Haloperidol for long‐term aggression in psychosis
Abstract
Background
Psychotic disorders can lead some people to become agitated. Characterised by restlessness, excitability and irritability, this can result in verbal and physically aggressive behaviour ‐ and both can be prolonged. Aggression within the psychiatric setting imposes a significant challenge to clinicians and risk to service users; it is a frequent cause for admission to inpatient facilities. If people continue to be aggressive it can lengthen hospitalisation. Haloperidol is used to treat people with long‐term aggression.
Objectives
To examine whether haloperidol alone, administered orally, intramuscularly or intravenously, is an effective treatment for long‐term/persistent aggression in psychosis.
Search methods
We searched the Cochrane Schizophrenia Group Trials Register (July 2011 and April 2015).
Selection criteria
We included randomised controlled trials (RCT) or double blind trials (implying randomisation) with useable data comparing haloperidol with another drug or placebo for people with psychosis and long‐term/persistent aggression.
Data collection and analysis
One review author (AK) extracted data. For dichotomous data, one review author (AK) calculated risk ratios (RR) and their 95% confidence intervals (CI) on an intention‐to‐treat basis based on a fixed‐effect model. One review author (AK) assessed risk of bias for included studies and created a 'Summary of findings' table using GRADE.
Main results
We have no good‐quality evidence of the absolute effectiveness of haloperidol for people with long‐term aggression. One study randomising 110 chronically aggressive people to three different antipsychotic drugs met the inclusion criteria. When haloperidol was compared with olanzapine or clozapine, skewed data (n=83) at high risk of bias suggested some advantage in terms of scale scores of unclear clinical meaning for olanzapine/clozapine for 'total aggression'. Data were available for only one other outcome, leaving the study early. When compared with other antipsychotic drugs, people allocated to haloperidol were no more likely to leave the study (1 RCT, n=110, RR 1.37, CI 0.84 to 2.24, low‐quality evidence). Although there were some data for the outcomes listed above, there were no data on most of the binary outcomes and none on service outcomes (use of hospital/police), satisfaction with treatment, acceptance of treatment, quality of life or economics.
Authors' conclusions
Only one study could be included and most data were heavily skewed, almost impossible to interpret and oflow quality. There were also some limitations in the study design with unclear description of allocation concealment and high risk of bias for selective reporting, so no firm conclusions can be made. This review shows how trials in this group of people are possible ‐ albeit difficult. Further relevant trials are needed to evaluate use of haloperidol in treatment of long‐term/persistent aggression in people living with psychosis.
PICOs
Plain language summary
Haloperidol for long‐term aggression in psychosis
Background
People experiencing distressing delusions and hallucinations can often become agitated and aggressive. The antipsychotic drug haloperidol is widely used for the treatment of schizophrenia and psychosis‐induced agitation despite the possibility it can cause a number of serious side effects such as nausea, vomiting, dizziness, restlessness and muscle spasms.
Study characteristics
The Cochrane Schizophrenia Group ran an electronic search for clinical trials involving the use of haloperidol for psychosis‐induced aggression in July 2011 and April 2015. We found one study with 110 participants, diagnosed with schizophrenia or schizoaffective disorder. Participants had been physically aggressive during recent hospitalisation and involved in at least one other aggressive event. The study randomised participants to receive either haloperidol, clozapine or olanzapine.
Key results
Most data presented were impossible to use and it is unclear if haloperidol is effective for reducing aggression or improving mental state for people who are aggressive due to psychosis. There were no data regarding side effects. The number of people leaving the study early from each treatment group was similar.
Quality of evidence
The quality of evidence available is low, only one study with a high risk of selective reporting of results provided data. No firm conclusions can be made until further good‐quality data are available.
Authors' conclusions
Summary of findings
| Haloperidol versus other antipsychotic drugs for long‐term aggression in psychosis | ||||||
| Patient or population: people with long‐term aggression in psychosis | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | haloperidolaloperidol versus other antipsychotic drugs | |||||
| Specific behaviour: aggression ‐ important decrease in aggression | We identified no trials reporting important decrease in aggression. When we compared haloperidol with olanzapine or clozapine, skewed data (n=83) from 1 small trial at high risk of bias suggested some advantage in terms of scores of unclear clinical meaning for olanzapine/clozapine for 'total aggression'. | |||||
| Specific behaviour: repeated need for tranquillisation | We identified no trials reporting repeated need for tranquillisation. | |||||
| Specific behaviours ‐ threat or injury to others/self | We identified no trials reporting threat or injury to others/self. When we compared haloperidol with olanzapine or clozapine, skewed data (n=83) from 1 small trial at high risk of bias suggested some advantage in terms of scores of unclear clinical meaning for olanzapine/clozapine for 'aggression against property' and 'aggression ‐ physical'. Verbal aggression was not changed. | |||||
| Adverse effects ‐ any serious, specific adverse effects | We identified no trials reporting any serious, specific adverse effects. | |||||
| Global outcomes ‐ overall improvement | We identified no trials reporting overall improvement. When we compared haloperidol with olanzapine or clozapine, skewed data (n=83) from 1 small trial at high risk of bias suggested some advantage in terms of scores of unclear clinical meaning for olanzapine/clozapine for a series of mental state ratings. | |||||
| Leaving the study early | Low1 | RR 1.37 | 110 | ⊕⊕⊝⊝ | ‐ | |
| 100 per 1000 | 137 per 1000 | |||||
| Moderate1 | ||||||
| 300 per 1000 | 411 per 1000 | |||||
| High1 | ||||||
| 500 per 1000 | 685 per 1000 | |||||
| Economic outcomes | We identified no trials reporting economic outcomes. | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Moderate control risk equivalent to that of people in the included study. | ||||||
Background
Description of the condition
Psychosis is associated with a number of mental disorders, including schizophrenia and bipolar disorder. Symptoms of psychosis include delusions and hallucinations, which can lead some people to become confused, frightened, agitated or a combination of these (APA 2004). Agitation is characterised by restlessness, excitability and irritability, and for some people, this can result in verbal and physical aggressive behaviour (Mohr 2005). Aggression is a frequent cause for admission to inpatient facilities and detention in restrictive environments, and if people continue to exhibit aggressive behaviour, this can prolong hospitalisation and interfere with discharge (Volavka 2002). Aggression within the psychiatric setting imposes a significant challenge to clinicians who have to manage the risk that the service user may present to themselves, other service users and staff (NICE 2005), whilst attempting to make an accurate diagnosis and formulation (Schleifer 2011).
Aggression can be acute or persistent. The former is often to do with the acute reaction to the so called 'positive' symptoms of the illness whilst persistent aggresssion may be to do with chronically untreated illness, partially treated illness, partially responsive illness or problems not directly a function of illness at all (Hodgins 2008).
Description of the intervention
Haloperidol was developed in 1958 by the Belgian company (Janssen Pharmaceutica), and, along with the earlier development of chlorpromazine, was considered a "psychopharmacological revolution" for the treatment of schizophrenia (López‐Munoz 2009; Figure 1). Newer antipsychotic medication has been developed for the day‐to‐day management of symptoms of schizophrenia, however, haloperidol continues to be in wide use, particularly for the management of psychosis‐induced agitation (Pratt 2008).

Haloperidol structure.
The Lundbeck Institute recommend a dosage of 5 mg/day to 15 mg/day, and under clinical supervision up to 100 mg/day. The plasma half‐life after oral dosage is about 17.5 hours; after intravenous (IV) injection it is approximately 15 hours. The release half‐life of the depot is about three weeks, with a steady state after three months. The therapeutic window of serum concentration in schizophrenia and schizoaffective disorder is 5 ng/mL to 17 ng/mL, and the maximal therapeutic effect occurs at 10 ng/mL (Psychotropics 2003).
Cardiac problems and adverse extrapyramidal effects (e.g. dystonia and akathisia) are associated with conventional antipsychotics such as haloperidol. Experiencing distressing adverse effects may act as a barrier for future engagement with services or treatment (Pratt 2008).
How the intervention might work
Haloperidol is mainly indicated for schizophrenia or other psychosis, mania, violent or dangerously impulsive behaviour, excitement and for short‐term adjunctive management of psychomotor agitation (British National Formulary 2011). Haloperidol is one of the butyrophenone family of antipsychotics (neuroleptics) (López‐Munoz 2009). It is thought that haloperidol prevents the occurrence of delusions and hallucinations by blocking the dopamine D2 receptors in the meso‐cortico‐limbic system; it is less clear how exactly haloperidol may affect aggression. In one study, whilst the dopamine D2 receptor binding was high in the temporal cortex for both haloperidol and atypical antipsychotics, haloperidol induced a significantly higher binding index in the thalamus and striatum than atypical antipsychotics. It is hypothesised that this antidopaminergic activity in the dorsolateral striatum may contribute to the adverse extrapyramidal effects that are associated with typical antipsychotics such as haloperidol (Xiberas 2001).
Why it is important to do this review
Any indication as to whether haloperidol as an intervention will alleviate long‐term aggression in psychosis is important in terms of managing the problem. This review is one of a series covering different treatments used for people whose psychosis is associated with aggression but the majority of these reviews are to do with acute aggression (Table 1).
| Focus of review | Reference |
| 'As required' medication | |
| Benzodiazepines | |
| Chlorpromazine | |
| Containment strategies | |
| Haloperidol (rapid tranquillisation) | |
| Haloperidol + promethazine | |
| Olanzapine IM | |
| Seclusion and restraint | |
| Zuclopenthixol acetate |
Objectives
To examine whether haloperidol alone, administered orally, intramuscularly or intravenously, is an effective treatment for long‐term aggression in psychosis.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials (RCT) or double blind trials (implying randomisation). We excluded quasi‐randomised studies, such as those allocating by alternate days of the week.
Types of participants
People exhibiting chronic/persistent (i.e. not acute episodes requiring rapid tranquillisation) agitation or aggression, or both concurrently with psychotic illness, regardless of age or sex, and definition used of 'chronic/persistent'.
Types of interventions
1. Haloperidol
Alone, any dose via any route of administration.
Compared with:
a. Other antipsychotic
Any dose via any route of administration.
b. Benzodiazepine
Any dose via any route of administration.
c. Anticonvulsant alone
Any dose via any route of administration.
d. Placebo or no intervention
Types of outcome measures
We planned to divide, where possible, outcomes into short‐term (up to three months), medium‐term (over three and up to six months) and long‐term (over six months).
Primary outcomes
1. Specific behaviours
1.1 Aggression ‐ important decrease in aggression ‐ medium‐term.
1.2 Repeated need for tranquillisation.
2. Adverse effects ‐ any serious, specific adverse effects ‐ medium‐term
Secondary outcomes
1. Specific behaviours
1.1 Self harm, including suicide
1.2 Injury to others
1.3 Aggression
1.3.1 Another episode of aggression ‐ other time periods
1.3.2 Clinically important change in aggression
1.3.3 No change in aggression
1.3.4 Average endpoint in aggression score
1.3.5 Average change in aggression scores
2. Global outcomes
2.1 Overall improvement
2.2 Use of restraints/seclusion
2.3 Relapse ‐ as defined by each study
2.4 Recurrence of violent incidents
2.5 Needing extra visits from the doctor
2.6 Refusing oral medication
2.7 Average endpoint in global score
2.8 Average change in global scores
2.9 Average dose of drug
3. Service outcomes
3.1 Duration of hospital stay
3.2 Re‐admission
3.3 Clinically important engagement with services
3.4 Engagement with services
3.5 Average endpoint engagement score
3.6 Average change in engagement scores
4. Mental state
4.1 Clinically important change in general mental state
4.2 Any change in general mental state
4.3 Average endpoint general mental state score
4.4 Average change in engagement scores
5. Adverse effects
5.1 Death
5.2 Any non‐serious general adverse effects
5.3 Any serious, specific adverse effects ‐ other time periods
5.4 Average endpoint general adverse effect score
5.5 Average change in general adverse effect scores
5.6 Clinically important change in specific adverse effects
5.7 Any change in specific adverse effects
5.8 Average endpoint specific adverse effects
5.9 Average change in specific adverse effects
6. Leaving the study early
6.1 For specific reasons
6.2 For general reasons
7. Satisfaction with treatment
7.1 Recipient of treatment not satisfied with treatment
7.2 Recipient of treatment average satisfaction score
7.3 Recipient of treatment average change in satisfaction scores
7.4 Informal treatment provider not satisfied with treatment
7.5 Informal treatment providers' average satisfaction scores
7.6 Informal treatment providers' average change in satisfaction scores
7.7 Professional providers not satisfied with treatment
7.8 Professional providers' average satisfaction score
7.9 Professional providers' average change in satisfaction scores
8. Acceptance of treatment
8.1 Accepting treatment
8.2 Average endpoint acceptance score
8.3 Average change in acceptance scores
9. Quality of life
9.1 Clinically important change in quality of life
9.2 Any change in quality of life
9.3 Average endpoint quality of life score
9.4 Average change in quality of life score
9.5 Clinically important change in specific aspects of quality of life
9.6 Any change in specific aspects of quality of life
9.7 Average endpoint specific aspects of quality of life
9.8 Average change in specific aspects of quality of life
10. Economic outcomes
10.1 Direct costs
10.2 Indirect costs
11. 'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2011) and GRADE profiler (GRADEPRO) to import data from Review Manager 5 (RevMan 2012) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes we rated as important to patient‐care and decision making. We aimed to select the following main outcomes for inclusion in the 'Summary of findings' table if obtainable.
1. Specific behaviour: aggression ‐ important decrease in aggression ‐ medium‐term.
2. Specific behaviour: repeated need for tranquillisation.
3. Specific behaviours ‐ threat or injury to others/self.
4. Adverse effects ‐ any serious, specific adverse effects.
5. Global outcomes ‐ overall improvement.
6. Economic outcomes.
Search methods for identification of studies
Electronic searches
The Information Specialist searched the Cochrane Schizophrenia Group's Study‐Based Register of Trials, July 2011 and 1 April 2015 using the phrase:
(*haloperi* or *R‐1625* or *haldol* or *alased* or *aloperidi* or *bioperido* or *buterid* or *ceree* or *dozic* or *duraperido* or *fortuna* or *serena* or *serenel* or *seviu* or *sigaperid* or *sylad* or *zafri* in intervention of STUDY) AND (*aggress* or *violen* or *agitat* or *tranq* in title, abstract, index terms of REFERENCE or intervention of STUDY)
The Cochrane Schizophrenia Group's Register of Trials is compiled by systematic searches of major resources (including MEDLINE, CINAHL, Embase, AMED, BIOSIS, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature and conference proceedings (see Group Module). There are no language, date, document type, or publication status limitations for inclusion of records into the register
Searching other resources
1. Reference searching
We inspected references of all included and excluded studies for further relevant studies.
2. Personal contact
We contacted the first author of each included study for information regarding unpublished trials.
Data collection and analysis
Selection of studies
One review author (MP) independently inspected citations from the searches and identified relevant abstracts. A second review author (AK) independently re‐inspected the majority to ensure reliability. Where disputes arose, we acquired the full report for more detailed scrutiny. We obtained full reports of the abstracts meeting the review criteria and one review author (AK) inspected these. One review author (MP) re‐inspected the reports in order to ensure reliable selection. Where it was not possible to resolve disagreement by discussion, we attempted to contact the authors of the study for clarification.
Data extraction and management
1. Extraction
One review author (AK) extracted data from the included study and extracted data presented only in graphs and figures whenever possible. If necessary, we attempted to contact authors through an open‐ended request in order to obtain missing information or for clarification whenever necessary.
2. Management
2.1 Forms
One review author (AK) extracted data onto standard, simple forms.
2.2 Scale‐derived data
We did not have any continuous outcomes but if we had come across them we would only have included continuous data from rating scales only if:
a. the psychometric properties of the measuring instrument were described in a peer‐reviewed journal (Marshall 2000); and
b. the measuring instrument had not been written or modified by one of the trialists for that particular trial.
Ideally the measuring instrument would be either i. a self report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly, in 'Description of studies' we would have noted if this was the case or not.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. However, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided to primarily use endpoint data, and only use change data if the endpoint data were not available. We aimed to combine endpoint and change data in the analysis as we would have preferred to use mean differences (MD) rather than standardised mean differences (SMD) throughout (Higgins 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we planned to apply the following standards to relevant continuous data before inclusion.
For endpoint data from studies including fewer than 200 participants:
a. when a scale starts from the finite number zero, we would subtract the lowest possible value from the mean, and divide this by the standard deviation (SD). If this value is lower than one, it strongly suggests that the data are skewed and we would exclude these data. If this ratio is higher than one but less than two, there is suggestion that the data are skewed: we would enter these data and test whether their inclusion or exclusion would change the results substantially. Finally, if the ratio is larger than two, we would include these data, because it is less likely that they are skewed (Altman 1996; Higgins 2011).
b. if a scale starts from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), which can have values from 30 to 210 (Kay 1986)), we would modify the calculation described above to take the scale starting point into account. In these cases, skewed data are present if 2 SD > (S ‐ S min), where S is the mean score and 'S min' is the minimum score.
Please note: we would enter all relevant data from studies of more than 200 participants in the analysis irrespective of the above rules, because skewed data pose less of a problem in large studies. We would also enter all relevant change data, as when continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether or not data are skewed.
2.5 Common measure
To facilitate comparison between trials, we would have converted variables that could be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary
Where possible, we aimed to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS (Kay 1986), this could be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we would have used the primary cut‐off presented by the original authors.
Assessment of risk of bias in included studies
One review author (AK) independently assessed risk of bias by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
Where advice was sought, if there was any disagreement the final rating was made by consensus, with the involvement of the second review author. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted the authors of the studies in order to obtain further information. Non‐concurrence in quality assessment was reported, but if disputes arose as to which category a trial was to be allocated, again, resolution was made by discussion.
We noted the level of risk of bias in both the text of the review and in the summary of findings Table for the main comparison.
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). For statistically significant results, we used 'Summary of findings' tables to calculate the number needed to treat for an additional beneficial (NNTB) or harmful (NNTH) outcome and its 95% CI.
2. Continuous data
We found no continuous outcomes in the included study. If we had found continuous outcomes, we would have estimated the MD between groups, preferring not to calculate effect size measures (SMD). However, if scales of very considerable similarity had been used, we would have presumed that there was a small difference in measurement, and calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. First, authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
We had one included study, which was not a cluster trial. If we had found cluster trials, we would have assessed whether clustering was accounted for in primary studies. Where clustering had been incorporated into the analysis of primary studies, we would have presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect. Where it was not incorporated, we would have attempted to contact the first authors of studies to obtain intra‐class correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). If the ICC was not reported, we would have assumed it to be 0.1 (Ukoumunne 1999).
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a washout phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). Our included study was not a cross‐over trial, but if we had found cross‐over trials we would only have used data from the first phase.
3. Studies with multiple treatment groups
Our included study did not involve more than two treatment arms. If we had found studies with more than two treatment arms we would have, if relevant, presented the additional treatment arms in comparisons. If data were binary, we would have simply added and combined them within the two‐by‐two table. If data were continuous, we would have combined data following the formula in Section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Where the additional treatment arms were not relevant, we would not have used these data.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, data must lose credibility (Xia 2009). In our included study, more than 50% of data were accounted for. For any particular outcome, if more than 50% of data had been unaccounted for, we would not have reproduced these data or used them within analyses (except for the outcome 'leaving the study early').
2. Binary
Our included study clearly described attrition for binary outcomes. If we had found a case where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we would have presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis). Participants leaving the study early would have all been assumed to have the same rates of negative outcome as participants who completed, with the exception of the outcome of death and adverse effects. For these outcomes, the rate of participants who stayed in the study ‐ in that particular arm of the trial ‐ would have been used for participants who did not. We did not undertake a sensitivity analysis to test how prone the primary outcome measures were to change when data only from people who completed the study to that point were compared to the intention‐to‐treat analysis using the above assumptions, as there was only one included study.
3. Continuous
3.1 Attrition
If we had found any case where attrition for a continuous outcome was between 0% and 50%, and reported data only from people who complete the study to that point, we would have presented and used these data.
3.2 Standard deviations
If we had found any case where SDs were not reported for continuous data, we would first try to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data, but an exact standard error (SE) and CIs available for group means, and either a P value or t value available for differences in mean, we would have calculated them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). When only the SE was reported, we would have calculated SDs using the formula SD=SE × square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions present detailed formula for estimating SDs from P values, t or F values, CIs, ranges or other statistics (Higgins 2011). Where this formula did not apply, we would have calculated the SDs according to a validated imputation method that was based on the SDs of the other included studies (Furukawa 2006).
3.3 Last observation carried forward
We anticipated that some studies would employ the method of last observation carried forward (LOCF) within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, where LOCF data were used in the trial, if less than 50% of the data were assumed, we would have reproduced these data and indicated that they were the product of LOCF assumptions.
Assessment of heterogeneity
1. Clinical heterogeneity
If we had included more than one study, we would have considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We would have inspected all studies for clearly outlying people or situations that we had not predicted would arise. If such situations or participant groups arose, we would have discussed these fully.
2. Methodological heterogeneity
If we had included more than one study, we would have considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We would have inspected all studies for clearly outlying methods that we had not predicted would arise. If such methodological outliers arose, we would have discussed these fully.
3. Statistical heterogeneity
3.1 Visual inspection
We would have visually inspected graphs to investigate the possibility of statistical heterogeneity.
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in Section 10.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We tried to locate protocols of included randomised trials. If the protocol was available, we compared outcomes in the protocol and in the published report. If the protocol was not available, we compared outcomes listed in the methods section of the trial report with actually reported results.
2. Funnel plot
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes because we included one study.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. However, there is a disadvantage to the random‐effects model. It puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose a fixed‐effect model for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
We did not anticipate any subgroup analyses.
2. Investigation of heterogeneity
If inconsistency had been high, we would have reported it. First, we would have investigated whether data were entered correctly. Second, if data were correct, we would have visually inspected the graph and successively removed studies outside of the company of the rest to see if homogeneity was restored. For this review, we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would have presented the data. If not, data would not be pooled and issues would be discussed. We know of no supporting research for this 10% cut‐off but are investigating use of prediction intervals as an alternative to this unsatisfactory state.
If unanticipated clinical or methodological heterogeneity were obvious, we would have simply stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
We did not undertake a sensitivity analysis as there was only one included trial.
Results
Description of studies
For substantive descriptions of studies, see Characteristics of included studies, Characteristics of excluded studies, and Characteristics of studies awaiting classification tables.
Results of the search
The search (July 2011) yielded 665 references of potentially eligible studies plus four additional references identified through other sources, of which we obtained five for second assessment. We excluded two papers for not meeting the inclusion criteria (due to wrong population) and one that awaits assessment as the full text is not obtainable (Lewis 2006). Finally, we included one study (two reports) in the present review (Krakowski 2006; Figure 2). The update search of April 2015 yielded no new studies but 19 more reports of the trials we had already identified (Figure 3).

Study flow diagram.

Study flow diagram (pre‐submission search of 1 April 2015).
Included studies
The Characteristics of included studies table provided details of the study that met the inclusion criteria (Krakowski 2006).
1. Length of study
The duration of the study was 12 weeks.
2. Participants
2.1 Clinical state
Participants were "physically assaultive" inpatients in state psychiatric facilities required to have "a clearly confirmed episode of physical assault directed at another person" and "some persistence of aggression, as evidenced by the presence of some other aggressive event, whether physical or verbal or against property."
2.2 Diagnosis
Participants were diagnosed with "schizophrenia or schizoaffective disorder (using diagnostic criteria form the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition)."
2.3 Exclusions
Exclusion criteria included people with a history of non‐response or intolerance to interventions, people with medical conditions adversely affected by interventions, people who received a depot antipsychotic drug within 30 days before randomisation, and people hospitalised for over one year.
2.4 Age
Participants ranged from 18 to 60 years.
3. Study size
The study initially randomised 110 participants; 70 participants completed the study.
4. Interventions
The study compared haloperidol with olanzapine and clozapine.
4.1 Dosing
4.1.1 Haloperidol: first six weeks, target endpoint 20 mg/day (mean dose 19.6 mg/day); last six weeks, range 10 mg/day to 30 mg/day (mean dose 23.3 mg/day).
4.1.2 Olanzapine: first six weeks, target endpoint 20 mg/day (mean dose 19.8 mg/day); last six weeks, range 10 mg/day to 35 mg/day (mean dose 24.7 mg/day).
4.1.3 Clozapine: first six weeks, target endpoint 500 mg/day (mean dose 457.1 mg/day); last six weeks, range 200 mg/day to 800 mg/day (mean dose 565.5 mg/day).
5. Leaving the study early
Sixteen people allocated to haloperidol discontinued the study, mainly due to clinical deterioration (n=7), but also due to adverse effects (n=4), withdrawn consent (n=4), or other reasons (n=1). Eleven people allocated to olanzapine discontinued the study, due to withdrawn consent (n=4), clinical deterioration (n=2), adverse effects (n=1) or other reasons (n=4). Thirteen people allocated to clozapine discontinued the study, mainly due to withdrawn consent (n=6), but also due to adverse effects (n=3), clinical deterioration (n=2) or other reasons (n=2).
6. Outcomes
We were able to use the outcome 'leaving the study early'.
6.1 Symptom scales
The study used the Modified Overt Aggression Scale (MOAS) and PANSS to assess treatment effects; however, data from these scales were skewed.
a. Aggression: Modified Overt Aggression Scale (Kay 1986)
The MOAS uses three categories of external aggression, that is, physical aggression against other people, verbal aggression and physical aggression against objects. The score for each type of incident represents the number of incidents over time as well as their severity. The overall score assigns different weightings to each category of aggression, thus represents the number of incidents over time, their severity and the type of aggression (a high score is poor).
b. Mental state: PANSS (Kay 1986)
The PANSS contains three subscales; positive, negative and a general psychopathology subscale. The study used change in score rather than endpoint scores, so a positive score is an improvement in clinical symptoms.
Excluded studies
Two studies did not meet all the inclusion criteria (Volavka 2002, 24 reports; Singh 1981, one report). We had to exclude these because it was clear that participants had not been specifically selected for persistence of aggression as well as psychotic illness, rather they were people with schizophrenia or schizoaffective disorder on whom hostility and aggression was being measured.
1. Awaiting classification
One study is awaiting classification as it is a conference proceeding. This did not report the full characteristics or all the outcome data necessary (Lewis 2006, 1 report). We will search for a full‐text publication of this study at the next review update but admit that it may never be published since the abstract was published in 2006.
2. Ongoing
We identified no ongoing studies.
Risk of bias in included studies
Overall, we judged the risk of bias for the included study to be unclear. It is possible that the study slightly overestimated any true positive effects and slightly underestimated any true negative effects (see Figure 4; Figure 5).

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
The method of randomisation was not explicit (other than stating block randomisation) and there was no further information on how randomisation and allocation concealment were undertaken. We rated selection bias as unclear.
Blinding
The study was conducted under double‐blind conditions but no details were given of how the blinding was ensured. It was stated that raters who performed all the clinical assessment were blind to treatment group. We rated performance and detection bias as unclear.
Incomplete outcome data
There was a low risk of bias as the study clearly reported attrition data in a table and performed an intention‐to‐treat analysis.
Selective reporting
The study did not report data on adverse effects or numerical data for the additional medications used to control adverse effects. We rated this to be a high risk of bias.
Other potential sources of bias
Eli Lilly and Company provided supplemental funding for "encapsulation of the medications", but the authors stressed that "the overall experimental design, data acquisition, statistical analyses, and interpretation of the results were implemented with no input from any of the pharmaceutical companies". Overall, we rated the risk to be unclear.
Effects of interventions
We categorised data from the one study (n=110) into three comparisons (Krakowski 2006, 12 reports).
1. Comparison 1: haloperidol versus olanzapine
There were two outcomes for haloperidol versus olanzapine.
1.1 Aggression: average scores (MOAS, high=poor, data skewed)
These continuous data had such large SDs as to suggest that analysis within Review Manager 5 would be inadvisable. Therefore, we presented them in a data table (Analysis 1.1).
1.2 Mental state: average change score (PANSS, positive=improvement, data skewed)
These continuous data had such large SDs as to suggest that analysis within Review Manager 5 would be inadvisable. Therefore, we presented them in a data table (Analysis 1.2).
2. Comparison 2: haloperidol haloperidol versus clozapine
There were two outcomes for haloperidol versus clozapine.
2.1 Aggression: average scores (MOAS, high=poor, data skewed)
These continuous data (one RCT) had such large SDs as to suggest that analysis within Review Manager 5 would be inadvisable. We presented data in a data table (Analysis 2.1).
2.2 Mental state: average change score (PANSS, positive=improvement, data skewed)
These continuous data were from one small study and were very skewed. We presented them in a data table (Analysis 2.2).
3. Comparison 3: haloperidolhaloperidol versus other antipsychotic drugs
There was one outcome for haloperidol versus other antipsychotic drug.
3.1 Leaving the study early
We found no evidence of a clear difference between haloperidol and other antipsychotic drugs for leaving the study early (1 RCT, n=110, RR 1.37, 95% CI 0.84 to 2.24, Analysis 3.1).
4. Missing outcomes
Although the study provided data for some outcomes, there were no data on most of the binary outcomes and none on service outcomes (use of hospital/police), satisfaction with treatment, acceptance of treatment, quality of life or economic outcomes.
Discussion
Summary of main results
This review compared haloperidol versus any other intervention for people with psychosis exhibiting long‐term aggression/agitation. There were only limited data available and these are summarised in summary of findings Table for the main comparison.
1. Aggression ‐ important decrease in aggression
We identified no trials reporting an important decrease in aggression. For the comparison of haloperidol versus olanzapine or clozapine, one small trial provided continuous but skewed data that was at high risk of bias suggesting some advantage in terms of scores of unclear clinical meaning for olanzapine/clozapine for 'total aggression' (Krakowski 2006). We do recognise the very great problems undertaking trials with this participant group. This review, its included study and at least one other (Lewis 2006) illustrate that randomisation is possible. This group of people is sufficiently prevalent, and this or other interventions used frequently enough, to merit investment of time and effort in evaluation of possible means of treatment.
2. Repeated need for tranquillisation
We identified no trials reporting repeated need for tranquillisation.
3. Specific behaviours ‐ threat or injury to others/self
We identified no trials reporting threat or injury to others/self. For the comparison of haloperidol versus olanzapine or clozapine, one small trial provided skewed data at high risk of bias suggesting some advantage in terms of scores of unclear clinical meaning for olanzapine/clozapine for 'aggression against property' and 'aggression ‐ physical' (Krakowski 2006). Verbal aggression was not changed. Data were limited making drawing of conclusions ill‐advised.
4. Adverse effects ‐ any serious, specific adverse effects
We identified no trials reporting any serious, specific adverse effects.
5. Global outcomes ‐ overall improvement
We identified no trials reporting overall improvement. For the comparison haloperidol versus olanzapine or clozapine, one small trial provided skewed data at high risk of bias suggesting some advantage in terms of scores of unclear clinical meaning for olanzapine/clozapine for a series of mental state ratings (Krakowski 2006). These outcomes were as close as we could find for 'global state' and were not informative.
6. Economic outcomes
We identified no trials reporting economic outcomes.
7. Leaving the study early
The Krakowski 2006 study (total n=110) reported leaving the study early and there was no clear evidence of a difference between haloperidol and other antipsychotic drugs (about 50% versus about 33%, RR 1.37, CI 0.84 to 2.24, Analysis 3.1). Recognising that this was a small study, there was no evidence that, across three months, haloperidol was clearly less well tolerated than the other more modern drugs. Although attrition was high, most people in both groups continued on their allocated drugs ‐ and, with this difficult group of people, this could be seen as important.
Overall completeness and applicability of evidence
1. Completeness
The study reported method and data mostly with clarity, enabling extraction of fairly reliable information. However, this study did not address all the outcomes specified for the review and the sample size was small. Initially it seemed that the Krakowski 2001 report of Krakowski 2006 estimated that 210 people were to be randomised but this seems to have proceeded to the smaller Krakowski 2006 report (n=110). The Krakowski 2006 report did not have aggression measured by the Buss Durkee Hostility scale or the Nurses' Observation Scale for In‐patient Evaluation (NOSIE), neither did it report mental state using Clinical Global Impression, Barratt Impulsivity Scale (trait impulsivity) or Wisconsin Card Sorting Test (cognitive) as had been listed in the Krakowski 2001 report. This does suggest that there was selective reporting in the Krakowski 2006 trial.
2. Applicability
The included study was highly relevant to the review question ‐ the participants, interventions and outcomes were directly comparable to the objectives of the review (Krakowski 2006). One study awaiting classification investigated haloperidol versus risperidone for aggression in male inmates with psychosis, using the MOAS and Barratt Impulsivity Scale, but could not be used in this review because the conference proceeding reported no data and we have not identified a full paper (Lewis 2006). The study was small (n=40) and specific to a certain population (prison inmates), so it might not be possible to generalise the findings to other populations. However, such a trial shows that more evidence is potentially there to incorporate into the questions asked by this review.
Quality of the evidence
Data were limited, low quality and difficult to interpret (summary of findings Table for the main comparison).
Potential biases in the review process
We are not aware of any biases in the review process. It is possible that we did not identify small studies due to a degree of publishing bias (Egger 1997), but we consider that we have identified all larger relevant studies.
Agreements and disagreements with other studies or reviews
We know of no other systematic reviews for the use of haloperidol for treating long‐term aggression in people with psychosis.
Study flow diagram (pre‐submission search of 1 April 2015).
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
| Study | Intervention | Mean | SD | N |
| total | ||||
| Krakowski 2006 | Haloperidol | 40.9 | 68.88 | 36 |
| Krakowski 2006 | Olanzapine | 32.7 | 69.83 | 37 |
| aggression ‐ physical | ||||
| Krakowski 2006 | Haloperidol | 20.7 | 30.61 | 36 |
| Krakowski 2006 | Olanzapine | 14.1 | 31.03 | 37 |
| aggression ‐ against property | ||||
| Krakowski 2006 | Haloperidol | 4.7 | 9.18 | 36 |
| Krakowski 2006 | Olanzapine | 2.7 | 6.21 | 37 |
| aggression ‐ verbal | ||||
| Krakowski 2006 | Haloperidol | 15.6 | 35.20 | 36 |
| Krakowski 2006 | Olanzapine | 16.0 | 29.48 | 37 |
Comparison 1 HALOPERIDOL versus OLANZAPINE, Outcome 1 Aggression: average scores (Modified Overt Aggression Scale (MOAS), high=poor, data skewed).
| Study | Intervention | Mean | SD | N |
| general psychopathology | ||||
| Krakowski 2006 | Haloperidol | 0.64 | 8.2 | 36 |
| Krakowski 2006 | Olanzapine | 2.69 | 5.5 | 37 |
| negative symptoms | ||||
| Krakowski 2006 | Haloperidol | 0.44 | 4.6 | 36 |
| Krakowski 2006 | Olanzapine | 0.72 | 3.0 | 37 |
| positive symptoms | ||||
| Krakowski 2006 | Haloperidol | ‐0.5 | 5.3 | 36 |
| Krakowski 2006 | Olanzapine | 1.41 | 3.6 | 37 |
| total | ||||
| Krakowski 2006 | Haloperidol | 0.58 | 1.52 | 36 |
| Krakowski 2006 | Olanzapine | 4.83 | 9.7 | 37 |
Comparison 1 HALOPERIDOL versus OLANZAPINE, Outcome 2 Mental state: average change score (Positive and Negative Syndrome Scale (PANSS), positive=improvement, data skewed).
| Study | Intervention | Mean | SD | N |
| total | ||||
| Krakowski 2006 | Haloperdiol | 40.9 | 68.88 | 36 |
| Krakowski 2006 | Clozapine | 25.1 | 43.45 | 37 |
| aggression ‐ physical | ||||
| Krakowski 2006 | Haloperdiol | 20.7 | 30.61 | 36 |
| Krakowski 2006 | Clozapine | 10.3 | 24.83 | 37 |
| aggression ‐ against property | ||||
| Krakowski 2006 | Haloperdiol | 4.7 | 9.18 | 36 |
| Krakowski 2006 | Clozapine | 2.6 | 3.10 | 37 |
| aggression ‐ verbal | ||||
| Krakowski 2006 | Haloperdiol | 15.6 | 35.20 | 36 |
| Krakowski 2006 | Clozapine | 12.2 | 37 | |
Comparison 2 HALOPERIDOL versus CLOZAPINE, Outcome 1 Aggression: average scores (Modified Overt Aggression Scale (MOAS), high=poor, data skewed).
| Study | Intervention | Mean | SD | N |
| general psychopathology | ||||
| Krakowski 2006 | Haloperidol | 0.64 | 8.2 | 36 |
| Krakowski 2006 | Clozapine | 1.43 | 37 | |
| negative symptoms | ||||
| Krakowski 2006 | Haloperidol | 0.44 | 4.6 | 36 |
| Krakowski 2006 | Clozapine | ‐0.56 | 4.9 | 37 |
| positive symptoms | ||||
| Krakowski 2006 | Haloperidol | ‐0.5 | 5.3 | 36 |
| Krakowski 2006 | Clozapine | 1.54 | 5.0 | 37 |
| total | ||||
| Krakowski 2006 | Haloperidol | 0.58 | 1.52 | 36 |
| Krakowski 2006 | Clozapine | 2.39 | 14.2 | 37 |
Comparison 2 HALOPERIDOL versus CLOZAPINE, Outcome 2 Mental state: average change score (Positive and Negative Syndrome Scale (PANSS), positive=improvement, data skewed).
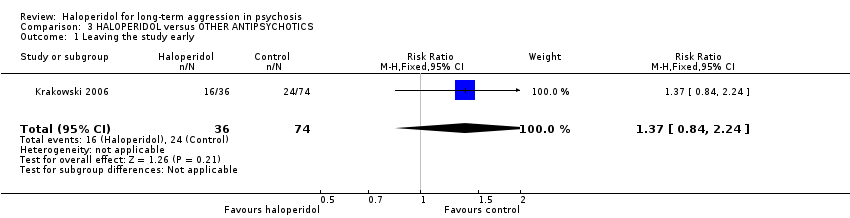
Comparison 3 HALOPERIDOL versus OTHER ANTIPSYCHOTICS, Outcome 1 Leaving the study early.
| Methods | Allocation: randomised, clearly described. Blinding: double, described and tested. Duration: 52 weeks. |
| Participants | Diagnosis: have psychoses. N=300*. Age: any. Sex: both. History: persistently aggressive. |
| Interventions | 1. Haloperidol: dose flexible within recommended limits. N=150. 2. Other antipsychotic drug: dose flexible within recommended limits. N=150. |
| Outcomes | Outcomes grouped into short term (3 months or less), medium term (> 3 and up to 6 months or less) and long‐term (> 6 months). Aggression: events. Adverse effects. Specific behaviours ‐ self harm including suicide, injury to others, aggression. Global outcomes ‐ overall improvement, use of additional medication, use of restraints/seclusion. Service outcomes ‐ duration of hospital stay, re‐admission. Mental state ‐ no clinically important change in general mental state. Leaving the study early ‐ why. Satisfaction with and acceptance of treatment. Quality of life ‐ no clinically important change in quality of life. Economic outcomes. |
| Notes | * Powered to be able to identify a difference of ˜ 20% between groups for primary outcomes with adequate degree of certainty. |
| N: number of participants. | |
| Haloperidol versus other antipsychotic drugs for long‐term aggression in psychosis | ||||||
| Patient or population: people with long‐term aggression in psychosis | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | haloperidolaloperidol versus other antipsychotic drugs | |||||
| Specific behaviour: aggression ‐ important decrease in aggression | We identified no trials reporting important decrease in aggression. When we compared haloperidol with olanzapine or clozapine, skewed data (n=83) from 1 small trial at high risk of bias suggested some advantage in terms of scores of unclear clinical meaning for olanzapine/clozapine for 'total aggression'. | |||||
| Specific behaviour: repeated need for tranquillisation | We identified no trials reporting repeated need for tranquillisation. | |||||
| Specific behaviours ‐ threat or injury to others/self | We identified no trials reporting threat or injury to others/self. When we compared haloperidol with olanzapine or clozapine, skewed data (n=83) from 1 small trial at high risk of bias suggested some advantage in terms of scores of unclear clinical meaning for olanzapine/clozapine for 'aggression against property' and 'aggression ‐ physical'. Verbal aggression was not changed. | |||||
| Adverse effects ‐ any serious, specific adverse effects | We identified no trials reporting any serious, specific adverse effects. | |||||
| Global outcomes ‐ overall improvement | We identified no trials reporting overall improvement. When we compared haloperidol with olanzapine or clozapine, skewed data (n=83) from 1 small trial at high risk of bias suggested some advantage in terms of scores of unclear clinical meaning for olanzapine/clozapine for a series of mental state ratings. | |||||
| Leaving the study early | Low1 | RR 1.37 | 110 | ⊕⊕⊝⊝ | ‐ | |
| 100 per 1000 | 137 per 1000 | |||||
| Moderate1 | ||||||
| 300 per 1000 | 411 per 1000 | |||||
| High1 | ||||||
| 500 per 1000 | 685 per 1000 | |||||
| Economic outcomes | We identified no trials reporting economic outcomes. | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Moderate control risk equivalent to that of people in the included study. | ||||||
| Focus of review | Reference |
| 'As required' medication | |
| Benzodiazepines | |
| Chlorpromazine | |
| Containment strategies | |
| Haloperidol (rapid tranquillisation) | |
| Haloperidol + promethazine | |
| Olanzapine IM | |
| Seclusion and restraint | |
| Zuclopenthixol acetate |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Aggression: average scores (Modified Overt Aggression Scale (MOAS), high=poor, data skewed) Show forest plot | Other data | No numeric data | ||
| 1.1 total | Other data | No numeric data | ||
| 1.2 aggression ‐ physical | Other data | No numeric data | ||
| 1.3 aggression ‐ against property | Other data | No numeric data | ||
| 1.4 aggression ‐ verbal | Other data | No numeric data | ||
| 2 Mental state: average change score (Positive and Negative Syndrome Scale (PANSS), positive=improvement, data skewed) Show forest plot | Other data | No numeric data | ||
| 2.1 general psychopathology | Other data | No numeric data | ||
| 2.2 negative symptoms | Other data | No numeric data | ||
| 2.3 positive symptoms | Other data | No numeric data | ||
| 2.4 total | Other data | No numeric data | ||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Aggression: average scores (Modified Overt Aggression Scale (MOAS), high=poor, data skewed) Show forest plot | Other data | No numeric data | ||
| 1.1 total | Other data | No numeric data | ||
| 1.2 aggression ‐ physical | Other data | No numeric data | ||
| 1.3 aggression ‐ against property | Other data | No numeric data | ||
| 1.4 aggression ‐ verbal | Other data | No numeric data | ||
| 2 Mental state: average change score (Positive and Negative Syndrome Scale (PANSS), positive=improvement, data skewed) Show forest plot | Other data | No numeric data | ||
| 2.1 general psychopathology | Other data | No numeric data | ||
| 2.2 negative symptoms | Other data | No numeric data | ||
| 2.3 positive symptoms | Other data | No numeric data | ||
| 2.4 total | Other data | No numeric data | ||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Leaving the study early Show forest plot | 1 | 110 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.37 [0.84, 2.24] |