Carbamazepine versus phenytoin monotherapy for epilepsy: an individual participant data review
Abstract
Background
This is an updated version of the original Cochrane Review published in Issue 2, 2002 and its subsequent updates in 2010 and 2015.
Epilepsy is a common neurological condition in which recurrent, unprovoked seizures are caused by abnormal electrical discharges from the brain. It is believed that with effective drug treatment, up to 70% of individuals with active epilepsy have the potential to become seizure‐free and go into long‐term remission shortly after starting drug therapy with a single antiepileptic drug in monotherapy.
Worldwide, carbamazepine and phenytoin are commonly‐used broad spectrum antiepileptic drugs, suitable for most epileptic seizure types. Carbamazepine is a current first‐line treatment for partial onset seizures in the USA and Europe. Phenytoin is no longer considered a first‐line treatment due to concerns over adverse events associated with its use, but the drug is still commonly used in low‐ to middle‐income countries because of its low cost. No consistent differences in efficacy have been found between carbamazepine and phenytoin in individual trials, although the confidence intervals generated by these studies are wide. Differences in efficacy may therefore be shown by synthesising the data of the individual trials.
Objectives
To review the time to withdrawal, six‐ and 12‐month remission, and first seizure with carbamazepine compared to phenytoin, used as monotherapy in people with partial onset seizures (simple partial, complex partial, or secondarily generalised tonic‐clonic seizures), or generalised tonic‐clonic seizures, with or without other generalised seizure types.
Search methods
For the latest update we searched the Cochrane Epilepsy Group’s Specialised Register (1st November 2016), the Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies Online (CRSO, 1st November 2016), MEDLINE (Ovid, 1946 to 1 November 2016), ClinicalTrials.gov (1 November 2016), and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP, 1st November 2016). Previously we also searched SCOPUS (1823 to 16th September 2014) as an alternative to Embase, but this is no longer necessary, because randomised and quasi‐randomised controlled trials in Embase are now included in CENTRAL. We handsearched relevant journals, contacted pharmaceutical companies, original trial investigators and experts in the field.
Selection criteria
Randomised controlled trials (RCTs) in children or adults with partial onset seizures or generalised onset tonic‐clonic seizures, comparing carbamazepine monotherapy versus phenytoin monotherapy.
Data collection and analysis
This is an individual participant data (IPD) review. Our primary outcome was time to withdrawal of allocated treatment, and our secondary outcomes were time to six‐month remission, time to 12‐month remission, and time to first seizure post‐randomisation. We used Cox proportional hazards regression models to obtain study‐specific estimates of hazard ratios (HRs) with 95% confidence intervals (CIs) and the generic inverse variance method to obtain the overall pooled HR and 95% CI.
Main results
IPD were available for 595 participants out of 1192 eligible individuals, from four out of 12 trials (i.e. 50% of the potential data). For remission outcomes, HR greater than 1 indicates an advantage for phenytoin; and for first seizure and withdrawal outcomes, HR greater than 1 indicates an advantage for carbamazepine. The methodological quality of the four studies providing IPD was generally good and we rated it at low risk of bias overall in the analyses.
The main overall results (pooled HR adjusted for seizure type) were time to withdrawal of allocated treatment: 1.04 (95% CI 0.78 to 1.39; three trials, 546 participants); time to 12‐month remission: 1.01 (95% CI 0.78 to 1.31; three trials, 551 participants); time to six‐month remission: 1.11 (95% CI 0.89 to 1.37; three trials, 551 participants); and time to first seizure: 0.85 (95% CI 0.70 to 1.04; four trials, 582 participants). The results suggest no overall statistically significant difference between the drugs for these outcomes. There is some evidence of an advantage for phenytoin for individuals with generalised onset seizures for our primary outcome (time to withdrawal of allocated treatment): pooled HR 0.42 (95% CI 0.18 to 0.96; two trials, 118 participants); and a statistical interaction between treatment effect and epilepsy type (partial versus generalised) for this outcome (P = 0.02). However, misclassification of seizure type for up to 48 individuals (32% of those with generalised epilepsy) may have confounded the results of this review. Despite concerns over side effects leading to the withdrawal of phenytoin as a first‐line treatment in the USA and Europe, we found no evidence that phenytoin is more likely to be associated with serious side effects than carbamazepine; 26 individuals withdrew from 290 randomised (9%) to carbamazepine due to adverse effects, compared to 12 out of 299 (4%) randomised to phenytoin from four studies conducted in the USA and Europe (risk ratio (RR) 1.42, 95% CI 1.13 to 1.80, P = 0.014). We rated the quality of the evidence as low to moderate according to GRADE criteria, due to imprecision and potential misclassification of seizure type.
Authors' conclusions
We have not found evidence for a statistically significant difference between carbamazepine and phenytoin for the efficacy outcomes examined in this review, but CIs are wide and we cannot exclude the possibility of important differences. There is no evidence in this review that phenytoin is more strongly associated with serious adverse events than carbamazepine. There is some evidence that people with generalised seizures may be less likely to withdraw early from phenytoin than from carbamazepine, but misclassification of seizure type may have impacted upon our results. We recommend caution when interpreting the results of this review, and do not recommend that our results alone should be used in choosing between carbamazepine and phenytoin. We recommend that future trials should be designed to the highest quality possible, with considerations of allocation concealment and masking, choice of population, choice of outcomes and analysis, and presentation of results.
PICOs
Plain language summary
Carbamazepine versus phenytoin (given as a single drug treatment) for epilepsy
Background
Epilepsy is a common neurological disorder in which recurrent seizures are caused by abnormal electrical discharges from the brain. We studied two types of epileptic seizures in this review: generalised onset seizures in which electrical discharges begin in one part of the brain and move throughout the brain, and partial onset seizures in which the seizure is generated in and affects only one part of the brain (the whole hemisphere of the brain or part of a lobe of the brain). For around 70% of people with epilepsy, generalised onset or partial onset seizures can be controlled by a single antiepileptic drug. Worldwide, phenytoin and carbamazepine are commonly used antiepileptic drugs, although carbamazepine is used more commonly in the USA and Europe due to concerns over side effects associated with phenytoin. Phenytoin is still commonly used in low‐ and middle‐income countries in Africa, Asia and South America, because of the low cost of the drug.
Objective
For this updated review, we looked at the evidence from 12 randomised controlled clinical trials comparing phenytoin and carbamazepine based on how effective the drugs were at controlling seizures (i.e. whether people went back to having seizures or had long periods of freedom from seizures (remission)), and how tolerable any related side effects of the drugs were.
Main results
We were able to combine data for 595 people from four of the 12 trials; for the remaining 597 people from eight trials, information was not available to use in this review. The evidence is current to November 2016.
Results of this review suggest that people with generalised seizures are more likely to withdraw from carbamazepine treatment earlier than from phenytoin treatment, due to seizure recurrence, side effects of the drug, or both, but for people with partial seizures there was no difference in times of withdrawal from treatment between the two drugs. Even though phenytoin is thought to cause more and worse side effects than carbamazepine, we found that twice as many people withdrew from treatment with carbamazepine due to side effects than from treatment with phenytoin.
Results of the review show no difference between carbamazepine and phenytoin for people achieving long periods of seizure freedom (six‐ or 12‐month remission of seizures), or experiencing more seizures after starting treatment.
We judge the evidence from this review to be of low to moderate quality. We recommend that caution is used when interpreting the results of this review, as we were unable to combine the data for all people treated in trials comparing carbamazepine to phenytoin. Also, up to 30% of people in the trials used in our results may have been wrongly classified as having generalised seizures; this may have affected the results of our review.
We recommend that any future trials comparing these drugs, or any other antiepileptic drugs, should be designed using high‐quality methods, and that the seizure types of people included in trials should be classified very carefully, to ensure results are of high quality.
Authors' conclusions
Summary of findings
| Carbamazepine compared with phenytoin for epilepsy | ||||||
| Patient or population: adults and children with new‐onset partial or generalised epilepsy Settings: outpatients Intervention: carbamazepine Comparison: phenytoin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenytoin | Carbamazepine | |||||
| Time to withdrawal of allocated treatment Range of follow‐up (all participants): 1 day to 4403 days | 37 per 100 | 35 per 100 (28 to 44) | HR 1.04 (0.78 to 1.39) | 546 (3 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical advantage for carbamazepine |
| Time to withdrawal of allocated treatment Range of follow‐up (all participants): 1 day to 4064 days | 42 per 100 | 37 per 100 (29 to 47) | HR 1.18 (0.87 to 1.60) | 428 (3 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical |
| Time to withdrawal of allocated treatment Range of follow‐up (all participants): 1 day to 4403 days | 14 per 100 | 30 per 100 (15 to 57) | HR 0.42 (0.18 to 0.96) | 118 (2 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical |
| Proportion of withdrawals due to adverse effects Range of follow‐up (all participants): 1 day to 4403 days | 4 per 100 | 6 per 100 (5 to 7) | RR 1.42 (1.13 to 1.80) | 546 (3 studies) | ⊕⊕⊕⊝ moderate2 | RR < 1 indicates a clinical advantage for carbamazepine |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The assumed risk is calculated as the event rate in the phenytoin treatment group. The corresponding risk in the carbamazepine treatment group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). The corresponding risk is calculated as the assumed risk x the relative risk (RR) of the intervention where RR = (1 ‐ exp(HR x ln(1 ‐ assumed risk)) ) / assumed risk | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Pooled HR for all participants adjusted for seizure type. | ||||||
| Carbamazepine compared with phenytoin for epilepsy | ||||||
| Patient or population: adults and children with new‐onset partial or generalised epilepsy Settings: outpatients Intervention: carbamazepine Comparison: phenytoin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenytoin | Carbamazepine | |||||
| Time to 12‐month remission Range of follow‐up (all participants): 0 days to 4222 days | 55 per 100 | 55 per 100 (46 to 65) | HR 1.01 (0.78 to 1.31) | 551 (3 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical |
| Time to 12‐month remission Range of follow‐up (all participants):0 days to 4222 days | 47 per 100 | 45 per 100 (36 to 55) | HR 0.94 (0.71 to 1.25) | 430 (3 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical |
| Time to 12‐month remission Range of follow‐up (all participants): 7 days to 4163 days | 85 per 100 | 88 per 100 (63 to 99) | HR 1.174 (0.53 to 2.57) | 121 (2 studies) | ⊕⊕⊝⊝ low2,3,4 | HR > 1 indicates a clinical |
| Time to 6‐month remission Range of follow‐up (all participants): 0 days to 4222 days | 63 per 100 | 67 per 100 (59 to 75) | HR 1.11 (0.89 to 1.37) | 551 (3 studies) | ⊕⊕⊕⊝ moderate2,3 | HR >1 indicates a clinical |
| Time to 6‐month remission Range of follow‐up (all participants): 0 days to 4222 days | 56 per 100 | 56 per 100 (47 to 66) | HR 1.02 (0.79 to 1.33) | 430 (3 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical |
| Time to 6‐month remission Range of follow‐up (all participants): 7 days to 4163 days | 93 per 100 | 97 per 100 (91 to 99) | HR 1.30 (0.89 to 1.92) | 121 (2 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The assumed risk is calculated as the event rate in the Phenytoin treatment group The corresponding risk in the carbamazepine treatment group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). The corresponding risk is calculated as the assumed risk x the relative risk (RR) of the intervention where RR = (1 ‐ exp(HR x ln(1 ‐ assumed risk)) ) / assumed risk | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Pooled HR for all participants adjusted for seizure type. | ||||||
| Carbamazepine compared with phenytoin for epilepsy | ||||||
| Patient or population: adults and children with new‐onset partial or generalised epilepsy Settings: outpatients Intervention: carbamazepine Comparison: phenytoin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenytoin | Carbamazepine | |||||
| Time to first seizure Range of follow‐up (all participants): 0 days to 4589 days | 65 per 100 | 71 per 100 (63 to 77) | HR 0.85 (0.70 to 1.04) | 582 (4 studies) | ⊕⊕⊝⊝ low2,3,4 | HR > 1 indicates a clinical |
| Time to first seizure Range of follow‐up (all participants): 0 days to 4589 days | 63 per 100 | 68 per 100 (60 to 77) | HR 0.86 (0.68 to 1.08) | 432 (4 studies) | ⊕⊕⊝⊝ low2,3,4 | HR > 1 indicates a clinical |
| Time to first seizure Range of follow‐up (all participants): 2 days to 4070 days | 69 per 100 | 75 per 100 (61 to 87) | HR 0.84 (0.57 to 1.24) | 150 (3 studies) | ⊕⊕⊝⊝ low2,3,4 | HR > 1 indicates a clinical |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The assumed risk is calculated as the event rate in the Phenytoin treatment group The corresponding risk in the carbamazepine treatment group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). The corresponding risk is calculated as the assumed risk x the relative risk (RR) of the intervention where RR = (1 ‐ exp(HR x ln(1 ‐ assumed risk)) ) / assumed risk | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Pooled HR for all participants adjusted for seizure type. 2Risk of bias unclear for one element of all of the three studies included in the analysis. De Silva 1996 and Heller 1995 are open‐label and it is unclear whether the lack of masking impacted upon the results; and we do not know how allocation was concealed in Mattson 1985. 348 adult participants in Heller 1995 and Ogunrin 2005 may have had their seizure type wrongly classified as generalised onset; sensitivity analyses show misclassification is unlikely to have had an impact on results and conclusions. 4Ogunrin 2005 is a short study (12 weeks) and has a small sample size of 37 compared to the other three studies of duration 3 ‐ 10 years and sample sizes of around 100 to 300 participants (De Silva 1996; Heller 1995; Mattson 1985). Ogunrin 2005 is less precise with wide CIs, and there is evidence that the treatment effect in this study changes over time. | ||||||
Background
This review is an update of a previously published review in the Cochrane Database of Systematic Reviews (see Other published versions of this review).
Description of the condition
Epilepsy is a common neurological condition in which recurrent, unprovoked seizures are caused by abnormal electrical discharges from the brain. Epilepsy is a disorder of many heterogeneous seizure types, with an estimated incidence of 33 to 57 per 100,000 person‐years worldwide (Annegers 1999; Hirtz 2007; MacDonald 2000; Olaffsson 2005; Sander 1996), accounting for approximately 1% of the global burden of disease (Murray 1994). The lifetime risk of epilepsy onset is estimated to be 1300 to 4000 per 100,000 person‐years (Hauser 1993; Juul‐Jenson 1983), and the lifetime prevalence could be as large as 70 million people worldwide (Ngugi 2010). It is believed that with effective drug treatment, up to 70% of individuals with active epilepsy have the potential to become seizure‐free and go into long‐term remission shortly after starting drug therapy (Cockerell 1995; Hauser 1993; Sander 2004), and that around 70% of individuals can achieve seizure freedom using a single antiepileptic drug in monotherapy (Cockerell 1995); current National Institute for Health and Clinical Excellence (NICE) guidelines recommend that both adults and children with epilepsy should be treated by monotherapy wherever possible (NICE 2012). The remaining 30% of individuals experience refractory or drug‐resistant seizures which often require treatment with combinations of antiepileptic drugs, or alternative treatments such as epilepsy surgery (Kwan 2000).
We study two seizure types in this review: generalised onset seizures (generalised tonic‐clonic seizures with or without other generalised seizure types), in which electrical discharges begin in one part of the brain and move throughout the brain; and partial onset seizures, in which the seizure is generated in and affects only one part of the brain (the whole hemisphere of the brain or part of a lobe of the brain).
Description of the intervention
Carbamazepine and phenytoin are among the most commonly used and earliest drugs licensed for the treatment of epileptic seizures; phenytoin has been used as monotherapy for partial seizures and generalised tonic‐clonic seizures for over 50 years (Gruber 1962) and carbamazepine for over 30 years (Shakir 1980). Current NICE guidelines (NICE 2012) for adults and children recommend carbamazepine as a first‐line treatment for partial onset seizures and as a second‐line treatment for generalised tonic‐clonic seizures if first‐line treatments sodium valproate and lamotrigine are deemed unsuitable; however, there is evidence that carbamazepine may exacerbate some other generalised seizure types such as myoclonic and absence seizures (Liporace 1994; Shields 1983; Snead 1985). Phenytoin is no longer considered a first‐line treatment in the USA and most of Europe, due to concerns over adverse events (Wallace 1997; Wilder 1995), but phenytoin is still used as a first‐line drug in low‐ to middle‐income countries (Ogunrin 2005; Pal 1998).
Both carbamazepine and phenytoin have been shown to have teratogenic effects where the risk is estimated to be two to three times that of the general population (Gladstone 1992; Meador 2008; Morrow 2006; Nulman 1997). Carbamazepine is associated particularly with neural tube defects (Matlow 2012) and phenytoin is associated with fetal hydantoin syndrome (Scheinfeld 2003), low folic acid levels and megaloblastic anaemia (Carl 1992). Both carbamazepine and phenytoin are associated with an allergic rash (Tennis 1997) in 5% to 10% of users, which on rare occasions may be life‐threatening, and phenytoin is also associated with long‐term cosmetic changes including gum hyperplasia, acne and coarsening of the facial features (Mattson 1985; Scheinfeld 2003).
How the intervention might work
Antiepileptic drugs suppress seizures by reducing neuronal excitability. Phenytoin and carbamazepine are broad‐spectrum treatments suitable for many seizure types and both have an anticonvulsant mechanism through blocking ion channels, binding with neurotransmitter receptors or through inhibiting the metabolism or reuptake of neurotransmitters (Ragsdale 1991; Willow 1985) and the modulation of gamma‐aminobutyric acid‐A (GABA‐A) receptors (Granger 1995).
Why it is important to do this review
The aim of this review is to summarise efficacy and tolerability data from existing trials comparing carbamazepine and phenytoin when used as monotherapy treatments. The adverse event profiles of the two drugs are well documented (see example references from Description of the intervention), but no consistent differences in efficacy have been found between the two drugs from a number of randomised controlled trials (RCTs) individually (for example: De Silva 1996; Heller 1995; Mattson 1985; Ramsay 1983). Although no clear difference in efficacy has been found from individual studies, the confidence intervals generated by these studies are wide. We cannot exclude important differences in efficacy, which may be shown by synthesising the data of the individual trials.
There are difficulties in undertaking a systematic review of epilepsy monotherapy trials, as the important efficacy outcomes require analysis of time‐to‐event data (for example, time to first seizure after randomisation). Although methods have been developed to synthesise time‐to‐event data using summary information (Parmar 1998; Williamson 2002), the appropriate statistics are not commonly reported in published epilepsy trials (Nolan 2013a). Furthermore, although seizure data have been collected in most epilepsy monotherapy trials, there has been no uniformity in the definition and reporting of outcomes. For example, trials may report time to 12‐month remission but not time to first seizure or vice versa, or some trials may define time to first seizure from the date of randomisation, while others use date of achieving a maintenance dose. Trial investigators have also adopted differing approaches to the analysis, particularly with respect to the censoring of time‐to‐event data. For these reasons, we performed this review using individual participant data (IPD), which helps to overcome these problems. This review is one in a series of Cochrane IPD Reviews investigating pair‐wise monotherapy comparisons. These data have also been included in a network meta‐analysis (Tudur Smith 2007), undertaken following a previous version of this review.
Objectives
To review the time to withdrawal, six‐ and 12‐month remission, and first seizure with carbamazepine compared to phenytoin, used as monotherapy in people with partial onset seizures (simple partial, complex partial, or secondarily generalised tonic‐clonic seizures) or generalised tonic‐clonic seizures, with or without other generalised seizure types.
Methods
Criteria for considering studies for this review
Types of studies
-
Studies must be randomised controlled trials (RCTs) using either an adequate method of allocation concealment (e.g. sealed opaque envelopes) or a quasi‐randomised method of allocation (e.g. allocation by date of birth).
-
Studies must be of parallel design; cross‐over studies are not an appropriate design for measuring the long‐term outcomes of interest in this review (see Types of outcome measures).
-
Studies must include a comparison of carbamazepine monotherapy with phenytoin monotherapy in individuals with epilepsy; cluster‐randomised studies are therefore not an eligible design.
We included studies regardless of blinding method (unblinded, single‐blind or double‐blind).
Types of participants
-
We included trials recruiting children or adults with partial onset seizures (simple partial, complex partial, or secondarily generalised tonic‐clonic seizures) or generalised onset tonic‐clonic seizures (as a primary generalised seizure type), with or without other generalised seizure types (e.g. absence, myoclonic, etc.).
-
We excluded studies that recruited only individuals with other generalised seizure types, without generalised tonic‐clonic seizures (such as studies recruiting only individuals with a diagnosis of absence seizures or juvenile myoclonic epilepsy, etc.) due to differences in first‐line treatment guidelines (NICE 2012).
-
We included individuals who had a new diagnosis of epilepsy or who had experienced a relapse following antiepileptic monotherapy withdrawal only, due to differences in first‐line treatment guidelines for individuals with refractory epilepsy (NICE 2012).
Types of interventions
Carbamazepine versus phenytoin (any doses) as monotherapy.
Types of outcome measures
We present the outcomes investigated in this review. Reporting of these outcomes in the original trial report was not an eligibility requirement for this review:
Primary outcomes
-
Time to withdrawal of allocated treatment (retention time) is the primary outcome. This is a combined outcome, reflecting both efficacy and tolerability, as treatment may be withdrawn due to continued seizures, side effects, non‐compliance or if additional add‐on treatment was initiated (i.e. allocated treatment had failed). This is an outcome to which the participant makes a contribution, and is the primary outcome measure recommended by the Commission on Antiepileptic Drugs of the International League Against Epilepsy (Commission 1998; ILAE 2006).
Secondary outcomes
-
Time to achieve 12‐month remission (seizure‐free period).
-
Time to achieve six‐month remission (seizure‐free period).
-
Time to first seizure post‐randomisation.
-
Adverse events (including those relating to treatment withdrawal)
Search methods for identification of studies
Electronic searches
We conducted searches for the original review in 1999, and subsequently in 2001, 2003, 2005, July 2007, November 2009, November 2011, October 2013, and September 2014. For the latest update we searched the following databases, applying no language restrictions:
-
The Cochrane Epilepsy Group’s Specialized Register (1st November 2016), using the search strategy outlined in Appendix 1.
-
The Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies Online (CRSO, 1st November 2016), using the search strategy outlined in Appendix 2.
-
MEDLINE (Ovid, 1946 to 1st November 2016), using the search strategy outlined in Appendix 3.
-
ClinicalTrials.gov (1st November 2016), using the search terms 'carbamazepine and phenytoin and epilepsy | Studies received on or after 09/16/2014’.
-
World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP, 1st November 2016), using the search terms ’carbamazepine and phenytoin and epilepsy not NCT*’ (new items selected manually).
Previously we also searched SCOPUS (1823 to 16th September 2014), using the search strategy outlined in Appendix 4, as an alternative to Embase, but this is no longer necessary, because randomised and quasi‐randomised controlled trials in Embase are now included in CENTRAL.
Searching other resources
In addition, we handsearched relevant journals, reviewed the reference lists of retrieved studies to search for additional reports of relevant studies, contacted Novartis (manufacturers of carbamazepine), Parke‐Davis (manufacturers of phenytoin), and experts in the field for information on any ongoing studies, and original investigators of relevant trials found.
Data collection and analysis
Selection of studies
Two review authors (SJN and AGM) independently assessed trials for inclusion, resolving any disagreements by discussion.
Data extraction and management
We requested the following individual participant data (IPD) for all trials meeting our inclusion criteria:
Trial methods:
-
method of generation of random list
-
method of concealment of randomisation
-
stratification factors
-
blinding methods
Participant covariates:
-
gender
-
age
-
seizure types
-
time between first seizure and randomisation
-
number of seizures prior to randomisation (with dates)
-
presence of neurological signs
-
electroencephalographic (EEG) results
-
computerised tomography/magnetic resonance imaging (CT/MRI) results
Follow‐up data:
-
treatment allocation
-
date of randomisation
-
dates of follow‐up
-
dates of seizures post‐randomisation or seizure frequency data between follow‐up visits
-
dates of treatment withdrawal and reasons for treatment withdrawal
-
dose
-
dates of dose changes
For each trial for which we did not obtain IPD, we carried out an assessment to see whether any relevant aggregate‐level data had been reported.
In one study (Mattson 1985), seizure data were provided in terms of the number of seizures recorded between each follow‐up visit rather than specific dates of seizures. To enable us to calculate time‐to‐event outcomes, we applied linear interpolation to approximate dates of seizures between follow‐up visits, assuming a uniform seizure rate. For example, if four seizures were recorded between two visits which occurred on 1st March 1990 and 1st May 1990 (an interval of 61 days), then the date of first seizure would be approximately 13th March 1990 (i.e. 61 days divided by number of seizures plus 1 rounded to the next day, i.e. 13 days). This allowed us to compute an estimate of the time to six‐month remission, 12‐month remission, and the time to first seizure.
We calculated time to six‐month and 12‐month remission from the date of randomisation to the date (or estimated date) the individual had first been free of seizures for six or 12 months respectively. If the person had one or more seizures in the titration period, a six‐month or 12‐month seizure‐free period could also occur between the estimated date of the last seizure in the titration period and the estimated date of the first seizure in the maintenance period.
We calculated time to first seizure from the date of randomisation to the date that their first seizure was estimated to have occurred. If seizure data were missing for a particular visit, we censored these outcomes at the previous visit. We also censored these outcomes if the individual died or if follow‐up ceased prior to the occurrence of the event of interest. These methods had been used in the remaining three trials (De Silva 1996; Heller 1995; Ogunrin 2005) for which outcome data (dates of seizures after randomisation) were provided directly.
In one trial (Ogunrin 2005), all participants completed the 12‐week trial duration without withdrawing from the study. For three trials (De Silva 1996; Heller 1995; Mattson 1985) we extracted dates and reason for treatment withdrawal from trial case report forms for the original review. Two review authors (SJN and CT) independently extracted data from all case report forms, resolving disagreements by reconsidering the case report forms at conference. For the remaining trials, data on length of time spent in trial and reason for withdrawal of allocated treatment were provided directly. For the analysis of time to event, we defined an 'event' as either the withdrawal of the allocated treatment due to poor seizure control, or adverse events, or both. We also classified non‐compliance with the treatment regimen or the addition of another antiepileptic drug as 'events'. We censored the outcome if treatment was withdrawn because the individual achieved a period of remission, or if the individual was still on allocated treatment at the end of follow‐up.
Assessment of risk of bias in included studies
Two review authors (SJN and JW) independently assessed all included studies for risks of bias (Higgins 2011), resolving any disagreements by discussion. The domains assessed as being at low, high or unclear risk of bias were random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting, and other potential sources of bias. We took into account all available information for an included study when making risk of bias judgements, including multiple publications of the study and additional information provided from study authors with IPD.
Measures of treatment effect
We measured all outcomes in this review as time‐to‐event outcomes with the hazard ratio (HR). We calculated outcomes from IPD provided where possible or extracted summary statistics from published studies.
Unit of analysis issues
We did not have any unit of analysis issues. The unit of allocation and analysis was the individual participant for all included studies and no studies were of a repeated measure, (longitudinal) nature, or of a cross‐over design.
Dealing with missing data
For each trial where IPD were supplied, we reproduced information from trial results where possible, and performed the following consistency checks:
-
We cross‐checked trial details against any published report of the trial and contacted original trial authors if we found missing data, errors or inconsistencies.
-
We reviewed the chronological randomisation sequence, and checked the balance of participant characteristics, taking account of factors stratified for in the randomisation procedure.
Assessment of heterogeneity
We assessed heterogeneity statistically using the Q test (P value < 0.10 for significance) and the I2 statistic (Higgins 2003) (greater than 50% indicating considerable heterogeneity), output produced using the generic inverse variance approach in Metaview, and visually by inspecting forest plots.
Assessment of reporting biases
Two review authors (SJN and JP) undertook all full quality and risk of bias assessments. In theory, a review using IPD should overcome issues of reporting biases, as unpublished data can be provided and unpublished outcomes calculated. Any selective reporting bias detected could be assessed with the ORBIT classification system (Kirkham 2010).
Data synthesis
We carried out our analysis on an intention‐to‐treat basis (that is, analysing participants in the group to which they were randomised, irrespective of which treatment they actually received). For the time‐to‐event outcomes 'Time to six‐month remission', 'Time to 12‐month remission' and 'Time to first seizure post‐randomisation', participants were therefore not censored if the treatment initially assigned was withdrawn.
For all outcomes, we investigated the relationship between the time‐to‐event and treatment effect of the antiepileptic drugs. We used Cox proportional hazards regression models to obtain study‐specific estimates of log (HR) or treatment effect and associated standard errors in statistical software SAS version 9.2 (Copyright, SAS Institute Inc. SAS and all other SAS Institute Inc. product or service names are registered trademarks or trademarks of SAS Institute Inc., Cary, NC, USA.). The model assumes that the ratio of hazards (risks) between the two treatment groups is constant over time (i.e. hazards are proportional). We tested this proportional hazards assumption of the Cox regression model for each outcome of each study by testing the statistical significance of a time‐varying covariate in the model. We also inspected Kaplan‐Meier plots for overlapping of curves, which can indicate departures from proportional hazards. We evaluated overall estimates of HRs (with 95% confidence intervals (CIs)) using the generic inverse variance method. We expressed results as a hazard ratio (HR) with 95% CIs.
By convention, a HR greater than 1 indicates that an event is more likely to occur earlier with carbamazepine than with phenytoin. Hence, for time to withdrawal of allocated treatment or time to first seizure, a HR greater than 1 indicates a clinical advantage for phenytoin (e.g. HR = 1.2 would suggest a 20% increase in the risk of withdrawal from carbamazepine compared to phenytoin) and for time to six‐month and 12‐month remission a HR greater than 1 indicates a clinical advantage for carbamazepine.
We used GRADE (GRADE 2004) quality assessment criteria in the 'Summary of findings' tables.
Subgroup analysis and investigation of heterogeneity
Due to the strong clinical belief that some antiepileptic drugs are more effective in some seizure types than others (see Description of the intervention and How the intervention might work), we have stratified all analyses by seizure type (partial onset versus generalised onset), according to the classification of main seizure type at baseline. We classified partial seizures (simple or complex) and partial secondarily generalised seizures as partial epilepsy. We classified primarily generalised seizures as generalised epilepsy. To statistically assess an association between treatment and seizure type we conducted a Chi2 test of interaction between treatment and epilepsy type.
If we found significant statistical heterogeneity to be present, we performed meta‐analysis with a random‐effects model in addition to a fixed‐effect model, presenting the results of both models and performing sensitivity analyses to investigate differences in study characteristics.
Sensitivity analysis
Misclassification of seizure type is a recognised problem in epilepsy, whereby some people with generalised seizures have been mistakenly classed as having partial onset seizures, and vice versa. There is clinical evidence that individuals with generalised onset seizures are unlikely to have an 'age of onset' greater than 25 to 30 years (Malafosse 1994). In a previous review, in our series of pair‐wise reviews for monotherapy in epilepsy, misclassification impacted upon the results (Nolan 2013b). Given the overlap with studies contributing to this review and the phenytoin versus sodium valproate review (Nolan 2013b), we suspected that misclassification of seizure type could also be likely in this review, and so we examined the distribution of age at onset for individuals with generalised seizures.
De Silva 1996 was a paediatric study and Mattson 1985 recruited participants with partial seizures only, so there were no participants with new‐onset generalised seizures over the age of 30 in these studies. Twenty‐nine out of 72 individuals (42%) with generalised onset seizures were over the age of 30 in Heller 1995, and 19 out of 29 individuals (66%) with generalised onset seizures were over the age of 30 in Ogunrin 2005. Therefore out of 150 participants from the four studies providing IPD, 48 (32%) may have been wrongly classified as having new‐onset generalised seizures.
We undertook the following two sensitivity analyses to investigate misclassification for each outcome:
-
We reclassified the 48 individuals with generalised seizure types and age at onset greater than 30 into an 'uncertain seizure type' group.
-
We reclassified the 48 individuals with generalised seizures and age of onset greater than 30 as having partial seizures.
Results
Description of studies
Results of the search
We identified 655 records from the databases and search strategies outlined in Electronic searches. We found three further records by handsearching and checking reference lists of included studies. We removed 265 duplicate records and screened 393 records (title and abstract) for inclusion in the review. We excluded 354 records based on title and abstract and assessed 39 full‐text articles for inclusion in the review. We excluded 14 studies (reported in 20 full‐text articles) from the review (see Excluded studies below) and included 12 trials (reported in 18 full‐text articles) in the review (see Included studies below). One study is awaiting classification following translation (Rysz 1994). See Figure 1 for PRISMA study flow diagram (Moher 2009).
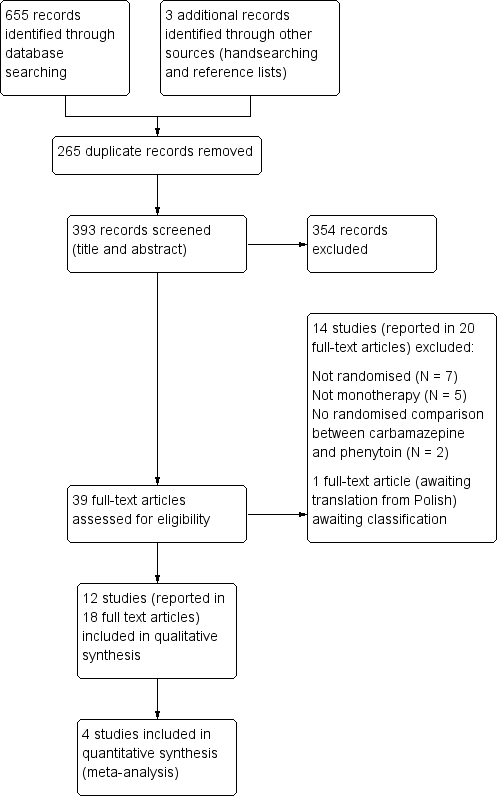
Study flow diagram.
Included studies
We included 12 trials in this review (Callaghan 1985; Cereghino 1974; Czapinski 1997; De Silva 1996; Forsythe 1991; Heller 1995; Mattson 1985; Miura 1993; Ogunrin 2005; Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995). One trial was available in abstract form only (Czapinski 1997).
One trial recruited individuals of all ages (Callaghan 1985), three trials recruited children only (defined as under the age of 16 in De Silva 1996, and under the age of 14 in Forsythe 1991 and Miura 1993); and the remaining eight trials recruited adults only. Four trials defined adults as individuals above the age of 18 (Cereghino 1974; Czapinski 1997; Mattson 1985; Ramsay 1983), one trial classed adults as older than 13 years (Heller 1995), two trials classed adults as older than 14 years (Ogunrin 2005; Ravi Sudhir 1995) and one trials classed adults as older than 15 years (Pulliainen 1994).
Ten trials recruited individuals with partial onset seizures and generalised onset seizures (Callaghan 1985; Cereghino 1974; De Silva 1996; Forsythe 1991; Heller 1995; Miura 1993; Ogunrin 2005; Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995), and two trials recruited individuals with partial onset seizures only (Czapinski 1997; Mattson 1985). Ten trials recruited individuals with new‐onset seizures or previously untreated seizures, or both (Callaghan 1985; Czapinski 1997; De Silva 1996; Forsythe 1991; Heller 1995; Miura 1993; Ogunrin 2005; Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995). One trial recruited institutionalised participants with uncontrolled seizures (Cereghino 1974), and one trial recruited "previously untreated or under treated" individuals (Mattson 1985).
Six trials were conducted in Europe (Callaghan 1985; Czapinski 1997; De Silva 1996; Forsythe 1991; Heller 1995; Pulliainen 1994), three in the USA (Cereghino 1974; Mattson 1985; Ramsay 1983), one in Nigeria (Ogunrin 2005), one in India (Ravi Sudhir 1995), and one in Japan (Miura 1993).
Individual participant data (IPD) could not be supplied for eight trials (Callaghan 1985; Cereghino 1974; Czapinski 1997; Forsythe 1991; Miura 1993; Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995), in which 597 individuals had been randomised to either phenytoin or carbamazepine. None of these eight trials reported the specific time‐to‐event outcomes chosen for this systematic review.
Forsythe 1991 presented times at which the allocated drug was withdrawn and the reason for withdrawal in the trial publication for each individual. Hence, we were able to incorporate this trial into the analysis of 'Time to withdrawal of allocated treatment’. For each participant, 'withdrawal and time of occurrence by month’ was presented and therefore to calculate 'Time to withdrawal of allocated treatment’ we assumed that, for example, if withdrawal occurred during the fifth month, that withdrawal occurred halfway between the fifth and sixth month (i.e. participants spent 167 full days on treatment before withdrawal).
We could not extract sufficient aggregate data from the trial publication in any other trial, and we therefore could not include them in data synthesis. Full details of outcomes considered and a summary of results in each eligible trial for which IPD were not available can be found in Table 1.
| Trial | Outcomes reported | Summary of results |
| 1. Seizure control: excellent (seizure‐free) | 1. PHT (n = 58); CBZ (n = 59) PHT: 39 (67%); CBZ: 22 (37%) | |
| 1. Behaviour measured with rating scale modified from the Ward Behaviour Rating Scale 2. Seizure control 3. Side effects 4. Withdrawals | 1. Behavioural scores were similar on both drugs 2. No difference between CBZ and PHT in terms of seizure control 3. Gastrointestinal and “impaired function” side effects were more common on CBZ than PHT in the first few study days. Side effects of both drugs were minimal in later stages of the study 4. PHT: 21 withdrawals out of 45 participants (47%); CBZ: 27 withdrawals out of 45 participants (60%) | |
| 1. Proportion achieving 24‐month remission at 3 years 2. Proportion excluded after randomisation due to adverse effects or no efficacy | 1. PHT: 59%; CBZ: 62% 2. PHT: 23%; CBZ: 30% | |
| 1. Cognitive assessments 2. Withdrawals from randomised drug | 1. No significant differences between the two treatment groups on any cognitive tests | |
| 1. Proportion of all randomised participants with seizure recurrence (by seizure type) 2. Proportion of participants with optimum plasma levels with seizure recurrence (by seizure type) | PHT (n = 51); CBZ (n = 66) 1. PHT (partial): 10/31 (32%); PHT (generalised): 7/20 (35%); 2. PHT (partial): 4/17 (24%); PHT (generalised): 1/8 (13%); | |
| 1. Cognitive assessments (visual motor speed, co‐ordination, attention and concentration, verbal and visuospatial learning, visual and recognition memory, reasoning, mood, handedness) 2. Harmful side effects | 1. Compared to CBZ, participants on PHT became slower (motor speed of the hand) and their visual memory decreased. There was an equal decrease in negative mood (helplessness, irritability, depression) on PHT and CBZ 2. Three participants taking PHT complained of tiredness, and 1 participant taking CBZ complained of facial skin problems, another tiredness and memory problems | |
| 1. Side effects (major and minor) 3. Laboratory results | 1. Incidence of:
2. Treatment failures among analysed participants: Seizure control (among analysed participants with no major side effects): PHT: 23/27 participants (86%); CBZ: 22/27 participants (82%) 3. Significantly lower mean LDH level at 24 weeks in CBZ participants than PHT participants (P < 0.01). Other laboratory results similar across treatment groups | |
| 1. Cognitive measures (verbal, performance, memory, visuomotor, perceptomotor organisation, visual organisation, dysfunction) | 1. No significant differences between any tests of cognitive function taken before treatment and after 10 ‐ 12 weeks for both treatment groups |
CBZ = carbamazepine, LDH = lactate dehydrogenase, PHT= phenytoin
IPD were provided by trial authors for the four remaining trials which recruited 595 participants, representing 49.9% of individuals from 1192 individuals in all eligible trials (De Silva 1996; Heller 1995; Mattson 1985; Ogunrin 2005). Two trials (Mattson 1985; Ogunrin 2005) directly provided computerised data, and the authors of the other two trials (Heller 1995; De Silva 1996) supplied a combination of both computerised and paper‐based (although mostly computerised) data.
Data were available for the following subject characteristics (percentage of 595 participants with data available): sex (100%), seizure type (100%), drug randomised (99% ‐ data missing for six participants in De Silva 1996), sex (99% ‐ data missing for eight participants), age at randomisation (98% ‐ data missing for nine participants), number of seizures in six months prior to randomisation (98% ‐ data missing for 11 participants), time since first seizure to randomisation (98% ‐ data missing for 10 participants). The results of neurological examinations were provided for 326 participants (55%) from three trials (De Silva 1996; Heller 1995; Ogunrin 2005), electroencephalographic (EEG) results were provided for 316 participants (53%) from one trial (Mattson 1985) and computerised tomography/magnetic resonance imaging (CT/MRI) results were provided for 324 participants (54%) in two trials (Mattson 1985; Ogunrin 2005).
Excluded studies
We excluded five studies which were not RCTs (Bird 1966; Kuzuya 1993; Sabers 1995; Shorvon 1978; Zeng 2010). We excluded seven trials which did not use carbamazepine and phenytoin in monotherapy (Bittencourt 1993; Canadian Study 1998; Hakami 2012; Kosteljanetz 1979; Rajotte 1967; Simonsen 1976; Troupin 1975), and we excluded two trials which did not make a randomised comparison between carbamazepine and phenytoin monotherapy (Kaminow 2003; Shakir 1980). See Characteristics of excluded studies for further details.
Risk of bias in included studies
For further details see Characteristics of included studies, Figure 2 and Figure 3.
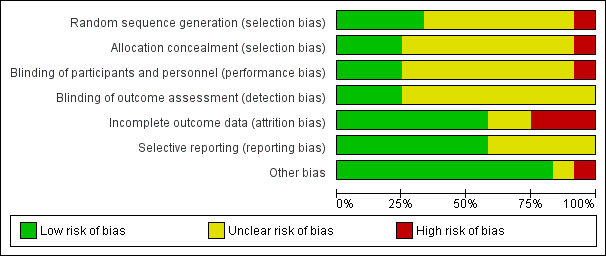
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
(1) Trials for which individual participant data (IPD) were provided
Three trials reported adequate methods of randomisation and allocation concealment; two trials used permuted blocks to generate a random list and concealed allocation by using sealed opaque envelopes (De Silva 1996; Heller 1995), and one trial used number tables to generate a random list and concealed allocation by allocating the randomised drug on a different site from where participants were randomised (Ogunrin 2005). One trial reported only that participants were randomised with stratification for seizure type (Mattson 1985); no further information was provided in the study publication or from the authors about the methods of generating the random list and concealment of allocation.
(2) Trials for which no IPD were available
One trial reported an adequate method of randomisation: random‐number tables (Cereghino 1974), but no details were provided on concealment of allocation. Two trials reported inadequate methods of randomisation and allocation concealment; Forsythe 1991 reported a method of quota allocation and did not report how allocation was concealed, and Callaghan 1985 reported a method of randomisation and allocation concealment based on two Latin squares which seems to take into account the drug preference of participants (the “drug of first preference” was selected from the randomisation list on a sequential basis). The remaining five trials (Czapinski 1997; Miura 1993; Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995) reported that the participants were "randomised" or "randomly allocated" etc., but did not provide information of the method of generation of the random list or of allocation concealment.
Blinding
(1) Trials for which IPD were provided
One trial double‐blinded participants and personnel using an additional blank tablet (Mattson 1985), but it is unclear if the outcome assessor was blinded in this trial. One trial blinded participants and the outcome assessors who performed cognitive testing, but a research assistant recruiting participants and providing counselling on medication adherence was not blinded (Ogunrin 2005). Two trials were unblinded for “practical and ethical reasons” (De Silva 1996; Heller 1995), but it is unclear whether the outcomes of these trials were influenced by the lack of masking.
(2) Trials for which no IPD were available
One trial double‐blinded participants and personnel using an additional blank tablet (Ramsay 1983), but it is unclear if the outcome assessor was blinded in this trial. Two trials single‐blinded the outcome assessor who performed cognitive testing; in one of these trials (Forsythe 1991) the participants and personnel were unblinded, and in the other (Pulliainen 1994), it was unclear if the participants and personnel were blinded or not. The remaining five trials (Callaghan 1985; Cereghino 1974; Czapinski 1997; Miura 1993; Ravi Sudhir 1995) did not provide any information on masking of participants, personnel or outcome assessors.
Incomplete outcome data
(1) Trials for which IPD were provided
In theory, a review using IPD should overcome issues of attrition bias, as unpublished data can be provided, unpublished outcomes calculated and all randomised participants can be analysed by an intention‐to‐treat approach. All four trials (De Silva 1996; Heller 1995; Mattson 1985; Ogunrin 2005) provided IPD for all randomised individuals and reported the extent of follow‐up for each individual. We queried any missing data with the original study authors. From the information provided by the authors, we deemed the small amount of missing data (Included studies) to be missing at random and that they did not have an effect on our analysis.
(2) Trials for which no IPD were available
Three trials reported attrition rates and analysed all randomised participants using an intention‐to‐treat approach (Callaghan 1985; Forsythe 1991; Miura 1993). Two trials reported attrition rates, but it was unclear if all participants were analysed (Cereghino 1974; Czapinski 1997). Three studies excluded between 20% and 35% of participants from the final analysis for “non‐compliance,” loss to follow‐up or uncontrolled seizures, and included only those who completed the analysis. This approach is not intention‐to‐treat, so we deemed these three studies to be at high risk of bias (Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995)
Selective reporting
We requested study protocols in all IPD requests, but protocols were not available for any of the 12 included trials, so we made a judgement of the risk of bias based on the information included in the publications, or from the IPD we received (see Characteristics of included studies for more information).
Trials for which IPD were provided
In theory, a review using IPD should overcome issues of reporting biases, as unpublished data can be provided and unpublished outcomes calculated. We acquired sufficient IPD to calculate the four outcomes ('Time to withdrawal of allocated treatment', 'Time to six‐month remission','Time to 12‐month remission' and 'Time to first seizure’) for three of the four trials (De Silva 1996; Heller 1995; Mattson 1985). The study duration of Ogunrin 2005 was 12 weeks and all randomised participants completed the study without withdrawing, so we could only calculate 'Time to first seizure' for this study.
Trials for which no IPD were available
Seizure outcomes or adverse events, or both, were fully reported in four trials (Callaghan 1985; Cereghino 1974; Miura 1993; Ramsay 1983). Two trials reported cognitive outcomes and adverse events, but no seizure outcomes (Forsythe 1991; Pulliainen 1994), and one trial reported cognitive outcomes only, but no adverse events or seizure outcomes (Ravi Sudhir 1995); however, as no protocols were available for these three trials, we do not know whether seizure outcomes or recording of adverse events, or both, were planned a priori. One trial was in abstract form only and did not provide sufficient information to assess selective reporting bias (Czapinski 1997).
Other potential sources of bias
We detected another source of bias in one of the included studies which has a cross‐over design (Cereghino 1974). Such a design is unlikely to be appropriate for monotherapy treatment, due to carry‐over effects from one treatment period into another (participants were also treated during washout periods with their "regular medication"), and such a design does not allow long‐term outcomes such as the time‐to‐event outcomes of interest to us in this review. For future updates of this review we will exclude studies of a cross‐over design.
Effects of interventions
See: Summary of findings for the main comparison Summary of findings ‐ Time to withdrawal of allocated treatment; Summary of findings 2 Summary of findings ‐ Time to 12‐ and 6‐month remission of seizures; Summary of findings 3 Summary of findings ‐ Time to first seizure after randomisation
A summary of the outcomes reported in trials for which no IPD were available are reported in Table 1. Details regarding the number of individuals (with IPD) contributing to each analysis are given in Table 2 and results are summarised in summary of findings Table for the main comparison for our primary outcome 'Time to withdrawal of allocated treatment', summary of findings Table 2 for the secondary outcomes 'Time to six‐ and 12‐month remission' and summary of findings Table 3 for the secondary outcome 'Time to first seizure'. Survival curve plots (cumulative incidence) are shown in Figure 4; Figure 5; Figure 6; Figure 7; Figure 8; Figure 9; Figure 10 and Figure 11. We produced all cumulative incidence plots in Stata software version 11.2 (Stata 2009), using data from all trials providing IPD combined. We would have liked to adjust for individual trials in survival curve plots but we do not know of any software which allows for this; we hope that such software may have been developed for future updates of this review.

Time to withdrawal of allocated treatment
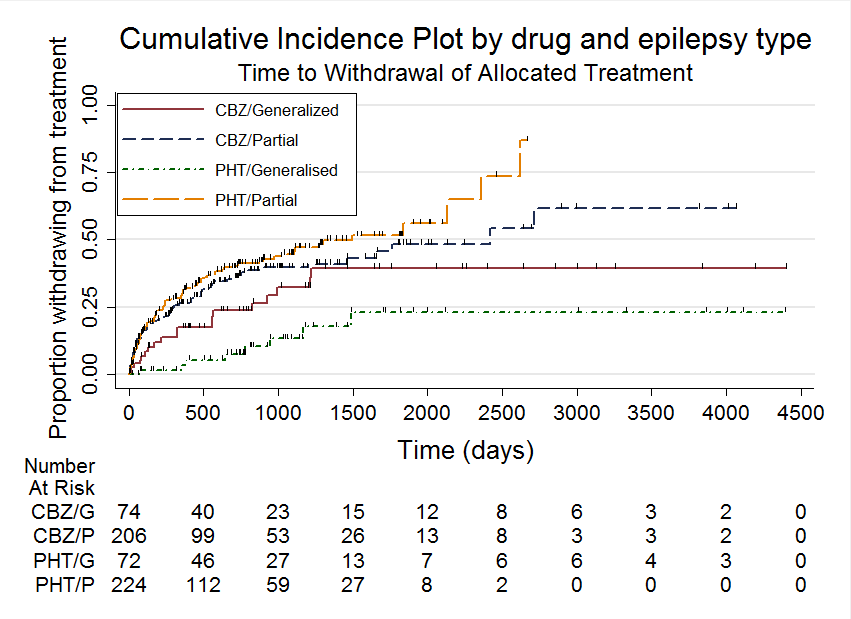
Time to withdrawal of allocated treatment, stratified by epilepsy type
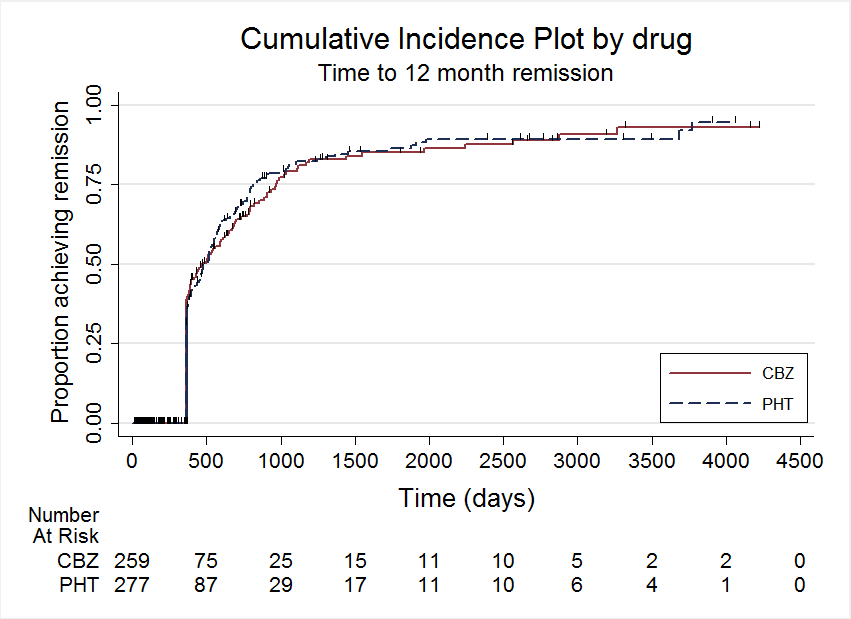
Time to 12 month remission

Time to 12 month remission, stratified by epilepsy type

Time to 6 month remission

Time to 6 month remission, stratified by epilepsy type

Time to first seizure

Time to first seizure, stratified by epilepsy type
| Trial | Number randomised | Time to withdrawal of allocated treatment | Time to 12‐month remission | Time to 6‐month remission | Time to first seizure | ||||||||||
| PHT | CBZ | Total | PHT | CBZ | Total | PHT | CBZ | Total | PHT | CBZ | Total | PHT | CBZ | Total | |
| 54 | 54 | 108 | 53 | 53 | 106 | 54 | 54 | 108 | 54 | 54 | 108 | 54 | 54 | 108 | |
| 63 | 61 | 124 | 61 | 60 | 121 | 63 | 61 | 124 | 63 | 61 | 124 | 63 | 61 | 124 | |
| 165 | 155 | 320 | 165 | 154 | 319 | 165 | 154 | 319 | 165 | 154 | 319 | 162 | 151 | 313 | |
| 20 | 23 | 43 | 20 | 23 | 43 | Information not available | Information not available | Information not available | |||||||
| 18 | 19 | 37 | Information not available | Information not available | Information not available | 18 | 19 | 37 | |||||||
| Total | 320 | 312 | 632 | 299 | 290 | 589 | 282 | 269 | 551 | 282 | 269 | 551 | 297 | 285 | 582 |
CBZ = carbamazepine, PHT= phenytoin
1Individual participant data (IPD) supplied for 114 participants recruited in De Silva 1996; randomised drug not recorded in six participants. Reasons for treatment withdrawal not available for two participants (one randomised to CBZ and one to PHT); these participants are not included in analysis of Time to treatment withdrawal.
2Reasons for treatment withdrawal not available for three participants (one randomised to CBZ and two to PHT) in Heller 1995; these participants are not included in analysis of Time to treatment withdrawal.
3No follow‐up data after randomisation available for one participant randomised to CBZ in Mattson 1985. Data on seizure recurrence not available for six additional participants (three randomised to CBZ and three to PHT); these participants are not included in the analysis of Time to first seizure.
4IPD for Time to treatment withdrawal available in the study publication of Forsythe 1991. Data for other outcomes not available.
5Study duration of Ogunrin 2005 is 12 weeks, so six‐ and 12‐month remission of seizures could not be achieved and cannot therefore be calculated. All randomised participants completed the study without withdrawing from treatment, so time to treatment withdrawal cannot be analysed.
All hazard ratios (HRs) presented below are calculated by generic inverse variance fixed‐effect meta‐analysis unless otherwise stated.
Time to withdrawal of allocated treatment
For this outcome, a HR greater than one indicates a clinical advantage for carbamazepine.
Time to withdrawal of allocated treatment and reason for withdrawal were available for 546 participants from three of the four trials providing IPD (99% of 558 participants from De Silva 1996, Heller 1995 and Mattson 1985 (see Included studies), and 45.8% of the 1192 participants from the 12 included studies). Although two participants withdrew from allocated treatment (one in each group) in De Silva 1996, a reason for withdrawal was not available and could not be determined from the case notes. Similarly in Heller 1995, for one participant taking carbamazepine, the reason for withdrawal was not available and could not be determined from case notes. Also in Heller 1995, two participants (both on phenytoin) had reasons for treatment withdrawal recorded but no date of withdrawal. We have not included the five participants with missing reasons for withdrawal or withdrawal dates from the two trials in analysis of time to withdrawal of allocated treatment. Sufficient IPD were available in the published report for a further 43 participants from one trial (Forsythe 1991). Therefore, 589 participants from four trials were available for the analysis of this outcome (see Table 2).
350 participants prematurely withdrew from treatment (59%): 172 out of 290 participants randomised to carbamazepine (59%) and 178 out of 299 participants randomised to phenytoin (60%). See Table 3 for reasons for premature termination of allocated treatment (by treatment) and how we classified these withdrawals in analysis. We deemed 210 participants (36%) to have withdrawn for reasons related to the study drug, 103 (36%) on carbamazepine and 107 (36%) on phenytoin, and we classified these withdrawals as 'events' in analysis. We classified the other 140 withdrawals as not related to the study drug and censored these participants in analysis, in addition to those who completed the study without withdrawing.
| Reason for early termination | Classification | Heller 19952,3 | Total1 | ||||||||
| CBZ n = 53 | PHT n = 53 | CBZ n = 23 | PHT n = 20 | CBZ n = 60 | PHT n = 63 | CBZ n = 154 | PHT n = 165 | CBZ n = 290 | PHT n = 299 | ||
| Adverse events | Event | 3 | 2 | 4 | 1 | 8 | 1 | 11 | 8 | 26 | 12 |
| Seizure recurrence | Event | 12 | 10 | 2 | 1 | 5 | 8 | 3 | 6 | 22 | 25 |
| Both seizure recurrence and adverse events | Event | 6 | 5 | 0 | 0 | 4 | 2 | 31 | 33 | 31 | 40 |
| Non‐compliance/participant choice | Event | 0 | 0 | 3 | 4 | 0 | 0 | 11 | 26 | 14 | 30 |
| Participant went into remission | Censored | 18 | 24 | 0 | 0 | 6 | 14 | 0 | 0 | 24 | 38 |
| Lost to follow‐up | Censored | 0 | 0 | 0 | 0 | 0 | 0 | 26 | 19 | 26 | 19 |
| Death4 | Censored | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 5 | 4 | 5 |
| Other5 | Censored | 0 | 0 | 0 | 0 | 0 | 0 | 16 | 11 | 16 | 11 |
| Completed the study (did not withdraw) | Censored | 14 | 12 | 14 | 14 | 37 | 38 | 53 | 57 | 118 | 121 |
n = number of individuals contributing to the outcome 'Time to withdrawal of allocated treatment’
1All participants in Ogunrin 2005 completed the study without withdrawing, so this study did not contribute to 'Time to withdrawal of allocated treatment'.
2One participant for Heller 1995 (CBZ) and two for De Silva 1996 (one PHT and one CBZ) have missing reasons for treatment withdrawal.
3Two participants from Heller 1995 (both PHT) had missing withdrawal times and did not contribute to analysis, but reasons for withdrawal are given.
4Death due to reasons not related to the study drug.
5Other reasons from Mattson 1985: participants developed other medical disorders including neurological and psychiatric disorders.
The overall pooled HR (for 589 participants in four trials) was 0.99 (95% confidence interval (CI) 0.75 to 1.30, P = 0.92), indicating no advantage for either drug. There was no evidence of statistical heterogeneity between trials (Chi2 = 2.73, degrees of freedom (df) = 3, P = 0.44, I2= 0%, see Analysis 1.1). The proportional hazards assumption of the Cox model was satisfied for all trials.
Phenytoin is no longer considered as a first‐line treatment in much of the USA and Europe, due to concerns around adverse events (see Description of the intervention). Table 3 shows that 26 out of 172 participants (15%) who withdrew from carbamazepine and 12 out of 178 participants (7%) who withdrew from phenytoin withdrew from the study due to adverse events; in other words, statistically significantly more participants withdrew from carbamazepine than from phenytoin due to adverse events in four studies conducted in the USA and Europe ( P = 0.012).
Withdrawal data for 43 participants extracted from Forsythe 1991 did not distinguish between epilepsy type (partial onset or generalised onset) and therefore could not be included in the meta‐analysis stratified by epilepsy type.
The overall pooled HR (adjusted by epilepsy type for 546 participants in three trials) was 1.04 (95% CI 0.78 to 1.39, P = 0.79), again indicating no clear advantage for either drug, and there was no evidence of statistical heterogeneity between trials (Chi2 = 5.86, df = 4, P = 0.21, I2= 32%, see Analysis 1.2). This result is similar to the unadjusted pooled HR (Analysis 1.1) and conclusions remain unchanged following the exclusion of 43 individuals (Forsythe 1991) in the stratified analysis.
For participants with partial onset seizures (n = 428, three trials), the pooled HR was 1.18 (95% CI 0.87 to 1.60, P = 0.30), indicating an advantage for carbamazepine, which is not statistically significant. For participants with generalised onset seizures (n = 118, two trials), the pooled HR was 0.42 (95% CI 0.18 to 0.96, P = 0.04), indicating a statistically significant advantage for phenytoin. We found a statistically significant interaction between seizure type (generalised versus partial onset) and treatment effect (Chi2 = 5.18, df = 1, P = 0.02, I2= 80.7%).
We conducted a sensitivity analysis to examine the impact of potential seizure misclassification on results for the 29 participants aged 30 years or older with new‐onset generalised seizures in Heller 1995 (see Sensitivity analysis). Following reclassification, for the remaining participants with generalised onset seizures (89) the pooled HR was 0.51 (95% CI 0.21 to 1.24, P = 0.14), which still indicates an advantage for phenytoin, but this advantage is no longer statistically significant. Reclassifying these 29 participants as having new‐onset partial seizures, the pooled HR for 517 participants is 1.11 (95% CI 0.82 to 1.50, P = 0.50), indicating a slight advantage for carbamazepine, which is not statistically significant. Following reclassification, the interaction between seizure type (generalised versus partial onset) and treatment effect is no longer statistically significant (Chi2 = 2.16, df = 1, P = 0.10, I2= 62.3%). Results were similar when the 29 participants were reclassified as uncertain seizure type (see Table 4).
| Analysis | Time to withdrawal | Time to six‐month remission | Time to 12‐month remission* | Time to first seizure |
| Original analysis | P: 1.18 (0.87, 1.60) G: 0.42 (0.18, 0.96) O: 1.04 (0.78, 1.39) | P: 1.02 (0.79, 1.33) G: 1.30 (0.89, 1.92) O: 1.11 (0.89, 1.37) | P: 0.94 (0.71, 1.25) G: 1.17 (0.53, 2.57) O: 1.01 (0.78, 1.31) | P: 0.86 (0.68, 1.08) G: 0.84 (0.57, 1.24) O: 0.85 (0.70, 1.04) |
| Test for interaction | Chi2 = 5.18; df = 1 P = 0.02; I2 = 80.7% | Chi2 = 1.03; df = 1 P = 0.31; I2 = 3.4% | Chi2 = 0.25; df = 1 P = 0.62; I2 = 0% | Chi2 = 0.01; df = 1 P = 0.93; I2 = 0% |
| Generalised and age at onset > 30 (classified as uncertain epilepsy type) | P: 1.18 (0.87, 1.60) G: 0.51 (0.21, 1.24) U: 0.19 (0.02, 2.14) O: 1.05 (0.79, 1.40) | P: 1.02 (0.79, 1.33) G: 1.69 (1.07, 2.27) U: 0.84 (0.35, 1.98) O: 1.13 (0.91, 1.41) | P: 0.94 (0.71, 1.25) G: 1.44 (0.90, 2.31) U: 0.52 (0.20, 1.34) O: 1.01 (0.80, 1.28) | P: 0.86 (0.68, 1.08) G: 0.91 (0.57, 1.46) U: 0.97 (0.43, 2.18) O: 0.88 (0.72, 1.07) |
| Test for interaction | Chi2 = 4.99; df = 2 P = 0.08; I2 = 59.9% | Chi2 = 4.01; df = 2 P = 0.13; I2 = 50.2% | Chi2 = 4.32; df = 2 P = 0.12; I2 = 53.7% | Chi2 = 0.12; df = 2 P = 0.94; I2 = 0% |
| Generalised and age at onset > 30 (reclassified as partial epilepsy) | P: 1.11 (0.82, 1.50) G: 0.51 (0.21, 1.24) O: 1.02 (0.77, 1.36) | P: 1.02 (0.80, 1.31) G: 1.69 (1.07, 2.27) O: 1.15 (0.92, 1.42) | P: 0.91 (0.69, 1.19) G: 1.44 (0.90, 2.31) O: 1.02 (0.81, 1.29) | P: 0.86 (0.69, 1.08) G: 0.91 (0.57, 1.46) O: 0.87 (0.71, 1.07) |
| Test for interaction | Chi2 = 2.65; df = 1 P = 0.10; I2 = 62.3% | Chi2 = 3.63; df = 1 P = 0.06; I2 = 72.5% | Chi2 = 2.79; df = 1 P = 0.09; I2 = 64.2% | Chi2 = 0.04; df = 1 P = 0.83; I2 = 0% |
df = degrees of freedom of Chi² distribution, G = generalised epilepsy, O = overall (all participants), P = partial epilepsy, U = uncertain seizure type
Results are presented as pooled hazard ratio (HR) (95% confidence interval (CI)) with fixed‐effect.
P < 0.05 is classified as statistically significant.
29 participants from Heller 1995 reclassified to partial epilepsy or uncertain epilepsy type for outcomes 'Time to treatment withdrawal', 'Time to 12‐month remission' and 'Time to 6‐month remission.'
48 participants from Heller 1995 and Ogunrin 2005 reclassified to partial epilepsy or uncertain epilepsy type for outcome 'Time to first seizure.'
See Analysis 1.2; Analysis 1.4; Analysis 1.6; and Analysis 1.8 for original analyses of 'Time to treatment withdrawal', 'Time to 12‐month remission', 'Time to 6‐month remission' and 'Time to first seizure', all stratified by epilepsy respectively.
* Original analysis calculated with random‐effects model due to substantial heterogeneity (see Analysis 1.4). Sensitivity analyses calculated with fixed‐effect model as no heterogeneity is present following reclassification of 29 participants in Heller 1995.
Given that subgroup sizes are unbalanced (118 with generalised seizures and 428 with partial seizures (as classified by the studies)) and that results may be confounded by misclassification of seizure type in up to 29 participants, we cannot draw any firm conclusions about an association between treatment and seizure type (i.e. that participants with partial seizures are less likely to withdraw from phenytoin and participants with generalised seizures are less likely to withdraw from carbamazepine). We require more evidence, particularly from individuals with correctly classified generalised onset seizures to inform this analysis.
We judged evidence for 'Time to withdrawal of allocated treatment' to be of moderate quality according to GRADE criteria, due to the potential impact of misclassification of seizure type on the results (summary of findings Table for the main comparison).
Time to achieve 12‐month remission
For this outcome, a HR greater than one indicates a clinical advantage for phenytoin.
Data for 551 participants (99% of 558 randomised participants from De Silva 1996, Heller 1995 and Mattson 1985 (see Included studies) and 45.8% of the 1192 participants from the 12 included studies) from three out of four trials providing IPD were available for the analysis of this outcome. Individuals were followed up for a maximum of 12 weeks in Ogunrin 2005, so it could not contribute to this outcome.
Two hundred and eighty‐nine out of 551 participants (52%) achieved 12‐month remission: 155 out of 282 (55%) on phenytoin and 134 out of 269 (50%) on carbamazepine. The overall pooled HR (for 551 participants, three trials) was 0.99 (95% CI 0.79 to 1.25, P = 0.95), suggesting no advantage for either drug. There was no evidence of statistical heterogeneity between trials (Chi2 = 1.49, df = 2, P = 0.47, I2= 0%, see Analysis 1.3).
Substantial statistical heterogeneity was present between the trials for generalised onset seizures (I2= 73%, P = 0.06), so we calculated HRs using the random‐effects model. For participants with partial onset seizures (n = 430, three trials), the pooled HR was 0.94 (95% CI 0.71 to 1.25, P = 0.68, random‐effects), indicating no clear advantage for either drug. For participants with generalised onset seizures (n = 121, two trials), the pooled HR was 1.17 (95% CI 0.53 to 2.57, P = 0.70, random‐effects), indicating an advantage for phenytoin, which is not statistically significant. Overall, the pooled HR (adjusted for seizure type for 551 participants, three trials) was 1.01 (95% CI 0.78 to 1.31, P = 0.93, random‐effects), suggesting no clear advantage for either drug (see Analysis 1.4). The test for interaction between seizure type (generalised versus partial onset) and treatment effect was not significant (Chi2 = 0.25, df = 1, P = 0.62, I2= 0%).
Following reclassification of the 29 participants aged 30 years or older with new‐onset generalised seizures in Heller 1995 (see Sensitivity analysis), the pooled HR for 92 participants with generalised onset seizures was 1.44 (95% CI 0.90 to 2.31, P = 0.32, I2= 0%, calculated with fixed‐effect model), showing that all of the heterogeneity in Analysis 1.4 is explained by misclassification of participants with generalised onset seizures. The pooled estimate for individuals with partial onset seizures and the overall estimate for all participants stratified by seizure type were similar to the original analysis, and our conclusions remain unchanged (see Table 4).
In De Silva 1996, there is an indication that the proportional hazards assumption may be violated (see Data synthesis); the P value of time‐varying covariate is 0.051 and visual inspection of the cumulative incidence plot (Figure 12) shows crossing of the curves at around 2500 days. In other words, up to 2500 days, participants on phenytoin seem to be achieving 12‐month remission quicker than those on carbamazepine, but this changes after 2500 days; however, participant numbers are small (15 participants at risk out of 108 randomised), so small changes may be magnified at this time.
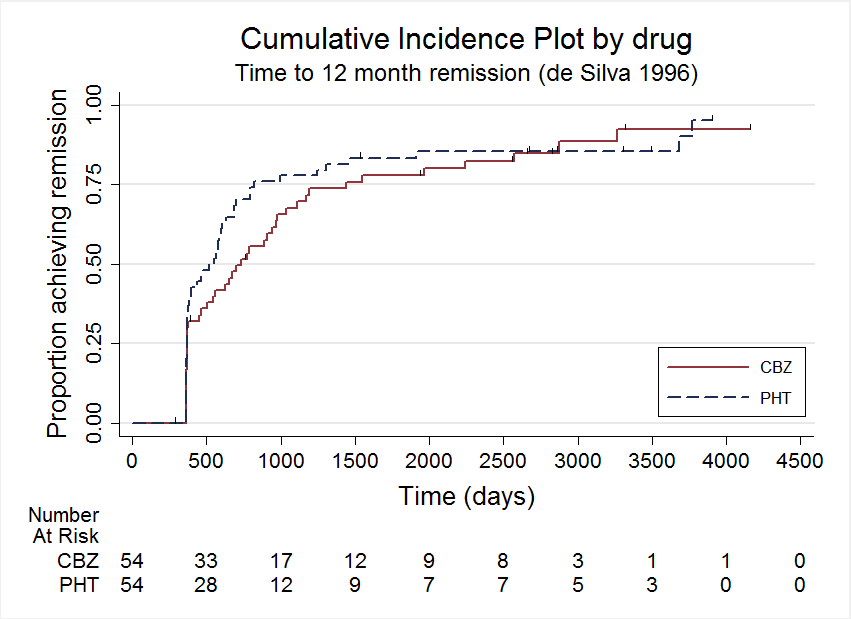
Time to 12 month remission, deSilva 1996
As a sensitivity analysis, we fitted a piecewise Cox regression model to investigate any change in treatment effect over time, assuming proportional hazards within each interval. From the visual inspection of Figure 12, the follow‐up period of De Silva 1996 is split into two intervals; 0 to 2500 days and over 2500 days (maximum follow‐up was 4163 days). We can estimate separate HRs for each interval as follows:
-
For the interval 0 to 2500 days (88 events in 108 participants at risk) the HR is 1.29 (95% CI 0.84 to 1.96, P = 0.23), suggesting an advantage for phenytoin, which is not statistically significant.
-
For the interval over 2500 days (five events in 15 participants at risk) the HR is 0.63 (95% CI 0.25 to 1.57, P = 0.32), suggesting an advantage for carbamazepine, which is not statistically significant.
These results suggest some indication of a change in treatment effect over time, with an advantage for phenytoin earlier on in the study, changing to an advantage for carbamazepine later in the study. However, CIs of estimates are wide, particularly for the HR after 2500 days due to small numbers of events and participants at risk, so we do not have statistically significant evidence to support the hypothesis of a change in treatment effect over time for De Silva 1996, and conclude that the change of direction in effect at around 2500 days is likely to be due to small participant numbers after this time.
We judged the evidence for 'Time to 12‐month remission' to be of low to moderate quality according to GRADE criteria, due to the potential impact of misclassification of seizure type on the results and heterogeneity between studies (summary of findings Table 2).
Time to achieve six‐month remission
For this outcome, a HR greater than one indicates a clinical advantage for phenytoin.
Data for 551 participants (99% of 558 randomised participants from De Silva 1996, Heller 1995 and Mattson 1985 (see Included studies) and 45.8% of the 1192 participants from the 12 included studies) from three out of four trials providing IPD were available for the analysis of this outcome. Individuals were followed up for a maximum of 12 weeks in Ogunrin 2005, so it could not contribute to this outcome.
Three hundred and thirty‐eight out of 551 participants (61%) achieved six‐month remission: 179 out of 282 (63%) on phenytoin and 159 out of 269 (59%) on carbamazepine. The overall pooled HR (for 551 participants, three trials) was 1.08 (95% CI 0.87 to 1.34, P = 0.46), suggesting no clear advantage for either drug. There was no evidence of statistical heterogeneity between trials (Chi2 = 0.35, df = 2, P = 0.84, I2= 0%, see Analysis 1.5).
For the participants with partial onset seizures (n = 430, three trials), the pooled HR was 1.02 (95% CI 0.79 to 1.33, P = 0.85), indicating no clear advantage for either drug. For the participants with generalised onset seizures (n = 121, two trials), the pooled HR was 1.30 (95% CI 0.89 to 1.92, P = 0.18), indicating an advantage for phenytoin, which is not statistically significant. Less statistical heterogeneity was present between the trials for generalised onset seizures compared to the analysis of time to 12‐month remission (I2 = 39%, P = 0.20), so we present HRs from a fixed‐effect model. Overall, the pooled HR (adjusted for epilepsy type for 551 participants, three trials) was 1.11 (95% CI 0.89 to 1.37, P = 0.36, three trials), suggesting a slight advantage for phenytoin, which is not statistically significant. The test for interaction between seizure type (generalised versus partial onset) and treatment effect was not significant (Chi2 = 1.03, df = 1, P = 0.31, I2 = 3.4%, see Analysis 1.6).
Following reclassification of the 29 participants aged 30 years or older with new‐onset generalised seizures in Heller 1995 (see Sensitivity analysis), the pooled HR for 92 participants with generalised onset seizures was 1.69 (95% CI 1.07 to 2.27, P = 0.02, I2 = 0%), showing a larger and statistically significant advantage for phenytoin. Reclassifying these 29 participants as having new‐onset partial seizures, the pooled HR for 517 participants is 1.02 (95% CI 0.80 to 1.31), similar to Analysis 1.6, indicating no clear advantage for either drug. Following reclassification, the test for interaction between seizure type (generalised versus partial onset) and treatment effect was borderline statistically significant (Chi2 = 3.63, df = 1, P = 0.06, I2 = 72.5%). Results were similar when the 29 participants were reclassified as uncertain seizure type (see Table 4).
However, as in the analysis of our primary outcome 'Time to withdrawal of allocated treatment', as subgroup sizes are unbalanced (118 with generalised seizures and 428 with partial seizures, as classified by the studies) and as results may be confounded by misclassification of seizure type in up to 29 participants, we cannot draw any firm conclusions about an association between treatment and seizure type (i.e. that participants achieve six‐month remission quicker on phenytoin than on carbamazepine). Again, we require more evidence, particularly from individuals with correctly classified generalised onset seizures to inform this analysis.
In De Silva 1996, there is an indication that the proportional hazards assumption may be violated (see Data synthesis); the P value of time‐varying covariate is 0.066 and visual inspection of the cumulative incidence plot (Figure 13) shows crossing of the curves at several points at around 1000 days, 1750 days and 3500 days, suggesting several changes in treatment effect over time. As in the sensitivity analysis of De Silva 1996 in 'Time to 12‐month remission', after 1000 days participant numbers are small (18 participants at risk out of 108 randomised), so small changes may be magnified in the later stages of study follow‐up.

Time to 6 month remission, deSilva 1996
As a sensitivity analysis, we fitted a piecewise Cox regression model to investigate any change in treatment effect over time, assuming proportional hazards within each interval. From the visual inspection of Figure 13, the follow‐up period of De Silva 1996 is split into three intervals; 0 to 1000 days, 1000 to 1750 days, and over 1750 days (maximum follow‐up is 4163 days). We did not consider an interval of 3500 days to the end of the study, due to very small participant numbers at this time (three participants at risk). We can estimate separate HRs for each interval as follows:
-
For the interval 0 to 1000 days (87 events in 108 participants at risk) the HR is 1.18 (95% CI 0.77 to 1.80, P = 0.44), suggesting an advantage for phenytoin, which is not statistically significant.
-
For the interval 1000 to 1750 days (three events in 18 participants at risk) the HR is 1.26 (95% CI 0.37 to 4.18, P = 0.71), again suggesting an advantage for phenytoin, which is not statistically significant.
-
For intervals over 1750 days (five events in 14 participants at risk) the HR is 0.76 (95% CI 0.41 to 1.39), suggesting an advantage for carbamazepine, which is not statistically significant.
As above, these results suggest some indication of a change in treatment effect over time, with an advantage for phenytoin earlier on in the study, changing to an advantage for carbamazepine later in the study. However, CIs of estimates are again wide, due to small participant numbers in the later two intervals, so we do not have statistically significant evidence to support the hypothesis of a change in treatment effect over time for De Silva 1996, and conclude that the apparent changes of direction in effect at later stages of the study are likely to be due to small participant numbers.
We judged evidence for 'Time to six‐month remission' to be of moderate quality according to GRADE criteria, due to the potential impact of misclassification of seizure type on the results (summary of findings Table 2).
Time to first seizure post‐randomisation
For this outcome, a HR greater than one indicates a clinical advantage for carbamazepine.
Data for 582 participants (99% of 558 randomised participants from De Silva 1996, Heller 1995 and Mattson 1985 (see Included studies), 100% from Ogunrin 2005, and 49% of the 1192 participants from the 12 included studies) from all four trials providing IPD were available for the analysis of this outcome.
Three hundred and eighty‐three out of 582 participants (66%) experienced a recurrence of seizures: 192 out of 297 (64%) on phenytoin and 191 out of 285 on carbamazepine (67%). The overall pooled HR (for 582 participants, four trials) was 0.88 (95% CI 0.72 to 1.08, P = 0.21), suggesting a slight advantage for phenytoin, which is not statistically significant. There was little evidence of statistical heterogeneity between trials (Chi2 = 4.53, df = 3, P = 0.21, I2 = 34%, see Analysis 1.7).
For the participants with partial onset seizures (n = 432, four trials), the pooled HR was 0.86 (95% CI 0.68 to 1.08, P = 0.20), indicating a slight advantage for phenytoin, which is not statistically significant. For the participants with generalised onset seizures (n = 150, three trials), the pooled HR was 0.84 (95% CI 0.57 to 1.24, P = 0.38), again indicating a slight advantage for phenytoin, which is not statistically significant. Again, there was some statistical heterogeneity between trials for generalised onset seizures (I2 = 45%, P = 0.16). Overall, the pooled HR (adjusted for epilepsy type for 582 participants, four trials) was 0.85 (95% CI 0.70 to 1.04, P = 0.38), suggesting a slight advantage for phenytoin, which is not statistically significant. The test for interaction between seizure type (generalised versus partial onset) and treatment effect was not significant (Chi2 = 0.01, df = 1, P = 0.93, I2 = 0%, see Analysis 1.8).
Following reclassification of the 48 participants aged 30 years or older with new‐onset generalised seizures in Heller 1995 and Ogunrin 2005 (see Sensitivity analysis), results were very similar and conclusions were unchanged (see Table 4). Unlike in analysis of 'Time to 12‐month remission', heterogeneity for participants with generalised onset seizures in Analysis 1.8 is barely reduced following reclassification of seizure type (I2 is reduced from 45% to 42% following reclassification).
Following visual inspection of the forest plot in Analysis 1.8 (generalised epilepsy type), there is a difference in the direction of effects of the three studies, with De Silva 1996 and Ogunrin 2005 showing an advantage for phenytoin, which is not statistically significant, and Heller 1995 showing a slight advantage for carbamazepine, which is not statistically significant).
From correspondence with the study authors, we know that De Silva 1996 and Heller 1995 were conducted under the same protocol and therefore trial characteristics should be homogeneous; the only difference between the two studies is within the age groups recruited (De Silva 1996 recruited children only and Heller 1995 recruited adults only). We therefore performed a further subgroup analysis by adult versus paediatric studies (Ogunrin 2005 also recruited adults only). For 101 adults with generalised onset seizures, the pooled HR was 1.02 (95% CI 0.62 to 1.68, P = 0.94), indicating no clear advantage for either drug, and for 49 children with generalised onset seizures in De Silva 1996 the HR was 0.63 (95% CI 0.34 to 1.16, P = 0.14), indicating an advantage for phenytoin, which is not statistically significant.
The test for interaction between age groups recruited (adults versus children) and treatment effect was not significant (Chi2 = 1.45, df = 1, P = 0.23, I2 = 30.9%). However, participant numbers with generalised onset seizures are quite limited in this review, so we may not have had the power to detect a difference between age groups.
In Ogunrin 2005 there is an indication that the proportional hazards assumption may be violated (see Data synthesis); the P value of time‐varying covariate is 0.02 and visual inspection of the cumulative incidence plot (Figure 14) shows clear crossing of the curves at around 10 days. In other words, up to 10 days, more participants seem to be having seizure recurrence on phenytoin, but this changes to those on carbamazepine after 10 days.
Time to first seizure, Ogunrin 2005
As a sensitivity analysis, we fitted a piecewise Cox regression model to investigate any change in treatment effect over time, assuming proportional hazards within each interval. From the visual inspection of Figure 14, the follow‐up period of Ogunrin 2005 is split into two intervals; 0 to 10 days and over 10 days (maximum follow‐up is 84 days). We can estimate separate HRs for each interval as follows:
-
For the interval 0 to 10 days (13 events in 37 participants at risk) the HR is 1.49 (95% CI 0.45 to 4.88, P = 0.51), suggesting an advantage for carbamazepine, which is not statistically significant.
-
For intervals over 10 days (eight events in 24 participants at risk) the HR is 32 (95% CI 0.11 to 0.91, P = 0.03), suggesting a large statistically significant advantage for phenytoin. Visual inspection of Figure 14 also shows a clear advantage for phenytoin after 10 days.
These results suggest some indication of a change in treatment effect over time, with a slight early advantage for carbamazepine, changing to a large statistically significant advantage for phenytoin later in the study, and support the hypothesis of a change in treatment effect over time for Ogunrin 2005. Ogunrin 2005 is by far the shortest of the studies for which we have IPD (maximum follow‐up was 84 days in Ogunrin 2005 compared to maximum follow‐up of 3995 days in Heller 1995, 4589 days in De Silva 1996 and 1838 days in Mattson 1985), and we did not find statistically significant evidence of a difference between carbamazepine and phenytoin for 'Time to first seizure after randomisation' in any of the three studies with a longer duration (see Analysis 1.7). The apparent large advantage for phenytoin from 10 to 84 days in Ogunrin 2005, may therefore have reduced in size or even changed direction to favour carbamazepine if this study had continued for a longer duration.
We judged evidence for 'Time to first seizure after randomisation' to be of low to moderate quality according to GRADE criteria, due to the potential impact of misclassification of seizure type on the results and imprecision of the effect sizes (summary of findings Table 3).
Adverse events
We extracted all reported information related to adverse events from the study publications. Miura 1993 and Ravi Sudhir 1995 did not report any information on adverse events and we are uncertain without access to protocols if these data were collected (see Selective reporting (reporting bias)). See Table 5 for details of all adverse event data provided in the other 10 studies included in this review. In summary, the adverse events reported by two or more studies in this review are as follows:
| Trial | Adverse event data1 | Summary of reported results | |
| Carbamazepine (CBZ) | Phenytoin (PHT) | ||
| All adverse events according to drug (note: no participants withdrew due to adverse events) | CBZ (n = 59): drowsiness (n = 2), rash (n = 3) | PHT (n = 58): gum hypertrophy (n = 2), rash (n = 2), ataxia (n = 2) | |
| Most frequently observed side effects | Gastrointestinal side effects and “impaired function” (general malaise). Frequency not clearly stated | Gastrointestinal side effects and “impaired function” (general malaise). Frequency not clearly stated | |
| “Exclusions” due to adverse events or no efficacy” | Proportion “excluded”: CBZ: 30% (out of 30 randomised to CBZ) | Proportion “excluded”: PHT: 23.3% (out of 30 randomised to PHT) | |
| “Unacceptable” adverse events leading to drug withdrawal5 | CBZ (n = 54): drowsiness (n = 1), blood dyscrasia (n = 1) | PHT (n = 54): drowsiness (n = 2), skin rash (n = 1), blood dyscrasia (n = 1), hirsutism (n = 1) | |
| Withdrawal due to adverse events (no other adverse event data reported) | 4 participants out of 23 randomised to CBZ withdrew for the following reasons (some withdrew for more than adverse event): slowing of mental function, headache, anorexia, nausea, abdominal pain, fatigue and drowsiness2 | 1 participant out of 20 randomised to PHT withdrew from the study due to depression and anorexia | |
| “Unacceptable” adverse events leading to drug withdrawal5 | CBZ (n = 61): drowsiness (n = 3), rash (n = 2), headache (n = 1), depression (n = 1) | PHT (n = 63): myalgia (n = 1), irritability (n = 1) | |
| Narrative report of ‘Adverse effects’ and ‘Serious side effects’ | CBZ (n = 155): motor disturbance (ataxia, incoordination, nystagmus, tremor: 33%); dysmorphic and idiosyncratic side effects (gum hypertrophy, hirsutism, acne and rash: 14%); gastrointestinal problems (27%); decreased libido or impotence (13%); No serious side effects | PHT (n = 165); motor disturbance (ataxia, incoordination, nystagmus, tremor: 28%); dysmorphic and idiosyncratic side effects (gum hypertrophy, hirsutism, acne and rash: 22 %); gastrointestinal problems (24%); decreased libido or impotence (11%) 1 serious side effect – 1 participant has confirmed lymphoma, rash improved rapidly following discontinuation of PHT | |
| No adverse events reported | N/A | N/A | |
| Participant reported symptomatic complaints (provided as IPD) | CBZ (n = 19): memory impairment (n = 9) psychomotor retardation (n = 1) inattention (n = 1) transient rash (n = 1) CBZ‐induced cough (n = 1) | PHT (n = 18): memory impairment (n = 7) psychomotor retardation (n = 1) inattention (n = 2) transient rash (n = 1) | |
| Participant‐reported adverse events | 1 participant on CBZ complained of facial skin problems; 1 participant on CBZ complained of tiredness and memory problems | 3 participants on PHT complained of tiredness | |
| Major and minor side effects | CBZ (n = 35): Major side effects: rash (n = 1), pruritus (n = 1), impotence (n = 2), dizziness (n = 1), headaches (n = 1), impaired cognition (n = 1), elevated liver enzymes (n = 1) Mild side effects: nausea (33%), headaches (24%), cognitive impairment (33%), nystagmus (52%), sedation (33%), fine tremor (20%) | PHT (n = 35): Major side effects: rash (n = 4), exfoliative dermatitis (n = 1), impotence (n = 1), dizziness (n = 1), nausea/vomiting (n = 1) Mild side effects: nausea (38%), gingival hyperplasia (12%), headaches (32%), cognitive impairment (15%), nystagmus (40%), sedation (15%), fine tremor (28%) | |
| No adverse events reported | N/A | N/A | |
CBZ = carbamazepine, N/A = not available, PHT= phenytoin
1Adverse event data are recorded as reported narratively in the publications, so exact definition of a symptom may vary. Adverse event data supplied as IPD for Ogunrin 2005. Adverse event data were not requested in original IPD requests (De Silva 1996; Heller 1995; Mattson 1985) but will be for all future IPD requests. For numbers of withdrawals due to adverse events in studies for which IPD were provided (De Silva 1996; Heller 1995; Mattson 1985) see Table 3.
2Participants may report more than one adverse event.
3Note that the recruited participants in Cereghino 1974 were institutionalised, so “precise nature of side effects was not always determinable.” The two most frequently occurring side effects were reported as the frequency of participants reporting the side effect on each day of the treatment period, but overall totals of participants reporting each side effect was not reported.
4Czapinski 1997 is an abstract only, so very little information is reported.
5Participants may have withdrawn due to adverse event alone or a combination of adverse events and poor efficacy (seizures).
For carbamazepine
-
Gastrointestinal side effects including abdominal pain, nausea and vomiting: (Cereghino 1974; Forsythe 1991; Mattson 1985; Ramsay 1983).
-
Drowsiness/tiredness/fatigue/sedation: (Callaghan 1985; De Silva 1996; Forsythe 1991; Heller 1995; Pulliainen 1994; Ramsay 1983).
-
Rash: (Callaghan 1985; Heller 1995; Mattson 1985; Ogunrin 2005).
-
Decreased libido, or impotence, or both: (Mattson 1985; Ramsay 1983).
-
Headaches: (Forsythe 1991; Heller 1995; Ramsay 1983).
-
Motor disturbance (including ataxia, incoordination, nystagmus, tremor, slowing of mental function, inattention, psychomotor retardation): (Forsythe 1991; Mattson 1985; Ogunrin 2005; Ramsay 1983).
-
Dysmorphic and idiosyncratic side effects (gum hypertrophy, hirsutism, acne, other skin problems): (Mattson 1985; Pulliainen 1994; Ramsay 1983).
-
Cognitive side effects and impairments, including depression and memory problems: (Heller 1995; Ogunrin 2005; Pulliainen 1994; Ramsay 1983).
For phenytoin
-
Gastrointestinal side effects including abdominal pain, nausea and vomiting: (Cereghino 1974; Mattson 1985; Ramsay 1983).
-
Drowsiness/tiredness/fatigue/sedation: (De Silva 1996; Pulliainen 1994; Ramsay 1983).
-
Rash: (Callaghan 1985; De Silva 1996; Mattson 1985; Ogunrin 2005).
-
Decreased libido, or impotence, or both: (Mattson 1985; Ramsay 1983).
-
Motor disturbance (including ataxia, incoordination, nystagmus, tremor, slowing of mental function, inattention, psychomotor retardation): (Callaghan 1985; Mattson 1985; Ogunrin 2005; Ramsay 1983).
-
Dysmorphic and idiosyncratic side effects (gum hypertrophy, hirsutism, acne, other skin problems): (Callaghan 1985; De Silva 1996; Mattson 1985; Ramsay 1983).
-
Cognitive side effects and impairments, including depression and memory problems: (Forsythe 1991; Ogunrin 2005; Ramsay 1983).
Because of the differences in methods of reporting adverse event data across the studies (see Table 5), it is difficult to summarise the 'most common' adverse events overall across the 12 studies, or to deduce whether carbamazepine or phenytoin are most associated with specific adverse events. Adverse event data for individuals were not included in the original IPD requests for earlier versions of this review, but will be sought in all future IPD requests.
Discussion
Summary of main results
The results of this review demonstrate a statistically significant advantage for phenytoin over carbamazepine for the 118 individuals with new‐onset generalised tonic‐clonic seizures for the primary global outcome 'Time to withdrawal of allocated treatment'; however, this result is likely to have been confounded by misclassification of seizure type for 29 individuals and when this misclassification is taken into account in sensitivity analysis, the advantage for phenytoin is no longer statistically significant. Results for 428 individuals with new‐onset partial seizures suggest an advantage for carbamazepine, which is not statistically significant. Overall, for the 546 individuals contributing withdrawal data to this review, we found no statistically significant evidence for a difference between carbamazepine and phenytoin.
Results of this review also show that among 589 participants recruited in the USA and Europe, carbamazepine is around twice as likely to be withdrawn than phenytoin for adverse events, despite concerns about serious adverse events leading to the replacement of phenytoin with carbamazepine or lamotrigine as a first‐line drug for partial onset seizures across much of the USA and Europe (NICE 2012).
Our primary outcome is a measure of effectiveness influenced by both the relative efficacy of the two drugs and differences in tolerability and safety, so a difference in efficacy in one direction may be confounded by a difference in tolerability in the other. It may therefore not be surprising that any estimated differences are small, and the results of this review cannot exclude clinically important differences between the drugs and between seizure types. Furthermore, the largest study, contributing over 60% of participants to the analysis of our primary outcome, recruited participants with partial onset seizures only, so the subgroups of participants by seizure type are unbalanced in size (428 participants with partial seizures versus 118 with generalised seizures), resulting in less precise results and wide CIs for individuals with generalised onset seizures.
Similarly for the secondary outcomes 'Time to 12‐month remission', 'Time to six‐month remission', and 'Time to first seizure', we found no statistically significant differences between phenytoin and carbamazepine, for participants overall or by seizure type. However, subgroups of participants by seizure type are again unbalanced in size, and misclassification of seizure types may have confounded analyses. More evidence is needed, particularly from individuals with correctly classified generalised seizures, to inform all of the outcomes of this review.
For all outcomes in this review we would recommend caution in the interpretation of the results (see Overall completeness and applicability of evidence), and we would not recommend basing a choice between these two drugs on the results of this review alone.
Overall completeness and applicability of evidence
We believe our systematic electronic searches identified all relevant evidence for this review. We have gratefully received individual participant data (IPD) for 595 individuals (50% of individuals from all eligible trials) from the authors of four trials (De Silva 1996; Heller 1995; Mattson 1985; Ogunrin 2005), which included a comparison of phenytoin versus carbamazepine for the treatment of epilepsy. However, 574 individuals (48%) from seven relevant trials (Callaghan 1985; Cereghino 1974; Czapinski 1997; Miura 1993; Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995) could not be included in any analysis as IPD were not available and outcomes of interest were not reported in the published reports. Sufficient data for 23 individuals (2%) were published in one trial (Forsythe 1991) to contribute to analysis for the primary outcome 'Time to withdrawal of allocated treatment', but insufficient data were available to include these individuals in the analyses by seizure type and the analyses of other outcomes. Having to exclude data from half of eligible participants due to lack of IPD and insufficient reporting in study publications is likely to have impacted on the applicability of the evidence, but it is difficult to quantify exactly how large this impact was on the results of this review (see Potential biases in the review process).
Three trials contributing around 80% of the participant data to this review recruited adults only (Heller 1995; Mattson 1985; Ogunrin 2005); the remaining study was a paediatric trial (De Silva 1996). Also, the largest single trial contributing over half of the participant data to this review (Mattson 1985) recruited individuals with partial onset seizures only, so that only around 25% of participants included in this review were experiencing generalised onset seizures. Furthermore, there is evidence within this review to suggest that up to 30% of individuals with new‐onset generalised seizures may have had their seizure type misclassified. For these reasons, the results of this review may not be fully generalisable to children or to individuals with generalised onset seizures, and more evidence is required from participants with generalised seizure types.
Quality of the evidence
The four trials for which IPD were available were generally at low risk of bias (see Figure 3). Three of the trials contributing around half of the participant data to this review described adequate methods of randomisation and allocation concealment (De Silva 1996; Heller 1995; Ogunrin 2005), but the largest single trial contributing 54% of participant data (Mattson 1985) did not describe the method of randomisation and allocation concealment used, and this information was not available from study authors. We are uncertain whether this lack of information has impacted on the results of this review. See summary of findings Table for the main comparison; summary of findings Table 2; summary of findings Table 3 for GRADE assessments of the quality of the evidence.
Two of the trials providing IPD blinded participants and outcome assessors (Mattson 1985; Ogunrin 2005) and the other two trials (De Silva 1996; Heller 1995) were designed as pragmatic open‐label trials, as masking of treatment would not be “practicable or ethical", would “undermine compliance” and would have “introduced bias due to a very large drop‐out rate.” For the three trials providing withdrawal information, the withdrawal rate in the double‐blinded trial (Mattson 1985) was 40%, and withdrawal rates were 36% and 24% in De Silva 1996 and Heller 1995 respectively (29.5% withdrawal rate overall in the two open‐label studies, which is statistically significantly lower than the withdrawal rate in the double‐blind study; P = 0.009). It is therefore debatable whether a double‐blind design is the most appropriate for trials of monotherapy in epilepsy of long duration and whether such a design does have an impact upon the dropout rate and therefore on the results of the trial.
Further differences between the studies were in the population recruited (age of participants and seizure types). We discuss these differences below in Overall completeness and applicability of evidence.
Trials for which no IPD were available were generally of poorer quality than those for which we were had IPD, with two studies describing inadequate methods of randomisation or allocation concealment (Callaghan 1985; Forsythe 1991), three trials presenting incomplete outcome data following exclusion of participants (Pulliainen 1994; Ramsay 1983; Ravi Sudhir 1995), one study using an inadequate cross‐over design for investigating monotherapy treatments (Cereghino 1974), and two trials providing very limited information on trial methodology, available only in abstract or summary form (Czapinski 1997; Miura 1993).
Potential biases in the review process
We were provided with IPD for 595 out of 1192 eligible participants (50%) from four out of 12 studies included; we conducted all analyses as IPD analyses. Such an approach has many advantages, such as allowing us to standardise definitions of outcomes across trials, and attrition and reporting biases being reduced as we can perform additional analyses and calculate additional outcomes from unpublished data. For the outcomes we used in this review which are of a time‐to‐event nature, an IPD approach is considered to be the ‘gold standard’ approach to analysis (Parmar 1998).
However, despite the advantages of this approach, for reasons out of our control we were not able to obtain IPD for 597 participants from eight eligible studies and no aggregate data were available for our outcomes of interest in study publications. We therefore had to exclude around half of eligible participants from our analyses, which may have introduced bias into the review.
From the results reported in these eight studies (see Table 1 for narrative description of the results of each study), only one study showed a statistically significant difference in efficacy between carbamazepine and phenytoin for participants with generalised onset seizures (73% seizure‐free with phenytoin versus 39% seizure‐free with carbamazepine, Callaghan 1985). There was no difference between treatments for participants with partial onset seizures (P = 0.006). Some significant differences between carbamazepine and phenytoin in terms of specific adverse events and cognitive adverse events were also reported (see Table 1). However, no consistent differences in efficacy or tolerability were reported in these eight studies, so it is unclear whether the exclusion of these studies from our meta‐analysis has impacted upon our results and conclusions. Furthermore, six of the eight studies that we could not include in meta‐analysis were at high risk of bias for at least one methodological aspect (see Figure 3), so inclusion of these data may have introduced bias into our results.
We have good evidence from previous reviews conducted by the Cochrane Epilepsy Group (Marson 2000; Nolan 2013b) that misclassification of seizure type is an important issue in epilepsy trials. We believe that the results of the original trials and hence the results of this meta‐analysis may have been confounded by classification bias, particularly the 48 individuals from two trials (Heller 1995; Ogunrin 2005) classified with new‐onset generalised seizures over the age of 30 (Malafosse 1994). Sensitivity analysis to investigate potential misclassification of these 48 individuals changes our conclusion for two outcomes ('Time to withdrawal of allocated treatment' and 'Time to six‐month remission'), and explains all heterogeneity among individuals with generalised onset seizures for the outcome 'Time to 12‐month remission'. Both studies with potentially misclassified participants used the International League Against Epilepsy (ILAE) classification of 1981 (Commission 1981) to classify generalised onset and partial onset seizures. Heller 1995 was initiated before the publication of the revised ILAE classification in 1989 (Commission 1989), so some individuals in Heller 1995 may have been classified correctly according to Commission 1981 but misclassified by the revised Commission 1989. Ogunrin 2005 was initiated around 10 years after the publication of Commission 1989, but this study was conducted in Nigeria, a low‐income country without access to the same facilities as trials conducted in the USA and Europe; seizure types were therefore classified clinically, and electroencephalographs (EEGs)/magnetic resonance imaging (MRI) were not required for diagnosis of epilepsy. Clinical classification may have contributed to potential misclassification in this study.
Finally, we made some assumptions in the statistical methodology used in this review. Firstly, when we received only follow‐up dates and seizure frequencies, we used linear interpolation to estimate seizure times. We are aware that an individual's seizure patterns may be non‐linear; we therefore recommend caution when interpreting the numerical results of the seizure‐related outcomes.
Further, the statistical methodology used in this review made an assumption that the treatment effect for each outcome did not change over time (proportional hazards assumption, see Data synthesis). For three of the outcomes, there was evidence that this assumption may have been violated for one of the trials. Sensitivity analysis showed that changes in treatment effect tended to occur in the later stages of the studies when small participant numbers were being followed up, so small changes in treatment effect would be magnified. However, we are aware that in studies of long duration (De Silva 1996; Heller 1995; Mattson 1985 followed up participants for between three and 10 years), the assumption of treatment effect remaining constant over time is unlikely to be appropriate, so if more data can be made available to us for updates of this review, we would like to perform statistical analyses which allow for treatment effects to vary over time.
Agreements and disagreements with other studies or reviews
No single trial included in this review has found convincing differences between phenytoin and carbamazepine with respect to seizure control or seizure type. However, CIs around estimates have been wide and equivalence cannot be inferred. Furthermore, this systematic review and meta‐analysis has not found any statistically significant differences between phenytoin and carbamazepine for any of the outcome measures for all included participants. The results of this review suggest a potential advantage for phenytoin over carbamazepine for our primary global outcome 'Time to withdrawal of allocated treatment' for individuals with generalised onset seizures, but this result may have been confounded by misclassification of seizure type.
To our knowledge, this is the only systematic review and meta‐analysis which compares phenytoin and carbamazepine monotherapies for partial onset seizures and generalised onset tonic‐clonic seizures. A network meta‐analysis has been published (Tudur Smith 2007), comparing all direct and indirect evidence from phenytoin, carbamazepine and other standard and new antiepileptic drugs licensed for monotherapy, and found no statistically significant differences between phenytoin and carbamazepine for the outcomes specified in this review; this agrees with the findings of this review. The network meta‐analysis is currently being updated to include more recently published studies such as Ogunrin 2005, so we will compare the results of this review with the updated network meta‐analysis.
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Time to withdrawal of allocated treatment
Time to withdrawal of allocated treatment, stratified by epilepsy type
Time to 12 month remission
Time to 12 month remission, stratified by epilepsy type
Time to 6 month remission, stratified by epilepsy type
Time to first seizure, stratified by epilepsy type
Time to 12 month remission, deSilva 1996
Time to 6 month remission, deSilva 1996
Time to first seizure, Ogunrin 2005

Comparison 1 Carbamazepine versus phenytoin, Outcome 1 Time to withdrawal of allocated treatment.

Comparison 1 Carbamazepine versus phenytoin, Outcome 2 Time to withdrawal of allocated treatment ‐ stratified by epilepsy type.
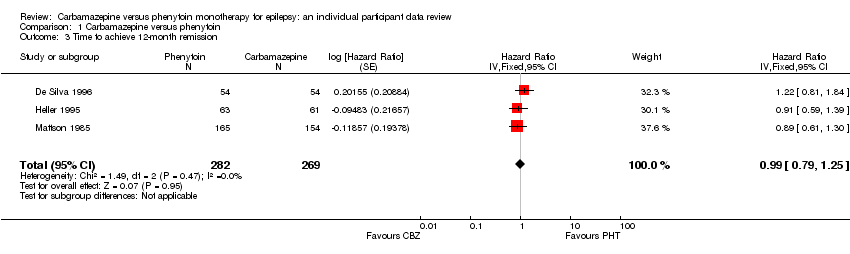
Comparison 1 Carbamazepine versus phenytoin, Outcome 3 Time to achieve 12‐month remission.
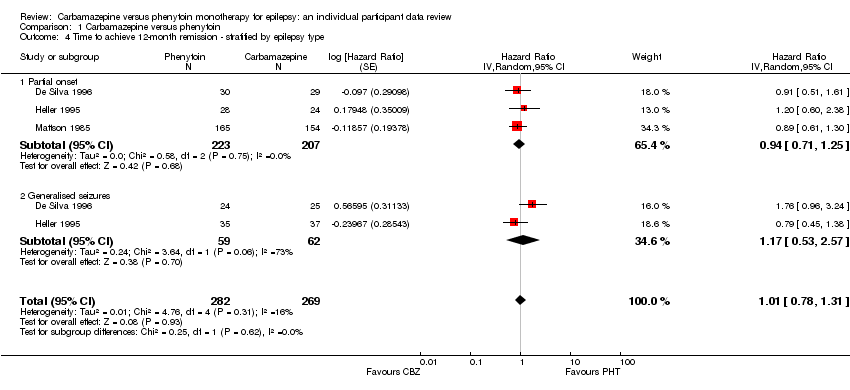
Comparison 1 Carbamazepine versus phenytoin, Outcome 4 Time to achieve 12‐month remission ‐ stratified by epilepsy type.
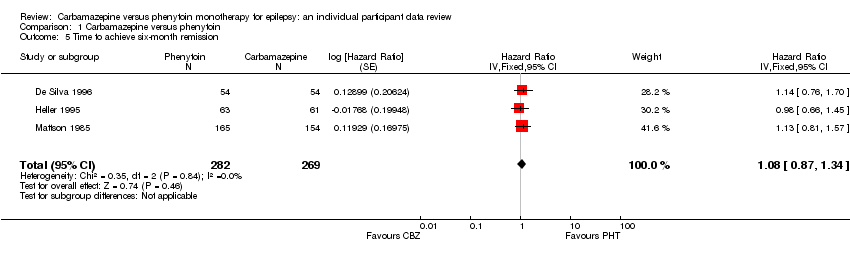
Comparison 1 Carbamazepine versus phenytoin, Outcome 5 Time to achieve six‐month remission.

Comparison 1 Carbamazepine versus phenytoin, Outcome 6 Time to achieve six‐month remission ‐ stratified by epilepsy type.
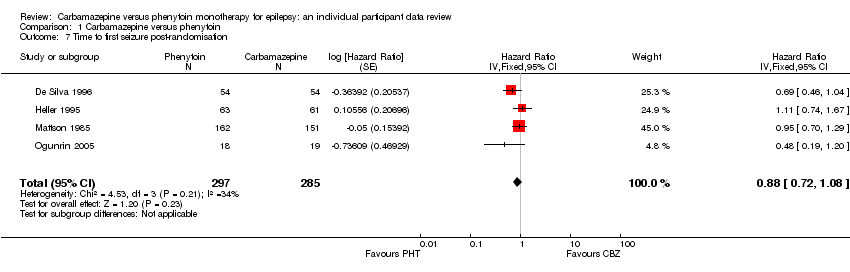
Comparison 1 Carbamazepine versus phenytoin, Outcome 7 Time to first seizure post‐randomisation.
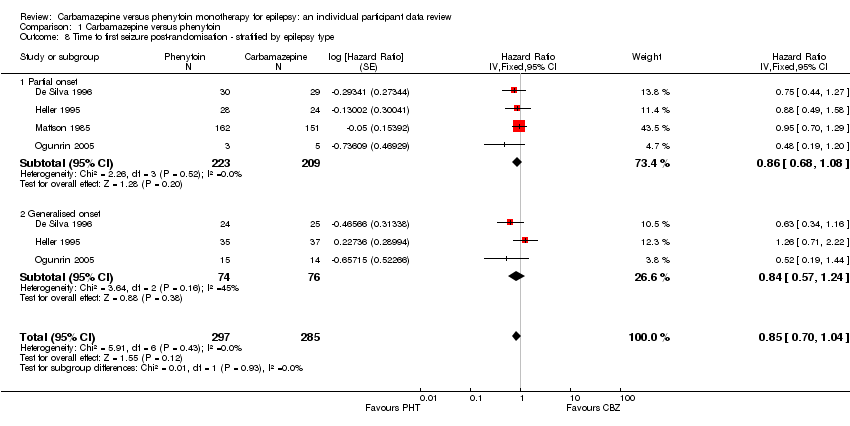
Comparison 1 Carbamazepine versus phenytoin, Outcome 8 Time to first seizure post‐randomisation ‐ stratified by epilepsy type.
| Carbamazepine compared with phenytoin for epilepsy | ||||||
| Patient or population: adults and children with new‐onset partial or generalised epilepsy Settings: outpatients Intervention: carbamazepine Comparison: phenytoin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenytoin | Carbamazepine | |||||
| Time to withdrawal of allocated treatment Range of follow‐up (all participants): 1 day to 4403 days | 37 per 100 | 35 per 100 (28 to 44) | HR 1.04 (0.78 to 1.39) | 546 (3 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical advantage for carbamazepine |
| Time to withdrawal of allocated treatment Range of follow‐up (all participants): 1 day to 4064 days | 42 per 100 | 37 per 100 (29 to 47) | HR 1.18 (0.87 to 1.60) | 428 (3 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical |
| Time to withdrawal of allocated treatment Range of follow‐up (all participants): 1 day to 4403 days | 14 per 100 | 30 per 100 (15 to 57) | HR 0.42 (0.18 to 0.96) | 118 (2 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical |
| Proportion of withdrawals due to adverse effects Range of follow‐up (all participants): 1 day to 4403 days | 4 per 100 | 6 per 100 (5 to 7) | RR 1.42 (1.13 to 1.80) | 546 (3 studies) | ⊕⊕⊕⊝ moderate2 | RR < 1 indicates a clinical advantage for carbamazepine |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The assumed risk is calculated as the event rate in the phenytoin treatment group. The corresponding risk in the carbamazepine treatment group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). The corresponding risk is calculated as the assumed risk x the relative risk (RR) of the intervention where RR = (1 ‐ exp(HR x ln(1 ‐ assumed risk)) ) / assumed risk | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Pooled HR for all participants adjusted for seizure type. | ||||||
| Carbamazepine compared with phenytoin for epilepsy | ||||||
| Patient or population: adults and children with new‐onset partial or generalised epilepsy Settings: outpatients Intervention: carbamazepine Comparison: phenytoin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenytoin | Carbamazepine | |||||
| Time to 12‐month remission Range of follow‐up (all participants): 0 days to 4222 days | 55 per 100 | 55 per 100 (46 to 65) | HR 1.01 (0.78 to 1.31) | 551 (3 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical |
| Time to 12‐month remission Range of follow‐up (all participants):0 days to 4222 days | 47 per 100 | 45 per 100 (36 to 55) | HR 0.94 (0.71 to 1.25) | 430 (3 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical |
| Time to 12‐month remission Range of follow‐up (all participants): 7 days to 4163 days | 85 per 100 | 88 per 100 (63 to 99) | HR 1.174 (0.53 to 2.57) | 121 (2 studies) | ⊕⊕⊝⊝ low2,3,4 | HR > 1 indicates a clinical |
| Time to 6‐month remission Range of follow‐up (all participants): 0 days to 4222 days | 63 per 100 | 67 per 100 (59 to 75) | HR 1.11 (0.89 to 1.37) | 551 (3 studies) | ⊕⊕⊕⊝ moderate2,3 | HR >1 indicates a clinical |
| Time to 6‐month remission Range of follow‐up (all participants): 0 days to 4222 days | 56 per 100 | 56 per 100 (47 to 66) | HR 1.02 (0.79 to 1.33) | 430 (3 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical |
| Time to 6‐month remission Range of follow‐up (all participants): 7 days to 4163 days | 93 per 100 | 97 per 100 (91 to 99) | HR 1.30 (0.89 to 1.92) | 121 (2 studies) | ⊕⊕⊕⊝ moderate2,3 | HR > 1 indicates a clinical |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The assumed risk is calculated as the event rate in the Phenytoin treatment group The corresponding risk in the carbamazepine treatment group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). The corresponding risk is calculated as the assumed risk x the relative risk (RR) of the intervention where RR = (1 ‐ exp(HR x ln(1 ‐ assumed risk)) ) / assumed risk | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Pooled HR for all participants adjusted for seizure type. | ||||||
| Carbamazepine compared with phenytoin for epilepsy | ||||||
| Patient or population: adults and children with new‐onset partial or generalised epilepsy Settings: outpatients Intervention: carbamazepine Comparison: phenytoin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Phenytoin | Carbamazepine | |||||
| Time to first seizure Range of follow‐up (all participants): 0 days to 4589 days | 65 per 100 | 71 per 100 (63 to 77) | HR 0.85 (0.70 to 1.04) | 582 (4 studies) | ⊕⊕⊝⊝ low2,3,4 | HR > 1 indicates a clinical |
| Time to first seizure Range of follow‐up (all participants): 0 days to 4589 days | 63 per 100 | 68 per 100 (60 to 77) | HR 0.86 (0.68 to 1.08) | 432 (4 studies) | ⊕⊕⊝⊝ low2,3,4 | HR > 1 indicates a clinical |
| Time to first seizure Range of follow‐up (all participants): 2 days to 4070 days | 69 per 100 | 75 per 100 (61 to 87) | HR 0.84 (0.57 to 1.24) | 150 (3 studies) | ⊕⊕⊝⊝ low2,3,4 | HR > 1 indicates a clinical |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The assumed risk is calculated as the event rate in the Phenytoin treatment group The corresponding risk in the carbamazepine treatment group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). The corresponding risk is calculated as the assumed risk x the relative risk (RR) of the intervention where RR = (1 ‐ exp(HR x ln(1 ‐ assumed risk)) ) / assumed risk | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Pooled HR for all participants adjusted for seizure type. 2Risk of bias unclear for one element of all of the three studies included in the analysis. De Silva 1996 and Heller 1995 are open‐label and it is unclear whether the lack of masking impacted upon the results; and we do not know how allocation was concealed in Mattson 1985. 348 adult participants in Heller 1995 and Ogunrin 2005 may have had their seizure type wrongly classified as generalised onset; sensitivity analyses show misclassification is unlikely to have had an impact on results and conclusions. 4Ogunrin 2005 is a short study (12 weeks) and has a small sample size of 37 compared to the other three studies of duration 3 ‐ 10 years and sample sizes of around 100 to 300 participants (De Silva 1996; Heller 1995; Mattson 1985). Ogunrin 2005 is less precise with wide CIs, and there is evidence that the treatment effect in this study changes over time. | ||||||
| Trial | Outcomes reported | Summary of results |
| 1. Seizure control: excellent (seizure‐free) | 1. PHT (n = 58); CBZ (n = 59) PHT: 39 (67%); CBZ: 22 (37%) | |
| 1. Behaviour measured with rating scale modified from the Ward Behaviour Rating Scale 2. Seizure control 3. Side effects 4. Withdrawals | 1. Behavioural scores were similar on both drugs 2. No difference between CBZ and PHT in terms of seizure control 3. Gastrointestinal and “impaired function” side effects were more common on CBZ than PHT in the first few study days. Side effects of both drugs were minimal in later stages of the study 4. PHT: 21 withdrawals out of 45 participants (47%); CBZ: 27 withdrawals out of 45 participants (60%) | |
| 1. Proportion achieving 24‐month remission at 3 years 2. Proportion excluded after randomisation due to adverse effects or no efficacy | 1. PHT: 59%; CBZ: 62% 2. PHT: 23%; CBZ: 30% | |
| 1. Cognitive assessments 2. Withdrawals from randomised drug | 1. No significant differences between the two treatment groups on any cognitive tests | |
| 1. Proportion of all randomised participants with seizure recurrence (by seizure type) 2. Proportion of participants with optimum plasma levels with seizure recurrence (by seizure type) | PHT (n = 51); CBZ (n = 66) 1. PHT (partial): 10/31 (32%); PHT (generalised): 7/20 (35%); 2. PHT (partial): 4/17 (24%); PHT (generalised): 1/8 (13%); | |
| 1. Cognitive assessments (visual motor speed, co‐ordination, attention and concentration, verbal and visuospatial learning, visual and recognition memory, reasoning, mood, handedness) 2. Harmful side effects | 1. Compared to CBZ, participants on PHT became slower (motor speed of the hand) and their visual memory decreased. There was an equal decrease in negative mood (helplessness, irritability, depression) on PHT and CBZ 2. Three participants taking PHT complained of tiredness, and 1 participant taking CBZ complained of facial skin problems, another tiredness and memory problems | |
| 1. Side effects (major and minor) 3. Laboratory results | 1. Incidence of:
2. Treatment failures among analysed participants: Seizure control (among analysed participants with no major side effects): PHT: 23/27 participants (86%); CBZ: 22/27 participants (82%) 3. Significantly lower mean LDH level at 24 weeks in CBZ participants than PHT participants (P < 0.01). Other laboratory results similar across treatment groups | |
| 1. Cognitive measures (verbal, performance, memory, visuomotor, perceptomotor organisation, visual organisation, dysfunction) | 1. No significant differences between any tests of cognitive function taken before treatment and after 10 ‐ 12 weeks for both treatment groups | |
| CBZ = carbamazepine, LDH = lactate dehydrogenase, PHT= phenytoin | ||
| Trial | Number randomised | Time to withdrawal of allocated treatment | Time to 12‐month remission | Time to 6‐month remission | Time to first seizure | ||||||||||
| PHT | CBZ | Total | PHT | CBZ | Total | PHT | CBZ | Total | PHT | CBZ | Total | PHT | CBZ | Total | |
| 54 | 54 | 108 | 53 | 53 | 106 | 54 | 54 | 108 | 54 | 54 | 108 | 54 | 54 | 108 | |
| 63 | 61 | 124 | 61 | 60 | 121 | 63 | 61 | 124 | 63 | 61 | 124 | 63 | 61 | 124 | |
| 165 | 155 | 320 | 165 | 154 | 319 | 165 | 154 | 319 | 165 | 154 | 319 | 162 | 151 | 313 | |
| 20 | 23 | 43 | 20 | 23 | 43 | Information not available | Information not available | Information not available | |||||||
| 18 | 19 | 37 | Information not available | Information not available | Information not available | 18 | 19 | 37 | |||||||
| Total | 320 | 312 | 632 | 299 | 290 | 589 | 282 | 269 | 551 | 282 | 269 | 551 | 297 | 285 | 582 |
| CBZ = carbamazepine, PHT= phenytoin 1Individual participant data (IPD) supplied for 114 participants recruited in De Silva 1996; randomised drug not recorded in six participants. Reasons for treatment withdrawal not available for two participants (one randomised to CBZ and one to PHT); these participants are not included in analysis of Time to treatment withdrawal. | |||||||||||||||
| Reason for early termination | Classification | Heller 19952,3 | Total1 | ||||||||
| CBZ n = 53 | PHT n = 53 | CBZ n = 23 | PHT n = 20 | CBZ n = 60 | PHT n = 63 | CBZ n = 154 | PHT n = 165 | CBZ n = 290 | PHT n = 299 | ||
| Adverse events | Event | 3 | 2 | 4 | 1 | 8 | 1 | 11 | 8 | 26 | 12 |
| Seizure recurrence | Event | 12 | 10 | 2 | 1 | 5 | 8 | 3 | 6 | 22 | 25 |
| Both seizure recurrence and adverse events | Event | 6 | 5 | 0 | 0 | 4 | 2 | 31 | 33 | 31 | 40 |
| Non‐compliance/participant choice | Event | 0 | 0 | 3 | 4 | 0 | 0 | 11 | 26 | 14 | 30 |
| Participant went into remission | Censored | 18 | 24 | 0 | 0 | 6 | 14 | 0 | 0 | 24 | 38 |
| Lost to follow‐up | Censored | 0 | 0 | 0 | 0 | 0 | 0 | 26 | 19 | 26 | 19 |
| Death4 | Censored | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 5 | 4 | 5 |
| Other5 | Censored | 0 | 0 | 0 | 0 | 0 | 0 | 16 | 11 | 16 | 11 |
| Completed the study (did not withdraw) | Censored | 14 | 12 | 14 | 14 | 37 | 38 | 53 | 57 | 118 | 121 |
| n = number of individuals contributing to the outcome 'Time to withdrawal of allocated treatment’ 1All participants in Ogunrin 2005 completed the study without withdrawing, so this study did not contribute to 'Time to withdrawal of allocated treatment'. | |||||||||||
| Analysis | Time to withdrawal | Time to six‐month remission | Time to 12‐month remission* | Time to first seizure |
| Original analysis | P: 1.18 (0.87, 1.60) G: 0.42 (0.18, 0.96) O: 1.04 (0.78, 1.39) | P: 1.02 (0.79, 1.33) G: 1.30 (0.89, 1.92) O: 1.11 (0.89, 1.37) | P: 0.94 (0.71, 1.25) G: 1.17 (0.53, 2.57) O: 1.01 (0.78, 1.31) | P: 0.86 (0.68, 1.08) G: 0.84 (0.57, 1.24) O: 0.85 (0.70, 1.04) |
| Test for interaction | Chi2 = 5.18; df = 1 P = 0.02; I2 = 80.7% | Chi2 = 1.03; df = 1 P = 0.31; I2 = 3.4% | Chi2 = 0.25; df = 1 P = 0.62; I2 = 0% | Chi2 = 0.01; df = 1 P = 0.93; I2 = 0% |
| Generalised and age at onset > 30 (classified as uncertain epilepsy type) | P: 1.18 (0.87, 1.60) G: 0.51 (0.21, 1.24) U: 0.19 (0.02, 2.14) O: 1.05 (0.79, 1.40) | P: 1.02 (0.79, 1.33) G: 1.69 (1.07, 2.27) U: 0.84 (0.35, 1.98) O: 1.13 (0.91, 1.41) | P: 0.94 (0.71, 1.25) G: 1.44 (0.90, 2.31) U: 0.52 (0.20, 1.34) O: 1.01 (0.80, 1.28) | P: 0.86 (0.68, 1.08) G: 0.91 (0.57, 1.46) U: 0.97 (0.43, 2.18) O: 0.88 (0.72, 1.07) |
| Test for interaction | Chi2 = 4.99; df = 2 P = 0.08; I2 = 59.9% | Chi2 = 4.01; df = 2 P = 0.13; I2 = 50.2% | Chi2 = 4.32; df = 2 P = 0.12; I2 = 53.7% | Chi2 = 0.12; df = 2 P = 0.94; I2 = 0% |
| Generalised and age at onset > 30 (reclassified as partial epilepsy) | P: 1.11 (0.82, 1.50) G: 0.51 (0.21, 1.24) O: 1.02 (0.77, 1.36) | P: 1.02 (0.80, 1.31) G: 1.69 (1.07, 2.27) O: 1.15 (0.92, 1.42) | P: 0.91 (0.69, 1.19) G: 1.44 (0.90, 2.31) O: 1.02 (0.81, 1.29) | P: 0.86 (0.69, 1.08) G: 0.91 (0.57, 1.46) O: 0.87 (0.71, 1.07) |
| Test for interaction | Chi2 = 2.65; df = 1 P = 0.10; I2 = 62.3% | Chi2 = 3.63; df = 1 P = 0.06; I2 = 72.5% | Chi2 = 2.79; df = 1 P = 0.09; I2 = 64.2% | Chi2 = 0.04; df = 1 P = 0.83; I2 = 0% |
| df = degrees of freedom of Chi² distribution, G = generalised epilepsy, O = overall (all participants), P = partial epilepsy, U = uncertain seizure type Results are presented as pooled hazard ratio (HR) (95% confidence interval (CI)) with fixed‐effect. See Analysis 1.2; Analysis 1.4; Analysis 1.6; and Analysis 1.8 for original analyses of 'Time to treatment withdrawal', 'Time to 12‐month remission', 'Time to 6‐month remission' and 'Time to first seizure', all stratified by epilepsy respectively. * Original analysis calculated with random‐effects model due to substantial heterogeneity (see Analysis 1.4). Sensitivity analyses calculated with fixed‐effect model as no heterogeneity is present following reclassification of 29 participants in Heller 1995. | ||||
| Trial | Adverse event data1 | Summary of reported results | |
| Carbamazepine (CBZ) | Phenytoin (PHT) | ||
| All adverse events according to drug (note: no participants withdrew due to adverse events) | CBZ (n = 59): drowsiness (n = 2), rash (n = 3) | PHT (n = 58): gum hypertrophy (n = 2), rash (n = 2), ataxia (n = 2) | |
| Most frequently observed side effects | Gastrointestinal side effects and “impaired function” (general malaise). Frequency not clearly stated | Gastrointestinal side effects and “impaired function” (general malaise). Frequency not clearly stated | |
| “Exclusions” due to adverse events or no efficacy” | Proportion “excluded”: CBZ: 30% (out of 30 randomised to CBZ) | Proportion “excluded”: PHT: 23.3% (out of 30 randomised to PHT) | |
| “Unacceptable” adverse events leading to drug withdrawal5 | CBZ (n = 54): drowsiness (n = 1), blood dyscrasia (n = 1) | PHT (n = 54): drowsiness (n = 2), skin rash (n = 1), blood dyscrasia (n = 1), hirsutism (n = 1) | |
| Withdrawal due to adverse events (no other adverse event data reported) | 4 participants out of 23 randomised to CBZ withdrew for the following reasons (some withdrew for more than adverse event): slowing of mental function, headache, anorexia, nausea, abdominal pain, fatigue and drowsiness2 | 1 participant out of 20 randomised to PHT withdrew from the study due to depression and anorexia | |
| “Unacceptable” adverse events leading to drug withdrawal5 | CBZ (n = 61): drowsiness (n = 3), rash (n = 2), headache (n = 1), depression (n = 1) | PHT (n = 63): myalgia (n = 1), irritability (n = 1) | |
| Narrative report of ‘Adverse effects’ and ‘Serious side effects’ | CBZ (n = 155): motor disturbance (ataxia, incoordination, nystagmus, tremor: 33%); dysmorphic and idiosyncratic side effects (gum hypertrophy, hirsutism, acne and rash: 14%); gastrointestinal problems (27%); decreased libido or impotence (13%); No serious side effects | PHT (n = 165); motor disturbance (ataxia, incoordination, nystagmus, tremor: 28%); dysmorphic and idiosyncratic side effects (gum hypertrophy, hirsutism, acne and rash: 22 %); gastrointestinal problems (24%); decreased libido or impotence (11%) 1 serious side effect – 1 participant has confirmed lymphoma, rash improved rapidly following discontinuation of PHT | |
| No adverse events reported | N/A | N/A | |
| Participant reported symptomatic complaints (provided as IPD) | CBZ (n = 19): memory impairment (n = 9) psychomotor retardation (n = 1) inattention (n = 1) transient rash (n = 1) CBZ‐induced cough (n = 1) | PHT (n = 18): memory impairment (n = 7) psychomotor retardation (n = 1) inattention (n = 2) transient rash (n = 1) | |
| Participant‐reported adverse events | 1 participant on CBZ complained of facial skin problems; 1 participant on CBZ complained of tiredness and memory problems | 3 participants on PHT complained of tiredness | |
| Major and minor side effects | CBZ (n = 35): Major side effects: rash (n = 1), pruritus (n = 1), impotence (n = 2), dizziness (n = 1), headaches (n = 1), impaired cognition (n = 1), elevated liver enzymes (n = 1) Mild side effects: nausea (33%), headaches (24%), cognitive impairment (33%), nystagmus (52%), sedation (33%), fine tremor (20%) | PHT (n = 35): Major side effects: rash (n = 4), exfoliative dermatitis (n = 1), impotence (n = 1), dizziness (n = 1), nausea/vomiting (n = 1) Mild side effects: nausea (38%), gingival hyperplasia (12%), headaches (32%), cognitive impairment (15%), nystagmus (40%), sedation (15%), fine tremor (28%) | |
| No adverse events reported | N/A | N/A | |
| CBZ = carbamazepine, N/A = not available, PHT= phenytoin 1Adverse event data are recorded as reported narratively in the publications, so exact definition of a symptom may vary. Adverse event data supplied as IPD for Ogunrin 2005. Adverse event data were not requested in original IPD requests (De Silva 1996; Heller 1995; Mattson 1985) but will be for all future IPD requests. For numbers of withdrawals due to adverse events in studies for which IPD were provided (De Silva 1996; Heller 1995; Mattson 1985) see Table 3. | |||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Time to withdrawal of allocated treatment Show forest plot | 4 | 589 | Hazard Ratio (Fixed, 95% CI) | 0.99 [0.75, 1.30] |
| 2 Time to withdrawal of allocated treatment ‐ stratified by epilepsy type Show forest plot | 3 | 546 | Hazard Ratio (Fixed, 95% CI) | 1.04 [0.78, 1.39] |
| 2.1 Partial onset | 3 | 428 | Hazard Ratio (Fixed, 95% CI) | 1.18 [0.87, 1.60] |
| 2.2 Generalised seizures | 2 | 118 | Hazard Ratio (Fixed, 95% CI) | 0.42 [0.18, 0.96] |
| 3 Time to achieve 12‐month remission Show forest plot | 3 | 551 | Hazard Ratio (Fixed, 95% CI) | 0.99 [0.79, 1.25] |
| 4 Time to achieve 12‐month remission ‐ stratified by epilepsy type Show forest plot | 3 | 551 | Hazard Ratio (Random, 95% CI) | 1.01 [0.78, 1.31] |
| 4.1 Partial onset | 3 | 430 | Hazard Ratio (Random, 95% CI) | 0.94 [0.71, 1.25] |
| 4.2 Generalised seizures | 2 | 121 | Hazard Ratio (Random, 95% CI) | 1.17 [0.53, 2.57] |
| 5 Time to achieve six‐month remission Show forest plot | 3 | 551 | Hazard Ratio (Fixed, 95% CI) | 1.08 [0.87, 1.34] |
| 6 Time to achieve six‐month remission ‐ stratified by epilepsy type Show forest plot | 3 | 551 | Hazard Ratio (Fixed, 95% CI) | 1.11 [0.89, 1.37] |
| 6.1 Partial onset | 3 | 430 | Hazard Ratio (Fixed, 95% CI) | 1.02 [0.79, 1.33] |
| 6.2 Generalised seizures | 2 | 121 | Hazard Ratio (Fixed, 95% CI) | 1.30 [0.89, 1.92] |
| 7 Time to first seizure post‐randomisation Show forest plot | 4 | 582 | Hazard Ratio (Fixed, 95% CI) | 0.88 [0.72, 1.08] |
| 8 Time to first seizure post‐randomisation ‐ stratified by epilepsy type Show forest plot | 4 | 582 | Hazard Ratio (Fixed, 95% CI) | 0.85 [0.70, 1.04] |
| 8.1 Partial onset | 4 | 432 | Hazard Ratio (Fixed, 95% CI) | 0.86 [0.68, 1.08] |
| 8.2 Generalised onset | 3 | 150 | Hazard Ratio (Fixed, 95% CI) | 0.84 [0.57, 1.24] |