Pharmacothérapie pour le trouble d'anxiété sociale
Referencias
References to studies included in this review
References to studies excluded from this review
References to studies awaiting assessment
References to ongoing studies
Additional references
References to other published versions of this review
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks and 6 days of dose‐tapering Post‐treatment: no follow‐up Placebo run‐in: 1‐week assessment period without medication | |
| Participants | Sample size: 92 randomised to paroxetine and placebo Mean age: 41 years Sex: 48 men and 44 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Aged 18‐65 years with previously untreated and incapacitating social anxiety; DSM‐IV social anxiety disorder causing substantial impairment and with a duration of at least 1 year; DSM‐IV diagnoses of generalised anxiety, dysthymia or a cluster C personality disorder were the only concurrent psychiatric disorders allowed". Exclusion criteria: quote: "No psychoactive medications were permitted, including beta‐receptor‐blocking agents; the blood and urine of all subjects was screened for substance abuse" Dropouts: 27/92 (8/44 in the paroxetine group and 19/48 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "The subjects were randomly allocated at baseline to double‐blind treatment for three months with paroxetine 20‐50 mg daily administered in 10‐mg weekly increments, or placebo. One dose reduction was allowed in case of adverse events." | |
| Outcomes | Primary outcomes: LSAS (for reduction of anxiety) and CGI (for treatment efficacy) Secondary outcomes: BSPS (for reduction of anxiety), SDI (for reduction of functional disability), FNES (for reduction of anxiety) and VAS scores (reflecting self‐confidence in social interactions, anticipatory anxiety, acute anxiety reactions in social situations, and dysphoria following anxiety reactions) Time points: Quote: "Assessments were made after 1, 2, 4, 6, 8 and 12 weeks, and after 6 days of dose‐tapering" | |
| Notes | Industry funded: yes. Quote: "This study was funded by Novo Nordisk Pharma, Sweden." Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was performed at the hospital pharmacy using tabulated random numbers" |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "The randomization and blinding and packaging of study materials were undertaken by our hospital pharmacy and by Wyeth" |
| Blinding of participants and personnel (performance bias) | Unclear risk | No description of blinding is provided in the study report. Quote: "Patients were randomized to double‐blind treatment with paroxetine 20‐50 mg daily or placebo for 3 months" |
| Blinding of outcome assessment (detection bias) | Unclear risk | No mention is made of whether the outcome assessors were indeed blinded and independent. Quote: "To reduce variability in assessments, all subjects were treated and assessed in one centre by the author and a research nurse" |
| Incomplete outcome data (attrition bias) | High risk | A larger proportion of participants discontinued the study in the paroxetine (8/44; 18%) group compared to the placebo group (19/48; 39%). Reasons for treatment withdrawal were provided for the treatment group with only reasons given for 5 participants in the placebo group. No information was provided on sample characteristics at endpoint. Overall 29% of the participants dropped out of the study. All analyses were intention‐to‐treat (ITT) |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. Quote: "Comparatively high response rate of subjects on paroxetine and the low response rate of those on placebo in this study may be due to the low variability in assessments in a single centre, the use of self‐rating instruments, or the fact that only previously untreated cases were included" |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group trial Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo lead‐in period | |
| Participants | Sample size: 434 were randomised to venlafaxine, paroxetine, or placebo (2 individuals excluded; 389 ITT population) Mean age (SD): 38.8 (10.97) years Sex: 183 men and 206 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Study participants were adult (18 years of age) outpatients who met DSM‐IV criteria for generalized SAD for at least 6 months prior to study day 1. Participants were eligible if they had a score 54 on item 1 (severity of illness) of the clinical global impression severity (CGI‐S) scale; a minimum total score of at least 50 on the Liebowitz social anxiety scale (LSAS), with 430% decrease between the prestudy and baseline visits (i.e. during the placebo lead‐in period); a prestudy Raskin depression total score 49, and a 17‐item Hamilton rating scale for depression (HAM‐D17) score <15; and provided informed consent". Exclusion criteria: quote: "Patients were excluded if they had been treated with venlafaxine immediate release or venlafaxine ER within 6 months of study day 1 or had concurrent disorders that confounded the evaluation of treatment, including substance use disorders, personality disorders (except avoidant personality disorder), depression or other primary anxiety disorders, diagnosed by clinical interview. While patients who had not responded to previous treatment with paroxetine were not prohibited from participating in the study, ongoing psychotherapy and recent treatment with psychoactive medications precluded entry into the study". Dropouts: 26/434 (6/144 in the paroxetine group, 7/144 in the venlafaxine group, and 13/146 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "After a 1‐week, single‐blind, placebo lead‐in period to eliminate subjects with situational anxiety and ascertain generalized social anxiety disorder, patients symptomatic at baseline were randomly assigned to receive flexible doses of venlafaxine ER (75–225 mg/day), paroxetine (20–50 mg/day), or placebo for up to 84 days". | |
| Outcomes | Primary outcome: LSAS (for reduction of anxiety) Secondary outcomes: CGI‐S (for reduction of anxiety), CGI‐I (for treatment efficacy), SPIN (for reduction of anxiety), the fear/anxiety and avoidance subscales of the LSAS (for reduction of anxiety), SDI (for reduction of functional disability) and the WPAI questionnaire (for reduction of functional disability) Time points: Quote: "Patient evaluations occurred at baseline and on days 7, 14, 21, 28, 42, 56, 70 and 84. Final efficacy evaluations were performed on the last day that the patient received a full dose of study medication or within 3 days thereafter" | |
| Notes | Industry funded: yes. Quote: "Contract/grant sponsor: Wyeth Research" Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Authors report that participants were randomised; however, no mention is made of the method of randomisation. Quote: "Adult outpatients with generalized SAD (n=434) were randomized to receive capsules of venlafaxine ER 75 mg to 225 mg/day, paroxetine 20 mg to 50 mg/day, or placebo for 12 weeks ... At the baseline visit, after the investigator had ascertained that the patient was qualified to enter the study, the patient was also given a randomization number and the accompanying treatment supplies". |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "The randomization and blinding and packaging of study materials were undertaken by our hospital pharmacy and by Wyeth". |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "study medication was provided in identically appearing capsules containing venlafaxine ER 75 mg, paroxetine 10 mg, paroxetine 20 mg, or placebo, and the number of capsules was identical for all treatments". |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "In order to ensure that the assessor (i.e. the investigator) was unaware of the treatment group to which a patient was assigned, study medication was provided in identically appearing capsules containing venlafaxine ER 75 mg, paroxetine 10 mg, paroxetine 20 mg, or placebo, and the number of capsules was identical for all treatments". |
| Incomplete outcome data (attrition bias) | Low risk | More participants withdrew from the placebo group (13/132; 10%) compared to the paroxetine (6/128; 5%) and venlafaxine (7/129; 5%) group. The most common reasons for withdrawal were adverse events and unsatisfactory response. No information was provided on whether participants differed in terms of characteristics by group at week 12, however. Nevertheless, the total proportion of dropouts (7%) is relatively low, suggesting that dropout rates may not have biased the outcomes. Quote: "A total of 363 (84%) patients completed the 12‐week double‐blind treatment period (119 in the placebo group, 122 in the venlafaxine ER group and 122 in the paroxetine group) ... the most common reasons for withdrawal were adverse events and unsatisfactory response. Significantly more participants in the placebo group withdrew due to unsatisfactory response than in the venlafaxine ER group or the paroxetine group". Overall 7% of the participants dropped out of the study. Baseline analysis were intention‐to‐treat (ITT) whereas the analysis of the primary and secondary outcomes were last observation carried forward (LOCF). |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias was identified. |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled study Duration of intervention: 10 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 273 randomised to fluvoxamine or placebo (273 randomised: 2 excluded; 271 ITT population) Mean age (SD): 38.6 (11.25) years Sex: 179 men and 86 women (265 randomised in the efficacy analysis population) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Eligible patients were aged 18–65 yr and were required to meet the DSM‐IV criteria for GSAD, have a minimum score of > 60 on the Liebowitz Social Anxiety Scale – Japanese Version, have no serious medical history, and to have taken no psychotropic medications for at least 14 d prior to randomization. The diagnosis of GSAD was made according to DSM‐IV criteria by well‐trained research psychiatrists. Patients were required, in addition to meeting DSM‐IV criteria for SAD, to exhibit fear and/or avoidance of at least four social situations (at least two involving interpersonal interactions)". Exclusion criteria: quote: "Patients were excluded if they had any Axis I psychiatric disorder (e.g. schizophrenia, bipolar disorder, major depressive disorder, dysthymic disorder, panic disorder, alcohol abuse/dependence), or medical or neurological disorder. Other exclusion criteria were any clinically significant abnormal laboratory or electrocardiogram (ECG) findings at the screening visit. Women who were pregnant, lactating, or not using an acceptable method of contraception were also ineligible". Dropouts: 6/271 (4/93 and 2/89 in the fluvoxamine groups and 0/89 in the placebo group; the additional two were excluded prior to the allocation of treatment). | |
| Interventions | Pharmacological intervention: quote: "Eligible patients were randomly assigned to either fluvoxamine (at an initial dose of 50 mg/d fluvoxamine in two divided doses) or placebo in a 2:1 ratio. Fluvoxamine‐treated patients were randomly divided into two subgroups; a daily dose was increased by 50mg increments per week to a maintenance dose of 150 mg/d in one subgroup and to that of 300 mg/d in the other subgroup". | |
| Outcomes | Primary outcome: LSAS‐J (for reduction of anxiety) Secondary outcomes: CGI (for treatment efficacy) and SDS (for reduction of functional disability) Time points: Quote: "Patients were evaluated at nine study visits (baseline and weeks 1, 2, 3, 4, 5, 6, 8, and 10)" | |
| Notes | Industry funded: yes. Quote: "This study was sponsored by Solvay Seiyaku K.K. and Meiji Seika Kaisha, Ltd". Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "This was a double‐blind study, meaning both the subjects and the investigators were blinded to the randomization scheme by double‐dummy method". |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "An independent third party (CRO) randomly allocated the packages of investigational drug using SAS procedure, as a set consisted of investigational drug for 4 cases for fluvoxamine group and 2 cases for placebo group, and sealed the packages. They held the key code during the course of the study and were to break the blind after all CRFs were collected and all CRF data had been entered into the database and the database locked". |
| Blinding of participants and personnel (performance bias) | Low risk | Author correspondence: quote: "This was a double‐blind study, meaning both the subjects and the investigators were blinded to the randomization scheme by double‐dummy method". |
| Blinding of outcome assessment (detection bias) | Low risk | Author correspondence: quote: "This was a double‐blind study, meaning both the subjects and the investigators were blinded to the randomization scheme by double‐dummy method". |
| Incomplete outcome data (attrition bias) | Low risk | A small proportion of participants withdrew from the fluvoxamine groups (4/93, 2/89) with no reported dropouts for the placebo group (0/89). Patients withdrew due to adverse events and protocol deviations. No information was provided on whether participants differed by group characteristics at at week 10, however. Nevertheless, the total proportion of dropouts (2%) is relatively low, suggesting that dropout rates may not have biased the outcomes. Quote: "The efficacy analysis population was composed of 265 patients (176 receiving fluvoxamine and 89 receiving placebo), excluding six patients for whom no valid post‐baseline efficacy evaluation was obtained due to premature discontinuation [four withdrew due to adverse events and two withdrew due to protocol deviations (inappropriate concomitant medications)]". Overall 2% of the participants dropped out of the study. Quote: "Efficacy data are presented for the last observation carried forward (LOCF) dataset. The LOCF dataset used the last available on‐treatment observation for each patient to estimate missing data‐points". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. There was a difference in gender proportions, but there is no reason to believe that this may have biased the study. Quote: "Our findings on the gender ratio confirmed that men were predominant in this study. However, it is unknown whether the finding represents the status of gender ratio in Japanese SAD patients. There seems to be no clear sex predominance for this disorder". |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: one‐week, single blind, placebo run‐in phase | |
| Participants | Sample size: 290 randomised to paroxetine or placebo Mean age (SD): 36 (11.5) years Sex: 133 men and 157 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Male or female out‐patients, aged 18 years or over, patients with a primary diagnosis of social phobia according to the DSM‐IV criteria were included in the study following the provision of written informed consent". Exclusion criteria: quote: "Patients were excluded at the screening visit if they had a primary diagnosis of any other Axis I disorder within the past six months, if they had been diagnosed as having body dysmorphic disorder or if they had a history of schizophrenia or bipolar affective disorder. Patients were excluded if they had a past history of seizure disorders or any serious medical disorder that could preclude the administration of paroxetine. In addition, patients requiring concomitant therapy with beta‐adrenergic blockers, monoamine oxidase inhibitors, benzodiazepines or other psychoactive medications were not included. Patients were also not included if: they had taken psychotropic drugs or antidepressants within the past two weeks or depot neuroleptics within 12 weeks; they had been previously unresponsive or intolerant to paroxetine, or they had used an investigational drug during the past month; they had undergone previous treatment for social phobia with an SSRI at a dose and duration that would have been adequate to show a response, or undergone electroconvulsive therapy (within three months) or psychotherapy (except ongoing stabilised therapies of six months or more). Other exclusion criteria included pregnancy (or a likelihood of becoming pregnant), lactation and alcohol substance misuse (within the past three months) or dependence (within the past six months). Patients were also excluded if they posed a current serious risk of suicide or homicide". Dropouts: 77/290 (35/139 in the paroxetine and 42/151 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "Patients initially received 20 mg/day paroxetine or placebo for two weeks, followed by 10 mg/day at weekly intervals to a maximum dose of 50 mg/day according to clinical response and tolerability". | |
| Outcomes | Primary outcomes: LSAS (for reduction of anxiety) and CGI‐I (for treatment efficacy) Secondary outcomes: SADS (for reduction of anxiety), SDS (for reduction of functional disability) and CGI‐S (for reduction of anxiety) Time points: Quote: "Efficacy and safety assessments were made at weeks 1,2,3,4,6,8 and 12 and additional, further safety assessments were made at week 15. At week 12, or on early withdrawal from the study, a physical examination, laboratory tests, body weight determination and HAM‐D (17‐item) assessments were performed. After the week 12 visit, the dose of study medication was reduced during a thno week tapering period; safety assessments (but not efficacy assessments) were made during this period. Patients also attended a follow‐up visit when safety pammetcrs were assessed if they had withdrawn from the study prematurely owing to an adverse event, or if they had completed the study with an ongoing adverse event" | |
| Notes | Industry funded: yes. Quote: "Smith Kline Beecharn Pharmaceuticals provided financial support for this study". Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "Block randomisation" |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "Randomisation was performed in the pharmacy at a distant site to the clinical site where the research team were based". |
| Blinding of participants and personnel (performance bias) | Low risk | Author correspondence: quote: "All patients and research staff were blinded to treatment allocation at all centres throughout the study". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | A similar proportion of participants withdrew from the paroxetine group (35/139, 25%) compared to the placebo group (42/151; 28%). Common withdrawals in the paroxetine group were adverse experience and lack of efficacy in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "There was no overall difference between the treatment groups in the number of patients who withdrew during the study: 35 (25%) patients in the paroxetine group v. 42 (28%) in the placebo group ... The number of patients lost to follow‐up, although comparable between the groups was high and is probably characteristic of the patient population under study; owing to the nature of the disorder". Overall 27% of the participants dropped out of the study. Quote: "Outcome measures were performed on the intent‐to‐treat (lTT) efficacy population. Last on‐therapy observations were carried forward for patients with missing data points". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 8 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week single‐blind placebo lead‐in | |
| Participants | Sample size: 12 randomised to olanzapine or placebo Participant age range: 18‐65 years Sex: not specified Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Subjects included men and women aged 18–65 years with a diagnosis of SAD and a minimum Brief Social Phobia Scale (BSPS) score of 20". Exclusion criteria: quote: "At the initial visit, blood samples were obtained from eligible subjects for serum chemistry, haematology, and serum beta‐human chorionic gonadotropin for women of childbearing potential ... No concomitant psychotropic medications were permitted during the study". Dropouts: 5/12 (3/7 in the olanzapine group and 2/5 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "Olanzapine was begun at a dose of 5 mg/day (or placebo equivalent) and was titrated upwards as tolerated and clinically indicated at the rate of 5 mg per week to a maximum of 20 mg/day". | |
| Outcomes | Primary and secondary outcomes: LSAS (for reduction of anxiety), BSPS (for reduction of anxiety), SPIN (for reduction of anxiety), SDS (for reduction of functional disability), CGI‐I (for treatment efficacy) and BAS, AIMS and SSS (side effects) Time points: Quote: "The BSPS, SPIN and SDS were performed at weeks 2, 3, 4, 6 and 8, and the LSAS at weeks 4 and 8. Global improvement was measured by the Clinical Global Impression‐Improvement scale (CGI‐I) at all post‐baseline visits (including week 1). Safety was assessed by recording adverse events using the Severity of Symptoms Scale, weight and vital signs, the Barnes Akathisia Scale (BAS) and the Abnormal Involuntary Movements Scale | |
| Notes | Industry funded: yes. Quote: "This study was supported by a grant from Eli Lilly and Company to Dr R. T. Davidson" Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method of randomisation was not reported. Quote: "Subjects were then randomised in a 2:1 ratio to receive flexible‐dose olanzapine or placebo, respectively, for 8 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | A similar proportion of participants withdrew from the olanzapine group (3/7; 43%) compared to the placebo group (2/5; 40%). Participants withdrew due to adverse experience or were lost to follow‐up. The 2 groups did not differ by sample characteristics at baseline. Quote: "Data analysis was performed on the intent‐to‐treat (ITT) population using the last‐observation‐carried‐forward (LOCF) method for missing data ... Missing data has been imputed using appropriate methods ... Of the 12 randomized subjects, seven received olanzapine and five received placebo. Demographic characteristics did not differ significantly between groups. Seven subjects (four olanzapine and three placebo) completed the study through week 8 ... Reasons for early discontinuation were similar in both groups and included loss to follow‐up and adverse experience. Adverse experiences associated with subject discontinuation included gastrointestinal distress (placebo) and sedation (olanzapine). Overall 42% of the subjects dropped out of the study." |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias was identified. |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled trial, maintenance study Duration of intervention: 12 weeks Post‐treatment: 6 months follow‐up Placebo run‐in: no | |
| Participants | Sample size: 84 randomised to phenelzine sulfate or placebo (166 randomised: 45 phenelzine sulfate, 40 cognitive behavioural group therapy, 42 combination therapy, 39 placebo) Mean age (SD): 31.35 (8.36) years Sex: 63 men and 21 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "The inclusion criteria were (1) a primary DSM‐IV diagnosis of SAD and (2) age 18 to 65 years". Exclusion criteria: quote: "The exclusion criteria were (1) a comorbid anxiety disorder more clinically salient for the patient; (2) a lifetime history of schizophrenia, bipolar disorder, or mental disorder due to a general medical condition; (3) major depressive disorder or substance use disorder in the past 6 months; (4) previous failure of treatment with phenelzine or CBT, defined as nonresponse to 60 mg or more of phenelzine (or the equivalent dose of another monoamine oxidase inhibitor) for at least 4 weeks or to 6 sessions of CBT for SAD; (5) concurrent psychiatric or psychological treatment; and (6) pregnancy, lactation, or inability or unwillingness to use contraceptive measures for the duration of the study". Dropouts: 18/84 (13/45 in the phenelzine sulfate group and 5/39 in the placebo group; 40 dropouts across all 4 groups: 22 participants withdrew before receiving treatment, 18 withdrew after receiving treatment) | |
| Interventions | Pharmacological intervention: quote: "Pharmacotherapy patients began with phenelzine sulfate, 15 mg/d, or matching placebo for 3 days, then 30 mg/d for 4 days, 45 mg/d for week 2, and 60 mg/d for weeks 3 and 4. Depending on clinical progress and adverse effects, the dosage could be raised to 75 mg for week 5 and to 90 mg for weeks 6 to 12". | |
| Outcomes | Primary and secondary outcomes: LSAS (for reduction of anxiety), ADIS (diagnostic measure), CGI‐S (for reduction of anxiety), HRSD (for reduction of depression), CGI‐I (for treatment efficacy), FQ (for reduction of anxiety), SIAS (for reduction of anxiety), SPS (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "The study had 4 phases. The first phase (acute treatment) lasted 12 weeks. Medication visits occurred weekly for 4 weeks, then every 2 weeks during this phase" The second, third and fourth phase was also 12 weeks each | |
| Notes | Industry funded: no. Quote: "This study was supported in part by grants DA023200 (Dr Blanco), MH44119 (Dr Heimberg), and MH57148 (Dr Liebowitz) from the National Institutes of Health; by the New York State Psychiatric Medication provided by industry: no Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized according to a table of pseudorandom numbers by the New York site data manager (A.B.S.), who had no patient contact". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether the participants were blinded. Quote: "Pharmacotherapy patients began with phenelzine sulfate, 15 mg/d, or matching placebo for 3 days ... Patient allocation was concealed from all other research personnel at both sites before randomization and from independent evaluators providing the clinician administered ..." |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Measures administered by independent evaluators blinded to treatment condition". |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportion of participants withdrew from the phenelzine sulfate group (13/45; 29%) compared to the placebo group (5/27; 19%). No information was provided regarding the reasons for treatment withdrawal. Nevertheless, participants did not differ by group characteristics at baseline. Quote: "Of the 166 individuals randomized, 12 from the placebo group and 10 from the phenelzine group withdrew from the study before receiving any treatment and were excluded from the analyses ... Groups did not differ significantly in demographic characteristics ... Rates of discontinuation were 37.1% (13 of 35) in the phenelzine group and 18.5% (5 of 27) in the placebo group. Those rates were not significantly different when examining all groups jointly or in pairwise treatment comparisons". Overall 25% of the participants dropped out of the study. Quote: "Using linear mixed‐effects models ... Response and remission rates were compared between groups using 2 tests of independence, using the last observation carried forward for individuals who dropped out before the endpoint". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias was identified. Quote: "There were some baseline differences across treatment groups and sites. However, the results remained significant after appropriate statistical adjustments, suggesting the robustness of the findings". |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind, maintenance study Duration of intervention: 24 weeks Post‐treatment: no follow‐up Placebo run‐in: 1 week single blind placebo period | |
| Participants | Sample size: 196 randomised to sertraline or placebo (387 randomised: 98 sertraline, 98 sertraline and exposure therapy, 93 exposure therapy, 98 placebo) Mean age (SD): 40.4 (10.4) years for all 4 groups, not specified for the sertraline and placebo separately Sex: 153 men and 234 women (for all 4 groups, not specified for the sertraline and placebo separately) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Subjects aged 18‐65 years with GSP according to DSM‐IV criteria study of at least 1 years duration and rated as moderately ill were included in the study ... Patients with comorbid dysthymia or specific phobias were allowed to enter the study". Exclusion criteria: quote: "Patients with panic disorder with onset before social phobia or any other current anxiety, major depression, substance use or eating disorder were not eligible. In addition, patients with a lifetime history of bipolar disorder or psychosis were excluded". Dropouts: 16/196 (9/98 in the sertraline and 7/98 in the placebo groups) | |
| Interventions | Pharmacological intervention: all participants received either 1 tablet of sertraline 50 mg or placebo once daily, the dose was increased to 100 mg at 4 weeks and 150 mg at 8 and 12 weeks | |
| Outcomes | Primary outcomes: CGI (for treatment efficacy) and SPS (for reduction of anxiety) Secondary outcomes: BSPS (for reduction of anxiety), MFQ (for reduction of anxiety), FNES (for reduction of anxiety), SDS (for reduction of functional disability) and SF‐36 (quality‐of‐life measure) Time points: Quote: "Investigators made intermediate efficacy ratings after 4, 8, 12, and 16 weeks, and final efficacy assessment after 24 weeks of treatment". | |
| Notes | Industry funded: yes. Quote: "Funding was obtained from Pfizer Inc". Medication provided by industry: yes Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Three hundred and eighty‐seven patients were randomly assigned by a computer to receive double‐blind sertraline or placebo in blocks of eight subjects so that four patients in each block were randomised to each of the treatments". |
| Allocation concealment (selection bias) | Low risk | Quote: "Sealed envelopes of allocations from this list were kept by the investigators and opened after the inclusion of the patient into the study ... Tablets were packaged and numbered by the sponsor and personally delivered to each investigator". |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in sealed envelopes. However, it is not clear whether both participants and personnel were blinded. Quote: "Sealed envelopes of allocations from this list were kept by the investigators and opened after the inclusion of the patient into the study". |
| Blinding of outcome assessment (detection bias) | High risk | Outcome assessors were not blinded to treatment. Quote: "Since many of the general practitioners included as investigators worked in single practices, it was not possible to obtain blinded efficacy assessment". |
| Incomplete outcome data (attrition bias) | Low risk | A small proportion of participants withdrew from the sertraline (9/98; 9%) and placebo groups (7/98; 7%). Reasons for withdrawal were not clearly stated by treatment group. Participants did not differ by group characteristics at week 24. Quote: "Two hundred and fifty‐three patients completed 24 weeks of treatment (65%). Three hundred and fifty‐four patients were included in the intent‐to‐treat efficacy population (93%) ... In individual analyses, no interaction was observed between response and each of the variables gender, age, country, recruitment method, medication or exposure therapy". Overall 8% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication and study funded by industry. No other sources of bias was identified. |
| Methods | Design: single‐centre, double‐blind, placebo‐controlled study (NCT00246441) Duration of intervention: 16 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 42 randomised to paroxetine or placebo Mean age (SD): 29 (7.4) years Sex: 22 men and 20 women Diagnostic measure: Structured Clinical Interview for DSM‐IV (SCID). Inclusion criteria: quote: "To be included in the study, individuals had to (1) be 18–65 years old; (2) have sufficiently severe social anxiety disorder, as defined by a total score of at least 60 on the Liebowitz Social Anxiety Scale; (3) report using alcohol to cope with social anxiety; and (4) consume at least 15 standard drinks in the previous 30‐day period". Exclusion criteria: quote: "Medical exclusion factors included: (1) history of prior medical detoxification from alcohol; (2) current use of psychotropic medications; (3) seeking treatment for alcohol problems; (4) urine drug screen positive for illicit drugs other than marijuana; and (5) liver enzymes greater than three times normal levels. History of prior medical detoxification or treatment seeking for alcohol problems was exclusionary for ethical reasons since no explicit alcohol intervention was provided ... They were excluded if they had current bipolar disorder, schizophrenia, substance abuse or dependence other than alcohol, nicotine, marijuana, or presence of significant suicidality". Dropouts: 4 (insufficient information to determine dropout rates for the 2 groups separately) | |
| Interventions | Pharmacological intervention: quote: "All subjects were initiated at a dose of 10 mg per day of paroxetine or matching placebo. Active medication and placebo were over‐encapsulated by the investigational pharmacy with 100 mg of riboflavin, a biomarker used to measure medication compliance. The titration plan in the protocol was to increase the dose weekly over 4 weeks from 10 to 20 to 40 to 60 mg daily, pending tolerability". | |
| Outcomes | Primary outcome: LSAS (for reduction of anxiety) Secondary outcomes: LSAS (for reduction of anxiety), CGI‐I (for treatment efficacy), and SPIN (for reduction of anxiety) Time points: Quote: "At weekly visits throughout the trial the clinician also rates improvement in social anxiety severity as compared to baseline on the same 1–7 point scale (CGI‐I)" | |
| Notes | Industry funded: no. Quote: "This work was supported by grants R01 AA013379 (CLR), K24 AA013314 (CLR), P50 AA010761, and K23 AA014430 (SWB) from the National Institute on Alcohol Abuse and Alcoholism". Medication provided by GlaxoSmithKline Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised using a computerised urn randomisation programme. Quote: "Following determination of eligibility, subjects were randomized to either paroxetine or matching capsule placebo, using a computerized urn randomization program". |
| Allocation concealment (selection bias) | Low risk | Quote: "Group assignment was maintained by an investigational pharmacist, who also prepared each week’s supply of study medication". |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether the participants were blinded. Quote: "Subjects were randomized to either paroxetine or matching capsule placebo ... All individuals involved in direct care or evaluation of study subjects, or who were involved in study supervision, were blind to group assignment". |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "All individuals involved in direct care or evaluation of study subjects, or who were involved in study supervision, were blind to group assignment ... Clinical and research ratings were collected independently". |
| Incomplete outcome data (attrition bias) | Low risk | 38 (90%) of the 42 participants completed the study. No information was provided on the reasons for study withdrawal. Participants did not differ by group characteristics at baseline. Nevertheless, the total proportion of dropouts (10%) is relatively low, suggesting that dropout rates may not have biased the outcomes. Quote: "All but four participants provided week 16 (end of trial) data, for a 90% research data completion rate ... There were no significant differences at baseline between groups, including age, gender, ethnicity, social anxiety severity, and alcohol use severity ... There were no significant differences between groups, all p values >.05 ... The number of subjects who dropped out of the trial because of side effects were 1 and 0 for the paroxetine and placebo group, respectively" Quote: "Using a mixed model analysis ... Data from all subjects who were randomised to treatment were included in the analysis, according to intent to treat (ITT) standards". |
| Selective reporting (reporting bias) | High risk | Pre‐specified secondary outcomes (i.e. for quality of life and depression) were not mentioned or measured in the study. |
| Other bias | Unclear risk | Medication was provided by industry. No other sources of bias was identified. |
| Methods | Design: single‐centre, randomised, placebo‐controlled, flexible dose, double‐blind Duration of intervention: 20 weeks or to undergo discontinuation treatment every 2 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 36 randomised to clonazepam or placebo Mean age (SD): 40.05 (7.6) years Sex: 23 men and 13 women Diagnostic measure: DSM‐III‐R. Inclusion criteria: quote: "Subjects entered the study if they fulfilled DSM‐III‐R criteria for a principal diagnosis of social phobia, granted informed consent, and were between the ages of 18 and 55". Exclusion criteria: quote: "Exclusion criteria were as follows: a history of schizophrenia, bipolar disorder, organic brain syndrome, antisocial personality disorder, mental retardation, major depression within the past 12 months, panic disorder, alcohol or substance abuse; the concomitant need for other psychotropic drugs; or any ongoing psychotherapy". Dropouts: 8/36 (2/17 in the clonazepam and 6/19 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "At week 24, all subjects exhibiting good clinical response on the CGI‐Improvement scale were randomly assigned to receive either continuation treatment (CT) at the same clonazepam dose for 5 additional months, or discontinuation treatment (DT), which required a fixed‐dose taper of 0.25 mg every 2 weeks. Therefore, 6 weeks of tapered doses were required for the group receiving 1.0 mg/day to reach 0.0 mg, 10 weeks for the 1.5‐mg group, 14 weeks for the 2‐mg group, and 18 weeks for the 2.5‐mg group". | |
| Outcomes | Primary and secondary outcomes: CGI‐S (for reduction of anxiety), BSPS (for reduction of anxiety), MSPSS (for reduction of anxiety), BWC (side effects measure), and fear was measured on a 0‐10 scale, and avoidance was measured along a 5‐point scale. Time points: Quote: "After patients were randomly assigned at week 24, all scales were administered at 2‐week intervals until study completion" | |
| Notes | Industry funded: yes. Quote: "This work was supported by a grant from Hoffmann‐LaRoche to Dr. Jonathan Davidson". Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "All subjects exhibiting good clinical response on the CGI‐Improvement scale were randomly assigned to receive either continuation treatment (CT) at the same clonazepam dose for 5 additional months, or discontinuation treatment (DT)". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Participants were blinded to treatment however there was insufficient evidence to determine if personnel were blinded. Quote: "Subjects received the same number of pills at each visit, with the diminishing dose supplemented by means of matching placebo. From weeks 24 to 26, all subjects received their usual dosage in double‐blind packaging to allow for adjustment to the double‐blind form of medication, having received the regular, marketed brand of the drug up to that time". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | A larger proportion of participants discontinued the study in the placebo group (6/19; 32%) compared to the clonazepam (2/17; 12%) group. Similiar withdrawals were reported across groups. The 2 groups did not differ by treatment characteristics at baseline. Quote: "Within the group of 36 subjects providing discontinuation data, no significant differences were observed between subjects assigned to CT vs. DT groups in age, gender, or ethnic status ... Two subjects in the CT and six in the DT group dropped out of the study for reasons either related to relapse or to other circumstances. The two CT dropouts were a result of side effects and loss to follow‐up. The six DT dropouts were a result of relapse, marital problems that became aggravated during the time of taper, and work obligations". Overall 22% of the participants dropped out of the study. Quote: "In the event of occasional missing measurement points, the immediately prior observation was carried forward (LOCF)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other source of bias was identified for this study. |
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 10 weeks Post‐treatment: no follow‐up Placebo run‐in: 2 week medication washout period | |
| Participants | Sample size: 75 randomised to clonazepam or placebo Mean age (SD): 37.2 (8.45) years Sex: 43 men and 32 women Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "To be eligible for the study, subjects were required to fulfil DSM‐III‐R criteria for social phobia, with absence of major depression or panic disorder in the last 6 months. Additionally, at least 12 months absence of alcohol or substance abuse was required". Dropouts: 19/75 (10/39 in the clonazepam and 9/36 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "Subjects were assigned to receive either clonazepam or placebo ... The initial dose was 0.25 mg per day for days 1 to 3, increasing to 0.25 mg twice daily on days 4 to 7.05 mg twice daily from days 8 to 14, 0.5 mg in the morning and 1mg at bedtime on days 15 to 17.1 mg twice daily on days 18 to 21, 1 mg in the morning and 1.5mg at bedtime on days 22 to 25, and 1.5 mg twice daily after day 25". | |
| Outcomes | Primary and secondary outcomes: CGI‐S (for reduction of anxiety), LSAS (for reduction of anxiety), FQ (for reduction of anxiety), FNES (for reduction of anxiety), HAMD (for reduction of depression) and Marks‐Kelly Disability Scale (SDS) (for reduction of functional disability) Time points: Quote: "All scales were administered at baseline and at weeks 2, 4, 6, 8, and 10, except for the FQ, which was administered at baseline and at weeks 6 and 10, and the Hamilton Rating Scale for Depression, which was administered at baseline and at week 10" | |
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "The randomization was determined by a list of computer‐generated numbers". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Low risk | Author correspondence: quote: "For the clonazepam trial, I can say that the only person who was not blinded was the statistician, and we never had any talk or contact with him about the matter of blinding during the trial. Neither patients, staff nor raters were unblinded". |
| Blinding of outcome assessment (detection bias) | Low risk | Author correspondence: quote: "For the clonazepam trial, I can say that the only person who was not blinded was the statistician, and we never had any talk or contact with him about the matter of blinding during the trial. Neither patients, staff nor raters were unblinded". |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the clonazepam (10/39; 26%) and placebo group (9/36; 25%). Common withdrawals were a result of poor response. No information was provided on sample characteristics and how groups differed at end point. Quote: "The numbers of subjects remaining in treatment with clonazepam and placebo were at week 19, n=29 and n=27. 75% of clonazepam subjects and 75% of placebo subjects competed the full course of treatment ... Dropout rates at week 8 were generally the result of poor response". Overall 25% of the participants dropped out of the study. Both intention‐to‐treat and last observation carried forward (LOCF) was carried out. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other source of bias was identified for this study. |
| Methods | Design: multicentre, randomised, placebo‐controlled, double‐blind, parallel group study Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no, however, quote: "patients taking psychotropic medications were required to discontinue medication 14 days (fluoxetine 30 days) prior to baseline". | |
| Participants | Sample size: 279 randomised to fluvoxamine or placebo Mean age (SD): 37.25 (0.95) years Sex: 179 men and 100 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Patients had to meet the following inclusion criteria: male or female aged 18 to 70 years, Diagnostic and Statistical Manual, 4th Edition (DSM‐IV) diagnosis of GSAD according to the modified Structured Clinical Interview for the DSM‐IV, minimum score of 60 on the Liebowitz Social Anxiety Scale (LSAS) at the screening visit, a score of less than 18 on the Montgomery‐Asberg Depression Rating Scale at the screening visit, and fluency in English. Women with less than 1 year postmenopausal were required to use an acceptable form of birth control. Pregnant or lactating women were not eligible". Dropouts: 119/279 (66/139 in the fluvoxamine and 53/140 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "Patients randomized to receive fluvoxamine CR began treatment at 100 mg/d at day 1 (baseline). The dose could be increased, based on efficacy and tolerability, in increments of 50 mg/d at 1‐week intervals up to a maximum dose of 300 mg/d. The dose remained constant during weeks 6 through 12. The minimum dose allowed at any time during the study was 100 mg/d". | |
| Outcomes | Primary outcome: LSAS (for reduction of anxiety) Secondary outcomes: CGI (for treatment efficacy), SDS (for reduction of functional disability), PGI (for treatment efficacy), ASEX (assesses sexual experiences), and MADRS (for reduction of depression) Time points: Quote: "Efficacy measures were assessed at baseline, weeks 2, 4, 6, 8, 10, and 12, or upon early termination" | |
| Notes | Industry funded: yes. Quote: "This study was supported by a grant from Solvay Pharmaceuticals, Inc". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "For fluvoxamine, randomization was determined for each site by the sponsor (Solvay) from a central source". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Low risk | Author correspondence: quote: "All site study personnel and the patients remained blind throughout the study". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | Similar proportions of participants withdrew from the fluvoxamine CR (66/139; 47%) and placebo group (53/140; 39%). Reasons for withdrawal were similar between groups except for lack of efficacy and adverse events. No information was provided on sample characteristics at endpoint, although groups did not differ significantly on demographics and clinical history at baseline. Quote: "Of these 279 patients, 73/139 (53%) in the fluvoxamine CR treatment group and 87/140 (62%) in the placebo treatment group completed the study, a non statistically significant difference. The reasons for withdrawal were similar between treatment groups with the exception of lack of efficacy (8% of the placebo group compared with <1% of the fluvoxamine CR group) and adverse events (26% of the fluvoxamine CR group vs. 1% of the placebo group)". Overall 43% of the participants dropped out of the study. Quote: "All analyses of response refer to the conventional last observation carried forward algorithm for all patients who had at least 1 dose of study medication, evaluable efficacy data at baseline and at least 1 post baseline efficacy assessment (intent‐to‐treat efficacy population, fluvoxamine CR = 121 patients; placebo = 126 patients)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias was identified. |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled parallel group trial Duration of intervention: 14 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 117 randomised to fluoxetine or placebo (295 randomised: 57 fluoxetine, 60 comprehensive cognitive behaviour therapy group, 59 combinations of comprehensive cognitive behaviour therapy and fluoxetine, 59 combinations of comprehensive cognitive behaviour therapy and placebo, 60 placebo group) Mean age (SD): 36.6 (10.65) years Sex: 66 men and 51 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Inclusion criteria were: (1) DSM‐IV diagnosis of GSP; (2) age between 18 and 65 years; (3) fluency in English; and (4) provision of written informed consent". Dropouts: 33/117 (13/57 in the fluoxetine and 20/60 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "Fluoxetine was started at 10 mg/d, increasing on day 8 to 20 mg/d, on day 15 to 30 mg/d, and on day 29 to 40 mg/d". | |
| Outcomes | Primary outcomes: CGI (for treatment efficacy) and BSPS (for reduction of anxiety) Secondary outcome: SPAI (for reduction of anxiety) Time points: Quote: "Independent evaluator ratings were conducted at baseline and at weeks 4, 8, and 14" | |
| Notes | Industry funded: no. Quote: "This study was supported by grant R10‐ MH49339‐05A1 from the National Institute of Mental Health, Bethesda, Md (Drs Davidson and Foa)". Medication provided by industry: quote: "Medication and matching placebo were provided by Eli Lilly, Indianapolis, Ind". Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Subjects were assigned to treatment by block randomisation, which was generated by computer program". |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "The fluoxetine study ‐ I believe we provided medication in bottles which carried a pre‐numbered label based on the randomization". |
| Blinding of participants and personnel (performance bias) | Low risk | Author correspondence: quote: "Patients, raters and medical staff were blind as to whether drug or placebo was given". |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "An independent rater, blinded to treatment assignments, conducted the primary outcome assessments". |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the clonazepam (13/57; 23%) and placebo group (20/60; 33%). Reasons for withdrawals were similar across groups (i.e. adverse effects, unclear, depression, not improving, treatment too difficult etc). No information was provided on sample characteristics at end point. Quote: "The overall significance for rate of dropout by treatment type was not statistically significant". Overall 28% of the participants dropped out of the study. Quote: "Linear mixed‐effect model analyses included all randomised subjects and were conducted using pretreatment and posttreatment behavioral measures, with the behavioral measure as the dependent variable". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication and study funded by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, fixed dose, double‐ blind Duration of intervention: 12 weeks Post‐treatment: 6, 9 and 12 months follow‐up Placebo run‐in: no | |
| Participants | Sample size: 77 randomised to brofaromine or placebo Mean age (SD): 37.8 (10.3) years Sex: 45 men and 32 women Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "Patients with comorbid DSM‐III‐R generalized anxiety disorder, simple phobia or dysthymia were accepted in the study". Exclusion criteria: quote: "Patients with a history of DSM‐III‐R major depressive episode, a total score of 15 or more on the HDRS, those with other Axis I disorders, suicidal ideation, severe sleep disturbances, organic brain diseases, alcohol or drug abuse within the last 5 years, pregnancy or lactation or some other clinically relevant medical condition that might interfere with the study were excluded". Dropouts: 8/77 (5/37 in the brofaromine and 3/40 in the placebo group). | |
| Interventions | Pharmacological intervention: quote: "Brofaromine or placebo was given twice daily; the first week 2 x 25 mg, the second week 2 x 75 mg to the dose of 150 mg/day". | |
| Outcomes | Primary outcomes: LSAS (for reduction of anxiety) and CGI (for treatment efficacy) Secondary outcomes: HRSD (for reduction of depression), MADRS (for reduction of depression), HARS (for reduction of anxiety), STAI (for reduction of anxiety) and SCL‐90 (for reduction of anxiety) Time points: Quote: "Assessments were made before treatment and at weeks 1, 2, 4, 6, 8 and 12. The HRSD was administered before treatment and at week 12 or when the patient prematurely discontinued the trial (end‐point). The Montgomery‐Asberg Depression Rating Scale (MADRS) was used at every visit" | |
| Notes | Industry funded: yes. Quote: "Grants for the study were given by Ciba, Pharmaceuticals Division, Sweden". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "The study was double blind and patients were randomised (1:1) to brofaromine or placebo and treated for 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | A small proportion of participants withdrew from the brofaromine (5/37; 14%) and placebo group (3/40; 8%). More participants in the brofaromine group withdrew due to side effects compared to the placebo group. However, the 2 groups did not differ significantly. Quote: "Five patients in the brofaromine group and 3 in the placebo group withdrew prematurely from the study. One brofaromine patient withdrew because of untolerable side effects, 1 after 3 days (increased anxiety) and 3 between week 2 and 10 (sleep disturbance, nausea and diarrhoea, and irritability and hyperactivity). The 3 placebo patients withdrew for administrative reasons, poor compliance and unsatisfactory therapeutic effect, respectively ... In all 35 different adverse symptoms were reported. The total number of such reports was 192 in the brofaromine group (n=36) and 94 in the placebo group (n=40). Most of the reported symptoms did not differ significantly between groups". Overall 10% of the participants dropped out of the study. ITT population was assessed and LOCF was used for missing data. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias was identified. |
| Methods | Design: multicentre, randomised, double‐blind, fixed dose study Duration of intervention: 11 weeks, with a 6‐day titration period and 1‐week taper period Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 329 randomised to pregabalin or placebo Mean age (SD): 35.4 (5.68) years Sex: 195 men and 134 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Patients were enrolled if they were at least 18 years of age and met the DSM‐IV criteria for SAD, generalized subtype, confirmed using the Mini‐International Neuropsychiatric Interview, with a LSAS score of at least 50 at both screening and baseline. Women were enrolled if they were using a hormonal or barrier method of contraception, or were menopausal or surgically sterilized, and had a negative pregnancy test at the screening visit, and were not lactating". Exclusion criteria: quote: "Patients were excluded from the study for any of the following reasons: a current DSM‐IV diagnosis of panic disorder, with or without agoraphobia, GAD, anorexia, bulimia, delirium, dementia, or any other clinically significant cognitive disorders, major depressive disorder, obsessive‐compulsive disorder, posttraumatic stress disorder, or borderline or antisocial personality disorder; a current or past history of schizophrenic or psychotic disorder, bipolar disorder, or factitious disorder; a diagnosis of substance abuse/dependence unless in full remission for at least 6 months or a positive urine drug screen; a score of at least 3 on item 1 (depressed mood) at screening of the Hamilton Depression Rating Scale (HAM‐D); a creatinine clearance of less than 60 ml/min; any clinically significant or unstable hematological, autoimmune, endocrine, cardiovascular, renal, hepatic, gastrointestinal, or neurological disorder; electrocardiogram (ECG) changes indicating acute ischemia; a recent history of seizure disorder, and any need for treatment with anti‐convulsants; any previous treatment with pregabalin, or use of gabapentin or benzodiazepines within 2 weeks of baseline; or current use of any psychotropic medications". Dropouts: 96/329 (25/78, 25/86, 26/82 in the pregabalin and 20/82 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "Patients who continued to meet eligibility criteria at the end of the screening phase were randomized to double‐blind, parallel‐group treatment with one of three fixed daily doses of pregabalin, 300 mg [administered 100 mg three times daily (TID)], 450 mg (administered 150 mg TID), 600 mg (administered 200 mg TID), or matching placebo". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: HARS (for reduction of anxiety), HAM‐D (for reduction of depression), CGI‐I (for treatment efficacy), MFQ (for reduction of anxiety) and the SF‐36 Health Survey (measure of health status) Time points: Quote: "The LSAS and the MFQ were administered at screening, baseline, and weeks 1, 2, 4, 6, 8, and 10 (the LSAS was also administered at a follow‐up visit). The CGI‐I was administered on the same schedule starting at week 1. Other secondary measures were obtained at screening, baseline, and at week 10 (or the time of early termination)" | |
| Notes | Industry funded: yes. Quote: "This study was funded by Pfizer Inc". Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients who continued to meet eligibility criteria at the end of the screening phase were randomized to double‐blind, parallel‐group treatment with one of three fixed daily doses of pregabalin...". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Patients who continued to meet eligibility criteria at the end of the screening phase were randomized to double‐blind, parallel‐group treatment with one of three fixed daily doses of pregabalin, 300 mg [administered 100 mg three times daily (TID)], 450 mg (administered 150 mg TID), 600 mg (administered 200 mg TID), or matching placebo". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | A similar proportion of participants withdrew from the pregabalin groups (25/78 300mg; 25/86 450mg; 26/82 600mg, 32%, 29%, and 31% respectively) and placebo group (20/82; 24%). More participants in the pregabalin groups withdrew due to side effects. Other reasons for withdrawal were similar across groups (i.e. discontinued, adverse events, lack of efficacy, withdrew consent, lost to follow‐up and miscellaneous). No information was provided on sample characteristics at endpoint; however, the groups were comparable on baseline characteristics. Quote: "The proportion of patients completing study treatment was slightly lower for patients in the pregabalin 300 (67.9%), 450 (70.9%), and 600 mg (68.3%) dosage groups compared with the placebo group (75.65) ... Baseline demographic and clinical characteristics were comparable among the four treatment groups ... The majority of patients in all treatment groups experienced adverse events during the double‐blind treatment phase. The proportion of patients experiencing at least one adverse event was higher in the pregabalin treatment groups than in the placebo group, though the rates among the three pregabalin dose groups were similar". Overall 29% of the participants dropped out of the study. Quote: "Efficacy measures were analysed using the intent‐to‐treat (ITT) population ... Endpoint was defined as last observation carried forward (LOCF)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. Quote: "The relatively high attrition rate in the pregabalin treatment groups (29–32%) may have biased this analysis". |
| Methods | Design: multicentre, randomised, double‐blind, placebo controlled, experimental trial Duration of intervention: 6 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 36 randomised to NK1 antagonist GR205171, citalopram or placebo Mean age (SD): 31.6 (7.7) years Sex: 17 men and 19 women Diagnostic measure: DSM‐IV Inclusion criteria: DSM‐IV criteria for social phobia with no other serious psychiatric disorders Exclusion criteria: quote: "Main criteria for exclusion were treatment of social anxiety in the past 6 months, current serious or dominant psychiatric disorder other than social phobia (e.g., psychosis, major depressive or bipolar disorder), neurological disorders, somatic disease, chronic use of prescribed medication, abuse of alcohol/narcotics, pregnancy, menopause, left handedness, previous PET examination, and positive family history of cancer". Dropouts: 0 | |
| Interventions | Pharmacological intervention: quote: "The NK1 group received a daily oral dose of 5 mg GR205171, which started after 14 days of placebo because of limited available safety data on repeated dosing. GR205171 was taken as 4 mL solution made up to 100 mL in orange juice. The SSRI group was treated with 40 mg citalopram (one tablet), starting with 20 mg (half tablet) during the first week". | |
| Outcomes | Primary outcome measures: CGI‐I (for treatment efficacy), STAI‐S (for reduction of anxiety) and LSAS‐SR (for reduction of anxiety) Secondary outcome measures: CGI‐S (for reduction of anxiety), CGI‐I (for treatment efficacy), SPSQ (for reduction of anxiety), SPS (for reduction of anxiety), SIAS (for reduction of anxiety), GAF (for reduction of functional disability), PRCS (for performance anxiety), and SDI (for reduction of functional disability) Time points: Quote: "Response rate was determined by the Clinical Global Impression improvement item (CGI‐I) administered by a psychiatrist (K.W.) at weeks 2, 4, and 6 and at follow‐ups" | |
| Notes | Industry funded: yes. Quote: "This research was funded by GlaxoSmithKline, with additional support from the Swedish Research Council (MF and TF), the Bank of Sweden Tercentenary Foundation (MF), and the Swedish Brain Foundation (TF)". Medication provided by industry: quote: "GlaxoSmithKline (Verona, Italy) supplied the study drugs for a 6‐week treatment period". Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "Randomization was performed by the statisticians at their Verona (Italy) research unit. Only the randomization list was provided to us, in my recollection a blocked randomization as the sample sizes were equal across the three arms". |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "The study codes (opaque envelopes) were locked in and kept safe by a Quintiles confederate during the whole study period until the study was unblinded". |
| Blinding of participants and personnel (performance bias) | Low risk | Author correspondence: quote: "Personnel involved in the study only had access to a randomization list containing randomization numbers. The randomization list was created in Verona and the allocation was kept secret there in accordance with GSK research standards, also see above regarding the study codes. All participants and personnel involved in the study (planning, treatment, data collection, imaging, analyses, CRO activities etc) were blinded". |
| Blinding of outcome assessment (detection bias) | Low risk | Author correspondence: quote: "All participants and personnel involved in the study (planning, treatment, data collection, imaging, analyses, CRO activities etc) were blinded". |
| Incomplete outcome data (attrition bias) | Low risk | There were no dropouts reported during this study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication and study funded by industry. No other sources of bias was identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: 6 months follow‐up Placebo run‐in: no | |
| Participants | Sample size: 64 randomised to phenelzine sulfate or pill‐placebo (133 randomised: 36 CBGT, 31 phenelzine sulfate, 33 pill‐placebo, 33 educational‐supportive group therapy) Mean age (SD): 34.1 (9.3) years Sex: 36 men and 28 women Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "For study inclusion, prospective patients had to meet criteria for social phobia and had to be between 18 and 65 years old, fluent in English, willing to provide written informed consent, and able to participate responsibly in treatment". Dropouts: 11/26 of 64 (5/31 in the phenelzine sulfate and 6/33 in the placebo groups, the additional dropouts were found in the CBGT (n=8) and ES group (n=7)) | |
| Interventions | Pharmacological intervention: quote: "Patients received 15 mg phenelzine sulfate tablets (n=31) or matching placebo (n=33) in 1 morning dose; dosages of 60 mg/d and greater were split between morning and noontime. Dosage started at 15 mg/d and increased to 30 mg/d on day 4, to 45 mg/d on day 8, and to 60 mg/d on day 15. After 4 weeks dosages could be raised to 75 mg/d depending on symptoms and adverse effects. After 5 weeks dosages could be raised to 90 mg/d". | |
| Outcomes | Primary and secondary outcome measures: CGI‐I (SPDS) (for treatment efficacy), LSAS (for reduction of anxiety), ADIS‐R (for reduction of anxiety), FNES (for reduction of anxiety), FQ (for reduction of anxiety), SIAS (for reduction of anxiety), SPS (for reduction of anxiety) and SCL‐90 (for reduction of anxiety) Time points: Quote: "Assessments were repeated after 6 (interviews and questionnaires only) and 12 weeks of treatment" | |
| Notes | Industry funded: no. Quote: "Supported by grant MH44 119 and MH40 121 from the National Institute of Mental Health, Bethesda, Md, and grant PO5 MH30906 from the New York State Psychiatric Institute Mental Health Clincal Research Center, New York". Medication provided by industry: quote: "Parke‐Davis Pharmaceuticals, Morris Plains, NJ, supplied Nardil and matching placebo". Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "The randomization sequence was generated by using a printed random numbers table from a statistics text book and was prepared before the study began, separately for each of the two study sites. The last digit of each number sequence was used to determine treatment allocation for cohorts of approximately 6 patients at a time (this was a study of group psychotherapy versus phenelzine)". |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "Cohorts of approximately 6 patients included both phenelzine and placebo patients randomly intermixed". |
| Blinding of participants and personnel (performance bias) | Low risk | Author correspondence: quote: "Blinding was carried out by separating of functions between personnel, by separation of location of offices used for different purposes, and by the mixing of drug and placebo patients in the same cohort. Regarding medication/placebo status, patients, physicians, and assessors were blinded". |
| Blinding of outcome assessment (detection bias) | Low risk | Author correspondence: quote: "Regarding medication/placebo status, patients, physicians, and assessors were blinded. We conducted regular assessments of the integrity of blinding, and on the few occasions when it appeared necessary, we switched patients to different assessors. Regarding psychotherapy status, this was clearly known to patients and therapists". |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the phenelzine sulfate (5/31; 16%) and pill‐placebo group (6/33; 18%). Reasons for withdrawal were similar across groups (i.e. noncompliance, lack of efficacy, adverse effects, non treatment effects, unknown reasons, and positive effects) and groups did not differ by sample characteristics at week 12. Quote: "Attrition (n=26) did not differ across conditions. Eight patients discontinued CBGT, 5 discontinued phenelzine sulfate therapy, 6 discontinued placebo use, and 7 discontinued ES. Five patients were noncompliant, 5 patients discontinued therapy because of positive treatment effects, 3 because of lack of efficacy, 5 because of adverse effects, 2 because of non treatment‐related events, and 6 because of unknown reasons. There were no severe adverse effects ... Completers and dropouts did not differ on demographic or pretreatment clinical measures or group cohesion". Overall 17% of the participants dropped out of the study. All analyses were ITT with LOCF for dropouts. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication was provided by industry. No other sources of bias was identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: 1 month follow‐up Placebo run‐in: 1‐week, single‐blind, placebo lead‐in period | |
| Participants | Sample size: 358 randomised to escitalopram or placebo Mean age (SD): 38 (11) years Sex: 195 men and 163 women Diagnostic measure: DSM‐IV and MINI Inclusion criteria: quote: "The patient population comprised female and male out‐patients with a primary diagnosis of generalised social anxiety disorder established by means of a diagnostic interview following DSM–IV criteria, using the MINI to assist in the exclusion of disallowed comorbidity. At the screening visit, patients 18–65 years old were selected if they had a total score of at least 70 on the LSAS; with exhibited fear or avoidance traits in at least four social situations, and were otherwise healthy based on a physical examination". Exclusion criteria: quote: "Patients were excluded if they had another Axis I disorder that was considered the primary diagnosis within the previous 6 months, if the investigator diagnosed a serious risk of suicide or if the MADRS total score was higher than 19. Patients were also excluded if they had a DSM–IV diagnosis of alcohol or drug misuse during the past 6 months, or if they had taken a psychoactive drug (including any type of antidepressant, beta‐blocker, benzodiazepine, narcotic, analgesic, antipsychotic, or herbal remedy) within 2 weeks (5 weeks for fluoxetine and 6 months for depot neuroleptics) before screening, or if the patient had a positive urine drug screen for opiates, methadone, cocaine, amphetamines or benzodiazepines. The only allowed concomitant use of a psychotropic drug during the study was chloral hydrate taken as a hypnotic but not for more than three consecutive nights. Furthermore, patients with a diagnosis of mania or hypomania, body dysmorphic disorder, schizophrenia/other psychotic disorder, eating disorders, mental retardation or any Axis II cluster diagnosis were also excluded. Patients with a known drug (including citalopram) allergy or hypersensitivity or a known lack of therapeutic response to an adequate trial with citalopram were also excluded. Patients participating in a formal psychotherapy programme that went beyond medical counselling were not included". Dropouts: 68/358 (36/181 in the escitalopram and 32/177 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "The initial dosage of escitalopram was 10 mg per day. The dosage could be increased to 20 mg per day after 4, 6 or 8 weeks of treatment in case of an unsatisfactory response, judged as a score above 5 on the CGI–S rating for severity or no decrease in CGI–S score since baseline. The mean daily dose of escitalopram was 17.6mg at week 12". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: LSAS subscales (for reduction of anxiety), CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), SDS (for reduction of functional disability), and MADRS (for reduction of depression) Time points: Quote: "Efficacy and tolerability were assessed at baseline and after 1, 2, 3, 4, 6, 8 and 12 weeks of treatment" | |
| Notes | Industry funded: yes. Quote: "The study was sponsored by H. Lundbeck A/S." Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients with generalised social anxiety disorder were randomised to receive placebo (n=177) or 10‐20 mg escitalopram (n=181) in a 12‐week, double‐blind trial". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Patients who met selection criteria entered a 1‐week, single‐blind, placebo lead‐in period before being randomised to 12 weeks of double‐blind treatment with escitalopram or matched placebo capsules". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the escitalopram (36/181; 20%) and placebo group (32/177; 18%). Withdrawals differed slightly, although not significantly, across groups (i.e. withdrew treatment, adverse effects, lack of efficacy, consent withdrawn, protocol violation, other administrative forms). More participants in the escitalopram group withdrew due to adverse effects, and more participants withdrew due to lack of efficacy in the placebo. The 2 groups did not differ by sample characteristics at baseline. Quote: "A total of 68 patients (19%) withdrew from the study, with no overall between group difference (18% in the placebo group and 20% in the escitalopram group). However, numerically more patients in the escitalopram group (8.8%) than in the placebo group (4.5%) withdrew because of adverse events and numerically more patients in the placebo group (6.2%) than in the escitalopram group (2.2%) withdrew because of lack of efficacy, with the latter difference approaching statistical significance ... There were slightly more men than women in both treatment groups. Baseline characteristics were similar for the two treatment groups with the exception of age and duration of the disorder, both of which were slightly higher in the escitalopram group". Overall 19% of the participants dropped out of the study. All analyses were ITT with LOCF for missing data. Observed cases analysed. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias was identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, fixed dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week placebo run‐in period | |
| Participants | Sample size: 578 randomised to moclobemide or placebo Mean age (SD): 36.4 (9.9) years Sex: 329 men and 249 women Diagnostic measure: SCID‐Ro (adapted from the DSM‐IV) Inclusion criteria: quote: "Patients to be included in the study were adult men and non‐pregnant, non‐lactating women who satisfied the DSM IV criteria for social phobia". Dropouts: 133/578 (insufficient information to determine dropout rates for the 2 groups separately) | |
| Interventions | Pharmacological intervention: quote: "After a 1‐week placebo run‐in period, patients fulfilling the entry criteria were randomly assigned to one of the three treatment groups to receive either 300 mg moclobemide, 600 mg moclobemide, or placebo in two divided daily doses for a 12‐week period. Patients were to take their tablets in the morning and in the evening after a meal. The patients of the 600 mg treatment group started with a reduced daily dose of 300 mg for the first 3 days, increasing to 600 mg on the 4th day". | |
| Outcomes | Primary and secondary outcome measures: LSPS (for reduction of anxiety), CIC‐SP (for treatment efficacy), SDS (for reduction of functional disability), CIS‐SP (for reduction of anxiety), PIC‐SP (for reduction of anxiety), HAM‐A (for reduction of anxiety), and the MADRS (for reduction of depression) Time points: Quote: "Assessments were performed at screen, on baseline and on weeks 1, 2, 3, 4, 6, 8, 10, and 12" | |
| Notes | Industry funded: no Medication provided by industry: medication was supplied by F Hoffmann‐La Roche Ltd Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "After a 1‐week placebo run‐in period, patients fulfilling the entry criteria were randomly assigned to one of the three treatment groups to receive either 300 mg moclobemide, 600 mg moclobemide, or placebo in two divided daily doses for a 12‐week period". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. Quote: "Moclobemide was supplied by F. Hoffmann‐La Roche Ltd as an oval, cylindrical, biconvex, film‐ coated tablet light yellow in colour and scored on one side, containing 150 mg moclobemide. Placebo tablets were identical both in appearance and composition". |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Moclobemide was supplied by F. Hoffmann‐La Roche Ltd as an oval, cylindrical, biconvex, film‐ coated tablet light yellow in colour and scored on one side, containing 150 mg moclobemide. Placebo tablets were identical both in appearance and composition". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | There was insufficient information to determine dropout rates for the 2 groups separately, although the study reported that dropouts were similar across groups. Common reasons for withdrawal across group were insufficient therapeutic response, withdrawal of consent, and adverse events. Quote: "The ITT population comprised 578 patients, who had received treatment and had at least one assessment after baseline; 445 patients completed the study through week 12. The most frequently cited reasons for discontinuation were insufficient therapeutic response (63 patients), withdrawal of consent (22 patients), and adverse events (19 patients). Attrition rates were similar among the three treatment groups (< 30%). The groups did not differ with respect to reasons for early termination, except that insufficient therapeutic response was somewhat more frequent in the placebo group (26 patients) than in the moclobemide 300 mg (18 patients) or 600 mg groups (19 patients). The three treatment groups were similar with respect to their demographic data and baseline characteristics of social phobia and concurrent psychiatric illnesses". Quote: "Demographic results presented in this paper are based on the ITT population. Efficacy results of weeks 8 and 12 were analysed with the last observation carried forward in the case of missing observations (LOCF analysis)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication was provided by industry. No other sources of bias were identified. |
| Methods | Design: single‐centre, randomised, placebo‐controlled, crossover, flexible dose, double‐blind Duration of intervention: 10 weeks Post‐treatment: 3 and 4 months follow‐up Placebo run‐in: no | |
| Participants | Sample size: 12 randomised to sertraline or placebo Mean age (SD): 42.62 (7.54) years Sex: 8 men and 4 women Diagnostic measure: DSM‐III‐R Inclusion criteria: men and women with a DSM‐III‐R social phobia diagnosis. Exclusion criteria: not specified Dropouts: 2/12 (2/6 in the sertraline and 0/6 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "The subjects were randomly assigned to receive either sertraline (N=6) (50‐200 mg/day, flexible dosing) or placebo (N=6) for 10 weeks, followed by taper and no treatment for 2 weeks. The subjects were then crossed over to the other treatment for a further 10 weeks. The sertraline dose was begun at 50 mg/day and was increased 50 mg/day every 2 weeks if there was no treatment response, except if the drug was not tolerated". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: BSPS (for reduction of anxiety), FQ (for reduction of anxiety), SDS (for reduction of functional disability), SF‐36 (MOS) (to measure general health), MADRS (for reduction of depression), and the Liebowitz Social Phobic Disorders Rating Form (for reduction of anxiety) change and severity scales Time points: Quote: "The patients were seen for administration of the outcome measures at baseline and at the end of weeks 2, 6, 10, 12, 14, 18, and 22" | |
| Notes | Industry funded: yes. Quote: "Supported in part by a grant from Pfizer Pharmaceuticals". Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "The subjects were randomly assigned to receive either sertraline (N=6) (50‐200 mg/day, flexible dosing) or placebo (N=6) for 10 weeks ...". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | High risk | A larger proportion of participants discontinued the study in the sertraline (2/6; 33%) group compared to the placebo group (0/6; 0%). One participant withdrew because of an adverse effect and the other withdrew because of lack of efficacy. No information was provided on whether groups differed by sample characteristics at week 10. Quote: "Overall, sertraline was well tolerated. Only one patient left the study early because of adverse events (e.g., queasiness, anxiety, and insomnia); that patient did so after treatment with sertraline, before crossover to placebo. The only other patient to discontinue early dropped out 1 week after crossover to placebo, because of a lack of efficacy after a substantial clinical response to sertraline during the first half of the study". Overall 17% of the participants dropped out of the study. All analyses were intention‐to‐treat. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 14 weeks Post‐treatment: 1 month follow‐up Placebo run‐in: 1‐week, single‐blind, placebo lead‐in | |
| Participants | Sample size: 60 randomised to fluoxetine or placebo Mean age (SD): 39.47 (12.84) years Sex: 25 men and 35 women. Diagnostic measure: DSM‐IV Inclusion criteria: quote: "The sample consisted of 60 subjects with a primary DSM‐IV diagnosis of generalized social phobia with a duration of at least 6 months". Exclusion criteria: quote: "Patients were excluded from the study for the following reasons: other concurrent Axis I disorders within the past 12 months; intolerance or nonresponse to previous fluoxetine treatment; previous participation in a fluoxetine study; concurrent use of psychotropic or centrally acting drugs, anticonvulsants, corticosteroids, or tryptophan; pregnancy or lactation; serious suicide risk; serious medical illness or abnormal lab or electrocardiogram results; history of severe allergies, multiple adverse drug reactions, or seizure disorder (with seizure during the last 12 months); or treatment with any form of psychotherapy during the trial". Dropouts: 12/60 (5/30 in the fluoxetine and 7/30 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "During the first 8 weeks of the 14‐week initial treatment phase, patients were started on either placebo or a fixed dose of fluoxetine 20 mg/day. During this time, a patient’s dose could be decreased to 10 mg/day at any time if an adverse event occurred that would have caused the patient to withdraw from the study. The patient could be increased back to 20 mg/day at the investigator’s discretion if the patient did not experience adverse events while receiving 10 mg/day and was not improving. During the last 6 weeks of the 14‐week initial phase, the patient’s dose could be increased every 2 weeks in 20 mg/day increments to a maximum of 60 mg/day, except for patients who were on 10 mg/day, who must first have been increased to 20 mg/day. A patient’s dose could also be reduced in decrements of 20 mg/day (or from 20 to 10 mg/day) at any time at the investigator’s discretion if an adverse event occurred that would have caused the patient to discontinue the study. Subsequently, a patient’s dose could be increased in increments of 20 mg/day (at scheduled visits only) back to a maximum of 60 mg/day (or from 10 to 20 mg/day followed by increments of 20/mg day to a maximum of 60 mg/day) if the patient did not experience adverse events at a subsequent visit and was not improving. This dosing schedule was used in order to evaluate whether patients would respond to 20 mg (an effective dose for depression) after 8 weeks, and if not, whether increasing the dose to as much as 60 mg would result in a positive response" | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: the Social Phobia Subscale of the FQ (for reduction of anxiety), CGI‐S (for reduction of anxiety), CGI‐I (for treatment efficacy), PGI (for treatment efficacy), HAM‐A (for reduction of anxiety), BSPS (for reduction of anxiety), HAM‐D (for reduction of depression), GAF (for reduction of functional disability) and SF‐36 (for quality of life) Time points: Quote: "The LSAS, CGI‐S, FQ, and BSPS were administered at all visits. The CGI‐I and PGI scales were administered at visits 2 through 17. The HAM‐A, HAM‐D, GAF, and SF‐36 were administered at visits 2, 5, 7, 10, and 17 (or final visit if discontinued early). The HAM‐A and HAM‐D were also administered at visit 1" | |
| Notes | Industry funded: yes. Quote: "This work was supported by a grant from Eli Lilly & Co". Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "randomly assigned to one of three treatment conditions". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the fluoxetine (5/30; 17%) and placebo groups (7/30; 23%). The proportions of women to men was 35:25. Common withdrawals included: participant moved, protocol violation, clinical relapse and adverse events. No information was provided on sample characteristics at end point, however. Quote: "Five (16%) of the 30 fluoxetine patients and 7 (23%) of the 30 placebo patients discontinued before completion of the 14‐week, double‐blind initial therapy phase. Fluoxetine was discontinued because the patient moved (1) or there was a protocol violation (2), clinical relapse (1), or adverse event (1) (palpitations). Placebo was discontinued because the patient moved (1) or there was a lack of efficacy (2), clinical relapse (1), or adverse event (3) (diarrhea, depression, and rash, respectively)". Overall 23% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. There was a high placebo response in this trial. Quote: "By comparison, the change on placebo in the current trial (23.37) was greater than any other individual trial and much greater than the mean placebo change found in the previous studies (11.13). The reason for the high placebo response in this trial is unknown". |
| Methods | Design: multicentre randomised, placebo‐controlled, fixed‐dose, active‐reference study Duration of intervention: 12 and 24 weeks Post‐treatment: 1 month follow‐up Placebo run‐in: 1‐week, single‐blind, placebo lead‐in period | |
| Participants | Sample size: 839 randomised to escitalopram, paroxetine, or placebo Mean age (SD): 36.98 (11.2) years Sex: 394 men and 445 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "The selection criteria were chosen to select physically healthy female and male outpatients with a primary diagnosis of generalised SAD according to DSM‐IV criteria. At the screening visit, patients between 18–65 years of age were included if they had a total score > 70 on the Liebowitz Social Anxiety Scale (LSAS), demonstrable fear and avoidance traits in at least four social situations, and a score > 5 on one or more of the Sheehan Disability Scale (SDS) subscales". Exclusion criteria: quote: "Patients were excluded if: a) they had another Axis I disorder designated the primary diagnosis within the previous 6 months; b) they had a MADRS total score > 18; c) they had a DSM‐IV diagnosis of schizophrenia/other psychotic disorder, mania or hypomania or history thereof, or were currently suffering from alcohol or drug abuse, eating disorders, MDD, panic disorders (patients with panic attacks not due to panic disorders could be included), obsessive compulsive disorders (OCD), body dysmorphic disorder; d) they had an Axis II Cluster B diagnosis; e) they had learning difficulties or had other cognitive disorder; f) the investigator detected a serious risk of suicide or the patient had a score > 5 in the MADRS item 10 (suicidal tendencies); g) they had a known lack of therapeutic response to any SSRI; h) they had a known hypersensitivity to citalopram or escitalopram or a history of severe drug allergy or hypersensitivity; i) they had taken a psychoactive drug (including antidepressants, beta‐blockers, benzodiazepines, antipsychotics, and psychoactive herbal remedies), monoamine oxidase inhibitors (MAOI), or prophylactic treatment (lithium, valproate, or carbamazepine) within 2 weeks (5 weeks for fluoxetine) before screening, an investigational drug (within 3 months before), or triptans; or j) they were receiving (or planning to initiate) formal psychotherapy". Dropouts: 242/839 (42/167, 56/167, and 49/170 in the escitalopram groups, 45/169 in the paroxetine group, and 50/166 in the placebo group, 24 weeks). | |
| Interventions | Pharmacological intervention: quote: "After screening, patients entered a 1‐week, single‐blind, placebo lead‐in period before being randomised equally to 24 weeks of double‐blind treatment with fixed doses of escitalopram (5, 10, or 20 mg/day), paroxetine (20 mg/day), or placebo. Patients who completed double‐blind treatment | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: LSAS (fear/anxiety, avoidance) scores (for reduction of anxiety), CGI‐S (for reduction of anxiety), CGI‐I (for treatment efficacy), SDS (for reduction of functional disability) and DESS (assess side effects) Time points: Quote: "Efficacy and tolerability were assessed at baseline and after 1, 2, 4, 6, 8, 10, 12, 16, 20, 24, 25, and 26 weeks of treatment; tolerability was also assessed 30 days after the last double‐blind dose of study product. Adverse events were assessed at all visits and the clinical assessments were made at the screening visit, and at Weeks 12 and 24" | |
| Notes | Industry funded: yes. Quote: "Contract grant sponsor: H. Lundbeck A/S" Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participantubjects were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "After screening, patients entered a 1‐week, single‐blind, placebo lead‐in period before being randomised equally to 24 weeks of double‐blind treatment with fixed doses of escitalopram". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the escitalopram (42/167, 25.1%; 56/167, 33.5%; 49/170, 28.8%) paroxetine (45/169, 26.6%) and placebo groups (50/166, 30.1%). Primary reasons for withdrawals were adverse events, lack of efficacy, withdrawal of consent and lost to follow‐up. The groups did not differ by sample characteristics at baseline. Quote: "There were no clinically relevant differences in patient demographics or baseline values between the five treatment groups. The treatment groups did not differ significantly in age of SAD onset or duration of SAD, baseline height, weight, or BMI ... Two hundred forty‐two patients (29%) withdrew from the study during the 24‐week, double‐blind period, with similar withdrawal rates in all treatment groups; 22% of all patients had withdrawn by Week 12. Withdrawals due to adverse events were lowest in the 5 mg escitalopram group, whereas withdrawals due to lack of effect were highest in the placebo group. Withdrawal of consent and loss to follow‐up each accounted for < 7% of withdrawals in any treatment group. Because most of the withdrawals occurred in the first 12 weeks of the study, the remaining 12 weeks was too long a period to carry observations forward, so most of the efficacy analyses are based on observed case (OC) analysis". Overall 29% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, flexible‐dose (GSK protocol ID: 29060/790) Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo run‐in period | |
| Participants | Sample size: 370 randomised to paroxetine or placebo (375 randomised: 5 participants withdrew prior to treatment and were excluded from the study, the ITT sample comprised of 370 participants) Mean age (SD): 38.85 (11) years Sex: 270 men and 100 women Diagnostic measure: MINI (according to DSM‐IV criteria) Inclusion criteria: quote: "The Mini‐International Neuropsychiatric Interview (Version 5.0; MINI) was used to screen for social anxiety disorder according to DSM‐IV criteria. Outpatients (≥ 18 years of age) who met the criteria as their primary diagnosis were enrolled. Patients older than 65 years were included if they did not have renal or hepatic impairment". Exclusion criteria: quote: "Patients with a Clinical Global Impressions (CGI)‐Global Improvement score of 1 or 2 at baseline (following the placebo run‐in period) or a score on the 17‐item Hamilton Rating Scale for Depression (HAM‐D) of ≥ 15 at baseline were excluded. Patients evaluated with the MINI who met DSM‐IV criteria for Axis I disorders such as major depressive disorder, obsessive‐compulsive disorder, or panic disorder as a primary diagnosis currently or within 6 months prior to the screening visit were excluded. Also excluded were patients with substance abuse within 3 months of screening or substance dependence within 6 months of screening and patients considered a current homicidal or suicidal risk. Patients with a history of seizures (except febrile seizures), schizophrenia, or bipolar disorder or a current diagnosis of body dysmorphic disorder or a serious medical illness were excluded. In addition, patients who had been treated with psychotropic medications or antidepressants within 14 days of screening, monoamine oxidase inhibitors or fluoxetine within 4 weeks of screening, depot neuroleptics within 12 weeks of screening, or electroconvulsive therapy within the past 3 months were excluded. Patients requiring concomitant therapy with β‐adrenergic blockers, monoamine oxidase inhibitors, benzodiazepines, or other psychoactive medications were excluded. Women who were pregnant, lactating, or of childbearing potential and not practicing a clinically accepted method of contraception were ineligible". Dropouts: 77/370 (30/186 in the paroxetine group and 47/184 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "All patients randomly assigned to paroxetine CR began therapy at 12.5 mg and remained at this daily dose for the first 2 weeks of treatment. Dose elevation was permitted in 12.5‐mg/day increments no more frequently than every 7 days to a maximum of 37.5 mg/day. One dose reduction was permitted only when made necessary by the development of an adverse event. Patients completing the study (or withdrawing prematurely) at doses of 37.5 mg/day received 1 week of taper phase medication at a daily dose of 25 mg before stopping treatment". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) and CGI‐I (for treatment efficacy) Secondary outcome measures: CGI‐S (for reduction of anxiety), SADS (for reduction of anxiety), SDS (for reduction of functional disability) and HAM‐D (for reduction of depression) Time points: Quote: "After the initial screening visit, these efficacy assessments were administered at baseline and weeks 1, 2, 3, 4, 6, 8, and 12 (or at the time of early withdrawal from the study). In addition, the 17‐item HAM‐D was administered by a clinician at baseline and at week 12 (or at the time of early withdrawal)" | |
| Notes | Industry funded: yes. Quote: "This study was funded by GlaxoSmithKline, Research Triangle Park, N.C". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Eligible patients were then randomly assigned at baseline to receive paroxetine CR (paroxetine hydrochloride) (flexible dose range of 12.5–37.5 mg/day) or placebo once daily in a 1:1 ratio for a treatment duration of 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Patients randomly assigned to placebo medication received placebo throughout the study and were dosed in an identical manner to patients randomly assigned to paroxetine CR". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the paroxetine (30/186, 16%) and placebo groups (47/184, 26%). Five participants withdrew prior to treatment and were excluded from the ITT analysis. Common withdrawals include adverse events, protocol deviation, loss to follow‐up and other. More participants in the placebo group discontinued due to lack of efficacy. The 2 groups did not differ by sample characteristics a baseline. Quote: "The treatment groups were generally comparable with respect to age, gender, and race ... A total of 156 patients (83.9%) in the paroxetine CR group and 137 patients (74.5%) in the placebo group completed the 12‐week study. Dropout rates due to adverse events were low and comparable in the 2 treatment groups (2.7% in the paroxetine CR group and 1.6% in the placebo group). A greater proportion of patients in the placebo group withdrew from the study prematurely due to lack of efficacy (2.2% in the paroxetine CR group and 15.8% in the placebo group)". Overall 21% of the participants dropped out of the study. The ITT population was assessed and LOCF and observed cases were used for primary and or secondary missing data. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported. |
| Other bias | Unclear risk | Funding for study provided by industry. The study had various limitations with regards to comparisons of paroxetine CR and IR, as well as dosage ranges different to prior studies conducted. The high percentage of dropouts in the placebo group due to lack of efficacy, however, raises the question of whether blinding of participants was broken. Quote: "It must be emphasized that this study did not include a comparison of paroxetine CR and paroxetine IR. Moreover, the dose ranges studied in the current study were not identical to the dose ranges employed in prior studies, hence conclusions regarding their relative tolerability and efficacy profiles cannot be drawn from these trials ... A greater proportion of patients in the placebo group withdrew from the study prematurely due to lack of efficacy (2.2% in the paroxetine CR group and 15.8% in the placebo group)". |
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 8 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo run‐in | |
| Participants | Sample size: 85 randomised to phenelzine, atenolol or placebo (data however is reported for 74 completers) Mean age (SD): 34.3 (8.5) years Sex: 51 men and 23 women Diagnostic measure: DSM‐III Inclusion criteria: quote: "Medically healthy patients aged 18 to 50 with a DSM‐III criteria for social phobia". Exclusion criteria: quote: "Current major depression or substance abuse, a history of schizophrenia, organicity, or bipolar disorder, avoidant personality disorder, and other medical conditions (e.g. benign essential tumours), as well as prior treatment of phenelzine or atenolol". Dropouts: 11/85 (4/29 in the phenelzine, 5/28 in the atenolol and 2/28 in the placebo groups) | |
| Interventions | Pharmacological intervention: "Treatment with atenolol began at 50 mg/d given in the morning and raised to 100 mg/d, if tolerated, after 2 weeks. Treatment with phenelzine sulfate was begun at 15mg/d and increased to 39 mg/d on day 4, to 45 mg/d on day 8, and to 60 mg/d on day 15. After 4 weeks, depending on clinical state and side effects, the dose of phenelzine sulfate could be optimally raised to 75 mg/d, and to 90 mg/d after 5 weeks. In addition to the 8 week short term treatment phase, the study had maintenance (8 weeks) and discontinuation phases (8 weeks)". | |
| Outcomes | Primary and secondary outcome measures: self‐report versions of the CGI‐S (for reduction of anxiety), LSPS (for reduction of anxiety), CGI‐I (for treatment efficacy), HSC (for reduction of anxiety), SADS (for reduction of anxiety), FNES (for reduction of anxiety), FQ (for reduction of anxiety), WPI (personality measure), HAM‐D (for reduction of depression), HAM‐A (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "These occurred before placebo washout, before randomisation, and at 4‐week intervals thereafter. Patients also underwent weekly physician and self‐ratings" | |
| Notes | Industry funded: no. Quote: "This study was supported in part by grant MH 40121 from the National Institute of Mental Health, Bethesda, Md". Medication provided by industry: yes. Quote: "Parke‐Davis Pharmaceuticals Co, Morris Plains, NJ, kindly supplied phenelzine, and Stuart Pharmaceuticals, Wilmington, Del, kindly supported atenolol for the study". Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "randomised to an 8 week short term comparison" |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | High risk | A larger proportion of participants discontinued the study in the phenelzine (4/25; 16%) and atenolol groups (5/23; 22%) compared to the placebo group (2/26; 8%). Common withdrawals included rash and other sides effects and change of mind. Groups did not differ by sample characteristics at end point, however. Quote: "Of the 85 patients randomised, 74 completers met prospectively determined criteria for inclusion in end‐phase analyses of at least four weeks of randomised treatment with 2 weeks of phenelzine or atenolol. Eleven other, including two placebo‐, four phenelzine‐, and five atenolol‐blind treatment failed to complete at least four weeks of double‐blind treatment. Reasons for doing so included rash, and cheese rash, change of mind, rediagnosis of schizophrenia,and painful erection, rash and other side effects and non compliance with study procedures ... There were no significant difference in demographic or baseline scores amongst the 74 completers". Overall 15% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication was provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, fixed dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1 week, single blind, placebo run‐in | |
| Participants | Sample size: 384 randomised to paroxetine or placebo Mean age (SD): 36.95 (40.2) years Sex: 225 men and 159 women Diagnostic measure: SCID (modified version of the DSM‐IV) Inclusion criteria: quote: "Adult outpatients (>18 years of age) who met the criteria for the generalised sub‐type of social anxiety disorder were enrolled; patients older than 65 years were permitted if they did not have renal or hepatic impairment and could tolerate paroxetine starting dose of at least 20 mg/day". Exclusion criteria: quote: "Patients who scored 1 or 2 on the CGI‐I scale at baseline or who had a score greater than or equal to 15 at baseline on the HAM‐D scale were excluded. Patients with comorbid psychiatric disorders such as major depression, OCD, generalised anxiety disorder, and panic disorder were excluded using the SCID if comorbid disorder occurred within the past 6 months and was predominant. Also excluded were patients with substance abuse or dependence within 6 months of baseline, body dysmorphic disorder, schizophrenia, bipolar disorder, homicidal/suicidal tendencies, serious medical illness, or a history of seizures, as well as patients who had started psychotherapy within 6 months of baseline or who had been treated with other psychotropic medications or antidepressants within 14 days of baseline, fluoxetine within 5 weeks of baseline, electroconvulsive therapy within the past 3 months. Patients requiring concomitant therapy with beat‐adrenergic medications, warfarin, anticoagulants, digitalis glycosides, phenytoin, cimetidine, or sumatriptan were not included. Women who were pregnant, lactating, or of child‐bearing potential and not practicing a clinically accepted method of contraception were ineligible". Dropouts: 142/384 (31/97, 40/95, 43/97 in the paroxetine and 28/95 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "All patients randomly assigned to paroxetine began therapy at 20 mg/day. Patients were instructed to take 2 capsules each morning irrespective of treatment assignment. Those randomly assigned to paroxetine 20 mg, remained at that dose for the duration of the study. At week 1, patients randomly assigned to the 40 mg paroxetine group were titrated to that daily dose. Doses for the 60 mg paroxetine group were titrated to 40 mg at week 1 and 60 mg at week 2". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) and CGI (for treatment efficacy) Secondary outcome measures: LSAS (fear and anxiety subscales) (for reduction of anxiety), CGI‐S (for reduction of anxiety), SADS (for reduction of anxiety), SDS (for reduction of functional disability) and HAM‐D (for reduction of depression) Time points: Quote: "After the initial screening visit, all other tests were administered to patients at baseline and weeks 1, 2, 3, 4, 6, 8, and 12 (at the time of discontinuation). Safety was assessed by monitoring adverse experiences and vital signs at weeks 1, 2, 3, 4, 6, 8, and 12 (or at discontinuation)" | |
| Notes | Industry funded: yes. Quote: "[s]upported by SmithKline Beecham Pharmaceuticals" Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "After a one week placebo run‐in: "Eligible patients then began a 12 week double blind treatment phase and were randomly assigned at baseline to receive paroxetine, 20, 40, or 60mg or placebo once daily in a 1:1:1:1 ratio". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | The medication and placebo groups did not differ significantly in attrition rate nor by sample characteristics at baseline. The proportion of dropouts increased with increasing dose of paroxetine (20 mg/d: 31/97, 31.9%; 40 mg/d: 40/95, 42.1%; 60 mg/d: 43/97, 44.3%), with the dropout rate in the arm receiving the lowest dose of medication similar to that observed amongst participants receiving placebo (28/95, 29.5%). The proportion of dropouts were higher in the paroxetine group due to adverse experience compared to the placebo group due to lack of efficacy. Withdrawals similar across groups included withdrawn, protocol violation, loss to follow‐up, and other reasons. Quote: "The demographic characteristics of the 4 treatment groups were well matched ... Approximately 63% of the randomly assigned subjects (242/384) completed the 12‐week study. There were no significant differences between the 20‐,40‐, or 60‐mg paroxetine groups and the placebo group in the overall attrition rates ... patients remaining in the study beyond week 2, discontinuation rates were comparable between the paroxetine (8%‐23%) and placebo (20%) groups. The primary reason for early discontinuation in any of the paroxetine groups was adverse experiences (17.5%, 21.1% and 23.7%) , whereas the primary reason for early discontinuation in the placebo group was lack of efficacy (10%) ... However there was no difference in the overall incidence of reported adverse experiences between 20‐mg (92%), 40 ‐mg (91%) and 60‐mg (88%) paroxetine groups ane the placebo group (83%)". Overall 37% of the participants dropped out of the study. Efficacy data are presented at both LOCF and OC for missing points. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1 week, single blind, placebo lead‐in period | |
| Participants | Sample size: 415 randomised to sertraline or placebo Mean age (SD): 35.05 (10.6) years Sex: 247 men and 168 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Study entry criteria required patients to be aged 18 years or over with a primary diagnosis of generalised social phobia of at least 2 years duration and a LSAS score > 68 at baseline. Social phobia was diagnosed using the DSM‐IV. In addition to meeting DSM‐IV criteria for social phobia, patients were required to exhibit fear and/or avoidance of at least 4 social situations. Women of childbearing potential were required to have negative results on a serum beta‐human chorionic gonadotropin pregnancy test and to be using a medically accepted form of contraception". Exclusion criteria: quote: "Patients were excluded if they met DSM‐IV criteria in the previous 6 months for substance dependence, body dysmorphic disorder, major depressive disorder, dysthymia, panic disorder, post‐traumatic stress disorder, or an eating disorder; if they reported any current or past diagnosis of schizophrenia, psychotic disorder, bipolar disorder, or obsessive‐compulsive disorder (OCD); or if they met criteria for a primary diagnosis of generalised anxiety disorder. Patients were also excluded for the following reasons: (1) HAM‐D score of 14 or items 1 rating moderate or greater in severity; (2) currently reporting serious suicidal or homicidal risk; (3) currently receiving specific behavioural or supportive therapy for social phobia or another anxiety disorder; (4) any history of seizure disorder; (5) any serious or uncontrolled medical illness or condition that preludes sertraline use; (6) women who were pregnant, nursing, or lactating; (7) receiving any concomitant therapy with any psychotropic drug or with any drug with a psychotropic component, except zolpidem for insomnia". Dropouts: 122/415 (59/211 in the sertraline and 63/204 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "Sertraline treatment was initiated at a daily dose of 25 mg, which was increased at week 1 to 50 mg. After 2 weeks at a dally dose of 50 mg, patients with sufficient clinical response but good tolerability were permitted to increase to 100 mg, and then by 50 mg increments per week to a maximum dose of 200 mg/day. | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety) and SPI (for reduction of anxiety) Time points: Quote: "Patients were evaluated for medication safety and efficacy at weeks 1, 2, 3, 4, 6, 8, and 12" | |
| Notes | Industry funded: yes. Quote: "Funded by Pfizer Inc, New York, N.Y". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients who continued to meet all inclusion and exclusion criteria were then randomly assigned in a double blind fashion to 12 weeks of double blind treatment with flexible doses of sertraline or matching placebo". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Patients who continued to meet all inclusion and exclusion criteria were then randomly assigned in a double blind fashion to 12 weeks of double blind treatment with flexible doses of sertraline or matching placebo". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of patients withdrew from the sertraline (59/211; 28%) and placebo groups (63/204; 31%). More participants in the sertraline group withdrew due to adverse events compared to others reasons in the placebo groups. Quote: "Patient characteristics were similar in both groups at baseline ... One hundred fifty‐two (72%) of the 211 patients treated with sertraline and 141 (69%) of the patients treated with placebo completed 12 weeks of double‐blind treatment. Reasons for premature discontinuation during treatment with sertraline and placebo, respectively, included the following: withdrawal of consent, 11 (5.2%) versus 17 (8.3%); lost to follow‐up, 17 (8.1%) versus 10 (4.9%); adverse events, 16 (7.6%) versus 6 (2.9%); insufficient clinical response, 5 (2.4%) versus 9 (4.4%); protocol violation, 3 (1.4%) versus 3 (1.5%); and miscellaneous other reasons, 7 (3.3%) versus 18 (8.8%)". Overall 29% of the participants dropped out of the study. All analyses were ITT and LOCF. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, double blind, placebo controlled, parallel group, flexible dose study Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1 week, single blind, placebo lead‐in period | |
| Participants | Sample size: 279 randomised to venlafaxine ER or placebo Mean age (SD): 35.4 (11.55) years (271 randomised, LOCF scores) Sex: 148 men and 123 women (271 randomised, LOCF scores) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Outpatients at least 18 years of age who met DSM‐IV criteria for social anxiety disorder for a least 6 months before study initiation were eligible for screening. Inclusion was dependent on a CGI‐S baseline score > 50 with a decrease of < 30% between prestudy and baseline, and a Covi Anxiety Scale score greater than Raskin Depression Scale score". Exclusion criteria: quote: "Individuals with a history of hepatic or medical disease, mental disorder due to a general medical condition, psychotic disorder or organic brain disorder, seizure disorder, or head trauma were excluded from enrolment. Patients with clinically important Axis I or Axis II comorbidities were excluded from study participation if the disorder was current or was predominant with 6 months of the start of the study. Also excluded were patients with a history of alcohol abuse within 1 year of the study, those who regularly used alcohol, and those with a urine drug screen positive for drugs of abuse. Those with multiple drug allergies or a clinically meaningful abnormality in vital signs and findings from physical examination, ECG, laboratory tests, or urine drug screen were not included. Individuals who used the investigational drugs, antipsychotics, sedative hypnotic drugs, antidepressants, anxiolytics, or migraine medication or received electroconvulsive therapy within 6 months before the study or formal psychotherapy within 30 days of the study day 1 were excluded, as were women of childbearing potential who were pregnant or breastfeeding or who did not utilise a medically acceptable form of contraception". Dropouts: 106/279 (51/139 in the venlafaxine ER and 55/140 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "The venlafaxine ER regimen was potentially 3‐step dose‐escalation process with an initial 75 mg/day dose during the first week ± 3 days. During the second week, the dose was increased to 150 mg/day if clinically indicated to enhance response. The dose was increased to 225 mg/day if clinically indicated on study day 15 ± 3 days. At study completion (or early termination), patients who had been taking more than 1 capsule daily for more than 1 week had their dose tapered". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐S (for reduction of anxiety), CGI‐I (for treatment efficacy), SPIN (for reduction of anxiety), LSAS subscales (for reduction of anxiety), SDS (for reduction of functional disability) and HAM‐D (for reduction of depression) Time points: Quote: "The primary efficacy variable was the LSAS Total score, which was assessed at weeks 1, 2, 3, 4, 6, 8, 10, and 12. The LSAS, CGI‐S, CGI‐I and SPIN assessments were performed at baseline and on study days 7, 14, 21, 28, 42, 56, 70, and 84; the SDS was administered at baseline and on study days 28 and 84; and the HAM‐D and Covi‐Raskin scales were administered at the prestudy visit and on study days 42 and 84 as ancillary evaluations. Final ratings for efficacy were obtained on the last day of full dose administration before tapering or within 3 days of the last full dose" | |
| Notes | Industry funded: yes. Quote: "This study was supported by Wyeth research, Collegeville, Pa". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "eligible patients were randomly assigned to receive either venlafaxine ER or placebo for up to 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the venlafaxine ER (51/139; 37%) and placebo groups (55/140; 39%). More participants in the venlafaxine group discontinued due to adverse events compared to unsatisfactory response in the placebo group. No information was provided on the other reasons for withdrawal, however the 2 groups did not differ by sample characteristics at baseline or at week 12. Quote: "A comparable number of patients in each treatment group completed the study, i.e., 88 in the venlafaxine group and 85 in the placebo group. Significantly more patients in the placebo group (15%) than in the venlafaxine group (2%) discontinued treatment because of unsatisfactory response, while significantly more patients in the venlafaxine group discontinued treatment because of adverse events (17%) ... There were no significant differences between treatment groups for any of the demographic or baseline characteristics, nor did the demographic and baseline characteristics of the ITT patient population differ appreciably from those of the safety population". Overall 38% of the participants dropped out of the study. All analyses were ITT and LOCF. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group comparison Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1 week, single blind, placebo lead‐in period | |
| Participants | Sample size: 440 randomised to venlafaxine ER, paroxetine or placebo Mean age (SD): 36.27 (11.3) years (for 413 participants) Sex: 221 men and 192 women (for 429 participants) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Outpatients 18 years and older who fulfilled the DSM‐IV criteria for SAD for 6 months or longer at screening were eligible to participate in the study. In addition, patients were required to have a LSAS score of 50 or more at prestudy and baseline evaluations; a score of 4 or more on the CGI‐S scale; a prestudy Covi Anxiety Scale total score greater than the Raskin Depression Scale total score; and a prestudy HAM‐D score of less than 15, with a score of 2 or less on the depressed mood items". Exclusion criteria: quote: "Patients with a clinically important Axis I or Axis II disorder other than SAD or avoidant personality disorder were excluded, as were those with a history or current diagnosis of any psychotic illness, patients who were suicidal, and those with a history of drug or alcohol dependence within 1 year of study start. In addition, patients were ineligible if they had used any psychopharmacologic medications within 7 days before study day 1; used antidepressants, anxiolytics, or herbal products intended to treat anxiety or depression within 14 days of the study; received electroconvulsive therapy within 6 months of the study; or used antipsychotic medication or fluoxetine or received treatment with formal psychotherapy within 30 days of the study. Patients with clinically significant abnormal findings on laboratory tests, electrocardiograms, or physical examinations; those with abnormal vital signs; those with a history or presence of clinically important medical conditions; and women of childbearing potential who were pregnant, breastfeeding, or not using a medically acceptable form of contraception were prohibited from participating". Dropouts: 111/440 (insufficient information to determine dropout rates for the three groups separately) | |
| Interventions | Pharmacological intervention: quote: "After a 7 day, single‐blind, placebo lead‐in period, eligible patients were randomly assigned to receive flexible doses of venlafaxine ER (75‐225 mg/d), paroxetine (20‐50 mg/d), or placebo for up to 12 weeks, followed by a taper period of up to 2 weeks. Starting doses were 75 mg/d for venlafaxine ER and 20 mg/d for paroxetine if clinically indicated to improve response, daily doses could be titrated upward each week by 75 mg for venlafaxine ER or 20 mg for paroxetine to a maximum 225 mg/d of venlafaxine ER and 50 mg/d of paroxetine. The dosage could be reduced at any time to improve tolerance; however, the minimum allowed dosages after day 7 were 75 mg/d for venlafaxine ER and 20 mg/d for paroxetine". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), and SPI (for reduction of anxiety) Time points: Quote: "The primary efficacy time point was the final on‐therapy (defined as observations that occurred within 3 days of the patient’s last full dose of study medication) LSAS total score. The week 12 last‐observation‐carried‐forward values were the final on‐therapy observations" | |
| Notes | Industry funded: yes. Quote: "This study was supported by Wyeth research, Collegeville, Pa". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "...patients were randomly assigned to receive flexible doses of venlafaxine hydrochloride ER (75‐225 mg/d), paroxetine (20‐50 mg/d), or placebo for up to 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | There was insufficient information to determine the proportion of dropouts for the three treatment groups. Nevertheless, the three groups did not differ by sample characteristics at baseline nor did they significantly differ by withdrawals. More participants in the venlafaxine and paroxetine group discontinued due to adverse events compared to unsatisfactory response in the placebo group. Quote: "There were no statistically significant differences between the treatment groups for any of the demographic or baseline characteristics ... Three hundred eighteen patients (74.1%) completed the 12‐week double‐blind treatment period and 111 (25.9%) withdrew from the study. Significantly more patients in the active treatment groups (venlafaxine ER and paroxetine) withdrew because of adverse events than in the placebo group, while significantly more patients in the placebo group withdrew because of lack of efficacy; however, there were no significant differences between the active treatment groups. Statistical analyses were performed using last‐observation carried‐forward values for the intent‐to‐treat population". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 10 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week single‐blind placebo washout | |
| Participants | Sample size: 106 randomised to brofaromine or placebo Mean age (SD): 36.5 (9.8) years Sex: 62 men and 40 women (for 102 ITT participants) Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "Participants were 102 outpatients (62 men and 40 women) with a primary DSM‐III‐R diagnosis of social phobia of at least 6 months' duration. Primary social phobia was defined as dominating the clinical picture and temporally preceding any secondary diagnosis ... A minimum score of 8 was required on the Avoidance Subscale of the LSAS ... A minimum score of 4 (moderately ill) was required on the Liebowitz Social Phobia Global Severity Rating Scale. A maximum score of 15 was allowed on the MADRS". Exclusion criteria: quote: "Patients were excluded for the following reasons: alcohol or drug abuse in the past 6 months; a DSM‐III‐R diagnosis of organic mental syndrome, organic anxiety syndrome, or caffeinism; clear and immediate suicide risk; personality pathology that might interfere with trial compliance; previous participation in another brofaromine trial; a previous failure to respond to an adequate trial of an SRI or MAO inhibitor; clinically significant comorbid medical disease; concurrent use of other psychotropic drugs, opiates, or certain dietary or prescription amines (patients were allowed to take chloral hydrate, diphenhydramine, and doxylamine); and patients who in the judgment of the investigator were likely to be noncompliant". Dropouts: 31/106 (14/52 in the brofaromine and 17/54 in the placebo groups). | |
| Interventions | Pharmacological intervention: quote: "Brofaromine dosage began at 50 mg/day and was titrated up to a maximum of 150 mg/day, depending on response to treatment. Patient visits were scheduled at weekly intervals for the first 2 weeks of double‐blind treatment and biweekly thereafter. After 10 weeks of double‐blind treatment (or at early termination), patients entered a 1‐to 2‐week double‐blind weaning period during which the trial drug dose was tapered and discontinued". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI (for treatment efficacy), GAF (for reduction of functional disability), HAM‐A (for reduction of anxiety), MADRS (for reduction of depression), FONE (for reduction of anxiety), LSAS (for reduction of anxiety), CSPS (for reduction of anxiety), and SDS (for reduction of functional disability) Time points: Quote: "Efficacy measures were obtained at bassline and at end of 2, 6, and 10 week of double‐blind treatment" | |
| Notes | Industry funded: yes. Quote: "This study was supported by the Ciba‐Geigy Corporation, Summit, NJ". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "patients who still met inclusion criteria and and did not respond to placebo ... were randomly assigned to 10 weeks of treatment with either brofaromine or place". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the brofaromine (14/52; 27%) and placebo groups (17/50; 34%). More participants in the brofaromine group discontinued due to adverse events compared to withdrawal of consent in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "Fourteen brofaromine patients and 17 placebo patients discontinued the trial prematurely. Eleven of the 14 brofaromine early terminators discontinued because of adverse experiences, as did four of the 17 placebo early terminators. Other reasons for early termination included unsatisfactory therapeutic effect (N = 3, placebo); did not meet protocol criteria (N = 1, placebo); patient noncompliance (N = 2, placebo); patient withdrew consent (N = 1, brofaromine; N = 6 placebo); and lost to follow‐up (N = 2, brofaromine; N = 1, placebo) ... No significant baseline differences were found between treatment groups on race, gender, age, or LSAS total score". Overall 30% of the participants dropped out of the study. Quote: "An intent‐to‐treat analysis was employed, with the last evaluable visit past baseline carried forward as endpoint". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: single‐centre, quasi‐experimental, randomised‐controlled, wait‐list study Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 30 randomised to sertraline or placebo (the additional 15 participants were randomised to psychotherapy) Mean age (SD): 24.5 (2.2) years Sex: 30 men Diagnostic measure: DSM‐IV‐TR (based on SCID) Inclusion criteria: quote: "SPIN score ≥ 24, age between 18 to 50 years, and meeting the DSM‐IV‐TR criteria for social phobia based on SCID" Exclusion criteria: quote: "being psychotic, or obsessive‐compulsive; having bipolar, or organic brain disorders; drug and alcohol dependency; having impulse control disorders, cluster A and B personality disorders, active disorder on axis III, a history of suicidal thoughts and actions, a history of violent behavior; being on psychotropic medications or receiving psychotherapy for the treatment of social phobia during the last 6 months, or experiencing the symptoms of social phobia as part of other psychiatric disorders". Dropouts: not specified | |
| Interventions | Pharmacological intervention: quote: "Members of the MED group received pharmacotherapy (sertraline) for 12 weeks. Patients in the WL group received no intervention. However, after the waiting period, they received preferred treatment services. Each group was evaluated 4 times during the study". | |
| Outcomes | Primary outcome measure: SPIN (for reduction of anxiety) Secondary outcome measures: GAF (for reduction of functional disability), CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety) Time points: Quote: "In this randomized‐controlled trial study, 13 male students were treated with short‐term dynamic psychotherapy (McCullough method) lasting 25 sessions, 11 students received sertraline for 12 weeks, and 14 students, as the waiting list, received no intervention for 8 weeks. Participants completed the Social Phobia Inventory (SPIN) as primary efficacy variable 4 times, and were rated with Clinical Global Impression scale (CGI) and Global Assessment of Functioning (GAF) as secondary efficacy variables". Time points were not specified | |
| Notes | Industry funded: no. Quote: "This research was funded by Tehran University of Medical Sciences and Mental Health Research Network (MHRN)". Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "The participants were randomly assigned into 3 groups: 1‐psychotherapy (STDP), 2‐ medical therapy (MED) and 3‐ waiting list (WL)". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | High risk | No information was provided to determine if participants and personnel were blinded. |
| Blinding of outcome assessment (detection bias) | High risk | No information was provided to determine if outcome assessors were blinded. |
| Incomplete outcome data (attrition bias) | High risk | No dropout rates were reported. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported on as specified in the protocol. |
| Other bias | Low risk | No other sources of bias were identified. |
| Methods | Design: single‐centre, randomised, double‐blind, fixed dose, placebo‐controlled study Duration of intervention: 10 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 66 randomised to mirtazepine or placebo Mean age (SD): 24 (3.45) years Sex: 66 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Women aged 18 or older who have social phobia were included in the study". Exclusion criteria: quote: "Exclusion criteria were psychotic symptoms, a severe major depressive episode (according to the DSM‐IV criteria), the current use of mirtazepine or other psychotropic medication, and psychotherapy. Potential subjects were also excluded if they currently were pregnant (or planning to be or not using contraception), severely somatically ill, currently suicidal, or abusing alcohol or drugs". Dropouts: 7/66 (insufficient information to determine dropout rates for the 2 groups separately) | |
| Interventions | Pharmacological intervention: quote: "Subjects received blinded capsule per day, either 30 mg of mirtazepine or matching placebo. The dosage of mirtazepine stayed constant". | |
| Outcomes | Primary outcome measures: SPI (for reduction of anxiety), LSAS (for reduction of anxiety) and SF‐36 (for quality of life) Secondary outcome measures: none Time points: Weekly examinations. | |
| Notes | Industry funded: no. Quote: "This study was not funded and was not influenced by outside economic interests". Medication provided by industry: no Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "We used an excel sheet with random number generator". |
| Allocation concealment (selection bias) | Low risk | Quote: "Subjects received blinded capsule per day, either 30 mg of mirtazepine or matching placebo. The dosage of mirtazepine stayed constant. Tablets were supplied in numbered boxes". |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Both subjects and clinicians were blinded to mirtazepine/placebo assignment". |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Both subjects and clinicians were blinded to mirtazepine/placebo assignment". |
| Incomplete outcome data (attrition bias) | Low risk | There was insufficient information to determine the proportion of dropouts for the three treatment groups, though a relatively small number dropped out overall (N = 7, 11%). No information was provided on the reasons for study withdrawal. There was also no information on group characteristics. Quote: "Seven patients who failed to appear more than twice for the weekly evaluations dropped out of the study". All analyses were ITT. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | Self reported measures were used but No other sources of bias were identified. Quote: "The questionnaires were filled out by the patients both independently and anonymously". |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, parallel, forced‐dose titration study (NKF100110) Duration of intervention: 12 weeks Post‐treatment: follow‐up not specified Placebo run‐in: not specified | |
| Participants | Sample size: 107 randomised to paroxetine or placebo (the additional participants were randomised to NCE, the results will be available once NCE is approved by the FDA). Mean age (SD): 34.4 (10.83) years Sex: 60 men and 44 women (for 104 participants) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Male or female subjects between 18‐65 years of age; A primary diagnosis of Generalized Social Anxiety Disorder/Social Phobia (DSM‐IV, 300.23)". Exclusion criteria: quote: "CGI‐I item score of 1 or 2 at baseline; Montgomery‐Asberg Depression Rating Scale (MADRS) score of 18 or more at screening; DSM‐IV criteria for any other Axis I disorder as a current primary disorder or within 6 months prior to the screening visit; Subjects with Body Dysmorphic Disorder; History of Schizophrenia, Schizoaffective Disorder, or a Bipolar Disorder; Current, serious suicidal or homicidal risk or suicide attempt within the past 6 months or have ever been homicidal; Unstable medical disorder; or a disorder that would interfere with the action, absorption, distribution, metabolism, or excretion of the NCE or paroxetine; may pose a safety concern; or interfere with the accurate assessment of safety or efficacy". Dropouts: 44/107 (14/36 in the paroxetine and 30/71 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "After completion of a screening period, subjects fulfilling the inclusion/exclusion criteria were randomised (2:2:2:1) to two dose ranges of an NCE, placebo, or paroxetine (20‐30 mg/day). A forced‐flexible dose titration scheme was employed". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), LSAS Fear and Avoidance subscales (for reduction of anxiety), SDS (for reduction of functional disability), MOS‐12 (assess changes in sleep quality), and LSEQ (assess changes in sleep quality) Time points: Week 1 and 12. Quote: "Adverse events (AEs) and Serious Adverse Events (SAEs) were reported during treatment weeks 1 through 12, the taper visit, and at the 14‐day follow‐up visit or early withdrawal visit. This summary includes data for the paroxetine and placebo groups. Results for the unmarketed NCE will be added, if and when the NCE is approved and marketed" | |
| Notes | Industry funded: not specified Medication provided by industry: not specified Any of the authors work for industry: not specified | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "After completion of a screening period, subjects fulfilling the inclusion/exclusion criteria were randomized (2:2:2:1) to two dose ranges of an NCE, placebo, or paroxetine (20‐30 mg/day). A forced‐flexible dose titration scheme was employed". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the paroxetine (14/36; 39%) and placebo groups (30/71; 42%). No information was provided on the reasons for study withdrawal and how participants differed by sample characteristics at week 12. Overall 41% of the participants dropped out of the study. ITT population was assessed and LOCF was used for missing data. |
| Selective reporting (reporting bias) | Unclear risk | Not all the required information is presented in the protocol to determine if selective reporting occurred. |
| Other bias | Unclear risk | It is unclear if any other bias occurred and if the study was funded. |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group, comparative study (PIR104776) Duration of intervention: 12 weeks Post‐treatment: follow‐up not specified Placebo run‐in: 2 weeks placebo run in | |
| Participants | Sample size: 400 randomised to paroxetine or placebo Mean age (SD): 36.96 (9.98) years Sex: 186 men and 209 women (for 395 participants) Diagnostic measure: DSM‐IV‐TR Inclusion criteria: quote: "Diagnosis of Social Anxiety Disorder (SAD) according to DSM‐IV‐TR criteria; Must have given written informed consent. But if the subject was under 20 years of age, both the subject and his/her proxy consenter had to have given written informed consent; Outpatients; Age: ≥18 years, < 65 years (at the time of informed consent); Sex: no restriction; Had to have a LSAS total score of ≥60 at baseline". Exclusion criteria: quote: "Subjects diagnosed with Axis I disorders other than SAD (e.g., major depression, dysthymic disorder, specific phobia (simple phobia), obsessive compulsive disorder, panic disorder) as a primary diagnosis according to DSM‐IV‐TR within 24 weeks prior to the Week ‐2 visit; Subjects with a history of or concurrent schizophrenia or bipolar disorder; Subjects with concurrent body dysmorphic disorder; Subjects who met the DSM‐IV‐TR criteria for substance abuse (alcohol or drugs) or substance dependence within 24 weeks prior to the Week ‐2 visit; Subjects who started psychotherapy, other than supportive psychotherapy, or cognitive‐behavioural therapy within 24 weeks prior to the Week ‐2 visit; Subjects who received electro‐convulsive therapy (ECT) within 12 weeks prior to the Week ‐2 visit; Subjects who were pregnant, lactating, might be pregnant, or were planning a pregnancy during the study period; Subjects who scored 3 or more on HAM‐D Item No.3 or, who, in the investigator's clinical judgement, were at acute risk of suicide attempt; Subjects with a history of or concurrent cancer or malignant tumor; Subjects who received MAO inhibitors (FP®) within 14 days prior to the scheduled Week 0 visit". Dropouts: 44/107 (43/267 in the paroxetine and 19/133 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "Subjects received paroxetine or placebo once daily after an evening meal for 12 weeks. Subjects randomized to paroxetine 20mg or 40mg were started on 10mg/day for the first week and the dose was then increased according to a fixed dosing schedule. Subjects in the paroxetine 20mg group remained on 20mg/day for 11 weeks. Subjects in the paroxetine 40mg group received 20mg/day for 1 week, 30mg/day for 1 week, and then 40mg/day for 9 weeks. Subjects randomized to placebo received the same number of placebo tablets in the same manner as for those randomized to paroxetine treatment. Taper phase: participants who completed the treatment phase underwent a 3‐week taper phase, and participants who withdrew prematurely underwent a taper phase of 1 to 3 weeks depending upon their final level of study medication. The dose was decreased by 10mg/day weekly until 10mg/day was reached". ITT population was assessed and LOCF was used for missing data. | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) Secondary outcome measures: change from baseline in LSAS total score (for reduction of anxiety), CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), and HAM‐D (for reduction of depression) Time points: Quote: "Change from baseline in LSAS total score (at Weeks 2, 3, 4, 6, 8 and 10). Proportion of CGI Global Improvement responders (i.e., subjects rated as either "very much improved" or "much improved") at Week 12. Change from baseline in LSAS Fear/Anxiety and Avoidance subscale scores (at Weeks 2, 3, 4, 6, 8, 10 and 12). Change from baseline in CGI Severity of Illness score (at Weeks 2, 3, 4, 6, 8, 10 and 12). Change from baseline in HAM‐D total score at Weeks 12 (score at Week 12 minus Score at Week 0)" | |
| Notes | Industry funded: not specified Medication provided by industry: not specified Any of the authors work for industry: not specified | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the paroxetine (43/267; 16%) and placebo groups (19/133; 14%). No information was provided on the reasons for study withdrawal and how participants differed by sample characteristics at week 12. Overall 16% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol. |
| Other bias | Unclear risk | It is unclear if any other bias occurred and if the study was funded. |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group, active comparator study (CRH103390) Duration of intervention: 12 weeks Post‐treatment: follow‐up not specified Placebo run‐in: not specified | |
| Participants | Sample size: 294 randomised to paroxetine, GW876008 or placebo Mean age (SD): 37.35 (11.86) years Sex: 155 men and 139 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Male and female subjects 18‐64 years of age inclusive with a primary diagnosis of SocAD/Social Phobia diagnosed using criteria established in the DSM‐V participated in the study". Exclusion criteria: quote: "Subjects who scored 1 or 2 on the Clinical Global Impression‐Global Improvement (CGI‐I) score item at the randomisation visit, or who met the DSM‐V criteria for major depressive disorder, or who scored ≥ 15 on the 17‐item Hamilton Rating Scale for Depression (HAMD‐17), at the screening visit were excluded from the study". Dropouts: 44/107 (13/42 in the paroxetine, 36/164 in the GW876008 and 25/88 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "Subjects were randomised to a 2:2:2:1 ratio such that 81 subjects received GW876008 25‐50mg/day, 83 received GW876008 100‐125 mg/day, 88 received placebo and 42 received paroxetine 20‐30 mg/day". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety), SADS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy) Time points: Baseline and week 12. | |
| Notes | Industry funded: not specified Medication provided by industry: not specified Any of the authors work for industry: not specified | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Subjects were randomised to a 2:2:2:1 ratio such that 81 subjects received GW876008 25‐50mg/day, 83 received GW876008 100‐125 mg/day, 88 received placebo and 42 received paroxetine 20‐30 mg/day". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the paroxetine (13/42; 31%), GW876008 (36/164; 22%) and placebo groups (25/88; 28%). No information was provided on the reasons for study withdrawal and how participants differed by sample characteristics at week 12. Overall 25% of the participants dropped out of the study. Quote: "Key efficacy analyses were intention to treat (ITT) using the mixed‐effects model repeated‐measure analysis for comparisons". |
| Selective reporting (reporting bias) | Unclear risk | Not all the required information is presented in the protocol to determine if selective reporting occurred. |
| Other bias | Unclear risk | It is unclear if any other bias occurred and if the study was funded. |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, fixed dose, parallel group study (NKP103401). Duration of intervention: 12 weeks Post‐treatment: follow‐up not specified Placebo run‐in: not specified | |
| Participants | Sample size: 133 randomised to paroxetine or placebo (the additional participants were randomised to NCE, the results will be available once NCE is approved by the FDA) Mean age (SD): 40.3 (11.21) years Sex: 54 men and 74 women (for 128 participants) Diagnostic measure: DSM‐IV and MINI Inclusion criteria: quote: "Male and female subjects, 18‐65 years of age, with a primary diagnosis of Generalised Social Anxiety Disorder/Social Phobia as defined in the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM‐IV), diagnosed using psychiatric confirmation of diagnosis in conjunction with the Mini International Neuropsychiatric Interview (MINI), Clinician Rated version 5.0". Exclusion criteria: quote: "Subjects were excluded if they scored 1 or 2 on the CGI‐I item at baseline; subjects with a score 15 or more on the Hamilton Depression (HAMD) Rating Scale 17 (HAMD‐17) item at screening were also excluded. In addition, subjects were excluded if they had a history of myocardial infarction within one year prior to the screening visit, or had body dysmorphic disorder or had a history of schizophrenia, schizoaffective disorder, or a bipolar disorder". Dropouts: 42/133 (23/68 in the paroxetine and 19/65 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "NCE/paroxetine combination, paroxetine monotherapy (7.5 mg/day, fixed dose) or placebo for a period of 12 weeks". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) Secondary outcome measures: LSAS (for reduction of anxiety), CGI‐I (for treatment efficacy), SADS (for reduction of anxiety), SDS (for reduction of functional disability) Time points: Baseline and week 12. | |
| Notes | Industry funded: not specified Medication provided by industry: not specified Any of the authors work for industry: not specified | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "A 12‐week randomised, multicentre, double‐blind, placebo‐controlled, fixed dose, parallel group study". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the paroxetine (23/68; 34%) and placebo groups (19/65; 29%). No information was provided on the reasons for study withdrawal and how participants differed by sample characteristics at week 12. Overall 32% of the participants dropped out of the study. ITT population was assessed and LOCF was used for missing data. |
| Selective reporting (reporting bias) | Unclear risk | Not all the required information is presented in the protocol to determine if selective reporting occurred. |
| Other bias | Unclear risk | It is unclear if any other bias occurred and if the study was funded. |
| Methods | Design: single‐centre, double‐blind, randomised, placebo‐controlled, parallel group (TMT106386) Duration of intervention: 8 weeks Post‐treatment: follow‐up not specified Placebo run‐in: not specified | |
| Participants | Sample size: 33 randomised to paroxetine or placebo (the additional participants were randomised to no treatment (HVT)) Mean age: 22.9 years Sex: 5 men and 28 women Diagnostic measure: DSM‐IV (using the SCID‐I) Inclusion criteria: quote: "Male or female subjects of 18 to 60 years of age, with a primary diagnosis of SAD (DSM‐IV, 300.23) diagnosed using psychiatric confirmation of diagnosis in conjunction with the structured clinical diagnostic interview (SCID‐I) were included into the study. Healthy participants free from significant cardiac, pulmonary, gastrointestinal, hepatic, renal, haematological, neurological and psychiatric disease as determined by history, physical examination, MRI and clinical laboratory test results (for healthy volunteers only) were also included in to the study". Exclusion criteria: not specified Dropouts: 1/33 (0/17 in the paroxetine and 1/16 in the placebo group) | |
| Interventions | Pharmacological intervention: quote: "Each eligible subject was assigned to receive following treatments in a 1:1 ratio as per the randomisation schedule: | |
| Outcomes | Primary and secondary outcome measures: STAI‐S (for reduction of anxiety), CGI (for treatment efficacy), LSAS (for reduction of anxiety) Time points: Week 2, 4, 6, 8. | |
| Notes | Industry funded: not specified Medication provided by industry: not specified Any of the authors work for industry: not specified | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Each eligible subject was assigned to receive following treatments in a 1:1 ratio as per the randomisation schedule". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | More participants withdrew from the placebo group (1/16; 6%) compared to the paroxetine (0/17; 0%) and venlafaxine (7/129; 5%) group. No information was provided on whether participants differed in terms of characteristics by group at week 12, however. Nevertheless, the total proportion of dropouts (6%) is relatively low, suggesting that dropout rates may not have biased the outcomes. Overall 0.6% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | Not all the required information is presented in the protocol to determine if selective reporting occurred. |
| Other bias | Unclear risk | It is unclear if any other bias occurred and if the study was funded. |
| Methods | Design: single‐centre, randomised, double‐blind, placebo‐controlled, comparative, flexible dose study Duration of intervention: 12 weeks Post‐treatment: 12‐month follow‐up Placebo run‐in: no | |
| Participants | Sample size: 52 randomised to paroxetine or placebo (102 randomised: 26 paroxetine, 24 CT, 26 combination, 26 placebo) Mean age (range): 30.85 (18‐65) years Sex: 26 men and 26 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Inclusion criteria were as follows: age of 18–65 years, fulfillment of DSM‐IV criteria for SAD, and symptoms present for at least 6 months". Exclusion criteria: quote: "Exclusion criteria were any form of physical disease, psychotic illness, acute suicidality, a primary diagnosis of major depressive disorder, diagnosis of body dysmorphic disorder, drug or alcohol dependence, and cluster A or cluster B personality disorders. Subjects not willing to accept random allocation were also excluded. We excluded patients who had been exposed to CT or to SSRIs previously in order to eliminate any bias of negative expectations to the treatment offered. Participants who were pregnant or were planning to become pregnant during the next 6 months were excluded due to the drug condition". Dropouts: 8/52 (5/26 in the paroxetine and 3/26 in the placebo group; 12 month follow‐up 4 dropouts) | |
| Interventions | Pharmacological intervention: quote: "Following the clinical guideline by Stein et al. [25] , drug treatment | |
| Outcomes | Primary outcome measure: FNES (for reduction of anxiety) Secondary outcome measures: LSAS (for reduction of anxiety), BAI (for reduction of anxiety), IIP‐64 (for reduction of functional disability) Time points: Quote: "Patients were assessed pretreatment, posttreatment at 12 weeks (post‐acute), and at the 12‐month follow‐up" | |
| Notes | Industry funded: no. Quote: "The study was financially supported by the Departments of Psychology and Neuroscience at the Norwegian University of Science and Technology (NTNU), Trondheim". Medication provided by industry: yes. Quote: "It was administered as capsules manufactured by the pharmaceutical laboratory at St. Olav’s University Hospital to make them identical to the placebo". Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The participants were randomly assigned to 1 of 4 conditions. The randomization used gender and diagnosis of APD as stratification variables in blocks of 10 to ensure equal distribution". |
| Allocation concealment (selection bias) | Low risk | Quote: "The medication used was paroxetine (paroxetine hydrochloride). It was administered as capsules manufactured by the pharmaceutical laboratory at St. Olav’s University Hospital to make them identical to the placebo. The placebo capsules contained lactose. The paroxetine and the placebo were identical in size, color, smell, taste, and appearance. The pharmaceutical laboratory at St. Olav’s University Hospital provided the medication to the psychiatrists". |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "The participants, independent diagnosticians, psychiatrists, and the principal investigator remained blinded to the paroxetine alone and pill placebo conditions. In addition, specific instructions were given to all participants to avoid disclosing information about their treatment to the evaluators". |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Blinding was conducted for the treatment conditions using medication or placebo and achieved for the primary outcome measures by using independent evaluators who were blinded to the treatment assignment". |
| Incomplete outcome data (attrition bias) | Low risk | A similar proportion of participants withdrew from the paroxetine group (5/26, 19%) and placebo group (3/26; 12%). Quote: "All data were analyzed based on an intention‐to‐treat approach, and missing data were treated using last observation carried forward on the primary measure. We used a linear mixed |
| Selective reporting (reporting bias) | High risk | Secondary outcomes where not specified in the protocol for LSAS, IIP‐64 and BAI measures as well as for relapse |
| Other bias | Unclear risk | Medication was provided by industry. Quote: "We are not sure whether a different setting of the interviews could bias the data, but this must be considered ... There is a possibility that drug treatment in our study may have been adversely affected by bias produced by the blinding process. Specifically, in completing self‐report measures, those in the drug arm may have doubted that they received the active treatment rather than placebo". |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, fixed dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo run‐in | |
| Participants | Sample size: 523 randomised to moclobemide or placebo Mean age (SD): 38.1 (10.4) years (ITT sample of 506) Sex: 290 men and 216 women (ITT sample of 506) Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "Subjects were screened using the Structured Clinical Interview for DSM‐III‐R, Roche version and were required to meet DSM‐III‐R criteria for social phobia (primary diagnosis). Subjects were included who had coexisting generalized anxiety disorder or avoidant personality disorder, but those with coexisting panic disorder, agoraphobia, or obsessive‐compulsive disorder were excluded. Subjects were required to achieve a score of 4 (moderate) or more on the Clinical Impression of Severity‐Social Phobia scale". Exclusion criteria: quote: "Any subjects who had mental retardation, organic mental disorders including dementia, psychoses including schizophrenia, bipolar disorder, or borderline personality disorder were excluded. Subjects were also excluded who had major depressive disorder within 6 months of the study, suicidal ideation, substance abuse within 6 months of the study, or positive urine drug screening. Also excluded were subjects with uncontrolled physical disease or significant laboratory abnormalities. Women of childbearing potential were required to take adequate contraceptive precautions". Dropouts: 158/523 (25/84 in the moclobemide and 33/85 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "Subjects were then randomly assigned to placebo or one of five doses of moclobemide (75 mg, 150 mg, 300 mg, 600 mg, or 900 mg). Those assigned to 75 mg and 150 mg received the full dose from the time of randomization, and those assigned to 300 mg, 600 mg, and 900 mg received 150 mg initially followed by increments of 150 mg every 4 days until the full dose was achieved". | |
| Outcomes | Primary outcome measure: CGI‐I (for treatment efficacy) Secondary outcome measures: CGI‐SP (for reduction of anxiety), BSPS (for reduction of anxiety), LSAS (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "The primary measure of efficacy was the global rating of improvement made on the Clinical Impression of Change scale at week 12. Efficacy measures also included patient‐rated scales completed at 4, 6, and 12 weeks. Adverse events, as observed or elicited by the clinician, were recorded at baseline and at each subsequent visit" | |
| Notes | Industry funded: yes. Quote: "This study was sponsored by Hoffmann‐La Roche, Inc., Nutley, NJ ". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Subjects were then randomly assigned to placebo or one of five doses of moclobemide". |
| Allocation concealment (selection bias) | Unclear risk | The study was described as "double‐blind" however there was insufficient evidence to determine if study medication was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the moclobemide (25/84; 29.7%) and placebo groups (33/85; 38.8%). No information was provided on the reasons for study withdrawal and how participants differed by sample characteristics at week 12. Nevertheless, the study reported that 158 participants dropped out of the study and these participants did not differ significantly. Quote: "One hundred fifty‐eight (31.2%) subjects dropped out before week 12. The proportion of placebo‐treated subjects who dropped out (38.8%) was higher than, but not significantly different from, the percentage of moclobemide‐treated subjects who failed to complete the trial (29.7%)". Overall 34% of the participants dropped out of the study. Quote: "The main analyses involved intent‐to‐treat subjects defined as those who received at least one dose of study medication and had at least one efficacy assessment after baseline. For these analyses, last observations were carried forward to replace missing values". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 15 weeks Post‐treatment: 2 and 15 months follow‐up Placebo run‐in: no | |
| Participants | Sample size: 54 randomised to moclobemide or placebo (82 out of 86 randomised: 27 moclobemide, 28 cognitive therapy, 27 placebo, four participants refused participation after randomisation because they were not allocated to the condition they preferred) Mean age (SD): 36.5 (11) years (for the moclobemide or placebo groups; 82 randomised) Sex: 31 men and 23 women (for the moclobemide or placebo groups; 82 randomised) Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "After a general diagnostic interview by an experienced clinician, a main diagnosis of social phobia was confirmed by a semi‐structured interview ... Patients were included when aged 18 to 65 years, and had no serious medical problems as revealed by medical history and laboratory screening tests". Exclusion criteria: quote: "Patients with comorbid panic disorder with or without agoraphobia, obsessive‐compulsive disorder, major depressive disorder, psychotic and organic mental disorder were excluded, as were patients suffering from psychoactive substance‐use disorder and borderline personality disorder". Dropouts: 11/54 for the moclobemide or placebo groups (3/27 in the moclobemide and 8/27 in the placebo groups). | |
| Interventions | Pharmacological intervention: quote: "In the pharmacotherapy condition, patients started either with placebo or moclobemide 450 mg/day, which was also the target dose. After 2 weeks, the dose was increased to 600 mg/day in case of insufficient efficacy and good tolerability or decreased to 300 mg/day in case of severe side effects. Moclobemide and placebo were supplied in matching tablets of 150 mg by Hoffman–La Roche Ltd., Basel, Switzerland". | |
| Outcomes | Primary and secondary outcome measures: LSAS (for reduction of anxiety), ADS (modified) (for reduction of anxiety), a 4‐point scale for the tolerability of moclobemide and placebo, MADRS (for reduction of depression), CIC (for treatment efficacy), IIS (for treatment efficacy), SCI (for reduction of functional disability), FQ (for reduction of anxiety), SDS (for reduction of functional disability), and SCL‐90 (for reduction of anxiety) Time points: Quote: "Measurements took place at pre‐test and post‐test (week 0 and 15), 2‐month follow‐up (week 23) as well as 15 months after completion of the trial" | |
| Notes | Industry funded: yes. Quote: "This study was sponsored by Hoffmann‐La Roche, Ltd., Basel, Switzerland". Medication provided by industry: yes. Quote: "Moclobemide and placebo were supplied in matching tablets of 150mg by Hoffman – LaRoche Ltd., Basel, Switzerland". Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "patients were randomly assigned to one of the three treatment conditions". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Pharmacotherapy was administered under double‐blind conditions ... Moclobemide and placebo were supplied in matching tablets of 150mg by Hoffman – LaRoche Ltd., Basel, Switzerland ... To check double‐blindness, patients and therapists were asked at post‐test to estimate whether moclobemide or placebo had been administered. Correct classifications did not differ from chance. These results indicated that in our study double‐blindness was maintained throughout". |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Independent raters, blind to therapeutic conditions, assessed social anxiety and avoidance with the LSAS ... and MADRS ..." |
| Incomplete outcome data (attrition bias) | Low risk | More participants discontinued in the placebo group (8/27; 30%) compared to the moclobemide group (3/27; 11%), largely due to non‐compliance with the protocol and inadequate treatment response. The 2 groups did not differ by sample characteristics at baseline and endpoint nor did the dropout rates differ significantly across groups. Quote: "A total of 67 patients completed the 15 weeks of treatment. Completers and dropouts were divided into the three treatment groups as follows: cognitive therapy, 24 completers and four dropouts; moclobemide, 24 completers and three dropouts; placebo, 19 completers and eight dropouts. The proportion of dropouts did not differ significantly between these conditions. All four patients in the cognitive therapy condition dropped out due to insufficient therapeutic response compared with time‐investment. In the moclobemide group reasons for dropping out were: extreme fatigue (n = 1) and insufficient therapeutic response (n = 2). Reasons for dropping out in the placebo group were: increase of migraine complaints (n = 1); insufficient therapeutic response (n = 3); increase of social phobic complaints (n = 1) and non‐compliance to the treatment protocol (n = 3). No other significant differences between completers and dropouts emerged on other demographic variables or baseline clinical scores. Insomnia was the only side‐effect that was reported significantly more often in the moclobemide |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication and study funded by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo controlled, parallel, flexible dose, double‐blind Duration of intervention: 14 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind placebo lead‐in | |
| Participants | Sample size: 69 randomised to gabapentin or placebo Mean age (SD): 35.6 (9.6) years Sex: 40 men and 29 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "The study sample consisted of outpatients of either gender who were aged 18 years or older and who had received a diagnosis of social phobia according to DSM‐IV criteria ... To exclude those with mild social anxiety limited to few situations that clinicians may or may not consider for drug therapy, study patients were required to have a minimum score of 50 on the LSAS at entry into the study". Exclusion criteria: quote: "Patients were excluded if they suffered from uncontrolled medical illnesses, had prominent depressive symptoms measured by a HAM‐D Depressed Mood subscale score of > 3 at baseline, met criteria for a current diagnosis of alcohol or substance abuse, or were taking other psychotropic agents". Dropouts: 30/69 (13/34 in the gabapentin and 17/35 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "Gabapentin was dispensed as 300 mg capsules. All patients initiated the randomized portion of the study with one capsule twice a day (either placebo or gabapentin 300 mg) and had to reach a dose of 1 capsule three times a day by the end of the first week Thereafter, as long as symptoms of social phobia were present and there were no limiting adverse effects, the dose was required to be escalated in increments of no more than 300 mg each day, up to the maximum of 3,600 mg/day. At the conclusion of the double‐blind phase of the trial, treatment was discontinued by reducing the dose by two capsules daily". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: BSPS (for reduction of anxiety), MMFQ (for reduction of anxiety), SPIN (for reduction of anxiety), CGIC (for treatment efficacy), HAM‐D (for reduction of depression) and HAM‐A (for reduction of anxiety) Time points: Quote: "Patients were evaluated at weekly visits for the first 4 weeks, biweekly for the next 4 weeks, and monthly thereafter. Various efficacy and safety assessments were made during each visit according to a predetermined schedule" | |
| Notes | Industry funded: yes. Quote: "This study was supported by the Parke‐Davis Division of Warner‐Lambert Company". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "After a 1‐week, single‐blind placebo lead‐in during which patients had to continue to meet entry criteria, they were randomly assigned to receive double‐blind treatment with either gabapentin or placebo for 14 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | A smaller proportion of participants withdrew from the gabapentin (13/34; 38%) than the placebo group (17/35; 49%). The primary reason for withdrawal in the gabapentin was adverse events compared to lack of efficacy and adverse events in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "More patients on gabapentin (62%) completed the study than did those on placebo (51%). Adverse events were the primary reason for early withdrawal from the gabapentin group during the study. Patients on placebo withdrew early for a variety of reasons, including adverse events and lack of efficacy. Overall, the rate of withdrawal was gradual over the 14‐week study (averaging two patients per visit per treatment group) and was similar for both treatment groups ... Patients in each treatment group were comparable with respect to demographics". Overall 43% of the participants dropped out of the study. Quote: "For all analyses, the last observation carried forward (LOCF) was used as the endpoint measurement". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, multiple fixed dose, clinical trial Duration of intervention: 10 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo lead‐in | |
| Participants | Sample size: 135 randomised to pregabalin or placebo Mean age (SD): 38.4 (11.5) years Sex: 79 men and 56 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "The study sample consisted of outpatient men and women, 18 years or older, suffering from social anxiety disorder ... Patients were also required to have a total score of 50 or greater on the LSAS to be included in the study". Exclusion criteria: quote: "Patients with any of the following axis I DSM‐IV diagnoses were excluded from the study: delirium, dementia, amnestic, or other cognitive disorders; schizophrenia; bipolar or schizoaffective disorder; substance abuse disorder active in the last 6 months; and panic disorder, agoraphobia, or obsessive‐compulsive disorders. Patients with a secondary diagnosis of major depressive disorder (based on DSM‐IV criteria) were not excluded, however, patients with a HAM‐D Rating Scale Item 1 (depressed mood) score 3 at screening were excluded. Patients with borderline or antisocial personality disorder, or ongoing psychodynamic or behavioral psychotherapy for social anxiety disorder were excluded from the study. In addition, patients were excluded if they suffered from uncontrolled medical illnesses, or if they were taking other psychotropic agents". Dropouts: 41/135 (20/47 and 11/42 in the pregabalin and 10/46 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "The study had 3 phases: screening, double‐blind treatment, and taper. Following a 1‐week, single‐blind, placebo lead‐in, patients who continued to meet entry criteria were randomized to double‐blind treatment with either pregabalin 600 mg/d (200 mg TID), pregabalin 150 mg/d (50 mg TID), or placebo. Study medication was titrated to the full assigned dose over the first 6 days of the double‐blind phase. Following 10 weeks of double‐blind treatment, the final efficacy assessments were made (termination visit). Patients then entered the taper phase, study medication dose was tapered off over 6 days, and a final follow‐up visit was conducted". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: BSPS (for reduction of anxiety), HAM‐A (for reduction of anxiety), SPI (for reduction of anxiety), FQ (for reduction of anxiety), CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety) and the SF‐3 (for quality of life) Time points: Quote: "Patients were evaluated every 1 to 2 weeks for the duration of the study" | |
| Notes | Industry funded: yes. Quote: "This study was supported by Parke‐Davis Pharmaceutical Research Division of Warner‐Lambert". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "The safety and efficacy of pregabalin for the treatment of social anxiety disorder was evaluated in a double‐blind, multicenter clinical trial in which 135 patients were randomized to 10 weeks of double‐blind treatment with either pregabalin 150 mg/d, pregabalin 600 mg/d, or placebo". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | A higher proportion of participants withdrew from the 150 mg/d and 600 mg/d (16/45; 36%) groups compared to the placebo group (10/46; 22%). Most participants dropped out of the study in the 600 mg/d pregabalin group because of adverse events and lack of compliance compared to the 150 mg/d pregabalin and placebo groups. No information was provided on if the 2 groups differed by sample characteristics at week 10. Quote: "Of the 135 randomized patients, 94 completed the study. One patient receiving pregabalin 600 mg/d had no postrandomisation efficacy assessment and was excluded from the efficacy analysis ... More patients receiving pregabalin 600 mg/d (n = 11, 23.4%) withdrew due to adverse events than patients receiving either pregabalin 150 mg/d (n = 4, 9.5%) or placebo (n = 4, 8.7%)". Overall 29% of the participants dropped out of the study. Quote: "The primary efficacy measure was changed in the LSAS total score from baseline (randomization visit) to end point (termination visit or last observation carried forward during the 10‐week double‐blind phase)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 8 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 15 randomised to paroxetine or placebo (18 randomised: 3 participants were excluded due to various reasons) Mean age (SD): 35.5 (8.4) years Sex: 13 men and 2 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "All individuals were required to meet DSM‐IV criteria for both current social anxiety disorder (social phobia) as well as alcohol abuse or dependence criteria. Other inclusion criteria included an age range between 18 and 70 years, willingness to attend 8 weekly outpatient study visits, and consumption of at least 15 standard drinks in the past 30 days". Exclusion criteria: quote: "An individual was excluded if (s)he had a primary Axis I DSM‐IV diagnosis other than alcohol abuse/dependence and social anxiety disorder (including dependence on another drug of abuse excepting nicotine). Other exclusion criteria included the presence of significant medical problems, current use of any prescribed psychotropic medicine on a regular basis, transportation problems, abnormal electrocardiogram, or elevated liver enzymes". Dropouts: 2/15 (1/6 in the paroxetine and 1/9 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "Paroxetine or visually matched placebo was delivered in 20‐mg pills for 8 weeks. Patients were instructed to take 1 pill per day in week 1, 2 pills per day in week 2, and 3 pills per day thereafter, unless there were dose‐limiting side effects. The targeted maintenance dosage was 60 mg/d". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety), CGI‐I (for treatment efficacy) and SPIN with TLFB drinking measures (for reduction of anxiety) Secondary outcome measures: BDI (for reduction of depression), ASI (for addiction) and ADS (for alcohol dependence) Time points: Quote: "After randomization and the meeting with the physician, the patient met weekly with the research | |
| Notes | Industry funded: yes. Quote: "This work was supported by an investigator‐initiated award from SmithKline Beecham (to J.R.D.)". Medication provided by industry: yes. Quote: "SmithKline Beecham supplied the drug and matched placebo". Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients were randomized according to a predetermined order prepared by the pharmaceutical company". |
| Allocation concealment (selection bias) | Low risk | Quote: "Paroxetine or visually matched placebo was delivered in 20‐mg pills for 8 weeks ... The institutional research pharmacy maintained the blind and dispensed all study medications". |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Paroxetine or visually matched placebo was delivered in 20‐mg pills for 8 weeks". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | Similar proportions of participants withdrew from the paroxetine (1/6; 17%) and placebo groups (1/9; 11%). No information was provided on the reasons for withdraws for the 2 groups and the groups did not differ by sample characteristics at baseline. Quote: "Treatment groups were similar at baseline on alcohol use measures, social anxiety severity, and demographic variables ... Week 8 SPIN data were available only for 12 of the 15 patients". Overall 13% of the participants dropped out of the study. Quote: "Two patients (1 from each treatment group) had missing data for 1 or more of the weekly assessments, and a last‐point‐carried‐forward approach was used to provide values for the missing data". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication and study funded by industry. The study sample comprised of more men than women. Quote: "Most patients were male (87%), about 36 years of age, and all were of white ethnicity". |
| Methods | Design: single‐centre, randomised, double‐blind, flexible dose, placebo‐controlled trial (NCT00260533) Duration of intervention: 10 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 27 randomised to atomoxetine (ATM) or placebo Mean age (SD): 42.05 (10.15) years Sex: 21 men and 6 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Medically healthy outpatients aged 18 to 65 years, with a clinically predominant diagnosis of DSM‐IV GSAD ... The subjects were required to have a score of 60 or higher on the LSAS and a total score of 14 or lower on the HAM‐D to be included in the study". Exclusion criteria: quote: "Other exclusion criteria included the presence of comorbid ADHD or another primary axis‐I disorder (including DSM‐IV diagnosis of another anxiety, eating, or substance use disorder in the previous 6 months), failure to respond to prior adequate trials of 2 or more medications to treat GSAD, and the subjects were on concomitant psychotropic medications in the previous 2 weeks (fluoxetine in the previous 4 weeks) or those receiving concurrent formal psychotherapy targeted at GSAD". Dropouts: 6/27 (3/14 in the atomoxetine and 3/13 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "The study medication was titrated on a fixed schedule over the first 2 weeks starting with 20 mg per day for 1 week and 40 mg for another week and then flexibly titrated based on response and tolerability in 20‐mg increments every 2 weeks up to a maximum dose of 100 mg. For the final 4 weeks of the study, the dose of medication was held stable". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) Secondary outcome measures: change in LSAS (for reduction of anxiety), CGI‐C (for treatment efficacy), HAM‐D (for reduction of depression) and SDS (for reduction of functional disability) Time points: Quote: "All measures were completed at each study visit (baseline and weeks 2, 4, 6, 8, and 10)" | |
| Notes | Industry funded: yes. Quote: "This study was supported by an investigator‐initiated research grant from Eli Lilly and Company". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Twenty‐seven outpatients with clinically prevailing diagnoses of GSAD by the DSM‐IV were randomized in a 1:1 ratio to 10 weeks of double‐blind flexible‐dose treatment with either ATM 40‐100 mg per day (n = 14) or placebo (n = 13)". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Within 1 week of the screening visit, eligible subjects were then randomized to receive a double‐blind treatment with either ATM or a matching placebo in a 1:1 ratio". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the atomoxetine (3/14; 21%) and placebo groups (3/13; 23%). Participants withdrew for the following reasons: adverse events, poor compliance, worsening depression, and lost to follow‐up. The 2 groups did not differ by sample characteristics at baseline nor did they significantly differ by attrition rates. Quote: "Overall, 21 of the 27 randomised subjects completed the study (n = 11 [78.6%], ATM; n = 10 [76.9%], placebo), with no significant difference in attrition rates for each group (ATM, 21.4%; placebo, 23.1%). No significant differences were found in any of the other baseline demographic or clinical characteristics between treatment groups. Reasons for early termination included adverse events (n = 1, ATM), poor compliance (n = 1, ATM), worsening depression (n = 1, placebo), and 3 participants lost to follow‐up (n = 1, ATM; n = 2, placebo)". Overall 22% of the participants dropped out of the study. Quote: "Analyses of results were performed for the efficacy on intention‐to‐treat (ITT) population (all randomised subjects who took any study medication and returned for 1 or more post baseline evaluation) using the last observation carried forward (LOCF), and for observed cases, a completer analysis was performed". |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol. |
| Other bias | Unclear risk | Funding for study provided by industry. Their were more men than women in the study sample (21:6). |
| Methods | Design: multicentre, double‐blind, randomised, flexible dose, placebo‐controlled trial Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1 week single‐blind, placebo treatment period | |
| Participants | Sample size: 272 randomised to venlafaxine ER or placebo Mean age (SD): 41.5 (12.25) years (for 261 participants) Sex: 150 men and 111 women (for 261 participants) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Patients could be included in the study if they were a male or female outpatient at least 18 years of age, had met the DSM‐IV criteria for (generalized) SAD for at least 6 months before the commencement of the study, had anxiety severe enough to warrant anxiolytic therapy, had a CGI‐S score of 4, and had a minimum score of 50 on the LSAS at the prestudy evaluation and on day 1 of the study, with a decrease of 30% between prestudy and baseline tests. In addition, the Covi Anxiety scale score had to be greater than the Raskin Depression total score (with a Raskin total score of not more than 9) at the prestudy evaluation". Exclusion criteria: quote: "Patients were excluded from the study if they had a clinically important medical condition, seizure disorder, mental disorder due to medical condition, myocardial infarction during the previous 6 months, laboratory or electrocardiographic abnormality, positive pregnancy test result at prestudy evaluation, pregnancy or lactation during the study, prestudy HAM‐D score of 15, Axis I or Axis II disorder current or predominant during last 6 months, alcohol or substance abuse within last year, suicidality, regular alcohol use, or drug abuse. Patients who failed to respond to prior treatment were eligible to participate; however, those who had received venlafaxine (ER or IR) within 6 months of study day 1 or had a known hypersensitivity to venlafaxine or related compounds were excluded. In addition, psychotherapy, electroconvulsive therapy, investigational drugs or procedures, sedative hypnotic agents, sumatriptan, naratriptan or zolmitriptan or similar agents, herbal remedies, monoamine oxidase inhibitors, benzodiazepines, anxiolytics, and antidepressants and antipsychotics were not permitted during the study and for a specified period before the start of the study". Dropouts: 100/272 (insufficient information to determine dropout rates for the 2 groups separately) | |
| Interventions | Pharmacological intervention: quote: "Patients were randomly assigned to receive venlafaxine ER (75 to 225 mg/d) or placebo for a 12‐week period, followed by a 2‐week (±3 days) taper period ...Patients were randomly assigned to take either venlafaxine ER 75 mg (1 capsule) or placebo on study days 1 to 7 (±3 days). On study days 8 (±3 days) to 14, the dose was increased to 2 capsules (placebo or 150 mg venlafaxine ER). If clinically indicated, on day 15 (±3 days), the dose was increased to 225 mg (3 capsules) or placebo (3 capsules). The | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐S (for reduction of anxiety), CGI‐I (for treatment efficacy), SPIN (for reduction of anxiety), LSAS fear and avoidance subscale (for reduction of anxiety) Time points: Quote: "Safety and efficacy measures as well as vital signs (including supine pulse rate, and supine and standing blood pressure) were recorded at baseline and weeks 1, 2, 3, 4, 6, 8, 10, and 12; adverse events were recorded at a poststudy evaluation. Patients were also evaluated 4 to 10 days after the last dose of medication" | |
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Two hundred seventy‐two outpatients were randomly assigned to receive either a flexible dose of venlafaxine ER (75 to 225 mg/d) or placebo for 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | The proportion of dropouts per treatment group could not be determined due to the lack of information provided by the study. More participants in the venlafaxine group withdrew to adverse events compared to those in the placebo group. The two group differed by sample characteristics at baseline and at week 12. Quote: "No significant differences were observed between treatment groups in any of the characteristics or baseline scores, and the characteristics were similar to those of the safety population ... Thus, 261 patients were included in the intent‐to‐treat efficacy analysis. There were 100 discontinuations. One hundred seventy‐two patients completed the study. Significantly more patients in the venlafaxine ER group (15%) than the placebo group (4%) withdrew because of adverse events". Quote: "Analyses of observed cases and last observation carried forward (LOCF) data were performed". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified, although there were more men than women who participated in the study. Quote: "The group was predominantly White, had more men than women, and the mean duration of the current episode of SAD was 25.9 to 28.6 years, underscoring the chronic nature of untreated SAD". |
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 8 weeks Post‐treatment: 1 month follow‐up Placebo run‐in: one week of single‐blind placebo | |
| Participants | Sample size: 78 randomised to moclobemide or placebo (77 ITT sample, due to data missing for one participant) Mean age (SD): 34.95 (17.3) years (for 77 ITT sample) Sex: 46 men and 31 women (for 77 ITT sample) Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "Out‐patients aged 18‐65 years, with a principal diagnosis of social phobia, absence of major depression, psychotic disorders, substance misuse (in the past six months) and other major psychiatric disorders were included in the study". Exclusion criteria: quote: "Past history of major depression or substance misuse, and current dysthymia were permitted". Dropouts: 20/77 (10/40 in the moclobemide and 10/37 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "Moclobemide was initiated at 100 mg twice daily and increased over a period of two weeks to a maximum dose of 400 mg twice daily. Dosage could be adjusted as clinically indicated to manage adverse effects". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety), LSPDS (for reduction of anxiety), CGI (for treatment efficacy), SADS (for reduction of anxiety) and FNES (for reduction of anxiety) Secondary outcome measures: FQ (for reduction of anxiety), SDS (for reduction of functional disability), BDI (for reduction of depression), HAM‐D (for reduction of depression), HAM‐A (for reduction of anxiety) and SCL‐90 (for reduction of anxiety) Time points: Quote: "Major evaluations were done at randomisation and after 4, 8 and 16 weeks of randomised treatment" | |
| Notes | Industry funded: no. Quote: "This study was supported by National Institute of Mental Health grant 5 R29 MH 47831‐04 to F.R.S". Medication provided by industry: yes. Quote: "Hofmann‐LaRoche, Nutley, NJ kindly supplied moclobemide and matching placebo for the study". Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author correspondence: quote: "A random number sequence, generated by a data manager with no patient contact, was used to generate the randomization sequence". |
| Allocation concealment (selection bias) | Low risk | Author correspondence: quote: "Yes, allocation sequence was concealed by keeping codes in sealed opaque envelopes". |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "Integrity of the blind design was assessed at week 8 by questionnaires which asked patients and independent evaluators to make a forced choice regarding their belief about which treatment each patient had received ... Neither patients nor independent evaluators identified the treatment condition correctly at a rate greater than chance ... Hofmann‐LaRoche, Nutley, NJ kindly supplied moclobemide and matching placebo for the study". |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "Assessment instruments included patient self‐ratings and ratings by an independent evaluator who, in addition to being blind to randomisation status, was blind to adverse effects and dosage adjustments". |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the moclobemide (10/40; 25%) and placebo groups (10/37; 27%). Reasons for withdrawal were similar across groups. The 2 groups did not differ by sample characteristics at baseline and no statistically significant difference was found between attrition rates. Quote: "The treatment groups did not differ in demographic features, social phobia subtype, comorbidity or rates of prior drug treatment. In the moclobemide group, 33 patients (82.5%) completed at least four weeks of randomised treatment, and 30 (75.0%) completed at least eight weeks. In the placebo group, 32 (86.5%) and 27 (73.0%) completed at least four and at least eight weeks, respectively. Rates of dropout by week 4 and by week 8 did not differ between moclobemide and placebo groups. A comparison of patients who dropped out to week 4 versus patients who completed at least 4 weeks yielded no significant differences between the 2 groups on any baseline variables ... Number of dropouts in each group, by reason for dropout, were: adverse effects (four moclobemide, three placebo), unknown (two moclobemide, three placebo), non‐compliance with appointments (one moclobemide), lack of efficacy (one moclobemide, two placebo), non‐compliance with medication (two moclobemide), personal reasons unrelated to the study (two placebo)". Overall 26% of the participants dropped out of the study. Quote: "Outcome over the first eight weeks of treatment was analysed for the following three samples: (a) intention‐to‐treat, including all subjects randomised and carrying last observations forward for dropouts ..." |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication was provided by industry. No other sources of bias were identified. |
| Methods | Design: single‐centre, randomised, double‐blind, parallel, flexible dose, placebo‐controlled study Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 60 randomised to mirtazepine or placebo Mean age (SD): 38.6 (10.5) years Sex: 26 men and 34 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Men and women aged between 18 and 65 years of age with generalized SAD according to the DSM‐IV classification and MINI". Exclusion criteria: quote: "Patients were excluded from the study if they had a current comorbid axis I diagnosis according to the DSM‐IV classification; in case of an actual risk for suicide according to the investigator, pregnancy or instable chronic physical conditions as assessed by the medical history and physical examination at screening. The HAM‐D was used to screen for comorbid depressive symptoms and patients were excluded if they had a score of 15 or more. ... Psychotropic medication or any psychotherapeutic interventions in the last month preceding or during the trial period were not allowed". Dropouts: 3/60 (2/30 in the mirtazepine and 1/30 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "The patients received an initial dose of 30mg/day of mirtazepine or an identical looking and tasting placebo. From day 15 onwards, the dose was increased to 45mg of mirtazepine or placebo ODT". | |
| Outcomes | Primary outcome measures: LSAS (for reduction of anxiety) and CGI (for treatment efficacy) Secondary outcome measures: FNES (for reduction of anxiety), SDS (for reduction of functional disability), and ASEX (for sexual functioning) Time points: Quote: "The patients were evaluated at screening, baseline, week 2, week 4, week 8 and week 12 with the LSAS, the FNES, the SDS, and the CGI" | |
| Notes | Industry funded: yes. "Quote: This study was funded by an unrestricted grant by Organon". Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients were randomly assigned to the double‐blind treatment with mirtazepine or placebo for 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "The patients received an initial dose of 30mg/day of mirtazepine or an identical looking and tasting placebo". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar, low proportions of participants withdrew from the mirtazepine (2/30; 7%) and placebo groups (1/30; 3%). More participants in the mirtazepine group withdrew due to adverse events compared to lost for continuation in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "Three patients (0.05%) did not complete the study, two patients from the mirtazepine group dropped out because of side effects (mainly sedation) and one patient from the placebo group was lost for continuation (reason unknown). There were no significant differences between the two groups in age, sex and age of onset". Overall 10% of the participants dropped out of the study. Quote: "Last observation carried forward (LOCF) efficacy analyses were conducted on all the patients who had received any double‐blind medication and from whom at least one valid post baseline efficacy evaluation was obtained". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: single‐centre, open‐label trial, followed by randomised, parallel, flexible dose, double‐blind placebo‐controlled discontinuation, and a relapse prevention phase Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 16 randomised to paroxetine or placebo (36 in the open label phase, 16 of which was randomised in the 12 week phase) Mean age: not specified Sex: 24 men and 6 women (for the 30 completers in the open label phase) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Patients who met DSM‐IV criteria for social phobia, generalized type, according to a semi‐structured interview derived from the Structured Clinical Interview for DSM‐III‐R ... The presence of current (past 6 months) comorbid major depression was an exclusion criterion, although patients with past histories of major depression were included". Exclusion criteria: quote: "Patients with comorbid panic disorder were excluded, unless the panic disorder was felt to be clearly secondary to the social phobia in terms of severity and current impact on functioning. Patients who were currently abusing substances, including alcohol, were excluded from the study". Dropouts: 6/16 (1/8 in the paroxetine and 5/8 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "Eligible subjects began treatment with 10 mg of paroxetine at bedtime. Treating clinicians (M.B.S., C.D.L.K., and R.A.C.) followed a protocol that mandated 10‐mg weekly increments to a maximum of 50 mg/day. If subjects experienced side effects, a once‐only dosage reduction of 10 mg/day was permitted, with the option of reinstituting the prior dose if clinically indicated 1 week later. Only one such dosage adjustment was allowed per patient. Patients who could not tolerate a minimum of 20 mg/day were withdrawn from the study. Treatment lasted for a total of 11 weeks ... Patients were then randomized to either continue paroxetine with no change in dose or to taper and then discontinue paroxetine with placebo substitution for a total of 12 weeks. Regardless of the starting dose, the rate of taper consisted of 1 week at 20 mg and then discontinuation. The taper and all subsequent dosing were conducted under double‐blind conditions wherein all subjects took the same number of turquoise‐colored tablets for the duration of the study". | |
| Outcomes | Primary and secondary outcome measures: MADRS (for reduction of depression), LSAS (for reduction of anxiety), DSPS (for reduction of anxiety), CGI‐S (for reduction of anxiety), CGI (patient‐rated) (for treatment efficacy), FNES (for reduction of anxiety), SIAS (for reduction of anxiety), SPS (for reduction of anxiety), and SDS (for reduction of functional disability) Time points: Quote: "The following ratings were conducted by an experienced clinician at baseline, week 4, week 8, and week 12 in the double‐blind discontinuation study: Montgomery‐Asberg Depression Rating Scale, Liebowitz Social Anxiety Scale, Duke Social Phobia Scale, and the Clinician‐Rated Clinical Global Impressions Scale (CGI; severity version at baseline and change version subsequently). Self‐ratings conducted pre‐ and posttreatment included a patient‐rated version of the CGI, the Fear of Negative Evaluation Scale, the Social Interactional Anxiety Scale, and the Social Performance Scale; the Sheehan Disability Scale (which has three subscales: work, social, and family disability) was completed at baseline, weeks 4, 8, and 11. In the double‐blind discontinuation study, a subject was considered "relapsed" if the clinician‐rated CGI was rated as "no improvement" or "worse" (compared to pre‐treatment) on two consecutive visits. At each visit, subjects were systematically questioned about possible side effects' | |
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Sixteen responders were randomized to an additional 12 weeks of either paroxetine (with no dosage change) or placebo (after a taper period) on a double‐blind basis". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "The taper and all subsequent dosing were conducted under double‐blind conditions wherein all subjects took the same number of turquoise‐colored tablets for the duration of the study". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | Information was not provided for number of dropouts during the randomised relapse‐prevention phase of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind (GSK protocol ID: 29060/382). Duration of intervention: 12 weeks. Post‐treatment: no follow‐up. Placebo run‐in: 1 week, single‐blind, placebo, run‐in period. | |
| Participants | Sample size: 187 randomised to paroxetine or placebo (184 efficacy population, lost to follow‐up before efficacy evaluation). Mean age (range): 36.3 (18‐76) years Sex: 81 men and 106 women. Diagnostic measure: DSM‐IV. Inclusion criteria: quote: "Eligible patients were 18 years of age or older; adults older than 65 years were permitted if they were able to tolerate a starting paroxetine dose of at least 20 mg/d". Exclusion criteria: quote: "Patients who required concurrent psychoactive medication (except chloral hydrate for insomnia), narcotic analgesics, warfarin sodium, digitalis glycosides, phenytoin, cimetidine, or sulfonylurea derivatives were excluded. Patients who had taken any psychotropic agent or beta‐blockers within 14 days prior to the study were ineligible, as were those who had received depot neuroleptics within the previous 12 weeks. Also excluded were patients with any other Axis I diagnosis that was considered to be clinically predominant within the previous 6 months. Patients who met DSM‐IV criteria for substance abuse or dependence within 3 or 6 months prior to this study, respectively, were excluded, as were those judged to be serious suicidal or homicidal risks. Additional reasons for exclusion were body dysmorphic disorder, history of seizure disorder, schizophrenia or bipolar affective disorder, any serious or uncontrolled medical illness or condition that precluded paroxetine use, electroconvulsive therapy within the previous 3 months, investigational drug use or participation in a clinical trial within the previous 12 months, and previous intolerance or lack of response to paroxetine (no subject was excluded on this basis). Women who were pregnant, lactating, or not using a clinically acceptable method of birth control also were ineligible. Finally patients with clinically significant abnormal laboratory or electrocardiographic findings that could not be resolved prior to baseline evaluations were not included". Dropouts: 53/187 (32/94 in the paroxetine and 21/93 in the placebo groups). | |
| Interventions | Pharmacological intervention: quote: "For the purposes of blinding, dosage was referred to as level 1 (20 mg), level 2 (30 mg), level 3 (40 mg), or level 4 (50 mg). Patients received an initial dose of level 1 medication once daily. After 2 weeks, the dosage could be increased to the next level (i.e. 50 mg/d) based on clinical response as determined by the treating physician. Dosage could be reduced to a minimum of level 1 (i.e. 20 mg/d) at any time if the physician felt that adverse effects warranted this adjustment". | |
| Outcomes | Primary outcome measures: CGI‐I (for treatment efficacy) and LSAS (for reduction of anxiety) Secondary outcome measures: SADS (for reduction of anxiety), SDI (for reduction of functional disability) and LSAS (for reduction of anxiety) Time points: Quote: "Patients were evaluated for safety and efficacy at weeks 1, 2, 3, 4, 6, 8, and 12. Adverse events were also assessed telephone at week 10" | |
| Notes | Industry funded: yes. Quote: "Financial support for this study was provided by SmithKline Beecham Pharmaceuticals. Collegeville, Pa". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned in a balanced fashion to the 2 treatment groups (in blocks of 4) using a computer‐generated randomisation code for up to 300 patients". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Patients who were rated no more than minimally improved and who continued to meet all inclusion criteria were randomised to receive paroxetine or placebo (identical in appearance) ... For the purposes of blinding, dosage was referred to as level 1 (20 mg), level 2 (30 mg), level 3 (40 mg) or level 4 (50 mg)". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Higher proportions of participants withdrew from the paroxetine (32/94; 34%) than the placebo groups (21/93; 23%). More participants in the paroxetine group withdrew due to adverse events, whereas being lost to follow‐up was cited as the being reasons for study dropout in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "Of the 187 patients randomised in this study, 4 patients received the drug but were lost to follow‐up prior to the first efficacy evaluation. Therefore, the efficacy data for the remaining 183 patients (i.e., the efficacy population) are reported ... No statistically significant differences between groups were detected with regards to demographic characteristics or mean baseline rating scale scores ... In the paroxetine group, 62 (66%) of 94 patients completed the 12‐week trial. The most common reasons for patient withdrawal were adverse events (15%; 14/94) or lost to follow‐up (13%, 12/94). In the placebo group, 72 (77%) of 93 patients completed the trial. The most common reason for discontinuation was lack of efficacy (11%, 10/93)". Overall 28% of the participants dropped out of the study. Quote: "For patients who did not complete the entire study, the last evaluation during treatment was used as an estimate of the missing data (i.e. last observation carried forward)". |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 92 randomised to fluvoxamine or placebo Mean age (SD): 39.4 (10.55) years Sex: 59 men and 32 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Eligible participants, in addition to meeting the DSM‐IV criteria for social phobia and having a minimum score of 20 on the Brief Social Phobia Scale, were required to be between 18 and 65 years of age and to have no serious medical conditions (and be taking no medications medications) that might put them at risk were they to receive fluvoxamine". Exclusion criteria: quote: "Patients taking psychotropic medications within the 7 days before the study were precluded from participating, as were patients with other psychiatric disorders that were deemed to be primary in terms of clinical significance (e.g., bipolar disorder, schizophrenia) or patients judged to be at serious suicidal or homicidal risk. Patients receiving a specific form of psychotherapy for social phobia were ineligible. Women who were pregnant, lactating, or not using an acceptable method of birth control were ineligible. Patients with clinically significant abnormal laboratory or ECG findings were also ineligible". Dropouts: 24/92 (14/48 in the fluvoxamine and 10/44 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "After the screening procedures, each patient was randomly assigned to either placebo or fluvoxamine (at an initial dose of 50 mg of fluvoxamine per day). After 1 week the daily dose could be increased by 50 mg each week to a maximum of 300 mg/day and could be reduced at any time if side effects necessitated this (i.e., flexible dosing)". | |
| Outcomes | Primary outcome measure: CGI‐I (for treatment efficacy) Secondary outcome measures: BSPS (for reduction of anxiety), SPI (for reduction of anxiety), LSAS (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "The patients were evaluated at weeks 1, 2, 3, 4, 6, 8, 10, and 12" | |
| Notes | Industry funded: yes. Quote: "Sponsored by Solvay Pharmaceuticals, Inc., Marietta, Ga.; funding provided by The Pharmacia & Upjohn Co., Kalamazoo, Mich". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "After the screening procedures, each patient was randomly assigned to either placebo or fluvoxamine". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | Similar proportions of participants withdrew from the fluvoxamine (14/48; 29%) and placebo groups (10/44; 23%). More participants in the fluvoxamine group withdrew due to adverse events compared to those in the placebo group. No other information was provided the reasons for treatment withdrawal nor was information reported on differences between sample characteristics at week 12. Quote: "Six patients did not return for at least one subsequent assessment, leaving 86 patients (42 taking fluvoxamine and 44 taking placebo) in the evaluable study group ... Fluvoxamine was generally well tolerated by the patients in the study, although more fluvoxamine‐treated patients (12, 25.0%) than placebo treated patients (N=4, 9.1%) discontinued the study early because of adverse events ... Final assessment was conducted at week 12 or earlier if the patient dropped out prematurely. Scores were available for 34 patients taking fluvoxamine and 34 patients taking placebo". Overall 26% of the participants dropped out of the study. Quote: "Analyses of response refer to the last observation carried forward for all subjects who had evaluable efficacy data at baseline and with treatment and who had taken at least one dose of study medication". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. Not all the participants in the sample had generalised disorder. Quote: "Although this was not an a priori criterion for entry into the study, nearly all of the patients (91.3%) suffered from the generalised type of the disorder". |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind placebo run‐in | |
| Participants | Sample size: 390 randomised to moclobemide or placebo (390 participants were randomised, of whom data was available for 384; seven participants randomised did not receive medication, so the ITT population for the 12‐week treatment trial comprised 377 participants (188 moclobemide, 189 placebo) Mean age (SD): 34.4 (10.5) years (for 384 participants) Sex: 204 men and 180 women (for 384 participants) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "All subjects met Diagnostic and Statistical Manual (DSM‐IV) diagnostic criteria for social anxiety disorder as the primary diagnosis, had a minimum score of 4 (moderate) on the Clinical Global Impressions scale severity item (CIS‐SP) and were aged 18–65 years. Subjects with comorbid panic disorder, generalized anxiety disorder, agoraphobia or other phobias, were eligible". Exclusion criteria: quote: "Subjects were to be excluded on a number of grounds, including: (i) comorbid psychiatric conditions, i.e. if there were current mood disorders (excluding dysthymia), obsessive–compulsive disorder, or substance use disorder (excluding nicotine); if there was a history of psychosis, schizophrenia, or bipolar disorder (type I or II); an organic mental disorder, dementia or mental retardation; or if there was suicidality; (ii) comorbid medical conditions, i.e. if there was hyperthyroidism, thyrotoxicosis, pheochromocytoma, neuroblastoma, epilepsy, significant liver disease or any significant unstable or uncontrolled medical disease; or if there were any clinically significant physical examination, laboratory, or electrocardiogram findings that would put the patient at risk or obscure the effects of treatment; (iii) treatment requirements and history, i.e. if there was treatment with moclobemide during the past 6 months; treatment with classical MAOIs in the 5 weeks preceding baseline; concomitant use of other psychotropic medications in the 2 weeks preceding baseline; or any investigational drug or experimental procedure in the 4 weeks preceding baseline; and (iv) response during placebo run‐in, i.e. if between the screening and baseline visits, clinical response was rated as 1 (very much improved) or 2 (much improved) on the Clinical Global Impression scale change item (CIC‐SP)". Dropouts: 64/390 (insufficient information to determine dropout rates for the 2 groups separately) | |
| Interventions | Pharmacological intervention: quote: "The moclobemide was initiated at 600 mg/day (300 mg a.m. +300 mg p.m.) for 1 week. This dose could be titrated downward to 450 mg/day (300 mg a.m. +150 mg p.m.) (minimum dose) if tolerability was less than moderate. Alternatively, dosage could be titrated upwards to 750 mg/day (450 mg a.m. +300 mg p.m.) (maximum dose) if tolerability was moderate or better and CIC‐SP was 2 or more. This dose range is at the higher end of the range suggested in most moclobemide package inserts (which describe initiation of medication at 300 mg daily and indicate that medication may be raised to 600 mg daily where necessary)". | |
| Outcomes | Primary outcome measure: CIC‐SP (for treatment efficacy) Secondary outcome measures: CIS‐SP (for reduction of anxiety), LSAS (for reduction of anxiety), MADRS (for reduction of depression), HAM‐A (for reduction of anxiety), SDS (for reduction of functional disability), PIC‐SP (for treatment efficacy), SAS (for reduction of anxiety), and MMFQ (for reduction of anxiety) Time points: Quote: "Clinic visits then took place at weeks 1, 2, 4, 8 and 12 for assessments of efficacy, tolerability, concurrent medications and compliance with study procedures. After an initial 12 weeks, it was at the discretion of study clinicians to invite patients to complete an additional 6 months of treatment. Monitoring visits to each site were performed in order to ensure investigator adherence to the protocol" | |
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "At the baseline visit, non‐responders (i.e. CIC‐SP of 3 or more) were randomised to moclobemide or placebo". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Moclobemide and placebo were provided as identical tablets". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | The proportion of dropouts per treatment group could not be determined due to the lack of information provided by the study. Reasons for withdrawal were not reported on for 4 participants, nor were the groups divided to determine the proportion of withdrawals for others reasons for discontinuation. It appears that more participants in the moclobemide group withdrew to adverse events compared to those in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "Three hundred and ninety patients were randomised, of whom data was available for 384. The two treatment groups were well matched with regard to demographic characteristics ... Seven patients randomised did not receive medication, so the ITT population for the 12‐week treatment trial comprised 377 subjects (188 moclobemide, 189 placebo) ... Sixty‐four patients discontinued during the 12‐week treatment trial; the most frequent reasons were insufficient response (11 in the placebo group, eight in the moclobemide group) and adverse events (10 in the moclobemide group, five in the placebo group). Other reasons for withdrawal included failure to return to follow‐up (n=9), not cooperating with the study (n=7), withdrawal of consent (n=6) and protocol violation (n=4)". Overall 18% of the participants dropped out of the study. Quote: "The intent‐to‐treat (ITT) sample, defined as patients randomized and having received post randomization medication, was used for the efficacy analyses, with last observation carried forward for the 12‐week acute treatment study". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind, maintenance study Duration of intervention: 24 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind placebo run‐in | |
| Participants | Sample size: 323 randomised to paroxetine or placebo Mean age (SD): 38.15 (11.45) years Sex: 128 men and 195 women Diagnostic measure: MINI and DSM‐IV Inclusion criteria: quote: "Eligible patients were aged at least 18 years and had a primary diagnosis of social anxiety disorder, as assessed by psychiatrists and other qualified health care professionals who received training using the MINI for DSM‐IV. No attempt was made to categorise patients as having generalised vs nongeneralised social anxiety disorder. Those older than 65 years had to be able to tolerate a paroxetine starting dosage of at least 20 mg/d and to be without renal or hepatic impairment". Exclusion criteria: quote: "Patients were excluded from the study if they had any Axis I disorder other than generalized anxiety disorder or agoraphobia during the 6 months before screening, a primary diagnosis of panic disorder during the previous 6 months, or a history of schizophrenia or bipolar affective disorder. Subscale or dependence according to DSM‐IV criteria also excluded patients from the study. Concomittant therapy with beta‐adrenergic blockers, monoamine oxidase inhibitors, benzodiazepines, or other psychoactive medication or scleroatrophic or antidepressant therapy within 14 days of baseline also precluded patients from entering the study. Patients who had previously received a therapeutic dosage of an SSRI for social anxiety disorder from an adequate duration to achieve a clinical response or who had received paroxetine for any indication but had not responded were also prevented from participating in the study". Dropouts: 66/323 (26/162 in the paroxetine and 40/161 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "All patients entering the 12‐week single blind acute phase received paroxetine for 12 weeks. The initial dosage was 20 mg/d with food, and patients continued receiving this dosage for at least 2 weeks. Therafter, dose titration (2‐8 weeks, by 10 mg increments) up to a maximum of 50 mg/d was permitted at the investigators discretion ... Responders at week 12 then continued with paroxetine or gradually switched to placebo for a further 24 weeks, depending on their randomization. Patients receiving paroxetine were to remain at the same dosage level as that of week 12. However, a single dosage reduction was permitted in the event of adverse experiences. Patients receiving placebo were dispensed medication to reduce their paroxetine dosage gradually during a 3‐week down titration period, depending on their level of medication at week 12, and then received placebo for the remainder of the study. The daily dosage was reduced to 10 mg each week to 20 mg/d and was maintained at this dosage until week 15, after which all patients randomised to placebo received medication for the remainder of the study". | |
| Outcomes | Primary outcome measure: CGI‐S (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), LSAS (for reduction of anxiety), SCL‐90 (for reduction of anxiety), EuroQol (for quality of life) and HAM‐D (for reduction of depression), Time points: Quote: "Assessments using the CGI improvement scale, LSAS, and Social phobia Inventory were made at baseline; at weeks 1, 2, 3, 4, 8, and 12 during the acute phase of the study; and at weeks 16, 20, 24, 28, 32, and 36 during the 24‐week maintenance treat phase. In addition, an evaluation using the Sheehan Disability Scale was made at each visit. Symptom Checlist‐90 (SCL‐90) and EuroQol (EQ‐5D) questionnaires were completed at baseline, at week 12 (end of acute phase), and during the maintenance phase at weeks 24 and 36 (study end point). Hamilton Depression Rating Scale assessments were performed at baseline, at the end of the acute phase (week 12), and at the end of the study (week 36) to evaluate the presence of depressive symptoms. To assess tolerability, adverse events were monitored throughout the study by asking patients non leading questions such as “Have you felt different in any way since your last visit?” | |
| Notes | Industry funded: yes. Quote: "This study was supported by SmithKline Beecham Pharmaceuticals". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A computer‐generated randomisation list was used to randomise patients in a 1:1 ratio to receive either paroxetine or placebo". |
| Allocation concealment (selection bias) | Low risk | Quote: "Each investigator and centre was allocated a block of consecutively numbered treatment packs, which were dispensed in strict sequential order. The paroxetine and placebo capsules were identical in appearance and packaged to maintain blinding". |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "The paroxetine and placebo capsules were identical in appearance and packaged to maintain blinding". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from the paroxetine (26/162; 16%) and placebo groups (40/161; 25%). Withdrawals were similar across groups (i.e. adverse events, lack of efficacy, deviation from protocol, lost to follow‐up and other. No information was provided on differences between sample characteristics at week 12. Quote: "Of the 323 patients continuing into the double‐blind maintenance phase of the study, 257 (136 paroxetine‐treated [84%] and 121 placebo‐treated [75%] patients) completed the study. Sixty‐six patients withdrew (26 paroxetine‐treated [16%] and 40 placebo‐treated [25%] patients). There were 26 withdrawals (8 in the paroxetine group and 18 in the placebo group) because of lack of efficacy. Withdrawals because of adverse events were more common in the placebo group (8 [5%]) than in the paroxetine (3 [2%]). Further reasons for withdrawal included deviation from protocol (4 paroxetine‐treated [3%] and 7 placebo‐treated [4%] patients, lost to follow‐up (6 paroxetine treated [4%] and 3 placebo‐treated [2%] patients), and other (5 paroxetine‐treated [3%] and 4 placebo‐treated [3%] patients). This last group comprised patients who moved away, those who withdrew consent, and those suspected to being pregnant". Overall 20% of the participants dropped out of the study. Quote: "Primary inferences were based on intention‐to‐treat last observation carried forward". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind, maintenance study Duration of intervention: 12 weeks Post‐treatment: 4, 5 and 6 months follow‐up Placebo run‐in: no | |
| Participants | Sample size: 112 randomised to fluvoxamine CR or placebo Mean age (SD): 37.15 (1.5) years (109 ITT sample) Sex: 58 men and 51 women (109 ITT sample) Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Subjects were outpatients, aged 18–70 yr, had a predominant DSM‐IV diagnosis of GSAD according to the modified SCID, and a minimum score of 60 on the LSAS at screening. Women of childbearing potential or less than 1 yr post‐menopausal were required to use a medically acceptable method of birth control, while pregnant and lactating women were not eligible". Exclusion criteria: quote: "Subjects were excluded if they met any of the following criteria: Subjects with psychiatric disorders deemed to be predominant in the last 6 months including major depressive disorder, dysthymic disorder, or panic disorder; subjects with history or current diagnosis of schizophrenia, other psychotic disorders, bipolar affective disorder, borderline personality, or obsessive–compulsive disorder; subjects who had a score of 18 on the MADRS at screening, and subjects at serious suicidal risk; subjects with evidence of substance abuse disorder or dependence within the past 6 months, and subjects with positive results on a urine drug screen; subjects with unstable or serious medical conditions; subjects who required formal CBT to treat social anxiety symptoms within the previous month; and subjects taking psychotropic medications". Dropouts: 22/112 (10/57 in the fluvoxamine CR and 12/55 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "In the acute phase, dosage was titrated weekly during | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "In the extension phase, these measures were administered every 4 wk (i.e. weeks 12, 16, 20 and 24), or on early termination. Safety assessments comprised adverse‐event monitoring, concomitant medication monitoring, and vitalsign measurement (at weeks 12, 16, 20, 24 or early termination) as well as physical examination, 12‐lead electrocardiogram, and clinical laboratory evaluation (haematology, serum chemistry, urinalysis, and urine drug screening, serum b‐HCG in females of childbearing potential) (at weeks 12 and endpoint)" | |
| Notes | Industry funded: yes. Quote: "This study was supported by a grant from Solvay Pharmaceuticals, Inc". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Subjects were randomized to fluvoxamine CR or placebo according to a centrally generated random allocation sequence, with concealment of the sequence at participating sites, which were provided with numbered packs of fluvoxamine CR or placebo that were identical in appearance". |
| Allocation concealment (selection bias) | Low risk | Quote: "Subjects were randomized to fluvoxamine CR or placebo according to a centrally generated random allocation sequence, with concealment of the sequence at participating sites, which were provided with numbered packs of fluvoxamine CR or placebo that were identical in appearance". |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Subjects were randomized to fluvoxamine CR or placebo according to a centrally generated random allocation sequence, with concealment of the sequence at participating sites, which were provided with numbered packs of fluvoxamine CR or placebo that were identical in appearance". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | Similar proportions of participants withdrew from fluvoxamine CR (10/57; 18%) and placebo groups (12/55; 22%). Reasons for withdrawal were not provided by the study. The 2 groups did not differ by sample characteristics at baseline. Quote: "Of the 112 subjects who enrolled, 47 (82 %) in the fluvoxamine CR‐treatment group and 43 (78%) in the placebo‐treatment group completed the extension phase ... At baseline (day 1 of the acute phase), most demographic (age, gender, ethnicity, marital status, years in school, occupational status) and clinical (GSAD duration, LSAS score, presence of Axis II disorders, family history of psychiatric disorder) variables did not significantly differ in subjects in the fluvoxamine CR (n=56) and placebo (n=53) groups". Overall 20% of the participants dropped out of the study. Quote: "Statistical analyses were nevertheless performed on the primary and secondary efficacy parameters for the intent‐to‐treat (ITT) population, using both last observation carried forward (LOCF) and observed cases". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, fixed and flexible dose, double‐blind study Duration of intervention: 28 weeks Placebo run‐in: yes | |
| Participants | Sample size: 395, with 261 receiving venlafaxine ER (131 fixed dose, 130 flexible dose), and 134 randomised to placebo Mean age (SD): 36.9 (11.6) years (364 ITT sample) Sex: 212 men and 152 women (364 ITT sample) Diagnostic measure: DSM‐IV, as assessed using the MINI Inclusion criteria: quote: "Eligible outpatients were those aged 18 years and older who fulfilled DSM‐IV criteria for GSAD for ≥6 months before the study. Additional inclusion criteria included a Liebowitz Social Anxiety Scale (LSAS) (Heimberg et al. 1999) score ≥50, with a decrease of ≤30% between pre‐study and baseline evaluations; a Clinical Global Impressions (CGI) Scale (Guy 1976) (severity of illness item 1) score ≥4; and pre‐study 17‐item Hamilton Depression (HAM‐D‐17) (Hamilton 1960) score <15, with a score ≤2 on the depressed mood item". Exclusion criteria: quote: "Patients were excluded if they had comorbid major depression, panic disorder with or without agoraphobia, or generalized anxiety disorder, had a history or current diagnosis of any psychotic illness, were acutely suicidal, used alcohol regularly (>24 oz of beer/day or the equivalent), or had a history of drug or alcohol dependence within 1 year of the study. In addition, patients who used psychopharmacologic medications within the 7 days before the study, used antidepressants or herbal products intended to treat anxiety or depression within 14 days of the study, received venlafaxine or ECT within 6 months of the study, or received cognitive behavioral therapy within 30 days of the study were ineligible. Also prohibited from participating were patients with clinically significant abnormal findings on laboratory tests, ECG, vital signs, or physical examination; those with a history or presence of clinically important medical conditions (including head trauma and seizure disorders); and women of childbearing potential who were pregnant, lactating, or not using a medically acceptable form of contraception". Dropouts: 218/368 (133/239 in the venlafaxine ER and 85/129 in the placebo groups) | |
| Interventions | Pharmacological intervention: a fixed dose (75 mg/d) and flexible dose (150 mg/d ‐ 225 mg/d) intervention compared to placebo x 28 weeks | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), SPIN (for reduction of anxiety), LSAS fear and avoidance subscales (for reduction of anxiety), SDS (for reduction of functional disability), WPAI (for reduction of functional disability), clinical response (CGI‐I < 3) (for treatment efficacy), remission (LSAS total score < 30) (for treatment efficacy) Time points: Quote: "Patients were evaluated at baseline (study day −1), and on days 7, 14, 21, 28, 42, 56, 84, 112, 140, 168, and 196. Safety assessments were based on reports of adverse events and measurements of vital signs that were recorded at each visit; laboratory determinations and ECGs, which were assessed at the prestudy (or baseline), week 12, and final visits; and routine physical examinations performed at the prestudy (or baseline) and final visits" | |
| Notes | Industry funded: yes. This study was supported by a grant from Wyeth. Medication provided by industry: yes Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information was provided on how the random sequence was generated. |
| Allocation concealment (selection bias) | Low risk | It appears as if a random number, and the individual package of medication to which it corresponded, were both supplied by the study sponsor. Quote: "Each participating center was pre‐assigned a specified block of patient numbers. A number was given to a patient at the screening visit after the informed consent was signed. At the baseline visit, once eligibility was confirmed, the patient was given a randomization number and the accompanying treatment supplies. The study sponsor supplied study medication as identical appearing capsules, packaged individually and label‐coded for each subject." |
| Blinding of participants and personnel (performance bias) | Low risk | The medication and placebo was provided in identical capsules. Quote: "The study sponsor supplied study medication as identical appearing capsules, packaged individually and label‐coded for each subject." |
| Blinding of outcome assessment (detection bias) | High risk | The clinician rated both outcome and side effects, with the high proportion of drug‐related adverse events in the combined venlfaxine groups compared to the placebo group (e.g. approximately 36% versus 10% for nausea) increasing the risk that the clinician could guess treatment allocation. Quote: "A second limitation of the present study is the possibility that raters may have been unblinded by the occurrence of particular medication‐related side effects (e.g. nausea or somnolence)". |
| Incomplete outcome data (attrition bias) | High risk | A greater proportion of participants withdraw from placebo, primarily due to lack of effect (possibly resulting from the placebo run‐in component of the study design). Though the demographic and symptom severity characteristics of participants in the treatment arms were comparable at baseline, no information on the composition of the participants at endpoint was provided. Quote: "The first 12 weeks of the protocol were completed by 234 (60.6% of) patients; the entire 28 weeks treatment protocol was completed by 164 (43%). The proportion of patients who withdrew from the protocol for any reason was significantly greater in the placebo group (66%) than in either active treatment group (48% and 56%, respectively; both P<0.05); this difference was primarily attributable to the greater number of placebo patients withdrawing due to lack of effect (26% of placebo patients versus 9% and 10% of the two venlafaxine treated groups). At week 28, the mean daily dose (taking into account missed doses) of venlafaxine ER was 72.2 mg. Withdrawals due to adverse events were significantly more likely to occur in the venlafaxine ER 75 mg group (15%) and the venlafaxine 150–225 mg group (21%) than in the placebo group (6%; P=0.026 and P=0.001, respectively)". Quote: "Statistical analyses were performed using last‐observation carried‐forward (LOCF) values". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, 2 arm, parallel group study (NCT00612859) Duration of intervention: 12 weeks Post‐treatment: 4, 5 and 6 months follow‐up Placebo run‐in: 1 week, single blind, placebo‐run‐in period | |
| Participants | Sample size: 216 randomised to levetiracetam or placebo (217 randomised, one participant dropped out of the levetiracetam group prior to medication intake and is not included in the analyses) Mean age (SD): 38.5 (11.75) years Sex: 133 men and 83 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Following the consent process, subjects were screened for study eligibility. The screening visit involved confirmation of GSAD diagnosis and evaluation of other psychiatric diagnoses with the MINI. In addition, medical history, physical exam including vital signs, and routine blood and urine tests were also conducted for safety monitoring and to ensure participants did not suffer from clinically significant medical conditions. Subjects were also required to have a score of > 60 on the LSAS and a total score of < 17 to be included in the study". Exclusion criteria: quote: "Female patients of childbearing potential were required to have a negative serum pregnancy test at screening and negative urine pregnancy tests administered periodically throughout the study. Other exclusion criteria included the presence of another primary Axis I disorder, failure to respond to adequate trials of > 2 medications to great GSAD, and concomitant psychotropic medications in the previous week". Dropouts: 70/216 (34/111 in the levetiracetam and 36/106 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "Study medication was titrated on a fixed schedule over the first 2 weeks from 250 mg/d up to 500 mg bid and then flexibly titrated over the next 4 weeks up to a maximum of 3,000 mg daily (1,500 mg bid). The dosage was then held stable for the remaining 6 weeks. Follow‐up was weekly for 2 weeks and then 2‐week intervals until the end of the study (week 12)". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐C (for treatment efficacy), HDRS (for reduction of depression), and SDS (for reduction of functional disability) Time points: Quote: "Follow‐ups was weekly for 2 weeks and then at 2‐week intervals until the end of the study (week 12)" | |
| Notes | Industry funded: yes. Quote: "This study was funded in its entirety by UCB Pharma, USA". Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Those who maintained an LSAS score > 60 and a CGI‐C >2 (score range: 0‐7) on their return visit (baseline‐week 0) were then randomly assigned to double‐blind treatment with either levetiracetam or matching placebo in a 1:1 ratio. Randomization was stratified according to LSAS scores at baseline (≤ 80, > 80) and age (≤ 40 years, > 40 years)". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Those who maintained an LSAS score > 60 and a CGI‐C >2 (score range: 0‐7) on their return visit (baseline‐week 0) were then randomly assigned to double‐blind treatment with either levetiracetam or matching placebo in a 1:1 ratio". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from levetiracetam (34/111; 31%) and placebo groups (36/106; 34%). With exception of adverse events, and lack of efficiency the reasons for dropouts were similar between groups. The 2 groups did not differ by sample characteristics at baseline, nor did they differ significantly in attrition rates. Quote: "Overall, 148 of the 217 randomly assigned patients (N = 77 [70%], levetiracetam; N=71 [67%], placebo) completed the treatment period, with no statistically significant difference in attrition rates for each group. There was no differences in demographic or clinical characteristics of subjects who terminated the study prematurely. Reasons for early termination included adverse events (n=11, levetiracetam; n‐6, placebo), lack/loss of efficacy (n‐5, levetiracetam; n=4, placebo), withdrawal of consent not related to adverse events/lack of efficacy (n=5, levetiracetam; n=1, placebo), and other reasons (n=7, levetiracetam; n=5, placebo)". Overall 32% of the participants dropped out of the study. The ITT sample was assessed using the LOCF and OCs. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, double‐blind, placebo‐controlled, fixed dose, parallel group trial (NCT00191022) Duration of intervention: 12 weeks Post‐treatment: 2 week follow‐up Placebo run‐in: 1 week, single blind, placebo‐run‐in period | |
| Participants | Sample size: 189 randomised to LY686017, paroxetine or placebo Mean age: not specified Sex: not specified Diagnostic measure: DSM IV‐TR Inclusion criteria: quote: "Patients meeting DSM‐IV‐TR diagnostic criteria for Generalized SAD as confirmed by the MINI were included in the study if they were outpatients between 18 and 65 years of age and presented with a CGI‐Severity score of ≥4". Exclusion criteria: quote: "Patients were excluded from the study if they exhibited any comorbid Axis I disorders within the last 6 months; or suffered from any Axis II disorder, except avoidant personality disorder. Further exclusion criteria were a history of substance or alcohol abuse or dependence within the past year and prior non‐responders to either selective serotonin reuptake inhibitors (SSRI) or serotonin norepinephrine reuptake inhibitors (SNRI)". Dropouts: 69/189 (insufficient information to determine dropout rates for the 2 groups separately) | |
| Interventions | Pharmacological intervention: quote: "At Visit 3, all patients were randomized to treatment with LY686017, 50 mg QD, placebo, or paroxetine, 10 mg QD for up to 12 weeks ... One week after the beginning of the acute therapy phase (at Visit 4), patients randomized to paroxetine received a dose escalation from 10 mg QD [4 times daily] to 20 mg QD. At the end of the acute therapy phase (Visit 9), patients in the LY686017 and placebo arms received a 2‐week supply of placebo. Patients in the paroxetine arm received paroxetine 10 mg QD for one week, followed by placebo for 1 week. All patients were discontinued at Visit 10". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: LSAS (for reduction of anxiety), CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), HAM‐A (for reduction of anxiety), and SDS (for reduction of functional disability) Time points: Time points were not specified | |
| Notes | Industry funded: yes. Quote: "Funding for this study was provided by Eli Lilly and Company (“Lilly”), Indianapolis, IN". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "189 outpatients suffering from SAD were randomly assigned to 12‐weeks treatment with 50 mg/d LY686017 (N=77), placebo (N=74), or 20 mg/d paroxetine (N=38)". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | The proportion of dropouts per treatment group could not be determined due to the lack of information provided by the study. The most common reasons for early discontinuation were adverse events and personal conflict or other participant decisions. This, however, was not specified by group. The 2 groups did not differ by sample characteristics at baseline. Quote: "189 patients were randomized to treatment and received at least one dose of the study drug, with 120 patients completing the study. The most common reasons for early discontinuation were adverse events (21 patients) and personal conflict or other patient decisions (21 patients). There were no statistically significant differences between treatment groups in patients who discontinued early ... The last‐observation‐carried‐forward (LOCF) method was used to impute missing data for the ANCOVA model". |
| Selective reporting (reporting bias) | High risk | Information was only provided for the primary outcome (i.e. LSAS) |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind (NCT00191022) Duration of intervention: 12 weeks Post‐treatment: 6‐month follow‐up Placebo run‐in: no | |
| Participants | Sample size: 46 randomised to atenolol or placebo (72 randomised: 25 atenolol, 26 flooding and 21 placebo) Mean age (range): 35.4 (18‐56) years Sex: 28 men and 34 women (for all three groups). Diagnostic measure: Initial Evaluation Form and Anxiety Disorders Interview Schedule‐Revised Inclusion criteria: quote: "Patients had to have a primary diagnosis of social phobia and could not have a secondary Axis I diagnosis other than generalized anxiety disorder, simple phobia, or dysthymia. In these cases, the additional diagnoses clearly had to be secondary to the social phobia with respect to chronology of onset and degree of impairment of daily functioning". Exclusion criteria: quote: "Patients who had an Axis II diagnosis of schizotypal, schizoid, borderline, paranoid, or antisocial personality disorder were excluded. Fifty‐five patients were excluded because of the presence of exclusionary Axis I or Axis II diagnoses, or medical conditions contraindicating the use of atenolol". Dropouts: 5/46 (4/25 in the atenolol and 1/21 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "Patients took 25 mg/day (in the morning) during the first week, 50 mg during the second and third weeks and 100 mg during the fourth and subsequent weeks". | |
| Outcomes | Primary and secondary outcome measures: SPAI (for reduction of anxiety), SAD (for reduction of anxiety), FNE (for reduction of anxiety), FQ (for reduction of anxiety), STAI (for reduction of anxiety), ISPI (for reduction of anxiety), and SPEFI (for reduction of functional disability) Time points: Quote: "Self‐report instruments were administered at pre‐ and posttreatment and at each follow‐up" | |
| Notes | Industry funded: no. Quote: "This study was supported in part by Grant MH 41852 from the National Institute of Mental Health and was conducted in the Anxiety Disorders Clinic, Western Psychiatric Institute and Clinic, Pittsburgh, Pennsylvania". Medication provided by industry: yes. Quote: "Also, we extend our appreciation to Stuart Pharmaceutical Company for supplying the atenolol and placebo medication that was used". Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients were assigned randomly to one of three groups: flooding, atenolol, or placebo". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Medications were given in double‐blinded fashion ... Atenolol tablets of different doses and placebo tablets had an identical appearance ... The monitoring physician received all assessment data independent of the treating nurse clinician and made all medication decisions independently". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. Quote: "Multiple measures of outcome were used, including self‐report, clinician ratings (including assessment by independent evaluators), behavioral assessment, and performance on composite indexes". |
| Incomplete outcome data (attrition bias) | Unclear risk | A larger proportion of participants discontinued the study in the atenolol (4/25; 16%) group compared to the placebo group (1/21; 5%), though a Chi2 test reported that the difference in dropout proportions across all 3 treatment arms was not significant (Chi2(2, N = 71) = 2.19, P > 0.05). No information was provided regarding the reasons for treatment withdrawal nor was there information reported on if the 2 groups differed by sample characteristics at week 12. Quote: "Nine patients (12.1%) dropped out during the course of the 12‐week treatment; 5 from flooding, 3 from atenolol, and 1 from placebo. In addition, 1 patient was removed from atenolol treatment because of orthostatic hypotension. Thus, there were 62 patients who completed the study". Overall 11% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication was provided by industry. No other sources of bias were identified. |
| Methods | Design: single‐centre, randomised, double‐blind, flexible dose, placebo‐controlled treatment trial (NCT00215254) Duration of intervention: 8 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 15 randomised to quetiapine or placebo Mean age (SD): 32.93 (8.64) years Sex: 7 men and 8 women Diagnostic measure: DSM IV Inclusion criteria: quote: "The inclusion criteria were as follows: (1) adult outpatients 18–65 years of age, (2) a primary diagnosis of SAD using DSM‐IV criteria, (3) a minimum Clinical Global Impression Severity score (CGI‐S) of 4 and minimum Brief Social Phobia Scale (BSPS) score of 20 at baseline, (4) written informed consent, and (5) a negative serum pregnancy test for women of childbearing potential". Exclusion criteria: quote: "The exclusion criteria were as follows: (1) current DSM IV diagnosis of bipolar disorder, schizophrenia or other psychotic disorder, mental retardation or other pervasive developmental disorder, or cognitive disorder due to a general medical condition, (2) any current primary anxiety disorder other than SAD, (3) current primary diagnosis of major depressive disorder, (4) history of substance abuse or dependence within the last 6 months, (5) suicidal risk or serious suicide attempt within the last year, (6) clinically significant medical condition or laboratory abnormality, (7) women of childbearing potential who are unwilling to practice an acceptable method of contraception, (8) concomitant use of medication with psychotropic effects, and (9) history of hypersensitivity to quetiapine". Dropouts: not specified | |
| Interventions | Pharmacological intervention: quote: "The titration schedule of quetiapine or matching placebo was a flexibly‐dosed regimen as follows: 25 mg twice a day for the first 3 days, 50 mg twice a day after that until the end of the first week, 100 mg twice a day for the second week, 150 mg twice a day for the third week, and 200 mg twice a day for the fourth week, with the actual doses prescribed dependent on the tolerability for each patient. Upon completion of the study, patients were tapered off the medication over 3 days. Patients who had a significant worsening of symptoms (i.e., increase in CGI‐S of 2 or more compared to baseline at 2 consecutive visits) were removed from the study and referred for treatment as clinically indicated. Patients who missed 5 consecutive days of treatment were discontinued from the trial". | |
| Outcomes | Primary outcome measures: BSPS (for reduction of anxiety) and CGI‐I (for treatment efficacy) Secondary outcome measures: SPIN (for reduction of anxiety) and SDI (for reduction of functional disability), as well as the CGI‐S (for reduction of anxiety), SOSS (for reduction of anxiety), BAS (for reduction of anxiety), and SAS (for reduction of anxiety) Time points: Quote: "At baseline and weeks 1, 3, and 5, efficacy assessments (BSPS, SPIN, CGI) and safety measures (vital signs, BAS, SAS, and SOSS ) were performed. At week 3, SDI was repeated. The full battery of assessments were performed at week 8 (the final visit) and, for women of childbearing potential, a serum pregnancy test was repeated" | |
| Notes | Industry funded: yes. Quote: "Funding was provided to the last author by AstraZeneca to conduct the study". Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were assessed for eligibility at a screening visit, with eligible patients returning for a baseline assessment in approximately 1 week, at which time they were randomized 2:1 to either quetiapine (10 patients) or placebo (5 patients); this was done by utilizing a computer code generated by a study statistician who did not have contact with subjects". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "This was an eight week, randomized, double‐blind, placebo controlled treatment trial of SAD with quetiapine (50–400 mg/day) or matching placebo." |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Unclear risk | The study did not report attrition. Quote: "Data analysis was performed on the intent‐to‐treat (ITT) population using the last‐observation‐carried‐forward (LOCF) method for missing data". |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported as specified in the protocol. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 20 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo run‐in | |
| Participants | Sample size: 204 randomised to sertraline or placebo Mean age (range): 35.65 (19‐56) years Sex: 114 men and 90 women Diagnostic measure: DSM IV Inclusion criteria: quote: "Inclusion criteria for the study required patients to meet DSM‐IV criteria for primary generalized social phobia of at least 1‐year duration at screening. Patients had to have a CGI severity rating of 4 or less (i.e., moderately ill or worse) and to be between 18 and 60 years of age without any serious or uncontrolled medical illness or condition that precluded sertraline use. Patients with an additional diagnosis of avoidant personality were allowed to participate. Patients with comorbid major depression were permitted to enter the study provided their diagnosis was secondary to social phobia, their baseline Montgomery Åsberg Depression Rating Scale score was 19 or less, the onset of social phobia predated onset of the current episode of depression by 5 years or more, and the patient did not represent a substantial suicide risk". Exclusion criteria: quote: "Patients were excluded if they had another primary axis I disorder or fulfilled criteria in the previous 6 months for panic disorder, agoraphobia, obsessive‐compulsive disorder, eating disorders, body dysmorphic disorder, or substance abuse. Other exclusion criteria included concomitant use before study Dropouts: 46/204 (31/135 in the sertraline and 15/69 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "Patients received an initial dose of 50 mg/day of sertraline or matching placebo. After 4 weeks, the dose could be increased by 50 mg/day every 3 weeks in the absence of satisfactory response (CGI improvement score indicating much or very much improved) up to a maximum allowable dose of 200 mg/day by week 10. The dose could be reduced to a minimum of 50 mg/day if required by the presence of intolerable side effects". | |
| Outcomes | Primary outcome measures: CGI‐I (for treatment efficacy), FQ (for reduction of anxiety), and BSPS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), Liebowitz Panic and Social Phobic Disorders Rating Form (for reduction of anxiety), SPAI (for reduction of anxiety), SADS (for reduction of anxiety), FNE (for reduction of anxiety), MADRS (for reduction of depression), CAS (for reduction of anxiety), and SDS (for reduction of functional disability) Time points: Quote: "Subjects were evaluated at weeks 1, 2, 4, 7, 10, 13, 16, and 20" | |
| Notes | Industry funded: yes. Quote: "Financial support for this study was provided by Pfizer Inc". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients who continued to meet all inclusion criteria and did not have a CGI severity score decline of 2 points or more were randomly assigned to receive sertraline or placebo in a ratio of 2:1". |
| Allocation concealment (selection bias) | Unclear risk | Study medication was identical; however, the method of how this concealment took place was not discussed. Quote: "Patients received an initial dose of 50 mg/day of sertraline or matching placebo". |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Patients received an initial dose of 50 mg/day of sertraline or matching placebo". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from sertraline (31/135; 23%) and placebo groups (15/69; 22%). Similar reasons for withdrawal were found across the 2 groups, although more participants withdrew in the sertraline group because of adverse events compare to those participants in the placebo groups. The 2 groups did not differ by sample characteristics at baseline. Quote: "There were no statistically significant differences between groups in demographic characteristics or mean baseline rating scale scores ... In the sertraline group, 104 (77%) of 135 patients completed the 20‐week trial. In the placebo group, 54 (78%) of 69 patients completed the trial. The reasons for patient discontinuation in the sertraline and placebo groups, respectively, were adverse events (N=16 versus N=1), lack of efficacy (N=4 versus N=4), withdrew consent (N=4 versus N=7), lost to follow‐up (N=3 versus N=1), protocol violation (N=1 versus N=0) and administrative reasons (N=3 versus N=2)". Overall 22% of the participants dropped out of the study. Quote: "Random regression was used to compare improvement slopes because it makes fewer assumptions about missing data while optimizing the use of available data". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, flexible dose, double‐blind, placebo‐controlled, parallel‐group trial Duration of intervention: 14 weeks Post‐treatment: no follow‐up Placebo run‐in: 1‐week, single‐blind, placebo run‐in | |
| Participants | Sample size: 105 randomised to nefazodone or placebo Mean age (SD): 35.8 (10.65) years Sex: 50 men and 55 women Diagnostic measure: DSM IV Inclusion criteria: quote: "Inclusion criteria for the study required subjects to be psychiatric outpatients between the ages of 18 and 65 years, to fulfil DSM‐IV criteria for GSP for more than 1 year, and to be of at least moderate illness severity on the basis of the CGI‐S rating. Patients with comorbid secondary major depression were permitted to participate in the study provided that their baseline score on the MADRS was 19 or less, there was no risk to suicidality on the basis of mental status examination, and the onset of their social phobia predated the major depressive disorder by at least 5 years". Exclusion criteria: quote: "Current comorbid Axis I disorders such a panic disorder with agoraphobia, obsessive‐compulsive disorder, body dysmorphic disorder, or alcohol/substance abuse were excluded from this study. Those with a lifetime history of bipolar affective disorder, schizophrenia, psychoses, delirium, dementia, or other cognitive disorders were also excluded, as were individual reporting 2 previous treatment failures for GSP". Dropouts: 22/102 (15/51 in the nefazodone and 7/51 in the placebo groups; three participants were randomly assigned to treatment but did not take at least 1 dose of study medication). | |
| Interventions | Pharmacological intervention: quote: "Nefazodone or placebo was started at an initial dose of 100 mg/day in divided doses. Doses were increased to 200 mg/day week 2, and up to 300 mg/day by week 4. Further increments of 100 mg were added every 2 weeks, until a maximum dose of 600 mg/ day was reached". | |
| Outcomes | Primary outcome measures: CGI‐I (for treatment efficacy) and LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐S (for reduction of anxiety), SPI (for reduction of anxiety), SPS (for reduction of anxiety), SIAS (for reduction of anxiety), BDI (for reduction of depression), BAS (for reduction of anxiety), SDS (for reduction of functional disability), and the RAND 36‐Item Health Survey (for general health) Time points: Quote: "Patients were evaluated at weeks 1, 2, 3, 5, 7, 9, 12, and 16" | |
| Notes | Industry funded: yes. Quote: "Partial funding for this study was provided by an investigator‐initiated research grant from Bristol‐Myers Squibb, Montreal, Quebec, Canada". Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Those subjects who continued to meet inclusion criteria were randomly assigned on a 1:1 basis to receive either nefazodone or placebo for 14 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | High risk | Quote: "Because of the distinct side effect profiles of placebo and any active medication, it is quite possible that the raters were not blind to experimental condition". |
| Incomplete outcome data (attrition bias) | High risk | A larger proportion of participants discontinued the study in the nefazodone (15/52; 29%) group compared to the placebo group (7/53; 13%). No information was provided regarding the reasons for treatment withdrawal except for adverse events. More participants in the nefazodone compared to the placebo group withdrew because of reported adverse events. No information was reported on if the 2 groups differed by sample characteristics at week 14. Quote: "Thirty‐six (70.6%) of 51 patients in the nefazodone group completed the trial compared with 44 (86.3%) of 51 in the placebo group". Overall 21% of the participants dropped out of the study. Quote: "All efficacy analyses were carried out on the intention‐to‐treat sample using the last‐observation carried forward method" . |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, fixed dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: 3 months follow‐up Placebo run‐in: no | |
| Participants | Sample size: 30 randomised to brofaromine or placebo Mean age (SE): 32.8 (2.0) years Sex: 9 men and 21 women. Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "Included in the study were patients suffering from social phobia according to DSM‐III‐R criteria". Exclusion criteria: quote: "Excluded were patients with another anxiety disorder, major affective disorder or psychotic disorder, alcohol abuse and those patients suffering from medical problems on the basis of a complete medical evaluation". Dropouts: 1/30 (0/15 in the brofaromine and 1/15 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "The dose of brofaromine was gradually increased from 50 to 150 mg daily (75 mg b.i.d.) in 3 weeks". | |
| Outcomes | Primary and secondary outcome measures: SCL‐90 (for reduction of anxiety), HAM‐D (for reduction of depression), SPS (for reduction of anxiety), STAI (for reduction of anxiety), and HAM‐A (for reduction of anxiety) Time points: Quote: "At the onset and the end of the study period. Adverse events were assessed by open questioning at week 1, 2, 4, 8 and 12" | |
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients were randomly allocated to one of the two treatment groups". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | A single participant of the 30 discontinued the study, in the placebo condition (1/15; 7%). |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified. |
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, fixed dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 30 randomised to fluvoxamine or placebo Mean age (SD): 35.2 (9.5) years Sex: 13 men and 17 women Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "Included in the study were patients suffering from social phobia according to DSM‐III‐R criteria". Exclusion criteria: quote: "Excluded were patients with another anxiety disorder, major affective disorder or psychotic disorder, alcohol or drug abuse, those patients suffering from medical problems on the basis of a complete medical evaluation and patients who were pregnant or lactating. A score of 15 or higher on the HAM‐D Scale was an exclusion criterion. Patients with personality disorders according to DSM‐III‐R criteria were also excluded". Dropouts: 2/30 (1/15 in the fluvoxamine and 1/15 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "The dose of fluvoxamine was gradually increased from 50 mg to 150 mg daily (50 mg t.i.d. [3 times daily]) in 3 weeks" | |
| Outcomes | Primary and secondary outcome measures: SAS (for reduction of anxiety and performance), HAS (for reduction of anxiety), SCL‐90 (for reduction of anxiety), and HDS (for reduction of depression) Time points: Quote: "Efficacy of the treatment was assessed using the Social Anxiety Scale (SAS) and the Hamilton Anxiety Scale (HAS) on baseline and at weeks 1, 2, 4, 8, and 12. At the outset and the end of the study period, patients completed the 90‐Item Symptom Checklist (SCL‐90). The Hamilton Depression Scale (HDS) was completed on baseline and at the end of treatment. Averse events were assessed by open questioning at weeks 1, 2, 4, 8 and 12" | |
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients were randomly allocated to one of the two treatment groups". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar, low proportions of participants withdrew from fluvoxamine (1/16; 6%) and placebo groups (1/14; 7%). One participant withdrew due to a side effect in the fluvoxamine group compared to lack of efficacy in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "From the patients who were recruited, one dropped out in the second week due to severe side effects (treated with fluvoxamine); another patient dropped out in week 8 due to lack of efficacy (treated with placebo) ... The two treatment groups did not differ in mean age, mean age of onset and sex". Overall 7% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified. |
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, fixed dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 30 randomised to buspirone or placebo Mean age (SD): 37.25 (8.85) years Sex: 19 men and 11 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Included in the study were patients suffering from social phobia, specific or generalised subtype, according to DSM‐IV criteria". Exclusion criteria: quote: "Excluded were patients with another anxiety disorder, major affective disorder or psychotic disorder, alcohol or drug abuse; and pregnancy or lactation and those patients suffering from medical evaluation. Patients with a personality disorder according to the DSM‐IV were also excluded. A score of 15 or higher on the HAM‐D was an exclusion criterion". Dropouts: 3/30 (0/15 in the buspirone and 3/15 in the placebo groups) | |
| Interventions | Pharmacological intervention: quote: "The dose of buspirone was gradually increased from 15 mg in the first week to 30 mg from the third week on (10 mg t.i.d. [3 times daily])". | |
| Outcomes | Primary and secondary outcome measures: SPS (for reduction of anxiety), HAM‐A (for reduction of anxiety), SCL‐90 (for reduction of anxiety) and HAM‐D (for reduction of depression) Time points: Quote: "Efficacy of the treatment was assessed using the Social Phobia Scale (SPS) and the Hamilton Rating Scale for Anxiety (HAM‐A) at baseline and at Weeks 1, 2, 4, 8, and 12. At the outset and the end of the study period, patients completed the 90‐item Symptom Checklist (SCL‐90). The HAM‐D was completed at baseline and at the end of treatment. Averse events were assessed by open questioning at weeks 1, 2, 4, 8, and 12" | |
| Notes | Industry funded: no Medication provided by industry: yes. Quote: "The authors thank Bristol‐Myers Squibb for their technical support and providing the trial medication". Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Patients were randomly allocated to one of the two treatment groups". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | More participants withdrew from the placebo group (3/15; 20%) compared to the buspirone (0/15; 0%) group. Participants dropped out for reasons of inefficacy or distance to the hospital. No information was provided on whether participants differed in terms of characteristics by group at week 12, however. Nevertheless, the total proportion of dropouts (10%) is relatively low, suggesting that dropout rates may not have biased the outcomes. Quote: "Of the 15 patients randomly assigned to receive placebo, 3 patients dropped out for reasons of inefficacy or distance to the hospital ... There were no dropouts among the 15 patients randomly assigned to receive buspirone". Overall 10% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Medication provided by industry. No other sources of bias were identified. Quote: "One third of the total patient sample used alcoholic beverages to reduce social phobic anxiety and symptoms in social situations". |
| Methods | Design: single‐centre, randomised, placebo‐controlled, parallel, flexible dose, double‐blind, cross‐over trial Duration of intervention: 8 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 78 randomised to phenelzine, moclobemide and placebo Mean age: not specified Sex: not specified Diagnostic measure: DSM‐III‐R (SCID) Inclusion criteria: quote: "The patients were of either sex, and aged 19‐60 years. The disorder had to meet the following criteria: by CGI severity score of > 4; (ii) global score on the SDSof 3; and clinical judgement that a drug treatment was indicated. All patients met the DSM‐III‐R criteria for social phobia, as diagnosed, by the Structured Clinical Interview for DSM‐III‐R. They had to have been free from any psychotropic medication for at least one month". Exclusion criteria: quote: "Patients were excluded if they had, or had a history of, any other DSM‐III‐R diagnoses to which social phobia could have been secondary. These included organic mental disorders, abuse of psychoactive substances, other anxiety disorders except generalised anxiety disorder, panic disorder (with a more stringent criterion than those of DSM‐III‐R, i.e. history of a single unexpected panic attack), and psychosis. Patients with significant medical illness e.g. essential tremor or Parkinson's disease that could mimic certain social phobic symptoms were also excluded. Inability to fill in self‐rating scales or to adhere to the study requirements, as well as concomitant psychotherapy or lack of protection against pregnancy, were other exclusion criteria". Dropouts: 4/78 (1/26 in the phenelzine, 0/26 in the moclobemide and 3/26 in the placebo groups; these rates are for phase I) | |
| Interventions | Pharmacological intervention: quote: "Medication was provided in capsules of identical appearance containing either moclobemide (100mg), phenelzine (15mg), or placebo. The initial dose was one capsule twice daily, morning, and afternoon; if tolerated, this dose was increased on day 4 to four capsules a day ‐ two in the morning, one in the afternoon, and one at bedtime. This dose was maintained until the end of week 4. At week5, if the dose was tolerated, it was increased again to five capsules per day ‐ two in the morning, two in the afternoon, and one at bedtime. At week 6, there was a further option to increase the dose to two capsules thrice daily; attempts were made to reach this maximum dose (600 mg/day moclobemide, 90 mg/day phenelzine) in all cases, irrespective of the degree of improvement". | |
| Outcomes | Primary outcome measures: CGI (for treatment efficacy), CGI‐S (for reduction of anxiety), WPI (for reduction of anxiety and measure of personality), SADS (for reduction of anxiety), and FNE (for reduction of anxiety) Secondary outcome measures: HRSD (for reduction of depression), HAM‐A (for reduction of anxiety), SCL‐90 (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "The CGI, the Social Phobia Scale, and a form to assess and record side‐effects were administered each week during phase I, and thereafter every four weeks until week 24. The other rating scales were administered at baseline and then every four weeks throughout the study. A battery of laboratory tests and an electrocardiogram were performed in all patients, immediately before inclusion and at weeks 8 and 16" | |
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "Blindness was maintained throughout by using capsules of identical appearance; these were administered according to a randomisation list". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Medication was provided in capsules of identical appearance containing either moclobemide (100mg), phenelzine (15mg), or placebo". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | A larger proportion of participants discontinued the study in the placebo group (3/26; 12%) compared to the phenelzine (1/26; 4%) and moclobemide group (0/26; 0%). Participants in the placebo group dropped out due to lack of efficacy compared to adverse events in the phenelzine group. Participants did not differ by group characteristics at baseline. Quote: "Seventy‐eight patients, 26 in each treatment group, entered phase I of the trial, while 45 patients (7 in the placebo group; 17 in the moclobemide group; 21 in the phenelzine group) entered phase II. Seventeen patients in the moclobemide group and 20 patients in the phenelzine group entered phase III ... There were no significant differences between the three treatment groups regarding demographic characteristics or diagnostic features. In phase I, during the first week, two patients refused to continue the study and were replaced. During weeks 4 to 8, four patients left the study three from the placebo group (for lack of efficacy) and one from the phenelzine group (for side‐effects ‐ dizziness, loss of libido, and headache). At the end of phase I, 21 non‐responders (16 from the placebo group, and 5 from the moclobemide group) were withdrawn from the study". Overall 8% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified. |
| Methods | Design: single‐centre, placebo‐controlled, parallel, flexible dose, double‐blind Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 60 randomised to bromazepam and placebo Mean age (SD): 36.7 (9.9) years Sex: 39 men and 21 women Diagnostic measure: DSM‐III‐R Inclusion criteria: quote: "The patients were of either sex and aged 19‐60 years. The disorder had to meet the following criteria: a CGI Severity score equal to or greater than 4 and a Sheehan Govla Disabilities score of at least 3. All patients met criteria for social phobia as diagnosed by the structured clinical interview for DSM‐III‐R. They had to have been free from any psychotropic medication for at least one month". Exclusion criteria: quote: "Patients were excluded if they had a history of any other DSM‐III‐R diagnoses to which social phobia could have been secondary. These excluded organic mental disorders, abuse of psychoactive substances, other anxiety disorders, except generalised anxiety disorder, panic disorder and psychotic disorders. Relative to mood disorders only past major depression or secondary dysthymia were allowed. Personailty disorders of cluster A or cluster B were excluded. Significant medical illnesses were also excluded. Inability to fill in self‐rating scales or to adhere to the study requirements were also reasons for exclusion". Dropouts: 3/60 (1/30 in the bromazepam and 2/30 in the placebo groups). | |
| Interventions | Pharmacological intervention: 3‐9 mg bromazepam or placebo | |
| Outcomes | Primary and secondary outcome measures: CGI (for treatment efficacy), LSAS (for reduction of anxiety), WPI (for reduction of anxiety and measure of personality), SADS (for reduction of anxiety), FNE (for reduction of anxiety), HAM‐D (for reduction of depression), HAM‐A (for reduction of anxiety), SCL‐90 (for reduction of anxiety) and SDS (for reduction of functional disability) Time points: Quote: "Medical visits were performed every week for dosage adjustments, assessments, recording of viral signs and evaluation and recording of treatment emergent adverse events" | |
| Notes | Industry funded: no Medication provided by industry: no Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not report on if the participants were randomly assigned to treatment and comparison nor was the procedure specified. |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "Blindnesss was maintained throughout by using tablets of identical appearance". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) | Low risk | Similar, low proportions of participants withdrew from bromazepam (1/30; 3%) and placebo groups (2/30; 7%). Pateients in the bromazepam group withdrew due to lack of efficacy and sedation compared to lack of efficacy and nausea in the placebo group. No information was provided on if groups differed by sample characteristics at week 12. Quote: "There were three dropouts. One in the placebo group at week 6, was due to lack of efficacy. Another in the placebo group was due to lack of efficacy and nausea, at week 5. One dropout occurred in the bromazepam group, at week 5, due to lack of efficacy and sedation". Overall 10% of the participants dropped out of the study. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Low risk | No other sources of bias were identified. |
| Methods | Design: multicentre, double‐blind, placebo‐controlled, parallel, flexible dose, relapse prevention study Duration of intervention: 24 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 50 randomised to sertraline continuation and placebo switch (65 eligible) Mean age (range): 36.6 (21‐57) years Sex: 32 men and 18 women Diagnostic measure: DSM‐IV Inclusion criteria: quote: "Inclusion criteria for the study required patients to meet DSM‐IV criteria for primary generalized social phobia of at least 1 year's duration at screening with a CGI‐S score of 4 or greater and to be between 18 and 60 years of age without any serious or uncontrolled medical illness or condition that precluded sertraline use. Patients with an additional diagnosis of avoidant personality were allowed to participate. Patients with comorbid major depression were permitted to enter the study provided that their diagnosis was the result of social phobia; their baseline MADRS score had to be 19 or less; the onset of social phobia had to predate the onset of the current episode of depression by 5 years or more; and they did not represent a significant suicide risk". Exclusion criteria: quote: "Patients were excluded if they had another primary axis I disorder or if they fulfilled criteria in the previous 6 months for panic disorder, agoraphobia, obsessive‐compulsive disorder, eating disorders, body dysmorphic disorder, or substance abuse. Other exclusion criteria included concomitant use prior to study screening of psychotropic medication within a period of five half‐lives, neuroleptics within 7 months, serotonergic antidepressants or an antianxiety medication for 3 or more weeks within 3 months, and cognitive‐behavior therapy within 4 weeks. Patients receiving benzodiazepines were permitted to enter the study after completing a minimum of 2 to 4 weeks of tapered discontinuation. Additional reasons for exclusion included a urinary screen positive for benzodiazepines at baseline; treatment with [beta]‐blockers, methyldopa, guanethidine, or clonidine; and participation in a clinical trial within the previous 12 months. Women who were pregnant, lactating, or not using an acceptable method of contraception were excluded, as were patients who had had a major life event in the last 3 months that, in the investigator's opinion, was influencing their current condition". Dropouts: 18/50 (3/25 in the sertraline continuation and 15/25 in the placebo switch groups) | |
| Interventions | Pharmacological intervention: quote: "In the initial short‐term treatment study, patients who met all inclusion criteria were randomly assigned to receive flexible‐dose sertraline (50‐200 mg/day) or placebo in a ratio of 2:1. After completion of the 20‐week double‐blind study, patients who had responded (Clinical Global Impression Scale of Improvement [CGI‐I] score of much or very much improved) were eligible to enter the 24‐week study. Patients who had been receiving sertraline were randomly assigned again in a double‐blind fashion in a ratio of 1:1 to either continue sertraline or switch to placebo for another 24 weeks. Patients who had been receiving placebo continued to receive double‐blind placebo. The only sleep medications permitted during the study were chloral hydrate (500‐1,000 mg per night) or zopiclone (3.75‐7.5 mg per night). Patients who developed persistent side effects could have their study medication dosage reduced to the next lower level". | |
| Outcomes | Primary outcome measures: CGI‐S (for reduction of anxiety), MFQ (for reduction of anxiety) and BSPS (for reduction of anxiety) Secondary outcome measures: SPAI (for reduction of anxiety), SADS (for reduction of anxiety), FNE (for reduction of anxiety), MADRS (for reduction of depression), CAS (for reduction of anxiety) and SDI (for reduction of functional disability) Time points: Quote: "The final visit of the initial 20‐week study served as the baseline visit of the continuation study. Subjects were also evaluated at weeks 24, 28, 32, 36, 40, and 44. Safety assessments included the evaluation, at each visit, of vital signs (weight, blood pressure, and heart rate) and the recording of spontaneously reported or observed adverse events. In addition, the use of concomitant medication was recorded and compliance was monitored by pill counts of returned medication" | |
| Notes | Industry funded: yes. Quote: "Financial support for this study was provided by Pfizer Inc". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author response: quote: "We know that there was random assignment but we did not take care of that. As patients were recruited for the study they were assigned the next study number in the sequence and received the medication over the course of the study assigned to that sequence number". |
| Allocation concealment (selection bias) | Low risk | Author response: quote: "The pharmaceutical firm provided the placebo and medication to us and these were taken to the hospital pharmacy for storage. The pharmacy dispensed the packaged medication from the group of medicine bottles for the participants ‐ week 1, week 2, and so on. The dose was monitored by the treating psychiatrist and patients returned unused medications". |
| Blinding of participants and personnel (performance bias) | Low risk | Author response: quote: "All of these persons were blinded". |
| Blinding of outcome assessment (detection bias) | Low risk | Author response: quote: "All of these persons were blinded". |
| Incomplete outcome data (attrition bias) | High risk | A larger proportion of participants discontinued the study in the placebo switch (15/25; 60%) group compared to the sertraline continuation group (3/25; 12%). The most common reason for treatment discontinuation in the placebo group was due to lack of efficacy and adverse events. The 2 groups did not differ by sample characteristics at baseline. Quote: "3 patients in the sertraline continuation group, 15 in the placebo switch group, and 9 in the placebo responder groups failed to complete the study (see table 3 page 641) ... There were no statistically significant differences between groups in demographic characteristics or mean baseline rating scale scores". Overall 36% of the participants dropped out of the study. ITT was assessed for efficacy outcomes. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: multicentre, randomised, double‐blind, flexible dose, placebo‐controlled study Duration of intervention: 12 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 300 randomised to fluvoxamine CR and placebo Mean age (SE): 32.95 (0.9) years Sex: 143 men and 157 women Diagnostic measure: DSM‐IV and DSM‐IV Axis I Disorders (SCID‐I) Inclusion criteria: quote: "To be eligible to participate in this study, outpatients had to meet the following inclusion criteria: male or female aged 18 to 70 years, a predominant DSM‐IV diagnosis of GSAD according to the modified SCID‐I, and a minimum score of 60 on the LSAS at screening. Women of childbearing potential or less than 1 year postmenopausal were required to use a medically acceptable method of birth control. Pregnant or lactating women were not eligible". Exclusion criteria: quote: "Subjects were excluded from the study if they met any of the following criteria: subjects with psychiatric disorders other than GSAD deemed to be predominant in the last 6 months including major depressive disorder, dysthymic disorder, or panic disorder were excluded. Subjects with history or current diagnosis of schizophrenia, other psychotic disorders, bipolar affective disorder, borderline personality, or obsessive compulsive disorder were excluded. Subjects who had a score 18 on the MADR Scale at screening were excluded. Subjects with evidence of substance abuse disorder or dependence with the past 6 months, subjects with positive results on a urine drug screen, subjects at serious suicidal risk, subjects with unstable or serious medical conditions, and subjects who required formal cognitive‐behavioral therapy to treat social anxiety symptoms within the previous month were also excluded". Dropouts: 101/300 (57/149 in the fluvoxamine CR and 44/151 in the placebo groups). | |
| Interventions | Pharmacological intervention: quote: "Subjects randomized to receive, fluvoxamine CR began at a bedtime dose of 100 mg at day 1. The dose was titrated weekly in 50 mg increments based on clinical judgment of response and tolerance up to a maximum of 300 mg/d, once daily, over the first 5 weeks of treatment. Thereafter, the dose was to remain constant for the duration of the double‐blind period. One decrease of 50 mg/d was permitted after week 1 and through the end of week 5. The minimum dose allowed at any time during the study was 100 mg/d". | |
| Outcomes | Primary outcome measure: LSAS (for reduction of anxiety) Secondary outcome measures: CGI‐I (for treatment efficacy), CGI‐S (for reduction of anxiety), SDS (for reduction of functional disability), PGI (for treatment efficacy) and ASEX (to measure sexual experience) Time points: Quote: "The LSAS was administered at screening, baseline, and weeks 2, 4, 6, 8, 10, and 12; the CGI‐S, SDS, and ASEX were administered at baseline, weeks 2, 4, 6, 8, 10, and 12; the CGI‐I and PGI were administered at weeks 2, 4, 6, 8, 10, and 12. Safety measures obtained at every visit included vital signs, weight, adverse events, and concomitant medications. A 12‐lead ECG and physical examination were performed at the screening visit and week 12; laboratory testing was performed at screening, baseline (if the screening period was more than 10 days in length), and week 12. All week 12 assessments were performed upon early discontinuation" | |
| Notes | Industry funded: yes. Quote: "This study was supported by a grant from Solvay Pharmaceuticals, Inc". Medication provided by industry: unclear Any of the authors work for industry: yes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "A total of 300 subjects with GSAD were randomly assigned to receive either fluvoxamine CR (N = 149) or placebo (N = 151) for 12 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on which parties were blinded and how blinding was achieved. |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | Low risk | Similar proportions of participants withdrew from fluvoxamine CR (57/149; 38%) and placebo groups (44/151; 29%). The reasons for withdrawal were similar across treatment groups. More participants in the fluvoxamine CR group withdrew due to adverse events compared to lack of efficacy in the placebo group. The 2 groups did not differ by sample characteristics at baseline. Quote: "Of the subjects who were randomized, 92 subjects (62%) in the fluvoxamine CR treatment group and 107 subjects (71%) in the placebo treatment group completed the study. The reasons for withdrawal were similar across treatment groups except for withdrawal due to lack of efficacy and withdrawal due to adverse experience. Fourteen subjects (9%) in the placebo treatment group but no subjects in the fluvoxamine CR treatment group withdrew due to lack of efficacy. A higher percentage of subjects in the fluvoxamine CR treatment group (38 subjects, 26%) than in the placebo treatment group (8 subjects, 5%) discontinued due to adverse events. Most subjects in the fluvoxamine CR group (20 subjects) discontinued within the first 3 weeks of treatment. There were no statistically significant differences between the 2 groups in demographics or disorder characteristics at baseline". Overall 34% of the subjects dropped out of the study. Quote: "Statistical analyses were performed using the last observation carried forward (LOCF) and observed case (OC) algorithms". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
| Methods | Design: single‐centre, randomised, double blind, flexible dose placebo controlled pilot study Duration of intervention: 7 weeks Post‐treatment: no follow‐up Placebo run‐in: no | |
| Participants | Sample size: 19 randomised to levetiracetam and placebo Mean age (SD): 37.5 (12.7) years Sex: 9 men and 10 women Diagnostic measure: DSM‐IV and MINI Inclusion criteria: quote: "Inclusion criteria were as follows: (1) age 18–65; (2) social anxiety disorder according to DSM‐IV criteria; (3) minimum baseline score of 20 on the BSPS; (4) medically stable; and (5) provision of written, informed consent". Exclusion criteria: quote: "Subjects who met the following criteria were excluded from the study: (1) history of psychosis or bipolar disorder; (2) substance use disorder or other primary anxiety disorder in the past six months; (3) primary diagnosis of major depression in the last year; (4) the use of other psychotropic medication in the previous week (14 days for MAOI and 28 days for fluoxetine); (5) need for ongoing use of psychotropic medications; or (6) pregnancy or lactation". Dropouts: 4/18 (4/11 in the levetiracetam and 0/7 in the placebo groups; one participant dropped out immediately after baseline for unrelated medical reasons, presenting a protocol violation, and was therefore replaced to provide 18 appropriately enrolled participants as per the study design). | |
| Interventions | Pharmacological intervention: quote: "Study medication was started at 500 mg at bedtime for 4 days, and increased as tolerated at the rate of 500mg every 3–4 days, to 2000 mg/day by day 14, and to a maximum daily dose of 3000 mg (1500 mg BID)". | |
| Outcomes | Primary outcome measure: BSPS (for reduction of anxiety) and CGI‐I (for treatment efficacy) Secondary outcome measures: LSAS (for reduction of anxiety) and SPIN (for reduction of anxiety) Time points: Quote: "Following the screening assessment, eligible subjects returned for a baseline visit at which they were randomly assigned to double‐blind treatment with either LEV or matching PBO. Subjects returned for followup at weeks 1, 2, 3, 5 and 7" | |
| Notes | Industry funded: yes. Quote: "Acknowledgement is due to a grant from UCB Pharma, Inc to Dr Davidson". Medication provided by industry: unclear Any of the authors work for industry: no | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants were randomly assigned to treatment and comparison. However, the procedure was not specified. Quote: "The study design called for 18 subjects to randomly receive double‐blind treatment with either LEV or matching PBO, in a 2:1 ratio, for 7 weeks". |
| Allocation concealment (selection bias) | Unclear risk | There was insufficient information provided to determine if study medication allocation was concealed. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Medication and placebo was provided in identical capsules. However, it is not clear whether both participants and personnel were blinded. Quote: "The study design called for 18 subjects to randomly receive double‐blind treatment with either LEV or matching PBO, in a 2:1 ratio, for 7 weeks". |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study was described as "double‐blind", though no information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) | High risk | More participants withdrew from the levetiracetam group (4/11; 36%) compared to the placebo (0/7; 0%) group. The 2 groups did not differ by sample characteristics at baseline. Quote: "Twenty‐four subjects were screened and 19 subjects were randomized to double‐blind treatment. Reasons for screen failure included voluntary withdrawal after reading the informed consent (n=3), a primary diagnosis of post‐traumatic stress disorder (n=1), and not meeting criteria for social anxiety disorder (n=1). Among those who were enrolled, one subject dropped out immediately after baseline for unrelated medical reasons, presenting a protocol violation, and was therefore replaced to provide 18 appropriately enrolled subjects as per the study design. Two subjects dropped out following the baseline visit due to early side effects from LEV (muscle spasms and pain, n=1; severe headache, n=1) and failed to return for efficacy ratings, thus leaving 16 subjects (n=9 LEV, n=7 PBO) available for the ITT LOCF analysis. Fourteen subjects (n=7 each LEV and PBO) completed the 7‐week treatment period ... No between treatment differences were observed in the baseline demographic characteristics". Overall 22% of the participants dropped out of the study. Quote: "Analyses were performed on the intention to treat (ITT) sample, which included all subjects who returned for at least one post‐baseline assessment, using the last observation carried forward (LOCF)". |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was not available for this study. |
| Other bias | Unclear risk | Funding for study provided by industry. No other sources of bias were identified. |
ADIS‐R: Anxiety Disorder Interview Schedule ‐ Revised; AIMS: Abnormal Involuntary Movements Scale; APD: avoidant personality disorder; ASEX: Arizona Sexual Experience Scale; ATM: atomoxetine; BAI: Beck Anxiety Inventory; BAS: Brief Assessment Scale; BSPS: Brief Social Phobia Scale; CAS: Clinical Anxiety Scale; CGI‐I: Clinical Global Impressions Scale ‐ Improvement item; CGI‐S: Clinical Global Impressions Scale ‐ Severity item; CIC‐SP: Clinical Impression of Change ‐ Social Phobia Scale; CIS‐SP: The Clinical Impression of Severity ‐ Social Phobia; CR: controlled release; CSPS: Cornell Social Phobia Scale; CT: Cognitive Therapy; DESS: Discontinuation Emergent Signs and Symptoms; DSM: Diagnostic and Statistical Manual of Mental Disorders; DSPS: Duke Social Phobia Scale; ECG: electrocardiogram; ECT: electroconvulsive therapy; ER: extended release; FONE/FNES: Fear of Negative Evaluation Scale; FQ: Fear Questionnaire; GAF: Global Assessment of Functioning; GSAD: generalised social anxiety disorder; GSK: Glaxo‐Smith Kline; GSP: generalised social phobia; HAM‐A/HARS: Hamiton Anxiety Rating Scale; HAM‐D/HDS/HDRS: Hamilton Depression Rating Scale; HSC: Hopkins Symptoms Checklist; IEs: Independent evaluator’s; IIP‐64: Inventory of Interpersonal Problems; IIS: Inventory of Interpersonal Situations; ISPI: Index of Social Phobia Improvement; ITT: intention‐to‐treat; LOCF: last observation carried forward; LSAS: Liebowitz Social Anxiety Scale; LSEQ: Leeds Sleep Evaluation Questionnaire; LSPDS: Liebowitz Social Phobic Disorders Scale; LSPS: The Liebowitz Social Phobia Symptom Scale; MADRS: Montgomery‐Asberg Depression Rating Scale; MFQ: Marks Fear Questionnaire; MINI: Mini‐International Neuropsychiatric Interview; MMFQ: Marks‐Mathews' Fear Questionnaire; MOS‐12: Medical Outcomes Study, 12‐item sleep module; NCE: New Chemical Entity; OC: observed case; PGI: Patient Global Impression of Improvement scale; PIC‐SP: The Patient's Impression of Change ‐Social Phobia; PRCS: Personal Report of Confidence as a Speaker Questionnaire; SADS: Social Avoidance and Distress Scale; SAS: symptom assessment scale; SCI: Social Cognitions Inventory; SCID: Structured Clinical Interview for DSM; SCID‐P: Structured Clinical Interview for DSM, Patient edition; SCL‐90: symptom checklist; SDI: social difficulties inventory; SDS: Sheehan Disability Scale; SF‐36: 36 Short‐Form Health Survey; SIAS: Social Interaction Anxiety Scale; SOSS: Severity of Symptoms Scale; SPAI: Social Phobia and Anxiety Inventory; SPEFI: Social Phobia Endstate Functioning Index; SPIN: Social Phobia Inventory; SPS: Social Phobia Scale; SPW: scale of psychological well‐being; SSRI: selective serotonin reuptake inhibitor; SSS: Severity of Symptoms Scale; STAI: State‐Trait Anxiety Inventory; TLFB: timeline follow‐back; VAS: Visual Analogue Scale; WPAI: Work Productivity and Impairment Questionnaire; WPI: WiIloughby Personality Inventory.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| No placebo group | |
| No placebo group | |
| No placebo control | |
| Open‐label pilot study, social phobia participants not separately analysed | |
| No placebo control | |
| No placebo control | |
| No diagnosis of social phobia | |
| No placebo control | |
| Concomitant behavioural therapy | |
| Measures performance anxiety | |
| Main focus is on panic attacks | |
| Secondary analysis of a previous study | |
| Focus on measuring regions of the brain through imaging | |
| Concomitant exposure therapy | |
| Open‐label pilot study not controlled from beginning | |
| No placebo group | |
| Concomitant behavioral therapy | |
| Measures performance anxiety | |
| Assessment of brain function using fMRI | |
| Augmentation study | |
| Commentary | |
| No diagnosis of social phobia | |
| Concomitant behavioral therapy | |
| Measures performance anxiety with brain imaging | |
| Variety of anxiety disorders measured | |
| Measures GAD not GSAD | |
| Concomitant exposure therapy | |
| No diagnosis of social phobia | |
| Concomitant exposure therapy. | |
| Not specific to social phobia diagnosis | |
| Augmentation design (of exposure therapy) | |
| Adolescent population | |
| No diagnosis of social phobia | |
| Measures performance anxiety | |
| No diagnosis of social phobia | |
| Examination anxiety and no placebo control | |
| Combination psychotherapy and pharmacotherapy compared to psychotherapy, no placebo control | |
| No diagnosis of social phobia | |
| No placebo control (continuation study of Heimberg 1998) | |
| Measures performance anxiety | |
| Variety of social anxiety disorders | |
| Review | |
| Concomitant individual and group cognitive therapy | |
| Social phobia participants not separately analysed | |
| Herbal medication | |
| Participants with social phobia, GAD and or panic disorder | |
| CBT for the treatment of GAD | |
| Assessment of brain function using fMRI | |
| Measures performance anxiety | |
| No diagnosis of social phobia | |
| No placebo control | |
| No placebo control | |
| Augmentation design (clomipramine + tryptophan vs clomipramine + placebo) | |
| Brain imaging study | |
| Social phobia participants not separately analysed | |
| Moclobemide + supportive guidance versus CBT + placebo pills versus combination of CBT + moclobemide | |
| Augmentation study, medication added to psychotherapy | |
| No placebo control | |
| Measured treatment‐emergent effects | |
| Social phobia participants not separately analysed | |
| Augmentation trial of pindolol as adjunctive treatment to the SSRI paroxetine and incomplete results (cross‐over data not reported separately for first leg of treatment) | |
| Measures neuroendocrine and behavioural responses | |
| No diagnosis of social phobia | |
| Concomitant behavioural therapy | |
| No placebo control | |
| Combined population and combined intervention (psychotherapy and medication) | |
| Combined population | |
| No placebo control | |
| Social phobia participants not separately analysed | |
| Measures performance anxiety and is an augmentation study – medication added to psychotherapy |
CBT: cognitive behavioural therapy; GAD: generalised anxiety disorder; GSAD: generalised social anxiety disorder; fMRI: functional magnetic resonance imaging.
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | Randomized, double‐blind placebo‐controlled study, 12 weeks |
| Participants | Quote: "Patients aged 18–64 years with a primary diagnosis of DSM‐IV‐TR defined SAD, a Liebowitz Social Anxiety Scale Japanese version (LSAS‐J) total score ≥60 and a Clinical Global Impression–Severity (CGI‐S) score ≥4 at baseline were randomly assigned (1:1:1) to placebo, escitalopram 10 mg or escitalopram 20 mg" |
| Interventions | Placebo Escitalopram 10 mg or 20 mg |
| Outcomes | Quote: "The primary endpoint was change from baseline to Week 12 in the LSAS‐J total score for both escitalopram 10 mg and 20 mg versus placebo (ANCOVA, FAS, LOCF), using a hierarchical testing procedure. Pre‐specified secondary endpoints included LSAS‐J sensitivity analyses' |
| Notes | Clinical trial identifier: JapicCTI‐121842 |
| Methods | Quote: "The study was a 12‐week double‐blind, placebo‐controlled, flexible‐dose trial; daily doses of vilazodone 20 mg/d to 40 mg/d or matching placebo were administered in a 1:1 ratio. Data were collected between November 2012 and April 2014" |
| Participants | Quote: "Enrollment was planned for 30 subjects who achieved the prospectively determined minimum adequate treatment of at least 6 consecutive weeks on ≥ 20 mg/d of vilazodone or the placebo equivalent. Subjects included men and women, aged 18–75 years, who met DSM‐IV‐TR criteria for generalized social anxiety disorder and had a minimum total Liebowitz Social Anxiety Scale (LSAS) score at screening and baseline of 70 and a minimum Clinical Global Impressions–Severity scale (CGI‐S) score of 4 (moderately ill). Subjects also had to agree to practice effective contraception methods. Exclusion criteria included lifetime bipolar disorder, schizophrenia, and body dysmorphic disorder, as well as posttraumatic stress disorder, obsessive‐compulsive disorder, panic disorder, and substance dependence within the past 24 weeks. Comorbid major depression, dysthymia, generalized anxiety disorder, and specific phobias were allowed if generalized social anxiety disorder was the primary disorder (the major clinical problem for which the subjects sought treatment). Subjects who were suicidal, who were medically unstable, who had a history of cancer or treatment‐refractory generalized social anxiety disorder (failure to respond to adequate trials of 2 effective agents), or who were in active cognitive‐behavioral therapy or were currently pregnant or lactating were excluded. Zolpidem as needed was allowed for insomnia if not taken more than 3 times per week. Other psychotropic drugs had to be discontinued at least 2 weeks before the baseline visit" |
| Interventions | Quote: "Subjects started at baseline on vilazodone 10 mg/d or placebo, taken in the morning with food, and increased to 20 mg/d or placebo after 1 week and to 40 mg/d or placebo after the second week. Dose increases could be delayed or reversed for problems of tolerability; however, attempts were made to raise all subjects to 40 mg/d. Noncompliance was defined as < 80% or > 120% of prescribed drug taken during any evaluation period. Subjects who were noncompliant at more than 2 consecutive study visits could be terminated" |
| Outcomes | Quote: "The MINI was used for diagnostic assessment of DSM‐IV disorders. The CGI‐S, CGI‐I, PGIC, and LSAS were administered to assess social anxiety disorder severity and change and global improvement. The LSAS is a 24‐item instrument developed by Liebowitz that assesses anxiety and avoidance in a variety of commonly encountered performance and social situations and was found by Heimberg et al to be reliable and valid. The HDRS‐17 and HARS were used to quantify depressive and anxiety symptoms at baseline and endpoint. Safety measures included routine laboratory tests, ECGs, and physical examinations. Subjects were asked about adverse events and concomitant medications at each study visit" |
| Notes | ClinicalTrials.gov identifier: NCT01712321 Funding/support: Quote: "This study was supported by an investigator‐initiated grant from Forest Research Institute, Inc, Jersey City, New Jersey, to The Medical Research Network, LLC" |
| Methods | Double‐blind controlled study |
| Participants | Patients with social phobia |
| Interventions | Clonazepam and placebo |
| Outcomes | — |
| Notes | — |
| Methods | Randomised, double‐blind treatment for 6 weeks |
| Participants | 18 SAD patients. Serotonin synthesis rate capacity was assessed before and after treatment in the patients and 17 age and sex‐matched healthy controls (HC; only scanned once) using positron emission tomography imaging with the radiotracer [11C]5‐HTP. |
| Interventions | Experimental: SSRI Citalopram or NK1R antagonist GR205171 Control: placebo |
| Outcomes | Primary outcome: Liebowitz Social Anxiety Scale (LSAS) was used to index symptom severity |
| Notes | Funding/Support: Quote: "This study was supported by the Swedish Research Council, the Swedish Brain Foundation, Riksbankens Jubileumsfond–the Swedish Foundation for Humanities and Social Sciences, and the Swedish Research Council for Health,Working Life, andWelfare. Ligand production of 5‐hydroxytryptophan for the patients was supported by GlaxoSmithKline" |
| Methods | Double‐blind, parallel group trial |
| Participants | 66 participants with social phobia |
| Interventions | Alprazolam, buspirone, or placebo |
| Outcomes | — |
| Notes | — |
| Methods | Study type: intervention Study design: randomised allocation |
| Participants | Enrollment: 28 Ages eligible for study: > 18 years (adult, senior). Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "Male or female outpatients > 18 years of age with a primary psychiatric diagnosis of generalized social anxiety disorder as defined by DSM‐IV criteria and an LSAS score > 50; physical examination, electrocardiogram, and laboratory findings without clinically significant abnormalities; willingness and ability to comply with the requirements of the study protocol". Exclusion criteria: quote: "Patient has a history of intolerance or lack of response to a treatment trial of duloxetine at highest tolerated dose (<120mg/day); patients with acute narrow angle glaucoma; pregnant women, lactating women, and women of childbearing potential who are not using medically accepted forms of contraception (e.g., IUD, oral contraceptives, barrier devices, condoms and foam, or implanted progesterone rods stabilized for at least 3 months); concurrent use of other psychotropic medications. Patients must discontinue regular benzodiazepine or antidepressant therapy at least one week (5 weeks for fluoxetine) prior to baseline. Concomitant beta‐blockers are proscribed unless prescribed for a medical indication (e.g., hypertension, at a stable daily dose for > 1 month); patients with a history of failure to satisfactorily respond to >2 prior adequate treatment trials; significant personality dysfunction likely to interfere with study participation; serious medical illness or instability for which hospitalization may be likely within the next year; Seizure disorders with the exception of a history of febrile seizures if they occurred during childhood, were isolated, and did not recur in adulthood; Concurrent psychotherapy initiated within 2 months of baseline is prohibited. Ongoing psychotherapy of any duration directed specifically toward treatment of the social anxiety disorder is excluded. Prohibited psychotherapy includes cognitive behavioural therapy or psychodynamic therapy that focuses on exploring specific, dynamic causes of the phobic symptomatology and provides skills for their management. General supportive individual, couples, or family therapy greater than 2 months duration is acceptable; diagnosis of any of the following mental disorders as defined by the DSM‐IV: a lifetime history of schizophrenia or any other psychosis, mental retardation, organic medical disorders or bipolar disorder; eating disorders in the past 6 months; alcohol or substance abuse in the past 3 months or dependence within the past 6 months; entry of patients with major depression, dysthymia, panic disorder, generalized anxiety disorder, post‐traumatic stress disorder or obsessive‐compulsive disorder will be permitted if the social anxiety disorder is judged to be the predominant disorder, in order to increase accrual of a clinically relevant sample; patients with significant suicidal ideation (MADRS item 10 score > 3) or who have enacted suicidal behaviours within 6 months prior to intake will be excluded from study participation and referred for appropriate clinical intervention. |
| Interventions | Active comparator: duloxetine 60 mg/day for 6 weeks; in phase 1 all participants entered an open trial Active comparator: duloxetine 120 mg for 18 weeks; in phase 2 participants were randomised to 60 mg duloxetine + placebo or 120 mg duloxetine Placebo comparator: duloxetine 60 mg + placebo for 18 weeks; in phase 2 participants were randomised to 60 mg duloxetine + placebo or 120 mg duloxetine |
| Outcomes | Primary outcome measures: anxiety symptoms as assessed by Liebowitz Social Anxiety Scale (time frame: 6 months) Baseline collected for phase 1 at week 0 and for phase 2 at week 6 |
| Notes | Responsible party: Naomi M Simon, Director, Center for Anxiety and Traumatic Stress Disorders, Massachusetts General Hospital ClinicalTrials.gov Identifier: NCT00114127 Other study ID numbers: 2004‐P‐001384 Study first received: 13 June 2005 Last updated: 5 June 2014 Locations: Massachusetts General Hospital, Boston, Massachusetts 02114, USA Sponsors and collaborators: Massachusetts General Hospital. Investigators: principal investigator: Naomi M Simon, MD, Massachusetts General Hospital. |
| Methods | Study type: intervention Study design: randomised allocation Intervention model: single group assignment Masking: double‐blind Primary purpose: treatment |
| Participants | Estimated enrollment: 50 Ages eligible for study: 18‐65 years (adult) Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "primary diagnosis of SAD; CGI (S) ≥ 4 at screen; LSAS ≥ 50 at baseline; Covi Anxiety Scale score greater than the Raskin depression Scale total score at screen". Exclusion criteria: quote: "non‐responsive to adequate trials of two or more treatment medications, if previously treated for SAD; HAM‐D ≥15 or a score of >2 on Item 1 at baseline; serious or unstable medical condition; alcohol or substance use disorder within 6 months prior to study". |
| Interventions | This study consists of two parts. The first part consists of 12 weeks of open‐label treatment with Gabitril. If the study doctor determines that the patients condition has improved and they have completed the initial 12 weeks of treatment they may be eligible for the second part of the study. This part is a 24‐week double‐blind treatment period with either Gabitril or placebo (inactive medication). There will also be a follow‐up visit about 1 to 3 weeks after they have completed taking the study medication. Altogether study participation is expected to last approximately 37 weeks. |
| Outcomes | Primary outcome measures: Liebowitz Social Anxiety Scale (LSAS); Clinical Global Impression‐Change (CGI‐C) Secondary outcome measures: Hamilton Anxiety Scale (HAM‐A); Social Phobia Inventory (SPIN); Pittsburgh Sleep Quality Index (PSQI); 36‐Item Short‐Form Health Survey (SF‐36); Clinical Global Impression‐S (CGI‐S) |
| Notes | Responsible Party: Emory University ClinicalTrials.gov Identifier: NCT00208741 Other study ID numbers: 0337‐2002 Study first received: 13 September 2005 Last updated: 8 November 2013 Locations: Emory University School of Medicine, Atlanta, GA 30329, USA Hillside Hospital of the North Shore‐Long Island Jewish Health System, Long Island, NY 10032, USA Columbia/New York State Psychiatric Institute, New York, NY 10032, USA Sponsors and collaborators: Emory University, Cephalon |
| Methods | Study type: intervention Study design: randomised allocation |
| Participants | Ages eligible for study: 18‐65 years (adult) Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "adult outpatients 18‐65 years of age, primary diagnosis of social anxiety disorder, using DSM‐IV criteria; minimum CGI severity score of 4 at baseline; minimum BSPS score of 20 at baseline; written informed consent; negative serum pregnancy test for women of childbearing potential". Exclusion criteria: quote: "current DSM‐IV diagnosis of bipolar disorder, schizophrenia or other psychotic disorder, or cognitive disorder due to a general medical condition, any current primary anxiety disorder other than SAD or current primary depression; history of substance abuse or dependence with the last 6 months; suicide risk or serious suicide attempt within the last year; clinically significant medical condition or laboratory abnormality; women of childbearing potential who are unwilling to practice an acceptable method of contraception; concomitant medication use for psychotropic purposes, history of hypersensitivity to quetiapine". |
| Interventions | Quote: "This is an eight week, randomized, double‐blind, placebo‐controlled trial of quetiapine (100‐400 mg/day) in social anxiety disorder". |
| Outcomes | Primary outcome measures: Brief Social Phobia Scale (BSPS) |
| Notes | ClinicalTrials.gov Identifier: NCT00215254 Other study ID numbers: 5639‐04‐3R0 Study first received: 20 September 2005 Last updated: 18 December 2006 Locations: Duke University Medical Center, Durham, North Carolina, 27705, USA Sponsors and collaborators: Duke University, AstraZeneca Investigators: principal investigator: Jonathan Davidson, MD, Duke University |
| Methods | Study type: intervention Study design: randomised allocation Intervention model: parallel assignment Masking: double‐blind (participant, caregiver, investigator) Primary purpose: treatment |
| Participants | Enrollment: 42 Ages eligible for study: 18‐65 years (adult) Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "Meets DSM‐IV criteria for current social anxiety disorder; reports social anxiety in most situations (generalized type); treatment seeking for relief of social anxiety; meets DSM‐IV criteria for current alcohol use disorder; reads at the 6th grade level or above; endorses using alcohol to cope with social anxiety either "very often" or "always"; reports no prior medical alcohol detoxification; willingness to be randomized to the placebo group; willingness to attend 16 weekly medication management visits and one alcohol‐related therapy session; Liebowitz Social Anxiety Scale Total score (modified version) of at least 60; endorses drinking at least 15 standard drinks in a typical 30 day period or reports drinking heavily (defined as greater‐than‐or‐equal‐to 4 standard drinks on one occasion for women; greater‐than‐or‐equal‐to 5 standard drinks on one occasion for men, respectively) on at least 2 days in a typical 30 day period". Exclusion criteria: quote: "Abuse or dependence on drugs other than nicotine or marijuana in last 90 days; current or past diagnosis of bipolar disorder or schizophrenia; significant suicide risk as assessed by the SCID; current use of psychotropic medications; treatment seeking for alcohol problems; any unstable medical condition that might interfere with safe participation in the trial; elevated liver enzymes (3 x greater than normal levels); history of adverse reaction to paroxetine; history of failure to respond to adequate trial or dose of paroxetine for social phobia (60 mg/day for at least 6 weeks); history of heart problems or abnormal ECG recording; pregnancy, nursing, or refusal to use effective birth control if sexually active and premenopausal; history of one or more alcohol detoxifications". |
| Interventions | Drug: paroxetine 16 weeks treatment; dosing will start at 20 mg/day paroxetine and will increase gradually to a maximum dose of 60 mg/day Drug: placebo treatment phase will last 16 weeks; dosing will start at 20 mg/day (placebo) and will increase gradually to a maximum dose of 60 mg/day |
| Outcomes | Primary outcome measures: social anxiety severity; alcohol use, quantity and frequency; drinking to cope, quantity and frequency Secondary outcome measures: quality of life, depressive symptoms Time frame: 16 weeks treatment; 6 month and 12 month follow‐up interviews |
| Notes | Responsible Party: Medical University of South Carolina ClinicalTrials.gov Identifier: NCT00246441 Other study ID numbers: NIAAARAN013379; R01AA013379; NIH Grant R01 AA013379 Study first received: 28 October 2005 Last updated: 1 December 2016 Locations: Medical University of South Carolina, Institute of Psychiatry; Charleston, South Carolina, 29425, USA Sponsors and collaborators: Medical University of South Carolina, National Institute on Alcohol Abuse and Alcoholism (NIAAA) Investigators: principal investigator: Carrie L Randall, PhD, Medical University of South Carolina |
| Methods | Study type: intervention Study design: randomised allocation |
| Participants | Estimated enrollment: 180 Ages eligible for study: 18‐65 years (adult) Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "The subject is male or female, 18 ‐ 65 years of age (inclusive); The subject meets current DSM‐IV‐TR (American Psychiatric Association, 2000) criteria for social phobia (300.23), generalized subtype, as confirmed by the Mini‐International Neuropsychiatric Interview at Screening (visit 1); The subject has had symptoms of SAD (social phobia) present for at least 6 months prior to screening (visit 1); The subject has a total score ≥ 60 on the LSAS at both screening (visit 1) and baseline (visit 2); The subject has a score ≥ 4 on the Clinical Global Impression ‐ Severity (CGI‐S) scale at both screening (visit 1) and baseline (visit 2); The subject has a score ≤ 15 on the 17‐item Hamilton Rating Scale for Depression (HAM‐D) at screening; The subject, if female and of child‐bearing potential (not 2 years post‐menopausal or surgically sterilized), must have a negative serum pregnancy test at screening (Visit 1) and be willing to avoid pregnancy and practice adequate birth control from the time of study enrollment until 30 days after the last dose of study medication. Adequate methods of birth control are: oral contraception, intrauterine device, implantable contraceptive device, depot contraceptive, or a barrier method plus spermicide. Additional serum pregnancy tests will be administered at visit 6, visit 8, and visit 9; The subject, if engaged in ongoing psychotherapy for SAD or any other mental health condition, must have been attending therapy regularly for at least 3 months prior to screening (Visit 1) and must agree to continue the same type and frequency of psychotherapy throughout the course of the study; The subject agrees to refrain from blood donation during the course of the study; The subject has written and oral fluency in English or Spanish; The subject is willing to participate in the study, as evidenced by a signed and dated written Informed Consent Form (ICF)". Exclusion criteria: quote: "The subject has a decrease >15 points on the LSAS total score between screening (visit 1) and baseline (visit 2); The subject has a clinically significant abnormality or clinically significant unstable medical condition as indicated by medical history, physical examination, ECG results, clinical laboratory testing, or the investigator's judgment at screening (visit 1) or baseline (visit 2); The subject has a QTc interval of 450 msec or greater at screening (visit 1) if male or a QTc interval of 470 msec or greater at screening (visit 1) if female; The subject has current hypothyroidism or hyperthyroidism or laboratory findings consistent with thyroid dysfunction. Subjects who are being treated for thyroid disorder are eligible if they have been on stable doses of thyroid hormone for at least 6 months and are currently euthyroid; The subject has any history of schizophrenia or other psychotic disorder, bipolar disorder, post‐traumatic stress disorder, borderline personality disorder, or antisocial personality disorder; The subject has a history within the previous 5 years of obsessive‐compulsive disorder or an eating disorder; The subject exhibits evidence of a clinically predominant DSM‐IV‐TR Axis I or II disorder other than social phobia or avoidant personality disorder within the 6 months prior to screening (visit 1); The subject, in the opinion of the investigator, presents a significant risk of doing harm to himself, herself, or others; The subject has met DSM‐IV‐TR criteria for alcohol or substance dependence (other than nicotine or caffeine dependence) within 6 months of screening (visit 1); The subject has met DSM‐IV‐TR criteria substance abuse (other than alcohol, nicotine or caffeine abuse) within 3 months of screening (visit 1); The subject tests positive on the urine drug screen conducted at screening (visit 1) for illicit drugs, including opiates, barbiturates, amphetamines, cocaine, and phencyclidine; The subject is a pregnant or lactating female; The subject has previously participated in a clinical trial for AV608 (previously identified as NKP608 and CGP608); The subject has used any prohibited medications, or has any anticipated need or intended use of these medications during the study; The subject has used any investigational drugs, products, or devices in the 3 months prior to screening (visit 1); The subject is a member of the investigative site staff or an immediate family member; The subject has any other condition that the investigator believes would jeopardize the safety or rights of the subject or would render the subject unable to comply with the trial protocol". |
| Interventions | Quote: "The purpose of this study is to look at the safety and effectiveness of an investigational drug (AV608) when used in subjects who have Social Anxiety Disorder. AV608 is an NK‐1 receptor antagonist that exhibits central nervous system activity after oral administration. The study will compare AV608 to placebo (a medically inactive substance) to see if AV608 helps the symptoms of Social Anxiety Disorder. Eligible subjects will be assigned by chance to take either AV608 or placebo for 12 weeks. During the study, subjects will be asked about their overall health and mood and their Social Anxiety Disorder". |
| Outcomes | Primary outcome measures: Liebowitz Social Anxiety Scale (LSAS) |
| Notes | Study start date: February 2006 Study completion date: December 2006 Locations: Alabama, Arizona, California, Florida, Maryland, New York, Ohio, Oklahoma, Oregon, Texas (USA) Sponsors and collaborators: Avera Pharmaceuticals ClinicalTrials.gov Identifier: NCT00294346 Other study ID numbers: AV608‐105 Study first received: 17 February 2006 Last updated: 15 February 2008 |
| Methods | Allocation: randomised |
| Participants | Enrollment: 71 Age: 18‐75 years (both sexes) Inclusion criteria: quote: "The patient has provided signed informed consent. Outpatients aged 18‐65 (extremes included). Patients with a primary diagnosis of Social Phobia according to DSM IV (300.23) criteria (diagnosis to be made using the Mini International Neuropsychiatric Interview (MINI)). On the basis of a physical examination, medical history and basic laboratory screening, the patient is, in the investigators opinion, in a suitable condition. Willing and able to attend study appointments in the correct time windows". Exclusion criteria: quote: "Any other axis I diagnosis that was a primary disorder in the previous six months. Continuation or commencement of formal psychotherapy. Alcohol or drug abuse as defined in the DSM IV within the last six months. Mania or hypomania as defined in the DSM IV. Current use of or commencement of antidepressant and anxiolytic medications. Patients, who have been on an antidepressant or other anxiolytic prior to the study, will have discontinued it more than two weeks prior to entry into the study. Those who have been on fluoxetine, will have been off of it for at least 5 weeks. Patients who have been on an herbal or alternative treatment judged to be potentially anxiolytic or with psychobiological activity, will have terminated usage of the agent more than two weeks prior to entering the study. Previous reaction to niacin administration, use of a non‐steroidal anti‐inflammatory, any psychotic disorder. Eating disorders as defined in the DSM IV. Mental retardation or other cognitive disorder. Clinical interpretation of apparent suicide risk. Laboratory values at screening or in medical history that may be considered through clinical interpretation to be significant. Diseases which could, through clinical interpretation, interfere with the assessments of safety, tolerability and efficacy. Serious illness: liver or renal insufficiency, cardiac, vascular, pulmonary, gastrointestinal, endocrine, neurological, infectious, neoplastic or metabolic disturbance. The patient is, in the opinion of the investigator, unlikely to be able to comply with the clinical trial protocol, or is unsuitable for any other reasons". |
| Interventions | Drug: cipralex |
| Outcomes | Primary outcome measures: changes in intensity of the vasodilatory response to 10 mM topical m‐N over 16 weeks. (time frame: 20 weeks) (designated as safety issue: no). |
| Notes | Sponsors and collaborators: START Clinic for Mood and Anxiety Disorders, H Lundbeck A/S. Principal investigator: Martin A Katzman, MD; START Clinic for the Mood and Anxiety Disorders |
| Methods | Study type: intervention Study design: randomised allocation |
| Participants | Enrollment: 217 Ages eligible for study: 18‐70 years (adult, senior) Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "Male or female outpatients between 18 and 70 years old inclusive; symptoms of social anxiety disorders (generalized type) present for at least 1 year prior to Selection Visit; had a score of ≥60 on the LSAS at the Selection Visit and at the Randomization Visit. Additionally, the clinician's global impression of change score must have been ≥ 2 at the randomization visit; had a telephone where they could be directly contacted". Exclusion criteria: quote: "History of autism or Asperger's disease; had another primary axis I disorder or fulfilled diagnostic and statistical manual of mental disorders‐4th edition (DSM‐IV) criteria in the 6 months prior to Screening; major depression as measured by a Hamilton Depression Rating Scale (HAM‐D‐17 items) total score of > 17 and/or a suicide subscale score on the HAM‐D‐17 items of > 2 at the selection or randomization visit; history of electroconvulsive therapy within the prior 3 months; history of psychotherapy which was not stable and ongoing for at least 6 months prior to visit 1; clinical history of significantly impaired renal function with an estimated creatinine clearance below 80 mL/min; clinically significant medical condition; history of any clinically significant allergic condition or allergy to LEV or pyrrolidone derivatives; neutrophil count of less than 1800/µL" |
| Interventions | Quote: "A multicenter, randomized, double‐blind, PBO‐controlled, parallel group study to assess the efficacy and safety of levetiracetam versus PBO for the treatment of social anxiety disorder (generalized type)". |
| Outcomes | Primary outcome measures: change in Liebowitz Social Anxiety Scale (LSAS) score from Visit 2 to the last evaluation period visit attended using last observation carried forward (LOCF) methods; safety: monitoring of AEs, clinical laboratory tests, physical examination and vital signs. |
| Notes | Study start date: September 2003 Study completion date: June 2004 Sponsors and collaborators: UCB Pharma Investigators: study director: UCB Clinical Trial Call Center +1 877 822 9493 (UCB) ClinicalTrials.gov Identifier: NCT00612859 Other study ID numbers: N01086 Study first received: 14 January 2008 Last updated: 25 November 2013 |
| Methods | Study type: intervention Study design: randomised Intervention model: parallel assignment Masking: double‐blind (participant, investigator, outcomes assessor). Primary purpose: treatment |
| Participants | Enrollment: 63 Ages eligible for study: 18‐75 years (adult, senior) Genders eligible for study: all Accepts healthy volunteers: no Inclusion criteria: quote: "Subjects must give written informed consent prior to any study procedures. Diagnosis of Social Anxiety Disorder (SAD) (300.23 Social Phobia/Social Anxiety Disorder, Generalized Subtype) according to DSM‐IV‐TR criteria, as determined by psychiatric evaluation with the Principal Investigator. A minimum score of 60 on the LSAS total score at both screening and baseline visits. A total HAM‐D score of less than 15 at the screening visit. CGI Severity score of 4 or greater at both screening and baseline visits. Female participants of childbearing potential must commit to an effective form of contraception for the duration of the trial. Effective forms of contraception include: condoms with spermicide, diaphragm with spermicide, hormonal contraceptive agents (oral, transdermal, or injectable), and implantable contraceptive devices". Exclusion criteria: quote: "An Axis I disorder other than SAD (e.g., post‐traumatic stress disorder, obsessive compulsive disorder, panic disorder) within 24 weeks of the baseline visit. Subjects with co‐morbid MDD, GAD, dysthymia, or specific phobias will be allowed if GSAD is the primary disorder in terms of clinical severity, as determined by the investigator; Any history or complication of schizophrenia or bipolar disorder; any complication of body dysmorphic disorder; substance dependence, as defined by DSM‐IV‐TR criteria, within 24 weeks of the baseline visit; subjects who are currently pregnant, lactating, or of childbearing potential and not practicing an effective method of contraception; Subjects scoring >2 on item #3 of the HAM‐D, or who, in the opinion of the PI, are at a clinically significant risk for suicide; systolic blood pressure ≥165 and/or diastolic blood pressure ≥95; positive urine drug screen at the screening visit; any current unstable and/or clinically significant medical condition, based on history or as evidenced in screening laboratory and ECG assessments; Any history or complication of cancer or malignant tumor; fluoxetine within 28 days of baseline; MAO inhibitors within 14 days of baseline ‐ any other psychotropics (including SSRIs, SNRIs, and benzodiazepines) within 14 days of baseline. Zolpidem (Ambien®) PRN is allowed for insomnia if not taken more than 3 times per week; subjects who started psychotherapy or cognitive‐behavioural therapy within 24 weeks of the baseline visit, except for supportive psychotherapy; electro‐convulsive therapy (ECT) within 12 weeks of the baseline visit; treatment refractory GSAD." |
| Interventions | Drug: desvenlafaxine (Pristiq); flexible dose, 50‐100 mg 4 times daily for 12 weeks Drug: matching placebo, taken 4 times daily for 12 weeks |
| Outcomes | Primary outcome measures: Change in the LSAS total score (time frame: baseline to study endpoint (week 12)) LSAS measuring social anxiety symptoms, possible total scores ranging from 0‐144, with higher scores indicating greater severity of symptoms Clinical Global Impression of Improvement Scale (CGI‐I) (time frame: baseline to week 12); CGI‐I: one item, measuring overall improvement of illness; possible scores range from 1‐7, with lower scores representing greater improvement CGI‐I responders: defined as having a CGI‐I scores of 1 or 2 at week 12/study endpoint; Patient Global Impression of Change (time frame: baseline to week 12) Participant‐rated global outcome scale, people who rated themselves as 1 (very much improved) or 2 (much improved) on the PGIC were considered self‐rated responders |
| Notes | Locations: the Medical Research Network, LLC New York, New York, USA, 10128 Sponsors and collaborators: the Medical Research Network, Pfizer Investigators: principal investigator Michael R Liebowitz, MD, the Medical Research Network |
AEs: Adverse events; ANCOVA: Analysis of covariance; BAI: Beck Anxiety Inventory; BPS: Propensity Scale; BSPS: Brief Social Phobia; BTS‐Q: Blushing, Trembling, and Sweating Questionnaire; CGI: Clinical Global Impression; CGI‐S: Clinical Global Impression – Severity Scale; DSM: Diagnostic and Statistical Manual for Mental Health Disorders; ECG: electrocardiogram; Euroquol SF‐36: Euroquol (quality of life) and the Medical Outcomes Study Short‐Form‐36 (SF‐36); FAS: full analysis set; GAD: Generalised Anxiety Disorder; GSAD: Generalised Social Anxiety Disorder; HDRS‐17: Hamilton Depression Rating Scale, 17 items; HAM‐A/HARS: Hamilton Anxiety Rating Scale; HAM‐D: Hamilton Rating Scale for Depression; Inc.: Incorporation; LOCF: Last Observation Carried Forward; LSAS: Liebowitz Social Anxiety Scale;LSAS‐J: Liebowitz Social Anxiety Scale ‐ Japanese Version; MADRS: Montgomery–Åsberg Depression Rating Scale; MAO: Monoamine oxidase; MDD: Major Depressive Disorder; mg/d: Milligram per day;MINI: Mini‐International Neuropsychiatric Interview; PGIC: Patient Global Impression of Change; PSWQ: Penn State Worry Questionnaire; SAD: Social Anxiety Disorder; SF‐36: 36‐item Short‐Form Health Survey; SIAS: Social Interaction Anxiety Scale; SNRIs: Serotonin–norepinephrine reuptake inhibitors; SPIN: Social Phobia Inventory; SPS: Social Phobia Scale; SSRIs: Selective serotonin re‐uptake inhibitors.
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Sertraline in the treatment of generalized social phobia with comorbidity |
| Methods | Randomised placebo control trial |
| Participants | Expected enrolment: 170 Inclusion criteria: ‐ outpatient with primary Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM‐IV) GSP plus at least one of the following comorbid DSM‐IV anxiety disorders: panic disorder with agoraphobia, obsessive compulsive disorder, major depressive disorder, generalised anxiety disorder ‐ score on LSAS > 50 ‐ score on MADRS < 25. Locations: Canada |
| Interventions | Sertraline and placebo |
| Outcomes | Primary outcome: Clinical Global Impression ‐ Improvement = 2 Liebowitz Social Anxiety Scale (LSAS) (mean change from baseline) Secondary outcome: mean change from baseline on the following scales: Quality of Life and Employment Satisfaction Questionnaire, Sheehan Disability Scale, Social Phobia Scale, Brief Social Phobia Scale, Penn State Worry Questionnaire, Panic and Agoraphobia Scale, Davidson Trauma Scale, Social Anxiety Spectrum Self‐Report (SHY‐SR), Yale‐Brown Obsessive Compulsive Scale, Montgomery‐Asberg Depression Rating Scale (MADRS) |
| Starting date | Expected completion August 2006 |
| Contact information | Van Ameringen 2006 |
| Notes | The recruitment status of this study is unknown because the information has not been verified recently. |
| Trial name or title | Vilazodone in the treatment of social anxiety disorder: a double blind study |
| Methods | 12 week double‐blind, placebo‐controlled trial |
| Participants | 30 outpatients aged 18‐75 years with SAD, generalised subtype who return for at least one postrandomisation visit where efficacy evaluations are conducted Inclusion criteria: diagnosis of social anxiety disorder, generalised subtype; LSAS total score of 70 at visits 1 and 2. Exclusion criteria: lifetime history of bipolar disorder or schizophrenia; current suicidal risk; current unstable medical condition |
| Interventions | Daily doses of vilazodone 20 mg/d to 40 mg/day or matching placebo |
| Outcomes | All participants randomised to drug or placebo and returning for at least one subsequent visit will be included in the primary efficacy analyses. Primary outcome measures: Change in Liebowitz Social Anxiety Scale (LSAS) ‐ total score (time frame: change from baseline to final study visit: minimum 1 week ‐ maximum 12 weeks). Randomised participants taking minimum target dose (20 mg or matching placebo daily) for at least six consecutive weeks will be considered a minimum adequate trial for the purposes of secondary analyses. Secondary outcome measures (time frame: study endpoint: minimum 6 weeks ‐ maximum 12 weeks): Responder rate, as defined by Clinical Global Impression of Improvement score of 1 or 2 Change in the Clinical Global Impression of Severity of Illness score Change on the LSAS anxiety and avoidance subscales Change in Hamilton Depression scale total Change in Hamilton Anxiety scale total Participant‐assessed responder rate, as defined by a Patient Global Impression of Change score of 1 (very much improved) or 2 (much improved) at study endpoint |
| Starting date | Date of registration: 18 October 2012 |
| Contact information | The Medical Research Network and Forest Laboratories |
| Notes | This study is ongoing, but not recruiting participants. |
| Trial name or title | Ketamine infusion for social anxiety disorder |
| Methods | Randomised, placebo‐controlled crossover study |
| Participants | Ages eligible for study: 18‐65 years Genders eligible for study: both Accepts healthy volunteers: no Inclusion criteria:
Exclusion criteria:
|
| Interventions | Pharmacological intervention: Experimental: ketamine (ketamine will be given at a dose of 0.5 mg/kg over 40 minutes. This dose is identical to that used in previous anti‐depressant studies of ketamine) Placebo comparator: saline (saline will be given at a dose of 0.5 mg/kg over a 40 minute period) |
| Outcomes | Primary outcome measures (time frame: first 2 weeks following infusion), at screening, 1 hour prior to infusion, 1, 2 and 3 hours after infusion, 1, 2, 3, 5, 7, 10, and 14 days following a single ketamine/saline infusion: Visual analogue scale (VAS) of anxiety states Secondary outcome measures (time frame: first 2 weeks following infusion): Anxiety severity, according to Beck Anxiety Inventory (BAI) at screening, 1 hour prior to infusion, 3 hours after infusion, 1, 2, 3, 5, 7, 10, and 14 days following a single ketamine/saline infusion Depression severity, according to Hamilton Depression Rating scale (HAM‐D) at screening, 1 hour prior to infusion, 3 hours after infusion, 1, 2, 3, 5, 7, 10, and 14 days following a single ketamine/saline infusion Clinical Global Impressions, according to Clinical Global Impressions (CGI) ratings of overall severity of SAD symptoms at screening, 1 hour prior to infusion, 3 hours after infusion, 1, 7 and 14 days following a single ketamine/saline infusion Brief Psychiatric Rating Scale, Positive Symptom Subscale (BPRS‐PS), with regard to thought content, conceptual disorganisation, hallucinatory behavior, and grandiosity at screening, 1 hour prior to infusion,1‐3 hours after infusion, 1 day following a single ketamine/saline infusion Clinician‐Administered Dissociative States Scale, according to Clinician‐Administered Dissociative States Scales (CADSS) ratings of dissociative symptoms at screening, 1 hour prior to infusion, 3 hours after infusion, 1 day following a single ketamine/saline infusion Self‐Statement During Public Speaking Scale (SPSS) (time frame: first week following infusion), according to Self‐Statement During Public Speaking Scale (SPSS) ratings of cognitions that occurred during a speech 1 hour prior to infusion, 3 hours after infusion, 1, 7, and days following a single ketamine/saline infusion Impromptu Speech Behavioral Assessment Test, according to the Impromptu Speech Behavioral Assessment Test (BAT) of social anxiety symptoms during public speaking at 1 hour prior to infusion, 1 and 7 days following a single ketamine/saline infusion Attention bias, according to the dot‐probe paradigm 1 hour before infusion, 3 hours after infusion, 1, 2, 3, 5, 7, 10 and 14 days following a single ketamine/saline infusion SAD severity, according to the Liebowitz Social Anxiety Scale (LSAS) ratings of SAD severity at screening, 1 hour before infusion, 3 hours after infusion, 1, 2, 3, 5, 7, 10 and 14 days following a single ketamine/saline infusion Positive and negative affect symptoms, according to positive and negative affect schedule (PANAS) ratings of positive and negative symptoms at screening, 1 hour before infusion, 1 and 7 days following a single ketamine/saline infusion State‐Trait Anxiety Inventory, at screening, 1 hour before infusion, 3 hours after infusion, 1, 2, 3, 5, 7, 10 and 14 days following a single ketamine/saline infusion |
| Starting date | Study start date: March 2014 Estimated study completion date: March 2018 |
| Contact information | Contact: Angeli Landeros, MD 203‐737‐4809. |
| Notes | This study is currently recruiting participants. |
| Trial name or title | Vortioxetine versus placebo in major depressive disorder comorbid with social anxiety disorder |
| Methods | 12 week randomised, double‐blind, parallel, placebo‐controlled study |
| Participants | Estimated enrollment: 40 Ages eligible for study: 18‐70 years Genders eligible for study: both Accepts healthy volunteers: no Inclusion criteria: quote: "Male and female adults between 18 and 70 years of age (inclusive); subjects must give written informed consent prior to any study procedures; Diagnosis of Major Depressive Disorder (MDD), single episode (296.2) or recurrent (296.3), according to Diagnostic and Statistical Manual of Mental Disorders, version 5 (DSM‐5) criteria, as determined by psychiatric evaluation with the investigator and confirmed by the Mini‐International Neuropsychiatric Interview (MINI); duration of current major depressive episode must be at least 4 weeks; diagnosis of social anxiety disorder (SAD) (300.23 social phobia) according to DSM‐5 criteria, as determined by psychiatric evaluation with the investigator and confirmed by the MINI; duration of current SAD must be at least 6 months, and SAD should be observable in subjects' lives when they are not suffering from MDD, if such periods have occurred. Subjects must have a minimum total score of 60 on the Liebowitz Social Anxiety Scale (LSAS) at both screening and baseline visits; subjects must have a minimum total Montgomery Asberg Depression Rating Scale (MADRS) score of 20 at both screening and baseline visits; subjects must have a Clinical Global Inventory (CGI) Severity score of 4 or greater at both screening and baseline visits, where the CGI is based on a composite of MDD and SAD; male and female subjects of childbearing potential must commit to an effective form of contraception for the duration of the trial (screening/visit 1 through follow‐up/visit 10); effective forms of contraception include: condoms with spermicide, diaphragm with spermicide, hormonal contraceptive agents (oral, transdermal, or injectable), or implantable contraceptive devices; true abstinence will also be considered an effective form of contraception". Exclusion criteria: quote: "Subjects with any lifetime history of bipolar disorder, schizophrenia, schizoaffective disorder, obsessive compulsive disorder, eating disorders, or body dysmorphic disorder. Subjects with comorbid generalized anxiety disorder, dysthymia, or specific phobias can be included in the study provided that MDD and SAD are considered to be the primary clinical conditions in terms of need for treatment; subjects with substance abuse, panic disorder, or post‐traumatic stress disorder, in the past 6 months before screening; subjects who started psychotherapy for SAD or MDD or had electroconvulsive therapy (ECT) in the past 6 months before screening. Subjects who have been receiving psychotherapy or cognitive behavioral therapy for more than 24 weeks prior to the baseline visit are eligible provided that the therapy continues at the same frequency for the duration of the trial; subjects who are currently pregnant or lactating, or who are of childbearing potential and not able and willing to practice an effective method of contraception (as outlined in Inclusion criterion #10) for the duration of the trial (screening/visit 1 through follow‐up/visit 10; subjects who, in the opinion of the investigator, are at a clinically significant risk for suicide. This would include prominent suicidal ideation or suicidal behavior in the past 6 months before screening; systolic blood pressure ≥165 and/or diastolic blood pressure ≥95, as measured at screening and baseline visits; positive urine drug screen at the screening visit, unless due to prescribed medication; any current unstable and/or clinically significant medical condition, based on history or as evidenced in screening laboratory or electrocardiogram (ECG) assessments; subjects with a history or complication of cancer or malignant tumor not in remission for at least 5 years. Basal cell skin cancers are not exclusionary; subjects receiving fluoxetine within 28 days of the baseline visit; subjects receiving monoamine oxidase inhibitors (MAOIs) within 14 days of the baseline visit; subjects receiving any other psychotropic medication (including selective serotonin re‐uptake inhibitors (SSRIs), serotonin‐norepinephrine reuptake inhibitors (SNRIs), and benzodiazepines) within 14 days of the baseline visit. Zolpidem (Ambien) PRN is allowed for insomnia if not taken more than 3 times per week for the duration of the trial; treatment refractory SAD: subjects who have a history of two or more failed treatment trials with an FDA‐approved SAD treatment, each given for at least 6 weeks, during which the subject received an adequate dosage; treatment refractory MDD: subjects who have a history of two or more failed treatment trials with an FDA‐approved MDD treatment in the current episode" |
| Interventions | Experimental: vortioxetine Vortioxetine 10 to 20 mg orally 4 times daily for 12 weeks Placebo comparator: placebo Placeboorally 4 times daily for 12 weeks |
| Outcomes | Primary outcome measures: CGI‐I responder rate (time frame: 12 weeks) Secondary outcome measures: Change in total MADRS score (time frame: baseline and 12 weeks); change in total LSAS score (time frame: baseline and 12 weeks) |
| Starting date | Study start date: December 2014 Estimated primary completion date: September 2015 |
| Contact information | Contact: Ann E Draine 212 595‐5012 ext 222 Contact: Michael R. Liebowitz, MD 212 595‐5012 |
| Notes | This study is not yet open for participant recruitment. |
| Trial name or title | A safety and efficacy study of JNJ‐42165279 in participants with social anxiety disorder |
| Methods | 12 week randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study |
| Participants | Estimated enrollment: 122 Ages eligible for study: 18‐64 years Genders eligible for study: both. Accepts healthy volunteers: no Inclusion criteria: quote: "must have a primary DSM‐5 diagnosis of Social anxiety disorder (SAD) except those with performance only as a specifier; participants with a diagnosis of comorbid generalized anxiety disorder (GAD) or major depressive disorder (MDD) may be included if the investigator considers SAD to be the predominant diagnosis; participants with current or lifetime history of attention deficit hyperactivity disorder (ADHD) and specific phobia may be included as well. Must have a Liebowitz Social Anxiety Scale score greater than or equal (≥) 70 at screening and baseline; participants with a current episode of MDD must have a HDRS17 total score less than or equal to (≤) 18. Must have a body mass index (BMI) between 18 and 35 kilogram per meter square (kg/m2), inclusive, at screening; female participants must be either postmenopausal or surgically sterile". Exclusion criteria: quote: "Participants who have performance only SAD are excluded. Participants with other current significant psychiatric condition(s) (Axis 1 under DSM‐IV), including, but not limited to, MDD with psychotic features (lifetime), bipolar disorder (including lifetime diagnosis), obsessive‐compulsive disorder, borderline personality disorder, eating disorder (e.g., bulimia, anorexia nervosa), autism spectrum disorders, post‐traumatic stress disorder (PTSD) or schizophrenia are excluded. Participants with a diagnosis of comorbid GAD or MDD may be included; participants currently receiving specific psychotherapy for SAD; has a history of more than two unsuccessful adequate pharmacological treatment trials for SAD, defined as lack of response to at least 10 weeks of treatment at adequate doses (e.g., paroxetine ≥ 40 milligram per day (mg/day) or its equivalent; or clonazepam ≥ 2.5 mg/day or its equivalent); concurrent use of psychotropic medications; has a history of or current thyroid disease, thyroid dysfunction and is currently untreated for it". |
| Interventions | Experimental: JNJ‐42165279. Participants will receive 25 milligram (mg) JNJ‐42165279 orally once daily from day 1 up to 12 weeks Placebo comparator: placebo. Participants will receive a matching placebo orally once daily from day 1 up to 12 weeks |
| Outcomes | Primary outcome measures: Change from baseline in the Liebowitz Social Anxiety Scale (LSAS) total score at week 12 Secondary outcome measures: Change from baseline in Liebowitz Social Anxiety Scale (LSAS) fear/anxiety and avoidance subscales at week 12 Number of participants who are responders and remitters on Liebowitz Social Anxiety Scale (LSAS) total score at week 12 Percentage of participants who are responders and remitters on Liebowitz Social Anxiety Scale (LSAS) total score at week 12 (time frame: baseline and week 12) (designated as safety issue: no) Change from baseline in structured interview guide for the Hamilton Anxiety Rating Scale (SIGH‐A) total score at week 12 Change from baseline in Hamilton Anxiety Rating scale (HAM‐A6) score at week 12 Change from baseline in Hamilton Depression Rating Scale (HDRS17) total score at week 12 Change from baseline in HDRS17 anxiety/somatisation factor total score at week 12 Change from baseline in 6‐Item Hamilton Depression Scale (HAM‐D6) score at week 12 Clinical Global Impression ‐ Improvement (CGI‐I) score from baseline at week 12 Number of participants who are responders on SIGH‐A total score at Week 12 Percentage of participants who are responders on SIGH‐A total score at week 12 |
| Starting date | Study start date: June 2015 Estimated study completion date: February 2017 |
| Contact information | Janssen Research & Development, LLC |
| Notes | This study has suspended participant recruitment. |
ADHD: attention deficit hyperactivity disorder; BAI: Beck Anxiety Inventory; BAT: Behavioural Assessment Test; BMI: body mass index; BPRS‐PS: Brief Psychiatric Rating Scale, Positive Symptom Subscale; CADSS: Clinician‐Administered Dissociative States Scales; CGIC/CGI‐I: Clinical Global Impressions Improvement Scale; CGI‐S: Clinical Global Impression of Severity of Illness; DSM: Diagnostic and Statistical Manual of Mental Disorders; DSM 5: Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition; DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition; DSM‐IV‐TR: Diagnostic and Statistical Manual of Mental Disorders; FDA: Food and Drug Administration (USA); GAD: Generalised Anxiety Disorder; GSP: Generalised Social Phobia; HAM‐A6: 6‐item Hamilton Anxiety Rating scale; HDRS17: Hamilton Rating Scale for Depression (17 items); LSAS: Liebowitz Social Anxiety Scale; MADRS: Montgomery Asberg Depression Rating Scale; MAOI: monoamine oxidase inhibitor; MDD: Major Depressive Disorder; MINI: Mini‐International Neuropsychiatric Interview; PANAS: positive and negative affect schedule; PO: Placebo; PRN: as needed; PTSD: post‐traumatic stress disorder; SAnD: Social Anxiety Disorder; SCID: structured clinical interview for DSM; SIGH‐A: Structured Interview Guide for the Hamilton Anxiety Rating Scale; SNRI: serotonin‐norepinephrine reuptake inhibitor; SPSS: Self‐Statement During Public Speaking Scale; SSRI: selective serotonin re‐uptake inhibitor; STAI: State‐Trait Anxiety Inventory.
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 1.1 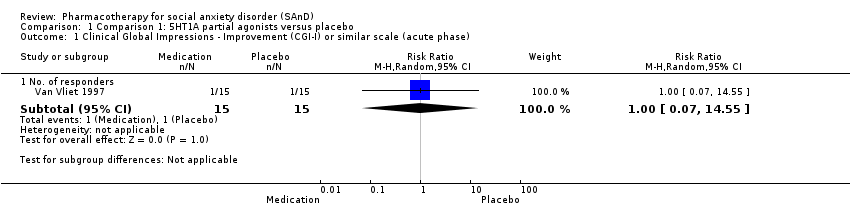 Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 14.55] |
| 2 Adverse events (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 1.2  Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 2 Adverse events (acute phase). | ||||
| 2.1 Dropout rate | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 68.26] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 1.3 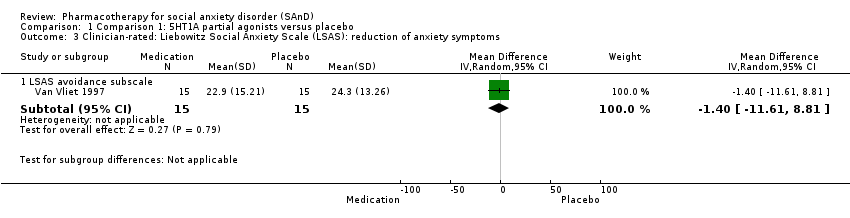 Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 3.1 LSAS avoidance subscale | 1 | 30 | Mean Difference (IV, Random, 95% CI) | ‐1.40 [‐11.61, 8.81] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 1.4 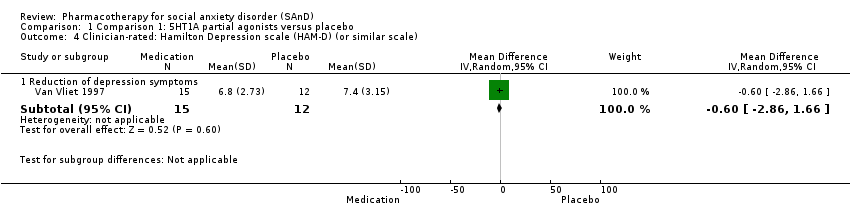 Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale). | ||||
| 4.1 Reduction of depression symptoms | 1 | 27 | Mean Difference (IV, Random, 95% CI) | ‐0.60 [‐2.86, 1.66] |
| 5 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 1.5 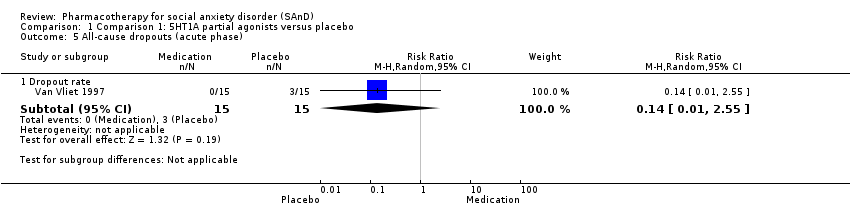 Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 5 All‐cause dropouts (acute phase). | ||||
| 5.1 Dropout rate | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.55] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 2.1  Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.16, 2.20] |
| 2 Adverse events (acute phase) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 2.2  Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 2 Adverse events (acute phase). | ||||
| 2.1 Dropout rate | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 2.90 [0.92, 9.14] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 2.3 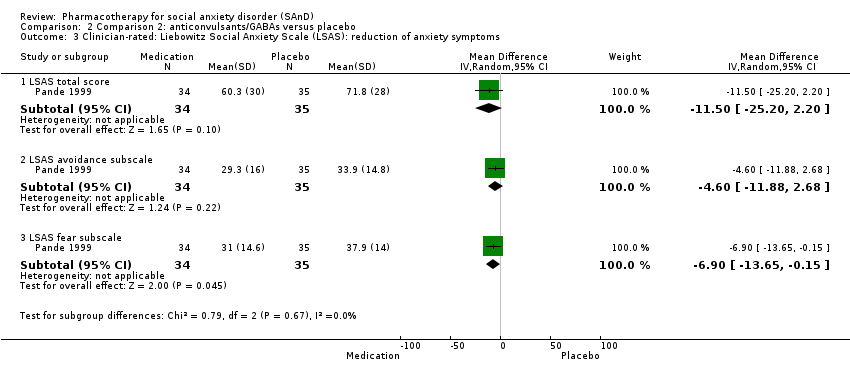 Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 3.1 LSAS total score | 1 | 69 | Mean Difference (IV, Random, 95% CI) | ‐11.5 [‐25.20, 2.20] |
| 3.2 LSAS avoidance subscale | 1 | 69 | Mean Difference (IV, Random, 95% CI) | ‐4.60 [‐11.88, 2.68] |
| 3.3 LSAS fear subscale | 1 | 69 | Mean Difference (IV, Random, 95% CI) | ‐6.90 [‐13.65, ‐0.15] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 2.4  Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale). | ||||
| 4.1 Reduction of depression symptoms | 1 | 69 | Mean Difference (IV, Random, 95% CI) | ‐2.3 [‐4.78, 0.18] |
| 5 All‐cause dropouts (acute phase) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 2.5  Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 5 All‐cause dropouts (acute phase). | ||||
| 5.1 Dropout rate | 3 | 488 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.78, 1.70] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 3.1  Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 2 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.70, 1.37] |
| 2 Adverse events (acute phase) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 3.2  Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 2 Adverse events (acute phase). | ||||
| 2.1 Dropout rate | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.81, 4.94] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 3.3 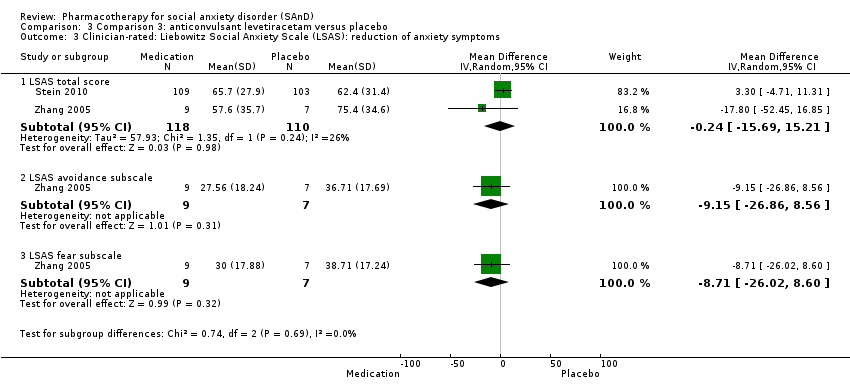 Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 3.1 LSAS total score | 2 | 228 | Mean Difference (IV, Random, 95% CI) | ‐0.24 [‐15.69, 15.21] |
| 3.2 LSAS avoidance subscale | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐9.15 [‐26.86, 8.56] |
| 3.3 LSAS fear subscale | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐8.71 [‐26.02, 8.60] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 3.4  Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale). | ||||
| 4.1 Reduction of depression symptoms | 1 | 212 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐1.33, 1.73] |
| 5 All‐cause dropouts (acute phase) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 3.5  Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 5 All‐cause dropouts (acute phase). | ||||
| 5.1 Dropout rate | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.30, 6.76] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 4.1 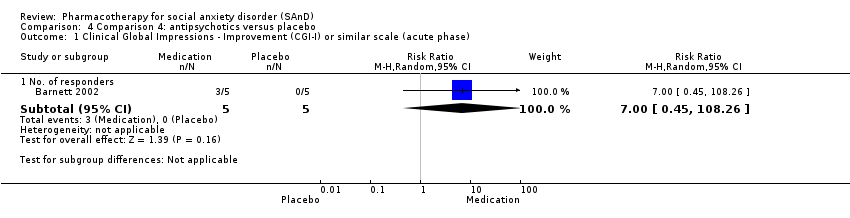 Comparison 4 Comparison 4: antipsychotics versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.45, 108.26] |
| 2 Adverse events (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 4.2  Comparison 4 Comparison 4: antipsychotics versus placebo, Outcome 2 Adverse events (acute phase). | ||||
| 2.1 Dropout rate | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.06, 8.90] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 4.3 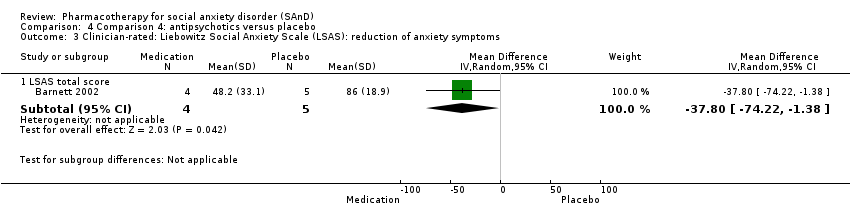 Comparison 4 Comparison 4: antipsychotics versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 3.1 LSAS total score | 1 | 9 | Mean Difference (IV, Random, 95% CI) | ‐37.8 [‐74.22, ‐1.38] |
| 4 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 4.4  Comparison 4 Comparison 4: antipsychotics versus placebo, Outcome 4 All‐cause dropouts (acute phase). | ||||
| 4.1 Dropout rate | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.27, 4.23] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 5.1 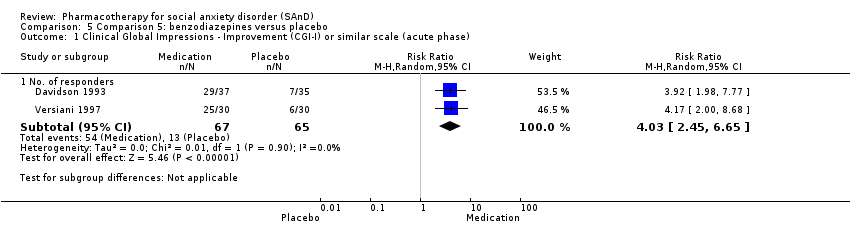 Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 2 | 132 | Risk Ratio (M‐H, Random, 95% CI) | 4.03 [2.45, 6.65] |
| 2 Relapse rate Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 5.2  Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 2 Relapse rate. | ||||
| 2.1 No. relapsed | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 0.12 [0.01, 2.14] |
| 3 Adverse events (acute phase) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 5.3  Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 3 Adverse events (acute phase). | ||||
| 3.1 Dropout rate | 2 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 1.68 [0.21, 13.13] |
| 4 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 5.4  Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 4 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 4.1 LSAS total score | 2 | 135 | Mean Difference (IV, Random, 95% CI) | ‐39.75 [‐71.11, ‐8.39] |
| 4.2 LSAS avoidance subscale | 1 | 75 | Mean Difference (IV, Random, 95% CI) | ‐10.40 [‐16.08, ‐4.72] |
| 4.3 LSAS fear subscale | 1 | 75 | Mean Difference (IV, Random, 95% CI) | ‐10.80 [‐16.62, ‐4.98] |
| 5 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 5.5  Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 5 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale). | ||||
| 5.1 Reduction of depression symptoms | 1 | 75 | Mean Difference (IV, Random, 95% CI) | ‐1.60 [‐3.96, 0.76] |
| 6 Reduction of functional disability Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 5.6 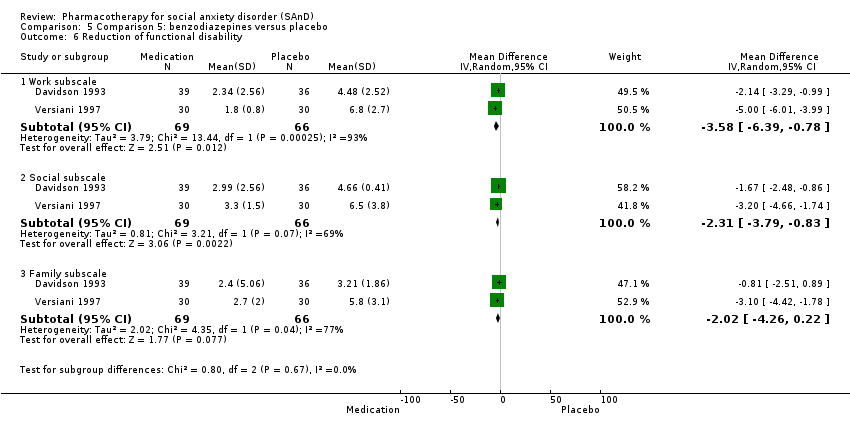 Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 6 Reduction of functional disability. | ||||
| 6.1 Work subscale | 2 | 135 | Mean Difference (IV, Random, 95% CI) | ‐3.58 [‐6.39, ‐0.78] |
| 6.2 Social subscale | 2 | 135 | Mean Difference (IV, Random, 95% CI) | ‐2.31 [‐3.79, ‐0.83] |
| 6.3 Family subscale | 2 | 135 | Mean Difference (IV, Random, 95% CI) | ‐2.02 [‐4.26, 0.22] |
| 7 All‐cause dropouts (acute phase) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 5.7 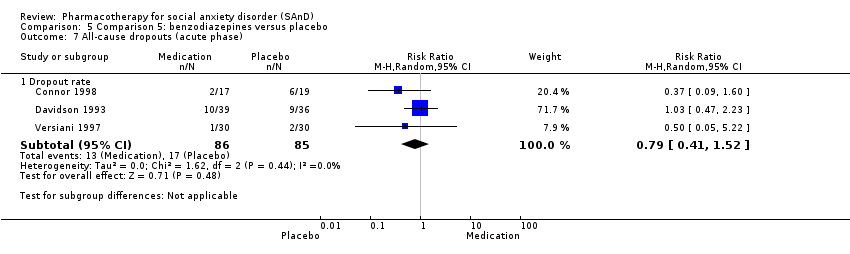 Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 7 All‐cause dropouts (acute phase). | ||||
| 7.1 Dropout rate | 3 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.41, 1.52] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 6.1  Comparison 6 Comparison 6: beta‐blockers versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 2 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.63, 1.88] |
| 2 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 6.2  Comparison 6 Comparison 6: beta‐blockers versus placebo, Outcome 2 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale). | ||||
| 2.1 Reduction of depression symptoms | 1 | 46 | Mean Difference (IV, Random, 95% CI) | 1.82 [‐1.38, 5.02] |
| 3 Reduction of functional disability Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 6.3 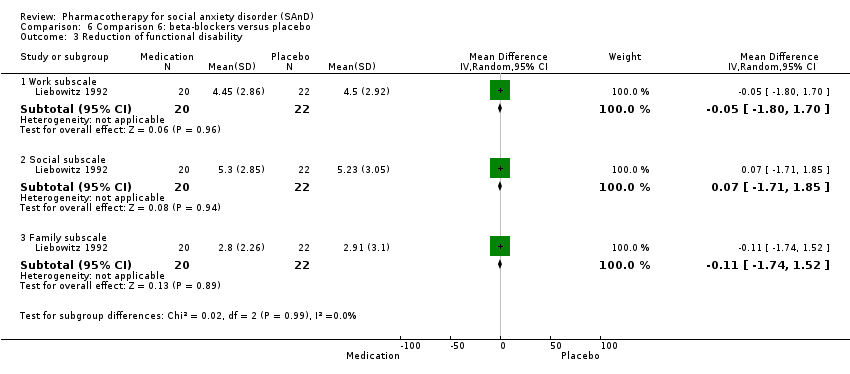 Comparison 6 Comparison 6: beta‐blockers versus placebo, Outcome 3 Reduction of functional disability. | ||||
| 3.1 Work subscale | 1 | 42 | Mean Difference (IV, Random, 95% CI) | ‐0.05 [‐1.80, 1.70] |
| 3.2 Social subscale | 1 | 42 | Mean Difference (IV, Random, 95% CI) | 0.07 [‐1.71, 1.85] |
| 3.3 Family subscale | 1 | 42 | Mean Difference (IV, Random, 95% CI) | ‐0.11 [‐1.74, 1.52] |
| 4 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 6.4  Comparison 6 Comparison 6: beta‐blockers versus placebo, Outcome 4 All‐cause dropouts (acute phase). | ||||
| 4.1 Dropout rate | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 3.36 [0.41, 27.80] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 7.1  Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 4 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 2.36 [1.48, 3.75] |
| 2 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 4 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 7.2  Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 2 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 2.1 LSAS total score | 4 | 218 | Mean Difference (IV, Random, 95% CI) | ‐16.39 [‐32.27, ‐0.51] |
| 2.2 LSAS avoidance subscale | 1 | 51 | Mean Difference (IV, Random, 95% CI) | ‐5.42 [‐14.69, 3.85] |
| 2.3 LSAS fear subscale | 1 | 51 | Mean Difference (IV, Random, 95% CI) | ‐5.23 [‐13.97, 3.51] |
| 3 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 7.3 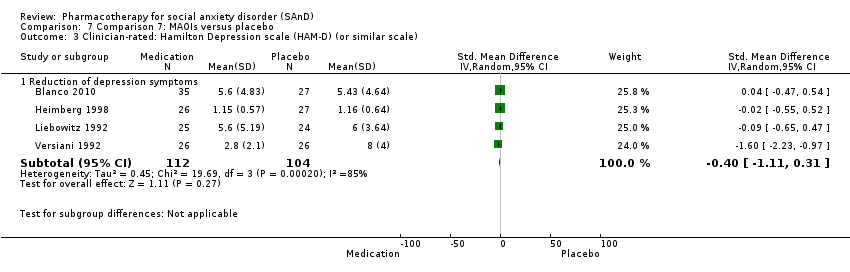 Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 3 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale). | ||||
| 3.1 Reduction of depression symptoms | 4 | 216 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐1.11, 0.31] |
| 4 Reduction of functional disability Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 7.4 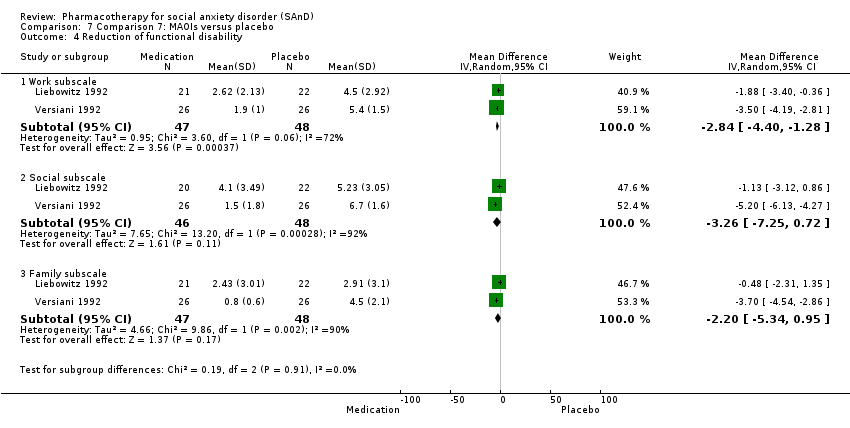 Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 4 Reduction of functional disability. | ||||
| 4.1 Work subscale | 2 | 95 | Mean Difference (IV, Random, 95% CI) | ‐2.84 [‐4.40, ‐1.28] |
| 4.2 Social subscale | 2 | 94 | Mean Difference (IV, Random, 95% CI) | ‐3.26 [‐7.25, 0.72] |
| 4.3 Family subscale | 2 | 95 | Mean Difference (IV, Random, 95% CI) | ‐2.20 [‐5.34, 0.95] |
| 5 All‐cause dropouts (acute phase) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 7.5  Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 5 All‐cause dropouts (acute phase). | ||||
| 5.1 Dropout rate | 4 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.71, 2.48] |
| 6 Clinical Global Impressions scale change item (CGI‐I) (long term) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 7.6  Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 6 Clinical Global Impressions scale change item (CGI‐I) (long term). | ||||
| 6.1 No. of responders | 2 | 113 | Risk Ratio (M‐H, Random, 95% CI) | 1.84 [1.02, 3.33] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 8.1  Comparison 8 Comparison 8: NARIs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.19, 2.54] |
| 2 Adverse events (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 8.2  Comparison 8 Comparison 8: NARIs versus placebo, Outcome 2 Adverse events (acute phase). | ||||
| 2.1 Dropout rate | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 2.8 [0.12, 63.20] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 8.3  Comparison 8 Comparison 8: NARIs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 3.1 LSAS total score | 1 | 26 | Mean Difference (IV, Random, 95% CI) | 2.60 [‐15.43, 20.63] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 8.4  Comparison 8 Comparison 8: NARIs versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale). | ||||
| 4.1 Reduction of depression symptoms | 1 | 26 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐2.73, 2.53] |
| 5 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 8.5  Comparison 8 Comparison 8: NARIs versus placebo, Outcome 5 All‐cause dropouts (acute phase). | ||||
| 5.1 Dropout rate | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.23, 3.81] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 9.1 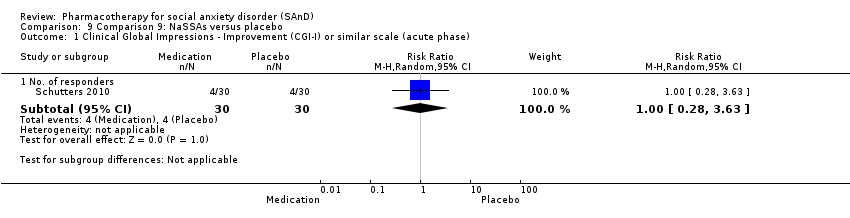 Comparison 9 Comparison 9: NaSSAs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.28, 3.63] |
| 2 Adverse events (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 9.2  Comparison 9 Comparison 9: NaSSAs versus placebo, Outcome 2 Adverse events (acute phase). | ||||
| 2.1 Dropout rate | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 99.95] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 9.3  Comparison 9 Comparison 9: NaSSAs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 3.1 LSAS total score | 2 | 126 | Mean Difference (IV, Random, 95% CI) | ‐15.37 [‐28.10, ‐2.63] |
| 3.2 LSAS avoidance subscale | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐3.90 [‐9.90, 2.10] |
| 3.3 LSAS fear subscale | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐3.70 [‐9.42, 2.02] |
| 4 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 9.4  Comparison 9 Comparison 9: NaSSAs versus placebo, Outcome 4 All‐cause dropouts (acute phase). | ||||
| 4.1 Dropout rate | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 20.90] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 10.1  Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 8 | 1270 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [1.32, 2.55] |
| 2 Adverse events (acute phase) Show forest plot | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 10.2 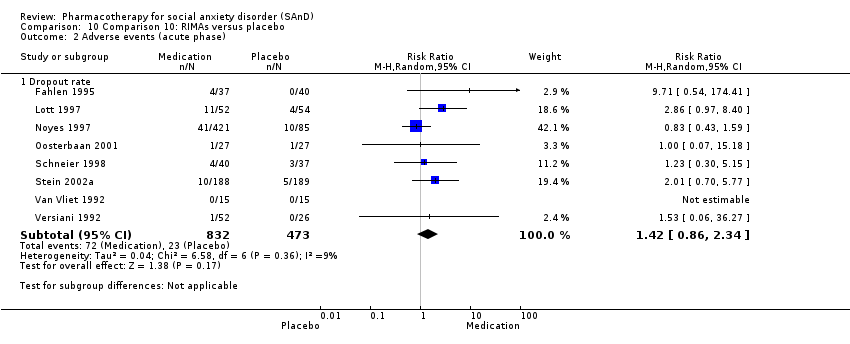 Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 2 Adverse events (acute phase). | ||||
| 2.1 Dropout rate | 8 | 1305 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.86, 2.34] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 9 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 10.3 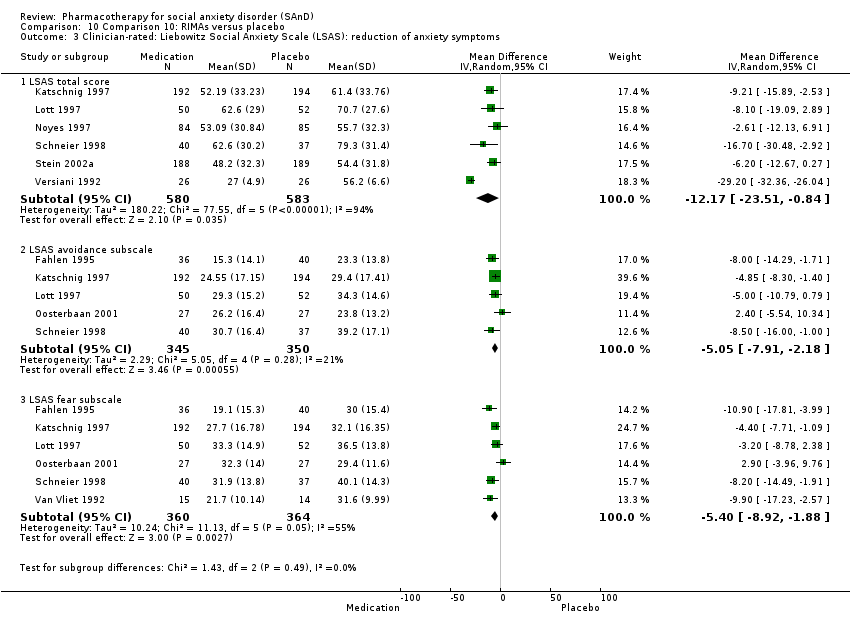 Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 3.1 LSAS total score | 6 | 1163 | Mean Difference (IV, Random, 95% CI) | ‐12.17 [‐23.51, ‐0.84] |
| 3.2 LSAS avoidance subscale | 5 | 695 | Mean Difference (IV, Random, 95% CI) | ‐5.05 [‐7.91, ‐2.18] |
| 3.3 LSAS fear subscale | 6 | 724 | Mean Difference (IV, Random, 95% CI) | ‐5.40 [‐8.92, ‐1.88] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 7 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 10.4 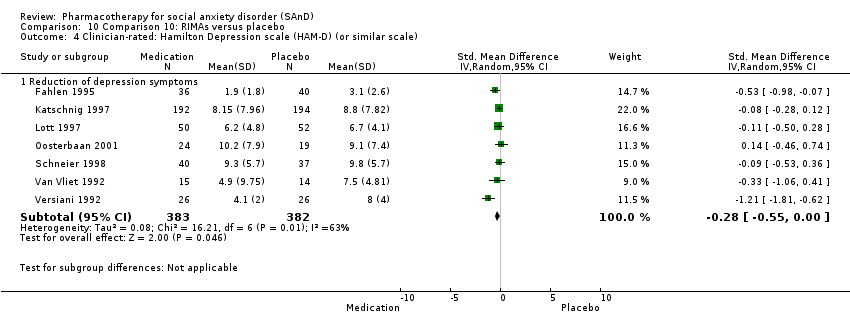 Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale). | ||||
| 4.1 Reduction of depression symptoms | 7 | 765 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.55, ‐0.00] |
| 5 Reduction of functional disability Show forest plot | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 10.5 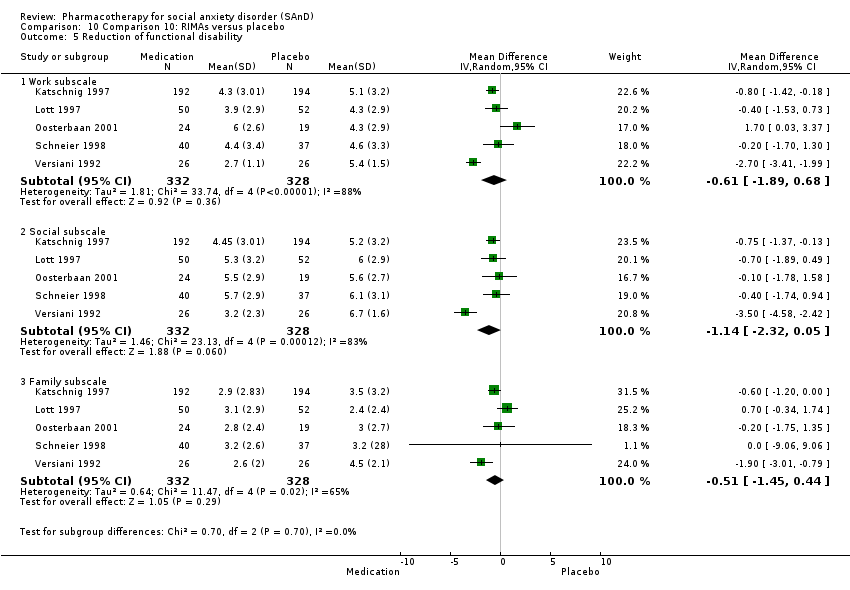 Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 5 Reduction of functional disability. | ||||
| 5.1 Work subscale | 5 | 660 | Mean Difference (IV, Random, 95% CI) | ‐0.61 [‐1.89, 0.68] |
| 5.2 Social subscale | 5 | 660 | Mean Difference (IV, Random, 95% CI) | ‐1.14 [‐2.32, 0.05] |
| 5.3 Family subscale | 5 | 660 | Mean Difference (IV, Random, 95% CI) | ‐0.51 [‐1.45, 0.44] |
| 6 All‐cause dropouts (acute phase) Show forest plot | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 10.6 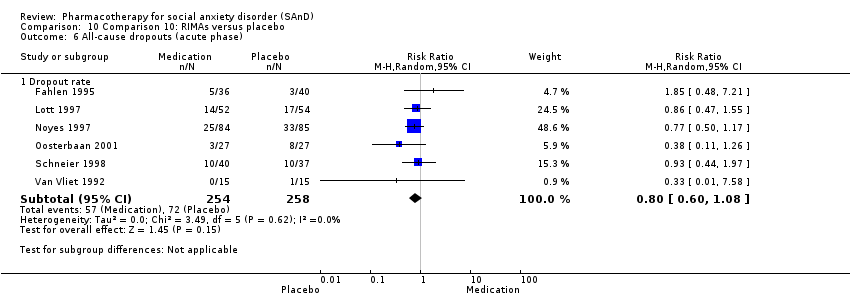 Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 6 All‐cause dropouts (acute phase). | ||||
| 6.1 Dropout rate | 6 | 512 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.60, 1.08] |
| 7 Clinical Global Impressions ‐ Improvement (CGI‐I) (long term) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 10.7  Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 7 Clinical Global Impressions ‐ Improvement (CGI‐I) (long term). | ||||
| 7.1 No. of responders | 1 | 90 | Risk Ratio (M‐H, Random, 95% CI) | 1.50 [1.12, 2.00] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 11.1 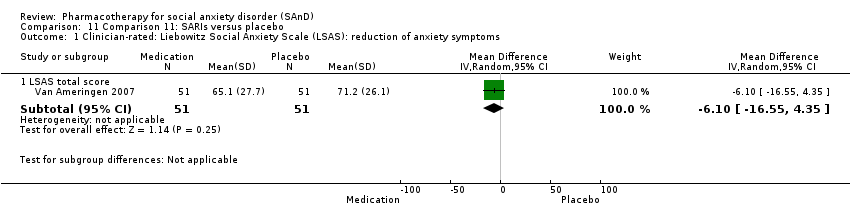 Comparison 11 Comparison 11: SARIs versus placebo, Outcome 1 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 1.1 LSAS total score | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐6.10 [‐16.55, 4.35] |
| 2 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 11.2  Comparison 11 Comparison 11: SARIs versus placebo, Outcome 2 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale). | ||||
| 2.1 Reduction of depression symptoms | 1 | 102 | Mean Difference (IV, Random, 95% CI) | 0.80 [‐2.10, 3.70] |
| 3 Reduction of functional disability Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 11.3 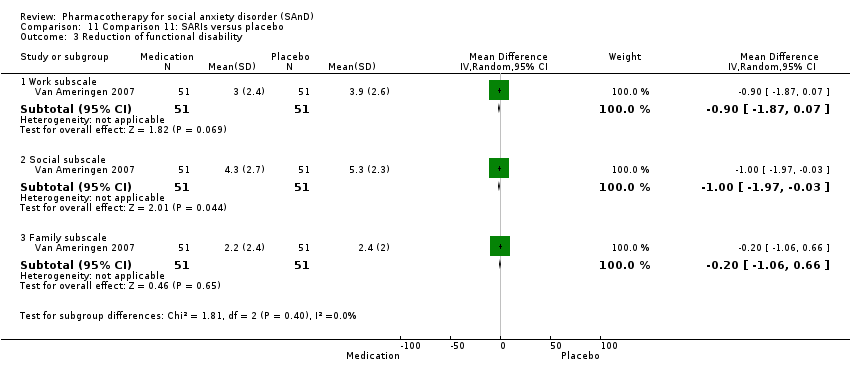 Comparison 11 Comparison 11: SARIs versus placebo, Outcome 3 Reduction of functional disability. | ||||
| 3.1 Work subscale | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐0.90 [‐1.87, 0.07] |
| 3.2 Social subscale | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐1.0 [‐1.97, ‐0.03] |
| 3.3 Family subscale | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐1.06, 0.66] |
| 4 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 11.4  Comparison 11 Comparison 11: SARIs versus placebo, Outcome 4 All‐cause dropouts (acute phase). | ||||
| 4.1 Dropout rate | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 2.18 [0.97, 4.92] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 12.1  Comparison 12 Comparison 12: SNRIs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 4 | 1173 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.85, 1.99] |
| 2 Adverse events (acute phase) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 12.2 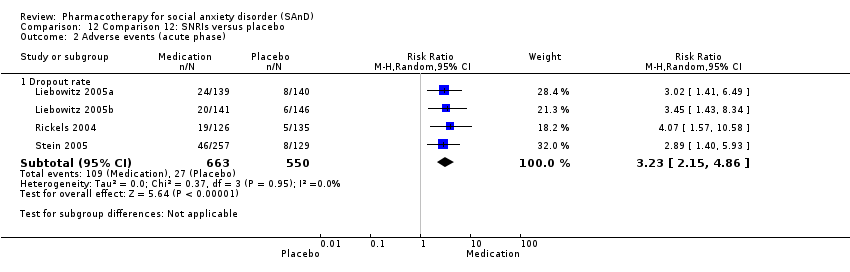 Comparison 12 Comparison 12: SNRIs versus placebo, Outcome 2 Adverse events (acute phase). | ||||
| 2.1 Dropout rate | 4 | 1213 | Risk Ratio (M‐H, Random, 95% CI) | 3.23 [2.15, 4.86] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 12.3  Comparison 12 Comparison 12: SNRIs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 3.1 LSAS total score | 3 | 902 | Mean Difference (IV, Random, 95% CI) | ‐11.91 [‐16.06, ‐7.76] |
| 3.2 LSAS avoidance subscale | 1 | 261 | Mean Difference (IV, Random, 95% CI) | ‐4.30 [‐8.14, ‐0.46] |
| 3.3 LSAS fear subscale | 1 | 261 | Mean Difference (IV, Random, 95% CI) | ‐4.0 [‐7.68, ‐0.32] |
| 4 All‐cause dropouts (acute phase) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 12.4  Comparison 12 Comparison 12: SNRIs versus placebo, Outcome 4 All‐cause dropouts (acute phase). | ||||
| 4.1 Dropout rate | 4 | 1224 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.76, 1.07] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 24 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 13.1  Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 24 | 4984 | Risk Ratio (M‐H, Random, 95% CI) | 1.65 [1.48, 1.85] |
| 2 Relapse rate Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 13.2  Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 2 Relapse rate. | ||||
| 2.1 No. relapsed | 3 | 389 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.22, 0.50] |
| 3 Adverse events (acute phase) Show forest plot | 24 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 13.3  Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 3 Adverse events (acute phase). | ||||
| 3.1 Dropout rate | 24 | 5131 | Risk Ratio (M‐H, Random, 95% CI) | 2.59 [1.97, 3.39] |
| 4 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 15 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 13.4  Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 4 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 4.1 LSAS total score | 14 | 1990 | Mean Difference (IV, Random, 95% CI) | ‐10.14 [‐14.05, ‐6.22] |
| 4.2 LSAS avoidance subscale | 7 | 1173 | Mean Difference (IV, Random, 95% CI) | ‐7.01 [‐10.21, ‐3.80] |
| 4.3 LSAS fear subscale | 7 | 1173 | Mean Difference (IV, Random, 95% CI) | ‐7.28 [‐10.86, ‐3.71] |
| 5 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 13.5  Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 5 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale). | ||||
| 5.1 Reduction of depression symptoms | 6 | 960 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐0.48, ‐0.03] |
| 6 Reduction of functional disability Show forest plot | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 13.6 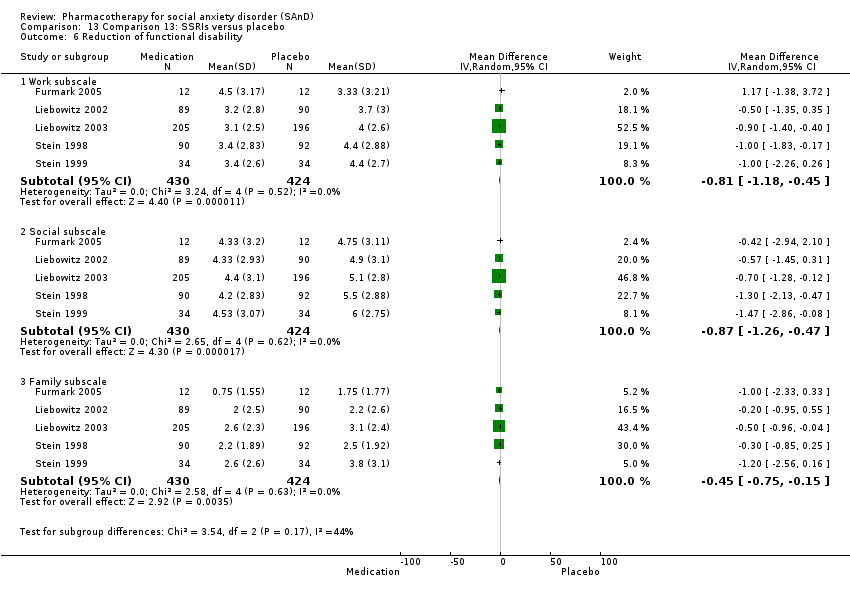 Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 6 Reduction of functional disability. | ||||
| 6.1 Work subscale | 5 | 854 | Mean Difference (IV, Random, 95% CI) | ‐0.81 [‐1.18, ‐0.45] |
| 6.2 Social subscale | 5 | 854 | Mean Difference (IV, Random, 95% CI) | ‐0.87 [‐1.26, ‐0.47] |
| 6.3 Family subscale | 5 | 854 | Mean Difference (IV, Random, 95% CI) | ‐0.45 [‐0.75, ‐0.15] |
| 7 All‐cause dropouts (acute phase) Show forest plot | 26 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 13.7  Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 7 All‐cause dropouts (acute phase). | ||||
| 7.1 Dropout rate | 26 | 5208 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.90, 1.14] |
| 8 Clinical Global Impressions scale change item (CGI‐I) (long term) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 13.8 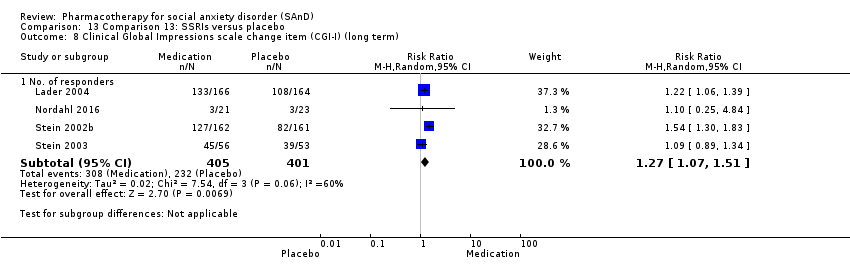 Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 8 Clinical Global Impressions scale change item (CGI‐I) (long term). | ||||
| 8.1 No. of responders | 4 | 806 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [1.07, 1.51] |
| 9 Adverse events (long term) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 13.9  Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 9 Adverse events (long term). | ||||
| 9.1 Dropout rate | 3 | 1274 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.43, 3.18] |
| 10 All‐cause dropouts (long term) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 13.10  Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 10 All‐cause dropouts (long term). | ||||
| 10.1 Dropout rate | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.37, 2.70] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 14.1  Comparison 14 Comparison 14: GW876008 versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 1 | 250 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.19] |
| 2 Adverse events (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 14.2  Comparison 14 Comparison 14: GW876008 versus placebo, Outcome 2 Adverse events (acute phase). | ||||
| 2.1 Dropout rate | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 2.95 [0.67, 13.02] |
| 3 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 14.3  Comparison 14 Comparison 14: GW876008 versus placebo, Outcome 3 All‐cause dropouts (acute phase). | ||||
| 3.1 Dropout rate | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.50, 1.20] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 15.1 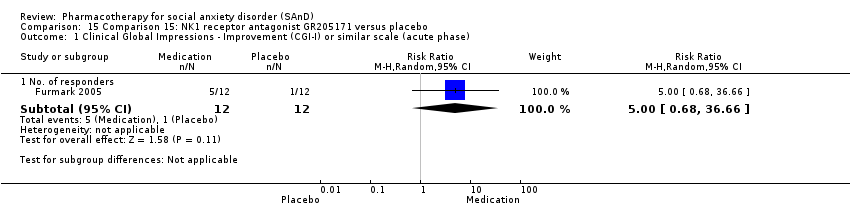 Comparison 15 Comparison 15: NK1 receptor antagonist GR205171 versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase). | ||||
| 1.1 No. of responders | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.68, 36.66] |
| 2 Adverse events (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 15.2 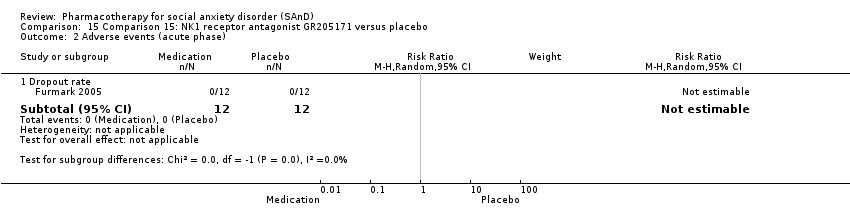 Comparison 15 Comparison 15: NK1 receptor antagonist GR205171 versus placebo, Outcome 2 Adverse events (acute phase). | ||||
| 2.1 Dropout rate | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 15.3 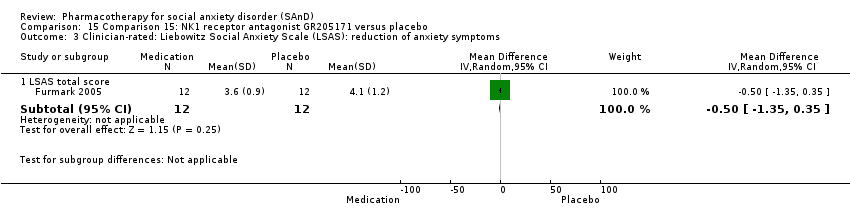 Comparison 15 Comparison 15: NK1 receptor antagonist GR205171 versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 3.1 LSAS total score | 1 | 24 | Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐1.35, 0.35] |
| 4 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 15.4  Comparison 15 Comparison 15: NK1 receptor antagonist GR205171 versus placebo, Outcome 4 All‐cause dropouts (acute phase). | ||||
| 4.1 Dropout rate | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 16.1  Comparison 16 Comparison 16: LY686017 versus placebo, Outcome 1 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms. | ||||
| 1.1 LSAS total score | 1 | 99 | Mean Difference (IV, Random, 95% CI) | 1.80 [‐6.92, 10.52] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar) scale: no. of responders (acute phase) Show forest plot | 51 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 17.1  Comparison 17 Comparison 17: total effect of medication compared to placebo for the treatment of SAnD, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar) scale: no. of responders (acute phase). | ||||
| 1.1 5HT1A partial agonists | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 14.55] |
| 1.2 Anticonvulsants/GABAs | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.16, 2.20] |
| 1.3 Anticonvulsant levetiracetam | 2 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.70, 1.37] |
| 1.4 Antipsychotics | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.45, 108.26] |
| 1.5 Benzodiazepines | 2 | 132 | Risk Ratio (M‐H, Random, 95% CI) | 4.03 [2.45, 6.65] |
| 1.6 Beta‐blockers | 2 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.63, 1.88] |
| 1.7 MAOIs | 3 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 2.37 [1.26, 4.45] |
| 1.8 NARIs | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.19, 2.54] |
| 1.9 NaSSAs | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.28, 3.63] |
| 1.10 RIMAs | 8 | 1270 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [1.32, 2.55] |
| 1.11 SNRIs | 4 | 1173 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.85, 1.99] |
| 1.12 SSRIs | 21 | 4553 | Risk Ratio (M‐H, Random, 95% CI) | 1.65 [1.46, 1.87] |
| 1.13 NK1 receptor antagonist GR205171 | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.68, 36.66] |
| 1.14 GW876008 | 1 | 250 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.19] |
| 2 Dropout rate: adverse events (acute phase) Show forest plot | 45 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 17.2  Comparison 17 Comparison 17: total effect of medication compared to placebo for the treatment of SAnD, Outcome 2 Dropout rate: adverse events (acute phase). | ||||
| 2.1 5HT1A partial agonists | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 68.26] |
| 2.2 Anticonvulsants/GABAs | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 2.90 [0.92, 9.14] |
| 2.3 Anticonvulsant levetiracetam | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.81, 4.94] |
| 2.4 Antipsychotics | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.06, 8.90] |
| 2.5 Benzodiazepines | 2 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 1.68 [0.21, 13.13] |
| 2.6 NARIs | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 2.8 [0.12, 63.20] |
| 2.7 NaSSAs | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 99.95] |
| 2.8 RIMAs | 8 | 1305 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.86, 2.34] |
| 2.9 SNRIs | 4 | 1213 | Risk Ratio (M‐H, Random, 95% CI) | 3.23 [2.15, 4.86] |
| 2.10 SSRIs | 22 | 4965 | Risk Ratio (M‐H, Random, 95% CI) | 2.56 [1.94, 3.38] |
| 2.11 NK1 receptor antagonist GR205171 | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2.12 GW876008 | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 2.95 [0.67, 13.02] |
| 3 Dropout rate: all‐cause dropouts (acute phase) Show forest plot | 54 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 17.3  Comparison 17 Comparison 17: total effect of medication compared to placebo for the treatment of SAnD, Outcome 3 Dropout rate: all‐cause dropouts (acute phase). | ||||
| 3.1 5HT1A partial agonists | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.55] |
| 3.2 Anticonvulsants/GABAs | 3 | 488 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.78, 1.70] |
| 3.3 Anticonvulsant levetiracetam | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.30, 6.76] |
| 3.4 Antipsychotics | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.27, 4.23] |
| 3.5 Benzodiazepines | 3 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.41, 1.52] |
| 3.6 Beta‐blockers | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 3.36 [0.41, 27.80] |
| 3.7 MAOIs | 4 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.71, 2.48] |
| 3.8 NARIs | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.23, 3.81] |
| 3.9 NaSSAs | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 20.90] |
| 3.10 RIMAs | 6 | 512 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.60, 1.08] |
| 3.11 SARIs | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 2.18 [0.97, 4.92] |
| 3.12 SNRIs | 4 | 1224 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.76, 1.07] |
| 3.13 SSRIs | 25 | 5078 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.89, 1.14] |
| 3.14 NK1 receptor antagonist GR205171 | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.15 GW876008 | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.50, 1.20] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase) Show forest plot | 53 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 18.1 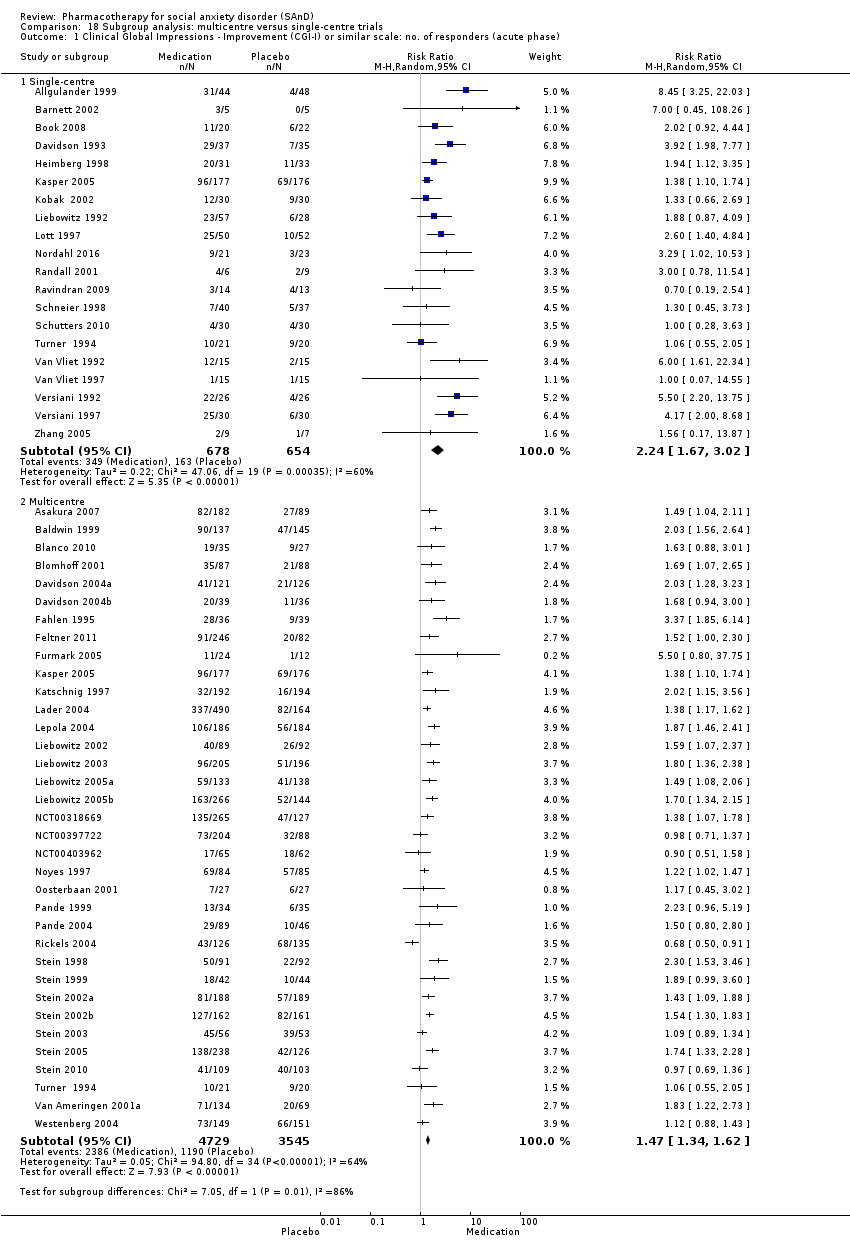 Comparison 18 Subgroup analysis: multicentre versus single‐centre trials, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase). | ||||
| 1.1 Single‐centre | 20 | 1332 | Risk Ratio (M‐H, Random, 95% CI) | 2.24 [1.67, 3.02] |
| 1.2 Multicentre | 35 | 8274 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [1.34, 1.62] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase) Show forest plot | 53 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 19.1  Comparison 19 Subgroup analysis: generalised SAnD compared to inclusive SAnD, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase). | ||||
| 1.1 Generalised | 26 | 5522 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [1.31, 1.69] |
| 1.2 Inclusive | 27 | 3712 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [1.54, 2.18] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase) Show forest plot | 50 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 20.1 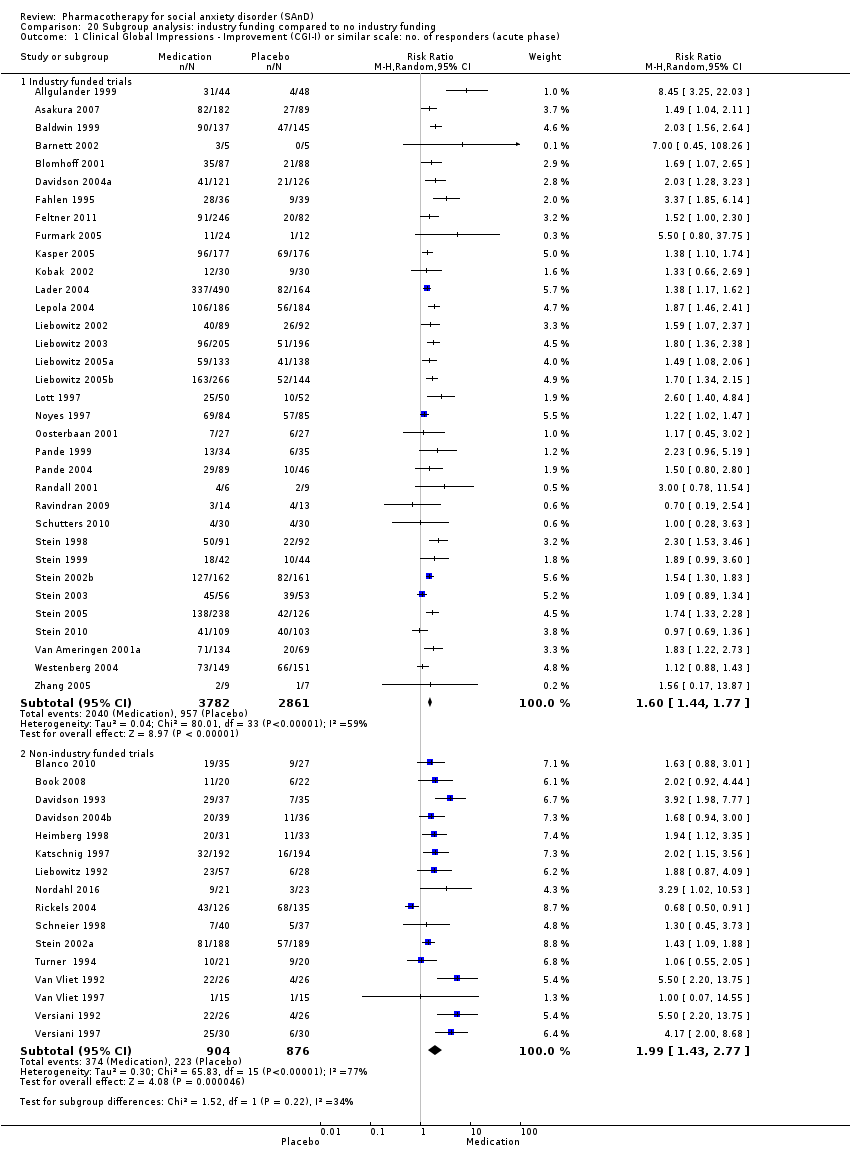 Comparison 20 Subgroup analysis: industry funding compared to no industry funding, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase). | ||||
| 1.1 Industry funded trials | 34 | 6643 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.44, 1.77] |
| 1.2 Non‐industry funded trials | 16 | 1780 | Risk Ratio (M‐H, Random, 95% CI) | 1.99 [1.43, 2.77] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase) Show forest plot | 53 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 21.1  Comparison 21 Subgroup analysis: trials that included MDD compared to no MDD, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase). | ||||
| 1.1 Trials including MDD participants | 20 | 2654 | Risk Ratio (M‐H, Random, 95% CI) | 1.77 [1.44, 2.18] |
| 1.2 Trials excluding MDD participants | 34 | 6765 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [1.35, 1.70] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 'Worst case' lost‐to‐follow‐up analysis Show forest plot | 25 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 22.1 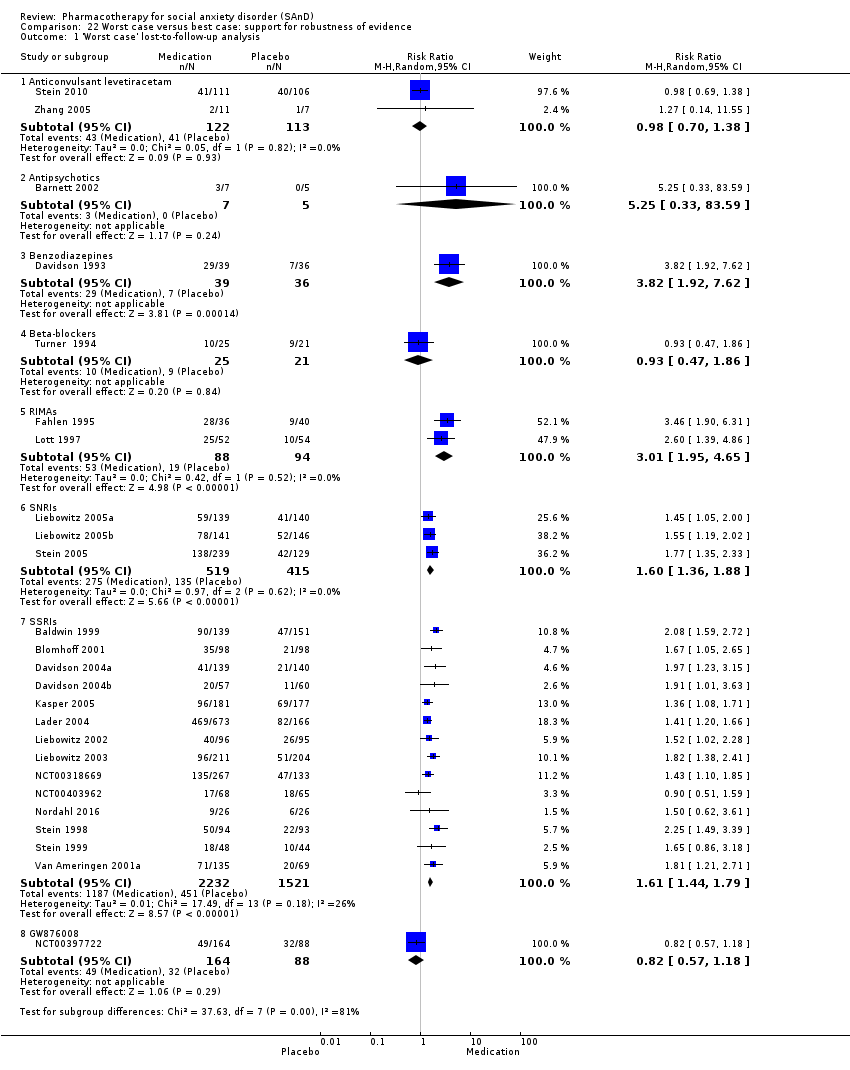 Comparison 22 Worst case versus best case: support for robustness of evidence, Outcome 1 'Worst case' lost‐to‐follow‐up analysis. | ||||
| 1.1 Anticonvulsant levetiracetam | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.70, 1.38] |
| 1.2 Antipsychotics | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 5.25 [0.33, 83.59] |
| 1.3 Benzodiazepines | 1 | 75 | Risk Ratio (M‐H, Random, 95% CI) | 3.82 [1.92, 7.62] |
| 1.4 Beta‐blockers | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.47, 1.86] |
| 1.5 RIMAs | 2 | 182 | Risk Ratio (M‐H, Random, 95% CI) | 3.01 [1.95, 4.65] |
| 1.6 SNRIs | 3 | 934 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.36, 1.88] |
| 1.7 SSRIs | 14 | 3753 | Risk Ratio (M‐H, Random, 95% CI) | 1.61 [1.44, 1.79] |
| 1.8 GW876008 | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.57, 1.18] |
| 2 'Best case' lost‐to‐follow‐up analysis Show forest plot | 25 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 22.2  Comparison 22 Worst case versus best case: support for robustness of evidence, Outcome 2 'Best case' lost‐to‐follow‐up analysis. | ||||
| 2.1 Anticonvulsant levetiracetam | 2 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.70, 1.37] |
| 2.2 Antipsychotics | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.45, 108.26] |
| 2.3 Benzodiazepines | 1 | 72 | Risk Ratio (M‐H, Random, 95% CI) | 3.92 [1.98, 7.77] |
| 2.4 Beta‐blockers | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.55, 2.05] |
| 2.5 RIMAs | 2 | 177 | Risk Ratio (M‐H, Random, 95% CI) | 2.97 [1.93, 4.58] |
| 2.6 SNRIs | 3 | 912 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [1.39, 1.91] |
| 2.7 SSRIs | 14 | 3577 | Risk Ratio (M‐H, Random, 95% CI) | 1.64 [1.46, 1.85] |
| 2.8 GW876008 | 1 | 250 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.19] |
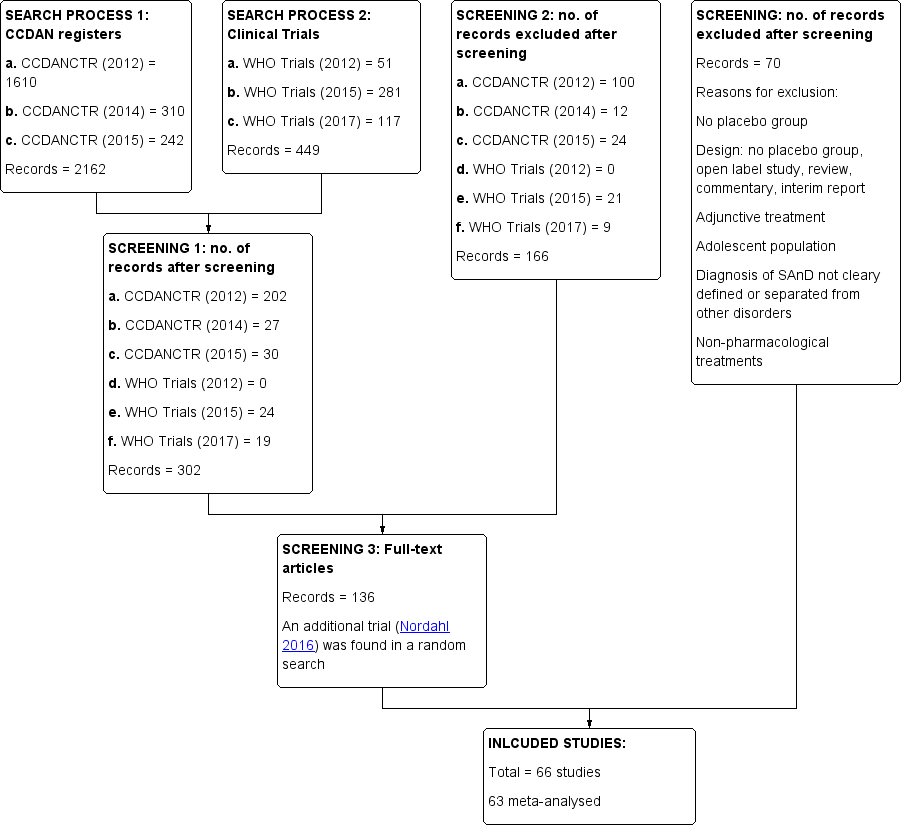
Study flow diagram.
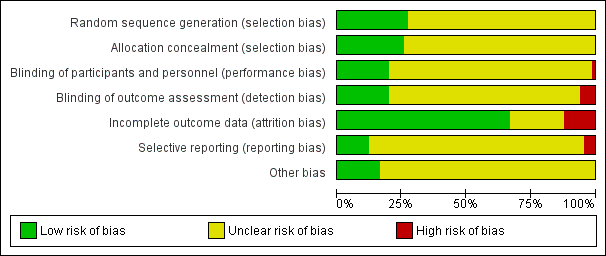
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
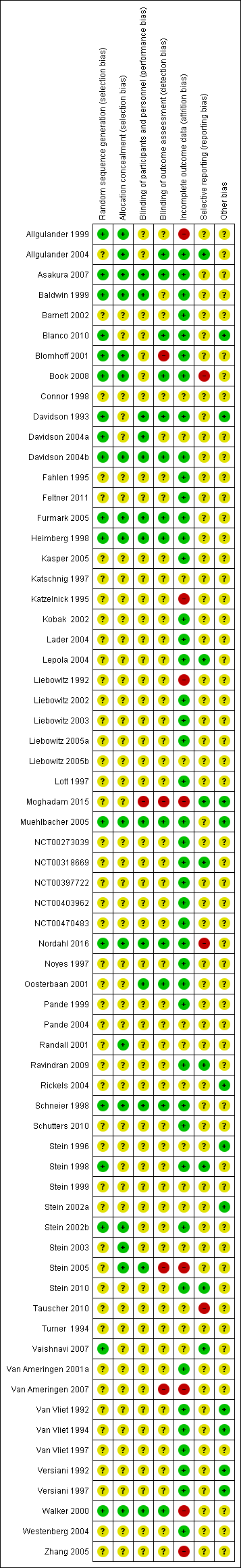
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Contour enhanced funnel plot for treatment response on the CGI‐I for all medication trials
Filled circles = trial effect estimates; empty circles = trim and fill generated missing study estimates
Contours in light‐gray, gray and dark gray correspond to increasing stringent statistical boundaries (alpha = 0.1, 0.05, and 0.01, respectively). White corresponds to regions of statistical non‐significance.

Contour enhanced funnel plot for treatment response on the CGI‐I for SSRI trials
Filled circles = trial effect estimates; empty circles = trim and fill generated missing study estimates
Contours in light‐gray, gray and dark gray correspond to increasing stringent statistical boundaries (alpha = 0.1, 0.05, and 0.01, respectively). White corresponds to regions of statistical non‐significance.

Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).

Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 2 Adverse events (acute phase).
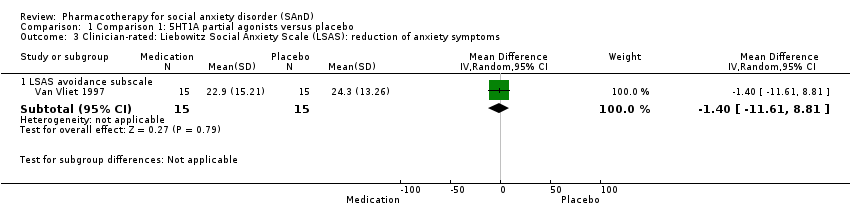
Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.

Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).

Comparison 1 Comparison 1: 5HT1A partial agonists versus placebo, Outcome 5 All‐cause dropouts (acute phase).

Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
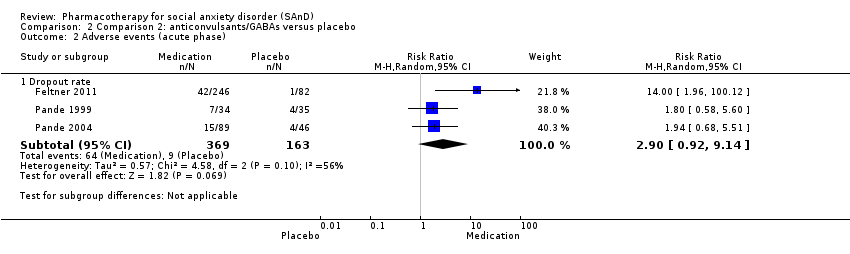
Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 2 Adverse events (acute phase).
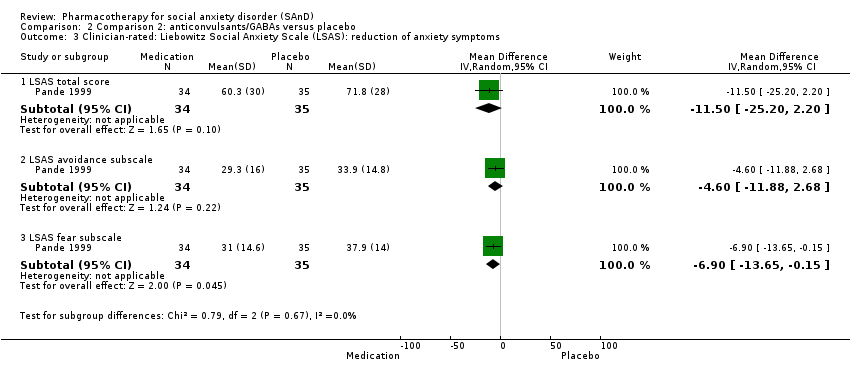
Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.

Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
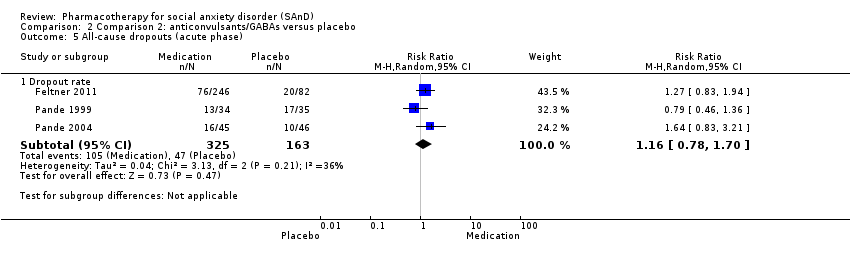
Comparison 2 Comparison 2: anticonvulsants/GABAs versus placebo, Outcome 5 All‐cause dropouts (acute phase).

Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
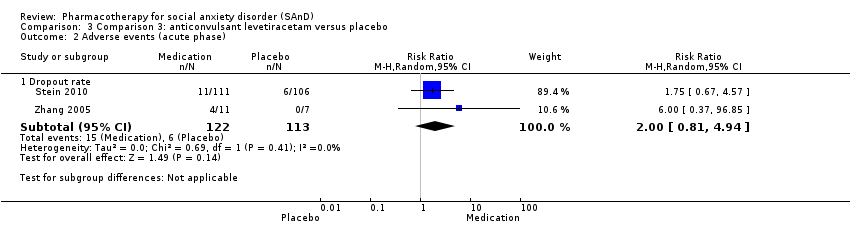
Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 2 Adverse events (acute phase).
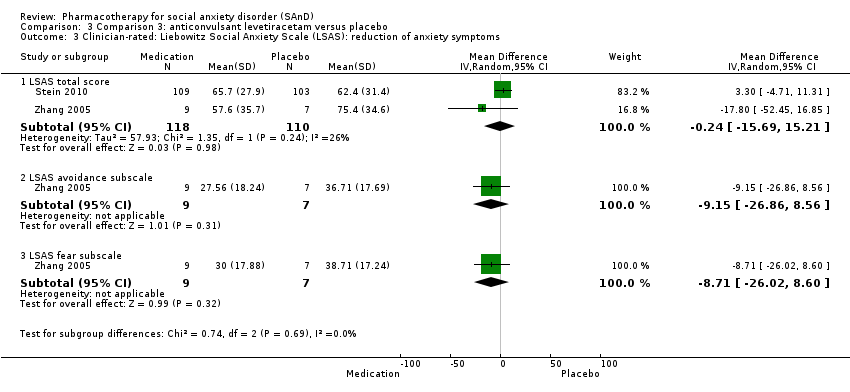
Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
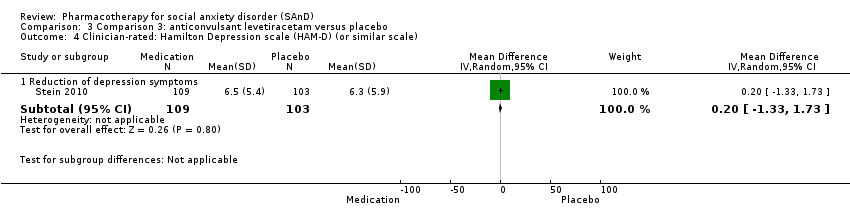
Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
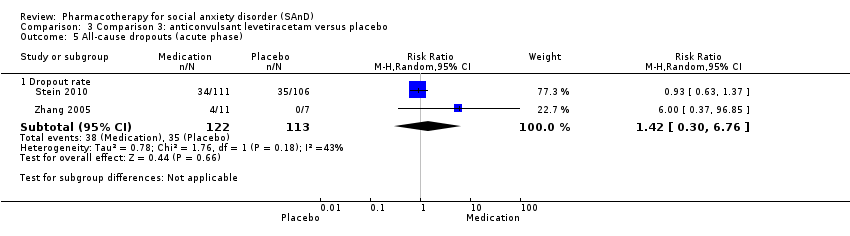
Comparison 3 Comparison 3: anticonvulsant levetiracetam versus placebo, Outcome 5 All‐cause dropouts (acute phase).

Comparison 4 Comparison 4: antipsychotics versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
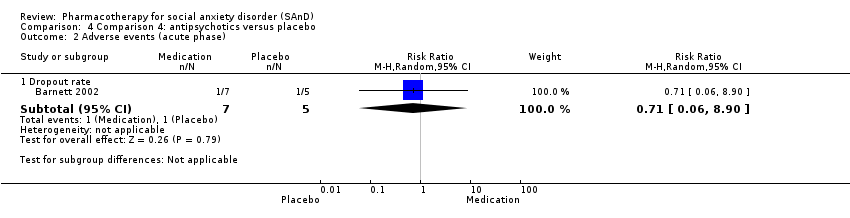
Comparison 4 Comparison 4: antipsychotics versus placebo, Outcome 2 Adverse events (acute phase).
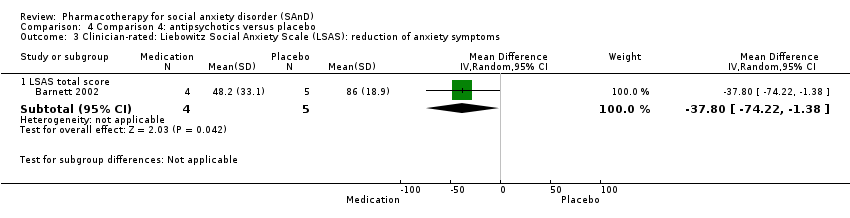
Comparison 4 Comparison 4: antipsychotics versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.

Comparison 4 Comparison 4: antipsychotics versus placebo, Outcome 4 All‐cause dropouts (acute phase).

Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).

Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 2 Relapse rate.

Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 3 Adverse events (acute phase).
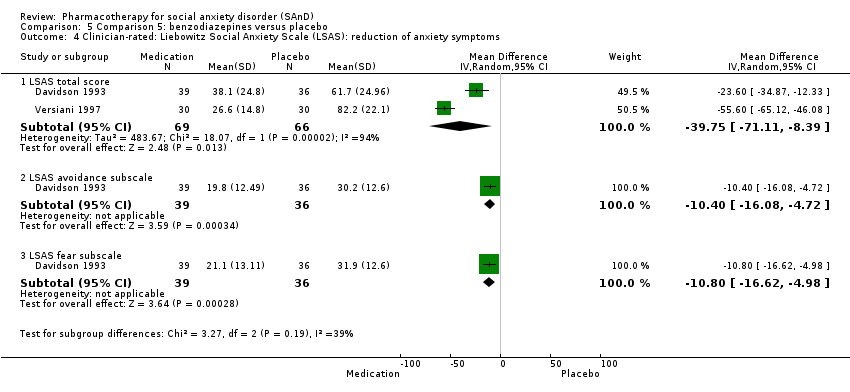
Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 4 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
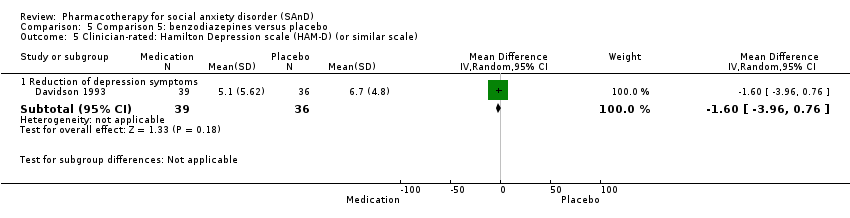
Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 5 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
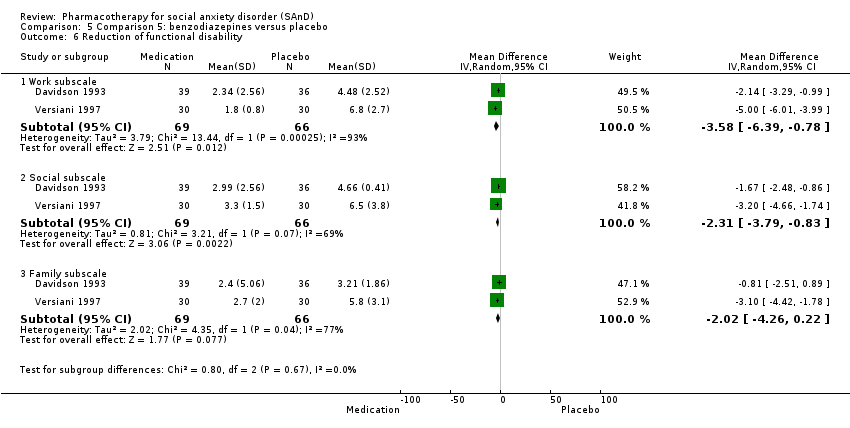
Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 6 Reduction of functional disability.

Comparison 5 Comparison 5: benzodiazepines versus placebo, Outcome 7 All‐cause dropouts (acute phase).

Comparison 6 Comparison 6: beta‐blockers versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).

Comparison 6 Comparison 6: beta‐blockers versus placebo, Outcome 2 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).
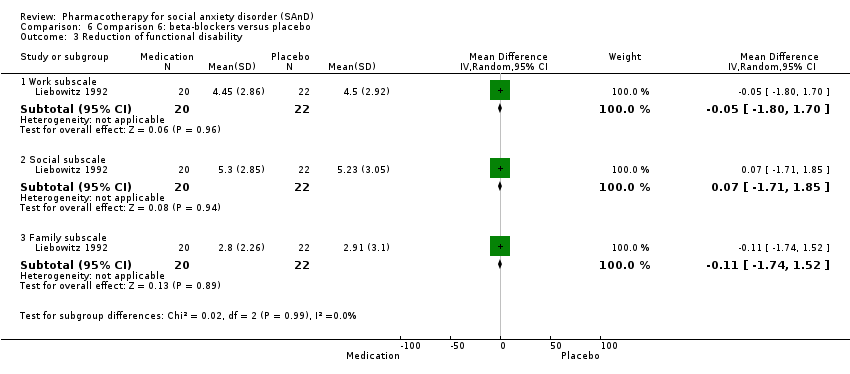
Comparison 6 Comparison 6: beta‐blockers versus placebo, Outcome 3 Reduction of functional disability.

Comparison 6 Comparison 6: beta‐blockers versus placebo, Outcome 4 All‐cause dropouts (acute phase).
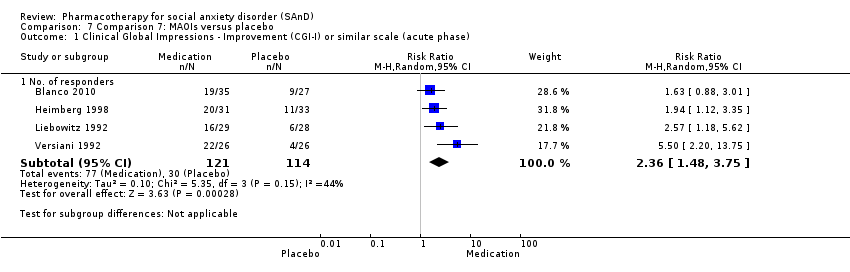
Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).

Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 2 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.
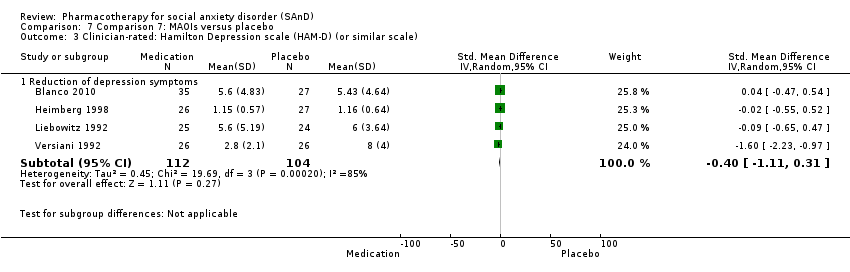
Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 3 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).

Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 4 Reduction of functional disability.

Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 5 All‐cause dropouts (acute phase).
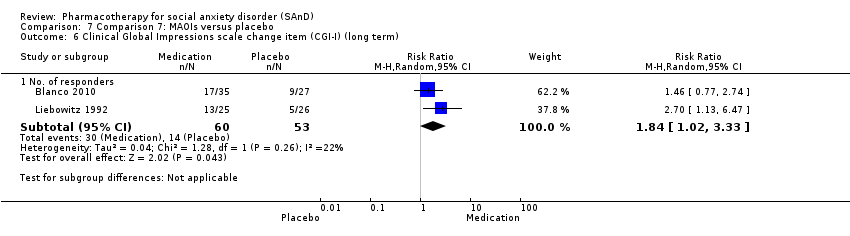
Comparison 7 Comparison 7: MAOIs versus placebo, Outcome 6 Clinical Global Impressions scale change item (CGI‐I) (long term).

Comparison 8 Comparison 8: NARIs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).

Comparison 8 Comparison 8: NARIs versus placebo, Outcome 2 Adverse events (acute phase).

Comparison 8 Comparison 8: NARIs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.

Comparison 8 Comparison 8: NARIs versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).

Comparison 8 Comparison 8: NARIs versus placebo, Outcome 5 All‐cause dropouts (acute phase).

Comparison 9 Comparison 9: NaSSAs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).

Comparison 9 Comparison 9: NaSSAs versus placebo, Outcome 2 Adverse events (acute phase).
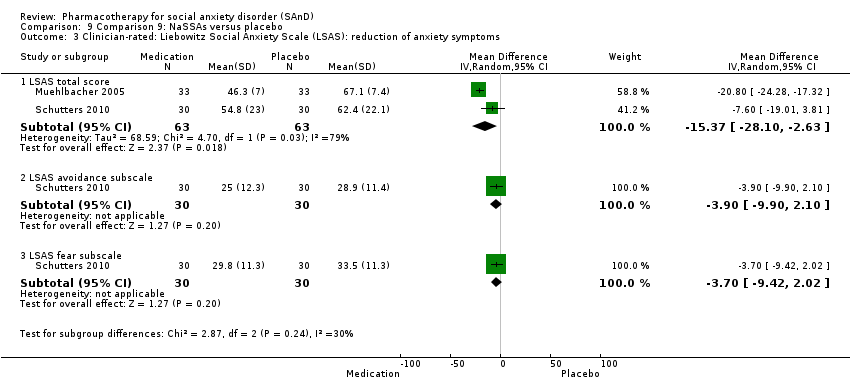
Comparison 9 Comparison 9: NaSSAs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.

Comparison 9 Comparison 9: NaSSAs versus placebo, Outcome 4 All‐cause dropouts (acute phase).

Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
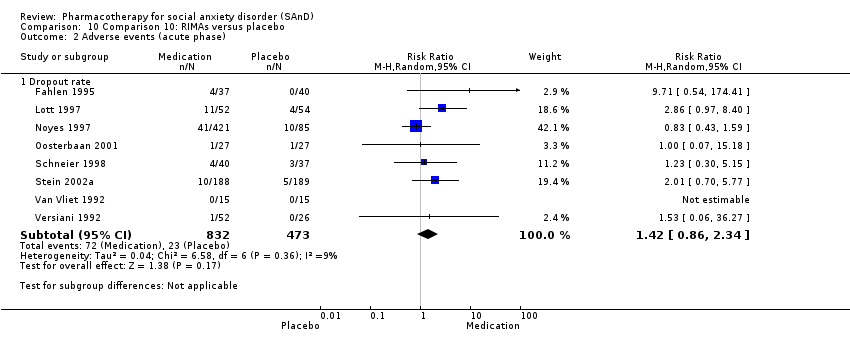
Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 2 Adverse events (acute phase).

Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.

Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).

Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 5 Reduction of functional disability.

Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 6 All‐cause dropouts (acute phase).
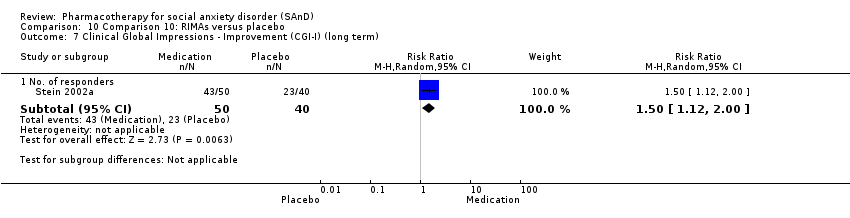
Comparison 10 Comparison 10: RIMAs versus placebo, Outcome 7 Clinical Global Impressions ‐ Improvement (CGI‐I) (long term).

Comparison 11 Comparison 11: SARIs versus placebo, Outcome 1 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.

Comparison 11 Comparison 11: SARIs versus placebo, Outcome 2 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).

Comparison 11 Comparison 11: SARIs versus placebo, Outcome 3 Reduction of functional disability.

Comparison 11 Comparison 11: SARIs versus placebo, Outcome 4 All‐cause dropouts (acute phase).

Comparison 12 Comparison 12: SNRIs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).

Comparison 12 Comparison 12: SNRIs versus placebo, Outcome 2 Adverse events (acute phase).
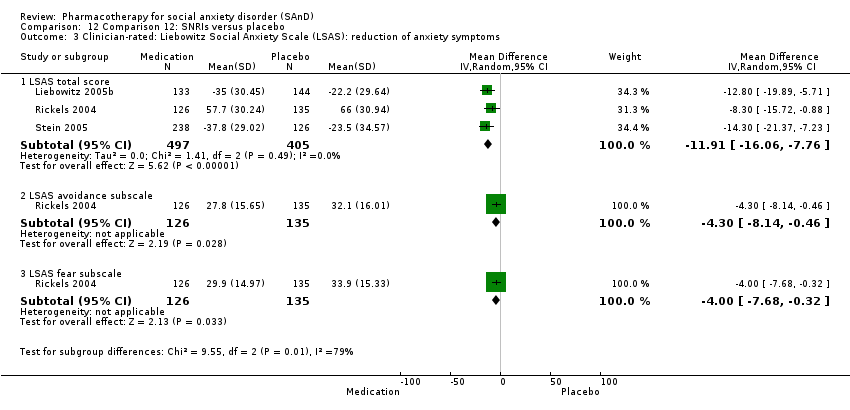
Comparison 12 Comparison 12: SNRIs versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.

Comparison 12 Comparison 12: SNRIs versus placebo, Outcome 4 All‐cause dropouts (acute phase).
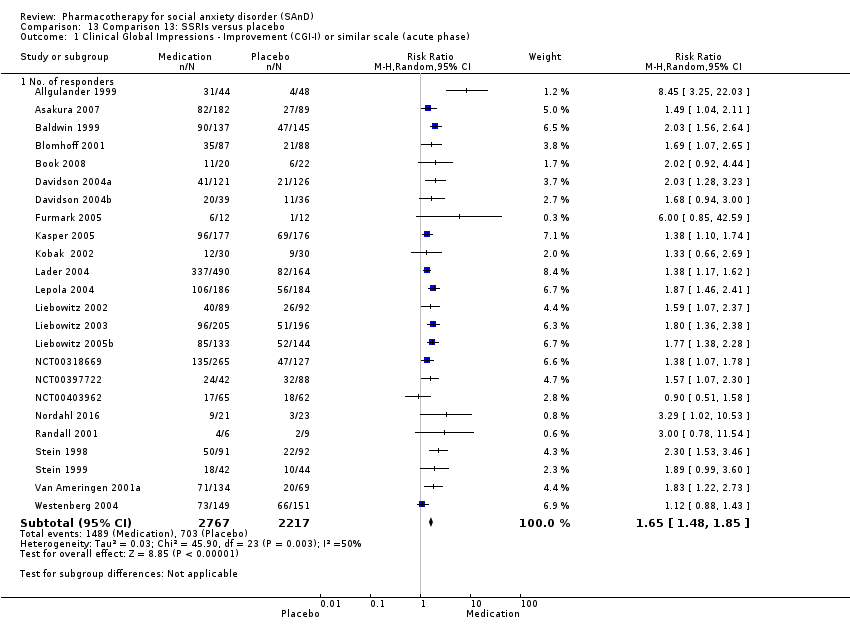
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
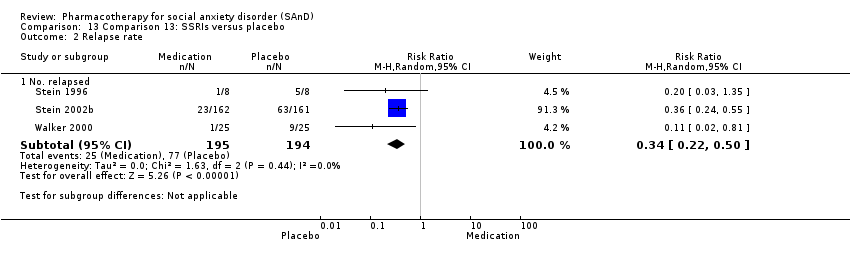
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 2 Relapse rate.

Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 3 Adverse events (acute phase).

Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 4 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.

Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 5 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale).

Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 6 Reduction of functional disability.

Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 7 All‐cause dropouts (acute phase).

Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 8 Clinical Global Impressions scale change item (CGI‐I) (long term).

Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 9 Adverse events (long term).
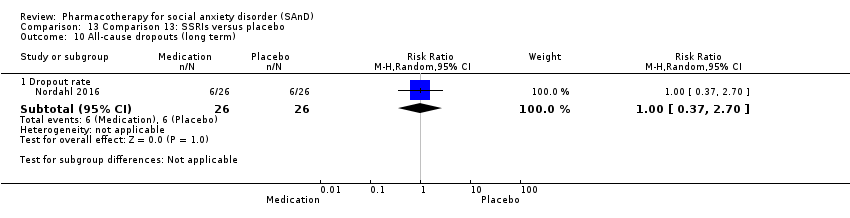
Comparison 13 Comparison 13: SSRIs versus placebo, Outcome 10 All‐cause dropouts (long term).
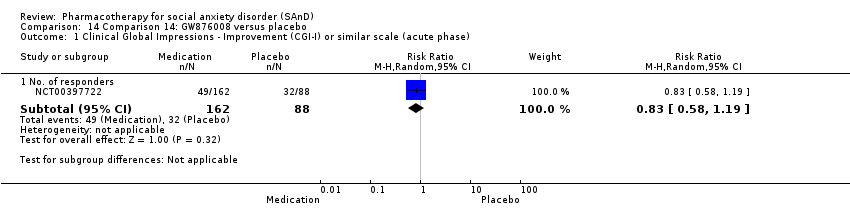
Comparison 14 Comparison 14: GW876008 versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).
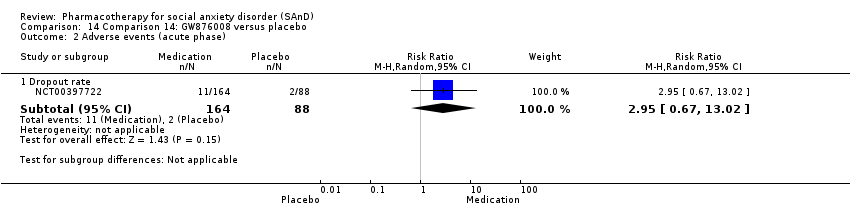
Comparison 14 Comparison 14: GW876008 versus placebo, Outcome 2 Adverse events (acute phase).

Comparison 14 Comparison 14: GW876008 versus placebo, Outcome 3 All‐cause dropouts (acute phase).
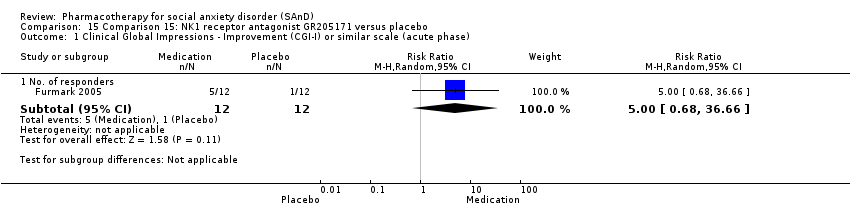
Comparison 15 Comparison 15: NK1 receptor antagonist GR205171 versus placebo, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase).

Comparison 15 Comparison 15: NK1 receptor antagonist GR205171 versus placebo, Outcome 2 Adverse events (acute phase).

Comparison 15 Comparison 15: NK1 receptor antagonist GR205171 versus placebo, Outcome 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.

Comparison 15 Comparison 15: NK1 receptor antagonist GR205171 versus placebo, Outcome 4 All‐cause dropouts (acute phase).

Comparison 16 Comparison 16: LY686017 versus placebo, Outcome 1 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms.

Comparison 17 Comparison 17: total effect of medication compared to placebo for the treatment of SAnD, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar) scale: no. of responders (acute phase).
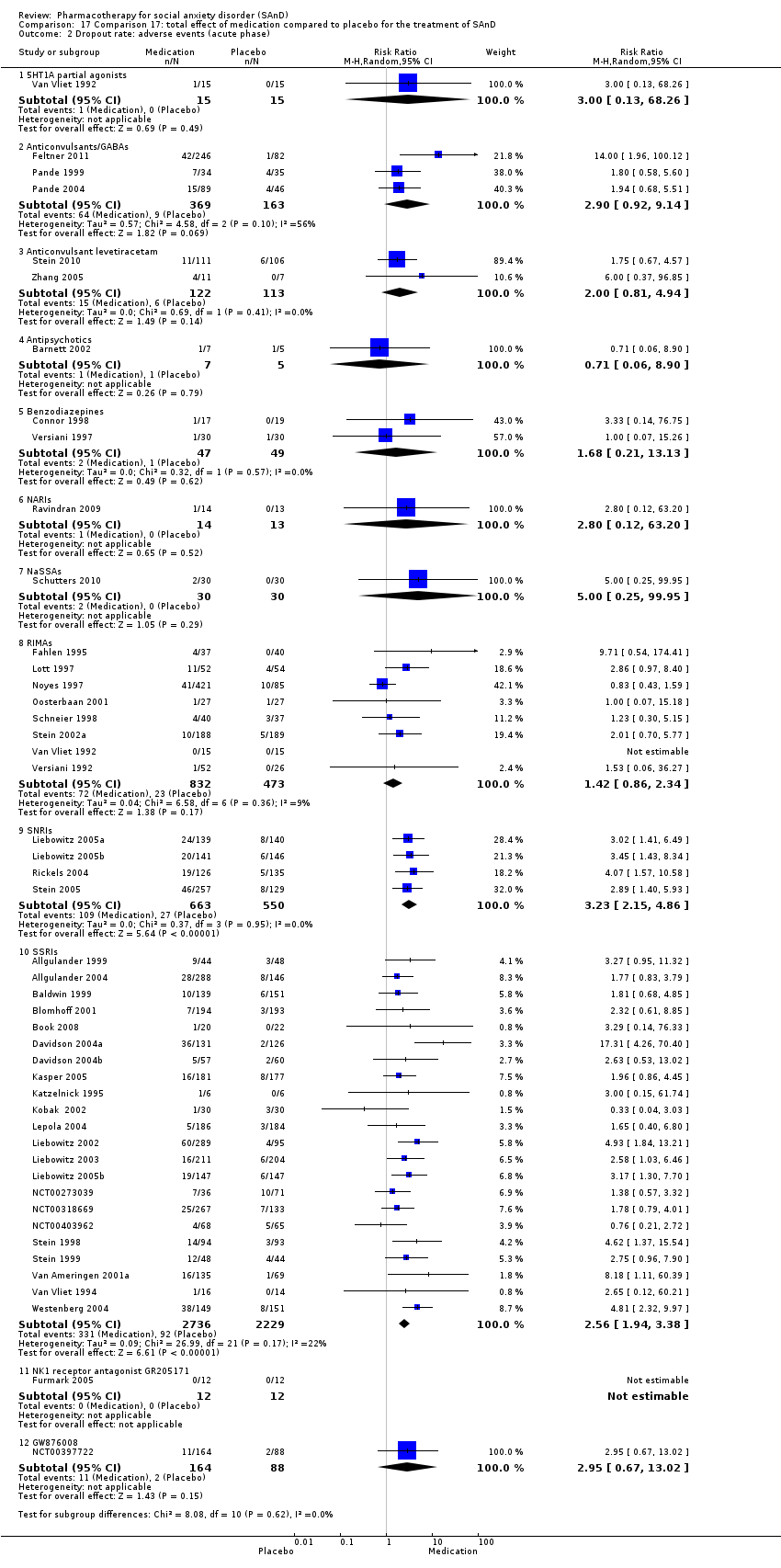
Comparison 17 Comparison 17: total effect of medication compared to placebo for the treatment of SAnD, Outcome 2 Dropout rate: adverse events (acute phase).

Comparison 17 Comparison 17: total effect of medication compared to placebo for the treatment of SAnD, Outcome 3 Dropout rate: all‐cause dropouts (acute phase).

Comparison 18 Subgroup analysis: multicentre versus single‐centre trials, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase).
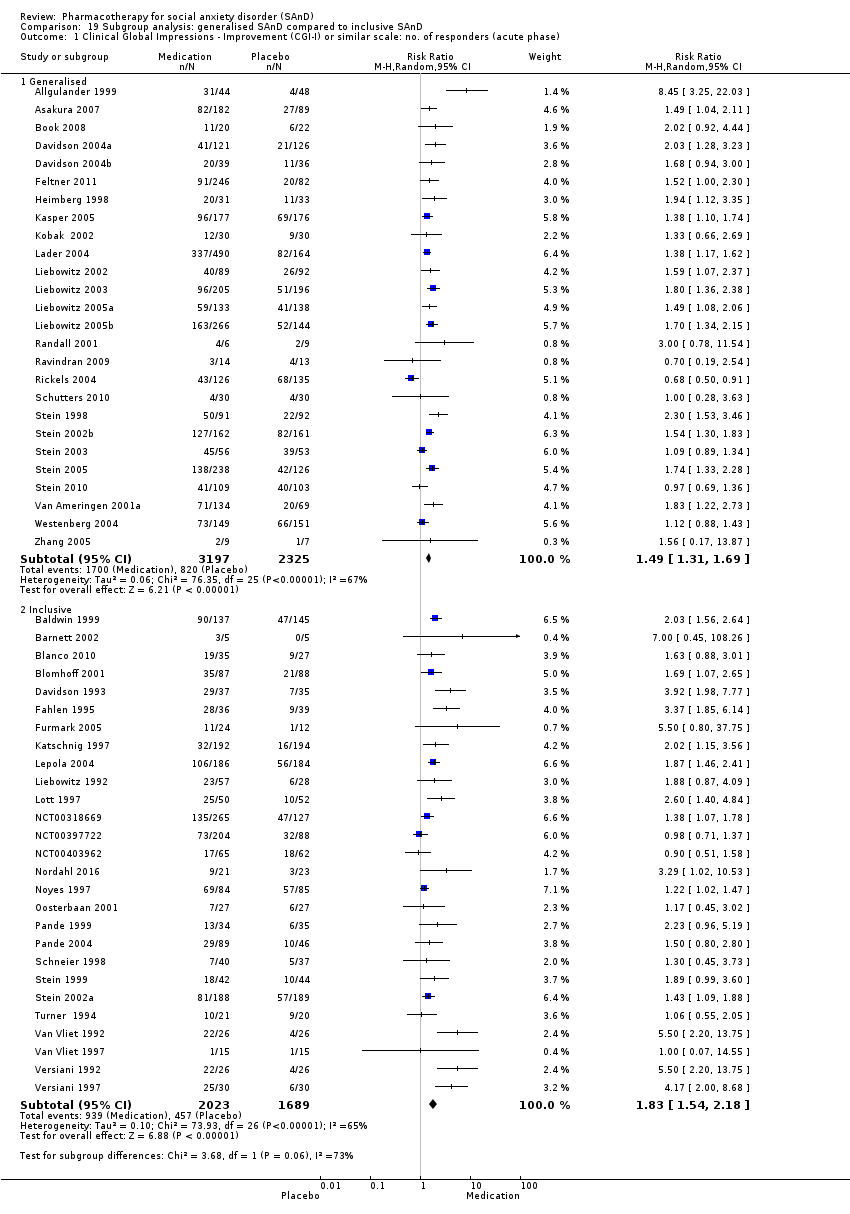
Comparison 19 Subgroup analysis: generalised SAnD compared to inclusive SAnD, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase).

Comparison 20 Subgroup analysis: industry funding compared to no industry funding, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase).

Comparison 21 Subgroup analysis: trials that included MDD compared to no MDD, Outcome 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase).
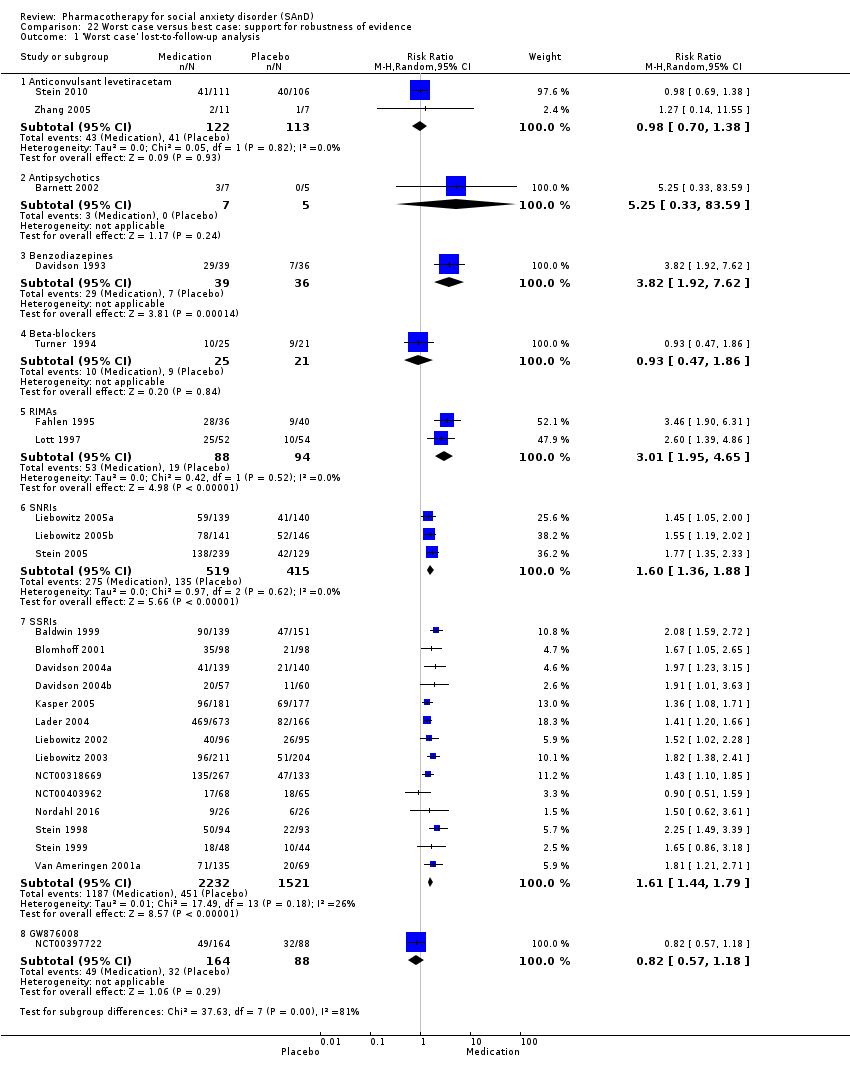
Comparison 22 Worst case versus best case: support for robustness of evidence, Outcome 1 'Worst case' lost‐to‐follow‐up analysis.

Comparison 22 Worst case versus best case: support for robustness of evidence, Outcome 2 'Best case' lost‐to‐follow‐up analysis.
| Comparison 1: 5HT1A partial agonists versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With 5HT1A partial agonists | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.00 | 30 | ⊕⊝⊝⊝ | There was no evidence of an effect on the number of participants in the 5HT1A partial agonist group compared to the placebo group who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 1.00). | |
| 67 per 1000 | 67 per 1000 | |||||
| Moderate | ||||||
| 67 per 1000 | 67 per 1000 | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 3.00 | 30 | ⊕⊝⊝⊝ | Dropout rates due to adverse events were low in the 5HT1A partial agonist group (1/30, 3%). No participants withdrew from the placebo group. | |
| 0 per 1000 | 0 per 1000 | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 24.3 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 1.4 points lower (11.61 lower to 8.81 higher) | 30 | ⊕⊝⊝⊝ | The mean LSAS avoidance anxiety score for the 5HT1A partial agonist intervention group was 22.9 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 2: anticonvulsants/GABAs versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With anticonvulsants/GABAs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.60 | 532 | ⊕⊕⊕⊝ | There was evidence of benefit on the number of participants with SAnD who responded to treatment (P = 0.004). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the anticonvulsant/GABA groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 221 per 1000 | 353 per 1000 | |||||
| Moderate | ||||||
| 217 per 1000 | 347 per 1000 | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 2.90 | 532 | ⊕⊝⊝⊝ | Dropout rates due to adverse events were high in the anticonvulsant/GABA groups (64/369, 17%) relative to placebo (9/163, 6%). | |
| 55 per 1000 | 160 per 1000 | |||||
| Moderate | ||||||
| 87 per 1000 | 252 per 1000 | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score for the control group was 71.8 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 11.50 lower | 69 | ⊕⊕⊝⊝ | The mean LSAS total anxiety score for the anticonvulsant/GABA intervention group was 60.3 which suggests 'moderate' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 33.9 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 4.60 points lower (11.88 lower to 2.68 higher) | 69 | ⊕⊕⊝⊝ | The mean LSAS avoidance anxiety score for the anticonvulsant/GABA intervention group was 29.3 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score for the control group was 37.9 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 6.90 points lower (13.65 lower to 0.15 lower) | 69 | ⊕⊕⊝⊝ | The mean LSAS fear anxiety score for the anticonvulsant/GABA intervention group was 31.0 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 3: levetiracetam versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | Wtih levetiracetam | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 0.98 | 228 | ⊕⊕⊕⊝ | There was no evidence of an effect on the number of participants in the anticonvulsant levetiracetam groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.90). | |
| 373 per 1000 | 365 per 1000 | |||||
| Moderate | ||||||
| 266 per 1000 | 261 per 1000 | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 2.00 | 235 | ⊕⊝⊝⊝ | The proportion of dropouts due to adverse events was high in participants receiving the anticonvulsant levetiracetam (15/122, 12%) relative to placebo (6/113, 5%). There was no evidence of a difference between the number of participants that dropped out due to adverse events (P = 0.14). | |
| 53 per 1000 | 106 per 1000 | |||||
| Moderate | ||||||
| 28 per 1000 | 56 per 1000 | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from 62.4 to 75.4 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 0.24 points lower (15.69 lower to 15.21 higher) | 228 | ⊕⊝⊝⊝ | The mean LSAS total anxiety score for the anticonvulsant levetiracetam intervention groups ranged from 55 to 65 which suggests 'moderate' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 36.71 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 9.15 points lower | 16 | ⊕⊝⊝⊝ | The mean LSAS avoidance anxiety score for the anticonvulsant levetiracetam intervention group was 27.56 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score for the control group was 38.71 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was | 16 | ⊕⊝⊝⊝ | The mean LSAS fear anxiety score for the anticonvulsant levetiracetam intervention group was 30.0 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 4: antipsychotics versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With antipsychotics | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 7.00 | 10 | ⊕⊝⊝⊝ | There was no evidence of an effect on the number of participants in the antipsychotic group compared to the placebo group who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.16). | |
| 0 per 1000 | 0 per 1000 | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 0.71 | 12 | ⊕⊝⊝⊝ | There was no difference on the number of participants who withdrew due to adverse events (P = 0.79). | |
| 200 per 1000 | 142 per 1000 | |||||
| Moderate | ||||||
| 200 per 1000 | 142 per 1000 | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score for the control group was 86.0 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 37.80 points lower | — | 9 | ⊕⊝⊝⊝ | The mean LSAS total anxiety score for the antipsychotic intervention group was 48.2 which suggests 'low' social phobia. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 5: benzodiazepines versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With benzodiazepines | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 4.03 | 132 | ⊕⊕⊝⊝ | There was evidence of a large effect on the number of participants with SAnD who responded to treatment (P < 0.00001). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the benzodiazepine groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 200 per 1000 | 806 per 1000 | |||||
| Moderate | ||||||
| 200 per 1000 | 806 per 1000 | |||||
| Relapse rate ‐ no. relapsed | Study population | RR 0.12 | 36 | ⊕⊕⊝⊝ | There was no evidence on the number of treatment responders who subsequently relapsed (P = 0.15). | |
| 211 per 1000 | 25 per 1000 | |||||
| Moderate | ||||||
| 211 per 1000 | 25 per 1000 | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 1.68 | 96 | ⊕⊝⊝⊝ | Dropout rates due to adverse events were low in the benzodiazepine groups (2/47; 4%) and did not differ from the placebo groups (P = 0.62). | |
| 20 per 1000 | 34 per 1000 | |||||
| Moderate | ||||||
| 17 per 1000 | 29 per 1000 | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from 61.7 to 82.2 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 39.75 points lower | 135 | ⊕⊝⊝⊝ | The mean LSAS total anxiety score for the benzodiazepine intervention groups ranged from 26.6 to 38.1 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 30.2 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 10.40 points lower (16.08 lower to 4.72 lower) | 75 | ⊕⊕⊝⊝ | The mean LSAS avoidance anxiety score for the benzodiazepine intervention group was 19.8 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score for the control group was 31.9 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 10.80 points lower (16.62 lower to 4.98 lower) | 75 | ⊕⊕⊝⊝ | The mean LSAS fear anxiety score for the benzodiazepine intervention group was 21.1 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 6: beta‐blockers versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With beta‐blockers | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.09 | 97 | ⊕⊝⊝⊝ | There was no evidence of an effect on the number of participants in the beta‐blocker groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.75). | |
| 312 per 1000 | 341 per 1000 | |||||
| Moderate | ||||||
| 332 per 1000 | 362 per 1000 | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 7: MAOIs versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With MAOIs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 2.36 | 235 | ⊕⊕⊝⊝ | There was evidence of an effect on the number of participants with SAnD who responded to treatment (P = 0.0003). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the MAOI groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 263 per 1000 | 621 per 1000 | |||||
| Moderate | ||||||
| 274 per 1000 | 647 per 1000 | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from 4.05 to 63.29 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 16.39 points lower | 218 | ⊕⊝⊝⊝ | The mean LSAS total anxiety score for the MAOI intervention groups ranged from 14.0 to 47.8 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 24.54 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 5.42 points lower | 51 | ⊕⊝⊝⊝ | The mean LSAS avoidance anxiety score for the MAOI intervention group was 19.12 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score for the control group was 27.31 | The mean reduction of of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 5.23 points lower (13.97 lower to 3.51 higher) | 51 | ⊕⊝⊝⊝ | The mean LSAS fear anxiety score for the MAOI intervention group was 22.08 which suggests 'low' social phobia. | |
| Clinical Global Impressions scale change item (CGI‐I):no. of responders (long term) Post‐treatment: 6 months | Study population | RR 1.84 | 113 | ⊕⊝⊝⊝ | There was data indicating greater efficacy over the long term on the number of participants with SAnD who responded to treatment (P = 0.04). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the MAOI groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 264 per 1000 | 486 per 1000 | |||||
| Moderate | ||||||
| 263 per 1000 | 484 per 1000 | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 8: NARIs versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With NARIs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 0.70 | 27 | ⊕⊝⊝⊝ | There was no evidence of an effect on the number of participants in the NARI group compared to the placebo group who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.58). | |
| 308 per 1000 | 215 per 1000 | |||||
| Moderate | ||||||
| 308 per 1000 | 216 per 1000 | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 2.80 | 27 | ⊕⊝⊝⊝ | Only 1 of 14 (7%) participants withdrew during 10 weeks of NARI treatment. There was no evidence for a difference in the number of participants who withdrew due to adverse events (P = 0.52). | |
| 0 per 1000 | 0 per 1000 | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score for the control group was 70.8 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 2.60 points higher (15.43 lower to 20.63 higher) | 26 | ⊕⊝⊝⊝ | The mean LSAS total anxiety score for the NARI intervention group was 73.4 which suggests 'marked' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 9: NaSSAs compared to placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With NaSSAs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.00 | 60 | ⊕⊝⊝⊝ | An equal number of participants (4/30, 13%) responded to treatment in both the NaSSA and placebo groups, however this difference was not statistically significant (P = 1.00). | |
| 133 per 1000 | 133 per 1000 | |||||
| Moderate | ||||||
| 133 per 1000 | 133 per 1000 | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 5.00 | 60 | ⊕⊝⊝⊝ | A small proportion of participants receiving NaSSAs dropped out due to adverse events (2/30, 7%). Although, the difference was not statistically significant (P = 0.29). | |
| 0 per 1000 | 0 per 1000 | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from 62.4 to 67.1 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 15.37 points lower | 126 | ⊕⊝⊝⊝ | The mean LSAS total anxiety score for the NaSSA intervention groups ranged from 46.3 to 54.8 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 28.9 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 3.90 points lower (9.90 lower to 2.10 higher) | 60 | ⊕⊕⊝⊝ | The mean LSAS avoidance anxiety score for the NaSSA intervention group was 25.0 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score for the control group was 33.5 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 3.70 points lower (9.42 lower to 2.02 higher) | 60 | ⊕⊕⊝⊝ | The mean LSAS fear anxiety score for the NaSSA intervention group was 29.8 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 10: RIMAs versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With RIMAs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.83 | 1270 | ⊕⊕⊝⊝ | There was evidence of benefit on the number of participants with SAnD who responded to treatment (P = 0.0003). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the RIMA groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 254 per 1000 | 465 per 1000 | |||||
| Moderate | ||||||
| 207 per 1000 | 379 per 1000 | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 1.42 | 1305 | ⊕⊝⊝⊝ | Dropout rates due to adverse events were low in the RIMA groups and equivalent to those observed in the placebo groups (72/83; 9%). | |
| 49 per 1000 | 69 per 1000 | |||||
| Moderate | ||||||
| 32 per 1000 | 45 per 1000 | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from 54.4 to 79.3 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 12.17 points lower (23.51 lower to 0.84 lower) | 1163 | ⊕⊝⊝⊝ | The mean LSAS total anxiety score for the RIMA intervention groups ranged from 27.0 to 62.6 which suggests low to 'moderate' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score ranged across control groups from 23.3 to 39.2 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was | 695 | ⊕⊕⊝⊝ | The mean LSAS avoidance anxiety score for the RIMA intervention groups ranged from 15.3 to 30.7 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score ranged across control groups from 29.4 to 40.1 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 5.40 points lower (8.92 lower to 1.88 lower) | 724 | ⊕⊝⊝⊝ | The mean LSAS fear anxiety score for the RIMA intervention groups ranged from 19.1 to 33.3 which suggests 'low' social phobia. | |
| Clinical Global Impressions scale change item (CGI‐I): no. of responders (long term) Post‐treatment: 1‐15 months | Study population | RR 1.50 | 90 | ⊕⊕⊕⊝ | There was evidence of a long‐term effect on treatment efficacy in participants with SAnD who responded to treatment (P = 0.006). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the RIMA group compared to the placebo group who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 575 per 1000 | 862 per 1000 | |||||
| Moderate | ||||||
| 575 per 1000 | 862 per 1000 | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 11: SARIs versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With SARIs | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score for the control group was 71.2 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 6.10 points lower (16.55 lower to 4.35 higher) | 102 | ⊕⊝⊝⊝ | The mean LSAS total anxiety score for the SARI intervention group was 65.1 which suggests 'marked' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 12: SNRIs compared to placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With SNRIs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.30 | 1173 | ⊕⊕⊝⊝ | There was no evidence of an effect on the number of participants in the SNRI groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.22). | |
| 374 per 1000 | 486 per 1000 | |||||
| Moderate | ||||||
| 347 per 1000 | 451 per 1000 | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 3.23 | 1213 | ⊕⊕⊕⊝ | The proportion of dropouts due to adverse events was more than three times as high in participants receiving SNRIs (109/663, 16%) compared to placebo (27/550, 5%), a statistically significant difference (P < 0.00001). | |
| 49 per 1000 | 159 per 1000 | |||||
| Moderate | ||||||
| 49 per 1000 | 158 per 1000 | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from ‐22.2 to 66.0 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 11.91 points lower (16.06 lower to 7.76 lower) | 902 | ⊕⊕⊕⊝ | The mean LSAS total anxiety score for the SNRI intervention groups ranged from ‐35.0 to 57.7 which suggests 'low' to 'moderate' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score for the control group was 32.1 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 4.30 points lower (8.14 lower to 0.46 lower) | 261 | ⊕⊕⊝⊝ | The mean LSAS avoidance anxiety score for the SNRI intervention group was 27.8 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score for the control group was 33.9 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 4.00 points lower (7.68 lower to 0.32 lower) | 261 | ⊕⊕⊝⊝ | The mean LSAS fear anxiety score for the SNRI intervention group was 29.9 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 13: SSRIs versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| With placebo | With SSRIs | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 1.65 | 4984 | ⊕⊝⊝⊝ | There was evidence of benefit on the number of participants with SAnD who responded to treatment (P < 0.00001). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the SSRI groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 317 per 1000 | 523 per 1000 | |||||
| Moderate | ||||||
| 290 per 1000 | 476 per 1000 | |||||
| Relapse rate ‐ no. relapsed | Study population | RR 0.34 | 389 | ⊕⊕⊕⊝ | There was evidence that SSRIs prevented relapse compared to placebo (P < 0.00001). | |
| 397 per 1000 | 135 per 1000 | |||||
| Moderate | ||||||
| 391 per 1000 | 133 per 1000 | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 2.59 | 5131 | ⊕⊕⊝⊝ | The proportion of dropouts due to adverse events was approximately three times higher amongst participants receiving SSRIs compared to placebo ((P < 0.00001). | |
| 40 per 1000 | 105 per 1000 | |||||
| Moderate | ||||||
| 37 per 1000 | 96 per 1000 | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score ranged across control groups from ‐7.8 to 69.88 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS total score) in the intervention groups was 10.14 points lower (14.05 to 6.22 lower) | 1990 | ⊕⊕⊝⊝ | The mean LSAS total anxiety score for the SSRI intervention groups ranged from 14.7 to 60.3 which suggests 'low' to 'moderate' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS avoidance subscale | The mean anxiety score ranged across control groups from 24.2 to 34.8 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS avoidance) in the intervention groups was 7.01 points lower (10.21 to 3.80 lower) | 1173 | ⊕⊕⊝⊝ | The mean LSAS avoidance anxiety score for the SSRI intervention groups ranged from 17.09 to 26.11 which suggests 'low' social phobia. | |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS fear subscale | The mean anxiety score ranged across control groups from 29.83 to 37.4 | The mean reduction of anxiety symptoms (clinician‐rated: LSAS fear) in the intervention groups was 7.28 points lower (10.86 to 3.71 lower) | 1173 | ⊕⊕⊝⊝ | The mean LSAS fear anxiety score for the SSRI intervention groups ranged from 19.84 to 32.79 which suggests 'low' social phobia. | |
| Clinical Global Impressions scale change item (CGI‐I): no. of responders (long term) Post‐treatment: 1‐4 months | Study population | RR 1.27 | 806 | ⊕⊕⊝⊝ | There was evidence for a response to long‐term treatment compared to placebo in participants with SAnD (P = 0.007). A RR score greater than 1 and 95% CI that does not overlap with 1 indicates that there were a statistically significantly greater number of people in the SSRI groups compared to the placebo groups who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale. | |
| 579 per 1000 | 735 per 1000 | |||||
| Moderate | ||||||
| 584 per 1000 | 742 per 1000 | |||||
| Dropouts due to adverse events (long term) Post‐treatment: 1‐4 months | Study population | RR 1.17 | 1274 | ⊕⊝⊝⊝ | We found no difference in dropout rates due to adverse events between the SSRI and control groups (P = 0.76). | |
| 52 per 1000 | 61 per 1000 | |||||
| Moderate | ||||||
| 50 per 1000 | 58 per 1000 | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| aDowngraded one level due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Comparison 14: GW876008 versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Comparison 14: GW876008 versus placebo | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 0.83 | 250 | ⊕⊕⊕⊝ | There was no evidence of an effect on the number of participants in the GW876008 group compared to the placebo group who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.32). | |
| 364 per 1000 | 302 per 1000 | |||||
| Moderate | ||||||
| 364 per 1000 | 302 per 1000 | |||||
| Dropouts due to adverse events (acute phase) | Study population | RR 2.95 | 252 | ⊕⊕⊝⊝ | The proportion of dropouts due to adverse events was approximately three times higher amongst participants receiving GW876008 (11/164, 7%) compared to placebo (2/88, 2%), though this difference was not statistically significant (P = 0.15). | |
| 23 per 1000 | 67 per 1000 | |||||
| Moderate | ||||||
| 23 per 1000 | 68 per 1000 | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| a Downgraded one level due to serious risk of bias (concerns with randomisation procedures). c Response is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar. | ||||||
| Comparison 15: NK1 receptor antagonist GR205171 versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Comparison 15: NK1 receptor antagonist GR205171 versus placebo | |||||
| Global Impressions scale change item (CGI‐I or similar): no. of responders (acute phase) | Study population | RR 5 | 24 | ⊕⊝⊝⊝ | There was no evidence of an effect on the number of participants in the NK1 receptor antagonist GR205171 group compared to the placebo group who responded 'Very Much Improved' or 'Much Improved' on the CGI‐I scale (P = 0.11). | |
| 83 per 1000 | 417 per 1000 | |||||
| Moderate | ||||||
| 83 per 1000 | 415 per 1000 | |||||
| Dropouts due to adverse events (acute phase) | See comment | See comment | Not estimable | 24 | ⊕⊕⊕⊝ | No participants withdrew due to adverse events. |
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score for the control group was 4.1 | The mean clinician‐rated: liebowitz social anxiety scale (lsas): reduction of anxiety symptoms ‐ lsas total score in the intervention groups was 0.5 points lower (1.35 lower to 0.35 higher) | 24 | ⊕⊝⊝⊝ | The mean LSAS total anxiety score for the NK1 receptor antagonist GR205171 intervention group was 3.6 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| a Downgraded one level due to serious risk of bias (concerns with randomisation procedures). c Response is defined as the number of participants with SAnD who responded to treatment, as assessed by the CGI‐I or similar. | ||||||
| Comparison 16: LY686017 versus placebo for social anxiety disorder (SAnD) | ||||||
| Patient or population: adults with SAnD | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Comparison 16: LY686017 versus placebo | |||||
| Reduction of of anxiety symptoms ‐ Clinician‐rated: LSAS total score | The mean anxiety score for the control group was ‐22.59 | The mean clinician‐rated: liebowitz social anxiety scale (lsas): reduction of anxiety symptoms ‐ lsas total score in the intervention groups was 1.8 points higher (6.92 lower to 10.52 higher) | 99 | ⊕⊝⊝⊝ | The mean LSAS total anxiety score for the LY686017 intervention group was ‐20.79 which suggests 'low' social phobia. | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| a Downgraded two levels due to serious risk of bias (concerns with randomisation procedures). | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 14.55] |
| 2 Adverse events (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 68.26] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS avoidance subscale | 1 | 30 | Mean Difference (IV, Random, 95% CI) | ‐1.40 [‐11.61, 8.81] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Reduction of depression symptoms | 1 | 27 | Mean Difference (IV, Random, 95% CI) | ‐0.60 [‐2.86, 1.66] |
| 5 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Dropout rate | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.55] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.16, 2.20] |
| 2 Adverse events (acute phase) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 2.90 [0.92, 9.14] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 1 | 69 | Mean Difference (IV, Random, 95% CI) | ‐11.5 [‐25.20, 2.20] |
| 3.2 LSAS avoidance subscale | 1 | 69 | Mean Difference (IV, Random, 95% CI) | ‐4.60 [‐11.88, 2.68] |
| 3.3 LSAS fear subscale | 1 | 69 | Mean Difference (IV, Random, 95% CI) | ‐6.90 [‐13.65, ‐0.15] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Reduction of depression symptoms | 1 | 69 | Mean Difference (IV, Random, 95% CI) | ‐2.3 [‐4.78, 0.18] |
| 5 All‐cause dropouts (acute phase) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Dropout rate | 3 | 488 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.78, 1.70] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 2 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.70, 1.37] |
| 2 Adverse events (acute phase) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.81, 4.94] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 2 | 228 | Mean Difference (IV, Random, 95% CI) | ‐0.24 [‐15.69, 15.21] |
| 3.2 LSAS avoidance subscale | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐9.15 [‐26.86, 8.56] |
| 3.3 LSAS fear subscale | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐8.71 [‐26.02, 8.60] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Reduction of depression symptoms | 1 | 212 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐1.33, 1.73] |
| 5 All‐cause dropouts (acute phase) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Dropout rate | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.30, 6.76] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.45, 108.26] |
| 2 Adverse events (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.06, 8.90] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 1 | 9 | Mean Difference (IV, Random, 95% CI) | ‐37.8 [‐74.22, ‐1.38] |
| 4 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Dropout rate | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.27, 4.23] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 2 | 132 | Risk Ratio (M‐H, Random, 95% CI) | 4.03 [2.45, 6.65] |
| 2 Relapse rate Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 No. relapsed | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 0.12 [0.01, 2.14] |
| 3 Adverse events (acute phase) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Dropout rate | 2 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 1.68 [0.21, 13.13] |
| 4 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 LSAS total score | 2 | 135 | Mean Difference (IV, Random, 95% CI) | ‐39.75 [‐71.11, ‐8.39] |
| 4.2 LSAS avoidance subscale | 1 | 75 | Mean Difference (IV, Random, 95% CI) | ‐10.40 [‐16.08, ‐4.72] |
| 4.3 LSAS fear subscale | 1 | 75 | Mean Difference (IV, Random, 95% CI) | ‐10.80 [‐16.62, ‐4.98] |
| 5 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 Reduction of depression symptoms | 1 | 75 | Mean Difference (IV, Random, 95% CI) | ‐1.60 [‐3.96, 0.76] |
| 6 Reduction of functional disability Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 Work subscale | 2 | 135 | Mean Difference (IV, Random, 95% CI) | ‐3.58 [‐6.39, ‐0.78] |
| 6.2 Social subscale | 2 | 135 | Mean Difference (IV, Random, 95% CI) | ‐2.31 [‐3.79, ‐0.83] |
| 6.3 Family subscale | 2 | 135 | Mean Difference (IV, Random, 95% CI) | ‐2.02 [‐4.26, 0.22] |
| 7 All‐cause dropouts (acute phase) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Dropout rate | 3 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.41, 1.52] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 2 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.63, 1.88] |
| 2 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Reduction of depression symptoms | 1 | 46 | Mean Difference (IV, Random, 95% CI) | 1.82 [‐1.38, 5.02] |
| 3 Reduction of functional disability Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Work subscale | 1 | 42 | Mean Difference (IV, Random, 95% CI) | ‐0.05 [‐1.80, 1.70] |
| 3.2 Social subscale | 1 | 42 | Mean Difference (IV, Random, 95% CI) | 0.07 [‐1.71, 1.85] |
| 3.3 Family subscale | 1 | 42 | Mean Difference (IV, Random, 95% CI) | ‐0.11 [‐1.74, 1.52] |
| 4 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Dropout rate | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 3.36 [0.41, 27.80] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 4 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 2.36 [1.48, 3.75] |
| 2 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 4 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 LSAS total score | 4 | 218 | Mean Difference (IV, Random, 95% CI) | ‐16.39 [‐32.27, ‐0.51] |
| 2.2 LSAS avoidance subscale | 1 | 51 | Mean Difference (IV, Random, 95% CI) | ‐5.42 [‐14.69, 3.85] |
| 2.3 LSAS fear subscale | 1 | 51 | Mean Difference (IV, Random, 95% CI) | ‐5.23 [‐13.97, 3.51] |
| 3 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Reduction of depression symptoms | 4 | 216 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐1.11, 0.31] |
| 4 Reduction of functional disability Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Work subscale | 2 | 95 | Mean Difference (IV, Random, 95% CI) | ‐2.84 [‐4.40, ‐1.28] |
| 4.2 Social subscale | 2 | 94 | Mean Difference (IV, Random, 95% CI) | ‐3.26 [‐7.25, 0.72] |
| 4.3 Family subscale | 2 | 95 | Mean Difference (IV, Random, 95% CI) | ‐2.20 [‐5.34, 0.95] |
| 5 All‐cause dropouts (acute phase) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Dropout rate | 4 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.71, 2.48] |
| 6 Clinical Global Impressions scale change item (CGI‐I) (long term) Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 No. of responders | 2 | 113 | Risk Ratio (M‐H, Random, 95% CI) | 1.84 [1.02, 3.33] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.19, 2.54] |
| 2 Adverse events (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 2.8 [0.12, 63.20] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 1 | 26 | Mean Difference (IV, Random, 95% CI) | 2.60 [‐15.43, 20.63] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Reduction of depression symptoms | 1 | 26 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐2.73, 2.53] |
| 5 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Dropout rate | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.23, 3.81] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.28, 3.63] |
| 2 Adverse events (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 99.95] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 2 | 126 | Mean Difference (IV, Random, 95% CI) | ‐15.37 [‐28.10, ‐2.63] |
| 3.2 LSAS avoidance subscale | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐3.90 [‐9.90, 2.10] |
| 3.3 LSAS fear subscale | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐3.70 [‐9.42, 2.02] |
| 4 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Dropout rate | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 20.90] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 8 | 1270 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [1.32, 2.55] |
| 2 Adverse events (acute phase) Show forest plot | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 8 | 1305 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.86, 2.34] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 9 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 6 | 1163 | Mean Difference (IV, Random, 95% CI) | ‐12.17 [‐23.51, ‐0.84] |
| 3.2 LSAS avoidance subscale | 5 | 695 | Mean Difference (IV, Random, 95% CI) | ‐5.05 [‐7.91, ‐2.18] |
| 3.3 LSAS fear subscale | 6 | 724 | Mean Difference (IV, Random, 95% CI) | ‐5.40 [‐8.92, ‐1.88] |
| 4 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 7 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Reduction of depression symptoms | 7 | 765 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.55, ‐0.00] |
| 5 Reduction of functional disability Show forest plot | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 Work subscale | 5 | 660 | Mean Difference (IV, Random, 95% CI) | ‐0.61 [‐1.89, 0.68] |
| 5.2 Social subscale | 5 | 660 | Mean Difference (IV, Random, 95% CI) | ‐1.14 [‐2.32, 0.05] |
| 5.3 Family subscale | 5 | 660 | Mean Difference (IV, Random, 95% CI) | ‐0.51 [‐1.45, 0.44] |
| 6 All‐cause dropouts (acute phase) Show forest plot | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Dropout rate | 6 | 512 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.60, 1.08] |
| 7 Clinical Global Impressions ‐ Improvement (CGI‐I) (long term) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 No. of responders | 1 | 90 | Risk Ratio (M‐H, Random, 95% CI) | 1.50 [1.12, 2.00] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 LSAS total score | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐6.10 [‐16.55, 4.35] |
| 2 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Reduction of depression symptoms | 1 | 102 | Mean Difference (IV, Random, 95% CI) | 0.80 [‐2.10, 3.70] |
| 3 Reduction of functional disability Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Work subscale | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐0.90 [‐1.87, 0.07] |
| 3.2 Social subscale | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐1.0 [‐1.97, ‐0.03] |
| 3.3 Family subscale | 1 | 102 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐1.06, 0.66] |
| 4 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Dropout rate | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 2.18 [0.97, 4.92] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 4 | 1173 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.85, 1.99] |
| 2 Adverse events (acute phase) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 4 | 1213 | Risk Ratio (M‐H, Random, 95% CI) | 3.23 [2.15, 4.86] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 3 | 902 | Mean Difference (IV, Random, 95% CI) | ‐11.91 [‐16.06, ‐7.76] |
| 3.2 LSAS avoidance subscale | 1 | 261 | Mean Difference (IV, Random, 95% CI) | ‐4.30 [‐8.14, ‐0.46] |
| 3.3 LSAS fear subscale | 1 | 261 | Mean Difference (IV, Random, 95% CI) | ‐4.0 [‐7.68, ‐0.32] |
| 4 All‐cause dropouts (acute phase) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Dropout rate | 4 | 1224 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.76, 1.07] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 24 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 24 | 4984 | Risk Ratio (M‐H, Random, 95% CI) | 1.65 [1.48, 1.85] |
| 2 Relapse rate Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 No. relapsed | 3 | 389 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.22, 0.50] |
| 3 Adverse events (acute phase) Show forest plot | 24 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Dropout rate | 24 | 5131 | Risk Ratio (M‐H, Random, 95% CI) | 2.59 [1.97, 3.39] |
| 4 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 15 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 LSAS total score | 14 | 1990 | Mean Difference (IV, Random, 95% CI) | ‐10.14 [‐14.05, ‐6.22] |
| 4.2 LSAS avoidance subscale | 7 | 1173 | Mean Difference (IV, Random, 95% CI) | ‐7.01 [‐10.21, ‐3.80] |
| 4.3 LSAS fear subscale | 7 | 1173 | Mean Difference (IV, Random, 95% CI) | ‐7.28 [‐10.86, ‐3.71] |
| 5 Clinician‐rated: Hamilton Depression scale (HAM‐D) (or similar scale) Show forest plot | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 Reduction of depression symptoms | 6 | 960 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐0.48, ‐0.03] |
| 6 Reduction of functional disability Show forest plot | 5 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 Work subscale | 5 | 854 | Mean Difference (IV, Random, 95% CI) | ‐0.81 [‐1.18, ‐0.45] |
| 6.2 Social subscale | 5 | 854 | Mean Difference (IV, Random, 95% CI) | ‐0.87 [‐1.26, ‐0.47] |
| 6.3 Family subscale | 5 | 854 | Mean Difference (IV, Random, 95% CI) | ‐0.45 [‐0.75, ‐0.15] |
| 7 All‐cause dropouts (acute phase) Show forest plot | 26 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Dropout rate | 26 | 5208 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.90, 1.14] |
| 8 Clinical Global Impressions scale change item (CGI‐I) (long term) Show forest plot | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 No. of responders | 4 | 806 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [1.07, 1.51] |
| 9 Adverse events (long term) Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 Dropout rate | 3 | 1274 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.43, 3.18] |
| 10 All‐cause dropouts (long term) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 Dropout rate | 1 | 52 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.37, 2.70] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 1 | 250 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.19] |
| 2 Adverse events (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 2.95 [0.67, 13.02] |
| 3 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Dropout rate | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.50, 1.20] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 No. of responders | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.68, 36.66] |
| 2 Adverse events (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dropout rate | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 LSAS total score | 1 | 24 | Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐1.35, 0.35] |
| 4 All‐cause dropouts (acute phase) Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Dropout rate | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinician‐rated: Liebowitz Social Anxiety Scale (LSAS): reduction of anxiety symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 LSAS total score | 1 | 99 | Mean Difference (IV, Random, 95% CI) | 1.80 [‐6.92, 10.52] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar) scale: no. of responders (acute phase) Show forest plot | 51 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 5HT1A partial agonists | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 14.55] |
| 1.2 Anticonvulsants/GABAs | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.16, 2.20] |
| 1.3 Anticonvulsant levetiracetam | 2 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.70, 1.37] |
| 1.4 Antipsychotics | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.45, 108.26] |
| 1.5 Benzodiazepines | 2 | 132 | Risk Ratio (M‐H, Random, 95% CI) | 4.03 [2.45, 6.65] |
| 1.6 Beta‐blockers | 2 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.63, 1.88] |
| 1.7 MAOIs | 3 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 2.37 [1.26, 4.45] |
| 1.8 NARIs | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.19, 2.54] |
| 1.9 NaSSAs | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.28, 3.63] |
| 1.10 RIMAs | 8 | 1270 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [1.32, 2.55] |
| 1.11 SNRIs | 4 | 1173 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.85, 1.99] |
| 1.12 SSRIs | 21 | 4553 | Risk Ratio (M‐H, Random, 95% CI) | 1.65 [1.46, 1.87] |
| 1.13 NK1 receptor antagonist GR205171 | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.68, 36.66] |
| 1.14 GW876008 | 1 | 250 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.19] |
| 2 Dropout rate: adverse events (acute phase) Show forest plot | 45 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 5HT1A partial agonists | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 68.26] |
| 2.2 Anticonvulsants/GABAs | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 2.90 [0.92, 9.14] |
| 2.3 Anticonvulsant levetiracetam | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.81, 4.94] |
| 2.4 Antipsychotics | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.06, 8.90] |
| 2.5 Benzodiazepines | 2 | 96 | Risk Ratio (M‐H, Random, 95% CI) | 1.68 [0.21, 13.13] |
| 2.6 NARIs | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 2.8 [0.12, 63.20] |
| 2.7 NaSSAs | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 5.0 [0.25, 99.95] |
| 2.8 RIMAs | 8 | 1305 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.86, 2.34] |
| 2.9 SNRIs | 4 | 1213 | Risk Ratio (M‐H, Random, 95% CI) | 3.23 [2.15, 4.86] |
| 2.10 SSRIs | 22 | 4965 | Risk Ratio (M‐H, Random, 95% CI) | 2.56 [1.94, 3.38] |
| 2.11 NK1 receptor antagonist GR205171 | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2.12 GW876008 | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 2.95 [0.67, 13.02] |
| 3 Dropout rate: all‐cause dropouts (acute phase) Show forest plot | 54 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 5HT1A partial agonists | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.55] |
| 3.2 Anticonvulsants/GABAs | 3 | 488 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.78, 1.70] |
| 3.3 Anticonvulsant levetiracetam | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.30, 6.76] |
| 3.4 Antipsychotics | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.27, 4.23] |
| 3.5 Benzodiazepines | 3 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.41, 1.52] |
| 3.6 Beta‐blockers | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 3.36 [0.41, 27.80] |
| 3.7 MAOIs | 4 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.71, 2.48] |
| 3.8 NARIs | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.23, 3.81] |
| 3.9 NaSSAs | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 20.90] |
| 3.10 RIMAs | 6 | 512 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.60, 1.08] |
| 3.11 SARIs | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 2.18 [0.97, 4.92] |
| 3.12 SNRIs | 4 | 1224 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.76, 1.07] |
| 3.13 SSRIs | 25 | 5078 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.89, 1.14] |
| 3.14 NK1 receptor antagonist GR205171 | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.15 GW876008 | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.50, 1.20] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase) Show forest plot | 53 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Single‐centre | 20 | 1332 | Risk Ratio (M‐H, Random, 95% CI) | 2.24 [1.67, 3.02] |
| 1.2 Multicentre | 35 | 8274 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [1.34, 1.62] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase) Show forest plot | 53 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Generalised | 26 | 5522 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [1.31, 1.69] |
| 1.2 Inclusive | 27 | 3712 | Risk Ratio (M‐H, Random, 95% CI) | 1.83 [1.54, 2.18] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase) Show forest plot | 50 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Industry funded trials | 34 | 6643 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.44, 1.77] |
| 1.2 Non‐industry funded trials | 16 | 1780 | Risk Ratio (M‐H, Random, 95% CI) | 1.99 [1.43, 2.77] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Clinical Global Impressions ‐ Improvement (CGI‐I) or similar scale: no. of responders (acute phase) Show forest plot | 53 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Trials including MDD participants | 20 | 2654 | Risk Ratio (M‐H, Random, 95% CI) | 1.77 [1.44, 2.18] |
| 1.2 Trials excluding MDD participants | 34 | 6765 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [1.35, 1.70] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 'Worst case' lost‐to‐follow‐up analysis Show forest plot | 25 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Anticonvulsant levetiracetam | 2 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.70, 1.38] |
| 1.2 Antipsychotics | 1 | 12 | Risk Ratio (M‐H, Random, 95% CI) | 5.25 [0.33, 83.59] |
| 1.3 Benzodiazepines | 1 | 75 | Risk Ratio (M‐H, Random, 95% CI) | 3.82 [1.92, 7.62] |
| 1.4 Beta‐blockers | 1 | 46 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.47, 1.86] |
| 1.5 RIMAs | 2 | 182 | Risk Ratio (M‐H, Random, 95% CI) | 3.01 [1.95, 4.65] |
| 1.6 SNRIs | 3 | 934 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.36, 1.88] |
| 1.7 SSRIs | 14 | 3753 | Risk Ratio (M‐H, Random, 95% CI) | 1.61 [1.44, 1.79] |
| 1.8 GW876008 | 1 | 252 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.57, 1.18] |
| 2 'Best case' lost‐to‐follow‐up analysis Show forest plot | 25 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Anticonvulsant levetiracetam | 2 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.70, 1.37] |
| 2.2 Antipsychotics | 1 | 10 | Risk Ratio (M‐H, Random, 95% CI) | 7.0 [0.45, 108.26] |
| 2.3 Benzodiazepines | 1 | 72 | Risk Ratio (M‐H, Random, 95% CI) | 3.92 [1.98, 7.77] |
| 2.4 Beta‐blockers | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.55, 2.05] |
| 2.5 RIMAs | 2 | 177 | Risk Ratio (M‐H, Random, 95% CI) | 2.97 [1.93, 4.58] |
| 2.6 SNRIs | 3 | 912 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [1.39, 1.91] |
| 2.7 SSRIs | 14 | 3577 | Risk Ratio (M‐H, Random, 95% CI) | 1.64 [1.46, 1.85] |
| 2.8 GW876008 | 1 | 250 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.58, 1.19] |