatment to each code. Two copies of the randomisation code and two of the randomised list of patients were obtained" |
| Allocation concealment (selection bias) | Low risk | Quote: "A duly closed and sealed copy of each document was kept in the Quality Assurance Unit and in the Production Quality Management Department of J. Uriach y Compañía S.A. A third closed and sealed copy randomised list was prepared for the CRO MDS PS Pharma Services. In addition, once the study was concluded, all the individual envelopes were returned to the monitor, who checked that none of them had been opened for an unjustified reason. After the lock of study database, the copy kept by the Quality Assurance Unit was opened and filed in the master file of the study" (further information obtained from study investigator) |
| Blinding of participants and personnel (performance bias) | Low risk | Quote from study: ''... double‐blind, placebo‐controlled... study..'' Further information from study investigator: "This was a double‐blind study so that neither the investigator nor the patient knew treatment assignation. To preserve double blinding, medication was packaged identically for both types of treatments, with identical outside appearance of the strips and boxes. Individual envelopes identified with the patient assignation number were prepared. Each one included the identity of the treatment assigned to each patient. These envelopes, duly closed and sealed, were submitted to the investigator" "All medication was given in a two tablets scheme, that is, the 10 mg dose was given in two (10 mg plus placebo) tablets, the 20 mg dose was given in two (10 mg plus 10 mg) tablets, and the placebo was given in two (placebo plus placebo) tablets" Further information from study investigator |
| Blinding of outcome assessment (detection bias) | Low risk | ''... double‐blind, placebo‐controlled... study..'' Comment: as above |
| Incomplete outcome data (attrition bias) | Unclear risk | No ITT; no actual scores given for DLQI, even at baseline 41/334 lost to follow‐up (12.2%) with 293 evaluable participants Reasons for withdrawal: rupatadine 10 mg: participant decision n = 4; loss to follow‐up n = 1; exclusion criteria n = 2; treatment failure n = 7; non‐attendance at scheduled visits n = 2; other n = 1 Rupatadine 20 mg: loss to follow up n = 1; exclusion criteria n = 3; treatment failure n = 4; lack of compliance n = 1 Placebo: participant decision n = 2; loss to follow‐up n = 1; serious adverse event n = 1; treatment failure n = 11 |
| Selective reporting (reporting bias) | Low risk | SAF population stands for safety population (i.e. all randomly assigned participants who received any study drug). Figure 2 in the study report is done with the ITT population, as 5 patients did not present the efficacy variables; this is why the intention‐to‐treat analysis was performed in 329 participants In Figure 1, the number of participants completing the trial from the placebo group is 98, not 88 (as confirmed by study investigator) DLQI scores not stated in published report (percentages only) |
| Other bias | Unclear risk | Funder: J Uriach y Compania, Spain |
| Methods | Design: double‐blind randomised 3‐arm RCT comparing cetirizine, terfenadine and placebo in a cross‐over study Duration: cross‐over study lasting for 6 weeks, subdivided into 3 periods of 2 weeks | |
| Participants | Number of participants randomly assigned: 30 Sex: not stated Age of participants, years: 15 to 69 (mean 48.8), but included at least 1 participant ineligible by age according to exclusion criteria Unit of allocation: cross‐over Country and setting: Netherlands, Belgium; setting not stated Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
No washout between treatments Participants could elect to finish particular treatment before 2 weeks was up and to move to next treatment in sequence Duration of intervention: short‐term (2 weeks per intervention) Length of follow‐up: 6 weeks (i.e. follow‐up extended beyond cessation of therapy) | |
| Outcomes | Timing of outcome assessment: at 2 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: both | |
| Notes | Study investigators concluded as follows: By investigator measures, cetirizine significantly was better than placebo in all outcomes; terfenadine findings were only borderline or were not significant By participant measures, both drugs were equally (and statistically) superior to placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | No allocation concealment methods described
|
| Blinding of participants and personnel (performance bias) | Low risk | Study states that it was double‐blind Quote: ‘products were given as identical capsules bid, with placebo as the morning intake in the cetirizine sequence’ |
| Blinding of outcome assessment (detection bias) | Unclear risk | Study states that it was double‐blind. Unclear how outcome assessors were blinded |
| Incomplete outcome data (attrition bias) | Unclear risk | 2/30 were excluded after placebo sequence (refusal to participate) (6%) Serious adverse events were unclear for all 3 treatments 10/30 early withdrawals from treatment: inefficacy in placebo group n = 3; adverse events: cetirizine n = 1; placebo n = 1; terfenadine n = 3; unspecified reason: placebo: n = 2. No ITT (although low dropout rate) |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported. Mixed dichotomous and continuous outcome reporting |
| Other bias | Unclear risk | Funder: UCB (makers of cetirizine) |
| Methods | Design: randomised double‐blind placebo‐controlled parallel multi‐centre study comparing efficacy of fexofenadine and levocetirizine Duration: up to 1 month | |
| Participants | Number of participants: 40 (20 in each group) Sex: 50% male, 50% female Age of participants, years: 14 to 70 Unit of allocation: participant Country and setting: India; setting hospital clinic Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
For relief of symptoms up to 1 month Duration of intervention: intermediate‐term (up to 1 month) Length of follow‐up: up to 4 weeks | |
| Outcomes | Timing of outcome assessment: 2 week and 4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician | |
| Notes | Study investigators concluded that fexofenadine was superior to levocetirizine | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: ''All participants were divided into two groups,'' described as randomised but no details given |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) | High risk | 2/20 participants in levocetirizine group were lost to follow‐up (no reasons given) and were replaced by 2 new participants. No ITT |
| Selective reporting (reporting bias) | High risk | Small study; no raw data; means and SDs of scores only; results not expressed as participant numbers. States placebo controlled, but no placebo results reported |
| Other bias | Unclear risk | Sponsor: Sanofi‐Aventis and Systopic Laboratories (provided drugs) |
| Methods | Design: cross‐over study comparing cetirizine with placebo Duration: 1 week | |
| Participants | Number of participants randomly assigned: 32 Sex: 50% female Age of participants, years: range 18 to 46 (mean 30.4 ± 8.2) Unit of allocation: cross‐over Country and setting: Singapore; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Length of follow‐up: 1 week Duration of intervention: short‐term (1 week per intervention) | |
| Outcomes | Timing of outcome assessment: 1 week Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: both | |
| Notes | Study investigators concluded that cetirizine yielded significantly better scores than placebo on physician and participant VAS and in participant diaries for itch and weals | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not stated |
| Allocation concealment (selection bias) | High risk | No allocation concealment |
| Blinding of participants and personnel (performance bias) | Low risk | Probably done |
| Blinding of outcome assessment (detection bias) | Unclear risk | Investigators’ assessment of severity on VAS |
| Incomplete outcome data (attrition bias) | High risk | No ITT. No mention of serious adverse events or dropouts due to them. 4 participants (12.5%) lost to follow‐up |
| Selective reporting (reporting bias) | High risk | Results derived from visual analogue scales; not stated whether groups were comparable at baseline |
| Other bias | Unclear risk | Funder: none No washout between treatments, but no sequential effect shown |
| Methods | Design: double‐blind multi‐centre 3‐arm trial of terfenadine, chlorpheniramine and placebo Duration: 6 weeks | |
| Participants | Number of participants randomly assigned: 136 Age group and gender: not stated Unit of allocation: participant Country and setting: USA; setting recruited from medical practices of principal investigators at 10 university research clinics (assumed that the practices were the settings) Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration Participants entered a single‐blind placebo period for a week, and if hives of moderate severity were present for 3 days during the week, participants were assigned to
Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: up to 6 weeks | |
| Outcomes | Timing of outcome assessment: weekly for 6 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigators and participants | |
| Notes | Study investigators concluded that chlorpheniramine was not statistically significantly different from placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation: Quote (page 575): ''participants were randomly assigned to one of three groups of equal size...'' |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Method of blinding unclear: Quote (page 575): ''Acceptable participants entered the first phase of the study and were administered placebo in a single‐blind fashion for a week. Those who developed moderately severe hives for at least 3 days that were actually observed by the investigators were then enrolled in the double‐blind phase'' Also, unclear if blinding adequate: quote: "In order to limit the number of drop‐outs, diphenhydramine 25mg capsules were offered as relief medication" |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unclear method of blinding outcome assessors |
| Incomplete outcome data (attrition bias) | Low risk | 122/136 who were randomly assigned completed the study (of an undisclosed number, 'more than half' were initially screened but excluded) 14 (10.2%) were lost to follow‐up Withdrawals due only to treatment failure: terfenadine n = 1; chlorpheniramine n = 4; placebo n = 9. Unbalanced between groups but unlikely to introduce bias |
| Selective reporting (reporting bias) | High risk | Results presented graphically only, no participant numbers or means with SD |
| Other bias | Unclear risk | Participants allowed a different antihistamine if uncontrolled: 22/42 (52%) of placebo group took diphenhydramine; 12/46 (26%) of chlorpheniramine group and 4/46 (9%) of terfenadine group already a subgroup, as participants unresponsive to antihistamines were excluded Funder: none stated |
| Methods | Design: multi‐centre (4 centres) randomised double‐blind parallel 2‐arm study of desloratadine vs loratadine Duration: 28 days | |
| Participants | Number of participants randomly assigned: 158 Sex: male 45%, female 64% Age of participants, years: 18 to 65, desloratadine mean 38.6; loratadine mean 39.1 Unit of allocation: participant Country and setting: China; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
Duration of disease in desloratadine group 26.9 weeks; loratadine group 26.1 weeks Groups comparable at baseline with t value and P value in Chart 2 | |
| Interventions | Interventions, dose, duration
Duration of intervention: 28 days (intermediate) Length of follow‐up: seen before treatment, at 14 days, at 28 days Concomitant/rescue treatment: not mentioned | |
| Outcomes | Timing of outcome assessment: 14 days and 28 days Primary outcomes of the trial
Secondary outcomes of the trial
Timing of outcome assessment: seen before treatment, at 14 days and at 28 days Clinician or participant report: clinician and participant | |
| Notes | Study investigators concluded that desloratadine is safe and effective in the treatment of CSU. No significant differences between the 2 groups, no serious adverse events. Desloratadine safe and effective Main study report in Chinese | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated to be randomised |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Participants and personnel were blinded to treatment group Not mentioned clearly |
| Blinding of outcome assessment (detection bias) | Unclear risk | Outcome assessors were blinded to treatment group Not mentioned clearly |
| Incomplete outcome data (attrition bias) | Unclear risk | Unclear, no details about dropout in translation of published report. ITT analysis carried out: unclear |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes were reported, but reporting of adverse events is unclear. Severity of adverse advents and whether or not these were likely to have been caused by trial medication were specified as an outcome, but reported results are unclear and state only that no serious adverse events occurred |
| Other bias | Unclear risk | Funder: not stated Assessment of compliance undertaken but not clearly stated, apart from exclusion criteria |
| Methods | Design: randomised double‐blind parallel‐group comparison of mizolastine vs loratadine Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 47; n = 24 mizolastine, n = 23 loratadine Sex: 36% male, 55% female Age of participants, years: 17 to 53, mizolastine mean 34.9; loratadine mean 33.0 Unit of allocation: participant Country and setting: China; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: 7, 14 and 28 days Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician and participant | |
| Notes | Study investigators concluded that no difference in side effects was found between the 2 groups. Findings indicated that the effect of mizolastine was much better than that of loratadine, and it could be selected as the priority treatment for CSU Main study reported in Chinese | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Double‐blind randomisation stated. Quote (page 482): "divided into two groups by randomised method" |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote (page 482): "double‐blind," but no further details given |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote (page 482): "double‐blind," but no further details given |
| Incomplete outcome data (attrition bias) | High risk | Stated 47 cases, but analyses include 23 in each group; data analysis was based on 46 cases; no reason given for this dropout |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Low risk | None detected Funder: not stated |
| Methods | Design: randomised 2‐arm double‐blind study of cetirizine vs fexofenadine Duration: 28 days | |
| Participants | Number of participants randomly assigned: 116; n = 59 cetirizine; n = 57 fexofenadine Sex: not stated Age of participants, years: range of 17 to 65 Unit of allocation: participant Country and setting: India; dermatology institute clinic Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (28 days) Length of follow‐up: 28 days | |
| Outcomes | Timing of outcome assessment: days 14 and 28 Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and investigator | |
| Notes | Study investigators concluded that cetirizine seemed to have therapeutic advantage over fexofenadine | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Method of randomisation not stated, described as randomised. Further information from study investigator states: "For the study we generated a randomisation list using a random number table" |
| Allocation concealment (selection bias) | Low risk | Study investigator states: "The code was kept with the central authority—not directly involved with the study and assessment of endpoints. Both the investigators and patients were blinded since the central authority provided the patients with similar looking sealed envelopes containing the medication and labelled only as A or B" |
| Blinding of participants and personnel (performance bias) | Low risk | Study investigator states: "Both the investigators and patients were blinded since the central authority provided the patients with similar looking sealed envelopes containing the medication and labelled only as A or B. The blinding was opened after assessing the results and the statistical analysis of the two groups A and B. It was done by inquiring from the central authority which was dispensing the drugs to the patient" |
| Blinding of outcome assessment (detection bias) | Low risk | Study investigator states: "Both the investigators and patients were blinded since the central authority provided the patients with similar looking sealed envelopes containing the medication and labelled only as A or B. The blinding was opened after assessing the results and the statistical analysis of the two groups A and B. It was done by inquiring from the central authority which was dispensing the drugs to the patient" |
| Incomplete outcome data (attrition bias) | Unclear risk | High dropout rate, no ITT (no ITT confirmed by study investigator); 19/116 lost to follow‐up in total (13%): 7 in cetirizine group and 12 in fexofenadine group within 2 weeks, ''the most common reason being treatment failure.'' Unclear if this contributes to bias |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes were reported, but other outcomes were reported as well (e.g. duration of weals, diurnal variation, intensity of itching, presence of weals). Symptoms during the day were not reported by groups |
| Other bias | Low risk | Funder: not stated. No indication of comparability of groups at baseline |
| Methods | Design: randomised multi‐centre double‐blind 2‐arm parallel‐group comparison of desloratadine vs loratadine Duration: 28 days | |
| Participants | Number of participants randomly assigned: 217; desloratadine n = 108; loratadine n = 109 Sex: male 43.8%, female 56.2% Age of participants, years: 18 to 65 Unit of allocation: participant Country and setting: China, secondary care, Southern Hospital, 3rd Military Medical University. Chongqing, China Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (28 days) Length of follow‐up: 28 days | |
| Outcomes | Timing of outcome assessment: 7, 14 and 28 days Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician and participant | |
| Notes | Main study report in Chinese Study investigators concluded that desloratadine is an effective and safe agent for CSU | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated to be randomised |
| Allocation concealment (selection bias) | Unclear risk | No details given |
| Blinding of participants and personnel (performance bias) | Unclear risk | Stated to be double‐blind, no further details given about blinding |
| Blinding of outcome assessment (detection bias) | Unclear risk | Stated to be double‐blind, no further details given about blinding |
| Incomplete outcome data (attrition bias) | Unclear risk | 3/105 dropped out or were lost from the desloratadine group; 3/106 dropped out or were lost from the loratadine group; no reasons given |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Low risk | None detected. Funder: not stated |
| Methods | Design: double‐blind randomised cross‐over study in which participants were treated sequentially (5‐arm comparison of hydroxyzine plus placebo, hydroxyzine plus terbutaline, hydroxyzine plus chlorpheniramine, hydroxyzine plus cimetidine) Duration: 7 to 10 days | |
| Participants | Number of participants randomly assigned: 23 Sex: 79% female Age of participants, years: mean 37 (range 24‐64) Unit of allocation: cross‐over participants Country and setting: USA; setting University of Colorado Health Sciences Center Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration Cross‐over study in which participants were treated sequentially with 5 regimens in double‐blind random sequence
Duration of intervention: short‐term (7‐10 days each intervention) Length of follow‐up: unclear | |
| Outcomes | Timing of outcome assessment: 7 to 10 days Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and investigator | |
| Notes | Study investigators concluded that hydroxyzine plus cimetidine was significantly better than other combinations. Hydroxyzine had transient soporific effects but was well tolerated. Unclear whether soporific effects were due to hydroxyzine and not to one of the other sedating antihistamines | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not stated: ''participants were treated orally in a double‐blind randomised serial fashion...'' |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | ''participants were treated orally in a double‐blind randomised serial fashion...'' Method of blinding unclear |
| Blinding of outcome assessment (detection bias) | Unclear risk | ''participants were treated orally in a double‐blind randomised serial fashion...'' Method of blinding unclear |
| Incomplete outcome data (attrition bias) | High risk | 19/23 completed. 4 removed because of non‐compliance (17% lost to follow‐up). One participant not accounted for Extreme tremulousness (n = 2) in terbutaline necessitated "abbreviated treatment course” ITT unclear (some terbutaline participants did not complete the course, but non‐compliant participants were excluded from analysis) |
| Selective reporting (reporting bias) | High risk | Adverse events not clearly reported |
| Other bias | Unclear risk | Funder: NIH Allergic Disease Center Grants Study duration not clearly defined (''7‐10 days'') No stated exclusions, and physical urticaria may have been included. Two groups were given a combination of 2 first‐generation antihistamines; concomitant treatment with hydroxyzine allowed in all groups |
| Methods | Design: double‐blind randomised 2‐arm cross‐over study of terfenadine vs clemastine Study 1: clemastine vs placebo Study 2: terfenadine vs placebo Duration: 2 weeks | |
| Participants | Number of participants randomly assigned: 60; 30 per group in each cross‐over phase Sex: 33% male, 67% female Age of participants, years: 18‐72 (mean 37) Unit of allocation: cross‐over, no washout period described Country and setting: Denmark; setting unclear Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: short‐term (2 weeks per intervention) Length of follow‐up: 14 days | |
| Outcomes | Timing of outcome assessment: 14 days Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and clinician | |
| Notes | Study author concluded that terfenadine was more efficacious than clemastine or placebo No review outcomes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "in random order, participants were assigned...''; no further details given |
| Allocation concealment (selection bias) | Unclear risk | Unclear, no details given |
| Blinding of participants and personnel (performance bias) | Unclear risk | Study is stated to be double‐blind; unclear how this was achieved |
| Blinding of outcome assessment (detection bias) | Unclear risk | Study is stated to be double‐blind; unclear how this was achieved, as only 1 study author/single investigator was involved |
| Incomplete outcome data (attrition bias) | Unclear risk | Number of participants who dropped out from each group not stated. Withdrawals not mentioned |
| Selective reporting (reporting bias) | High risk | Results incompletely reported, no numerical data (graphs only); able to rate efficacy only according to number of weals, as symptom score used in results was not defined |
| Other bias | Unclear risk | Funder: not stated Diagnosis of CSU not clearly defined; unclear whether concomitant medications allowed; single study author |
| Methods | Design: double‐blind randomised controlled 3‐arm study comparing levocetirizine vs desloratadine vs placebo Duration: not stated | |
| Participants | Number of participants randomly assigned: 107; levocetirizine group: 37; desloratadine group: 34; placebo: 36 Sex: not stated Age of participants, years: 18 to 60 Unit of allocation: participant Country and setting: Albania; tertiary centre Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
(each increasing weekly to 10 mg, then 20 mg) Duration of intervention: unclear Length of follow‐up: not stated | |
| Outcomes | Primary outcomes of the trial
| |
| Notes | Study investigators concluded that Increasing the dosage of levocetirizine and desloratadine up to 4‐fold improved chronic urticaria symptoms without compromising safety | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study states randomly assigned, but not clear how done |
| Allocation concealment (selection bias) | Unclear risk | Not specified |
| Blinding of participants and personnel (performance bias) | Unclear risk | Study says double–blind but no other details |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) | Unclear risk | All participants accounted for, but 9 from active arms left the study (reason not stated), and all participants from placebo arm (36) dropped out from the study |
| Selective reporting (reporting bias) | Unclear risk | No timing of assessment stated |
| Other bias | Unclear risk | Funder: not specified This study was available as a short conference abstract only; we were unable to identify a published report or to obtain further information from the study investigator |
| Methods | Design: randomised double‐blind 3‐arm cross‐over placebo‐controlled study of acrivastine and clemastine Duration: 5 days each cross‐over period | |
| Participants | Number of participants randomly assigned: 18 Sex: 33% male, 67% female Age of participants, years: 14 to 75 (mean 43.2) Unit of allocation: cross‐over Country and setting: UK; setting unclear Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Cycle through each regimen with 2‐day breaks in between with no relevant treatment Duration of intervention: short‐term (5 days per intervention) Length of follow‐up: unclear (at end of each 5‐day treatment period?) | |
| Outcomes | Timing of outcome assessment: 5 days Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator and participant questionnaire | |
| Notes | Study investigators concluded no significant differences in efficacy were noted between acrivastine and clemastine; both were significantly preferred by participants over placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Described as randomised, but randomisation method was unclear |
| Allocation concealment (selection bias) | Unclear risk | No allocation concealment described |
| Blinding of participants and personnel (performance bias) | Unclear risk | ''A double‐blind, placebo‐controlled study.'' Mentioned only in title; no other details given |
| Blinding of outcome assessment (detection bias) | Unclear risk | ''A double‐blind, placebo‐controlled study.'' Mentioned only in title; no other details given |
| Incomplete outcome data (attrition bias) | Low risk | 18/18 completed. None lost to follow‐up |
| Selective reporting (reporting bias) | High risk | Although both doctor questionnaire and participant self‐assessment were carried out, only simple participant perceptions reported; no objective data provided |
| Other bias | Unclear risk | Funder: Sponsorship: Wellcome Foundation Ltd No mention of whether concomitant treatments were permitted Underpowered |
| Methods | Design: randomised double‐blind cross‐over 3‐arm trial comparing 10 or 20 mg cetirizine vs placebo Duration: 15 days | |
| Participants | Number of participants randomly assigned: 30 Sex: 27% male, 63% female Age of participants, years: range 15 to 70 (mean 43) Unit of allocation: cross‐over Country and setting: Sweden; setting unclear Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
After 15 days, participants were allowed to cross over with no washout period. Non‐responders were allowed to increase to twice‐daily dosing Duration of intervention: short‐term (15 days) Length of follow‐up: end of each 15‐day treatment period | |
| Outcomes | Timing of outcome assessment: 15 days. Visits at baseline and at end of each treatment phase Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant | |
| Notes | Study investigators concluded that cetirizine was significantly more effective than placebo in reducing incidence of erythema, weals and pruritus Study 2 in the published report was a laboratory study on the effects of certain agonists; the results were not included in this analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Unclear method of randomisation. Participants 'randomly assigned' |
| Allocation concealment (selection bias) | Unclear risk | Unclear. Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind study. Unclear how this was done |
| Blinding of outcome assessment (detection bias) | Unclear risk | Double‐blind study. Unclear how this was done |
| Incomplete outcome data (attrition bias) | Unclear risk | Number of participants who dropped out from the study (if any) unclear |
| Selective reporting (reporting bias) | High risk | Extensive laboratory tests were carried out (for adverse effects) but were not reported. Measures of incidence and severity of weals were reported |
| Other bias | Low risk | None detected. Funder: not stated |
| Methods | Design: randomised multi‐centre parallel‐group 3‐arm double‐blind placebo‐controlled study of cetirizine vs hydroxyzine vs placebo Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 219; cetirizine: 69, hydroxyzine: 69, placebo: 73 Sex: not stated Age of participants, years: 12+, no further details Unit of allocation: participant Country and setting: USA; centres included medical and science centres and private practice Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Intermediate dose of cetirizine not stated Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: baseline, 3 days, 1 week, 2 weeks, 3 weeks, 4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant | |
| Notes | Investigator concludes that cetirizine and hydroxyzine have equivalent efficacy, and both are superior to placebo; no significant differences in adverse effects were noted, except for somnolence and nausea; no significant differences between cetirizine and placebo were observed in terms of somnolence | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants "randomly assigned"; quote (page 1015), no further information |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | High risk | Stated to be double‐blinded but unclear how this was achieved; in addition, participants and personnel do not appear blinded Quote (page 1015): ''All patients receiving active drugs began by taking the lowest daily dose... The dose was titrated up as necessary in two steps to respective allowable maximums..." |
| Blinding of outcome assessment (detection bias) | High risk | Stated to be double‐blinded but unclear how this was achieved; in addition, participants and personnel do not appear blinded Quote (page 1015): ''All patients receiving active drugs began by taking the lowest daily dose... The dose was titrated up as necessary in two steps to respective allowable maximums..." |
| Incomplete outcome data (attrition bias) | High risk | Losses to follow‐up not clearly described 31/219 randomly assigned lost to follow‐up (14%) Each table provides different numbers of included participants; not clear why and when participants were lost to follow‐up; study authors note in discussion that 10% of cetirizine group, 7% of hydroxyzine group and 24% of placebo group withdrew because of lack of efficacy Adverse events (serious adverse events requiring treatment withdrawal):
|
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Low risk | None detected. Funder: not stated |
| Methods | Design: multi‐centre randomised double‐blind parallel‐group placebo‐controlled 2‐arm study of fexofenadine vs placebo Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 259 Sex: 26% male, 74% female Age of participants, years: 247 younger than 65; 8 older than 65 Unit of allocation: participant Country and setting: US; setting secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Single‐blind placebo run‐in phase for 2 to 5 days At next visit, participants had to qualify for entry into randomised portion of study, then were randomly assigned 2:1 to fexofenadine:placebo Duration of intervention: intermediate‐term (28 days) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: seen weekly for 4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
| |
| Notes | Same study as Spector 2007 Study investigators concluded that fexofenadine at 180 mg once daily offered effective well‐tolerated relief of the symptoms of urticaria | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 662): ''Randomisation in a 2:1 manner to receive either fexofenadine hydrochloride,180mg, or placebo once daily'' |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Low risk | Study stated as double‐blind. Initial single‐blind run‐in with placebo for 2 to 5 days Quote (page 663): "Patients received double blind study medications packaged in bottles with 40 tablets" (enough for course of treatment) Comment: probably done |
| Blinding of outcome assessment (detection bias) | Low risk | Study stated as double‐blind. Initial single‐blind run‐in with placebo for 2 to 5 days. Participants received study medication in bottles with 40 tablets (enough for course of treatment) Comment: probably done, but unclear how outcome assessors were blinded |
| Incomplete outcome data (attrition bias) | Unclear risk | Did not include participants lacking postbaseline result. Unclear how many dropped out at each time point along with reasons for dropout Losses to follow‐up of 259 randomly assigned: fexofenadine 12/167 (7%); 13/92 (14%): 7% dropped out of fexofenadine group, 14% from placebo group (lack of efficacy) Adverse events: Most common reason for loss to follow‐up was lack of efficacy (6 of 13 from placebo group, and 1 of 12 from fexofenadine group). Other reasons not stated Asthma requiring hospitalisation: n = 1 in fexofenadine group; headache occurred in 5% of fexofenadine group and 3% of placebo group; not stated whether any of these required treatment withdrawal Numbers of participants in each study were very unclear at each time point |
| Selective reporting (reporting bias) | High risk | Study authors combined unadjusted mean DLQI scores from 2 weeks and 4 weeks |
| Other bias | Unclear risk | Funder: Aventis Inc (Sanofi‐Aventis Group) |
| Methods | Design: double‐blind randomised multiple (3)‐arm cross‐over study comparing cetirizine, terfenadine and placebo Duration: 6 weeks | |
| Participants | Number of participants randomly assigned: 30 in sequence Sex: 44% male, 56% female Age of participants, years: 41.2 ± 14.7 (range 21‐74) Country and setting: Belgium; secondary care Unit of allocation: cross‐over Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Unclear which treatments were compared at each phase Duration of intervention: short‐term (2 weeks) Length of follow‐up: 2 weeks (for each phase) No washout between treatments; if intolerable, symptoms due to lack of response could progress early to next in sequence (after clinic visit). Seen at each change in treatment 5 instances of rescue medications used in placebo group only (antihistamines n = 4, dexamethasone n = 1) (against protocol) | |
| Outcomes | Timing of outcome assessment: end of weeks 1 and 3 at each phase Primary outcomes of the trial
Secondary outcomes of the trial
| |
| Notes | Study investigators concluded that no significant differences in efficacy were found between the 2 active treatments; both were better than placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | ''Random sequence''; multiple sequence defined by Latin square design |
| Allocation concealment (selection bias) | Unclear risk | Unclear, no apparent allocation concealment |
| Blinding of participants and personnel (performance bias) | Low risk | Identical capsules |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unclear how outcome assessors were blinded to treatment allocation |
| Incomplete outcome data (attrition bias) | Low risk | 3/30 participants lost to follow‐up (10%). 2 dropouts due to lack of efficacy during first sequence (cetirizine n = 1 and terfenadine n = 1); 1 dropout in second sequence for same (cetirizine) |
| Selective reporting (reporting bias) | High risk | Results not clearly reported; placebo sequence had rescue medication. Study investigators conclude... ''thus only for wheals were borderline or significant differences from placebo recorded by investigator'' 5 instances of rescue medication used in placebo group only (antihistamines n = 4, dexamethasone n = 1) (against protocol) |
| Other bias | Unclear risk | Funder: UCB Braine‐L’Alleud |
| Methods | Design: randomised double‐blind 4‐arm cross‐over study to investigate efficacy of acrivastine at 2 doses vs clemastine and placebo Duration: 5 days in each arm | |
| Participants | Number of participants randomly assigned: 20 Sex: 60% female Age of participants, years: 18 to 72 (mean 41.3) Country and setting: Lubeck; secondary care Unit of allocation: cross‐over Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Unclear which treatments were compared at each phase 3‐day washout initially, then 2‐day break between treatments Duration of intervention: short‐term (5 days per intervention) Length of follow‐up: 5 days | |
| Outcomes | Timing of outcome assessment: 5 days Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator and participant (self‐assessment form) | |
| Notes | Study investigators concluded that assessment showed all 3 active drugs were better than placebo; no statistically significantly difference was noted between them | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding of participants and personnel (performance bias) | Unclear risk | Study stated to be double‐blind, but no details given |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unclear if blinded. As investigators appear to have made subjective decision on best treatment, it is possible that they were not blinded to treatment allocation |
| Incomplete outcome data (attrition bias) | Unclear risk | Only 20 participants; no dropouts; no serious adverse events noted. Raw figures not shown (means only) |
| Selective reporting (reporting bias) | Unclear risk | Incomplete reporting of data (mean scores only, without SD of self‐assessment for wealing, itching and overall discomfort). Percentage scores of physician assessment and overall improvement |
| Other bias | Unclear risk | Funder: Wellcome Research Laboratories |
| Methods | Design: randomised double‐blind parallel‐group 2‐arm study of mizolastine vs loratadine Duration: 28 days | |
| Participants | Number of participants randomly assigned: 61 Sex: 64% male, 46% female Age of participants, years: 40 ± 13 Country and setting: France; secondary care Unit of allocation: participant Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Not stated whether this was with placebo (although placebo seems likely) Duration of intervention: short‐term (28 days) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: 4 visits in total: at screening (day 3 or 7), at inclusion (day 10), after 14 and 28 days of treatment Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator and participant | |
| Notes | Study investigators concluded that no statistically significant differences in efficacy were found between drugs, as measured by number of urticarial episodes and discomfort from symptoms; duration of episodes shorter with mizolastine (not statistically significant) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ''Patients were randomly allocated...''; no details given about method of randomisation |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind study. Unclear how blinding was achieved |
| Blinding of outcome assessment (detection bias) | Unclear risk | Double‐blind study. Unclear how blinding was achieved |
| Incomplete outcome data (attrition bias) | High risk | Number of participants completing the study and evaluable does not match corresponding information given about non‐completers 7/61 lost to follow‐up (11%): mizolastine group: 3/26, loratadine group: 4/35 Withdrawals: adverse events: serious leading to withdrawal: mizolastine: painful erythema of hands n = 1 Minor events: mizolastine: fatigue n = 2; drowsiness n = 1; loratadine: drowsiness n = 1; dizziness n = 1; rhinitis n = 1 A figure of n = 56 is given as the number of evaluable participants in the published report, but after withdrawals and losses to follow‐up, the number of participants remaining in the trial is 54 |
| Selective reporting (reporting bias) | Unclear risk | All outcomes reported but presented only as number of participants free of symptoms, which decreased significantly in both groups (numbers not given; results expressed graphically and related to number of participants free of individual symptom (e.g. pruritus), but not possible to tell how many participants were symptom‐free overall |
| Other bias | Unclear risk | Number of participants free of symptoms decreased significantly in both groups, but numbers not given; figures given relate to numbers of participants free of individual symptom (e.g. pruritus), but not possible to tell how many participants were symptom‐free overall Funder: none |
| Methods | Design: 5‐centre double‐blind parallel‐group randomised trial of mizolastine vs loratadine | |
| Participants | Number of participants randomly assigned: 213 enrolled: 104 mizolastine, 102 loratadine, 206 completed the trial Sex: mizolastine: male 46 (42.6%), female 62 (57.4%); loratadine: male 50 (47.6%), female 55 (52.4%) Age of participants, years: mizolastine 39.32; loratadine 37.9 Unit of allocation: participant Country and setting: Beijing, China; multi‐centre secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (28 days) Length of follow‐up: 28 days | |
| Outcomes | Timing of outcome assessment: 7, 14, 28 days Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and clinician. Outcomes reported by clinician apart from VAS score (participant rated severity of itch by visual analogue scale) | |
| Notes | Study investigators concluded that mizolastine is quicker in action than loratadine, with similar efficacy. The incidence of adverse effects is similar in the 2 groups Main study report is written in Chinese | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 306): "randomly divided" |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote (page 306): "double blind" |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote (page 306): "double blind" |
| Incomplete outcome data (attrition bias) | Unclear risk | 206/213 completed, but study stated that 12 participants dropped out; it is unclear if they dropped out from the number enrolled or the number completing |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes were reported |
| Other bias | Low risk | None detected. Funder: not stated |
| Methods | Design: double‐blind 2‐arm cross‐over trial comparing nifedipine (calcium channel blocker) with chlorpheniramine Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 22 Sex: 13% male, 27% female Age of participants, years: 36 (range 21‐54) Unit of allocation: cross‐over Country and setting: Taiwan; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration Washout of at least 12 hours, then randomised:
Participants had one or another active drug, then 2 days of placebo, then the other active drug; all capsules identical in appearance Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: unclear (daily scores) Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator and participant | |
| Notes | Study investigators concluded that nifedipine not a first‐line drug for chronic urticaria, and that it is helpful for only some selected patients | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation method unclear |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind study. All medication in capsules and identical in appearance; participants blinded. Probably done but no details given |
| Blinding of outcome assessment (detection bias) | Unclear risk | Double‐blind study. All medication in capsules and identical in appearance; participants blinded. Probably done but no details given |
| Incomplete outcome data (attrition bias) | Low risk | 18/22 participants completed the study (18% lost to follow‐up): n = 3 military personnel posted elsewhere, n = 1 severe nausea on nifedipine ×1 Minor adverse events: nifedipine: n = 3 dizziness, n = 1 drowsiness, n = 2 mild hypotension Comment: Substantial losses to follow‐up may have introduced a source of bias to the study. No ITT |
| Selective reporting (reporting bias) | High risk | Duration of follow‐up not clearly defined Unclear whether concomitant medications were permitted or whether participants were compliant Blood pressure results not reported in full, only in terms of adverse events |
| Other bias | Unclear risk | Underpowered Funder: none |
| Methods | Design: topical treatment of urticaria; randomised controlled 2‐arm study of oxatomide gel vs dechlorpheniramine cream Duration:15 days | |
| Participants | Number of participants randomly assigned: 27; 12 in oxatomide group and 15 in dechlorpheniramine group Sex: 50% female oxatomide group, 33% female dechlorpheniramine group Age of participants, years: oxatomide group: mean 39.6 ± 5.2; dechlorpheniramine group: mean 38.3 ± 2.6 Country and setting: Italy; secondary care Unit of allocation: participant Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: short‐term (15 days) Length of follow‐up: 15 days | |
| Outcomes | Timing of outcome assessment: 15 days Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician and participant | |
| Notes | Study investigators concluded that similar statistically significant improvements were observed in erythema and number of weals. Study authors concluded that both treatments worked | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: ''According to a controlled experimental design completely randomised between patients, half were treated with oxatomide (O) and half with dechlorpheniramine (D)''; no details about method of randomisation |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | High risk | No blinding described Comment: open to bias as 2 different formulations (gel and cream preparations) |
| Blinding of outcome assessment (detection bias) | High risk | No blinding described Comment: open to bias as 2 different formulations (gel and cream preparations) |
| Incomplete outcome data (attrition bias) | Unclear risk | No ITT; 2/27 (13%) lost to follow‐up (did not attend) in dechlorpheniramine group (2/15). No reasons given Comment: unclear whether this degree of loss to follow‐up in a small study introduced a source of bias |
| Selective reporting (reporting bias) | Unclear risk | All outcomes reported but presented graphically; improvement in symptoms expressed as percentage (change in mean score and SD), not by participant number Included some participants with localised urticaria. No measurement of actual dose applied Comment: unclear |
| Other bias | Low risk | None detected. Funder: not stated |
| Methods | Design: randomised single‐blinded single‐centred parallel‐group 2‐arm trial comparing rupatadine with levocetirizine Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 70 (rupatadine n = 35; levocetirizine n = 35) Sex: rupatadine group: male 15 (43%), female 20 (57% ); levocetirizine group: male 16 (45%), female 19 (60%) Age of participants, years: 12 to 60 Unit of allocation: participant Country and setting: India; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: 4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: both | |
| Notes | Study investigators concluded that the incidence of adverse drug reactions was found to be less in the rupatadine group. Rupatadine is a better choice in CSU in comparison with levocetirizine because of a better efficacy and safety profile | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Unclear which group was blinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unclear which group was blinded |
| Incomplete outcome data (attrition bias) | Unclear risk | 54/70 completed (10 did not report for follow‐up, 6 were non‐compliant). No participant stopped treatment because of adverse effects |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes were reported |
| Other bias | Low risk | None detected. Funder: not stated |
| Methods | Design: randomised 3‐arm open study Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 97 (olopatadine 10 mg group 35; olopatadine 5 mg group 30; no medication group 32) Sex: olopatadine 10 mg group: male 31.4%, female 68.6%; olopatadine 5 mg group: male 16.7%, female 83.3%; no medication group: male 28.1%, female 71.9% Age of participants, years (mean): olopatadine 10 mg group 55.2 ± 14.9; olopatadine 5 mg group 55.0 ± 13.5; no medication group 59.1 ± 15.1 Unit of allocation: participant Washout period: none, but run‐in phase of 4 to 6 weeks with all participants receiving 10 mg olopatadine; no cross‐over Country and setting: Japan; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
At least moderate to severe disease Duration of disease greater than 6 weeks | |
| Interventions | Interventions, dose, duration All participants with CSU with a VAS itch score higher than 50 were treated with 10 mg olopatadine hydrochloride daily for 4 to 6 weeks. Of these, participants having a VAS itch score less than 20 were randomly allocated to 1 of 2 groups:
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks Concomitant/rescue treatment not permitted | |
| Outcomes | Timing of outcome assessment: Efficacy end point was defined as length of time the VAS itch score remained less than 50 with no additional treatment and ongoing up to 4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician | |
| Notes | Study investigators concluded that both 10 mg olopatadine and 5 mg olopatadine were effective and were better than no treatment but were not significantly different from each other | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, but no details |
| Allocation concealment (selection bias) | High risk | Allocation not apparently concealed, as the trial was open |
| Blinding of participants and personnel (performance bias) | High risk | No |
| Blinding of outcome assessment (detection bias) | High risk | No |
| Incomplete outcome data (attrition bias) | Unclear risk | Withdrawals noted were 12 out of 35 in 10 mg group, 6 out of 30 in 5 mg group and 22 out of 32 in no medication group. Reasons for all withdrawals were unclear. ITT analysis was carried out |
| Selective reporting (reporting bias) | Low risk | Outcomes reported |
| Other bias | Unclear risk | Funder: not stated |
| Methods | Design: randomised double‐blind 3‐arm cross‐over study of chlorpheniramine vs placebo Duration: not stated | |
| Participants | Number of participants randomly assigned: 24. Numbers in each group not stated Sex: not stated Age of participants: not stated Unit of allocation: cross‐over, chlorpheniramine vs placebo arm data used in this review Country and setting: Australia; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: for all 3 interventions, duration of treatment was not stated Length of follow‐up: not stated | |
| Outcomes | Timing of outcome assessment: not stated Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator | |
| Notes | Study investigators concluded that significant improvement in urticaria was seen with chlorpheniramine and with chlorpheniramine plus cimetidine compared with placebo; no difference was noted between efficacy of active treatments | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation method not stated |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Unclear, no details given |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unclear, no details given |
| Incomplete outcome data (attrition bias) | High risk | Numbers in each group not stated. No adverse events described; 2/24 randomly assigned (8%) lost to follow‐up because of irregular tablet intake |
| Selective reporting (reporting bias) | Unclear risk | Write‐up published as item of correspondence, unclear whether all prespecified outcomes were reported |
| Other bias | Unclear risk | Funder: none Very short report, no further information available |
| Methods | Design: double‐blind randomised multi‐centre 2‐arm trial of loratadine vs placebo Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: unclear; 169 evaluated for safety, 153 evaluated for efficacy, numbers randomly assigned to each group unclear Sex: not stated Unit of allocation: participant Age of participants: not stated Country and setting: USA (primary and secondary) Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: at baseline, then weekly for 4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator | |
| Notes | Study investigators concluded that loratadine is efficacious and safe | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation method unclear |
| Allocation concealment (selection bias) | Unclear risk | No allocation concealment described |
| Blinding of participants and personnel (performance bias) | Unclear risk | No details given about blinding, described as double‐blind |
| Blinding of outcome assessment (detection bias) | Unclear risk | No details given about blinding, described as double‐blind |
| Incomplete outcome data (attrition bias) | High risk | Numbers randomly assigned and number in each group unclear (not stated). 169 evaluated for safety, 153 for efficacy. Treatment failure: loratadine group n = 1; placebo group n = 10; other numbers of and reasons for withdrawal not stated |
| Selective reporting (reporting bias) | High risk | Raw figures not given—only percentages and P values. Outcome measures unclear; statistics not specified. Raw figures not stated; origin of P values not stated (i.e. appropriateness of statistical methods unclear) |
| Other bias | Unclear risk | Funder: none Very short report |
| Methods | Design: randomised double‐blind 3‐arm trial of loratadine vs hydroxyzine vs placebo Duration: 1 week | |
| Participants | Number of participants randomly assigned: 59 total, 18 for urticaria (disaggregated data obtained from study authors for CSU and atopic dermatitis participants) Sex: 29% male, 71% female Age of participants, years: 18 to 63 Unit of allocation: participant Country and setting: USA; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: short‐term (1 week) Length of follow‐up: 1 week | |
| Outcomes | Timing of outcome assessment: 1 week Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator and participant (diary). Diary cards including size and number of weals, erythema and pruritus; scored between 0 and 3; summed for total symptoms score plus global estimation of effect at end | |
| Notes | Study investigators concluded that loratadine is as effective as hydroxyzine in the treatment of urticaria | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not clear; ''randomly assigned'' |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Low risk | No details about blinding given, described as double‐blind |
| Blinding of outcome assessment (detection bias) | Low risk | No details about blinding given, described as double‐blind |
| Incomplete outcome data (attrition bias) | Unclear risk | Number of dropouts from 18 randomly assigned with urticaria not stated. Results expressed as percentages with P values. No outcomes expressed as participant numbers; no baseline values. Unclear how many withdrew with reasons Comment: The report does not contain sufficient information to allow a judgement about whether incomplete outcome data introduced a source of bias |
| Selective reporting (reporting bias) | Unclear risk | Laboratory tests conducted at start, unclear whether these were monitored as an outcome. Erythema not reported as an outcome (but included in overall symptom score) Comment: unclear whether outcomes were fully reported. No further information was available from investigators |
| Other bias | Unclear risk | Funder: none. Short report with insufficient information to allow a judgement about risk of other bias |
| Methods | Design: randomised double‐blind multi‐centre parallel 2‐arm placebo‐controlled study of desloratadine 5 mg vs placebo Duration: 6 weeks | |
| Participants | Number of participants randomly assigned: 226 (116 desloratadine; 110 placebo) Sex: 27% male, 73% female desloratadine group; 24% male, 76% female placebo Age of participants, years: desloratadine 41.8 (range 13‐80); placebo 39.2 (range 13‐84) Unit of allocation: participant Country and setting: US, Chile, Canada, Venezuela, Germany, Norway, Sweden, Belgium; setting unclear Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: 6 weeks | |
| Outcomes | Timing of outcome assessment: 1, 2, 4 and 6 weeks (visits at screening, at baseline (day 1), on day 4, at weeks 1, 2, 4 and 6. Efficacy and safety assessments made at visit day 4 and weeks 1, 2, 4 and 6) Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator and participant | |
| Notes | Study investigators concluded that at week 1: mean improvement from baseline in reflective pruritus score significantly greater in desloratadine group; overall more effective than placebo; significant improvement in total symptoms score and interference in sleep and daily activities in desloratadine group; reduction in number and size of largest hive significantly better in desloratadine group Statistically significant improvements noted by day 2 of study Week 6: statistically significant improvement in pruritus from baseline in desloratadine group compared with placebo; desloratadine‐treated participants had significantly greater control of morning instantaneous total symptoms score compared with placebo patients. Overall, desloratadine was statistically significantly better than placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 536): "1:1 ratio according to computer generated schedule" "Blocks of 4 using random numbers generated by SAS function UNIFORM with seed based on clock‐time" Comment: adequate randomisation |
| Allocation concealment (selection bias) | Low risk | Quote (page 537): "A set of sealed envelopes containing the identification of test drug corresponding to each subject was provided to each center. This enabled the investigator to identify the treatment assignment of individual subjects in the event of an emergency without compromising the blinding of other subjects. The randomisation schedule for blinding of treatments was disclosed only after study completion" Comment: Sealed envelopes indicate that this was probably done |
| Blinding of participants and personnel (performance bias) | Low risk | Quote (page 537): "Desloratadine, in 5 mg tablets, and placebo were identical in appearance and packaged identically in sealed coded envelopes. All study personnel were blinded to the identity of medication. A set of sealed envelopes containing the identification of test drug corresponding to each subject was provided to each center. This enabled the investigator to identify the treatment assignment of individual subjects in the event of an emergency without compromising the blinding of other subjects. The randomisation schedule for blinding of treatments was disclosed only after study completion" |
| Blinding of outcome assessment (detection bias) | Low risk | Quote (page 537): "Desloratadine, in 5 mg tablets, and placebo were identical in appearance and packaged identically in sealed coded envelopes. All study personnel were blinded to the identity of medication. A set of sealed envelopes containing the identification of test drug corresponding to each subject was provided to each center. This enabled the investigator to identify the treatment assignment of individual subjects in the event of an emergency without compromising the blinding of other subjects. The randomisation schedule for blinding of treatments was disclosed only after study completion" |
| Incomplete outcome data (attrition bias) | High risk | Lost to follow‐up: desloratadine 19/116 (19%); placebo 35/110 (31%) Treatment failure: desloratadine n = 14, placebo n = 29; other adverse events: desloratadine n = 3, placebo n = 2, non‐compliance each group n = 1, loss to follow‐up: desloratadine n = 1, placebo n = 2, lack of desire to continue placebo n = 1 Adverse events: serious (requiring withdrawal): desloratadine: 3 (1 bronchitis/sinusitis, 1 URTI, 1 nausea); placebo: 2 (1 vomiting, 1 sedation); minor adverse events: desloratadine: headache n = 18, nausea n = 7, dry mouth n = 6; placebo: headache n = 11, nausea n = 2, dry mouth n = 5 Actual participant numbers missing from results (percentages of improvement in scores and statistical significance only given); ITT stated by authors but unclear because of lack of provided data Comment: judged as high risk because of high dropout rate |
| Selective reporting (reporting bias) | Unclear risk | Prespecified outcomes were reported, other than compliance Excluded participants previously non‐responsive to antihistamines; may involve selective reporting of positive results |
| Other bias | Unclear risk | Funder: Schering‐Plough Research Institute |
| Methods | Design: randomised double‐blind parallel‐group study; desloratadine 5 mg, 10 mg, 20 mg Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 314 Country and setting: Germany; setting unclear Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Intermediate duration of intervention (4 weeks) | |
| Outcomes | Timing of outcome assessment: 4 weeks Primary outcomes of the trial
Clinician or participant report: UAS participant reported | |
| Notes | Study investigators provided no conclusions Study number P04849; also indexed as EudraCT: 2006‐001449‐33. Results available online | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no details given |
| Allocation concealment (selection bias) | Unclear risk | No details given |
| Blinding of participants and personnel (performance bias) | Low risk | Participants and personnel were blinded to treatment group |
| Blinding of outcome assessment (detection bias) | Low risk | Outcome assessors were blinded to treatment group |
| Incomplete outcome data (attrition bias) | Low risk | Group 5 mg 12/106
Group 10 mg: 9/104
Group 20 mg: 10/104
Withdrawal and losses accounted for and balanced between groups ITT analysis carried out |
| Selective reporting (reporting bias) | Low risk | All outcomes reported |
| Other bias | Unclear risk | Underpowered because of poor recruitment; thus study is inconclusive Funder: Schering‐Plough |
| Methods | Design: multi‐centre double‐blind randomised placebo‐controlled 5‐arm parallel study with 4 different doses of fexofenadine vs placebo Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 468 Sex: 30% male, 70% female placebo; 31% male, 69% female 20 mg; 29% male, 71% female 60 mg; 33% male, 68% female 120 mg; 27% male, 73% female 240 mg Age of participants, years: range 12 to 65 Country and setting: USA; secondary care Unit of allocation: participant Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: 1, 2, 3, 4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator and participant | |
| Notes | Study investigators concluded that all fexofenadine groups were significantly better than placebo groups re pruritus, reduction in weal score and reduced interference with sleep/activities | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation unclear, no details given; described as randomised |
| Allocation concealment (selection bias) | Unclear risk | No details given |
| Blinding of participants and personnel (performance bias) | Unclear risk | Described as 24‐hour single‐blind lead‐in, followed by 4‐week double‐blind treatment, but no details given |
| Blinding of outcome assessment (detection bias) | Low risk | Described as 24‐hour single‐blind lead in, followed by 4‐week double‐blind treatment, but no details given about blinding of outcome assessors |
| Incomplete outcome data (attrition bias) | High risk | 186/468 randomly assigned lost to follow‐up as follows: 437 had at least 1 postbaseline adverse event assessment; 418 had at least 1 postbaseline 12‐hour mean pruritus score assessment (i.e. for safety analysis, losses of n = 31 (6%); for efficacy assessments, losses of n = 50 (10.6%)) In text, study authors state that only 282 participants completed the study (losses n = 186 (40%)) Serious adverse events present in all groups but not specified. Most common adverse events reported (not specified which led to withdrawal of treatment) were as follows: headache ˜ 25%, URTI ˜ 8%, nausea 5%, dyspepsia 5%, diarrhoea 2%, gastroenteritis 2%, "pain" 4%, abdominal pain 2%, myalgia 4% Frequencies similar across all groups including placebo Incorrectly stated ITT; large number of withdrawals not accounted for Table implies that figures states 325 were evaluated at end of study (4 weeks). This is not resolved in text Study authors state ITT analysis but did not include all participants randomly assigned at the beginning of the study (31 with no data and not included in the analysis) Comment: Discrepancies in number of dropouts and large number of withdrawals suggest that attrition bias could have been introduced |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes were reported but excluded persons known to be unresponsive to antihistamines; serious adverse events present in all groups but not specified Comment: unclear whether selective reporting and participant selection introduced a source of bias |
| Other bias | Unclear risk | Funder: Hoechst Marion Roussel |
| Methods | Design: randomised double‐blind placebo‐controlled 3‐arm study of desloratadine alone vs desloratadine with montelukast (H1‐ and H2‐antagonists) vs placebo Duration: 6 weeks | |
| Participants | Number of participants randomly assigned: 81 (27 in each group) Sex: 24% male, 76% female desloratadine; 15.4% male, 84.6% female desloratadine plus montelukast; 40% male, 60% female placebo Age of participants, years: 37.5 ± 10.9 (desloratadine plus placebo); 35.6 ± 12.8 (desloratadine plus montelukast); 36.8 ± 10.7 (placebo) Country and setting: Italy; secondary care Unit of allocation: participant Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
1‐week single‐blind placebo run‐in ended with 1‐week single‐blind placebo washout. Concomitant medications not allowed during course of trial Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: 6 weeks | |
| Outcomes | Timing of outcome assessment: baseline, 3 weeks and 6 weeks (participants examined by physician 4 times over 8‐week period: first after 1‐week placebo run‐in, second after 3 weeks’ active treatment, third at end of treatment, final at end of placebo washout week) Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator and participant | |
| Notes | Study investigators concluded that both treatments were significantly more effective than placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation unclear |
| Allocation concealment (selection bias) | Unclear risk | Methods of allocation concealment not stated. Sealed envelopes in pharmacy Comment: Not entirely clear whether sealed envelopes related to blinding of medication or allocation concealment, judged as unclear |
| Blinding of participants and personnel (performance bias) | Low risk | 1‐week single‐blind placebo run‐in and run‐out. Double‐blind study, adequately blinded Quote (page 1402): ''Patients were not informed that the treatment would be divided into specific periods'' "The tablets were encapsulated in double blind fashion and sealed in envelopes by a pharmacist along with the instruction sheets at the beginning of the trial. All treatments were dispensed by a third party" |
| Blinding of outcome assessment (detection bias) | Low risk | Outcome assessors were blinded as above Comment: probably done, inferred from text |
| Incomplete outcome data (attrition bias) | Low risk | 5/81; 5 lost to follow‐up (6%) Number and reasons for withdrawal:
Comment judged as low risk, as withdrawals with reasons were given and were balanced between the 2 groups No ITT; most results reported as number of participants showing any improvement, with no indication of effect size |
| Selective reporting (reporting bias) | Unclear risk | Attempt at QoL measurement, but DLQI into non‐validated scale; HRQoL results unclear—presented only graphically; y‐axis (if consistent with other graphs) indicates only % participants showing any improvement (no indication of effect size) Baseline data unclear: Symptom severity scale appears to be out of 12, yet at baseline, mean is stated as about 60 in each group |
| Other bias | Unclear risk | Funder: none |
| Methods | Design: randomised double‐blind 2‐arm placebo‐controlled study of levocetirizine 5 mg vs placebo Duration: 6 weeks | |
| Participants | Number of participants randomly assigned:106, n = 53 levocetirizine, n = 53 placebo Sex: 33% male, 67% female levocetirizine; 41% male, 59% female placebo Age of participants, age: mean 41.1 (SD 22‐71) levocetirizine; mean 39 (SD 22‐69) placebo Country and setting: Italy; secondary care Unit of allocation: participant Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
1‐week placebo run‐in (single‐blind), then treatment for 6 weeks, then 1‐week placebo washout at end of study Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: 8 weeks i.e. follow‐up extended after cessation of therapy. | |
| Outcomes | Timing of outcome assessment: screening, after placebo run‐in, after 3 weeks of active treatment, after 1 week of washout (at 6 weeks) Examined by physician 4 times over 8‐week period: first after placebo run‐in (1 week); second after 3 weeks’ active treatment; third after 6 weeks’ active treatment; fourth after final week of placebo—participants complete visual analogue scale for overall severity of urticaria since last visit (0 = none, 10 = worst) Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: both | |
| Notes | Study investigators concluded that mean total symptoms score decreased by 81% vs 1% by end of study period in levocetirizine vs placebo group, respectively Treatment group had statistically significant decrease in number of weals at all visits (overall 79% reduction in score); also statistically significant decrease in urticarial episodes and size of weals (75% reduction); pruritus also (85% reduction) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation unclear (''randomly assigned'') |
| Allocation concealment (selection bias) | Unclear risk | Sealed envelopes in pharmacy. Comment: not entirely clear whether sealed envelopes related to blinding of medication or allocation concealment, judged as unclear |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study; participants not informed that treatments would be divided up into specific periods: 1‐week placebo run‐in (single‐blind), then treatment for 6 weeks, then 1‐week placebo washout at end of study. Double‐blind study: ''tablets were encapsulated in a double blind fashion and sealed in envelopes by a pharmacist together with instruction sheets.'' 1‐week placebo run‐in (single‐blind), then treatment for 6 weeks, then 1 week placebo washout at end of study. Medications dispensed by third party |
| Blinding of outcome assessment (detection bias) | Low risk | Outcome assessors were blinded as above Comment: probably done, inferred from text |
| Incomplete outcome data (attrition bias) | Low risk | 100/106 randomly assigned participants completed Withdrawals: levocetirizine n = 2, n = 4 in placebo group, dropped out in placebo run‐in phase because of non‐compliance n = 2, heart attack n = 1, needed to take oral steroids for aggravated urticaria n = 3 (2 in placebo group and 1 in levocetirizine group) No ITT Comment: low number of dropouts, evenly balanced between groups, not thought to contribute bias |
| Selective reporting (reporting bias) | Unclear risk | Results not clearly reported. Attempt at measuring quality of life, but not with validated scale; results of this inadequately reported. Quality of life scores statistically significantly improved from baseline in levocetirizine group but not in placebo group (no scores given) |
| Other bias | Low risk | Funder: none |
| Methods | Design: randomised 2‐arm multi‐centre (10 centres) parallel‐group comparison of mizolastine vs placebo Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 78 (39 each in mizolastine and placebo) Sex: 40% male, 60% female Age of participants, years: 40 ± 13 Unit of allocation: participant Country and setting: Germany; research clinic Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration Placebo run‐in periods (i.e. variable washout period of any other medications (depending on previous medication type) before study commenced)
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: at days 0, 14 and 28 Primary outcomes of the trial:
Secondary outcomes of the trial
Clinician or participant report: clinician and participant | |
| Notes | Study report written in German Study investigators concluded that mizolastine demonstrates clinical and statistical superiority over placebo in the treatment of CSU, and showed a good safety profile | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated to be randomised, unclear as method not stated |
| Allocation concealment (selection bias) | Unclear risk | Unclear, no details given |
| Blinding of participants and personnel (performance bias) | Unclear risk | Stated to be double‐blind, identical placebos given |
| Blinding of outcome assessment (detection bias) | Unclear risk | Stated to be double‐blind, but no details given |
| Incomplete outcome data (attrition bias) | Low risk | Stated to be ITT analysis. 19 participants dropped out: 11/39 mizolastine, 8/39 placebo. Reasons given: 2 in total for undesirable effects, 12 in total for lack of effect, 5 in total for other reasons (page S26, table 2) |
| Selective reporting (reporting bias) | Unclear risk | Unclear; not all physiological measures were reported in full in the study report |
| Other bias | Low risk | None detected. Funder: not stated |
| Methods | Design: randomised parallel‐group 2‐arm study of desloratadine vs placebo Duration: 42 days | |
| Participants | Number of participants randomly assigned: 137 (desloratadine n = 65, placebo n = 72) Sex: gender not stated Age of participants, years: 18, mean age not stated Unit of allocation: participant Country and setting: France (multicentre), research clinics Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: 42 days | |
| Outcomes | Timing of outcome assessment: 14 days and 42 days Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and clinician | |
| Notes | Study investigators concluded that desloratadine was effective from the first dose and throughout 6 weeks in CSU | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated, described as randomised |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not stated |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) | Unclear risk | Unclear how many out of 137 randomly assigned participants completed the study |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Unclear risk | Funder: Schering‐Plough. Short conference abstract |
| Methods | Design: multi‐centre 2‐arm randomised double‐blind trial of desloratadine vs placebo Duration: 42 days | |
| Participants | Number of participants randomly assigned: 142 (desloratadine n = 65, placebo n = 77) Age, years: > 18; desloratadine 41.2 ± 15.4; placebo 41.5 ± 15.2 Gender: desloratadine 36.9% male, 63.1% female; placebo 40.3% male, 59.7% female Unit of allocation: participant Country and setting: France; set in 40 dermatology centres Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: up to 6 weeks | |
| Outcomes | Timing of outcome assessment: days 7, 14, 42 (patient diaries collected at these times or at time of early termination, if applicable) Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant diaries, general (non‐directive) questions by investigators, or clinical examination | |
| Notes | Study investigators concluded that desloratadine was shown to be significantly superior to placebo in improving severity of pruritus as measured by reflective pruritus scores measured between days 0 and 14 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 39): "following screening (visit 1), a computer‐generated allocation code was used to randomly assign patients" |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not stated |
| Blinding of participants and personnel (performance bias) | Low risk | Described as double‐blind, medication was provided to participants in a numbered container based on their randomisation code |
| Blinding of outcome assessment (detection bias) | Unclear risk | Described as double‐blind but method of investigator blinding not stated |
| Incomplete outcome data (attrition bias) | Low risk | 85/142 randomly assigned participants completed the study Figure 1 of the study report provides an account of dropouts from each group. 142 randomly assigned (65 desloratadine, 77 placebo). 5 withdrawn as received no treatment or had no baseline data. Of remaining 137 (65 desloratadine, 72 placebo), 85 completed (49 desloratadine, 36 placebo). 16 withdrew from desloratadine group (12 lack of efficacy, 1 adverse event (pregnancy, not treatment‐related), 1 withdrew consent, 1 was lost to follow‐up, 1 other reason). 36 withdrew from placebo group (34 lack of efficacy, 2 lost to follow‐up described as 'loss of sight' in fact lost to follow‐up) |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes were reported, but results are presented as percentages or as graphs with statistical significance Comment: endpoint at 6 weeks is not clear |
| Other bias | Unclear risk | Funder: Schering‐Plough Research Institute |
| Methods | Design: randomised double‐blind 2‐arm parallel‐group study of loratadine vs cetirizine Duration: 2 weeks | |
| Participants | Number of participants randomly assigned: 46 (22 loratadine, 18 cetirizine) Sex: not stated Age of participants: not stated Unit of allocation: participant Country and setting: not stated; investigators located in USA and Canada Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: short‐term (2 weeks) Length of follow‐up: 2 weeks | |
| Outcomes | Timing of outcome assessment: baseline and days 7 and 14 Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician and participant diary card | |
| Notes | Study investigators concluded that loratadine and cetirizine were well tolerated with comparable efficacy | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated to be randomised, methods not described |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Stated to be double‐blind, no details given |
| Blinding of outcome assessment (detection bias) | Unclear risk | Stated to be double‐blind, no details given |
| Incomplete outcome data (attrition bias) | Low risk | 40/46 complete (22 loratadine, 18 cetirizine). These non‐completers had fewer than 7 days of therapy or lack of valid follow‐up visit or both |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Unclear risk | Funder: Schering‐Plough, USA |
| Methods | Design: multi‐centre double‐blind randomised parallel‐group study comparing fexofenadine 60 mg, 120 mg, 180 mg, 240 mg or placebo, each once daily Duration: 6 weeks | |
| Participants | Number of participants randomly assigned: 222 (details unclear) Age of participants, years: placebo 43 ± 13; fexofenadine 60 mg 44 ± 17; fexofenadine 120 mg 45 ± 12; fexofenadine 180 mg 43 ± 15; fexofenadine 240 mg 44 ± 14 Overall age, years: 44 ± 14 Sex (% female): placebo 54%, fexofenadine 60 mg 58%, fexofenadine 120 mg 47%, fexofenadine 180 mg 68%, fexofenadine 240 mg 59% Total female: 58% Duration of symptoms: 3 years ± 5 Severity of urticaria: unclear Unit of allocation: participant Country and setting: France, UK, Germany; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
(each once daily) Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: 6 weeks | |
| Outcomes | Timing of outcome assessment: weekly assessment Total symptoms score (TSS) (0‐4 for number of weals; 0‐3 for itching intensity; 0‐7 combined TSS) Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: both | |
| Notes | Study investigators concluded that good or excellent response in 60 mg group = 63%, 120 mg group 50%, 180 mg group 64%, 240 mg group 55% and placebo group 41%; not clear whether timing for this result but may be 6 weeks Fexofenadine is effective at 120 mg and above from week 1 as compared with placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants non‐responsive to other antihistamine treatment excluded Method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not stated how |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not stated how |
| Incomplete outcome data (attrition bias) | Low risk | No ITT. 76 participants withdrew before completion: 30 lost from placebo, 14 from 120 mg group, 16 from 180 mg group and 8 from 240 mg group Treatment group reason for withdrawal: lack of effect 13%, adverse events 7%, patient request 6% Placebo group: lack of effect 33% and adverse events 20% |
| Selective reporting (reporting bias) | Unclear risk | No ITT; study says weekly as well as fortnightly; all active drug groups put together and reported only selectively |
| Other bias | Unclear risk | Funder: Hoechst Marion Roussel |
| Methods | Design: randomised double‐blind multi‐centre placebo‐controlled 2‐arm study of ebastine 10 mg daily vs placebo Duration: 14 days | |
| Participants | Number of participants randomly assigned: 204 (ebastine 100, placebo 104) Sex: not stated Age of participants: not stated Unit of allocation: participant Country and setting: Spain; secondary care (outpatient clinics) Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: short‐term (14 days) Length of follow‐up: 14 days | |
| Outcomes | Timing of outcome assessment: baseline, 7 days, 14 days Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: both | |
| Notes | Study investigators concluded that a significantly greater reduction in weal size and weal number was seen in ebastine group over placebo. Pruritus significantly less in ebastine group | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind study; matching placebo capsules; unclear whether blinding was adequate for both participants and personnel |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unclear, no details given |
| Incomplete outcome data (attrition bias) | Low risk | Losses to follow‐up: 27/204 (13%), ebastine 9/100 (9%), placebo 18/104 (17%) Withdrawals due to lack of efficacy: ebastine n = 3 and placebo n = 13; poor tolerability: ebastine n = 1; lack of efficacy plus poor tolerability: ebastine n = 1, placebo n = 3; other reasons not due to treatment: ebastine n = 4, placebo n = 2 Withdrawals with reasons stated and balanced between the 2 groups No ITT |
| Selective reporting (reporting bias) | Unclear risk | No clear definition of outcomes, unclear how outcome assessments were used to generate assessments of (Quote) (page 52): "cure, improvement, no change, worsening" Age and sex of participants not known |
| Other bias | Unclear risk | Funder: Almirall Note: Groups were well matched for age, sex, duration of urticaria, previous treatment and response to prior antihistamine therapy |
| Methods | Design: double‐blind randomised placebo‐controlled 2‐arm study of ketotifen (mast cell stabiliser) vs placebo Duration: 2 weeks | |
| Participants | Number of participants randomly assigned: 30 (16 ketotifen and 14 placebo) Sex: 25% male, 75% female ketotifen; 26% male, 64% female placebo Age of participants, years: mean 30.4 Unit of allocation: participant Country and setting: Thailand; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
2‐week run‐in: participants allowed to take chlorpheniramine 4 mg prn up to 6‐hourly; then randomly assigned to ketotifen or placebo and still allowed to take chlorpheniramine concomitantly; numbers of chlorpheniramine tablets taken recorded Duration of intervention: short‐term (2 weeks) Length of follow‐up: 2 weeks | |
| Outcomes | Timing of outcome assessment: baseline and 2 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator and participant diaries | |
| Notes | Study investigators concluded that ketotifen was significantly better than placebo. Chlorphenamine requirement was dropped in significantly more participants taking ketotifen than placebo (94% vs 7%) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to 2 groups, method not described |
| Allocation concealment (selection bias) | Unclear risk | Third party sealed envelopes with code number, containing active treatment or placebo prepared and sealed by a third person, but unclear if this refers to blinding or to concealment of allocation |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study; identical white tablets provided to participants, blinded to participants, as supplied in sealed coded envelopes prepared by a third party |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unclear whether outcome assessors were blinded and how |
| Incomplete outcome data (attrition bias) | Low risk | No losses to follow‐up: 30 (16 ketotifen and 14 placebo) |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes were reported Comment: The 'slightly effective' and 'ineffective' evaluation of patient score put together as ineffective |
| Other bias | Unclear risk | Funder: Sandoz Allowed concomitant treatment with chlorpheniramine such that positive results of ketotifen group might be due to this alone or to the combination of ketotifen plus chlorpheniramine; very small numbers; sponsored by manufacturer |
| Methods | Design: randomised double‐blind multi‐centre 2‐arm trial comparing emedastine and loratadine Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 192 (emedastine n = 84, loratadine n = 77) Sex: loratadine: 34.5% male, 65.5% female; emedastine: 25% male, 75% female Age of participants, years: loratadine: 42.6 ± 14.7; emedastine:43.4 ± 13.3 Country and setting: Italy, France, Hungary, Czech Republic; secondary care Unit of allocation: participant Inclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks (plus optional visits 2 weeks after cessation of therapy). | |
| Outcomes | Timing of outcome assessment: 1, 2 and 4 weeks (plus optional visits at week 2 and 2 weeks after discontinuation of treatment)
Secondary outcomes of the trial
Clinician or participant report: both | |
| Notes | Study investigators concluded that no significant differences between treatments were noted at 4 weeks according to investigator and participant scores; mean symptom scores improved significantly from baseline in both groups | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer list prepared by sponsor |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes containing code breaks were given to each centre |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind, double‐dummy study; 2 identical capsules; placebo run‐in |
| Blinding of outcome assessment (detection bias) | Low risk | Double‐blind, double‐dummy study; 2 identical capsules; placebo run‐in, but unclear whether outcome assessors were specifically blinded to allocation Comment: probably done |
| Incomplete outcome data (attrition bias) | Unclear risk | 152/192 completed 12 participants excluded after randomisation, as they took prohibited antihistamines. 19 more excluded after randomisation, as they failed to report scores for hives/itching during placebo run‐in period. Only the remaining 161 participants were included in ITT. 31 lost to follow‐up (16%). Per‐protocol (PP) analysis included only 153 participants—8 had major protocol violations or dropped out Two were withdrawn because of serious adverse events: 1 suicide attempt, 1 fracture following a fall in emedastine group No ITT Comment: All dropouts accounted for but not clearly by group |
| Selective reporting (reporting bias) | Low risk | No selective reporting Comment: excluded participants unresponsive to antihistamines |
| Other bias | Unclear risk | Funder: Saluc‐Pharma S.A. Study drug manufactured and packaged by sponsor; no power calculation |
| Methods | Design: randomised multi‐centre randomised parallel‐group double‐blind 2‐arm study comparing levocetirizine 5 mg and desloratadine 5 mg Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 886 (levocetirizine n = 438, desloratadine n = 448) Sex: levocetirizine 35.2% male, 64.8% female; desloratadine 36.2% male, 68.3% female Age of participants, years (range): levocetirizine 43.36 (18‐79.2); desloratadine 42.85 (18.1‐81.3) Unit of allocation: participant Country and setting: multi‐centre Germany and UK; secondary Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 5 weeks i.e. follow up extended after cessation of therapy. | |
| Outcomes | Timing of outcome assessment: 4 scheduled visits over a period of 5 weeks: screening visit 1 (V1; week ‐1), randomisation visit (V2; week 0), control visit (V3; week 1) and final visit (V4; week 4) Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and clinician | |
| Notes | Study investigators concluded that levocetirizine 5 mg was significantly more efficacious than desloratadine 5 mg in the treatment of CSU symptoms | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 597): "Randomization to the study drug was achieved by allocation of a unique study number to each subject and a computer‐generated sequential randomisation number provided by the Biostatistics Department of the study sponsor (UCB S.A., Brussels, Belgium)" |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote (page 597): "capsules identical in shape, size and colour to allow a double‐blind design" |
| Blinding of outcome assessment (detection bias) | Unclear risk | Unclear how outcome assessors were blinded |
| Incomplete outcome data (attrition bias) | Unclear risk | 832/886 completed
In NCT00264303, reasons for withdrawal were given but do not correspond with the number of participants analysed—n = 25 levocetirizine and n = 29 desloratadine—because of adverse event, lack of efficacy, loss to follow‐up, participant preference to withdraw, other reasons. However, number of participants analysed was n = 434 in levocetirizine group and n = 443 (877 total) in desloratadine group (only 9 losses out of 886 specified, reasons unclear). Judged as unclear risk |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Unclear risk | Funder: UCB |
| Methods | Design: randomised double‐blind placebo‐controlled parallel‐group 2‐arm study of desloratadine vs placebo Duration: 6 weeks | |
| Participants | Number of participants randomly assigned: 190 (95 in each group) Sex: 29% male, 71% female desloratadine; 22% male, 78% female placebo Age of participants, years: 12 to 79 Country and setting: USA and Germany; secondary care Unit of allocation: participant Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Once daily for 6 weeks (sufficient time for washout of any medications before study was employed) Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: 6 weeks | |
| Outcomes | Timing of outcome assessment: days 1 and 4, then weeks 1, 2, 4, 6 Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator and participant | |
| Notes | Study investigators concluded that results from week 1 were maintained throughout study duration Desloratadine significantly superior to placebo in reducing average mean reflective pruritus score (56% vs 21%) and total symptoms score (51.6% vs 19.3%). Interference with sleep and daily activities, number of weals, size of largest weal all significantly reduced by desloratadine vs placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated schedule, randomly assigned |
| Allocation concealment (selection bias) | Unclear risk | Unclear, no details given |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study; matched placebo |
| Blinding of outcome assessment (detection bias) | Unclear risk | Double‐blind study; unclear how outcome assessors were blinded to treatment allocation |
| Incomplete outcome data (attrition bias) | Unclear risk | Losses to follow‐up: desloratadine: 19/95 (20%); placebo: 32/95 (34%) Treatment failure: desloratadine n = 13, placebo n = 21; non‐compliance: desloratadine n = 3, placebo n = 6; adverse events: desloratadine n = 3, placebo n = 2; other losses to follow‐up: desloratadine n = 0, placebo n = 2; did not wish to continue: placebo n = 1 Adverse events: serious adverse events (requiring treatment withdrawal): desloratadine: 3 not specified (not life threatening); placebo: 2 not specified (not life threatening) Comment: high level of loss to follow‐up; more losses in the placebo group; unclear whether this contributed to bias |
| Selective reporting (reporting bias) | Unclear risk | Desloratadine significantly superior to placebo in reducing average mean reflective pruritus score (74% vs 49%). Numbers for total symptoms score not given at 6 weeks, but said to be significant. Actual reductions in scores not stated, only percentages; unclear how clinically significant these reductions are |
| Other bias | Unclear risk | Funder: Schering‐Plough Research Institute |
| Methods | Design: randomised double‐blind cross‐over 3‐arm study comparing acrivastine vs hydroxyzine vs placebo Duration: 5 days each treatment | |
| Participants | Number of participants randomly assigned: 21 Sex: 47% female Age of participants, years: 18‐70, mean 38.3 Country and setting: Finland and UK; secondary Unit of allocation: cross‐over participants Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
3‐day washout initially; then 2‐day washout period between treatments Duration of intervention: short‐term (5 days per intervention) Length of follow‐up: 5 days for each treatment | |
| Outcomes | Timing of outcome assessment: at 5 days for each treatment Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: investigator and participant (daily diary) | |
| Notes | Study investigators concluded that participant data showed no differences between active treatments; both better than placebo (P value < 0.05) Physician data showed active treatment better than placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation unclear, described as randomised |
| Allocation concealment (selection bias) | Unclear risk | Unclear. Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind study. Method of blinding not stated |
| Blinding of outcome assessment (detection bias) | Unclear risk | Double‐blind study. Method of blinding not stated |
| Incomplete outcome data (attrition bias) | High risk | Incomplete reporting of data; reasons for withdrawal not stated. No ITT 3/21 lost to follow‐up (34%) |
| Selective reporting (reporting bias) | High risk | Investigators appear to have made subjective decision on best treatment. No raw data—mean scores only. Adverse effects of drowsiness significantly more prevalent with hydroxyzine than with placebo, but numbers of participants experiencing this not stated |
| Other bias | Unclear risk | Funder: Wellcome Research Laboratories |
| Methods | Design: randomised 2‐arm parallel‐group study comparing ketotifen and fluoxetine Duration: 6 weeks | |
| Participants | Number of participants randomly assigned: 60 (30 in each group) Sex: 41% female, 59% male Unit of allocation: participant Age of participants, years (SD): 19‐74 (42.08 ± 19.24) Country and setting: Turkey, research clinic Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (6 weeks) Length of follow‐up: 6 weeks | |
| Outcomes | Timing of outcome assessment: baseline, weekly and at 6 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: unclear | |
| Notes | Study investigators concluded that significantly greater improvement in symptom score was seen in the ketotifen group (P value < 0.001); fluoxetine led to significant improvement in number of lesions, degree of itch and angio‐oedema | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation unclear ("randomly divided") |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | High risk | No blinding (1 treatment twice daily and the other treatment 4 times daily) |
| Blinding of outcome assessment (detection bias) | High risk | No blinding (1 treatment twice daily and the other treatment 4 times daily) |
| Incomplete outcome data (attrition bias) | Unclear risk | No raw data; adverse events not mentioned, nor withdrawals from study |
| Selective reporting (reporting bias) | Unclear risk | Limited report of outcomes, as reported only as abstract for poster presentation |
| Other bias | Unclear risk | Funder: not stated. Very short poster abstract |
| Methods | Design: prospective randomised double‐blind cross‐over trial Duration: 5 days | |
| Participants | Number of participants randomly assigned: 24 Sex: 75% female Age of participants, years: mean 45, range 19 to 68 Unit of allocation: cross‐over (first phase only considered) Country and setting: Bulgaria; tertiary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration Initial in‐hospital treatment, assessment of effectiveness and tolerability of levocetirizine 10 and 20 mg vs hydroxyzine 100 and 200 mg. This was done in a double‐blind fashion on alternate‐day regimens
Length of follow‐up: 5 days on each treatment Short duration of intervention (5 days) Length of follow‐up: 5 days | |
| Outcomes | Timing of outcome assessment: day 5 Primary outcomes of the trial
Secondary outcomes of the trial
Quality of life measures: median CU‐Q2oL scores Clinician or participant report: physicians calculated weal scores and severity of pruritus | |
| Notes | Study investigators concluded that higher than standard doses of cetirizine can improve quality of life in participants discontinuing steroid treatment. Addition of hydroxyzine does not seem to provide benefit but causes increased daytime somnolence Study ID NCT01250652 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stated to be randomised. Randomisation, ensuring balanced numbers of cases in the 2 treatment arms, was performed pair‐wise at a specialised website (http://www.randomizer.org/) Comment: Exposure to study medication after randomisation to determine tolerability of medications on alternate‐day regimens may have compromised randomisation through a potential carry‐over effect (insufficient washout period between phases) |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind: “Medication was given morning and evening in opaque gelatine capsules that were prepared by a technician who was not aware of the clinical work” |
| Blinding of outcome assessment (detection bias) | Low risk | “The investigators, who were blinded to the treatment groups” |
| Incomplete outcome data (attrition bias) | Unclear risk | 25 participants were initially randomly assigned. 24 participants completed the study—1 withdrawal after randomisation (personal reasons) |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Unclear risk | Funder: UCB Pharma. Short report, conference abstract |
| Methods | Design: randomised multi‐centre double‐blind 2‐arm placebo‐controlled parallel study of fexofenadine vs placebo Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 160 Sex, fexofenadine: 18% male, 72% female; placebo: 30% male, 70% female Age of participants, years: fexofenadine: 40 ± 11; placebo: 38 ± 13 Country and setting: US; secondary care Unit of allocation: participant Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 to 6 weeks i.e. follow‐up extended after the cessation of therapy. | |
| Outcomes | Timing of outcome assessment: 4 or 6 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: Participants completed self‐administered questionnaire, DLQI (score range 0‐30), work productivity and activity impairment questionnaire (WPAI—0‐100%, high = greater productivity) at entry, interim visit (15 ± 2 days’ treatment), final visit (30 ± 4 days) or early termination | |
| Notes | Study investigators concluded that significant differences were demonstrated in only 1 study out of 2. Increase in "work productivity," "overall work productivity," regular daily activities significantly higher in fexofenadine group compared with placebo group (both studies); no differences between groups re time missed from class/work | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: ''Patients were randomised to receive 60 mg fexofenadine HCl twice daily or placebo twice daily...'' |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: ''24 hour single‐blind placebo lead‐in, and a subsequent 4‐week double blind treatment period.'' Unclear how blinding was achieved |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote: ''24 hour single‐blind placebo lead‐in, and a subsequent 4‐week double blind treatment period.'' Unclear how blinding was achieved |
| Incomplete outcome data (attrition bias) | Unclear risk | No mention of whether there were dropouts; therefore unable to corroborate ITT analyses as reported in Study 1 |
| Selective reporting (reporting bias) | Unclear risk | Ambiguity re duration of trial, as reported methods say 4 weeks and results say 4 to 6 weeks. DLQI measures reported but not as proportions of participants with 50% or greater improvement in quality of life measurements whilst taking H1‐antihistamines; therefore not possible to include DLQI data |
| Other bias | Unclear risk | Funder: Hoechst Marion Roussel. Study investigators report 2 identical studies in this paper; if studies identical, unclear why not combined as a single study |
| Methods | Design: randomised multi‐centre double‐blind 2‐arm placebo‐controlled parallel study of fexofenadine vs placebo Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 165 Sex: fexofenadine: 26% male, 74% female; placebo: 27% male, 77% female Age of participants, years: fexofenadine: 38 ± 13; placebo:40 ± 13 Country and setting: USA; secondary care Unit of allocation: participant Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 to 6 weeks i.e. follow‐up extended after the cessation of therapy. | |
| Outcomes | Timing of outcome assessment: 4 or 6 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant (for QoL questionnaires) and clinician | |
| Notes | Study investigators reported that significantly better improvement in fexofenadine was achieved in both studies in symptoms/feelings, daily activities, work or school and personal relations; for leisure and treatment, significant difference was demonstrated in 1 study out of 2 Increase in "work productivity," "overall work productivity," regular daily activities significantly higher in fexofenadine group compared with placebo group (both studies); no difference between groups re time missed from class/work | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: ''Patients were randomised to receive 60 mg fexofenadine HCl twice daily or placebo twice daily...'' |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: ''24 hour single‐blind placebo lead‐in, and a subsequent 4‐week double blind treatment period.'' Unclear how blinding was achieved |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote: ''24 hour single‐blind placebo lead‐in, and a subsequent 4‐week double blind treatment period.'' Unclear how blinding was achieved |
| Incomplete outcome data (attrition bias) | High risk | Study 2: number randomly assigned stated in text to be 165; in tables, total number adds up to 167; no mention of whether there were dropouts. Unable to confirm ITT numbers in study report for Study 2 |
| Selective reporting (reporting bias) | Unclear risk | Ambiguity regarding duration of trial methods: says 4 weeks; results say 4 to 6 weeks. DLQI measures reported but not as proportions of participants with 50% or greater improvement in quality of life measurements whilst taking H1‐antihistamines; therefore not possible to include DLQI data |
| Other bias | Unclear risk | Funder: Hoechst Marion Roussel Study authors report 2 identical studies in this paper; if studies identical, unclear why not combined as a single study |
| Methods | Design: randomised single‐blind 4‐arm trial comparing a combination of sedating H1‐antihistamine and non‐sedating H1‐antihistamine (hydroxyzine plus cetirizine); combination of H1‐antihistamine and H2‐antihistamine (hydroxyzine plus famotidine); and combination of H1‐antihistamine and LRA (hydroxyzine plus montelukast) vs placebo Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 120 Sex: 38% male, 62% female Age of participants, years: 31 (18–45); 36.4 (20–52); 34.8 (20–54); 33.2 (18–48) Unit of allocation: participant Country and setting: Taiwan; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration 1‐week ‘run‐in’ period to wash out previous antihistamine used for treatment. Randomly assigned to receive:
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: 4 weeks | |
| Outcomes | Timing of outcome assessment: baseline and after 4 weeks of treatment Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant and clinician | |
| Notes | Study investigators concluded that the combination of H1‐ and H2‐receptor antagonists provided the greatest treatment efficacy according to the measures used in this small study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Participants were not blinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote (page 195): ''The same investigating physician who was blinded to the treatment regimens saw the patient'' |
| Incomplete outcome data (attrition bias) | Unclear risk | 107/120 randomly assigned completed the study (13 of 30 participants from placebo group dropped out after experiencing no real benefit following therapy for 1–2 weeks) Comment: All dropouts were from the placebo group, but as the reasons for this are given, we have judged the risk of bias as low |
| Selective reporting (reporting bias) | Unclear risk | All prespecified outcomes were reported |
| Other bias | Unclear risk | Funder: Montelukast provided by manufacturer Merck Sharp Dohme |
| Methods | Design: randomised controlled trial of mizolastine 10 mg daily for 4 weeks followed by 10 mg every other day for 4 weeks and followed by 10 mg per 3 days in the last 4 weeks for comparison with long‐term mizolastine 10 mg daily for 12 weeks Duration: 12 weeks | |
| Participants | Number of participants randomly assigned: 100 (experimental decremental dose group); control n = 50, long‐term 10 mg mizolastine n = 50 Sex: intervention: men 27, women 23 (46% female); control group: men 28; women 22 (44% female) Age of participants, mean in years: experimental group 32.66; control group 30.86 Unit of allocation: single participants Country and setting: China; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: not mentioned Concomitant/rescue treatment permitted: not mentioned | |
| Outcomes | Timing of outcome assessment: 4, 8, 12 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician | |
| Notes | Study investigators concluded that long‐term decrement in mizolastine therapy is effective, safe and convenient in the treatment of chronic urticaria | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised, no details given |
| Allocation concealment (selection bias) | Unclear risk | No details given |
| Blinding of participants and personnel (performance bias) | High risk | Not blinded |
| Blinding of outcome assessment (detection bias) | High risk | Not blinded |
| Incomplete outcome data (attrition bias) | Low risk | 3/50 participants in each group were lost to follow‐up, no reasons given No ITT analysis carried out |
| Selective reporting (reporting bias) | Low risk | All outcomes reported |
| Other bias | Low risk | Funder: not stated |
| Methods | Design: randomised double‐blind parallel‐group single‐dose study of desloratadine 5 mg vs 20 mg desloratadine Duration: 5 hours (short‐term) | |
| Participants | Number or participants randomly assigned: n = 29 (5 mg desloratadine n = 13, 20 mg desloratadine n = 16) Sex: 5 mg desloratadine group: 9 women/4 men (64% female); 20 mg desloratadine group: 7 women/9 men (43% female) Age of participants, mean years: 5 mg desloratadine group 43.5 ± 12.9; 20 mg desloratadine group: 41.7 ± 11.3 Unit of allocation: selected body area in single participants Country and setting: Germany; Allergie‐Centrum‐Charité, a tertiary referral centre for allergies and urticaria Inclusion criteria of the trial
Exclusion criteria of the trial
Mild to severe disease, duration of disease longer than 6 weeks | |
| Interventions | Interventions, dose, duration
short‐term (5 hours) Length of follow‐up: after 5 hours | |
| Outcomes | Timing of outcome assessment (state which time points): 5 hours Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician | |
| Notes | Study investigators concluded that a direct comparison between 5 mg and 20 mg of desloratadine showed no difference in weal area, weal volume or number of weals. "In contrast, a comparison of the reduction in the total weal number after 5 hours during treatment with 5 mg desloratadine minus no treatment versus treatment with 20 mg minus no treatment showed significant differences (p < 0.01)" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation scheme |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding of participants and personnel (performance bias) | Low risk | All participants and clinical staff, for example, study nurses and study physicians involved in the study, were blinded until the end of the trial |
| Blinding of outcome assessment (detection bias) | Low risk | All participants and clinical staff, for example, study nurses and study physicians involved in the study, were blinded until the end of the trial |
| Incomplete outcome data (attrition bias) | Unclear risk | Unclear whether any were lost to follow‐up and any reasons No ITT analysis carried out |
| Selective reporting (reporting bias) | Low risk | No selective outcome reporting |
| Other bias | Unclear risk | Funder: This study was financially supported by Schering‐Plough (Essex Pharma GmbH, Germany). In addition, the study medication was provided by Schering‐Plough |
| Methods | Design: randomised 3‐arm trial of azelastine 2 mg/d; azelastine 4 mg/d vs combined azelastine and cimetidine 2 mg/d Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 103 Sex: male 52%, female 48% (from 100 participants who were available for analysis) Unit of allocation: participant Country and setting: China; secondary care Age between 16‐81 years, mean age 39.16 years Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: not specified but assumed to be at endpoint (i.e. 4 weeks) | |
| Outcomes | Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician | |
| Notes | Main study report written in Chinese Study investigators concluded that all 3 groups have similar efficacy, but azelastine 4 mg/d and combined azelastine and cimetidine 2 mg had greater efficacy than azelastine 2 mg alone The difference in this comparison was statistically significant (P < 0.05) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Unclear, described as open randomisation |
| Allocation concealment (selection bias) | High risk | No concealment |
| Blinding of participants and personnel (performance bias) | High risk | Not blinded |
| Blinding of outcome assessment (detection bias) | High risk | Not blinded |
| Incomplete outcome data (attrition bias) | High risk | 103 were randomly assigned; 100 are included in the analyses with no reasons given for dropout |
| Selective reporting (reporting bias) | High risk | No clear definition of outcomes; unclear whether assessment of compliance was carried out |
| Other bias | Low risk | None detected. Funder: not stated |
| Methods | Design: randomised double‐blind parallel‐group comparing cetirizine and levocetirizine Duration: 28 days | |
| Participants | Number of participants randomly assigned: 44 (22 in each group) Sex: levocetirizine: 54% (female); cetirizine: 63% (female) Age of participants (between 18 and 65), mean in years: levocetirizine 36.27; cetirizine 36.73 Unit of allocation: participant Country and setting: China; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (28 days) Length of follow‐up: 7, 14, 28 days | |
| Outcomes | Timing of outcome assessment: 7, 14, 28 days Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician | |
| Notes | Study investigators concluded that no significant difference in curative effect was noted between the 2 groups. No serious adverse effects were found. Levocetirizine is effective and safe in the treatment of CSU | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 477): ‘randomised, double blinded’ |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: ‘randomised, double blinded,’ but no further details given |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote: ‘randomised, double blinded,’ but no further details given |
| Incomplete outcome data (attrition bias) | Unclear risk | Unclear whether any dropped out; not stated |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Other bias | Low risk | None detected. Funder: not stated |
| Methods | Design: randomised open‐label parallel‐group 3‐arm comparison of mizolastine vs cetirizine vs loratadine Duration: 28 days | |
| Participants | Number of participants randomly assigned: 96; mizolastine n = 30, cetirizine n = 34, loratadine n = 32 Sex: 60% male, 40% female Age of participants (18 to 72), mean age in years: mizolastine 45, cetirizine 38, loratadine 36.5 Unit of allocation: participant Country and setting: China; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (28 days) Length of follow‐up: 5 weeks i.e. follow‐up extended after the cessation of therapy. | |
| Outcomes | Timing of outcome assessment: 14, 28 and a further follow up at 7 days post intervention Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: clinician | |
| Notes | Study investigators concluded that all 3 antihistamines have high clinical efficacy and safety in the treatment of CSU. No statically significant difference was noted among the 3 groups | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Open‐label. randomised |
| Allocation concealment (selection bias) | High risk | No concealment |
| Blinding of participants and personnel (performance bias) | High risk | Open label, no blinding |
| Blinding of outcome assessment (detection bias) | High risk | Open label, no blinding |
| Incomplete outcome data (attrition bias) | Unclear risk | No dropouts reported |
| Selective reporting (reporting bias) | Low risk | All outcomes reported |
| Other bias | Low risk | None detected. Funder: not stated |
| Methods | Design: randomised 2‐arm parallel trial of desloratadine vs loratadine Duration: 4 weeks | |
| Participants | Number of participants randomly assigned: 41 (desloratadine n = 21, loratadine n = 20) Sex: 49% female, 51% male: desloratadine 42% female (12 men/9 women); loratadine 55% female (9 men/11 women) Age of participants, mean in years: desloratadine 32.7; loratadine 31.8 Unit of allocation: participant Country and setting: China; secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
Duration of intervention: intermediate‐term (4 weeks) Length of follow‐up: not mentioned Concomitant rescue treatment not permitted | |
| Outcomes | Timing of outcome assessment: assumed to be at end of intervention period—4 weeks Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: participant | |
| Notes | Study investigators concluded that no significant difference in curative effect was noted between the 2 groups. No serious adverse effects were found. Desloratadine was found to be effective and safe in treating patients with CSU | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Double‐blind randomised controlled trial |
| Allocation concealment (selection bias) | Unclear risk | Not mentioned in this study |
| Blinding of participants and personnel (performance bias) | Unclear risk | Participants and personnel were blinded to treatment group Not clear about this, as it was not mentioned clearly in the method of assessment |
| Blinding of outcome assessment (detection bias) | Unclear risk | Outcome assessors were blinded to treatment group Not clear, as it was not mentioned in the method of assessment |
| Incomplete outcome data (attrition bias) | Unclear risk | No dropouts reported |
| Selective reporting (reporting bias) | Low risk | None, all prespecified outcomes were reported |
| Other bias | Unclear risk | Funder: not stated. Severity of disease was not clear. Duration of disease with desloratadine ranged from 6 weeks to 6 years, and with loratadine from 6 weeks to 6.5 years |
| Methods | Design: international multi‐centre double‐blind randomised placebo and active treatment‐controlled parallel‐group 2‐arm study comparing bilastine 20 mg vs levocetirizine 5 mg once daily and placebo Duration: 28 days | |
| Participants | Number of participants randomly assigned: 525 (bilastine n = 173, levocetirizine n = 166, placebo n = 184, unclear numbers) Sex: bilastine: 63% male, 27% female; levocetirizine: 54% male, 46% female; placebo: 40% male, 60% female Age of participants, years: bilastine 41.7, levocetirizine 39.8, placebo 39.4 Unit of allocation: participants Country and setting: Argentina, Belgium, France, Germany, Poland, Romania, Spain (46 centres); secondary care Inclusion criteria of the trial
Exclusion criteria of the trial
| |
| Interventions | Interventions, dose, duration
(once daily) Duration of intervention: intermediate‐term (28 days) Length of follow‐up: 28 days | |
| Outcomes | Timing of outcome assessment: 0, 14 and 28 days (or at early discontinuation visit in cases of withdrawal from the study) Primary outcomes of the trial
Secondary outcomes of the trial
Clinician or participant report: both | |
| Notes | Study investigators concluded that no significant difference in efficacy was noted between the 2 active groups; both were better than placebo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Probably done; quote (page 517): "Randomization to treatment was achieved according to a computer‐generated randomisation code provided by the study sponsor (FAES FARMA, SA, Spain)" |
| Allocation concealment (selection bias) | Unclear risk | Quote (page 518): "treatments were allocated to each patient in their chronological order of entry into the study" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote (page 518): "The study medications were supplied as identical over‐encapsulated tablets in individually coded aluminium blister packs to ensure blinding of both the investigators and the patients to treatment'' |
| Blinding of outcome assessment (detection bias) | Unclear risk | As above Comment: unclear whether investigators had access to the randomisation list, but this is unlikely given that randomisation was carried out offsite, so we have judged this as low risk |
| Incomplete outcome data (attrition bias) | Unclear risk | 457/525 completed. Withdrawals as follows: placebo (n = 35): lack of efficacy 24, adverse event 1, participant decision 3, poor compliance with protocol 3, loss to follow‐up 2, "other" 2; bilastine (n = 15): lack of efficacy 5, adverse event 3, participant decision 4, loss to follow‐up 1, "other" 1; levocetirizine (n = 15): lack of efficacy 7; participant decision 4, poor compliance with protocol 1, loss to follow‐up 2, "other" 1 Comment: Although the study report states ITT, the analysis did not include 3 participants who were randomly assigned but did not receive any medication; another 6 participants were not included in the analysis |
| Selective reporting (reporting bias) | Low risk | All outcomes reported |
| Other bias | Unclear risk | Funder: FAES Farma (bilastine makers). MDS Pharma Services Inc for technical assistance for development of study, data management and statistical analysis |
AE: adverse event.
AEQLQ: Aerius Quality of Life Questionnaire.
ALAT: alanine transaminase.
ASAT: aspartate transaminase.
BMI: body mass index.
CIU: chronic idiopathic urticaria.
CPK: creatine phosphokinase.
CSU: chronic spontaneous urticaria.
DLQI: Dermatology Life Quality Index.
ECG: electrocardiogram.
FBC: full blood count.
ITT: intention‐to‐treat.
K+: potassium ion.
LFT: liver function test.
MNW: mean number of weals.
MPS: mean pruritus score.
MTSS: mean total symptoms score.
Na2+: sodium ion.
NSAID: non‐steroidal anti‐inflammatory drug.
QoL: quality of life.
RCT: randomised controlled trial.
SAF: safety population.
SIWS: scale for interference of wheals with sleep
SD: standard deviation.
SSRI: symptom scores reduction index.
TSS: total symptoms score.
UAS: Urticaria Activity Score.
U/E: upper extremity.
URTI: upper respiratory tract infection.
VAS: visual analogue scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| Letter to editor, no study results, not an RCT | |
| CIU mentioned only in abstract, not defined further, no further information available | |
| RCT of astemizole vs cetirizine with no placebo group, astemizole excluded as withdrawn | |
| Quote: ''...randomly divided into two 15 participant groups'' Unclear if this is true randomisation. Terfenadine vs cetirizine, terfenadine no longer in use. | |
| RCT on astemizole | |
| No chronic ordinary urticaria; does not meet inclusion criteria. Only terfenadine: not in use | |
| CIU not defined | |
| CIU not defined, described only as urticaria | |
| Included acute urticaria (less than 1 month's duration) | |
| Acute urticaria, not CIU | |
| Not proper randomisation ('divididos em 2 grupos de 10') | |
| Chronic urticaria unspecified; other causes of pruritus included | |
| Included physical urticaria | |
| Astemizole (not in use) | |
| All participants given same combination of antihistamines | |
| Not CSU by our definition | |
| All participants given same H1‐antihistamine | |
| Included participants with allergic rhinitis ('All patients had allergic rhinitis or CSU') | |
| Chronic urticaria unspecified; did not exclude physical urticaria/vasculitis | |
| Astemizole and terfenadine: not in use | |
| No definition of chronic idiopathic urticaria; methods do not state any diagnosis | |
| Study of ciclosporin | |
| RCT on terfenadine | |
| RCT on terfenadine | |
| RCT, included other forms of urticaria | |
| Chronic urticaria, not further specified | |
| No relevant data, described participants allocated to double‐blind treatment in random order. Not randomised. Compared chlorpheniramine plus cimetidine (no other active treatment). No information about dose | |
| Chronic urticaria, not CSU | |
| No clinical trials; not relevant to study | |
| No clinical trials; review on pharmacokinetics and pharmacodynamics—not relevant to study | |
| Chronic urticaria treatment described, CSU not defined (duration of urticaria history not stated) | |
| No P values; outcome measures not meaningful | |
| Included acute urticaria | |
| Quote: ''Patients were divided into two 75 person‐groups.'' Not an RCT | |
| Terfenadine only (not in use) | |
| Astemizole only (not in use) | |
| Included conditions other than CSU | |
| RCT, included other forms of urticaria | |
| Not a comparison of H1‐antihistamines | |
| Included acute urticaria (1 month's duration) | |
| Included conditions other than CSU | |
| Montelukast vs cetirizine. Chronic urticaria not fully defined in terms of duration | |
| Chronic urticaria, not CSU | |
| Terfenadine vs placebo | |
| Chronic urticaria unspecified, random order administration, unclear whether randomised, self‐assessment questionnaire | |
| Included other causes of urticaria (e.g. vasculitis) | |
| Compared regular vs prn desloratadine in participants known to be responsive to the drug. All participants given desloratadine, no comparison | |
| Terfenadine only (not in use) | |
| CSU not defined | |
| Review article | |
| RCT. Multi‐centre parallel‐group 2‐arm double‐blind study of loratadine vs astemizole. Astemizole no longer in use, excluded | |
| Rhinitis and urticaria put together, no separate data | |
| Terfenadine only (not in use); looking at hives, not chronic ordinary urticaria | |
| CSU not defined, no efficacy data | |
| Astemizole only (not in use) | |
| Acupuncture combined with bloodletting and regular Western medication (loratadine), loratadine arm not compared with active pharmacological intervention | |
| Included acute urticaria | |
| Included acute urticaria | |
| Included acute urticaria | |
| Included acute urticaria | |
| Included acute urticaria | |
| Included acute urticaria | |
| Included acute urticaria | |
| Terfenadine only (not in use); did not use proper randomisation | |
| Review article specific to effects of interventions on driving | |
| Included participants with acute and chronic urticaria with no separate data (CSU not defined) | |
| Included physical urticaria ('1 had cold urticaria') | |
| Not randomised | |
| Included acute urticaria | |
| Astemizole | |
| Included physical urticaria | |
| Included physical urticaria | |
| Included physical, cholinergic urticaria; urticaria less than 6 weeks' duration | |
| Included acute urticaria (less than 1 month's duration) | |
| Included urticaria less than 6 weeks' duration | |
| Included autoimmune urticaria | |
| Included physical urticaria | |
| Cost‐effectiveness study | |
| Included physical urticaria | |
| Chronic urticaria not further specified | |
| Phase III study of TAU‐284 (bepotastine besilate) on chronic urticaria: a multi‐centre double‐blind comparative study with placebo. Included urticaria of less than 1 month's duration | |
| Both groups received same antihistamine | |
| RCT, compared only cetirizine vs terfenadine with no placebo group; therefore excluded, as terfenadine not in use | |
| Retrospective observational single‐centre study of participants with CSU in Korea; 'divided patients into two study groups' | |
| Terfenadine only (not in use) | |
| Duration of disease less than 4 weeks (acute urticaria) | |
| Duration of disease less than 4 weeks (acute urticaria) | |
| Included acute urticaria | |
| Included acute urticaria | |
| Included acute urticaria | |
| Terfenadine only (not in use) | |
| Included physical urticaria | |
| Included physical urticaria | |
| Included physical urticaria | |
| Included acute urticaria | |
| Astemizole vs terfenadine (not in use) | |
| Included acute urticaria | |
| Included acute urticaria | |
| Included acute urticaria with CSU, separate data not available | |
| Validation study of DLQI | |
| CSU not defined (abstract only) | |
| RCT of astemizole vs loratadine, no placebo group. Excluded, as astemizole now withdrawn | |
| Included physical urticaria | |
| Randomised double‐blind placebo‐controlled study of safety and efficacy of miltefosine (not an H1‐antihistamine) in antihistamine‐resistant chronic spontaneous urticaria | |
| Randomised, omalizumab, not an H1‐antihistamine | |
| CSU not defined, rhinitis and urticaria combined, no separate data | |
| Combined H1 and H2 therapy | |
| CSU not defined, abstract only, no other details available | |
| States randomly divided, but not randomised | |
| Abstract states 'randomly divided,' but translation of methods indicates that participants were 'put into groups numbered 1 to 4,' without mention of randomisation | |
| Included acute urticaria | |
| Included acute urticaria (4 weeks' duration) | |
| Included acute urticaria | |
| All participants taking same antihistamine, cetirizine | |
| RCT, but compared terfenadine with brompheniramine; terfenadine no longer in use, therefore excluded | |
| CSU not defined, mentions only chronic urticaria | |
| Chronic urticaria unspecified; no useful data | |
| Chronic urticaria unspecified | |
| Terfenadine only (not in use) | |
| Terfenadine only (not in use) | |
| Terfenadine only (not in use) | |
| Terfenadine only (not in use) | |
| Included acute urticaria | |
| RCT terfenadine | |
| Review article: treatment in children | |
| Included physical urticaria, excluded on that basis | |
| RCT, compared only terfenadine with cetirizine, no placebo group, terfenadine no longer in use, therefore excluded | |
| Lack of data; study authors contacted; data not available | |
| Included acute urticaria | |
| Included participants with allergic rhinitis | |
| Management of aspirin‐exacerbated urticaria | |
| Review article on updosing | |
| Chronic urticaria unspecified | |
| Chronic urticaria, not CSU | |
| CCT, not RCT | |
| Included acute urticaria (4 weeks' duration) | |
| Randomised, but pharmacology study only, no clinical outcomes | |
| Not properly randomised: medications dispensed ''in random order,'' but unclear whether this was true randomisation. No details on number of participants in each group | |
| Included acute urticaria (less than 1 month) | |
| Included delayed pressure urticaria | |
| Included physical urticaria | |
| Participants were divided, not randomly assigned | |
| Chronic urticaria, not CSU | |
| Chronic urticaria, CSU not defined | |
| Allergic disorders; not chronic ordinary urticaria | |
| Included acute urticaria (4 weeks' duration or less) | |
| Outcomes are histochemical only | |
| Double‐blind but not an RCT | |
| RCT on astemizole | |
| Included acute urticaria | |
| Test of domestic vs imported cetirizine, randomised, but no control or placebo group. No CSU, chronic urticaria only | |
| CCT and included cholinergic urticaria | |
| Included acute urticaria | |
| Other dermatoses included; not an RCT; clinical observation study | |
| Included chronic spontaneous urticaria; CSU not defined | |
| Inadequate data and reporting | |
| Not an RCT; included other dermatoses | |
| Combination therapy H1 and H2; all participants took same combination | |
| Combined H1‐ and H2‐antihistamines | |
| Included physical urticaria, duration not specified | |
| Chronic urticaria unspecified | |
| Included acute urticaria (4 weeks' duration) | |
| Chronic urticaria unspecified | |
| RCT taking terfenadine | |
| Included acute urticaria, no separate data | |
| RCT of loratadine vs astemizole (not in use), no placebo group | |
| CSU duration of less than 4 weeks included (i.e. acute urticaria included) | |
| Described as 'chronic urticaria' but CIU not defined; also included acute urticaria with no separate data | |
| Cholinergic urticaria |
CCT: controlled clinical trial.
CIU: chronic idiopathic urticaria.
DLQI: Dermatology Life Quality Index.
RCT: randomised controlled trial.
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | Double‐blind randomised dose‐ranging trial in 4 parallel groups |
| Participants | CSU, number randomly assigned unclear |
| Interventions | 10, 20 and 30 mg bilastine once daily vs placebo |
| Outcomes | Unclear |
| Notes | Unable to locate a copy at this time for further assessment |
| Methods | Double‐blind (CCT?) |
| Participants | Unclear |
| Interventions | H1‐ plus H2‐blockers |
| Outcomes | Unclear, included pruritus |
| Notes | Unable to locate a copy at this time for further assessment |
| Methods | RCT (double‐blind randomised placebo‐controlled parallel study) |
| Participants | 37 participants |
| Interventions | Terfenadine, 60 mg twice daily, vs placebo vs hydroxyzine, 25 mg four times daily |
| Outcomes | Adverse effects including somnolence, therapeutic use |
| Notes | Unable to locate a copy at this time for further assessment |
| Methods | RCT |
| Participants | Urticaria |
| Interventions | Astemizole and loratadine |
| Outcomes | Unclear |
| Notes | Unable to locate a copy at this time for further assessment |
| Methods | Design: unclear, states 'random allocation' |
| Participants | Unclear, no abstract available |
| Interventions | Unclear, includes cetirizine but no details about comparator arms |
| Outcomes | Unclear |
| Notes | Unable to locate a copy at this time for further assessment |
| Methods | RCT (randomly divided into 3 groups) |
| Participants | One hundred and twenty cases of chronic urticaria |
| Interventions | 38 participants in the combination therapy group orally received half pack of compound FKS decocted in water twice daily and mizolastine tablet 10 mg once daily; 39 participants orally received mizolastine tablet 10 mg once daily; 38 patients in the combination therapy group orally received half pack of compound FKS decocted in water twice daily and mizolastine tablet 10 mg once daily; 39 participants orally received mizolastine tablet 10 mg once daily for 4 weeks |
| Outcomes | Efficacy, scores of symptoms and signs, improvement. |
| Notes | Chinese language study; awaiting copy of paper and translation |
| Methods | RCT (randomly divided) |
| Participants | 209 participants were randomly divided into 2 groups: experimental group (106 cases) and control group (103 cases) |
| Interventions | One arm given mizolastine and ketotifen with gradual dose reduction for 10 weeks, while the other participants were given mizolastine alone with gradual dose reduction for 10 weeks |
| Outcomes | Total effective rates for experimental group and control group were 76.1% and 43.5%, respectively (P value < 0.05). 4 weeks after treatment, recurrence rates for experimental group and control group were 10.4% and 22.8%, respectively (P value < 0.05). Adverse effects included dry mouth and drowsiness |
| Notes | Chinese language study awaiting copy of paper and translation |
| Methods | RCT or CCT, double‐blind |
| Participants | 121 participants with chronic urticaria |
| Interventions | Azatadine maleate 2 mg/d (1 mg/tablet) was administered for 5 days to 61 participants with chronic urticaria, and clemastine fumarate 2.68 mg/d (1.34 mg/tablet) was given to a matched control group of 60 participants |
| Outcomes | Efficacy, itching, adverse effects |
| Notes | Unable to locate a copy at this time for further assessment |
| Methods | RCT |
| Participants | 203 participants with chronic idiopathic urticaria |
| Interventions | Loratadine, hydroxyzine and placebo |
| Outcomes | Efficacy and safety |
| Notes | Unable to locate a copy at this time for further assessment |
| Methods | Prospective randomised non‐blinded comparative clinical study and assessment of quality of life |
| Participants | n = 51 |
| Interventions | Cetirizine 10 mg once daily to 51 participants with urticaria. Participants with inadequate symptom control were randomly assigned to cetirizine 20 mg once daily (dose‐increase group) or olopatadine 5 mg twice daily (drug‐change group) |
| Outcomes | Severity of weal and itching and quality of life (QoL) were measured by Skindex—16 were evaluated |
| Notes | Awaiting interlibrary loan and, if available, full translation |
| Methods | Observer‐blind RCT, single centre |
| Participants | Chronic urticaria, characterised by frequent appearance of weals for > 6 weeks |
| Interventions | Olopatadine (5 mg twice daily) or levocetirizine (5 mg/d) for 9 weeks, continuously for first 4 weeks and then on demand basis for last 5 weeks |
| Outcomes | Primary outcome measures were Urticaria Activity Score (UAS) and urticaria total severity score (TSS). Routine haematological and biochemical tests and treatment‐emergent adverse events were monitored for safety |
| Notes | Awaiting interlibrary loan |
| Methods | Unclear |
| Participants | CSU and healthy participants |
| Interventions | Conventional and double doses of fexofenadine HCl on CSU (and on histamine‐induced skin responses by iontophoresis using visual and laser Doppler imaging scales in healthy donors) |
| Outcomes | Cutaneous manifestations in CSU (and histamine‐induced flare and itch in healthy donors) |
| Notes | Awaiting interlibrary loan |
| Methods | RCT |
| Participants | Urticaria |
| Interventions | Mizolastine vs cyproheptadine |
| Outcomes | Unclear |
| Notes | Unable to locate a copy at this time for further assessment |
| Methods | RCT |
| Participants | 136 participants with chronic idiopathic urticaria |
| Interventions | Randomly assigned to 3 groups: mizolastine 10 mg daily; loratadine 10 mg daily; mizolastine 10 mg daily combined with compound glycyrrhizin 75 mg 3 times a day |
| Outcomes | Symptom score reduction index (SSRI) |
| Notes | Awaiting interlibrary loan and, if available, full translation |
| Methods | RCT |
| Participants | 92 participants with refractory urticaria |
| Interventions | Montelukast combined with cetirizine, and control group with Tripterygium glycoside combined with cetirizine |
| Outcomes | Total symptoms scores were calculated for participants at baseline and at week 2 and week 4 during treatment based on individual scores for weal and pruritus; incidence of adverse events was recorded |
| Notes | Chinese language study awaiting translation |
| Methods | RCT |
| Participants | 120 mixed group of acute/chronic urticaria with allergic rhinitis |
| Interventions | Mizolastine vs cetirizine |
| Outcomes | Therapeutic effect and adverse reactions |
| Notes | Awaiting interlibrary loan and, if available, full translation |
CCT: controlled clinical trial.
CSU: chronic spontaneous urticaria.
QoL: quality of life.
RCT: randomised controlled trial.
SSRI: symptom score reduction index.
TSS: total severity score.
UAS: Urticaria Activity Score.
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Comparison of efficacy, safety and cost‐effectiveness of rupatadine and olopatadine, antihistaminics, in study participants with urticaria |
| Methods | To compare efficacy, safety and cost‐effectiveness of rupatadine and olopatadine in participants with chronic idiopathic urticaria: a randomised double‐blind comparative parallel‐group study Method of generating randomisation sequence: random numbers table Method of allocation concealment: prenumbered or coded identical containers Blinding and masking: participant and Investigator blinded |
| Participants | Target sample size 60, with CSU Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes | Primary outcomes assessed at 1, 3 and 6 weeks
Secondary outcomes assessed at 1, 3 and 6 weeks
|
| Starting date | 17 April 2014 |
| Contact information | Dr Ganesh N Dakhale Department of Pharmacology, Government Medical College, Nagpur. 440003 Nagpur, MAHARASHTRA India 8308833593 Government Medical College Nagpur, India |
| Notes | Other study ID CTRI/2014/04/004545, ongoing, sponsored by Indira Gandhi Government Medical College, India |
| Trial name or title | A 6‐week multi‐centre double‐blind randomised placebo‐controlled parallel‐group study to assess the efficacy and safety of rupatadine 10 and 20 mg in the treatment of chronic idiopathic urticaria (CIU): a phase III clinical trial |
| Methods | RCT |
| Participants | Male or female 12 to 65 years of age |
| Interventions |
|
| Outcomes | Primary outcomes: Primary endpoint will be based on daily subjective assessment of the severity of each symptom of CSU, as recorded by participants in their diaries:
Secondary outcomes
|
| Starting date | 2004‐10‐28 |
| Contact information | J Uriach y Compañía, S.A. |
| Notes | Ongoing |
| Trial name or title | A randomised double‐blind placebo‐controlled exploratory trial to evaluate 1‐week oral treatment with R129160 (60 mg twice daily) in participants with chronic idiopathic urticaria—oral treatment with R129160 (60 mg twice daily) in participants with chronic idiopathic urticaria |
| Methods | RCT |
| Participants | Male and female 18 to 64 years of age with CSU, Czech Republic, Belgium, Netherlands |
| Interventions |
|
| Outcomes | Primary outcomes
|
| Starting date | 18 July 2007 |
| Contact information | Barrier Therapeutics nv |
| Notes | Not recruiting, no results posted |
| Trial name or title | Study of optimal treatment duration with antihistamine in idiopathic urticaria patients |
| Methods | Parallel cluster‐randomised by institution, open. Allocation concealment by centralised registration |
| Participants | 120 (target sample size). Male or female 20 years of age or older Inclusion criteria
Exclusion criteria
|
| Interventions | 4 week treatment with fexofenadine (120 mg/d) vs no treatment |
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | 23 May 2008 |
| Contact information | Motoaki Inoue Clinical Research Support Center Kyusyu Secretariat 3‐1‐1 Umade, Higashi‐ku Fukuoka, 812‐8582 +81‐92‐631‐2920 inoue@cres‐kyushu.or.jp |
| Notes | Also known as SOLIDARITIE. Closed, no results posted Sponsored by West Japan Urticaria Therapy Study Group, Clinical Research Support Center Kyusyu |
| Trial name or title | A study of the antipruritic effect and onset of sleepiness with oral antihistamines. Comparison of sedative and non‐sedative antihistamine |
| Methods | Randomised cross‐over open allocation concealment by numbered containers |
| Participants | Male and female 16 years of age and older Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | 3 May 2005 |
| Contact information | Hiramatsu Yusunari Future Medical Research Institute LLC Japan Telephone: 03‐5777‐1001 EBMs Co, Ltd Clinical Business Division |
| Notes | Sponsored by Non‐Profit Organization Health Institute Research of Skin Closed, no results posted |
| Trial name or title | A study evaluating efficacy of non‐sedative antihistamine up‐titration in patients with chronic urticaria who did not respond to standard therapy |
| Methods | Parallel randomised |
| Participants | Male and female 16 years of age and older Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | 1 June 2012 |
| Contact information | Sayumi Hasegawa World Trade Center Bldg 24F 2‐4‐1, Hamamatsu‐cho, Minato‐ku, Tokyo 105‐6124 Japan, Japan EBMs Co, Ltd Business Strategy Division |
| Notes | Completed, no results posted Funder: non‐profit organisation, Health Institute Research of Skin |
| Trial name or title | A study on the optimal treatment for chronic idiopathic urticaria insufficient for the second‐generation antihistamines |
| Methods | Parallel, randomised, open |
| Participants | Male and female 20 years of age or older. Inclusion criteria
Exclusion criteria
|
| Interventions | Antihistamines (details unclear) |
| Outcomes | Primary outcomes
|
| Starting date | 1 April 2011 |
| Contact information | Tamotsu Ebihara 35 Shiannomachi Shinjuku‐ku Tokyo, JAPAN, Japan Keio University School of Medicine, Department of Dermatology |
| Notes | Ongoing, recruiting Sponsored by Keio University School of Medicine |
| Trial name or title | Preliminary trial of the increase in antihistamines in dose and a combination of different antihistamines for refractory spontaneous urticaria |
| Methods | Open parallel‐group randomised allocation concealed by numbered containers |
| Participants | Target of 32 participants with idiopathic urticaria (definition and duration not stated), male or female 20 years of age or older Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | 1 April 2013 |
| Contact information | Takuma Kohsaka Hiroshima University Hospital Department of Dermatology 1‐2‐3, Kasumi, Minami‐ku, Hiroshima, 734‐8551 +81‐82‐257‐5237 |
| Notes | Sponsored by Hiroshima Univeristy, and funded by GlaxoSmithKline, ongoing, completing 31 March 2015 |
| Trial name or title | A 4‐week dose‐finding multi‐centre double‐blind randomised placebo‐controlled parallel‐group trial to assess the efficacy and safety of different doses of rupatadine compared with placebo in the treatment of chronic idiopathic urticaria |
| Methods | RCT |
| Participants | Male and female 12 to 65 years of age with CSU |
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | October 2002 |
| Contact information | Eva Arnaiz, PhD, J Uriach y Compania |
| Notes | Terminated, no study results posted |
| Trial name or title | A 6‐week multi‐centre double‐blind randomised placebo‐controlled parallel‐group study to assess the efficacy and safety of rupatadine 10 and 20 mg in the treatment of chronic idiopathic urticaria (CIU): a phase III clinical trial |
| Methods | RCT |
| Participants | Male and female 12 to 65 years of age with CSU |
| Interventions |
|
| Outcomes | Primary outcomes
Secondary outcomes
|
| Starting date | April 2004 |
| Contact information | J Uriach and Company |
| Notes | Terminated, no study results posted Other study ID IC010RUP304 |
| Trial name or title | Double‐blind, randomised, placebo‐controlled, phase III study comparing the efficacy and safety of bilastine 20 mg once daily and levocetirizine 5 mg for the treatment of chronic idiopathic urticaria |
| Methods | RCT |
| Participants | Male and female 18 to 70 years of age with CSU |
| Interventions |
|
| Outcomes | Primary outcomes
|
| Starting date | July 2006 |
| Contact information | Faes Farma, S.A. Principal Investigator: Olmos, MD Hosp Clinico San Carlos, Servicio Dermatologia (Madrid, Spain) |
| Notes | Completed July 2007 (final data collection date for primary outcome measure), no study results posted Other study IDs EudraCT 2006‐001245‐33, BILA 2006/UCI |
| Trial name or title | A comparative double‐blind double‐dummy study of desloratadine (dl) 5 mg once daily, cetirizine 10 mg once daily and placebo once daily in patients with chronic idiopathic urticaria (CIU) |
| Methods | RCT |
| Participants | Male and female 12 to 70 years of age with CSU |
| Interventions |
|
| Outcomes | Primary outcomes
|
| Starting date | March 2004 |
| Contact information | Schering‐Plough |
| Notes | Terminated May 2005, no study results posted. Study terminated, as could not be resupplied with study medication in a timely manner Other study ID P03736 |
| Trial name or title | A comparative double‐blind double‐dummy study of desloratadine (dl) 5 mg once daily, cetirizine 10 mg once daily and placebo once daily in patients with chronic idiopathic urticaria (CIU). |
| Methods | RCT |
| Participants | Male and female 12 to 70 years of age with CSU |
| Interventions |
|
| Outcomes | Primary outcomes
|
| Starting date | May 2004 |
| Contact information | No contacts or locations posted, Schering‐Plough |
| Notes | Terminated August 2005, no study results posted Other study ID P03735 |
| Trial name or title | A pilot multi‐centre double‐blind randomised study for comparison of Aerius® "Continuous Treatment" versus Aerius® "PRN Regimen" on chronic idiopathic urticaria patient quality of life |
| Methods | RCT |
| Participants | Male and female over 18 years of age with CSU |
| Interventions |
|
| Outcomes | Primary outcomes
|
| Starting date | April 2003 |
| Contact information | Head, Clinical Trials Registry & Results Disclosure Group, Schering‐Plough |
| Notes | Known as ATTITUD. Completed April 2004, no study results posted Other study ID P03147 |
CIU: chronic idiopathic urticaria.
CSU: chronic spontaneous urticaria.
DLQI: Dermatology Life Quality Index.
MAO: monoamine oxidase.
MNW: mean number of weals.
MPS: mean pruritus scale.
MTSS: mean total symptoms score.
NRS: numerical rating scale
OPD: Out‐patient Department.
QoL: quality of life.
SIWS: scale for interference of wheals with sleep
VAS: visual analogue scale.
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines Show forest plot | 2 | 124 | Risk Ratio (M‐H, Random, 95% CI) | 1.86 [0.91, 3.79] |
| Analysis 1.1  Comparison 1 Loratadine 10 mg versus placebo, Outcome 1 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines. | ||||
| 1.1 Short‐term duration of intervention (10 mg) | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.42, 21.30] |
| 1.2 Intermediate‐term duration of intervention (10 mg) | 1 | 112 | Risk Ratio (M‐H, Random, 95% CI) | 1.73 [0.81, 3.72] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 2 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.76, 1.43] |
| Analysis 2.1 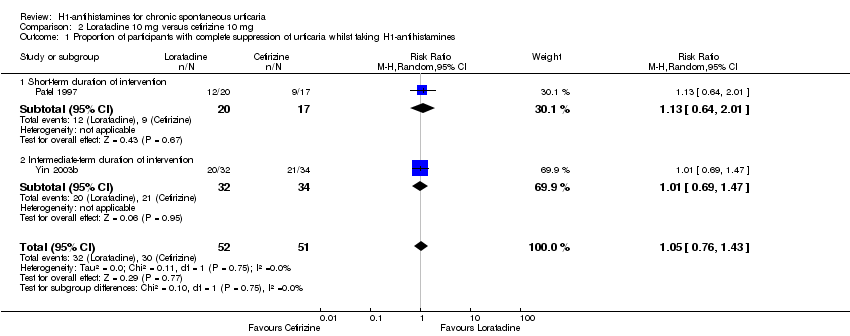 Comparison 2 Loratadine 10 mg versus cetirizine 10 mg, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines. | ||||
| 1.1 Short‐term duration of intervention | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.64, 2.01] |
| 1.2 Intermediate‐term duration of intervention | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.69, 1.47] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 3.1 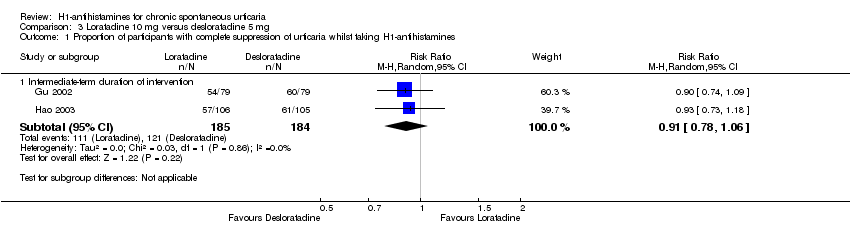 Comparison 3 Loratadine 10 mg versus desloratadine 5 mg, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines. | ||||
| 1.1 Intermediate‐term duration of intervention | 2 | 369 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.78, 1.06] |
| 2 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 3.2  Comparison 3 Loratadine 10 mg versus desloratadine 5 mg, Outcome 2 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines. | ||||
| 2.1 Intermediate‐term duration of intervention | 3 | 410 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.64, 1.71] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 4.1  Comparison 4 Loratadine 10 mg versus mizolastine 10 mg, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines. | ||||
| 1.1 Intermediate‐term duration of intervention | 3 | 316 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.64, 1.16] |
| 2 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 4.2  Comparison 4 Loratadine 10 mg versus mizolastine 10 mg, Outcome 2 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines. | ||||
| 2.1 Intermediate‐term duration of intervention | 3 | 314 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.55, 1.42] |
| 3 Proportion of participants with at least 50% improvement in QoL whilst taking H1‐antihistamines Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 4.3 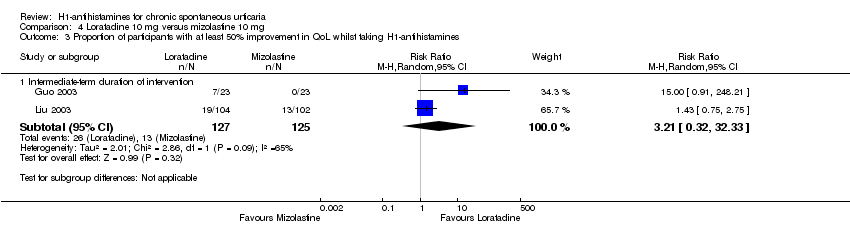 Comparison 4 Loratadine 10 mg versus mizolastine 10 mg, Outcome 3 Proportion of participants with at least 50% improvement in QoL whilst taking H1‐antihistamines. | ||||
| 3.1 Intermediate‐term duration of intervention | 2 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 3.21 [0.32, 32.33] |
| 4 Serious adverse events (i.e. serious enough to require withdrawal of treatment) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 4.4 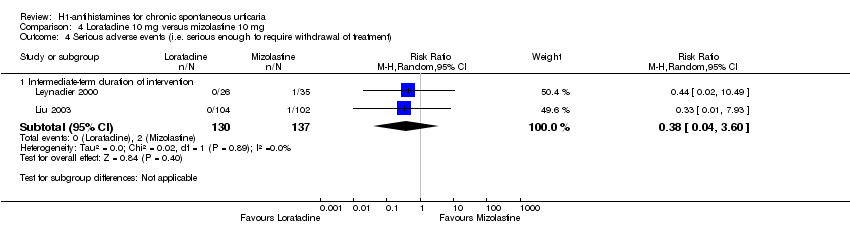 Comparison 4 Loratadine 10 mg versus mizolastine 10 mg, Outcome 4 Serious adverse events (i.e. serious enough to require withdrawal of treatment). | ||||
| 4.1 Intermediate‐term duration of intervention | 2 | 267 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.04, 3.60] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 5.1  Comparison 5 Loratadine 10 mg versus emedastine 2 mg, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines. | ||||
| 1.1 Intermediate‐term duration of intervention | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Proportion of participants with good or excellent response whilst taking H1‐antihistamines Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 5.2  Comparison 5 Loratadine 10 mg versus emedastine 2 mg, Outcome 2 Proportion of participants with good or excellent response whilst taking H1‐antihistamines. | ||||
| 2.1 Intermediate‐term duration of intervention | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Serious adverse events (i.e. serious enough to require withdrawal of treatment) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 5.3  Comparison 5 Loratadine 10 mg versus emedastine 2 mg, Outcome 3 Serious adverse events (i.e. serious enough to require withdrawal of treatment). | ||||
| 3.1 Intermediate‐term duration of intervention | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 6.1  Comparison 6 Loratadine 10 mg versus hydroxyzine 25 mg, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines. | ||||
| 1.1 Short‐term duration of intervention | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 2 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 2.72 [1.51, 4.91] |
| Analysis 7.1 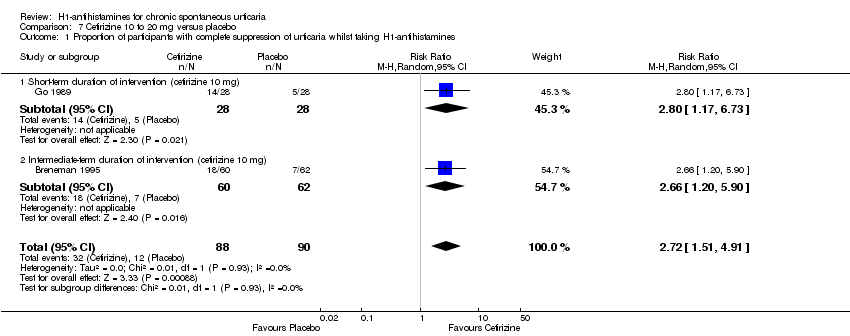 Comparison 7 Cetirizine 10 to 20 mg versus placebo, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines. | ||||
| 1.1 Short‐term duration of intervention (cetirizine 10 mg) | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 2.8 [1.17, 6.73] |
| 1.2 Intermediate‐term duration of intervention (cetirizine 10 mg) | 1 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 2.66 [1.20, 5.90] |
| 2 Serious adverse events (i.e. serious enough to require withdrawal of treatment) Show forest plot | 3 | 389 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.68, 13.22] |
| Analysis 7.2  Comparison 7 Cetirizine 10 to 20 mg versus placebo, Outcome 2 Serious adverse events (i.e. serious enough to require withdrawal of treatment). | ||||
| 2.1 Intermediate‐term duration of intervention (cetirizine 10 mg) | 2 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 4.60 [0.79, 26.67] |
| 2.2 Intermediate‐term duration of intervention (cetirizine 10 to 20 mg) | 1 | 142 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.07, 16.59] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Serious adverse events (i.e. serious enough to require withdrawal of treatment) Show forest plot | 2 | 261 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.25, 2.45] |
| Analysis 8.1 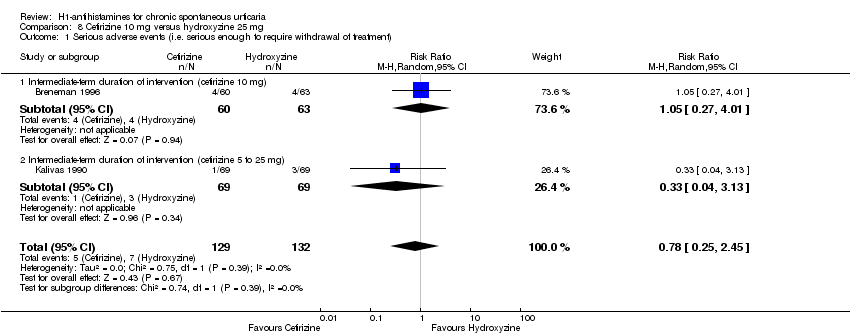 Comparison 8 Cetirizine 10 mg versus hydroxyzine 25 mg, Outcome 1 Serious adverse events (i.e. serious enough to require withdrawal of treatment). | ||||
| 1.1 Intermediate‐term duration of intervention (cetirizine 10 mg) | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.27, 4.01] |
| 1.2 Intermediate‐term duration of intervention (cetirizine 5 to 25 mg) | 1 | 138 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.04, 3.13] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 9.1 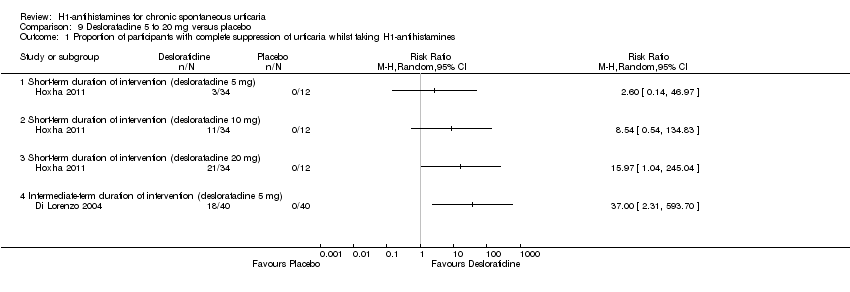 Comparison 9 Desloratadine 5 to 20 mg versus placebo, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines. | ||||
| 1.1 Short‐term duration of intervention (desloratadine 5 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Short‐term duration of intervention (desloratadine 10 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Short‐term duration of intervention (desloratadine 20 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Intermediate‐term duration of intervention (desloratadine 5 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Serious adverse events (i.e. serious enough to require withdrawal of treatment) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 9.2  Comparison 9 Desloratadine 5 to 20 mg versus placebo, Outcome 2 Serious adverse events (i.e. serious enough to require withdrawal of treatment). | ||||
| 2.1 Intermediate‐term duration of 5 mg of intervention | 3 | 466 | Risk Ratio (M‐H, Random, 95% CI) | 1.46 [0.42, 5.10] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Serious adverse events (i.e. serious enough to require withdrawal of treatment to withdrawal) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 10.1  Comparison 10 Hydroxyzine 25 mg versus placebo, Outcome 1 Serious adverse events (i.e. serious enough to require withdrawal of treatment to withdrawal). | ||||
| 1.1 Intermediate‐term duration of intervention | 2 | 270 | Risk Ratio (M‐H, Random, 95% CI) | 3.64 [0.77, 17.23] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 11.1  Comparison 11 Levocetirizine 5 to 20 mg versus placebo, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines. | ||||
| 1.1 Short‐term duration of intervention (levocetirizine 5 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Short‐term duration of intervention (levocetirizine 10 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Short‐term duration of intervention (levocetirizine 20 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Intermediate‐term duration of intervention (levocetirizine 5 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines Show forest plot | 1 | 245 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [1.03, 1.77] |
| Analysis 12.1  Comparison 12 Rupatadine 10 to 20 mg versus placebo, Outcome 1 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines. | ||||
| 1.1 Intermediate‐term duration of intervention (rupatadine 10 mg) | 1 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.86, 1.91] |
| 1.2 Intermediate‐term duration of intervention (rupatadine 20 mg) | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.98, 2.06] |

Study flow diagram.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
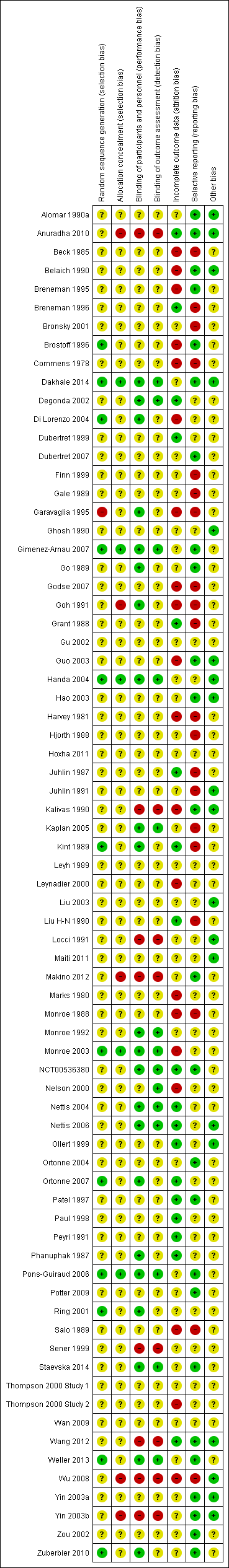
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Comparison 1 Loratadine 10 mg versus placebo, Outcome 1 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines.

Comparison 2 Loratadine 10 mg versus cetirizine 10 mg, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines.

Comparison 3 Loratadine 10 mg versus desloratadine 5 mg, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines.

Comparison 3 Loratadine 10 mg versus desloratadine 5 mg, Outcome 2 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines.

Comparison 4 Loratadine 10 mg versus mizolastine 10 mg, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines.
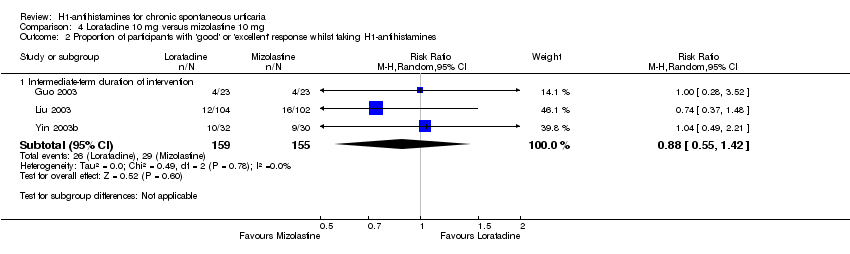
Comparison 4 Loratadine 10 mg versus mizolastine 10 mg, Outcome 2 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines.
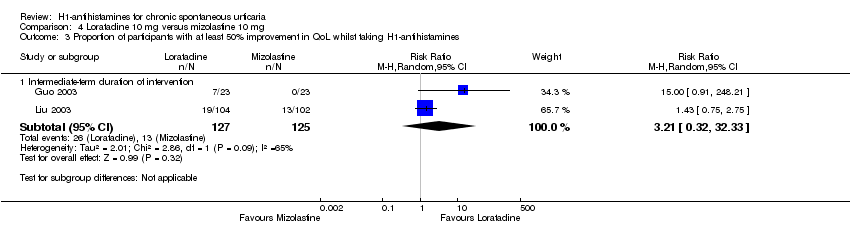
Comparison 4 Loratadine 10 mg versus mizolastine 10 mg, Outcome 3 Proportion of participants with at least 50% improvement in QoL whilst taking H1‐antihistamines.
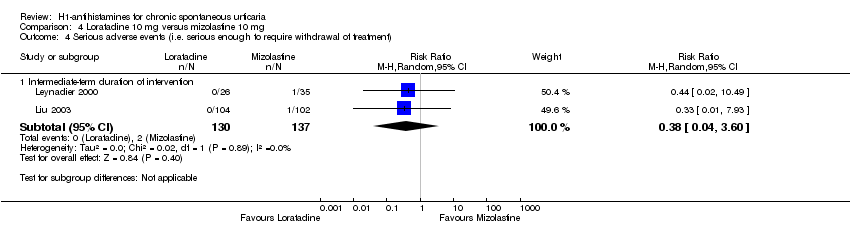
Comparison 4 Loratadine 10 mg versus mizolastine 10 mg, Outcome 4 Serious adverse events (i.e. serious enough to require withdrawal of treatment).
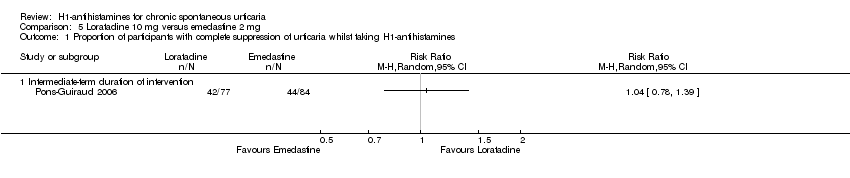
Comparison 5 Loratadine 10 mg versus emedastine 2 mg, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines.
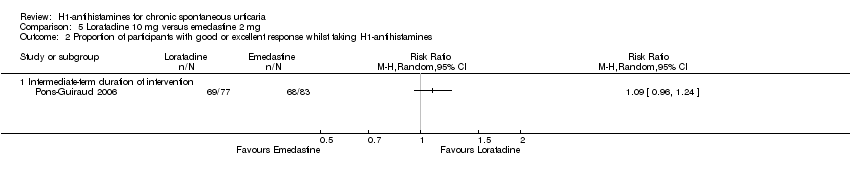
Comparison 5 Loratadine 10 mg versus emedastine 2 mg, Outcome 2 Proportion of participants with good or excellent response whilst taking H1‐antihistamines.
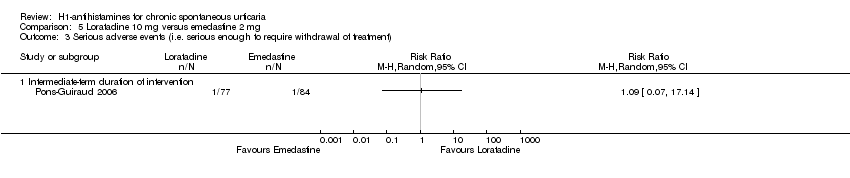
Comparison 5 Loratadine 10 mg versus emedastine 2 mg, Outcome 3 Serious adverse events (i.e. serious enough to require withdrawal of treatment).
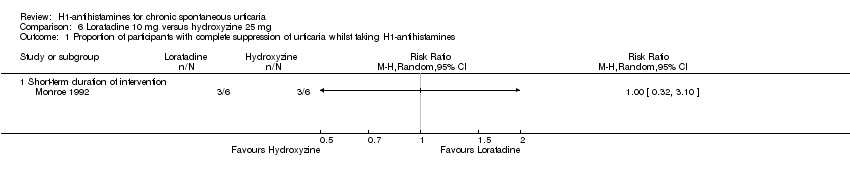
Comparison 6 Loratadine 10 mg versus hydroxyzine 25 mg, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines.

Comparison 7 Cetirizine 10 to 20 mg versus placebo, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines.

Comparison 7 Cetirizine 10 to 20 mg versus placebo, Outcome 2 Serious adverse events (i.e. serious enough to require withdrawal of treatment).

Comparison 8 Cetirizine 10 mg versus hydroxyzine 25 mg, Outcome 1 Serious adverse events (i.e. serious enough to require withdrawal of treatment).
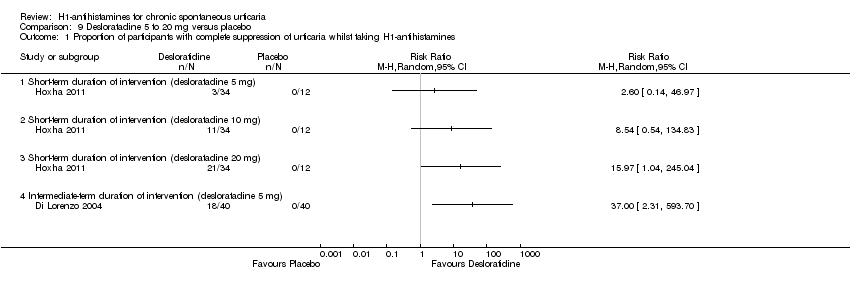
Comparison 9 Desloratadine 5 to 20 mg versus placebo, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines.

Comparison 9 Desloratadine 5 to 20 mg versus placebo, Outcome 2 Serious adverse events (i.e. serious enough to require withdrawal of treatment).

Comparison 10 Hydroxyzine 25 mg versus placebo, Outcome 1 Serious adverse events (i.e. serious enough to require withdrawal of treatment to withdrawal).

Comparison 11 Levocetirizine 5 to 20 mg versus placebo, Outcome 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines.
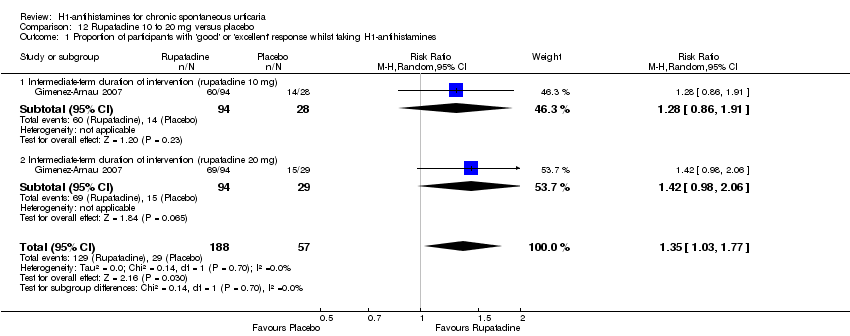
Comparison 12 Rupatadine 10 to 20 mg versus placebo, Outcome 1 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines.
| Cetirizine 10 to 20 mg versus placebo for chronic spontaneous urticaria | ||||||
| Patient or population: patients with chronic spontaneous urticaria | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (placebo) | Cetirizine 10 to 20 mg | |||||
| Complete suppression of urticaria | Study population | RR 2.72 | 178 | ⊕⊕⊝⊝ | Favours cetirizine | |
| 133 per 1000 | 363 per 1000 | |||||
| Moderate | ||||||
| 146 per 1000 | 397 per 1000 | |||||
| Adverse events leading to withdrawal | Study population | RR 3 | 389 | ⊕⊕⊝⊝ | Favours neither intervention nor control | |
| 10 per 1000 | 30 per 1000 | |||||
| Moderate | ||||||
| 14 per 1000 | 42 per 1000 | |||||
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDesign limitation (risk of bias). | ||||||
| Desloratadine 5 to 20 mg versus placebo for chronic spontaneous urticaria | ||||||
| Patient or population: patients with chronic spontaneous urticaria | ||||||
| Outcomes | Illustrative comparative risks* | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (placebo) | Desloratadine 5 to 20 mg | |||||
| Complete suppression of urticaria: short‐term duration of intervention (desloratadine 5 mg) | See comment | See comment | Not estimable | 46 | ⊕⊕⊝⊝ | Favours neither intervention nor control Only 1 study, a conference abstract (Hoxha 2011) |
| Complete suppression of urticaria: short‐term duration of intervention (desloratadine 10 mg) | See comment | See comment | Not estimable | 46 | ⊕⊕⊝⊝ | Favours neither intervention nor control Only 1 study, a conference abstract (Hoxha 2011) |
| Complete suppression of urticaria: short‐term duration of intervention (desloratadine 20 mg) | See comment | See comment | Not estimable | 46 | ⊕⊕⊝⊝ | Favours desloratadine Only 1 study, a conference abstract (Hoxha 2011) |
| Complete suppression of urticaria: intermediate‐term duration of intervention (desloratadine 5 mg) | See comment | See comment | Not estimable | 80 | ⊕⊕⊝⊝ | Favours desloratadine Only 1 study (Di Lorenzo 2004) |
| Adverse effects leading to withdrawal: intermediate‐term duration of 5 mg of intervention | Study population | RR 1.46 | 466 | ⊕⊕⊝⊝ | Favours neither intervention nor control | |
| 17 per 1000 | 25 per 1000 | |||||
| Moderate | ||||||
| 18 per 1000 | 26 per 1000 | |||||
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDesign limitation (risk of bias). | ||||||
| Levocetirizine 5 to 20 mg versus placebo for chronic spontaneous urticaria | ||||||
| Patient or population: patients with chronic spontaneous urticaria | ||||||
| Outcomes | Illustrative comparative risks* | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (placebo) | Levocetirizine 5 to 20 mg | |||||
| Complete suppression of urticaria: short‐term duration of intervention (levocetirizine 5 mg) | See comment | See comment | Not estimable | 49 | ⊕⊕⊝⊝ | Favours neither intervention nor control Only 1 study, a conference abstract (Hoxha 2011) |
| Complete suppression of urticaria: short‐term duration of intervention (levocetirizine 10 mg) | See comment | See comment | Not estimable | 49 | ⊕⊕⊝⊝ | Favours neither intervention nor control Only 1 study, a conference abstract (Hoxha 2011) |
| Complete suppression of urticaria: short‐term duration of intervention (levocetirizine 20 mg) | See comment | See comment | Not estimable | 49 | ⊕⊕⊝⊝ | Favours levocetirizine Only 1 study, a conference abstract (Hoxha 2011) |
| Complete suppression of urticaria: intermediate‐term duration of intervention (levocetirizine 5 mg) | See comment | See comment | Not estimable | 100 | ⊕⊕⊝⊝ | Favours levocetirizine Only 1 study (Nettis 2006) |
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDesign limitation (risk of bias). | ||||||
| Rupatadine 10 to 20 mg versus placebo for chronic spontaneous urticaria | ||||||
| Patient or population: patients with chronic spontaneous urticaria | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (placebo) | Rupatadine 10 to 20 mg | |||||
| Good or excellent response | Study population | RR 1.35 | 245 | ⊕⊕⊝⊝ | Favours rupatadine | |
| 509 per 1000 | 687 per 1000 | |||||
| Moderate | ||||||
| 509 per 1000 | 687 per 1000 | |||||
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDesign limitation (risk of bias). | ||||||
| Loratadine 10 mg versus placebo for chronic spontaneous urticaria | ||||||
| Patient or population: patients with chronic spontaneous urticaria | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (placebo) | Loratadine 10 mg | |||||
| Good or excellent response | Study population | RR 1.86 | 124 | ⊕⊕⊝⊝ | Favours neither intervention nor control | |
| 155 per 1000 | 289 per 1000 | |||||
| Moderate | ||||||
| 160 per 1000 | 298 per 1000 | |||||
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDesign limitation (risk of bias). | ||||||
| Loratadine 10 mg versus cetirizine 10 mg for chronic spontaneous urticaria | ||||||
| Patient or population: patients with chronic spontaneous urticaria | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (cetirizine 10 mg) | Loratadine 10 mg | |||||
| Complete cessation of urticaria | Study population | RR 1.05 | 103 | ⊕⊕⊝⊝ | Combined short and intermediate‐term duration of intervention. Favours neither intervention nor control | |
| 588 per 1000 | 618 per 1000 | |||||
| Moderate | ||||||
| 574 per 1000 | 603 per 1000 | |||||
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDesign limitation (risk of bias). | ||||||
| Loratadine 10 mg versus desloratadine 5 mg for chronic spontaneous urticaria | ||||||
| Patient or population: patients with chronic spontaneous urticaria | ||||||
| Outcomes | Illustrative comparative risks* | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (desloratadine | Loratadine 10 mg | |||||
| Complete suppression of urticaria: intermediate‐term duration of intervention | Study population | RR 0.91 | 369 | ⊕⊕⊝⊝ | Favours neither intervention nor control | |
| 658 per 1000 | 598 per 1000 | |||||
| Moderate | ||||||
| 670 per 1000 | 610 per 1000 | |||||
| Good or excellent response: intermediate‐term duration of intervention | Study population | RR 1.04 | 410 | ⊕⊕⊝⊝ | Favours neither intervention nor control No participants reported a good or excellent response in the loratadine group in Zou 2002 We found low levels of statistical heterogeneity in this analysis I2 = 40%) | |
| 263 per 1000 | 274 per 1000 | |||||
| Moderate | ||||||
| 228 per 1000 | 237 per 1000 | |||||
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDesign limitation (risk of bias). | ||||||
| Loratadine 10 mg compared to mizolastine 10 mg for chronic spontaneous urticaria | ||||||
| Patient or population: patients with chronic spontaneous urticaria | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (mizolastine | Loratadine 10 mg | |||||
| Complete cessation of urticaria: intermediate‐term duration of intervention | Study population | RR 0.86 | 316 | ⊕⊝⊝⊝ | Overall, favours neither loratadine nor mizolastine In Guo 2003, more participants in mizolastine group had complete cessation of urticaria than in the other 2 studies (Liu 2003 and Yin 2003b) | |
| 675 per 1000 | 581 per 1000 | |||||
| Moderate | ||||||
| 667 per 1000 | 574 per 1000 | |||||
| Good or excellent response: intermediate‐term duration of intervention | Study population | RR 0.88 | 314 | ⊕⊕⊝⊝ | Favours neither loratadine nor mizolastine | |
| 187 per 1000 | 165 per 1000 | |||||
| Moderate | ||||||
| 174 per 1000 | 153 per 1000 | |||||
| Adverse events leading to withdrawal: intermediate‐term duration of intervention | Study population | RR 0.38 | 267 | ⊕⊕⊝⊝ | Favours neither loratadine nor mizolastine | |
| 15 per 1000 | 6 per 1000 | |||||
| Moderate | ||||||
| 19 per 1000 | 7 per 1000 | |||||
| Proportion of participants with at least 50% improvement in QoL: intermediate‐term duration of intervention | Study population | RR 3.21 | 252 | ⊕⊝⊝⊝ | Favours neither loratadine nor mizolastine No participants in the mizolastine group in Guo 2003 reported at least 50% improvement in QoL | |
| 104 per 1000 | 334 per 1000 | |||||
| Moderate | ||||||
| 64 per 1000 | 205 per 1000 | |||||
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDesign limitation (risk of bias). | ||||||
| Loratadine 10 mg versus emedastine 2 mg for chronic spontaneous urticaria | ||||||
| Patient or population: patients with chronic spontaneous urticaria | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (emedastine 2 mg) | Loratadine 10 mg | |||||
| Complete cessation of urticaria: intermediate‐term duration of intervention | Study population | RR 1.04 | 161 | ⊕⊕⊕⊝ | Favours neither loratadine nor emedastine Only 1 study (Pons‐Guiraud 2006) | |
| 524 per 1000 | 545 per 1000 | |||||
| Moderate | ||||||
| 524 per 1000 | 545 per 1000 | |||||
| Good or excellent response: intermediate‐term duration of intervention | Study population | RR 1.09 | 160 | ⊕⊕⊕⊝ | Favours neither loratadine nor emedastine Only 1 study (Pons‐Guiraud 2006) | |
| 819 per 1000 | 893 per 1000 | |||||
| Moderate | ||||||
| 819 per 1000 | 893 per 1000 | |||||
| Adverse events leading to withdrawal: intermediate‐term duration of intervention | Study population | RR 1.09 | 161 | ⊕⊕⊕⊝ | Favours neither loratadine nor emedastine Only 1 study (Pons‐Guiraud 2006) | |
| 12 per 1000 | 13 per 1000 | |||||
| Moderate | ||||||
| 12 per 1000 | 13 per 1000 | |||||
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aRelatively few participants and few events and/or wide confidence intervals. | ||||||
| Loratadine 10 mg versus hydroxyzine 25 mg for chronic spontaneous urticaria | ||||||
| Patient or population: patients with chronic spontaneous urticaria | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (hydroxyzine 25 mg) | Loratadine 10 mg | |||||
| Complete suppression of urticaria: short‐term duration of intervention | Study population | RR 1 | 12 | ⊕⊕⊝⊝ | Favours neither intervention or control Only 1 study (Monroe 1992) | |
| 500 per 1000 | 500 per 1000 | |||||
| Moderate | ||||||
| 500 per 1000 | 500 per 1000 | |||||
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDesign limitation (risk of bias). | ||||||
| Cetirizine 10 mg versus hydroxyzine 25 mg for chronic spontaneous urticaria | ||||||
| Patient or population: patients with chronic spontaneous urticaria | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (hydroxyzine 25 mg) | Cetirizine 10 mg | |||||
| Adverse events leading to withdrawal | Study population | RR 0.78 | 261 | ⊕⊕⊝⊝ | Favours neither cetirizine nor hydroxyzine | |
| 53 per 1000 | 41 per 1000 | |||||
| Moderate | ||||||
| 54 per 1000 | 42 per 1000 | |||||
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDesign limitation (risk of bias). | ||||||
| Hydroxyzine 25 mg versus placebo for chronic spontaneous urticaria | ||||||
| Patient or population: patients with chronic spontaneous urticaria | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | Number of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control (placebo) | Hydroxyzine 25 mg | |||||
| Adverse events leading to withdrawal: intermediate‐term duration of intervention | Study population | RR 3.64 | 270 | ⊕⊕⊝⊝ | Favours neither intervention nor control | |
| 14 per 1000 | 53 per 1000 | |||||
| Moderate | ||||||
| 15 per 1000 | 55 per 1000 | |||||
| *The basis for the assumed risk (e.g. median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence. | ||||||
| aDesign limitation (risk of bias). | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines Show forest plot | 2 | 124 | Risk Ratio (M‐H, Random, 95% CI) | 1.86 [0.91, 3.79] |
| 1.1 Short‐term duration of intervention (10 mg) | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.42, 21.30] |
| 1.2 Intermediate‐term duration of intervention (10 mg) | 1 | 112 | Risk Ratio (M‐H, Random, 95% CI) | 1.73 [0.81, 3.72] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 2 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.76, 1.43] |
| 1.1 Short‐term duration of intervention | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.64, 2.01] |
| 1.2 Intermediate‐term duration of intervention | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.69, 1.47] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Intermediate‐term duration of intervention | 2 | 369 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.78, 1.06] |
| 2 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Intermediate‐term duration of intervention | 3 | 410 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.64, 1.71] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Intermediate‐term duration of intervention | 3 | 316 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.64, 1.16] |
| 2 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Intermediate‐term duration of intervention | 3 | 314 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.55, 1.42] |
| 3 Proportion of participants with at least 50% improvement in QoL whilst taking H1‐antihistamines Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Intermediate‐term duration of intervention | 2 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 3.21 [0.32, 32.33] |
| 4 Serious adverse events (i.e. serious enough to require withdrawal of treatment) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Intermediate‐term duration of intervention | 2 | 267 | Risk Ratio (M‐H, Random, 95% CI) | 0.38 [0.04, 3.60] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Intermediate‐term duration of intervention | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Proportion of participants with good or excellent response whilst taking H1‐antihistamines Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Intermediate‐term duration of intervention | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Serious adverse events (i.e. serious enough to require withdrawal of treatment) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Intermediate‐term duration of intervention | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Short‐term duration of intervention | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 2 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 2.72 [1.51, 4.91] |
| 1.1 Short‐term duration of intervention (cetirizine 10 mg) | 1 | 56 | Risk Ratio (M‐H, Random, 95% CI) | 2.8 [1.17, 6.73] |
| 1.2 Intermediate‐term duration of intervention (cetirizine 10 mg) | 1 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 2.66 [1.20, 5.90] |
| 2 Serious adverse events (i.e. serious enough to require withdrawal of treatment) Show forest plot | 3 | 389 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.68, 13.22] |
| 2.1 Intermediate‐term duration of intervention (cetirizine 10 mg) | 2 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 4.60 [0.79, 26.67] |
| 2.2 Intermediate‐term duration of intervention (cetirizine 10 to 20 mg) | 1 | 142 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.07, 16.59] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Serious adverse events (i.e. serious enough to require withdrawal of treatment) Show forest plot | 2 | 261 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.25, 2.45] |
| 1.1 Intermediate‐term duration of intervention (cetirizine 10 mg) | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.27, 4.01] |
| 1.2 Intermediate‐term duration of intervention (cetirizine 5 to 25 mg) | 1 | 138 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.04, 3.13] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Short‐term duration of intervention (desloratadine 5 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Short‐term duration of intervention (desloratadine 10 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Short‐term duration of intervention (desloratadine 20 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Intermediate‐term duration of intervention (desloratadine 5 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Serious adverse events (i.e. serious enough to require withdrawal of treatment) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Intermediate‐term duration of 5 mg of intervention | 3 | 466 | Risk Ratio (M‐H, Random, 95% CI) | 1.46 [0.42, 5.10] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Serious adverse events (i.e. serious enough to require withdrawal of treatment to withdrawal) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Intermediate‐term duration of intervention | 2 | 270 | Risk Ratio (M‐H, Random, 95% CI) | 3.64 [0.77, 17.23] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with complete suppression of urticaria whilst taking H1‐antihistamines Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Short‐term duration of intervention (levocetirizine 5 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Short‐term duration of intervention (levocetirizine 10 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Short‐term duration of intervention (levocetirizine 20 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Intermediate‐term duration of intervention (levocetirizine 5 mg) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Proportion of participants with 'good' or 'excellent' response whilst taking H1‐antihistamines Show forest plot | 1 | 245 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [1.03, 1.77] |
| 1.1 Intermediate‐term duration of intervention (rupatadine 10 mg) | 1 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.86, 1.91] |
| 1.2 Intermediate‐term duration of intervention (rupatadine 20 mg) | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.98, 2.06] |