Metilfenidato de liberación inmediata para el trastorno de déficit de atención e hiperactividad (TDAH) en adultos
Resumen
Antecedentes
El trastorno por déficit de atención e hiperactividad (TDAH) se caracteriza por síntomas de falta de atención o impulsividad o ambos, y por la hiperactividad, que afecta a niños, adolescentes y adultos. En algunos países, el metilfenidato es la primera opción para tratar a los adultos que presentan TDAH moderado o grave. Sin embargo, la evidencia sobre la eficacia y los efectos adversos del metilfenidato de liberación inmediata en el tratamiento del TDAH en adultos es limitada y controvertida.
Objetivos
Evaluar la eficacia y los efectos perjudiciales (eventos adversos) del metilfenidato de liberación inmediata en el tratamiento del TDAH en adultos.
Métodos de búsqueda
En enero de 2020 se hicieron búsquedas en CENTRAL, MEDLINE, Embase, ocho bases de datos adicionales y tres registros de ensayos. También se buscaron informes internos en los sitios web de la Agencia Europea de Medicamentos y de la US Food and Drug Administration. Se verificaron las citas de los ensayos incluidos para identificar los ensayos adicionales no registrados por las búsquedas electrónicas.
Criterios de selección
Ensayos controlados aleatorizados (ECA) que compararon el metilfenidato de liberación inmediata, a cualquier dosis, con un placebo u otras intervenciones farmacológicas (incluidas las formulaciones de liberación prolongada de metilfenidato) para el TDAH en adultos. Los desenlaces principales incluyeron los cambios en los síntomas del TDAH (eficacia) y los efectos perjudiciales. Los desenlaces secundarios incluyeron cambios en la impresión clínica de gravedad y mejoría, el nivel de funcionalidad, la depresión, la ansiedad y la calidad de vida. Los desenlaces podían haber sido evaluados por los investigadores o los participantes.
Obtención y análisis de los datos
Dos autores de la revisión, de forma independiente, extrajeron los datos sobre las características de los ensayos, los participantes, las intervenciones, los desenlaces y los conflictos de intereses económicos. Los desacuerdos se resolvieron mediante debate o mediante consulta a un tercer autor de la revisión. Se obtuvo información adicional, no publicada, de los autores de un ensayo incluido que había informado los datos de eficacia en un gráfico. Se calcularon las diferencias de medias (DM) o las DM estandarizadas (DME) con intervalos de confianza (IC) del 95% para los datos continuos informados en la misma o en diferentes escalas, respectivamente. Las variables dicotómicas se resumieron como razones de riesgos (RR) con IC del 95%.
Resultados principales
Se incluyeron diez ensayos publicados entre 2001 y 2016 con a 497 adultos con TDAH. Tres ensayos se realizaron en Europa y uno en Argentina; los ensayos restantes no informaron el lugar de realización. Los ECA compararon el metilfenidato de liberación inmediata con el placebo, un sistema oral de liberación osmótica (OROS) de metilfenidato (una formulación de liberación prolongada), una formulación de liberación prolongada de bupropión, litio y Pycnogenol® (extracto de corteza de pino marítimo). Los participantes incluyeron pacientes ambulatorios, pacientes en tratamiento de adicciones y adultos dispuestos a asistir a un programa intensivo ambulatorio para la dependencia de la cocaína. La duración del seguimiento varió entre seis y 18 semanas.
Metilfenidato de liberación inmediata versus placebo
Se encontró evidencia de certeza muy baja de que, en comparación con el placebo, el metilfenidato de liberación inmediata podría reducir los síntomas del TDAH cuando se midió con escalas evaluadas por el investigador (DM ‐20,70; IC del 95%: ‐23,97 a ‐17,43; un ensayo, 146 participantes; puntuaciones finales; Adult ADHD Investigator Symptom Report Scale (AISRS), con una puntuación de 0 a 54), pero la evidencia es incierta. El efecto del metilfenidato de liberación inmediata sobre los síntomas del TDAH cuando se midió con escalas evaluadas por los participantes fue moderado, pero la certeza de la evidencia es muy baja (DME ‐0,59; IC del 95%: ‐1,25 a 0,06; I2 = 69%; dos ensayos, 138 participantes; puntuaciones finales).
Hay evidencia de certeza muy baja de que, en comparación con el placebo, el metilfenidato de liberación inmediata podría reducir la impresión clínica de la gravedad de los síntomas del TDAH (DM ‐0,57; IC del 95%: ‐0,85 a ‐0,28; dos ensayos, 139 participantes; I2 = 0%; puntuaciones de cambio y finales; escala Clinical Global Impression [CGI]‐Severity [puntuación desde 1 (muchísimo mejor) hasta 7 (muchísimo peor)]). Hay evidencia de certeza baja de que, en comparación con el placebo, el metilfenidato de liberación inmediata podría tener un ligero impacto en la impresión clínica de una mejoría en los síntomas del TDAH (DM ‐0,94; IC del 95%: ‐1,37 a ‐0,51; un ensayo, 49 participantes; puntuaciones finales; escala CGI‐Improvement [puntuación desde 1 (muchísimo mejor) hasta 7 (muchísimo peor)]). No hay evidencia clara de un efecto sobre la ansiedad (DM ‐0,20; IC del 95%: ‐4,84 a 4,44; un ensayo, 19 participantes; puntuaciones de cambio; Hamilton Anxiety Scale (HAM‐A; puntuación desde 0 hasta 56); evidencia de certeza muy baja) o la depresión (DM 2,80; IC del 95%: ‐0,09 a 5,69; un ensayo, 19 participantes; puntuaciones de cambio; Hamilton Depression Scale (HAM‐D; puntuación desde 0 hasta 52); evidencia de certeza muy baja) en los análisis que compararon el metilfenidato de liberación inmediata con el placebo.
Metilfenidato de liberación inmediata versus litio
En comparación con el litio, no se sabe con certeza si el metilfenidato de liberación inmediata aumenta o disminuye los síntomas del TDAH (DM 0,60; IC del 95%: ‐3,11 a 4,31; un ensayo, 46 participantes; puntuaciones finales; Conners' Adult ADHD Rating Scale (puntuación desde 0 hasta 198); evidencia de certeza muy baja); la ansiedad (DM ‐0,80; IC del 95%: ‐4,49 a 2,89; un ensayo, 46 participantes; puntuaciones finales; HAM‐A; evidencia de certeza muy baja) o la depresión (DM ‐1,20; IC del 95%: ‐3,81 a 1,41, un ensayo, 46 participantes; puntuaciones finales; escala HAM‐D; evidencia de certeza muy baja). Ninguno de los ensayos incluidos evaluó los cambios evaluados por los participantes en los síntomas del TDAH, ni la impresión clínica de la gravedad o la mejoría en los participantes tratados con metilfenidato de liberación inmediata en comparación con el litio.
Los efectos adversos se evaluaron e informaron de manera deficiente. Todos los ensayos se consideraron con alto riesgo de sesgo debido al informe selectivo de los efectos perjudiciales y al enmascaramiento de los evaluadores de los desenlaces (no cegar al evaluador de los desenlaces para medir los eventos adversos). En general, cuatro ensayos con 203 participantes que recibieron metilfenidato de liberación inmediata y 141 participantes que recibieron placebo describieron la aparición de efectos perjudiciales. El uso de metilfenidato de liberación inmediata en estos ensayos aumentó el riesgo de complicaciones gastrointestinales (RR 1,96; IC del 95%: 1,13 a 2,95) y la pérdida del apetito (RR 1,77; IC del 95%: 1,06 a 2,96). Los eventos adversos cardiovasculares se informaron de manera incongruente, lo que impidió un análisis exhaustivo. Un ensayo en que comparó el metilfenidato de liberación inmediata con el litio informó cinco y nueve episodios adversos, respectivamente.
Se consideró que en cuatro ensayos hubo preocupaciones notables de intereses adquiridos que influyeron en la evidencia, y los autores de dos ensayos omitieron información relacionada con las fuentes de financiación y los conflictos de intereses.
Conclusiones de los autores
No se encontró evidencia segura de que el metilfenidato de liberación inmediata comparado con el placebo o el litio pudiera reducir los síntomas del TDAH en adultos (evidencia de certeza baja y muy baja). Los adultos tratados con metilfenidato de liberación inmediata tienen un mayor riesgo de presentar efectos perjudiciales gastrointestinales y metabólicos en comparación con el placebo. Los médicos deben considerar si es apropiado prescribir metilfenidato de liberación inmediata, debido a su limitada eficacia y el mayor riesgo de efectos perjudiciales. En los ECA futuros se debería estudiar la eficacia y los riesgos a largo plazo del metilfenidato de liberación inmediata, y la influencia de los conflictos de intereses en los efectos informados.
PICO
Resumen en términos sencillos
Metilfenidato de liberación inmediata para el trastorno de déficit de atención e hiperactividad (TDAH) en adultos
¿Cuál es el objetivo de esta revisión?
Se revisó la evidencia sobre los efectos de tratar a los adultos con TDAH con un medicamento estimulante llamado metilfenidato de liberación inmediata.
Mensajes clave
En comparación con el placebo (una píldora falsa), el metilfenidato de liberación inmediata puede provocar una pequeña reducción de los síntomas del TDAH y puede aumentar la percepción del médico de una mejoría de los síntomas. El metilfenidato de liberación inmediata aumentó el riesgo de efectos adversos como pérdida del apetito, sequedad de la boca, náuseas y dolores de estómago.
En comparación con el litio (un fármaco para tratar la hiperactividad y la excitación), el metilfenidato de liberación inmediata puede provocar pocos o ningún cambio en los síntomas del TDAH, la ansiedad y la depresión.
Estos resultados no están claros y no se sabe si se puede confiar en ellos.
¿Qué se estudió en esta revisión?
El TDAH es un deterioro de la salud mental. El problema se diagnostica en adultos que muestran signos de falta de atención (p.ej., problemas de concentración), hiperactividad (p.ej., incapacidad de permanecer sentados) e impulsividad (p.ej., hacer cosas sin pensar).
Se buscaron los ensayos que compararan el metilfenidato de liberación inmediata, a cualquier dosis, con otros medicamentos (incluidas las formulaciones de liberación prolongada de metilfenidato en las que el fármaco se libera lentamente a lo largo del tiempo) o con un placebo, para tratar el TDAH en adultos. Se quería saber el efecto del metilfenidato de liberación inmediata sobre los síntomas del TDAH y si las personas presentaban episodios adversos. También se quería saber si las personas tratadas con el medicamento o sus médicos percibían cambios en los síntomas (que empeoraban o mejoraban), en la salud mental (depresión, ansiedad) o en la calidad de vida.
¿Cuáles son los principales resultados de la revisión?
Se encontraron diez ensayos con 497 adultos. Tres ensayos se realizaron en Europa y uno en Argentina; los ensayos restantes no informaron el lugar de realización. Seis ensayos compararon el metilfenidato de liberación inmediata con el placebo. En los demás ensayos, el metilfenidato de liberación inmediata se comparó con una forma de liberación prolongada de bupropión (un antidepresivo), litio, una forma de metilfenidato de liberación prolongada denominada sistema oral de liberación osmótica (OROS), y Pycnogenol® (un medicamento derivado de la corteza de pino). Las personas fueron tratadas durante seis a 18 semanas. Los participantes fueron principalmente pacientes ambulatorios; algunos participantes fueron pacientes hospitalizados para el tratamiento de adicciones o personas dispuestas a asistir a un programa intensivo ambulatorio para la dependencia de la cocaína.
Metilfenidato de liberación inmediata versus placebo
Un ensayo con 146 participantes informó que el metilfenidato de liberación inmediata podría reducir los síntomas del TDAH cuando los evalúan los médicos. Cuando los participantes evalúan sus propios síntomas, podría haber un efecto positivo moderado. Sin embargo, no hay seguridad acerca de estos resultados y podrían cambiar con el agregado de más datos. El metilfenidato de liberación inmediata parece tener poco o ningún efecto en la reducción de los síntomas de la ansiedad y la depresión. Preocupan los métodos y los conflictos de intereses de este ensayo y de los otros nueve ensayos que se evaluaron.
Metilfenidato de liberación inmediata versus litio
El metilfenidato de liberación inmediata podría tener poco o ningún efecto sobre los síntomas del TDAH (evaluados por los médicos), o la ansiedad y la depresión, pero los resultados no están claros. Ninguno de los ensayos incluidos evaluó los cambios en los síntomas del TDAH evaluados por los participantes, ni la impresión clínica de la gravedad o la mejoría en los participantes tratados con metilfenidato de liberación inmediata en comparación con el litio.
Episodios adversos
Los episodios adversos (efectos secundarios) se evaluaron e informaron de manera deficiente en todos los ensayos. En general, cuatro ensayos con 203 participantes que recibieron metilfenidato de liberación inmediata y 141 participantes que recibieron placebo describieron la aparición de efectos perjudiciales. En esos ensayos se informó que el uso de metilfenidato de liberación inmediata aumentó el riesgo de complicaciones digestivas y de pérdida del apetito. Se informaron efectos perjudiciales en el corazón y la circulación, pero de manera limitada e incongruente. Un ensayo en que comparó el metilfenidato de liberación inmediata con el litio informó cinco y nueve episodios adversos, respectivamente.
En casi todos los ensayos hubo preocupaciones importantes relacionadas con las fuentes de financiación y los conflictos de intereses.
¿Cuál es el grado de actualización de esta revisión?
La evidencia está actualizada hasta el 3 de enero de 2020.
Authors' conclusions
Summary of findings
| Immediate‐release methylphenidate versus placebo for attention deficit hyperactivity disorder (ADHD) in adults | |||||
| Patient or population: adults with ADHD (available evidence for participants aged between 25 to 53 years old) | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
|---|---|---|---|---|---|
| Assumed risk with placebo | Corresponding risk with immediate‐release methylphenidate | ||||
| Efficacy (changes in symptoms of ADHD): investigator‐rated | The mean efficacy score in the control group was 33.8 points | The mean efficacy score in the intervention group was 20.70 points lower (23.97 lower to 17.43 lower) | 146 (1 RCT) | ⊕⊝⊝⊝ | End scores. IR methylphenidate may reduce symptoms of ADHD when rated by investigators but the evidence is very uncertain. |
| Efficacy (changes in symptoms of ADHD): participant‐rated Follow‐up: range = 7 weeks to 12 weeks | The mean efficacy score in the intervention group was 0.59 points lower (1.25 lower to 0.06 higher) | 138 (2 RCTs) | ⊕⊝⊝⊝ Very lowa,b | End scores. IR methylphenidate may have a moderate to no effect on symptoms of ADHD when rated by participants but the evidence is very uncertain. The effect would represent a moderate difference between the control and the intervention group. As a rule of thumb, 0.2 points represents a small difference, 0.5 a moderate and 0.8 a large effect. | |
| Clinical impression: severity | ‐ | The mean clinical impression of symptom severity score in the intervention groups was 0.57 points lower (0.85 lower to 0.28 lower) | 139 (2 RCTs) | ⊕⊝⊝⊝ Very lowb,c | End scores and Change scores. IR methylphenidate may reduce clinicians' impressions of the severity of ADHD symptoms but the evidence is very uncertain. |
| Clinical impression: improvement | The mean clinical impression of improvement score in the control group was 3.54 points | The mean clinical impression of improvement score in the intervention group was0.94 points lower (1.37 lower to 0.51 lower) | 49 (1 RCT) | ⊕⊕⊝⊝ | End scores. IR methylphenidate may slightly increase clinicians' impressions of improvement in ADHD symptoms. |
| Anxiety: investigator‐rated | ‐ | The mean anxiety score in the intervention group was 0.20 points lower (4.84 lower to 4.44 higher) | 19 (1 RCT) | ⊕⊝⊝⊝ | Change scores. There is no clear evidence of an effect, but the evidence is very uncertain. |
| Depression: investigator‐rated | ‐ | The mean depression score in the intervention group was 2.80 points higher (0.09 lower to 5.69 higher) | 19 (1 RCT) | ⊕⊝⊝⊝ | Change scores. There is no clear evidence of an effect, but the evidence is very uncertain. |
| Harms: adverse events (poorly assessed and reported) | Among participants experiencing at least 1 adverse event, the use of IR methylphenidate increased the risk of gastrointestinal complications (RR 1.96, 95% CI 1.13 to 2.95) and loss of appetite (RR 1.77, 95% CI 1.06 to 2.96). Cardiovascular adverse events were reported inconsistently, preventing a comprehensive analysis. | ‐ | ‐ | IR methylphenidate may increase the risk of gastrointestinal adverse events and loss of appetite. It is unclear whether IR methylphenidate induces cardiovascular adverse events. Overall, adverse events were poorly assessed and reported in all included studies. We considered all studies to be at high risk of bias due to selective outcome reporting of harms and masking of the outcome assessor (failure to blind outcome assessor to measure harms). | |
| *The basis for the assumed risk was the median control group risk across studies. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ADHD: Attention deficit hyperactivity disorder; CI: Confidence interval; IR: immediate‐release; IV: Fourth version; MD: Mean difference; RCT: Randomised controlled trial; RR: risk ratio | |||||
| GRADE Working Group grades of evidence | |||||
| aDowngraded twice due to high and unclear risk of bias in multiple criteria (random sequence generation, allocation concealment, blinding of outcome assessors, incomplete outcome data, and selective outcome reporting). | |||||
| Immediate‐release methylphenidate versus lithium for attention deficit hyperactivity disorder (ADHD) in adults | |||||
| Patient or population: adults with ADHD (available evidence for participants aged between 25 to 53 years old) | |||||
| Outcomes | Anticipated absolute effects* (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
|---|---|---|---|---|---|
| Assumed risk with lithium | Assumed risk with immediate‐release methylphenidate | ||||
| Efficacy (changes in symptoms of ADHD): investigator‐rated | The mean efficacy score in the control group was 28.4 points | The mean efficacy score in the intervention group was 0.60 points higher (3.11 lower to 4.31 higher) | 46 (1 RCT) | ⊕⊝⊝⊝ | End scores. It is uncertain whether IR methylphenidate is more effective than lithium. |
| Efficacy (changes in symptoms of ADHD): participant‐rated ‐ not reported | ‐ | ‐ | ‐ | ‐ | Not reported |
| Clinical impression: severity | ‐ | ‐ | ‐ | ‐ | Not reported |
| Clinical impression: improvement | ‐ | ‐ | ‐ | ‐ | Not reported |
| Anxiety: investigator‐rated | The mean anxiety score in the control group was 6.2 points | The mean anxiety score in the intervention group was 0.80 points lower (4.49 lower to 2.89 higher) | 46 (1 RCT) | ⊕⊝⊝⊝ | End scores. IR methylphenidate may have little to no effect on anxiety but the evidence is very uncertain. |
| Depression: investigator‐rated | The mean depression score in the control group was 7.8 points | The mean depression score in the intervention group was 1.20 points lower (3.81 lower to 1.41 higher) | 46 (1 RCT) | ⊕⊝⊝⊝ | End scores. IR methylphenidate may have little to no effect on depression but the evidence is very uncertain. |
| Harms: adverse events (poorly assessed and reported) | 1 trial comparing IR methylphenidate to lithium reported 5 and 9 adverse events, respectively. | ‐ | ‐ | Adverse events were poorly assessed and reported in all included studies. We considered all studies to be at high risk of bias due to selective outcome reporting of harms and masking of the outcome assessor (failure to blind outcome assessor to measure harms). | |
| *The basis for the assumed risk was the median control group risk across studies. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ADHD: Attention deficit hyperactivity disorder; CI: Confidence interval; IR: immediate‐release; MD: Mean difference; RCT: Randomised controlled trial. | |||||
| GRADE Working Group grades of evidence | |||||
| aDowngraded twice due to unclear risk of bias in several domains (random sequence generation, allocation concealment, blinding of outcome assessment, incomplete and selective outcome reporting) and high risk of bias due to incomplete outcome data. | |||||
Background
Description of the condition
Attention deficit hyperactivity disorder (ADHD) is defined as a mental health disability, which usually begins before 12 years of age, and is characterized by three main symptoms: inattention, impulsivity, and hyperactivity. The intensity of the symptoms tends to decrease with ageing, but in 40% to 50% of people diagnosed with ADHD in childhood, symptoms may persist during adolescence and adulthood (NIMH 2016; Sibley 2016). Recent studies have shown that symptoms of ADHD may appear only in adulthood (Agnew‐Blais 2016; Caye 2016; Moffitt 2015), yet it is a controversial issue (Franke 2018; Moncrieff 2011). In some cases, ADHD remains undiagnosed until adulthood because it is not recognized during childhood, or it presents in a mild form (NIMH 2017). Symptoms may also be associated with the onset and persistence of secondary disorders or diseases (Cheng 2017; Fayyad 2017; NIMH 2016), which reinforces the discussion of whether this is a different clinical condition (Moncrieff 2011). The persistence of symptoms of inattention, hyperactivity and impulsivity may negatively affect the individual's social, academic or professional activities (APA 2013).
The diagnosis of ADHD is based on the presence of at least six (in children and adolescents) or five (in adults older than 17 years) of the 18 symptoms that are indicative of inattention, hyperactivity and impulsivity. This core list of symptoms was developed by the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM‐5; APA 2013), to be applied for the diagnosis of ADHD in children; it is also listed in the International Classification of Diseases 10th and 11th editions (ICD‐10 and ICD‐11; WHO 1992; WHO 2018, respectively) as Attention Deficit Hyperactivity Disorder. The symptoms should be observed in different circumstances of the individual's daily life and must represent a negative disruption to regular activities and tasks related to one or more contexts of life. In addition, the symptoms may be recognized in a variety of degrees of intensity, depending on the specific characteristics of each individual, their overall behavior, and on the predominance of one symptom or another. Considering the predominance of one symptom or another, ADHD may be classified into three presentations/subtypes: predominantly inattentive; predominantly hyperactive or impulsive; or combined (in which all symptoms are present, but there is no clear predominance among them), which can change over time. Inattentive presentation is characterized by becoming distracted or struggling to concentrate when performing tasks, combined with a lack of persistence, a lack of a sense of planning and an inability to organize tasks or things. Hyperactivity is a pattern of excessive motor activity in children and restlessness in adults. Finally, the impulsiveness presentation is manifested when the individual takes actions or has attitudes with no judgment or awareness of the possible consequences or associated risks (APA 2013). The Adult ADHD Self‐Report Scale (ASRS) was developed to support the diagnosis of ADHD in adults; it consists of a set of structured questions, based on the DSM‐5 criteria (APA 2013), and has been demonstrated to have high sensitivity and specificity in detecting ADHD symptoms in adults (Ustun 2017).
The diagnostic criteria for ADHD in the DSM‐5 was amended to include the criteria for a diagnosis of ADHD in adults (Wakefield 2016). In this revision, the age of onset of the first symptoms was increased to 12 years, which reduced the diagnostic threshold for individuals aged 17 or older, and the description of some situations used in the diagnosis was modified to make it appropriate for adults (Epstein 2013; Wakefield 2016). Furthermore, Autism Spectrum Disorder (ASD) was no longer an exclusionary diagnosis, allowing the comorbid diagnosis of ADHD and ASD (Epstein 2013). Adults with ADS experience high rates of comorbidities, the most common being mood disorders, anxiety disorder and ADHD (Hofvander 2009). Epidemiological and clinical data on this psychiatric disorder in adulthood are limited, since the diagnostic criteria in the DSM‐5's predecessors precluded a dual diagnosis of ADHD and ASD (Pehlivanidis 2020). Thus, trials that used diagnostic criteria available prior to the DSM‐5 may not be directly applicable to the clinical practice of patients with both ADHD and ASD.
The prevalence of ADHD in adults is lower than the prevalence in children and adolescents, which ranges between 3% and 7% (Polanczyk 2007; Thomas 2015). The variation in the estimates of the prevalence of ADHD in children is probably due to the diagnostic criteria used (Polanczyk 2014; Thomas 2015). Overall, prevalence estimates using the third, revised edition of the DSM (APA 1987) are 2.4% to 3% lower than prevalence estimates using the third (APA 1980) or fourth (APA 1994) editions of the DSM (Thomas 2015). Similarly, prevalence estimates using the ICD‐10 are 4.1% lower in comparison with prevalence estimates using the DSM‐IV (Thomas 2015). Nevertheless, prevalence rates of ADHD in children have remained stable in the last 30 years (Polanczyk 2014). The average prevalence of ADHD in adults is estimated to be at 2.8% and appears to be associated with the economic development of the country, with higher resource‐rich settings presenting higher prevalence estimates (average of 3.3%) (Fayyad 2017). Differences in the prevalence of prescribing and dispensing medicines are observed also between regions within the same country, which have different socioeconomic characteristics of access to healthcare services and medications (Perini 2014).
ADHD is more frequent in males than in females, with a ratio varying from 2:1 to 5:1 in children and from 1:1 to 6:1 in adults (APA 2013). However, symptoms of inattention tend to appear much later in males than in females, while the inattentive presentation is most prevalent in adults with ADHD (APA 2013; Cheng 2017). The presence of multimorbidity in individuals with ADHD is extremely common, and the manifestation of the condition in childhood often overlaps with the occurrence of other disorders ( e.g. challenging disorder and conduct disorder), imposing an additional layer of complexity to the diagnosis of the spectrum of individuals' problems (APA 2013; NICE 2018). In adulthood, ADHD commonly co‐exists with other psychiatric conditions such as anxiety, depression, nervous tic, and intellectual disability (Cheng 2017; Kessler 2006).
Description of the intervention
Psychostimulant medications, such as amphetamines, have been used in the treatment of ADHD in children and adolescents since the 1930s (Bradley 1937). Currently, methylphenidate, dexamphetamine, and atomoxetine are recommended treatments for individuals with ADHD (Kolar 2008; NICE 2018). There is some evidence suggesting that stimulants are effective in reducing ADHD symptoms, contributing to better productivity at work and a decrease in suicidal behavior (Chen 2014; Mészáros 2009; Wigal 2010). However, some authors have been unable to establish whether the benefits of immediate‐release methylphenidate (IR methylphenidate) in the treatment of ADHD in children and adolescents would be more significant than the associated harms (i.e. adverse events), in comparison with placebo or no treatment (Storebø 2015). A recent systematic review and network meta‐analysis including children, adolescents and adults with ADHD concluded that short‐term treatment with IR methylphenidate is more efficacious and more tolerable than placebo in children, and more efficacious and less well tolerated in adults (Cortese 2018).
In Europe, pharmacological treatment is considered the first‐line treatment for adults with moderate or severe ADHD, with lisdexamfetamine or methylphenidate being the first choice (NICE 2018). The second line of pharmacological treatment is atomoxetine, a non‐stimulant drug with lower potential for abuse than stimulant drugs; atomoxetine is also recommended as a first‐line treatment option in people with comorbid substance‐use disorder (DynaMed Plus 2016). A third‐line option includes bupropion, modafinil and desipramine. Cognitive behavioral therapy is an option for people who do not tolerate drug therapy or choose not to use medications, and this approach can also be used in combination with pharmacological treatment (DynaMed Plus 2016). For instance, the Canadian ADHD Resource Alliance recommends a multimodal approach, including psychosocial treatment combined with medications when appropriate (CADDRA 2020). In the USA, psychostimulant compounds, such as methylphenidate and amphetamines, are the most widely used medications for the management of ADHD symptoms in adults. With the exception of atomoxetine, non‐stimulant medications have generally been considered second‐line medications (Wolraich 2019). Behavior‐management strategies to minimize distractions and increase organization are encouraged as part of the treatment (APA 2017; Urion 2020).
Methylphenidate is available in different formulations: immediate‐release and extended‐ or sustained‐release preparations. Immediate‐release formulations are absorbed instantly after the tablet or capsule is ingested. A maximum concentration of the medication in the blood is achieved in a short period, and the onset of action is fast. Extended‐release formulations are absorbed more slowly. The concentration in the blood increases gradually, and the drug's effect is maintained for a more extended period (Perrie 2012).
Factors such as dose, type of formulation, and the presence of comorbid substance‐use disorders appear to modify the efficacy of methylphenidate in the treatment of ADHD in adults (Castells 2011). An individualized approach is extremely important in the treatment of adults, with special consideration given to conditions co‐existing with ADHD. The ideal dose of IR methylphenidate varies between individuals, and treatment should be initiated in small doses with weekly increments. This allows for an optimal dosage to control symptoms and manage adverse effects (NICE 2018).
It is recommended that initial treatment begins with doses of 5 mg, two or three times daily for immediate‐release preparations, and equivalent doses for other preparations. The dosages can be increased until the maximum doses are reached that offer the optimum dose of the medicine for each person, with maximum treatment benefits and the lowest risk of harms, i.e. the lowest risk of adverse events (NICE 2018). The recommended Defined Daily Dose (DDD) of methylphenidate by the World Health Organization is 30 mg/day for adults (WHO 2017).
How the intervention might work
Methylphenidate is a central nervous system stimulant of indirect sympathomimetic action. Although its mechanism of action has not yet been fully elucidated, it is thought to present a mode of action similar to dexamphetamine (Sweetman 2014). It facilitates dopaminergic and noradrenergic transmission by inhibiting dopamine and norepinephrine transporters, decreasing receptivity and consequently increasing the extracellular concentration of neurotransmitters (Engert 2008; Schabram 2014; Volkow 2001).
Research findings suggest that individuals with ADHD have a higher number of dopamine transporter binding sites. Methylphenidate binds to these transporters and prevents re‐uptake of dopamine. The decrease in the availability of these receivers for connection is directly related to a clinical improvement in ADHD symptoms (Dresel 2000). The increase of dopamine in the synaptic cleft as a function of methylphenidate action results in improved attention and decreased distraction, modulating the sense of motivation and interest in performing tasks that consequently improve performance (Volkow 2002). In animal models, it has been observed that the inhibition of norepinephrine re‐uptake by methylphenidate is more prominent than that seen in previous studies, and may result in persistent improvements in ADHD symptoms in those treated from adolescence to adulthood (Somkuwar 2015). This sympathomimetic activity is linked to one of the greatest current concerns about the use of methylphenidate: the risk of cardiovascular adverse effects associated with the drug. The inhibition of norepinephrine re‐uptake is the most likely cause of an increase in blood pressure and heart rate in people using methylphenidate (Heal 2006). Furthermore, at low doses, the use of stimulants may result in an increase in wakefulness, attention, ability to sustain focus and vigor. This can further explain the effects observed with the use of these substances for increasing focused attention and reducing hyperactivity (Wood 2013). Regarding the pharmacokinetic profile, the oral bioavailability of methylphenidate ranges from 11% to 53%, with the maximum concentration given by the immediate‐release formulation approximately two hours after the administration of the drug; the terminal half‐life of the drug is two hours (Chan 1983; Wargin 1983).
Why it is important to do this review
Several clinical trials have been conducted to investigate the efficacy and harms of IR methylphenidate for treating ADHD in children and adolescents. A number of systematic reviews and meta‐analyses have also been published, evaluating the effect of IR methylphenidate in this population (Charach 2011; Charach 2013; Hanwella 2011; Kambeitz 2014; Maia 2017; Punja 2013; Reichow 2013; Storebø 2015). Fewer studies have focused on the use of IR methylphenidate in adults with ADHD; as a result, many countries contraindicate its use in this age group (EMA 2009).
Currently, the available evidence for the likely efficacy and harms of using IR methylphenidate to treat adults with ADHD is controversial and incomplete, which precludes firm conclusions (Maidment 2003; Wilens 2003). For instance, in a narrative review that included six controlled clinical trials, three suggested treatment efficacy, while two studies failed to show efficacy, and the results from one study were considered conflicting (Maidment 2003). Another narrative review suggested that IR methylphenidate was more efficacious than placebo in the treatment of ADHD in adults (Fredrikesen 2013); the conclusions were based on five randomized controlled trials (RCTs), and 10 open‐label extension studies of initial short‐term RCTs. Adults with childhood‐onset of symptoms have been observed with significant improvements in their symptoms of ADHD, with therapeutic response as high as 78% when receiving IR methylphenidate compared with 4% improvement when receiving placebo (Spencer 1995). However, another study found no significant difference between IR methylphenidate and placebo (Kuperman 2001).
Systematic reviews and network meta‐analyses of the efficacy and harms of using IR methylphenidate to treat adults with ADHD have reached different conclusions. A systematic review and network meta‐analysis of placebo‐controlled trials compared shorter‐acting stimulant drugs (including IR methylphenidate, mixed amphetamine and dextroamphetamine), longer‐acting stimulant drugs and longer‐acting forms of bupropion (Peterson 2008). The study authors found a higher rate of clinical response (30% in the reduction of ADHD symptoms) among adults receiving shorter‐acting stimulant drugs in direct comparison to placebo and in indirect comparisons to longer‐acting stimulant drugs and longer‐acting forms of bupropion. People treated with shorter‐acting stimulant drugs were found to have a higher risk of appetite loss and sleep disturbances compared with people treated with placebo. Conversely, a higher risk of appetite loss was demonstrated among participants receiving longer‐acting stimulant drugs compared with people treated with shorter‐acting stimulant drugs (Peterson 2008). Additional research did not demonstrate differences in efficacy between osmotic‐controlled release oral delivery system (OROS) methylphenidate and atomoxetine (indirect comparison), although both drugs were shown to be more efficacious than placebo (Bushe 2016). Another systematic review with a network meta‐analysis (Cortese 2018) showed that IR methylphenidate is more efficacious than placebo, but not so for amphetamines, in the short term. Methylphenidate was less acceptable than placebo and increased weight loss and systolic and diastolic blood pressure (Cortese 2018). However, it is important to consider some limitations of this review that included double‐blind RCTs (parallel group, cross‐over, or cluster) published and unpublished until 2017 that assessed treatments for ADHD as oral monotherapy of at least one week's duration. First, the searches are current to April 2017, and therefore there is a value in updating the review, particularly considering that there is still controversy about the efficacy of methylphenidate to treat ADHD. Secondly, although cross‐over trials were included in the review, data from the pre‐cross‐over phase were included in the analysis. It has been shown that cross‐over trials and parallel‐group trials provided similar relevant data in the context of ADHD (Greenhill 2001; Krogh 2019; Stein 1996), and it is relevant to include this additional information in an updated systematic review. Third, the authors of that review assessed the overall evidence contributing to the indirect comparisons as low and very low certainty of evidence. Indirect or mixed comparisons may have biases similar to those in observational studies and may therefore be downgraded to a lower evidence level similar to those studies (Cipriani 2013). Finally, some of the authors of that review declared receiving funding from pharmaceutical industries. The way in which a review is conducted is an important issue to consider when assessing its results. A systematic review may be carried out with methodological rigor and yet its results may be biased if they are influenced by conflicts of interest, particularly those pertaining to research or individual sponsorship (Barnes 1998; Bes‐Rastrollo 2013; Dunn 2014).
The reasons for the variability in the available evidence are not clearly documented in the literature, but they appear to be related to factors such as dose, type of formulation and treatment regimen (Castells 2011), as well as the comparison methods used (Cortese 2018). The inconsistency of this evidence and the absence of systematic reviews of methodological rigor might have a negative impact on clinical decision‐making (Maidment 2003; Wilens 2003). Our systematic review therefore evaluates the efficacy and harms of IR methylphenidate as reported in RCTs. The contribution of this systematic review is to examine the benefit and harm profile of IR methylphenidate for the treatment of ADHD in adults, in accordance with a rigorous methodological approach (Higgins 2020), and the PRISMA guidelines (Liberati 2009; Moher 2015). A Cochrane Review evaluating extended‐release formulations of methylphenidate for adults with ADHD is also in progress (Boesen 2017).
Objectives
To evaluate the efficacy and harms (adverse events) of IR methylphenidate for treating ADHD in adults.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials (RCTs) of parallel and cross‐over designs.
Types of participants
Adults aged 18 years or older with a diagnosis of ADHD according to the Diagnostic and Statistical Manual of Mental Disorders (DSM) Third Edition (DSM‐III; APA 1980), Third Edition Revised (DSM‐III‐R; APA 1987), Fourth Edition (DSM‐IV; APA 1994) or Fifth Edition (DSM‐5; APA 2013); or with a diagnosis of hyperkinetic disorders according to the International Statistical Classification of Diseases and Related Health Problems Ninth Revision (ICD‐9), and Tenth Revision (ICD‐10) (WHO 1992).
Types of interventions
IR methylphenidate administered at any dosage as part of any treatment regimen, compared with placebo or other pharmacological interventions (including methylphenidate extended‐release formulations).
An extended‐release formulation refers to the different extended‐release drug‐delivery systems, including OROS, which is a specific osmotic type of extended‐release system. Extended‐release formulations are a type of pharmacological intervention and were therefore considered eligible for inclusion when compared with IR methylphenidate.
Types of outcome measures
We addressed the following outcomes in this review.
Primary outcomes
-
Efficacy: changes in symptoms of ADHD (hyperactivity, impulsivity, and inattentiveness), based on clinical assessment by a physician or by self‐report, and measured by any validated clinical scale reported in the trials (e.g. Adult ADHD Self‐Report Screening Scale (Ustun 2017)).
-
Harms: all adverse events, classified as serious or non‐serious, including but not restricted to: cardiovascular, neurological, gastrointestinal, metabolic events, and psychiatric disorders. Serious events were defined as any adverse effect that resulted in death or was life‐threatening, required hospital admission or prolonged hospitalization, caused persistent or significant disability or incapacity, or required intervention to prevent permanent damage to a body structure or impairment of a body function (ICH 2016). See Differences between protocol and review.
Secondary outcomes
-
Changes in the clinical impression of severity or improvement, level of functioning, depression and anxiety, based on a clinical assessment by a physician (e.g. Clinical Global Impressions Scale; Guy 1976) or participant self‐report.
-
Quality of life, measured by validated psychometric instruments (e.g. the World Health Organization Quality of Life: Brief version (WHOQOL‐BREF; Skevington 2004), or the 12‐Item Short‐Form Health Survey (Ware 1996).
We considered outcomes according to the follow‐up durations reported in the included studies.
Search methods for identification of studies
Electronic searches
We searched the following electronic databases up to January 2020.
-
Cochrane Central Register of Controlled Trials (CENTRAL; 2020, Issue 1) in the Cochrane Library, which includes the Cochrane Developmental, Psychosocial and Learning Problems Specialised Register (searched 6 January 2020).
-
MEDLINE Ovid (1946 to 3 January 2020).
-
MEDLINE In‐Process & Other Non‐Indexed Citations Ovid (searched 3 January 2020).
-
MEDLINE Epub Ahead of Print Ovid (searched 3 January 2020).
-
Embase Ovid (1980 to 10 January 2020).
-
PsycINFO Ovid (1806 to 13 January 2020).
-
Cumulative Index to Nursing and Allied Health Literature (CINAHL EBSCOhost); 1980 to 6 January 2020).
-
Science Citation Index Web of Science, Clarivate (SCI; 1970 to 7 January 2020).
-
Social Sciences Citation Index Web of Science (SSCI; 1970 to 7 January 2020).
-
Conference Proceedings Citation Index ‐ Science Web of Science Web of Science, Clarivate (CPCI‐S; 1990 to 7 January 2020).
-
Conference Proceedings Citation Index ‐ Social Science & Humanities Web of Science, Clarivate (CPCI‐SS&H; 1990 to 7 January 2020).
-
Cochrane Database of Systematic Reviews (CDSR; 2020, Issue 1) part of the Cochrane Library (searched 6 January 2020).
-
Database of Abstracts of Reviews of Effects (DARE; Final Issue: 2015, Issue 2) part of the Cochrane Library (searched 6 January 2020).
-
ClinicalTrials.gov (clinicaltrials.gov; searched 13 January 2020).
-
World Health Organization International Clinical Trials Registry Platform (WHO ICTRP; apps.who.int/trialsearch; searched 13 January 2020).
-
Drug Industry Documents (www.industrydocumentslibrary.ucsf.edu/drug; searched 13 January 2020).
SG ran the searches, adapting the MEDLINE search strategy published in the protocol (Cândido 2018) for the remaining databases; see Appendix 1 for detailed search strategies.
Searching other resources
We searched for internal reports on the websites of the European Medicines Agency (EMA; www.ema.europa.eu/ema), and the US Food and Drug Administration (FDA; www.fda.gov). We checked citations of included RCTs to identify additional trials not captured by the electronic searches.
Data collection and analysis
We were not able to use all of the planned methods in the review protocol (Cândido 2018). In the following sections, we report only methods applied. See Differences between protocol and review and Table 1 for unused methods.
| Method section | Description | Reason for non‐use |
|---|---|---|
| Types of outcomes | We planned to measure the primary efficacy outcome over the short term (within six months) and long term (longer than 6 months) | These data were not available as no study lasted more than 4.5 months |
| We planned to measure serious adverse events as the primary outcome of harms. | None of the included studies assessed serious adverse events according to the definition of the International Council for Harmonisation (ICH 2016) | |
| Selection of studies | We planned to contact the authors of studies whenever there was insufficient information available to decide whether a study was eligible for inclusion. | This action was not necessary for the ongoing studies |
| Assessment of risk of bias | We planned to revise the 'Risk of bias' domains for randomized controlled trials if new guidelines were released during the development of this review. | When we came to the completion of this review, Cochrane had released a new 'risk of bias' tool but had not yet made it mandatory for all Cochrane Reviews; it is being piloted with only a few reviews within Cochrane. Future updates of this review will take any related updates into account |
| Measures of treatment effect > Continuous outcomes | For studies reporting outcome values other than the mean and standard deviation, we planned to apply standard errors, CI, t values and P values to estimate the results, whenever possible | None of these additional data were reported and thus we were not able to estimate any missing results. We contacted study authors to request the additional information but have not received response |
| If skewed data were detected, we planned to consult a statistician on the best data transformation approach | We did not encounter this situation; therefore, it was not necessary to consult a statistician | |
| Unit of analysis issues > Cross‐over trials | We planned to estimate within‐participant differences between the intervention groups at the end of the study follow‐up period, using the MD and standard deviation to conduct a paired analysis and avoid a unit‐of‐analysis error (Elbourne 2002; Higgins 2020) | No trial reported participant‐level differences between intervention groups |
| Assessment of heterogeneity | We planned to investigate clinical heterogeneity through subgroup analyses | There were insufficient data evaluated and reported in the included studies to allow us to undertake subgroup analyses |
| Assessment of reporting bias | We planned to use funnel plots to investigate the presence of publication bias (the selective publication of trials with positive findings), and other small‐study effects, among the studies included in the review (Page 2020). We planed to use Egger's test to assess for funnel plot asymmetry (Egger 1997), providing 10 or more trials were included in a meta‐analysis. We planned to consult a statistician in situations where we were unable to interpret the asymmetries objectively and when we might have considered alternative statistical tests (Page 2020) | We could not assess reporting bias due to there being an insufficient number of trials (fewer than 10) in the quantitative analyses |
| Data synthesis | Where considerable heterogeneity (I2 statistic greater than 75%) was detected, particularly in the presence of high inconsistency in the direction of effect, we planned not to calculate the average effect of the intervention through a meta‐analysis | Only 1 study contributed data for most of the outcomes and comparisons; therefore, the insufficient data prevented an appropriate assessment of heterogeneity |
| Subgroup analysis and investigation of heterogeneity | We planned to explore potential sources of heterogeneity if the available data from the studies allowed us to stratify participant subgroups by the following characteristics.
We planned to calculate a pooled effect size for each subgroup | There were insufficient data evaluated and reported in the included studies to allow us to undertake subgroup analyses |
| Sensitivity analysis | If there were an adequate number of studies (2 or more), we planned to perform a sensitivity analysis to explore the causes of heterogeneity and test the robustness of the results to decisions made during the development of the review. Specifically, we planned to reanalyze the data:
| There were insufficient studies included in the meta‐analyses to perform any of our preplanned sensitivity analyses |
| Summary of findings and assessment of the certainty of the evidence | We planned to describe the outcomes of interest over short (within 6 months) and long (longer than 6 months) periods of treatment | None of the trials investigated the effects of treatment with IR methylphenidate in adults for a period longer than 18 weeks (4.5 months) |
CI: Confidence interval
IR: immediate release
MD: Mean difference
Selection of studies
We used the reference manager software EndNote (EndNote 2017), to merge records returned from the searches and remove any duplicates. Working in pairs, three review authors independently screened titles and abstracts to remove clearly irrelevant records. Next, we screened the full texts of potentially relevant reports for eligibility, in accordance with the aforementioned inclusion criteria (Criteria for considering studies for this review); note that outcomes measurement and reporting were not used as eligibility criteria. At this stage, we linked together multiple reports of the same study. We resolved disagreements in the selection process by consensus or by consulting a third review author. We recorded the selection process in a PRISMA diagram (Moher 2009).
Data extraction and management
Two review authors (DJ, RC) independently extracted data from each included trial using a standardized data extraction form. We resolved disagreements in the data extraction process by discussion or by consulting a third review author (CP). Our data extraction form was piloted and tailored to record data on the:
-
Characteristics of the studies;
-
Characteristics of the participants;
-
Characteristics of the treatment and comparator interventions;
-
Methods used to measure the outcomes and follow‐up duration;
-
Outcomes measurements (any measures related to primary or secondary outcomes, as described under Types of outcome measures); and
-
Disclosure of financial conflict of interests.
We obtained additional information from the authors of the one included trial that had efficacy data reported only in a graphic illustration.
Assessment of risk of bias in included studies
We assessed the risks of bias of the included studies across the following six domains, as described in Cochrane’s 'Rsk of bias' tool (Higgins 2011):
-
Sequence generation (selection bias);
-
Allocation sequence concealment (selection bias);
-
Blinding of participants and personnel (performance bias);
-
Blinding of outcome assessment (detection bias);
-
Incomplete outcome data (attrition bias); and
-
Selective outcome reporting (reporting bias).
Two review authors (DJ, RC) independently assessed the risks of bias, resolving any disagreements by discussion or by consulting a third review author (CP).
We assessed the risk of bias resulting from some domains for different groups or outcomes separately, in accordance with the instructions outlined by Cochrane (Higgins 2011). Specifically, we assessed:
-
Blinding of participants and personnel separately for a) participants and b) personnel;
-
Blinding of outcome assessment separately for a) beneficial outcomes and b) harmful outcomes; and
-
Selective outcome data separately for a) primary beneficial outcomes and b) harmful outcomes.
We rated the risk of bias in each domain as high, low or unclear, and accompanied each rating by a statement to support our judgments.
Conflicts of interest
Some author teams have included information on financial conflict of interest as a 'risk of bias' domain (Jorgensen 2016). However, this element does not reflect an independent methodological domain and its inclusion is considered inappropriate according to Cochrane standards (Higgins 2011). Specific 'Risk of bias' domains recognized as being influenced by financial conflicts of interest are already included in the tool; for example, incomplete outcome data and selective outcome reporting. We also considered the 'selective outcome reporting' domain when evaluating the certainty of the evidence (see 'Summary of findings' table under Data synthesis).
Complying with Cochrane guidelines, we extracted data about the trials' sources of funding and conflicts of interest and judged whether there were reasons for concern about their impact on the results analyzed from the included trials (Boutron 2020). More specifically, we considered there to be 'no concerns' when study authors did not receive funding or declared receiving funding from research grants, 'notable concerns' when study authors declared receiving grants from companies with a vested interest, or 'unclear concerns' when there was insufficient information to support a judgment of 'no concerns' or 'notable concerns'.
Measures of treatment effect
Continuous outcomes
To summarize results measured as continuous variables and reported using the same rating scales, we calculated the mean difference (MD) and presented it with a 95% confidence interval (CI). When outcomes were measured and reported on different rating scales, we used standard deviations to standardize the MD and calculated a standardized mean difference (SMD). We selected change scores rather than endpoint scores when both results were available in the same trial. If change scores were not reported, we extracted data on endpoint scores.
Significant heterogeneity in outcomes measurements and incomplete reporting of the results prevented the conduction of meta‐analyses for most of the outcomes assessed in this review. See Data synthesis. When combining outcome data in the meta‐analysis, we conducted analyses of the MD of change scores and endpoint scores when information was available for both measures (Deeks 2020). When combining outcome data in the meta‐analysis using the SMD, we analyzed change scores and endpoint scores reported on different scales separately (Deeks 2020).
To calculate the above described estimators from cross‐over trials, we needed to extract data on a paired analysis of within‐participant differences (Elbourne 2002). This analysis was not reported in any of the cross‐over trials included in this review. We therefore adopted an approach to treat cross‐over trials as parallel trials, since were able to summarize data comparing all measures of the intervention groups from all treatment periods (Higgins 2020).
Dichotomous outcomes
To summarize results measured as dichotomous outcomes, we calculated the risk ratio (RR) with a 95% CI. To calculate the RR, we needed to extract data on absolute numbers related to the sample size and the frequency of each specific outcome.
For harms outcomes, in addition to the above we categorized reported adverse events according to organ system and calculated the absolute risks of each individual event. As one person can experience more than one adverse event, the sample size of the trials could not be pooled in an analysis. We therefore calculated the total of adverse events reported in each treatment group and calculated the RR of experiencing an adverse event among participants who had experienced at least one event.
Incomplete and narrative reports of outcomes
We included and described in the review trials reporting results of effect measures and measures of uncertainty, and trials providing a narrative description of the results; however, trials providing only narrative descriptions are not included in quantitative syntheses.
Unit of analysis issues
RCTs with parallel design
We recorded loss to follow‐up data for risk of bias purposes and analyzed beneficial data according to an intention‐to‐treat (ITT) analysis whenever the data were available. This means that the unit of analysis in this review is the participant, and their outcomes were considered in the intervention group to which they were randomized, regardless of whether they received the intervention or not.
RCTs with cross‐over design
We did not anticipate any major concern about a carry‐over effect in relation to the treatment of ADHD, considering that it is mostly a stable condition and the treatment effects of IR methylphenidate and other pharmacological interventions are expected to be reversible and short‐lived. Our assumptions have been confirmed in studies assessing the possible occurrence of carry‐over effects with IR methylphenidate (Greenhill 2001; Krogh 2019; Stein 1996). Nevertheless, a unit‐of‐analysis error can occur in cross‐over data, if the analysis overlooks issues with correlation among the participants' measurements during the different treatment periods. Paired analyses took into consideration issues with correlation, but were not available in the included studies. We therefore treated the treatment periods of the cross‐over trials as treatment groups in a parallel design, to include the results in the analysis (Higgins 2020). This approach could lead to a unit‐of‐analysis error, although it is considered a conservative approach, as each cross‐over trial receives less weight in the analysis (Higgins 2020).
Harms
For data on harms, we accepted a modified ITT analysis, where the participants and their outcome data would be included in the analysis for those who received at least one dose of the tested interventions. Additionally, as one participant can experience more than one adverse event during a treatment period, we recorded data on all participants experiencing each reported event.
Dealing with missing data
Whenever possible, we based the analysis on ITT data from the individual clinical trials, accounting for dropout data. We attempted to access trial registries, when available, and contacted the authors of the most recent trials to obtain complete information about missing outcome data not fully covered in the reports of the included trials. We took missing data into consideration in the 'Risk of bias' analysis.
Assessment of heterogeneity
We avoided excessive methodological heterogeneity by combining data, whenever appropriate, only among trials with similar designs (i.e. RCTs of parallel design were not pooled with RCTs of cross‐over design). Where we deemed it possible and appropriate to combine trials of different designs, we conducted a sensitivity analysis (Sensitivity analysis), to assess the robustness of the results. We assessed statistical heterogeneity between trials using the I2 statistic for quantification of variability and reported Tau2, Chi2 and P values (Deeks 2020).
Due to insufficient trials, we were not able to investigate clinical heterogeneity through subgroup analyses (see Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
We could not assess reporting bias due to the insufficient number of trials included in the quantitative analyses. Ten or more trials need to be included in the meta‐analysis to allow us to use Egger’s test to assess for funnel plot asymmetry (Egger 1997); a funnel plot with fewer studies would not have the power to distinguish chance from real asymmetry.
Data synthesis
Efficacy
For most of the outcome data and comparisons, only one trial contributed data and a meta‐analysis was not possible. Whenever possible, we combined the efficacy outcomes in a meta‐analysis using the generic inverse variance technique. The inverse variance method is a “common and simple version of the meta‐analysis” and is the method implemented in the software where Cochrane Reviews are developed (Deeks 2020). We performed meta‐analysis using a random‐effects model to account for the heterogeneity between trials (Deeks 2020). We reported the heterogeneity using the I2 statistic; however, the insufficient number of trials and available data contributing to the quantitative analysis prevented an appropriate assessment of heterogeneity.
Effect size multiplicity
Problems with multiplicity of effect size can happen whenever an included trial reports on more than two arms or on more than one scale to measure the same outcome (López‐López 2018). To avoid introducing statistical dependency into the results estimated in the meta‐analysis, we selected the comparison of main interest to the review research question and combined the treatment effects of this comparison only. Consequently, we considered the effect sizes of trials with more than one arm (i.e. comparing IR methylphenidate with placebo and other interventions), whenever appropriate, in a meta‐analysis comparing IR methylphenidate with placebo.
The rationale for the application of the above‐described method considers that the following interventions identified in this review are not standard options in the treatment of adults with ADHD: bupropion, lithium and Pycnogenol®.
Harms
We recorded adverse events reported by participants receiving IR methylphenidate, placebo and other interventions. We then classified the reported adverse events according to organ systems. We calculated the RR with its 95% CI of the number of events according to organ systems and the intervention groups, whenever appropriate. Finally, we plotted the RRs with the 95% CI of the events according to organ systems and intervention groups in a forest plot.
Subgroup analysis and investigation of heterogeneity
There were insufficient data evaluated and reported in the included trials to allow us to undertake any subgroup analyses.
Sensitivity analysis
There were not enough trials (two or more) included in the meta‐analyses to perform most of the sensitivity analysis preplanned in the review protocol (Cândido 2018). We were able to evaluate the impact of the meta‐analysis model (fixed‐effect model or random‐effects model) and the different RCT designs (parallel versus cross‐over) in one analysis of the efficacy of IR methylphenidate as rated by the participants compared with placebo.
Summary of findings and assessment of the certainty of the evidence
Two review authors (DJ, RC) independently assessed the results of the review for the certainty of the evidence, the magnitude of the effect of the interventions examined, and the sum of available data on the main outcomes using the GRADE approach (Schünemann 2020a). We resolved disagreements by discussion or by consulting a third review author (CP). The GRADE approach consists of five judgment considerations on the certainty of a body of evidence: risk of bias, inconsistency, indirectness, imprecision and publication bias. We presented the reconciled main findings of the certainty of the evidence analyzed in this review in a 'Summary of findings' table, according to four levels of the certainty of the evidence: high, moderate, low and very low (Schünemann 2020a).
We presented the certainty ratings, along with the magnitude of the effect of the interventions examined, and the sum of available data on the outcomes listed below in a 'Summary of findings' table for the following comparisons: IR methylphenidate versus placebo and IR methylphenidate versus lithium.
-
Efficacy: changes in symptoms of ADHD assessed by the investigator
-
Efficacy: changes in symptoms of ADHD assessed by the participant
-
Harms (adverse events)
-
Clinical impression‐severity
-
Clinical impression‐improvement
-
Anxiety
-
Depression
The outcomes were measured in the included trials at different follow‐up time points, and we therefore describe the means or ranges of the follow‐up period in the 'Summary of findings' tables. We selected change scores rather than endpoint scores when data were available for the same outcome and could not be combined in a meta‐analysis. Results calculated using SMDs were interpreted according to the rule of thumb described by Cohen 1988, which suggests that a SMD of 0.2 represents a “small” difference, an SMD of 0.5 represents a “medium” difference, and an SMD of 0.8 represents a “large” difference (Schünemann 2020b; Takeshima 2014).
Results
Description of studies
Results of the search
Our searches identified a total of 20,796 records (Appendix 2). We screened 9808 records after duplicates were removed. After screening titles and abstracts, 72 full‐text reports were considered to be potentially relevant. Assessment of the full‐text reports led to the inclusion of 10 RCTs (from 10 reports) that met our inclusion criteria (Criteria for considering studies for this review). One study is awaiting classification (Studies awaiting classification), and one study is ongoing (Ongoing studies). See Figure 1.
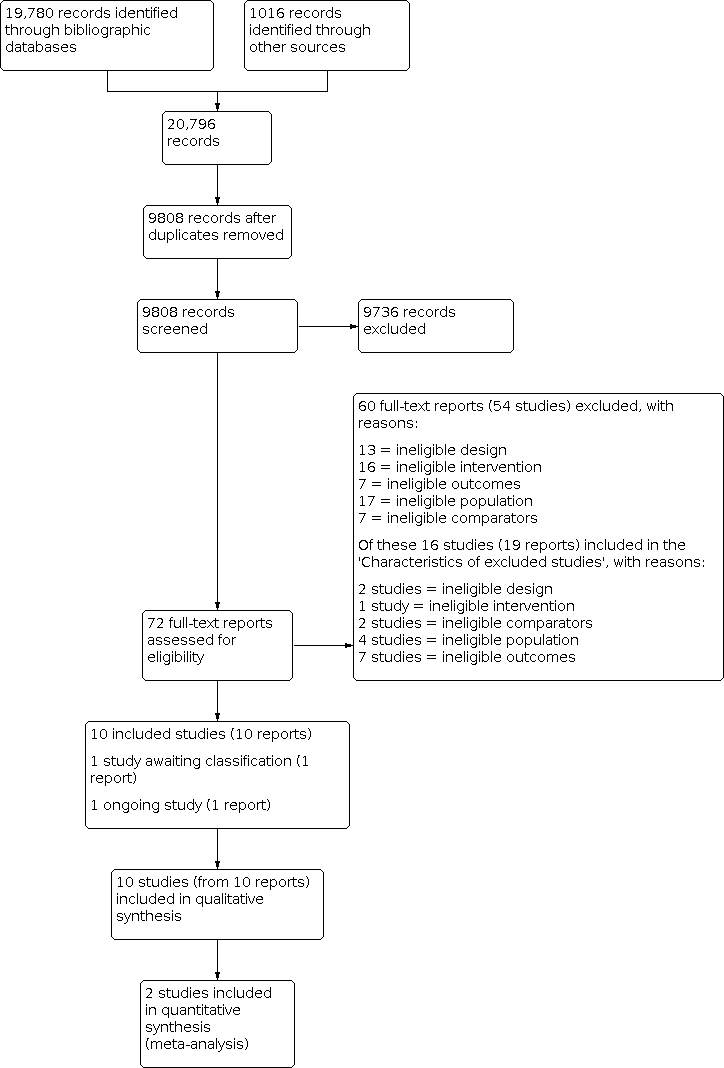
Flow diagram illustrating the results of the study selection process.
Included studies
We included 10 RCTs in this review. Below, we summarize the key characteristics of the included trials. Further details of the trials' methods, participants, interventions, and outcomes are shown in the Characteristics of included studies tables.
Study design
The included RCTs comprised five trials with a parallel design (Kuperman 2001; Schrantee 2016; Schubiner 2002; Spencer 2005; Spencer 2011) and five trials with a cross‐over design (Bouffard 2003; Carpentier 2005; Dorrego 2002; Kooij 2004; Tenenbaum 2002), published between 2001 and 2016.
Location and setting
Three trials reported having been conducted in Europe (Carpentier 2005; Kooij 2004; Schrantee 2016) and one in Argentina (Dorrego 2002); the remaining studies did not report the location where they were conducted. Most trials recruited outpatient participants but one was conducted in an addiction treatment facility and included participants receiving concomitant inpatient treatment for various substance‐use disorders (Carpentier 2005). An additional trial assessed participants with ADHD and substance‐use disorder if they were willing to enter an intensive outpatient program to treat their cocaine dependence (Schubiner 2002).
Samples size
The included trials randomized a total of 497 participants. Among the trials with a parallel design, 173 people were randomized to receive IR methylphenidate, 142 to receive placebo, and 11 to receive sustained‐release bupropion (SR bupropion) formulations. Among the trials with a cross‐over design, 147 participants were randomized to sequential treatment with IR methylphenidate and placebo, and 24 people to sequential treatment with IR methylphenidate, Pycnogenol® and placebo. The number of participants included in each of the studies is described in the Characteristics of included studies tables.
Follow‐up and attrition
The duration of the treatment and follow‐up periods varied significantly among the trials. Among those with a parallel design, treatment duration and follow‐up were six weeks (Spencer 2005; Spencer 2011), eight weeks (Kuperman 2001), 12 weeks (Schubiner 2002), and 16 weeks (Schrantee 2016). Among the trials with a cross‐over design, treatment duration and follow‐up were five weeks (Bouffard 2003), seven weeks with one week washout between treatments (Kooij 2004), eight weeks (Carpentier 2005), 17 weeks with one week washout between treatments (Tenenbaum 2002), and 18 weeks with two weeks washout between treatments (Dorrego 2002).
Attrition rates varied and were taken into account in the 'Risk of bias' assessment (Risk of bias in included studies). Table 2 details the follow‐up time points of each of the included RCTs.
Interventions and comparators
Six two‐arm trials compared IR methylphenidate with placebo (Bouffard 2003; Carpentier 2005; Kooij 2004; Schrantee 2016; Schubiner 2002; Spencer 2005). One trial tested the continuous efficacy of switching from IR methylphenidate to osmotic release oral system (OROS) methylphenidate in adults who responded to treatment with IR methylphenidate (Spencer 2011). Treatment with IR methylphenidate was compared with lithium in one trial (Dorrego 2002).
One three‐arm trial compared IR methylphenidate with SR bupropion and placebo (Kuperman 2001); another compared IR methylphenidate with Pycnogenol® and placebo (Tenenbaum 2002). Pycnogenol® is the US registered trademark name for a commercially‐available maritime pine bark extract (MedlinePlus 2019).
We did not identify eligible RCTs that compared IR methylphenidate with extended‐release formulations of methylphenidate other than OROS.
In most trials, IR methylphenidate was administered following a dose‐titration scheme, starting at small doses (5 to 10 mg) given two or three times daily, and increasing to a maximum of 40 to 60 mg daily.
Table 2 describes the comparators investigated in each of the included RCTs.
| Study | Comparator | Time points | Investigator measurements | Self‐report measurements | Measurements with complete data |
|---|---|---|---|---|---|
| RCTs with parallel design | |||||
|
| 8 weeks following a single‐blind, 7‐day placebo lead‐in | None | Adult ADHD Symptom Checklist Severity Scale (ADHDRS); updated and validated for DSM‐IV ADHD criteria | Change scores and end scores | |
| Placebo | 16 weeks | None | Adult ADHD Symptom Checklist Severity Scale (ADHDRS); updated and validated for the DSM‐IV ADHD criteria | Data not reported | |
| Placebo | 12 weeks of treatment (13 weeks in total, including 1 week of baseline testing and 12 weeks of treatment) | Barkley's ADHD Rating Scale | Barkley's ADHD Rating Scale | End scores for self‐reported measurements; investigator‐rated scores reported narratively | |
| Placebo | 6 weeks | Adult ADHD Investigator Symptom Report Scale (AISRS) | None | End scores | |
| OROS methylphenidate | 6 weeks | Adult ADHD Investigator System Symptom Report Scale (AISRS) | None | Data reported narratively | |
| RCTs with cross‐over design | |||||
| Placebo | 5 weeks | None |
| None. Only incomplete data (end means with P value and range) | |
| Placebo | 8 weeks (4 treatment phases of 2 weeks each) |
| None | None. Only incomplete data (t‐test values) | |
| Lithium | 18 weeks with a 2‐week washout period in between treatment periods | Hyperactivity, Impulsivity, Learning Problems, Conduct Disorder, Restlessness, and Antisocial Behavior subscales of the Conners’ Adult ADHD Rating Scale | None | End scores | |
| Placebo | 7 weeks, with 1 week of washout in between treatments periods | None | ADHD Rating Scale ‐ IV | End scores of the first period of treatment | |
|
| 17 weeks (2 weeks in each treatment separated by 1 washout week) |
|
| None. Only incomplete data (initial scores) | |
ADHD: Attention deficit hyperactivity disorder
OROS: osmotic release oral system
SR: Sustained‐release
aMeasurements reported by the individual’s significant other, not the trial investigator.
Characteristics of the participants
All included trials used DSM‐IV criteria, alone or in combination with other scales, to diagnose ADHD in the adults recruited into the trials.
The mean age of the participants ranged from 25 to 40 years old in nine studies; one trial reported only the age range of the participants, which varied from 17 to 51 years old (Bouffard 2003). Most trials included individuals of both sexes (range = 8% to 75%). One trial included only male participants (Schrantee 2016).
Two trials reported the subtypes of ADHD that characterized the participants recruited: predominantly inattentive, predominantly hyperactive‐impulsive, or combined (Carpentier 2005; Schrantee 2016). In Schrantee 2016, participants presenting with the combined subtype comprised 54% of those receiving IR methylphenidate and 79% of those receiving placebo. Participants presenting with the predominantly inattentive subtype were 46% and 21% in the IR methylphenidate and placebo groups, respectively. In Carpentier 2005, 76% of participants presented with the combined ADHD subtype, 20% with the predominantly inattentive subtype, and 4% with the hyperactive‐impulsive subtype.
Outcomes and outcome measurements
Primary outcomes
Efficacy: changes in the symptoms of ADHD
Changes in the symptoms of ADHD (the primary outcome of efficacy) were assessed using different scales; commonly, the trials applied multiple symptom‐rating scales to measure the effects of the treatments under investigation. Nevertheless, complete and extractable data were available from only a few of the trials (Dorrego 2002; Kuperman 2001; Spencer 2005). Data from one trial, Schrantee 2016, were provided following email correspondence with the contact author (Junqueira 2019 [pers comm]).
The scales used to assess the primary outcome of efficacy (changes in the symptoms of ADHD) measure the frequency or severity of ADHD symptoms, with higher scores generally indicating an increase in symptoms occurrence or illness severity.
-
The Adult ADHD Symptom Checklist Severity Scale (ADHDRS); updated and validated for the DSM‐IV ADHD criteria (scores range from 0 to 54): used in two trials (Kuperman 2001; Schrantee 2016);
-
Barkley's ADHD Rating Scale (scores range from one to seven): used in one trial (Schubiner 2002);
-
The Adult ADHD Investigator Symptom Report Scale (AISRS; scores range from 0 to 54): used in two trials (Spencer 2005; Spencer 2011);
-
Conners’ Adult ADHD Rating Scale (scores range from 0 to 198): used in two trials (Bouffard 2003; Dorrego 2002);
-
Barkley's ADHD Problem Behaviours Scale (scores range from 0 to 42): used in three trials (Bouffard 2003; Carpentier 2005; Tenenbaum 2002);
-
The ADHD Rating Scale‐IV (scores range from zero to three): used in two trials (Carpentier 2005; Kooij 2004);
-
Attention Deficit Scale for Adults (ADSA; scores range from 0 to 54): used in one trial (Tenenbaum 2002);
-
Barratt Impulsiveness Scale (30 items scored on a four‐point Likert scale, ranging from one (rarely/never) to four (almost always/always)): used in one trial (Tenenbaum 2002);
-
Copeland Symptom Checklist for Adult Attention Deficit Disorders (eight categories scored on a three‐point Likert scale, ranging from zero (not at all) to three (very much); percentages are computed for each category): used in one trial (Tenenbaum 2002).
Table 2 describes the measures used in the included RCTs to assess the primary outcome of efficacy.
Harms
With the exception of Tenenbaum 2002, nine included studies reported some data on harms; however, among these trials, only four described the measurement methods planned or implemented to detect adverse events occurring among participants receiving treatment (Kooij 2004; Schubiner 2002; Spencer 2005; Spencer 2011). Four trials reported information on the specific time points when adverse events were assessed (Kuperman 2001; Schubiner 2002; Spencer 2005; Spencer 2011). Data on adverse events were reported as a general statement in one trial (Carpentier 2005), while the remaining RCTs provided data on a subset of the total events (Table 3).
| Study | Reporting of harms outcomes |
|---|---|
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
|
ADHD: attention deficit hyperactivity disorder
IR: immediate release
RCTs: randomized controlled trials
Table 4 provides a comprehensive description of the harms assessed and reported in the included RCTs.
| Study | Comparator | Harms assessed? | Measurement method | Time points | What results were reported? |
|---|---|---|---|---|---|
| RCTs with parallel design | |||||
|
| Yes | Not reported | At each return visit |
| |
| Placebo | Yes | Not reported | Not reported | No data reported, only a general statement included in the paper | |
| Placebo | Yes |
| Weekly |
| |
| Placebo | Yes |
| At each visit |
| |
| OROS methylphenidate | Yes |
|
|
| |
| RCTs with cross‐over design | |||||
| Placebo | Yes | Not reported | Not reported |
| |
| Placebo | Yes | Not reported | Not reported | No data reported, only a general statement included in the paper | |
| Lithium | Yes | Not reported | Not reported |
| |
| Placebo | Yes | Modified Side Effects Rating Scale from Barkley 1998 | Not reported |
| |
|
| The trial did not access outcomes of harms | N/A | N/A | N/A | |
N/A: not applicable
OROS: osmotic release oral system
SR: sustained release
Secondary outcomes
The included trials used multiple scales to assess the secondary efficacy outcomes, as described below. The scales measure the frequency or severity of the symptoms, and generally higher scores indicate an increase in symptom occurrence or illness severity, unless otherwise specified.
Changes in the clinical impression measures of severity or improvement
-
Clinical Global Impression (CGI) ‐ Severity (‐S) or ‐ Improvement (‐I) scale (scores range from one (very much improved) to seven (very much worse)): used in six trials (Carpentier 2005; Kooij 2004; Kuperman 2001; Schrantee 2016; Spencer 2005; Spencer 2011).
Changes in the level of functioning
-
Global Assessment of Functioning (GAF; scores range from 0 to 100): used in four trials (Kooij 2004; Schrantee 2016; Spencer 2005; Spencer 2011).
Changes in depression
-
Hamilton Depression Scale (HAM‐D; scores range from 0 to 52): used in six trials (Dorrego 2002; Kooij 2004; Kuperman 2001; Schrantee 2016; Spencer 2005; Spencer 2011).
-
Beck Depression Inventory (BDI; scores range from 0 to 63): used in four trials (Bouffard 2003; Kuperman 2001; Schrantee 2016; Tenenbaum 2002).
Changes in anxiety
-
Hamilton Anxiety Scale (HAM‐A; scores range from 0 to 56): used in six trials (Bouffard 2003; Dorrego 2002; Kooij 2004; Kuperman 2001; Schrantee 2016; Spencer 2011).
-
Beck Anxiety Inventory (BAI; scores range from 0 to 63): used in one trial (Tenenbaum 2002).
Quality of life
None of the included studies assessed quality of life.
Table 5 describes the measures used in each of the included RCTs to assess the secondary outcomes.
| Study | Comparator | Secondary outcomes | Measurement methods | What results were reported? | |
|---|---|---|---|---|---|
| RCTs with parallel design | |||||
|
|
|
|
| ||
| Placebo |
|
| Change scores, end scores and baseline scores: CGI‐I and CGI‐Sa | ||
| Placebo | No information | None | None | ||
| Placebo |
|
| Narrative report with general statements: GAF | ||
| OROS methylphenidate |
|
| No data reported | ||
| RCTs with cross‐over design | |||||
| Placebo |
|
| Data reported narratively with general statements for both outcomes | ||
| Placebo |
|
| Data reported incompletely for all outcomes | ||
| Lithium |
|
| Data on end scores for both outcomes | ||
| Placebo |
|
| Data on end scores for CGI‐S | ||
|
|
|
| Data on initial scores for both outcomes | ||
ADHD: attention deficit hyperactivity disorder
OROS: osmotic release oral system
SR: Sustained‐release
aFull dataset shared by the study authors.
Clinical trials registration
With the exception of one trial (Schrantee 2016), none of the included trials appeared to be registered in a clinical trial registry. The trial reports did not mention registration, and a dedicated search could not locate them in any trial registry.
Conflicts of interest
Funding sources and potential conflict of interests were reported in all but two included trials (Carpentier 2005; Dorrego 2002). Amongst the eight trials reporting sources of funding: two reported support from research grants and several companies with a vested interest, and also disclosed that the trials' authors received consulting fees and sat on the advisory boards of pharmaceutical companies (Spencer 2005; Spencer 2011); two reported support from research grants and a company with a vested interest (Schrantee 2016; Tenenbaum 2002); one reported being fully supported by a pharmaceutical company (Kuperman 2001); and three reported being supported entirely by research grants from public and private institutions (Bouffard 2003; Kooij 2004; Schubiner 2002). Additional details are described at the Characteristics of included studies tables.
Based on the sources of funding and conflict of interests disclosed in the trial publications, we have concerns about the impact of the conflicts of interest on the results analyzed from the included trials (Boutron 2020) (Table 6). We detected notable reasons for concern in four trials (Kuperman 2001; Spencer 2005; Spencer 2011; Tenenbaum 2002); reasons for concern were unclear in two RCTs (Carpentier 2005; Dorrego 2002), since sources of funding and conflict of interests were not reported in these trials.
| Study | Judgment | Rationale |
|---|---|---|
| RCTs with parallel design | ||
| Notable concerns |
| |
| No concerns |
| |
| No concerns |
| |
| Notable concerns |
| |
| Notable concerns |
| |
| RCTs with cross‐over design | ||
| No concerns |
| |
| Unclear |
| |
| Unclear |
| |
| No concerns |
| |
| Notable concerns |
| |
Excluded studies
We excluded 60 reports (54 studies) at the full‐text screening stage. We documented the reasons for excluding these reports, which comprised:
-
Study design not eligible (13 reports);
-
Interventions investigated not eligible (16 reports);
-
Treatment comparisons not eligible (7 reports);
-
Population included in the primary study not eligible (17 reports); and
-
Studies investigating effects on IR methylphenidate in diverse outcomes (e.g. participants' performance on the Continuous Paired‐Associate Learning Test (7 reports).
We selected 16 studies (from 19 reports) to report in more detail in the Characteristics of excluded studies tables. These are studies which at first sight appeared to be eligible for inclusion, but proved ineligible on closer inspection. We documented the reasons for excluding these reports, which comprised:
-
Study design not eligible (2 studies);
-
Interventions investigated not eligible (1 study);
-
Treatment comparisons not eligible (2 studies);
-
Population included in the primary study not eligible (4 studies); and
-
Studies investigating effects on IR methylphenidate in diverse outcomes (7 studies).
Seven studies investigated research questions and outcomes not related to the objectives of this review.
-
Kinsbourne 2001 sought to assess participants' performance on the Continuous Paired‐Associate Learning Test (CPALT) after a single administration of three doses (5, 10, and 20 mg) of methylphenidate and placebo.
-
Ni 2013 compared the long‐term efficacy of methylphenidate and atomoxetine in improving executive functions in drug‐naïve adults with ADHD.
-
Ni 2016 evaluated the effects of methylphenidate in intra‐individual variability in reaction time (IIV‐RT) in people with ADHD.
-
Vansickel 2011 evaluated the acute effects of methylphenidate on cigarette‐smoking behavior in individuals diagnosed with ADHD.
-
Verster 2008 evaluated the effects of methylphenidate on driving performance of adults with ADHD.
-
Bouziane 2019 evaluated whether methylphenidate modulates human brain white matter in an age‐dependent manner.
-
NCT02477280 sought to examine the effects of methylphenidate on objective and self‐rated task performance during the Quantified Behavior Test.
Studies awaiting classification
We were unable to access the abstract or full text of one trial (IRCT20110802007202N15); this trial is awaiting classification (Characteristics of studies awaiting classification). The author team made extensive efforts to retrieve this study by searching several databases, the libraries of two different institutions and requesting the support of the Information Specialist of the Cochrane Developmental, Psychosocial and Learning Problems.
Studies ongoing
One clinical trial was classified as 'Ongoing' according to the record in the European Union Clinical Trial Register (EU‐CTR): EU CTR 2012‐005246‐38. See the Characteristics of ongoing studies table for more information.
Risk of bias in included studies
We judged most of the included trials to be at unclear risk of selection, performance and detection bias. We considered most trials to be at high risk of attrition and reporting bias for both the efficacy (symptoms of ADHD) and harms (adverse events) outcomes. For harms, we rated all the included trials at high risk of reporting bias.
A comprehensive description of the risk of bias for each trial across each domain is provided in the 'Risk of bias' tables (beneath the Characteristics of included studies tables). We summarize the risks of bias below and in Figure 2 and Figure 3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Random sequence generation (selection bias)
We judged three trials at low risk of selection bias for this domain (Bouffard 2003; Kooij 2004; Schrantee 2016), as they described adequate methods to generate the randomization sequence. Two trials did not report the method used to generate the randomization sequence of allocation and had high imbalance in the numbers and baseline characteristics of the participants between intervention groups. We judged these RCTs to be at high risk of bias (Schubiner 2002; Spencer 2011). Five trials provided insufficient information to permit a judgement so we rated them at unclear risk of bias (Carpentier 2005; Dorrego 2002; Kuperman 2001; Spencer 2005; Tenenbaum 2002).
Allocation concealment (selection bias)
We deemed one trial to be at low risk of selection bias for this domain, as allocation concealment was ensured by the use of a central clinical unit to generate the sequence of allocation (Schrantee 2016). We judged the nine remaining trials to be at unclear risk of bias as they provided insufficient information to permit a judgment (Bouffard 2003; Carpentier 2005; Dorrego 2002; Kooij 2004; Kuperman 2001; Schubiner 2002; Spencer 2005; Spencer 2011; Tenenbaum 2002).
Blinding
Blinding of participants and personnel (performance bias)
We assessed performance bias separately for participants and personnel.
Blinding of participants
Six trials described adequate methods to blind participants, mostly by the use of identical‐looking tablets, and we judged these trials to be at low risk of performance bias for the blinding of participants (Dorrego 2002; Kooij 2004; Schrantee 2016; Schubiner 2002; Spencer 2005; Tenenbaum 2002). Three trials were described as single‐blinded but it was not clear who was blinded; we judged these trials to be at high risk of performance bias (Carpentier 2005; Kuperman 2001; Spencer 2011). One trial provided insufficient information to permit a judgement, so we rated it at unclear risk of bias (Bouffard 2003).
Blinding of personnel
Four trials described adequate methods to blind personnel, mostly by the use of identical‐looking tablets; we rated these trials at low risk of performance bias for the blinding of personnel (Kooij 2004; Schrantee 2016; Spencer 2005; Tenenbaum 2002). Three trials were described as single‐blinded, but it was not clear who was blinded; we judged these trials to be at high risk of performance bias (Carpentier 2005; Kuperman 2001; Schubiner 2002). Three trials provided insufficient information to permit a judgment, so we rated them at unclear risk of bias (Bouffard 2003; Dorrego 2002; Spencer 2011).
Blinding of outcome assessment (detection bias)
We assessed blinding of outcome assessment separately for the primary outcomes of efficacy (symptoms of ADHD) and harms (adverse events).
Efficacy
Two trials reported that the investigators rating the symptoms of ADHD were blinded to the treatment allocation of the participants; we rated these trials at low risk of detection bias for symptoms of ADHD (Spencer 2005; Spencer 2011). The remaining eight trials provided insufficient information to permit a judgment and were rated at unclear risk of detection bias (Bouffard 2003; Carpentier 2005; Dorrego 2002; Kooij 2004; Kuperman 2001; Schrantee 2016; Schubiner 2002; Tenenbaum 2002).
Harms
One trial reported that the assessment of harms was performed by unblinded clinicians; we judged this trial (Spencer 2011) at high risk of detection bias for adverse events. The remaining nine trials provided insufficient information to permit a judgment, so we rated them at unclear risk of detection bias (Bouffard 2003; Carpentier 2005; Dorrego 2002; Kooij 2004; Kuperman 2001; Schrantee 2016; Schubiner 2002; Spencer 2005; Tenenbaum 2002).
Incomplete outcome data
In three trials, reasons for missing outcome data appeared unlikely to be related to the true outcome; we considered these trials to be at low risk of attrition bias (Carpentier 2005; Kuperman 2001; Schrantee 2016).
We detected imbalances in dropout rates and retention rates at follow‐up that we considered likely to be related to the true outcome in the reports of five trials; we rated these trials at high risk of attrition bias (Dorrego 2002; Schubiner 2002; Spencer 2005; Spencer 2011; Tenenbaum 2002). Two trials provided insufficient information to permit a judgment, so we rated these RCTs at unclear risk of bias (Bouffard 2003; Kooij 2004).
Selective reporting
We assessed selective reporting (reporting bias) for the primary outcomes of efficacy (symptoms of ADHD) and harms (adverse events) separately.
Efficacy
We deemed one RCT (Kuperman 2001) to be at low risk of reporting bias for symptoms of ADHD, as the outcomes planned in the Methods section of the study report were fully covered in the Results section. The results related to the outcomes of efficacy were incompletely reported in eight trials; we judged these trials to be at high risk of selective reporting bias (Bouffard 2003; Carpentier 2005; Kooij 2004; Schrantee 2016; Schubiner 2002; Spencer 2005; Spencer 2011; Tenenbaum 2002). One trial (Dorrego 2002) provided insufficient information to permit judgment, so we rated it at unclear risk of reporting bias.
Harms
In all trials, the methods used to identify outcomes of harms were not reported or were deemed inappropriate. We also judged that the assessment of harms was likely to differ between intervention groups and could be influenced by knowledge of the intervention received. For these reasons, we rated all 10 trials at high risk of selective reporting bias for adverse events (Bouffard 2003; Carpentier 2005; Dorrego 2002; Kooij 2004; Kuperman 2001; Schrantee 2016; Schubiner 2002; Spencer 2005; Spencer 2011; Tenenbaum 2002).
Other potential sources of bias
We did not assess other potential sources of bias in the included trials (See Differences between protocol and review).
Effects of interventions
See: Summary of findings 1 Immediate‐release methylphenidate versus placebo for attention deficit hyperactivity disorder (ADHD) in adults; Summary of findings 2 Immediate‐release methylphenidate versus lithium for attention deficit hyperactivity disorder (ADHD) in adults
See summary of findings Table 1; summary of findings Table 2.
IR methylphenidate versus placebo
Four trials reported data on changes in the symptoms of ADHD: one reported results on investigator‐rated scales (Spencer 2005) and three on participant‐rated scales (Kooij 2004; Kuperman 2001; Schubiner 2002).
Primary outcomes
Efficacy: changes in symptoms of ADHD
The trials assessed the outcomes using different rating scales and reported results using change scores and end scores, which prevented us from pooling the data in a meta‐analysis.
The available data suggest that IR methylphenidate might reduce symptoms of ADHD when measured by investigator‐rated scales (MD −20.70, 95% CI −23.97 to −17.43; Analysis 1.1; very low‐certainty evidence), namely the AISRS (scores range from 0 to 54). This analysis was based on one RCT with parallel design and 146 participants (Spencer 2005).
One trial (Schubiner 2002) measured changes in symptoms of ADHD rated by the investigators and the participant; however, the trial enrolled a small sample of participants (n = 24) and data were incompletely reported. Overall, after 12 weeks, 33% (8/24) of the participants treated with IR methylphenidate were classified by physicians as showing moderate improvement (a reduction of one or two points on a scale ranging from one (very much improved) to seven (very much worse), compared with 46% (11/24) of participants treated with placebo. When participants rated their own symptoms, 73% (18/24) of those treated with IR methylphenidate reported moderate improvement compared with 42% (10/24) of those treated with placebo.
When measured by participant‐rated scales, it was unclear whether IR methylphenidate could reduce or increase the symptoms of ADHD. For this analysis, two sets of trials contributed data; the available data were derived from different scales and were reported for different point measures (change scores and end scores), thus preventing pooling. Kuperman 2001 (a RCT with a parallel design) reported change scores from 19 participants on the ADHDRS (scores range from 0 to 54) (MD 2.30, 95% CI −6.20 to 10.80; Analysis 1.2; very low‐certainty evidence); Kooij 2004 and Schubiner 2002 (trials with parallel and cross‐over designs, respectively) provided end scores from 138 participants (SMD −0.59, 95% CI −1.25 to 0.06; Tau2 = 0.16; Chi2 = 3.26 (P = 0.07); I2 = 69%; Analysis 1.3; Figure 4; very low‐certainty evidence).

Comparison 1 IR methylphenidate versus Placebo, Outcome: Symptom of ADHD
We observed slightly different results when repeating the analyses using a fixed‐effect model (SMD −0.51, 95% CI −0.85 to −0.17; I2 = 69%), and with the cross‐over trial excluded (SMD −0.97, 95% CI −1.57 to −0.37; versus SMD −0.59, 95% CI −1.25 to 0.06 (with cross‐over trial): Analysis 1.3; Figure 4).
Harms
None of the included trials reported ascertaining or evaluating participants for serious adverse events as defined by the ICH 2016 (see Types of outcome measures). One trial reported that "no serious adverse events were noted in any of the individuals studied" (Schrantee 2016), although the method of assessing adverse events was not described in the trial report. It is therefore not clear how these events were defined.
One trial comparing IR methylphenidate with placebo provided data on the total number of participants experiencing at least one adverse event (Kooij 2004). The available data did not allow a conclusion of the difference between participants receiving IR methylphenidate compared with placebo on the risk of harms (RR 1.19, 95% CI 0.94 to 1.52; Analysis 1.4). Three trials reported data on discontinuation due to adverse events (Kooij 2004; Schubiner 2002; Spencer 2005), with one participant discontinuing treatment with placebo and seven discontinuing treatment while receiving IR methylphenidate.
Data on the occurrence of harms, as reported by four trials (Bouffard 2003; Kooij 2004; Schubiner 2002; Spencer 2005) were described for a total of 203 participants who received IR methylphenidate and for a total of 141 participants who received placebo. These participants experienced 651 adverse events in total: 438 events were experienced by people receiving IR methylphenidate (median number of events experienced by people who had events = 12, interquartile range (IQR) = 8 to 28), while 213 were experienced by people receiving placebo (median number of events experienced by people who had events = 8, IQR = 5 to 11).
Amongst the adverse events reported, participants receiving IR methylphenidate had an increased risk of developing gastrointestinal complications (e.g. dry mouth, nausea and stomach aches; RR 1.96, 95% CI 1.13 to 2.95) and metabolism and nutrition complications (e.g. decreased appetite or loss of appetite; RR 1.77, 95% CI 1.06 to 2.96) (Figure 5). Table 7 summarizes the reported harms according to organ systems and individual events.
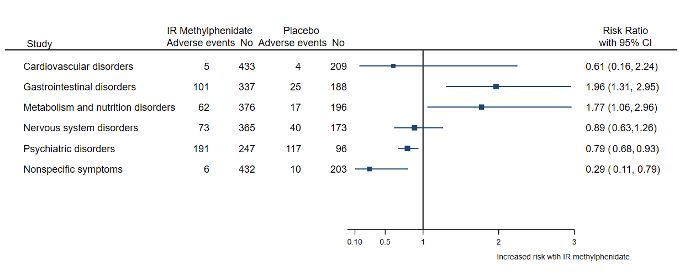
Comparison 2 IR methylphenidate vs Placebo, Outcome: Harms (total of events among patients who had experienced at least one event)
| Participants treated with IR methylphenidate | Participants treated with placebo | ||||||
|---|---|---|---|---|---|---|---|
| Organ system category | Event | n | % | Organ system category | Event | n | % |
| Cardiovascular disorders | Tachycardia | 4 | 0.9 | Cardiovascular disorders | Tachycardia | 1 | 0.4 |
| Elevated blood pressure | 1 | 0.2 | Chest pain | 2 | 0.9 | ||
| Palpitations | 1 | 0.5 | |||||
| Gastrointestinal disorders | Dry mouth | 47 | 10.7 | Gastrointestinal disorders | Abdominal complaints | 2 | 0.8 |
| Abdominal complaints | 6 | 1.4 | Constipation | 1 | 0.5 | ||
| Constipation | 2 | 0.5 | Diarrhea | 2 | 0.9 | ||
| Diarrhea | 5 | 1.1 | Gastrointestinal problem | 3 | 1.4 | ||
| Gastrointestinal problem | 7 | 1.6 | Nausea | 3 | 1.4 | ||
| Nausea | 15 | 3.4 | Nausea or upset stomach | 5 | 2.3 | ||
| Nausea or upset stomach | 8 | 1.8 | Stomach aches | 6 | 2.8 | ||
| Metabolism and nutrition disorders | Decreased in appetite/Loss of appetite | 62 | 14.2 | Metabolism and nutrition disorders | Decreased in appetite/Loss of appetite | 17 | 8.0 |
| Nervous system disorders | Dizziness | 9 | 2.1 | Nervous system disorders | Dizziness | 4 | 1.9 |
| Headache | 64 | 14.6 | Headache | 36 | 16.9 | ||
| Psychiatric disorders | Anxious | 19 | 4.3 | Psychiatric disorders | Anxious | 15 | 7.0 |
| Disorientation, insomnia, and anxiety lasting several hours | 1 | 0.2 | Euphoric, unusually happy | 7 | 3.3 | ||
| Euphoric, unusually happy | 10 | 2.3 | Insomnia or trouble sleeping | 8 | 3.8 | ||
| Irritability | 14 | 3.2 | Irritability | 13 | 6.1 | ||
| Moody | 31 | 7.1 | Moody | 2 | 0.9 | ||
| Sadness | 15 | 3.4 | Sadness | 9 | 4.2 | ||
| Stare a lot or daydream | 12 | 2.7 | Stare a lot or daydream | 17 | 8.0 | ||
| Talk less with others | 11 | 2.5 | Talk less with others | 12 | 5.6 | ||
| Tics or nervous movements | 4 | 0.9 | Tics or nervous movements | 5 | 2.3 | ||
| Tics | 3 | 0.7 | Tics | 1 | 0.5 | ||
| Sleeping problems (difficulty/trouble sleeping, insomnia, nightmares) | 71 | 16.2 | Sleeping problems (difficulty/trouble sleeping, insomnia, nightmares) | 28 | 13.2 | ||
| Nonspecific symptoms | Drowsiness | 6 | 1.4 | Nonspecific symptoms | Drowsiness | 10 | 4.7 |
| Total events | 438 | 100 | Total events | 213 | 100 | ||
IR: Immediate release
There was no difference in the reported cardiovascular adverse events between participants receiving IR methylphenidate and those receiving placebo; however, data on cardiovascular adverse events were reported inconsistently, which prevents a comprehensive analysis. The available reports are described in Table 8.
| Study | Electrocardiogram | Heart rate | Diastolic blood pressure | Systolic blood pressure | Blood pressure |
|---|---|---|---|---|---|
| ‐ | ‐ | "No significant increase of diastolic blood pressure between baseline and methylphenidate" | Increased in participants receiving IR methylphenidate (mean = 123 mmHg) compared with placebo (mean 128 = mmHg) (P < 0.05) | ‐ | |
| "Mean heart rate was 4.8 beats/min higher (p=0.002) after IR‐MPH" | "The diastolic blood pressure remained virtually unchanged" | "The systolic blood pressure was 0.13 mmHg higher after methylphenidate but this was not statistically significant (p=0.954)" | |||
| ‐ | ‐ | ‐ | ‐ | 1 participant receiving IR methylphenidate with elevated blood pressure, worst occurrence reported only | |
|
| "Small but statistically significant increases in pulse (83 ± 13 vs. 76 ± 13 bpm, t=4.4, df(77), p<.001" | No increase in participants receiving IR methylphenidate (mean = 78 mmHg, SD = 9 mmHg) in comparison with placebo (mean = 76 mmHg, SD = 9 mmHg) | No increase in participants receiving IR methylphenidate (mean = 128 mmHg, SD = 12 mmHg) in comparison with placebo (mean = 126 mmHg, SD = 14 mmHg) | ‐ |
bpm: beats of the heart per minute
df: degrees of freedom
IR: immediate‐release
SD: standard deviation
Additional data were also reported narratively for body weight‐related events, although inconsistent reporting prevented further analysis:
-
in Bouffard 2003, "There was no significant weight loss"; and
-
in Kooij 2004, "Mean body weight was 1.7 kg lower (p < 0.001) after methylphenidate treatment compared to placebo".
Secondary outcomes
Changes in the clinical impression measures of severity
Two trials (Kooij 2004; Schrantee 2016) reported data from 139 participants on the clinical impression of ADHD symptom severity, measured by the CGI‐S scale (scores range from one (very much improved) to seven (very much worse)). The trials reported change scores and end scores, and the available data were pooled in a meta‐analysis. The results demonstrate a decrease in the severity of ADHD symptoms in participants treated with IR methylphenidate compared to those treated with placebo (MD −0.57, 95% CI −0.85 to −0.28; I2 = 0%; Analysis 1.5; very low‐certainty evidence). We did not observe changes in the results after excluding the cross‐over trial from the analysis (MD −0.60, 95% CI −0.92 to −0.28).
One trial (Kuperman 2001) provided a narrative description of the results: "Response rates based on the primary outcome of CGI response were 64% for bupropion, 50% for IR methylphenidate, and 27% for placebo. There was not a significantly greater response rate observed in the active treatment groups than in the placebo group (p = 0.14)."
Changes in the clinical impression measures of improvement
One trial (Schrantee 2016) provided data from 49 participants on the clinical impression of improvement of ADHD symptoms, measured by the CGI‐I scale (scores range from one (very much improved) to seven (very much worse)). The results were available for the scores measured at the end of the follow‐up period. The data suggest that IR methylphenidate increased the clinical impression of improvement in ADHD symptoms (MD −0.94, 95% CI −1.37 to −0.51; Analysis 1.6; low‐certainty evidence).
Level of functioning
Outcome measurement on level of functioning on the GAF scale (0 to 100) was reported narratively by one trial: "The mean difference between IR methylphenidate and placebo response of completers constitutes an 11% difference from baseline (end point placebo – end point IR methylphenidate/baseline IR methylphenidate; t = 3.4, df = 94, p < .01)" (Spencer 2005). The remaining trials accessing level of functioning did not report results (Kooij 2004; Schrantee 2016; Spencer 2011).
Anxiety and depression
One RCT with a parallel design (Kuperman 2001) provided data from 19 participants on changes in symptoms of anxiety and depression in adults with ADHD. It is uncertain whether, compared with placebo, IR methylphenidate impacts symptoms of anxiety (MD −0.20, 95% CI −4.84 to 4.44; Analysis 1.7; very low‐certainty evidence). Compared with placebo, the effects of IR methylphenidate on symptoms of depression in adults with ADHD were also uncertain (MD 2.80, 95% CI −0.09 to 5.69; Analysis 1.8; very low‐certainty evidence). The results were based on scores on the HAM‐A (scores range from 0 to 56) and HAM‐D (scores range from 0 to 52) scales, respectively.
Two other trials provided a narrative description of the results for anxiety and depression.
-
Kooij 2004: "Methylphenidate was associated with higher symptom levels of depression and anxiety than placebo, as was apparent from higher HAM‐D and HAM‐A scores: 2.4 (p=0.002) and 2.9 (p=0.002) points, respectively. When defined as a HAM‐D >16, 11% (n=5) had depression after methylphenidate compared to 9% (n=4) after placebo. When defined as a HAM‐A >21, 7% (n=3) had anxiety after methylphenidate compared to 4% (n=2) after placebo".
-
Bouffard 2003: "There was a reduction in anxiety and depression scores with both placebo and methylphenidate. However, methylphenidate was significantly superior to placebo in reducing anxiety scores (P < 0.05), and there was a trend for it to be superior in reducing depression scores".
IR methylphenidate versus OROS methylphenidate
One trial (Spencer 2011) compared the treatment effects of OROS methylphenidate in adults who were respondents to treatment with the IR formulation of methylphenidate and switched to treatment with the OROS formulation of methylphenidate.
Primary outcomes
Efficacy: changes in symptoms of ADHD
Spencer 2011 reported only narrative results on changes in symptoms of ADHD from an analysis of 53 participants: "There was no clinically or statistically significant difference, F(1, 52) = 0.1, p = .7, between the treatment groups in the AISRS rating scale through 6 weeks of treatment".
Harms
Spencer 2011 reported data on changes in cardiac parameters. Additional data on spontaneously‐reported emergent adverse events were described in a figure with non‐extractable data.
A total of eight participants reported discontinuing treatment while receiving IR methylphenidate and none while receiving OROS methylphenidate. There were a total of seven adverse events experienced by people receiving IR methylphenidate (median number of events experienced by people who had events = 2, IQR = 1 to 4), while 21 adverse events were experienced by people receiving OROS methylphenidate (median number of events experienced by people who had events = 6, IQR = 1 to 14). Table 9 summarizes the reported harms according to organ systems and individual events.
| Participants treated with IR methylphenidate | Participants treated with OROS methylphenidate | ||||||
|---|---|---|---|---|---|---|---|
| Organ system category | Event | n | % | Organ system category | Event | n | % |
| Cardiovascular disorders | Diastolic blood pressure > 90 mm | 1 | 14 | Cardiovascular disorders | Diastolic blood pressure > 90 mm | 1 | 5 |
| Pulse > 90 bpm | 4 | 57 | Pulse > 90 bpm | 14 | 67 | ||
| Systolic blood pressure > 140 mm Hg | 2 | 29 | Systolic blood pressure >140 mm Hg | 6 | 29 | ||
| Total events | 7 | 100 | Total events | 21 | 100 | ||
bpm: beats per minute
IR: immediate release
OROS: osmotic release oral system
Secondary outcomes
Spencer 2011 did not measure any of the secondary outcomes.
IR methylphenidate versus lithium
One trial with a cross‐over design (Dorrego 2002) investigated treatment with IR methylphenidate versus lithium.
Primary outcomes
Efficacy: changes in symptoms of ADHD
Dorrego 2002 assessed changes in the symptoms of ADHD using an investigator‐rated scale and reported end scores. Based on the available data from 46 participants, it is uncertain whether IR methylphenidate increases or decreases symptoms of ADHD (MD 0.60, 95% CI −3.11 to 4.31; Analysis 2.1; very low‐certainty evidence) assessed by the CAARS scale (scores range from 0 to 198), compared with lithium.
Harms
Dorrego 2002 provided data on the number of participants experiencing adverse events. Three participants were reported to have discontinued treatment while receiving IR methylphenidate and one while receiving lithium.
There were a total of five adverse events experienced by people receiving IR methylphenidate (median number of events experienced by people who had events = 1, IQR = 0 to 2), while nine adverse events were experienced by people receiving lithium (median number of events experienced by people who had events = 2, IQR = 1 to 4). Table 10 summarizes the reported harms according to organ systems and individual events.
| Participants treated with IR methylphenidate | Participants treated with lithium | ||||||
|---|---|---|---|---|---|---|---|
| Organ system category | Event | n | % | Organ system category | Event | n | % |
| Cardiovascular disorders | Orthostatic hypotension | 1 | 20 | Cardiovascular disorders | Chest discomfort | 1 | 8 |
| Gastrointestinal disorders | Diarrhea | 3 | 60 | Gastrointestinal disorders | Diarrhea | 3 | 28 |
| Nausea | 1 | 10 | |||||
| Nervous system disorders | Headache | 1 | 20 | Nervous system disorders | Headache | 5 | 54 |
| Total events | 5 | 100 | Total events | 9 | 100 | ||
IR: immediate release
Secondary outcomes
Anxiety and depression
Data from 46 participants recruited by Dorrego 2002 were unclear on the effects of IR methylphenidate versus lithium for improving symptoms of anxiety (MD −0.80, 95% CI −4.49 to 2.89; Analysis 2.2), assessed by the HAM‐A (scores range from 0 to 56), and depression (MD −1.20, 95% CI −3.81 to 1.41; Analysis 2.3) assessed by the HAM‐D ( scores range from 0 to 52) scales. We rated the certainty of this evidence as very low.
Dorrego 2002 did not report data on changes in the clinical impression of severity and improvement, or assess measures on level of functioning.
IR methylphenidate versus extended‐release bupropion
One trial (Kuperman 2001) investigated treatment with IR methylphenidate versus extended‐release (SR) bupropion. The RCT implemented a parallel design and three comparison arms: IR methylphenidate versus bupropion versus placebo. To avoid multiplicity of the effect size, we selected the comparison between IR methylphenidate and placebo as the most relevant to answer our review question and omitted the comparison between IR methylphenidate and bupropion (see Data synthesis). The IR methylphenidate and the placebo arms contributed data to the comparison between IR methylphenidate and placebo, previously explored in this section.
IR methylphenidate versus Pycnogenol®
One trial (Tenenbaum 2002) compared the treatment of adults with ADHD with Pycnogenol® versus placebo in a three‐arm cross‐over trial (IR methylphenidate versus Pycnogenol® versus placebo). The trial reported incomplete data on initial scores from 24 participants on symptoms of ADHD, anxiety and depression, thereby not allowing further analysis. Changes in the clinical impression of severity and improvement, and level of functioning were not assessed or mentioned in the trial report.
Discussion
Summary of main results
In this review, we evaluated the evidence for the treatment of adults with ADHD with IR methylphenidate in comparison with placebo, OROS‐methylphenidate, lithium, SR‐bupropion, and Pycnogenol®.
We found very low‐certainty evidence that IR methylphenidate could be more efficacious than a placebo in reducing ADHD symptoms when symptoms were rated by the trial’s investigators (Spencer 2005). However, when participants rated their own symptoms (Kooij 2004; Kuperman 2001; Schubiner 2002), there was no certain evidence that IR methylphenidate reduced the symptoms of ADHD. Overall, there was only low‐certainty evidence that IR methylphenidate might slightly improve the global clinical impression scores when compared to placebo, and uncertain evidence about its effect on ADHD symptoms when compared with lithium (Dorrego 2002).
Compared with placebo, there was low‐certainty evidence that treatment with IR methylphenidate might result in a reduction in the clinical impression of the severity of ADHD symptoms (Kooij 2004; Schrantee 2016). Low‐certainty evidence suggests that, compared with placebo, adults treated with IR methylphenidate could have an increase in the clinical impression of improvement of ADHD symptoms (Schrantee 2016). However, we considered this potential treatment benefit at high risk of imprecision caused by single‐study results with small sample sizes (Schünemann 2020a). There was very low‐quality evidence for the remaining outcomes across the comparisons among IR methylphenidate, placebo and lithium. Overall, incomplete and under‐reported data precluded further analysis and accurate assessment of treatment effects on the prespecified secondary outcomes among the remaining treatment comparisons of IR methylphenidate, OROS methylphenidate, SR‐bupropion, and Pycnogenol®.
We found only narrative data for the comparison between IR and OROS methylphenidate; no clinically relevant difference between the treatment groups was reported (Spencer 2011). Also, the data for the comparison between IR methylphenidate and Pycnogenol® were incomplete, thus precluding any analysis (Tenenbaum 2002).
Finally, we did not undertake a pair‐wise comparison between IR methylphenidate and SR bupropion, in order to avoid multiplicity of effect size. The trial providing data on this comparison evaluated eight participants receiving IR methylphenidate in comparison with 11 participants receiving SR bupropion and 11 participants receiving placebo (Kuperman 2001). We judged that to include data on the IR methylphenidate group, it would be better to preserve the sample size for this comparison group. We therefore chose to select only the most relevant comparison to answer the review question (Higgins 2020). Bupropion is not considered an option to treat adults with ADHD, so we focused on exploring whether IR methylphenidate could demonstrate any benefits to patients in comparison with treatment with placebo.
In four trials, there was a two‐fold increase in the occurrence of adverse events among adults treated with IR methylphenidate in comparison with those treated with placebo. There was evidence that gastrointestinal (e.g. dry mouth, nausea and stomach aches) and metabolism and nutrition (appetite/weight‐related events) complications were notably more common in the group of participants treated with IR methylphenidate. Cardiovascular complications, which are one of the main concerns among people treated with methylphenidate (CADDRA 2020), were reported inconsistently and mainly as vague narrative reports. Overall, the information about adverse events is unclear. In all cases, we cannot be sure whether adverse events were not measured, were sought but not detected, or were measured but not included in the full publication. The absence of reporting should not automatically be assumed to equate to the absence of harms (Loke 2015). The lack of data on harms compromises risk assessments of the efficacy of IR methylphenidate for ADHD in adults.
Of note, none of the included trials assessed quality of life and few studies included participants with comorbid disorders, which contrasts with the high prevalence of other psychiatric conditions diagnosed in people with ADHD (Cheng 2017; Kessler 2006).
All the above evidence should be considered in light of the low certainty of the evidence, the limited amount of trials evaluating each comparison, particularly change in symptoms, and the significant number of studies for which we had 'notable concerns' for a potential impact of vested interests on the results analyzed and reported (Boutron 2020). Authors from four of the trials declared receiving funding from companies with a financial interest in the findings of IR methylphenidate for the treatment of ADHD in adults; authors from two studies omitted information related to the sources of funding and conflict of interests.
Overall completeness and applicability of evidence
The overall completeness and applicability of the evidence relating to the efficacy and harms of IR methylphenidate to treat adults with ADHD is limited by a number of factors. First, while several clinical trials, systematic reviews and meta‐analyses have been published on the effect of treating ADHD in children and adolescents with IR methylphenidate (Charach 2011; Charach 2013; Hanwella 2011; Kambeitz 2014; Maia 2017; Punja 2013; Reichow 2013; Storebø 2015), there is a dearth of such data on adults. Second, the limited data available do not include the analysis of the efficacy and harms of IR methylphenidate in people with psychiatric comorbidities. This is problematic, considering the high prevalence of psychiatric comorbidities in adults with ADHD (Cheng 2017; Kessler 2006).
Additionally, despite the high prevalence observed for the co‐occurrence of ADHD and ASD in adults (14% to 78%) (Muit 2020), the dual diagnosis of ADHD and ASD or other psychiatric disorders was only introduced in the DSM‐5 (Epstein 2013; Pehlivanidis 2020). All included trials in our review, however, used diagnostic criteria developed prior to the DSM‐5. This means that the evidence from our review may not apply to the treatment of people with both ADHD and ASD or other concomitant psychiatric disorders. Our findings should also be interpreted with caution, given the context of the Research Domain Criteria (RDoC) framework and Comparative Effectiveness Research (CER) approaches (NIMH 2020), which operate under the framework of a larger spectrum of mental‐related disorders when exploring mental health and illness.
The small sample sizes and the short duration of the available RCTs are other limitations of the trials included in this review. The small sample sizes limit the reliability of the findings, and the short durations of the RCTs are contradictory, given the chronic characteristic of ADHD and the need for long‐term treatment. Finally, the high heterogeneity among the clinical rating scales used to ascertain the effects of IR methylphenidate on the treatment of ADHD in adults impacts the interpretation of the available evidence. Obtaining standardized, comparable results from clinical trials is essential to allow for a proper synthesis and appraisal of the evidence.
Quality of the evidence
We judged the certainty of the evidence for most outcomes assessed in this review as very low, due to critical concerns over the 'Risk of bias' profiles of the individual studies, with high or unclear risk of bias in most domains (selection, performance, detection, attrition and reporting biases).
Information on random sequence generation was available for only half of the trials; among those, we deemed a proportion (40%) to be at high risk of selection bias. Performance bias for masking of personnel was also inadequate in half of the available trials; it was rated at high risk of bias in three trials and three trials provided insufficient information to permit a judgment. Performance bias related to blinding of participants was the one domain in which we judged most trials (six) to be at low risk of bias. Most trials provided insufficient information to permit a judgment of detection bias, and most were also at high risk of attrition and selective reporting bias.
Another reason to downgrade our certainty in the evidence was imprecision. Single‐study results with small sample sizes provided most of the evidence. The maximum number of participants providing data on the assessed outcomes was 157, significantly below the optimal information size (Schünemann 2020a). Considering that we have notable concerns about vested interests influencing the results of the included trials, publication bias can be suspected as present and highly influencing the missing information on treatment harms.
Potential biases in the review process
We undertook a systematic and comprehensive search that, to the best of our knowledge, permitted us to identify all IR methylphenidate trials performed in adults with ADHD. We were also able to obtain additional missing data from one of the included trials. We did not, however, contact pharmaceutical industries and corresponding authors of the included publications to enquire about additional studies that we may have missed. We therefore cannot be assured that we found all relevant studies on the topic.
Methylphenidate has been associated with potentially harmful effects (EMA 2009; US FDA 2007). However, methods to detect adverse events were not consistently described in the included trials and most (five out nine trials) did not report these methods adequately. All trials lacked adequate reporting of harms experienced by participants. In general, the emphasis of RCT designs to assess treatment efficacy leads to an inadequate and biased assessment of harms (Golder 2011; Ioannidis 2009; Loke 2015; Saini 2014; Schroll 2016). We are aware that the systematic evaluation of adverse drug events requires the inclusion of data from other study designs, mainly non‐randomized studies, and we are developing an additional systematic review to explore this question (Cândido 2018).
In addition to the domains used to assess risks of bias, the way a study is conducted, the publication of its full results, the consistency between the comparisons made, and the format in which the results are presented are additional issues that are important to consider when assessing the risks of bias of a study (Bero 2017). A study may be carried out with methodological rigor, and thus present at low risk of bias, and yet its results may be biased (Bero 2017). In this context, conflicts of interest, including research and individual sponsorship, can be a relevant source of bias in a trial’s results (Lundh 2017a; Lundh 2017b).
In this review, funding sources and potential conflict of interests were reported in all but two included trials (Carpentier 2005; Dorrego 2002). Among the studies that received funding from pharmaceutical industries (Kuperman 2001; Spencer 2005; Spencer 2011; Tenenbaum 2002), two showed favorable results for the use of IR methylphenidate, and two demonstrated no benefit in the use of the drug. Notably, the trials demonstrating no benefits of IR methylphenidate were designed to evaluate the efficacy of a different drug as a therapeutic alternative for the treatment of ADHD in adults. Caution is therefore recommended in the analysis of the results synthesized from the studies included in this review; besides methodological biases, they also presented with important potential influences from conflicts of interest favoring beneficial results associated with the use of a drug of interest.
Agreements and disagreements with other studies or reviews
Two previous systematic reviews with network meta‐analyses have assessed the effects of methylphenidate for treating ADHD in adults (Cortese 2018, Peterson 2008). Cortese 2018 estimated the efficacy of different medicines for the improvement of ADHD symptoms in children, adolescents, and adults in double‐blinded RCTs of pharmacological treatment of ADHD. Amphetamines were considered a better option to treat ADHD in adults compared with placebo, followed by methylphenidate (Cortese 2018). Peterson 2008 compared shorter‐acting stimulant drugs (including IR methylphenidate), longer‐acting stimulant drugs and longer‐acting forms of bupropion. They found a higher rate of clinical response of shorter‐acting stimulant drugs compared to placebo, longer‐acting stimulant drugs and longer‐acting forms of bupropion. Methylphenidate was also found to cause appetite loss, sleep disturbances (Peterson 2008), weight loss and increased systolic and diastolic blood pressure (Cortese 2018). Differences in the methodological and analytical approaches undertaken by Cortese 2018, Peterson 2008 and our review limit comparison among the results. However, there are similarities among the adverse events found to be induced by treatment with methylphenidate.
We rated the certainty of the evidence in this review mainly as very low, which is comparable with the certainty‐of‐evidence ratings reported by other Cochrane Reviews assessing the effects of methylphenidate treatment for ADHD in children and adolescents (Castells 2018; Punja 2016; Storebø 2015). The validity and certainty of the evidence for the effects of pharmacological treatments for ADHD are limited by factors that include attrition bias, failure to blind participants, personnel, and outcome detection, selection bias, reporting bias and statistical heterogeneity (Castells 2018; Punja 2016; Storebø 2015). Improving the validity and the quality of primary studies investigating the efficacy and harms of pharmacological treatments for ADHD is essential to increase the reliability of the findings of this and other systematic reviews.
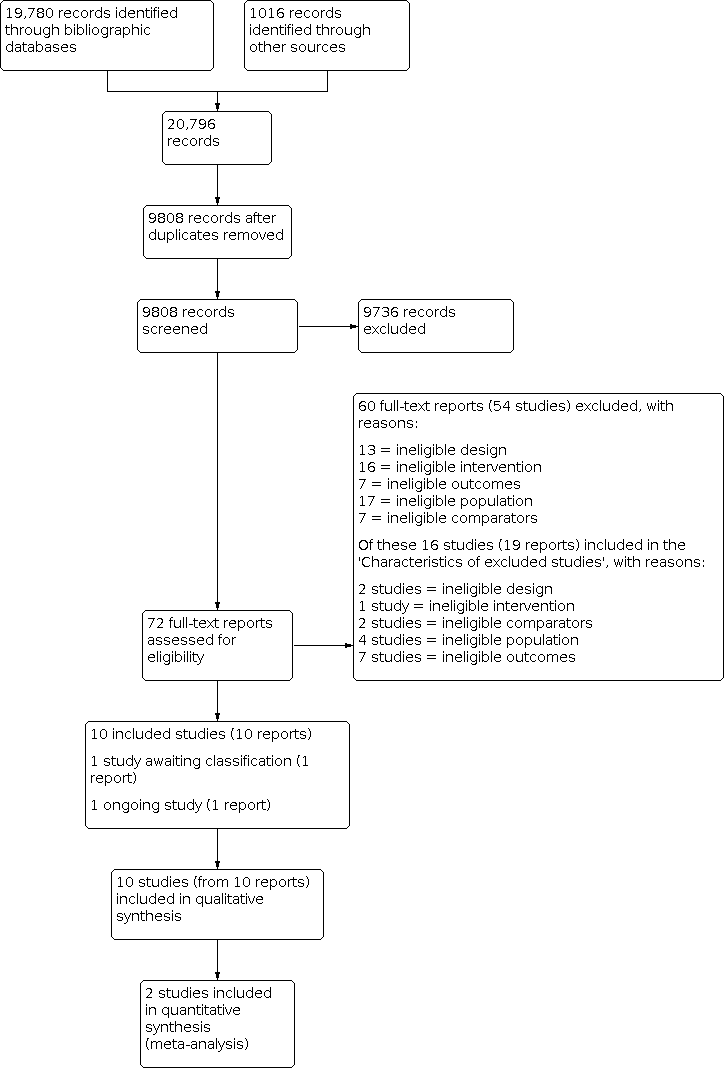
Flow diagram illustrating the results of the study selection process.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.

Comparison 1 IR methylphenidate versus Placebo, Outcome: Symptom of ADHD

Comparison 2 IR methylphenidate vs Placebo, Outcome: Harms (total of events among patients who had experienced at least one event)

Comparison 1: IR methylphenidate versus placebo, Outcome 1: Efficacy: investigator‐rated (end scores)

Comparison 1: IR methylphenidate versus placebo, Outcome 2: Efficacy: participant‐rated (change scores)

Comparison 1: IR methylphenidate versus placebo, Outcome 3: Efficacy: participant‐rated (end scores)

Comparison 1: IR methylphenidate versus placebo, Outcome 4: Harms: patients experiencing at least one adverse event

Comparison 1: IR methylphenidate versus placebo, Outcome 5: Clinical impression: severity (change scores)

Comparison 1: IR methylphenidate versus placebo, Outcome 6: Clinical impression: improvement (end scores)

Comparison 1: IR methylphenidate versus placebo, Outcome 7: Anxiety: investigator‐rated (change scores)

Comparison 1: IR methylphenidate versus placebo, Outcome 8: Depression: investigator‐rated (change scores)

Comparison 2: IR methylphenidate versus lithium, Outcome 1: Efficacy: investigator‐rated (end scores)

Comparison 2: IR methylphenidate versus lithium, Outcome 2: Anxiety: investigator‐rated (end scores)

Comparison 2: IR methylphenidate versus lithium, Outcome 3: Depression: investigator‐rated (end scores)
| Immediate‐release methylphenidate versus placebo for attention deficit hyperactivity disorder (ADHD) in adults | |||||
| Patient or population: adults with ADHD (available evidence for participants aged between 25 to 53 years old) | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
|---|---|---|---|---|---|
| Assumed risk with placebo | Corresponding risk with immediate‐release methylphenidate | ||||
| Efficacy (changes in symptoms of ADHD): investigator‐rated | The mean efficacy score in the control group was 33.8 points | The mean efficacy score in the intervention group was 20.70 points lower (23.97 lower to 17.43 lower) | 146 (1 RCT) | ⊕⊝⊝⊝ | End scores. IR methylphenidate may reduce symptoms of ADHD when rated by investigators but the evidence is very uncertain. |
| Efficacy (changes in symptoms of ADHD): participant‐rated Follow‐up: range = 7 weeks to 12 weeks | The mean efficacy score in the intervention group was 0.59 points lower (1.25 lower to 0.06 higher) | 138 (2 RCTs) | ⊕⊝⊝⊝ Very lowa,b | End scores. IR methylphenidate may have a moderate to no effect on symptoms of ADHD when rated by participants but the evidence is very uncertain. The effect would represent a moderate difference between the control and the intervention group. As a rule of thumb, 0.2 points represents a small difference, 0.5 a moderate and 0.8 a large effect. | |
| Clinical impression: severity | ‐ | The mean clinical impression of symptom severity score in the intervention groups was 0.57 points lower (0.85 lower to 0.28 lower) | 139 (2 RCTs) | ⊕⊝⊝⊝ Very lowb,c | End scores and Change scores. IR methylphenidate may reduce clinicians' impressions of the severity of ADHD symptoms but the evidence is very uncertain. |
| Clinical impression: improvement | The mean clinical impression of improvement score in the control group was 3.54 points | The mean clinical impression of improvement score in the intervention group was0.94 points lower (1.37 lower to 0.51 lower) | 49 (1 RCT) | ⊕⊕⊝⊝ | End scores. IR methylphenidate may slightly increase clinicians' impressions of improvement in ADHD symptoms. |
| Anxiety: investigator‐rated | ‐ | The mean anxiety score in the intervention group was 0.20 points lower (4.84 lower to 4.44 higher) | 19 (1 RCT) | ⊕⊝⊝⊝ | Change scores. There is no clear evidence of an effect, but the evidence is very uncertain. |
| Depression: investigator‐rated | ‐ | The mean depression score in the intervention group was 2.80 points higher (0.09 lower to 5.69 higher) | 19 (1 RCT) | ⊕⊝⊝⊝ | Change scores. There is no clear evidence of an effect, but the evidence is very uncertain. |
| Harms: adverse events (poorly assessed and reported) | Among participants experiencing at least 1 adverse event, the use of IR methylphenidate increased the risk of gastrointestinal complications (RR 1.96, 95% CI 1.13 to 2.95) and loss of appetite (RR 1.77, 95% CI 1.06 to 2.96). Cardiovascular adverse events were reported inconsistently, preventing a comprehensive analysis. | ‐ | ‐ | IR methylphenidate may increase the risk of gastrointestinal adverse events and loss of appetite. It is unclear whether IR methylphenidate induces cardiovascular adverse events. Overall, adverse events were poorly assessed and reported in all included studies. We considered all studies to be at high risk of bias due to selective outcome reporting of harms and masking of the outcome assessor (failure to blind outcome assessor to measure harms). | |
| *The basis for the assumed risk was the median control group risk across studies. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ADHD: Attention deficit hyperactivity disorder; CI: Confidence interval; IR: immediate‐release; IV: Fourth version; MD: Mean difference; RCT: Randomised controlled trial; RR: risk ratio | |||||
| GRADE Working Group grades of evidence | |||||
| aDowngraded twice due to high and unclear risk of bias in multiple criteria (random sequence generation, allocation concealment, blinding of outcome assessors, incomplete outcome data, and selective outcome reporting). | |||||
| Immediate‐release methylphenidate versus lithium for attention deficit hyperactivity disorder (ADHD) in adults | |||||
| Patient or population: adults with ADHD (available evidence for participants aged between 25 to 53 years old) | |||||
| Outcomes | Anticipated absolute effects* (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
|---|---|---|---|---|---|
| Assumed risk with lithium | Assumed risk with immediate‐release methylphenidate | ||||
| Efficacy (changes in symptoms of ADHD): investigator‐rated | The mean efficacy score in the control group was 28.4 points | The mean efficacy score in the intervention group was 0.60 points higher (3.11 lower to 4.31 higher) | 46 (1 RCT) | ⊕⊝⊝⊝ | End scores. It is uncertain whether IR methylphenidate is more effective than lithium. |
| Efficacy (changes in symptoms of ADHD): participant‐rated ‐ not reported | ‐ | ‐ | ‐ | ‐ | Not reported |
| Clinical impression: severity | ‐ | ‐ | ‐ | ‐ | Not reported |
| Clinical impression: improvement | ‐ | ‐ | ‐ | ‐ | Not reported |
| Anxiety: investigator‐rated | The mean anxiety score in the control group was 6.2 points | The mean anxiety score in the intervention group was 0.80 points lower (4.49 lower to 2.89 higher) | 46 (1 RCT) | ⊕⊝⊝⊝ | End scores. IR methylphenidate may have little to no effect on anxiety but the evidence is very uncertain. |
| Depression: investigator‐rated | The mean depression score in the control group was 7.8 points | The mean depression score in the intervention group was 1.20 points lower (3.81 lower to 1.41 higher) | 46 (1 RCT) | ⊕⊝⊝⊝ | End scores. IR methylphenidate may have little to no effect on depression but the evidence is very uncertain. |
| Harms: adverse events (poorly assessed and reported) | 1 trial comparing IR methylphenidate to lithium reported 5 and 9 adverse events, respectively. | ‐ | ‐ | Adverse events were poorly assessed and reported in all included studies. We considered all studies to be at high risk of bias due to selective outcome reporting of harms and masking of the outcome assessor (failure to blind outcome assessor to measure harms). | |
| *The basis for the assumed risk was the median control group risk across studies. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ADHD: Attention deficit hyperactivity disorder; CI: Confidence interval; IR: immediate‐release; MD: Mean difference; RCT: Randomised controlled trial. | |||||
| GRADE Working Group grades of evidence | |||||
| aDowngraded twice due to unclear risk of bias in several domains (random sequence generation, allocation concealment, blinding of outcome assessment, incomplete and selective outcome reporting) and high risk of bias due to incomplete outcome data. | |||||
| Method section | Description | Reason for non‐use |
|---|---|---|
| Types of outcomes | We planned to measure the primary efficacy outcome over the short term (within six months) and long term (longer than 6 months) | These data were not available as no study lasted more than 4.5 months |
| We planned to measure serious adverse events as the primary outcome of harms. | None of the included studies assessed serious adverse events according to the definition of the International Council for Harmonisation (ICH 2016) | |
| Selection of studies | We planned to contact the authors of studies whenever there was insufficient information available to decide whether a study was eligible for inclusion. | This action was not necessary for the ongoing studies |
| Assessment of risk of bias | We planned to revise the 'Risk of bias' domains for randomized controlled trials if new guidelines were released during the development of this review. | When we came to the completion of this review, Cochrane had released a new 'risk of bias' tool but had not yet made it mandatory for all Cochrane Reviews; it is being piloted with only a few reviews within Cochrane. Future updates of this review will take any related updates into account |
| Measures of treatment effect > Continuous outcomes | For studies reporting outcome values other than the mean and standard deviation, we planned to apply standard errors, CI, t values and P values to estimate the results, whenever possible | None of these additional data were reported and thus we were not able to estimate any missing results. We contacted study authors to request the additional information but have not received response |
| If skewed data were detected, we planned to consult a statistician on the best data transformation approach | We did not encounter this situation; therefore, it was not necessary to consult a statistician | |
| Unit of analysis issues > Cross‐over trials | We planned to estimate within‐participant differences between the intervention groups at the end of the study follow‐up period, using the MD and standard deviation to conduct a paired analysis and avoid a unit‐of‐analysis error (Elbourne 2002; Higgins 2020) | No trial reported participant‐level differences between intervention groups |
| Assessment of heterogeneity | We planned to investigate clinical heterogeneity through subgroup analyses | There were insufficient data evaluated and reported in the included studies to allow us to undertake subgroup analyses |
| Assessment of reporting bias | We planned to use funnel plots to investigate the presence of publication bias (the selective publication of trials with positive findings), and other small‐study effects, among the studies included in the review (Page 2020). We planed to use Egger's test to assess for funnel plot asymmetry (Egger 1997), providing 10 or more trials were included in a meta‐analysis. We planned to consult a statistician in situations where we were unable to interpret the asymmetries objectively and when we might have considered alternative statistical tests (Page 2020) | We could not assess reporting bias due to there being an insufficient number of trials (fewer than 10) in the quantitative analyses |
| Data synthesis | Where considerable heterogeneity (I2 statistic greater than 75%) was detected, particularly in the presence of high inconsistency in the direction of effect, we planned not to calculate the average effect of the intervention through a meta‐analysis | Only 1 study contributed data for most of the outcomes and comparisons; therefore, the insufficient data prevented an appropriate assessment of heterogeneity |
| Subgroup analysis and investigation of heterogeneity | We planned to explore potential sources of heterogeneity if the available data from the studies allowed us to stratify participant subgroups by the following characteristics.
We planned to calculate a pooled effect size for each subgroup | There were insufficient data evaluated and reported in the included studies to allow us to undertake subgroup analyses |
| Sensitivity analysis | If there were an adequate number of studies (2 or more), we planned to perform a sensitivity analysis to explore the causes of heterogeneity and test the robustness of the results to decisions made during the development of the review. Specifically, we planned to reanalyze the data:
| There were insufficient studies included in the meta‐analyses to perform any of our preplanned sensitivity analyses |
| Summary of findings and assessment of the certainty of the evidence | We planned to describe the outcomes of interest over short (within 6 months) and long (longer than 6 months) periods of treatment | None of the trials investigated the effects of treatment with IR methylphenidate in adults for a period longer than 18 weeks (4.5 months) |
| CI: Confidence interval | ||
| Study | Comparator | Time points | Investigator measurements | Self‐report measurements | Measurements with complete data |
|---|---|---|---|---|---|
| RCTs with parallel design | |||||
|
| 8 weeks following a single‐blind, 7‐day placebo lead‐in | None | Adult ADHD Symptom Checklist Severity Scale (ADHDRS); updated and validated for DSM‐IV ADHD criteria | Change scores and end scores | |
| Placebo | 16 weeks | None | Adult ADHD Symptom Checklist Severity Scale (ADHDRS); updated and validated for the DSM‐IV ADHD criteria | Data not reported | |
| Placebo | 12 weeks of treatment (13 weeks in total, including 1 week of baseline testing and 12 weeks of treatment) | Barkley's ADHD Rating Scale | Barkley's ADHD Rating Scale | End scores for self‐reported measurements; investigator‐rated scores reported narratively | |
| Placebo | 6 weeks | Adult ADHD Investigator Symptom Report Scale (AISRS) | None | End scores | |
| OROS methylphenidate | 6 weeks | Adult ADHD Investigator System Symptom Report Scale (AISRS) | None | Data reported narratively | |
| RCTs with cross‐over design | |||||
| Placebo | 5 weeks | None |
| None. Only incomplete data (end means with P value and range) | |
| Placebo | 8 weeks (4 treatment phases of 2 weeks each) |
| None | None. Only incomplete data (t‐test values) | |
| Lithium | 18 weeks with a 2‐week washout period in between treatment periods | Hyperactivity, Impulsivity, Learning Problems, Conduct Disorder, Restlessness, and Antisocial Behavior subscales of the Conners’ Adult ADHD Rating Scale | None | End scores | |
| Placebo | 7 weeks, with 1 week of washout in between treatments periods | None | ADHD Rating Scale ‐ IV | End scores of the first period of treatment | |
|
| 17 weeks (2 weeks in each treatment separated by 1 washout week) |
|
| None. Only incomplete data (initial scores) | |
| ADHD: Attention deficit hyperactivity disorder aMeasurements reported by the individual’s significant other, not the trial investigator. | |||||
| Study | Reporting of harms outcomes |
|---|---|
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
| ADHD: attention deficit hyperactivity disorder | |
| Study | Comparator | Harms assessed? | Measurement method | Time points | What results were reported? |
|---|---|---|---|---|---|
| RCTs with parallel design | |||||
|
| Yes | Not reported | At each return visit |
| |
| Placebo | Yes | Not reported | Not reported | No data reported, only a general statement included in the paper | |
| Placebo | Yes |
| Weekly |
| |
| Placebo | Yes |
| At each visit |
| |
| OROS methylphenidate | Yes |
|
|
| |
| RCTs with cross‐over design | |||||
| Placebo | Yes | Not reported | Not reported |
| |
| Placebo | Yes | Not reported | Not reported | No data reported, only a general statement included in the paper | |
| Lithium | Yes | Not reported | Not reported |
| |
| Placebo | Yes | Modified Side Effects Rating Scale from Barkley 1998 | Not reported |
| |
|
| The trial did not access outcomes of harms | N/A | N/A | N/A | |
| N/A: not applicable | |||||
| Study | Comparator | Secondary outcomes | Measurement methods | What results were reported? | |
|---|---|---|---|---|---|
| RCTs with parallel design | |||||
|
|
|
|
| ||
| Placebo |
|
| Change scores, end scores and baseline scores: CGI‐I and CGI‐Sa | ||
| Placebo | No information | None | None | ||
| Placebo |
|
| Narrative report with general statements: GAF | ||
| OROS methylphenidate |
|
| No data reported | ||
| RCTs with cross‐over design | |||||
| Placebo |
|
| Data reported narratively with general statements for both outcomes | ||
| Placebo |
|
| Data reported incompletely for all outcomes | ||
| Lithium |
|
| Data on end scores for both outcomes | ||
| Placebo |
|
| Data on end scores for CGI‐S | ||
|
|
|
| Data on initial scores for both outcomes | ||
| ADHD: attention deficit hyperactivity disorder aFull dataset shared by the study authors. | |||||
| Study | Judgment | Rationale |
|---|---|---|
| RCTs with parallel design | ||
| Notable concerns |
| |
| No concerns |
| |
| No concerns |
| |
| Notable concerns |
| |
| Notable concerns |
| |
| RCTs with cross‐over design | ||
| No concerns |
| |
| Unclear |
| |
| Unclear |
| |
| No concerns |
| |
| Notable concerns |
| |
| Participants treated with IR methylphenidate | Participants treated with placebo | ||||||
|---|---|---|---|---|---|---|---|
| Organ system category | Event | n | % | Organ system category | Event | n | % |
| Cardiovascular disorders | Tachycardia | 4 | 0.9 | Cardiovascular disorders | Tachycardia | 1 | 0.4 |
| Elevated blood pressure | 1 | 0.2 | Chest pain | 2 | 0.9 | ||
| Palpitations | 1 | 0.5 | |||||
| Gastrointestinal disorders | Dry mouth | 47 | 10.7 | Gastrointestinal disorders | Abdominal complaints | 2 | 0.8 |
| Abdominal complaints | 6 | 1.4 | Constipation | 1 | 0.5 | ||
| Constipation | 2 | 0.5 | Diarrhea | 2 | 0.9 | ||
| Diarrhea | 5 | 1.1 | Gastrointestinal problem | 3 | 1.4 | ||
| Gastrointestinal problem | 7 | 1.6 | Nausea | 3 | 1.4 | ||
| Nausea | 15 | 3.4 | Nausea or upset stomach | 5 | 2.3 | ||
| Nausea or upset stomach | 8 | 1.8 | Stomach aches | 6 | 2.8 | ||
| Metabolism and nutrition disorders | Decreased in appetite/Loss of appetite | 62 | 14.2 | Metabolism and nutrition disorders | Decreased in appetite/Loss of appetite | 17 | 8.0 |
| Nervous system disorders | Dizziness | 9 | 2.1 | Nervous system disorders | Dizziness | 4 | 1.9 |
| Headache | 64 | 14.6 | Headache | 36 | 16.9 | ||
| Psychiatric disorders | Anxious | 19 | 4.3 | Psychiatric disorders | Anxious | 15 | 7.0 |
| Disorientation, insomnia, and anxiety lasting several hours | 1 | 0.2 | Euphoric, unusually happy | 7 | 3.3 | ||
| Euphoric, unusually happy | 10 | 2.3 | Insomnia or trouble sleeping | 8 | 3.8 | ||
| Irritability | 14 | 3.2 | Irritability | 13 | 6.1 | ||
| Moody | 31 | 7.1 | Moody | 2 | 0.9 | ||
| Sadness | 15 | 3.4 | Sadness | 9 | 4.2 | ||
| Stare a lot or daydream | 12 | 2.7 | Stare a lot or daydream | 17 | 8.0 | ||
| Talk less with others | 11 | 2.5 | Talk less with others | 12 | 5.6 | ||
| Tics or nervous movements | 4 | 0.9 | Tics or nervous movements | 5 | 2.3 | ||
| Tics | 3 | 0.7 | Tics | 1 | 0.5 | ||
| Sleeping problems (difficulty/trouble sleeping, insomnia, nightmares) | 71 | 16.2 | Sleeping problems (difficulty/trouble sleeping, insomnia, nightmares) | 28 | 13.2 | ||
| Nonspecific symptoms | Drowsiness | 6 | 1.4 | Nonspecific symptoms | Drowsiness | 10 | 4.7 |
| Total events | 438 | 100 | Total events | 213 | 100 | ||
| IR: Immediate release | |||||||
| Study | Electrocardiogram | Heart rate | Diastolic blood pressure | Systolic blood pressure | Blood pressure |
|---|---|---|---|---|---|
| ‐ | ‐ | "No significant increase of diastolic blood pressure between baseline and methylphenidate" | Increased in participants receiving IR methylphenidate (mean = 123 mmHg) compared with placebo (mean 128 = mmHg) (P < 0.05) | ‐ | |
| "Mean heart rate was 4.8 beats/min higher (p=0.002) after IR‐MPH" | "The diastolic blood pressure remained virtually unchanged" | "The systolic blood pressure was 0.13 mmHg higher after methylphenidate but this was not statistically significant (p=0.954)" | |||
| ‐ | ‐ | ‐ | ‐ | 1 participant receiving IR methylphenidate with elevated blood pressure, worst occurrence reported only | |
|
| "Small but statistically significant increases in pulse (83 ± 13 vs. 76 ± 13 bpm, t=4.4, df(77), p<.001" | No increase in participants receiving IR methylphenidate (mean = 78 mmHg, SD = 9 mmHg) in comparison with placebo (mean = 76 mmHg, SD = 9 mmHg) | No increase in participants receiving IR methylphenidate (mean = 128 mmHg, SD = 12 mmHg) in comparison with placebo (mean = 126 mmHg, SD = 14 mmHg) | ‐ | |
| bpm: beats of the heart per minute | |||||
| Participants treated with IR methylphenidate | Participants treated with OROS methylphenidate | ||||||
|---|---|---|---|---|---|---|---|
| Organ system category | Event | n | % | Organ system category | Event | n | % |
| Cardiovascular disorders | Diastolic blood pressure > 90 mm | 1 | 14 | Cardiovascular disorders | Diastolic blood pressure > 90 mm | 1 | 5 |
| Pulse > 90 bpm | 4 | 57 | Pulse > 90 bpm | 14 | 67 | ||
| Systolic blood pressure > 140 mm Hg | 2 | 29 | Systolic blood pressure >140 mm Hg | 6 | 29 | ||
| Total events | 7 | 100 | Total events | 21 | 100 | ||
| bpm: beats per minute | |||||||
| Participants treated with IR methylphenidate | Participants treated with lithium | ||||||
|---|---|---|---|---|---|---|---|
| Organ system category | Event | n | % | Organ system category | Event | n | % |
| Cardiovascular disorders | Orthostatic hypotension | 1 | 20 | Cardiovascular disorders | Chest discomfort | 1 | 8 |
| Gastrointestinal disorders | Diarrhea | 3 | 60 | Gastrointestinal disorders | Diarrhea | 3 | 28 |
| Nausea | 1 | 10 | |||||
| Nervous system disorders | Headache | 1 | 20 | Nervous system disorders | Headache | 5 | 54 |
| Total events | 5 | 100 | Total events | 9 | 100 | ||
| IR: immediate release | |||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1.1 Efficacy: investigator‐rated (end scores) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 1.2 Efficacy: participant‐rated (change scores) Show forest plot | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.3 Efficacy: participant‐rated (end scores) Show forest plot | 2 | 138 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.59 [‐1.25, 0.06] |
| 1.3.1 RCT with parallel design | 1 | 48 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.97 [‐1.57, ‐0.37] |
| 1.3.2 RCT with cross‐over design | 1 | 90 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐0.71, 0.12] |
| 1.4 Harms: patients experiencing at least one adverse event Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.5 Clinical impression: severity (change scores) Show forest plot | 2 | 139 | Mean Difference (IV, Random, 95% CI) | ‐0.57 [‐0.85, ‐0.28] |
| 1.5.1 RCTs with parallel design | 1 | 49 | Mean Difference (IV, Random, 95% CI) | ‐0.60 [‐0.92, ‐0.28] |
| 1.5.2 RCTs with cross‐over design | 1 | 90 | Mean Difference (IV, Random, 95% CI) | ‐0.44 [‐1.05, 0.17] |
| 1.6 Clinical impression: improvement (end scores) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 1.7 Anxiety: investigator‐rated (change scores) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 1.8 Depression: investigator‐rated (change scores) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 2.1 Efficacy: investigator‐rated (end scores) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2.2 Anxiety: investigator‐rated (end scores) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2.3 Depression: investigator‐rated (end scores) Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |