Gabapentin for fibromyalgia pain in adults
Abstract
Background
This review replaces part of an earlier review that evaluated gabapentin for both neuropathic pain and fibromyalgia, now split into separate reviews for the two conditions. This review will consider pain in fibromyalgia only.
Fibromyalgia is associated with widespread pain lasting longer than three months, and is frequently associated with symptoms such as poor sleep, fatigue, depression, and reduced quality of life. Fibromyalgia is more common in women.
Gabapentin is an antiepileptic drug widely licensed for treatment of neuropathic pain. It is not licensed for the treatment of fibromyalgia, but is commonly used because fibromyalgia can respond to the same medicines as neuropathic pain.
Objectives
To assess the analgesic efficacy of gabapentin for fibromyalgia pain in adults and the adverse events associated with its use in clinical trials.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies Online, MEDLINE via Ovid and Embase via Ovid from inception to 24 May 2016. We also searched the reference lists of retrieved studies and reviews, and searched online clinical trial registries.
Selection criteria
Randomised, double‐blind trials of eight weeks' duration or longer for treating fibromyalgia pain in adults, comparing gabapentin with placebo or an active comparator.
Data collection and analysis
Two independent review authors extracted data and assessed trial quality and risk of bias. We planned to use dichotomous data to calculate risk ratio and number needed to treat for one additional event, using standard methods. We assessed the evidence using GRADE (Grading of Recommendations Assessment, Development and Evaluation) and created a 'Summary of findings' table.
Main results
Two studies tested gabapentin to treat fibromyalgia pain. One was identified in previous versions of the review and is included here. We identified another study as a conference abstract, with insufficient detail to determine eligibility for inclusion; it is awaiting assessment. The one included study of 150 participants was a 12‐week, multi‐centre, randomised, double‐blind, placebo‐controlled, parallel‐group study using last‐observation‐carried‐forward imputation for withdrawals. The maximum dose was 2400 mg daily. The overall risk of bias was low, except for attrition bias.
At the end of the trial, the outcome of 50% reduction in pain over baseline was not reported. The outcome of 30% or greater reduction in pain over baseline was achieved by 38/75 participants (49%) with gabapentin compared with 23/75 (31%) with placebo (very low quality). A patient global impression of change any category of "better" was achieved by 68/75 (91%) with gabapentin and 35/75 (47%) with placebo (very low quality).
Nineteen participants discontinued the study because of adverse events: 12 in the gabapentin group (16%) and 7 in the placebo group (9%) (very low quality). The number of serious adverse events were not reported, and no deaths were reported (very low quality).
Authors' conclusions
We have only very low quality evidence and are very uncertain about estimates of benefit and harm because of a small amount of data from a single trial. There is insufficient evidence to support or refute the suggestion that gabapentin reduces pain in fibromyalgia.
PICOs
Plain language summary
Gabapentin for pain in adults with fibromyalgia
Bottom Line
There is no good evidence to support or contradict the suggestion that gabapentin at daily doses of 1200 to 2400 mg reduces pain in fibromyalgia.
Background
Fibromyalgia is a complex disorder characterised by widespread pain, fatigue, poor sleep, low mood, and other bodily symptoms. Common pain‐relieving medicines such as paracetamol and ibuprofen are not usually considered effective. Antiepileptic drugs are commonly used to treat fibromyalgia, but there is uncertainty about how good they are.
Gabapentin is a medicine used to treat pain caused by nerves that are not working properly. Gabapentin changes the way that the nerves send messages to the brain. It can be taken in a tablet or a liquid, with or without food. Doses are usually 1200 mg to 2400 mg each day. At the start of treatment low doses are used to minimise side effects, but the dose is usually increased after a few weeks.
Study characteristics
In May 2016 we searched for clinical trials where gabapentin was used to treat pain due to fibromyalgia in adults. We found one study that met the requirements for this review. The study tested 1200 to 2400 mg/day of gabapentin compared with a placebo over 12 weeks, in 150 people.
Key results
The study did not report the number of people with pain reduced by half at the end of week 12. At that time 5 in 10 people taking gabapentin and 3 in 10 taking the placebo had their pain reduced by at least one third. A report of feeling better to any degree was reported by 9 in 10 taking gabapentin and 5 in 10 taking placebo.
About 2 in 10 people taking gabapentin stopped taking the medicine because of side effects, compared with 1 in 10 taking the placebo. The study did not report the number of people with serious side effects, but did report that there were no deaths.
Quality of the evidence
We rated the quality of the evidence as very low because there was only a single small study with important study limitations. Several factors reduced our confidence in the result. Very low quality evidence means that we are very uncertain about the results.
Authors' conclusions
Summary of findings
| Gabapentin compared with placebo for fibromyalgia | ||||||
| Patient or population: adults with fibromyalgia Settings: community Intervention: gabapentin Comparison: placebo | ||||||
| Outcomes | Assumed risk ‐ probable outcome with intervention | Corresponding risk ‐ probable outcome with control | Relative effect | Number of | Quality of the evidence | Comments |
| gabapentin | placebo | |||||
| 30% pain reduction at 12 weeks | 38/75 | 23/75 | Not calculated | 1 study, 150 participants | very low | One included study of fewer than 200 participants; LOCF imputation. Downgraded three levels because of small numbers and study limitations |
| 50% pain reduction at 12 weeks | No data | No data | ‐ | ‐ | very low | Outcome not reported |
| PGIC ‐ any category of "better" at 12 weeks | 68/75 | 35/75 | Not calculated | 1 study, 150 participants | very low | One included study of fewer than 200 participants; LOCF imputation; non‐standard outcome ‐ usually top two categories of better, not top three, used Downgraded three levels because of small numbers and study limitations |
| Withdrawals due to adverse events | 12/75 | 7/75 | Not calculated | 1 study, 150 participants | very low | One included study of fewer than 200 participants; few events Downgraded three levels because of small numbers |
| Serious adverse events | "No significant group differences" | ‐ | 1 study, 150 participants | very low | ‐ | |
| Deaths | None reported | ‐ | 1 study, 150 participants | very low | ‐ | |
| CI: Confidence interval; LOCF: last observation carried forward; PGIC: Patient Global Impression of Change | ||||||
| Descriptors for levels of evidence (EPOC 2015): † Substantially different: a large enough difference that it might affect a decision. | ||||||
Background
This Cochrane review is based on a template for reviews of drugs used to relieve fibromyalgia. The aim is for all reviews to use the same methods, based on new criteria for what constitutes reliable evidence in chronic pain (Moore 2010a; Appendix 1).
This Cochrane review has been split from an earlier review on fibromyalgia and neuropathic pain (Moore 2011a), and will consider only fibromyalgia, due to the pathogenesis of the pain being different from neuropathic pain. The most recent version of the review is being amended and updated to focus solely on neuropathic pain (Moore 2014a).
The history of earlier versions of this Cochrane review is available in Figure 1 and Appendix 2.

History of Earlier Reviews
Description of the condition
Fibromyalgia symptoms can be assessed by patient self‐report using the fibromyalgia criteria and severity scales for clinical and epidemiological studies, a modification of the American College of Rheumatology (ACR) Preliminary Diagnostic Criteria for Fibromyalgia (so‐called Fibromyalgia Symptom Questionnaire; Wolfe 2011). Fibromyalgia was previously defined by the ACR 1990 classification criteria as widespread pain lasting for longer than three months with tenderness on palpation at 11 or more of 18 specified tender points (Wolfe 1990). For a clinical diagnosis, the ACR 1990 classification criteria and the ACR 2010 preliminary diagnostic criteria can both be used (Wolfe 1990 and Wolfe 2010 respectively). As there is no specific laboratory test, diagnosis is established by obtaining a history of the key symptoms and the exclusion of somatic diseases sufficiently explaining the key symptoms (Wolfe 2010). The indexing of fibromyalgia within the international classification of diseases is under debate. While some rheumatologists have thought of it as a specific pain disorder and central sensitivity syndrome (Clauw 2014; Yunus 2008), recent research points at small fibre pathology in a subgroup of fibromyalgia patients that may be of pathophysiological importance (Oaklander 2013; Üçeyler 2013a). In psychiatry and psychosomatic medicine, fibromyalgia symptoms are categorised as a functional somatic syndrome, a bodily distress syndrome, a somatic physical symptom disorder, or a somatoform disorder (Häuser 2014).
Fibromyalgia is a heterogenous condition. The definite aetiology (causes) of this syndrome remains unknown. A model of interacting biological and psychosocial variables in the predisposition, triggering, and development of the chronicity of fibromyalgia symptoms has been suggested (Sommer 2012). Depression (Forseth 1999), genetics (Arnold 2013; Lee 2012), obesity combined with physical inactivity (Mork 2010), physical and sexual abuse in childhood (Häuser 2011), sleep problems (Mork 2010), and smoking (Choi 2010) are associated with future development of fibromyalgia. Psychosocial stress (working place and family conflicts) and physical stress (infections, surgery, accidents) might trigger the onset of chronic widespread pain and fatigue (Clauw 2014; Sommer 2012). Depression and post‐traumatic stress disorder worsen fibromyalgia symptoms (Häuser 2013a; Lange 2010).
Several factors are associated with the pathophysiology (functional changes associated with or resulting from disease) of fibromyalgia, but the relationship is unclear. The functional changes include alteration of sensory processing in the brain, reduced reactivity of the hypothalamus‐pituitary‐adrenal axis to stress, increased pro‐inflammatory and reduced anti‐inflammatory cytokine profiles (produced by cells involved in inflammation), and disturbances in neurotransmitters such as dopamine and serotonin (Oaklander 2013; Sommer 2012; Üçeyler 2013a). Prolonged exposure to stress, as outlined above, may contribute to these functional changes in predisposed individuals (Bradley 2009). There are similarities to, and differences from, neuropathic pain (Koroschetz 2011).
Patients often report high disability levels and poor quality of life along with extensive use of medical care (Häuser 2015). Many people with fibromyalgia are significantly disabled, and experience moderate or severe pain for many years (Bennett 2007). Chronic painful conditions comprised five of the 11 top‐ranking conditions for years lived with disability in 2010 (Vos 2012), and are responsible for considerable loss of quality of life, employment, and increased health costs (Moore 2014a).
Fibromyalgia is common. One component of fibromyalgia, chronic widespread pain, is not only associated with other symptoms such as poor sleep, fatigue, and depression (Wolfe 2014), but is also estimated to affect 11% of the general population (Mansfield 2016). Numerous studies have investigated prevalence of fibromyalgia in different settings and countries. One review gives a global mean prevalence of potential cases of fibromyalgia of 2.7% (range 0.4% to 9.3%), with a mean of 3.1% in the Americas, 2.5% in Europe, and 1.7% in Asia (Queiroz 2013). Changes in diagnostic criteria do not appear to have significantly affected estimates of prevalence (Wolfe 2013). A large US survey using a modification of the 2010 ACR criteria found a prevalence of 1.8%, but 73% of these patients were given a different diagnosis by their physician (Walitt 2015). Estimates of prevalence in specific populations vary greatly, but have been reported to be as high as 9% in female textile workers in Turkey and 10% in metalworkers in Brazil (59% in those with repetitive strain injury; Queiroz 2013). When the 1990 ACR criteria are used for clinical surveys, women are more frequently diagnosed with the disorder. Using these criteria, the women to men ratio has ranged from 8:1 to 30:1 in patients who were studied in clinical institutions and surveys. However, with criteria that do not use tender point examination, the sex ratio can be close to equal. The sex ratio has ranged from 4:1 to 1:1 in studies that were conducted in the general population using the research criteria for fibromyalgia (Häuser 2015; Queiroz 2013).
Fibromyalgia pain is known to be difficult to treat effectively, with only a minority of individuals experiencing a clinically relevant benefit from any intervention. A multidisciplinary approach is recommended by recent evidence‐based guidelines, with pharmacological treatment being combined with physical or cognitive training, or both. Interventions aim to reduce the key symptoms of fibromyalgia (pain, sleep problems, fatigue) and the associated symptoms (depression, disability) and to improve daily functioning (Eich 2012; Fitzcharles 2013). Conventional analgesics are usually not effective. Antidepressants such as serotonin and noradrenaline reuptake inhibitors (Häuser 2013b; Lunn 2014), tricyclic agents such as amitriptyline (Moore 2012a), or antiepileptics like gabapentin or pregabalin (Moore 2014b; Üçeyler 2013b; Wiffen 2013) are often offered as treatment. The proportion of patients who achieve worthwhile pain relief (typically at least 50% reduction in pain intensity) is small (Moore 2013b), and generally reaches only 10% to 25% more than with placebo, with number needed to treat for an additional beneficial outcome (NNT) between 9.8 and 14 in fibromyalgia (Wiffen 2013); somewhat higher (worse) than for neuropathic pain (Kalso 2013; Wiffen 2013). Those who do experience good levels of pain relief, however, also benefit from substantial reductions in other symptoms, such as fatigue, depression, and anxiety, with significant improvement in ability to function, sleep, work, and quality of life (Moore 2010c; Straube 2011). Fibromyalgia is not particularly different from other chronic pain with regard to a small proportion of trial participants having a good response to analgesic treatment (Moore 2013b).
Description of the intervention
Gabapentin, whilst licensed for the treatment of neuropathic pain in adults in many parts of the world, is not licensed for the treatment of fibromyalgia in any country. As fibromyalgia can respond to the same medicines as neuropathic pain, it has been used off‐license for the treatment of fibromyalgia. Pregabalin, which is closely related to gabapentin, is licensed for treatment of fibromyalgia in the USA.
Gabapentin is given orally, usually as tablets or capsules, but sometimes as an oral solution (50 mg/mL). Guidance suggests that gabapentin treatment can be started at a dose of 300 mg per day for treating neuropathic pain, which is replicated in its use for treating fibromyalgia pain. Based on individual patient response and tolerability, the dosage may be increased by 300 mg per day until the patient experiences satisfactory pain relief or adverse effects make taking the drug intolerable (EMC 2009). Gabapentin is marketed under various trade names, including NeurontinTM, and is also available as generic products in some parts of the world.
Gabapentin has a half‐life of five to seven hours. It is absorbed through a saturable transport system, so that absorption is not linear, and the transporter is found only in the proximal small bowel. This means that the drug needs to be administered at least three times daily, and may result in plasma trough levels. Two new formulations have attempted to improve the availability of the drug. The first is an extended release, gastro‐retentive formulation, designed to provide continuous delivery at the optimal site of absorption over 8 to 10 hours (Sang 2013). The second uses an extended‐release prodrug (gabapentin encarbil) that is absorbed through a high capacity transport system found throughout the intestine, and then undergoes rapid hydrolysis to gabapentin. It is claimed to provide sustained, dose‐proportional gabapentin exposure (Backonja 2011), and can be administered twice daily.
Gabapentin can also be formulated as an aqueous solution for injection. This formulation is not available commercially.
How the intervention might work
Gabapentin's mechanism of action is primarily attributed to its effect on calcium channels located throughout the peripheral and central nervous systems, which modify the release of neurotransmitters and reduce excitability of nerve cells (Boyle 2014; Chang 2014). This mode of action confers antiepileptic, analgesic, and sedative effects. Research also indicates that gabapentin acts by blocking new synapse formation (Eroglu 2009).
Why it is important to do this review
Gabapentin is used off‐license to treat fibromyalgia. The earlier Cochrane review considered fibromyalgia alongside neuropathic pain (Moore 2011a), but an editorial decision was made to split the review by condition, which necessitated a new review and update of the evidence.
Like the earlier Cochrane review, this new review assesses evidence in ways that make both statistical and clinical sense, and uses developing criteria for what constitutes reliable evidence in chronic pain (Appendix 1; Moore 2010a). We have followed the standards of the Cochrane Pain, Palliative and Supportive Care (PaPaS) Group, as set out in the Cochrane PaPaS Group Author and Referee Guidance for pain studies (PaPaS 2012).
Objectives
To assess the analgesic efficacy of gabapentin for fibromyalgia pain in adults and the adverse events associated with its use in clinical trials.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) with double‐blind assessment of outcomes reporting outcomes after eight weeks of treatment or longer. We required full journal publication, with the exception of extended abstracts with sufficient data for analysis and online clinical trial results summaries of otherwise unpublished clinical trials. We excluded short abstracts (usually meeting reports), non‐randomised studies, studies of experimental pain, case reports, and clinical observations.
Types of participants
Studies included participants aged 18 years and above, with fibromyalgia diagnosed using the 1990 or 2010 criteria (Wolfe 1990; Wolfe 2010), and with initial pain of at least moderate intensity.
Types of interventions
Gabapentin at any dose, by any route, administered for the relief of pain in fibromyalgia, and compared to placebo or any active comparator. We excluded studies that used gabapentin to treat pain resulting from the use of other drugs.
Types of outcome measures
We anticipated that studies would use a variety of outcome measures, with most studies using standard subjective scales (numerical rating scale (NRS) or visual analogue scale (VAS)) for pain intensity or pain relief, or both. We were particularly interested in Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) definitions for moderate and substantial benefit in chronic pain studies (Dworkin 2008). These were defined as:
-
at least 30% pain relief over baseline (moderate);
-
at least 50% pain relief over baseline (substantial);
-
much or very much improved on Patient Global Impression of Change (PGIC; moderate);
-
very much improved on PGIC (substantial).
These outcomes differ from those used in most earlier reviews (Wiffen 2005), as they concentrate on dichotomous outcomes where pain ratings do not follow a normal (Gaussian) distribution. People with chronic pain desire high levels of pain relief, ideally more than 50%, and ideally leaving them with no worse than mild pain (Moore 2013a; O'Brien 2010).
Primary outcomes
-
Participant‐reported pain relief of 30% or greater.
-
Participant‐reported pain relief of 50% or greater.
-
PGIC much or very much improved.
-
PGIC very much improved.
Secondary outcomes
-
Any pain‐related outcome indicating some improvement.
-
Withdrawals due to lack of efficacy or adverse events, or for any cause.
-
Any adverse event.
-
Any serious adverse event. Serious adverse events typically include any untoward medical occurrence or effect that results in death, is life‐threatening, requires hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, is a congenital anomaly or birth defect, is an 'important medical event' that may jeopardise the patient, or that may require an intervention to prevent one of the above characteristics or consequences.
-
Specific adverse events, particularly somnolence and dizziness.
These outcomes are not eligibility criteria for this review, but are outcomes of interest within the studies that meet the inclusion criteria of the review.
Search methods for identification of studies
Two review authors (TC and PW) independently performed literature searches for eligible studies. We resolved any disagreements or uncertainties by discussion with a third review author where necessary.
Electronic searches
We searched the following databases, without language restrictions.
-
Cochrane Central Register of Controlled Trials (CENTRAL; via the Cochrane Register of Studies Online) to 24 May 2016.
-
MEDLINE (via Ovid) from 1946 to 24 May 2016.
-
Embase (via Ovid) from 1974 to 24 May 2016.
The individual search strategies for CENTRAL, MEDLINE, and Embase are shown in Appendix 4.
Searching other resources
We reviewed the bibliographies of all relevant RCTs and review articles, and searched the following clinical trial databases: ClinicalTrials.gov (ClinicalTrials.gov) and the World Health Organization International Clinical Trials Registry Platform (apps.who.int/trialsearch/), to identify additional published or unpublished data. We did not contact study investigators or study sponsors.
Data collection and analysis
Selection of studies
In order to determine study eligibility, two authors (TC and PW) independently read the abstract of each study identified by the search. We eliminated studies that clearly did not satisfy the inclusion criteria, and obtained full‐text copies of the remaining study reports. TC and PW independently read these reports and reached agreement regarding inclusion by discussion. We did not anonymise the studies in any way before assessment. We have included a Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) flow chart (Figure 2).
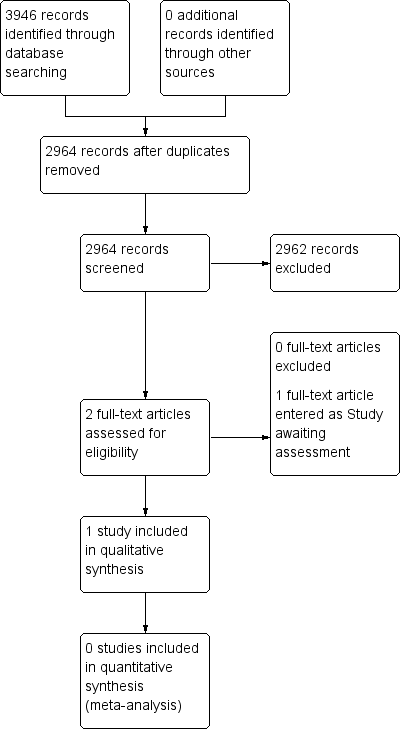
Study flow diagram.
Data extraction and management
Two review authors (TC and PW) independently extracted the data using a standard form and confirmed agreement before entering the data into Review Manager 5 (RevMan 2014), or any other analysis tool. We included information about the pain condition and number of participants treated, drug and dosing regimen, study design (placebo or active control), study duration and follow‐up, analgesic outcome measures and results, withdrawals, and adverse events (participants experiencing any adverse event or serious adverse event).
Assessment of risk of bias in included studies
We used the Oxford Quality Score as the basis for inclusion (Jadad 1996), limiting inclusion to studies that were randomised and double‐blind as a minimum.
Two review authors (TC and PW) independently assessed the risk of bias for each included study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), and adapted from those used by the Cochrane Pregnancy and Childbirth Group. We resolved any disagreements by discussion. We assessed the following for each included study.
-
Random sequence generation (checking for possible selection bias): We assessed the method used to generate the allocation sequence as being at either low risk of bias (any truly random process, for example random number table or computer random number generator) or unclear risk of bias (when the method used to generate the sequence is not clearly stated). We excluded studies at high risk of bias that use a non‐random process (for example, odd or even date of birth; hospital or clinic record number).
-
Allocation concealment (checking for possible selection bias): The method used to conceal allocation to interventions prior to assignment determines whether intervention allocation could have been foreseen in advance of, or during, recruitment, or changed after assignment. We assessed the methods as being at either low risk of bias (for example, telephone or central randomisation; consecutively numbered, sealed, opaque envelopes) or unclear risk of bias (when the method is not clearly stated). We excluded studies that did not conceal allocation and were therefore at high risk of bias (for example, an open list).
-
Blinding of participants, personnel, and outcome assessment (checking for possible performance and detection bias): We assessed the methods used to blind study participants and outcome assessors from knowledge of which intervention a participant received. We assessed the methods as being at either low risk of bias (authors state that it was blinded and describe the method used to achieve blinding, for example, identical tablets, matched in appearance and smell) or unclear risk of bias (authors state that it was blinded but do not provide an adequate description of how blinding was achieved). We excluded studies at high risk of bias that were not double‐blind.
-
Incomplete outcome data (checking for possible attrition bias due to the amount, nature, and handling of incomplete outcome data): We assessed the methods used to deal with incomplete data as being at low risk of bias (less than 10% of participants did not complete the study, or authors used 'baseline observation carried forward' analysis, or both), unclear risk of bias (used 'last observation carried forward' (LOCF) analysis), or high risk of bias (used 'completer' analysis).
-
Size of study (checking for possible biases confounded by small size): We assessed studies as being at low risk of bias (200 participants or more per treatment arm), unclear risk of bias (50 to 199 participants per treatment arm), or high risk of bias (fewer than 50 participants per treatment arm).
Measures of treatment effect
We used dichotomous data to calculate the risk ratio (RR) with 95% confidence interval (CIs) using a fixed‐effect model unless we found evidence of significant statistical heterogeneity (see Assessment of heterogeneity).
We calculated NNTs as the reciprocal of the absolute risk reduction (McQuay 1998). For unwanted effects, the NNT becomes the number needed to treat for an additional harmful outcome (NNTH) and is calculated in the same manner. Where the unwanted effect is less common with treatment than control, we used the term Number Needed to Treat to Prevent (NNTp).
We did not use continuous data in analyses because it is inappropriate where there is an underlying skewed distribution, as is usually the case with analgesic response.
Unit of analysis issues
We accepted randomisation to the individual participant only. In the event of a study having more than one active treatment arm in which data were not combined for analysis, we planned to split the control treatment arm between active treatment arms.
Dealing with missing data
We used intention‐to‐treat (ITT) analysis where the ITT population consists of participants who were randomised, took at least one dose of the assigned study medication, and provided at least one post‐baseline assessment. We assigned missing participants zero improvement wherever possible.
Assessment of heterogeneity
We planned to deal with clinical heterogeneity by combining studies that examine similar conditions, and to assess statistical heterogeneity visually (L'Abbé 1987), and with the I² statistic. When the I² statistic value was greater than 50%, we would consider the possible reasons for this.
Assessment of reporting biases
The aim of this Cochrane review was to use dichotomous outcomes of known utility and of value to patients (Hoffman 2010; Moore 2010b; Moore 2010c; Moore 2010d; Moore 2013a). The review does not depend on what the authors of the original studies chose to report or not, though clearly difficulties would arise if included studies failed to report relevant dichotomous results.
We planned to assess publication bias using a method designed to detect the amount of unpublished data with a null effect required to make any result clinically irrelevant (usually taken to mean a NNT of 10 or higher in this condition; Moore 2008). In the event, this was not possible.
Data synthesis
We planned to use a fixed‐effect model for meta‐analysis. We would have used a random‐effects model if there was significant clinical heterogeneity and it was considered appropriate to combine studies.
We planned to analyse data in three tiers, according to outcome and freedom from known sources of bias.
-
The first tier would use data meeting current best standards, where studies report the outcome of at least 50% pain intensity reduction over baseline (or its equivalent), without the use of LOCF analysis or other imputation method for dropouts, report an ITT analysis, last eight or more weeks, have a parallel‐group design, and have at least 200 participants (preferably at least 400) in the comparison (Moore 1998; Moore 2010a; Moore 2012a; Moore 2012b). We planned to report these top‐tier results first.
-
The second tier would use data from at least 200 participants but where one or more of the first‐tier conditions above was not met (for example, reporting at least 30% pain intensity reduction, using LOCF or a completer analysis, or lasting four to eight weeks).
-
The third tier of evidence related to data from fewer than 200 participants, or where there were expected to be significant problems because, for example, of very short duration studies of less than four weeks, where there was major heterogeneity between studies, or where there were shortcomings in allocation concealment, attrition, or incomplete outcome data. For this third tier of evidence, no data synthesis is reasonable and may be misleading, but an indication of beneficial effects might be possible.
Quality of the evidence
We used the GRADE approach to assess the quality of evidence related to each of the key outcomes, and report our judgement on the quality of the evidence in the 'Summary of findings' table (Chapter 12, Appendix 6; Higgins 2011). Two review authors independently rated the quality of evidence for each outcome.
In addition, there may be circumstances where the overall rating for a particular outcome needs to be adjusted as recommended by GRADE guidelines (Guyatt 2013a). For example, if there are so few data that the results are highly susceptible to the random play of chance (Moore 2008b), or if a studies use LOCF imputation in circumstances where there are substantial differences in adverse event withdrawals (Moore 2012b), one would have no confidence in the result, and would need to downgrade the quality of the evidence by 3 levels, to very low quality. In circumstances where there were no data reported for an outcome, we would report the level of evidence as very low quality (Guyatt 2013b).
Summary of findings table
We have included a 'Summary of findings' table as set out in the PaPaS author guide (PaPaS 2012), and recommended in the Cochrane Handbook (Chapter 11, Higgins 2011). The table includes, where possible, outcomes equivalent to moderate or substantial benefit of at least 30% and at least 50% pain intensity reduction, PGIC (possibly at least substantial improvement and at least moderate improvement) (Dworkin 2008), withdrawals due to lack of efficacy, withdrawals due to adverse events, serious adverse events, and death (a particular serious adverse event).
For the 'Summary of findings' table we used the following descriptors for levels of evidence (EPOC 2015):
High: This research provides a very good indication of the likely effect. The likelihood that the effect will be substantially different† is low.
Moderate: This research provides a good indication of the likely effect. The likelihood that the effect will be substantially different† is moderate.
Low: This research provides some indication of the likely effect. However, the likelihood that it will be substantially different† is high.
Very low: This research does not provide a reliable indication of the likely effect. The likelihood that the effect will be substantially different† is very high.
† Substantially different: a large enough difference that it might affect a decision.
Subgroup analysis and investigation of heterogeneity
We did not plan to perform any subgroup analyses since the experience of previous reviews indicated that there would be too few data for any meaningful subgroup analysis.
Sensitivity analysis
We did not plan to perform any sensitivity analyses.
Results
Description of studies
For this review we made no attempt to contact authors or manufacturers of gabapentin, as in previous versions of this review (Moore 2011a; Moore 2014a). Clinical trial reports or synopses from previously unpublished studies had became available as a result of legal proceedings in the USA for the previous update.
Results of the search
Searching identified two studies using gabapentin to treat pain in fibromyalgia. One was completed and could be included (Arnold 2007); this study was identified in a previous version of the review. Another was available only as a conference abstract, with insufficient detail to determine eligibility for inclusion (Mouzopoulos 2014); this study has been put into Studies awaiting classification. A flow diagram of the search results is shown in Figure 2.
Included studies
Arnold 2007 investigated 150 participants in a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Participants had diagnosis made using ACR (1990) criteria and had a pain score of 4/10 or greater on an NRS (moderate or severe pain) at randomisation; the mean initial pain score was 5.8/10. The median age was 48 years and 90% were women. The dose of gabapentin was titrated over the first six weeks of treatment, kept stable for a further six weeks, then tapered over one week. During the stable treatment phase, participants were required to take between 1200 and 2400 mg daily, administered in three doses. The median dosage was 1800 mg daily at endpoint.
Participants were excluded if they had previously been treated with gabapentin or pregabalin. Results were analysed using LOCF imputation for withdrawals.
See Characteristics of included studies table.
Excluded studies
We did not exclude any studies after reading the full article.
Risk of bias in included studies
We judged the included study to be at low or unclear risk of bias for all domains except for incomplete outcome reporting (attrition), for which we judged the risk of bias to be high. There was an unclear risk of bias for selection (both random sequence generation and allocation concealment). There was a low risk of bias for performance and detection bias (blinding), selective reporting, and size. See Figure 3 and the Characteristics of included studies table for our reasoning.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Effects of interventions
See: Summary of findings for the main comparison Gabapentin compared with placebo for fibromyalgia
Efficacy
There was no first or second tier evidence.
Third tier evidence came from one study with fewer than 200 participants. We did not carry out any data synthesis because any results would potentially be misleading. See summary of findings Table for the main comparison.
Participant‐reported pain relief of 30% or greater
This outcome was reported in 38/75 (49%) of participants with gabapentin and 23/75 (31%) with placebo, using LOCF.
Participant‐reported pain relief of 50% or greater
This outcome was not reported.
PGIC very much improved, or much or very much improved
These outcomes were not reported, but the study did report that 68% of participants described their condition as "better" at the end of treatment with gabapentin, as did 35% with placebo. These results are estimated from a graph, and use LOCF.
Any pain‐related outcomes
The Brief Pain Inventory average pain severity score assesses average pain over the previous 24 hours. After 12 weeks of treatment, the average score was 3.2 (standard deviation (SD) 2.0) with gabapentin and 4.6 (SD 2.6) with placebo, and the estimated difference (by longitudinal analysis) was ‐0.92 (95% CI ‐1.75 to ‐0.71).
Withdrawals
Lack of efficacy
One participant taking gabapentin and two taking placebo withdrew from the study due to lack of efficacy.
Adverse events
Twelve participants taking gabapentin and seven taking placebo withdrew from the study due to adverse events.
Any cause
Eighteen participants withdrew from treatment with gabapentin and 13 with placebo. Reasons for withdrawal other than lack of efficacy or adverse events were loss to follow up, withdrawal of consent, and patient decision (gabapentin 5, placebo 4).
Adverse events
Any adverse event
The number of participants experiencing any adverse event was not reported.
Any serious adverse event
The study reported that there were "no significant group differences in the percentage of serious treatment‐emergent adverse events", but not the number of participants experiencing any serious adverse event.
Specific adverse events
Treatment‐emergent adverse events occurring in at least 5% of participants in the gabapentin group were reported. Dizziness, lightheadedness, and sedation were reported significantly more often with gabapentin than placebo.
Discussion
Summary of main results
Limited evidence from a single trial over 12 weeks suggested that a small number of people with fibromyalgia may have a useful degree of pain relief with gabapentin, at a maximum dose of 2400 mg daily, compared with placebo. Other drugs ‐ pregabalin, duloxetine, milnacipran – have better evidence to support their use, but they also only provide benefits to around 10% or so of people with fibromyalgia (Cording 2015; Lunn 2014; Moore 2009). More and larger studies are obviously required to determine whether gabapentin is effective in fibromyalgia, and just what proportion of people would benefit.
Overall completeness and applicability of evidence
The evidence is very weak. A single study with small numbers of participants and events, and with other possible quality issues, means that no reliance can be placed in the results we have for both efficacy and harm.
Quality of the evidence
We downgraded the evidence to very low because of the sparseness of evidence from this single trial (Guyatt 2013a), and because the use of LOCF imputation in the face of greater adverse event withdrawals for active than placebo can lead to overestimation of treatment effects (Moore 2012b). Very low quality evidence means that we are very uncertain about the results.
Potential biases in the review process
We carried out extensive searches of major databases using broad search criteria, and also searched two large clinical trial registries. We feel that it is unlikely that we have missed significant studies.
Agreements and disagreements with other studies or reviews
Two previous systematic reviews found the same single study for gabapentin in fibromyalgia (Häuser 2009; Tzellos 2010), as did previous versions of this review (Moore 2011a; Moore 2014a). Guidelines for treatment of fibromyalgia recommend a multidisciplinary approach with an emphasis on development of coping strategies and exercise, but pharmacological interventions may be useful for some people; these may include anticonvulsants, but most of the evidence supporting their use relates to pregabalin (Fitzcharles 2013; Macfarlane 2016).
History of Earlier Reviews
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
| Gabapentin compared with placebo for fibromyalgia | ||||||
| Patient or population: adults with fibromyalgia Settings: community Intervention: gabapentin Comparison: placebo | ||||||
| Outcomes | Assumed risk ‐ probable outcome with intervention | Corresponding risk ‐ probable outcome with control | Relative effect | Number of | Quality of the evidence | Comments |
| gabapentin | placebo | |||||
| 30% pain reduction at 12 weeks | 38/75 | 23/75 | Not calculated | 1 study, 150 participants | very low | One included study of fewer than 200 participants; LOCF imputation. Downgraded three levels because of small numbers and study limitations |
| 50% pain reduction at 12 weeks | No data | No data | ‐ | ‐ | very low | Outcome not reported |
| PGIC ‐ any category of "better" at 12 weeks | 68/75 | 35/75 | Not calculated | 1 study, 150 participants | very low | One included study of fewer than 200 participants; LOCF imputation; non‐standard outcome ‐ usually top two categories of better, not top three, used Downgraded three levels because of small numbers and study limitations |
| Withdrawals due to adverse events | 12/75 | 7/75 | Not calculated | 1 study, 150 participants | very low | One included study of fewer than 200 participants; few events Downgraded three levels because of small numbers |
| Serious adverse events | "No significant group differences" | ‐ | 1 study, 150 participants | very low | ‐ | |
| Deaths | None reported | ‐ | 1 study, 150 participants | very low | ‐ | |
| CI: Confidence interval; LOCF: last observation carried forward; PGIC: Patient Global Impression of Change | ||||||
| Descriptors for levels of evidence (EPOC 2015): † Substantially different: a large enough difference that it might affect a decision. | ||||||