Bifosfonatos para la enfermedad de Paget del hueso en adultos
Referencias
Referencias de los estudios incluidos en esta revisión
Referencias de los estudios excluidos de esta revisión
Referencias de los estudios en curso
Referencias adicionales
Referencias de otras versiones publicadas de esta revisión
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | RCT. Randomisation ratio: 9 (placebo): 7 (etidronate 1 mg/kg/day): 7 (etidronate 2.5 mg/kg/day): 9 (etidronate 5 mg/kg/day): 8 (etidronate 10 mg/kg/day): 7 (etidronate 20 mg/kg/day). Superiority design | |
| Participants | Diagnostic criteria: x‐ray evidence of Paget's disease of bone. Inclusion criteria: Pain at one or more sites of Paget's bone involvement and elevated serum alkaline phosphatase or urinary hydroxyproline at least twice the upper limit of normal (ULN) range. Exclusion criteria: not stated. Number screened: not stated. Number randomised: 50. Number analysed: 47 (65 years, 60% male; percentages monostotic, symptomatic and previously treated for Paget's disease of bone not stated) | |
| Interventions | 6 parallel treatment groups; placebo and etidronate in 5 different doses (1 mg, 2.5 mg, 5 mg, 10 mg and 20 mg/kg/day). Co‐interventions: not stated | |
| Outcomes | Outcomes reported in abstract:
Time points for measurement: 6 months. How were the outcomes measured: prospectively | |
| Setting and date | One hospital bone unit (Miami, FL, USA). Period when the study was conducted: Not stated | |
| Follow up period | 6 months | |
| Publication details and funding source | Language of publication: English. Funding source: Supported in part by the General Clinical Research Center and a grant from the National Institute of Health (5MO1‐RR‐261‐08). Etidronate was supplied by the Procter and Gamble Company, Cincinnati, OH, USA Declarations of interest among primary researchers: Not stated | |
| Notes | Etidronate groups data were pooled for meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | There were no data about how participants were selected. The authors did not use the term 'randomization', but the study was double‐blinded, so the study design should have had some way of randomising participants to administer treatment. However, the risk of bias should be likely high because no information was provided for the clinical characteristics of groups before starting the study (gender, age…) and the data on mean alkaline phosphatase or urinary hydroxyproline are clearly different at the start of the study for all groups |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "double‐blind study". |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote: "Scans were coded and read blind for qualitative change by at least two observers". Comment: Outcome assessment was blinded for scans, but there was insufficient information to permit judgement about other outcomes |
| Incomplete outcome data (attrition bias) | High risk | Data from the original manuscript (Altman 1973) did not agree with data in the extension study (Khairi 1974). Altman 1973: "Fifty patients entered the study". Khairi 1974: "Fifty‐four patients started in this study". Numbers of participants included in the treatment groups differed for Altman 1973 and Khairi 1974. In Altman 1973 23 participants were randomised to placebo plus 1 mg/kg and 2.5 mg/kg dose groups; this number was 24 in Khairi 1974. Altman 1973 reported randomising 9, 8 and 7 participants respectively to 5 mg/kg, 10 mg/kg and 10 mg/kg groups; in Khairi 1974 8, 10 and 8 participants were reported to have been randomised to these respective treatment arms |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement. Methods and results sections were consistent. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting was insufficient to permit judgement. |
| Methods | RCT. Randomisation ratio: 1:2 (pamidronate: olpadronate). Superiority design | |
| Participants | Diagnostic criteria: Not stated. Inclusion criteria: active Paget's disease of bone despite previous therapies. Exclusion criteria: not stated. Number screened: not stated. Number randomised: 27. Number analysed: 21 (age and sex not stated; percentages monostotic, symptomatic and previously treated for Paget's disease of bone were not stated) | |
| Interventions | Two parallel treatment groups; pamidronate 400 mg/day for 4 months vs. olpadronate 200 mg/day for 12 days. Co‐interventions: Not reported | |
| Outcomes | Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints: Number of participants who experienced adverse events related to use of bisphosphonates. Time points for measurement: 6 months. How were the outcomes were measured: Insufficient information. Likely prospectively | |
| Setting and date | Multicentre; Argentina. Period when the study was conducted: Not stated | |
| Follow up period | 6 months | |
| Publication details and funding source | Language of publication: English. Funding source: Not stated. Declarations of interest among primary researchers: Not stated | |
| Notes | Data only from abstracts | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomized in a 1:2 schedule." Randomisation method not reported |
| Allocation concealment (selection bias) | Unclear risk | Randomisation method not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Both formulations were double dummy with placebos" Comment: Probably done. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Insufficient information to permit judgement |
| Incomplete outcome data (attrition bias) | Unclear risk | Insufficient information to permit judgement. Only an abstract of the trials was published. Many data were lacking; there were no data on attrition, exclusions or adverse events |
| Selective reporting (reporting bias) | Unclear risk | Insufficient reporting to permit judgement. Methods and results sections were poorly described. It was not possible to assess if they were consistent. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting was insufficient to permit judgement. |
| Methods | RCT. Randomisation ratio: 1:1:1:1:1 (placebo: zoledronate 50 µg: 100 µg; 200 µg; 400 µg) Superiority design | |
| Participants | Diagnostic criteria: x‐ray evidence of Paget's disease of bone Inclusion criteria: Paget's disease of bone and serum alkaline phosphatase concentrations at least twice the ULN Exclusion criteria: Treatment with bisphosphonates in the previous 6 months or calcitonin or plicamycin in the previous 3 months. Creatinine clearance < 60 mL/min. Abnormal liver function Number screened: not stated Number randomised: 176 Number analysed: 172 (71 years, 61% male; percentages monostotic, symptomatic or previously treated for Paget's disease of bone were not stated) | |
| Interventions | 5 parallel treatment groups; placebo and zoledronate in 4 doses (50 µg, 100 µg, 200 µg and 400 µg in a single intravenous infusion) Co‐interventions: Not reported | |
| Outcomes | Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity Secondary endpoints: Numbers of participants who experienced adverse events related to use of bisphosphonates Time points for measurement: Baseline, 5 days, 10 days, 30 days, 45 days, 60 days and 90 days. How were the outcomes measured: prospectively | |
| Setting and date | 20 sites in USA and UK. Period when the study was conducted: Not stated | |
| Follow up period | 3 months | |
| Publication details and funding source | Language of publication: English. Funding source: Funding source not reported in the manuscript, but one author, P Richardson, indicated "Ciba Pharmaceuticals" (abstract) and "Clinical Research, Novartis Pharmaceuticals, East Hanover, NJ, USA" (primary reference) affiliations Declarations of interest among primary researchers: Not stated | |
| Notes | Zoledronate groups data were pooled for meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The patients were randomized in a double‐blind manner". The allocation method was not described |
| Allocation concealment (selection bias) | Unclear risk | The allocation method was not described |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Randomized in a double blind manner". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Insufficient information to permit judgement. The risk of bias was likely to be low for serum data because assays for bone‐specific alkaline phosphatase, urinary calcium, creatinine, hydroxyproline, pyridinoline and deoxypiridinoline were conducted in a central laboratory (Nichols Institute, San Juan Capistrano, CA, USA) |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "All efficacy analyses were based on all randomised patients who had at least one post baseline measurement (intention‐to‐treat analysis). Four of the 176 patients did not complete all post baseline evaluations". Comment: Attrition data were provided |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement. Methods and results sections were consistent. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting was insufficient to permit judgement |
| Methods | RCT. Randomisation ratio: 12 (placebo): 7 (etidronate 2.5 mg/kg/day): 8 (etidronate 5 mg/kg/day): 11 (etidronate 10 mg/kg/day): 10 (etidronate 20 mg/kg/day). Superiority design | |
| Participants | Diagnostic criteria: x‐ray evidence of Paget's disease of bone. Inclusion criteria: symptoms referable to the disorder, elevation of serum alkaline phosphatase and urinary hydroxyproline (with at least twice the normal value of one these indices). Exclusion criteria: No co‐existing condition that would complicate the interpretation of therapeutic results. No other specific therapy for Paget's disease for at least 6 months before entry into the study. Number screened: not stated. Number randomised: 48. Number analysed: 48 (age not stated, 58% male; percentages monostotic, symptomatic or previously treated for Paget's disease of bone were not stated) | |
| Interventions | 5 parallel treatment groups; placebo, and etidronate in 4 doses (2.5 mg, 5 mg, 10 mg and 20 mg/kg/day). Co‐interventions: Not reported | |
| Outcomes | Outcomes reported in abstract Primary endpoint: mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints: change in bone pain (assessment tool used not reported); radiologically‐confirmed clinical fracture. Time points for measurement: Baseline, 1 month, 2 months, 3 months, 4.5 months, 6 months. How were the outcomes measured: prospectively | |
| Setting and date | Participants admitted to 4 New York City Hospitals. Period when the study was conducted: Not stated | |
| Follow up period | 6 months | |
| Publication details and funding source | Language of publication: English. Funding source: Supported by National Institute of Health grants (MO1.RR00645, 5.MO1‐RR‐96, AM‐09579, TIAM‐05397 and TIAM‐05531). Etidronate was supplied by the Procter and Gamble Company, Cincinnati, OH, USA. Declarations of interest among primary researchers: Not stated | |
| Notes | Etidronate groups data were pooled for meta‐analysis. Data from 27 additional participants treated with etidronate in a non‐blinded, non‐randomised basis were included in the manuscript | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "each patient… was assigned randomly" |
| Allocation concealment (selection bias) | Unclear risk | Randomisation method was not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "for all the patients in treatment group A neither the investigator nor the patient knew the identity of the treatment". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Incomplete outcome data (attrition bias) | Unclear risk | No losses to follow‐up reported. However, it was not clear how many participants were included |
| Selective reporting (reporting bias) | High risk | We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. Likely selective reporting risk is high due to vague data on adverse events. There are no specific number of patients but only sentences as "some patients… showed a rise in serum phosphate" |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting was insufficient to permit judgement |
| Methods | RCT. Randomisation ratio: 1:1:1:1 (placebo: tiludronate 200 mg, 400 mg, 600 mg). Superiority design | |
| Participants | Diagnostic criteria: Paget's disease confirmed by scintigraphy or radiography or both Inclusion criteria: Age ≥ 18 years; serum alkaline phosphate concentration at least twice the ULN Exclusion criteria:
Number screened: not stated. Number randomised: 112. Number analysed: 112 (70 years, 54% male, 30% monostotic, 63% symptomatic, percentage previously treated for Paget's disease of bone not stated) | |
| Interventions | 4 parallel treatment groups; placebo and tiludronate in 3 doses (200 mg, 400 mg, 600 mg/day) for 12 weeks. Co‐interventions: Not reported | |
| Outcomes | Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Treatment success was defined as 50% reduction in serum alkaline phosphatase concentration compared with baseline after 12 weeks' treatment. (Concentration decreased by 25% or less compared with week 0 reading was defined as resistant). Secondary endpoints
Time points for measurement: Baseline, 2 weeks, 8 weeks, 12 weeks, 24 weeks. How were the outcomes measured: prospectively | |
| Setting and date | 16 hospitals in the UK. Period when the study was conducted: Not stated | |
| Follow up period | 6 months. An additional follow‐up to assess the need for re‐treatment was carried out 18 months after the last participant had completed the 6 month trial. Follow‐up was via a postal questionnaire with participants who had completed at least 11 weeks of treatment | |
| Publication details and funding source | Language of publication: English. Funding source: Research supported by Sanofi Winthrop Ltd. Declarations of interest among primary researchers: Not stated | |
| Notes | Tiludronate groups data were pooled for meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were allocated sequential study numbers and randomly assigned to one of the four treatment groups". |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "double‐blind randomised study". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "Data were analysed on an 'intention to treat' basis". Comment: Reasons for missing outcome data (lost to follow‐up and withdrawals) are reported |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. Quote: "A visual analogue scale was used for patients to assess their Pagetic pain". Comment: Although stated that "there were no significant differences between any of the treatment groups" data for pain assessment were only provided for placebo and tiludronate higher dose groups |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting was insufficient to permit judgement |
| Methods | RCT. Randomisation ratio: 1:1 (symptomatic treatment: intensive treatment). Superiority design | |
| Participants | Diagnostic criteria: diagnosis confirmed on plain radiograph of at least one skeletal site according standard criteria from UK guidelines (Selby 2002). Inclusion criteria: Paget's disease of bone and life expectancy > 1 year. Exclusion criteria: No specific exclusion criteria were applied on the basis of treatment history, baseline alkaline phosphatase or co‐existing diseases. Number assessed: 2110. Number randomised: 1331. Number analysed: 1159 (74 years, 51% male, 35% monostotic, 69% symptomatic, 90% previously treated with bisphosphonates for Paget's disease, 52% had normal alkaline phosphatase at baseline) | |
| Interventions | Two parallel treatment groups; symptomatic vs. intensive treatment. Symptomatic treatment: Philosophy; treat bone pain, not alkaline phosphatase.
Intensive treatment: Philosophy; maintain normal alkaline phosphatase.
Co‐interventions: Analgesics and nonsteroidal anti‐inflammatory drugs for symptomatic and intensive treatment group | |
| Outcomes | Outcomes reported in abstract Primary endpoint: Radiologically‐confirmed clinical fracture. Secondary endpoints:
Time points for measurement: Visits scheduled at 4 monthly intervals. How were the outcomes measured: prospectively | |
| Setting and date | 39 secondary referral centres in the UK. Period when the study was conducted: December 2001 to June 2004 | |
| Follow up period | Median 3 years (range 2 to 5 years) | |
| Publication details and funding source | Language of publication: English. Funding source: Supported by grants from the Arthritis Research Campaign UK (Ref. 13627), National Association for Relief of Paget’s Disease, Procter & Gamble Pharmaceuticals and Sanofi‐Aventis. Declarations of interest among primary researchers:
| |
| Notes | The study was described by the authors as a "pragmatic randomised controlled trial designed to compare the effects of two management strategies". The study was not blinded | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were assigned to the treatment groups by an integrated telephone and web‐based randomization system" |
| Allocation concealment (selection bias) | Low risk | Quote: "Treatment allocation employed minimization to ensure that the treatment group were balanced with respect to key prognostic variables" |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "The study was not blinded". Comment: |
| Blinding of outcome assessment (detection bias) | Low risk | Information provided by authors: The assessments of events were performed by individuals who were unaware of the treatment allocation |
| Incomplete outcome data (attrition bias) | Low risk | Information provided by authors: The study was an event‐driven trial, with fracture as the primary endpoint. Data on all fracture events were available even in subjects who had withdrawn from the study. Comment: The risk of attrition bias was low for data at 2 years (minimum duration of follow‐up). The rate of losses to follow‐up at this point was 12% and 13% respectively. The risk of attrition bias is high for 36 or 48 months |
| Selective reporting (reporting bias) | Low risk | Study protocol available. All proposed outcomes were reported |
| Other bias | Low risk | Comment: The study design allowed the attending clinicians to choose different drugs within a specified treatment strategy. Evidence that the attending clinicians adhered to the allocated strategy came from the observation that alkaline phosphatase levels differed significantly between groups |
| Methods | RCT. Randomisation ratio: 1:1:1 (placebo: tiludronate 200 mg: tiludronate 400 mg). Superiority design | |
| Participants | Diagnostic criteria: Diagnosis confirmed by radiographic and clinical criteria. Inclusion criteria: participants aged > 32 years and with serum alkaline phosphatase values at least twice ULN. Exclusion criteria:
Number screened: not stated. Number randomised: 139. Number analysed: 134 (70 years, 54% male; percentages monostotic, symptomatic or previously treated for Paget's disease of bone not stated) | |
| Interventions | Three parallel treatment groups; placebo and tiludronate in two different doses (200 mg, 400 mg) nightly for 12 weeks. Co‐interventions: Calcium‐containing food and supplements were avoided around the time the study medication was taken | |
| Outcomes | Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: Baseline, 2 weeks, 4 weeks, 8 weeks, 12 weeks, 16 weeks, 20 weeks, 24 weeks. How were the outcomes measured: prospectively | |
| Setting and date | 20 centres in the USA and Canada. Period when the study was conducted: Not stated | |
| Follow up period | 6 months | |
| Publication details and funding source | Language of publication: English. Funding source: Research supported by Sanofi Winthrop Ltd. Declarations of interest among primary researchers: Not stated | |
| Notes | Data on the tiludronate groups were pooled for meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "In a randomized, double‐blind, placebo‐controlled study... were enrolled at 20 centers throughout the USA and Canada" Comment: Likely that the risk of bias was low because for a multicentre design, the allocation process should be centralized |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) | Low risk | "Double‐blind" |
| Blinding of outcome assessment (detection bias) | Unclear risk | Insufficient information to permit judgement. The risk is probably low for laboratory data because two laboratories were involved:
|
| Incomplete outcome data (attrition bias) | Low risk | Reasons for attrition and exclusion were reported. 2/91 (2%) participants were lost from the interventions group and 3/48 (65) participants from the placebo group |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. Comment: Data for pain assessment were poorly reported |
| Other bias | Unclear risk | No other risks of bias found, but reporting was insufficient to permit judgement. |
| Methods | RCT. Randomisation ratio: 2:1 (pamidronate: zoledronate). Superiority design | |
| Participants | Diagnostic criteria: confirmed by bone scintigraphy and x‐ray of areas of increased isotope uptake. Inclusion criteria: Presence of serum total alkaline phosphatase above ULN (298 IU/L) on 2 consecutive measurements and no treatment with bisphosphonates or other drugs affecting bone metabolism for at least 6 months before the study. Exclusion criteria:
Number screened: not stated. Number randomised: 90. Number analysed: 89 (70 years, 64% male; percentage monostotic not stated, 99% symptomatic, 66.7% previously treated for Paget's disease of bone) | |
| Interventions | 2 parallel treatment groups; 4 mg single infusion of zoledronate or 30 mg infusion of pamidronate for 2 consecutive days every 3 months. Co‐interventions: 1 g calcium and 800 IU cholecalciferol per day | |
| Outcomes | Outcomes reported in abstract Primary endpoint: Rate of therapeutic response (defined as normalisation of alkaline phosphatase levels or a reduction of at least 75% in total alkaline phosphatase excess). Secondary endpoints:
Time points for measurement: Baseline and 6 months. How were the outcomes measured: prospectively | |
| Setting and date | One centre in Siena, Italy. Period when the study was conducted: Not stated | |
| Follow up period | 6 months | |
| Publication details and funding source | Language of publication: English. Funding source: Not stated. Declarations of interest among primary researchers: No authors had conflicts of interest | |
| Notes | The study was subdivided:
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomized study... randomization was stratified according to baseline alkaline phosphatase levels and previous bisphosphonate treatment (as a binary variable: yes or no)". Comment: Random sequencing was probably computer‐generated; the randomisation was stratified |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) | High risk | Open‐label |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "29 of 30 patients receiving zoledronate and 60 of 60 patients receiving pamidronate completed the follow‐up at 6 mo". Comment: Only one participant was lost to follow‐up at 6 months |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | No other risks of bias found, but reporting insufficient to permit judgement |
| Methods | RCT. Randomisation ratio: 1:1 (risedronate: etidronate). Superiority design | |
| Participants | Diagnostic criteria: confirmed by bone scintigraphy or x‐ray. Inclusion criteria: participants aged 18 to 85 years, with serum alkaline phosphatase concentration of at least twice the ULN. Women had to be at least 1 year postmenopause or using contraception. Exclusion criteria:
Number screened: 179. Number randomised: 123. Number analysed: 103 (66 years, 69% males, 24% monostotic, 91% symptomatic, 72% previously treated for Paget's disease of bone) | |
| Interventions | Two parallel treatment groups; oral risedronate 30 mg daily for 2 months and oral etidronate 400 mg for 6 months. Co‐interventions: Not reported | |
| Outcomes | Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: Baseline and 1 through 6, 8, 10, 12, and 18 months for laboratory measures. Baseline, 2, 6 and 12 months for quality of life (including pain). How were the outcomes measured: prospectively | |
| Setting and date | 12 study centres in the USA and Canada. Period when the study was conducted: Not stated | |
| Follow up period | 12 months plus an optional 6 month extended follow‐up | |
| Publication details and funding source | Language of publication: English Funding source: Supported by Procter & Gamble Pharmaceuticals, Cincinnati, OH, USA. Declarations of interest among primary researchers: Not stated | |
| Notes | None | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were assigned unique, sequential identification numbers according to the chronological order of entry at each of the 12 centers and were stratified into those who had, and those who had not, received previous etidronate treatment. Within each center, and within each stratum of previous etidronate use, patients were assigned to one of the two treatment groups according to a randomisation schedule generated using SAS Version 6.07 PLAN procedure (SAS Institute, Cary, NC)". |
| Allocation concealment (selection bias) | Low risk | Computerised random number generator was used |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Double blind design". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Incomplete outcome data (attrition bias) | High risk | Comment: Although the authors stated that the study included an intention‐to‐treat analysis:
|
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting insufficient to make judgement |
| Methods | RCT. Randomisation ratio: 2:1 (alendronate: placebo). Superiority design | |
| Participants | Diagnostic criteria: diagnosis established by finding hyperphosphatasia and characteristic radiographic and scintigraphic features. Inclusion criteria: participants diagnosed as active Paget's disease of bone. Exclusion criteria:
Number screened: not stated. Number randomised: 15. Number analysed: 15 (67 years, 60% male, % monostotic not stated, 87% symptomatic, 66% previously treated for Paget's disease of bone) | |
| Interventions | Two parallel treatment groups: intravenous alendronate 10 mg/day for 5 days and placebo. Co‐interventions: Not reported | |
| Outcomes | Outcomes reported in abstract: Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: Baseline and 2, 3 and 4 weeks from the start of treatment and monthly thereafter for 6 months. How were the outcomes measured: prospectively | |
| Setting and date | One centre in Sheffield, UK. Period when the study was conducted: Not stated | |
| Follow up period | 6 months. Comment: The blind was broken 4 weeks after the start of treatment. The 10 participants who received treatment with alendronate were additionally followed for 6 months. The 5 participants who received placebo were allowed to withdraw from the study after the blind was broken | |
| Publication details and funding source | Language of publication: English Funding source: Supported by a Programme Grant from the Medical Research Council and by Merck, Sharp and Dohme Research Laboratories, Woodbridge, NJ, USA and Harlow, UK. Declarations of interest among primary researchers: Not stated | |
| Notes | None | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "A group of 15 patients with active Paget’s disease of bone were randomised prospectively". Comment: Insufficient information provided to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "receive 5 days of treatment with either alendronate (10 mg/day) or placebo under double‐blind conditions". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Incomplete outcome data (attrition bias) | High risk | Quote: "The blind was broken 4 weeks after the start of treatment. The 10 participants who received treatment with alendronate were additionally followed for 6 months. The 5 participants who received placebo were allowed to withdraw from the study after the blind was broken." Comment: The follow‐up was different for participants in the placebo and experimental groups (e.g. data on urinary hydroxyproline and serum alkaline phosphatase (Figure 1) were registered to different weeks from the start of treatment for alendronate or placebo) |
| Selective reporting (reporting bias) | High risk | We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. Comment: Pain data were reported, but pain was not defined as an outcome. There was no reporting of how pain was assessed. There were no data reported on adverse effects. |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting was insufficient to permit judgement |
| Methods | RCT. Randomisation ratio: 1:1 (etidronate: etidronate plus calcitonin). Superiority design | |
| Participants | Diagnostic criteria: Iliac crest bony biopsy, skeletal radiography and scintigraphy were performed to confirm the diagnosis of Paget’s disease. Inclusion criteria: participants with biochemically active Paget’s disease whose bone pain was unresponsive to simple analgesia or nonsteroidal anti‐inflammatory agents. Exclusion criteria: Not stated. Number screened: not stated. Number randomised: 44. Number analysed: 44 (age and sex not stated; percentage monostotic not stated, 100% symptomatic, 10% previously treated for Paget's disease of bone) | |
| Interventions | Two parallel treatment groups: oral etidronate 400 mg and oral etidronate 400 mg in combination with subcutaneous calcitonin 100 MRC units thrice weekly for six months. Co‐interventions: Participants were on a constant calcium low gelatin diet. | |
| Outcomes | Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: Baseline, monthly during treatment, and every 2 to 3 months for an additional 6 months period. How were the outcomes measured: prospectively | |
| Setting and date | One centre in Nottingham, UK. Period when the study was conducted: Not stated | |
| Follow up period | 12 months | |
| Publication details and funding source | Language of publication: English Funding source: Not stated. Declarations of interest among primary researchers: Not stated | |
| Notes | The study randomised a group of participants to be treated with etidronate or etidronate plus calcitonin. The authors also compared these two groups with data accumulated from participants who received calcitonin alone prior to the introduction of etidronate in the metabolic unit (28 participants); this group was excluded because participants were not randomised | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Forty‐four sequential patients were randomised to treatment". Comment: Insufficient information provided to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "patients were randomised to treatment with either EHDP 400 mg daily alone (21 patients) or in combination with SCT 100 MRC units thrice weekly (23 patients)". Comment: Because there was no mention of placebo, it is likely that one group was treated with oral drugs alone and the other with a combination of oral and subcutaneous drugs; hence, blinding was not possible |
| Blinding of outcome assessment (detection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "Two patients treated with EHDP defaulted from follow‐up but the other 70 were reevaluated in the metabolic unit after 6 months". Comment: Attrition rate was low; only two participants were lost to follow‐up. The proportion of missing outcomes compared with observed event risk was insufficient to have a clinically‐relevant impact on the intervention effect estimate |
| Selective reporting (reporting bias) | High risk | We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. Comment: There are some contradictions between methods and results sections.
|
| Other bias | Unclear risk | No other risks of bias found, but reporting insufficient to make judgement |
| Methods | RCT. Randomisation ratio: 1:1:1 (etidronate plus 1α‐hydroxyvitamin D: etidronate plus placebo: placebo plus placebo). Superiority design | |
| Participants | Diagnostic criteria: Radiological and bone scan evidence of Paget's disease of bone. Inclusion criteria: Symptomatic participants. Exclusion criteria: Treatment for Paget's disease of bone in the 12 months before study commencement. Number screened: not stated. Number randomised: 32. Number analysed: 29 (age and sex not reported; 31% monostotic, 100% symptomatic, 37.5% previously treated for Paget's disease of bone) | |
| Interventions | Three parallel treatment groups: oral etidronate 400 mg plus 1α‐hydroxyvitamin D 0.5 μg, oral etidronate 400 mg plus placebo and placebo plus placebo for three months. Co‐interventions: participants received other medications according to routine practice (e.g. analgesics, nonsteroidal anti‐inflammatory drugs) (information provided by authors). | |
| Outcomes | Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity Secondary endpoints: change in bone pain (linear analogue technique with a scale ranging from 0 (no pain) to 30 (very severe pain)). Time points for measurement: Baseline, and after 2, 8 and 12 weeks on drug therapy. How were the outcomes measured: prospectively | |
| Setting and date | One centre in Glasgow, UK. Period when the study was conducted: not stated | |
| Follow up period | 3 months | |
| Publication details and funding source | Language of publication: English Funding source: Partially supported by a grant to BFB from the Scottish Hospital Endowments Research Trust and a grant to ITB from Brocades (UK) Ltd. The 1α‐hydroxyvitamin D and placebo tablets were supplied by Leo Laboratories Ltd. Declarations of interest among primary researchers: not stated | |
| Notes | Data on etidronate plus 1α‐hydroxyvitamin D and etidronate plus placebo groups were pooled for meta‐analysis. Transiliac bone biopsies were obtained before therapy in all cases and after therapy in 28 participants. Bone histomorphometry data were reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "On entry to the trial patients were randomised". Information provided by authors: Allocation by referring to random numbers |
| Allocation concealment (selection bias) | Low risk | Information provided by authors: Investigators were blinded to treatment allocation |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "double blind" Information provided by authors: A double dummy was used with placebo/placebo, etidronate/placebo and etidronate/1α‐hydroxyvitamin D |
| Blinding of outcome assessment (detection bias) | Low risk | Information provided by authors: Investigators were blinded to treatment when assessing outcome |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "There were three withdrawals from the trial after randomisation". Comment: The rate of withdrawals (and reasons) were reported |
| Selective reporting (reporting bias) | High risk | We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. The authors reported symptoms and side effects and described only one adverse event ("one patient who developed increasing pain and radiological deterioration at the site of an asymptomatic pseudofracture of the tibia"). However, at the start of the results section the authors described another adverse event which was not included in the "symptoms and side effects" paragraph ("1 patient in Group A stopped medication because of dyspepsia"). Data on hypercalcaemia are on biochemistry ("One patient... developed mild hypercalcaemia"). There were no other data on adverse events. Information provided by authors: Adverse events were not systematically recorded |
| Other bias | Unclear risk | No other risks of bias found, but reporting insufficient to make judgement |
| Methods | RCT. Randomisation ratio: 1:1:1:1:1 (placebo: tiludronate 100 mg: tiludronate 200 mg: tiludronate 400 mg: tiludronate 800 mg). Superiority design | |
| Participants | Diagnostic criteria: Paget's disease of bone with characteristic radiologic lesions and/or with 99mTc bisphosphonates scintigraphic evidence of local increase in bone turnover. Inclusion criteria: Serum alkaline phosphatase levels were at least twice the ULN. Exclusion criteria:
Number screened: not stated. Number randomised: 149. Number analysed: 149 (69 years, 54 % male, % monostotic not stated, % symptomatic participants not stated, 82% previously treated for Paget's disease of bone) | |
| Interventions | The study was divided into 2 periods. In the first period there were five parallel treatment groups: placebo (4 x placebo capsules), tiludronate 100 mg (2 x placebo and 2 x 50 mg tiludronate capsules), tiludronate 200 mg (4 x capsules 50 mg tiludronate), tiludronate 400 mg (4 x capsules 100 mg), tiludronate 800 mg (4 x capsules 200 mg tiludronate). All participants took 2 capsules at 10.00 am and 2 capsules at 4.00 pm daily for 3 months. In the second period all participants received 4 placebo capsules. The investigators initiated tiludronate therapy during the second period for participants experiencing a lack of efficacy (participants were considered discontinued from the study). Co‐interventions: Not reported | |
| Outcomes | Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: Baseline and monthly intervals. How were the outcomes measured: prospectively | |
| Setting and date | 29 centres in Belgium, France and USA. Period when the study was conducted: Not stated | |
| Follow up period | 6 months | |
| Publication details and funding source | Language of publication: English Funding source: Supported by Sanofi Research, Montpellier, France. Declarations of interest among primary researchers: Not stated | |
| Notes | Data on tiludronate groups were pooled for meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "double‐blind, randomised, placebo‐controlled study". Comment: Insufficient information provided to permit judgement, but probably low risk because the allocation process should have been centralized because the study involved 29 centres in different countries |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "double‐blind, randomised, placebo‐controlled study". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote: "All biochemical determinations were performed at Comment: Insufficient information provided, but likely low risk because it is unlikely that the blinding was broken in a central location |
| Incomplete outcome data (attrition bias) | High risk | Quote: "Follow up assessments were available for all 149 patients." Quote: "Thirty‐five patients withdrew prematurely from the study. The reasons were lack of efficacy in 9, adverse events in 15, at the patient’s request in 4, lost to follow‐up in 4, and miscellaneous reasons unrelated to treatment in 3. There was no relationship noted between the dosage of tiludronate and the number of study withdrawals." Comment: Although the reasons for missing outcome data were explained and withdrawals balanced between groups, the proportion of missing outcomes (25%) compared with observed event risk is enough to induce clinically‐relevant bias in intervention effect estimate |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | There were no data on exposure to co‐interventions other than bisphosphonates. No other risks of bias found, but reporting insufficient to permit judgement |
| Methods | RCT. Randomisation ratio: 1:1 (placebo: alendronate 40 mg). Superiority design | |
| Participants | Diagnostic criteria: Paget’s disease of bone documented by standard clinical, radiological or scintigraphic methods or both. Inclusion criteria:
Exclusion criteria:
Number screened: not stated. Number randomised: 55. Number analysed: 53 (70 years, 56 % male, percentage monostotic or symptomatic not stated, 35% previously treated for Paget's disease of bone) | |
| Interventions | Two parallel treatment groups: placebo and oral alendronate 40 mg daily for 6 months. Co‐interventions: Calcium (450 mg to 500 mg) and vitamin D (400 IU to 600 IU) | |
| Outcomes | Outcomes reported in abstract: Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: baseline, 3 and 6 months. How were the outcomes measured: prospectively | |
| Setting and date | Three sites in Australia, New Zealand, and UK. Period when the study was conducted: Not stated | |
| Follow up period | 6 months | |
| Publication details and funding source | Language of publication: English. Funding source: Supported by Merck Research Laboratories, Rahway, NJ, USA. Declarations of interest among primary researchers: Not stated | |
| Notes | Bone biopsy conducted for 28 participants. Bone histomorphometry data reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned". Comment: Probably done because the study was multicentre, involving 3 countries |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "The study was double‐blinded". Comment: Insufficient information provided to permit judgement, but probably done |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Radiographs were all evaluated by one skeletal radiologist, who was blinded with respect to film sequence and treatment allocation". "Bone biopsy: Each specimen was coded, so that the reader was unaware of the subjects' drug therapy..." Adverse events that were considered by the blinded investigator to be possibly treatment related occurred in..." |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "No subjects withdrew from the study because of drug side effects. Comment: Attrition rate was low because only two participants did not complete the study in the placebo group but none in the alendronate group. The authors did not explain the reasons for not completing the trial for these two participants. However, both participants were from the placebo group and the proportion of missing outcomes compared with observed event risk were insufficient to have a clinically relevant impact on the intervention effect estimate |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes. |
| Other bias | Unclear risk | No other risks of bias found, but reporting was insufficient to permit judgement |
| Methods | RCT: Randomisation ratio: 1:1:1 (placebo: ibandronate 6 mg: ibandronate 12 mg). Superiority design | |
| Participants | Diagnostic criteria: Paget’s disease of bone confirmed radiologically. Inclusion criteria: Serum alkaline phosphatase activities at baseline at least twice the upper limit of the reference range. Exclusion criteria: Other medical conditions, or on medications that affect bone or calcium metabolism. Number screened: not stated. Number randomised: 25. Number analysed: 23 (73 years, 74 % male, percentages monostotic and symptomatic not stated, 64% previously treated for Paget's disease of bone) | |
| Interventions | Three parallel treatment groups: placebo (infusions of normal saline at baseline and at 1 month), ibandronate 6 mg (infusions of 6 mg of ibandronate at baseline and normal saline at 1 month), ibandronate 12 mg (infusions of 6 mg of ibandronate at both baseline and 1 month). Co‐interventions: None (information provided by authors) | |
| Outcomes | Outcomes reported in abstract: Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints: Number who achieved normalised alkaline phosphatase level. Time points for measurement: Not reported. At least at baseline, 3, 6 and 12 months. How were the outcomes measured: prospectively | |
| Setting and date | One centre in Auckland, New Zealand. Period when the study was conducted: Not stated | |
| Follow up period | 6 months (12 months, see notes) | |
| Publication details and funding source | Language of publication: English. Funding source: Supported by the Health Research Council of New Zealand and Roche Products (NZ) Ltd. Declarations of interest among primary researchers: not stated | |
| Notes | Ibandronate group data were pooled for meta‐analysis. Participants were followed for an additional 6 month period after finishing the first 6 months of the study. In this second part of the study, 6 placebo group participants (67%) were given ibandronate in a non‐randomised way. Data from this second part of the study were not included in this review. In 2017 the authors published a manuscript with data from original participants after 10 years follow up | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly allocated to one of three groups using a minimization algorithm to ensure balance between groups for alkaline phosphatase level". Quote: "Six subjects who had placebo at both of these time points subsequently were given ibandronate 6 mg at 6 months, so posttreatment data are shown for 20 subjects" Quote: "Because the focus of this study was on relative response of markers to ibandronate, the pre‐ and posttreatment data in the two ibandronate groups and in those patients from the ‘placebo’ group who went on to have ibandronate have been pooled, so that every subject in the study provides a pre‐ and 6 months post‐ibandronate value". Comment: Although initially participants were randomised, the authors pooled data from participants who were initially randomised to ibandronate with data from placebo participants who received ibandronate in a 6 month study extension. Note: Although the study general assessment of risk of bias for random sequence generation is high, we assessed risk as unclear because on we included data from the first part of the study only in the meta‐analysis |
| Allocation concealment (selection bias) | Unclear risk | Comment: As for assessment of random sequence generation, although for the first 6 months the authors probably concealed allocation, when finishing the first part of the study, they treated two thirds of placebo participants with ibandronate so they knew participant allocation at that time. Note: Although the study general assessment of risk of bias for allocation concealment is high, we assess the risk as unclear because, on meta‐analysis, we introduce only data from the first part of the study. |
| Blinding of participants and personnel (performance bias) | Low risk | Information provided by authors: Infusions were prepared by staff members who had no contact with the participants, so that study staff and participants were blinded to treatment received |
| Blinding of outcome assessment (detection bias) | Low risk | Information provided by authors: Outcome assessment was blinded |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "'Placebo' group (nine patients), received infusions of normal saline at baseline and at 1 month, the ‘6 mg’ group (nine patients, eight with follow‐up data) received ibandronate 6 mg at baseline and placebo at 1 month, and the ‘12 mg’ group (seven patients, six with follow‐up data) received ibandronate 6 mg at both baseline and 1 month". Comment: Follow‐up data were provided and missing outcome data are likely balanced; however, reasons for missing outcomes were not explained. The proportion of missing outcomes compared with observed event risk is not enough to have a clinically‐relevant impact on the intervention effect estimate |
| Selective reporting (reporting bias) | High risk | Information provided by authors: Adverse events were not systematically recorded |
| Other bias | Unclear risk | No other risks of bias found, but reporting was insufficient to permit judgement |
| Methods | RCT. Randomisation ratio: 1:1 (zoledronate acid: risedronate). Non inferiority design | |
| Participants | Diagnostic criteria: Paget’s disease of bone was confirmed by x‐ray, magnetic resonance imaging, computerized tomography, radio‐isotope imaging, etc. Inclusion criteria:
Exclusion criteria:
Number screened: 688 (371 + 317). Number randomised: 357 (185 + 172). Number analysed: 349 (70 years, 67.7% male; percentages monostotic or symptomatic not stated; 54% previously treated for Paget's disease of bone). An extension study of the core trial was published including participants who had therapeutic response defined as normalisation of the alkaline phosphatase level or a reduction of at least 75% in alkaline phosphatase excess (the difference from the midpoint of the reference range) at 6 months of treatment. Number screened for the extension study: 296 (169 responders from 182 participants from the zoledronate group + 127 responders from 175 participants from the risedronate group). Number analysed for the extension study: 267 (152 responders from the zoledronate group + 115 responders from the risedronate group) (70 years; sex, monostotic status and symptomatic participants not stated; 100% previously treated for Paget's disease of bone) | |
| Interventions | Two parallel treatment groups: zoledronate (single 5 mg infusion zoledronate at baseline followed by placebo tablets daily for 60 days), risedronate (saline infusion followed by 30 mg tables of risedronate daily for 60 days). Co‐interventions: All participants received 1 g calcium per day and 400 IU to 1000 IU calciferol per day. No further interventions were added after treatment in the core trial for the extension study | |
| Outcomes | Outcomes reported in abstract: Primary endpoint: Proportion of participants who had therapeutic response. Therapeutic response was defined as normalisation of the alkaline phosphatase level or a reduction of at least 75% in the alkaline phosphatase excess (the difference from the midpoint of the reference range) at 6 months. Secondary endpoints:
Time points for measurement: Not reported. Probably at baseline, 10 days, 1, 2, 3 and 6 months according to x‐axis graph data. Quote: "Physical examinations, hematologic tests, and serum chemical tests were performed regularly throughout the six‐month study. Serum creatinine and urinary protein were measured 9 to 11 days after intravenous dosing". How were the outcomes measured: prospectively. Relapse rate was the primary endpoint for the extension study. Relapse was defined as a return of alkaline phosphatase to within 20% of the pre‐treatment baseline value. Partial relapse was defined as an alkaline phosphatase level that was both 50% above its 6 month value and 1.25 times the ULN range. Outcomes were monitored at 6 monthly visits | |
| Setting and date | 76 centres in 10 countries (Australia, Belgium, Canada, France, Germany, New Zealand, South Africa, Spain, UK, USA). Period when the study was conducted: January 2002 to March 2004. Period when the extension study was conducted: Follow up covers the period from the end of the core trials to 29 January 2009 | |
| Follow up period | 6 months. 6.5 years observation period in the extension study: median follow‐up time from the beginning of the core study was 5.0 years for zoledronate and 3.3 years for risedronate (patient‐years of follow‐up were 574 years and 340 years respectively) | |
| Publication details and funding source | Language of publication: English. Funding source: Supported by a research grant from Novartis Pharma AG, Basel, Switzerland. Declarations of interest among primary researchers:
| |
| Notes | The authors reported pooled data from two identical RCTs. Participants identified as treatment responders were entered to an extended observation period including only those whose alkaline phosphatase was normal at completion of the core study. Bone biopsies were performed for 22 participants (12 zoledronate acid and 10 risedronate). Bone histomorphometry data were reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned in a double‐blind fashion through an interactive voice‐response system". Comment: Probably done; authors describe a telephone system for sequence generation |
| Allocation concealment (selection bias) | Low risk | Study was conducted using a central allocation system (telephone) |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Patients were randomly assigned in a double‐blind fashion". Comment: Probably done; placebo treatment is reported. The risk of bias was judged as high risk for the extension studies because much of the extended follow‐up was conducted with participants and doctors knowing what treatment they received in the core trial (information provided by authors). |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Measurements were made by Covance Central Laboratory Services" Information provided by authors: Outcome assessment was blinded |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "All efficacy variables, except the time to a therapeutic response, were analysed according to the modified intention‐to‐treat principle, which required patients to have a baseline and at least one post‐baseline measurement of alkaline phosphatase." Comment: Participant flow diagram including reasons for losses to follow‐up was included. MIssing outcome data were balanced between interventions and the proportion of missing outcomes compared with observed event risk was insufficient to have a clinically relevant impact on the intervention effect estimate. The risk of bias was judged as high risk for the extension studies because only participants who responded to treatment were included in the follow‐up. Participants who relapsed or whose physicians provided further treatment for Paget's disease were discontinued from the study during the follow‐up. |
| Selective reporting (reporting bias) | High risk | Comment: Brief Pain Inventory‐Short Form (BPI‐SF) was stated as a secondary outcome measure in the study characteristics in the trial registry record, but was not reported in the study publication. Data on fractures were not reported in the study publication but were proposed in the trial registry record |
| Other bias | Unclear risk | No other risks of bias found, but reporting was insufficient to permit judgement |
| Methods | RCT. Randomisation ratio: 1:1:1 (tiludronate 400 mg 3 months, tiludronate 400 mg 6 months; etidronate 400 mg 6 months). Superiority design | |
| Participants | Diagnostic criteria: Paget's disease of bone was confirmed by the presence of radiologically evident lesions characteristic of the disease. Inclusion criteria: Serum alkaline phosphatase concentration at least twice the ULN range. Exclusion criteria:
Number screened: Not stated. Number randomised: 234. Number analysed: 234 (69 years, 59% males, % monostotic not stated, 74% symptomatic participants, 71% previously treated for Paget's disease of bone) | |
| Interventions | Three parallel treatment groups: tiludronate 400 mg 3 months (2 x 200 mg tiludronate tablets and 2 x etidronate placebo capsules during the first 3 months followed by 2 x tiludronate placebo tablets and 2 x etidronate placebo capsules during the second 3 months), tiludronate 400 mg 6 months (2 x 200 mg tiludronate tablets and 2 x etidronate placebo capsules for 6 months) and etidronate 400 mg 6 months (2 x 200 mg etidronate capsules and 2 tiludronate placebo tablets for 6 months). Co‐interventions: Not reported | |
| Outcomes | Outcomes reported in abstract: Primary endpoint: Proportion of participants who had a response to treatment. Response to treatment was defined as a reduction of at least 50% in serum alkaline phosphatase concentration after a 3 month treatment period. Secondary endpoints:
Time points for measurement: Baseline, 3 months, 6 months. How were the outcomes measured: prospectively | |
| Setting and date | 85 centres in 6 countries (Belgium, France, Germany, Italy, Netherlands, Spain). Period when the study was conducted: February 1991 to September 1992 | |
| Follow up period | 6 months | |
| Publication details and funding source | Language of publication: English. Funding source: Supported by Sanofi Recherche, Montpellier, France. Declarations of interest among primary researchers: Not stated | |
| Notes | Data on tiludronate groups were pooled for meta‐analysis | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomly allocated into 1 of 3 treatment groups." Comment: Insufficient information provided. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "The study was a double‐blind, randomized, multicenter, parallel‐group comparison." "According to the randomisation, patients received 4 doses daily: 2 200 mg tiludronate tablets and 2 etidronate placebo capsules during the first 3 months followed by 2 tiludronate placebo tablets and 2 etidronate placebo capsules during the second 3 months, 2 200 mg tiludronate tablets and 2 etidronate placebo capsules for 6 months, or 2 200‐mg etidronate capsules and 2 tiludronate placebo tablets for 6 months." Comment: Probably done |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote: "All samples were analysed at a central laboratory (Cerba Laboratories, Cergy‐Pontoise, France)." Comment: Probably done for laboratory assessment. The information provided was insufficient to judge the blinding of outcome for clinical assessment |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "Of the 234 patients enrolled, 30 (12.8%) discontinued treatment before completing the 6 months of the study: 14 in the tiludronate 3‐month group, 11 in the tiludronate 6‐month group, and 5 in the etidronate group." "Intent‐to‐treat analyses were conducted." "For dichotomous variables, cases with missing data were considered as treatment failures." Comment: Proportion of missing data were reported. Although the number of participants who discontinued treatment from both tiludronate groups was twice the number in the etidronate group, the proportion of missing outcomes compared with observed event risk was insufficient to have a relevant impact of the effect estimate |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | No other risks of bias found, but reporting was insufficient to permit judgement |
| Methods | RCT: Randomisation ratio: 1:1 (alendronate 40 mg: etidronate 400 mg). Superiority design | |
| Participants | Diagnostic criteria: Paget's disease of bone was confirmed by standard clinical, radiological or scintigraphic imaging or both methods Inclusion criteria: Serum alkaline phosphatase at least twice the ULN if the participant had never been treated with bisphosphonates or plicamycin, or at least 4 times the ULN if the participant had received such therapy at any time in the past. Exclusion criteria:
Number screened: Not stated. Number randomised: 89. Number analysed: 88 (69 years, 67% male, percentages of monostotic and symptomatic participants not stated, 25% previously treated for Paget's disease of bone) | |
| Interventions | Two parallel treatment groups: alendronate 40 mg (1 x etidronate placebo tablet and 1 x 40 mg alendronate tablet for 6 months) and etidronate 400 mg (1 x 400 mg etidronate tablet and 1 x alendronate placebo tablet for 6 months). Co‐interventions: All participants received daily vitamin supplements containing 450 mg calcium carbonate and 400 IU vitamin D. Analgesics were available on demand and use was not balanced ("the regression analysis with adjustment for analgesics use at month 6 between the two treatment groups approached significance") | |
| Outcomes | Outcomes reported in abstract Primary endpoint: Mean percentage change from baseline in serum total alkaline phosphatase activity. Secondary endpoints:
Time points for measurement: Baseline, 1 month, 3 months, 6 months. How were the outcomes measured: prospectively | |
| Setting and date | 11 centres in USA. Period when the study was conducted: Not reported | |
| Follow up period | 6 months | |
| Publication details and funding source | Language of publication: English. Funding source: Not reported. Declarations of interest among primary researchers: Not reported | |
| Notes | Transiliac bone biopsy was conducted for 43 participants at month 6. Bone biopsies were obtained from 25 healthy volunteers as control | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "the study was randomised". Comment: insufficient information provided to permit judgement. Allocation was probably centralized because this was a multicentre study |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information provided to permit judgement |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "The study was randomised and double blind", "The etidronate tablets were purchased as Didronel, crushed, and recompressed into tablets identical to placebo for etidronate". Comment: Probably done. |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Baseline and 6 months radiographs of one or more sites were read by one radiologist, who remained blind with respect to both treatment allocation and sequence of films". "Each specimen (bone biopsy) was blinded, so that the reader was unaware of the subjects' drug therapy." |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Only one participant was excluded from the analysis (in the alendronate group). Reason for exclusion was reported. The proportion of missing outcomes compared with observed event risk was insufficient to have a relevant impact of the effect estimate |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | There were differences between groups in co‐interventions because analgesics were taken on demand and use was not balanced. However this was thought unlikely to have resulted in bias. No other risks of bias found, but reporting was insufficient to permit judgement |
| Methods | Interventional extension study including participants who complete a previous RCT (Langston 2010). The bisphosphonate of first choice in the intensive treatment arm was different from the original trial. Randomisation ratio: 1:1 (symptomatic treatment: intensive treatment). Superiority design | |
| Participants | Diagnostic criteria: participants with Paget's disease of bone confirmed by plain radiology of at least one skeletal site according standard criteria from UK guidelines (Selby 2002) Inclusion criteria: Participants who completed the PRISM trial (Langston 2010). Exclusion criteria: No specific exclusion criteria were applied on the basis of treatment history, baseline alkaline phosphatase or co‐existing diseases. Number invited to participate: 2110 Number enrolled: 502 Number analysed: 404 (76 years, 54% male, 62% symptomatic, 91% previously treated with bisphosphonates for Paget's disease, 70% had normal alkaline phosphatase at baseline) | |
| Interventions | Two parallel treatment groups; symptomatic vs. intensive treatment. Symptomatic treatment: Philosophy; treat bone pain, not alkaline phosphatase.
Intensive treatment: Philosophy; maintain normal alkaline phosphatase.
Co‐interventions: Analgesics and nonsteroidal anti‐inflammatory drugs | |
| Outcomes | Primary outcome: radiologically‐confirmed clinical fracture. Secondary outcomes:
Time points for measurement: Data on fractures, orthopaedic procedures and serious adverse events were collected on a continuous basis. Laboratory data, quality of life, bone pain and adverse events (based on participant diaries) data were measured annually. How were the outcomes measured: prospectively | |
| Setting and date | 30 secondary referral centres in the UK. Period the study was conducted: January 2007 to January 2012 | |
| Follow up period | 3 years | |
| Publication details and funding source | Language of publication: English. Funding source: The study was supported by grants from the Arthritis Research Campaign UK (Ref. 13627) and the National Association for Relief of Paget’s Disease. Declarations of interest among primary researchers:
| |
| Notes | The study was described by the authors as a "pragmatic randomised controlled trial designed to compare the effects of two management strategies". The study was not blinded | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "At the end of PRISM, patients were asked if they wanted to continue in the study for a further three years". Comment: The participants included in this trial had participated in a previous trial (Langston 2010). The risk of bias assessment should be the same as for the previous trial. However, as only participants who voluntarily agreed to continue in the study were included, it is difficult to verify if the balance among the trial groups created in the original trial by randomisation was kept in this extension study. At baseline serum alkaline phosphatase levels were lower in the intensive versus symptomatic group reflecting the fact that these participants already had been subjected to intensive treatment |
| Allocation concealment (selection bias) | High risk | Comment: The participants included in this trial had participated in a previous trial (Langston 2010). The risk of bias assessment is the same as for the previous trial |
| Blinding of participants and personnel (performance bias) | Low risk | Comment: The study was not blinded. The main outcome (fractures) was unlikely to be influenced by lack of blinding. The risk of bias was high for quality of life, adverse events or bone pain, but low for other secondary endpoints as orthopaedic procedures or alkaline phosphatase concentrations |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Patient‐reported fractures and orthopaedic procedures were validated against medical records and x‐ray reports at participating centres by assessors blinded to treatment allocation" |
| Incomplete outcome data (attrition bias) | Low risk | Comment: Although a similar proportion of participants were deceased, withdrew from the study, declined to participate or were lost to follow up in each group (41/232 (18%) in the symptomatic versus 57/270 (21%) in the intensive group) at 3 years time, the proportion of missing outcomes compared with observed event risk is enough to induce clinically relevant bias in intervention effect estimate (fracture). However, the assessment of attrition bias is not assessed as high but low as, according to authors, the study was an event driven trial, with fracture as the primary endpoint. Data on all fracture events were available even in subjects who had withdrawn from the study |
| Selective reporting (reporting bias) | Low risk | Study protocol available. There was a prespecified record of the studies outcomes and all of them were included in published manuscript |
| Other bias | Unclear risk | The study design permitted attending clinicians to choose between different drugs within a specified treatment strategy. Evidence that the attending clinicians adhered to this strategy was confirmed by the observed difference in alkaline phosphatase levels between groups |
| Methods | RCT. Randomisation ratio: 1:1 (alendronate 40 mg: pamidronate 60 mg). Superiority design | |
| Participants | Diagnostic criteria: Paget's disease of bone was confirmed by the presence of typical lesions of Paget’s disease on isotope bone scanning and radiographs. Inclusion criteria: Serum alkaline phosphatase above the upper limit of laboratory reference range. Exclusion criteria:
Number screened: 139. Number randomised: 72. Number analysed: 72 (70 years, 58% male, % monostotic not stated, 94% symptomatic participants, 39% participants previously treated for Paget's disease of bone) | |
| Interventions | Two parallel treatment groups: alendronate 40 mg daily in 3 months blocks and pamidronate four x 60 mg IV infusions a year (once every 3 months). Treatment was continued until biochemical remission was achieved or there were a no significant reduction on two consecutives measurements. Biochemical remission was defined as both, serum total alkaline phosphatase activity and urine deoxypyridinoline/creatinine ratio within the reference range. Co‐interventions: All participants with plasma baseline total alkaline phosphatase > 675 U/L were prescribed ergocalciferol 30,000 U weekly for 3 months and calcium carbonate 600 mg daily for 8 months to minimize bisphosphonate‐induced secondary hyperparathyroidism. Other participants were treated with calcium and vitamin D supplements at the treating clinician’s discretion | |
| Outcomes | Outcomes reported in abstract: Primary endpoint: Proportion of participants achieving biochemical remission (see above). Secondary endpoints: Numbers of participants; with change in bone pain (measured on VAS); with change on quality of life from baseline (assessed using the SF‐36 Australian version); who experienced severe side effects related to use of bisphosphonates; who withdrew due to adverse events, mean percentage change from baseline in serum total alkaline phosphatase activity; who normalised alkaline phosphatase level; and who relapsed due to recurrence of increased serum alkaline phosphatase level. Time points for measurement: Baseline, 3, 6, 9, 12, 18 and 24 months. How were the outcome measured: Prospectively | |
| Setting and date | Three centres in Western Australia. Period when the study was conducted: From May 1997 to October 2001 | |
| Follow up period | 1 year without protocol amendment (2 year with protocol amendment) | |
| Publication details and funding source | Language of publication: English. Funding source: Although the authors described the trial as an "investigator‐initiated study", the study was supported by research grants from Merck, Sharp & Dohme (Australia), Novartis Pharmaceuticals Australia and the Arthritis Foundation of Western Australia. Declarations of interest among primary researchers: Not stated | |
| Notes | The initial protocol was amended to cross participants over from pamidronate to alendronate treatment at 12 months. According to the author, it was apparent that some participants randomised to pamidronate were showing little or no biochemical response to treatment and for "ethical" reasons, they amended the protocol so that participants randomised to pamidronate who did not achieve biochemical remission at 12 months were crossed over to alendronate treatment for the second year of the study. We included data from the first year only in the meta‐analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "To ensure that the two treatment groups were well matched, randomisation was stratified (using an in‐house computer program) by two variables: baseline plasma alkaline phosphatase (in three strata: 136–270, 271–675, > 675 U/L) and previous bisphosphonate treatment (as a binary variable: yes or no)." "In the course of the study, it was apparent that some patients randomised to pamidronate (predominantly in the previously treated subgroup) were showing little or no biochemical response to treatment. For ethical reasons, the protocol was amended so that patients randomised to pamidronate who did not achieve biochemical remission at 12 months were crossed over to alendronate treatment for the second year of the study". Comment: The risk assessment was judged as low risk for first year data. For the second year, allocation was broken, and the risk of bias was assessed as high |
| Allocation concealment (selection bias) | Low risk | Authors describe using a computer program to generate the allocation |
| Blinding of participants and personnel (performance bias) | High risk | Open label trial |
| Blinding of outcome assessment (detection bias) | High risk | Open label trial |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "Proportions of patients achieving remission for each treatment (on an intention to treat basis) were compared by Fisher’s exact test". Comment: Data on screened participants, randomised participants and withdrawals (with reasons) are reported in a flow chart (Figure 1) |
| Selective reporting (reporting bias) | Unclear risk | Methods and results sections were consistent. Insufficient information to permit judgement. We did not have access to a trial register record or study protocol to know if there was a prespecified record of the studies outcomes |
| Other bias | Unclear risk | Likely high at 12 months due to amendment of the initial protocol. No other risks of bias found, but reporting was insufficient to permit judgement |
Abbreviations: IV ‐ intravenous; RCT ‐ randomised controlled trial; ULN ‐ upper limit of normal
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| Not a randomised clinical study. Study without a comparison of interest. The study compared alendronate (20 mg or 40 mg daily) for 3 to 6 months | |
| Randomised clinical study. Study without a comparison of interest. The study compared three doses of neridronate (12.5 mg, 25 mg, 50 mg or 100 mg daily for two consecutive days) | |
| Not a randomised clinical study. Study without a comparison of interest. The study describes the outcomes in a series of participants treated with different regimes of oral Etidronate (5 mg/kg daily for 6 months, 10 mg/kg daily for 3 months, or 20 mg/kg daily for 3 moths). | |
| Not a randomised clinical study. Study without an outcome of interest. The study was designed to analyse the effect of different doses of Etidronate (5 mg/kg or 20 mg/kg for 6 months) or clodronate (400 mg/day or 1600 mg/day for 6 months) over the serum acid level. | |
| Not a randomised clinical study. Study without a comparison of interest. The study compares three doses of the Neridronate (400 mg oral for one month, 25 mg daily infusion for 5 days and 50 mg daily infusion for 5 days). | |
| Randomised clinical study. Study without a comparison of interest. The study compared two regimes of administration of pamidronate (initial 30 mg infusion followed by three infusion of 60 mg and a final placebo infusion at fortnightly intervals or initial placebo infusion followed by three infusion of 60 mg and a final 30 mg infusion at fortnightly intervals), with the same total dose (210 mg). | |
| Not a randomised clinical study. Study without a comparison of interest. The study shows data from a cohort of 107 elderly participants (mean age 76 years) treated with intravenous zoledronate followed‐up for 10 years. | |
| Randomised clinical study. Study without a comparison of interest. The study compared four doses of clodronate (400, 800, 1600 or 2400 mg daily), with (the four doses) or without (only 400 and 1600 mg doses) vitamin D and calcium supplementation (elemental calcium 1g/day and vitamin D2 8000 IU/day). | |
| Data on a sample of participants diagnosed with Paget's disease of bone from a randomised clinical trial (Roux 1995). The study was designed to analyse radiological changes during treatment with bisphosphonates. | |
| Not a randomised clinical study. Study without a comparison of interest. Data from the Belgian Paget's Disease Registry of patients diagnosed as Paget's disease of bone and treated with a 5 mg intravenous infusion of zoledronate. | |
| Not a randomised clinical study. Study with a comparison of interest. "Open trial" (9 participants were randomised and 8 participants were put directly into one of the two treatment groups) comparing the effectiveness of two bisphosphonates (etidronate 20 mg/kg once daily for 3 months vs. pamidronate 4 to 5 mg/kg twice daily for 3 months). | |
| Case‐control study. Study without a comparison of interest. Cases were patients diagnosed as Paget's disease of bone with temporal bone involvement. Control were healthy individuals matched for age and sex. The study was designed to analyse the effectiveness of two bisphosphonates (pamidronate 30 mg daily infusion for 6 days or oral tiludronate 400 mg daily for 3 months). The study included audiometric assessment and hearing threshold examination. | |
| Not a randomised clinical study. Study with a comparison of interest. The study compares the effectiveness of two bisphosphonates (Clodronate 300 mg/daily intravenous infusion for 5 consecutive days vs. Alendronate 5 mg/ daily intravenous infusion for 5 consecutive days). | |
| Not a randomised clinical study. Study without a comparison of interest. The study compares three regimes of administration of pamidronate, two with a total dose of 180 mg (30 mg weekly infusions for 6 weeks or 45 mg infusions every 3 months for one year) and one with a total dose of 360 mg (30 mg weekly infusions for 6 weeks and 60 mg weekly infusions for three additional weeks). A random subgroup of 6 participants were given placebo infusions of 0.9% saline weekly for three weeks at the start of treatment in a single‐blind study in the group of 30 mg weekly doses for 6 weeks (data are not shown on manuscript). | |
| Data on a sample of participants diagnosed as Paget's disease of bone from a randomised clinical trial, and a case‐control study. For the case‐control study, cases were selected from three of five groups of a clinical trial on Paget's disease of bone (Schaffer 1996). Controls were healthy individuals matched for age and sex. The clinical trial included the cases and was designed to analyse the effect of zoledronate on bone turnover markers. | |
| Data on a sample of participants diagnosed with Paget's disease of bone from a randomised clinical trial and a case‐control study (selected from a clinical trial on Paget's disease of bone, Schaffer 1996). Controls were healthy age‐matched individuals. The clinical trial included the cases and was designed to analyse the effect of zoledronate on bone turnover markers | |
| Data from a sample of participants diagnosed with Paget's disease of bone from a randomised clinical trial (Altman 1973). The study was designed to assess the changes in radionuclide uptake after bisphosphonate treatment | |
| Not a randomised clinical study. Study without a comparison of interest. The study compared three different doses of ibandronate (2 mg, 4 mg or 6 mg intravenous infusions) for re‐treatment of participants previously treated with ibandronate after biochemical relapse (increase of alkaline phosphatase). | |
| Not a randomised clinical study. Study without a comparison of interest. The study compared three different doses of pamidronate. Participation allocation had two steps: three groups according to index of bone resorption (urinary hydroxyproline x plasma creatinine/urinary creatinine). The higher the index (< 5, 5 to 9.99, ≥ 10), the higher the pamidronate dose (120 mg, 180 mg, 240 mg). The three groups were then randomised to two subgroups; total dose in 30 mg infusions or total dose in 60 mg infusions | |
| Randomised clinical study. Study without a comparison of interest. The study compared administration of alendronate 280 mg weekly vs. 40 mg daily for 6 months | |
| Not a randomised clinical study. Study without a comparison of interest. The study compared administration of etidronate (7.5 mg/kg or 15 mg/kg daily and etidronate plus calcitonin simultaneously (etidronate 7.5 mg/kg daily together with calcitonin 0.5 mg once daily subcutaneous for 6 months) or sequentially (etidronate 15 mg/kg daily for 6 months followed by calcitonin 0.5 mg twice daily subcutaneous for 6 months) | |
| Extension study (Altman 1973) with a non‐randomised crossover design. The groups from the original study (placebo and etidronate doses of 1 mg/kg, 2.5 mg/kg, 5 mg/kg, 10 mg/kg, 20 mg/kg) were reassigned to different doses of etidronate for six additional months. Participants from placebo and 1 mg/kg and 2.5 mg/kg groups were reassigned to etidronate on doses of 5 mg/kg, 10 mg/kg or 20 mg/kg. Participants from the 5 mg/kg group were kept on the same dose. Participants from the 10 mg/kg or 20 mg/kg groups were reassigned to placebo or to remain on the same etidronate dose | |
| Not a randomised clinical study. Study without a comparison of interest. Data from a cohort of participants diagnosed with Paget's disease of bone who were treated with etidronate; 60/116 participants took part in a previous study (Khairi 1974) | |
| Randomised clinical study. Study without a comparison of interest. The study compared different doses (40 or 80 mg/daily) and regimens (3 or 6 months) of oral alendronate | |
| Review. Compilation of the results of two RCTs on alendronate for Paget's disease of bone (Reid 1996 and Siris 1996). | |
| Not a randomised clinical study. Study without a comparison of interest. The study compared two regimens of pamidronate infusion (1 infusion of 60 mg vs. 2 infusions of 60 mg over 24 hours) | |
| Randomised clinical study. Study without a comparison of interest. The study compared two regimens of neridronate (100 mg intravenous infusion for 2 consecutive days vs. 25 mg intramuscular infusion once a week for 2 months) | |
| Not a randomised clinical study. Study without a comparison of interest. The study compared three doses of intravenous alendronate (2.5 mg, 5 mg and 10 mg daily) for 5 consecutive days | |
| Not a randomised clinical study. Study without a comparison of interest. The study compared one oral and four intravenous regimens of pamidronate (600 mg daily for 6 months, 40 mg daily for five days, 20 mg daily for 10 days, 10 mg daily for 4 days and a single dose of 10 mg) | |
| Randomised clinical study. Study without a comparison of interest. The study compared three regimens of tiludronate (200 mg daily for 6 months, 400 mg daily for 6 months, 200 mg daily for 3 months and 400 mg daily for 3 additional months) | |
| Randomised clinical study. Study without a comparison of interest. The study compared three regimens of tiludronate (600 mg, 800 mg and 1200 mg daily) for five days | |
| Not a randomised clinical study. Study without a comparison of interest. The study compared four doses of etidronate (1 mg, 5 mg, 10 mg and 20 mg) for 6 months and placebo | |
| Extension study (of Canfield 1977). Only participants from the Columbia Presbyterian Medical Center (1 of the 4 hospitals in the original trial) were included in the study. All participants were treated with etidronate | |
| Not a randomised clinical study. Study without a comparison of interest. The study compared three regimens of pamidronate (15 mg, 30 mg, or 45 mg infusions) at 6 week intervals | |
| Randomised clinical study. Study without a comparison of interest. The study compared seven regimens of pamidronate in two formulations (3 dose levels of oral capsules of pamidronate; 300 mg, 600 mg and 900 mg and four dose levels of oral tables of dimethyl pamidronate; 50 mg, 100 mg, 200 mg and 400 mg). Each dose was administered for 15 days |
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Zoledronate in the Prevention of Paget's: the ZiPP study |
| Methods | Multisite double blind placebo controlled randomised trial. Two sub‐studies:
Follow up: 5 years. |
| Participants | Participant inclusion criteria Interventional study: Observational study: Relatives of patients with SQSTM1 mutations, aged between 30 years old or greater who on screening are found NOT to have SQSTM1 mutations Countries of recruitment: Australia, Belgium, Ireland, Italy, Spain, United Kingdom. |
| Interventions | Interventional study: Participants will be randomised to either infusions of zoledronate (Aclasta®) 5 mg by intravenous infusion over 15 minutes or placebo (0.9% saline) at baseline. |
| Outcomes | Interventional study: Primary outcome: Total number of subjects who develop new bone lesions between the baseline visit and the final follow up visit. Observational study: Primary outcome: Anxiety/depression, measured using the HADS scale. Both studies: Secondary outcomes: 1. Development of elevated bone turnover, as measured by alkaline phosphatase and other biochemical markers of bone turnover. 2. Quality of life, and anxiety and depression assessed by the SF‐36, BPI and HADS questionnaires. |
| Starting date | 12/01/2009 |
| Contact information | Mr Adam Wilson Clinical Trials Manager e‐mail: [email protected] |
| Ending date | 31/01/2020 |
| Target number of participants | Intervention study: 188 participants Observational study: 125 participants |
| Identifier | ISRCTN11616770 DOI 10.1186/ISRCTN11616770 |
| Notes | Sponsor information: University of Edinburgh (UK) Funder name: Medical Research Council (MRC) (UK) (ref: G0701625; 85281) |
| Trial name or title | Takeda Study Registration Call Center, post marketing Group Manager |
| Methods | Prospective cohort |
| Participants | Inclusion criteria: participants diagnosed with Paget's disease of bone treated with sodium risedronate tablets Exclusion criteria: No exclusions by age or gender Countries of recruitment: Japan |
| Interventions | Sodium risedronate tablets (Benet 17.5 mg) administered orally with a sufficient volume (approximately 180 mL) of water once daily after waking for 8 consecutive weeks. |
| Outcomes | Primary outcome:
Secondary outcomes:
|
| Starting date | September 2008 |
| Contact information | Telephone: 1‐800‐778‐2860 (USA & EU), email: [email protected] |
| Ending date | Estimated study completion date: July 2017. |
| Target number of participants | 2500 |
| Identifier | ClinicalTrials.gov Identifier: NCT02106455 |
| Notes | Sponsor information: Takeda |
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Number of participants whose bone pain disappeared completely Show forest plot | 2 | 205 | Risk Ratio (M‐H, Random, 95% CI) | 3.42 [1.31, 8.90] |
| Analysis 1.1  Comparison 1 Bisphosphonates versus placebo, Outcome 1 Number of participants whose bone pain disappeared completely. | ||||
| 2 Number of participants with change in bone pain Show forest plot | 7 | 481 | Risk Ratio (M‐H, Random, 95% CI) | 1.97 [1.29, 3.01] |
| Analysis 1.2  Comparison 1 Bisphosphonates versus placebo, Outcome 2 Number of participants with change in bone pain. | ||||
| 2.1 Etidronate vs. placebo | 3 | 124 | Risk Ratio (M‐H, Random, 95% CI) | 1.75 [0.79, 3.87] |
| 2.2 Tiludronate vs. placebo | 3 | 344 | Risk Ratio (M‐H, Random, 95% CI) | 1.98 [1.27, 3.08] |
| 2.3 Alendronate vs. placebo | 1 | 13 | Risk Ratio (M‐H, Random, 95% CI) | 10.00 [0.69, 144.38] |
| 3 Number of participants experiencing radiologically‐confirmed clinical fractures Show forest plot | 4 | 356 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.18, 4.51] |
| Analysis 1.3  Comparison 1 Bisphosphonates versus placebo, Outcome 3 Number of participants experiencing radiologically‐confirmed clinical fractures. | ||||
| 3.1 Etidronate vs. placebo | 2 | 95 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.09, 9.06] |
| 3.2 Tiludronate vs. placebo | 2 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 0.88 [0.09, 8.64] |
| 4 Number of participants who experienced adverse events related to use of bisphosphonates Show forest plot | 6 | 678 | Risk Difference (M‐H, Random, 95% CI) | 0.11 [‐0.00, 0.22] |
| Analysis 1.4  Comparison 1 Bisphosphonates versus placebo, Outcome 4 Number of participants who experienced adverse events related to use of bisphosphonates. | ||||
| 4.1 Etidronate vs. placebo | 1 | 47 | Risk Difference (M‐H, Random, 95% CI) | 0.02 [‐0.21, 0.25] |
| 4.2 Zoledronate vs. placebo | 1 | 176 | Risk Difference (M‐H, Random, 95% CI) | 0.27 [0.12, 0.42] |
| 4.3 Tiludronate vs. placebo | 3 | 400 | Risk Difference (M‐H, Random, 95% CI) | 0.07 [‐0.08, 0.22] |
| 4.4 Alendronate vs. placebo | 1 | 55 | Risk Difference (M‐H, Random, 95% CI) | 0.12 [‐0.11, 0.34] |
| 5 Number of participants who withdrew due to adverse events Show forest plot | 6 | 517 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.38, 2.69] |
| Analysis 1.5  Comparison 1 Bisphosphonates versus placebo, Outcome 5 Number of participants who withdrew due to adverse events. | ||||
| 5.1 Etidronate vs. placebo | 1 | 47 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 Tiludronate vs. placebo | 3 | 400 | Odds Ratio (M‐H, Random, 95% CI) | 0.96 [0.34, 2.67] |
| 5.3 Alendronate vs. placebo | 2 | 70 | Odds Ratio (M‐H, Random, 95% CI) | 1.74 [0.06, 50.43] |
| 6 Mean percentage change from baseline in serum total alkaline phosphatase level Show forest plot | 8 | 592 | Mean Difference (IV, Random, 95% CI) | ‐50.09 [‐67.72, ‐32.46] |
| Analysis 1.6  Comparison 1 Bisphosphonates versus placebo, Outcome 6 Mean percentage change from baseline in serum total alkaline phosphatase level. | ||||
| 6.1 Etidronate vs. placebo | 3 | 122 | Mean Difference (IV, Random, 95% CI) | ‐55.85 [‐66.50, ‐45.20] |
| 6.2 Zoledronate vs. placebo | 1 | 176 | Mean Difference (IV, Random, 95% CI) | ‐22.26 [‐27.99, ‐16.53] |
| 6.3 Tiludronate vs. placebo | 2 | 256 | Mean Difference (IV, Random, 95% CI) | ‐58.0 [‐64.25, ‐51.75] |
| 6.4 Alendronate vs. placebo | 1 | 15 | Mean Difference (IV, Random, 95% CI) | ‐39.9 [‐51.28, ‐28.52] |
| 6.5 Ibandronate vs. placebo | 1 | 23 | Mean Difference (IV, Random, 95% CI) | ‐96.1 [‐147.01, ‐45.19] |
| 7 Number of participants who achieved normalised alkaline phosphatase level Show forest plot | 8 | 580 | Risk Ratio (M‐H, Random, 95% CI) | 9.96 [3.74, 26.58] |
| Analysis 1.7  Comparison 1 Bisphosphonates versus placebo, Outcome 7 Number of participants who achieved normalised alkaline phosphatase level. | ||||
| 7.1 Etidronate vs. placebo | 3 | 121 | Risk Ratio (M‐H, Random, 95% CI) | 4.51 [0.90, 22.55] |
| 7.2 Tiludronate vs. placebo | 3 | 381 | Risk Ratio (M‐H, Random, 95% CI) | 13.79 [2.77, 68.61] |
| 7.3 Alendronate vs. placebo | 1 | 55 | Risk Ratio (M‐H, Random, 95% CI) | 27.96 [1.74, 448.28] |
| 7.4 Ibandronate vs. placebo | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 14.00 [0.92, 212.92] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean change from baseline in pain Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 2.1  Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 1 Mean change from baseline in pain. | ||||
| 1.1 Risedronate vs. etidronate | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Number of participants who experienced adverse events related to use of bisphosphonates Show forest plot | 2 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.72, 1.35] |
| Analysis 2.2  Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 2 Number of participants who experienced adverse events related to use of bisphosphonates. | ||||
| 2.1 Risedronate vs. etidronate | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.68, 1.43] |
| 2.2 Alendronate vs. etidronate | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.55, 1.76] |
| 3 Number of participants who withdrew due to adverse events Show forest plot | 2 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.25, 1.89] |
| Analysis 2.3  Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 3 Number of participants who withdrew due to adverse events. | ||||
| 3.1 Risedronate vs. etidronate | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.22, 2.79] |
| 3.2 Alendronate vs. etidronate | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.11, 2.90] |
| 4 Mean percentage change from baseline in serum total alkaline phosphatase level Show forest plot | 2 | 212 | Mean Difference (IV, Random, 95% CI) | ‐40.95 [‐49.09, ‐32.81] |
| Analysis 2.4  Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 4 Mean percentage change from baseline in serum total alkaline phosphatase level. | ||||
| 4.1 Risedronate vs. etidronate | 1 | 123 | Mean Difference (IV, Random, 95% CI) | ‐43.9 [‐48.06, ‐39.74] |
| 4.2 Alendronate vs. etidronate | 1 | 89 | Mean Difference (IV, Random, 95% CI) | ‐35.1 [‐45.85, ‐24.35] |
| 5 Number of participants who achieved normalised alkaline phosphatase level Show forest plot | 2 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 4.30 [2.72, 6.79] |
| Analysis 2.5  Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 5 Number of participants who achieved normalised alkaline phosphatase level. | ||||
| 5.1 Risedronate vs. etidronate | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 4.81 [2.58, 8.98] |
| 5.2 Alendronate vs. etidronate | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 3.78 [1.93, 7.38] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Number of participants with bone pain change Show forest plot | 2 | 436 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [1.15, 1.51] |
| Analysis 3.1  Comparison 3 Comparison of two aminobisphosphonates, Outcome 1 Number of participants with bone pain change. | ||||
| 1.1 Zoledronate (1) vs. pamidronate (2) | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [1.10, 1.53] |
| 1.2 Zolendronate (1) vs. risedronate (2) | 1 | 347 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.06, 1.74] |
| 2 Number of participants who experienced adverse events related to use of bisphosphonates Show forest plot | 3 | Risk Difference (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 3.2  Comparison 3 Comparison of two aminobisphosphonates, Outcome 2 Number of participants who experienced adverse events related to use of bisphosphonates. | ||||
| 2.1 Olpadronate (1) vs. pamidronate (2) | 1 | Risk Difference (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Zoledronate (1) vs. pamidronate (2) | 1 | Risk Difference (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Zolendronate (1) vs. risedronate (2) | 1 | Risk Difference (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Number of participants who withdrew due to adverse events Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 3.3  Comparison 3 Comparison of two aminobisphosphonates, Outcome 3 Number of participants who withdrew due to adverse events. | ||||
| 3.1 Zoledronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Zolendronate (1) vs. risedronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Alendronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Mean percentage change from baseline in serum total alkaline phosphatase level Show forest plot | 3 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| Analysis 3.4  Comparison 3 Comparison of two aminobisphosphonates, Outcome 4 Mean percentage change from baseline in serum total alkaline phosphatase level. | ||||
| 4.1 Olpadronate (1) vs. pamidronate (2) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Zolendronate (1) vs. risedronate (2) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Alendronate (1) vs. pamidronate (2) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Number of participants who achieved normalised alkaline phosphatase level Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 3.5  Comparison 3 Comparison of two aminobisphosphonates, Outcome 5 Number of participants who achieved normalised alkaline phosphatase level. | ||||
| 5.1 Olpadronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Zoledronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Zolendronate (1) vs. risedronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 Alendronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Number of participants who experienced biochemical relapse with increased alkaline phosphatase level Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 3.6  Comparison 3 Comparison of two aminobisphosphonates, Outcome 6 Number of participants who experienced biochemical relapse with increased alkaline phosphatase level. | ||||
| 6.1 Zoledronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Zoledronate (1) vs. risedronate (2) | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Alendronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |

Geometry of the network of randomised trials of bisphosphonates for Paget's disease of bone. The nodes of the network represent the treatments compared. The links reflect comparisons and the number of links is proportional to the number of comparisons

Study flow diagram

Risk of bias summary: review authors' judgements about each risk of bias item for each included study

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies

Forest plot of comparison: 1 Bisphosphonates versus placebo, outcome: 1.2 Number of participants with change in bone pain

Forest plot of comparison: 1 Bisphosphonates versus placebo, outcome: 1.4 Number of participants who experienced adverse events related to use of bisphosphonates

Comparison 1 Bisphosphonates versus placebo, Outcome 1 Number of participants whose bone pain disappeared completely.
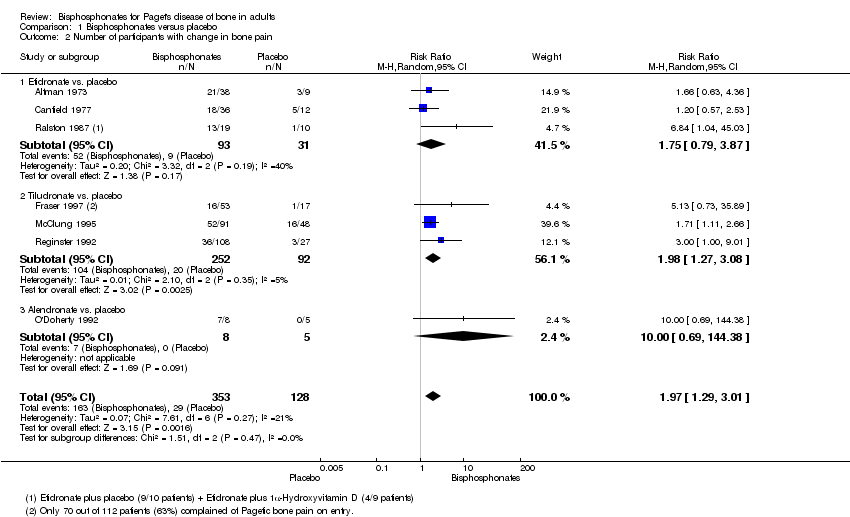
Comparison 1 Bisphosphonates versus placebo, Outcome 2 Number of participants with change in bone pain.

Comparison 1 Bisphosphonates versus placebo, Outcome 3 Number of participants experiencing radiologically‐confirmed clinical fractures.

Comparison 1 Bisphosphonates versus placebo, Outcome 4 Number of participants who experienced adverse events related to use of bisphosphonates.

Comparison 1 Bisphosphonates versus placebo, Outcome 5 Number of participants who withdrew due to adverse events.

Comparison 1 Bisphosphonates versus placebo, Outcome 6 Mean percentage change from baseline in serum total alkaline phosphatase level.

Comparison 1 Bisphosphonates versus placebo, Outcome 7 Number of participants who achieved normalised alkaline phosphatase level.

Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 1 Mean change from baseline in pain.

Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 2 Number of participants who experienced adverse events related to use of bisphosphonates.

Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 3 Number of participants who withdrew due to adverse events.

Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 4 Mean percentage change from baseline in serum total alkaline phosphatase level.

Comparison 2 Aminobisphosphonates versus non‐aminobisphosphonates, Outcome 5 Number of participants who achieved normalised alkaline phosphatase level.
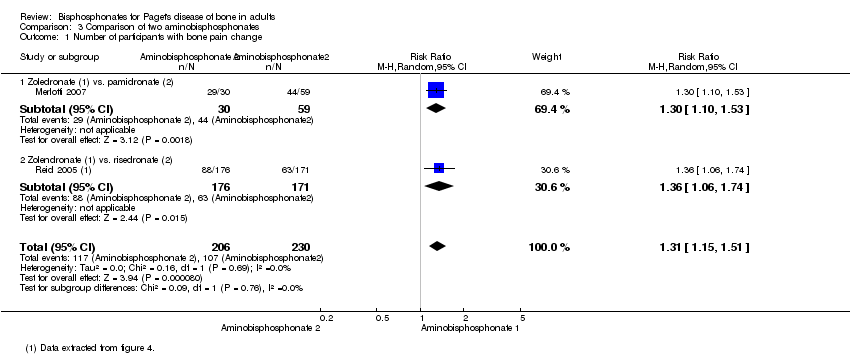
Comparison 3 Comparison of two aminobisphosphonates, Outcome 1 Number of participants with bone pain change.

Comparison 3 Comparison of two aminobisphosphonates, Outcome 2 Number of participants who experienced adverse events related to use of bisphosphonates.
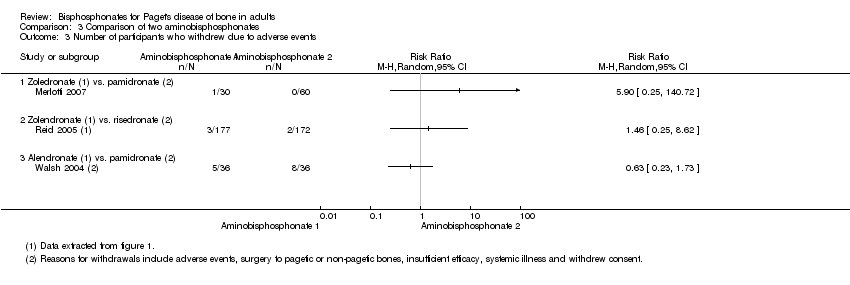
Comparison 3 Comparison of two aminobisphosphonates, Outcome 3 Number of participants who withdrew due to adverse events.

Comparison 3 Comparison of two aminobisphosphonates, Outcome 4 Mean percentage change from baseline in serum total alkaline phosphatase level.

Comparison 3 Comparison of two aminobisphosphonates, Outcome 5 Number of participants who achieved normalised alkaline phosphatase level.

Comparison 3 Comparison of two aminobisphosphonates, Outcome 6 Number of participants who experienced biochemical relapse with increased alkaline phosphatase level.
| Bisphosphonates versus placebo for Paget's disease of bone | ||||||
| Patient or population: Paget's disease of bone Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with placebo | Risk with bisphosphonates | |||||
| Number of participants with change in bone pain (disappearance of pain)¹ | Study population | RR 3.42 | 205 | ⊕⊕⊕⊝ | NNTB 5 (1 to 35) Absolute risk difference: 23% more (12% to 34%) Relative percent change: 242 % (31% to 790%) (Improvement) | |
| 91 per 1000 | 311 per 1000 | |||||
| Number of participants who experienced radiologically‐confirmed fractures | Low (study population)³ | RR 0.89 | 356 | ⊕⊝⊝⊝ | Absolute risk difference: 1% more (‐2% to 5%) Relative percentage change: 11% (‐82% to 331%) (improvement) Effect is uncertain due to very low quality evidence | |
| 0 per 1000 | 0 per 1000 | |||||
| Moderate³ | ||||||
| 9 per 1000 | 8 per 1000 (2 to 39) | |||||
| High³ | ||||||
| 52 per 1000 | 46 per 1000 (9 to 224) | |||||
| Number of participants who needed orthopaedic surgery (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) | See comments | Outcome not reported in the included studies |
| Number of participants with change in quality of life measures (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) | See comments | Outcome not reported in the included studies |
| Number of participants with change in hearing thresholds (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) | See comments | Outcome not reported in the included studies |
| Number of participants who experienced side effects related to use of bisphosphonates | Study population | RR 1.32 | 678 | ⊕⊕⊝⊝ | Absolute risk difference: 11% more (0% to 22%) Relative percent change: 32 % (‐10% to 92%) (worsening) Gastrointestinal side effects (diarrhoea, dyspepsia, vomiting, nausea, oesophagitis or gastritis) were the most common | |
| 483 per 1000 | 638 per 1000 | |||||
| Number of participants who withdrew due to adverse events | Study population | RR 1.01 | 517 | ⊕⊕⊝⊝ | Absolute risk difference: 0% (‐4% to 3%) Relative percent change: 1% (‐59% to 152%) (worsening) | |
| 41 per 1000 | 41 per 1000 | |||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| ¹ When pain was assessed as any pain reduction instead of disappearance of pain, the outcomes were consistent: 227 per 1000 in placebo vs. 446 per 1000 (292 to 682) in bisphosphonates group (RR 1.97, 95% CI 1.29 to 3.01), NNTD 4 (2 to 13), absolute risk difference 33% more (18% to 49%), relative percentage change 97% (29% to 201% improvement), based on results from seven RCTs (481 participants). Visual analogue scale ranging from 0 to 10 was used in four of the seven studies. One study classified pain in three groups; the tool used for pain assessment was not detailed in the other two studies. Moderate quality evidence: downgraded by one level; there was high risk for attrition bias in three studies and high risk for reporting bias in three studies. The outcome did not change in a sensitive analysis excluding high risk of bias studies. ² Downgraded by one level (imprecision). Few events, resulting in wide CI. ³ The 0% calculated assumed risk in the control group (no fractures in placebo group) is misleading. This outcome is likely due to the short follow‐up period of the studies. To give a more accurate data we have added two scenarios of moderate and high prevalence using data from a study with a longest follow‐up period, the PRISM‐EZ trial (Tan 2017). In summary there are three scenarios to calculate the assumed risk in the control group: 1) To calculate low prevalence we used data from the included studies (placebo groups). 2) To calculate moderate prevalence we used the percentage of fractures in bones affected by Paget's disease of bone in the symptomatic treatment arm of the PRISM‐EZ trial. 3) To calculate high prevalence we used the percentage of fractures in both affected and unaffected bones in the symptomatic treatment arm of the PRISM‐EZ trial. ⁴ Downgraded by two levels (limitation of studies). Most data were from studies assessed at high risk of bias; there was high risk for attrition bias in two studies and high risk for reporting bias in one study. ⁵ Downgraded by one level (indirectness). Long‐term impact on fractures was not assessed. ⁶ Downgraded by one level (limitation of studies). High risk for attrition bias in two studies. ⁷ Downgraded by one level (inconsistency). The side effects considered in the studies were heterogeneous. In addition, considerable heterogeneity was found when the six studies were meta‐analysed (I² = 75%, P = 0.001). However, only one study (McClung 1995) showed more adverse events in the placebo group than the bisphosphonates group. The heterogeneity could not be explained by differences in design of this study since it was similar to other studies. A sensitivity analysis excluding this study found low heterogeneity I² = 6% (RR 1.38, 95% CI 1.08 to 1.78). ⁸ Downgraded by two levels. Half of the included studies were assessed at high risk of bias; there was high risk for attrition bias in three studies and high risk for reporting bias in one study. ⁹ Rated down for imprecision. Optimal information size criterion was not met. The 95% CI is too wide. | ||||||
| Zoledronate versus pamidronate or risedronate for Paget's disease of bone | ||||||
| Patient or population: Paget's disease of bone | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect | № of participants | Quality of the evidence | Comments | |
| Risk with pamidronate or risedronate | Risk with zoledronate | |||||
| Number of participants with change in bone pain¹ | Study population | RR 1.31 | 436 | ⊕⊝⊝⊝ | NNTB: 7 (4 to 14) Absolute risk difference: 17% (8% to 26%) Relative percent change: 31% (15% to 51%) (improvement) Not pooled effects: Zoledronate vs. pamidronate (89 participants); RR 1.30 (1.10 to 1.53), NNTB 4 (3 to 13). Zoledronate vs. risedronate (347 participants); RR 1.36 (1.06 to 1.74), NNTB 8 (4 to 45). | |
| 465 per 1000 | 609 per 1000 | |||||
| Number of participants who experienced fractures (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) | See comments | Outcome not reported in the included studies |
| Number of participants who needed orthopaedic surgery (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) | See comments | Outcome not reported in the included studies |
| Number of participants with change in quality of life measures (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) | See comments | Effect is uncertain. Zoledronate showed a marginal improvement at 6 months in QoL when compared with risedronate. The physical component summary score of SF‐36 improved with zoledronate compared to risedronate (1.6 vs. 0.3 change from baseline score, on a 0 to 100 scale). This result is unlikely to be of clinical importance |
| Number of participants with change in hearing thresholds (not measured) | See comments | See comments | Not estimable | 0 (0 RCTs) | See comments | Outcome not reported in the included studies |
| Number of participants who experienced side effects related to use of bisphosphonates. | Study population | RR 1.05 | 437 | ⊕⊕⊝⊝ | Absolute risk difference: 4% (‐4% to 12%) Relative percent change: 5% (‐5% to 16%) (worsening) | |
| 745 per 1000 | 782 per 1000 | |||||
| Number of participants who withdrew due to adverse events (withdrawals) | Study population | RR 2.04 | 437 | ⊕⊝⊝⊝ | Absolute risk difference: 1% (‐2% to 3%) Relative percent change: 104% (‐57% to 859%) (worsening) | |
| 9 per 1000 | 18 per 1000 | |||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| ¹Unlike summary of findings Table for the main comparison (comparing bisphosphonates vs. placebo) when compared zoledronate vs pamidronate or risedronate we used any change of pain instead of disappearance of pain because data were not available. However, readers could find data on disappearance of pain in the original zoledronate vs. pamidronate manuscript (Merlotti 2007) [10/47 vs. 6/60, RR 2.12 95% IC 0.83‐5.43]. We did not include these data because we think they are misleading. They come from accumulate zoledronate effects in two different study phases (30 zoledronate patients from phase 1 + 17 patients no responders to pamidronate in phase 1 treated with zoledronate in phase 2) vs. pamidronate in only one study phase (60 patients). ² Downgraded by two levels (limitation of studies). Information is from studies at high risk of bias. High risk for performance bias in 1 study and high risk for reporting bias in 1 study. ³ Downgraded by one level (indirectness). In the risedronate study, the author assessed bodily pain but not bone pain associated directly with Paget's bone lesions (Change in bone pain was defined as"5‐point improvement from baseline" in SF‐36 bodily pain item). In the pamidronate study change in bone pain was defined as "subjects reported disappearance or decrease in pain". ⁴Downgraded by one level (imprecision). There were few events, resulting in wide CI, | ||||||
| Study ID | Intervention | Comparator | Alkaline phosphatase | Follow‐up | N | Age | Male | Symptomatic | Previously treated |
| Etidronate | Placebo | Yes | 6 m | 50 | 67 y | 60% | NA | NA | |
| Etidronate | Placebo | Yes | 6 m | 48 | NA | 58% | NA | NA | |
| Etidronate | Placebo | No | 3 m | 32 | NA | NA | 100% | 38% | |
| Tiludronate | Placebo | Yes | 6 m | 112 | 70 y | 54% | 63% | NA | |
| Tiludronate | Placebo | Yes | 6 m | 139 | 70 y | 54% | NA | NA | |
| Tiludronate | Placebo | Yes | 6 m | 149 | 69 y | 54% | NA | 82% | |
| Alendronate | Placebo | Yes | 6 m | 15 | 67 y | 60% | 87% | 66% | |
| Alendronate | Placebo | Yes | 6 m | 55 | 70 y | 56% | NA | 35% | |
| Zoledronate | Placebo | Yes | 3 m | 176 | 71 y | 61% | NA | NA | |
| Ibandronate | Placebo | Yes | 6 m | 25 | 73 y | 74% | NA | 64% | |
| Tiludronate | Etidronate | Yes | 6m | 234 | 69y | 59% | 74% | 71% | |
| Alendronate | Etidronate | Yes | 6m | 89 | 69y | 67% | NA | 25% | |
| Risedronate | Etidronate | Yes | 12 m | 123 | 66 y | 69% | 91% | 72% | |
| Alendronate | Pamidronate | Yes | 12 m | 72 | 70 y | 58% | 94% | 39% | |
| Olpadronate | Pamidronate | Yes | 6 m | 27 | NA | NA | NA | NA | |
| Zoledronate | Pamidronate | Yes | 6 m | 90 | 70 y | 69% | 99% | 67% | |
| Zoledronate | Risedronate | Yes | 6 m | 357 | 70 y | 68% | NA | 54% | |
| Etidronate + calcitonin | Etidronate | Yes | 12 m | 44 | NA | NA | 100% | 10% | |
| Intensive | Symptomatic | No | 3 y | 1331 | 74 y | 51% | 69% | NA | |
| Intensive | Symptomatic | No | 3 y | 502 | 76 y | 55% | 63% | 70% | |
| Alkaline phosphatase: Serum total alkaline phosphatase above the upper limit of normal as an inclusion criterion. Follow‐up: Extended follow‐up periods are shown in parentheses. N: Number of randomised participants. NA: Not available. SC: Sample size calculated before study. | |||||||||
| Study ID | Outcome | Zoledronate | Risedronate | RR (95% IC) | ||
| Events | N | Events | N | |||
| Radiologically‐confirmed clinical fracture | 2 | 177 | 2 | 172 | 0.97 (0.14 to 6.82) | |
| Reid 2011 (extension) | Radiologically‐confirmed clinical fracture | 3 | 152 | 1 | 115 | 2.30 (0.24 to 22.36) |
| Quality of life change from baseline | 48 | 176 | 36 | 171 | 1.30 (0.89 to 1.89) | |
| Reid 2011 (extension) | Clinical relapse | 14 | 152 | 29 | 115 | 0.30 (0.15 to 0.60) |
| Study ID | Outcome | Mean (SD) | N | Mean (SD) | N | Mean difference |
| Mean change from baseline in pain | ‐0.5 (1.75) | 101 | ‐0.4 (2.13) | 92 | ‐0.10 (‐0.65 to 0.45) | |
| Mean change from baseline in quality of life¹ | 1.5 (0.5) | 176 | 0.2 (0.6) | 171 | 1.30 (1.18 to 1.42) | |
| Reid 2011 (extension) | Mean change from baseline in total SF‐36 score² | 1.3 (3.1) | 152 | ‐2.5 (2.6) | 115 | 3.8 (3.12 to 4.49) |
| ¹Physical‐component summary (data extracted from Figure 4 in Reid 2005). ²Total SF‐36 scores to 54 months (data extracted from Figure 6 in Reid 2011 extension) (+1.3 ± 3.1 versus ‐2.5 ± 2.6) [D] | ||||||
| Ouctome | Etidronate plus calcitonin | Etidronate | RR (95% IC) | ||
| Events | N | Events | N | ||
| Change in bone pain | 10 | 21 | 15 | 23 | 0.73 (0.43 to 1.25) |
| Study ID | Outcome | Intensive | Symptomatic | RR (95% IC) | ||
| Events | N | Events | N | |||
| Improvement in bone pain | 78 | 295 | 96 | 311 | 0.86 (0.67 to 1.10) | |
| Radiologically‐confirmed fractures | 46 | 661 | 49 | 663 | 0.94 (0.64 to 1.39) | |
| Radiologically‐confirmed fractures*¹ | 22 | 270 | 12 | 232 | 1.58 (0.80 to 3.11) | |
| Radiologically‐confirmed fractures (pagetic bone) | 8 | 661 | 13 | 663 | 0.62 (0.25 to 1.49) | |
| Radiologically‐confirmed fractures (pagetic bone)¹ | 5 | 270 | 2 | 232 | 2.15 (0.42 to 10.96) | |
| Number of orthopaedic surgeries | 48 | 661 | 55 | 663 | 0.88 (0.60 to 1.27) | |
| Number of orthopaedic surgeries¹ | 15 | 270 | 7 | 232 | 1.84 (0.76 to 4.44) | |
| Number of orthopaedic procedures | 50 | 661 | 63 | 663 | 0.78 (0.53 to 1.15) | |
| Number of orthopaedic procedures | 16 | 270 | 9 | 232 | 1.52 (0.69 to 3.39) | |
| Change in hearing thresholds² | 134 | 505 | 133 | 486 | 0.97 (0.79 to 1.19) | |
| Hearing classification worse at study end (left ear)³ | 6 | 50 | 8 | 63 | 0.95 (0.35 to 2.55) | |
| Hearing classification worse at study end (right ear)³ | 4 | 51 | 8 | 60 | 0.58 (0.19 to 1.84) | |
| Serious adverse events | 345 | 661 | 359 | 663 | 0.96 (0.87 to 1.07) | |
| Serious adverse events | 87 | 270 | 66 | 232 | 1.13 (0.87 to 1.48) | |
| Withdrawal due to adverse events⁴ | 83 | 661 | 79 | 663 | 1.05 (0.79 to 1.41) | |
| Normalised alkaline phosphatase levels | 512 | 661 | 406 | 663 | 1.26 (1.18 to 1.36) | |
| Study ID | Outcome | Mean (SD) | N | Mean (SD) | N | Mean difference |
| Mean change from baseline in quality of life (bodily pain SF‐36)⁵ | ‐0.4 (8.9) | 479 | 0.3 (9.4) | 477 | ‐0.7 (‐1.8 to 0.5) | |
| Mean change from baseline in quality of life (bodily pain SF‐36)⁶ | 0.1 (9.3) | 149 | ‐1.0 (9.1) | 138 | ‐1.0 (‐3.0 to 1.1) | |
| Mean change from baseline in quality of life (physical summary SF‐36)*⁵ | ‐1.2 (8.1) | 408 | ‐1.1 (8.2) | 396 | ‐0.1 (‐1.3 to 1.0) | |
| Mean change from baseline in quality of life (physical summary SF‐36)⁶ | ‐1.0 (7.7) | 144 | ‐2.7 (7.7) | 126 | ‐1.6 (‐3.4 to 0.3) | |
| Mean change from baseline in quality of life (mental summary SF‐36)*⁵ | ‐1.7 (10.2) | 408 | ‐2.6 (10.9) | 396 | 0.9 (‐0.6 to 2.3) | |
| Mean change from baseline in quality of life (mental summary SF‐36)⁶ | ‐1.0 (10.0) | 144 | ‐0.4 (9.9) | 126 | ‐0.6 (‐1.7 to 3.1) | |
| Mean hearing loss (left ear)³ | 1.8 (14.6) | 50 | 0.0 (12.6) | 63 | 1.8 (‐3.4 to 7.0) | |
| Mean hearing loss (right ear)³ | 2.5 (5.7) | 51 | 2.1 (9.4) | 60 | 0.5 (‐2.4 to 3.3) | |
| Mean percentage change from baseline in serum total alkaline phosphatase activity | ‐40.5 (23.7) | 430 | ‐18 (71.2) | 424 | ‐22.5 (‐29.6 to ‐15.4) | |
| Mean percentage change from baseline in adjusted serum total alkaline phosphatase activity | ‐0.15 (0.72) | 203 | ‐0.05 (0.75) | 181 | ‐0.11 (‐0.03 to 0.25) | |
| Data at 24 months for Langston 2010. ¹Data shown for these outcomes are number of events, patient years of follow up, rate ratios and 95% CI calculated using the method described by Cohen 2011. ²Number of participants using hearing aids at the end of the study. ³Patients with baseline and end of the trial measurements. ⁴Serious adverse event: any untoward medical occurrence that: 1) results in death, 2) is life‐threatening, 3) requires inpatient hospitalisation or prolongation of existing hospitalisation, 4) results in persistent or significant disability/incapacity, or 5) is a congenital anomaly/birth defect. ⁵Data at 24 months. ⁶Difference between baseline and 36 months. | ||||||
| Outcome | Tiludronate | Etidronate | RR (95% IC) | ||
| Events | N | Events | N | ||
| Number of participants with change in bone pain | 32 | 120 | 10 | 52 | 1.39 (0.74 to 2.61) |
| Number of participants with radiologically‐confirmed fractures | 1 | 155 | 2 | 79 | 0.25 (0.02 to 2.77) |
| Number of participants with severe side effects | 75 | 155 | 27 | 79 | 1.42 (1.00 to 2.00) |
| Number of participants who withdrew due to adverse events | 10 | 155 | 2 | 79 | 2.55 (0.57 to 11.35) |
| Number of participants who normalised alkaline phosphatase levels | 40 | 155 | 9 | 79 | 2.27 (1.16 to 4.43) |
| Study ID | Comparison | Side effect | Bisphosphonate | Placebo | RR (95% CI) |
| Etidronate (38) vs. placebo (9) | Diarrhoea | 5 (13%) | 1 (11%) | 1.18 (0.16 to 8.93) | |
| Zoledronate (141) vs. placebo (35) | Fatigue Fever Arthralgia Pain, back Pain, skeletal Hypocalcaemia | 12 (9%) 7 (5%) 15 (11%) 14 (10%) 11 (8%) 3 (2%) | 0 (0%) 0 (0%) 3 (9%) 1 (3%) 2 (6%) 0 (0%) | 6.34 (0.38 to 104.5) 3.80 (0.50 to 28.88) 1.24 (0.89 to 1.77) 3.47 (0.53 to 23.02) 1.37 (0.85 to 2.19) 1.78 (0.74 to 4.24) | |
| Tiludronate (86) vs. placebo (26) | Nausea Vomiting Dyspepsia Diarrhoea Arthralgia Skeletal pain Raised liver enzymes Eosinophilia | 15 (17%) 7 (7%) 9 (10%) 14 (16%) 8 (9%) 5 (6%) 1 (1%) 0 (0%) | 2 (8%) 0 (0%) 0 (0%) 0 (0%) 2 (8%) 3 (12%) 0 (0%) 1 (4%) | 2.27 (0.65 to 7.86) 4.66 (0.45 to 48.06) 5.90 (0.40 to 87.16) 9.00 (0.32 to 252.8) 1.21 (0.91 to 1.61) 0.50 (0.13 to 1.97) 0.93 (0.04 to 22.20) 0.10 (0.01 to 2.47) | |
| Tiludronate (91) vs. placebo (48) | Gastrointestinal | 31 (34%) | 15 (31%) | 1.09 (0.66 to 1.81) | |
| Tiludronate (117) vs. placebo (32) | Gastralgia Nausea | 20 (17.1%) 11 (9.4%) | 5 (16.1%) 3 (9.6%) | 1.09 (0.45 to 2.69) 1.00 (0.30 to 3.38) | |
| Alendronate (27) vs. placebo (28) | Gastrointestinal Gastritis Duodenal ulcer Oesophagitis | 2 (7%) 0 (0%) 0 (0%) 1 (4%) | 5 (18%) 1 (4%) 1 (4%) 0 (0%) | 0.42 (0.09 to 2.00) 0.35 (0.02 to 8.13) 0.35 (0.02 to 8.13) 3.10 (0.13 to 73.10) |
| Study ID | Comparison | Adverse effect | Bisphosphonate 1 | Bisphosphonate 2 | RR (95% CI) |
| Tiludronate (155) vs. etidronate (79) | Gastrointestinal | 32 (20.8%) | 10 (12.7%) | 1.63 (0.85 to 3.14) | |
| Abdominal pain | 10 (6.5%) | 2 (2.5%) | 2.55 (0.57 to 11.35) | ||
| Nausea, vomiting | 8 (5.2%) | 2 (2.5%) | 2.04 (0.44 to 9.37) | ||
| Fracture | 1 (1%) | 2 (3%) | 0.25 (0.02 to 2.77) | ||
| Alendronate (42) vs. etidronate (47) | Gastrointestinal | 11 (26%) | 10 (21%) | 1.23 (0.58 to 2.60) | |
| Abdominal distention | 0 (%) | 1 (2%) | 0.37 (0.02 to 8.90) | ||
| Abdominal pain | 3 (7%) | 4 (9%) | 0.84 (0.2 to 3.54) | ||
| Acid regurgitation | 1 (2%) | 1 (2%) | 1.12 (0.07 to 17.34) | ||
| Dyspepsia | 0 (0%) | 1 (2%) | 0.37 (0.02 to 8.90) | ||
| Melena | 1 (2%) | 0 (0%) | 3.35 (0.14 to 80.05) | ||
| Nausea | 2 (5%) | 3 (6%) | 0.75 (0.13 to 4.25) | ||
| Leg pain | 1 (2%) | 9 (19%) | 0.12 (0.02 to 0.94) | ||
| Laboratory adverse experiences | 9 (21%) | 6 (13%) | 1.68 (0.65 to 4.32) | ||
| Risedronate (62) vs. etidronate (61) | Upper gastrointestinal | 12 (19%) | 12 (20%) | 0.98 (0.48 to 2.02) | |
| Olpadronate (14) vs. pamidronate (7) | Digestive | 9 (64%) | 7 (100%) | 0.68 (0.44 to 1.03) | |
| Zoledronate (47)* vs. pamidronate (60) | Influenza‐like illness | 4 (9%) | 5 (8%) | 1.02 (0.29 to 3.59) | |
| Myalgia | 3 (6%) | 4 (7%) | 0.96 (0.23 to 4.07) | ||
| Pyrexia | 3 (6%) | 4 (7%) | 0.96 (0.23 to 4.07) | ||
| Fatigue | 3 (6%) | 8 (13%) | 0.48 (0.13 to 1.71) | ||
| Headache | 4 (9%) | 5 (8%) | 1.02 (0.29 to 3.59) | ||
| Diarrhoea | 1 (2%) | 2 (3%) | 0.64 (0.06 to 6.83) | ||
| Bone pain | 3 (6%) | 6 (10%) | 0.64 (0.17 to 2.42) | ||
| Pain in arm or leg | 3 (6%) | 4 (7%) | 0.96 (0.23 to 4.07) | ||
| Hypocalcaemia | 3 (6%) | 1 (2%) | 3.83 (0.41 to 35.64) | ||
| Dermatitis | 1 (2%) | 0 (0%) | 0.42 (0.02 to 10.17) | ||
| Zoledronate (177) vs. risedronate (172) | Study days 1 to 3 | ||||
| Influenza‐like illness | 17 (9.6%) | 7 (4.1%) | 2.36 (1 to 5.55) | ||
| Myalgia | 13 (7.3%) | 6 (3.5%) | 2.11 (0.82 to 5.41) | ||
| Pyrexia | 13 (7.3%) | 1 (0.6%) | 12.63 (1.67 to 95.53) | ||
| Fatigue | 12 (6.8%) | 4 (2.3%) | 2.92 (0.96 to 8.86) | ||
| Headache | 12 (6.8%) | 7 (4.1%) | 1.67 (0.67 to 4.13) | ||
| Rigor | 12 (6.8%) | 1 (0.6%) | 11.66 (1.53 to 88.72) | ||
| Nausea | 11 (6.2%) | 3 (1.7%) | 3.56 (1.01 to 12.55) | ||
| Bone pain | 9 (5.1%) | 2 (1.2%) | 4.37 (0.96 to 19.95) | ||
| After study day 3 | |||||
| Pain in an arm or leg | 13 (7.3%) | 12 (7%) | 1.05 (0.49 to 2.24) | ||
| Arthralgia | 9 (5.1%) | 19 (11%) | 0.46 (0.21 to 0.99) | ||
| Dizziness | 9 (5.1%) | 5 (2.9%) | 1.75 (0.6 to 5.11) | ||
| Nasopharyngitis | 9 (5.1%) | 14 (8.1%) | 0.62 (0.28 to 1.41) | ||
| Diarrhoea | 8 (4.5%) | 9 (5.2%) | 0.86 (0.34 to 2.19) | ||
| Headache | 7 (4%) | 10 (5.8%) | 0.68 (0.26 to 1.75) | ||
| Back pain | 4 (2.3%) | 12 (7.0%) | 0.32 (0.11 to 0.98) | ||
| Symptomatic hypocalcaemia | 2 (1.1%) | 1 (0.6%) | 1.94 (0.18 to 21.24) | ||
| Reid 2011 (extension) | Zoledronate (152) vs. risedronate (115) | Atrial fibrillation | 1 (0.7%) | 1 (0.9%) | 0.76 (0.05 to 12.20) |
| Atrial flutter | 0 (0%) | 2 (1.7%) | 0.15 (0.01 to 3.13) | ||
| Osteonecrosis jaw | 0 (0%) | 0 (0%) | ‐ | ||
| Alendronate (36) vs. pamidronate (36) | Gastrointestinal | 16 (44%) | 4 (11%) | 4 (1.48 to 10.80) | |
| Fatigue | 0 (0%) | 23 (64%) | 0.02 (0.00 to 0.34) | ||
| General aches/pain | 4 (11%) | 16 (44%) | 0.25 (0.09 to 0.68) | ||
| Deteriorating kidney failure | 1 (3%) | 0 (0%) | 3.00 (0.12 to 71.28) | ||
| *Zoledronate group data were extracted from Table 3 in Merlotti 2007. In this table, the authors presented data from 30 participants who took part in the first part of the study (which was included in our systematic review) plus 17 participants from the second part of the study (which was not included in our systematic review). | |||||
| Study ID | Comparison | Side effect | Intensive | Symptomatic | RR (95% CI) |
| Intensive (661) vs. symptomatic (663) | All adverse events Serious adverse events Musculoskeletal Sensory Gastrointestinal Cardiovascular Arrythmia Cancer Renal Other | 3429 345 691 203 172 360 13 (1.9%) 55 98 1850 | 3471 359 734 196 157 327 7 (1%) 47 78 1932 | ‐ ‐ ‐ ‐ ‐ ‐ 1.86 (0.75 to 4.64) ‐ ‐ ‐ | |
| Intensive (270) vs. symptomatic (232) | All adverse events Serious adverse events Musculoskeletal Osteonecrosis of the jaw Delayed union of fracture Ophthalmic Uveitis Gastrointestinal Cardiovascular Arrythmia Cerebrovascular Central nervous system Endocrine Ear, nose or throat Genitourinary Haematological Respiratory Skin Miscellaneous | 226 87 123 1 2 34 1 54 67 14 4 28 28 28 41 10 48 41 33 | 196 66 104 0 1 41 0 46 49 8 3 28 21 26 39 9 43 33 32 | 0.99 (0.91 to 1.07) 1.13 (0.87 to 1.48) 1.02 (0.84 to 1.23) 2.58 (0.11 to 63.01) 1.72 (0.16 to 18.83) 0.71 (0.47 to 1.08) 2.58 (0.11 to 63.01) 1.01 (0.71 to 1.43) 1.18 (0.85 to 1.62) 1.50 (0.64 to 3.52) 1.14 (0.26 to 5.07) 0.86 (0.52 to 1.41) 1.15 (0.68 to 1.96) 0.93 (0.56 to 1.53) 0.90 (0.61 to 1.35) 0.96 (0.40 to 2.31) 0.95 (0.66 to 1.39) 1.07 (0.70 to 1.63) 0.89 (0.56 to 1.40) | |
| * Data represent total numbers of reported side effects regardless of numbers of participants who experienced them. The authors reported numbers of participants only for arrhythmia. | |||||
| Study ID | Outcome | Tiludronate | Placebo | RR (95% IC) | ||
| Events | N | Events | N | |||
| Number of participants who relapsed due to bone pain recurrence | 23 | 66 | 13 | 19 | 0.51 (0.32 to 0.80) | |
| Study ID | Outcome | Risedronate | Tiludronate | RR (95% IC) | ||
| Events | N | Events | N | |||
| Number of participants who relapsed due to recurrence of increased serum alkaline phosphatase level | 2 | 62 | 8 | 61 | 0.25 (0.05 to 1.11) | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Number of participants whose bone pain disappeared completely Show forest plot | 2 | 205 | Risk Ratio (M‐H, Random, 95% CI) | 3.42 [1.31, 8.90] |
| 2 Number of participants with change in bone pain Show forest plot | 7 | 481 | Risk Ratio (M‐H, Random, 95% CI) | 1.97 [1.29, 3.01] |
| 2.1 Etidronate vs. placebo | 3 | 124 | Risk Ratio (M‐H, Random, 95% CI) | 1.75 [0.79, 3.87] |
| 2.2 Tiludronate vs. placebo | 3 | 344 | Risk Ratio (M‐H, Random, 95% CI) | 1.98 [1.27, 3.08] |
| 2.3 Alendronate vs. placebo | 1 | 13 | Risk Ratio (M‐H, Random, 95% CI) | 10.00 [0.69, 144.38] |
| 3 Number of participants experiencing radiologically‐confirmed clinical fractures Show forest plot | 4 | 356 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.18, 4.51] |
| 3.1 Etidronate vs. placebo | 2 | 95 | Odds Ratio (M‐H, Random, 95% CI) | 0.90 [0.09, 9.06] |
| 3.2 Tiludronate vs. placebo | 2 | 261 | Odds Ratio (M‐H, Random, 95% CI) | 0.88 [0.09, 8.64] |
| 4 Number of participants who experienced adverse events related to use of bisphosphonates Show forest plot | 6 | 678 | Risk Difference (M‐H, Random, 95% CI) | 0.11 [‐0.00, 0.22] |
| 4.1 Etidronate vs. placebo | 1 | 47 | Risk Difference (M‐H, Random, 95% CI) | 0.02 [‐0.21, 0.25] |
| 4.2 Zoledronate vs. placebo | 1 | 176 | Risk Difference (M‐H, Random, 95% CI) | 0.27 [0.12, 0.42] |
| 4.3 Tiludronate vs. placebo | 3 | 400 | Risk Difference (M‐H, Random, 95% CI) | 0.07 [‐0.08, 0.22] |
| 4.4 Alendronate vs. placebo | 1 | 55 | Risk Difference (M‐H, Random, 95% CI) | 0.12 [‐0.11, 0.34] |
| 5 Number of participants who withdrew due to adverse events Show forest plot | 6 | 517 | Odds Ratio (M‐H, Random, 95% CI) | 1.01 [0.38, 2.69] |
| 5.1 Etidronate vs. placebo | 1 | 47 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 Tiludronate vs. placebo | 3 | 400 | Odds Ratio (M‐H, Random, 95% CI) | 0.96 [0.34, 2.67] |
| 5.3 Alendronate vs. placebo | 2 | 70 | Odds Ratio (M‐H, Random, 95% CI) | 1.74 [0.06, 50.43] |
| 6 Mean percentage change from baseline in serum total alkaline phosphatase level Show forest plot | 8 | 592 | Mean Difference (IV, Random, 95% CI) | ‐50.09 [‐67.72, ‐32.46] |
| 6.1 Etidronate vs. placebo | 3 | 122 | Mean Difference (IV, Random, 95% CI) | ‐55.85 [‐66.50, ‐45.20] |
| 6.2 Zoledronate vs. placebo | 1 | 176 | Mean Difference (IV, Random, 95% CI) | ‐22.26 [‐27.99, ‐16.53] |
| 6.3 Tiludronate vs. placebo | 2 | 256 | Mean Difference (IV, Random, 95% CI) | ‐58.0 [‐64.25, ‐51.75] |
| 6.4 Alendronate vs. placebo | 1 | 15 | Mean Difference (IV, Random, 95% CI) | ‐39.9 [‐51.28, ‐28.52] |
| 6.5 Ibandronate vs. placebo | 1 | 23 | Mean Difference (IV, Random, 95% CI) | ‐96.1 [‐147.01, ‐45.19] |
| 7 Number of participants who achieved normalised alkaline phosphatase level Show forest plot | 8 | 580 | Risk Ratio (M‐H, Random, 95% CI) | 9.96 [3.74, 26.58] |
| 7.1 Etidronate vs. placebo | 3 | 121 | Risk Ratio (M‐H, Random, 95% CI) | 4.51 [0.90, 22.55] |
| 7.2 Tiludronate vs. placebo | 3 | 381 | Risk Ratio (M‐H, Random, 95% CI) | 13.79 [2.77, 68.61] |
| 7.3 Alendronate vs. placebo | 1 | 55 | Risk Ratio (M‐H, Random, 95% CI) | 27.96 [1.74, 448.28] |
| 7.4 Ibandronate vs. placebo | 1 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 14.00 [0.92, 212.92] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Mean change from baseline in pain Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 1.1 Risedronate vs. etidronate | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Number of participants who experienced adverse events related to use of bisphosphonates Show forest plot | 2 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.72, 1.35] |
| 2.1 Risedronate vs. etidronate | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.68, 1.43] |
| 2.2 Alendronate vs. etidronate | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.55, 1.76] |
| 3 Number of participants who withdrew due to adverse events Show forest plot | 2 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.25, 1.89] |
| 3.1 Risedronate vs. etidronate | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.22, 2.79] |
| 3.2 Alendronate vs. etidronate | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.11, 2.90] |
| 4 Mean percentage change from baseline in serum total alkaline phosphatase level Show forest plot | 2 | 212 | Mean Difference (IV, Random, 95% CI) | ‐40.95 [‐49.09, ‐32.81] |
| 4.1 Risedronate vs. etidronate | 1 | 123 | Mean Difference (IV, Random, 95% CI) | ‐43.9 [‐48.06, ‐39.74] |
| 4.2 Alendronate vs. etidronate | 1 | 89 | Mean Difference (IV, Random, 95% CI) | ‐35.1 [‐45.85, ‐24.35] |
| 5 Number of participants who achieved normalised alkaline phosphatase level Show forest plot | 2 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 4.30 [2.72, 6.79] |
| 5.1 Risedronate vs. etidronate | 1 | 123 | Risk Ratio (M‐H, Random, 95% CI) | 4.81 [2.58, 8.98] |
| 5.2 Alendronate vs. etidronate | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 3.78 [1.93, 7.38] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Number of participants with bone pain change Show forest plot | 2 | 436 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [1.15, 1.51] |
| 1.1 Zoledronate (1) vs. pamidronate (2) | 1 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [1.10, 1.53] |
| 1.2 Zolendronate (1) vs. risedronate (2) | 1 | 347 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [1.06, 1.74] |
| 2 Number of participants who experienced adverse events related to use of bisphosphonates Show forest plot | 3 | Risk Difference (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Olpadronate (1) vs. pamidronate (2) | 1 | Risk Difference (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Zoledronate (1) vs. pamidronate (2) | 1 | Risk Difference (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Zolendronate (1) vs. risedronate (2) | 1 | Risk Difference (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Number of participants who withdrew due to adverse events Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Zoledronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Zolendronate (1) vs. risedronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Alendronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Mean percentage change from baseline in serum total alkaline phosphatase level Show forest plot | 3 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Olpadronate (1) vs. pamidronate (2) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Zolendronate (1) vs. risedronate (2) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Alendronate (1) vs. pamidronate (2) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Number of participants who achieved normalised alkaline phosphatase level Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Olpadronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Zoledronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Zolendronate (1) vs. risedronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 Alendronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Number of participants who experienced biochemical relapse with increased alkaline phosphatase level Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 Zoledronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Zoledronate (1) vs. risedronate (2) | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Alendronate (1) vs. pamidronate (2) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |