Estimulación nerviosa eléctrica transcutánea (ENET) para el control del dolor en la enfermedad de células falciformes
Resumen
Antecedentes
La enfermedad de células falciformes (ECF), uno de los trastornos hereditarios más comunes, se asocia con episodios de dolor vaso‐oclusivo y hemólisis que provocan morbilidad recurrente, ingresos hospitalarios y ausentismo laboral o escolar. Las crisis se tratan convencionalmente con opioides, no opioides y otros coadyuvantes con el riesgo de desarrollar complicaciones, adicciones y comportamiento de búsqueda de drogas. Se han utilizado diferentes tratamientos no farmacológicos, como la estimulación nerviosa eléctrica transcutánea (ENET) para controlar el dolor en otras afecciones dolorosas. Por lo tanto, debe examinarse la eficacia de la ENET para el tratamiento del dolor en la ECF.
Objetivos
Evaluar los efectos beneficiosos y perjudiciales de la ENET para el tratamiento del dolor en pacientes con ECF que experimentan crisis de dolor o dolor crónico (o ambos).
Métodos de búsqueda
Se realizaron búsquedas en el Registro de Ensayos de Hemoglobinopatías del Grupo Cochrane de Fibrosis Quística y Enfermedades Genéticas (Cochrane Cystic Fibrosis and Genetic Disorders Group Haemoglobinopathies Register), que incluyeron listas de referencias identificadas de búsquedas exhaustivas en bases de datos electrónicas o búsquedas manuales de revistas pertinentes y libros de resúmenes de actas de congresos. También se realizaron búsquedas en registros de ensayos en línea y en las listas de referencias de artículos y revisiones relevantes.
Fecha de la última búsqueda: 26 de febrero de 2020.
Criterios de selección
Se incluyeron ensayos controlados aleatorizados (ECA) y cuasialeatorizados, en los que se evaluó la ENET para el tratamiento del dolor en pacientes con ECF.
Obtención y análisis de los datos
Dos autores de la revisión evaluaron de forma independiente la elegibilidad de los ensayos identificados por las búsquedas en la literatura de acuerdo a los criterios de inclusión. Dos autores de la revisión de forma independiente extrajeron posteriormente los datos, evaluaron el riesgo de sesgo con la herramienta Cochrane estándar y calificaron la calidad de la evidencia con los criterios GRADE.
Resultados principales
Un ECA cruzado doble ciego con 22 participantes con ECF (de 12 a 27 años de edad) fue elegible para su inclusión. Después de la estratificación en cuatro grados de severidad de las crisis de dolor, los participantes fueron asignados al azar para recibir ENET o placebo (ENET falsa). El ensayo finalizó después de 60 episodios de tratamiento (30 episodios de tratamiento de cada grupo de tratamiento).
Hay una falta de claridad en cuanto al diseño del ensayo y el análisis de los datos cruzados. Cuando un participante había sido asignado al tratamiento con ENET debido a un episodio de dolor y posteriormente regresaba con un nuevo episodio de dolor de un grado similar, entonces recibiría el tratamiento con ENET falsa (diseño cruzado). En el caso de los que experimentaban un episodio de dolor de diferente gravedad, no está claro si fueron aleatorizados nuevamente o se les administró el tratamiento alternativo. El informe y el análisis se basaron en el número total de eventos de dolor y no en el número de participantes. No está claro en cuántos participantes se realizó el cruzamiento del grupo de ENET al grupo de ENET falsa y viceversa. El ensayo presentaba un riesgo alto de sesgo en relación con la generación de la secuencia aleatoria y la ocultación de la asignación; un riesgo poco claro en relación con el cegamiento de los participantes y el personal; y un riesgo bajo en relación con el cegamiento de los evaluadores de resultados y el informe de resultados selectivo.
El ensayo fue pequeño y de calidad muy baja; además, debido a la cuestión del diseño del ensayo no fue posible analizar los datos de forma cuantitativa. Por lo tanto, se presentó solo un resumen narrativo y se aconseja precaución en la interpretación de los resultados. En relación con los resultados primarios predefinidos, el ensayo incluido no informó sobre el alivio del dolor entre las dos y cuatro semanas después de la intervención. Los autores del ensayo informaron de que no se encontraron diferencias en los cambios en las calificaciones del dolor (registrados a una hora y cuatro horas después de la intervención) entre los grupos de ENET y de placebo. En relación con los resultados secundarios, el uso de analgésicos durante el ensayo tampoco mostró diferencias entre los grupos. Debido a la calidad de la evidencia, no existe seguridad en cuanto a si la ENET mejora la satisfacción general en comparación con la ENET falsa. No se evaluó la capacidad de hacer frente a las actividades de la vida diaria. En cuanto a los eventos adversos, aunque se informó de un caso de picazón en el grupo de ENET, el sitio y la naturaleza de la picazón no se indicaron de forma clara; por lo tanto, no puede atribuirse claramente a la ENET. Además, dos participantes que recibieron ENET «falsa» informaron de un empeoramiento del dolor con la intervención.
Conclusiones de los autores
Debido a que solo se incluyó un ensayo pequeño y de calidad muy baja, con un riesgo alto de sesgo en varios dominios, no es posible establecer la conclusión de si la ENET es perjudicial o beneficiosa para el tratamiento del dolor en pacientes con ECF. Se necesita un ECA bien diseñado y con el poder estadístico adecuado para evaluar la función de la ENET en el tratamiento del dolor en pacientes con ECF.
PICO
Resumen en términos sencillos
Estimulación nerviosa eléctrica transcutánea (ENET) para controlar el dolor en pacientes con enfermedad de células falciformes
Pregunta de la revisión
¿Cuál es la función de la estimulación nerviosa eléctrica transcutánea (ENET) en el tratamiento del dolor en pacientes con enfermedad de células falciformes (ECF)? ¿Cuáles son los efectos adversos de la ENET en pacientes con ECF?
Antecedentes.
La ECF es un trastorno hereditario que afecta los glóbulos rojos. Estas células contienen hemoglobina que transporta el oxígeno. Los glóbulos rojos enfermos bloquean los vasos sanguíneos y provocan una disminución del suministro de sangre, lo que hace que llegue menos oxígeno a los órganos afectados y provoca episodios de dolor y daños en los órganos. El dolor severo requiere medicación e incluso hospitalización. Los analgésicos prescritos pueden tener varios efectos secundarios, incluida la dependencia de fármacos. Por lo tanto, los investigadores están buscando otras opciones además de los tratamientos farmacológicos para los pacientes con ECF.
La ENET es un dispositivo electromédico pequeño a pilas que produce corriente de bajo voltaje y se utiliza para el alivio del dolor en varias afecciones dolorosas. Es segura, económica y fácil de usar. Debido a que se han observado respuestas contradictorias en diferentes afecciones dolorosas, surgió la necesidad de producir una revisión Cochrane con una búsqueda exhaustiva, para determinar el efecto de la ENET para el tratamiento del dolor en pacientes con ECF.
Fecha de la búsqueda
La evidencia está actualizada hasta el 26 de febrero 2020.
Características de los estudios
Se buscaron ensayos bien diseñados para observar el efecto de la ENET en comparación con la ENET «falsa» en pacientes con ECF para aliviar el dolor, reducir la intensidad del dolor, reducir la frecuencia de los episodios de dolor, lograr una diferencia en el uso de analgésicos, mejorar la calidad de vida y evaluar cualquier efecto adverso.
Solo se encontró un ensayo (22 participantes de entre 12 y 27 años de edad). Los participantes fueron clasificados en cuatro grupos según la gravedad del dolor. En la primera visita, los participantes de los diferentes grupos fueron elegidos al azar para recibir ENET o un tratamiento con ENET «falsa». Para una nueva crisis de la misma gravedad, los participantes recibieron la intervención alternativa a la primera. En el caso de los que experimentaron un episodio de dolor de diferente gravedad, no está claro qué tratamiento se les administró.
Ni el participante ni el investigador sabían qué tratamiento se administraba.
Resultados clave
Se analizaron 30 episodios (en 22 participantes) de tratamiento con ENET y 30 de tratamiento con ENET «falsa». Debido a la calidad baja de los datos y a los problemas con el diseño del ensayo, solo fue posible realizar un informe descriptivo sin ningún análisis formal. Se debe tener cuidado al interpretar estos resultados. En el ensayo incluido no se encontró ninguna diferencia en la calificación del dolor en una escala del 1 al 10 al final de una hora y cuatro horas entre los grupos de tratamiento con ENET y con ENET «falsa». No hubo diferencias entre los grupos en cuanto a la cantidad de analgésicos utilizados. Debido a la calidad muy baja de la evidencia, tampoco existe seguridad en cuanto a si la ENET mejora la satisfacción general en comparación con la ENET «falsa». Un único paciente que recibió ENET informó de un efecto adverso menor de picazón, mientras que dos pacientes que recibieron ENET «falsa» informaron de un empeoramiento del dolor con la intervención. Debido a que solo se incluyó un ensayo con evidencia de calidad muy baja, no es posible afirmar si la ENET logra alguna diferencia en el tratamiento del dolor en pacientes con ECF.
Calidad de la evidencia
La publicación del ensayo no informó con claridad cómo se generó la lista aleatorizada para la asignación de los participantes a los dos grupos de tratamiento, por lo que se evaluó como en riesgo alto de sesgo. El informe y el análisis se basaron únicamente en el número total de eventos de dolor y no en el número de pacientes que informaron de los mismos. No está claro a partir de este informe en cuántos participantes se realizó el cruzamiento del grupo de tratamiento con ENET al grupo de tratamiento con ENET «falsa». El primer tratamiento puede tener un efecto en el tratamiento subsiguiente y no hay datos claros sobre el proceso de cruzamiento de un grupo de tratamiento a otro. Por lo tanto, se establece la conclusión de que el ensayo presenta un riesgo alto de sesgo y los resultados son difíciles de interpretar.
Authors' conclusions
Summary of findings
| TENS compared with placebo (sham TENS) for SCD | ||||||
| Patient or population: children and young adults with SCD Settings: outpatients Intervention: TENS Comparison: sham TENS | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Sham TENS (placebo) | TENS | |||||
| Pain relief (percentage relief compared to baseline) Follow‐up: 2 to 4 weeks post intervention | This outcome was not measured. | |||||
| Pain intensity (visual analogue score from 0 to 10 where 10 is the most severe pain) Follow‐up: up to 4 hours post intervention | No significant difference in pain intensity was seen between the TENS group and sham TENS group after 1 hour (P = 0.30) or after 4 hours (P = 0.69). | 22 | ⊕⊝⊝⊝ | The unit of analysis for this outcome was pain crises so 1 participant may have had more than 1 episode. The total number of episodes included in the analysis was 25 in the TENS group and 30 in the placebo group. | ||
| Frequency of pain episodes | This outcome was not measured. | Each episode of pain was treated independently as a new episode. Total number of episodes was not reported. | ||||
| Change in consumption of analgesics (number of participants requiring analgesia) Follow‐up: up to 4 hours post intervention | There was no significant difference between the TENS group and the placebo group in the number of people requiring analgesia up to one hour post treatment (TENS 14 %; sham TENS 25 %, X² = 1.07, P = 0.30). There was no significant difference between the TENS group and the placebo group in the number of people requiring analgesia up to four hours post treatment (TENS 61 %; sham TENS 66 %, X² = 0.16, P = 0.69). | 22 | ⊕⊝⊝⊝ | The unit of analysis was pain episodes rather than participants. | ||
| Change in QoL | This outcome was not measured. | Participants were asked to rate whether the treatment was helpful or harmful but this related directly to the pain. | ||||
| Change in ability to cope with ADL | This outcome was not measured. | |||||
| Adverse effects Follow‐up: up to 1 hour and 4 hours post treatment. | There was one case of itching reported in the TENS group but it was unclear whether it was related to the treatment. 2 participants in the placebo group felt that the pain got worse with treatment. | 22 | ⊕⊝⊝⊝ | No data were provided for this outcome. Results were presented narratively. | ||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| a. Downgraded twice because of high risk of bias within the included trial. The randomisation process was unclear and when participants entered the trial for a second or third episode, they were allocated to the alternative treatment from their original allocation. Although the trial authors described their method of blinding participants to the intervention, it is unclear how successful this was given that the TENS would give a tingling sensation and the sham TENS would not. There were also issues around selective reporting of outcomes and a lack of clarity around allocation of each pain episode to a treatment arm. b. Downgraded once due to imprecision from a low number of participants and events. | ||||||
Background
Description of the condition
Please refer to the glossary for the explanation of clinical terms (Appendix 1).
Sickle cell disease (SCD) encompasses a host of genetically inherited disorders in which red blood cells become increasingly deformed and friable, causing vaso‐occlusion and haemolysis. This disease is one of the most common, severe, single gene mutation (monogenic) disorders (Weatherall 2001).
The disease is most prevalent in sub‐Saharan Africa, affecting an estimated 230,000 children born regionally every year ‐ accounting for 80% of the total global incidence (David 2010). In the USA, it affects around 100,000 people, predominantly those of African descent. The disease occurs in about one in every 500 African‐American births and one in every 1000 to 1400 Hispanic‐American births. Approximately two million Americans, or one in 12 African Americans, carry the sickle cell allele (WHO 2015). This geographical predominance corresponds to an adaptive advantage: heterozygous carriers (sickle cell trait) are naturally resistant to infection by the endemic Plasmodium falciparum (P falciparum) malarial parasite (Barbedo 1974).
The vaso‐occlusive crisis (VOC) is the hallmark of the disease and is often unpredictable, varies in intensity, duration, location and severity, and can be precipitated by known and unknown factors (Ballas 2005). Vaso‐occlusive pain episodes are the most common cause of recurrent morbidity, hospital admissions and work or school absenteeism in people with SCD (Platt 1991). Approximately 90% of hospital admissions of people with SCD are for treating acute pain (Brozovic 1987). Hypoxia, dehydration, acidosis, cold exposure and strenuous exercise also can lead to sickling of red blood cells leading to an acute painful episode.
Nociceptive sickle cell pain may be acute recurrent painful crises, chronic pain syndromes and neuropathic pain. The acute painful crisis evolves through prodromal, initial, established and resolving phases (Ballas 2012). Chronic sickle cell pain may be due to avascular necrosis and leg ulcers or intractable pain without any obvious signs. Chronic pain is usually associated with emotional distress, behavioural dysfunction, family stress, financial concern, frequent visits to healthcare providers, heavy use of analgesic medications and fear (Ballas 2005).
Treatment aims for SCD are to relieve pain, prevent infections and manage complications (Stinson 2003). Despite being the main source of discomfort for people with SCD, therapies for pain crises are not definitive. Pharmacotherapies involve opioids, non‐opioids and adjuvants. Non‐opioids (e.g. acetaminophen, nonsteroidal anti‐inflammatory drugs (NSAIDs) tramadol and corticosteroids) have a 'ceiling effect' beyond which they are no longer effective, and NSAIDs and corticosteroids have well‐known complications such as haemostasis, acute renal failure, congestive heart failure when overused (Niscola 2009). Thus opioids are frequently employed, as they have fewer systemic effects. However, their use is plagued with reports of addiction, tolerance and drug‐seeking behaviour (Neville 2011). Moreover, opioids may contribute to the development of acute chest syndrome during an acute sickle cell pain crisis (Buchanan 2005).
These factors have motivated clinicians and researchers worldwide to embrace a multidisciplinary approach towards pain management in people with SCD, with a focus on including non‐pharmacological interventions. According to the recommendations of the American Pain Society, pharmacological treatments for SCD should be complimented by psychological, behavioral, and physical modalities (American Pain Society Guidelines 1999).
Description of the intervention
As defined by the American Physical Therapy Association, TENS is the application of electrical stimulation to the skin for pain control. It is non‐invasive, inexpensive, safe, and easy to use; a small battery‐powered device applies an electric current via two or more non‐invasive skin electrodes to stimulate underlying nerves and thus reduce pain perception. It can be applied with different frequencies, varying from low (< 10 Hz) to high (> 50 Hz). Intensity can also vary with low‐intensity stimulation producing a sensation alone, while high‐intensity stimulation triggers muscle contraction, and hence movement. Low‐frequency TENS is usually given at high‐intensity (producing motor contraction and sensation), while high‐frequency TENS is given at lower intensities (producing both sensation and muscle contraction) (DeSantana 2008). Conventional TENS has a high‐stimulation frequency (40 Hz to 150 Hz) and low intensity between 10 mA to 30 mA. The pulse duration is short (up to 50 microseconds). The onset of analgesia with this setup is virtually immediate. Pain relief lasts while the stimulus is turned on, but it usually abates when the stimulation stops. In acupuncture like settings, the TENS unit delivers low frequency stimulus trains at 1 Hz to 10 Hz, at a high stimulus intensity, close to the tolerance limit of the individual. This method is uncomfortable and is often considered for those who do not respond to conventional TENS. Pulsed (burst) TENS uses low‐intensity stimuli firing in high‐frequency bursts, but does not have any added advantage over the conventional method.
Over the last 40 years, TENS has been evaluated for the management of pain in numerous conditions, including fibromyalgia (Sunshine 1996) rheumatoid arthritis, osteoarthritis, post‐caesarean pain, lower back pain (Milne 2004) neck pain and numerous other causes. While reviews report that evidence on the efficacy of TENS is inconclusive, magnetic resonance imaging (MRI) studies implicate central pain signal modulation indicating a central action (Kocyigit 2012). There are contraindications for the use of TENS. These include use during pregnancy, as it may induce premature labour, as well as application over the carotid sinuses, due to the risk of acute hypotension through a vasovagal reflex. In addition to these, it should not be placed over the anterior neck, because laryngospasm due to laryngeal muscle contraction may occur, the electrodes should not be placed in an area of sensory impairment, where the possibility of burns exists and a TENS unit should be used cautiously in individuals with a spinal cord stimulator or an intrathecal pump.
How the intervention might work
A variety of mechanisms for the analgesic action of TENS have been suggested, including presynaptic inhibition in the dorsal horn of the spinal cord, endogenous pain control via endorphins, and direct inhibition of nerve excitation and restoration of afferent input (Kaye 2015). These produce a host of responses, including sensation, movement (muscle contraction), and pain relief. In people with SCD, vaso‐occlusion leads to secondary tissue injury which generates several major pain mediators like interleukin‐1, bradykinin which in turn sensitise peripheral nerve endings and facilitate the transmission of painful stimuli along A‐δ and C fibres that reach the cerebral cortex via the spinal cord and the thalamus (Ballas 2005). This partly corresponds to the 'gate control theory' (Melzack 1965) for the mechanism of analgesia produced by TENS where the 'open gate' between C fibres and T cells which allow pain transmission centrally is closed by the electrical stimulation to the skin provided by the TENS instrument.
Why it is important to do this review
Over recent years, although considerable knowledge has been gained on the pathophysiological mechanisms of pain and the pharmacogenetics of analgesics (including opioids) there has been not much progress in the clinical management of sickle cell pain. Many adults with SCD still face accusations, assumptions and disbelief about their painful condition which is often wrongly perceived by some healthcare providers as drug‐seeking behaviour (Ballas 2014). Mismanagement of pain in SCD may lead to serious psychosocial and physiological consequences, such as depression, low self esteem, anxiety and reduced participation in social activities, resulting in chronic pain interspersed with episodes of acute exacerbations (Smith 2005). Hence, organisations such as the American Pain Society recommended that pharmacologic treatment for SCD should be complemented by complementary and alternative medicine.
Cochrane Reviews on TENS for specific chronic pain conditions such as rheumatoid arthritis, osteoarthritis, have suggested that TENS is more effective than placebo (sham) TENS, although methodological weaknesses in randomised controlled trials (RCTs) have sometimes hindered definitive conclusions (Bennett 2011; Brosseau 2003; Johnson 2010; Rutjes 2009). Studies carried out on the effect of TENS on neck pain suggested that active TENS may be more effective than placebo TENS (Kroeling 2013). A systematic review on the effect of TENS on cancer pain in adults suggested bone pain on movement may improve in a cancer population on application of TENS, but most of the results remained inconclusive due to a limited number of RCTs included in the review (Hurlow 2012).
Most reviews of TENS therapy are inconclusive at present, which is why a standardised, rigorous search and a Cochrane Review are required in this disease area. This issue is further compounded by the practical need to manage the acute pain crises, as well as chronic pain conditions, experienced by millions of people with SCD worldwide. Current pharmacological therapies produce too many unacceptable side effects, and as already stated, recommendations encourage the use of non‐pharmacological methods in SCD pain management. A Cochrane Review of the current evidence regarding the effectiveness of TENS as a complementary therapy for managing pain in people with SCD will allow health professionals and researchers make informed decisions about the use of this treatment.
Objectives
To assess the benefits and harms of TENS for managing pain in people with SCD who experience pain crises or chronic pain (or both).
Methods
Criteria for considering studies for this review
Types of studies
We planned to include RCTs and quasi‐RCTs (if sufficient evidence was provided to demonstrate that the treatment and control groups are similar at baseline).
In reference to cross‐over trials, as we were currently unaware of the long‐term effects of these interventions, we were unable to determine whether the effects of the first intervention will interfere with those of the second. In order to avoid introducing this bias into the analysis, we planned to include only first‐arm data from cross‐over trials, when available.
Types of participants
People with known SCD (SS, SC, Sβ⁺ and Sβ⁰, proven by electrophoresis and sickle solubility test, with family studies or DNA tests as appropriate) of all ages and both sexes, in any setting.
Types of interventions
We included all standard modes of TENS, biphasic or monophasic electrical current delivered in pulses in high intensity or high frequency, low intensity or low frequency, or other standard modes that produce perceptible sensation at the area of application. We excluded other modes of electrotherapy, e.g. TENS delivered in barely perceptible intensity.
Eligible comparisons are:
-
TENS with conventional treatment (e.g. analgesics) versus conventional treatment alone;
-
TENS versus placebo (sham TENS);
-
TENS versus other non‐pharmacological modalities for managing pain.
We did not plan to compare different intensities and frequencies of TENS.
Placebo (sham TENS) devices may look exactly the same as active TENS devices, but are deactivated and produce no current or may produce a brief period of stimulation at the beginning which fades out later. Due to the lack of perceptible stimulation in sham TENS, the blinding of participants to the mode of treatment is almost impossible (Sluka 2013) and this represents a risk of bias to all sham‐controlled trials of TENS (Gibson 2015).
Types of outcome measures
Primary outcomes
-
Pain relief *
-
Pain intensity (as assessed by visual analogue pain scale score (VAS) or other validated assessment tools for measuring pain intensity during the use of TENS)
-
Frequency of pain episodes
* We aimed to present data at two to four weeks and four weeks post intervention. For long‐term usage (e.g. for a period of one month or more) we planned to consider the outcome measures up to three months. We also considered pain relief as moderate (at least 30% pain relief over baseline) or substantial (at least 50% pain relief over baseline) as defined by IMMPACT guidelines (Dworkin 2008).
Secondary outcomes
-
Changes in consumption of analgesic and opioids during pain episodes
-
Changes in quality of life (QoL) (as measured by a validated scale)
-
Ability to cope with the activities of daily living (ADL)
-
Adverse effects of the intervention
Search methods for identification of studies
We searched for all relevant published and unpublished trials without restrictions on language, year or publication status.
Electronic searches
The Cochrane Cystic Fibrosis and Genetic Disorders Group's Information Specialist conducted a search of the Group's Cystic Fibrosis Trials Register for relevant trials using the following terms: (sickle cell OR (haemoglobinopathies AND general)) AND TENS).
The Haemoglobinopathies Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of the Cochrane Library) and weekly searches of MEDLINE. Unpublished work is identified by searching the abstract books of five major conferences: the European Haematology Association conference; the American Society of Hematology conference; the British Society for Haematology Annual Scientific Meeting; the Caribbean Health Research Council Meetings; and the National Sickle Cell Disease Program Annual Meeting. For full details of all searching activities for the register, please see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group's website.
Date of the most recent search of the Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register: 26 February 2020.
We searched the following databases (Appendix 2):
-
PubMed (www.ncbi.nlm.nih.gov/pubmed, 1946 to 04 April 2017);
-
Embase Ovid (1974 to 16 October 2017).
In addition, we also searched the following trial registries and other resources (Appendix 2):
-
US National Institutes of Health Ongoing Trials Register, ClinicalTrials.gov (clinicaltrials.gov/; searched on 04 September 2017);
-
International Standard Randomised Controlled Trials Number (www.isrctn.com/; searched on 04 September 2017);
-
World Health Organisation International Clinical Trials Registry Platform (apps.who.int/trialsearch/; searched on 04 September 2017);
-
Google Scholar (scholar.google.co.uk/; searched on 04 September 2017).
Regarding our use of Google Scholar, we performed a Google Scholar Advanced search, sorted the results based on relevance, and screened the first 100 search results for relevant trials. For details of our search strategies, please refer to the appendices (Appendix 2).
Searching other resources
Reference lists
We checked the bibliography of the included trial for further references to relevant trials. We also searched research papers which cannot be directly included in this review (observational studies, systematic and narrative reviews, conference reports, etc.) for the citations of relevant trials which may have been eligible for inclusion. These papers were then sought and assessed for possible inclusion in the review.
Authors and organisations
We contacted the trial authors who have conducted prominent research in the relevant field regarding their ongoing trials or other relevant papers which may be eligible for inclusion. We contacted the following organisations for further information:
-
NCCIH formerly known as NCCAM;
-
Foundation for Alternative and Integrative Medicine;
-
American Alternative Medical Association;
-
The Complementary and Natural Healthcare Council;
-
International Alternative Medical Association;
-
The Association for Applied Psychophysiology and Biofeedback (formerly the Biofeedback Society of America).
Grey literature
Regarding grey literature, research has shown that published studies often overestimate outcomes, compared to grey literature articles (Hopewell 2007). Moreover, exclusion of grey literature can result in systematic error, thus seriously threatening the validity of a systematic review (McAuley 2000). Therefore, to avoid this risk of publication bias, we searched grey literature databases in our attempt to identify relevant trials and authors from conference proceedings. We searched the following databases (recommended in the Cochrane Handbook For Systematic Reviews of Interventions) for unpublished reports or articles which may be appropriate for inclusion in this systematic review (Higgins 2011a):
-
OpenGrey (www.opengrey.eu/: searched on 04 September 2017);
-
PsychEXTRA (www.apa.org/psycextra/; searched on 04 September 2017);
-
the Grey Literature Report (http://greylit.org/; searched on 04 September 2017);
-
the Agency for Healthcare Research and Quality (www.ahrq.gov/; searched on 04 September 2017);
-
MedNar (http://mednar.com/mednar/desktop/en/search.html; searched on 04 September 2017).
For details of our search strategies, please refer to the appendices (Appendix 2).
Data collection and analysis
Selection of studies
Two review authors (SP, MG) independently assessed the eligibility of the trials identified by the literature searches by completing a trial selection form that was designed in accordance with the inclusion criteria. Each author independently evaluated each title for inclusion. If we did not find the relevant information in the abstract, we tried to retrieve the relevant full text, report(s). Both authors approved the inclusion of the trial in the review. In the case of any discrepancy related to eligibility, a third author (SKB) would have arbitrated, however, no such discrepancy occurred. We tabulated the excluded trials under 'Characteristics of excluded studies' and gave reasons for the exclusion. The review authors were not blinded to the identity of the trial authors, institutions or trial results during their assessments. We also produced a PRISMA chart to illustrate the trial selection process (Figure 1).

Study flow diagram.
Data extraction and management
Two review authors (SP, SM) independently and simultaneously extracted data from the selected articles and this was verified by two other review authors with an aim to resolve any disagreements by discussion and consensus.
We extracted data using a standardised, pre‐tested data extraction form. The review authors designed the data extraction form together and pilot tested it using a sample RCT and a quasi‐RCT to ensure practical functionality.
We extracted the following information from the included trial.
1. Trial characteristics and source
-
Trial identifier (ID) ‐ created by the review author
-
Report ID ‐ created by the review author
-
Citation
-
Contact details
2. Methodology
-
Trial design
-
Trial time and duration
-
Setting
-
Randomisation
-
Allocation sequence concealment
-
Type of blinding used
-
Concerns about bias
-
Intention‐to‐treat analysis
3. Participants
-
Total number
-
Eligibility criteria
-
Age and gender of participants
4. Interventions (TENS and variants)
-
Total number of intervention groups
For each intervention and comparison group of interest
-
Electrode position
-
Professional discipline of the clinician delivering TENS
-
Frequency and intensity of the electrical current applied through the TENS device
-
Frequency of administration
-
Duration of administration
-
Co‐intervention(s)
5. Outcome measures
-
Outcome definition
-
Units of measurement (if relevant)
-
For scales, state upper and lower limits, and the interpretation of the scale
6. Results
-
Number of participants allocated to each intervention group
For each outcome of interest
-
Sample size
-
Missing participants
-
Summary data for each intervention group (mean and standard deviation (SD) for continuous data, 2 x 2 contingency table for dichotomous data, etc.)
We had obtained two reports of the same trial by the same authors. However, since the outcomes were the same in both, we considered the later report.
Assessment of risk of bias in included studies
Two review authors (RD, SM) independently assessed the risk of bias of the included trial by using the criteria outlined in chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b).
We assessed each trial according to the following six components.
-
Random sequence generation (selection bias)
-
Allocation concealment (selection bias)
-
Masking (blinding) of participants and personnel (performance bias), and masking of outcome assessor.
-
Incomplete outcome data (attrition bias) through withdrawals, dropouts and protocol deviations; and
-
Selective reporting bias
-
Other bias
We also assessed for any other sources of bias as reported in the Cochrane Handbook for Systematic Reviews of Interventions (bias related to the specific trial design, early or abrupt end of trials, fraudulent trials) (Higgins 2011b). For each of the mentioned components, we assigned judgements of either low, unclear or high risk of bias.
We recorded the results in the standard table provided in the Review Manager software (RevMan 2014), and summarised the findings in a 'Risk of bias' table or graph. We resolved all concerns or issues by discussion with a third review author (ALA). We also addressed the implications of the 'Risk of bias' assessment in the discussion section.
Measures of treatment effect
For future updates of this review, if we include more trials, we plan to analyse and present dichotomous data (e.g. improvement in pain rating, analgesic usage, and overall satisfaction with TENS) using the risk ratios (RR) with 95% confidence intervals (CIs). We will report adverse effects of the intervention with 99% CIs to avoid errors due to multiple statistical testing. We will analyse continuous data (e.g. pain scores) using the mean difference (MD) and 95% CIs. We will also calculate the standardised MD (SMD) if different scales of measurement have been used.
We will present count data (e.g. frequency of pain episodes) as the MD, by comparing the difference in the mean number of events in the intervention group as compared to the control group and present them as rate ratio with 95% CI in case of rare events.
If we identify trials with multiple treatment arms, we will only include those treatment arms whose parameters are minimally different from the other included trials.
In this current version of the review, since no meta‐analyses were possible, we have presented a narrative summary.
Unit of analysis issues
We planned to use individuals with SCD as the unit of analysis in this review.
Regarding cross‐over trials, as we are currently unaware of the long‐term effects of these interventions, we are unable to determine whether the effects of the first intervention will interfere with those of the second. In order to avoid introducing this bias into the analysis, we aimed only to include first‐arm data from cross‐over trials, when available. The included trial did not present first‐arm data and we were unable to analyse any data from this trial, also whilst reporting on 22 participants, data on 60 treatment episodes were presented (30 for each treatment group). We have therefore only reported on the included trial in a narrative way.
For future updates, we intend to report any included cluster‐randomised trials separately.
Dealing with missing data
We contacted the trial authors and requested access to the missing data regarding methods, participants, interventions and outcomes. The lead investigator on the included trial provided the original protocol (Wang 1988).
Assessment of heterogeneity
For future updates, if more trials are included in the review, in order to deal with clinical heterogeneity, we will pool trials that examine similar interventions. We will perform separate analyses for TENS compared to conventional treatment and TENS compared to placebo TENS or other non‐pharmacological treatment. We will use the Chi² test for assessing heterogeneity (significance level: P < 0.1). We will quantify the degree of heterogeneity using the I² statistic (Deeks 2008). The guidelines for interpretation of the I² values will be as follows.
-
0% to 40% indicates unimportant levels of heterogeneity
-
30% to 60% indicates moderate heterogeneity
-
50% to 90% indicates substantial heterogeneity
-
75% to 100% indicates considerable heterogeneity
We will also consider a visual inspection of the forest plot to identify whether CIs overlap.
Assessment of reporting biases
For the included trial, we compared the protocol provided by the author with the published report (Wang 1988). We also requested and obtained the raw data for comparison with the published data. However, the raw data did not provide us with either the baseline pain score or the SD. In the absence of any other eligible trial, we could not impute the SD, hence we were unable to conduct any paired analysis.
Although this version of the review does not include trials from the grey literature, we do anticipate that these may be included in future updates. Therefore, it is particularly important for us to conduct an assessment of publication bias. For future updates, if we identify at least 10 trials for inclusion in a meta‐analysis, we will explore potential publication bias by generating a funnel plot and performing Egger’s test to determine the degree of asymmetry (Egger 1997). If the included trials differ in sample size then we will visually inspect the funnel plots to explore the possibility of reporting biases. We will interpret the results of Egger’s test cautiously due to the presence of many limitations in quantifying possible reporting biases (Moore 2008). We will address the implications of the publication bias assessment on the review findings in the discussion section.
Data synthesis
We synthesised the quantitative data compiled using the data extraction form but did not undertake any meta‐analyses due to issues of the trial design and reporting. We will calculate the I² statistic to determine statistical heterogeneity, and thus determine whether we will use a fixed‐effect model (unimportant heterogeneity), or a random‐effects model (at least moderate heterogeneity) for the meta‐analysis (DerSimonian 1986). The results of the meta‐analysis will be reported as pooled effect estimates, stated with 95% CIs and illustrated graphically in a forest plot.
Subgroup analysis and investigation of heterogeneity
In future, if we identify substantial (or higher) statistical heterogeneity we plan the following subgroup analysis investigating the possible effect of TENS based on:
-
age groups (below 18 years of age, 18 years and over);
-
gender (male, female);
-
stimulation parameters (e.g. high stimulation frequency with low intensity, low frequency with a high stimulus intensity);
-
frequency of administration of TENS (e.g. daily or not on a daily basis);
-
duration of treatment with TENS (e.g. 30‐minute sessions and under or more than 30 minutes).
Sensitivity analysis
If appropriate we will assess the robustness of our findings by performing sensitivity analyses according to Cochrane recommendations (Deeks 2011). If there are sufficient comparable trials, i.e. 10 or more, we will perform sensitivity analyses to study the effect of excluding trials with high risks of bias due to inadequate allocation of concealment, blinding, randomisation method or level of dropouts. Furthermore, we will also consider the impact of using a fixed‐effect model compared to a random‐effects model.
Summary of findings table
Even though there are limited data available from the included trial (with a high risk of bias across several domains), we have produced a summary of findings table (summary of findings Table for the main comparison).
We have used the GRADE approach to create a 'Summary of findings' table, as suggested in chapters 11 and 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011a; Schünemann 2011b). The table presents the trial population and setting, the type of intervention and control group, the outcome measures listed below and an assessment of the evidence quality (as measured by the GRADE approach).
We used the GRADE approach to rate the quality of the evidence as 'high', 'moderate', 'low', or 'very low' using the five GRADE considerations.
-
Risk of bias: serious or very serious
-
Inconsistency: serious or very serious
-
Indirectness: serious or very serious
-
Imprecision: serious or very serious
-
Publication bias: likely or very likely
We generated a 'Summary of findings' table for the 'TENS compared to placebo' comparison with the following outcomes (if data available). For future updates, if more comparisons are included, we plan to produce multiple tables.
-
Pain relief
-
Pain intensity as assessed by VAS or any other validated pain score
-
Frequency of pain episodes
-
Changes in consumption of analgesics and opioids during pain episodes
-
Changes in QoL (as measured by a validated scale)
-
Changes in ability to cope with the ADL
-
Adverse effects of the intervention
Results
Description of studies
Results of the search
In the search from the Cystic Fibrosis and Genetic Disorders Group’s Haemoglobinopathies Trials Register, the Group's Information Specialist identified two references, both pertained to the same trial. We further identified eight references from a search of PubMed; 98 references from Google Scholar (Advanced Search), one reference from a Clinical Trials Registry; eight references from a search of the Agency for Healthcare Research and Quality (AHRQ) database; and 180 references from a search of MedNar. We identified no relevant references from searches of online trial registries.
As illustrated in an additional figure, after removing duplicates, we screened 266 references (Title/Abstract), of which we excluded 243 references (Figure 1). We reviewed the full texts of 23 remaining references and excluded 21 trials and included one trial (two references) (Wang 1988).
Included studies
We included one trial in this review (Wang 1988).
Study design
The included trial was a double‐blind RCT; it was conducted in St. Jude Children's Research Hospital (SJCRH), University of Tennessee, Memphis, USA and approved by the Human Subjects Committee at SJHCRH. The trial objective was to assess the efficacy of TENS for managing pain in people with SCD. The trial was concluded after completion of 60 episodes of treatment (22 participants). The trial was described as cross‐over; however, at the first visit, participants were stratified on a self‐reported pain severity score (four categories) and randomised to treatment or control (within these blocks), subsequent pain crises of the same severity in a participant led to cross‐over by giving the opposite regimen of the previous one, whereas for those experiencing an episode of a different severity, it is not clear from the paper whether these were re‐randomised or given the alternate treatment (regardless of pain grade).
Participants
22 people with SCD (20 Hb SS, I Hb SC, I Hb Sβº thalassaemia) were included in this RCT. The age of the participants was between 12 to 27 years, with a median age of 17.5 years. Out of the 22 participants, 12 were female.
During initiation, participants were stratified into four groups according to pain severity grade based on their pain score rating (visual analogue scale from 0 to 10, with 10 being the severest pain the participant had ever experienced), relief from analgesic and the need for hospitalisations. If participants had pain at multiple sites, the site with the maximum pain was taken into consideration.
Following allocation of treatment group, the assigned treatment (either TENS or sham TENS) was initiated and continued for at least four hours. In the TENS group, at the one‐hour time point, there was one missing response regarding improvement in pain rating and at the four‐hour time point there were three missing responses. The control group (sham TENS) had only two missing responses at the end of four hours. At the end of the trial period, four responses were missing from the TENS group and five were missing from the placebo group regarding overall value of TENS.
Interventions
The intervention device was a battery‐powered Mentor 100 Dual Channel TENS apparatus (Mentor Corporation, Minneapolis, Minn.) using electrical impulses. The rate and width of each electrical impulse, along with the amplitude, was adjustable. The distal electrode was applied at the area of maximum pain within each painful area or over a trigger point in the involved area as indicated on a reference map of pain pathways (TENS Chart, Med General, Inc., Minneapolis, Minn.). If the pain was confined to an extremity or small area, the proximal electrode was placed over a trigger point or along a pain pathway within the involved dermatome. If the pain was reported in the back, chest or abdomen, the electrode was placed over the paravertebral dorsal nerve root corresponding to the pain dermatome(s). The TENS unit was switched on only after randomisation. If the participant was allocated to the TENS group then the settings were set at the original levels while for control it was set at zero. In order to ensure blinding for both the participant and the assessor, the assistant covered the machine's indicator light and control with tape.
Outcomes
Outcomes included change in pain rating (VAS) after one hour and four hours of either intervention or sham treatment. Other outcomes analysed were the use of analgesics up to one hour and up to four hours, as well as the participant's assessment of the overall value of TENS. Adverse outcomes were reported.
Excluded studies
Trials were excluded at the full‐text stage if they did not match the inclusion criteria, or if they matched specific exclusion criteria. Reasons for exclusion were as follows: 14 articles did not describe RCTs or quasi‐RCTs; five articles did not include TENS among the interventions tested; and three trials did not use an experimental study design.
Risk of bias in included studies
Please refer to the risk of bias graph,where we have included our judgements about each risk of bias item presented as percentages across all included studies (Figure 2).
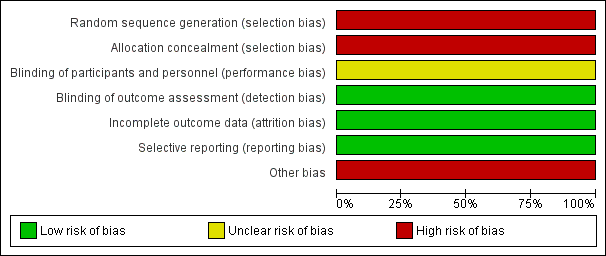
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Sequence generation
The trial authors reported that when a participant entered the trial, stratification by four severity 'grades’, was performed. Randomisation was carried out by an assistant using "randomisation cards", but it was not clear how the randomisation sequence was generated. It was reported that if individuals returned for a subsequent visit and were assessed as having the same severity grade of pain, then the participant was automatically crossed over to the alternative treatment group. It is not clear from the paper whether participants returning with a different severity grade of pain, were re‐randomised or given the alternate treatment (regardless of pain grade). We have therefore assessed the trial as having a high risk of bias for this domain.
Allocation concealment
The double‐blind randomisation was carried out by an 'assistant', using randomisation cards for each severity grade. We have therefore assessed the trial as having a high risk of bias for this domain.
Blinding
Participants and personnel
If the participant was assigned to ‘TENS’, the assistant reset the amplitude settings on the TENS device to their original levels. If the card indicated ‘placebo’, the settings were left at zero.The assistant covered the machine's indicator light and control with tape so that the participant remained unaware about the assignment. However, the assistant who was doing the allocation was also performing the intervention (resetting the TENS machine). Therefore, we assessed this domain as having an unclear risk of bias.
Outcome assessor
The assistant covered the machine’s indicator light and control with tape so that the investigator remained unaware about the assignment. We therefore assessed this domain as having a low risk of bias.
Incomplete outcome data
From the available data there was a less than 15% attrition rate among the participants. Therefore, we consider attrition bias to be of low risk.
Selective reporting
The trial protocol was supplied on request from the authors. Some outcome variables reported in the protocol (days of hospitalisation, days of missed school or work) are not reported in the final results, leading to selective reporting. Based on the largely null outcomes reported we assume that reporting of these missing outcomes may not have affected the conclusion about efficacy of TENS in managing pain in people with SCD. Thus we regarded that the risk of bias due to selective reporting was low.
Other potential sources of bias
The included trial is at a high risk for other potential sources of bias. The number of participants allocated to the control and intervention groups are not clearly stated. Although the trial is of cross‐over design, the number of participants crossed over during the trial is also not clearly stated. The unit of analysis in the trial is the pain episode rather than individual participant; whereas the analysis has been done in the terms of participants, making the statements conflicting.
Effects of interventions
See: Summary of findings for the main comparison
The included trial compared TENS with placebo (sham) TENS. Prior to randomisation of the participants the TENS machine was turned on to produce a tingling sensation followed by a mild discomfort when the amplitude was then reduced. Later, after randomisation, participants receiving the intervention received TENS within the recommended range (approximately 100 Hz pulses per second and the pulse width at 30 ps) while for those assigned to placebo the setting of the machine was turned to zero. To maintain participant blinding, they were briefed that a tingling sensation may or may not be felt.
The included cross‐over trial did not present first‐arm data and we were unable to analyse any data from this trial, also whilst reporting on 22 participants, data on 60 treatment episodes were presented (30 for each treatment group). We have therefore only provided a narrative report.
The quality of the evidence has been graded for those outcomes included in the summary of findings table. For the definitions of these gradings, please refer to the summary of findings tables (summary of findings Table for the main comparison).
Primary outcomes
1. Pain relief
We aimed to present data at two to four weeks and after four weeks post intervention. For long‐term usage (e.g. for a period of at least one month) we planned to consider the outcome measures up to three months. We also considered pain relief as moderate (at least 30% pain relief over baseline) or substantial (at least 50% pain relief over baseline) as defined by IMMPACT guidelines (Dworkin 2008)
Our included trial did not report pain relief at two to four weeks post intervention, there was also no long‐term usage reported (Wang 1988).
2. Pain intensity
The included trial recorded pain intensity by VAS at one and four hours post intervention (Wang 1988). No significant changes in the pain intensity was noted between the TENS and placebo group at the end of one hour (P = 0.30) and four hours (P = 0.69) (very low‐quality evidence).
3. Frequency of pain episodes
The unit of analysis was the pain crisis in a participant in this trial. More than one pain crisis in a participant was treated as an independent episode for analysis, hence the frequency of pain episodes were not reported in the included trial.
Secondary outcomes
1. Changes in consumption of analgesic and opioids during pain episodes
It was stated in the trial report that "the two groups were similar in analgesic usage. Twelve patients (20%) required narcotic analgesics before the 1‐hour rating with no significant difference between placebo (25%) and TENS (14%) (X² = 1.07, P = 0.30). Thirty‐eight patients (63% required pain medication before the 4‐hour rating, again with no significant difference between placebo (66%) and TENS (61%) (X2 = 0.16, p = 0.69)". We graded this as very‐low quality evidence.
2. Changes in QoL (as measured by a validated scale)
The included trial did not assess QoL using a validated scale. Only a subjective overall feeling of satisfaction with the intervention was reported and it was mainly related to the pain.
3. Ability to cope with the ADL
This was not assessed and not reported in the included trial.
4. Adverse effects of the intervention
Although one case of itching was reported in the TENS group in the included trial, the site and nature of itching was not clearly stated. Hence, it cannot be clearly attributed to adverse effect of TENS. Two people in the sham TENS group felt the intervention increased the pain. We graded this as very‐low quality evidence.
Discussion
Summary of main results
Considering the methodological deficiencies of the single included trial, we conclude the evidence level to be of very low quality, due to this, the true effect could be substantially different from that reported. There was no evidence of differences between TENS and sham TENS groups when they rated their pain on a VAS. There was also no evidence of a difference in analgesic use between the two intervention groups. No clear evidence of the adverse effects of TENS were identified. We had planned to assess pain relief at two and four weeks post intervention, however, there was no provision in the research design of the included trial to provide such information. Similarly, as part of our secondary outcomes, we wanted to include changes in QoL (assessed by a validated scale) and ADL, neither of which were assessed in the included trial. Therefore, we are uncertain whether TENS improves overall satisfactions compared to sham TENS (very low‐quality evidence).
Overall completeness and applicability of evidence
During the search, we found that there was a scarcity of RCTs using TENS for managing pain in people with SCD. Only one trial was identified. The trial included people diagnosed with SCD aged from 12 to 27 years. The quality of evidence of the trial was judged to be very low due to both methodological deficiencies and incomplete reporting. The trial evaluated one of our primary outcomes, pain intensity during intervention with TENS. However, other primary outcomes of this review, pain relief at the end of two and four weeks as well as pain frequency during intervention was not reported in this trial. Only one of our secondary outcomes of analgesic usage was reported. The remaining secondary outcomes were not evaluated. Therefore, we can conclude that there is limited applicability of available evidence from the included trial for our review.
Quality of the evidence
Overall, the quality of the evidence was rated as very low across different outcomes according to GRADE methodology (summary of findings Table for the main comparison). With only one included trial we cannot draw any robust conclusions in relation to the objectives of our review. The risk of bias assessments ranged from low to high. Random sequence generation and allocation concealment were judged to be of high risk due to incomplete information provided by the trial authors. Apart from these, other methodological limitations such as lack of paired data or first‐arm data for the cross‐over design, reporting of the analysis as a parallel design rather than a cross‐over design, results in further lowering the quality of evidence.
However, the blinding of participants, personnel and outcome assessors was performed adequately. The missing responses were also less than 15%. Hence, performance, detection and attrition bias of the included trial is concluded to be of low risk.
Potential biases in the review process
We carried out a comprehensive search under the expert guidance of the Cochrane Cystic Fibrosis and Genetic Disorders Review Group, across various databases including grey literature and clinical trial registries. However, the search resulted in trials published mainly in the English language only.
Two review authors independently assessed the trials for inclusion and extracted data from the included trial using the standard data collection form. Two other authors also assessed the risk of bias of the included trial independently (with a third author as adjudicator if necessary). Thus we tried to minimise bias in trial selection, data collection, analysis and assessment of the risk of bias. In order to reduce other potential sources of bias we requested and received the raw data from the trial authors for further analysis.
Agreements and disagreements with other studies or reviews
Our search revealed very few systematic reviews and individual studies on the effect of non‐pharmacological interventions for pain in SCD. However, TENS was not included in any one of them except the included trial. Thus, there is no scope for comparison with similar evidence.
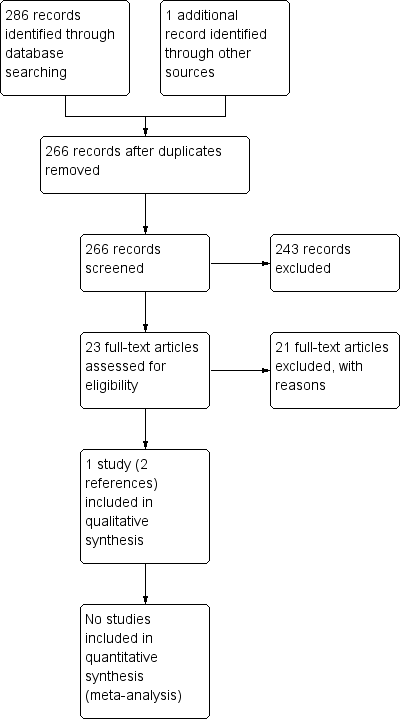
Study flow diagram.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
| TENS compared with placebo (sham TENS) for SCD | ||||||
| Patient or population: children and young adults with SCD Settings: outpatients Intervention: TENS Comparison: sham TENS | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of Participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Sham TENS (placebo) | TENS | |||||
| Pain relief (percentage relief compared to baseline) Follow‐up: 2 to 4 weeks post intervention | This outcome was not measured. | |||||
| Pain intensity (visual analogue score from 0 to 10 where 10 is the most severe pain) Follow‐up: up to 4 hours post intervention | No significant difference in pain intensity was seen between the TENS group and sham TENS group after 1 hour (P = 0.30) or after 4 hours (P = 0.69). | 22 | ⊕⊝⊝⊝ | The unit of analysis for this outcome was pain crises so 1 participant may have had more than 1 episode. The total number of episodes included in the analysis was 25 in the TENS group and 30 in the placebo group. | ||
| Frequency of pain episodes | This outcome was not measured. | Each episode of pain was treated independently as a new episode. Total number of episodes was not reported. | ||||
| Change in consumption of analgesics (number of participants requiring analgesia) Follow‐up: up to 4 hours post intervention | There was no significant difference between the TENS group and the placebo group in the number of people requiring analgesia up to one hour post treatment (TENS 14 %; sham TENS 25 %, X² = 1.07, P = 0.30). There was no significant difference between the TENS group and the placebo group in the number of people requiring analgesia up to four hours post treatment (TENS 61 %; sham TENS 66 %, X² = 0.16, P = 0.69). | 22 | ⊕⊝⊝⊝ | The unit of analysis was pain episodes rather than participants. | ||
| Change in QoL | This outcome was not measured. | Participants were asked to rate whether the treatment was helpful or harmful but this related directly to the pain. | ||||
| Change in ability to cope with ADL | This outcome was not measured. | |||||
| Adverse effects Follow‐up: up to 1 hour and 4 hours post treatment. | There was one case of itching reported in the TENS group but it was unclear whether it was related to the treatment. 2 participants in the placebo group felt that the pain got worse with treatment. | 22 | ⊕⊝⊝⊝ | No data were provided for this outcome. Results were presented narratively. | ||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| a. Downgraded twice because of high risk of bias within the included trial. The randomisation process was unclear and when participants entered the trial for a second or third episode, they were allocated to the alternative treatment from their original allocation. Although the trial authors described their method of blinding participants to the intervention, it is unclear how successful this was given that the TENS would give a tingling sensation and the sham TENS would not. There were also issues around selective reporting of outcomes and a lack of clarity around allocation of each pain episode to a treatment arm. b. Downgraded once due to imprecision from a low number of participants and events. | ||||||