Interventions pour l'hyponatrémie hypotonique non hypovolémique chronique
References
References to studies included in this review
References to studies excluded from this review
Additional references
References to other published versions of this review
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Jump to:
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'Eligible patients were randomised ... using an interactive voice recognition system programmed with a computer‐generated randomisation scheme. Randomization was stratified by study centre in blocks of 4.' Comment: the randomisation procedure produced groups in the overall study that were not entirely balanced in age and sex, but fairly well‐balanced in terms of medical history and characteristics related to the heart failure. However, randomisation was not stratified for serum sodium concentration. Despite fairly similar numbers of participants in both the experimental and the control group, no baseline data available specifically for subgroup with hyponatraemia, making judgement on risk of bias difficult. The sequence generation in the overall study was good and low risk. However this is a subgroup analysis with low numbers relative to the number of groups and the sequence did not stratify for them. |
| Allocation concealment (selection bias) | Low risk | Quote: 'Patients were screened within 72 hours and randomised within 96 hours of admission. Randomization occurred between 8 and 9 AM; the study drug was to be administered at 9 AM.' |
| Blinding of participants and personnel (performance bias) | High risk | Quote: '...patients...randomised to receive ... tolvaptan or placebo. ' and '...was a multicenter, randomised, double‐blind, placebo‐controlled... trial' Additional info provided by the sponsor: 'Both OPC‐41061 and placebo tablets were identical in appearance'. Comment: blinding attempted, although participants are unlikely to be fully blinded due to important increases in urine output. Also, co‐interventions were not reported in detail and included standard heart failure treatment left at discretion of investigator and may have differed between groups according to baseline serum sodium concentration. This could have influenced outcome measurement of serum sodium concentrations. |
| Blinding of outcome assessment (detection bias) | Low risk | Additional info provided by the sponsor: 'Did not have separate outcome assessors in this trial'. Comment: blinding outcome assessors attempted and measured outcome objective. |
| Incomplete outcome data (attrition bias) | Low risk | Comment: 10% attrition for outcome death at 60 days; only 1 participant (1%) excluded from analysis because of missing follow‐up serum sodium concentration measurement. |
| Selective reporting (reporting bias) | High risk | Comment: protocol available with matched primary and secondary outcomes for the overall study. However, results included in this review for the subgroup with hyponatraemia are based on post‐hoc analyses of the data, with reported outcomes not matching the primary and secondary outcomes of the original study. |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | treatment group 1
Treatment group 2
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'Eligible patient ... were stratified according to volume status and then randomly assigned in a 1:1:1 ratio...' Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: 'This double‐blind, placebo controlled ... study', and '...patients ... were...assigned to ... or placebo', and 'All patients were prescribed fluid restriction (not to exceed 500 mL in any 3‐hour period or 2.0 L in a 24‐hour period for both oral and parenteral fluids) prior to baseline and throughout the study, unless changes were necessary for safety reasons', and 'Four patients, 2 given conivaptan 80 mg/d and 2 given placebo, violated the 2‐L/d fluid restriction.' Comment: blinding attempted, and although participants and personnel are unlikely to be fully blinded due to important increases in urine output, all measured outcomes short‐term and generally objective. Fluid restriction was prescribed in both groups, violations reported and similar between groups |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding outcome assessors probably attempted and main outcomes objective. Unlikely for participants to have been fully blinded, and plausible 'high risk' of bias for self‐reported outcome of thirst |
| Incomplete outcome data (attrition bias) | Low risk | Comment: overall 13% attrition; reasons well documented, most occurred in the placebo group due to adverse events. Given <20% attrition and limited opportunity for introducing bias in such a short‐term study ‐ we judged it 'Low risk'. |
| Selective reporting (reporting bias) | Low risk | Comment: no protocol, short term study (≤1 week), primary outcome death and secondary outcomes related to serum sodium concentration and rapid increase in serum sodium concentration reported |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: '...subjects were randomised by an Interactive Voice Response System...'. Comment: central allocation occurred. |
| Allocation concealment (selection bias) | Low risk | Quote: '...subjects were randomised by an Interactive Voice Response System...'. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: '... double‐blind, placebo‐controlled phase 3 trial...' Comment: Blinding of participants and personnel probably attempted, but insufficient information to assess the extent to which it was achieved or to permit judgement of 'High risk' or 'Low risk'.. Co‐interventions were left at the discretion of the treating physician. |
| Blinding of outcome assessment (detection bias) | Low risk | 'The Clinical Endpoints Committee was to perform a review (blinded to treatment group and serum sodium levels) and adjudicate all events assessed by the investigators as clinical endpoints using predetermined criteria for each end‐point...'. Comment: blinding probably attempted and although un‐blinding likely to have occurred for serum sodium concentration, assessment of other outcomes done by independent blinded adjudication committee. |
| Incomplete outcome data (attrition bias) | Low risk | Quote: 'The FDA statistical reviewer, Dr Jialu Zhang, also performed a number of sensitivity analyses to assess the impact of missing values... All these sensitivity analyses showed consistent results: a statistically significant treatment effect on serum sodium with a modest effect size in the range 1.2 to 1.4 mEq/L.' Comment: overall 27% discontinuation at 30 days, < 1% of which never received treatment; 50% of discontinuations occurred in placebo group. Last observation carried forward method was used as imputation method for dealing with missing serum sodium concentration values, which could have caused overestimation of the effect of the study medication. However, sensitivity analyses conducted by the FDA statistical reviewer indicated this effect to be negligible. Cognitive function tests were taken at day 30 or the early termination visit. Given the better scores in the placebo group, any bias would only serve to increase the difference further in favour of the placebo group and would not change the conclusions drawn from the analysis. |
| Selective reporting (reporting bias) | Low risk | Comment: the study was registered with ClinicalTrials.gov, but no clear listing of predefined outcome measures and time‐points. Given early stopping of trial due to numerically more deaths in treatment group, selective outcome reporting unlikely to be an issue. |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Treatment group 2
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'randomised... study', and 'Patients ... were randomly assigned'. Comment: insufficient information to permit judgement of 'High risk' or 'Low risk'. |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk'. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: '...double‐blind, placebo‐controlled...study', and 'Patients...were... assigned to ... or placebo'. Comment: blinding probably attempted, but insufficient information to assess to what extent it was achieved or to permit judgement of 'High risk' or 'Low risk'. |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether assessors of non patient‐reported outcomes were blinded, but main measured outcomes objective. Unlikely for participants to have been fully blinded, and plausible 'high risk' of bias for self‐reported outcome of thirst. |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: overall 16% attrition, reasons not documented. Given < 20% attrition in short‐term study, we judged this not at high risk of bias per se, rather insufficient information to permit judgement of 'High risk' or 'Low risk'. |
| Selective reporting (reporting bias) | Low risk | Comment: no protocol, short‐term study (≤ 1 week), secondary outcomes related to serum sodium concentration and over‐correction of serum sodium concentration reported. |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Treatment group 2
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote:'...eligible patients were randomly allocated to...', and '4 patients were not randomised through the interactive voice recognition system'. Comment: Central allocation probably occurred. The randomisation produced groups well‐balanced in age and serum sodium concentration, less so in sex |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: '...patients were randomly allocated to ... or placebo', and 'The randomised, double‐blind, placebo‐controlled ...', and '...recommended fluid intake 1000‐1500 mL'. Comment: blinding attempted, and although participants and personnel are unlikely to be fully blinded due to important increases in urine output, all measured outcomes short‐term and fairly objective. Fluid restriction was prescribed in all groups |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding outcome assessors probably attempted and all measured outcomes objective |
| Incomplete outcome data (attrition bias) | Low risk | Comment: overall 11% attrition, due to treatment discontinuation, reasons fairly well documented, asymmetrically in experimental group receiving highest dose due to adverse events. Missing data dealt with using last observation carried forward method. Given < 20% attrition and limited opportunity for introducing bias in such a short‐term study ‐ we judged it 'Low risk' |
| Selective reporting (reporting bias) | Low risk | Comment: study registered with ClinicalTrials.gov in 2005. Primary outcome matches, more outcomes reported than originally registered ‐ predominantly outcomes related to harms. Short‐term study (≤ 1 week), secondary outcomes related to serum sodium concentration and rapid increase in serum sodium concentration reported |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'The patients were randomly divided in two groups...' Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | High risk | Comment: blinding not attempted |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding not attempted, but main outcomes objective such that un‐blinding of outcome detection unlikely to be an important source of bias |
| Incomplete outcome data (attrition bias) | Low risk | Comment: attrition or treatment discontinuation not specifically reported, but it appears as if all participants completed the treatment protocol |
| Selective reporting (reporting bias) | High risk | Comment: no protocol, short‐term study (≤ 1 week), secondary outcomes related to rapid increase in serum sodium concentration not reported |
| Other bias | Low risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: '...within each centre, patients were randomised to... according to a centralized, blocked randomization performed using a central Interactive Voice Response System'. Additional info provided by the sponsor: 'Subjects were not stratified by baseline serum sodium'. Comment: the randomisation procedure was adequate and produced well balanced groups in the overall study. Although randomisation was not stratified for serum sodium concentration, fairly similar numbers of participants in both the experimental and the control group, with similar ages and baseline serum sodium concentration |
| Allocation concealment (selection bias) | Low risk | Quote: '...within each centre, patients were randomised to... according to a centralized, blocked randomisation performed using a central Interactive Voice Response System' |
| Blinding of participants and personnel (performance bias) | High risk | Quote: 'Patients...assigned to receive oral tolvaptan 30 mg or matching placebo. ' and '...is and international, ..., double‐blind, placebo‐controlled study...'. Additional info provided by the sponsor: 'The trial drug was packaged so that each subject received an identical number of tablets regardless of the treatment group assigned'. Comment: blinding attempted, although participants are unlikely to be fully blinded due to important increases in urine output. Also, co‐interventions were not reported and may have differed between groups according to baseline serum sodium concentration. This could have influenced outcome measurement of serum sodium concentrations and length of hospital stay |
| Blinding of outcome assessment (detection bias) | Low risk | Additional info provided by the sponsor: 'Did not have separate outcome assessors in this trial'. Comment: blinding outcome assessors attempted and all measured outcomes objective |
| Incomplete outcome data (attrition bias) | High risk | Comment: 30% (33% in experimental group, 29% in control group) attrition from analysis of serum sodium concentration at day 7 |
| Selective reporting (reporting bias) | High risk | Comment: protocol available with matched primary and secondary outcomes for the overall study. However, results included in this review for the subgroup with hyponatraemia are based on post‐hoc analyses of the data, with reported outcomes not matching the primary and secondary outcomes of the original study |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Treatment group 2
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'A computer‐generated randomisation table was available at each participating hospital. A patient number was assigned when a patient had qualified during the pre‐study screening. Before administration of the first study medication, the patient was assigned a randomisation number by an unblinded site pharmacist. This unblinded dispenser did not administer the drug to patients, and the contact between him and the investigators and study nurses was kept to a minimum.' Comment: the randomisation procedure produced well balanced groups in terms of baseline serum sodium concentration, no information on other baseline characteristics |
| Allocation concealment (selection bias) | Low risk | Comment: the randomisation procedure produced well balanced groups in terms of baseline serum sodium concentration, no information on other baseline characteristics |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: '...randomised, double blind, placebo‐controlled study...', and '...patients were randomised to placebo or 50 mg twice a day...' Comment: blinding attempted, and although participants and personnel are unlikely to be fully blinded due to important increases in urine output, all measured outcomes short‐term and fairly objective. Co‐intervention of fluid restriction similar in all groups |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether assessors of non patient‐reported outcomes were blinded, but main measured outcomes objective. Unlikely for participants to have been fully blinded, and plausible 'high risk' of bias for self‐reported outcome of thirst |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: published in abstract form only, insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Selective reporting (reporting bias) | High risk | Comment: no protocol, short‐term study (≤ 1 week) but no reporting of primary outcomes death or health‐related quality of life or secondary outcomes related to rapid increase in serum sodium concentration in a way that permitted data extraction |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Treatment group 2
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'Eligible patients were stratified by volume status and assigned randomly in a 1:1:1 ratio...' Comment: insufficient information to permit judgement of 'High risk' or 'Low risk'. |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk'. |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: 'This double‐blind, placebo‐controlled ... study', and 'Fluid intake was limited to ..up to a maximum of 2.0 litres in 24h. ....and 24‐h sodium intake remained stable throughout the study'. Comment: blinding attempted, and although participants and personnel are unlikely to be fully blinded due to important increases in urine output, all measured outcomes short‐term and fairly objective. Fluid restriction and stable sodium intake were prescribed in all groups |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding outcome assessors probably attempted and all measured outcomes objective |
| Incomplete outcome data (attrition bias) | Low risk | Comment: overall 9% attrition, 5% exclusions for never having received the drug, 4% due to treatment discontinuation, reasons well‐documented. Missing data dealt with through analytic techniques for repeated measures using the last available measurement before discontinuation. Given <20% attrition and limited opportunity for introducing bias in such a short‐term study ‐ we judged it 'Low risk' |
| Selective reporting (reporting bias) | Low risk | Comment: no protocol, short term study (≤1 week), secondary outcomes related to serum sodium concentration and rapid increase in serum sodium concentration reported |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: '...central random assignment procedure ... by means of an Interactive Voice‐Response System' Comment: the randomisation procedure produced groups in the overall study that were not entirely balanced in age and sex, not in terms of medical history or characteristics related to the heart failure. The randomisation was not stratified for serum sodium concentration. The large number of groups relative the total number of participants would have made it likely for the groups to be unbalanced in prognostic characteristics. However, the absence of baseline data specifically for the subgroup with hyponatraemia, make it difficult to judge both magnitude and direction of possible bias. |
| Allocation concealment (selection bias) | Low risk | Quote: '...central random assignment procedure ... by means of an Interactive Voice‐Response System'. |
| Blinding of participants and personnel (performance bias) | High risk | Quote: '...were assigned to receive...or placebo...', and '...a double‐blind, randomised trial' Additional info provided by sponsor: 'Both OPC‐41061 and placebo tablets were identical in appearance'. Comment: blinding attempted, although participants are unlikely to be fully blinded due to important increases in urine output. Also, co‐interventions were not reported in detail for the subgroup with hyponatraemia and included standard heart failure treatment left at discretion of investigator and may have differed between groups according to baseline serum sodium concentration. This could have influenced outcome measurement of serum sodium concentrations |
| Blinding of outcome assessment (detection bias) | Low risk | Additional info provided by sponsor: 'Did not have separate outcome assessors in this trial'. Comment: blinding outcome assessors attempted, main outcome objective. Unlikely for participants to have been fully blinded, and plausible 'high risk' of bias for self‐reported outcome of thirst |
| Incomplete outcome data (attrition bias) | High risk | Comment: 34% attrition for outcomes related to serum sodium concentration measurements in comparison with numbers provided in the study report |
| Selective reporting (reporting bias) | High risk | Comment: no protocol available, long‐term study (> 1 week), primary outcomes and secondary outcomes not reported in a way that allowed extraction of raw data |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: '...subjects were randomised...', and 'This study was a multicenter, prospective, randomised.... trial.' Additional info provided by the sponsor: 'The study was conducted using local (per site) randomisation.' Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | High risk | Quote: 'This study was a multicenter, prospective, randomised, open‐label.... trial', and ...subjects were randomised ... to tolvaptan or fluid restriction plus placebo'. Additional info provided by the sponsor: 'The study drug was supplied as either 5 mg, 15 mg OPC‐4 1061 tablets or matching placebo. All tablets were identical in appearance.' and '(investigators were) not concealed as separate instructions were given to investigators for subjects on fluid‐restriction/placebo and those on active therapy.' Comment: blinding of participants attempted, and although participants are unlikely to have been fully blinded due to important increases in urine output, serum sodium concentration short‐term and generally objective. Personnel were not blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding of outcome assessors not attempted, but main measured outcomes objective. Participants not blinded, and plausible 'high risk' of bias for self‐reported outcome of thirst |
| Incomplete outcome data (attrition bias) | Low risk | Quote: 'Two of the 17 subjects assigned to the tolvaptan group and 3 of the 11 subjects assigned to the fluid restriction group were withdrawn during the run‐in phase because of protocol violations or because withdrawal criteria were met.' Comment: overall 17% attrition ‐12% in experimental group, 27% in placebo group ‐ due to protocol violation or because participants never received the study medication. Reasons not documented in detail. Given <20% attrition, short‐term study, we judged risk of bias to be 'Low' |
| Selective reporting (reporting bias) | High risk | Comment: no protocol, short‐term study (<1 week), secondary outcomes related to rapid increase in serum sodium concentration not reported. Treatment duration 12 days, yet primary outcome measured at 5 days; Thirst measured at last inpatient visit, which could have been anywhere between 5 and 12 days after study initiation and different for both groups |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'Patients...were randomised...'. Comment: insufficient information to permit judgement of 'High risk' or 'Low risk'. No baseline characteristics presented |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | High risk | Quote: 'Patients...were randomised into a multi‐centre, single‐blind study to receive in a 2:1 ratio either a flexible dose‐regimen of satavaptan (5‐50 mg once daily) or placebo'. Comment: blinding of participants probably attempted. However, insufficient information for assessing to what extent it was achieved, or to permit judgement of 'High risk' or 'Low risk'. Blinding of personnel not attempted. No information on how differential treatment and follow‐up was avoided |
| Blinding of outcome assessment (detection bias) | High risk | Quote: 'Patients...were randomised into a multi‐centre, single‐blind study...' Comment: outcome assessors not blinded and main outcome measure ‐ objective outcome measures |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Selective reporting (reporting bias) | High risk | Comment: no protocol and long term (> 1 week) study, primary outcomes and secondary outcomes related to rapid increase in serum sodium concentration not reported |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'The HARMONY study was a ... randomised .... study', and 'Subjects were initially randomised to ... '. Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: 'The HARMONY study was a ... double‐blind, placebo‐controlled ... study'. Comment: blinding probably attempted, although due to increase in urine output, un‐blinding likely to have occurred. Fluid restriction was largely left to the discretion of the investigator, but fewer participants receiving the study medication had fluid restriction initiated or tightened by day 30 (11% versus 21%) |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether assessors of non patient‐reported outcomes were blinded, but main measured outcomes objective. Unlikely for participants to have been fully blinded, and plausible 'high risk' of bias for self‐reported outcome of thirst |
| Incomplete outcome data (attrition bias) | High risk | Comment: overall 17% discontinuation at 8 weeks, numbers and reasons similarly distributed between experimental and placebo group. However, 67% discontinuation at 24 weeks, according to the FDA briefing document because, quote 'According to the applicant, because the primary and secondary efficacy endpoints targeted time points between the initial eight weeks from randomisation, therefore, when the last subject enrolled in the study had completed eight weeks of treatment, the applicant instructed the investigators to stop blinded therapy in all subjects in the study' |
| Selective reporting (reporting bias) | Low risk | Comment: the study was registered with ClinicalTrials.gov, but no clear listing of predefined outcome time‐points. That said, all preregistered outcomes and outcomes related to over correction reported. Some additional outcomes were insufficiently reported for contribution to meta‐analysis |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'Patients...randomised...'. Comment: insufficient information to permit judgement of 'High risk' or 'Low risk'. No baseline characteristics presented. |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk'. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk'. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk'. |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk'. |
| Selective reporting (reporting bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk'. |
| Other bias | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk'. |
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'Each participating centre received blocks of study treatment which contained equal numbers of each of the four therapies on a random sequence generated by a computer program', and 'The median number of patients included in the 30 participating centres was 3 (range 1 to 14)'. Comment: The randomisation procedure generated groups well‐balanced for age, sex and baseline serum sodium concentration. The severity of liver disease as assessed by the serum bilirubin concentration, Child‐Pugh and MELD score was greater for the placebo group in comparison with the other groups. We assumed the influence of the imbalance thus generated would be negligible in practice |
| Allocation concealment (selection bias) | Low risk | Comment: considering randomisation procedure and extent of attempting to blind, likely adequate |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: 'This was a ...double‐blind, placebo‐controlled efficacy study', and 'all...were maintained on a low‐sodium diet ... and daily fluid intake was limited to 1.5L unless patients were thirsty or there was a medical need to increase fluid intake' Comment: blinding attempted, and although participants and personnel are unlikely to be fully blinded due to important increases in urine output, all measured outcomes short‐term and fairly objective. Fluid restriction occurred in all groups, any increase in fluid intake likely to have occurred in the intervention groups, with possible bias favouring the placebo group for outcomes related to serum sodium concentration |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding outcome assessors probably attempted, main measured outcomes objective. Unlikely for participants to have been fully blinded, and plausible 'high risk' of bias for self‐reported outcome of thirst |
| Incomplete outcome data (attrition bias) | Low risk | Comment: overall 5% attrition, presumably due to treatment discontinuation, despite efforts to measure outcomes regardless. Reasons for treatment discontinuation well documented. Missing data dealt with through analytic techniques for repeated measures using the last available measurement before discontinuation. Given <20% attrition and limited opportunity for introducing bias in such a short‐term study ‐ we judged it 'Low risk' |
| Selective reporting (reporting bias) | High risk | Comment: protocol registered after completion of the study. Outcomes related to serum sodium concentration said to be measured at 14 days, yet only reported at day 5. Protocol also lists 'trail making test' and 'quality of life' as outcomes, which are not reported in the study publication |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'Subjects will be randomised...' Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | High risk | Quote: 'A pilot...double‐blind, placebo‐controlled ... study', and 'Subjects will be randomised to ... or matching placebo tablet', and 'during this period, fluid restrictions should be loosened or suspended, until the subject's response to therapy can be evaluated, typically over the first few days of therapy. Fluid restriction may be reinstated at any time in subjects whose sodium fails to improve or worsens with study therapy. Subjects entering ...with a serum sodium concentration less than 130 mEq/L may be fluid restricted if necessary at the discretion of the investigator'. Comment: blinding of both participants and personnel attempted. However, unlikely for participants to have been fully blinded due to likely important increases in urine output. Also, fluid restriction was not mandatory and could be adapted at the discretion of the investigator. This may have biased outcome measures at one month in favour of the study medication |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether assessors of non patient‐reported outcomes were blinded, but main measured outcomes objective. Unlikely for participants to have been fully blinded, and plausible 'high risk' of bias for self‐reported outcome of thirst |
| Incomplete outcome data (attrition bias) | Low risk | Comment: overall 9% attrition, 8% due to treatment discontinuation, asymmetrical due to adverse events in the experimental group. Given < 10% attrition in long‐term study, we judged opportunity for introducing attrition bias 'Low' |
| Selective reporting (reporting bias) | Low risk | Comment: study registered with ClinicalTrials.gov in 2007 before study initiation. Primary and secondary outcomes including time‐points match |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: '...a randomised clinical trial'. Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | High risk | Quote: 'treatment group 1: Fluid restriction (1.5 L)+Na restriction (80 mmol/d), Group 2: Human Salt Poor Albumin (40 gm/d) + Fluid (1.5 L) + Na restriction (80 mmol/d) Comment: attempts at blinding not mentioned, blinding unlikely to have happened |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether assessors of non patient‐reported outcomes were blinded, but main measured outcomes objective |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Selective reporting (reporting bias) | High risk | Comment: no protocol, short‐term study (≤ 1 week) but no reporting of primary outcomes death or health‐related quality of life, and no reporting of secondary outcomes related to rapid increase in serum sodium concentration in a way that permitted data extraction |
| Other bias | Unclear risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'This was a randomised, parallel‐group study..', and 'Eligible patients were... randomised to...' Comment: large number of groups relative to the number of included participants may have caused simple randomisation to fail: groups are not entirely balanced in terms of age and sex. Insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: 'The placebo ampoules and premixed bags contained all the components of the active formulation except conivaptan. For the ampoule formulation, the contents of the 4 mL ampoule was added to a bag containing 96 mL of 5% dextrose injection, so that the final volume was 100 mL for the loading dose or a bag containing 246 mL of 5% dextrose injection for the continuous infusion.' Comment: blinding of participants and personnel attempted and given all groups included active ingredient, un‐blinding due to differences in urine output not very likely |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding outcome assessors adequate |
| Incomplete outcome data (attrition bias) | Low risk | Comment: overall 17% attrition due to exclusion after randomisation (3%) and treatment discontinuation (14%); reasons well‐documented. Missing data dealt with through analytic techniques for repeated measures using the last available measurement before discontinuation. Given < 20% attrition and limited opportunity for introducing bias in such a short‐term study ‐ we judged it 'Low risk' |
| Selective reporting (reporting bias) | Low risk | Comment: no protocol and short term study (< 1 week), secondary outcomes related to both serum sodium concentration and rapid increase in serum sodium correction reported. Because a repeated measures analytic technique was used, unclear at what time point serum sodium concentration was measured for all participants, yet given short term study, bias thus introduced probably negligible in practice |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Treatment group 2
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'This randomised, double‐blind...' and 'Eligible patients were stratified by baseline SSC (serum sodium concentration) ... and then randomly assigned... Comment: methods for sequence generation not reported. Due to the large number of groups relative to the number of included participants, adequate simple randomisation may have failed to produce groups with similar baseline prognosis. E.g. there is an imbalance in age, sex and volume status across the groups. We assumed the influence of the imbalance thus generated would be negligible in practice |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: 'In each dosing regimen, conivaptan or placebo was administered via a 30‐minute i.v. infusion using a 100 mL plastic bag containing either 20 mg conivaptan hydrochloride in 5% dextrose injection or placebo (5% dextrose injection). In the once‐daily regimen, conivaptan was administered at 0 and 24 hours, and placebo was administered at 12 and 36 hours to maintain blinding' Comment: blinding attempted, and although participants and personnel are unlikely to be fully blinded due to important increases in urine output, all measured outcomes very short‐term and fairly objective |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding outcome assessors probably attempted and all measured outcomes objective |
| Incomplete outcome data (attrition bias) | Low risk | Comment: overall 18% attrition due to exclusion after randomisation (2%) and treatment discontinuation (16%); reasons fairly well‐documented. All save 1 were re‐included in the analysis for sodium concentration measurements using the analytic techniques for repeated measures using the last available measurement before discontinuation. Given < 20% attrition and limited opportunity for introducing bias in such a short term study, we judged it 'Low risk of bias' |
| Selective reporting (reporting bias) | Low risk | Comment: no protocol and short term study (≤ 1 week), secondary outcomes related to both serum sodium concentration and rapid increase in serum sodium correction reported |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'The LIBRA study was a ... randomised .... study', and 'Subjects were initially randomised to ... ' Comment: Overall insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: 'The Libra study was a ... double‐blind, placebo‐controlled ... study' Comment: blinding probably attempted, although due to increase in urine output, un‐blinding likely to have occurred. Fluid restriction was largely left to the discretion of the investigator, and more participants receiving the study medication had fluid restriction initiated or tightened by day 30 (32% versus 23%), but more participants in the placebo group were on fluid restriction at baseline (37% versus 65%) and at any time‐point throughout the study |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding insufficiently reported but main outcomes objective such that un‐blinding of outcome detection unlikely to be an important source of bias |
| Incomplete outcome data (attrition bias) | High risk | Comment: overall 13% discontinuation at 7 days, 5% of which never received treatment; 67% of discontinuations occurred in placebo group. 31% discontinuation at 30 days, with last observation carried forward method as imputation method for dealing with missing serum sodium concentration values, which may have caused overestimation of the effect of the study medication. Cognitive function tests were taken at day 30 or the early termination visit. We judged outcomes measured at 7 days to at 'Low risk', but those at 30 days to be at 'High risk' of attrition bias |
| Selective reporting (reporting bias) | Low risk | Comment: protocol first registered with ClinicalTrials.gov in 2008. Primary registered outcome was average daily area under the curve (AUC) of change from baseline in serum sodium concentrations up to 72 hrs, whereas the reported primary outcome is change in serum sodium concentration at 7 days. That said, all reregistered outcomes were reported and secondary outcomes related to over‐correction also reported |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: '...the patient was randomised by opening the lowest numbered, sealed envelope.' Comment: although methods for generating the sequence were not reported, we judged it likely for methods to have been adequate given researchers went to trouble of using sealed, numbered envelopes |
| Allocation concealment (selection bias) | Low risk | Quote: '...the patient was randomised by opening the lowest numbered, sealed envelope.' |
| Blinding of participants and personnel (performance bias) | High risk | Comment: participants nor personnel blinded to treatment allocation. Treatment consisted of the study drug added to standard care versus usual care alone. Usual care was left at the discretion of the treating physician with two participants treated with the study medication versus one participant treated with usual care receiving hypertonic saline. This differential treatment in the two groups could have influenced the outcome in favour of the study drug |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: from ClinicalTrials.gov 'Masking: single blind ‐ outcomes assessor' Comment: still unclear whether outcome assessors of non patient‐reported outcomes other than Glasgow Coma Scale and NHS Stroke Scale were blinded, but outcomes objective |
| Incomplete outcome data (attrition bias) | Low risk | Quote: 'all randomised patients completed the protocol' Comment: no attrition or exclusion |
| Selective reporting (reporting bias) | Low risk | Comment: protocol registered with ClinicalTrials.gov, reported outcomes match those originally registered |
| Other bias | Unclear risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'Les patients ont été randomisés en deux bras...' Additional information provided by the authors: 'Random sequence was managed as a single randomisation list managed by the sponsor' |
| Allocation concealment (selection bias) | Low risk | Additional information provided by the authors: 'Allocation was concealed using masking envelope'. Comment: Allocation concealment method considered adequate |
| Blinding of participants and personnel (performance bias) | High risk | Additional information provided by the authors: 'No measures were taken to prevent participants or personnel from knowing to which treatment participants had been allocated' Comment: blinding not attempted and difficult because of intervention type. There were no specific instructions concerning co‐interventions. Differences in co‐interventions may have influenced difference in outcome in favour of experimental treatment |
| Blinding of outcome assessment (detection bias) | Low risk | Additional information provided by the authors: 'No measures were taken to prevent outcome assessors from knowing to which treatment participants had been allocated' Comment: blinding of outcome assessors not attempted, but main measured outcomes objective. |
| Incomplete outcome data (attrition bias) | High risk | Comment: overall 26% incomplete outcome data at four weeks; 1 attrition in control group, due to being released from hospital before four weeks; 1 exclusion in experimental group for gastrointestinal bleed after withdrawing proton pump inhibitor; 3 in control group: 2 for stopping a drug possibly causing hyponatraemia ‐ hence possibly biasing results in favour of experimental treatment ‐ 1 due to death |
| Selective reporting (reporting bias) | Low risk | Comment: protocol available, long‐term study (> 1 week), and both primary outcome death and secondary outcomes related to serum sodium concentration reported. Given lowest serum sodium concentration equalled 129 mmol/L and the intervention consisted of medication withdrawal, potential for rapid increases in serum sodium concentration very limited |
| Other bias | Low risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'In a randomised, double‐blind, placebo‐controlled trial, 15...were treated...' Comment: insufficient information to permit judgement of ‘Low risk’ or ‘High risk’ |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of ‘Low risk’ or ‘High risk’ |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: 'In a randomised, double‐blind, placebo‐controlled trial, 15...were treated...' Comment: blinding of participants and personnel attempted. However, lack of detail in reporting of outcomes makes it difficult to assess to what extent it was actually achieved. insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether assessors of non patient‐reported outcomes were blinded, but main measured outcomes objective |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Selective reporting (reporting bias) | High risk | Comment: no protocol, long‐term study (> 1 week) but no reporting of primary outcomes death or health‐related quality of life, no reporting of secondary outcomes related to rapid increase in serum sodium concentration |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'This randomised double‐blind placebo‐controlled trial...' and 'Randomisation was stratified by underlying disease and whether the hyponatraemia was mild...or marked...' Comment: since the investigators went to the trouble to stratify allocation, it seems probable enough that sequence generation was adequate |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: 'This randomised double‐blind placebo‐controlled trial...' and 'Patients received tolvaptan or matching placebo..' Comment: blinding probably attempted, although due to increase in urine output, un‐blinding likely to have occurred. Fluid restriction was largely left to the discretion of the investigator, but fewer participants receiving the study medication had fluid restriction initiated or tightened by day 30 (11% versus 21%) |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether assessors of non patient‐reported outcomes were blinded, but main measured outcomes objective |
| Incomplete outcome data (attrition bias) | Low risk | Comment: No attrition for main outcomes |
| Selective reporting (reporting bias) | High risk | Comment: the study was registered with ClinicalTrials.gov, but only primary efficacy outcome clearly listed, short‐term study (≤1 week) death reported, but no reporting of secondary outcomes related to rapid increase in serum sodium concentration. Also study results ‐ incompletely/differentially ‐ reported for the subgroups of SIADH, heart failure and cirrhosis separately |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'This randomised double‐blind placebo‐controlled trial...' and 'Randomisation was stratified by underlying disease and whether the hyponatraemia was mild...or marked...' Comment: since the investigators went to the trouble to stratify allocation, it seems probable enough that sequence generation was adequate |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: 'This randomised double‐blind placebo‐controlled trial...' Comment: blinding of participants and personnel attempted. However, lack of detail in reporting of outcomes makes it difficult to assess to what extent it was actually achieved. insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether assessors of non patient‐reported outcomes were blinded, but main measured outcomes objective |
| Incomplete outcome data (attrition bias) | Low risk | Comment: No attrition for main outcomes |
| Selective reporting (reporting bias) | High risk | Comment: the study was registered with ClinicalTrials.gov, but only primary efficacy outcome clearly listed, short‐term study (≤ 1 week) but no reporting of primary outcomes death or health‐related quality of life, no reporting of secondary outcomes related to rapid increase in serum sodium concentration in a way that permitted data extraction. Also study results ‐ incompletely/differentially ‐ reported for the subgroups of SIADH, heart failure and cirrhosis separately |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'A multicenter, randomised, double‐blind,..', and 'Patients were randomised to receive...' Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: 'A multicenter,..., double‐blind, placebo‐controlled clinical trial...', and 'Patients were randomised to receive placebo or tolvaptan...' Comment: 'insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether assessors of non patient‐reported outcomes were blinded, but main measured outcomes objective |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: 'insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Selective reporting (reporting bias) | High risk | Comment: the study was registered with ClinicalTrials.gov, but only primary efficacy outcome clearly listed,, short‐term study (≤ 1 week) but no reporting of primary outcomes death or health‐related quality of life, and no reporting of secondary outcomes related to rapid increase in serum sodium concentration in a way that permitted data extraction. Also study results ‐ incompletely/differentially ‐ reported for the subgroups of SIADH, heart failure and cirrhosis separately |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: '...we chose Baysian adaptive randomisation for its several reported useful features.' and 'After obtaining the patient's consent, the research coordinator logged into the Clinical Trial Conduct website to register and randomise the patients.' Comment: valid randomisation methods produced imbalanced groups in terms of age due to small sample size, sex and baseline serum sodium concentration were well balanced and the imbalance in age was likely favouring the control group as they contained the younger participants |
| Allocation concealment (selection bias) | Low risk | Comment: 'After obtaining the patient's consent, the research coordinator logged into the Clinical Trial Conduct website to register and randomise the patients.' |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: 'All research personnel except therapy distributing pharmacy and biostatistician were blinded to the treatment allocation.' Comment: blinding probably attempted, and although participants are unlikely to be fully blinded due to important increases in urine output, measured outcomes were short‐term and mostly objective for the data used in meta‐analysis. Co‐interventions were similar between groups |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: 'All research personnel except therapy distributing pharmacy and biostatistician were blinded to the treatment allocation.' Comment: Blinding of outcome assessors probably done |
| Incomplete outcome data (attrition bias) | Low risk | Comment: overall 42% attrition for serum sodium concentration, 46% in placebo group, 29% in tolvaptan group, extensive sensitivity analysis including worst case imputation methods still resulted in statistically significant results. No attrition for death |
| Selective reporting (reporting bias) | High risk | Comment: study protocol registered with ClinicalTrials.gov in 2010. Only study's primary outcome serum sodium concentration registered. Long‐term study (>1 week) with primary outcome death reported. However secondary outcomes related to rapid increase in serum sodium concentration not reported |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'Patients...underwent central randomisation with the use of random permuted blocks and stratification according to whether the hyponatraemia was mild or marked...' Comment: the randomisation procedure produced well balanced groups |
| Allocation concealment (selection bias) | Low risk | Quote: 'Patients...underwent central randomisation with the use of random permuted blocks and stratification according to whether the hyponatraemia was mild or marked...' |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: '...trials were...randomised, double‐blind, placebo‐controlled efficacy studies...' and 'Patients were assigned...to receive oral tolvaptan...or matching placebo...', and 'Fluid restriction was not mandatory'. Additional info supplied by sponsor: 'Study medication was supplied for each subject in monthly kits per randomisation number, each labelled with a two‐panel double‐blind disclosure label specifying the treatment assignment (i.e., the compound name, strength, and lot number) in the concealed portion of the label. The study drug was packaged so that each subject received an identical number of tablets regardless of the treatment group assignment. All tablets were identical in appearance. The investigator was instructed to not open the treatment assignment code unless knowledge of the subject’s treatment was required for the subject’s clinical care and safety. In this event, the investigator was to contact OMRI by telephone with an explanation of the need for opening the treatment assignment code before or within 24 hours of opening the code.' Comment: blinding of both participants and personnel attempted. However, unlikely for participants to have been fully blinded due to important increases in urine output. We judged this would not have introduced important risk of bias for death and the outcomes related to serum sodium concentration, but may have biased health‐related quality of life measures in favour of the study medication. Also, fluid restriction was not mandatory and could be adapted by both participant and treating physician ‐ e.g. based on urine output. This may have resulted in underestimation of the risk of rapid increase in serum sodium concentration |
| Blinding of outcome assessment (detection bias) | Low risk | Additional info provided by the sponsor: 'Serum sodium samples were collected and analysed at the site’s local hospital laboratory. All serum sodium samples were collected and analysed at the same local hospital for purposes of consistency of efficacy analysis. Local laboratory results for serum sodium were captured in the electronic case report form (CRF). Study personnel measuring secondary endpoints were blinded as above and recorded results in the electronic CRF.' Comment: outcome assessors of non patient‐reported outcomes were blinded. However, unlikely for participants to have been fully blinded given important increases in urine output, which may have biased the self‐reported outcome health‐related quality of life |
| Incomplete outcome data (attrition bias) | High risk | Quote: 'Patients who received at least one dose of the study medication were included in the safety analyses. Patients whose serum sodium concentrations were evaluated at baseline and one or more times after baseline were included in the efficacy analysis'. And 'serum sodium concentrations were measured at days 2, 3, 4,11, 18, 25, 30, and 37' Comment: overall 26% attrition at 30 days, 25% in treatment group 1, 26% in the placebo group; reasons not reported in detail. Repeated measures analytic technique was used with all measurements of serum sodium concentration included until patient attrition. We judged this may likely have overestimated the change in serum sodium concentration in favour of the study medication. For the mental component score of the SF‐12 health survey, measured at 30 days, attrition reached 30% in treatment group 1 and 44% in the placebo group |
| Selective reporting (reporting bias) | High risk | Comment: study protocol registered with ClinicalTrials.gov. Outcome measures were only registered in September 2005. Health‐related quality of life not registered as one of the outcomes; both the mental and physical component of the SF‐12 health survey were reported separately and measured at day 4 and day 30 using two analytic techniques, but only data for the statistically significant mental component score at day 30 were available for analysis from the published paper. Also because a repeated measures analytic technique was used, unclear at what time point serum sodium concentration was measured for all participants |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'Patients...underwent central randomisation with the use of random permuted blocks and stratification according to whether the hyponitraemia was mild or marked...' Comment: the randomisation procedure produced well balanced groups |
| Allocation concealment (selection bias) | Low risk | Quote: 'Patients...underwent central randomisation with the use of random permuted blocks and stratification according to whether the hyponatraemia was mild or marked...' |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: '...trials were...randomised, double‐blind, placebo‐controlled efficacy studies...' and 'Patients were assigned...to receive oral tolvaptan...or matching placebo...', and 'Fluid restriction was not mandatory' Comment: blinding of both participants and personnel attempted. However, unlikely for participants to have been fully blinded due to important increases in urine output. We judged this would not have introduced important risk of bias for death and the outcomes related to serum sodium concentration, but may have biased health‐related quality of life measures in favour of the study medication. Also, fluid restriction was not mandatory and could be adapted by both participant and treating physician ‐ e.g. based on urine output. This may have resulted in underestimation of the risk of rapid increase in serum sodium concentration |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether outcome assessors of non patient‐reported outcomes were blinded, but outcomes fairly objective. However, unlikely for participants to have been fully blinded given important increases in urine output, which may have biased the self‐reported outcome health‐related quality of life |
| Incomplete outcome data (attrition bias) | High risk | Quote: 'Patients who received at least one dose of the study medication were included in the safety analyses. Patients whose serum sodium concentrations were evaluated at baseline and one or more times after baseline were included in the efficacy analysis'. And 'serum sodium concentrations were measured at days 2,3,4,11,18,25,30, and 37' Comment: overall 26% attrition at 30 days, 25% in treatment group 1, 26% in placebo group; reasons not reported in detail. Repeated measures analytic technique was used with all measurements of serum sodium concentration included until patient attrition. We judged this may likely have overestimated the change in serum sodium concentration. For the mental component score of the SF‐12 health survey, measured at 30 days, attrition reached 31% in treatment group 1 and 29% in the placebo group |
| Selective reporting (reporting bias) | High risk | Comment: study protocol registered with ClinicalTrials.gov. Outcome measures were only registered in September 2005. Health‐related quality of life not registered as one of the outcomes. Note that the study was registered as separate trial, but the health‐related quality of life outcomes and adverse events were reported in a joint analysis for the two studies together without data allowing separation for the purpose of this review. Also because a repeated measures analytic technique was used, unclear at what time point serum sodium concentration was measured for all participants |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Treatment group 2
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'subjects...were randomised'. Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: 'In a multi‐center...double blind trial'. Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Blinding of outcome assessment (detection bias) | Unclear risk | Quote: 'In a multi‐center...double blind trial'. Comment: insufficient information to permit judgement of 'High risk' or 'Low risk' |
| Incomplete outcome data (attrition bias) | Low risk | Comment: attrition or treatment discontinuation not specifically reported, but given single dose study, risk of bias seems low nonetheless |
| Selective reporting (reporting bias) | Low risk | Comment: no protocol, short‐term study (single dose), secondary outcomes related to over‐correction of serum sodium concentration reported |
| Other bias | Unclear risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'Patients in both groups were assigned randomly to receive...' and 'a randomisation list was prepared with help of random tables before initiation of the study.' Comment: valid randomisation methods may have produced imbalanced groups due to small sample size, but any imbalance was likely favouring the experimental group as control group contained the sicker children |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of ‘Low risk’ or ‘High risk’ |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: blinding of participants probably not relevant given setting, short‐term study and objective outcomes. Attempts to blind personnel not reported, probably not done, may have influenced administration of co‐interventions not reported in the paper |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding of outcome assessors probably not done, but unlikely this will have caused substantial bias given short‐term study and objectivity of outcomes |
| Incomplete outcome data (attrition bias) | Low risk | Comment: no attrition |
| Selective reporting (reporting bias) | Low risk | Comment: no protocol, but primary outcome and secondary outcome related to serum sodium concentration reported |
| Other bias | Low risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Treatment group 2
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: '...patients were randomly assigned to take placebo or a once‐daily dose...'. Comment: due to the large number of groups relative to the number of included participants, the randomisation procedure may have failed to produce groups with similar baseline prognosis. E.g. there is an imbalance in age, SCr, urinary sodium across the groups. We assumed the influence of the imbalance thus generated would be negligible in practice |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of ‘Low risk’ or ‘High risk’ |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: '...patients were randomly assigned to take placebo or a once‐daily dose of 25 or 50 mg of satavaptan during a double‐blind period of up to 5 days...', and 'The double‐blind period was followed by 23 d of open‐label treatment...' Comment: blinding probably attempted, and although participants are unlikely to be fully blinded due to important increases in urine output, all measured outcomes are short‐term and fairly objective. Co‐interventions similar for all groups |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether assessors of non patient‐reported outcomes were blinded, but main measured outcomes objective. Unlikely for participants to have been fully blinded, and plausible 'high risk' of bias for self‐reported outcome of thirst |
| Incomplete outcome data (attrition bias) | Low risk | Comment: only 1 participant (3%), in the placebo group, dropped out due to worsening of pre‐existing vasculitis |
| Selective reporting (reporting bias) | Low risk | Comment: no protocol, but short‐term study (≤ 1 week) and both secondary outcomes related to serum sodium concentration and rapid increase in serum sodium concentration reported |
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Treatment group 2
Treatment group 3
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: 'patients were randomised at a central site by the Computerized Randomization/Enrollment (CORE) system' Comment: due to the large number of groups relative to the number of included participants, randomisation procedure may have failed to produce groups with similar baseline prognosis. E.g. there is an imbalance in age across the groups. We assumed the influence of the imbalance thus generated would be negligible in practice |
| Allocation concealment (selection bias) | Low risk | Quote: 'patients were randomised at a central site by the Computerized Randomization/Enrollment (CORE) system' |
| Blinding of participants and personnel (performance bias) | High risk | Quote: '...randomised...to receive twice daily per os either placebo, ...' Comment: blinding probably attempted, but due to increase in urine output, un‐blinding likely to have occurred and as fluid intake was adjusted to urine output, this may likely have resulted in underestimation of the incidence of rapid increase in sodium concentration as well as influenced thirst measurements |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: unclear whether assessors of non patient‐reported outcomes were blinded, but main measured outcomes objective. Unlikely for participants to have been fully blinded, and plausible 'high risk' of bias for self‐reported outcome of thirst |
| Incomplete outcome data (attrition bias) | High risk | Comment: 27% attrition, due to treatment discontinuation (21%); reasons well‐documented. 'High risk' for sodium concentration measurements given re‐inclusion occurred using last available measurement before discontinuation |
| Selective reporting (reporting bias) | High risk | Comment: no protocol ‐ study duration 8 days without adequate reporting of secondary outcomes related serum sodium concentration permitting data
|
| Other bias | High risk |
|
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: '...they were randomly divided...' Comment: insufficient evidence to permit judgement of 'Low risk' or 'High risk' |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient evidence to permit judgement of 'Low risk' or 'High risk' |
| Blinding of participants and personnel (performance bias) | Unclear risk | Comment: insufficient evidence to permit judgement of 'Low risk' or 'High risk' |
| Blinding of outcome assessment (detection bias) | Unclear risk | Comment: insufficient evidence to permit judgement of 'Low risk' or 'High risk' |
| Incomplete outcome data (attrition bias) | Unclear risk | Comment: insufficient evidence to permit judgement of 'Low risk' or 'High risk' |
| Selective reporting (reporting bias) | High risk | Comment: no protocol, study duration > 1 week, primary and secondary outcomes insufficiently reported |
| Other bias | Unclear risk | Comment: insufficient evidence to permit judgement of 'Low risk' or 'High risk' |
| Methods |
| |
| Participants |
| |
| Interventions | Treatment group 1
Treatment group 2
Control group
Co‐interventions (all groups)
| |
| Outcomes |
| |
| Notes |
| |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: 'patients were stratified by volume status and randomly assigned to...' |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information to permit judgement of ‘Low risk’ or ‘High risk’ |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: '...double‐blind, placebo‐controlled study,...' and 'The placebo group received a 100‐ml loading dose of D5W on day 1 followed by continuous infusion of 250 ml D5W...' Comment: blinding probably attempted, and although participants are unlikely to be fully blinded due to important increases in urine output, all measured outcomes fairly objective. Co‐interventions Fluid‐restriction and salt intake were similar for all groups |
| Blinding of outcome assessment (detection bias) | Low risk | Comment: blinding outcome assessors for non patient‐reported outcomes probably attempted and main measured outcomes objective. Unlikely for participants to have been fully blinded, and plausible 'high risk' of bias for self‐reported outcome of thirst |
| Incomplete outcome data (attrition bias) | High risk | Comment: overall 25% attrition due to exclusion after randomisation (4%) and treatment discontinuation (21%); reasons well‐documented. 'Low risk' for death, but 'high risk' for sodium concentration measurements given reinclusion occurred using last available measurement before discontinuation. Although the opportunity for introducing bias is probably limited in such a short‐term study ‐ we judged it 'high risk' as matter of principle |
| Selective reporting (reporting bias) | Low risk | Comment: no protocol, but death, serum sodium concentration and outcomes related to rapid increase in serum sodium concentration reported. However, violation intention to treat analysis may be an issue. Quote: 'The efficacy analyses were conducted using a modified intent‐to‐treat approach and included all randomised patients who had at least 1 baseline serum [Na+] measurement, received at least 1 dose of study medication, and had at least 1 valid efficacy measurement.' Comment: 4 (4%) excluded after randomisation for not having received the study drug |
| Other bias | High risk |
|
AKI ‐ acute kidney injury; ALT ‐ alanine aminotransferase; AST ‐ aspartate aminotransferase; AVP ‐ Arginine vasopressin; BP ‐ blood pressure; BPM ‐ beats per minute; BUN ‐ blood urea nitrogen; CHF ‐ chronic heart failure; COPD ‐ chronic obstructive pulmonary disease; CrCl ‐ creatinine clearance; FDA ‐ food and Drug Administration; GFR ‐ glomerular filtration rate; Hb ‐ haemoglobin; INR ‐ international normalised ratio; MI ‐ myocardial infarction; NYHA ‐ New York Heart Association; PCW ‐ pulmonary capillary wedge; RCT ‐ randomised controlled trial; SCr ‐ serum creatinine; SD ‐ standard deviation; SF‐12 ‐ Short Form Health Survey; SIADH ‐ syndrome of inappropriate antidiuretic hormone; VAS ‐ visual analogue scale; WCC ‐ white cell count
Characteristics of excluded studies [ordered by study ID]
Jump to:
| Study | Reason for exclusion |
| Wrong population: RCT comparing lixivaptan versus placebo in participants with heart failure. Study population included both participants with and without hyponatraemia. We attempted to contact the authors on 24/10/2014 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT comparing fluid restriction versus usual care after discharge in patients hospitalised for acute decompensation of chronic heart failure. Inclusion criteria state hyponatraemia ‐ but defined as a serum sodium concentration ≤ 137 mmol/L. At discharge, and hence start of the intervention, the median serum sodium concentration in both groups equalled 135.5 mmol/L with interquartile ranges 134 to 137 mmol/L. This means hardly no participants had hyponatraemia as defined in this review. Consequently, any treatment given does not have the same harms (rapid increases in serum sodium concentration, osmotic demyelination syndrome) to consider. We judged the studied participants in this trial did not meet the projected characteristics of the patient group targeted by this review | |
| Wrong population: people with hyponatraemia due to primary psychogenic polydipsia are excluded from the review | |
| Wrong population: RCT comparing combined versus sequential diuretic treatment in participants with cirrhosis. Study population included both participants with and without hyponatraemia. We attempted to contact the authors on 23/10/2014 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong intervention: RCT in which in patients with liver cirrhosis and ascites two different salt‐restricted diets are evaluated in combination with step‐wise increase of diuretic treatment in both groups in order to reduce weight and ascites | |
| Wrong population: RCT comparing rituximab monotherapy versus the best available treatment in patients with mixed cryoglobulinaemia syndrome | |
| Wrong population: RCT in which a single dose of conivaptan was given to normonatraemic critically ill patients with severe traumatic brain injury | |
| Wrong population: RCT comparing lixivaptan versus placebo in participants with heart failure. Study population included both participants with and without hyponatraemia. We attempted to contact the authors on 23/10/2014 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Double counting of included participants. Study represents a second RCT built on top of a first included RCT (Gines 2008b) using the same study drug. Including the trial would give rise to double counting of participants | |
| Wrong population: RCT comparing satavaptan with placebo in participants with liver cirrhosis. Study population possibly included both participants with and without hyponatraemia. We attempted to contact the authors with a request for the outcome data for the subgroup of participants with hyponatraemia. We received a response from the sponsor with inclusion of the full text. However, no additional data were provided | |
| Wrong population: Post‐hoc analysis of RCT comparing tolvaptan versus placebo in participants with heart failure. Study population included both participants with and without hyponatraemia. We attempted to contact the authors on 04/12/2016 with a request for the outcome data for the subgroup of participants with hyponatraemia. As the primary study data have not yet been published, primary author unable to supply raw data | |
| Wrong population: RCT comparing high‐dose furosemide + low volume hypertonic saline versus high‐dose furosemide in participants with heart failure. Study population possibly included both participants with and without hyponatraemia. We attempted to contact the authors on 24/10/2014 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT comparing tolvaptan versus placebo in participants with heart failure. Study population possibly included both participants with and without hyponatraemia. We attempted to contact the authors on 24/10/2014 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT comparing tolvaptan versus placebo in participants with heart failure. Study population possibly included both participants with and without hyponatraemia. We attempted to contact the authors on 24/10/2014 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: study evaluating fludrocortisone for preventing hyponatraemia in people with subarachnoidal bleeding. Patients were not hyponatraemic at randomisation | |
| Wrong population: RCT comparing tolvaptan versus placebo in participants with liver cirrhosis and hepatic oedema. Study population included both participants with and without hyponatraemia. We attempted to contact the authors on 04/12/2016 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT in people with acute hyponatraemia after an ultra‐distance run | |
| Wrong population: RCT comparing high‐dose furosemide + low volume hypertonic saline versus high‐dose furosemide in participants with heart failure. Study population possibly included both participants with and without hyponatraemia. We attempted to contact the authors on 24/10/2014 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT on conivaptan in people with acute hyponatraemia | |
| Wrong population: RCT in people with congestive heart failure, but none had hyponatraemia at the start of the study | |
| Wrong population: RCT in people with exercise‐associated hyponatraemia, which is to be seen as a form of acute hyponatraemia present < 48 hours, requiring immediate intervention. Only chronic hyponatraemia, defined as present > 48 hours is included in the review | |
| Wrong population: RCT comparing tolvaptan versus placebo in participants with liver cirrhosis and hepatic oedema. Study population included both participants with and without hyponatraemia. We attempted to contact the authors on 04/12/2016 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT comparing intermittent infusion of small volumes of NaCl 3% versus slow continuous infusion of NaCl 3% in people with moderately severe or severe symptoms attributed to hyponatraemia. These people are considered requiring immediate intervention to increase the serum sodium concentration. Only those that did not require immediate treatment were considered in the review | |
| Wrong population: RCT comparing tolvaptan versus placebo in participants with heart failure. Study population included both participants with and without hyponatraemia. We attempted to contact the authors on 04/12/2016 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT comparing tolvaptan versus placebo in participants with heart failure. Abstract title implies all participants had hyponatraemia, but investigators actually included everyone with serum sodium concentration below 140 mmol/L. Study population included both participants with and without hyponatraemia according to our definition. We attempted to contact the authors on 04/12/2016 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT comparing tolvaptan versus carperitide in participants with heart failure. Study population possibly included both participants with and without hyponatraemia. We attempted to contact the authors on 24/10/2014 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT comparing tolvaptan versus placebo in participants with heart failure. Study population included both participants with and without hyponatraemia. We attempted to contact the authors on 04/12/2016 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT comparing tolvaptan versus placebo in participants with heart failure. Study population included both participants with and without hyponatraemia. We attempted to contact the authors on 04/12/2016 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT comparing tolvaptan versus placebo in participants with liver cirrhosis and ascites. Study population included both participants with and without hyponatraemia. We attempted to contact the authors on 04/12/2016 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT comparing satavaptan versus placebo in participants with liver cirrhosis and hepatic oedema. Study population included both participants with and without hyponatraemia. We attempted to contact the authors on 04/12/2016 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT comparing satavaptan with placebo in participants with liver cirrhosis. Study population possibly included both participants with and without hyponatraemia. We attempted to contact the authors with a request for the outcome data for the subgroup of participants with hyponatraemia. Dr Wong referred us to the sponsor, who was subsequently contacted on 24/10/2014. The sponsor agreed to provide the data but to date we received no further communication | |
| Wrong population: RCT comparing satavaptan versus placebo in participants with liver cirrhosis. Study population possibly included both participants with and without hyponatraemia. We attempted to contact the authors with a request for the outcome data for the subgroup of participants with hyponatraemia. Dr. Ginès referred us to the sponsor, who was subsequently contacted on 24/10/2014, but we received no response | |
| Wrong population: RCT comparing clonidine versus placebo in participants with liver cirrhosis. Study population possibly included both participants with and without hyponatraemia. We attempted to contact the authors on 24/10/2014 with a request for the outcome data for the subgroup of participants with hyponatraemia, but received no response | |
| Wrong population: RCT in people with CKD stage 3 or 4. This is not a group that likely contains a substantial number of people with hyponatraemia | |
| Wrong population: RCT comparingcyclooxygenase‐2 inhibitors versus non‐selective NSAID and the risk of developing hyponatraemia |
CKD ‐ chronic kidney disease; NSAID ‐ non‐steroidal anti‐inflammatory drugs; RCT ‐ randomised controlled trial
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Death at 6 months Show forest plot | 15 | 2330 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.92, 1.33] |
| Analysis 1.1 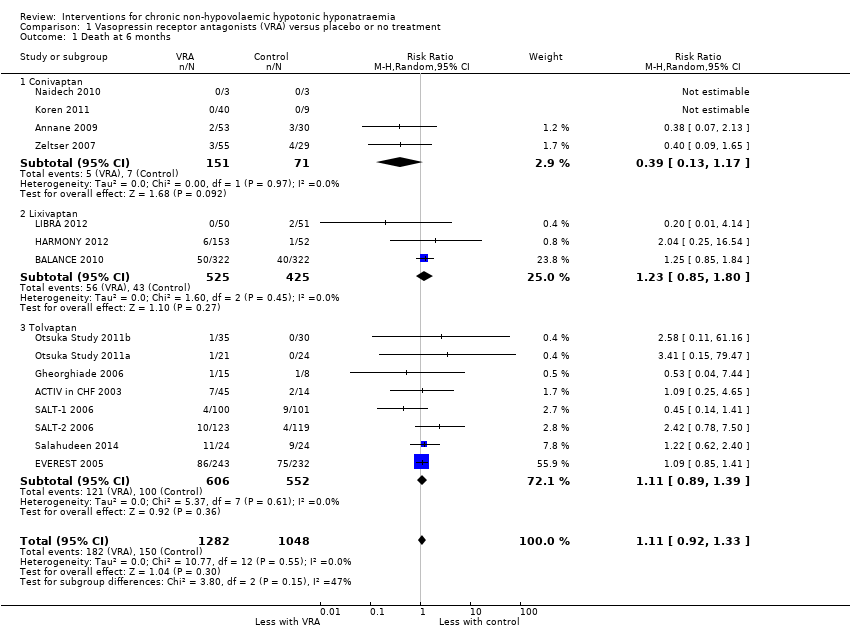 Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 1 Death at 6 months. | ||||
| 1.1 Conivaptan | 4 | 222 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.13, 1.17] |
| 1.2 Lixivaptan | 3 | 950 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.85, 1.80] |
| 1.3 Tolvaptan | 8 | 1158 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.89, 1.39] |
| 2 Health‐related quality of life Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 1.2  Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 2 Health‐related quality of life. | ||||
| 2.1 Mental component SF‐12 | 2 | 297 | Mean Difference (IV, Random, 95% CI) | 4.76 [0.11, 9.41] |
| 2.2 Physical component SF‐12 | 2 | 300 | Mean Difference (IV, Random, 95% CI) | 1.04 [‐1.81, 3.90] |
| 3 Cognitive function: trail making test Part B Show forest plot | 2 | 858 | Mean Difference (IV, Random, 95% CI) | 6.89 [‐6.34, 20.12] |
| Analysis 1.3  Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 3 Cognitive function: trail making test Part B. | ||||
| 4 Length of hospital stay Show forest plot | 3 | 610 | Mean Difference (IV, Random, 95% CI) | ‐1.63 [‐2.96, ‐0.30] |
| Analysis 1.4  Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 4 Length of hospital stay. | ||||
| 4.1 Satavaptan | 1 | 139 | Mean Difference (IV, Random, 95% CI) | ‐1.80 [‐3.80, 0.20] |
| 4.2 Tolvaptan | 2 | 471 | Mean Difference (IV, Random, 95% CI) | ‐1.50 [‐3.28, 0.29] |
| 5 Change from baseline serum sodium concentration Show forest plot | 21 | 2641 | Mean Difference (IV, Random, 95% CI) | 4.17 [3.18, 5.16] |
| Analysis 1.5  Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 5 Change from baseline serum sodium concentration. | ||||
| 5.1 Conivaptan | 5 | 292 | Mean Difference (IV, Random, 95% CI) | 5.17 [2.65, 7.69] |
| 5.2 Lixivaptan | 4 | 1070 | Mean Difference (IV, Random, 95% CI) | 2.24 [0.78, 3.70] |
| 5.3 Satavaptan | 3 | 257 | Mean Difference (IV, Random, 95% CI) | 4.91 [2.88, 6.94] |
| 5.4 Tolvaptan | 9 | 1022 | Mean Difference (IV, Random, 95% CI) | 4.22 [3.55, 4.89] |
| 6 Response in serum sodium concentration Show forest plot | 18 | 2104 | Risk Ratio (M‐H, Random, 95% CI) | 2.49 [1.95, 3.18] |
| Analysis 1.6  Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 6 Response in serum sodium concentration. | ||||
| 6.1 Conivaptan | 4 | 282 | Risk Ratio (M‐H, Random, 95% CI) | 2.48 [1.54, 4.01] |
| 6.2 Lixivaptan | 4 | 1024 | Risk Ratio (M‐H, Random, 95% CI) | 2.21 [1.19, 4.10] |
| 6.3 Satavaptan | 4 | 331 | Risk Ratio (M‐H, Random, 95% CI) | 3.33 [1.88, 5.89] |
| 6.4 Tolvaptan | 6 | 467 | Risk Ratio (M‐H, Random, 95% CI) | 2.36 [1.75, 3.18] |
| 7 Rapid increase in serum sodium concentration Show forest plot | 14 | 2058 | Risk Ratio (M‐H, Random, 95% CI) | 1.67 [1.16, 2.40] |
| Analysis 1.7  Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 7 Rapid increase in serum sodium concentration. | ||||
| 7.1 Conivaptan | 4 | 290 | Risk Ratio (M‐H, Random, 95% CI) | 3.77 [0.89, 15.98] |
| 7.2 Lixivaptan | 4 | 994 | Risk Ratio (M‐H, Random, 95% CI) | 1.39 [0.90, 2.17] |
| 7.3 Satavaptan | 4 | 331 | Risk Ratio (M‐H, Random, 95% CI) | 2.61 [0.73, 9.30] |
| 7.4 Tolvaptan | 2 | 443 | Risk Ratio (M‐H, Random, 95% CI) | 6.70 [0.82, 54.56] |
| 8 Hypernatraemia during treatment Show forest plot | 10 | 1592 | Risk Ratio (M‐H, Random, 95% CI) | 1.37 [0.63, 3.01] |
| Analysis 1.8 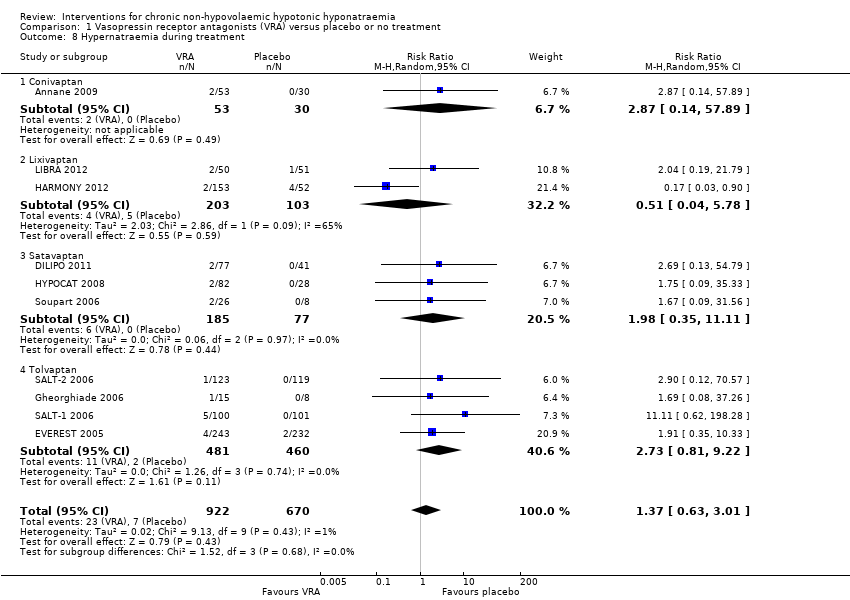 Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 8 Hypernatraemia during treatment. | ||||
| 8.1 Conivaptan | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 2.87 [0.14, 57.89] |
| 8.2 Lixivaptan | 2 | 306 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.04, 5.78] |
| 8.3 Satavaptan | 3 | 262 | Risk Ratio (M‐H, Random, 95% CI) | 1.98 [0.35, 11.11] |
| 8.4 Tolvaptan | 4 | 941 | Risk Ratio (M‐H, Random, 95% CI) | 2.73 [0.81, 9.22] |
| 9 Thirst Show forest plot | 13 | Odds Ratio (Random, 95% CI) | 2.77 [1.80, 4.27] | |
| Analysis 1.9 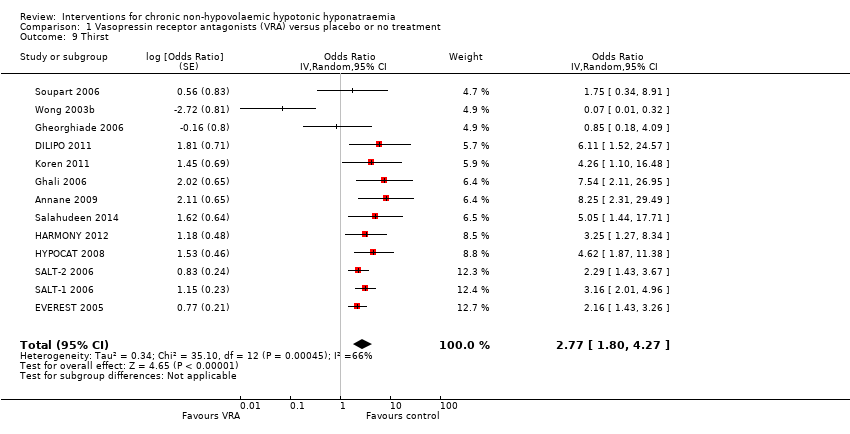 Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 9 Thirst. | ||||
| 10 Other adverse events Show forest plot | 17 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 1.10  Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 10 Other adverse events. | ||||
| 10.1 Polyuria | 6 | 1272 | Risk Ratio (M‐H, Random, 95% CI) | 4.69 [1.59, 13.85] |
| 10.2 Hypotension | 14 | 1748 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.75, 1.63] |
| 10.3 Acute kidney injury | 8 | 1920 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.67, 1.18] |
| 10.4 Liver function abnormalities | 3 | 811 | Risk Ratio (M‐H, Random, 95% CI) | 2.43 [0.88, 6.70] |
| 11 Injection‐site complications at 2 to 7 days Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| Analysis 1.11  Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 11 Injection‐site complications at 2 to 7 days. | ||||
| 11.1 Reactions | 1 | 49 | Risk Ratio (M‐H, Random, 95% CI) | 7.56 [0.49, 115.93] |
| 11.2 Phlebitis | 2 | 133 | Risk Ratio (M‐H, Random, 95% CI) | 3.52 [1.00, 12.41] |
| 11.3 Thrombosis | 2 | 133 | Risk Ratio (M‐H, Random, 95% CI) | 1.75 [0.21, 14.80] |
| 12 Treatment discontinuation Show forest plot | 14 | 2429 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.85, 1.00] |
| Analysis 1.12  Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 12 Treatment discontinuation. | ||||
| 12.1 Conivaptan | 4 | 288 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.39, 1.30] |
| 12.2 Lixivaptan | 4 | 1008 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.84, 1.03] |
| 12.3 Satavaptan | 2 | 145 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.17, 1.46] |
| 12.4 Tolvaptan | 4 | 988 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.66, 1.13] |
| 13 Death during follow‐up: sensitivity analysis Show forest plot | 16 | 2404 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.91, 1.32] |
| Analysis 1.13  Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 13 Death during follow‐up: sensitivity analysis. | ||||
| 13.1 Conivaptan | 5 | 296 | Risk Ratio (M‐H, Random, 95% CI) | 0.46 [0.19, 1.15] |
| 13.2 Lixivaptan | 3 | 950 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.37, 3.15] |
| 13.3 Tolvaptan | 8 | 1158 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.89, 1.39] |
| 14 Rapid increase in serum sodium concentration: sensitivity analysis Show forest plot | 14 | 2058 | Risk Ratio (M‐H, Random, 95% CI) | 1.67 [1.16, 2.40] |
| Analysis 1.14  Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 14 Rapid increase in serum sodium concentration: sensitivity analysis. | ||||
| 14.1 > 8 mmol/L/d | 4 | 257 | Risk Ratio (M‐H, Random, 95% CI) | 3.22 [0.65, 15.94] |
| 14.2 > 12 mmol/L/d | 10 | 1801 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [1.09, 2.43] |


Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Effect of baseline serum sodium concentration on change in natraemia: meta‐regression

Funnel plot of comparison: 1 Vasopressin receptor antagonists versus placebo or no treatment, outcome: 1.6 Response in serum sodium concentration.

Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 1 Death at 6 months.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 2 Health‐related quality of life.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 3 Cognitive function: trail making test Part B.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 4 Length of hospital stay.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 5 Change from baseline serum sodium concentration.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 6 Response in serum sodium concentration.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 7 Rapid increase in serum sodium concentration.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 8 Hypernatraemia during treatment.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 9 Thirst.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 10 Other adverse events.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 11 Injection‐site complications at 2 to 7 days.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 12 Treatment discontinuation.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 13 Death during follow‐up: sensitivity analysis.
Comparison 1 Vasopressin receptor antagonists (VRA) versus placebo or no treatment, Outcome 14 Rapid increase in serum sodium concentration: sensitivity analysis.
| Vasopressin receptor antagonists versus placebo or no treatment for chronic non‐hypovolaemic hypotonic hyponatraemia | ||||||
| Patient or population: chronic non‐hypovolaemic hypotonic hyponatraemia | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No. of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| placebo or no treatment | Vasopressin receptor antagonists | |||||
| Death Follow‐up: range 2 to 180 days | Study population | RR 1.11 | 2330 (15) | ⊕⊝⊝⊝ | Interpretation: effect uncertain; may both result in 11/1000 fewer to 47/1000 more deaths within 6 months | |
| 143 per 1000 1 | 159 per 1000 | |||||
| Health‐related quality of life (assessed with mental component score of SF‐124) Follow‐up: 30 days | The mean change from baseline in health‐related quality of life in the control group ranged between 0.75 and 2.39 on a 0 to 100 point scale (worst to best) 5 | The mean health‐related quality of life in the intervention group was 4.76 higher (0.11 higher to 9.41 higher) | ‐ | 297 (2) | ⊕⊝⊝⊝ | Physical component score also measured in both studies; RR 1.04; CI ‐1.81 to 3.90 Interpretation: anywhere from 0.1 to 9.5/100 points higher increase with treatment, but questionable tool for QoL measurement in hyponatraemia and unclear minimally important clinical difference |
| Length of hospital stay | The mean length of hospital stay in the control group was 6 to 11 days 5 | The mean length of hospital stay in the intervention group was 1.63 days lower (2.96 lower to 0.30 lower) | ‐ | 580 (2) | ⊕⊕⊝⊝ | |
| Cognitive function (assessed with various tools) Follow‐up: 1 to 6 months | Across five studies, confidence intervals spanned the line of no effect and did not include a clinically meaningful effect | ‐ | 1169 (5) | ⊕⊕⊝⊝ LOW 10 | Tools used to assess cognitive function: making test B; reaction time, psychomotor, processing speeds; Mini mental state exam; overall meta‐analysis including all five studies not meaningfully possible | |
| Change from baseline in serum sodium concentration Follow‐up: range 1 to 180 days | The mean change from baseline in serum sodium concentration in the control group was 0.3 to 4.8 mmol/L 5 | The mean change from baseline in serum sodium concentration in the intervention group was 4.17 mmol/L higher (3.18 higher to 5.16 higher) | ‐ | 2641 (21) | ⊕⊕⊕⊝ | |
| Serum sodium concentration (response) Follow‐up: range 4 to 180 days | Study population | RR 2.49 | 2104 (18) | ⊕⊕⊕⊝ | Response most commonly defined by investigators as > 5 to > 6 mmol/L increase or normalisation of serum sodium concentration | |
| 231 per 1000 1 | 576 per 1000 | |||||
| Rapid sodium increase Follow‐up: range 1 to 5 days | Study population | RR 1.67 | 2058 (14) | ⊕⊕⊕⊝ | Rapid increase most commonly defined as > 12 mmol/d | |
| 44 per 1000 1 | 73 per 1000 | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1 Source of assumed baseline risk was calculated as the unweighted summed event rate in the control groups of the trials included in the meta‐analysis 2 Downgraded one level because studies considered at serious risk of suffering from * selective reporting: 6/16 studies with treatment duration > 1 week did not report death and had no protocol, accounting for 16% of the total number of participants in those studies * commercial sponsorship; possible financial conflict of interest of the authors: all studies sponsored by pharmaceutical companies wanting to commercialize the treatment; all save one had author lists who featured people who had received money for presentations or consultancy, or were employed by the sponsor; only used as supporting reason for downgrading. 3 Downgraded two levels for imprecision. The 95% CI of the pooled estimate includes both important reduction (11/1000 fewer) and increase (47/1000 more) in death with vasopressin receptor antagonists. 4 Choice of the outcome‐measure based on the fact that this was the only one reported in any of the studies. There are concerns around the validity of the SF‐12 as a measure for health‐related quality of life in the field of hyponatraemia as it gauges domains and symptoms not directly attributable to hyponatraemia. 5 Source of the assumed baseline risk was the range of outcomes in the control groups of the trials included in the meta‐analysis 6 Downgraded one level because studies considered seriously at risk of suffering from * Performance bias: self‐reported outcome, participants likely unblinded to treatment due to polyuria as side effect * Selective reporting bias: both mental and physical component score of SF‐12 measured; at week 1 or 2 and day 30 using two different analytic techniques. Only data at day 30 available for analysis. * Attrition bias: overall 37% of data missing, unknown whether missing at random or not. * Commercial sponsorship or possible financial conflict of interest of authors: both studies were sponsored by the company seeking to commercialise the treatment; both had author lists who featured people who had received money for presentations or consultancy, or were employed by the sponsor; only using as supporting argument for downgrading 7 Downgraded one level for indirectness due to concerns around validity of the SF‐12 for measuring quality of life in the context of hyponatraemia and one level for imprecision: only studied in two studies. 8 Downgraded one level because studies considered seriously at risk of suffering from performance bias: participants and personnel likely unblinded to treatment due to polyuria as side effect; this could have influenced self‐reported and professional appreciation of clinical condition and so have influenced decision to discharge from hospital. 9 Downgraded one level for imprecision. The 95% CI of the pooled estimate includes both negligible shortening (0.3 days shorter) and clinically important shortening (3 days shorter) of hospital stay with vasopressin receptor antagonists. 10 Downgraded two levels for indirectness and imprecision. Only studied in 5/28 studies, with most data for lixivaptan, and other studies not reaching the optimal information size. 11 Downgraded one level because we considered studies seriously at risk of suffering from * Attrition bias: 7/21 studies, accounting for 53% of the total number of participants in those studies at high risk of bias either due to true attrition or because a repeated measures analytic technique was used with all measurements of serum sodium concentration included until patient attrition, which we judged would likely have overestimated the treatment effect. *Commercial sponsorship; only used as supporting argument. 12 Downgraded one level for indirectness; risks controlled in tightly organised randomised trial with several measurements of serum sodium concentration daily to avoid rapid correction. In real life, risk of rapid correction likely greater. Commercial sponsorship bias; only used as supporting argument. | ||||||
| Covariate | Number of studies included in meta‐regression | Scale | Absolute change in mean difference | P value |
| Baseline serum sodium concentration | 21 | Per 1 mmol/L increase | ‐0.33 (‐0.65 to ‐0.02) | 0.04 |
| Compound | 21 | Relative to conivaptan | ‐ | 0.17 |
| Conivaptan | 5 | ‐ | ‐ | ‐ |
| Lixivaptan | 4 | ‐ | ‐2.79 (‐5.47 to ‐0.10) | ‐ |
| Satavaptan | 3 | ‐ | ‐0,23 (‐3.34 to 2.87) | ‐ |
| Tolvaptan | 9 | ‐ | ‐0.82 (‐3.13 to 1.50) | ‐ |
| Cause of hyponatraemia | 21 | Relative to SIADH | ‐ | 0.18 |
| SIADH | 5 | ‐ | ‐ | ‐ |
| Combined SIADH ‐ Heart failure, cirrhosis | 10 | ‐ | 0.67 (‐1.77 to 3.13) | ‐ |
| Heart failure | 6 | ‐ | ‐1.19 (‐3.84 to 1.47) | ‐ |
| Treatment duration | 21 | Per day increase | ‐0.02 (‐0,04 to 0.00) | 0.1 |
| Risk of selection bias | 21 | Relative to low risk | ‐ | 0.86 |
| Low risk | 8 | ‐ | ‐ | ‐ |
| High risk | 13 | ‐ | 0.18 (‐1.90 to 2.27) | ‐ |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Death at 6 months Show forest plot | 15 | 2330 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.92, 1.33] |
| 1.1 Conivaptan | 4 | 222 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.13, 1.17] |
| 1.2 Lixivaptan | 3 | 950 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.85, 1.80] |
| 1.3 Tolvaptan | 8 | 1158 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.89, 1.39] |
| 2 Health‐related quality of life Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Mental component SF‐12 | 2 | 297 | Mean Difference (IV, Random, 95% CI) | 4.76 [0.11, 9.41] |
| 2.2 Physical component SF‐12 | 2 | 300 | Mean Difference (IV, Random, 95% CI) | 1.04 [‐1.81, 3.90] |
| 3 Cognitive function: trail making test Part B Show forest plot | 2 | 858 | Mean Difference (IV, Random, 95% CI) | 6.89 [‐6.34, 20.12] |
| 4 Length of hospital stay Show forest plot | 3 | 610 | Mean Difference (IV, Random, 95% CI) | ‐1.63 [‐2.96, ‐0.30] |
| 4.1 Satavaptan | 1 | 139 | Mean Difference (IV, Random, 95% CI) | ‐1.80 [‐3.80, 0.20] |
| 4.2 Tolvaptan | 2 | 471 | Mean Difference (IV, Random, 95% CI) | ‐1.50 [‐3.28, 0.29] |
| 5 Change from baseline serum sodium concentration Show forest plot | 21 | 2641 | Mean Difference (IV, Random, 95% CI) | 4.17 [3.18, 5.16] |
| 5.1 Conivaptan | 5 | 292 | Mean Difference (IV, Random, 95% CI) | 5.17 [2.65, 7.69] |
| 5.2 Lixivaptan | 4 | 1070 | Mean Difference (IV, Random, 95% CI) | 2.24 [0.78, 3.70] |
| 5.3 Satavaptan | 3 | 257 | Mean Difference (IV, Random, 95% CI) | 4.91 [2.88, 6.94] |
| 5.4 Tolvaptan | 9 | 1022 | Mean Difference (IV, Random, 95% CI) | 4.22 [3.55, 4.89] |
| 6 Response in serum sodium concentration Show forest plot | 18 | 2104 | Risk Ratio (M‐H, Random, 95% CI) | 2.49 [1.95, 3.18] |
| 6.1 Conivaptan | 4 | 282 | Risk Ratio (M‐H, Random, 95% CI) | 2.48 [1.54, 4.01] |
| 6.2 Lixivaptan | 4 | 1024 | Risk Ratio (M‐H, Random, 95% CI) | 2.21 [1.19, 4.10] |
| 6.3 Satavaptan | 4 | 331 | Risk Ratio (M‐H, Random, 95% CI) | 3.33 [1.88, 5.89] |
| 6.4 Tolvaptan | 6 | 467 | Risk Ratio (M‐H, Random, 95% CI) | 2.36 [1.75, 3.18] |
| 7 Rapid increase in serum sodium concentration Show forest plot | 14 | 2058 | Risk Ratio (M‐H, Random, 95% CI) | 1.67 [1.16, 2.40] |
| 7.1 Conivaptan | 4 | 290 | Risk Ratio (M‐H, Random, 95% CI) | 3.77 [0.89, 15.98] |
| 7.2 Lixivaptan | 4 | 994 | Risk Ratio (M‐H, Random, 95% CI) | 1.39 [0.90, 2.17] |
| 7.3 Satavaptan | 4 | 331 | Risk Ratio (M‐H, Random, 95% CI) | 2.61 [0.73, 9.30] |
| 7.4 Tolvaptan | 2 | 443 | Risk Ratio (M‐H, Random, 95% CI) | 6.70 [0.82, 54.56] |
| 8 Hypernatraemia during treatment Show forest plot | 10 | 1592 | Risk Ratio (M‐H, Random, 95% CI) | 1.37 [0.63, 3.01] |
| 8.1 Conivaptan | 1 | 83 | Risk Ratio (M‐H, Random, 95% CI) | 2.87 [0.14, 57.89] |
| 8.2 Lixivaptan | 2 | 306 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.04, 5.78] |
| 8.3 Satavaptan | 3 | 262 | Risk Ratio (M‐H, Random, 95% CI) | 1.98 [0.35, 11.11] |
| 8.4 Tolvaptan | 4 | 941 | Risk Ratio (M‐H, Random, 95% CI) | 2.73 [0.81, 9.22] |
| 9 Thirst Show forest plot | 13 | Odds Ratio (Random, 95% CI) | 2.77 [1.80, 4.27] | |
| 10 Other adverse events Show forest plot | 17 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 Polyuria | 6 | 1272 | Risk Ratio (M‐H, Random, 95% CI) | 4.69 [1.59, 13.85] |
| 10.2 Hypotension | 14 | 1748 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.75, 1.63] |
| 10.3 Acute kidney injury | 8 | 1920 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.67, 1.18] |
| 10.4 Liver function abnormalities | 3 | 811 | Risk Ratio (M‐H, Random, 95% CI) | 2.43 [0.88, 6.70] |
| 11 Injection‐site complications at 2 to 7 days Show forest plot | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 Reactions | 1 | 49 | Risk Ratio (M‐H, Random, 95% CI) | 7.56 [0.49, 115.93] |
| 11.2 Phlebitis | 2 | 133 | Risk Ratio (M‐H, Random, 95% CI) | 3.52 [1.00, 12.41] |
| 11.3 Thrombosis | 2 | 133 | Risk Ratio (M‐H, Random, 95% CI) | 1.75 [0.21, 14.80] |
| 12 Treatment discontinuation Show forest plot | 14 | 2429 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.85, 1.00] |
| 12.1 Conivaptan | 4 | 288 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.39, 1.30] |
| 12.2 Lixivaptan | 4 | 1008 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.84, 1.03] |
| 12.3 Satavaptan | 2 | 145 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.17, 1.46] |
| 12.4 Tolvaptan | 4 | 988 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.66, 1.13] |
| 13 Death during follow‐up: sensitivity analysis Show forest plot | 16 | 2404 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.91, 1.32] |
| 13.1 Conivaptan | 5 | 296 | Risk Ratio (M‐H, Random, 95% CI) | 0.46 [0.19, 1.15] |
| 13.2 Lixivaptan | 3 | 950 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.37, 3.15] |
| 13.3 Tolvaptan | 8 | 1158 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.89, 1.39] |
| 14 Rapid increase in serum sodium concentration: sensitivity analysis Show forest plot | 14 | 2058 | Risk Ratio (M‐H, Random, 95% CI) | 1.67 [1.16, 2.40] |
| 14.1 > 8 mmol/L/d | 4 | 257 | Risk Ratio (M‐H, Random, 95% CI) | 3.22 [0.65, 15.94] |
| 14.2 > 12 mmol/L/d | 10 | 1801 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [1.09, 2.43] |