Intervenciones farmacológicas para el tratamiento del síndrome de dolor pelviano crónico/prostatitis crónica
Referencias
Referencias de los estudios incluidos en esta revisión
Referencias de los estudios excluidos de esta revisión
Referencias de los estudios en espera de evaluación
Referencias de los estudios en curso
Referencias adicionales
Referencias de otras versiones publicadas de esta revisión
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Study design: Parallel‐group randomised controlled trial Study dates: not available Setting: outpatient ‐ possibly single centre ‐ international Country: Saudi Arabia | |
| Participants | Inclusion criteria: "with national Institutes of Health (NIH) diagnosis of category III CP/CPPS, but without erectile dysfunction" Exclusion criteria: Not available Sample size: 108 randomised participants. Age (mean in years ±SD): Group 1 41.3 (SD 4.2); Group 2 39.8 (SD 5.1) Baseline NIH‐CPSI score: Not available All participants were men | |
| Interventions | Group 1 (n = 54): levofloxacin 500 mg daily, 4 weeks Group 2 (n = 54): levofloxacin 500 mg daily, plus tadalafil 5 mg daily; 4 weeks Co‐interventions: no other co‐interventions described | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI questionnaire Time points measured: 4 weeks Time points reported: 4 weeks Sexual dysfunction How measured: IIEF score Time points measured: 4 weeks Time points reported: 4 weeks Adverse events How measured: Narratively | |
| Funding sources | None | |
| Declarations of interest | Not available | |
| Notes | Study available as abstract only | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not available (abstract only) |
| Allocation concealment (selection bias) | Unclear risk | Not available (abstract only) |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not available (abstract only) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not available (abstract only) |
| Incomplete outcome data (attrition bias) | Unclear risk | Not available (abstract only) |
| Selective reporting (reporting bias) | Unclear risk | Not available (abstract only) |
| Other bias | Unclear risk | Not available (abstract only) |
| Methods | Study design: Parallel‐group randomised controlled trial with factorial design Study dates: start date July 2001 ‐ end date June 2002abstract Setting: outpatient ‐ multicentre 10 tertiary medical centres ‐ international Country: USA and Canada | |
| Participants | Inclusion criteria: men with symptoms of discomfort or pain in the pelvic region for at least a 3‐month period within the previous 6 months. Candidates must have at least a “moderate” overall score on the NIH‐CPSI defined as 15 or more points of a potential of 0 to 43 points Exclusion criteria: any prostate, bladder, or urethral cancer, seizure disorder; concurrent Inflammatory bowel disease; active urethral stricture; neurologic disease or disorder affecting the bladder; liver disease; neurologic impairment or psychiatric disorder preventing understanding of consent and ability to comply with protocol. Prior 12 mo: Diagnosed with or treated for symptomatic genital herpes. Prior 3 mo: urinary tract infection with a urine culture value of 100,000 CFU/mL; clinical evidence of urethritis, including urethral discharge or positive culture, diagnostic of sexually transmitted diseases (including gonorrhoea, chlamydia, mycoplasma or trichomonas, but not including HIV/AIDS); symptoms of acute or chronic epididymitis. Prior treatments: any pelvic radiation, systemic chemotherapy; intravesical chemotherapy; intravesical BCG, transurethral procedures, balloon dilation of the prostate, open prostatectomy or any other prostate surgery or treatment such as cryotherapy or thermal therapy; prior treatment for orchialgia without pelvic symptoms; known allergy or sensitivity to ciprofloxacin hydrochloride, tamsulosin hydrochloride, or any of their known components. Prior 3 mo: Prostate biopsy Sample size: 196 randomised participants. Age (mean in years ±SD): Group 1 = 42.6 (SD 12.0); Group 2 = 45.9 (SD 11.7); Group 3 = 45.3 (SD 9.7); Group 4 = 44.5 (SD 11.4) Baseline NIH‐CPSI score: Group 1 = 25 (SD 5.1), Group 2 = 24.2 (SD 6.2), Group 3 = 24.6 (SD 6.2), Group 4 = 25.3 (SD 6.1) All participants were men | |
| Interventions | Group 1 (n = 49): ciprofloxacin placebo, 1 tablet, twice daily, plus tamsulosin placebo, 1 tablet daily; 6 weeks Group 2 (n = 49): ciprofloxacin 500 mg, 1 tablet, twice daily, plus tamsulosin placebo, 1 tablet daily; 6 weeks Group 3 (n = 49): ciprofloxacin placebo, 1 tablet, twice daily, plus tamsulosin 0.4 mg, 1 tablet daily; 6 weeks Group 4 (n = 49): ciprofloxacin 500 mg, 1 tablet, twice daily, plus tamsulosin 0.4 mg, 1 tablet daily; 6 weeks Co‐interventions: no co‐interventions described | |
| Outcomes | Prostatitis Symptoms How measured: with NIH‐CPSI questionnaire, total score and subscores measurements Time points measured: 2 at baseline screening (1 to 3 weeks apart), and every 3 weeks until 12 weeks. Time points reported: change from baseline at 6 weeks (Figure 2 shows means for week 0‐3‐6‐9‐12) Quality of life How measured:Medical Outcomes Study 12‐items Short‐Form Survey Time points measured: not stated Time points reported: change from baseline at 6 weeks Adverse events How measured: National Cancer Institute Common Toxicity Criteria classification, open‐ended questions Time points measured: "throughout the study" ‐ at each contact Time points reported: cumulative report at 6 weeks | |
| Funding sources | National Institute of Health cooperative agreement. Boehringer Ingelheim provided tamsulosin and matching placebo. Bayer Corporation provided ciprofloxacin and matching placebo | |
| Declarations of interest | Grant by National Institutes of Health cooperative agreements U01 DK53572, U01 DK53730, U01 DK53736, U01 DK53734, U01 DK53732, U01 DK53746, and U01 DK53738. Boehringer Ingelheim provided tamsulosin and matching placebo; Bayer Corporation provided ciprofloxacin and matching placebo. Authors: Nickel, Zeitlin, Alexander, Shoskes have received honoraria from the aforementioned Pharmaceuticals | |
| Notes | The study reported a 7‐point patient‐reported global response assessment at 6 weeks. Results are presented per group individually and per factor (group ciprofloxacin vs placebo; group tamsulosin vs placebo) Contact information: Dr. Alexander: Urology (112), Veterans Affairs Maryland Health Care System, 10 North Greene Street, Baltimore, MD 21201. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Each patient was randomly assigned by computer" |
| Allocation concealment (selection bias) | Low risk | Quote: "The research pharmacist at each site provided the blinded study drugs in 2 tamper‐evident bottles." |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: "All clinical investigators, research nurses, and patients were blinded to treatment assignments until all patients had completed follow‐up." |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "All clinical investigators, research nurses, and patients were blinded to treatment assignments until all patients had completed follow‐up." |
| Incomplete outcome data (attrition bias) | Low risk | Quote: "174 men (89%) were available for evaluation at 6 weeks [...]. Another 3 participants withdrew from study follow‐up between 6 and 12 week" |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes in the published protocol (Alexander 2004) were reported |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: 2008 (start and end date) Setting: outpatient, multicentre, national Country: Russia | |
| Participants | Inclusion criteria: age 18 ‐ 50; diagnosis of CP/CPPS (type IIIa) for 6 months and more; NIH‐CPSI baseline pain score ≥ 3 Exclusion criteria: postoperative patients in urogenital area; patients with neurogenic bladder dysfunctions, cervical sclerosis of urethra, urethral stricture, prostate hyperplasia, bladder cancer, prostate cancer, bladder diverticulosis; infection of urogenital tract, medication that is known to affect urination prior to study, diseases known to alternate biochemical indices twofold and more (decrease/increase) Sample size: 78 participants were enrolled and randomised Age (years): Group 1 mean = 34.8 (SD = 8.79); Group 2 mean = 39,0 (SD = 7.49) NIH‐CPSI baseline score (obtained from the graph): Group 1 mean = 20.0 (SE = 1.0) Group 2 mean = 25.0 (SE = 0.5) All participants were men | |
| Interventions | Group 1 (n = 55): Cernilton (pollen extract) 2 pills 3 times/day, orally, for 3 months Group 2 (n = 23): Cernilton (pollen extract) 1 pills 3 times/day, orally, for 3 months Co‐interventions: no co‐interventions described | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐total score Time points measured: baseline, 3 months, 6 months Time points reported: baseline, 3 months, 6 months | |
| Funding sources | none | |
| Declarations of interest | none | |
| Notes | The urinary symptoms as measured by IPSS scale were Intended according to Methods section but it was not possible to Identify the findings throughout the article. This article was written in Russian Contact information: [email protected] (the author provided the full text of the study) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | High risk | Participants and personnel were not blinded. After randomisation both groups were allotted 3‐month pack of Cernilton |
| Blinding of outcome assessment (detection bias) | High risk | Participants and personnel were not blinded. After randomisation both groups were allotted 3‐month pack of Cernilton |
| Incomplete outcome data (attrition bias) | Low risk | There are no missing outcome data (all outcomes) |
| Selective reporting (reporting bias) | High risk | The symptoms as measured by IPSS scale was Intended according to Methods section but It was not possible to Identify the findings throughout the article. The variance of resulting figures was only reported graphically |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date July 2000 ‐ end date August 2002 Setting: outpatient – single centre‐ national Country: United Kindom | |
| Participants | Inclusion criteria: all patients aged 18 – 65 years, with CPPS (inflammatory or non‐inflammatory) for ≥ 6 months and who had failed to improve with standard antibiotic therapy; negative infection screen, including the 4‐glass test and bacterial location studies; normal urological investigations; normal liver function tests, urea and electrolytes, full blood count and erythrocyte sedimentation ratio Exclusion criteria: contraindication to steroid therapy Sample size: 21 Age (years): Group 1 = 43.0 (10.7) Group 2 = 39.1 (10.1) Baseline NIH‐CPSI score: Group 1 = 25.5 (SD 7.6) Group 2 23.4 (SD 5.8) All participants were men | |
| Interventions | Group 1 (n = 9): prednisolone 20 mg (4 capsules) once daily for 7 days, then 15 mg (3 capsules) once daily for 7 days, 10 mg (2 capsules) once daily for 7 days, finishing with 5 mg (1 capsule) once daily for 7 days Group 2 (n = 12): matched placebo Co‐interventions: no information available | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score Time points measured: baseline, 1, 3, 6 and 12 months Time points reported: baseline and 3 months (P values) Quality of life How measured: General Health Questionnaire (GHQ‐30) Time points measured: baseline, 1, 3, 6 and 12 months Time points reported: not reported Anxiety and depression How measured: Hospital Anxiety and Depression Scale (HADS) Time points measured: baseline, 1, 3, 6 and 12 months Time points reported: baseline and 3 months (P values) | |
| Funding sources | Quote:“STH NHS FT Research Department – Small Grants award scheme. A grant for this study was provided by Central Sheffield University Hospitals Research Department, Royal Hallamshire Hospital, Sheffield, under the Pilot Project Research Scheme.” | |
| Declarations of interest | None declared | |
| Notes | The authors stated that the participants did not fill the GHQ‐30 at follow‐up as planned Contact information: [email protected]; [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Low risk | Quote: “Randomization was undertaken by the dispensing pharmacy, and hence both the participants and the investigator were unaware of the allocation.” |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: “both the participants and the investigator were unaware of the allocation.” |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: “both the participants and the investigator were unaware of the allocation.” |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: 3/9 patients were excluded from the experimental arm analysis due to the use of antibiotics or lack of improvement of symptoms (none in the control group). Not all participants filled the GHQ‐30 questionnaire |
| Selective reporting (reporting bias) | High risk | Only P values for most endpoints available. We wrote to study authors |
| Other bias | Low risk | No other sources of bias identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient, multicentre, national Country: Russia | |
| Participants | Inclusion criteria: diagnosis of CP/CPPS (type IIIb), sexual dysfunction symptoms as defined by a locally‐developed questionnaire Exclusion criteria: diabetes mellitus, arterial hypertension (stage 1 or 2), postoperative and post‐traumatic sexual dysfunction Sample size: 57 participants were enrolled and randomised Age (years): Group 1 not available; Group 2 not available NIH‐CPSI baseline score: Group 1 mean = 22.4; Group 2 mean = 21.6 All participants were men | |
| Interventions | Group 1 (n = 29): Prolit Super, 2 capsules at breakfast and lunchtime orally for 2 months, containing:a tablet of 600 mg strobilanthi folium, orthosiphonis folium, radix ginseng, sea horse, imperatae rhizoma, glycyrrhizae radix Group 2 (n = 28): placebo, 2 capsules at breakfast and lunchtime orally for 2 months Co‐interventions: no information available | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐total score and pain, urinary, and QoL domains Time points measured: baseline, 1 month, 2 months Time points reported: baseline, 1 month, 2 months Adverse event How measured: narratively | |
| Funding sources | none | |
| Declarations of interest | none | |
| Notes | This article was written in Russian. No contact information available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | No information available. Authors replied they used random numbers |
| Allocation concealment (selection bias) | Unclear risk | No information available. Authors did not provide further information |
| Blinding of participants and personnel (performance bias) | Unclear risk | No information available. Authors replied that participants were blinded, no information regarding study personnel |
| Blinding of outcome assessment (detection bias) | Low risk | No information available. Authors replied that participants were blinded |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: All 57 enrolled completed the trial follow‐up and had outcome data available |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. The information on variation (SD/SE) of the outcomes was not reported but the authors provided them |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date March 2012 ‐ end date October 2012 Setting: outpatient, single centre, national Country: Italy | |
| Participants | Inclusion criteria: presence of symptoms of pelvic pain for at least 3 months during the 6 months before study entry, a score in the pain domain of the NIH‑CPSI (14) of > 7 and a negative 4‑glass result in the Meares‑Stamey test Exclusion criteria: Patients < 18 and > 65 years of age, affected by major concomitant diseases, with known anatomical abnormalities of the urinary tract or with evidence of other urological diseases, and with residual urine volume > 50 ml resulting from bladder outlet obstruction were excluded. Men with a reported allergy to pollen extract, who had recently (< 4 weeks) undergone oral or parenteral treatment or who were currently using prophylactic antibiotic drugs were also excluded. Additionally, all patients with a history of gastrointestinal bleeding or duodenal or gastric ulcers were excluded. All patients positive to tests for chlamydia trachomatis (Ct), ureaplasma urealyticum, neisseria gonorrhoeae, herpes viruses (HSV 1/2) and human papillomavirus (HPV) were also excluded Sample size: 87 men Age (years): Group 1 = 33.8 years (SD 6.78); Group 2 = 33.7 (SD 5.44) Baseline NIH‐CPSI score: Group 1 = 24.9 (SD 2.1); Group 2 = 25.5 (SD 3.0) All participants were men | |
| Interventions | Group 1 (n = 41): DEPROX 500® (1 g pollen extract, 500 mg per tablet, and vitamins B1, B2, B6, B9, B12 and PP; 2 capsules in the evening every 24 hours) for 4 weeks Group 2 (n = 46): ibuprofen (600 mg, 1 tablet 3 times a day) for 4 weeks Co‐interventions: no information | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: 30 days Time points reported: 30 days Urinary Symptoms How measured: IPSS score Time points measured: 30 days Time points reported: 30 days Quality of life How measured: QoL Italian score Time points measured: 30 days Time points reported: 30 days Adverse events How measured: Narrative account | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | E‑mail: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “single sequence of random assignments (simple randomization) was performed using a pseudo‑random number generator software” |
| Allocation concealment (selection bias) | Unclear risk | No information on allocation concealment. We wrote to study authors. |
| Blinding of participants and personnel (performance bias) | High risk | Quote: “this was not a blinded study” |
| Blinding of outcome assessment (detection bias) | High risk | Quote: “this was not a blinded study” |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: missing data for 1/41 and 2/46 participants in each study arm due to lost to follow‐up |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date January 2015 ‐ end date December 2015 Setting: Outpatient ‐ single centre ‐ national Country: Italy | |
| Participants | Inclusion criteria: Men aged 18 ‐ 65 years old with the presence of pelvic pain symptoms for at least 3 months during the 6 months before study enrolment in accordance with the EUA guidelines, a score in the pain domain of the NIH‐CPSI of > 4 and a negative result for the Meares‐Stamey 4‐glass test Exclusion criteria: individuals affected by major concomitant diseases or with known anatomical abnormalities of the urinary tract or with evidence of other urological diseases with residual urine volume > 50 mL resulting from bladder outlet obstruction; men with a reported allergy to pollen extract who had undergone oral or parenteral treatment or who were currently using prophylactic antibiotic drugs (in the last 4 weeks), and all patients who tested positive for sexual transmitted diseases Sample size: 70 participants randomised Age (years): Group 1 = 32.4 (SD 4.3); Group 2 = 32.8 (SD 4.9) NIH‐CPSI baseline score: Group 1 = 25.1 (SD 2.1); Group 2 = 25.6 (SD 2.9) All participants were men | |
| Interventions | Group 1 (n = 36): 2 tables of pollen extract and vitamins in a single dose in the evening. Each tablet contained 1 g of pollen extract and vitamins B1, B2, B6, B9, B12, and PP. Duration: 3 months Group 2 (n = 34): 2 tables of bromelain in a single daily dose in the evening (extract obtained from the stem and fruit of the pineapple plant) Duration: 3 months. Co‐interventions: not available | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score and pain subscore Time points measured: baseline and 3 months Time points reported: baseline and 3 months Quality of life How measured: SF‐36 Time points measured: baseline and 3 months Time points reported: baseline and 3 months Quality of life How measured: Narrative | |
| Funding sources | Not available | |
| Declarations of interest | Quote: “No potential conflict of interest relevant to this article was reported.” | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “All patients who met the inclusion criteria were randomized to either the treatment or control group by using a computer‐generated allocation sequence” |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | High risk | Quote: “Neither the physicians nor the patients were blinded to the treatment type.” |
| Blinding of outcome assessment (detection bias) | High risk | Quote: “Neither the physicians nor the patients were blinded to the treatment type.” |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: outcome data were available for 32/36 participants in the pollen extract group and 33/34 in the bromelain group. Unbalanced attrition |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient ‐ single centre ‐ national Country: Italy | |
| Participants | Inclusion criteria: individuals with a diagnosis of chronic abacterial prostatitis/prostatodynia (after anamnesis, physical examination, prostatic ultrasound, urinalysis and urine culture, sperm analysis and culture, urethral swab, Stamey test and antibiogram) Exclusion criteria: none specified Sample size: 54 men Age (years): median 34 (range 28 ‐ 44) Baseline NIH‐CPSI score: not available. All participants were men | |
| Interventions | Group 1 (n = 22): mepartricin 40 mg per day during 60 days Group 2 (n = 20): vitamin C 500 mg per day during 60 days used as placebo Co‐interventions: not described | |
| Outcomes | Adverse events Narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | This study also measured spontaneous pain, palpatory pain, urinary frequency, nycturia and prostatic volume. Contact information not available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | Unclear risk | No blinding was mentioned. It is not clear that the placebo pills were similar to the experimental treatment |
| Blinding of outcome assessment (detection bias) | Unclear risk | No blinding was mentioned. It is not clear that the placebo pills were similar to the experimental treatment |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: 54 participants were randomised, 12 were lost to follow‐up (no reasons were reported for missing outcome data) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias identified |
| Methods | Study design: Parallel randomised trial Study dates: start date July 2006 ‐ end date March 2008 Setting: Outpatient ‐ single centre ‐ national Country: South Korea | |
| Participants | Inclusion criteria: men aged ≥ 20 and ≤ 50 years old with symptoms of discomfort or pain in the pelvic region for at least a 3‐month period Participants had a clinical diagnosis of CP/CPPS (III) Exclusion criteria: men with previous history of CP/CPPS, BPH, or neurogenic bladder. Men with a prostate volume > 30 g at digital rectal examination or positive culture in urine or EPS. Men with suspected prostate cancer Total number of participants randomly assigned: 103 Age (mean, SD): Group 1 = 39.70 ± 7.72; Group 2 = 38.46 ± 7.52; Group 3 = 39.64 ± 9.62 Baseline NIH‐CPSI score: Group 1 = 24.6 (SD 6.9); Group 2 = 24.3 (SD 8.3); Group 3 = 24.7 (SD 7.4) All participants were men | |
| Interventions | Group 1 (n = 36): Only co‐intervention ‐ 8 weeks Group 2 (n = 33): alfuzosin 10 mg daily for 8 weeks Group 3 (n = 34): terpene mixture (Rowatinex®) 1 capsule 3 times a day for 8 weeks Co‐interventions: all participants received levofloxacin 100 mg 3 times a day for 8 weeks. | |
| Outcomes | Prostatitis Symptoms How measured: NIH CPSI questionnaire Time points measured: baseline, 8 weeks after treatment Time points reported: baseline, 8 weeks after treatment Subgroups: no | |
| Funding sources | Not reported | |
| Declarations of interest | Not reported | |
| Notes | Language of publication: Korean Young Jin Seo. E‐mail: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. Wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. Wrote to study authors |
| Blinding of participants and personnel (performance bias) | Unclear risk | No information available. Wrote to study authors |
| Blinding of outcome assessment (detection bias) | Unclear risk | No information available. Wrote to study authors |
| Incomplete outcome data (attrition bias) | Unclear risk | No information available. Wrote to study authors |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available. |
| Other bias | Low risk | No other sources of bias detected. |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date April 2000 ‐ end date September 2001 Setting: outpatient, multicentre, international Country: Malaysia and USA | |
| Participants | Inclusion criteria: men aged 20 to 50 years old with diagnostic criteria for CP/CPPS with a score of 1 or more in the items pelvic pain and quality of life and 4 or more in item 9 of the NIH‐CPSI score, suffering from symptoms lasting more than 3 months and desired to be treated Exclusion criteria: criteria for bacterial infection of the prostate and urinary tract, significant medical problems, concurrent medication that could affect the lower urinary tract function and previous experience with alpha blockers; along with other exclusion criteria in a previous consensus Sample size: 100 randomised Age (years): Group 1 median 36 (range 24 ‐ 49); Group 2 median 35 (range 20 ‐ 50) NIH‐CPSI baseline score: Group 1 = 25.1 (SD 7.1); Group 2 = 27.2 (SD 7.7) All participants were men | |
| Interventions | Group 1 (n = 50): Terazosin 1 mg for 4 days, 2 mg during 10 days and then 5 mg during 12 weeks Group 2 (n = 50): Placebo of similar appearance in the same dosage Co‐interventions: They were not permitted to take any other medication for CP/CPPS or those that affected the lower urinary tract | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, week 2, 6 and 14 Time points reported: baseline, week 2, 6 and 14 Urinary Symptoms How measured: IPSS Time points measured: baseline, week 2, 6 and 14 Time points reported: baseline, week 2, 6 and 14 Adverse events How measured: Narratively | |
| Funding sources | Supported by an unrestricted grant from Abbott Laboratories, Malaysia | |
| Declarations of interest | The contact author has "financial interest and/or other relationship with Abbott Laboratories" | |
| Notes | Contact information: Department of Urological Surgery, University of Washington School of Medicine, VA Puget Sound Health Care System, 1660 S. Columbian Way, Seattle, Washington 98108 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Random number table” |
| Allocation concealment (selection bias) | Unclear risk | No information available. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind study. Blinding of personnel not described. Participants were blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded using placebo |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: 14% of participant in each group did not complete assessments |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date October 2007 ‐ end date August 2008 Setting: Outpatient ‐ single centre ‐ national Country: China | |
| Participants | Inclusion criteria: Individuals aged 20 to 50 years old, who meet the diagnostic criteria for type IIIB chronic prostatitis, with a course of disease 3 months to 4 years and had a NIH‐CPSI scoring of 10 or more. Individuals who did not take medications for chronic prostatitis or urination in the past week and who met the criteria of “chi and blood stasis syndrome” based on Traditional Chinese Medicine (TCM) theory of syndrome differentiation Exclusion criteria: Individuals with positive results of expressed prostate massage culture; individuals with pain and discomfort caused by other diseases; individuals with serious liver or kidney insufficiency and those who were allergic to the drugs used in this trial Sample size: 70 patients Age (years): Group 1 not available; Group 2 not available; Overall 20 ˜ 45, mean = 29.6 NIH‐CPSI baseline score: Group 1 = 25.3 (SD 3.8); Group 2 = 24.1 (SD 3.4) All participants were men | |
| Interventions | Group 1 (n = 36): Qiantongding decoction (oral) twice a day for 1 month Group 2 (n = 34): XiaoYanTong (oral) 25 mg three times a day for 1 month Co‐interventions: All other medications and treatment options were discontinued during the process | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI (total, pain, quality of life) Time points measured: pre‐treatment, post‐treatment Time points reported: pre‐treatment, post‐treatment Adverse Events How measured: Narratively | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese. Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “Patients were assigned to trial group (36 patients) and control group (34 patients) by simple random sampling” (p 90). Comment: However, it is unclear what the exact method was and how the allocation process was carried out. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not described. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Masking of participants and personnel was not described. However, considering the visible difference between the 2 drugs, one could tell which medication the participants was taking. Therefore, masking was unlikely |
| Blinding of outcome assessment (detection bias) | High risk | Blinding was unlikely (visibly different interventions) |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors |
| Other bias | Low risk | No other sources of bias identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date July 2003 ‐ end date March 2007 Setting: outpatient ‐ national ‐ single centre Country: China | |
| Participants | Inclusion criteria: Patients were included if they had pain or discomfort in the pelvic region for at least 3 months, a total score of at least 12 on the NIH‐CPSI and anticipated improvement of symptoms with therapy Exclusion criteria: Participants with previous treatment with tamsulosin or any other alpha‐adrenergic receptor blocker for any reason, participants with chronic bacterial prostatitis, previous urinary tract infection within the last year, history of genitourinary cancer, inflammatory bowel disease, urethral stricture, symptomatic genital herpes, neurologic disease affecting the bladder, those who were taking medication that could affect lower urinary tract function, and those with benign prostatic hyperplasia or elevated PSA and those with severe symptoms (total NIH‐CPSI score > 38) Sample size: 100 randomised Age (years)(mean +/‐ SD): Group 1 tamsulosin (35.3 ± 6.8); Group 2 placebo (33.3 ± 7.2) Baseline NIH‐CPSI score: Group 1 = 23.3 (SD 6.2); Group 2 = 22.5 (SD 5.6) All participants were men | |
| Interventions | Group 1 (n = 50): 0.2 mg of tamsulosin once daily for 6 months Group 2 (n = 50): identical‐looking placebo Co‐interventions: All participants were instructed to avoid spicy food, caffeine and alcohol during the study period | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI total score Time points measured: at the start of treatment (baseline) and at 3, 6, 12, 18 and 30 months after initiation of treatment Time points reported: at the start of treatment (baseline) and at 3, 6, 12, 18 and 30 months after initiation of treatment Sexual Dysfunction How measured: IIEF score Time points measured: at the start of treatment (baseline) and at 3, 6, 12, 18 and 30 months after initiation of treatment Time points reported: at the start of treatment (baseline) and at 3, 6, 12, 18 and 30 months after initiation of treatment Adverse events How measured: Narratively | |
| Funding sources | No information available | |
| Declarations of interest | Quote: “None.” | |
| Notes | Contact information: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Patients were randomly allocated using computer‐generated random numbers into two equal groups” |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors (email bounced) |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: “Study investigators and subjects were unaware of the treatment assignments.” |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: “Study investigators and subjects were unaware of the treatment assignments.” |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes at 6 months: outcome data were available in 46/50 in the tamsulosin group and 47/50 in the placebo group (low risk) All outcomes at 12 months: outcome data were available in 45/50 in the tamsulosin group and 47/50 in the placebo group (low risk) All outcomes at 30 months: outcome data were available in 35/50 in the tamsulosin group and 37/50 in the placebo group (high risk) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors (email bounced) |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: not available Setting: outpatient – single centre ‐ national Country: Taiwan | |
| Participants | Inclusion criteria: men with chronic non‐bacterial prostatitis Exclusion criteria: not available Sample size: 215 men Age (years): not available Baseline NIH‐CPSI score: not available All participants were men | |
| Interventions | Group 1 (n = 51): levofloxacin 500 mg daily for 6 weeks Group 2 (n = 53): ciprofloxacin 250 mg twice daily for 6 weeks Group 3 (n = 61): levofloxacin 500 mg twice daily for 6 weeks Group 1 (n = 50): control group Co‐interventions: Not available | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 3 and 6 weeks Time points reported: not available Co‐interventions: NSAIDs (diclofenac 500mg) twice daily and alpha blocker (tamsulosin 0.2 mg) twice daily were prescribed for all participants | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | This information was provided by two abstracts, no full text was available. No contact information available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available (abstract only) |
| Allocation concealment (selection bias) | Unclear risk | No information available (abstract only) |
| Blinding of participants and personnel (performance bias) | Unclear risk | No information available (abstract only) |
| Blinding of outcome assessment (detection bias) | Unclear risk | No information available (abstract only) |
| Incomplete outcome data (attrition bias) | Unclear risk | No information available (abstract only) |
| Selective reporting (reporting bias) | Unclear risk | No information available (abstract only) |
| Other bias | Unclear risk | No information available (abstract only) |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date January 2011 ‐ end date December 2011 Setting: Outpatient ‐ multicentre ‐ national Country: Republic of Korea | |
| Participants | Inclusion criteria. Participants with a diagnosis of category IIIa of IIIb chronic prostatitis and a NIH‐CPSI score ≥ 15 Exclusion criteria: The presence of urinary tract infection or uropathogen within the past 12 months, serious medical problems, NIH consensus exclusion criteria (Alexander 2004a), drug therapy that might affect lower urinary tract functions within the past 3 months Sample size: 75 participants Baseline NIH‐CPSI score: Group 1 = 20.5; Group 2 = 21.2; Group 3 = 22.5 Age (years): range 22 ‐ 42 years, mean = 29.1 ± 5.2 years All participants were men | |
| Interventions | Group 1 (n = 25) received 500 mg ciprofloxacin twice a day for 4 weeks Group 2 (n = 25) received 100 mg aceclofenac twice a day for 4 weeks Group 3 (n = 25) received 150 mg roxithromycin twice a day for 4 weeks Co‐intervention: none | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score and subscore Time points measured: baseline, 4, and 12 weeks after drug administration Time points reported: baseline, 4, and 12 weeks after drug administration. Some results were presented graphically Adverse events How measured: Narratively Urinary symptoms How measured: 7‐item International Prostate Symptom Score (IPSS) Comment: presumably only total IPSS score without QoL score Time points measured: baseline, 4, and 12 weeks after drug administration Time points reported: not reported | |
| Funding sources | Not available | |
| Declarations of interest | None | |
| Notes | ClinicalTrials.gov Identifier: NCT01843946 Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "To prevent the imbalance of subject number among the groups, which could occur upon general randomization, blocked randomization was used in this study. In addition, after the randomization table was prepared for each study center, the subjects were randomized to the treatment groups according to the randomization table.” Comment: Probably done |
| Allocation concealment (selection bias) | High risk | Randomisation table, “open‐label, randomized allocation” |
| Blinding of participants and personnel (performance bias) | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) | High risk | Open‐label study |
| Incomplete outcome data (attrition bias) | Unclear risk | No information available |
| Selective reporting (reporting bias) | High risk | The table of IPSS scores outlined in absolute numbers is missing. Instead, the authors provided the results of IPSS score change in the Results section NIH‐CPSI score do not include variability (SD or IQ range), some time points are presented graphically |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient, single centre, national Country: Russia | |
| Participants | Inclusion criteria: age 22 ‐ 60; diagnosis of abacterial CP/CPPS, erectile dysfunction symptoms Exclusion criteria: the following co‐morbidities: non‐compensated forms of endocrine, cardiovascular, and mental disorders; prostate cancer, prostate hyperplasia Sample size: 60 participants were enrolled and randomised Age (years): Group 1 (not available); Group 2 (not available) NIH‐CPSI baseline score: not available All participants were men | |
| Interventions | Group 1 (n = 30): Cytoflavin 10 ml intravenously for 10 days, then 2 pills of Cytoflavin twice a day for 20 days Group 2 (n = 30): no treatment Co‐intervention: α‐blockers ‐ 1 month, nonspecific anti‐inflammatory drugs – 2 weeks, prostate massage and vibrovacuum fallostimulation – 10 times, antibiotics – 10 days | |
| Outcomes | None of the outcomes relevant for this review (See Notes) | |
| Funding sources | none | |
| Declarations of interest | none | |
| Notes | Prostatitis symptoms were reported using a locally‐ developed scale; we have no information regarding its validation. Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | Unclear risk | No information available |
| Blinding of outcome assessment (detection bias) | Unclear risk | No information available |
| Incomplete outcome data (attrition bias) | Unclear risk | No information available |
| Selective reporting (reporting bias) | High risk | No protocol available. The information on variation (SD/SE) of the outcomes was reported |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date June 2001 ‐ end date November 2002 Setting: outpatient ‐ single centre ‐ national Country: Italy | |
| Participants | Inclusion criteria: men aged 18 ‐ 55 years with nonbacterial type III CP/CPPS for at least 1 year; suffering from pain, sexual dysfunction and difficulty voiding Exclusion criteria: evidence of infection in urethral swab, urine culture and semen culture in the participant and sexual partner, PSA > 4 ng/mL; HIV infection, diabetes mellitus, and stroke in the last 3 months. Participants had not taken antibiotics in the last 3 months Sample size: 30 randomised and 4 were excluded after randomisation Age (years): Group 1 median 32 (range 26 ‐ 49); Group 2 median 34 (range 26 ‐ 52) NIH‐CPSI baseline score: Group 1 median 25 (range 21 ‐ 34); Group 2 median 25 (range 18 ‐ 45) All participants were men | |
| Interventions | Group 1 (n = 13): mepartricin 40 mg daily for 60 days Group 2 (n = 13): placebo daily for 60 days Co‐interventions: not described | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline and 60 days Time points reported: baseline and 60 days Adverse events How measured: Narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | No email address available. Contact information: Aldo Franco De Rose, M.D., “Luciano Giuliani” | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “The 30 envelopes were sealed, mixed, and numbered in progressive order before being placed in a box” – random sorting. |
| Allocation concealment (selection bias) | Low risk | Quote: “Randomization was performed using 30 opaque envelopes” |
| Blinding of participants and personnel (performance bias) | Unclear risk | Not specified whether personnel were blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Placebo was used for the blinding of participants |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: 4 participants (2/15 in each study arm) were excluded after randomisation for the presence of positive cultures |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: Not available Setting: Outpatient, single centre, national Country: Germany | |
| Participants | Inclusion criteria: individuals with more than 12 months of diagnosis of chronic abacterial prostatitis with negative urine and sperm culture who were refractory to other treatments Exclusion criteria: not available Sample size: 40 NIH‐CPSI score: not available Age (years): Overall median 39 years old (range 21 ‐ 56) All participants were men | |
| Interventions | Group 1 (n = 20): phenoxybenzamine 10 mg twice daily for 6 weeks Group 2 (n = 20): placebo Co‐interventions: not available | |
| Outcomes | Adverse events How measured: Narratively | |
| Funding sources | Röhm Pharma GmbH, Weiterstadt | |
| Declarations of interest | Not available | |
| Notes | Email: not available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block‐randomisation, assumed random. |
| Allocation concealment (selection bias) | Unclear risk | No information available. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind, not specified who was blinded. |
| Blinding of outcome assessment (detection bias) | Unclear risk | Double‐blind, not specified who was blinded. |
| Incomplete outcome data (attrition bias) | High risk | Outcome data was available for 17/20 patients in the intervention arm, and 13/20 in the placebo arm. |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available. |
| Other bias | Low risk | No other sources of bias identified. |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient ‐ national ‐ single centre Country: USA | |
| Participants | Inclusion criteria: men aged 20 to 60 years old with a diagnosis of CP/CPPS type III prostatitis with no response to previous treatment Exclusion criteria: positive semen, prostatic fluid or urine culture; elevated PSA, abnormalities in cystoscopy or ultrasonography, abnormal cystometrography, benign prostatic hyperplasia with an AUA score exceeding 15, bladder neck obstruction, abnormal uroflowmetry results, > 3 to 5 red blood cells in urine sample Sample size: 60 randomised Age (years): overall mean 35 years (range 20 ‐ 55) NIH‐CPSI baseline score: not available All participants were men | |
| Interventions | Group 1 (n = 30): Prostat/Poltit containing “74 mg highly defined extract of pollen from selected Graminae species” during 6 months Group 2 (n = 30): Identical‐looking Placebo during 6 months Co‐interventions: the participants were told not to engage in other pharmacological and non‐pharmacological treatments | |
| Outcomes | Adverse events Narratively | |
| Funding sources | Quote: “The study medication was produced by Allergon AB” | |
| Declarations of interest | Not available | |
| Notes | The clinical outcome measure was a non‐validated symptom score. Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Using computer‐generated sequence |
| Allocation concealment (selection bias) | Low risk | Pharmacy central allocation |
| Blinding of participants and personnel (performance bias) | Low risk | Participants and personnel were blinded to the treatment arm |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: “The placebo tablets were identical in appearance to the active tablets but contained no pollen extract” |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: 2 participants (7%) were lost to follow‐up in the placebo group with no outcome data |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: not available Setting: outpatient ‐ single centre ‐ national Country: Egypt | |
| Participants | Inclusion criteria: clinically‐proven chronic prostatitis and chronic pelvic pain syndrome type III after failure of several courses of medical therapy were selected from patients referred to the neuro‐urology unit of the urology department for voiding dysfunction evaluation and management; “with urodynamically proven detrusor external sphincter dyssynergia (DESD)” Exclusion criteria: not available Sample size: 52 men Age (years): overall range 25 to 48 years Baseline NIH‐CPSI score: not available All participants were men | |
| Interventions | Group 1 (n = 26): Received 100 units of reconstituted Botulinum Toxin A (BTA) into the external urethral sphincter (EUS) endoscopically Group 2 (n = 26): Received combined injection in the EUS and prostate with 100 units of BTA for each location Co‐interventions: none | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 12 weeks, 6 and 12 months Time points reported: Not available (only P value) Adverse Events How measured: Narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | This trial was only available as an abstract. We attempted contact with study author: [email protected], [email protected] Quote: “55 % of GI [Group 1] patients were scheduled for second session of combined chemo‐denervation following the same protocol for GII at the end of the study period.” | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Abstract only. No information available |
| Allocation concealment (selection bias) | Unclear risk | Abstract only. No information available |
| Blinding of participants and personnel (performance bias) | Unclear risk | Abstract only. No information available |
| Blinding of outcome assessment (detection bias) | Unclear risk | Abstract only. No information available |
| Incomplete outcome data (attrition bias) | Unclear risk | Abstract only. No information available |
| Selective reporting (reporting bias) | Unclear risk | Abstract only. No information available |
| Other bias | Unclear risk | Abstract only. No information available |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date January 2004 ‐ end date December 2008 Setting: outpatient ‐ single centre ‐ national Country: Turkey | |
| Participants | Inclusion criteria: Type III chronic prostatitis Exclusion criteria: Neurologic diseases, urethral strictures, chronic diseases such chronic liver and kidney failure and diabetes mellitus, history of pelvic surgery, chronic antidepressant, anti‐psychotic and anxiolytic drug use with prior psychiatric disorder diagnosis and recent urinary tract infections Sample size: 87 Age (years): Group 1: 34.13 ± 7.39; Group 2: 34.76 ± 7.99; Group 3: 33.72 ± 8.25 Baseline NIH‐CPSI score: Group 1: 23.92 ± 5.3; Group 2: 24.11 ± 4.2; Group 3: 23.72 ± 3.2 All participants were men | |
| Interventions | Group 1 (n = 45): A quinolone 500 mg 4 ‐ 6 weeks + ibuprofen 400 mg 4 weeks + terazosin 5 mg 12 weeks. Possibly oral, once a day Group 2 (n = 17): Terazosin 5 mg 12 weeks. Possibly oral, once a day Group 3 (n = 25): A quinolone 500 mg 4 ‐ 6 weeks + ibuprofen 400 mg 4 weeks. Possibly oral, once a day Co‐interventions: None | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score (subscores) Time points measured: Not reported Time points reported: Only mean follow‐up times reported Group 1: 7.13 ± 2.51 months Group 2: 6.01 ± 1.52 months Group 3: 6.93 ± 2.12 months Subgroups: None | |
| Funding sources | Not reported | |
| Declarations of interest | Not reported | |
| Notes | "Improvement" was reported but this outcome was not defined in the report E‐mail: [email protected], [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) | High risk | Open‐label study |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | High risk | Only subscores were reported, not total NIH‐CPSI score |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date November 2011 ‐ end date January 2013 Setting: outpatient ‐ single centre ‐ national Country: Iran | |
| Participants | Inclusion criteria: men aged ≥ 18 years old with type III CP/CPPS, refractory to a 4‐ to 6‐week treatment with antibiotics, alpha blockers and muscle relaxants; with a pain and urinary score ≥10 and a pain score ≥8 using the NIH‐CPSI score Exclusion criteria: “previous or concurrent malignancy; inflammatory bowel disease; history of pelvic radiation and systemic or intravesical chemotherapy; previous pelvic and rectal surgery or trauma; presence of a neurological disorder affecting the bladder; bladder stones; urethral stricture; history of prostate surgery or biopsy; urinary tract infection or sexually transmitted disease in the last 3 months; neuromuscular junction disorder; consumption of anticongestives, anticholinergics or any medicine affecting the neuromuscular junction in the last 2 weeks; coagulopathy; intake of antiplatelet agents in the last 10 – 14 days, anticoagulants in the last 7 days or steroids or sex hormones in the last 4 months; opioid or drug or alcohol abuse during the last 5 years; serum PSA level > age‐specific range; abnormal digital rectal examination with family history of prostate cancer; serum creatinine > 2 mg/dl; abnormal urinary cytology with haematuria; post‐voiding urine residue > 200 mL; indwelling urinary catheter, cystostomy or nephrostomy; penile prosthesis or artificial urinary sphincter; previous injection of or hypersensitivity to botulinum toxin A" Sample size: 60 Age (years): Group 1 mean 42.67 (SD 11.24); Group 2 mean 38.17 (SD 11.77) Baseline NIH‐CPSI score: Group 1 = 34.97 (SD 4.81); Group 2 = 33.20 (SD 5.19) All participants were men | |
| Interventions | Group 1 (n = 30): 100 IU of Botulinum Toxin A (BTA) was diluted in 2 ml of normal saline and administrated for prostate volumes < 30 mL. For volumes 30 ‐ 60 ml, 200 UI were used Group 2 (n = 30): Placebo injections of 2 mL of normal saline injections Co‐interventions: all participants received 10 mL of lidocaine gel (2%) into the urethra 10 minutes before injections at 3 different sites of each lateral lobe, using a 23‐gauge endoscopic needle and rigid cystoscopy. A 16‐F Foley catheter was fixed for 12 hours after injection and oral ciprofloxacin 500 mg twice a day was administered for 7 days. All medications for CP/CPPS were discontinued from 1 month | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 1, 3 and 6 months Time points reported: baseline, 1, 3 and 6 months Urinary Symptoms How measured: AUA‐symptom score Time points measured: baseline, 1, 3 and 6 months Time points reported: baseline, 1, 3 and 6 months Adverse events How measured: Narratively | |
| Funding sources | “not available” | |
| Declarations of interest | None declared | |
| Notes | Clinical Trial registry:IRCT201208051853N8 Contact information: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “In a 1:1 randomization schedule, the patients were divided into two equal groups:” Comment: Unclear how the random sequence generation was generated |
| Allocation concealment (selection bias) | Low risk | Quote: "Concealed numbered envelopes were used for group assignment, which were opened after gathering the baseline values." Comment: Low risk of bias for allocation concealment |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: “One examiner, blinded to the randomization schedule and treatment arrangement, evaluated all patients at baseline and follow‐up visits.” Comment: Patients were blinded with the use of placebo |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: “One examiner, blinded to the randomization schedule and treatment arrangement, evaluated all patients at baseline and follow‐up visits.” Comment: Patients were blinded with the use of placebo |
| Incomplete outcome data (attrition bias) | Low risk | Quote: “All patients completed the follow‐up program and we had no missing data.” |
| Selective reporting (reporting bias) | Low risk | All outcomes matched those in the clinical trial registry |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date June 2014 ‐ end date January 2015 Setting: outpatient ‐ single centre ‐ national Country: Italy | |
| Participants | Inclusion criteria: men aged 22 to 61 years with complaints of chronic pelvic pain for at least 6 months; IPSS score > 13 at visit 1; pain domain of NIH‐CPSI higher than 1 at visit 1; total PSA lower than 4 ng/ml; with symptoms compatible with category III CP/CPPS according to the NIH criteria Exclusion criteria: men affected by major comorbidities, with known anatomical abnormalities of the urinary tract or with evidence of other urological diseases, and with residual urine volume > 50 ml resulting from bladder outlet obstruction; reported allergy to the drugs administered during the trial, who had recently undergone oral or parenteral treatment or who were currently using prophylactic antibiotic drugs or finasteride, or both; men with positive tests for sexually transmitted diseases Sample size: 44 participants Age (years): Overall mean 41.32 (SD 1.69) NIH‐CPSI baseline score: Not available All participants were men | |
| Interventions | Group 1 (n = 22): Palmitoylethanolamide 300 mg plus alpha‐lipoic acid 300 mg (Peanase®), 2 capsules daily for 12 weeks Group 2 (n = 22): Serenoa repens at 320 mg, 1 capsule daily for 12 weeks Co‐interventions: "At time‐point V2 (visit two), after complete clinical and microbiological assessments, patients received a full course of pharmacological therapy." (prior to the experimental drug administration) | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline and 12 weeks Time points reported: baseline and 12 weeks (graphically) Urinary Symptoms How measured: IPSS Time points measured: baseline and 12 weeks Time points reported: baseline and 12 weeks (graphically) Sexual Dysfunction How measured: IIEF5 Time points measured: baseline and 12 weeks Time points reported: baseline and 12 weeks (graphically) Adverse Events How measured: Narrative | |
| Funding sources | Not available | |
| Declarations of interest | “No conflict of interest declared” | |
| Notes | We contacted the study author for the missing information: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We contacted study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We contacted study authors |
| Blinding of participants and personnel (performance bias) | High risk | Single‐blind trial, it was not specified who was blinded. We contacted study authors |
| Blinding of outcome assessment (detection bias) | Unclear risk | Single‐blind trial, it was not specified who was blinded. We have no information regarding participant blinding. We contacted study authors |
| Incomplete outcome data (attrition bias) | Unclear risk | All outcomes: follow‐up data are not available. We contacted study authors |
| Selective reporting (reporting bias) | High risk | No protocol available, outcome data were only presented graphically. We contacted study authors |
| Other bias | Unclear risk | Baseline data poorly reported (narrative fashion). We contacted study authors |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date January 2009 ‐ end date December 2012 Setting: outpatient ‐ single centre ‐ national Country: Italy | |
| Participants | Inclusion criteria: men >18 years old with pelvic or perineal pain, or both, and sexual dysfunction during at least 3 of the previous 6 months, with a score of at least 15 on the NIH‐CPSI Exclusion criteria: men with bacterial prostatitis, urethritis, urethral stricture, neurogenic bladder, those previously treated with antidepressants, with hepatic insufficiency, a history of alcohol use or evidence of chronic liver disease, and severe orthostatic hypotension Sample size: 38 Age (years): Group 1 mean 47 (SD 13); Group 2 mean 46.6 (SD 12.2) Baseline NIH‐CPSI score: Group 1 mean 25.1 (SD 3.7); Group 2 mean 24.25 (SD 8.4) All participants were men | |
| Interventions | Group 1 (n = 20): oral duloxetine hydrochloride 60 mg/day. Duloxetine dose was escalated during the first 15 days. Duration of treatment: 16 weeks Group 2 (n = 18): only co‐interventions Co‐interventions: tamsulosin 0.4 mg/day, saw palmetto 320 mg/day during 16 weeks | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline and 16 weeks Time points reported: baseline and 16 weeks Sexual Dysfunction How measured: International Index of Erectile Function‐5 Time points measured: baseline and 16 weeks Time points reported: baseline and 16 weeks Anxiety and depression How measured: Hamilton Anxiety Rating Scale (HAM‐A) Time points measured: baseline and 16 weeks Time points reported: baseline and 16 weeks Anxiety and depression How measured: Hamilton Depression Rating Scale (HAM‐D) Time points measured: baseline and 16 weeks Time points reported: baseline and 16 weeks Adverse events How measured: Narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | E‐mail: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study reported “block randomization”, but there is no information about its sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | High risk | The study was not blinded (visibly different interventions) |
| Blinding of outcome assessment (detection bias) | High risk | The study was not blinded (visibly different interventions) |
| Incomplete outcome data (attrition bias) | Unclear risk | Quote: “Four patients in group 1 stopped taking duloxetine due to intolerable adverse effects (nausea, sleep disturbances, sedation) within 1 month after the beginning of the study". Comment: It is unclear if all outcome measures include these participants (unbalanced attrition) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient ‐ single centre ‐ national Country: UK | |
| Participants | Inclusion criteria: men aged 18 to 60 with symptoms suggestive of CP/CPPS Exclusion criteria: chronic bacterial prostatitis (4‐glass test), those with a diagnosis of > 2 years, who had taken medication in the past 4 weeks, HIV positive, prostatic carcinoma, allergic to zafirlukast or doxycycline, renal or hepatic impairment, men with a poor command of English and those with known eosinophilia or severe allergic rhinitis Sample size: 20 men Age (years): Group 1 mean 36.4 (SD 13.9); Group 2 mean 35.1 (SD 9.5) Baseline NIH‐CPSI score: Not available All participants were men | |
| Interventions | Group 1 (n = 10): Zafirlukast 20 mg twice a day for 4 weeks Group 2 (n = 7): Placebo tablets twice a day for 4 weeks Co‐interventions: doxycycline 100 mg twice daily for 4 weeks was administered to all participants | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score (subscores) Time points measured: baseline, 4 weeks Time points reported: baseline, 4 weeks Adverse events How measured: Narratively | |
| Funding sources | The Prostate Research Campaign UK and AstraZeneca, UK | |
| Declarations of interest | Not available | |
| Notes | Total score was not available Email: david.goldmeie@st‐marys.nhs.uk | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A randomisation list was used to create permuted blocks of different sizes |
| Allocation concealment (selection bias) | Low risk | Central allocation by pharmacy |
| Blinding of participants and personnel (performance bias) | Low risk | The participants and personnel were blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: 3/7 participants (30%) were lost at follow‐up (missing outcome data) in the placebo arm |
| Selective reporting (reporting bias) | Unclear risk | Protocol was not available. Total NIH‐CPSI score was not reported |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: not available Setting: outpatient ‐ single centre ‐ national Country: USA | |
| Participants | Inclusion criteria: Men aged 18 – 65 years with CPPS type IIIA or IIIB were identified from the prostatitis clinic; at least 15 in the NIH‐CPSI score; all men had extensive previous treatment Exclusion criteria: active urinary tract infection, genitourinary malignancy, suicidal or overtly psychotic behaviour, post‐surgical pain, pain from another source in the genito‐urinary tract (e.g. renal calculi), history of radiation therapy, or history of genitourinary tuberculosis Sample size: 29 randomised Age (years): Group 1 mean 47 (range 25 ‐ 77); Group 2 54 (range 25 ‐ 80) NIH‐CPSI baseline score: Group 1 = 27.4 (SD 5.5); Group 2 = 24.5 (SD 5.2) All participants were men | |
| Interventions | Group 1 (n = 13): onabotulinum toxin A was injected after sterile preparation of the perineal skin: 100 U BTX‐A was diluted in 2 ml normal saline and the injection was guided using an electromyography needle to instil the preparation into the bulbospongiosus muscle and the mid‐bulbospongiosus muscle Group 2 (n = 16): saline injection using the same procedure Co‐interventions: not described | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline and at 1 and 2 months Time points reported: baseline, 1 and 2 months (for the cross‐over phase) Adverse events How measured: narratively | |
| Funding sources | Not available | |
| Declarations of interest | Reported as “none” | |
| Notes | 10 participants in the saline injection group switched to open‐label Botulinum toxin A injection and these results are the one presented at 2 months C. C. Yang. E‐mail: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “Randomization took place via random number generation” |
| Allocation concealment (selection bias) | Low risk | “blinded packaging by the research pharmacy.” |
| Blinding of participants and personnel (performance bias) | High risk | No information provided about blinding. Participants in the placebo group chose Botulinum injection at 1 month and “Patients were noted to have decreased spasm of the perineal musculature following injection” |
| Blinding of outcome assessment (detection bias) | High risk | No information provided about blinding. Participants in the placebo group chose Botulinum injection at 1 month and “Patients were noted to have decreased spasm of the perineal musculature following injection” |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data was available for all participants. |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available |
| Other bias | Low risk | No other bias detected |
| Methods | Study design: parallel group randomised trial Study dates: start date May 1997 ‐ end date October 1999 Setting: outpatient, national, single centre Country: Turkey | |
| Participants | Inclusion criteria: diagnosed with CPPS Exclusion criteria: not specified Sample size: 91 recruited, probably 89 randomised Age (years): mean 39.6 (overall range 17‐48) NIH‐CPSI baseline score (PSSI score): Group 1 9.61 (SD 1.61) Group 2 9.27 (SD 1.88) All participants were men | |
| Interventions | Group 1 (n = 39): terazosin 2 mg daily for three months Group 2 (n = 30): placebo for three months Co‐interventions: preventive measures (frequent sexual intercourse, avoid spicy food and constipation) | |
| Outcomes | Prostatitis Symptoms How measured: PSSI Time points measured: before and after the treatment Time points reported: before and after treatment Adverse events How measured: Narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | Address for correspondence: Osman Gül, MD, Y. Ziraat Mahallesi 14. sokak No: 8/C (Ok‐Ya Tıp Merkezi), Dı¸skapı, Ankara, Turkey Phone: +90 312 342 34 94; Fax: +90 312 384 02 83 91 participants were recruited, but then the authors say that 42 and 47 (89 participants) were randomised in two groups. It is not stated the reason for the two missing participants. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | Unclear risk | No information available |
| Blinding of outcome assessment (detection bias) | Unclear risk | They used placebo tablets but there is not mention of blinding |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: 8/47 (17.0%) in terazosin and 12/42 (28.6%) in placebo lost to follow‐up |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias identified |
| Methods | Study design: parallel group randomised trial Study dates: start date August 2012 ‐ end date August 2013 Setting: single center in China, outpatient Country: China | |
| Participants | Inclusion criteria: Men aged 18‐56. Prostatic fluid examination: Lecithin body disappear or decrease in number, normal or raised WBC count, normal urine test, and prostatic fluid culture negative. Discomfort during urination. NIH‐CPSI pain subscore >= 10. Men who had not taken alpha‐blockers, steroids, or antibiotics recently. Exclusion criteria: Men aged below 18 or above 56. Patients who had serious primary diseases of the liver, kidney, cerebral blood vessels, cardiovascular system, or hematopoietic system. Patients who had BPH, prostate cancer, or serious psychiatric disorders. Patients who were allergic to the drugs concerning the trial. Patients who did not take medications according to trial protocol or whose treatment effect could not be evaluated. Patients who did no meet inclusion criteria. Sample size: 96 patients randomized Age (years): Intervention group: range 24‐48. Mean ± SD: 32.4 ‐ 5.8 Control group: range 25‐46. Mean ± SD: 31.9 ‐ 5.5 NIH‐CPSI baseline score: Intervention group: 27.63 ± 9.52 Control group: 27.16 ± 9.36 Sex (M/F): All participants were men. | |
| Interventions | Intervention group: bazheng decoction. 320mL daily (160mL in the morning and 160mL in the evening). The course of treatment was 7 days. Treatment effect was observed after 2 treatment courses. Control group: only the co‐intervention Co‐interventions:Tamsolusin 0.2mg daily | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score Time points measured: Baseline, week 2 Time points reported: Baseline, week 2 Subgroups:None. Adverse events Not reported. | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | A composite clinical and biochemical outcome (white blood cells) is reported to indicate “improvement”. No contact information available. This study was written in Chinese. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "(In Chinese) 96 patients were randomly allocated to control group (48 patients) and trial group (48 patients) using random number table method". |
| Allocation concealment (selection bias) | Unclear risk | No information available. |
| Blinding of participants and personnel (performance bias) | High risk | Visibly different intervention, blinding was unlikely |
| Blinding of outcome assessment (detection bias) | High risk | Visibly different intervention, blinding was unlikely |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date 2009 ‐ end date 2013 Setting: outpatient ‐ multicentre ‐ national Country: Japan | |
| Participants | Inclusion criteria: criteria (Propert) men aged 20 ‐ 80 with symptoms of pelvic pain for 3 months or more before study, with a total NIH‐CPSI score ≥ 15 points Exclusion criteria: documented urinary tract infection, urethritis, epididymitis and sexually transmitted disease, prostate surgery, urogenital cancer, treatment with phytotherapeutic agents, alpha blockers and antimicrobials and the presence of bladder outlet obstruction Sample size: 100 randomised Age (years): Group 1 mean 50.1 (SD 13.7); Group 2 mean 53 (SD 14.6) Baseline NIH‐CPSI score: Group 1 mean 22.3 (SD 4.7); Group 2 mean 20.3 (SD 5.8) All participants were men | |
| Interventions | Group 1 (n = 50): Eviprostat 2 capsules 3 times a day (umbellate wintergreen Chimaphila umbellate extract 0.5 mg, aspen Populus tremula extract 0.5 mg, small pasque flower Pulsatilla pratensis extract 0.5 mg, field horsetail Equisetum arvense extract 1.5 mg and germ oil from wheat Tritium aestivum 15.0 mg) for 8 weeks Group 2 (n = 50): pollen extract 2 capsules 3 times a day (60 mg Cernitin T60 and 3 mg Cernitin GBX) for 8 weeks Co‐interventions: none described | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score Time points measured: baseline, 4 and 8 weeks Time points reported: baseline, 4 and 8 weeks Adverse events How measured: Narratively | |
| Funding sources | not available | |
| Declarations of interest | Quote: “The authors declare that they have no competing interests.” | |
| Notes | Clinical Trial record: UMIN000019618. We wrote to study authors to obtain information for 'Risk of bias' judgements and standard deviations for the primary outcome. We wrote to [email protected]‐u.ac.jp, but the email bounced. We calculated the SD from exact P values | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “The allocation manager randomly determined” Comment: Not enough information to make judgement. We wrote to study authors |
| Allocation concealment (selection bias) | Low risk | Allocation using sealed numbered envelopes |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study – identical regimens |
| Blinding of outcome assessment (detection bias) | Low risk | Double‐blind study – identical regimens |
| Incomplete outcome data (attrition bias) | High risk | 9/50 were excluded after randomisation in the Eviprostat group, 11/50 were excluded after randomisation in the Pollen Extract group |
| Selective reporting (reporting bias) | Low risk | Protocol was published (UMIN000019618) and review outcomes were well described |
| Other bias | Low risk | No other sources of bias identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date January 2004 ‐ end date December 2004 Setting: outpatient ‐ single centre ‐ national Country: South Korea | |
| Participants | Inclusion criteria: men with a diagnosis of type III CP/CPPS, aged 20 ‐ 60 years old with experience of pelvic pain for > 3 months during the previous 6 months Exclusion criteria: benign prostatic hyperplasia, NIH consensus criteria (Propert), urinary tract surgery or previous treatment Sample size: 81 randomised Age (years): Group 1 mean 37.5 (range 30 ‐ 50); Group 2 mean 41.5 (range 23 ‐ 56) Group 3 mean 41.1 (range 27 ‐ 60) Baseline NIH‐CPSI score: Group 1 mean 22.6; Group 2 mean 22.4; Group 3 mean 24.1 All participants were men | |
| Interventions | Group 1 (n = 26): levofloxacin 100 mg twice daily for 6 weeks Group 2 (n = 26): doxazosin 4 mg once daily before sleep for 6 weeks Group 2 (n = 29): levofloxacin 100 mg twice daily plus doxazosin once daily for 6 weeks Co‐interventions: none described | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline and 6 weeks Time points reported: baseline and 6 weeks. No SD available (we wrote to study authors) Adverse events How measured: Narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | Contact information: Dr. Hyeon Jeong ([email protected]) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Visibly different intervention, blinding was unlikely |
| Blinding of outcome assessment (detection bias) | High risk | Visibly different intervention, blinding was unlikely |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date October 2007 ‐ end date October 2008 Setting: outpatient ‐ single centre ‐ national Country: China | |
| Participants | Inclusion criteria: individuals with pelvic pain for > 3 months, mostly in regions such as the perineum, penis, crissum, urethra, lumbosacral region, etc. Abnormal urination, such as urinary urgency, frequent urination, difficult urination, dysuria, nocturia, etc. Normal or elevated WBC in EPS or VB3, culture‐negative. NIH‐CPSI ≥ 10. Patients who had NOT been treated with NSAIDs or alpha blockers before Exclusion criteria: Patients with gonococcal urethritis or non‐gonococcal urethritis, prostate cancer, hyperplasia of prostate, narrowing of the urethra, urolithiasis, neurogenic bladder, neuropsychiatric disorders, etc., which affected the function of lower urinary tract Sample size: 115 Age (years): Group 1 Mean 31.64 (SD 5.96) Group 2 Mean 33.56 (SD 5.61) Group 3 Mean 32.23 (SD 4.41) NIH‐CPSI baseline score: Group 1 = 23.38 (SD 4.01); Group 2 = 21.58 (SD 3.96); Group 3 = 22.34 (SD 4.21) All participants were men | |
| Interventions | Group 1 (n = 40): Dexketoprofen trometamol (oral, 12.5 mg/tablet) 3 times a day Group 2 (n = 41): Indomethacin (oral, 25 mg) 3 times a day Group 3 (n = 42): only terazosin (see co‐interventions) Co‐interventions: Terazosin (oral, 2 mg) every night Participants in all 3 groups took medications for 4 weeks The second night, the dosage of terazosin for all groups was cut down to a half | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score and subscore Time points measured: before treatment, after treatment Time points reported: before treatment, after treatment (week 4) Subgroups: none Adverse events How measured: Narratively | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “115 patients were randomly allocated into 3 groups”, (p 826), but the method of randomisation was not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not described |
| Blinding of participants and personnel (performance bias) | High risk | Visibly different interventions. Blinding was unlikely |
| Blinding of outcome assessment (detection bias) | High risk | Visibly different interventions. Blinding was unlikely |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date January 2004 ‐ end date February 2005 Setting: outpatient ‐ single centre ‐ national Country: South Korea | |
| Participants | Inclusion criteria: men aged ≤ 45 years old with prostate volume ≤ 25 mL, with a clinical diagnosis of CP/CPPS (III) Exclusion criteria: not reported Total number of participants randomly assigned: 127 Age (years): not reported. Wrote to study authors NIH‐CPSI baseline score: Group 1 = 21.8 (SD 5.5); Group 2 = 21.5 (SD 7.4) All participants were men | |
| Interventions | Group 1 (n = 71): Terazosin 3 ‐ 4 mg/day during 12 weeks Group 2 (n = 56): No treatment Co‐interventions: levofloxacin 300 mg/day, tamiflunate 3 tablets/day for 12 weeks | |
| Outcomes | Prostatitis Symptoms How measured: NIH CPSI questionnaire score and subscores Time points measured: baseline and 12 weeks after treatment Time points reported: baseline and 12 weeks after treatment | |
| Funding sources | Ilyang Pharmaceutical | |
| Declarations of interest | Not reported | |
| Notes | Language of publication: Korean E‐mail: [email protected] (email bounced) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. Wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. Wrote to study authors |
| Blinding of participants and personnel (performance bias) | Unclear risk | No information available. Wrote to study authors |
| Blinding of outcome assessment (detection bias) | Unclear risk | No information available. Wrote to study authors |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available. Wrote to study authors |
| Other bias | Low risk | No other sources of bias detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: Not available Setting: outpatient ‐ single centre ‐ national Country: USA | |
| Participants | Inclusion criteria: (Consensus criteria Alexander 2004a) age > 18 years old suffering from pelvic pain or discomfort for at least 3 months during the 6 months before entry, NIH‐CPSI > 20 and pain subscore > 4 Exclusion criteria: urinary tract infection in the previous 6 months, a history of cancer, neurological disorders, previous urological surgery, diabetes, nephrolithiasis, urinary retention and no medical treatment recently prescribed (alpha blockers, antibiotics and anti‐inflammatory drugs for 4 weeks and 5‐alpha‐reductase inhibitors for at least 6 months) Sample size: 64 were randomised Age (years): Group 1 mean 42.9 (SD 11.2); Group 2 mean 43.5 (SD 10.1) Baseline NIH‐CPSI score: Group 1 mean 24.7 (5.3); Group 2 mean 23.9 (5.7) All participants were men | |
| Interventions | Group 1 (n = 32): Serenoa repens (saw palmetto) 325 mg daily for 12 months Group 2 (n = 32): finasteride 5 mg daily for 12 months Co‐interventions: not described | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 3, 6 and 12 months Time points reported: graphically; numerically in the abstract for total score Urinary Symptoms How measured: AUA score Time points measured: baseline, 3, 6 and 12 months Time points reported: subscore at 12 months Adverse events How measured: Narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | Main findings were presented graphically. The total number randomised and the available at follow‐up were available, but there is no information about how many were allocated to each group (we assumed 32 in each group). We wrote to study authors ([email protected]) but the email bounced | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | High risk | The study is described as single‐blind; but it is not specified who was blinded; participants or personnel could have been blinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | The study is described as single‐blind; but it is not specified who was blinded |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: there were 31, 29 and 28 participants evaluated at 3, 6 and 12 months, respectively, in the saw palmetto group, and 30, 28 and 28 were evaluated at 3, 6, 12 months in the finasteride group. Balanced attrition, at 3‐month follow‐up, missing outcome data < 10% |
| Selective reporting (reporting bias) | High risk | Most data were presented graphically, total NIH‐CPSI score in the abstract with no information on standard deviation. P values were selectively reported |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date June 2001 ‐ end date March 2002 Setting: Outpatient ‐ single centre ‐ national Country: South Korea | |
| Participants | Inclusion criteria: men aged ≥ 20 and ≤ 50 years old with symptoms of chronic prostatitis, with a clinical diagnosis of CP/CPPS (IIIb) Exclusion criteria: men with positive culture in urine or EPS, or positive culture in PCR (Chlamydia trachomatis, Trichomonas vaginalis, Mycoplasma hominis, Mycoplasma genitalium, Ureaplasma urealyticum) Total number of participants randomly assigned: 63 Age: not reported. Wrote to study authors NIH‐CPSI baseline score: Group 1 = 18.83 (SD 6.57); Group 2 = 18.00 (SD 5.81); Group 3 = 18.09 (SD 6.75) All participants were men | |
| Interventions | Group 1 (n = 18): tamsulosin (0.2 mg orally every day) Group 2 (n = 15): ibuprofen (600mg tid po) with Misoprostol (300 mcg orally 3 times a day) Group 3 (n = 22): tamsulosin (0.2 mg orally every day) + ibuprofen (600 mg orally 3 times a day) with Misoprostol (300 mcg orally 3 times a day) Duration: 8 weeks Co‐interventions: not described | |
| Outcomes | Prostatitis Symptoms How measured: NIH CPSI questionnaire Time points measured: baseline, 2 weeks, 4 weeks, 8 weeks, 12 weeks after treatment Time points reported: baseline, 12 weeks after treatment Adverse events: clinically significant (not defined) How measured: visit to outpatient clinic | |
| Funding sources | Not reported | |
| Declarations of interest | Not reported | |
| Notes | Language of publication: Korean Contact information. E‐mail: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomly assigned the patients consecutively”. Comment: The authors did not clarify the details in Korean. Wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. Wrote to study authors |
| Blinding of participants and personnel (performance bias) | Unclear risk | No information available. Wrote to study authors |
| Blinding of outcome assessment (detection bias) | Unclear risk | No information available. Wrote to study authors |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: missing outcome data in 8/63 due to follow‐up loss (no information on loss) |
| Selective reporting (reporting bias) | High risk | Analysis intentions are not available Favorable outcomes for the experimental intervention were likely to report in results (e.g. Qmax) and subdomains of NIH‐CPSI were omitted. Results of multiple measurements at different time points were omitted |
| Other bias | Unclear risk | No information of baseline characteristics |
| Methods | Study design: Parallel‐group randomised trial Study dates: Not reported Setting: Outpatient ‐ single centre ‐ national Country: South Korea | |
| Participants | Inclusion criteria: men aged ≥ 20 and ≤ 40 years old (mainly) with symptoms of discomfort or pain in the pelvic region for at least a 3‐month period, with negative urine or EPS culture. Participant has a clinical diagnosis of CP/CPPS (III). Participant with NIH‐CPSI has reported total score of 8 or more (pain domain must be 4 or more) Exclusion criteria: men with a history of neurogenic bladder or lower urinary tract surgery, or positive culture on urine or EPS culture. Men with prior treatment of chronic prostatitis or BPH. Men suspected prostate cancer or BPH on DRE Total number of participants randomly assigned: 46 Group 1 age (years): 40.8 ± 7.9 Group 2 age (years): 39.4 ± 8.2 NIH‐CPSI baseline score: Group 1 = 22.6 ± 4.2; Group 2 = 23.1 ± 5.2 All participants were men | |
| Interventions | Group 1 (n = 31): propiverine (20 mg, once daily) for 2 months Group 2 (n= 15): only co‐intervention for 2 months Co‐interventions: all participants received gatifloxacin 200 mg, twice a day | |
| Outcomes | Prostatitis Symptoms How measured: NIH CPSI questionnaire Time points measured: start, 1 month and 2 months Time points reported: start, 1 month and 2 months Subgroups: no Urinary Symptoms How measured: IPSS questionnaire Time points measured: start, 1 month and 2 months Time points reported: start, 1 month and 2 months Subgroups: no Adverse events How measured: Narratively | |
| Funding sources | Grant from Dankook University | |
| Declarations of interest | Not reported | |
| Notes | Language of publication: Korean Contact information: e‐mail: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. Wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. Wrote to study authors |
| Blinding of participants and personnel (performance bias) | Unclear risk | Single‐blinded: No information given, but usually participants were blinded in single‐blinded fashion. Wrote to study authors |
| Blinding of outcome assessment (detection bias) | Unclear risk | Single‐blinded: No information given, but usually outcome assessors were blinded in single‐blinded fashion. Wrote to study authors. |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Unclear risk | Analysis intentions are not available Clinically significant AEs were reported without the definition of which AEs counted as clinically significant. Clinically significant AEs were likely to be favourable outcomes to the experimental intervention. Wrote to study authors |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: 01 March 2008 to 28 February 2009 Setting: outpatient ‐ multicentre ‐ national Country: Republic of Korea | |
| Participants | Inclusion criteria: Age < 55 years. Diagnosis of chronic prostatitis/chronic pelvic pain syndrome; “pain or discomfort in the pelvic region for at least 3 months, a total score of at least 12 on the NIH‐CPSI and anticipated improvement of symptoms with therapy.” Exclusion criteria: "urinary tract infection determined by urine study, hypoechoic lesions on transrectal ultrasonography (TRUS), a serum PSA level of 3 ng/dL or more, or a history of disease that could have affected the results of this study” Sample size: 107 randomised Age (years): Group A: mean 45.7; Group B: mean 46.5; Group C: mean 46.1 Baseline NIH‐CPSI score: Group A: mean 23.6 ± 5.5; Group B: mean 23.9 ± 5.9; Group C: mean 23.17 ± 5.5 All participants were men | |
| Interventions | Group A (n = 40): Only co‐interventions Group B (n = 32): 50 mg of diclofenac twice daily for 12 weeks Group C (n = 28): 500 mg of ciprofloxacin twice daily for 12 weeks Co‐interventions: All participants underwent 2 weeks of wash‐out time from the first visit. All participants received tamsulosin 0.2 mg once daily for 12 weeks | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score and subscore Time points measured: baseline and 12 weeks after treatment Time points reported: baseline and 12 weeks after treatment Urinary symptoms How measured: International Prostate Symptom Score (IPSS) Time points measured: baseline and 12 weeks after treatment Time points reported: baseline and 12 weeks after treatment | |
| Funding sources | Not available | |
| Declarations of interest | none | |
| Notes | Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “Patients were enrolled to subgroup by the table of random sampling digit in each medical center.” Comment: No additional information provided. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information provided. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Quote: "We did not have placebo group, because the effects of each drugs such as a‐blocker, antibiotics and non‐steroidal anti‐inflammatory drugs were widely known to the standard drug for CP/CPPS treatment" |
| Blinding of outcome assessment (detection bias) | High risk | Open‐label study |
| Incomplete outcome data (attrition bias) | High risk | 5 participants excluded from analysis since they received “other anti‐inflammatory drugs or antibiotics while the study was in progress.” Additionaly, “Two patients opted for transurethral resection of the prostate.” Comment: No information available for what treatment arm they were assigned to |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors |
| Other bias | Unclear risk | Some baseline characteristics were missing. We wrote to study authors |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient ‐ multicentre ‐ national Country: South Korea | |
| Participants | Inclusion criteria: men aged 20 to 49 years old (to exclude benign prostate hyperplasia) diagnosed with CP/CPPS and with pelvic pain for 3 or more months, negative urine and prostate secretions cultures, post‐voiding residual volume < 100 mL, maximum urinary flow rate > 15 mL/sec or more and a total NIH‐CPSI score of 8 or more and an NIH‐CPSI pain score of 4 or more at baseline Exclusion criteria: prostate surgery, use of 5α‐reductase inhibitors for 3 or more months, anticholinergic use within 6 months of baseline, elevated PSA level (> 4.0 ng/mL), prostatic cancer, urethral stricture, diabetes mellitus, neurogenic bladder and hypersensitivity to ciprofloxacin or solifenacin Sample size: 96 randomised Age (years): Not available Baseline NIH‐CPSI score: Group 1 = 19.8 ± 6.4; Group 2 = 22.4 ± 6.0 All participants were men | |
| Interventions | Group 1 (n = 49): only co‐intervention Group 2 (n = 47): solifenacin 5 mg daily for 8 weeks Co‐interventions: ciprofloxacin 1000 mg daily for 8 weeks | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score and subscores Time points measured: baseline, 4 weeks and 8 weeks Time points reported: baseline, 4 weeks and 8 weeks Urinary Symptoms How measured: IPSS score Time points measured: baseline, 4 weeks and 8 weeks Time points reported: baseline, 4 weeks and 8 weeks Sexual Dysfunction How measured: IIEF score Time points measured: baseline, 4 weeks and 8 weeks Time points reported: baseline, 4 weeks and 8 weeks | |
| Funding sources | Not available | |
| Declarations of interest | Quote: “No potential conflict of interest relevant to this article was reported” | |
| Notes | Contact information: [email protected] (Hyung‐Jee Kim) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Single‐blind study. |
| Blinding of outcome assessment (detection bias) | High risk | Single‐blind study, it is not clear whether this included participants, however there were visibly different interventions. Blinding was unlikely (outcome assessors) |
| Incomplete outcome data (attrition bias) | High risk | The Methods section stated that they recruited 49 participants in the ciprofloxacin group and 47 in the ciprofloxacin+solifenacin group, but outcome data (all outcomes) were available for 40 and 47 participants respectively. The authors have not specified the reasons for missing outcome data. We wrote to them asking for information |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors |
| Other bias | Unclear risk | Baseline characteristics were not available. Small differences in baseline NIH‐CPSI score between groups |
| Methods | Study design: Parallel‐group randomised trial Study dates: Not available Setting: outpatient ‐ single centre ‐ national Country: South Korea | |
| Participants | Inclusion criteria: participants with CP/CPPS type III Exclusion criteria: symptoms lasting for < 3 months, proven urinary tract infection, invasive prostate‐related procedures (transurethral resection of the prostate, transurethral incision of the prostate, or transurethral needle ablation), LUTS without significant pain, significant signs and symptoms of obstructive voiding, or prostate volume of > 50 cm3 Sample size: 88 Age (years): Group 1 = 44.2 ± 6.9 years; Group 2 = 45.3 ± 7.0 years Baseline NIH‐CPSI score: Group 1 = 22.1 ± 1.5; Group 2 = 19.5 ± 1.6 All participants were men | |
| Interventions | Group 1 (n = 40): only co‐interventions Group 2 (n = 48): Mirodenafil 50 mg daily for 6 weeks Co‐interventions: All participants received levofloxacin 500 mg daily for w6eeks | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline and at 6 weeks Time points reported: baseline and at 6 weeks (change from baseline) Urinary Symptoms How measured: IPSS score Time points measured: baseline and at 6 weeks Time points reported: baseline and at 6 weeks (change from baseline) Sexual Dysfunction How measured: International Index of Erectile Function 5 (IIEF‐5) Time points measured: baseline and at 6 weeks Time points reported: baseline and at 6 weeks (change from baseline) Adverse Events How measured: Narratively only for group 2 | |
| Funding sources | Pusan National University Hospital Clinical Research Grants (2014) | |
| Declarations of interest | Not available | |
| Notes | Contact: Hyun Jun Park. E‐mail: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Single‐blind trial |
| Blinding of outcome assessment (detection bias) | Unclear risk | Single‐blind trial. No information about whether participants were blinded |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors |
| Other bias | Unclear risk | Small differences in baseline NIH‐CPSI score between groups |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date January 2004 ‐ end date November 2004 Setting: outpatient ‐ single centre ‐ national Country: Bosnia and Herzegovina | |
| Participants | Inclusion criteria: CP/CPPS was diagnosed after blood tests, urinary and sperm cultures Exclusion criteria: urinary tract infection, suspicion of prostate cancer, acute prostatitis, urethral disease, calculosis of the lower urinary tract, identified allergy to 1 of the medications, diagnosed psychiatric disorder, diabetes, kidney insufficiency, consumption of medications that affected voiding Sample size: 90 participants randomised Age (years): overall mean 40.3 (SD 8.23) NIH‐CPSI baseline score: overall mean 25.8 (SD 4.8) All participants were men | |
| Interventions | Group 1 (n not available): ciprofloxacin 2 doses of 500 mg daily for 30 days Group 2 (n not available): doxazosin 2 mg daily for 30 days Group 3 (n not available): ciprofloxacin + doxazosin daily for 30 days Co‐interventions: none described | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline and after treatment Time points reported: baseline and after treatment | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | Contact information: www.researchgate.net/scientific‐contributions/33546794_Benjamin_Kulovac | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “patients were distributed in the three groups at random” Comment: no other information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | Quote: “patients were distributed in the three groups at random” Comment: no other information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Quote: “Prospective, comparative, open, clinical, manipulative study” |
| Blinding of outcome assessment (detection bias) | High risk | Quote: “Prospective, comparative, open, clinical, manipulative study” |
| Incomplete outcome data (attrition bias) | Unclear risk | Quote: “Patient list was made for all the patients in whom all the results were included.” Comment: No follow‐up information was reported. We wrote to study authors |
| Selective reporting (reporting bias) | High risk | The number of participants in each group was not reported. Baseline subscores were measured but they were not reported after treatment |
| Other bias | Unclear risk | Limited baseline characteristics |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date May 1997 ‐ end date October 1998 Setting: outpatient ‐ single centre ‐ national Country: Italy | |
| Participants | Inclusion criteria: participants suffering from non‐bacterial prostatitis Exclusion criteria: patients with positive cultures of urine and prostate secretions; patients were evaluated to “exclude concurrent urinary diseases” Sample size: 80 patients randomised Age (years): mean 36.19 (SD 11.53) NIH‐CPSI baseline score: Not available All participants were men | |
| Interventions | Group 1 (n not available): terazosine 5 mg daily for 60 days Group 2 (n not available): Tamsulosin 0.4 mg daily for 60 days Group 3 (n not available): placebo daily for 60 days Co‐interventions: No antibiotics were administered. | |
| Outcomes | Prostatitis Symptoms How measured: PSSI (0 to 12 score) at 60 days Adverse events How measured: narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | Email not available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was done using a “computer‐generated list” |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | Low risk | Participants were blinded. Study doctors were blinded. Drugs were packaged in “anonymous boxes” |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded. Drugs were packaged in “anonymous boxes” |
| Incomplete outcome data (attrition bias) | Unclear risk | There is no information about how many participants had outcome data (all outcomes) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Unclear risk | There is no information about how many participants were in each group |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient ‐ single centre ‐ national Country: United Kindom | |
| Participants | Inclusion criteria: individuals with symptoms compatible with CPPS Exclusion criteria: individuals with urethritis or positive prostate culture, symptoms suggestive of benign prostatic hyperplasia or significant abnormalities on baseline bloods were excluded. Additionally, current treatment with an antidepressant or anxiolytic drug, history of seizures, or any history of hypersensitivity or intolerance to a selective serotonin reuptake inhibitor were exclusion criteria Sample size: 14 participants randomised Age (years): not available. We wrote to study authors Baseline NIH‐CPSI score (PSS score): Group 1 = 23.4; Group 2 = 28 All participants were men | |
| Interventions | Group 1 (n = 7): Sertraline 50 mg daily for 13 weeks. They were then unblinded and continued for a total of 26 weeks Group 2 (n = 7): Placebo. At 13 weeks they were offered the opportunity to cross over to sertraline Co‐interventions: Not described | |
| Outcomes | Prostatitis Symptoms How measured: validated prostatic symptom frequency (PSF) and prostatic symptom severity (PSS) score Time points measured: 6, 13, 26 weeks Time points reported: 13 weeks (blinded), 26 weeks (unblinded) Anxiety and Depression How measured: Hospital Anxiety and Depression (HAD) scale, and a psychosexual (Psex) questionnaire Time points measured: 6, 13, 26 weeks Time points reported: 13 weeks (blinded), 26 weeks (unblinded) Sexual Dysfunction How measured: Psychosexual (Psex) questionnaire (unclear validity) Time points measured: 6, 13, 26 weeks Time points reported: 13 weeks (blinded), 26 weeks (unblinded) Adverse Events How measured: Narratively | |
| Funding sources | Quote: “MSSVD for funding the manufacture of the matched placebo used in this study” | |
| Declarations of interest | None | |
| Notes | 14 participants were randomised and divided “equally”; we assume 7 in each group R A Lee, email: [email protected] (email bounced) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind study; it is not clear who was blinded. We wrote to study authors |
| Blinding of outcome assessment (detection bias) | Low risk | Participants seem to have been blinded due to the use of a matched placebo. |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: 1 participant was lost to follow‐up in the placebo group (very small trial) |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available. We wrote to study authors |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date October 2003 ‐ end date April 2004 Setting: outpatient ‐ single centre ‐ national Country: Korea | |
| Participants | Inclusion criteria: men aged 18 or older with CP/CPPS, NIH‐CPSI pain subscore of 4 or more, and a urinary subscore of 3 or less, with reported pain in the pelvic region for at least 3 months. Exclusion criteria: bacterial prostatitis, bacteriuria in the last three months, a history of cancer, neurological disorders, urological surgery, diabetes, abnormal digital rectal examination except for benign enlargement, nephrolithiasis, urinary retention and an abnormal peak urinary flow. Antibiotics, alpha blockers, a‐adrenergic agents, anticholinergic, antispasmodic or muscle relaxants, cimetidine, warfarin and herbal medications were interrupted 4 weeks prior the start of the study Sample size: 50 randomised Age (years): Group 1 mean 44.2 (SD 2.1); Group 2 mean 42.7 (SD 2.9) NIH‐CPSI (baseline): Group 1 mean 21.62 (SD 4.2); Group 2 mean 22.12 (SD 6.0) All participants were men | |
| Interventions | Group 1 (n = 25): terpene mixture rowatinex® 200 mg (pinene 31%, camphene 15%, anethol 4%, borneol 10%, cineol 3% and fenchone 4% in olive oil) 3 times a day during 6 weeks Group 2 (n = 25): ibuprofen 600 mg 3 times a day during 6 weeks Co‐interventions: all participants underwent a 2‐week wash‐out period from previous analgesic medications | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: during wash‐out period, “baseline”, 15 days and 6 weeks Time points reported: 6 weeks, other time points were reported graphically Adverse Events How measured: Narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | S. W. Kim. E‐mail: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | “Single blind”, at least participants or personnel were not blinded |
| Blinding of outcome assessment (detection bias) | High risk | “Single blind”, it’s not clear if participants were blinded. Visibly different interventions, blinding was unlikely |
| Incomplete outcome data (attrition bias) | Unclear risk | No information available. We wrote to study authors |
| Selective reporting (reporting bias) | High risk | Scores were presented graphically at most time points and in those who were reported, they did not include measure of variability (e.g. SD). We wrote to study authors |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group assignment randomised trial Study dates: not available Setting: outpatient ‐ single centre ‐ national Country: Finland | |
| Participants | Inclusion criteria: individuals who presented with subjective symptoms of pain and discomfort associated with the genitourinary tract and in the external genitalia, as well as irritative bladder symptoms; they were evaluated with urine sample after prostatic massage and included those who contained 10 or more white blood cells. All patients had received antibiotic regimens for their symptoms from their own doctors, with no relief Exclusion criteria: Urethritis, benign prostate hyperplasia, Evidence of prostate cancer, history of infection in the urinary tract Sample size: 41 Age (years): Placebo 46 years (range 33 ‐ 61); Finasteride 47 (range 32 ‐ 61) Baseline NIH‐CPSI score: not available All participants were men | |
| Interventions | Placebo (n = 10): placebo 12 months. Medication was taken once‐a‐day in the morning before breakfast Finasteride (n = 31): finasteride 5 mg daily for 12 months. Medication was taken once‐a‐day in the morning before breakfast Co‐interventions: ketoprofene was provided to both group for pain relief. All participated had a 4‐week placebo tablets run‐in period | |
| Outcomes | Prostatitis Symptoms How measured: PSSI Time points measured: −1 (baseline), 0, 1, 3, 6, 12 months Time points reported: −1 (baseline), 0, 1, 3, 6, 12 months Urinary Symptoms How measured: IPSS Time points measured: −1 (baseline), 0, 1, 3, 6, 12 months Time points reported: −1 (baseline), 0, 1, 3, 6, 12 months Adverse Events How measured: Narratively | |
| Funding sources | None | |
| Declarations of interest | None | |
| Notes | Randomisation was 3:1. No contact information available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | Unclear risk | “Double blind”, It was not specified who was blinded |
| Blinding of outcome assessment (detection bias) | Unclear risk | “Double blind”, It was not specified who was blinded |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: 12% attrition in finasteride group, 20% attrition in placebo group |
| Selective reporting (reporting bias) | High risk | No protocol available. The rate of sexual adverse events was reported only in the finasteride group (narratively) |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date June 2002 ‐ end date December 2002 Setting: outpatient ‐ single centre ‐ national Country: China | |
| Participants | Inclusion criteria: Age 20 to 50, patients who had symptoms of chronic prostatitis; course of disease: 3 months to 5 years, NIH‐CPSI ≥ 10, in the past 1 month, no history of antibiotics treatment because of infection of the urinary tract or any other reasons; in the past 1 week, patients did not take any drugs for treating chronic prostatitis or drugs that affect urination Exclusion criteria: Patients with neurogenic bladder, narrowing of the urethra, benign prostate hyperplasia, prostate cancer, urinary infection, tuberculosis, calculi, or other diseases that affect urination; Patients with serious diabetes, cardiovascular diseases, or insufficiency of the liver or kidney; Patients with psychiatric disorders, habitual diarrhoea, or inflammatory bowel disease Sample size: 125 participants, 76 with type III prostatitis Age (years): Overall 20 ˜ 50; mean 32.7 Baseline NIH‐CPSI score: Group 1 = 25.45 (SD 5.82); Group 2 = 22.87 (SD 5.79) All participants were men | |
| Interventions | Group 1 (n = 41 with type III prostatitis): QianLieAnShuan (Prostat), rectal administration, 1 tablet each time, administer once every night for 30 days Group 2 (n = 35 with type III prostatitis): Substrate for QianLieAnShuan (placebo), rectal administration, 1 tablet each time, administer once every night for 30 days Co‐interventions: A wash‐out period of 7 days for all participants. Discontinue any other medications for prostatitis, including antibiotics, alpha blockers, TCM drugs, herbal medications, etc. Discontinue any other treatment options | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI Time points measured: before treatment, Day 30 Time points reported: before treatment, Day 30 Adverse Events How measured: Narratively | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese. No contact information available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomized, double‐blinded, placebo‐controlled, and multi‐center clinical trial” and “patients were randomly divided into trial group and control group” Comment: the exact method of randomisation is unknown |
| Allocation concealment (selection bias) | Unclear risk | Quote: “randomized, double‐blinded, placebo‐controlled, and multi‐center clinical trial”. Comment: The exact method for allocation concealment is not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | Blinding of participants and personnel is not described |
| Blinding of outcome assessment (detection bias) | Unclear risk | Blinding of outcome assessment is not described |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: 1 patient in the active treatment group was lost to follow‐up, very low attrition |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date July 2004 ‐ end date January 2006 Setting: outpatient ‐ single centre ‐ national Country: China | |
| Participants | Inclusion criteria: Patients who meet the diagnostic criteria for Chronic Pelvic Pain Syndrome. 2‐glass test: urine microscopy and culture for bacteria negative. Prostate fluid negative for Chlamydia, mycoplasma, fungi, and trichomonas. WBC in prostate fluid < 10/HP AND seminal WBC < 5/HP Exclusion criteria: Age > 40. Patients with tuberculosis of the prostate. Patients with prostate cancer Sample size: 108 participants Age (years): Group 1 (20 ˜ 40, Mean ± SD: 30.6 ± 5.3); Group 2 (21 ˜ 44, Mean ± SD: 29.3 ± 5.1) Baseline NIH‐CPSI score: Group 1 mean 26.3 (SD 4.9); Group 2 mean 25.8 (SD 5.1) All participants were men | |
| Interventions | Group 1 (n = 56): Ingredients of 1 dose of Tiaoshen Tonglin decoction were decocted by machine, and packed into 2 bags (150 mL each), taking 1 bag (150mL) in the morning on an empty stomach and the other before going to bed, for 60 days Group 2 (n = 52): Terazosin hydrochloride tablet 2 mg once a day (before going to bed) for 60 days Co‐interventions: All other medications discontinued | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: before treatment, after treatment Time points reported: before treatment (60 days) Subgroups: none Adverse Events How measured: Narratively | |
| Funding sources | Key Project of the Science and Technology Department of Hebei Province (Project No. 052761143) | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese. Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “108 patients … were stratified by course of disease(≤ 3 months, 3˜12 months, ≥12 months) and Maximum Flow Rate(MFR, ≥15mL/s, <15mL/s), numbered according to date of admission, and randomly assigned to two groups by referring to random sequence chart” (p 252). Comment: We wrote to study authors. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not described. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Masking of participants and personnel was not described. However, considering the visible difference between the two interventions, masking was unlikely |
| Blinding of outcome assessment (detection bias) | High risk | Blinding was unlikely (visibly different interventions) |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date October 2010 ‐ end date March 2011 Setting: outpatient ‐ single centre ‐ national Country: China | |
| Participants | Inclusion criteria: Patients were included with reference to NIH‐CPSI, i.e. patients with long‐term and recurrent pain or discomfort of the pelvic area, with or without urinary symptoms or sexual dysfunction, course of disease > 3 months, “Two‐glass test” culture for bacteria negative Exclusion criteria: Patients with other diseases of the lower urinary tract, psychiatric disorders or other serious conditions Sample size: 257 patients (37 lost to follow‐up) Age (years): Total (18 ˜ 42, mean ± SD: 30.6 ± 6.4) Baseline NIH‐CPSI score (mean±SD): Group 1 = 25.9 ± 5.8; Group 2 = 23.7 ± 6.7; Group 3 = 24.8 ± 7.2 All participants were men | |
| Interventions | Group 1 (n = 98): Qianlieping capsule (2.0 g per capsule), by mouth, 3 times a day. Tamsulosin (0.2 mg), by mouth, once a day, for 6 weeks Group 2 (n = 56): Tamsulosin (0.2 mg), by mouth, once a day for 6 weeks Group 3 (n = 66 ): Qianlieping capsule (2.0 g per capsule), by mouth, 3 times a day for 6 weeks Co‐interventions: Not mentioned | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI, number of lecithin body in prostatic fluid Time points measured: before treatment, after treatment Time points reported: before treatment, after treatment (Week 6) Subgroups: none | |
| Funding sources | 1. Special project in Henan‐provincial Key Disciplines and Specialties of Traditional Chinese Medicine Academic Leaders Cultivation Program (2013ZY03032) 2. Zhengzhou Municipal Projects on Science and Technology Innovation Team (121PCXTD) | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese. Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “Patients were randomly allocated into 3 groups upon complete informed consent” (p 857) Comment: The method of randomisation was not reported. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not described. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Masking of participants and personnel was not described. However, it is easy for participants to know which intervention they are taking |
| Blinding of outcome assessment (detection bias) | High risk | Blinding could not be achieved when assessing participant‐reported outcomes |
| Incomplete outcome data (attrition bias) | High risk | Outcome data (all outcomes) of 37/257 patients were missing (14% attrition). Not specified study arm |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: not available Setting: outpatient ‐ single centre ‐ national Country: China | |
| Participants | Inclusion criteria: Patients met the diagnostic criteria for CP/CPPS, complicated by erectile dysfunction; NIH‐CPSI symptoms subscore ≥ 9, EPS culture negative; IIEF‐5 ≤ 21 Exclusion criteria: Prostate tumour, other urogenital diseases, diabetes, hyperlipidaemia, chronic insufficiency of the liver or the kidney, severe disorders of cardiovascular system, infectious diseases, severe neurologic or psychiatric disorders Sample size: 138 participants Age (years): Group 1, trial group: 24 ˜ 51, mean 36; Group 2, control group: 23 ˜ 53, mean 38 NIH‐CPSI baseline score: Group 1 = 28.3 ± 4.5; Group 2 = 27.4 ± 7.6 All participants were men | |
| Interventions | Group 1 (n = 70): Starting from Week 5: Vardenafil 10 mg once a day, taken orally, 30 mins before having sexual intercourse, for 8 weeks Group 2 (n = 68): Only co‐interventions Co‐interventions: Huafenqinutang orally twice a day; course of treatment: 8 weeks Discontinue any drugs for treating CP/CPPS and ED 2 weeks before and during the trial, including antibiotics, alpha blockers, TCM drugs, herbal medications, antidepressants, or neuropsychiatric medications | |
| Outcomes | Prostatitis Symptoms How measured: drop in NIH‐CPSI (significantly effective: 60% ‐ 80%; effective: 30% ‐ 60%; not effective: < 30%) Time points measured: the end of Week 4, the end of Week 8 Time points reported: the end of Week 4, the end of Week 8 Subgroups: None Sexual Dysfunction How measured: IIEF‐5 (significantly effective: > 21; effective: 15 ‐ 20; not effective: < 10 ‐ 15) Time points measured: the end of Week 4, the end of Week 8 Time points reported: the end of Week 4, the end of Week 8 Subgroups: None Adverse events How measured: Narratively | |
| Funding sources | Not reported | |
| Declarations of interest | Not reported | |
| Notes | This study was written in Chinese Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “138 patients were randomized to 2 groups” (in Chinese) Comment: It is unclear what method of randomisation was used. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Not described. However, considering the visible difference between the interventions of the 2 groups (2 drugs vs 1 drug), blinding could not be achieved |
| Blinding of outcome assessment (detection bias) | High risk | Blinding of outcome assessment is not described. In participant‐reported outcomes, however, participants are not blinded |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date October 2000 ‐ end date March 2001 Setting: outpatient ‐ single centre ‐ national Country: China | |
| Participants | Inclusion criteria: Patients who met the criteria of non‐bacterial prostatitis (NIH‐IIIa) according to the first International Prostatitis Collaborative Network Workshop (November 5 ‐ 6, 1998, Washington DC); Age > 20 AND at least 3 months after clinical diagnosis (pelvic or perineal pain or discomfort within the 3 months before inclusion is required) Exclusion criteria: Patients with infection of the urinary system or positive urine culture results, benign prostate hyperplasia, suspected prostate cancer (screening by PSA); Patients with other acute diseases or severe diseases of the heart, brain, liver, kidney or haematopoietic system Sample size: 60 participants Age (years): Group 1 “Bensylyt (Phenoxybenzamine Hydrochloride)” (22 ˜ 48, Mean ± SD: 39.8 ± 4.5) Group 2 “flavoxate hydrochloride” (23 ˜ 49, Mean ± SD: 38.5 ± 4.2) Group 3 “placebo” (23 ˜ 48, Mean ± SD: 39.1 ± 4.3) Baseline NIH‐CPSI score: Group 1 = 21.95 (SD 3.49); Group 2 = 21.75 (SD 4.27); Group 3 = 21.85 (SD 3.95) All participants were men | |
| Interventions | Group 1 (n = 20): Placebo tablet (compound tablet of starch and Vitamin C, looks the same as Bensylyt) orally, twice a day for 1 month Group 2 (n = 20): Phenoxybenzamine (Bensylyt) tablet (10 mg) orally, twice a day for 1 month Group 3 (n = 20): Flavoxate hydrochloride (200mg) orally, three times a day for 1 month Co‐interventions: During the trial, all other options or medications for treating prostatitis were discontinued, including physical therapy, antibiotics, traditional Chinese medicine, etc. | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI Time points measured: before treatment, week 2, week 4, after treatment Time points reported: before treatment, after treatment Subgroups: none Adverse Events Possible adverse events were described but not their incidence | |
| Funding sources | Special project of Ministry of Health (WKZ‐2000‐16) | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese No contact information available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “starting from one particular row, the numbers (which do not end with zero) on a random sequence chart were assigned to patients according to their date of admission; patients assigned to numbers that end with 1˜3, 4˜6, and 7˜9 were assigned to Group I, II, and III respectively.” (p 170) |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not described |
| Blinding of participants and personnel (performance bias) | High risk | In the Discussion, Quote: “when designing this trial, we intended to do a double‐blinded, randomized, placebo‐controlled prospective clinical trial. However, due to technical problems, the placebo used in this trial was a combination of starch and Vitamin C, with the same appearance and method of administration as phenoxybenzamine hydrochloride tablet. We failed to acquire a placebo which has the same appearance and usage as flavoxate HCI‐neptumus.” (p 171) |
| Blinding of outcome assessment (detection bias) | High risk | Blinding could not be achieved when assessing participant‐reported outcomes |
| Incomplete outcome data (attrition bias) | High risk | 3/20 participants in placebo group dropped out because of serious symptoms of the disease. Outcome data (all outcome) not available for these participants. (p 170) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date January 2016 ‐ end date August 2016 Setting: outpatient ‐ single centre ‐ national Country: Italy | |
| Participants | Inclusion criteria: participants with CP/CPPS Exclusion criteria: patients who received medical treatment for lower urinary tract symptoms, major concomitant diseases and with residual urine volume > 50 ml were not included in this study Sample size: total randomised not available Age (years): not available NIH‐CPSI baseline score: not available All participants were men | |
| Interventions | Group 1 (n = 29): Deprox 500 mg 2 tablets a day for 6 weeks Group 2 (n = 34): Serenoa repens 320 mg 1 tablet a day for 6 weeks Co‐interventions: not available | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 6 weeks Time points reported: baseline, 6 weeks Urinary Symptoms How measured: IPSS score Time points measured: baseline, 6 weeks Time points reported: baseline, 6 weeks | |
| Funding sources | None | |
| Declarations of interest | Not available | |
| Notes | Abstract only | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | High risk | The interventions were visibly different, blinding was unlikely |
| Blinding of outcome assessment (detection bias) | High risk | The interventions were visibly different, blinding was unlikely |
| Incomplete outcome data (attrition bias) | Unclear risk | Abstract only (no information available) |
| Selective reporting (reporting bias) | Unclear risk | Abstract only (no information available) |
| Other bias | Unclear risk | Abstract only (no information available) |
| Methods | Study design: parallel group randomized trial Study dates: start date March 2016 – end date June 2016 Setting: outpatient, national, single centre Country: Italy | |
| Participants | Inclusion criteria: the presence of persistent pelvic pain for at least 3 months, in the 6 months preceding the study according to the EAU guidelines; NIH‐CPSI pain score greater than 7 and a negative Meares‐Stamey test. Exclusion criteria: age < 18 or > 65 years old; presence of anatomical abnormalities of the urinary tract system; additional urologic diseases; post‐void residual urine volume > 50 cc; known allergy to the experimental treatment; participants who underwent (< 4 weeks) oral or parenteral antibiotics treatment or who were using prophylactic antibiotic treatment (< 4 weeks); positivity to Chlamydia Trachomatis test, Ureaplasma Urealyticum, Neisseria Gonorrhoeae, Herpes Virus (HSV 1/2) and Human Papillomavirus (HPV). Sample size: 54 participants randomized Age (years): Group 1 mean (SD) 34 ± 5.9 years Group 2 mean (SD) 33.7 ± 4.62 years NIH‐CPSI baseline score: Group 1 mean (SD) 25.67 ± 1.62 Group 2 mean (SD) 25.96 ± 1.63 Sex (M/F): All participants were men | |
| Interventions | Group 1 (n = 27): flower pollen extract, two tablets in a single daily dose for four weeks: each dose contained 1 g of pollen extract and B1, B6, B2, B 9, B12, and PP vitamins. Group 2 (n = 27): quercetin (500 mg) complex twice daily for four weeks Co‐interventions: none described | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score Time points measured: baseline, 4 weeks Time points reported: baseline, 4 weeks Urinary symptoms How measured: IPSS score Time points measured: baseline, 4 weeks Time points reported: baseline, 4 weeks Adverse events How measured: Narratively | |
| Funding sources | The study states “non‐sponsored”. | |
| Declarations of interest | The authors declare no conflict of interest. | |
| Notes | Contact information: Angela Maurizi, MD (Corresponding Author) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors. |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors. |
| Blinding of participants and personnel (performance bias) | High risk | Only the patients were blinded to the type of treatment. |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: "The patients enrolled were blinded to the type of treatment." |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants. |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available. |
| Other bias | Low risk | No other sources of bias were identified. |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient ‐ multicentre ‐ national Country: Finland | |
| Participants | Inclusion criteria: participants fulfilling the diagnostic criteria of type III CP/CPPS, NIH‐CPSI score > 11 and pain score > 4 Exclusion criteria: patients with a history of symptoms for < 3 months, proven urinary tract infection during the past year, antibiotic treatment during the preceding 6 months were excluded, those who had undergone invasive prostate‐related procedures (transurethral resection of the prostate, transurethral incision of the prostate, transurethral needle ablation), patients who reported lower urinary tract symptoms without significant pain, patients with significant signs and symptoms of obstructive voiding, and patients with prostate volumes > 40 cm3 were also excluded Sample size: 40 participants randomised Age (years): Group 1 mean 49; Group 2 mean 50 NIH‐CPSI baseline score: Group 1 mean 26; Group 2 mean 23 All participants were men | |
| Interventions | Group 1 (n = 19): alfuzosin 5 mg twice daily for 6 months Group 2 (n = 21): identical placebo in the same posology Co‐interventions: Quote: “Patients in the alfuzosin and placebo groups were allowed to take analgesics (ibuprofen, ketoprofen, diclofenac) up to three times daily for a maximum of 7 days during the 6‐month initial evaluation period, but antibiotics, 5‐alpha‐reductase inhibitors, and alpha‐blockers were not allowed in any of the three groups” | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 2, 4, 6, 9 and 12 months Time points reported: baseline, 6 months and 12 months, responders Adverse Events How measured: Narratively | |
| Funding sources | Partially funded by NIH‐NIDDK (R01 DK5374601) Quote: “The study drugs, including the placebo, were supplied by Leiras OY, Turku, Finland. The tablets were packed and blinded by the pharmacy of the University of Helsinki.” | |
| Declarations of interest | Quote: “J. C. Nickel is a paid consultant to, and study investigator funded by, Sanofi‐Synthelabo” | |
| Notes | This was a pilot study. A third group of 30 participants who refused randomisation was included in the study (data not included in this review). Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | No information available in the paper. The author communicated that the randomisation sequence was centrally generated |
| Allocation concealment (selection bias) | Low risk | No information available in the paper. The author communicated that the allocation was concealed |
| Blinding of participants and personnel (performance bias) | Low risk | Participants were blinded with the use of placebo. The author specified that personnel were blinded too |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded with the use of placebo |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: data were available: At 6 months: 17/19 participants in the alfuzosin group, 20/21 placebo At 12 months: 16/19 participants in the alfuzosin group, 20/21 placebo Unbalanced loss of outcome data |
| Selective reporting (reporting bias) | High risk | Some time points were not presented, and those presented had no standard deviation. Adverse events were presented as “No patients dropped out of the study because of an adverse event.” |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date Jan 2004 ‐ end date March 2005 Setting: outpatient ‐ single centre ‐ national Country: South Korea | |
| Participants | Inclusion criteria: men with symptoms of chronic prostatitis and without prior treatment of CP/CPPS. Participant has a clinical diagnosis of CP/CPPS (III). Participant with NIH‐CPSI has reported total score of 15 or more Exclusion criteria: Men with prior treatment of CP/CPPS or BPH, positive culture on urine or EPS culture, or prostate volume ≥ 30 gm on DRE Total number of participants randomly assigned: 54 Age: Group 1 age (years): 44.7 ± 7.5; Group 2 age (years): 45.8 ± 9.6 Baseline NIH‐CPSI score: Group 1 = 23.1 (SD 8.1); Group 2 = 23.9 (SD 8.3) All participants were men | |
| Interventions | Group 1: alfusozin (10 mg, once a day) for 2 months Group 2: only co‐interventions for 2 months Co‐interventions: levofloxacin alone (100 mg, 3 times a day) | |
| Outcomes | Prostatitis Symptoms How measured: NIH CPSI questionnaire Time points measured: baseline, 2 months after treatment Time points reported: baseline, 2 months after treatment Subgroups: no Adverse events How measured: Narratively | |
| Funding sources | Not reported | |
| Declarations of interest | Not reported | |
| Notes | Language of publication: Korean. Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of outcome assessment (detection bias) | Unclear risk | No information available. We wrote to study authors |
| Incomplete outcome data (attrition bias) | Unclear risk | No information available. We wrote to study authors |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available. We wrote to study authors |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date September 2006 ‐ end date March 2007 Setting: outpatient ‐ multicentre ‐ national Country: Italy | |
| Participants | Inclusion criteria: men with type IIIA CP/CPPS with NIH‐CPSI score > 15, WBC count > 10, age 20 ‐ 50 Exclusion criteria: proven urinary tract infection in the last year, antibiotic treatment or other medications (non‐steroidal anti‐inflammatory drugs or corticosteroids therapy, alfuzosin, tamsulosin, doxazosin, terazosin, anticholinergics, finasteride, or dutasteride) during the previous 6 months; prior prostate surgery; urogenital diseases; neurologic disease affecting the bladder Sample size: 102 patients randomised Age (years): Group 1 mean 40.96 (SD 8.33); Group 2 mean 35.9 (SD 6.76) NIH‐CPSI baseline score: Group 1 mean 27.45 (SD 5); Group 2 mean 27.76 (SD 3.3) All participants were men | |
| Interventions | Group 1 (n not available): a capsule of Profluss which consisted of 320 mg of S. repens oil extract (85%), 5 mg oil extracted lycopene (6%) 50 ug seleniated sodium for 8 weeks Group 2 (n not available): a capsule of 320 mg of S. repens for 8 weeks | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 4, 8 and 16 weeks Time points reported: baseline, 4, 8 and 16 weeks (graphically in the latter case) Urinary Symptoms How measured: IPSS score Time points measured: baseline and 16 weeks Time points reported: baseline and 16 weeks (graphically in the latter case) Adverse events How measured: Narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | Prof. Carlo Magno. E‐Mail [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Patients were divided by a computerized, centralized randomization into two equally sized groups” |
| Allocation concealment (selection bias) | Low risk | Quote: “Patients were divided by a computerized, centralized randomization into two equally sized groups” |
| Blinding of participants and personnel (performance bias) | Unclear risk | “Double‐Blind”, did not fully describe the masking process. We wrote to study authors |
| Blinding of outcome assessment (detection bias) | Unclear risk | “Double‐Blind”, did not fully describe the masking process. We wrote to study authors |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: “All enrolled patients completed this study.” |
| Selective reporting (reporting bias) | High risk | Primary outcome was presented without variability measurement; some time points were only presented graphically |
| Other bias | Unclear risk | Number of participants in each arm are not available. We wrote to study authors |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date June 2015 ‐ end date January 2016 Setting: outpatient ‐ single centre ‐ national Country: Italy | |
| Participants | Inclusion criteria: participants aged 20 to 50 with symptoms of pelvic pain for 3 months or more, negative 4‐glass test, Meares‐Stamley test and a total NIH‐CPSI score ≥ 15 Exclusion criteria: participants with urinary tract infections, sexually transmitted diseases, treatment with phytotherapeutic agents, alpha blockers, antibiotics and urogenital cancer Sample size: 55 participants Age (years): Group 1 median 32 (IQR 29 ‐ 38); Group 2 median 32 (IQR 28.75 ‐ 38.75) NIH‐CPSI baseline score: Group 1 median 20.5 (IQR 15 ‐ 24.5); Group 2 median 20 (IQR 15 ‐ 25) All participants were men | |
| Interventions | Group 1 (n = 27): rectal suppositories of curcumin extract 350 mg (95%) and calendula extract 80 mg, 1 suppository daily for 1 month Group 2 (n = 28): identical looking placebo suppositories in the same regimen Co‐interventions: not available | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline and 12 weeks Time points reported: baseline and 12 weeks Sexual Dysfunction How measured: IIEF score, PEDT score Time points measured: baseline and 12 weeks Time points reported: baseline and 12 weeks Adverse Events How measured: narrative, only in the placebo group | |
| Funding sources | None | |
| Declarations of interest | No conflict of interests declared | |
| Notes | None | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | High risk | Single‐blind study, only participants were blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: outcome data were available in 24/27 participants in group A and in 24/28 participants in group B |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient ‐ multicentre ‐ national Country: Canada | |
| Participants | Inclusion criteria: participants of 18 years of age who had experienced symptoms of discomfort or pain in the pelvic region for at least 3 months during the 6 months before study entry, and had no documented history of cystitis or positive cultures in the past 12 months and had not been receiving any antibiotic treatment for any reason in the previous month. In addition, all participants had to discontinue all analgesic medications for a minimum of 1 week and no changes in any prostatitis‐specific medication in the previous 4 weeks was allowed Exclusion criteria: see Alexander 2004 Sample size: 80 Age (years): Group 1 = 56,2 years (range 36 to 78); Group 2 = 56 years (39 to 77) Baseline NIH‐CPSI score: Group 1 = 21.3 (SD 7.2); Group 2 = 24.4 (SD 8.2) All participants were men | |
| Interventions | Group 1 (n = 35): placebo 1 tablet a day during 6 weeks Group 2 (n = 45): levofloxacin 500 mg a day during 6 weeks Co‐interventions: not described | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI Time points measured: baseline, 3, 6 weeks Time points reported: baseline, 3, 6 weeks Adverse Events How measured: Narratively | |
| Funding sources | Janssen‐Ortho Canada and the National Institutes of Health | |
| Declarations of interest | Not available | |
| Notes | Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “The medications were prepackaged according to a computer‐generated, randomized allocation schedule” |
| Allocation concealment (selection bias) | Low risk | Quote: “The medications were prepackaged according to a computer‐generated, randomized allocation schedule” |
| Blinding of participants and personnel (performance bias) | Unclear risk | “Double blind” study, but who was blinded is not defined (not specified whether personnel were blinded). We wrote to study authors and to the NIH/NIDDK but they could not provide additional data |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded with the use of placebo |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: 1 participant withdrew from the study (not specified study arm) |
| Selective reporting (reporting bias) | Low risk | Protocol not available. We wrote to study authors and to the NIH/NIDDK but they could not provide additional data |
| Other bias | Low risk | No other sources of bias detected |
| Methods | Study design: Parallel group randomised trial Study dates: study dates not available Setting: outpatient ‐ multicenter ‐ international Country: USA and Canada | |
| Participants | Inclusion criteria: patients > 18 years old, with symptoms of discomfort or pain in the pelvic region for at least 3 months during the 6 months before entry, with a score of 4 or more on the NIH‐CPSI average pain score (question 4) for baseline and week 1 Exclusion criteria: "1. Treatment with BCG, or has unilateral orchialgia without pelvic symptoms, active urethral stricture, neurological disease or disorder affecting the bladder, a history of prostate, bladder or urethral cancer, or has a history of pelvic radiation, systemic or intravesical chemotherapy. 6. Currently a user (including “recreational use”) of any illicit drugs, or has a history (within the past 5 years) of drug or alcohol abuse. Sample size: 161 participants randomised Age (years): Group 1 mean = 45.8 (SD 12); Group 2 mean = 44.1 (SD 12); Group 3 mean = 48 (SD 13.3) NIH‐CPSI baseline score: Group 1 mean = 22.9 (SD 5.2); Group 2 mean = 22.5 (SD 6.5); Group 3 mean = 20.5 (SD 6) All participants were men | |
| Interventions | Group 1 (n = 59): placebo (in the images of rofecoxib 25 and 50 mg) Group 2 (n = 53): rofecoxib 25 mg and placebo (image of 50 mg) Group 2 (n = 49): rofecoxib 50 mg and placebo (image of 25 mg) Co‐interventions: discontinue all previous medication 1 week before taking the study drug, with a 1 week of run‐in period with placebo. All participants were permitted to take up to 2.6 g of paracetamol for rescue analgesia (except the placebo run‐in period) | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: −1, 0, 3 and 6 weeks Time points reported: −1, 0, 3 and 6 weeks Adverse Events How measured: Narratively | |
| Funding sources | Supported by Merck Research Laboratories, White House Station, New Jersey | |
| Declarations of interest | Financial interest and/or other relationship with Merck, Pharmacia, Alza, Ortho‐McNeil, Janssen‐Ortho Canada, Bayer Canada, Sanofi Synthelabo Canada and Glaxo Smith Kline, Boehringer‐ Ingleheim, Zeneca, Abbott, Lilly, Sepracor, Pfizer, Vivus, Nexmed, Praecis, Watson, Unimed, Myriad, Macrochem, Interneuron, Yamanouchi | |
| Notes | Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Investigators received randomized blocks of study drug labeled with allocation numbers and balanced by treatment group” |
| Allocation concealment (selection bias) | Low risk | Quote: “Investigators received randomized blocks of study drug labeled with allocation numbers and balanced by treatment group” (Central allocation) |
| Blinding of participants and personnel (performance bias) | Low risk | “Double blind” study, but who was blinded is not defined (not specified whether personnel were blinded). We wrote to study authors: personnel were blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded with the use of placebo |
| Incomplete outcome data (attrition bias) | High risk | Quote: “Four patients were excluded from analysis of the primary end point, NIH‐CPSI pain score, because they provided no data during the 6‐week treatment period and the last value carried forward method was used to estimate data for 20 patients (6 to 7 per group)” |
| Selective reporting (reporting bias) | High risk | SD was not reported for the identified outcomes in the study |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient ‐ multicentre ‐ national Country: USA | |
| Participants | Inclusion criteria: men aged “≥ 18 years and had symptoms of discomfort or pain in the pelvic region for ≥ 3 months during the 6 months before entry.” With “presence of any white blood cells per high‐power microscopic field in the expressed prostatic secretion (EPS) or in the sediment of the post‐prostatic massage urine sample” Exclusion criteria: a history of cystitis with a positive culture of an uropathogen or previous positive culture of expressed prostatic secretion or semen; history of urinary tract cancer, genital herpes, inflammatory bowel disease. Participants who had received chemotherapy or radiation. Participants with unilateral orchialgia with no pelvic symptoms, with active urethral stricture, neurological disease or disorder affecting the bladder, with a neurological impairment or psychiatric disorder preventing his understanding of consent and his ability to comply with the protocol. Participants with a history of any sexually transmitted disease in the past 3 months, a history of TURP, or other transurethral intervention, balloon dilation of the prostate, open prostatectomy, or any other prostate surgery or treatment such as cryotherapy or thermal therapy. Patients who had previously or concurrently received 5a‐reductase therapy Sample size: 76 participants were randomised Age (years): Group 1 mean = 46.9 (SD 1.7, range 27 – 63); Group 2 mean = 41.7, (SD 2.1, range 26 – 71) NIH‐CPSI baseline score: Group 1 = 20.1 (SD 1.4); Group 2 = 22.5 (SD 1.6) All participants were men | |
| Interventions | Group 1 (n= 31): finasteride 5 mg daily for 6 months Group 2 (n= 33): matching placebo for 6 months Co‐interventions: Not available. | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 3 and 6 months Time points reported: baseline, 3 and 6 months Adverse Events How measured: Narratively | |
| Funding sources | Quote: “This study was funded by the University Grants Program (Merck Inc.) as an independent investigator initiated research project. All the authors Prostatitis Research Centers are funded by grants from the NIH/NIDDK” | |
| Declarations of interest | Quote: “J.C. Nickel is a study investigator/consultant; D.A. Shoskes is a study investigator. Source of funding: Investigator Initiated Independent Research Grant from Merck.” | |
| Notes | The report states that there were 76 participants, but only 64 were available for outcome assessment. There is no information about how many participants were lost to follow‐up in each study arm. Participants had a placebo run‐in period Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors and to the NIH/NIDDK but they could not provide additional data |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors and to the NIH/NIDDK but they could not provide additional data |
| Blinding of participants and personnel (performance bias) | Unclear risk | No information about blinding of personnel. Participants were blinded (placebo), We wrote to study authors and to the NIH/NIDDK but they could not provide additional data |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded with the use of placebo |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: the report states that there were 76 participants, but only 64 were available for outcome assessment. There is no information about how many participants were lost to follow‐up in each study arm |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors and to the NIH/NIDDK but they could not provide additional data |
| Other bias | Low risk | No other sources of bias identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date July 2000 ‐ end date May 2001 Setting: outpatient ‐ multicenter ‐ national Country: USA | |
| Participants | Inclusion criteria: men ≤ 55 years old with a diagnosis CP/CPPS; NIH‐CPSI score of 15 or more, pain subscore score of 8 or more at screening and baseline, and pain in the pelvic region for 3 or more months were required for study inclusion Exclusion criteria: participants with chronic or acute bacterial prostatitis, acute urinary retention within 4 weeks of screening, urethral stricture, abnormal digital rectal examination except for benign enlargement, bacteriuria within 3 months of screening, significant urogenital disease, prior prostate surgery or pelvic radiotherapy, abnormal serum chemistries or complete blood count, history of allergy to alpha‐adrenergic antagonists or hypersensitivity to tamsulosin, postural hypotension, cancer diagnosed within 5 years of baseline, finasteride use within 3 months, or significant cardiac, endocrine, or neurological disorders “Medications that could interfere with the study drug or influence symptoms of CP/CPPS were not permitted (such as alpha‐adrenergic blocking drugs, alpha‐adrenergic agents, drugs with anticholinergic activity, antispasmodics or muscle relaxants, parasympathomimetics or cholinomimetics, antibiotics, nonsteroidal anti‐inflammatory drugs, cimetidine, warfarin, herbal medications and intravesical bacillus Calmette‐Guerin)" Sample size: 58 participants randomised Age (years): Group 1 mean = 40.8 (range 21 ‐ 56); Group 2 mean = 40.9 (range 21 ‐ 54) NIH‐CPSI baseline score: Group 1 mean = 26.4 (SD 4.9); Group 2 mean = 26.2 (SD 6.5) All participants were men | |
| Interventions | Group 1 (n = 28): 0.4 mg tamsulosin HCl capsule daily for 6 weeks (days 1 to 45) after a 2‐week wash‐out period with placebo; 30 minutes after breakfast Group 2 (n = 30): matching placebo in the same posology Co‐interventions: Not available | |
| Outcomes | Prostatitis symptoms How measured: baseline, and 6 weeks (day 45) Time points measured: baseline, and 6 weeks (day 45) Adverse events How measured: narratively | |
| Funding sources | Personal communication with the author: Pfizer | |
| Declarations of interest | Not available | |
| Notes | Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “sequentially”, no other information available. We wrote to study authors and they confirmed it was generated at random |
| Allocation concealment (selection bias) | Low risk | No information available. We wrote to study authors and they confirmed that allocation was concealed |
| Blinding of participants and personnel (performance bias) | Low risk | Participants were blinded, not specified if study personnel were blinded. We wrote to study authors: personnel were blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded using placebo |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes (outcome assessment at 6 weeks) 5/30 participants did not complete study in the placebo group; 2 participants did not complete study in the tamsulosin group, but they included outcome data using "last observation carried forward" (LOCF) |
| Selective reporting (reporting bias) | Low risk | No protocol available. We wrote to study authors: all outcomes were prespecified and only short‐term data were obtained |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient ‐ multicentre ‐ international Country: Canada and USA | |
| Participants | Inclusion criteria: Men of 18 to 50 years old with a clinical diagnosis of CP/CPPS at least 3 months in duration, with a score > 6 on questions 1 to 4 and 3 or more on question 4 of the NIH‐ CPSI score Exclusion criteria: a history of cystitis with an associated positive urine culture, prostate, bladder or urethral cancer, genital herpes in the last year, “any sexually transmitted disease in the last 3 months, inflammatory bowel disease, pelvic radiation or chemotherapy, unilateral orchialgia without pelvic symptoms, active urethral stricture; neurological diseases or disorders of the bladder, surgery on or physical treatment of the prostate, renal failure, pelvic or rectal surgery other than hemorrhoidectomy, hemostasis disorders, occult blood in the stool, anticoagulant use, that is greater than 1 gm aspirin or opioids (United States Schedule II), medication for sexual dysfunction, sensitivity to PPS or capsule components and planned surgery during or within 4 weeks of the study.” Sample size: 100 participants randomised Age (years): Group 1 = 40.8 (range 21 ‐ 59); Group 2 = 37.5 (range 25 ‐ 55) NIH‐CPSI baseline score: Group 1 mean = 27.1 (SD 1.1); Group 2 mean = 25.8 (SD 1.13) All participants were men | |
| Interventions | Group 1 (n = 51): Oral pentosan polysulfate sodium capsules of 300 mg 3 times a day for 16 weeks Group 2 (n = 49): Identical‐looking placebo in the same doses for 16 weeks Co‐interventions: not available | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 4, 8, 12 and 16 weeks Time points reported: baseline, 4, 8, 12 and 16 weeks Adverse Events How measured: Narratively | |
| Funding sources | Ortho‐McNeil/Alza | |
| Declarations of interest | Financial interest and/or other relationship with Ortho‐McNeil, Bayer, Boehringer‐Ingelheim, Sanofi‐Synthelabo, Merck, Glaxo‐SmithKline, Farr Laboratories, Novartis, Abbott and Lilly/ICOS, Pfizer, and Abbott | |
| Notes | The study used stratified randomisation by disease severity but there was no subgroup analysis by disease severity. ClinicalTrial.gov identifier NCT00236990 refers to a similar study (see studies awaiting classification). Personal contact with the author (Dr. Nickel) could neither confirm nor reject that this is the registration record of the trial Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “Randomization was stratified according to symptom severity at screening, as measured by the SSI” Comment: no further information was provided |
| Allocation concealment (selection bias) | Unclear risk | Quote: “Randomization was stratified according to symptom severity at screening, as measured by the SSI” Comment: no further information was provided |
| Blinding of participants and personnel (performance bias) | Unclear risk | “double‐blind”, but it was not specified who was blinded (regarding personnel) |
| Blinding of outcome assessment (detection bias) | Low risk | “double‐blind”, but it was not specified who was blinded; however, identical‐looking placebo was used (participants were blinded) |
| Incomplete outcome data (attrition bias) | High risk | Quote: “Of the enrolled patients 73% completed the 16‐week study. Of 27 patients who did not complete the study 11 in the PPS group and 4 in the placebo group discontinued due to adverse events or intercurrent illnesses” |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available (See notes in Characteristics of study) |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial6 Study dates: start date February 2005 ‐ end date January 2008 Setting: outpatient ‐ multicentre ‐ internationalCountry: United States, Canada and Malaysia | |
| Participants | Inclusion criteria: participants who had signed the informed consent, male aged 18 years of older who had symptoms of discomfort or pain in the pelvic region for at least 6 weeks and symptoms being present in the last 2 years Exclusion criteria: Participants with positive gram‐negative or enterococcus culture of midstream urine, previous treatment with the study drug or other alpha blockers in the past 2 years, presence of a history of prostate, penile, testicular, bladder, or urethral cancer or has undergone pelvic radiation, systemic chemotherapy, or intravesical chemotherapy, moderate or severe hepatic impairment, severe renal sufficiency, severe or unstable cardiovascular, respiratory, hematological, endocrinological, neurological or other somatic disorders, those who had unilateral orchialgia without pelvic symptoms, active urethral stricture, or neurological disease or disorder affecting the bladder. Additional criteria were uninvestigated, significant haematuria, previous treatment with TURP, TUIP, TUIBN, TUMT, TUNA, balloon dilation of the prostate, open prostatectomy or any other prostate surgery or treatment such as cryotherapy or thermal therapy. Participants with neurological impairment or psychiatric disorder preventing his understanding of consent and his ability to comply with the protocol Due to medication interactions, those who were taking potent CYP3A4 inhibitors Sample size: 272 participants randomised Age (years): Group 1 mean = 40.1 (SD 12.3); Group 2 = 40.1 (SD 11.4) Baseline NIH‐CPSI score: Group 1 mean = 25.1 (SD 5.9); Group 2 = 23.8 (SD 6.3) All participants were men | |
| Interventions | Group 1 (n = 138): alfuzosin 10 mg once daily for 12 weeks Group 2 (n = 134): identical‐looking placebo of the same posology Co‐interventions: not specified | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline and 12 weeks Time points reported: baseline and 12 weeks Adverse events How measured: All were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Time points measured: baseline, 6 and 12 weeks Time points reported: baseline and 12 weeks Sexual Dysfunction How measured: IIEF and Male Sexual Health Questionnaire Time points measured: baseline and 12 weeks Time points reported: baseline and 12 weeks Quality of Life How measured: SF‐12 Time points measured: baseline and 12 weeks Time points reported: baseline and 12 weeks Depression and Anxiety How measured: Hospital Anxiety and Depression scale Time points measured: baseline and 12 weeks Time points reported: baseline and 12 weeks | |
| Funding sources | Quote: “sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases, and Sanofi‐Aventis provided the study drug and placebo at no cost. Sanofi‐Aventis was not involved in the design of the study, the analysis of the data, or the preparation of the manuscript” | |
| Declarations of interest | "Dr. Nickel reports receiving a lecture fee from Sanofi‐Aventis, consulting fees from Pfizer and Farr Labs, and research support from Allergan and American Medical Systems; Drs. O’Leary and Landis, receiving consulting and advising fees from Sanofi‐Aventis; Dr. Krieger, receiving consulting and advising fees from Pfizer; Dr. Alexander, receiving lecture fees from Boehringer Ingelheim; Dr. Shoskes, receiving consulting fees from Farr | |
| Notes | ClinicalTrials.gov number: NCT00103402. Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “with the use of a centrally controlled, web based data‐management system. A permuted‐block randomization procedure with randomly assigned block sizes of 4, 6, and 8 was used” |
| Allocation concealment (selection bias) | Low risk | Quote: “with the use of a centrally controlled, web based data‐management system. A permuted‐block randomization procedure with randomly assigned block sizes of 4, 6, and 8 was used” |
| Blinding of participants and personnel (performance bias) | Low risk | Participants received identical‐looking matched placebo and study investigators were unaware of the treatment assignments |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinding using placebo |
| Incomplete outcome data (attrition bias) | High risk | Loss of follow‐up CPSI Experimental 22 (15.9%) placebo 17/134 (13.6%)/ IIEF 28/138 (20.3%) placebo 25/134 (18.7%)/ SF12 Experimental 23/138 (16.7%) placebo 21/134 (15.7%)/ Depression Experimental 23/138 (16.7%) placebo 17/134 (12.7%) |
| Selective reporting (reporting bias) | Low risk | Outcomes matched those described in the clinical trial registry |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date September 2008 ‐ end date October 2009 Setting: outpatient ‐ multicentre ‐ national Country: Canada | |
| Participants | Inclusion criteria: Men at least 18 years of age, total NIH‐CPSI total score of 15, NIH‐CPSI pain score of 8 with pain in the pelvic region for at least 3 months prior to screening Exclusion criteria: previous experience with silodosin or alpha blockers or participated in an investigation in the last 30 days, ≥ 2 urinary tract infections within the previous 12 months, the presence of medical condition that in the opinion of the investigator precludes safe participation in the study or could confound the efficacy evaluation. Concurrent use of ketoconazole, or other known potent inhibitors of cytochrome P450 3A4 or any medication in the opinion of the investigator that precludes safe participation in the study or could confound the efficacy evaluation Sample size: 151 participants randomised Age (years): Group 1 mean = 49.2 (SD 13.3); Group 2 = 46.7 (SD 15.6); Group 3 = 49 (SD 11.6) NIH‐CPSI baseline score: Group 1 mean = 26 (SD 6.3); Group 2 mean = 26.8 (SD 5.9); Group 3 mean = 27.9 (SD 6.2) All participants were men | |
| Interventions | Group 1 (n = 52): Silodosin 4 mg daily for 12 weeks Group 2 (n = 45): Silodosin 8 mg daily for 12 weeks Group 3 (n = 54): Daily placebo Co‐interventions: Not specified | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 4, 8 and 12 weeks. Time points reported: baseline and 12 weeks Quality of life How measured: SF‐12 Time points measured: baseline, 4, 8 and 12 weeks. Time points reported: baseline and 12 weeks Adverse events How measured: Narratively | |
| Funding sources | Watson Laboratories, Inc | |
| Declarations of interest | Quote: “Financial interest and/or other relationship with GlaxoSmithKline, Johnson & Johnson, Pfizer, Watson, Taris Biomedical, Ferring, Farr Labs, Triton, Trillium Therapeutics and Astellas.” | |
| Notes | Clinical Trial Registry: NCT00740779 Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation schedule created by the sponsor. Not specified how it was generated. We wrote to study authors: the authors had no data on this topic |
| Allocation concealment (selection bias) | Low risk | Central pharmacy allocation |
| Blinding of participants and personnel (performance bias) | Low risk | Both participants and personnel were blinded using placebo |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded using placebo |
| Incomplete outcome data (attrition bias) | Unclear risk | All outcomes: 43/52, 31/45 and 41/54 of participants in each study arm completed the study. The results are reported as intention‐to‐treat, but it is unclear if all participants had outcome data at 12 weeks follow‐up. We wrote to study authors: the authors had no data on this topic |
| Selective reporting (reporting bias) | Low risk | Outcomes matched clinical trial registry |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date March 2009 ‐ start date March 2010 Setting: outpatient ‐ multicentre ‐ national Country: USA, Canada, France, Sweden and Switzerland | |
| Participants | Inclusion criteria: Diagnosis of chronic prostatitis, men at least 18 years of age, moderate to severe chronic prostatitis (total NIH‐CPSI score ≥ 15), with an average pain score above a predefined level; to use contraception Exclusion criteria: History of symptoms for < 3 of the last 6 months, recurrent urinary tract infections, or genito‐urinary cancer, use of finasteride or dutasteride within 6 months, history of hepatitis B, C or human immunodeficiency virus (HIV) Sample size: 62 participants randomised Age (years): Group 1 mean = 50.5 (SD 11.9); Group 2 mean = 43.2 (SD 13.5) NIH‐CPSI baseline score: Not available. All participants were men | |
| Interventions | Group 1 (n = 30): single dose of 20 mg of IV tanezumab (monoclonal antibody directed against the pain‐mediating neurotrophin, nerve growth factor) Group 2 (n = 32): single dose of IV placebo Co‐interventions: All other medications for CP/CPPS were permitted provided they had started using it > 3 months before screening | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 6 weeks, 16 weeks Time points reported: baseline, 6 weeks, 16 weeks Adverse events How measured: Narratively | |
| Funding sources | Quote: “This study was sponsored by Pfizer, Inc.; editorial/medical writing support was provided by Joseph Oleynek of UBC Scientific Solutions and was funded by Pfizer, Inc.” | |
| Declarations of interest | Quote: “J. C. Nickel is a consultant/investigator for GlaxoSmithKline, Johnson & Johnson, Pfizer, Inc., Watson Pharmaceuticals, Ferring Pharmaceuticals, Tocris Bioscience, Farr Laboratories, Astellas Pharma, Triton Pharma, Trillium Therapeutics, and Eli Lilly; M. Pontari is a consultant for Eli Lilly and Azcan; D. A. Shoskes is a consultant for Farr Laboratories and an investor in, and receives compensation from, Triurol; G. Atkinson, I. W. Mills, and T. J. Crook are employees of, and hold stock or options, in Pfizer, Inc. J. N. Krieger declares that he has no relevant financial interests” | |
| Notes | This is part of a series of 3 trials Study A4091010 (ClinicalTrials.gov identifier: NCT00601484) tanezumab vs placebo in patients with IC/BPS Study A4091035 (ClinicalTrials.gov identifier: NCT00999518) tanezumab in 200 patients with IC/BPS. Study A4091019 (ClinicalTrials.govidentifier: NCT00826514) tanezumab vs placebo in male patients with CP/CPPS (data was extracted for this trial from three journal articles and the clinical trial record) Participants had a run‐in period Quote: “Patients were stratified according to their baseline mean daily NRS “average” pain (moderate, 4 to <7; severe, ≥7)”, this was not reported in outcomes Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomization "The subject’s daily pain numeric ratings scale (NRS) was collected via interactive voice response system (IVRS) commencing from the evening of the first day of the screening assessment period, and was used to assess inclusion into the study and to determine stratification at randomization" (Clinical trial record) EUCTR2008‐004861‐25‐SE. |
| Allocation concealment (selection bias) | Low risk | Central allocation (see above) |
| Blinding of participants and personnel (performance bias) | Low risk | Participants and personnel were blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded (using placebo) |
| Incomplete outcome data (attrition bias) | High risk | Outcome data (NIH‐CPSI score) were available for 25/30 and 26/32 of participants in Groups 1 and 2 respectively For adverse events, the denominator in the table mentions complete outcome data (low risk of bias) |
| Selective reporting (reporting bias) | High risk | NIH‐CPSI scores are not reported; only change from baseline. The report states that the measurement of NIH‐CPSI score extended to 16 weeks, but the report only mentions 6 weeks |
| Other bias | High risk | Editorial support was provided by Pfizer (industry bias) |
| Methods | Study design: Parallel‐group randomised trial Study dates: March 1983 to August 1983 Setting: outpatient ‐ multicentre ‐ national Country: Japan | |
| Participants | Inclusion criteria: Non‐specific chronic prostatitis patients. Diagnosis was determined by subjective symptoms, observation of the prostate palpation site, urethral secretion after the prostatic gland muscle and urine findings comprehensively Exclusion criteria: No detail Sample size: 76 Age (years): No details Baseline NIH‐CPSI score: not available. All participants were men | |
| Interventions | Group 1 (n = 32): PPCI (aminoacid preparation) 6 capsules daily and placebo C agent 6 tablets daily divided in three doses, one after each meal, 2 capsules and 2 tablets were administered concomitantly Group 2 (n = 30): C agent (probably pollen extract) 6 tablets a day, PPC placebo 6 capsules daily divided in three doses, one after each meal, 2 capsules and 2 tablets were administered concomitantly Co‐interventions: not available. Interventions probably lasted 4 weeks (follow‐up period) | |
| Outcomes | None of the outcomes prespecified in this review | |
| Funding sources | No details | |
| Declarations of interest | No details | |
| Notes | No email available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details in the text. Insufficient information to make judgement |
| Allocation concealment (selection bias) | Low risk | Test drugs and control drugs are randomly allocated and distributed by a control roller (Hirakata citizen Role of pharmacist Sano Yukiro Hospital). Central allocation |
| Blinding of participants and personnel (performance bias) | Low risk | Each drug had a placebo version, identical in appearance |
| Blinding of outcome assessment (detection bias) | Low risk | Each drug had a placebo version, identical in appearance |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: outcome data available for: Group 1: n = 32 (84.2%), Group 2: n = 30 (78.9%), Dropout: n = 14 |
| Selective reporting (reporting bias) | Unclear risk | The protocol was not available |
| Other bias | Low risk | No other sources of bias identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date November 2002 ‐ end date September 2003 Setting: outpatients ‐ single institute ‐ national Country: South Korea | |
| Participants | Inclusion criteria: men with symptoms of discomfort or pain in the pelvic region for at least a 3‐month period. Participant has a clinical diagnosis of CP/CPPS (IIIb). After 8 weeks conventional treatment (run‐in), men with symptom improvement and WBC < 10/HPF in EPS were included Exclusion criteria: Before 8 weeks conventional treatment, men with WBC ≥ 10/HPF in EPS, a positive urine analysis or culture, or positive EPS culture Total number of participants randomly assigned: 50 Age: Group 1 (years): 36.2 (24 ‐ 45); Group 2 (years): 35.2 (23 ‐47) Baseline NIH‐CPSI score: Group 1 = 23.1 (SD 4.4); Group 2 = 22.4 (SD 3.7) All participants were men | |
| Interventions | Group 1: Cranberry Juice (Ocean Spray®) 150 mL twice a day for 12 weeks Group 2: No treatment during the same time Co‐interventions: all participants underwent an 8 week‐run‐in period with levofloxacin 100m g 3 times a day, NSAID, Alpha blocker, behaviour therapy, and hot sitz bath. For non‐responders, 4 weeks of same treatments were added | |
| Outcomes | Prostatitis Symptoms How measured: NIH CPSI questionnaire Time points measured: baseline, 12 weeks after treatment Time points reported: baseline, 12 weeks after treatment Subgroups: no Adverse events How measured: Narratively | |
| Funding sources | Not reported | |
| Declarations of interest | Not reported | |
| Notes | Language of publication: Korean E‐mail: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Participants in the control group did not receive placebo, while those in the treatment group received intervention |
| Blinding of outcome assessment (detection bias) | High risk | Participants in the control group did not receive placebo, while those in the treatment group received intervention |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors |
| Other bias | High risk | Active run‐in (After 8 weeks conventional treatment, men with symptom improvement and WBC < 10/HPF in EPS were included) |
| Methods | Study design: Parallel‐group randomised trial Study dates: not available Setting: outpatient ‐ single centre ‐ national Country: South Korea | |
| Participants | Inclusion criteria: not available Exclusion criteria: not available Sample size: 78 Age (years): Group 1 = 41.2 ± 6.7 years Group 2 = 43.3 ± 7.1 years Baseline NIH‐CPSI score: not available All participants were men | |
| Interventions | Group 1 (n = 40): Only co‐intervention Group 2 (n = 38): tadalafil 10 mg daily for 4 weeks Co‐interventions: Levofloxacin 500 mg daily for 4 weeks | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline and at 4 weeks Time points reported: baseline and at 4 weeks Urinary Symptoms How measured: IPSS Time points measured: baseline and at 4 weeks Time points reported: baseline and at 4 weeks Sexual Dysfunction How measured: International Index of Erectile Function 5 (IIEF‐5) Time points measured: baseline and at 4 weeks Time points reported: baseline and at 4 weeks Adverse events How measured: Narratively | |
| Funding sources | None | |
| Declarations of interest | Not available | |
| Notes | We extracted this information from an abstract presentation; We contacted the author: Dr Park ([email protected]) and he mentioned that there was no publication available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | Unclear risk | No information available |
| Blinding of outcome assessment (detection bias) | Unclear risk | No information available |
| Incomplete outcome data (attrition bias) | Unclear risk | No information available |
| Selective reporting (reporting bias) | Unclear risk | No information available |
| Other bias | Unclear risk | No information available |
| Methods | Study design: Paralle‐ group randomised trial Study dates: study dates not available Setting: outpatient ‐ single centre ‐ national Country: South Korea | |
| Participants | Inclusion criteria: men with NIH diagnosis of CP/CPPS Exclusion criteria: not available Sample size: 86 participants randomised Age (years): Group 1 = 49.2 (SD 6.7); Group 2 = 48.3 (SD 7.1) NIH‐CPSI baseline score: Not available All participants were men | |
| Interventions | Group 1 (n = 40): only co‐interventions Group 2 (n = 46): tadalafil 5 mg/day for 6 weeks Co‐interventions: Levofloxacin 500 mg daily for 6 weeks | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score Time points measured: baseline, 6 weeks Time points reported: change from baseline at 6 weeks Urinary symptoms How measured: IPSS score Time points measured: baseline, 6 weeks Time points reported: change from baseline at 6 weeks Sexual dysfunction How measured: IIEF score Time points measured: baseline, 6 weeks Time points reported: change from baseline at 6 weeks Adverse events How measured: narrative, only in tadalafil arm | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | From abstract of the Proceedings of the EAU | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not available (abstract only) |
| Allocation concealment (selection bias) | Unclear risk | Not available (abstract only) |
| Blinding of participants and personnel (performance bias) | High risk | The abstract states “single‐blind” |
| Blinding of outcome assessment (detection bias) | Unclear risk | The abstract states “single blind”, but it is unclear how participants were blinded (visibly different interventions) |
| Incomplete outcome data (attrition bias) | Unclear risk | Not available (abstract only) |
| Selective reporting (reporting bias) | Unclear risk | Not available (abstract only) |
| Other bias | Unclear risk | Not available (abstract only) |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date August 1999 ‐ end date June 2002 Setting: outpatient ‐ possibly single centre ‐ national Country: China | |
| Participants | Inclusion criteria: Not clearly stated. Common characteristics included: perineal pain or discomfort, frequent urination, secretion at the opening of urethra; WBC/EPS > 10/HP, lecithin body < + + /HP, urine and EPS bacterial culture‐negative Exclusion criteria: Patients with narrowing of the urinary tract, hyperplasia of the prostate, or prostate tumour; patients with serious primary diseases of the cardiovascular system, cerebral‐vascular system, liver, kidney, or haemopoietic system; patients with psychiatric disorders; patients who did not meet the diagnostic criteria of non‐bacterial prostatitis, OR patients who did not take medications as instructed Sample size: 160 participants Age (years): Overall: 23 ˜ 49, Age 20 ˜ 29: 61 participants; age 30 ˜ 39: 72 participants; Age 40 ˜ 49: 27 participants Baseline NIH‐CPSI score: not available All participants were men | |
| Interventions | Group A (n = 40): Oral antiphlogistic medicinal granules, 1 pack, 3 times a day; Duration: 30 days Group B (n = 40): Oral antiphlogistic medicinal granules, 1 pack, 3 times a day, plus enema using anti‐inflammatory mixture 100 ‐ 150 mL, 33 ℃, 15 cm into the anus; Duration: 30 days Group C (n = 40): Oral antiphlogistic medicinal granules, 1 pack, 3 times a day, plusrectal administration of anti‐inflammatory capsule; Duration: 30 days Group D (n = 40): Oral antiphlogistic medicinal granules, 1 pack, 3 times a day, plusrectal administration of anti‐inflammatory capsule, plus water bath and fumigation‐and‐washing using anti‐inflammatory mixture 250 mL, 20 ‐ 30 mins; after that, irradiate the lower abdomen using microwave for 15 mins; Duration: 30 days Co‐interventions: Participants were instructed to discontinue all other treatment options during the trial and observational period | |
| Outcomes | There was no report of outcomes relevant to this review | |
| Funding sources | Shantou Municipal Special project in Science and Technology (2001‐52) | |
| Declarations of interest | Not mentioned | |
| Notes | A combination of subjective clinical parameters alongside laboratory findings formed a composite outcome (not relevant for our review) This study was written in Chinese. Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “(160 patients with CNP) were randomly assigned to 4 groups: A, B, C, and D” (in Chinese) Comment: However it is not clear what method was used |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment is not described |
| Blinding of participants and personnel (performance bias) | High risk | Blinding of participants and personnel is not described. However, the difference among the 4 interventions is visible, thus blinding cannot be achieved |
| Blinding of outcome assessment (detection bias) | High risk | Blinding of outcome assessment is not described. Self‐reported outcomes, participants are not blinded |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: Outpatient ‐ single centre ‐ national Country: Sweden | |
| Participants | Inclusion criteria: participants with a diagnosis of CP/CPPS Exclusion criteria: not available. Sample size: 54 participants randomised Age (years): Not available NIH‐CPSI baseline score: Not available All participants were men | |
| Interventions | Group 1 (n = 20): Placebo twice a day Group 2 (n = 18): Allopurinol 300 mg in the morning and placebo in the evening Group 2 (n = 16): Allopurinol 600 mg (in 2 doses of 300 mg) Treatment duration: after the 240‐day visit or 8 months of treatment Co‐interventions: not available | |
| Outcomes | No outcomes relevant to this review were reported (see Notes) | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | There is a reported 12‐point symptom scale. This scale evaluated symptoms of prostate discomfort. It’s not clear that it’s validated and does not include the domains of the NIH‐CPSI score. Other outcomes were PSA levels and urine parameters. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomised using a spreadsheet |
| Allocation concealment (selection bias) | Low risk | Central allocation (in another country) |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind study. Not specified if personnel were blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Double‐blind study. Participants were blinded using placebo |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: Only 34 out of the 54 patients completed the study |
| Selective reporting (reporting bias) | High risk | Most of the data are presented graphically |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date 2006 ‐ end date 2007 Setting: outpatient ‐ multicentre ‐ national Country: USA | |
| Participants | Inclusion criteria: men aged > 18 years, with symptoms of discomfort or pain in the pelvic region during at least 3 of the previous 6 months, and had a total score of at least 15 of 43 on the NIH‐CPSI at screening and randomisation visits approximately 2 weeks apart Exclusion criteria: kidney insufficiency, thrombocytopenia, allergy to any anti‐seizure medication, known sensitivity to pregabalin, treatment with thiazolidinedione or antidiabetic agents, NYHA class III or IV congestive heart failure, a history of thrombocytopenia or bleeding diathesis, and a history of alcohol abuse. “Participants were not excluded if they had previous treatment for CP/CPPS or for taking analgesics for another condition if they continued to have pelvic pain despite the analgesic therapy and had a score of at least 15 on the NIH‐CPSI. Previous treatment with gabapentin or pregabalin was allowed if it was completed at least 2 weeks before study enrolment.” Sample size: 324 Age (years): Group 1 mean = 48 (SD 13); Group 2 mean = 45.2 (SD 12.2) NIH‐CPSI baseline score: Group 1 mean = 26.2 (SD 5.6); Group 2 mean = 25.9 (SD 6.1) All participants were men | |
| Interventions | Group 1 (n = 218): pregabalin 150 mg/day (50 mg orally 3 times daily) for 2 weeks, then 300 mg/day (100 mg orally 3 times daily) for 2 weeks, and then 600 mg/d (200 mg orally 3 times daily) for 2 weeks Group 2 (n = 106): same escalation procedure with placebo Co‐interventions: If a participant could not tolerate the dosage increase, he was allowed to remain at the previous tolerated dosage | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, and 6 weeks Time points reported: baseline, and 6 weeks Quality of Life How measured: SF‐12 Time points measured: baseline, and 6 weeks Time points reported: baseline, and 6 weeks Anxiety and Depression How measured: Hospital Anxiety and Depression Scale (HADS) Time points measured: baseline, and 6 weeks Time points reported: baseline, and 6 weeks Sexual Dysfunction How measured: IIEF‐Sexual Health Inventory for Men (SHIM) Time points measured: baseline, and 6 weeks Time points reported: baseline, and 6 weeks Adverse Events How measured: Narratively | |
| Funding sources | Quote: “No author received compensation for the performance of this study except as salary support from a grant from the National Institutes of Health. This study was supported by cooperative agreements U01DK65209,UO1DK65268, U01 DK65297, U01 DK65187, U01 DK65277, U01 DK65189, U01 DK65174, U01 DK65266, U01 DK65257, U01 DK65186, and U01 DK65287 from the National Institute of Diabetes and Digestive and Kidney Diseases and the National Center for Minority Health and Health Disparities. Pregabalin and matching placebo capsules were provided by Pfizer Inc.” | |
| Declarations of interest | Consulting fees from Sanofi‐Aventis, Pfizer, GlaxoSmithKline, Pfizer, Bioness Inc, Boston Scientific, Allergan, Astella, Merck, Ortho Women’s Health, Farr Labs, Watson, Medtronic, NeurAxon, and Genyous Biomed, American Medical Systems, Roche, Triuro, Boehringer‐Ingelheim, Decode Genetics, Alita Pharmaceuticals, NovaBay Pharmaceuticals, Regeneron Pharm Inc, IMS Health, Exoxemis Inc, CombinatoRx Inc, Monitor Company Group LP and Advanstar Communications | |
| Notes | Clinicaltrials.gov Identifier: NCT00371033 Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “randomly assigned 2:1 in each clinical site via a centrally controlled Web‐based data management system to receive treatment with either pregabalin or matching placebo using a permuted block randomization” |
| Allocation concealment (selection bias) | Low risk | Quote: “randomly assigned 2:1 in each clinical site via a centrally controlled Web‐based data management system to receive treatment with either pregabalin or matching placebo using a permuted block randomization” |
| Blinding of participants and personnel (performance bias) | Low risk | Quote: “Study investigators and participants were unaware of treatment assignment” |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: “Study investigators and participants were unaware of treatment assignment” |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all but 8/218 and 3/106 participants in each group |
| Selective reporting (reporting bias) | Low risk | All outcomes matched the clinical registry |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient ‐ multicentre ‐ national Country: Austria | |
| Participants | Inclusion criteria: men with category A CP/CPPS Exclusion criteria: not available Sample size: 142 randomised Age (years): not available NIH‐CPSI baseline score: not available All participants were men | |
| Interventions | Group 1 (n = 72): Permixon (Serenoa repens) Group 2 (n = 70): Placebo Co‐interventions: not described | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 6 weeks, 12 weeks, 6, 12 and 18 months Time points reported: none Urinary Symptoms How measured: IPSS score Time points measured: baseline, 6 weeks, 12 weeks, 6, 12 and 18 months Time points reported: none | |
| Funding sources | None | |
| Declarations of interest | Not available | |
| Notes | These characteristics were extracted from an abstract. No full text was available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | Unclear risk | No information available |
| Blinding of outcome assessment (detection bias) | Unclear risk | No information available |
| Incomplete outcome data (attrition bias) | Unclear risk | No information available |
| Selective reporting (reporting bias) | Unclear risk | No information available |
| Other bias | Unclear risk | No information available |
| Methods | Study design: Parallel‐group randomised trial Study dates: Not reported Setting: Out patients ‐ single institute ‐ national Country: South Korea | |
| Participants | Inclusion criteria: men aged ≥ 20 and ≤ 40 years old (mainly) with symptoms of discomfort or pain in the pelvic region for at least a 3‐month period, with negative urine or EPS culture. Participant has a clinical diagnosis of CP/CPPS (III). Participant with NIH‐CPSI has reported total score of 8 or more (pain domain must be 4 or more) Exclusion criteria: men with a history of neurogenic bladder or lower urinary tract surgery, or positive culture on urine or EPS culture. Men with prior treatment of chronic prostatitis or BPH. Men suspected prostate ca. or BPH on DRE Total number of participants randomly assigned: 57 Age (years) Group 1 = 41.6 ± 9.2 (13 ‐ 53); Group 2 = 38.5 ± 7.8 (31 ‐ 57) The lower range of age for group 1 is 13 in the text, but the correct number might be correct number might be 31 (digit inversion) Baseline NIH‐CPSI score: Group 1 = 21.9 (1.3); Group 2 = 19.5 (1.2) All participants were men | |
| Interventions | Group 1: tosulfoxacin (150 mg, 3 times a day) and alfuzosin (10 mg/day) for 2 months Group 2: tosulfoxacin (150 mg, 3 times a day) for 2 months Co‐interventions: not available | |
| Outcomes | Prostatitis Symptoms How measured: NIH CPSI questionnaire Time points measured: start, 1 month and 2 months of the study Time points reported: start, 1 month and 2 months of the study Subgroups: no Urinary Symptoms How measured: IPSS questionnaire Time points measured: start, 1 month and 2 months of the study Time points reported: start, 1 month and 2 months of the study Subgroups: no Sexual Dysfunction How measured: IIEF‐5 questionnaire Time points measured: start, 1 month and 2 months of the study Time points reported: start, 1 month and 2 months of the study Subgroups: no Adverse events How measured: Narratively | |
| Funding sources | Grant of Dankook University | |
| Declarations of interest | Not reported | |
| Notes | Language of publication: Korean Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | Unclear risk | Single‐blinded: No information available.. We wrote to study authors |
| Blinding of outcome assessment (detection bias) | Unclear risk | Single‐blinded: No information available. We wrote to study authors |
| Incomplete outcome data (attrition bias) | Unclear risk | No information available. We wrote to study authors |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available.. We wrote to study authors |
| Other bias | Low risk | No other sources of bias detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: in 1994, exact dates not mentioned Setting: outpatient ‐ may be single centre ‐ national Country: China | |
| Participants | Inclusion criteria: Not mentioned. According to the paper, participants included should at least be nonbacterial prostatitis Exclusion criteria: Not mentioned Sample size: 60 participants Age (years): not reported Baseline NIH‐CPSI score: not available. All participants were men | |
| Interventions | Group 1 (n = 30): QianLieAnWan was administered orally 2 ˜ 3 times a day, 9 g each time for 30 days Group 2 (n = 30): QianLieKang tablet was administered regularly combined with thermotherapy for 30 days Co‐interventions: Not mentioned | |
| Outcomes | There was no report of outcomes relevant to this review | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | A combination of subjective clinical parameters alongside laboratory findings formed a composite outcome (not relevant for our review). This study was written in Chinese. No contact information available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “patients were randomly assigned to two groups”(in Chinese) Comment: the method for randomisation is not mentioned |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment is not described |
| Blinding of participants and personnel (performance bias) | High risk | Blinding of participants and personnel is not described. However, considering the visible difference between the 2 interventions (with or without massage), blinding was unlikely |
| Blinding of outcome assessment (detection bias) | High risk | Blinding of participants and personnel is not described. However, considering the visible difference between the two interventions (with or without massage), blinding was unlikely. |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for al participants |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient ‐ single centre ‐ national Country: USA | |
| Participants | Inclusion criteria: participants with CPPS for at least 6 months (they might not have met the 1999 consensus criteria for CP/CPPS) Exclusion criteria: positive culture in urine, prostatic secretion, urethral swab, first voided and midstream urine Sample size: 30 Age (years): Group 1 mean = 43.5 (SD 3.7); Group 2 mean = 46.2 (SD 4) NIH‐CPSI baseline score: Group 1 mean = 20.2 (SD 1.1); Group 2 mean = 21.0 (SD 1.8) All participants were men | |
| Interventions | Group 1 (n = 15): quercetin capsules 500 mg orally twice daily for 1 month Group 2 (n = 15): placebo capsules orally twice daily for 1 month Co‐interventions: not available | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline and 1 month Time points reported: baseline and 1 month Adverse Events How measured: Narratively | |
| Funding sources | No information available. | |
| Declarations of interest | Quote: “D. A. Shoskes and J. Rajfer own stock in companies that will benefit from sales of the supplements reported in this study.” | |
| Notes | A second open‐label period was continued with 17 participants, these results were not included in this review due to insufficiencies in the report. No contact information available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “Patients were randomized 1:1 in a double‐blind fashion” Comment: no other information available |
| Allocation concealment (selection bias) | Unclear risk | Quote: “Patients were randomized 1:1 in a double‐blind fashion” Comment: no other information available |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: “Patients were randomized 1:1 in a double‐blind fashion” Comment: it is not clear if personnel were blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: “Patients were randomized 1:1 in a double‐blind fashion” and “Both quercetin and placebo capsules were identical in appearance.” |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: 2/15 participants in the placebo group left due to worsening symptoms (no outcome data) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient ‐ single centre ‐ national Country: India | |
| Participants | Inclusion criteria: men diagnosed with chronic prostatitis or chronic pelvic pain syndrome, aged 15 to 65 years Exclusion criteria: not available Sample size: 68 participants randomised Age (years): not available NIH‐CPSI baseline score: not available All participants were men | |
| Interventions | Group 1 (n = 36): tadalafil 5 mg in once‐daily dose for 6 weeks plus standard medical therapy Group 2 (n = 32): standard medical therapy Co‐interventions: standard medical therapy comprised: levofloxacin 500 mg for 6 weeks and alfuzosin 10 mg for 6 weeks | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score Time points measured: baseline, 6 weeks Time points reported: 6 weeks (change from baseline with P value) Urinary symptoms How measured: IPSS score Time points measured: baseline, 6 weeks Time points reported: 6 weeks (change from baseline with P value) Sexual dysfunction How measured: baseline, IIEF score Time points measured: baseline, 6 weeks Time points reported: 6 weeks (change from baseline with P value) Adverse events How measured: narratively, reported by the author in personal communication | |
| Funding sources | None | |
| Declarations of interest | Not available | |
| Notes | The information was combined from an abstract from a medical conference and the clinical trial registry (CTRI/2016/08/007208). We wrote to study authors to complete the missing information: [email protected]. The author replied with additional information | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Author replied that it was generated by a computer |
| Allocation concealment (selection bias) | Low risk | Author replied that it was a central allocation |
| Blinding of participants and personnel (performance bias) | High risk | Author replied that the study was not blinded |
| Blinding of outcome assessment (detection bias) | High risk | Author replied that the study was not blinded |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: author replied that there was a "5‐10% fall out" |
| Selective reporting (reporting bias) | Unclear risk | No information available (abstract only) |
| Other bias | Unclear risk | No information available (abstract only) |
| Methods | Study design: Parallel‐group randomised trial Study dates: study dates not available Setting: outpatient Country: Russia | |
| Participants | Inclusion criteria: men with a diagnosis of CP/CPPS (type IIIa) for 2 years and more; age 25 ‐ 45; absence of urinary/reproductive tract infections; altered prostate secretion which is commonly found in CP/CPPS cases; symptoms of CP/CPPS Exclusion criteria: not specified Sample size: 64 patients were randomised Age (years): not reported NIH‐CPSI baseline total score: Group 1 mean = 25.66 (SD 6.55); Group 2 mean = 25.73 (SD 4.97) All participants were men | |
| Interventions | Group 1 (n = 29): terazosin orally, gradually increasing from 1 to 5 mg/day for 2 weeks, and then 8 weeks of 5 mg/day terazosin Group 2 (n = 22): placebo orally, imitation of the gradual increase of the active drug for 2 weeks, and then 8 weeks of placebo | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐total score and pain, urinary, and QoL domains Time points measured: baseline, 12 months Time points reported: baseline, 12 months Adverse events How measured: narratively | |
| Funding sources | None | |
| Declarations of interest | None | |
| Notes | Prior to randomisation, all recruited volunteers (n = 64) were treated with placebo for 2 weeks to identify placebo responses. So in total 64 participants entered the study, but the follow‐up was documented for 29 and 22 men in active treatment group and placebo group, respectively. No contact information available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information provided other than "patients were randomized into 2 groups.." statement |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | Low risk | The packaging for the 2 alternative treatments was identical with exception of labels A1 and A2 (for active treatment)/ B1 and B2 (for placebo). Neither participants nor investigators were aware about the labelling |
| Blinding of outcome assessment (detection bias) | Low risk | The packaging for the 2 alternative treatment was identical with exception of labels A1 and A2 (for active treatment)/ B1 and B2 (for placebo). Neither patients nor investigators were aware about the labelling |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: missing outcome data in 3/29 from active group vs 10/22 from placebo group |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Unclear risk | Some of baseline parameters (age, duration of symptoms) were not reported |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date May 2007 ‐ end date February 2008 Setting: Outpatient ‐ possibly single centre ‐ national Country: China | |
| Participants | Inclusion criteria: men aged 18 to 55 years old with symptoms of chronic prostatitis, such as frequent urination, discomfort urination, or perineal pain or discomfort, which lasted for at least 4 weeks, WBC/EPS ≥ 10/HP, decreased counting or disappearance of lecithin bodies, bacteria culture negative; routine urine analysis normal. Patients who did not take any other medication for chronic prostatitis or drugs that interfere with urination Exclusion criteria: Acute prostatitis, benign hyperplasia of the prostate, prostate cancer, neurogenic bladder, urethral malformation or narrowing or serious neurosis, stones of the ureter or bladder, inguinal hernia, inflammation of the ulna, varicocoele, epididymitis, or diseases of the colon or rectum, serious primary diseases of the heart, brain, liver and haemopoietic system, patients with “allergic constitution” or allergic to multiple drugs, patients who could not co‐operate, say, patients with mental disorders, patients who had any of the following conditions which could influence the effectiveness of treatment or the assessment of safety: (1) patients who did not meet the inclusion criteria; (2) patients who did not take medications as instructed; (3) patients whose effectiveness of treatment could not be assessed, or whose information is incomplete Sample size: 115 participants Age (years): Overall: 19 ˜ 47, Mean: 31.6 NIH‐CPSI baseline total score: Group 1 = 24.46 (SD 5.38); Group 2 = 23.51 (SD 4.86) All participants were men | |
| Interventions | Group 1 (n = 73): QianLieAnTong tablet (0.38 g per tablet), by mouth 3 times a day, 4 tablets each time for 1 month Group 2 (n = 42): Only co‐interventions Co‐interventions: Terazosin hydrochloride tablet (2 mg per tablet), by mouth once a day, 1 tablet each time; taking the tablet before going to bed No other medications or physical therapies allowed during the trial | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI global and subscore Time points measured: before treatment, after treatment Time points reported: before treatment, after treatment Subgroups: none Adverse Events How measured: Narratively | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese No contact information available. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “we conducted a randomized controlled clinical trial”. Comment: However, the method for randomisation is not described |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment is not described |
| Blinding of participants and personnel (performance bias) | High risk | Blinding of participants and personnel is not described. However, considering the visible differences between the two interventions, blinding cannot be achieved |
| Blinding of outcome assessment (detection bias) | High risk | Blinding of outcome assessment is not described. Self‐reported outcomes, participants are not blinded |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date March 2006 ‐ end date December 2006 Setting: outpatient ‐ possibly single centre ‐ national Country: China | |
| Participants | Inclusion criteria: men aged 20 to 50 years old, with a course of disease > 3 months, diagnosed with type III chronic prostatitis according to NIH‐1999 criteria and volunteered to participate in this clinical trial Exclusion criteria: type I, II or IV prostatitis, patients with haemorrhoids, anal fissure, anal fistula or other anorectal diseases. Patients with diseases of the urinary system, such as hyperplasia of the prostate, prostate cancer, epididymitis, gonorrhoea, etc. Patients who could not tolerate the medications for this trial Sample size: 90 participants Age (years): Group 1: Trial group: 33.56 ± 8.43; Group 2: Control group: 33.00 ± 8.46 NIH‐CPSI baseline total score: Group 1 = 24.53 (SD 4.90); Group 2 = 25.07 (SD 4.19) All participants were men | |
| Interventions | Group 1 (n = 45): Tamsulosin tablet 0.2 mg, by mouth once a day; take the medication before going to bed. QianLie AnShuan (Prostat) 2 g once a day; participants took the lateral position and inserted the drug into the anus for about 3 ˜ 4 cm after stool each night before going to bed Group 2 (n = 43): Terazosin 2 mg by mouth once a day, take the medication before going to bed Co‐interventions: Duration of treatment: 6 weeks. Avoid alcohol and staying up late during the trial. A regular lifestyle is recommended during the trial | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI global and subscore Time points measured: before treatment, after treatment Time points reported: before treatment, after treatment Subgroups: none Adverse Events How measured: Narratively | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese No contact information available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “starting from a particular row, the numbers on a random sequence chart were assigned to the patients according to the date of admission. If the number was odd then patient was assigned to the trial group, if even then control group” |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment is not described |
| Blinding of participants and personnel (performance bias) | High risk | Blinding of participants and personnel is not described. However, considering the visible differences between the 2 interventions, blinding cannot be achieved |
| Blinding of outcome assessment (detection bias) | High risk | Blinding of outcome assessment is not described. Self‐reported outcomes, participants are not blinded |
| Incomplete outcome data (attrition bias) | Low risk | Missing outcome (all outcomes) data: Quote: “2 patients of the control group dropped out because of intolerable adverse effects and drug withdrawal” (2/45 = 4.4% of attrition) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: January 2003 ‐ February 2004 Setting: Hospital outpatient ‐ multicentre Country: Turkey | |
| Participants | Inclusion criteria: Type IIIB chronic prostatitis; NIH CPSI scores in articles 1. and 2. to be 1 or higher, and score in article 9 to be 4 or higher; symptoms for longer than 3 months Exclusion criteria: Diagnosed as chronic bacterial prostatitis by lower urinary system localisation test, history of urinary tract infection within last year, having important medical problems, having any of the NIH consensus exclusion criteria and history of treatment with alpha blockers Sample size: 45 Age (years): 34.1 ± 8.39 years. Groups : No group mean ages were reported NIH‐CPSI baseline total score: not available All participants were men | |
| Interventions | Group 1 (n = 23): Thiocolchicoside 120 mg/day and ibuprofen 1200 mg/day. For 6 months. Possibly oral, once a day Group 2 (n = 22): Only co‐intervention Co‐interventions: Both groups received terazosin 5 mg a day | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI subscores Time points measured: Pre‐treatment, end of treatment and 6 months after the end of treatment Time points reported: Pre‐treatment, end of treatment and 6 months after the end of treatment | |
| Funding sources | Not reported | |
| Declarations of interest | Not reported | |
| Notes | Dose of thiocolchicoside seems high, since it usually ranges from 8 to 16 mg a day, but this was extracted textually from the report Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) | High risk | Open‐label study |
| Incomplete outcome data (attrition bias) | High risk | Outcome data were not available for 6 participants, who were excluded from the study because of the side effects of the drug (hypotension 3 participants, gastric complaints 3 participants) |
| Selective reporting (reporting bias) | Unclear risk | Total NIH‐CPSI score not reported. No protocol available. We wrote to study authors |
| Other bias | Unclear risk | Participants' baseline characteristics were not described |
| Methods | Study design: Parallel‐group randomised trial Study dates: "study dates not available" Setting: outpatient ‐ single centre ‐ national Country: UK | |
| Participants | Inclusion criteria: male participants aged 18 or older, with perigenital pain of > 1 year, absence of local or systemic inflammation or infection Exclusion criteria: the presence of kidney or bladder stones, urethral stricture or bladder pathology, abnormal ultrasound; neuropsychiatric illness, suicide risk, previous treatment with fluvoxamine and current treatment with antidepressants Sample size: 42 participants were randomised Age (years): median age 41 (range 18 ‐ 72) NIH‐CPSI baseline score: not available. All participants were men | |
| Interventions | Group 1 (n = 21): fluvoxamine 50 mg daily for 8 weeks Group 2 (n = 21): matching placebo tablets of the same posology Co‐interventions: dose was duplicated in both arms if there was no improvement in symptoms. Compliance was checked | |
| Outcomes | Anxiety and depression How measured: Montgomery‐Asberg Depression Rating Scale, Hamilton Rating Scale for Anxiety, Hospital Anxiety and Depression Scale, General Health Questionnaire Time points measured: baseline and 8 weeks Time points reported: baseline and 8 weeks Adverse events How measured: Narratively | |
| Funding sources | Supported by a grant from Solvay/Duphar, Southampton, UK | |
| Declarations of interest | Dr. Rao has received honoraria from Lilly, Organon, Pfizer and Lundbeck | |
| Notes | Prostatitis symptoms were not assessed. A pain score was used as an outcome Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation |
| Allocation concealment (selection bias) | Unclear risk | Quote: “allocated in a double‐blind design”. Comment: No other information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind. Not specified if study personnel were blinded. We wrote to study authors |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded using placebo |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: outcome data were not available in 8/21 and 5/21 participants in each group, due to adverse events |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date September 2004 ‐ end date December 2005 Setting: outpatient ‐ possible single centre ‐ national Country: Turkey | |
| Participants | Inclusion criteria: a diagnosis of Category IIIB CPPS, aged 20 – 45 years old, with a NIH‐CPSI score of > 1 on items 1 and 2 (pain and discomfort); a score of > 4 on item 9 (quality of life), symptoms for > 3 months and a desire to be treated Exclusion criteria: criteria for chronic bacterial prostatitis, Category IIIA CPPS, history of a previous urinary tract infection or a uropathogen documented within the last year, those with significant medical problems, NIH consensus exclusion criteria and those who had been treated or were taking medications that could affect lower urinary tract function Sample size: 90 participants randomised Age (years): overall mean 29.1 (SD 5.2) NIH‐CPSI baseline score: Group 1 mean = 23.1 (SD 1.8); Group 2 = 21.9 (SD 1.5); Group 3 mean = 22.9 (SD 1.2) All participants were men | |
| Interventions | Group 1 (n = 30): only co‐interventions Group 2 (n = 30): ibuprofen 400 mg and thiocolchicoside 12 mg daily for 6 months + co‐interventions Group 3 (n = 30): 1 placebo tablet a day for 6 months Co‐interventions: Both groups 1 and 2 received doxazosin 4 mg a day | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score Time points measured: baseline, 6 and 12 months Time points reported: baseline, 6 and 12 months Adverse events How measured: Narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | We wrote to study author. We received a response through our Turkish collaborator (see Acknowledgements) Email: [email protected]. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “were randomised into three groups in order of appearance” Comment: no additional information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | Quote: “were randomised into three groups in order of appearance” Comment: no additional information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Quote: “Placebo tablets compounded of lactose had a similar, appearance to doxazosin tablets. Ibuprofen was given at a low dose, with an effort to avoid side‐effects during the long period of treatment”; Comment: the interventions in group 1 and 2 were visibly different. No information on blinding of personnel. Author stated that there was no blinding (personal communication) |
| Blinding of outcome assessment (detection bias) | High risk | The interventions in group 1 and 2 were visibly different. No information on blinding of participants. Author stated that there was no blinding (personal communication) |
| Incomplete outcome data (attrition bias) | Unclear risk | Quote: “Eighty‐three of the initial 90 patients were eligible for evaluation after 6 mo, and 79 patients were eligible at the end of 12 mo”, then the author states that at 6 months 1, 1 and 2 participants had dropped out in each study arm. It is unclear how many participants were evaluated at each time point and study arm. We wrote to study authors |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date December 1999 ‐ end date January 2004 Setting: Outpatient ‐ multicentre ‐ national Country: Germany | |
| Participants | Inclusion criteria: men aged 18 ‐ 65 with symptoms of pelvic pain for at least 3 months during the 6 months before the trial; pain domain score of NIH‐CPSI ≥ 7; leukocytes of ≥10 in VB3 (field of vision: x 400) Exclusion criteria: Quote: “(1) urinary tract infection; (2) acute bacterial or chronic bacterial prostatitis at study entry (bacteriuria ≥104 colony‐forming units (CFU)/ml in mid‐stream urine (VB2) or ≥103 CFU/ml in VB3); (3) history of urethritis, with discharge 4 week prior to study entry; (4) a history of epididymitis or sexually transmitted disease (STD); (5) residual urine volume >50 ml resulting from bladder outlet obstruction (BOO); (6) indication for or history of prostate surgery, including prostate biopsy; (7) history of urogenital cancer; (8) treatment with phytotherapeutic agents, a‐blocker agents, or antimicrobial substances with prostatic penetration 4 wk prior to study entry; (9) treatment with agents influencing intraprostatic hormone metabolism 6 months prior to study entry.” Sample size: 139 participants Age (years): Group Pollen extract: mean age = 39.7 ± 7.2 Group Placebo: mean age = 39.3 ± 9.1 Baseline NIH‐CPSI score: Group 1 = 19.3 (SD 5.1); Group 2 = 20.3 (SD 5.2) All participants were men | |
| Interventions | Group 1 Pollen extract (n = 70): "two capsules every 8 hours, with the active substance consisting of 60 mg Cernitin T60 (water soluble fraction) and 3 mg Cernitin GBX (fat soluble fraction)" Duration: 12 weeks Group 2 Placebo (n = 69): “two capsules every 8 hours, with identical capsulation and weight only containing the inactive substances in proportional doses as compared with the pollen extract". Duration: 12 weeks Co‐interventions: Patients included in the screening phase were pre‐treated with azithromycin (250 mg every 6 hours for 1 day) to eliminate atypical pathogens | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI total score and subscores in pain, micturition, and quality of life domains Time points measured: baseline, 6,and 12 weeks Time points reported: baseline, 6, and 12 weeks Urinary symptoms How measured: International Prostate Symptom Score (IPSS) Time points measured: baseline, 6, and 12 weeks Time points reported: baseline, 6, and 12 weeks Adverse events How measured: Narratively | |
| Funding sources | Quote: “This study was supported by an unrestricted grant by Strathmann AG&Co” | |
| Declarations of interest | “None” | |
| Notes | Sexual dysfunction was collected but not in a validated scale We contacted Dr. Wagenlehner for clarification but the author replied that he no longer has the study data | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Randomisation was carried out in blocks (n = 4) within the centre using a random number generator.” |
| Allocation concealment (selection bias) | Low risk | Quote: “The investigators were instructed to use the study drug in ascendant order of random numbers available in the respective trial centre.” |
| Blinding of participants and personnel (performance bias) | Low risk | Double‐blind study. Using “identical capsulation and weight” capsules |
| Blinding of outcome assessment (detection bias) | Low risk | Double‐blind study. Using “identical capsulation and weight” capsules |
| Incomplete outcome data (attrition bias) | High risk | Missing outcome data (all outcomes) Post‐randomisation exclusion of 9 patients due "to wrong allocation" and 12 participants due to outcome data not being available (no treatment arm specified) |
| Selective reporting (reporting bias) | High risk | Clinical trial record specified outcome measurements at week 6 (presented only graphically) |
| Other bias | High risk | The pharmaceutical company participated in the conduct of the study: Quote: “These sponsors contributed to the design and conduct of the study as well as data collection.” Additionally, there were post‐randomisation exclusions for “wrong allocation”. This could indicate that there were problems in the allocation concealment. We requested further explanations from the authors, but they no longer had the data set |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date July 2008 ‐ end date December 2010 Setting: outpatient ‐ multicentre ‐ international Country: Austria, Germany, Poland, Portugal | |
| Participants | Inclusion criteria: Men aged 30 to ≤ 60 years, presenting CP/CPPS (type II or type III): pain or discomfort in the pelvic region for at least 3 months in the previous 6 months; corresponding symptoms could be perineal, lower abdominal, testicular and/or penile, rectal and lower back, or suprapubic, and might be associated with ejaculatory discomfort or voiding (associated voiding symptoms are irritative or obstructive in nature, similar to symptoms associated with benign prostatic hyperplasia); with a total NIH‐CPSI ≥ 15; having signed a written informed consent Exclusion criteria: Consensus NIH criteria: 1. Any prostate, bladder, or urethral cancer, seizure disorder 2. Presence of a concurrent inflammatory bowel disease, disorder affecting the bladder, liver disease 3. Prior 12 months diagnosed with or treated for symptomatic genital herpes 4. Prior 3 months Urinary Tract Infection, with a urine culture value of >100,000 CFU/mL; clinical evidence of urethritis, sexually transmitted diseases, symptoms of acute or chronic epididymitis 5. Any pelvic radiation, systemic chemotherapy; intravesical chemotherapy; intravesical BCG, TURP, TUIP, TUIBN, TUMT, TUNA, any other prostate surgery or treatment such as cryotherapy or thermal therapy; prior treatment for orchialgia without pelvic symptoms to treatment 6. Prior 3 months prostate biopsy 7. Treatment by the following concomitant medication: immunosuppressive medication, e.g. systemic corticosteroids (>15 mg prednisolone); methotrexate; any other immunostimulant medication or live vaccine 8. Prior 6‐month treatment by the following medication: initiated or stopped finasteride or other androgen hormone inhibitors 9. Prior 4‐week treatment by the following medication: immunosuppressive medication or immunostimulant medication or live vaccine; antimicrobial agents (oral or parenteral); started, stopped, or changed dose level of any prostatitis‐specific medications 10. Prior 2‐week treatment by bioflavonoid agents, zinc or iron supplements 11. Inability to comply with the requirements of the protocol (e.g. psychiatric problems; knowledge of language, unable to complete a patient diary, etc...) 12. Known allergy or previous intolerance or known hypersensitivity to the trial drug 13. Participation in another clinical trial and/or treatment with an experimental drug within 3 months before study start and during the present trial Sample size: 185 participants randomised Age (years): Group 1 mean = 47.8 (SD 8.7); Group 2 mean = 47.6 (SD 8.1) NIH‐CPSI baseline score: Group 1 mean = 21.8 (SD 3.8); Group 2 mean = 23.0 (SD 5.6) All participants were men | |
| Interventions | Group 1 (n = 94): OM‐89 (OM Pharma SA., Geneva, Switzerland) containing 6 mg of E. coli lysate from 18 strains; 1 capsule daily for 3 months, then 3 months without treatment, followed by 1 capsule daily for 10 days a month for 3 months Group 2 (n = 91): Same posology of placebo (matched capsule) Co‐interventions: other medications were allowed in both groups (alpha blockers, antibiotics, analgesics, relaxants, sedatives, etc.) | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 3, 9, 12 months Time points reported: baseline, 3, 9, 12 months Adverse Events How measured: Narratively | |
| Funding sources | Quote: “The study was supported by Vifor Pharma/OM Pharma SA, Geneva. Data management and statistical analysis were performed by ICTA, Fontaines‐les‐Dijon (France), a contract research organization.” | |
| Declarations of interest | Quote: “Florian M.E. Wagenlehner has served as a paid consultant for Astellas, AstraZeneca, Bionorica, Cernelle, Cubist, OM‐Pharma, Lilly Pharma, Pierre Fabre, Rosen‐Pharma. He has received lecture honoraria from AstraZeneca, Bionorica, OM‐Pharma, Pierre Fabre, Rosen Pharma, Serag Wiessner, Zambon. He has been paid for performing clinical trials on behalf of Astellas, AstraZeneca, Calixa, Cerexa, Cernelle, Cubist, GSK, Merlion, OM‐Pharma, Janssen‐Cilag, Johnson & Johnson, Lilly Pharma, Pharmacia, Pierre‐Fabre, Rosen Pharma, Sanofi‐Aventis, Strathmann, Zambon. Stefania Ballarini is a Vifor Pharma/OM Pharma Global Medical Affairs employee. Kurt G. Naber has served as paid consultant for Basilea, Bionorica, Cubist, Galenus, MerLion, OM Pharma/Vifor, Paratek, Pierre Fabre, Rempex, Rosen Pharma, Zambon. He has received lecture honoraria from Angelini, Daiichi Sankyo, OM Pharma/Vifor, Pierre Fabre, Zambon. He has been paid for performing clinical trials on behalf of Basilea, Bionorica, MerLion, OM Pharma/Vifor, Rosen Pharma, Zambon.” | |
| Notes | Clinical Trial registry: EudraCT number: 2007‐004609‐85 Contact information: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | No information available in the paper but the author described random blocks in personal communication |
| Allocation concealment (selection bias) | Low risk | No information available in the paper but the author described concealed allocation in personal communication |
| Blinding of participants and personnel (performance bias) | Low risk | Participants were blinded with the use of placebo. No information about study personnel in the paper, but the author described blinding of personnel in personal communication |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded with the use of placebo |
| Incomplete outcome data (attrition bias) | High risk | At 3 months follow‐up outcome data (prostatitis symptoms) were available for 87/94 (active treatment) and 79/91 (placebo) participants, at 12 months, 81/94 (active treatment) and 73/91 (placebo). Unbalanced attrition For adverse events outcome data were available for all participants (low risk of bias) at complete follow‐up |
| Selective reporting (reporting bias) | Low risk | All outcomes planned in the clinical trial registry were reported |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date September 2002 ‐ end date March 2003 Setting: outpatient ‐ single centre ‐ national Country: China | |
| Participants | Inclusion criteria: According to Nickel 1998. NIH‐CPSI > 8 Exclusion criteria: According to Nickel 1998. Sample size: 38 participants Age (years): Overall 20 ˜ 48; mean 28 Baseline NIH‐CPSI score: not available. All participants were men | |
| Interventions | Group 1 (n = 24): 6 mL of mixed solution of Chuanshentong (4 mL) and 2% lidocaine (2 mL) was trans‐perineally injected into one lobe of the prostate, once a day for 6 days Group 2 (n = 14): 6 mL of lidocaine and saline solution was trans‐perineally injected into one lobe of the prostate, once a day for 6 days Co‐interventions: not available. | |
| Outcomes | Prostatitis Symptoms How measured: changes in NIH‐CPSI Time points measured: before treatment, Week 4 ‐ 12 Time points reported: Week 4, Week 6 Subgroups: none Adverse Events How measured: Narratively | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese Email: [email protected] (email bounced) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote:“Patient s were randomly divided into 2 groups…”, Comment: the exact method for randomisation is unknown. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment is not described. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Patients were not blinded |
| Blinding of outcome assessment (detection bias) | High risk | Blinding of outcome assessment is not described. Self‐reported outcomes, participants are not blinded |
| Incomplete outcome data (attrition bias) | Low risk | 1 participant in trial group (1/24 = 4.2%) and 1 participant in control group (1/14 = 7.1%) lost follow‐up at week 3 |
| Selective reporting (reporting bias) | High risk | It seemed that the authors had follow‐up data from Week 4 to Week 12, but only those of Week 4 and Week 6 were reported |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date January 2011 ‐ end date January 2014 Setting: outpatient ‐ possible single centre ‐ national Country: China | |
| Participants | Inclusion criteria: men suffering from urinary symptoms (frequent urination, dribbling urine sense) and pain or discomfort in the perineum, testicles, and lumbosacral region for over 3 months with negative results of urinary and prostatic secretions culture and no history of treatment with antibiotics and alpha receptor blocker Exclusion criteria: a history of urinary tract infection, benign prostatic hyperplasia and other pelvic organic diseases Sample size: 115 randomised Age (years): Group 1 mean = 36.6 (SD 8.1); Group 2 mean = 38.5 (SD 8.3); Group 3 mean = 37.8 (SD 7.9) NIH‐CPSI baseline score: Group 1 mean = 24.2 (SD 7.1); Group 2 = 22.3 (SD 6.6); Group 3 = 22.6 (SD 6.8) All participants were men | |
| Interventions | Group 1 (n = 38): levofloxacin 200 mg twice daily for 6 weeks Group 2 (n = 38): terazosin 2 mg once daily for 6 weeks Group 3 (n = 39): levofloxacin 200 mg twice daily and terazosin 2 mg once daily for 6 weeks Co‐interventions: All participants received dietary advice (to avoid spicy food and alcohol consumption, increase water intake), received prostatic massage once a week and were advised to take warm baths | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score Time points measured: baseline and 6 weeks Time points reported: baseline and 6 weeks Sexual Dysfunction How measured: IIEF‐5 Time points measured: baseline and 6 weeks Time points reported: baseline and 6 weeks Adverse Events How measured: Narratively | |
| Funding sources | None | |
| Declarations of interest | None | |
| Notes | Clinical Trial Registry: ChiCTR‐IPR‐15006189 This study was written in Chinese Email: Zhonghua Xu [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random blocks |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) | High risk | Open‐label study |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | Low risk | Predefined outcomes were reported (Clinical Trial Registry) |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date October 1984 ‐ end date February 1985 Setting: outpatient ‐ single centre ‐ national Country: Sweden | |
| Participants | Inclusion criteria: score with typical history > 9, score of signs and symptoms > 9, age between 20 and 50 years old, normal blood testing, no earlier isolation of bacteria in prostate secretion and urine and informed consent Exclusion criteria: other diseases except Reiter syndrome and ankylosing spondylitis (for their relationship with prostatitis), receiving other medication during the study period Sample size: 30 participants randomised Age (years): Group 1 mean = 37.3; Group 2 mean = 37.9 NIH‐CPSI baseline score: Not available All participants were men | |
| Interventions | Group 1 (n = 15): Elmiron® (Pentosan polysulphate) 100 mg, 2 capsules twice daily for 3 months Group 2 (n = 15): Placebo with the same posology for 3 months Co‐interventions: not available | |
| Outcomes | Adverse events How measured: Narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | The study included several outcomes on clinical improvement, but it was not measured on any validated scale. No contact information available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) | Unclear risk | The study is described as “double blind”, but it is not clear who was blinded (it is not clear whether personnel were blinded) |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: “the study is described as double blind” There was use of identical placebo |
| Incomplete outcome data (attrition bias) | High risk | 3 participants (2 in Pentosan and 1 in placebo) were excluded due to the presence of E. coli in the culture. 1 participant stopped treatment in the pentosan group due to adverse events. 2 participants in the Pentosan group were excluded because they decided to use antibiotics. In total, 5 participants dropped out in the Pentosan group and 1 in the placebo group |
| Selective reporting (reporting bias) | Unclear risk | Protocol not available |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date January 2006 ‐ end date December 2007 Setting: outpatient ‐ single centre ‐ national Country: China | |
| Participants | Inclusion criteria: (not specified clearly, drawn from participant’s characteristics) Diagnostic criteria: with reference to NIH standard (J Urol 2004) and Chinese standard (2007) [Refs 1,5]. Age 20 ˜ 50, first and second score of NIH‐CPSI > 1, course of disease > 3 months and patients actively sought medical treatment. Patients did not receive any treatment before admission, did not take any alpha blockers or cyclo‐oxygenase inhibitors. In an effort to exclude chronic bacterial prostatitis, all patients undertook routine urine test, urine culture, EPS routine test and EPS culture before and after prostate massage Exclusion criteria: Patients who received analgesics, had a history of acute urinary infection, had serious cardiovascular or cerebrovascular diseases, or were allergic to alpha blockers or cyclo‐oxygenase inhibitors Sample size: 123 participants Age (years): Group 1: doxazosin group: 34.5 ± 8.5; Group 2: diclofenac group: 35.0 ± 8.8; Group 3: combination group: 34.9 ± 8.4 Baseline NIH‐CPSI score: Group 1 = 24.1 ± 2.2; Group 2 = 23.95 ± 2.17; Group 3 = 23.82 ± 1.72 All participants were men | |
| Interventions | Group 1 (n = 43): Doxazosin 4 mg by mouth once a day for 12 weeks Group 2 (n = 39 ): Diclofenac 75 mg by mouth once a day for 12 weeks Group 3 (n = 41): Doxazosin 4 mg by mouth once a day + diclofenac 75 mg by mouth once a day for 12 weeks Co‐interventions: no information | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI global and subscore Time points measured: before treatment, after treatment Time points reported: before treatment, after treatment Subgroups: none Adverse Events How measured: Narratively | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese E‐mail: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “123 patients were randomly divided into 3 groups”. Comment: However, the exact method for randomisation is unknown. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment is not described. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Blinding of participants and personnel is not described. However, considering the visible differences between the interventions (single drug versus combination), blinding cannot be achieved |
| Blinding of outcome assessment (detection bias) | High risk | Blinding of outcome assessment is not described. Self‐reported outcomes, participants are not blinded |
| Incomplete outcome data (attrition bias) | Low risk | Quote: “8 patients lost follow‐up and the attrition is 6.5%”, and “3 of them lost follow‐up because they did not have adequate time for the treatment; the other 5 of them lost follow‐up for unknown reason” |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date November 2011 ‐ start date November 2012 Setting: outpatient ‐ possibly single centre ‐ national Country: China | |
| Participants | Inclusion criteria: Age > 18; Course of disease > 3 months; NIH‐CPSI > 15; The article reported that the participants included were already diagnosed with type III CP/CPPS Exclusion criteria: Patients who had a history of infection of the urinary system within 3 months; patients with history of tumour of the urinary system or the rectum; patients with diseases of the nervous system; patients with narrowing of the urinary tract or history of transurethral surgery; patients with acute diseases or other diseases which needed treatment; Age > 55 Sample size: 88 participants Age (years, mean, SD): Group 1: experimental group 33.88 ± 5.68; Group 2: control group 34.54 ± 6.45. Baseline NIH‐CPSi score: Group 1 = 28.32 ± 4.90; Group 2 = 28.54 ± 5.03 All participants were men | |
| Interventions | Group 1 (n = 44): Usual care (see Co‐interventions) + YuLeShu Oral Mixture 3 times a day, 20 mL each time Group 2 (n = 44): Usual care only (see co‐interventions) Co‐interventions: Common clinical practice for CP/CPPS in that setting, which includes: Anaibiotics: levofloxacin tablet 0.2 g twice a day, course of treatment is 4 weeks; alpha blockers: doxazosin hydrochloride tablet 2 mg once a day, course of treatment is 4 weeks; Chinese patent medicine: QianLieTongYu capsule 3 times a day, 4 capsules each time, course of treatment is 4 weeks; prostate massage: once a week Duration: 1 course of treatment (total: 4 weeks) | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI global and pain subscore Time points measured: before treatment, after treatment Time points reported: before treatment, after treatment Subgroups: none Sexual Dysfunction How measured: IIEF‐5 Time points measured: before treatment, after treatment Time points reported: before treatment, after treatment Subgroups: none Anxiety and depression How measured: HAM‐D, HAM‐A Time points measured: before treatment, after treatment Time points reported: before treatment, after treatment Subgroups: none | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The paper reported that the randomisation was achieved by using computer, but it is unclear what software was used and what the exact method of randomisation was. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment is not described. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Blinding of participants and personnel is not described. However, considering the visible difference between the 2 different interventions, blinding was unlikely |
| Blinding of outcome assessment (detection bias) | High risk | Blinding was unlikely (See above) |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date December 1998 ‐ end date May 1999 Setting: Outpatient Country: China | |
| Participants | Inclusion criteria: Not clearly specified. Patient characteristics: main clinical manifestation: suprapubic or perineal discomfort, frequent urination, discomfort during urination, etc. Tenderness during prostate massage, no special body sign otherwise. Routine urine test: WBC < 10/HP. Routine EPS test: WBC > 10/HP. Mid‐stream urine and EPS culture before, during and after the treatment negative. Ultrasound test revealed no bladder or ureter diseases Exclusion criteria: Hyperplasia of the prostate, prostate cancer, tuberculosis of the prostate, etc. Sample size: 60 participants Age (years): Group 1: Antibacterial drugs group (18 ˜ 64, mean: 27.8); Group 2: Prostat group (19 ˜ 63, mean: 28.7) Baseline NIH‐CPSI score: not available All participants were men | |
| Interventions | Group 1 (n = 30): Antibacterial treatment: Trimethoprim plus sulfamethoxazole 2 tablets, by mouth twice a day, 10 days, ofloxacin 0.2 g, by mouth twice a day, 10 days (some of the participants took norfloxacin levofloxacin instead 0.2 g, by mouth twice a day, for 10 days); minocycline 0.1 g, by mouth twice a day, for 10 days (a few of the participants took erythromycin instead 0.5 g, by mouth 4 times a day, for 10 days); sequential therapy of the 3 drugs; repeat every month, duration: 3 months Group 2 (n = 30): Prostat 0.375 g, twice a day, 90 days Co‐interventions: Bland diet; avoid alcohol or tobacco Hip bath with hot water | |
| Outcomes | None of the outcomes relevant to this review | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | Outcomes included: "self‐reported symptomatic relieves, changes in symptomatic scores (SFQ)" with no reference to a validated scale and WBC cell count in prostatic secretions This study was written in Chinese No contact information available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “60 patients were randomly assigned to 2 groups”. Comment: However, the method for randomisation is not described. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment is not described |
| Blinding of participants and personnel (performance bias) | High risk | Blinding of participants and personnel is not described. However, considering the visible differences between the 2 interventions, blinding cannot be achieved |
| Blinding of outcome assessment (detection bias) | High risk | Blinding of outcome assessment is not described. Self‐reported outcomes, participants are not blinded |
| Incomplete outcome data (attrition bias) | High risk | The paper reported that “in Antibacterial‐drugs Group, the numbers of patients who kept follow‐up at the end of the first, second and third month were 27, 26, and 23, respectively. In Prostat Group, the numbers were 29, 27, and 23 respectively.” 7 participants lost follow‐up in each group, which is 23% attrition |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date June 2007 ‐ end date December 2007 Setting: outpatient ‐ possibly single centre ‐ national Country: China | |
| Participants | Inclusion criteria: NIH‐CPSI > 10, OR CPSI > 6 plus WBC in EPS > 10/HP; symptoms lasting for at least 6 weeks, such as frequent urination, discomfort urination, perineal pain or discomfort, etc.; routine urine test normal, EPS bacteria culture negative; age 18 to 50 years; patients who did not take any antibiotics, alpha blockers or other herbal medications within 2 weeks; OR patients who took these medications, but agreed to take a 1‐week wash‐out period Exclusion criteria: Patients who have already undertaken intra‐prostate or intra‐vas deferens injection therapy; patients with benign prostate hyperplasia above moderate degree; patients with urethritis, cystitis, pyelonephritis or calculi of the urinary system; patients with organic urination disorders, such as neurogenic bladder; patients with suspected prostate tuberculosis, prostate tumour or eosinophilic granuloma; patients with mental diseases or tumour; patients with chronic or severe diseases of other systems; patients who could not continue treatment or refused examinations; patients who were considered not appropriate for this clinical trial by the researchers Sample size: 160 participants Age (years): Group 1: experimental group: Mean ± SD: 29. 4 ±7.7; Group 2: control group: Mean ± SD: 29.1 ± 7.0) Baseline NIH‐CPSI score: Group 1 = 22.79 (SD 2.56); Group 2 = 22.43 (SD 3.63) All participants were men | |
| Interventions | Group 1 (n = 80): For the first 2 weeks, prednisone acetate tablet 15 mg by mouth once a day For the next 2 weeks, levofloxacin 0.1 g by mouth twice a day for 4 weeks Group 2 (n = 80): For the first 2 weeks, placebo tablet (looks the same as prednisone acetate) by mouth once a day For the next 2 weeks, levofloxacin 0.1 g by mouth twice a day for 4 weeks Co‐interventions: Not specified | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI global and subscore Time points measured: before treatment, Week 2, Week 4 Time points reported: before treatment, Week 2, Week 4 Subgroups: none Adverse Events How measured: Narratively | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “randomized, placebo‐controlled and double‐blinded”, and that “patients were randomly divided into trial group and control group”. Comment: However, we do not know what the method for randomisation is. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | Quote: “randomized, placebo‐controlled and double‐blinded” Comment: Method for allocation concealment is not described. We wrote to study author. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Blinding of personnel not described. Participants were blinded. We wrote to study author. |
| Blinding of outcome assessment (detection bias) | Low risk | Quote: “the appearance, packaging design, and taste of the placebo tablet is not significantly different from those of the prednisone acetate tablet” |
| Incomplete outcome data (attrition bias) | Low risk | Quote: “missing 2 patients of the trial group because of lost follow‐up” Comment: 2.5% attrition in the trial group |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date June 2009 ‐ end date August 2009 Setting: outpatient ‐ possibly single centre ‐ national Country: China | |
| Participants | Inclusion criteria: NIH‐CPSI > 10, OR NIH‐CPSI > 6 together with WBC/EPS > 10/HP; frequent urination, discomfort during urination, perineal pain/discomfort, or other symptoms lasting for at least 6 weeks; routine urine test normal, EPS bacteria culture negative; age 18 ˜ 50 Exclusion criteria: Men who undertook intra‐prostate, intra‐urethra, or intra‐ureter therapy; men with benign prostate hyperplasia above moderate degree; urethritis, cystitis, pyelonephritis, or stones of the urinary system; patients with neurogenic bladder or other organic urinary dysfunction; patients with suspected prostate tuberculosis, prostate tumour or eosinophilic granuloma; patients who could not persist with the treatment or refused follow‐up; patients who were considered inappropriate for the trial by researchers Sample size: 156 participants Age (years): Group 1: Placebo: Mean ± SD: 30.1 ± 6.2; Group 2: Terazosin: Mean ± SD: 31.0 ± 6.6; Group 3: Tamsulosin ; Mean ± SD: 31.1 ± 6.4 Baseline NIH‐CPSI score: Group 1 = 25.38 (SD 3.73); Group 2 = 25.45 (SD 3.25); Group 3 = 25.49 (SD 3.11) All participants were men | |
| Interventions | Group 1 (n = 52): Placebo tablet (with the same appearance, packaging and taste as tamsulosin tablet) by mouth once a day, 12 weeks Group 2 (n = 52 ): terazosin 1 mg by mouth once a day, 12 weeks Group 3 (n = 52 ): tamsulosin 0.2 mg by mouth once a day, 12 weeks Co‐interventions: 1 week of wash‐out period | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI Time points measured: before treatment, Week 4, Week 8, Week 12 Time points reported: before treatment, Week 4, Week 8, Week 12 | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese No contact information available. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “we used random, double‐blinded measures to assign the patients included to 3 groups” (in Chinese) Comment: however it is unclear what the exact method is |
| Allocation concealment (selection bias) | Unclear risk | The paper reported that they used double‐blinded measures, but the exact method for allocation concealment is not described |
| Blinding of participants and personnel (performance bias) | Unclear risk | The paper reported that the placebo tablet shared the same appearance, packaging, taste, and usage as tamsulosin tablet. However it is not clear that participants and personnel were unaware of assignments |
| Blinding of outcome assessment (detection bias) | Unclear risk | Blinding of outcome assessment is not described |
| Incomplete outcome data (attrition bias) | Low risk | 2/52 in Group 1 because lost to follow‐up (3.8% attrition) 1/52 each in Groups 2 and 3 because lost to follow‐up (1.9%) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: From September 2005 to March 2006 Setting: Outpatient Country: China | |
| Participants | Inclusion criteria: Individuals between 18 and 50 years old who had a NIH‐CPSI > 10, OR NIH‐CPSI > 6 plus WBC in expressed prostatic secretion > 10/HP; symptoms lasting for > 6 weeks. Patients did not take antibiotics, alpha blockers, or other herbal medications within 2 weeks before the trial starts, OR patients who took medications but agreed to undertake a 1‐week wash‐out Exclusion criteria: Patients who have already undertaken intra‐prostate or intra‐vas deferens injection therapy. Patients with benign prostate hyperplasia above moderate degree. Patients with urethritis, cystitis, pyelonephritis or calculi of the urinary system. Patients with organic urination disorders, such as neurogenic bladder. Patients with suspected prostate tuberculosis, prostate tumour or eosinophilic granuloma. Patients with mental diseases or tumour. Patients with chronic or severe diseases of other systems. Patients who could not continue treatment or refused re‐examinations. Patients who were considered not appropriate for this clinical trial by the researchers Sample size: 160 participants Age (years): Group 1: trial group: Mean ± SD: 31.23 ± 8.03; Group 2: control group: Mean ± SD: 31.84 ± 6.39 Baseline NIH‐CPSI score: not available All participants were men | |
| Interventions | Group 1 (n = 80 ): For 8 weeks, Prostat® 1 tablet(0.375 g) by mouth twice a day (Serenoa repens) Group 2 (n = 80): For 8 weeks, Placebo 1 tablet by mouth twice a day Co‐interventions: Levofloxacin 1 tablet (0.1 g) by mouth twice a day for the first 4 weeks | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI subscore Time points measured: before treatment, Week 4 after treatment, Week 8 after treatment Time points reported: before treatment, Week 4 after treatment, Week 8 after treatment Subgroups: none Adverse events How measured: Narratively | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese Contact information not available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “multi‐center, double‐blinded, parallel randomized controlled trial”. Comment: We do not know what the method for randomisation is |
| Allocation concealment (selection bias) | Unclear risk | Quote: “randomized, placebo‐controlled and double‐blinded”, Comment: Method for allocation concealment is not described. |
| Blinding of participants and personnel (performance bias) | Unclear risk | Quote: “the appearance, packaging design, and taste of the placebo tablet is not significantly different from those of the Prostat®” |
| Blinding of outcome assessment (detection bias) | Low risk | Blinding of outcome assessment is not described. Self‐reported outcomes, participants are not blinded |
| Incomplete outcome data (attrition bias) | Low risk | Quote: “one patient from the trial group dropped out”, 1.25% attrition in the trial group, but we do not know the reason |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date December 2002 ‐ end date August 2004 Setting: outpatient ‐ possibly single centre ‐ national Country: China | |
| Participants | Inclusion criteria: men, aged 18 – 50 years, with a diagnosis of chronic nonbacterial prostatitis were eligible for inclusion, NIH‐CPSI score of > 10 or a total NIH‐CPSI score of > 6 with a WBC count of > 10 cells/high power field and continuous symptoms of prostatitis for 6 weeks Exclusion criteria: participants who had been treated with alpha‐adrenergic antagonists or antibiotics in the period 4 weeks prior to the start of the study Sample size: 105 participants randomised Age (years): Not available. NIH‐CPSI baseline score: Group 1 (n = 42, comprised 2 groups of 21 participants with type IIIA and type IIIB) 15.19 (SD 4.78) and 15.38 (SD 4.12); Group 2 (comprised 2 groups of 21 participants with type IIIA and type IIIB) 24.9 (SD3.16) and 17.38 (SD 3.22); Group 3 = 16.05 (SD 3.99) All participants were men | |
| Interventions | Participants were divided in 5 groups according their subtype of CP/CPPS (type A and type B). For this review, we group them in three categories according to their received treatment Group 1 (n = 42, comprised of two groups of 21 participants with type IIIA and type IIIB): tamsulosin 0.2 mg daily for 90 days Group 2 (n = 42, comprised of two groups of 21 participants with type IIIA and type IIIB): tamsulosin 0.2 mg daily and levofloxacin 0.2 g after breakfast daily for 90 days Group 3 (n = 21, only type IIIA participants): Levofloxacin 0.2 g after breakfast daily for 90 days Co‐interventions: Not available | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score Time points measured: baseline, 45 and 90 days Time points reported: baseline, 45 and 90 days Adverse events How measured: Narratively | |
| Funding sources | Quote: “This research was supported by funds from Astellas Pharma China Inc” | |
| Declarations of interest | Quote: “The authors had no conflicts of interest to declare in relation to this article.” | |
| Notes | Contact information: [email protected] (email bounced) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Quote: “Patients and healthcare workers were not blinded to the treatment regimens.” |
| Blinding of outcome assessment (detection bias) | High risk | Quote: “Patients and healthcare workers were not blinded to the treatment regimens.” |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: Quote: "All patients completed the treatments and were scored according to prostatitis symptoms in follow‐up checks" |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors |
| Other bias | High risk | Large baseline differences in NIH‐CPSI scores. No other baseline characteristics were reported |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date October 2005 ‐ end date May 2006 Setting: outpatient ‐ single institute ‐ national Country: South Korea | |
| Participants | Inclusion criteria: men with symptoms of chronic prostatitis, EPS WBC ≥ 10/HPF (IIIa). Participant with NIH‐CPSI has reported total score of 15 or more Exclusion criteria: Men with prior treatment of chronic prostatitis, positive culture on urine or EPS culture, or prostate volume ≥ 30 gm on DRE Sample size: 69 Age (years) Group 1 = 41.3 ± 9.5 (21 ‐ 57); Group 2 = 41.8 ± 9.2 (23 ‐ 60) Baseline NIH‐CPSI score: Group 1 = 24.0 ± 6.3; Group 2 = 24.7 ± 6.9 All participants were men | |
| Interventions | Group 1: gatifloxacin (200 mg, twice a day, GatifloⓇ) and doxazosin (CarduraⓇ, dose was not defined) for 6 weeks Group 2: gatifloxacin alone (200 mg, twice a day, GatifloⓇ) for 6 weeks Co‐interventions: no information | |
| Outcomes | Prostatitis Symptoms How measured: NIH CPSI questionnaire Time points measured: baseline, 6 weeks Time points reported: baseline, 6 weeks Subgroups: no | |
| Funding sources | Not reported | |
| Declarations of interest | Not reported | |
| Notes | Language of publication: Korean Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of outcome assessment (detection bias) | Unclear risk | No information available. We wrote to study authors |
| Incomplete outcome data (attrition bias) | Unclear risk | No information available. We wrote to study authors |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: Start date 15 June 2003 ‐ end date 26 August 2003 Setting: outpatient ‐ possibly single centre ‐ national Country: China | |
| Participants | Inclusion criteria: According to the diagnostic criteria of chronic prostatitis by NIH‐CPCRN (Alexander 2004a) Exclusion criteria: Patients who had suspected or confirmed urethritis or cystitis; Patients with suspected or confirmed prostate hyperplasia or prostate cancer by intra‐rectal ultrasound exam; 3‐cup test or PPMT test confirmed VB3 or bacterial infection; EPS immunologic test for chlamydia or mycoplasma positive; antibiotics treatment effective in the early stages; Patients complicated by other serious diseases which necessitate medication; Patients allergic to sulfanilamide drugs Sample size: 64 participants Age (years): Group 1: 21 ˜ 48, mean 34.4; Group 2: 19 ˜ 53, mean 36.8 Baseline NIH‐CPSI score: Group 1 = 29.6 (SD 1.8); Group 2 = 27.1 (SD 1.5) All participants were men | |
| Interventions | Group 1 (n = 32): For 6 weeks, celecoxib 200 mg by mouth once a day Group 2 (n = 32): For 6 weeks, celecoxib 200 mg by mouth twice a days Co‐interventions: Discontinue any medication for at least 1 week before the trial started. Not to take any other medication for prostatitis during the trial period | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI global and pain subscore Time points measured: 1 day before treatment, Weeks 2, 4, and 6 after taking medication Time points reported: 1 day before treatment, Weeks 2, 4, and 6 after taking medication Subgroups: none Adverse Events How measured: Narratively | |
| Funding sources | Not mentioned | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “64 patients were randomly assigned to two groups”. Comment: We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Blinding of participants and personnel is not described. However, the difference between the interventions of the 2 groups is visible |
| Blinding of outcome assessment (detection bias) | High risk | Blinding of outcome assessment is not described. Self‐reported outcomes, participants are not blinded |
| Incomplete outcome data (attrition bias) | Low risk | In Group A, 2 participants dropped out (2/30); In Group B, 1 participant dropped out (1/30); Reasons for dropping out are not adequately described |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. We wrote to study authors |
| Other bias | Low risk | No other sources of bias were detected |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date January 2005 ‐ end date May 2007 Setting: Outpatient ‐ possibly single centre ‐ national Country: China | |
| Participants | Inclusion criteria: Western Medicine criteria: patients who met the CP/CPPS criteria and PPMT criteria. (Krieger JN, Nyberg L Jr, Nickel JC. NIH consensus definition and classification of prostatitis. JAMA 1999] [Nickel JC. The Pre and Post Massage Test(PPMT): a simple screen for prostatitis. Tech Urol 1997) Traditional Chinese Medicine criteria: patients who met the “combination of TCM and Western Medicine criteria for chronic prostatitis” (www.adpycn.com, 2004) Exclusion criteria: Patients who had urinary infection, chronic epididymis, hyperplasia of the prostate, or suspected or confirmed prostate cancer; Patients with diseases of other systems or psychiatric disorders Sample size: 248 participants (30 participants dropped out) Age (years): Group A: 25 ˜ 50 (Mean ± SD: 32.42 ± 7.29); Group B: 23 ˜ 48 (Mean ± SD: 29.44 ± 5.28); Group C: 22 ˜ 50 (Mean ± SD: 31.23 ± 8.83); Group D: 22 ˜ 50 (Mean ± SD: 30.79 ± 7.64) Baseline NIH‐CPSI score: Group A = 22.98 ± 5.95; Group B = 20.98 ± 7.27; Group C = 21.61 ± 6.69; Group D = 20.05 ± 7.43 All participants were men | |
| Interventions | Group A (n = 58): For 4 weeks, Aike decoction twice a day, 1 pack each time Group B (n = 50): For 4 weeks, Bazhengsan decoction twice a day, 1 pack each time Group C (n = 62): For 4 weeks, Prostatitis decoction twice a day, 1 pack each time Group D (n = 48): For 4 weeks, Placebo capsule 3 times a day, 3 capsules each time Co‐interventions: No information | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI global and subscore Time points measured: before treatment, Week 2, Week 4 Time points reported: before treatment, Week 2, Week 4 Subgroups: none Adverse Events How measured: Narratively | |
| Funding sources | Subsidisation Programme of Department of Education of Fujian Province (No. JA02229) | |
| Declarations of interest | Not mentioned | |
| Notes | This study was written in Chinese Email:[email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “218 patients were randomly assigned to 4 groups according to random sequence chart” Comment: However, the exact method for using the random sequence chart is not clear. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | The article reported that there was allocation concealment, but the exact method for concealment is unknown. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | Quote: “the decoctions was uniformly packaged, shared the same appearance, and had a number printed on the package. The doctors gave out the drug to the patients according to the number of the drugs, and did not know what is contained in the package”. Comment: However, different decoctions could vary in taste; additionally, the placebo tablet used in Group D was visibly different from decoction, and therefore blinding was not possible |
| Blinding of outcome assessment (detection bias) | High risk | The article reported that there was blinding of outcome assessment. Self‐reported outcomes, however, participants are not blinded |
| Incomplete outcome data (attrition bias) | High risk | Quote: “30 patients dropped out in the middle of the trial and were not included in the statistics”(30/248), 12% attrition |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date January 2011 ‐ end date January 2012 Setting: outpatient ‐ possibly single centre ‐ national Country: China | |
| Participants | Inclusion criteria: > 18 years and diagnosed with CP/CPPS with discomfort or pain symptoms in the pelvic area for at least 3 months NIH‐CPSI score ≥ 15 points; pain score should be ≥ 4 points Exclusion criteria: previous treatment with doxazosin or other alpha‐adrenergic receptor blockers for CP/CPPS or for any other reason in the past; urinary tract infection; genital herpes in the past 3 months; 5‐alpha‐reductase inhibitor for 3 months in the past year; unilateral testicular pain without pelvic area symptoms; urinary or reproductive system cancer; inflammatory bowel disease; active urethral stricture; prostate or bladder operation history; neurogenic bladder; or the usage of a strong P‐3A4 enzyme inhibitor (ketoconazole, itraconazole, ritonavir, etc.) or erythromycin; additionally, patients with mania, bipolar disorder, psychosis, or who were identified as a suicide risk by researchers were excluded; plus, those patients who used monoamine oxidase inhibitors or who had liver disease or other serious diseases in the 2 weeks before screening were also excluded Sample size: 150 participants randomised Age (years): Group 1 = 32.51 (22.58); Group 2 = 33.59 (range 20 ‐ 67); Group = 32.78 (range 23 ‐ 62) NIH‐CPSI baseline score: Group 1 = 21.78; Group 2 = 22.18; Group 3 = 21.85 All participants were men | |
| Interventions | Group 1 (n = 50): sertraline 50 mg daily, adjusted to 25 or 100 mg according to efficacy and tolerability + doxazosin; 6 months Group 2 (n = 50): duloxetine 30 mg daily, titrated to 60 mg and then 120 mg daily if tolerated (otherwise 60 mg daily + doxazosin); 6 months Group 3 (n = 50): doxazosin 4 mg daily; 6 months Co‐interventions: not available | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score Time points measured: baseline, 1, 3 and 6 months Time points reported: baseline, 1, 3 and 6 months Anxiety and Depression How measured: Hospital Anxiety and Depression scale (HAD) Time points measured: baseline, 1, 3 and 6 months Time points reported: baseline, 1, 3 and 6 months Adverse events How measured: Narrative | |
| Funding sources | None | |
| Declarations of interest | None | |
| Notes | Phase I: run‐in period Phase II: 6‐month treatment period Phase III: withdrawal of treatment for 2 weeks Email: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | The interventions were visibly different, and personnel actively manipulated the interventions differently. Blinding was unlikely |
| Blinding of outcome assessment (detection bias) | High risk | The interventions were visibly different, and personnel actively manipulated the interventions differently. Blinding was unlikely |
| Incomplete outcome data (attrition bias) | High risk | All outcomes: outcome data were not available for 9/50 participants in the doxazosin group, 9/50 in the sertraline group and 6/50 in the duloxetine group |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Low risk | No other sources of bias were identified |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date August 2006 ‐ end date January 2008 Setting: outpatient ‐ possibly single centre ‐ national Country: China | |
| Participants | Inclusion criteria: Participants with CP/CPPS diagnosis refractory (patient unsatisfied with their clinical response) to standard conventional therapy (antibiotics, α‐blockers); aged 18 to 58 years, symptoms > 6 months with ≥ 1 month of treatment with appropriate antibiotics or/and alpha blockers; negative infection screening, including the 4‐glass test and bacterial localisation studies; total NIH‐CPSI score of 14 or more; NIH‐CPSI pain score of 8 or more at baseline Exclusion criteria: “presence of chronic bacterial prostatitis after a 4‐glass lower urinary tract localization test; previous urinary tract infection or a uropathogen documented within the last year; cancer of the genitourinary tract; a history of active genital herpes within the previous year; active urethral stricture; inflammatory bowel disease; a history of pelvic radiation or systemic chemotherapy; a history of intravesical chemotherapy; prostate or bladder surgery, and neurologic disease affecting the bladder” Sample size: 64 participants were randomised Age (years): not available NIH‐CPSI baseline score: not available All participants were men | |
| Interventions | This study included a 2‐week placebo run‐in. Group 1 (n = 32): Celecoxib 200 mg daily for 8 weeks Group 2 (n = 32): Matching placebo for 8 weeks Co‐interventions: not available | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score and responder analysis Time points measured: baseline and weekly for 8 weeks Time points reported: baseline and weekly for 6 (numerically) and 8 weeks (graphically) Adverse Events How measured: Narratively | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | Contact information: [email protected] (email bounced) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated (Excel spreadsheet) |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind study. Participants were blinded, no information about blinding of study personnel. We wrote to study authors |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded using placebo |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: outcome data were available for all participants |
| Selective reporting (reporting bias) | High risk | NIH‐CPSI scores were presented graphically. We wrote to study authors |
| Other bias | Unclear risk | Baseline characteristics not available. We wrote to study authors |
| Methods | Study design: parallel group randomized trial Study dates: not available Setting: outpatient, national, multicentre Country: China | |
| Participants | Inclusion criteria: participants with type IIIB prostatitis (following the 2014 Chinese Urological Association CUA guidelines: long‐term or repeated pelvic pain or discomfort with varying degrees of micturition symptoms and sexual dysfunction for a duration ≥3 months, normal prostatic fluid and negative VB3 testing) and premature ejaculation (lifelong premature ejaculation or a clinically significant reduction in latency time, often up to approximately 3 min or less (acquired premature ejaculation) and accompanied by associated distress such as annoyance, depression and/or avoidance of intimacy), aged between 18 and 45 years old, with a fixed sexual partner and a regular sex life for at least 6 months (and three or more sexual encounters in the past month) and had 1–2 sexual encounters per week during the treatment process. Exclusion criteria: participants with comorbid mental disorders, cardio‐cerebrovascular diseases, liver and kidney insufficiency, alcohol dependence, hepatitis B/HCV/HIV infection, etc.; erectile dysfunction; those who had received serotoninergic drugs, SSRIs, TCAs, PDE5i or P450 3A4 inhibitor in the last 4 weeks or received these drugs during the study period; and intolerance to dapoxetine or tamsulosin. Sample size: 251 participants randomized Age (years): Group 1 mean 33.9 (SD 6.56) years Group 2 mean 31.2 (SD 5.96) years NIH‐CPSI baseline score: Group 1 mean 24.29 (SD 5.92) Group 2 mean 26.15 (SD 7.76) Sex (M/F): All participants were men | |
| Interventions | Group 1 (n = 168): 30 mg dapoxetine before sexual activity and 0.2 mg tamsulosin once a day for 12 weeks. Group 2 (n = 83): only co‐interventions for the same period. Co‐interventions:tamsulosin hydrochloride sustained‐release capsule (0.2 mg, once a day) | |
| Outcomes | Prostatitis symptoms How measured: NIH‐CPSI score Time points measured: baseline, 4 and 8 weeks Time points reported: baseline, 4 and 8 weeks Subgroups: none Sexual dysfunction How measured: Premature Ejaculation Profile (PEP) Time points measured: baseline, 4, 8 and 12 weeks Time points reported: baseline, 4, 8 and 12 weeks Subgroups: none Adverse events How measured: Narratively | |
| Funding sources | National Key R&D Plan of China (2017YF1002003); National Natural Science Foundation of China (81671512, 81701524 and 8170060493). | |
| Declarations of interest | Not available | |
| Notes | Baseline characteristics were available for participants who completed the 12‐week follow‐up (see risk of bias judgements for details regarding attrition). Most outcome data were available graphically at 4 and 8 weeeks. Contact information: Shujie Xia and Zheng Li Emails: [email protected] (SX); [email protected] (ZL) Clinical Trial Registry: ChiCTR1800019441 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomly divided into two treatment groups in a ratio of 1:2." Insufficient information to make a judgement. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Patients were randomly divided into two treatment groups in a ratio of 1:2." Insufficient information to make a judgement. |
| Blinding of participants and personnel (performance bias) | High risk | The interventions were visibly different. Blinding was unlikely. |
| Blinding of outcome assessment (detection bias) | High risk | The interventions were visibly different. Blinding was unlikely. |
| Incomplete outcome data (attrition bias) | High risk | Quote: "Follow‐up data were available for 228, 182 and 114 participants at 4 weeks, 8 weeks and 12 weeks of treatment respectively". Attrition of outcome data (all outcomes) was 10%, 27% and 55% respectively. Reasons for withdrawal were not specified. |
| Selective reporting (reporting bias) | Unclear risk | The clinical trial registry offered insufficient information regarding timepoints and outcomes measured and reported in the trial. |
| Other bias | Low risk | No other sources of bias were identified. |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date October 2005 ‐ end date March 2007 Setting: outpatient ‐ possibly single centre ‐ national Country: China | |
| Participants | Inclusion criteria: Participants diagnosed with CPPS who had undergone conventional clinical treatment, including antibiotics, anti‐inflammatory agents, alpha blockers, phytotherapy, prostatic massage and transrectal ultrasonography of prostate and for whom the treatments had failed and who had no allergy to tetracycline Exclusion criteria: not available. Sample size: 48 participants randomised Age (years): range 29 to 50 NIH‐CPSI baseline score: available graphically All participants were men | |
| Interventions | Group 1 (n = 24): 500 mg tetracycline HCL orally plus 0.4 g vitamin C and 0.2 g co‐vitamin B daily for 3 months. Group 2 (n = 24): only vitamin B (as “placebo”) for the same period Co‐interventions: not available | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline and 3 months Time points reported: baseline and 3 months (graphically) | |
| Funding sources | Not available | |
| Declarations of interest | Not available | |
| Notes | This study focused on the detection of nano‐bacteria (this outcome was not included in our review) Contact information: song‐bo‐[email protected] (email bounced) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. We wrote to study authors |
| Blinding of participants and personnel (performance bias) | High risk | The study refers to the use of placebo in the control group, but the interventions were visibly different. Blinding was unlikely |
| Blinding of outcome assessment (detection bias) | High risk | The study refers to the use of placebo in the control group, but the interventions were visibly different. Blinding was unlikely |
| Incomplete outcome data (attrition bias) | Low risk | All outcome: outcome data were available for all participants |
| Selective reporting (reporting bias) | High risk | Data were presented graphically. NIH‐CPSI scores were available only in 1 group. We wrote to study authors |
| Other bias | Unclear risk | Baseline characteristics were missing. We wrote to study authors |
| Methods | Study design: Parallel‐group randomised trial Study dates: start date September 2002 ‐ end date September 2004 Setting: outpatient ‐ possibly single centre ‐ national Country: Iran | |
| Participants | Inclusion criteria: Participants with pain in penis, perineal region, supra‐pubic, testis and/or pelvis after ejaculation alongside voiding symptoms such as dysuria, frequency and sense of incomplete urination; symptoms duration > 1 year and NIH‐CPSI score > 14; aged 20 to 40 years; normal abdominal palpation; negative 4‐glass study Exclusion criteria: Men with a medical history of documented urinary tract infection, sexually transmitted disease, urethral stricture, neurological disease, drugs which mimic these symptoms (for example, anticholinergics and psychotropics), haematuria and pyuria; abnormal ultrasonography or other imaging; urinary system disease and genitourinary system surgery Sample size: 56 participants randomised Age (years): Group 1 mean = 33.28 (SD 6.4); Group 2 mean = 33.52 (SD 6.15) NIH‐CPSI baseline score: Group 1 mean = 26.28 (SD 5.5); Group 2 mean = 25.07 (SD 6.53) All participants were men | |
| Interventions | Group 1 (n = 29): allopurinol 100 mg 3 times a day for 3 months in addition to ofloxacin for the first 3 weeks Group 2 (n = 27): matching placebo and ofloxacin in the same posology Co‐interventions: not available | |
| Outcomes | Prostatitis Symptoms How measured: NIH‐CPSI score Time points measured: baseline, 1, 2 and 3 months Time points reported: baseline, 1, 2 and 3 months Adverse Events How measured: Narratively | |
| Funding sources | Not available | |
| Declarations of interest | None | |
| Notes | Dr. Amir Mohsen Ziaee: [email protected] | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available. Wrote to study authors |
| Allocation concealment (selection bias) | Unclear risk | No information available. Wrote to study authors |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double‐blind study. No information regarding the blinding of personnel. Participants were blinded. Wrote to study authors |
| Blinding of outcome assessment (detection bias) | Low risk | Participants were blinded using placebo |
| Incomplete outcome data (attrition bias) | Low risk | All outcomes: Quote: “All patients were followed to the end of the study (no loss to follow‐up)." |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. Wrote to study authors |
| Other bias | Low risk | No other sources of bias identified |
AUA: American Urological Association; BCG: bacillus Calmette‐Guerin; BPH: benign prostatic hyperplasia; DRE: digital rectal examination; EPS: expressed prostatic secretions IPSS: International Prostatic Symptom score;s LUTS: lower urinary tract symptom; TURP: transurethral resection of the prostate
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
| Controlled clinical trial. Participants were assigned to control by preference. | |
| Non‐randomised comparative study for Sabal serrulata plant extract (study in Russian) | |
| Non‐randomised controlled trial of thermobalancing therapy for CP/CPPS | |
| Includes participants with bacterial prostatitis. CP/CPPS definition does not include pain | |
| "Intraprostatic botulinum toxin A injection" trial. Personal contact with the author confirmed that the trial was stopped due to problems in recruiting (prospective participants with high levels of liver enzymes) | |
| The study included participants with type II, IIIA, and IIIB chronic prostatitis in their trials and did not present disaggregated results. | |
| Non‐randomised cross‐over comparative study for minocycline | |
| Non‐randomised controlled trial for a physiotherapeutic device and thermobalancing therapy | |
| Non‐controlled study for botulinum toxin A | |
| Quasi‐randomised study of doxazosin vs placebo | |
| This trial of nursing interventions included participants with bacterial prostatitis | |
| Prolexan trial. 60% had bacterial prostatitis, no disaggregated results were available | |
| Electrode plasmapheresis for chronic bacterial prostatitis | |
| Non‐randomised controlled trial for a "combined treatment including complex physical factors" | |
| Abdominal cluster needle, quasi‐randomised trial (randomisation is based on date of admission) | |
| This study was assessed by Cochrane Japan collaborators. It was found to be a non‐randomised study for kampo medicine | |
| Non‐randomised study for a "combined psycho‐ and physiotherapeutic treatment program for patients with chronic pelvic pain syndrome (CPPS)" | |
| Non‐randomised trial for a dietary supplement Prostatinol | |
| Non‐randomised trial for rectal suppositoria Vitaprost | |
| Non‐randomised controlled trial for "magnetolaser therapy" | |
| Observational study secondary to a myofascial trigger point release RCT (included in the twin review Non‐Pharmacological Interventions for treating chronic prostatitis/chronic pelvic pain syndrome) | |
| Non‐randomised study for "Uro‐Vaxom" | |
| Non‐randomised study for a combination of traditional Chinese medicine and Western medicine | |
| Trial for "combination (ciprofloxacin+doxazosin) vs monotherapy (ciprofloxacin)". This trial included participants with bacterial prostatitis | |
| Non‐randomised controlled study for vitaprost (translated from Russian) | |
| Non‐randomised controlled study for Gentos | |
| Quasi‐randomised study for "catgut embedding therapy" | |
| Report of a single arm of the osteopathy trial after 5 years (included in the twin review Non‐Pharmacological Interventions for treating chronic prostatitis/chronic pelvic pain syndrome) | |
| The definition of the study population was not according to NIH criteria | |
| Trial for viagra (Sildenafil). This study has been terminated due to illness of PI | |
| This study on botox has been terminated since re‐organisation of personnel forced termination | |
| This study on "Sympathetic Plexus Block" has suspended participant recruitment. (PI health issues) | |
| This study on thalidomide has been terminated. (Study closed. Difficult enrollment of patients with prostatitis) | |
| Trial of Botulinum Toxin Type A as a single intrasphincteric injection. This study was terminated due to slow accrual | |
| Personal contact with the author (Dr. Nickel): trial stopped for futility before endpoint, it was never published, "8 patients received botox as per protocol 1 patient had mild improvement". Based on slow enrollment, poor results, the trial was discontinued. No follow‐up or report other than to the Institutional Review Board | |
| This study on Gralise(R) has been terminated due to difficulties in recruitment and low enrollment | |
| This study was terminated due to difficulty in enrolling participants. Study for JALYN (dutasteride‐tamsulosin combination) | |
| This study has been withdrawn prior to enrolment | |
| Study on the effects of AQX‐1125. This study was terminated due to the decision of the Sponsor | |
| Analysis of the effects of dutasteride in participants with CP/CPPS symptoms. The included participants were not evaluated to reach a diagnosis of CP/CPPS. The randomised controlled trial's objective is the prevention of prostate cancer | |
| Single‐arm trial of tadalafil for CP/CPPS | |
| Non‐randomised study cross‐over trial of muscle relaxants | |
| Non‐randomised study of Serenoa repens for people with lower urinary tract symptoms (LUTS) | |
| The active treatment and the comparison groups comprised 27% (6 out of 22) of bacterial prostatitis patients. There were no disaggregated data for CP/CPPS participants | |
| Non‐randomised study of "combined physiotherapy" | |
| The definition of CP/CPPS does not match the current definition. In fact some participants had "non‐specific urethritis" and 6 participants were asymptomatic at the beginning of the study | |
| Quasi‐randomised study of antibiotic vs phytotherapeutic therapy | |
| This study was assessed by Cochrane Japan collaborators. It was found to be a non‐randomised study for levofloxacin and cernitin pollen extract | |
| Non‐randomised study for the comparison of "minocycline, trimethoprim, co‐trimoxazole or diazepam" | |
| Non‐randomised study for "Vitaprost" | |
| Non‐randomised study for "Vitaprost" as add‐on therapy to physiotherapy | |
| This study was a Phase II, dose‐finding study with an adaptive randomisation design. | |
| Non‐randomised study of combination therapy (antibiotics, alpha blockers, traditional Chinese medicine, etc.) | |
| he diagnosis of CP/CPPS was not according to the review definition. The authors referred to the presence of both "Chinese medicine (CM) Gan (肝)‐qi stagnancy syndrome type" and benign prostatic hyperplasia (BPH) |
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | A randomised, double‐blind, placebo‐controlled, parallel‐group study to determine the effect of BXL628 in patients with Chronic Non‐Bacterial Prostatitis category III Chronic Pelvic Pain Syndrome, CP/CPPS Method: Randomised controlled trial, double‐blind, phase II study Trial status: completed |
| Participants | Adults with CP/CPPS (no further information provided), estimated n = 120 |
| Interventions | Intervention: BXL628 150 µg Control: Placebo |
| Outcomes | Not available. |
| Notes | Sponsor: BIOXELL SPA Contact: [email protected], [email protected], [email protected], [email protected], [email protected] (no response) Data extracted from: www.clinicaltrialsregister.eu/ctr‐search/trial/2005‐001849‐42/IT |
| Methods | A randomised placebo‐controlled study of tamsulosin, voltarol and the combination in types IIIa and IIIb prostatitis using the newly developed and validated National Institutes of Health (NIH) symptom score Method: Randomised controlled trial Overall trial start date: 04 May 2000 Overall trial end date: 03 November 2003 |
| Participants | Participant inclusion criteria: 60 men aged between 30 and 50 Participant type: no further specifications Target number of participants: 60 Participant exclusion criteria: not provided at time of registration |
| Interventions | 1. Placebo only 2. Tamsulosin 3. Voltarol 4. Tamsulosin and Voltarol |
| Outcomes | Quote: "The main outcome measure will be a reduction in the National Institute of Health Chronic Prostatitis Symptom Index (NIHCPSI), which is a newly produced and validated symptom score for assessing men with this condition." |
| Notes | Funder name: Cambridge Consortium ‐ Addenbrooke's (UK) Contact: [email protected] (no response) Data extracted from: www.isrctn.com/ISRCTN46815629 |
| Methods | This study had two stages: The first stage is a descriptive study in patients with chronic prostatitis. The second stage was an open prospective randomized trial. of patients with CP/CPPS study was conducted. Two groups of patients with chronic nonbacterial prostatitis with signs of inflammation were formed. A control group (n=29) received standard treatment, rectal 0.5 methyluracil suppositories three times per week, 20 suppositories per course. In addition to the standard treatment, the patients of the study group (n=31) were administered Longidaza 3000 IU rectal suppositories also three times a week, 20 suppositories per course. |
| Participants | Participants with CP/CPPS |
| Interventions | Control group (n=29): "standard treatment" with rectal 0.5 methyluracil suppositories three times per week, 20 suppositories per course. Intervention group (n=31): longidaza 3000 IU rectal suppositories also three times a week, 20 suppositories per course. |
| Outcomes | Fibrous connective tissue formation Prostatitis symptoms |
| Notes | The report of this study included only the histopathological outcomes. We wrote to study authors: [email protected] (no response) |
| Methods | An effectiveness and safety study of ELMIRON (pentosan polysulfate sodium) for the treatment of chronic non‐bacterial inflammation of the prostate gland Study type: Interventional, randomised, parallel assignment, clinical trial Enrolment: 283 |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Quote: "The objective of this randomized, double‐blind, placebo‐controlled study is to determine the effectiveness and safety of ELMIRON® 100 mg three times per day for 12 weeks, as compared with placebo" |
| Outcomes | Primary outcome measures: Change in total NIH‐CPSI score from baseline to Week 12 |
| Notes | Sponsor: McNeil Consumer & Specialty Pharmaceuticals, a Division of McNeil‐PPC, Inc We contacted Dr. Nickel since the characteristics of the study were very similar to those of the trial Nickel 2005. However the number of participants randomised and the dose of pentosan are different. Dr. Nickel replied that this could be the trial registry for that study. Data extracted from ClinicalTrials.gov: NCT00236990 |
| Methods | Efficacy Study of Tamsulosin and Tolterodine Treatment for Chronic Prostatitis (ESTTFCP) Study Type: Interventional, randomised, parallel assignment, clinical trial |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: tolterodine 4 mg + tamsulosin 0.4 mg Active comparator: tamsulosin 0.4 mg + placebo |
| Outcomes | Primary outcome measures: National Institutes of Health Chronic Prostatitis Symptom Index: time frame: 4 months |
| Notes | Xiaohou Wu, MD 86‐2389011122 [email protected] (no response) Sponsors and Collaborators: Chongqing Medical University, Fuling Central Hospital of Chongqing City Data extracted from ClinicalTrials.gov: NCT00913315 This trial was also registered in ChiCTR‐TRC‐09000391 http://www.chictr.org.cn/showprojen.aspx?proj=9142 This registry indicated that sample size would be 160 (80 participants in each arm) |
| Methods | Treating urological chronic pelvic pain syndrome (UCPPS) pain (UCPPS) Study type: Interventional, randomised, parallel‐assignment, clinical trial |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: D‐Cycloserine 200 mg twice daily and acetominophen when necessary Placebo capsules (lactose)/twice a day and acetaminophen when necessary |
| Outcomes | Primary outcome measures: Visual Analog Scale (VAS): time frame: 18 weeks after baseline visit Secondary outcome measures: McGill Pain Questionnaire: time frame: 18 weeks after baseline visit Functional Magnetic Resonance Imaging (fMRI) connectivity: time frame: 12 weeks after baseline visit |
| Notes | Contact: a‐[email protected] (no response) Sponsor information not available Data extracted from ClinicalTrials.gov: NCT02385266 |
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Effect of saw palmetto extract in chronic prostatitis/chronic pelvic pain syndrome (CP/CPPS): a multicenter, randomized, double‐blind, placebo‐controlled trial |
| Methods | Study design: randomised parallel controlled trial Study dates: 01 January 2017 to 30 June 2018 Setting: outpatient, national/multicentre Country: China |
| Participants | Inclusion criteria: 1. Male 18 ‐ 50 years old; 2. The participants has pelvic area pain or discomfort at least 3 months prior to admission; 3. The NIH‐CPSI total scores > 10,and pain scores ≥ 4; 4. Men with clinically and laboratory‐diagnosed CP/CPPS; 5. The participants have sexual desire to live at least in the last 3 months; 6. Signed informed consent; 7. Participants could understand the requirements of this trial Exclusion criteria: 1. Urinary tract infection or urine culture positive, clinical diagnosis of urethritis (including urethral mouth secretions, the culture was positive), acute epididymitis patients, patients with suspected prostate cancer; 2. Patients with genital herpes diagnosed or treated within 12 months before admission; 3. Patients with diseases that could affect micturition, such as neurogenic bladder dysfunction or urethral stricture; 4. Patients who have a history of operation or trauma, which affect the evaluation of efficacy of the drug; 5. Patients with severe cardiovascular disease, sexually‐transmitted diseases (including gonorrhoea, chlamydia, mycoplasma or Trichomonas, but does not include any HIV/AIDS), malignant tumour, peptic ulcer, haemorrhage disease; 6. Patients are taking antibiotics, non‐steroidal anti‐inflammatory drugs, alpha receptor blockers, bioflavonoids, Chinese patent medicines and plant drugs for prostatitis; 7. Patients are taking drugs that affect function of bladder outlet and sexual; 8. Patients with insufficiency of liver or kidney, AST or ALT more than 1.5 times the upper limit of normal, creatinine more than the upper limit of normal and with clinical significance; 9. Have a history of allergic reactions of alcohol, study drug or similar drugs; 10. Patients with mental diseases, acrasia, alcohol dependence and drug abuse; 11. In 3 months participated in any other clinical trials, or have birth preparation; 12. Researchers recognised anyone who is not suitable to participate in this clinical study of patients Sample size: 240 participants |
| Interventions | Intervention (n = 160): Saw palmetto extract 160 mg orally twice daily (duration not available) Control (n = 80): Placebo orally twice daily (duration not available) |
| Outcomes | Prostatitis symptoms: NIH‐CPSI score (time points not available) Sexual dysfunction: IIEF score (time points not available) Co‐interventions: not described |
| Starting date | Not available (approved by the ethics committee 15 November 2016) |
| Contact information | |
| Notes | Funding: Peking University First Hospital Data extracted from clinical trial registry: ChiCTR‐IPR‐16010196 |
| Trial name or title | Effectiveness of pregabalin as an adjuvant therapy for chronic nonbacterial prostatitis |
| Methods | Study design: Randomised controlled trial, double‐blind, phase II study Study dates: 20 March 2016 ‐ 21 March 2017 (recruitment) Setting: outpatient, probably national and single centre Country: Iran |
| Participants | Inclusion criteria: 1) Age over 18 years; 2) no prior history of other urinary system disorders such as urinary tract stones, neurogenic bladder, and urinary infections; 3) no history of major underlying diseases such as cirrhosis, renal failure, diabetes, or hypertension; 4) normal levels of urine, fasting blood sugar (FBS), and creatinine; 5) normal results on the first neurological examination of the perineal area, lower limbs, lower limb reflexes, and bulbocavernosus reflex; 6) no use of other medicines; 7) no signs of psychological disorders; 8) confirmed diagnosis of chronic prostatitis; 9) non‐sensitivity to anti‐epileptic drugs, pregabalin, thiazolidinediones, or other anti‐diabetic agents. Exclusion criteria: 1) Unwillingness to continue the study, and 2) major side‐effects due to the use of medications. Target sample size: 288 |
| Interventions | Intervention group: Patients with chronic nonbacterial prostatitis use of terazosin tablets (2 mg) and pregabalin capsules (50 mg) on a daily basis for the first week, followed by the daily use of 100 mg pregabalin capsules for six weeks., measurement of pain, and urinary symptoms after therapy. Control group: Patients with chronic nonbacterial prostatitis use of Terazosin tablets (2 mg) and placebo capsules (50 mg) on a daily basis for the first week, followed by the daily use of 100 mg placebo capsules for six weeks, measurement of pain, and urinary symptoms after therapy. |
| Outcomes | Evaluation and comparison of pain, urinary symptoms, quality of life, and side‐effects of medications between the intervention and control groups, based on the scoring system introduced by the NIH‐CPSI score (at 6 weeks). |
| Starting date | 20 March 2016 |
| Contact information | Contact information: m‐[email protected] (Ali Serous) |
| Notes | Funding source: Vice Chancellor for Research of Arak University of Medical Sciences Data extracted from clinical trial registry: IRCT2016022225507N2 |
| Trial name or title | A clinical trial to comparison the effectiveness of melatonin and capsules containing starch (placebo) as an adjuvant therapy to control symptoms of chronic nonbacterial prostatitis patients |
| Methods | Study design: Randomised controlled trial, double‐blind, phase II study Study dates: 22 August 2016 ‐ 23 August 2017 (recruitment) Setting: outpatient, probably national and single centre Country: Iran |
| Participants | Inclusion criteria: Age over 18 years; no prior history of other urinary system disorders such as urinary tract stones, neurogenic bladder, and urinary infections; no history of underlying diseases such as cirrhosis, renal failure, diabetes, or hypertension; normal levels of urine, fasting blood sugar (FBS), and creatinine; normal results on the first neurological examination of the perineal area, lower limbs, lower limb reflexes, and bulbocavernosus reflex; having weight between 50 to 100 kg; no use of other medicines; no signs of psychological disorders; confirmed diagnosis of chronic prostatitis; creatinine equal to or lower than 1.2 mg; platelet count over 100000/mm3; non‐sensitivity to anti‐epileptic drugs, pregabalin, thiazolidinediones, or other anti‐diabetic agents. Exclusion criteria: Unwillingness to continue the study; major side‐effects due to the use of medications. |
| Interventions | Intervention group: patients in this group will receive 3mg of melatonin capsules daily for six weeks. Control group: patients in this group will receive 2mg of terazosin tablets (Razak Company) plus 3mg of starch capsules (placebo). |
| Outcomes | Evaluation and comparison of pain relief, urinary symptoms and side‐effects of medications between two groups, based on the scoring system introduced by the National Institute of Health‐Chronic Prostatitis Symptom Index (NIH‐CPSI) and global response assessment (GRA) two, four and six weeks after intervention. |
| Starting date | 22 August 2016 |
| Contact information | Contact information: m‐[email protected] (Ali Serous) |
| Notes | Funding source: Vice Chancellor for Research of Arak University of Medical Sciences Data extracted from clinical trial registry: IRCT2016071025507N4 |
| Trial name or title | The Effect of Cinnamon on Patients With Chronic Prostatitis/Chronic Pelvic Pain Syndrome; a Pilot Study |
| Methods | Study design: randomised parallel controlled trial Study dates: 01 January 2017 to 30 June 2018 Setting: outpatient, national/multicentre Country: China |
| Participants | Inclusion Criteria:
Exclusion Criteria:
|
| Interventions | Experimental: Each patient will receive sixty capsules, each capsule contained 1gm of cinnamon bark powder and instructed to use it twice daily for one month Placebo Comparator: Each patient will receive sixty capsules, each capsule contained placebo and instructed to use it twice daily for one month |
| Outcomes | Prostatitis symptoms Primary Outcome Measures: positive response [Time Frame: 1 month] A reduction in the NIH‐CPSI score of 6 or more points from the initial score A reduction in one or more the sub‐scores of the NIH‐CPSI |
| Starting date | February 1, 2018 Completion date: June 30, 2019 (study results not available) |
| Contact information | Principal Investigator: Tawfik J Al‐Marzooq, F.I.C.M.S. |
| Notes | We classified this study as "ongoing" since it was only recently completed. |
Data and analyses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms: short term Show forest plot | 18 | 1524 | Mean Difference (IV, Random, 95% CI) | ‐5.01 [‐7.41, ‐2.61] |
| Analysis 1.1  Comparison 1 Alpha‐blockers versus placebo, Outcome 1 Prostatitis symptoms: short term. | ||||
| 1.1 Terazosin | 5 | 436 | Mean Difference (IV, Random, 95% CI) | ‐5.73 [‐10.96, ‐0.50] |
| 1.2 Doxazosin | 4 | 262 | Mean Difference (IV, Random, 95% CI) | ‐5.17 [‐11.50, 1.16] |
| 1.3 Phenoxybenzamine | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐5.80 [‐7.91, ‐3.69] |
| 1.4 Tamsulosin | 4 | 302 | Mean Difference (IV, Random, 95% CI) | ‐5.89 [‐13.16, 1.38] |
| 1.5 Alfuzosin | 4 | 381 | Mean Difference (IV, Random, 95% CI) | ‐2.63 [‐4.55, ‐0.71] |
| 1.6 Silodosin | 1 | 103 | Mean Difference (IV, Random, 95% CI) | ‐3.60 [‐6.81, ‐0.39] |
| 2 Prostatitis symptoms: pain Show forest plot | 13 | 1243 | Mean Difference (IV, Random, 95% CI) | ‐2.39 [‐3.57, ‐1.22] |
| Analysis 1.2  Comparison 1 Alpha‐blockers versus placebo, Outcome 2 Prostatitis symptoms: pain. | ||||
| 2.1 Terazosin | 3 | 289 | Mean Difference (IV, Random, 95% CI) | ‐3.86 [‐6.00, ‐1.73] |
| 2.2 Doxazosin | 3 | 202 | Mean Difference (IV, Random, 95% CI) | ‐2.19 [‐4.56, 0.17] |
| 2.3 Phenoxybenzamine | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐2.25 [‐3.78, ‐0.72] |
| 2.4 Tamsulosin | 3 | 265 | Mean Difference (IV, Random, 95% CI) | ‐3.17 [‐6.31, ‐0.03] |
| 2.5 Alfuzosin | 3 | 344 | Mean Difference (IV, Random, 95% CI) | ‐0.59 [‐1.67, 0.49] |
| 2.6 Silodosin | 1 | 103 | Mean Difference (IV, Random, 95% CI) | ‐1.40 [‐2.97, 0.17] |
| 3 Prostatitis symptoms: urinary Show forest plot | 13 | 1243 | Mean Difference (IV, Random, 95% CI) | ‐1.48 [‐2.29, ‐0.66] |
| Analysis 1.3  Comparison 1 Alpha‐blockers versus placebo, Outcome 3 Prostatitis symptoms: urinary. | ||||
| 3.1 Terazosin | 3 | 289 | Mean Difference (IV, Random, 95% CI) | ‐2.07 [‐3.79, ‐0.34] |
| 3.2 Doxazosin | 3 | 202 | Mean Difference (IV, Random, 95% CI) | ‐1.71 [‐4.34, 0.92] |
| 3.3 Phenoxybenzamine | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐1.60 [‐2.28, ‐0.92] |
| 3.4 Tamsulosin | 3 | 265 | Mean Difference (IV, Random, 95% CI) | ‐1.63 [‐3.85, 0.59] |
| 3.5 Alfuzosin | 3 | 344 | Mean Difference (IV, Random, 95% CI) | ‐0.67 [‐1.05, ‐0.29] |
| 3.6 Silodosin | 1 | 103 | Mean Difference (IV, Random, 95% CI) | ‐0.90 [‐2.00, 0.20] |
| 4 Prostatitis symptoms: quality of life Show forest plot | 13 | 1243 | Mean Difference (IV, Random, 95% CI) | ‐1.61 [‐2.49, ‐0.73] |
| Analysis 1.4  Comparison 1 Alpha‐blockers versus placebo, Outcome 4 Prostatitis symptoms: quality of life. | ||||
| 4.1 Terazosin | 3 | 289 | Mean Difference (IV, Random, 95% CI) | ‐2.51 [‐4.62, ‐0.40] |
| 4.2 Doxazosin | 3 | 202 | Mean Difference (IV, Random, 95% CI) | ‐1.34 [‐3.24, 0.56] |
| 4.3 Phenoxybenzamine | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐2.2 [‐3.14, ‐1.26] |
| 4.4 Tamsulosin | 3 | 265 | Mean Difference (IV, Random, 95% CI) | ‐1.78 [‐4.96, 1.40] |
| 4.5 Alfuzosin | 3 | 344 | Mean Difference (IV, Random, 95% CI) | ‐0.53 [‐1.34, 0.29] |
| 4.6 Silodosin | 1 | 103 | Mean Difference (IV, Random, 95% CI) | ‐1.40 [‐2.49, ‐0.31] |
| 5 Prostatitis symptoms: responders rate Show forest plot | 7 | 721 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.94, 1.61] |
| Analysis 1.5  Comparison 1 Alpha‐blockers versus placebo, Outcome 5 Prostatitis symptoms: responders rate. | ||||
| 5.1 Doxazosin | 1 | 55 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.67, 1.20] |
| 5.2 Tamsulosin | 3 | 254 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [1.07, 2.06] |
| 5.3 Alfuzosin | 2 | 309 | Risk Ratio (M‐H, Random, 95% CI) | 1.82 [0.48, 6.80] |
| 5.4 Silodosin | 1 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.78, 1.52] |
| 6 Prostatitis symptoms: long term Show forest plot | 4 | 235 | Mean Difference (IV, Random, 95% CI) | ‐5.60 [‐10.89, ‐0.32] |
| Analysis 1.6  Comparison 1 Alpha‐blockers versus placebo, Outcome 6 Prostatitis symptoms: long term. | ||||
| 6.1 Terazosin | 1 | 51 | Mean Difference (IV, Random, 95% CI) | ‐7.76 [‐10.90, ‐4.62] |
| 6.2 Tamsulosin | 1 | 92 | Mean Difference (IV, Random, 95% CI) | ‐1.40 [‐2.73, ‐0.07] |
| 6.3 Alfuzosin | 1 | 35 | Mean Difference (IV, Random, 95% CI) | ‐3.4 [‐7.30, 0.50] |
| 6.4 Doxazosin | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐9.7 [‐10.25, ‐9.15] |
| 7 Prostatitis symptoms: long term Show forest plot | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [1.06, 2.32] |
| Analysis 1.7  Comparison 1 Alpha‐blockers versus placebo, Outcome 7 Prostatitis symptoms: long term. | ||||
| 7.1 Tamsulosin | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [1.06, 2.32] |
| 8 Adverse events Show forest plot | 19 | 1588 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.09, 2.34] |
| Analysis 1.8  Comparison 1 Alpha‐blockers versus placebo, Outcome 8 Adverse events. | ||||
| 8.1 Terazosin | 6 | 431 | Risk Ratio (M‐H, Random, 95% CI) | 2.03 [1.14, 3.61] |
| 8.2 Doxazosin | 3 | 195 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [0.75, 2.88] |
| 8.3 Phenoxybenzamine | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 35.00 [2.25, 544.92] |
| 8.4 Tamsulosin | 5 | 367 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [0.60, 3.03] |
| 8.5 Alfuzosin | 4 | 452 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.37, 3.78] |
| 8.6 Silodosin | 1 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 2.52 [1.15, 5.52] |
| 9 Sexual dysfunction Show forest plot | 4 | 452 | Mean Difference (IV, Random, 95% CI) | 0.26 [‐1.13, 1.65] |
| Analysis 1.9  Comparison 1 Alpha‐blockers versus placebo, Outcome 9 Sexual dysfunction. | ||||
| 9.1 Terazosin | 1 | 77 | Mean Difference (IV, Random, 95% CI) | 1.10 [‐0.84, 3.04] |
| 9.2 Tamsulosin | 1 | 99 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐4.84, 4.44] |
| 9.3 Alfuzosin | 2 | 276 | Mean Difference (IV, Random, 95% CI) | ‐0.70 [‐2.89, 1.48] |
| 10 Quality of life: mental Show forest plot | 3 | 421 | Mean Difference (IV, Random, 95% CI) | 0.15 [‐2.63, 2.92] |
| Analysis 1.10  Comparison 1 Alpha‐blockers versus placebo, Outcome 10 Quality of life: mental. | ||||
| 10.1 Tamsulosin | 1 | 90 | Mean Difference (IV, Random, 95% CI) | ‐1.00 [‐7.16, 1.16] |
| 10.2 Alfuzosin | 1 | 228 | Mean Difference (IV, Random, 95% CI) | 2.1 [‐0.64, 4.84] |
| 10.3 Silodosin | 1 | 103 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐3.09, 3.69] |
| 11 Quality of life: physical Show forest plot | 3 | 421 | Mean Difference (IV, Random, 95% CI) | 1.17 [‐0.97, 3.30] |
| Analysis 1.11  Comparison 1 Alpha‐blockers versus placebo, Outcome 11 Quality of life: physical. | ||||
| 11.1 Tamsulosin | 1 | 90 | Mean Difference (IV, Random, 95% CI) | 2.4 [‐0.52, 5.32] |
| 11.2 Alfuzosin | 1 | 228 | Mean Difference (IV, Random, 95% CI) | ‐0.5 [‐2.51, 1.51] |
| 11.3 Silodosin | 1 | 103 | Mean Difference (IV, Random, 95% CI) | 2.5 [‐0.81, 5.81] |
| 12 Anxiety and depression Show forest plot | 1 | 232 | Mean Difference (IV, Random, 95% CI) | ‐1.1 [‐2.54, 0.34] |
| Analysis 1.12  Comparison 1 Alpha‐blockers versus placebo, Outcome 12 Anxiety and depression. | ||||
| 12.1 Alfuzosin | 1 | 232 | Mean Difference (IV, Random, 95% CI) | ‐1.1 [‐2.54, 0.34] |
| 13 Urinary symptoms Show forest plot | 2 | 143 | Mean Difference (IV, Random, 95% CI) | ‐2.68 [‐5.90, 0.54] |
| Analysis 1.13  Comparison 1 Alpha‐blockers versus placebo, Outcome 13 Urinary symptoms. | ||||
| 13.1 Terazosin | 1 | 86 | Mean Difference (IV, Random, 95% CI) | ‐4.5 [‐6.64, ‐2.36] |
| 13.2 Alfuzosin | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐1.20 [‐1.76, ‐0.64] |
| 14 Prostatitis symptoms: responders rate (sensitivity analysis according to risk of bias) Show forest plot | 2 | 155 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.61, 2.48] |
| Analysis 1.14  Comparison 1 Alpha‐blockers versus placebo, Outcome 14 Prostatitis symptoms: responders rate (sensitivity analysis according to risk of bias). | ||||
| 14.1 Tamsulosin | 2 | 155 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.61, 2.48] |
| 15 Adverse events (sensitivity analysis according to risk of bias) Show forest plot | 2 | 153 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.63, 1.47] |
| Analysis 1.15  Comparison 1 Alpha‐blockers versus placebo, Outcome 15 Adverse events (sensitivity analysis according to risk of bias). | ||||
| 15.1 Tamsulosin | 2 | 153 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.63, 1.47] |
| 16 Prostatitis symptoms: short term (subgroup analysis by co‐interventions) Show forest plot | 18 | 1524 | Mean Difference (IV, Random, 95% CI) | ‐5.01 [‐7.41, ‐2.61] |
| Analysis 1.16  Comparison 1 Alpha‐blockers versus placebo, Outcome 16 Prostatitis symptoms: short term (subgroup analysis by co‐interventions). | ||||
| 16.1 Without co‐interventions | 7 | 688 | Mean Difference (IV, Random, 95% CI) | ‐3.56 [‐5.26, ‐1.86] |
| 16.2 With co‐interventions (analgesics, antibiotics) | 11 | 836 | Mean Difference (IV, Random, 95% CI) | ‐5.69 [‐8.90, ‐2.48] |
| 17 Adverse events (subgroup analysis by co‐interventions) Show forest plot | 19 | 1588 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.09, 2.34] |
| Analysis 1.17  Comparison 1 Alpha‐blockers versus placebo, Outcome 17 Adverse events (subgroup analysis by co‐interventions). | ||||
| 17.1 Without co‐interventions | 11 | 1089 | Risk Ratio (M‐H, Random, 95% CI) | 1.69 [1.10, 2.60] |
| 17.2 With co‐interventions (analgesics, antibiotics) | 8 | 499 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.46, 3.17] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 2.1  Comparison 2 5 alpha reductase inhibitors versus placebo, Outcome 1 Prostatitis symptoms. | ||||
| 2 Prostatitis symptoms: responder rate Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| Analysis 2.2  Comparison 2 5 alpha reductase inhibitors versus placebo, Outcome 2 Prostatitis symptoms: responder rate. | ||||
| 3 Adverse events Show forest plot | 2 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.33, 2.30] |
| Analysis 2.3  Comparison 2 5 alpha reductase inhibitors versus placebo, Outcome 3 Adverse events. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 5 | 372 | Mean Difference (IV, Random, 95% CI) | ‐2.43 [‐4.72, ‐0.15] |
| Analysis 3.1  Comparison 3 Antibiotic therapy versus placebo, Outcome 1 Prostatitis symptoms. | ||||
| 1.1 Ciprofloxacin | 3 | 215 | Mean Difference (IV, Random, 95% CI) | ‐1.69 [‐3.15, ‐0.23] |
| 1.2 Levofloxacin | 2 | 157 | Mean Difference (IV, Random, 95% CI) | ‐2.98 [‐9.41, 3.44] |
| 2 Prostatitis symptoms: pain Show forest plot | 4 | 319 | Mean Difference (IV, Random, 95% CI) | ‐0.92 [‐1.78, ‐0.06] |
| Analysis 3.2  Comparison 3 Antibiotic therapy versus placebo, Outcome 2 Prostatitis symptoms: pain. | ||||
| 2.1 Ciprofloxacin | 2 | 155 | Mean Difference (IV, Random, 95% CI) | ‐0.78 [‐1.86, 0.30] |
| 2.2 Levofloxacin | 2 | 164 | Mean Difference (IV, Random, 95% CI) | ‐0.72 [‐2.74, 1.30] |
| 3 Prostatitis symptoms: urinary Show forest plot | 4 | 319 | Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.65, 0.41] |
| Analysis 3.3  Comparison 3 Antibiotic therapy versus placebo, Outcome 3 Prostatitis symptoms: urinary. | ||||
| 3.1 Ciprofloxacin | 2 | 155 | Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐1.17, 0.58] |
| 3.2 Levofloxacin | 2 | 164 | Mean Difference (IV, Random, 95% CI) | 0.13 [‐0.85, 1.11] |
| 4 Prostatitis symptoms: quality of life Show forest plot | 4 | 319 | Mean Difference (IV, Random, 95% CI) | ‐0.45 [‐0.91, 0.02] |
| Analysis 3.4  Comparison 3 Antibiotic therapy versus placebo, Outcome 4 Prostatitis symptoms: quality of life. | ||||
| 4.1 Ciprofloxacin | 2 | 155 | Mean Difference (IV, Random, 95% CI) | ‐0.44 [‐1.04, 0.15] |
| 4.2 Levofloxacin | 2 | 164 | Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐1.52, 0.53] |
| 5 Prostatitis symptoms: responder rate Show forest plot | 2 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.73, 1.74] |
| Analysis 3.5  Comparison 3 Antibiotic therapy versus placebo, Outcome 5 Prostatitis symptoms: responder rate. | ||||
| 5.1 Ciprofloxacin | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.48, 2.09] |
| 5.2 Levofloxacin | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.70, 2.05] |
| 6 Adverse events Show forest plot | 4 | 336 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.66, 1.55] |
| Analysis 3.6  Comparison 3 Antibiotic therapy versus placebo, Outcome 6 Adverse events. | ||||
| 6.1 Ciprofloxacin | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.60, 1.57] |
| 6.2 Levofloxacin | 3 | 241 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.46, 2.97] |
| 7 Sexual dysfunction Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 3.7  Comparison 3 Antibiotic therapy versus placebo, Outcome 7 Sexual dysfunction. | ||||
| 7.1 Levofloxacin | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Quality of life: mental Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 3.8  Comparison 3 Antibiotic therapy versus placebo, Outcome 8 Quality of life: mental. | ||||
| 8.1 Ciprofloxacin | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Quality of life: physical Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 3.9  Comparison 3 Antibiotic therapy versus placebo, Outcome 9 Quality of life: physical. | ||||
| 9.1 Ciprofloxacin | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Urinary symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 3.10  Comparison 3 Antibiotic therapy versus placebo, Outcome 10 Urinary symptoms. | ||||
| 10.1 Ciprofloxacin | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Prostatitis symptoms: subgroup analysis (age) Show forest plot | 5 | 372 | Mean Difference (IV, Random, 95% CI) | ‐2.43 [‐4.72, ‐0.15] |
| Analysis 3.11  Comparison 3 Antibiotic therapy versus placebo, Outcome 11 Prostatitis symptoms: subgroup analysis (age). | ||||
| 11.1 Mean age > 50 years old | 1 | 80 | Mean Difference (IV, Random, 95% CI) | 0.60 [‐3.79, 4.99] |
| 11.2 Mean age < 50 years old | 4 | 292 | Mean Difference (IV, Random, 95% CI) | ‐2.92 [‐5.37, ‐0.46] |
| 12 Adverse events: subgroup analysis (age) Show forest plot | 4 | 336 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.66, 1.55] |
| Analysis 3.12  Comparison 3 Antibiotic therapy versus placebo, Outcome 12 Adverse events: subgroup analysis (age). | ||||
| 12.1 Mean age > 50 years old | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.46, 2.97] |
| 12.2 Mean age < 50 years old | 3 | 256 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.60, 1.57] |
| 13 Prostatitis symptoms: subgroup analysis (co‐interventions) Show forest plot | 5 | 372 | Mean Difference (IV, Random, 95% CI) | ‐2.43 [‐4.72, ‐0.15] |
| Analysis 3.13  Comparison 3 Antibiotic therapy versus placebo, Outcome 13 Prostatitis symptoms: subgroup analysis (co‐interventions). | ||||
| 13.1 Without co‐interventions | 2 | 167 | Mean Difference (IV, Random, 95% CI) | ‐1.57 [‐4.77, 1.63] |
| 13.2 With co‐interventions (alpha blockers) | 3 | 205 | Mean Difference (IV, Random, 95% CI) | ‐2.96 [‐6.28, 0.37] |
| 14 Adverse events: subgroup analysis (co‐interventions) Show forest plot | 4 | 336 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.66, 1.55] |
| Analysis 3.14  Comparison 3 Antibiotic therapy versus placebo, Outcome 14 Adverse events: subgroup analysis (co‐interventions). | ||||
| 14.1 Without co‐interventions | 2 | 175 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.66, 1.55] |
| 14.2 With co‐interventions (alpha blockers) | 2 | 161 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 7 | 585 | Mean Difference (IV, Random, 95% CI) | ‐2.50 [‐3.74, ‐1.26] |
| Analysis 4.1  Comparison 4 Antiinflammatories versus placebo, Outcome 1 Prostatitis symptoms. | ||||
| 1.1 Corticosteroids | 1 | 158 | Mean Difference (IV, Random, 95% CI) | ‐2.82 [‐4.10, ‐1.54] |
| 1.2 Nonsteroidal anti‐inflammatory drugs | 5 | 369 | Mean Difference (IV, Random, 95% CI) | ‐2.56 [‐4.50, ‐0.62] |
| 1.3 Thiocolchicoside and ibuprofen | 1 | 58 | Mean Difference (IV, Random, 95% CI) | ‐1.5 [‐2.10, ‐0.90] |
| 2 Prostatitis symptoms: pain Show forest plot | 6 | 497 | Mean Difference (IV, Random, 95% CI) | ‐2.28 [‐4.08, ‐0.48] |
| Analysis 4.2  Comparison 4 Antiinflammatories versus placebo, Outcome 2 Prostatitis symptoms: pain. | ||||
| 2.1 Corticosteroids | 1 | 158 | Mean Difference (IV, Random, 95% CI) | ‐1.67 [‐1.96, ‐1.38] |
| 2.2 Nonsteroidal anti‐inflammatory drugs | 3 | 265 | Mean Difference (IV, Random, 95% CI) | ‐1.80 [‐3.80, 0.20] |
| 2.3 Antileukotriene | 1 | 17 | Mean Difference (IV, Random, 95% CI) | 1.60 [‐5.79, 8.99] |
| 2.4 Thiocolchicoside and ibuprofen | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐5.2 [‐5.59, ‐4.81] |
| 3 Prostatitis symptoms: urinary Show forest plot | 6 | 497 | Mean Difference (IV, Random, 95% CI) | ‐0.75 [‐1.53, 0.03] |
| Analysis 4.3  Comparison 4 Antiinflammatories versus placebo, Outcome 3 Prostatitis symptoms: urinary. | ||||
| 3.1 Corticosteroids | 1 | 158 | Mean Difference (IV, Random, 95% CI) | ‐0.69 [‐0.88, ‐0.50] |
| 3.2 Nonsteroidal anti‐inflammatory drugs | 3 | 265 | Mean Difference (IV, Random, 95% CI) | ‐0.38 [‐0.64, ‐0.13] |
| 3.3 Antileukotriene | 1 | 17 | Mean Difference (IV, Random, 95% CI) | 0.60 [‐0.84, 2.04] |
| 3.4 Thiocolchicoside and ibuprofen | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐3.00 [‐3.47, ‐2.53] |
| 4 Prostatitis symptoms: quality of life Show forest plot | 6 | 497 | Mean Difference (IV, Random, 95% CI) | ‐1.25 [‐2.58, 0.08] |
| Analysis 4.4  Comparison 4 Antiinflammatories versus placebo, Outcome 4 Prostatitis symptoms: quality of life. | ||||
| 4.1 Corticosteroids | 1 | 158 | Mean Difference (IV, Random, 95% CI) | ‐0.5 [‐0.75, ‐0.25] |
| 4.2 Nonsteroidal anti‐inflammatory drugs | 3 | 265 | Mean Difference (IV, Random, 95% CI) | ‐0.45 [‐0.90, 0.01] |
| 4.3 Antileukotriene | 1 | 17 | Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐4.86, 4.06] |
| 4.4 Thiocolchicoside and ibuprofen | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐4.5 [‐5.02, ‐3.98] |
| 5 Prostatitis symptoms: responder rate Show forest plot | 2 | 82 | Risk Ratio (M‐H, Random, 95% CI) | 1.44 [0.68, 3.03] |
| Analysis 4.5  Comparison 4 Antiinflammatories versus placebo, Outcome 5 Prostatitis symptoms: responder rate. | ||||
| 5.1 Corticosteroids | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.25, 4.00] |
| 5.2 Nonsteroidal anti‐inflammatory drugs | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.67 [0.69, 4.04] |
| 6 Prostatitis symptoms: long term Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 4.6  Comparison 4 Antiinflammatories versus placebo, Outcome 6 Prostatitis symptoms: long term. | ||||
| 6.1 Thiocolchicoside and ibuprofen | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Adverse events Show forest plot | 7 | 540 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.81, 2.00] |
| Analysis 4.7  Comparison 4 Antiinflammatories versus placebo, Outcome 7 Adverse events. | ||||
| 7.1 Corticosteroids | 1 | 160 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.24, 102.53] |
| 7.2 Nonsteroidal anti‐inflammatory drugs | 4 | 303 | Risk Ratio (M‐H, Random, 95% CI) | 1.85 [0.79, 4.32] |
| 7.3 Antileukotriene | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.23, 1.37] |
| 7.4 Thiocolchicoside and ibuprofen | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.83, 2.43] |
| 8 Urinary symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 4.8  Comparison 4 Antiinflammatories versus placebo, Outcome 8 Urinary symptoms. | ||||
| 8.1 Nonsteroidal anti‐inflammatory drugs | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Prostatitis symptoms: subgroup analysis (co‐interventions) Show forest plot | 7 | 585 | Mean Difference (IV, Random, 95% CI) | ‐2.50 [‐3.74, ‐1.26] |
| Analysis 4.9  Comparison 4 Antiinflammatories versus placebo, Outcome 9 Prostatitis symptoms: subgroup analysis (co‐interventions). | ||||
| 9.1 Without co‐interventions | 1 | 64 | Mean Difference (IV, Random, 95% CI) | ‐3.62 [‐4.85, ‐2.39] |
| 9.2 With co‐interventions | 6 | 521 | Mean Difference (IV, Random, 95% CI) | ‐2.29 [‐3.67, ‐0.90] |
| 10 Adverse events Show forest plot | 7 | 540 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.81, 2.00] |
| Analysis 4.10  Comparison 4 Antiinflammatories versus placebo, Outcome 10 Adverse events. | ||||
| 10.1 Without co‐interventions | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 20.97] |
| 10.2 With co‐interventions | 6 | 476 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.73, 2.15] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 5 | 320 | Mean Difference (IV, Random, 95% CI) | ‐5.02 [‐6.81, ‐3.23] |
| Analysis 5.1  Comparison 5 Phytotherapy versus placebo, Outcome 1 Prostatitis symptoms. | ||||
| 1.1 Pollen extract vs. placebo | 1 | 137 | Mean Difference (IV, Random, 95% CI) | ‐2.5 [‐4.44, ‐0.56] |
| 1.2 Prolit Super Septo versus placebo | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐9.0 [‐16.81, ‐1.19] |
| 1.3 Calendula‐Curcuma versus placebo | 1 | 48 | Mean Difference (IV, Random, 95% CI) | ‐6.0 [‐7.28, ‐4.72] |
| 1.4 Quercetin (flavonoid) versus placebo | 1 | 28 | Mean Difference (IV, Random, 95% CI) | ‐5.80 [‐10.80, ‐0.80] |
| 1.5 Add‐on cranberry compared to standard care | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐5.40 [‐7.51, ‐3.29] |
| 2 Prostatitis symptoms: pain subscore Show forest plot | 5 | 431 | Mean Difference (IV, Random, 95% CI) | ‐1.42 [‐1.99, ‐0.85] |
| Analysis 5.2  Comparison 5 Phytotherapy versus placebo, Outcome 2 Prostatitis symptoms: pain subscore. | ||||
| 2.1 Pollen extract vs. placebo | 2 | 296 | Mean Difference (IV, Random, 95% CI) | ‐1.30 [‐2.05, ‐0.55] |
| 2.2 Prolit Super Septo versus placebo | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐4.0 [‐13.31, 5.31] |
| 2.3 Quercetin (flavonoid) versus placebo | 1 | 28 | Mean Difference (IV, Random, 95% CI) | ‐2.8 [‐5.41, ‐0.19] |
| 2.4 Add‐on cranberry compared to standard care | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐1.40 [‐2.34, ‐0.46] |
| 3 Prostatitis symptoms: urinary symptoms Show forest plot | 5 | 431 | Mean Difference (IV, Random, 95% CI) | ‐0.99 [‐1.75, ‐0.23] |
| Analysis 5.3  Comparison 5 Phytotherapy versus placebo, Outcome 3 Prostatitis symptoms: urinary symptoms. | ||||
| 3.1 Pollen Extract vs. Placebo | 2 | 296 | Mean Difference (IV, Random, 95% CI) | ‐0.49 [‐1.15, 0.16] |
| 3.2 Prolit Super Septo versus placebo | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐1.30 [‐6.15, 3.55] |
| 3.3 Quercetin (flavonoid) versus placebo | 1 | 28 | Mean Difference (IV, Random, 95% CI) | ‐1.5 [‐3.27, 0.27] |
| 3.4 Add‐on cranberry compared to standard care | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐1.80 [‐2.47, ‐1.13] |
| 4 Prostatitis symptoms: quality of life Show forest plot | 5 | 431 | Mean Difference (IV, Random, 95% CI) | ‐1.60 [‐2.30, ‐0.90] |
| Analysis 5.4  Comparison 5 Phytotherapy versus placebo, Outcome 4 Prostatitis symptoms: quality of life. | ||||
| 4.1 Pollen Extract vs. Placebo | 2 | 296 | Mean Difference (IV, Random, 95% CI) | ‐1.09 [‐1.66, ‐0.51] |
| 4.2 Prolit Super Septo versus placebo | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐3.70 [‐9.12, 1.72] |
| 4.3 Quercetin (flavonoid) versus placebo | 1 | 28 | Mean Difference (IV, Random, 95% CI) | ‐1.90 [‐3.97, 0.17] |
| 4.4 Add‐on cranberry compared to standard care | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐2.2 [‐2.75, ‐1.65] |
| 5 Prostatitis symptoms: responder rate Show forest plot | 3 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 1.78 [1.25, 2.52] |
| Analysis 5.5  Comparison 5 Phytotherapy versus placebo, Outcome 5 Prostatitis symptoms: responder rate. | ||||
| 5.1 Pollen extract vs. placebo | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [1.05, 2.05] |
| 5.2 Prolit Super Septo versus placebo | 1 | 57 | Risk Ratio (M‐H, Random, 95% CI) | 2.02 [1.23, 3.32] |
| 5.3 Quercetin (flavonoid) versus placebo | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.33 [1.14, 9.75] |
| 6 Adverse events Show forest plot | 7 | 540 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.54, 2.36] |
| Analysis 5.6  Comparison 5 Phytotherapy versus placebo, Outcome 6 Adverse events. | ||||
| 6.1 Pollen Extract vs. Placebo | 3 | 357 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.34, 3.79] |
| 6.2 Prolit Super Septo versus placebo | 1 | 57 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 6.3 Calendula‐Curcuma versus placebo | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.13, 70.16] |
| 6.4 Quercetin (flavonoid) versus placebo | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 1.73 [0.18, 16.99] |
| 6.5 Add‐on cranberry compared to standard care | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Sexual dysfunction Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 5.7  Comparison 5 Phytotherapy versus placebo, Outcome 7 Sexual dysfunction. | ||||
| 7.1 Calendula‐Curcuma versus placebo | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Urinary symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 5.8  Comparison 5 Phytotherapy versus placebo, Outcome 8 Urinary symptoms. | ||||
| 8.1 Pollen Extract vs. Placebo | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Prostatitis symptoms: subgroup analysis (co‐interventions) Show forest plot | 5 | 320 | Mean Difference (IV, Random, 95% CI) | ‐5.02 [‐6.81, ‐3.23] |
| Analysis 5.9  Comparison 5 Phytotherapy versus placebo, Outcome 9 Prostatitis symptoms: subgroup analysis (co‐interventions). | ||||
| 9.1 Without co‐interventions | 3 | 133 | Mean Difference (IV, Random, 95% CI) | ‐6.06 [‐7.28, ‐4.84] |
| 9.2 With co‐interventions | 2 | 187 | Mean Difference (IV, Random, 95% CI) | ‐3.92 [‐6.76, ‐1.08] |
| 10 Adverse events: subgroup analysis (co‐interventions) Show forest plot | 7 | 540 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.54, 2.36] |
| Analysis 5.10  Comparison 5 Phytotherapy versus placebo, Outcome 10 Adverse events: subgroup analysis (co‐interventions). | ||||
| 10.1 Without co‐interventions | 4 | 191 | Risk Ratio (M‐H, Random, 95% CI) | 2.09 [0.33, 13.30] |
| 10.2 With co‐interventions | 3 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.34, 3.79] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 6.1  Comparison 6 Botulinum toxin A versus placebo, Outcome 1 Prostatitis symptoms. | ||||
| 1.1 Intraprostatic | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐25.80 [‐30.15, ‐21.45] |
| 1.2 Pelvic floor muscles | 1 | 29 | Mean Difference (IV, Random, 95% CI) | ‐2.6 [‐5.59, 0.39] |
| 2 Prostatitis symptoms: pain subscore Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| Analysis 6.2  Comparison 6 Botulinum toxin A versus placebo, Outcome 2 Prostatitis symptoms: pain subscore. | ||||
| 2.1 Intraprostatic | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐14.63 [‐16.76, ‐12.50] |
| 2.2 Pelvic floor muscles | 1 | 29 | Mean Difference (IV, Random, 95% CI) | ‐1.70 [‐3.23, ‐0.17] |
| 3 Prostatitis symptoms: urinary subscore Show forest plot | 2 | 89 | Mean Difference (IV, Random, 95% CI) | ‐2.82 [‐7.40, 1.76] |
| Analysis 6.3  Comparison 6 Botulinum toxin A versus placebo, Outcome 3 Prostatitis symptoms: urinary subscore. | ||||
| 3.1 Intraprostatic | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐5.17 [‐6.72, ‐3.62] |
| 3.2 Pelvic floor muscles | 1 | 29 | Mean Difference (IV, Random, 95% CI) | ‐0.5 [‐1.85, 0.85] |
| 4 Prostatitis symptoms: quality of life Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 6.4  Comparison 6 Botulinum toxin A versus placebo, Outcome 4 Prostatitis symptoms: quality of life. | ||||
| 4.1 Intraprostatic | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Pelvic floor muscles | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse events Show forest plot | 2 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.25, 99.95] |
| Analysis 6.5  Comparison 6 Botulinum toxin A versus placebo, Outcome 5 Adverse events. | ||||
| 5.1 Intraprostatic | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.25, 99.95] |
| 5.2 Pelvic floor muscle | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 6 Urinary symptoms Show forest plot | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐9.67 [‐13.97, ‐5.37] |
| Analysis 6.6  Comparison 6 Botulinum toxin A versus placebo, Outcome 6 Urinary symptoms. | ||||
| 6.1 Intraprostatic | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐9.67 [‐13.97, ‐5.37] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 7.1  Comparison 7 Allopurinol versus placebo, Outcome 1 Prostatitis symptoms. | ||||
| 2 Prostatitis symptoms: pain subscore Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 7.2  Comparison 7 Allopurinol versus placebo, Outcome 2 Prostatitis symptoms: pain subscore. | ||||
| 3 Prostatitis symptoms: urinary subscore Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 7.3  Comparison 7 Allopurinol versus placebo, Outcome 3 Prostatitis symptoms: urinary subscore. | ||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 7 | 835 | Mean Difference (IV, Random, 95% CI) | ‐3.13 [‐4.99, ‐1.28] |
| Analysis 8.1  Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 1 Prostatitis symptoms. | ||||
| 1.1 Prostant versus placebo | 2 | 164 | Mean Difference (IV, Random, 95% CI) | ‐3.93 [‐7.76, ‐0.10] |
| 1.2 Qianlieping (add‐on) versus medical treatment alone | 1 | 154 | Mean Difference (IV, Random, 95% CI) | ‐3.40 [‐5.03, ‐1.77] |
| 1.3 Qianlieantong (add‐on) versus medical treatment alone | 1 | 115 | Mean Difference (IV, Random, 95% CI) | ‐6.67 [‐8.14, ‐5.20] |
| 1.4 Yuleshu (add‐on) versus medical care alone | 1 | 88 | Mean Difference (IV, Random, 95% CI) | ‐1.41 [‐2.57, ‐0.25] |
| 1.5 Aike Mixture versus placebo | 1 | 74 | Mean Difference (IV, Random, 95% CI) | ‐3.59 [‐8.24, 1.06] |
| 1.6 Bazhengsan versus placebo | 2 | 162 | Mean Difference (IV, Random, 95% CI) | ‐1.53 [‐9.28, 6.23] |
| 1.7 Prostatitis Decoction versus placebo | 1 | 78 | Mean Difference (IV, Random, 95% CI) | 1.07 [‐3.78, 5.92] |
| 2 Prostatitis symptoms: pain subscore Show forest plot | 5 | 585 | Mean Difference (IV, Random, 95% CI) | ‐1.49 [‐2.44, ‐0.53] |
| Analysis 8.2  Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 2 Prostatitis symptoms: pain subscore. | ||||
| 2.1 Prostant versus placebo | 2 | 164 | Mean Difference (IV, Random, 95% CI) | ‐1.42 [‐2.51, ‐0.34] |
| 2.2 Qianlieantong versus medical treatment alone | 1 | 115 | Mean Difference (IV, Random, 95% CI) | ‐2.54 [‐3.24, ‐1.84] |
| 2.3 Yuleshu versus medical care | 1 | 88 | Mean Difference (IV, Random, 95% CI) | ‐2.52 [‐3.64, ‐1.40] |
| 2.4 Aike Mixture versus placebo | 1 | 74 | Mean Difference (IV, Random, 95% CI) | ‐2.21 [‐4.60, 0.18] |
| 2.5 Bazhengsan versus placebo | 1 | 66 | Mean Difference (IV, Random, 95% CI) | 1.35 [‐1.22, 3.92] |
| 2.6 Prostatitis Decoction versus placebo | 1 | 78 | Mean Difference (IV, Random, 95% CI) | 1.02 [‐1.52, 3.56] |
| 3 Prostatitis symptoms: urinary subscore Show forest plot | 4 | 497 | Mean Difference (IV, Random, 95% CI) | ‐0.78 [‐1.50, ‐0.06] |
| Analysis 8.3  Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 3 Prostatitis symptoms: urinary subscore. | ||||
| 3.1 Prostant versus placebo | 2 | 164 | Mean Difference (IV, Random, 95% CI) | ‐1.62 [‐3.37, 0.13] |
| 3.2 Qianlieantong versus medical treatment alone | 1 | 115 | Mean Difference (IV, Random, 95% CI) | ‐0.39 [‐1.02, 0.24] |
| 3.3 Aike Mixture versus placebo | 1 | 74 | Mean Difference (IV, Random, 95% CI) | ‐0.84 [‐2.31, 0.63] |
| 3.4 Bazhengsan versus placebo | 1 | 66 | Mean Difference (IV, Random, 95% CI) | 0.25 [‐1.35, 1.85] |
| 3.5 Prostatitis Decoction versus placebo | 1 | 78 | Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐1.73, 1.21] |
| 4 Prostatitis symptoms: quality of life Show forest plot | 2 | 306 | Mean Difference (IV, Random, 95% CI) | ‐0.97 [‐2.14, 0.20] |
| Analysis 8.4  Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 4 Prostatitis symptoms: quality of life. | ||||
| 4.1 Prostant versus placebo | 1 | 88 | Mean Difference (IV, Random, 95% CI) | ‐1.91 [‐1.00, ‐0.82] |
| 4.2 Aike Mixture versus placebo | 1 | 74 | Mean Difference (IV, Random, 95% CI) | ‐1.10 [‐3.36, 1.16] |
| 4.3 Bazhengsan versus placebo | 1 | 66 | Mean Difference (IV, Random, 95% CI) | 0.66 [‐1.65, 2.97] |
| 4.4 Prostatitis Decoction versus placebo | 1 | 78 | Mean Difference (IV, Random, 95% CI) | ‐0.34 [‐2.59, 1.91] |
| 5 Adverse events Show forest plot | 4 | 584 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.22, 8.02] |
| Analysis 8.5  Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 5 Adverse events. | ||||
| 5.1 Prostant versus placebo | 2 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.03, 19.50] |
| 5.2 Qianlieping versus medical treatment alone | 1 | 154 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.3 Aike Mixture versus placebo | 1 | 74 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.4 Bazhengsan versus placebo | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 4.33 [0.26, 72.96] |
| 5.5 Prostatitis Decoction versus placebo | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 2.43 [0.14, 42.93] |
| 6 Sexual dysfunction Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 8.6  Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 6 Sexual dysfunction. | ||||
| 6.1 Yuleshu versus medical care | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Anxiety and depression: anxiety Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 8.7  Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 7 Anxiety and depression: anxiety. | ||||
| 7.1 Yuleshu versus medical care | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Anxiety and depression: depression Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Analysis 8.8  Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 8 Anxiety and depression: depression. | ||||
| 8.1 Yuleshu versus medical care | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |

Study flow diagram.
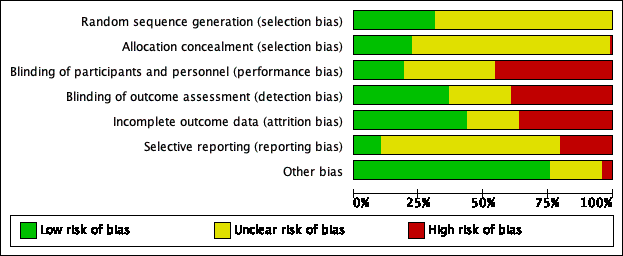
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
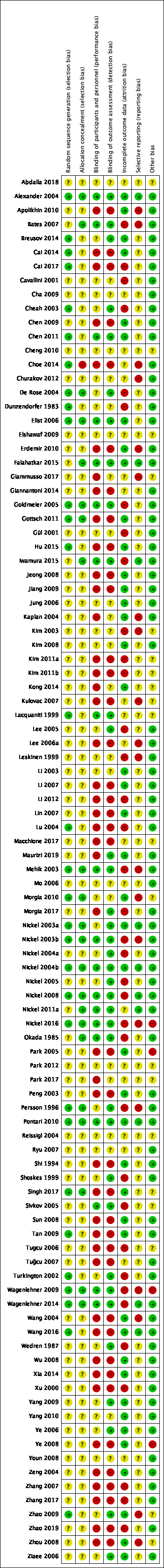
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
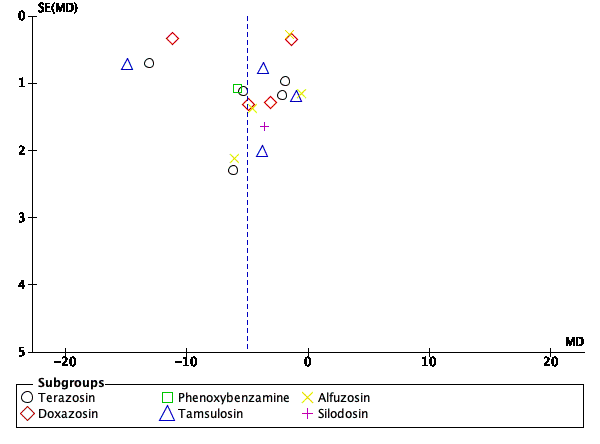
Funnel plot of comparison: 1 Alpha‐blockers versus placebo, outcome: 1.1 Prostatitis symptoms: short term.

Funnel plot of comparison: 1 Alpha‐blockers versus placebo, outcome: 1.8 Adverse events.
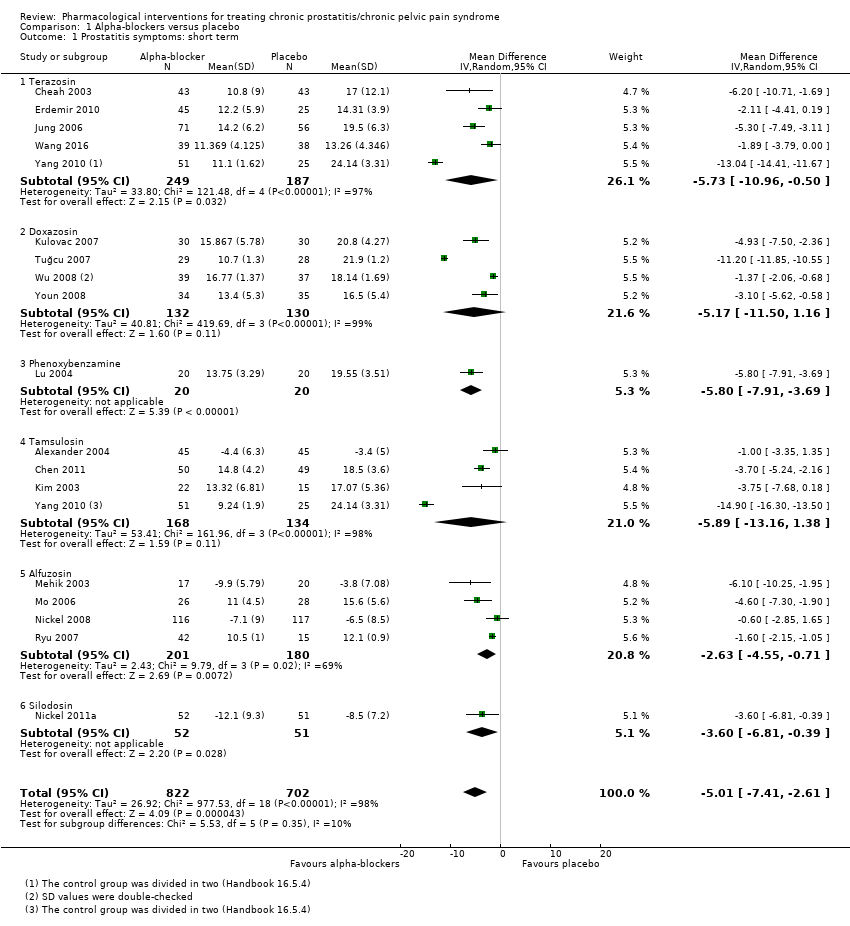
Comparison 1 Alpha‐blockers versus placebo, Outcome 1 Prostatitis symptoms: short term.

Comparison 1 Alpha‐blockers versus placebo, Outcome 2 Prostatitis symptoms: pain.

Comparison 1 Alpha‐blockers versus placebo, Outcome 3 Prostatitis symptoms: urinary.

Comparison 1 Alpha‐blockers versus placebo, Outcome 4 Prostatitis symptoms: quality of life.
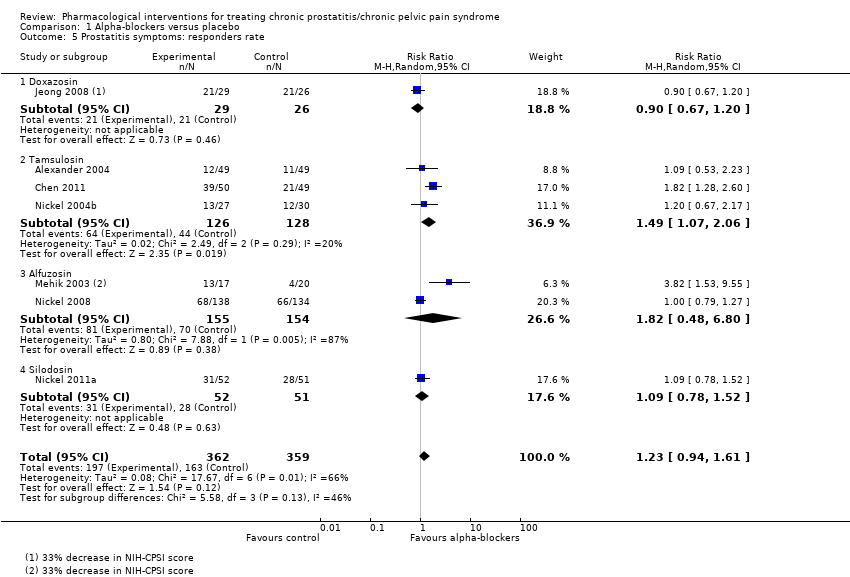
Comparison 1 Alpha‐blockers versus placebo, Outcome 5 Prostatitis symptoms: responders rate.
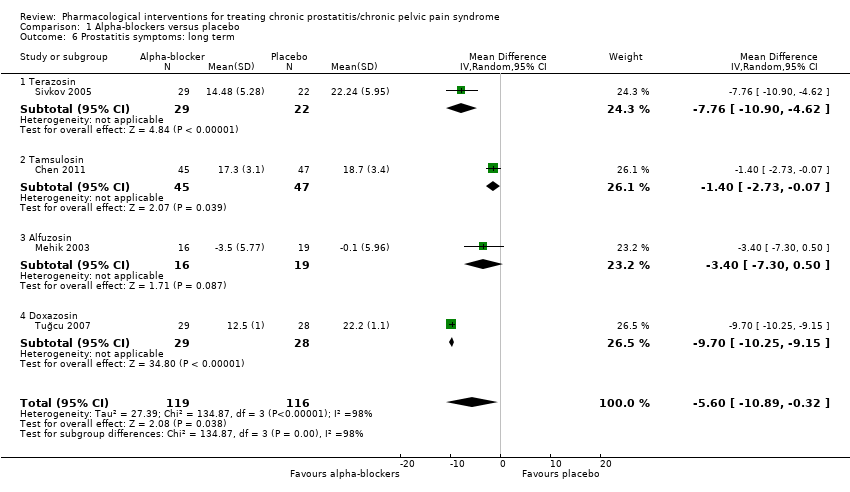
Comparison 1 Alpha‐blockers versus placebo, Outcome 6 Prostatitis symptoms: long term.
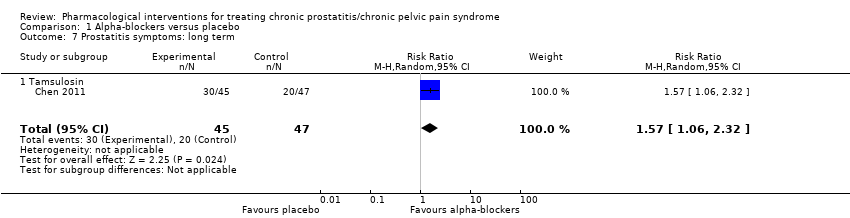
Comparison 1 Alpha‐blockers versus placebo, Outcome 7 Prostatitis symptoms: long term.

Comparison 1 Alpha‐blockers versus placebo, Outcome 8 Adverse events.

Comparison 1 Alpha‐blockers versus placebo, Outcome 9 Sexual dysfunction.
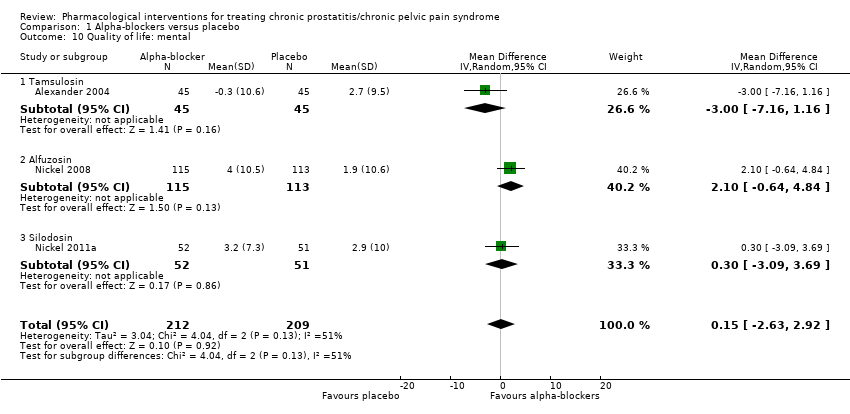
Comparison 1 Alpha‐blockers versus placebo, Outcome 10 Quality of life: mental.

Comparison 1 Alpha‐blockers versus placebo, Outcome 11 Quality of life: physical.
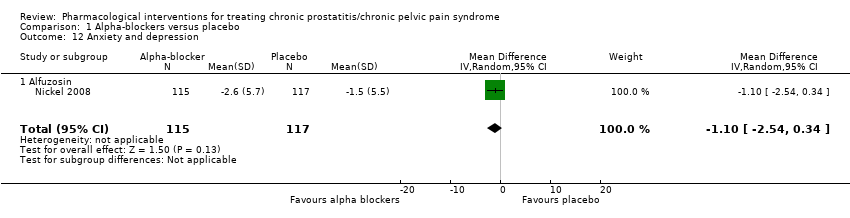
Comparison 1 Alpha‐blockers versus placebo, Outcome 12 Anxiety and depression.
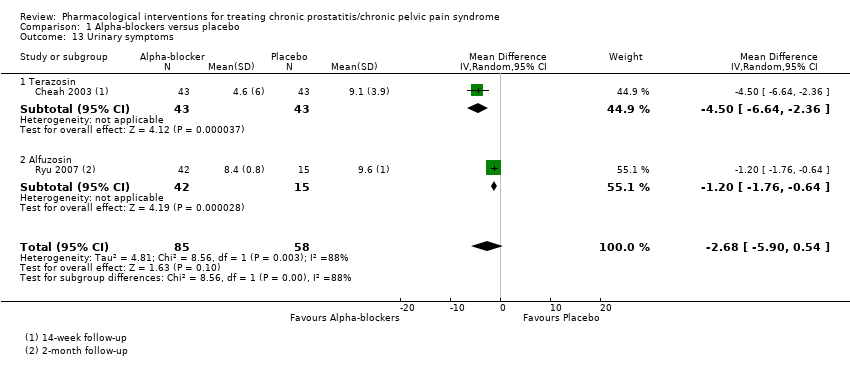
Comparison 1 Alpha‐blockers versus placebo, Outcome 13 Urinary symptoms.

Comparison 1 Alpha‐blockers versus placebo, Outcome 14 Prostatitis symptoms: responders rate (sensitivity analysis according to risk of bias).
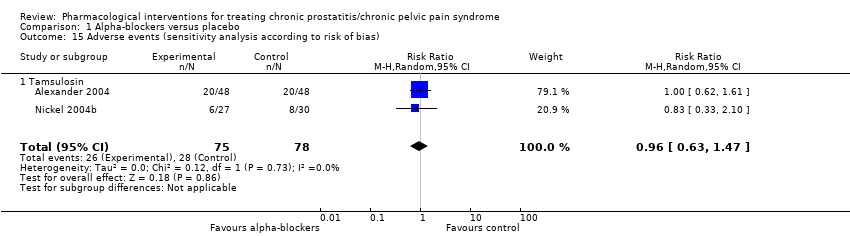
Comparison 1 Alpha‐blockers versus placebo, Outcome 15 Adverse events (sensitivity analysis according to risk of bias).

Comparison 1 Alpha‐blockers versus placebo, Outcome 16 Prostatitis symptoms: short term (subgroup analysis by co‐interventions).
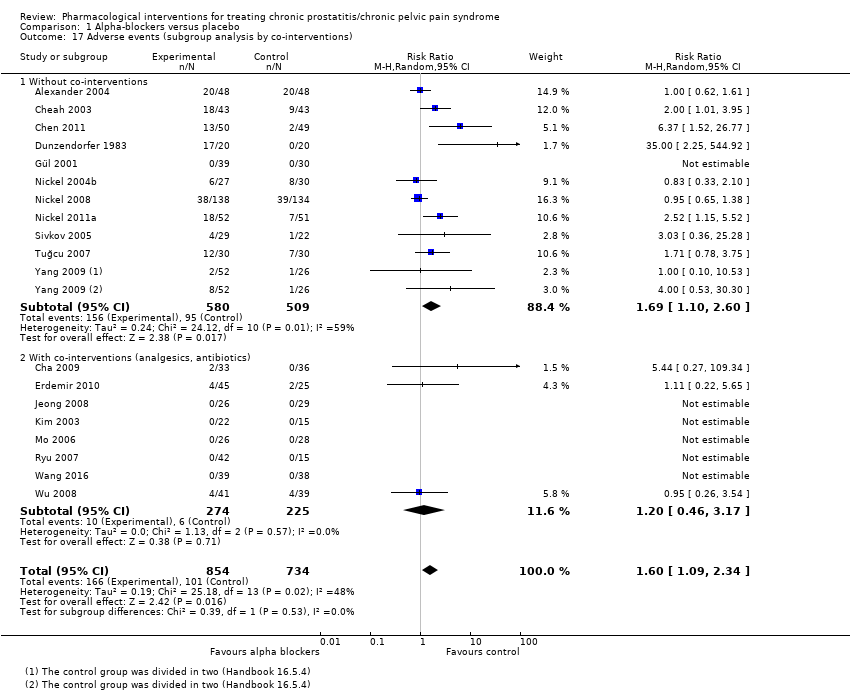
Comparison 1 Alpha‐blockers versus placebo, Outcome 17 Adverse events (subgroup analysis by co‐interventions).
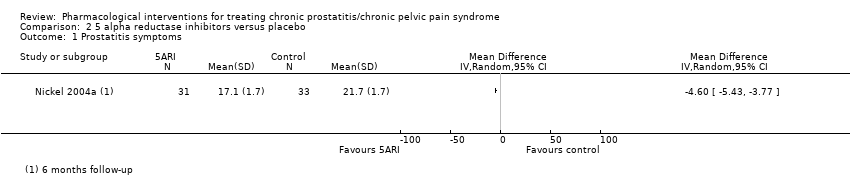
Comparison 2 5 alpha reductase inhibitors versus placebo, Outcome 1 Prostatitis symptoms.
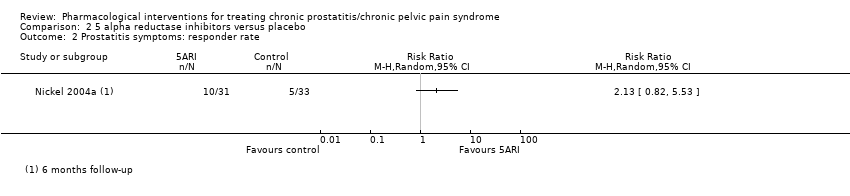
Comparison 2 5 alpha reductase inhibitors versus placebo, Outcome 2 Prostatitis symptoms: responder rate.

Comparison 2 5 alpha reductase inhibitors versus placebo, Outcome 3 Adverse events.

Comparison 3 Antibiotic therapy versus placebo, Outcome 1 Prostatitis symptoms.

Comparison 3 Antibiotic therapy versus placebo, Outcome 2 Prostatitis symptoms: pain.
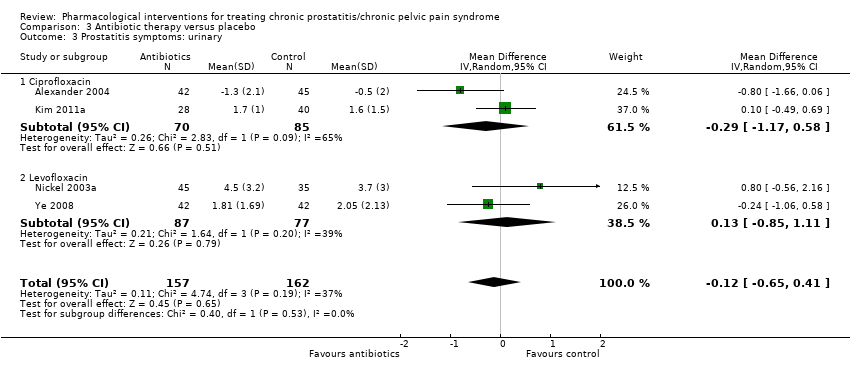
Comparison 3 Antibiotic therapy versus placebo, Outcome 3 Prostatitis symptoms: urinary.

Comparison 3 Antibiotic therapy versus placebo, Outcome 4 Prostatitis symptoms: quality of life.
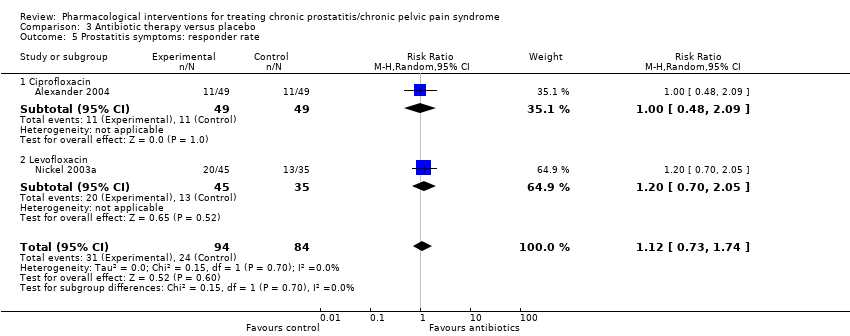
Comparison 3 Antibiotic therapy versus placebo, Outcome 5 Prostatitis symptoms: responder rate.

Comparison 3 Antibiotic therapy versus placebo, Outcome 6 Adverse events.
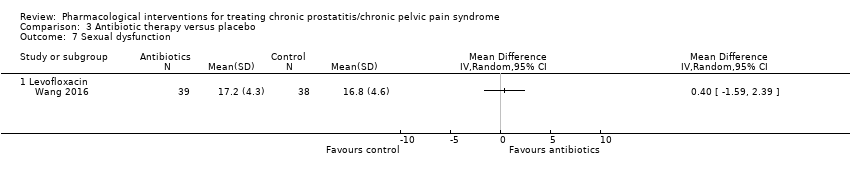
Comparison 3 Antibiotic therapy versus placebo, Outcome 7 Sexual dysfunction.
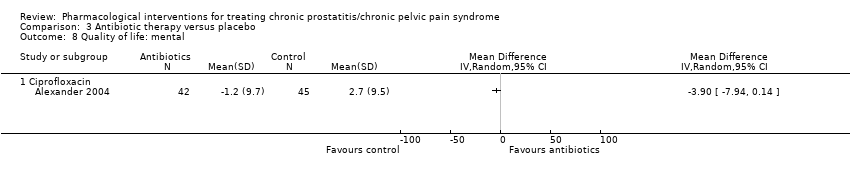
Comparison 3 Antibiotic therapy versus placebo, Outcome 8 Quality of life: mental.
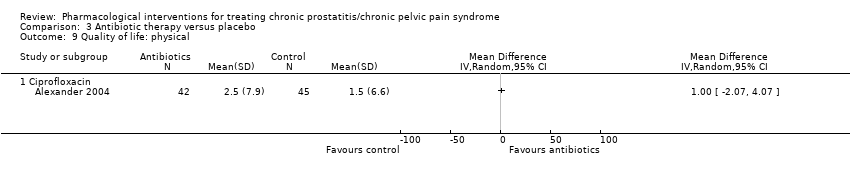
Comparison 3 Antibiotic therapy versus placebo, Outcome 9 Quality of life: physical.
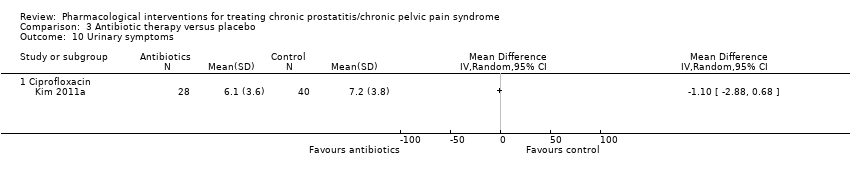
Comparison 3 Antibiotic therapy versus placebo, Outcome 10 Urinary symptoms.

Comparison 3 Antibiotic therapy versus placebo, Outcome 11 Prostatitis symptoms: subgroup analysis (age).

Comparison 3 Antibiotic therapy versus placebo, Outcome 12 Adverse events: subgroup analysis (age).
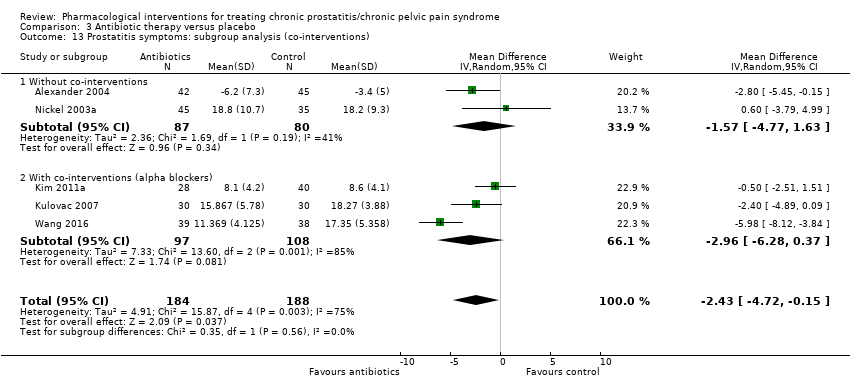
Comparison 3 Antibiotic therapy versus placebo, Outcome 13 Prostatitis symptoms: subgroup analysis (co‐interventions).
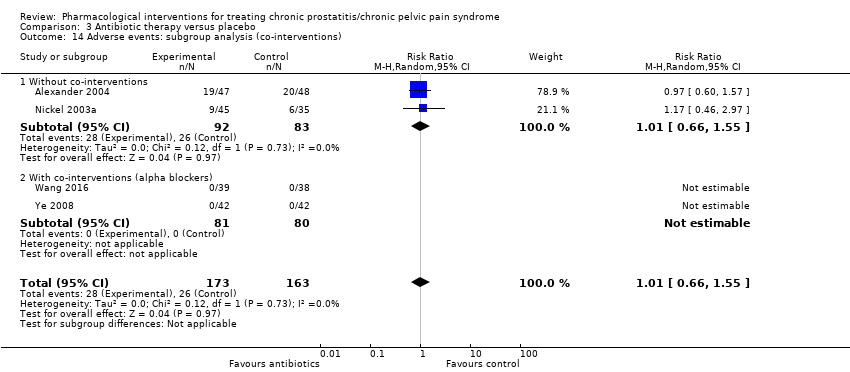
Comparison 3 Antibiotic therapy versus placebo, Outcome 14 Adverse events: subgroup analysis (co‐interventions).

Comparison 4 Antiinflammatories versus placebo, Outcome 1 Prostatitis symptoms.

Comparison 4 Antiinflammatories versus placebo, Outcome 2 Prostatitis symptoms: pain.
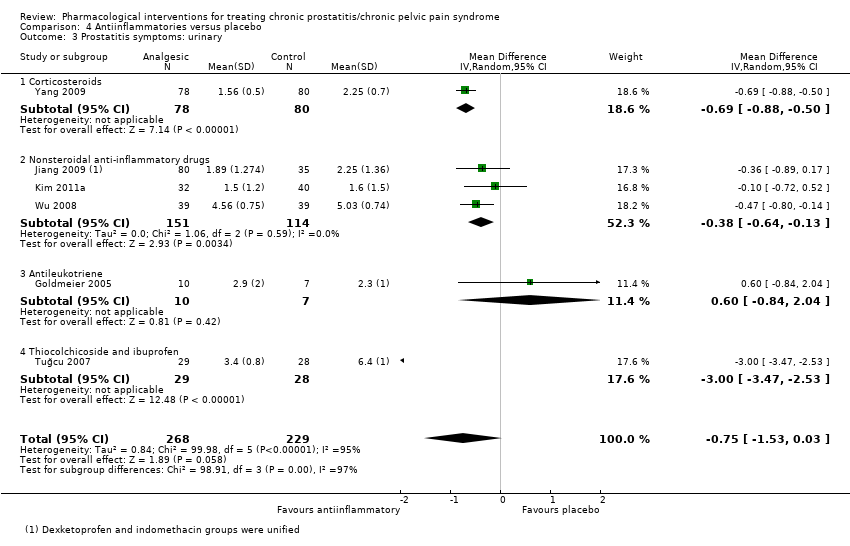
Comparison 4 Antiinflammatories versus placebo, Outcome 3 Prostatitis symptoms: urinary.

Comparison 4 Antiinflammatories versus placebo, Outcome 4 Prostatitis symptoms: quality of life.

Comparison 4 Antiinflammatories versus placebo, Outcome 5 Prostatitis symptoms: responder rate.
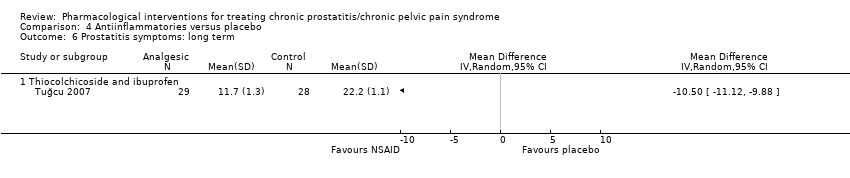
Comparison 4 Antiinflammatories versus placebo, Outcome 6 Prostatitis symptoms: long term.

Comparison 4 Antiinflammatories versus placebo, Outcome 7 Adverse events.
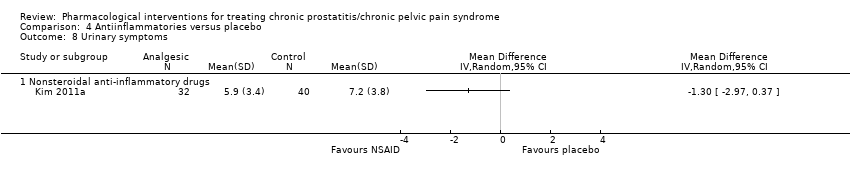
Comparison 4 Antiinflammatories versus placebo, Outcome 8 Urinary symptoms.
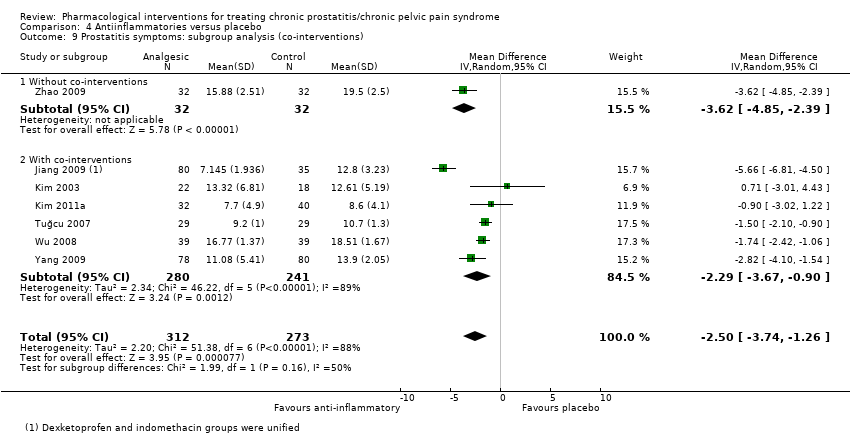
Comparison 4 Antiinflammatories versus placebo, Outcome 9 Prostatitis symptoms: subgroup analysis (co‐interventions).
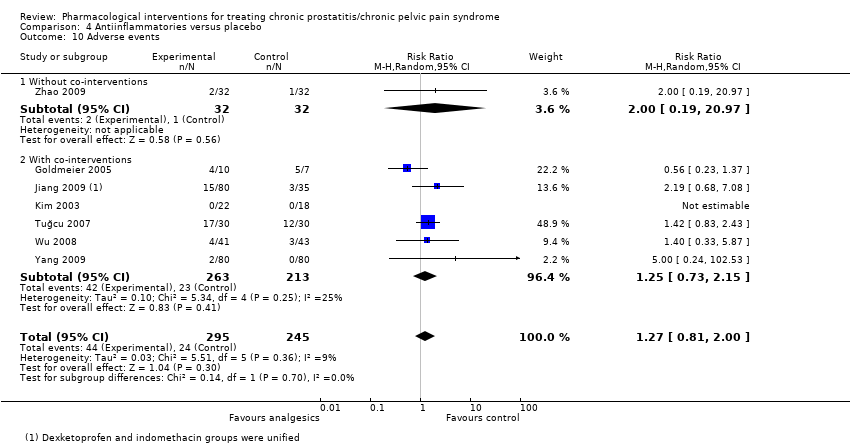
Comparison 4 Antiinflammatories versus placebo, Outcome 10 Adverse events.
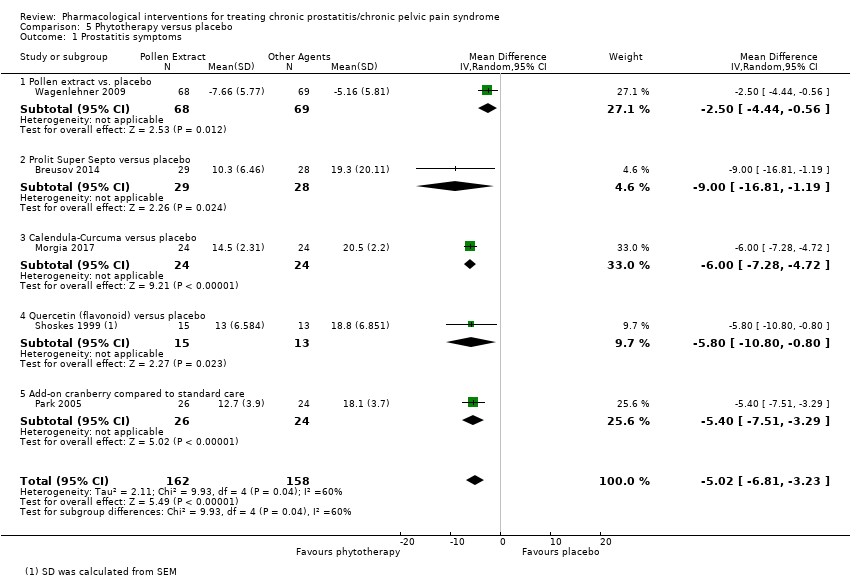
Comparison 5 Phytotherapy versus placebo, Outcome 1 Prostatitis symptoms.
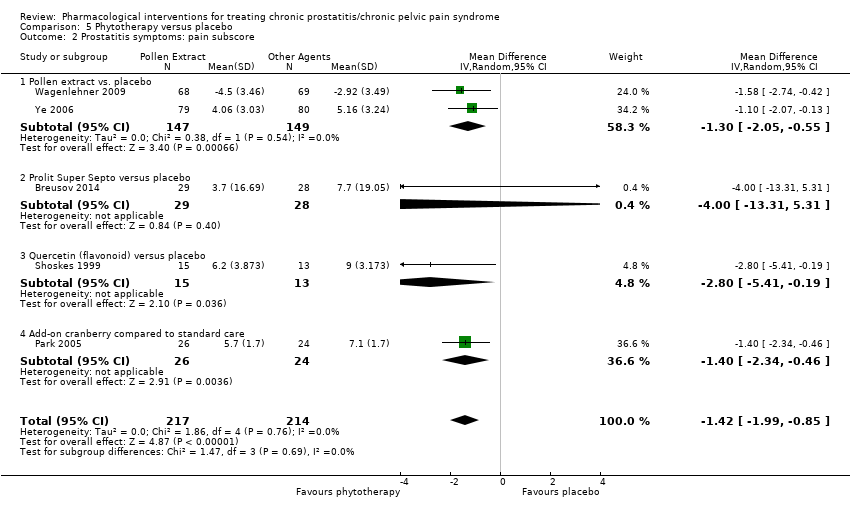
Comparison 5 Phytotherapy versus placebo, Outcome 2 Prostatitis symptoms: pain subscore.
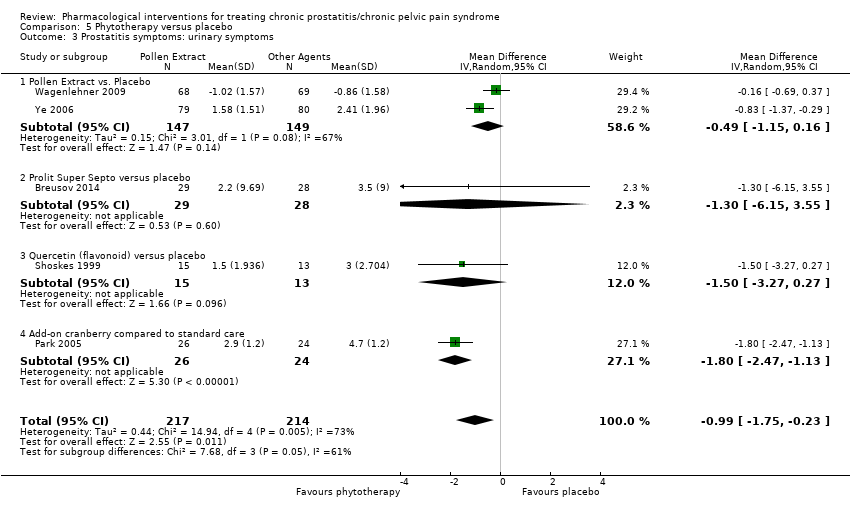
Comparison 5 Phytotherapy versus placebo, Outcome 3 Prostatitis symptoms: urinary symptoms.

Comparison 5 Phytotherapy versus placebo, Outcome 4 Prostatitis symptoms: quality of life.
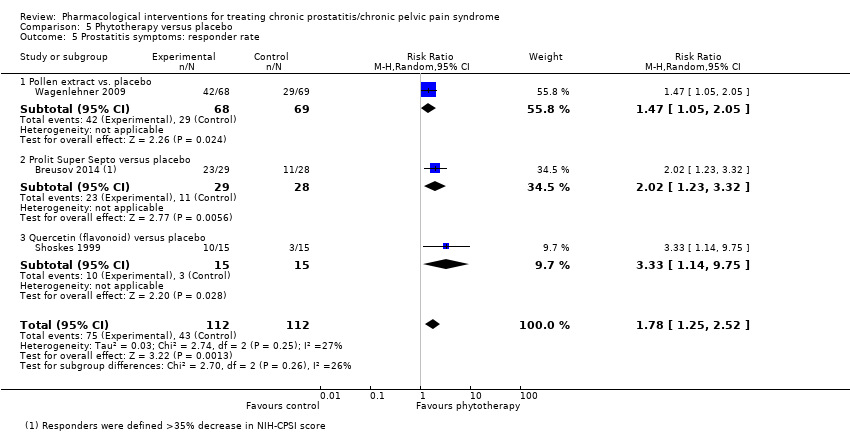
Comparison 5 Phytotherapy versus placebo, Outcome 5 Prostatitis symptoms: responder rate.
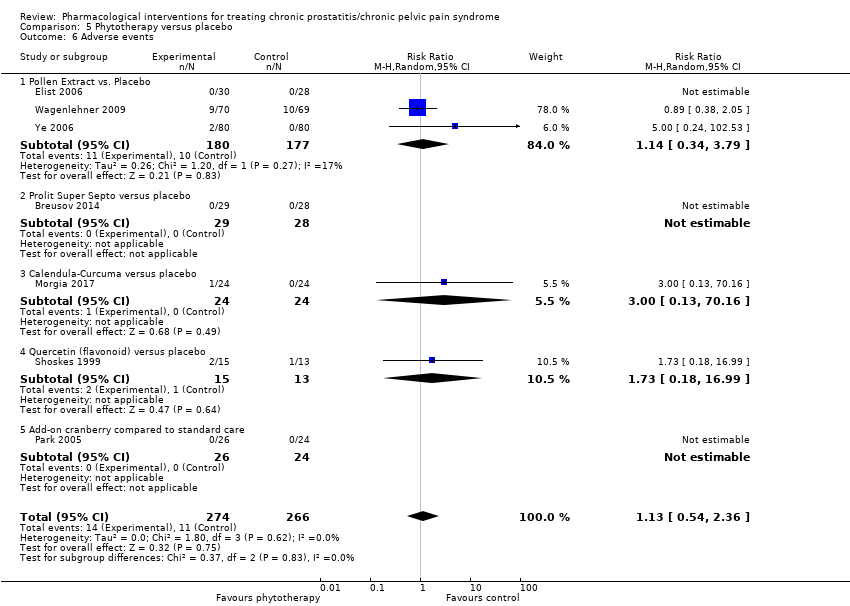
Comparison 5 Phytotherapy versus placebo, Outcome 6 Adverse events.
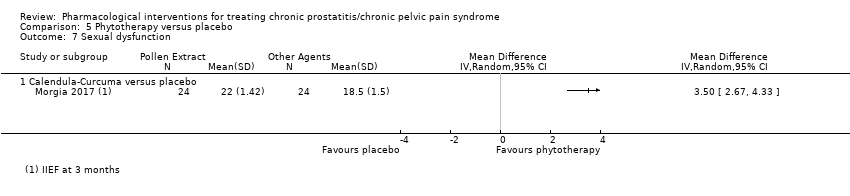
Comparison 5 Phytotherapy versus placebo, Outcome 7 Sexual dysfunction.
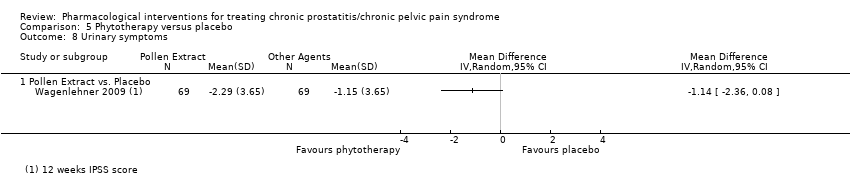
Comparison 5 Phytotherapy versus placebo, Outcome 8 Urinary symptoms.
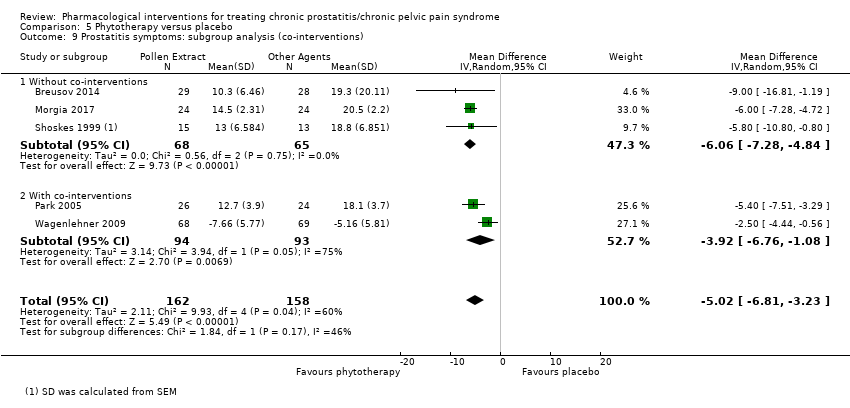
Comparison 5 Phytotherapy versus placebo, Outcome 9 Prostatitis symptoms: subgroup analysis (co‐interventions).

Comparison 5 Phytotherapy versus placebo, Outcome 10 Adverse events: subgroup analysis (co‐interventions).

Comparison 6 Botulinum toxin A versus placebo, Outcome 1 Prostatitis symptoms.
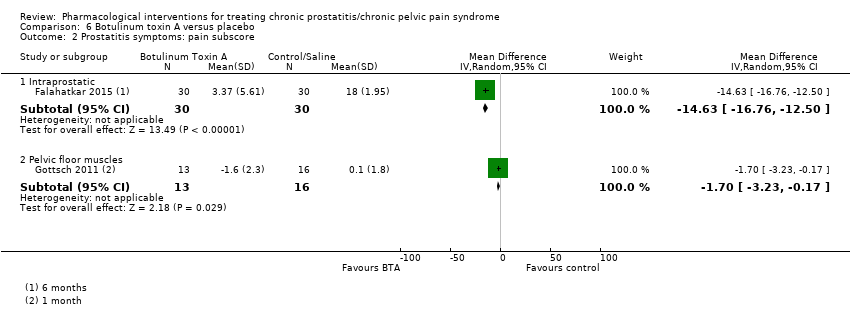
Comparison 6 Botulinum toxin A versus placebo, Outcome 2 Prostatitis symptoms: pain subscore.
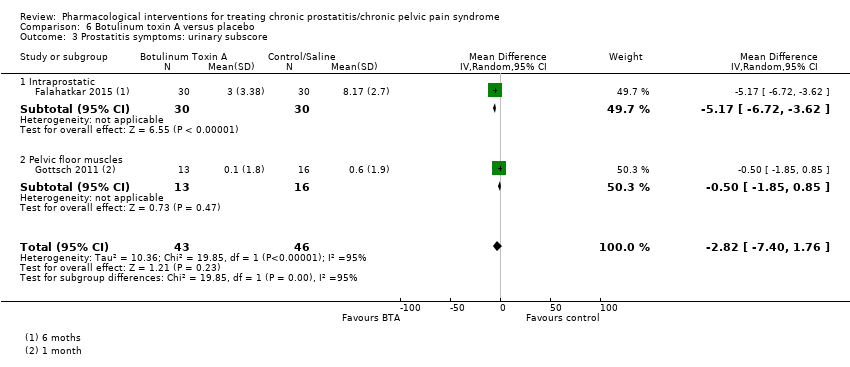
Comparison 6 Botulinum toxin A versus placebo, Outcome 3 Prostatitis symptoms: urinary subscore.

Comparison 6 Botulinum toxin A versus placebo, Outcome 4 Prostatitis symptoms: quality of life.

Comparison 6 Botulinum toxin A versus placebo, Outcome 5 Adverse events.
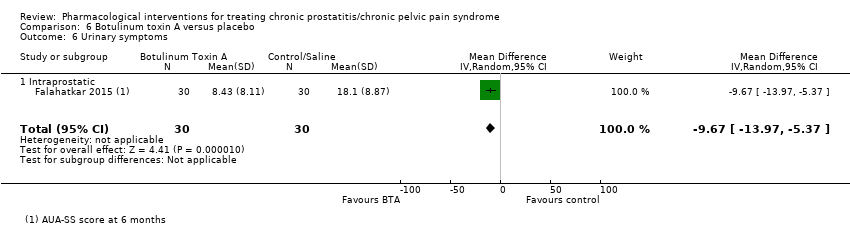
Comparison 6 Botulinum toxin A versus placebo, Outcome 6 Urinary symptoms.
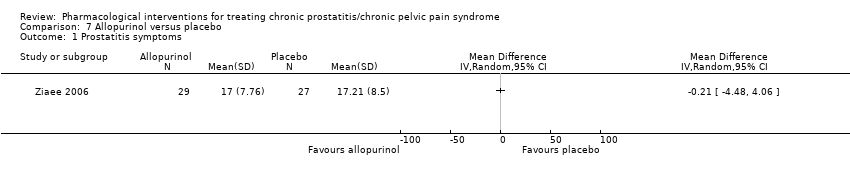
Comparison 7 Allopurinol versus placebo, Outcome 1 Prostatitis symptoms.
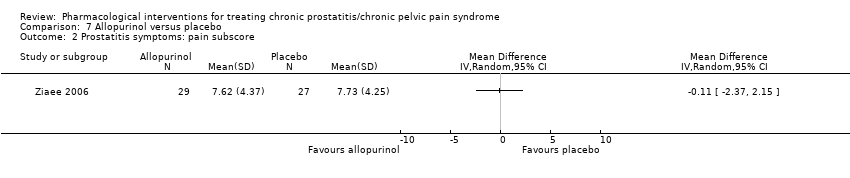
Comparison 7 Allopurinol versus placebo, Outcome 2 Prostatitis symptoms: pain subscore.

Comparison 7 Allopurinol versus placebo, Outcome 3 Prostatitis symptoms: urinary subscore.
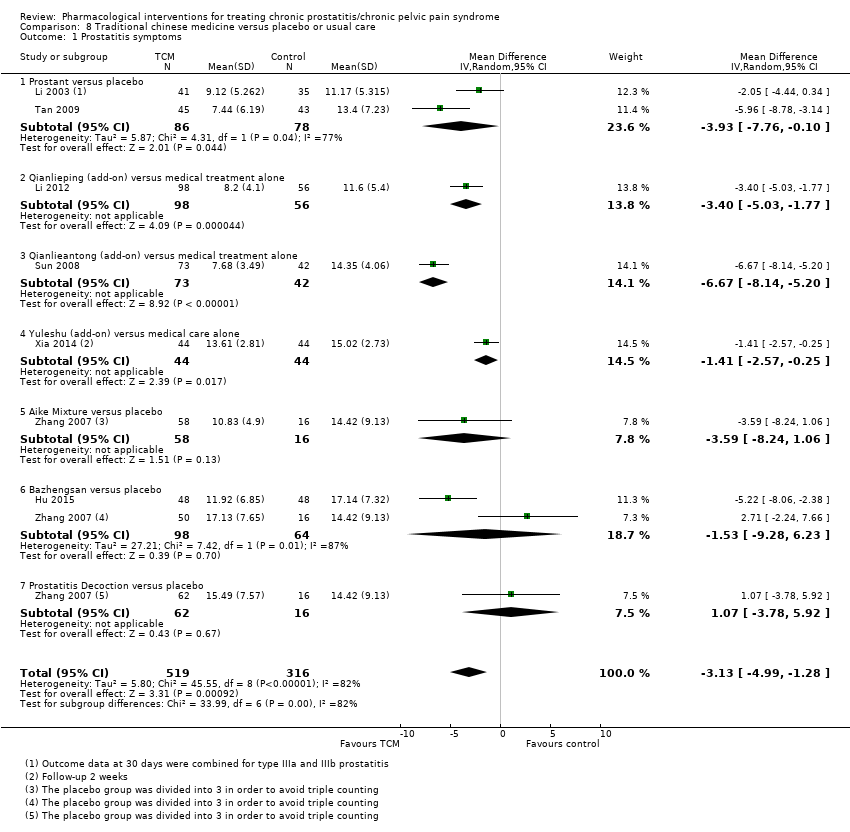
Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 1 Prostatitis symptoms.

Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 2 Prostatitis symptoms: pain subscore.

Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 3 Prostatitis symptoms: urinary subscore.
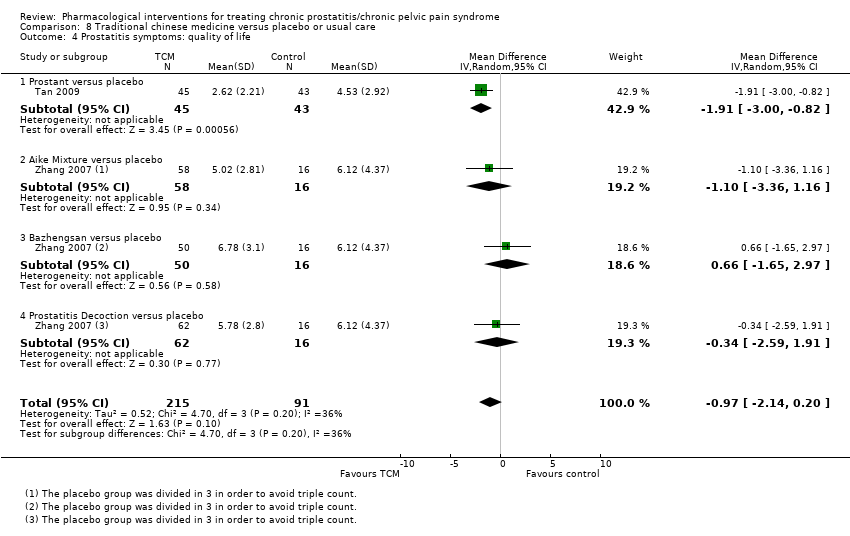
Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 4 Prostatitis symptoms: quality of life.
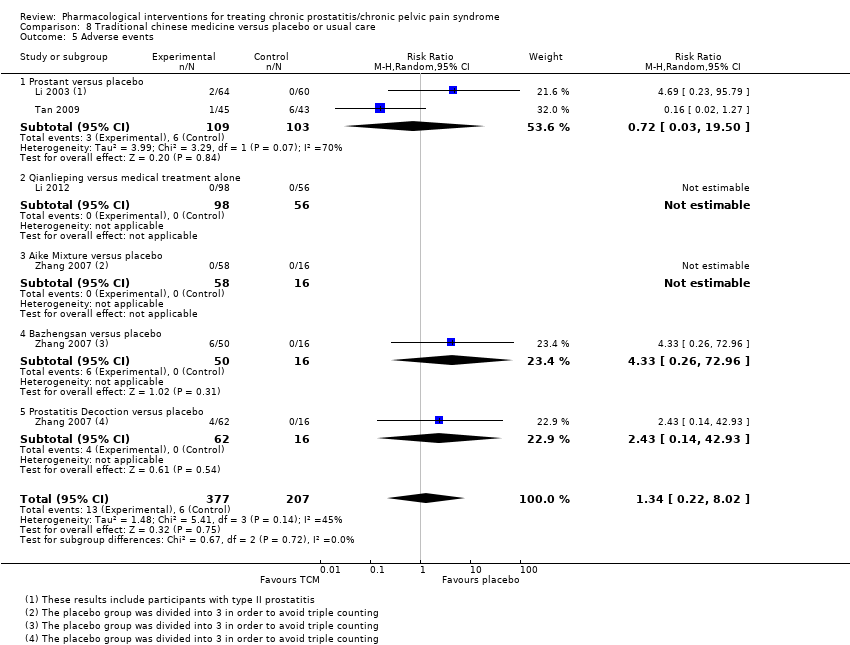
Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 5 Adverse events.
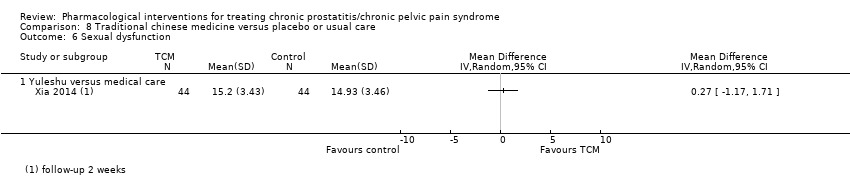
Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 6 Sexual dysfunction.

Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 7 Anxiety and depression: anxiety.
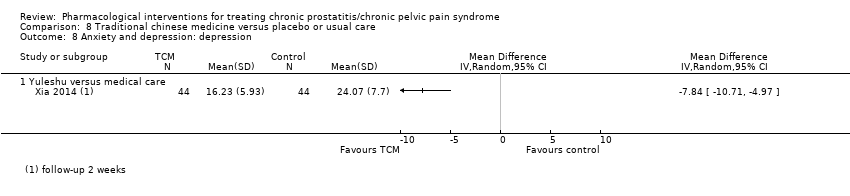
Comparison 8 Traditional chinese medicine versus placebo or usual care, Outcome 8 Anxiety and depression: depression.
| Alpha blockers compared to placebo for chronic prostatitis/chronic pelvic pain syndrome | |||||
| Patient or population: men with chronic prostatitis/chronic pelvic pain syndrome Some comparisons included alpha blockers as add‐on therapy to medical therapy (e.g. antibiotics) versus medical therapy alone. | |||||
| Outcomes | № of participants | Quality of the evidence | Relative effect | Anticipated absolute effects* (95% CI) | |
| Risk with placebo | Risk difference with alpha‐blockers | ||||
| Prostatitis symptoms A decrease of 25% or 6 points is considered an important improvement | 1524 | ⊕⊝⊝⊝ | ‐ | The mean prostatitis symptoms ranged from 12.1 to 24.14 | MD 5.01 lower |
| Prostatitis symptoms | 253 (4 RCTs) | ⊕⊝⊝⊝ | ‐ | The mean prostatitis symptoms ranged from 18.7 to 22.24 | MD 5.6 lower |
| Prostatitis symptoms: 'responders' | 721 | ⊕⊝⊝⊝ | RR 1.23 | Study population | |
| 477 per 1000 | 110 more per 1000 | ||||
| Adverse events Any adverse event | 1588 | ⊕⊕⊝⊝ | RR 1.60 | Study population | |
| 94 per 1000 | 56 more per 1000 | ||||
| Sexual dysfunction | 452 | ⊕⊕⊕⊝ | ‐ | The mean sexual dysfunction ranged from 16.1 to 18.4 | MD 0.26 higher |
| Quality of life | 421 | ⊕⊕⊕⊝ | ‐ | The mean quality of life ranged from 41 to 46 | MD 0.15 higher |
| Anxiety and depression | 232 | ⊕⊕⊝⊝ | ‐ | The mean anxiety and depression was 12.8 | MD 1.1 lower |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | |||||
| GRADE Working Group grades of evidence | |||||
| aDowngraded one level due to study limitations: unclear or high risk of bias in most domains in most studies. | |||||
| 5‐alpha reductase inhibitors compared to placebo for chronic prostatitis/chronic pelvic pain syndrome | |||||
| Patient or population: men with chronic prostatitis/chronic pelvic pain syndrome | |||||
| Outcomes | № of participants | Quality of the evidence | Relative effect | Anticipated absolute effects* (95% CI) | |
| Risk with placebo | Risk difference with 5 alpha reductase inhibitors | ||||
| Prostatitis symptoms A decrease of 25% or 6 points is considered an important improvement | 64 | ⊕⊕⊕⊝ | ‐ | The mean prostatitis symptoms was 21.7 | MD 4.6 lower |
| Prostatitis symptoms: 'responders' | 64 | ⊕⊕⊝⊝ | RR 2.13 | Study population | |
| 152 per 1000 | 171 more per 1000 | ||||
| Adverse events | 105 | ⊕⊕⊝⊝ | RR 0.87 | Study population | |
| 163 per 1000 | 21 fewer per 1000 | ||||
| Sexual dysfunction ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| Quality of life ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| Anxiety and depression ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | |||||
| GRADE Working Group grades of evidence | |||||
| aDowngraded one level due to high risk of bias: unclear or high risk of bias in most domains in the main study of this comparison. | |||||
| Antibiotic therapy compared to placebo for chronic prostatitis/chronic pelvic pain syndrome | |||||
| Patient or population: men with chronic prostatitis/chronic pelvic pain syndrome Some comparisons included antibiotics as add‐on therapy to medical therapy (e.g. alpha blockers) versus medical therapy alone | |||||
| Outcomes | № of participants | Quality of the evidence | Relative effect | Anticipated absolute effects* (95% CI) | |
| Risk with placebo | Risk difference with antibiotic therapy | ||||
| Prostatitis symptoms A decrease of 25% or 6 points is considered an important improvement | 372 | ⊕⊕⊝⊝ | ‐ | The mean prostatitis symptoms ranged from 8.6 to 18.2 | MD 2.43 lower |
| Prostatitis symptoms: 'responders' | 178 | ⊕⊕⊝⊝ | RR 1.12 | Study population | |
| 286 per 1000 | 34 more per 1000 | ||||
| Adverse events | 336 | ⊕⊕⊕⊝ | RR 1.01 | Study population | |
| 213 per 1000 | 2 more per 1000 | ||||
| Sexual dysfunction | 77 | ⊕⊕⊕⊝ | ‐ | The mean sexual dysfunction was 16.8 | MD 0.4 higher |
| Quality of life | 87 | ⊕⊕⊕⊝ | ‐ | The mean quality of life was 44.3 | MD 3.9 lower |
| Anxiety and depression ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | |||||
| GRADE Working Group grades of evidence | |||||
| aDowngraded one level due to inconsistency: statistical heterogeneity 75%. | |||||
| Anti‐inflammatories compared to placebo for chronic prostatitis/chronic pelvic pain syndrome | |||||
| Patient or population: men with chronic prostatitis/chronic pelvic pain syndrome | |||||
| Outcomes | № of participants | Quality of the evidence | Relative effect | Anticipated absolute effects* (95% CI) | |
| Risk with control | Risk difference with anti‐inflammatories | ||||
| Prostatitis symptoms A decrease of 25% or 6 points is considered an important improvement | 585 | ⊕⊕⊝⊝ | ‐ | The mean prostatitis symptoms ranged from 8.6 to 19.5 | MD 2.5 lower |
| Prostatitis symptoms: 'responders' | 82 | ⊕⊕⊝⊝ | RR 1.44 | Study population | |
| 91 per 1000 | 40 more per 1000 | ||||
| Adverse events | 540 | ⊕⊕⊝⊝ | RR 1.27 | Study population | |
| 98 per 1000 | 26 more per 1000 | ||||
| Sexual dysfunction ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| Quality of life ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| Anxiety and depression ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | |||||
| GRADE Working Group grades of evidence | |||||
| aDowngraded one level due to high risk of bias: unclear or high risk of bias in most domains in most studies. | |||||
| Phytotherapy compared to placebo for chronic prostatitis/chronic pelvic pain syndrome | |||||
| Patient or population: men with chronic prostatitis/chronic pelvic pain syndrome | |||||
| Outcomes | № of participants | Quality of the evidence | Relative effect | Anticipated absolute effects* (95% CI) | |
| Risk with placebo or other agents | Risk difference with phytotherapy | ||||
| Prostatitis symptoms A decrease of 25% or 6 points is considered an important improvement | 320 | ⊕⊕⊝⊝ | ‐ | The mean prostatitis symptoms ranged from 10.3 to 14.5 | MD 5.02 lower |
| Prostatitis symptoms: 'responders' | 224 | ⊕⊕⊕⊝ | RR 1.78 | Study population | |
| 384 per 1000 | 299 more per 1000 | ||||
| Adverse events | 540 | ⊕⊕⊝⊝ | RR 1.13 | Study population | |
| 41 per 1000 | 5 more per 1000 | ||||
| Sexual dysfunction | 48 | ⊕⊕⊝⊝ | ‐ | The mean sexual dysfunction was 18.5 | MD 3.5 higher |
| Quality of life ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| Anxiety and depression ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | |||||
| GRADE Working Group grades of evidence | |||||
| aDowngraded one level due to high risk of bias: unclear or high risk of bias in most domains in most studies. | |||||
| Botulinum toxin A compared to placebo for chronic prostatitis/chronic pelvic pain syndrome | |||||
| Patient or population: men with chronic prostatitis/chronic pelvic pain syndrome | |||||
| Outcomes | № of participants | Quality of the evidence | Relative effect | Anticipated absolute effects* (95% CI) | |
| Risk with placebo | Risk difference with botulinum toxin A | ||||
| Prostatitis symptoms Subgroup: Intraprostatic injection, participants age > 50 years old, basal NIH‐CPSI score > 30 Assessed with: NIH‐CPSI score. Benefit is indicated by lower scores A decrease of 25% or 6 points is considered an important improvement | 60 | ⊕⊕⊝⊝ | ‐ | The mean prostatitis symptoms ‐ Intraprostatic injection was 36.37 | MD 25.8 lower |
| Prostatitis symptoms Subgroup: Pelvic floor muscles injection, participants age < 50 years old, basal NIH‐CPSI score < 30 | 29 | ⊕⊕⊝⊝ | ‐ | The mean prostatitis symptoms ‐ Pelvic floor muscles injection was 27.8 | MD 2.6 lower |
| Adverse events | 89 | ⊕⊕⊝⊝ | RR 5.00 | Study population | |
| 22 per 1000 | 87 more per 1000 | ||||
| Sexual dysfunction ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| Quality of life ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| Anxiety and depression ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | |||||
| GRADE Working Group grades of evidence | |||||
| aDowngraded one level due to unclear risk of bias (random sequence generation). | |||||
| Allopurinol compared to placebo for chronic prostatitis/chronic pelvic pain syndrome | |||||
| Patient or population: men with chronic prostatitis/chronic pelvic pain syndrome | |||||
| Outcomes | № of participants | Quality of the evidence | Relative effect | Anticipated absolute effects* (95% CI) | |
| Risk with placebo | Risk difference with allopurinol | ||||
| Prostatitis symptoms A decrease of 25% or 6 points is considered an important improvement | 56 | ⊕⊕⊝⊝ | ‐ | The mean prostatitis symptoms was 17.21 | MD 0.21 lower |
| Adverse events Follow‐up: 3 months | 110 | ⊕⊕⊝⊝ | ‐ | No adverse events were observed in the included studies | |
| Sexual dysfunction ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| Quality of life ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| Anxiety and depression ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | |||||
| GRADE Working Group grades of evidence | |||||
| aDowngraded one level due to imprecision: confidence interval includes appreciable benefits and harms.˜ | |||||
| Traditional Chinese Medicine compared to placebo for chronic prostatitis/chronic pelvic pain syndrome | |||||
| Patient or population: men with chronic prostatitis/chronic pelvic pain syndrome Some comparisons included antibiotics, alpha blockers and other Western medications as co‐interventions | |||||
| Outcomes | № of participants | Quality of the evidence | Relative effect | Anticipated absolute effects* (95% CI) | |
| Risk with placebo or usual care | Risk difference with Traditional Chinese medicine | ||||
| Prostatitis symptoms A decrease of 25% or 6 points is considered an important improvement | 835 | ⊕⊕⊝⊝ | ‐ | The mean prostatitis symptoms ranged from 11.17 to 15.02 | MD 3.13 lower |
| Adverse events | 584 | ⊕⊕⊝⊝ | RR 1.34 | Study population | |
| 29 per 1000 | 10 more per 1000 | ||||
| Sexual dysfunction | 88 | ⊕⊕⊕⊝ | ‐ | The mean sexual dysfunction was 14.93 | MD 0.27 higher |
| Quality of life ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ |
| Anxiety and depression: anxiety | 88 | ⊕⊕⊝⊝ | ‐ | The mean anxiety and depression: anxiety was 23.3 | MD 9.5 lower |
| Anxiety and depression: depression | 88 | ⊕⊕⊝⊝ | ‐ | The mean anxiety and depression: depression was 24.07 | MD 7.84 lower |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | |||||
| GRADE Working Group grades of evidence | |||||
| aDowngraded one level due to inconsistency: high statistical heterogeneity (> 80%). Some of this inconsistency might be explained by the differences between the interventions under this comparison. | |||||
| Study | Intervention | Dose | Comparison | Dose for comparison Co‐interventions |
| Tadalafil study | 5 mg daily | Only co‐intervention | Both arms received levofloxacin 500 mg daily | |
| Ciprofloxacin + tamsulosin | Ciprofloxacin 500 mg twice daily, tamsulosin 0.4 mg once daily | C1: Ciprofloxacin C2: Tamsulosin C3: Placebo | They used double placebo (factorial design) | |
| Cernilton® (pollen extract) | 2 pills, 3 times a day | Cernilton® (pollen extract) | 1 pill, 3 times a day | |
| Prednisolone | 20 mg once daily and then tapered to 5 mg in 1 month | Placebo | Same regimen as active treatment | |
| Prolit super (plant extracts) | 4 capsules a day | Placebo | Same regimen as active treatment | |
| Deprox ® (pollen extract) | 2 capsules a day | Ibuprofen | 600 mg three times daily | |
| Deprox ® (pollen extract) | 2 capsules a day | Bromelain | 2 tablets a day | |
| Mepatricin | 40 mg daily | Vitamin C (as placebo) | 500 mg daily | |
| Alfusozin | 10 mg daily | C1: Terpene mixture 1 capsule three times a day C2: Only co‐intervention | All participants received levofloxacin 300 mg daily | |
| Terazosin | Titrated from 1 to 5 mg daily in two weeks | Placebo | Same regimen as active treatment | |
| Qiantongding decoction | Mix of herbal medications (oral infusion), twice a day | XiaoYanTong (Indometacin) | 25 mg tablet three times a day | |
| Tamsulosin | 0.2 mg daily | Placebo | Same regimen as active treatment | |
| Levofloxacin | 500 mg daily | C1: Ciprofloxacin 500 mg daily C2: Levofloxacin 1 g daily C3: No antibiotic | ‐ | |
| Roxithromycin | 300 mg daily | C1: Ciprofloxacin 1g daily C2: Aceclofenac 200 mg daily | ‐ | |
| Cytoflavin | 10 ml intravenously for 10 days, then 4 pills a day for 20 days | No cytoflavin | Both arms received: α‐blockers ‐ 1 month anti‐inflammatory drugs – 2 weeks prostate massage and vibrovacuum fallostimulation – 10 times antibiotics – 10 days | |
| Mepartricin | 40 mg daily for 60 days | Placebo | Same regimen as active treatment | |
| Phenoxybenzamine | 20 mg daily | Placebo | Same regimen as active treatment | |
| Pollen extract | 74 mg in Prolit® capsules | Placebo | Same regimen as active treatment | |
| Botulinum toxin A | 100 units applied in the external urethral sphincter | Botulinum toxin A | 100 units applied to the prostate and sphincter | |
| Quinolone + ibuprofen + terazosin | 500 mg, 400 mg and 5 mg respectively | C1: Terazosin 5 mg C2: Quinolone 500 mg + ibuprofen 400 mg | ‐ | |
| Botulinum toxin A | 200 units applied to the prostate | Placebo | Same regimen as active treatment | |
| Palmitoylethanolamide | 300 mg in Penease® capsules twice a day | Serenoa repens | 320 mg 1 capsule daily [All] "patients received a full course of pharmacological therapy" before allocation | |
| Duloxetin | 60 mg, escalated during the first 15 days | Only co‐interventions | Co‐interventions included Serenoa repens and tamsulosin | |
| Zafirlukast | 20 mg twice a day | Placebo | Same regimen as active treatment Both arms received doxycycline 100 mg twice daily for 4 weeks | |
| Botulinum toxin A | 100 units applied to pelvic floor muscles | Placebo | Same regimen as active treatment | |
| Terasozin | 2 mg daily | Placebo | Same regimen as active treatment | |
| Bazheng decoction | 320 mL daily | Only co‐intervention | Co‐intervention: tamsulosin 0.2 mg a day | |
| Pollen extract | 63 mg 3 times a day | Eviprostat® (herbal extract) | 1 capsule 3 times a day | |
| Levofloxacin | 200 mg daily | C1: Doxazosin C2: Levofloxacin + Doxazosin | 4 mg daily | |
| Desketoprofen | 12.5 mg 3 times a day | C1: Indomethacin 25 mg 3 times a day C2: Only co‐interventions | All participants received terazosin 2 mg daily for 4 weeks | |
| Terazosin | 3 ‐ 4 mg | No terazosin | All participants received levofloxacin 300 mg/day, tamiflunate 3 tablets/day for 12 weeks | |
| Serenoa repens | 325 mg daily | Finasteride | 5 mg daily | |
| Tamsulosin | 0.2 mg daily | C1: Ibuprofen + Misoprostol C2: Tamsulosin + ibuprofen + misoprostol | 600 mg ibuprofen three 3 times a day, 300 mcg misoprostol 3 times a day | |
| Propiverine | 20 mg daily | No propiverine | Both groups received gatifloxacin 200 mg twice daily | |
| Ciprofloxacin | 500 mg twice daily | C1: Diclofenac C2: Only co‐intervention | 50 mg twice daily Both groups received tamsulosin 0.2 mg daily for 12 weeks | |
| Solifenacin | 5 mg daily | No solifenacin | Both groups received ciprofloxacin 1000 mg daily for 8 weeks | |
| Mirodenafil | 50 mg daily | Only co‐intervention | Both groups received levofloxacin 500 mg daily for 6 weeks | |
| Doxazosin | 2 mg daily | C1: Ciprofloxacin C2: Doxazosin + ciprofloxacin | 500 mg twice a day | |
| Terazosin | 5 mg daily | C1: Tamsulosin C2: Placebo | 0.4 mg daily | |
| Sertraline | 50 mg daily | Placebo | Same regimen as active treatment | |
| Terpene mixture | Rowatinex® 200 mg | Ibuprofen | 600 mg 3 times a day | |
| Finasteride | 5 mg | Placebo | Same regimen as active treatment Ketoprofene was provided to both group for pain relief | |
| QianLieAnShuan (Prostat, 前列安栓) | Herbal suppository each night | Placebo suppository | Same regimen as active treatment | |
| Tiaoshen Tonglin | Mix of herbal medications (oral 150 ml infusion) daily | Terazosin | 2 mg daily | |
| Qianlieping | 2 g herbal capsule 3 times a day | C1: Tamsulosin C2: Qianlieping + Tamsulosin | 0.2 mg daily | |
| Vardenafil | 10 mg daily before sexual intercourse (from week 5 onwards) | Only co‐interventions | Both groups received traditional Chinese medicine (Huafenqinutang oral dose for 8 weeks) | |
| Phenoxybenzamine | 10 mg twice a day | C1: Flavoxate C2: Placebo | 200 mg 3 times a day | |
| Deprox ® (pollen extract) | 2 tablets a day | Serenoa repens | 320 mg daily | |
| Deprox ® (pollen extract) | 2 tablets a day | Quercetin | 500 mg twice daily | |
| Alfusozin | 5 mg twice a day | Placebo | Same regimen as active treatment Both groups "were allowed to take analgesics (ibuprofen, ketoprofen, diclofenac)" | |
| Alfusozin | 10 mg daily | No alfuzosin | Both groups received levofloxacin 100mg 3 times a day | |
| Profluss ® (Serenoa repens + lycopene + seleniated sodium) | 320 mg | Serenoa repens | 320 mg | |
| Curcumin‐calendula | 350 mg curcumin and 80 mg calendula in rectal suppositories daily | Placebo | Same regimen as active treatment | |
| Levofloxacin | 500 mg daily | Placebo | Same regimen as active treatment | |
| Rofecoxib | 50 mg and 25 mg doses daily | C1: Rofecoxib C2: Placebo | All participants had the same regimen All participants were permitted to take up to 2.6 g of paracetamol for rescue analgesia | |
| Finasteride | 5 mg daily | Placebo | Same regimen as active treatment | |
| Tamsulosin | 0.4 mg daily | Placebo | Same regimen as active treatment | |
| Pentosan polysulphate | 300 mg 3 times a day | Placebo | Same regimen as active treatment | |
| Alfusozin | 10 mg daily | Placebo | Same regimen as active treatment | |
| Silodosin | 8 mg daily | C1: Silodosin (4 mg) C2: Placebo | Same regimen as active treatment | |
| Tanezumab | 20 mg intravenous single dose | Placebo | Same regimen as active treatment | |
| PPC (aminoacid preparation) | 6 capsules a day | Pollen extract | Same regimen as active treatment | |
| Cranberry juice | 150 ml twice daily | No treatment | All participants underwent an 8 week‐run‐in period with levofloxacin 100mg 3 times a day, NSAID, alpha blocker, behaviour therapy, and hot sitz bath; for non‐responder, 4 additional weeks of same treatments were added | |
| Tadalafil | 10 mg daily | Only co‐intervention | Both groups received levofloxacin 500 mg daily for 4 weeks | |
| Tadalafil | 5 mg daily | Only co‐intervention | Both groups received levofloxacin 500 mg daily for six weeks | |
| Antiphlogistic agent | Mixture of herbal remedies 3 times a day | C1: Antiphlogistic + enema C2: Antiphlogistic + suppository C3: Antiphlogistic + "rectal fumigation" | ‐ | |
| Allopurinol | 300 mg twice a day | C1: Allopurinol 300 mg + placebo C2: Placebo twice a day | Both groups received the same number of pills | |
| Pregabalin | Titrated from 150 to 600 mg daily | Placebo | Same regimen as active treatment | |
| Serenoa repens | No description on dose or regimen | Placebo | No description on dose or regimen | |
| Alfusozin | 10 mg daily | Only co‐intervention | Both groups received tosufloxacin 150mg 3 times a day | |
| QianLieAnWan | Herbal ball‐shaped formulation, 9g 2 ‐ 3 times a day | C1: QianLieKang C2: Pollen extract capsule | ‐ | |
| Quercetin | 500 mg twice a day | Placebo | Same regimen as active treatment | |
| Tadalafil | 5 mg daily | Only co‐interventions | Both groups received levofloxacin 500 mg for 6 weeks and alfuzosin 10 mg for 6 weeks | |
| Terazosin | Titrated to 5 mg daily | Placebo | Same regimen as active treatment | |
| QianLieAnTong | Herbal capsule 0.38 g four tables 3 times a day | No QianLieAnTong | Both groups received terazosin 2 mg daily | |
| QianLieAnShuan (Prostat, 前列安栓) | Herbal suppository each night | No QuanLieAnShuan (Prostant) | Both groups received alpha blockers (tamsulosin or terazosin) | |
| Tiocolchicoside + ibuprofen | 120 (sic) and 1200 mg respectively, daily | Only co‐intervention | Both groups received terazosin 5 mg a day | |
| Tiocolchicoside + ibuprofen + doxazosin | 12 mg + 400 mg + 4 mg respectively, daily | C1: Doxazosin C2: Placebo | C1: Doxazosin 4 mg daily | |
| Fluvoxamine | 50 mg daily | Placebo | Same regimen as active treatment | |
| Pollen extract | 60 mg carnitine twice a day, daily | Placebo | Same regimen as active treatment Both groups were pretreated with azithromycin (250 mg every 6 hours for 1 day) | |
| OM‐89 (E. coli lysate) | 6 mg from 18 strains | Placebo | Same regimen as active treatment Other medications were allowed in both groups | |
| Chuanshentong | Sage and carrot‐family herbal extracts, injected in the prostate | Placebo injection | Same regimen as active treatment | |
| Terazosin | 2 mg daily | C1: Levofloxacin C2: Terazosin + Levofloxacin | Levofloxacin dose: 200 mg twice daily Both groups received dietary advice and prostatic massage once a week and were advised to take warm baths | |
| Pentosan polysulphate | 100 mg twice daily | Placebo | Same regimen | |
| Doxazosin | 4 mg daily | C1: Diclofenac C2: Doxazosin + diclofenac | 75 mg daily | |
| YuLeShu | Herbal mixture 20 ml 3 times a day | Only co‐interventions | Both groups received levofloxacin 0.2g twice a day, doxazosin 2 mg daily, QianLieTongYu 3 times a day, 4 capsules each time and weekly prostate massage | |
| Pollen extract (普适泰 = 舍尼通) | 0.375 g 3 times a day | Antibiotic treatment | Sulfamethoxazole 2 tablets, twice daily 10 days, ofloxacin 0.2 g, twice daily 10 days, minocycline 0.1 g twice daily, 10 days, sequentially, repeated monthly for 3 months | |
| Prednisone | 15 mg daily | Placebo | Both groups received levofloxacin 100 mg twice a day for 4 weeks | |
| Terazosin | 1 mg daily | C1: Tamsulosin C2: Placebo | 0.2 mg daily | |
| Pollen extract (普适泰 = 舍尼通) | 0.375 g twice a day | Placebo | Both groups received levofloxacin 100 mg twice a day for 4 weeks | |
| Tamsulosin | 0.2 mg daily | C1: Levofloxacin C2: Tamsulosin + levofloxacin | Levofloxacin 0.2 g daily | |
| Doxazosin | Dose was not defined | Only co‐intervention | Both groups received gatifloxacin 200mg twice daily | |
| Celecoxib | 200 mg daily | Celecoxib | 200 mg 3 times a day | |
| Aike decoction | Twice daily | C1: Bazhengsan decoction C2: Prostatitis decoction C3: Placebo | All decoctions were administered twice daily | |
| Sertraline | Titrated from 50 to 100 mg individually | C1:Duloxetine C2: Only co‐intervention | Duloxeine titrated from 30 to 120 mg individually All participants received doxazosin 4 mg daily | |
| Celecoxib | 200 mg daily | Placebo | Same regimen | |
| Tetracycline | 500 mg daily | Placebo | Placebo was "Vitamin B" | |
| Allopurinol | 100 mg 3 times a day | Placebo | Same regimen | |
| C1, C2, C3 were used to refer to multiple treatment arms. NSAID: Nonsteroidal anti‐inflammatory drugs. Blank cells indicate that dosing is described in the adjacent cell and co‐interventions were not described. | ||||
| Study | n randomised | n analysed | Duration ‐ Follow‐up | Mean age (years) Baseline | Previous treatments | Mean NIH‐CPSI score Baseline | Trial period | Country | Funding |
| 108 | 108 | 4 weeks | 40.55 | N/A | N/A | N/A | Saudi Arabia | None | |
| 196 | 174 | 12 weeks | 44.58 | N/A | 24.78 | 2001 ‐ 2002 | USA and Canada | Government + Industry | |
| 78 | 78 | 12 weeks | 36.90 | N/A | 22.50 | 2008 | Russia | None | |
| 21 | 18 | 8 weeks 52 weeks | 41.05 | N/A | 24.45 | 2000 ‐ 2002 | UK | Government | |
| 57 | 57 | 8 weeks | N/A | N/A | 22.00 | N/A | Russia | None | |
| 87 | 84 | 4 weeks | 33.75 | N/A | 25.20 | 2012 | Italy | N/A | |
| 70 | 65 | 12 weeks | 32.60 | N/A | 25.35 | 2015 | Italy | N/A | |
| 54 | 42 | 4 weeks | 34.00 | N/A | N/A | N/A | Italy | N/A | |
| 103 | 103 | 8 weeks | 39.27 | N/A | 24.53 | 2006 ‐ 2008 | South Korea | N/A | |
| 100 | 86 | 12 weeks 14 weeks | 35.50 | N/A | 26.15 | 2000 ‐ 2001 | Malaysia and US | Industry | |
| 70 | 70 | 4 weeks | 29.60 | No | 24.70 | 2007 ‐ 2008 | China | N/A | |
| 100 | 93 | 24 weeks 120 weeks | 34.30 | No | 22.90 | 2003 ‐ 2007 | China | N/A | |
| 215 | 215 | 6 weeks | N/A | Yes | N/A | N/A | Taiwan | N/A | |
| 75 | 75 | 4 weeks 12 weeks | 29.10 | N/A | 21.40 | 2011 | South Korea | N/A | |
| 60 | Not ANLZ | 4 weeks | N/A | N/A | N/A | N/A | Russia | None | |
| 30 | 26 | 8 weeks | 33.00 | N/A | 25.00 | 2001 ‐ 2002 | Italy | N/A | |
| 40 | 30 | 6 weeks | 39.00 | Yes | N/A | N/A | Germany | Industry | |
| 60 | 58 | 24 weeks | 35.00 | Yes | N/A | N/A | USA | Industry | |
| 52 | 52 | 52 weeks | 36.50 | Yes | N/A | N/A | Egypt | N/A | |
| 87 | 87 | 12 weeks 20 weeks | 34.20 | N/A | 23.92 | 2004 ‐ 2008 | Turkey | N/A | |
| 60 | 60 | 24 weeks | 40.42 | Yes | 34.09 | 2011 ‐ 2013 | Iran | N/A | |
| 44 | 44 | 12 weeks | 41.32 | N/A | N/A | 2014 ‐ 2015 | Italy | N/A | |
| 38 | 34 | 16 weeks | 46.80 | N/A | 24.68 | 2009 ‐ 2012 | Italy | N/A | |
| 20 | 17 | 4 weeks | 35.75 | N/A | N/A | N/A | UK | Industry | |
| 29 | 29 | 4 weeks | 50.50 | Yes | 25.95 | N/A | USA | N/A | |
| 91 | 69 | 12 weeks | 39.60 | N/A | PSSI 9.61/9.27 | 1997 ‐ 1999 | Turkey | N/A | |
| 96 | 96 | 2 weeks | 32.15 | N/A | 27.40 | 2012 ‐ 2013 | China | N/A | |
| 100 | 80 | 8 weeks | 51.55 | No | 21.30 | 2009 ‐ 2013 | Japan | N/A | |
| 81 | 81 | 6 weeks | 40.03 | No | 23.03 | 2004 | South Korea | N/A | |
| 115 | 115 | 4 weeks | 32.48 | No | 22.43 | 2007 ‐ 2008 | China | N/A | |
| 127 | 127 | 12 weeks | N/A | N/A | 21.65 | 2004 ‐ 2005 | South Korea | Industry | |
| 64 | 61 | 52 weeks | 43.20 | N/A | 24.30 | N/A | USA | N/A | |
| 63 | 55 | 8 weeks 12 weeks | N/A | N/A | 18.31 | 2001 ‐ 2002 | South Korea | N/A | |
| 46 | 46 | 2 months | 40.10 | N/A | 22.85 | N/A | South Korea | University | |
| 107 | 100 | 12 weeks | 46.10 | N/A | 23.56 | 2008 ‐ 2009 | South Korea | N/A | |
| 96 | 87 | 8 weeks | N/A | N/A | 21.10 | N/A | South Korea | N/A | |
| 88 | 88 | 6 weeks | 44.75 | N/A | 20.80 | N/A | South Korea | University | |
| 90 | 90 | 4 weeks | 40.30 | N/A | 25.80 | 2004 | Bosnia and Herzegovina | N/A | |
| 80 | 80 | 8 weeks | 36.19 | N/A | N/A | 1997 ‐ 1998 | Italy | N/A | |
| 14 | 13 | 13 weeks | N/A | N/A | PSS 23.4/28 | N/A | UK | NGO | |
| 50 | 50 | 6 weeks | 43.45 | N/A | 21.87 | 2003 ‐ 2004 | South Korea | N/A | |
| 41 | 35 | 52 weeks | 46.50 | Yes | N/A | N/A | Finland | None | |
| 76 | 75 | 4 weeks | 32.70 | N/A | 48.32 | 2002 | China | N/A | |
| 108 | 108 | 8 weeks | 29.95 | N/A | 26.05 | 2004 ‐ 2006 | China | Government | |
| 257 | 220 | 6 weeks | 30.60 | N/A | 24.80 | 2010 ‐ 2011 | China | Government | |
| 138 | 138 | 8 weeks | 37.00 | N/A | 27.85 | N/A | China | N/A | |
| 60 | 57 | 4 weeks | 39.13 | N/A | 21.85 | 2000 ‐ 2001 | China | Government + Industry | |
| N/A | 63 | 6 weeks | N/A | N/A | N/A | 2016 | Italy | None | |
| 54 | 54 | 4 weeks | 33.85 | N/A | 25.82 | 2016 | Italy | None | |
| 40 | 36 | 24 weeks 52 weeks | 49.50 | N/A | 24.50 | N/A | Finland | Government | |
| 54 | 54 | 8 weeks | 45.25 | N/A | 23.50 | 2004 ‐ 2005 | South Korea | N/A | |
| 102 | 102 | 8 weeks 16 weeks | 38.43 | No | 27.61 | 2006 ‐ 2007 | Italy | N/A | |
| 55 | 48 | 4 weeks 12 weeks | 32.00 | No | 20.25 | 2015 ‐ 2016 | Italy | None | |
| 80 | 79 | 6 weeks | 56.10 | N/A | 22.85 | N/A | Canada | Government + Industry | |
| 161 | 157 | 6 weeks | 45.97 | N/A | 21.97 | N/A | USA and Canada | Industry | |
| 76 | 64 | 24 weeks | 44.30 | N/A | 21.30 | N/A | USA | Government + Industry | |
| 58 | 58 | 6 weeks | 40.85 | N/A | 26.30 | 2000 ‐ 2001 | USA | Industry | |
| 100 | 73 | 16 weeks | 39.15 | N/A | 26.45 | N/A | USA and Canada | Industry | |
| 272 | 233 | 12 weeks | 40.10 | N/A | 24.45 | 2005 ‐ 2008 | USA, Canada and Malaysia | Government + Industry | |
| 151 | 115 | 12 weeks | 48.30 | N/A | 26.90 | 2008 ‐ 2009 | Canada | Industry | |
| 62 | 51 | 16 weeks | 46.85 | N/A | N/A | 2009 ‐ 2010 | USA, Canada, France, Sweden and Switzerland | Industry | |
| 76 | Not ANLZ | 4 weeks | N/A | N/A | N/A | 1983 | Japan | N/A | |
| 50 | 50 | 12 weeks | 35.85 | Yes | 22.75 | 2002 ‐ 2003 | South Korea | N/A | |
| 78 | 78 | 4 weeks | N/A | N/A | N/A | N/A | South Korea | None | |
| 86 | 86 | 6 weeks | 48.75 | N/A | N/A | N/A | South Korea | N/A | |
| 160 | Not ANLZ | 4 weeks | 36.00 | N/A | N/A | 1999 ‐ 2002 | China | Government | |
| 54 | 34 | 32 weeks | N/A | N/A | N/A | N/A | Sweden | N/A | |
| 324 | 313 | 6 weeks | 46.60 | Yes | 26.05 | 2006 ‐ 2007 | USA | Government + Industry | |
| 142 | 142 | 72 weeks | N/A | N/A | N/A | N/A | Austria | None | |
| 57 | N/A | 8 weeks | 40.05 | N/A | 20.70 | N/A | South Korea | University | |
| 60 | Not ANLZ | 4 weeks | N/A | N/A | N/A | 1994 | China | N/A | |
| 30 | 28 | 4 weeks | 44.85 | N/A | 20.60 | N/A | USA | N/A | |
| 68 | 61‐65 | 6 weeks | N/A | N/A | N/A | N/A | India | None | |
| 64 | 51 | 8 weeks 52 weeks | N/A | N/A | 25.70 | N/A | Russia | None | |
| 115 | 115 | 4 weeks | 31.60 | No | 23.99 | 2007 ‐ 2008 | China | N/A | |
| 90 | 88 | 6 weeks | 36.01 | N/A | 24.80 | 2006 | China | N/A | |
| 45 | 39 | 24 weeks | 34.10 | N/A | N/A | 2003 ‐ 2004 | Turkey | N/A | |
| 90 | 79 | 24 weeks 52 weeks | 29.10 | No | 22.63 | 2004 ‐ 2005 | Turkey | N/A | |
| 42 | 29 | 8 weeks | 41.00 | N/A | N/A | N/A | UK | Industry | |
| 139 | 118 | 12 weeks | 39.50 | N/A | 19.80 | 1999 ‐ 2004 | Germany | Industry | |
| 185 | 154 | 24 weeks 52 weeks | 47.70 | N/A | 22.40 | 2008 ‐ 2010 | Austria, Germany, Poland and Portugal | Industry | |
| 38 | 36 | 12 weeks | 28.00 | N/A | N/A | 2002 ‐ 2003 | China | N/A | |
| 115 | 115 | 6 weeks | 37.63 | No | 23.03 | 2011 ‐ 2014 | China | None | |
| 30 | 24 | 12 weeks | 37.60 | N/A | N/A | 1984 ‐ 1985 | Sweden | N/A | |
| 123 | 115 | 12 weeks | 34.80 | No | 23.96 | 2006 ‐ 2007 | China | N/A | |
| 88 | 88 | 4 weeks | 34.21 | N/A | 28.43 | 2011 ‐ 2012 | China | N/A | |
| 60 | Not ANLZ | 12 weeks | 28.25 | N/A | N/A | 1998 ‐ 1999 | China | N/A | |
| 160 | 158 | 4 weeks | 29.25 | N/A | 22.61 | 2007 | China | N/A | |
| 156 | 153 | 12 weeks | 30.73 | N/A | 25.44 | 2009 | China | N/A | |
| 160 | 159 | 8 weeks | 31.54 | N/A | N/A | 2005 ‐ 2006 | China | N/A | |
| 105 | 105 | 12 weeks | N/A | N/A | 20.05 | 2002 ‐ 2004 | China | Industry | |
| 69 | N/A | 6 weeks | 41.55 | N/A | 24.35 | 2005 ‐ 2006 | South Korea | N/A | |
| 64 | 61 | 6 weeks | 35.60 | N/A | 28.35 | 2003 | China | N/A | |
| 248 | 218 | 4 weeks | 30.97 | N/A | 21.41 | 2005 ‐ 2007 | China | Government | |
| 150 | 126 | 24 weeks | 32.96 | No | 21.94 | 2011 ‐ 2012 | China | None | |
| 64 | 64 | 8 weeks | N/A | No | N/A | 2006 ‐ 2008 | China | N/A | |
| 251 | 114 | 12 weeks | 32.55 | N/A | 25.22 | N/A | China | Government | |
| 48 | 48 | 12 weeks | 39.50 | Yes | N/A | 2005 ‐ 2007 | China | N/A | |
| 56 | 56 | 12 weeks | 33.40 | N/A | 25.68 | 2002 ‐ 2004 | Iran | N/A | |
| N/A: not applicable; NGO: non‐governmental organization; NIH‐CPSI: National Institutes of Health Chronic Prostatitis Symptom Index; PSS: Prostatitis Severity Score; PSSI: Prostatitis Symptoms Severity Index; USA: United States of America. Duration and follow‐up are differentiated when data were available. | |||||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms: short term Show forest plot | 18 | 1524 | Mean Difference (IV, Random, 95% CI) | ‐5.01 [‐7.41, ‐2.61] |
| 1.1 Terazosin | 5 | 436 | Mean Difference (IV, Random, 95% CI) | ‐5.73 [‐10.96, ‐0.50] |
| 1.2 Doxazosin | 4 | 262 | Mean Difference (IV, Random, 95% CI) | ‐5.17 [‐11.50, 1.16] |
| 1.3 Phenoxybenzamine | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐5.80 [‐7.91, ‐3.69] |
| 1.4 Tamsulosin | 4 | 302 | Mean Difference (IV, Random, 95% CI) | ‐5.89 [‐13.16, 1.38] |
| 1.5 Alfuzosin | 4 | 381 | Mean Difference (IV, Random, 95% CI) | ‐2.63 [‐4.55, ‐0.71] |
| 1.6 Silodosin | 1 | 103 | Mean Difference (IV, Random, 95% CI) | ‐3.60 [‐6.81, ‐0.39] |
| 2 Prostatitis symptoms: pain Show forest plot | 13 | 1243 | Mean Difference (IV, Random, 95% CI) | ‐2.39 [‐3.57, ‐1.22] |
| 2.1 Terazosin | 3 | 289 | Mean Difference (IV, Random, 95% CI) | ‐3.86 [‐6.00, ‐1.73] |
| 2.2 Doxazosin | 3 | 202 | Mean Difference (IV, Random, 95% CI) | ‐2.19 [‐4.56, 0.17] |
| 2.3 Phenoxybenzamine | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐2.25 [‐3.78, ‐0.72] |
| 2.4 Tamsulosin | 3 | 265 | Mean Difference (IV, Random, 95% CI) | ‐3.17 [‐6.31, ‐0.03] |
| 2.5 Alfuzosin | 3 | 344 | Mean Difference (IV, Random, 95% CI) | ‐0.59 [‐1.67, 0.49] |
| 2.6 Silodosin | 1 | 103 | Mean Difference (IV, Random, 95% CI) | ‐1.40 [‐2.97, 0.17] |
| 3 Prostatitis symptoms: urinary Show forest plot | 13 | 1243 | Mean Difference (IV, Random, 95% CI) | ‐1.48 [‐2.29, ‐0.66] |
| 3.1 Terazosin | 3 | 289 | Mean Difference (IV, Random, 95% CI) | ‐2.07 [‐3.79, ‐0.34] |
| 3.2 Doxazosin | 3 | 202 | Mean Difference (IV, Random, 95% CI) | ‐1.71 [‐4.34, 0.92] |
| 3.3 Phenoxybenzamine | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐1.60 [‐2.28, ‐0.92] |
| 3.4 Tamsulosin | 3 | 265 | Mean Difference (IV, Random, 95% CI) | ‐1.63 [‐3.85, 0.59] |
| 3.5 Alfuzosin | 3 | 344 | Mean Difference (IV, Random, 95% CI) | ‐0.67 [‐1.05, ‐0.29] |
| 3.6 Silodosin | 1 | 103 | Mean Difference (IV, Random, 95% CI) | ‐0.90 [‐2.00, 0.20] |
| 4 Prostatitis symptoms: quality of life Show forest plot | 13 | 1243 | Mean Difference (IV, Random, 95% CI) | ‐1.61 [‐2.49, ‐0.73] |
| 4.1 Terazosin | 3 | 289 | Mean Difference (IV, Random, 95% CI) | ‐2.51 [‐4.62, ‐0.40] |
| 4.2 Doxazosin | 3 | 202 | Mean Difference (IV, Random, 95% CI) | ‐1.34 [‐3.24, 0.56] |
| 4.3 Phenoxybenzamine | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐2.2 [‐3.14, ‐1.26] |
| 4.4 Tamsulosin | 3 | 265 | Mean Difference (IV, Random, 95% CI) | ‐1.78 [‐4.96, 1.40] |
| 4.5 Alfuzosin | 3 | 344 | Mean Difference (IV, Random, 95% CI) | ‐0.53 [‐1.34, 0.29] |
| 4.6 Silodosin | 1 | 103 | Mean Difference (IV, Random, 95% CI) | ‐1.40 [‐2.49, ‐0.31] |
| 5 Prostatitis symptoms: responders rate Show forest plot | 7 | 721 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.94, 1.61] |
| 5.1 Doxazosin | 1 | 55 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.67, 1.20] |
| 5.2 Tamsulosin | 3 | 254 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [1.07, 2.06] |
| 5.3 Alfuzosin | 2 | 309 | Risk Ratio (M‐H, Random, 95% CI) | 1.82 [0.48, 6.80] |
| 5.4 Silodosin | 1 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.78, 1.52] |
| 6 Prostatitis symptoms: long term Show forest plot | 4 | 235 | Mean Difference (IV, Random, 95% CI) | ‐5.60 [‐10.89, ‐0.32] |
| 6.1 Terazosin | 1 | 51 | Mean Difference (IV, Random, 95% CI) | ‐7.76 [‐10.90, ‐4.62] |
| 6.2 Tamsulosin | 1 | 92 | Mean Difference (IV, Random, 95% CI) | ‐1.40 [‐2.73, ‐0.07] |
| 6.3 Alfuzosin | 1 | 35 | Mean Difference (IV, Random, 95% CI) | ‐3.4 [‐7.30, 0.50] |
| 6.4 Doxazosin | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐9.7 [‐10.25, ‐9.15] |
| 7 Prostatitis symptoms: long term Show forest plot | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [1.06, 2.32] |
| 7.1 Tamsulosin | 1 | 92 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [1.06, 2.32] |
| 8 Adverse events Show forest plot | 19 | 1588 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.09, 2.34] |
| 8.1 Terazosin | 6 | 431 | Risk Ratio (M‐H, Random, 95% CI) | 2.03 [1.14, 3.61] |
| 8.2 Doxazosin | 3 | 195 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [0.75, 2.88] |
| 8.3 Phenoxybenzamine | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 35.00 [2.25, 544.92] |
| 8.4 Tamsulosin | 5 | 367 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [0.60, 3.03] |
| 8.5 Alfuzosin | 4 | 452 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.37, 3.78] |
| 8.6 Silodosin | 1 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 2.52 [1.15, 5.52] |
| 9 Sexual dysfunction Show forest plot | 4 | 452 | Mean Difference (IV, Random, 95% CI) | 0.26 [‐1.13, 1.65] |
| 9.1 Terazosin | 1 | 77 | Mean Difference (IV, Random, 95% CI) | 1.10 [‐0.84, 3.04] |
| 9.2 Tamsulosin | 1 | 99 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐4.84, 4.44] |
| 9.3 Alfuzosin | 2 | 276 | Mean Difference (IV, Random, 95% CI) | ‐0.70 [‐2.89, 1.48] |
| 10 Quality of life: mental Show forest plot | 3 | 421 | Mean Difference (IV, Random, 95% CI) | 0.15 [‐2.63, 2.92] |
| 10.1 Tamsulosin | 1 | 90 | Mean Difference (IV, Random, 95% CI) | ‐1.00 [‐7.16, 1.16] |
| 10.2 Alfuzosin | 1 | 228 | Mean Difference (IV, Random, 95% CI) | 2.1 [‐0.64, 4.84] |
| 10.3 Silodosin | 1 | 103 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐3.09, 3.69] |
| 11 Quality of life: physical Show forest plot | 3 | 421 | Mean Difference (IV, Random, 95% CI) | 1.17 [‐0.97, 3.30] |
| 11.1 Tamsulosin | 1 | 90 | Mean Difference (IV, Random, 95% CI) | 2.4 [‐0.52, 5.32] |
| 11.2 Alfuzosin | 1 | 228 | Mean Difference (IV, Random, 95% CI) | ‐0.5 [‐2.51, 1.51] |
| 11.3 Silodosin | 1 | 103 | Mean Difference (IV, Random, 95% CI) | 2.5 [‐0.81, 5.81] |
| 12 Anxiety and depression Show forest plot | 1 | 232 | Mean Difference (IV, Random, 95% CI) | ‐1.1 [‐2.54, 0.34] |
| 12.1 Alfuzosin | 1 | 232 | Mean Difference (IV, Random, 95% CI) | ‐1.1 [‐2.54, 0.34] |
| 13 Urinary symptoms Show forest plot | 2 | 143 | Mean Difference (IV, Random, 95% CI) | ‐2.68 [‐5.90, 0.54] |
| 13.1 Terazosin | 1 | 86 | Mean Difference (IV, Random, 95% CI) | ‐4.5 [‐6.64, ‐2.36] |
| 13.2 Alfuzosin | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐1.20 [‐1.76, ‐0.64] |
| 14 Prostatitis symptoms: responders rate (sensitivity analysis according to risk of bias) Show forest plot | 2 | 155 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.61, 2.48] |
| 14.1 Tamsulosin | 2 | 155 | Odds Ratio (M‐H, Random, 95% CI) | 1.23 [0.61, 2.48] |
| 15 Adverse events (sensitivity analysis according to risk of bias) Show forest plot | 2 | 153 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.63, 1.47] |
| 15.1 Tamsulosin | 2 | 153 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.63, 1.47] |
| 16 Prostatitis symptoms: short term (subgroup analysis by co‐interventions) Show forest plot | 18 | 1524 | Mean Difference (IV, Random, 95% CI) | ‐5.01 [‐7.41, ‐2.61] |
| 16.1 Without co‐interventions | 7 | 688 | Mean Difference (IV, Random, 95% CI) | ‐3.56 [‐5.26, ‐1.86] |
| 16.2 With co‐interventions (analgesics, antibiotics) | 11 | 836 | Mean Difference (IV, Random, 95% CI) | ‐5.69 [‐8.90, ‐2.48] |
| 17 Adverse events (subgroup analysis by co‐interventions) Show forest plot | 19 | 1588 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [1.09, 2.34] |
| 17.1 Without co‐interventions | 11 | 1089 | Risk Ratio (M‐H, Random, 95% CI) | 1.69 [1.10, 2.60] |
| 17.2 With co‐interventions (analgesics, antibiotics) | 8 | 499 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.46, 3.17] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Prostatitis symptoms: responder rate Show forest plot | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Adverse events Show forest plot | 2 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.33, 2.30] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 5 | 372 | Mean Difference (IV, Random, 95% CI) | ‐2.43 [‐4.72, ‐0.15] |
| 1.1 Ciprofloxacin | 3 | 215 | Mean Difference (IV, Random, 95% CI) | ‐1.69 [‐3.15, ‐0.23] |
| 1.2 Levofloxacin | 2 | 157 | Mean Difference (IV, Random, 95% CI) | ‐2.98 [‐9.41, 3.44] |
| 2 Prostatitis symptoms: pain Show forest plot | 4 | 319 | Mean Difference (IV, Random, 95% CI) | ‐0.92 [‐1.78, ‐0.06] |
| 2.1 Ciprofloxacin | 2 | 155 | Mean Difference (IV, Random, 95% CI) | ‐0.78 [‐1.86, 0.30] |
| 2.2 Levofloxacin | 2 | 164 | Mean Difference (IV, Random, 95% CI) | ‐0.72 [‐2.74, 1.30] |
| 3 Prostatitis symptoms: urinary Show forest plot | 4 | 319 | Mean Difference (IV, Random, 95% CI) | ‐0.12 [‐0.65, 0.41] |
| 3.1 Ciprofloxacin | 2 | 155 | Mean Difference (IV, Random, 95% CI) | ‐0.29 [‐1.17, 0.58] |
| 3.2 Levofloxacin | 2 | 164 | Mean Difference (IV, Random, 95% CI) | 0.13 [‐0.85, 1.11] |
| 4 Prostatitis symptoms: quality of life Show forest plot | 4 | 319 | Mean Difference (IV, Random, 95% CI) | ‐0.45 [‐0.91, 0.02] |
| 4.1 Ciprofloxacin | 2 | 155 | Mean Difference (IV, Random, 95% CI) | ‐0.44 [‐1.04, 0.15] |
| 4.2 Levofloxacin | 2 | 164 | Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐1.52, 0.53] |
| 5 Prostatitis symptoms: responder rate Show forest plot | 2 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.73, 1.74] |
| 5.1 Ciprofloxacin | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.48, 2.09] |
| 5.2 Levofloxacin | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.70, 2.05] |
| 6 Adverse events Show forest plot | 4 | 336 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.66, 1.55] |
| 6.1 Ciprofloxacin | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.60, 1.57] |
| 6.2 Levofloxacin | 3 | 241 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.46, 2.97] |
| 7 Sexual dysfunction Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 7.1 Levofloxacin | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Quality of life: mental Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8.1 Ciprofloxacin | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Quality of life: physical Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 9.1 Ciprofloxacin | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Urinary symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 10.1 Ciprofloxacin | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Prostatitis symptoms: subgroup analysis (age) Show forest plot | 5 | 372 | Mean Difference (IV, Random, 95% CI) | ‐2.43 [‐4.72, ‐0.15] |
| 11.1 Mean age > 50 years old | 1 | 80 | Mean Difference (IV, Random, 95% CI) | 0.60 [‐3.79, 4.99] |
| 11.2 Mean age < 50 years old | 4 | 292 | Mean Difference (IV, Random, 95% CI) | ‐2.92 [‐5.37, ‐0.46] |
| 12 Adverse events: subgroup analysis (age) Show forest plot | 4 | 336 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.66, 1.55] |
| 12.1 Mean age > 50 years old | 1 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.46, 2.97] |
| 12.2 Mean age < 50 years old | 3 | 256 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.60, 1.57] |
| 13 Prostatitis symptoms: subgroup analysis (co‐interventions) Show forest plot | 5 | 372 | Mean Difference (IV, Random, 95% CI) | ‐2.43 [‐4.72, ‐0.15] |
| 13.1 Without co‐interventions | 2 | 167 | Mean Difference (IV, Random, 95% CI) | ‐1.57 [‐4.77, 1.63] |
| 13.2 With co‐interventions (alpha blockers) | 3 | 205 | Mean Difference (IV, Random, 95% CI) | ‐2.96 [‐6.28, 0.37] |
| 14 Adverse events: subgroup analysis (co‐interventions) Show forest plot | 4 | 336 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.66, 1.55] |
| 14.1 Without co‐interventions | 2 | 175 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.66, 1.55] |
| 14.2 With co‐interventions (alpha blockers) | 2 | 161 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 7 | 585 | Mean Difference (IV, Random, 95% CI) | ‐2.50 [‐3.74, ‐1.26] |
| 1.1 Corticosteroids | 1 | 158 | Mean Difference (IV, Random, 95% CI) | ‐2.82 [‐4.10, ‐1.54] |
| 1.2 Nonsteroidal anti‐inflammatory drugs | 5 | 369 | Mean Difference (IV, Random, 95% CI) | ‐2.56 [‐4.50, ‐0.62] |
| 1.3 Thiocolchicoside and ibuprofen | 1 | 58 | Mean Difference (IV, Random, 95% CI) | ‐1.5 [‐2.10, ‐0.90] |
| 2 Prostatitis symptoms: pain Show forest plot | 6 | 497 | Mean Difference (IV, Random, 95% CI) | ‐2.28 [‐4.08, ‐0.48] |
| 2.1 Corticosteroids | 1 | 158 | Mean Difference (IV, Random, 95% CI) | ‐1.67 [‐1.96, ‐1.38] |
| 2.2 Nonsteroidal anti‐inflammatory drugs | 3 | 265 | Mean Difference (IV, Random, 95% CI) | ‐1.80 [‐3.80, 0.20] |
| 2.3 Antileukotriene | 1 | 17 | Mean Difference (IV, Random, 95% CI) | 1.60 [‐5.79, 8.99] |
| 2.4 Thiocolchicoside and ibuprofen | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐5.2 [‐5.59, ‐4.81] |
| 3 Prostatitis symptoms: urinary Show forest plot | 6 | 497 | Mean Difference (IV, Random, 95% CI) | ‐0.75 [‐1.53, 0.03] |
| 3.1 Corticosteroids | 1 | 158 | Mean Difference (IV, Random, 95% CI) | ‐0.69 [‐0.88, ‐0.50] |
| 3.2 Nonsteroidal anti‐inflammatory drugs | 3 | 265 | Mean Difference (IV, Random, 95% CI) | ‐0.38 [‐0.64, ‐0.13] |
| 3.3 Antileukotriene | 1 | 17 | Mean Difference (IV, Random, 95% CI) | 0.60 [‐0.84, 2.04] |
| 3.4 Thiocolchicoside and ibuprofen | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐3.00 [‐3.47, ‐2.53] |
| 4 Prostatitis symptoms: quality of life Show forest plot | 6 | 497 | Mean Difference (IV, Random, 95% CI) | ‐1.25 [‐2.58, 0.08] |
| 4.1 Corticosteroids | 1 | 158 | Mean Difference (IV, Random, 95% CI) | ‐0.5 [‐0.75, ‐0.25] |
| 4.2 Nonsteroidal anti‐inflammatory drugs | 3 | 265 | Mean Difference (IV, Random, 95% CI) | ‐0.45 [‐0.90, 0.01] |
| 4.3 Antileukotriene | 1 | 17 | Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐4.86, 4.06] |
| 4.4 Thiocolchicoside and ibuprofen | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐4.5 [‐5.02, ‐3.98] |
| 5 Prostatitis symptoms: responder rate Show forest plot | 2 | 82 | Risk Ratio (M‐H, Random, 95% CI) | 1.44 [0.68, 3.03] |
| 5.1 Corticosteroids | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.25, 4.00] |
| 5.2 Nonsteroidal anti‐inflammatory drugs | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.67 [0.69, 4.04] |
| 6 Prostatitis symptoms: long term Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 6.1 Thiocolchicoside and ibuprofen | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Adverse events Show forest plot | 7 | 540 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.81, 2.00] |
| 7.1 Corticosteroids | 1 | 160 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.24, 102.53] |
| 7.2 Nonsteroidal anti‐inflammatory drugs | 4 | 303 | Risk Ratio (M‐H, Random, 95% CI) | 1.85 [0.79, 4.32] |
| 7.3 Antileukotriene | 1 | 17 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.23, 1.37] |
| 7.4 Thiocolchicoside and ibuprofen | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.83, 2.43] |
| 8 Urinary symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8.1 Nonsteroidal anti‐inflammatory drugs | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Prostatitis symptoms: subgroup analysis (co‐interventions) Show forest plot | 7 | 585 | Mean Difference (IV, Random, 95% CI) | ‐2.50 [‐3.74, ‐1.26] |
| 9.1 Without co‐interventions | 1 | 64 | Mean Difference (IV, Random, 95% CI) | ‐3.62 [‐4.85, ‐2.39] |
| 9.2 With co‐interventions | 6 | 521 | Mean Difference (IV, Random, 95% CI) | ‐2.29 [‐3.67, ‐0.90] |
| 10 Adverse events Show forest plot | 7 | 540 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.81, 2.00] |
| 10.1 Without co‐interventions | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.19, 20.97] |
| 10.2 With co‐interventions | 6 | 476 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.73, 2.15] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 5 | 320 | Mean Difference (IV, Random, 95% CI) | ‐5.02 [‐6.81, ‐3.23] |
| 1.1 Pollen extract vs. placebo | 1 | 137 | Mean Difference (IV, Random, 95% CI) | ‐2.5 [‐4.44, ‐0.56] |
| 1.2 Prolit Super Septo versus placebo | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐9.0 [‐16.81, ‐1.19] |
| 1.3 Calendula‐Curcuma versus placebo | 1 | 48 | Mean Difference (IV, Random, 95% CI) | ‐6.0 [‐7.28, ‐4.72] |
| 1.4 Quercetin (flavonoid) versus placebo | 1 | 28 | Mean Difference (IV, Random, 95% CI) | ‐5.80 [‐10.80, ‐0.80] |
| 1.5 Add‐on cranberry compared to standard care | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐5.40 [‐7.51, ‐3.29] |
| 2 Prostatitis symptoms: pain subscore Show forest plot | 5 | 431 | Mean Difference (IV, Random, 95% CI) | ‐1.42 [‐1.99, ‐0.85] |
| 2.1 Pollen extract vs. placebo | 2 | 296 | Mean Difference (IV, Random, 95% CI) | ‐1.30 [‐2.05, ‐0.55] |
| 2.2 Prolit Super Septo versus placebo | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐4.0 [‐13.31, 5.31] |
| 2.3 Quercetin (flavonoid) versus placebo | 1 | 28 | Mean Difference (IV, Random, 95% CI) | ‐2.8 [‐5.41, ‐0.19] |
| 2.4 Add‐on cranberry compared to standard care | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐1.40 [‐2.34, ‐0.46] |
| 3 Prostatitis symptoms: urinary symptoms Show forest plot | 5 | 431 | Mean Difference (IV, Random, 95% CI) | ‐0.99 [‐1.75, ‐0.23] |
| 3.1 Pollen Extract vs. Placebo | 2 | 296 | Mean Difference (IV, Random, 95% CI) | ‐0.49 [‐1.15, 0.16] |
| 3.2 Prolit Super Septo versus placebo | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐1.30 [‐6.15, 3.55] |
| 3.3 Quercetin (flavonoid) versus placebo | 1 | 28 | Mean Difference (IV, Random, 95% CI) | ‐1.5 [‐3.27, 0.27] |
| 3.4 Add‐on cranberry compared to standard care | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐1.80 [‐2.47, ‐1.13] |
| 4 Prostatitis symptoms: quality of life Show forest plot | 5 | 431 | Mean Difference (IV, Random, 95% CI) | ‐1.60 [‐2.30, ‐0.90] |
| 4.1 Pollen Extract vs. Placebo | 2 | 296 | Mean Difference (IV, Random, 95% CI) | ‐1.09 [‐1.66, ‐0.51] |
| 4.2 Prolit Super Septo versus placebo | 1 | 57 | Mean Difference (IV, Random, 95% CI) | ‐3.70 [‐9.12, 1.72] |
| 4.3 Quercetin (flavonoid) versus placebo | 1 | 28 | Mean Difference (IV, Random, 95% CI) | ‐1.90 [‐3.97, 0.17] |
| 4.4 Add‐on cranberry compared to standard care | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐2.2 [‐2.75, ‐1.65] |
| 5 Prostatitis symptoms: responder rate Show forest plot | 3 | 224 | Risk Ratio (M‐H, Random, 95% CI) | 1.78 [1.25, 2.52] |
| 5.1 Pollen extract vs. placebo | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [1.05, 2.05] |
| 5.2 Prolit Super Septo versus placebo | 1 | 57 | Risk Ratio (M‐H, Random, 95% CI) | 2.02 [1.23, 3.32] |
| 5.3 Quercetin (flavonoid) versus placebo | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.33 [1.14, 9.75] |
| 6 Adverse events Show forest plot | 7 | 540 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.54, 2.36] |
| 6.1 Pollen Extract vs. Placebo | 3 | 357 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.34, 3.79] |
| 6.2 Prolit Super Septo versus placebo | 1 | 57 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 6.3 Calendula‐Curcuma versus placebo | 1 | 48 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.13, 70.16] |
| 6.4 Quercetin (flavonoid) versus placebo | 1 | 28 | Risk Ratio (M‐H, Random, 95% CI) | 1.73 [0.18, 16.99] |
| 6.5 Add‐on cranberry compared to standard care | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 7 Sexual dysfunction Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 7.1 Calendula‐Curcuma versus placebo | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Urinary symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8.1 Pollen Extract vs. Placebo | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Prostatitis symptoms: subgroup analysis (co‐interventions) Show forest plot | 5 | 320 | Mean Difference (IV, Random, 95% CI) | ‐5.02 [‐6.81, ‐3.23] |
| 9.1 Without co‐interventions | 3 | 133 | Mean Difference (IV, Random, 95% CI) | ‐6.06 [‐7.28, ‐4.84] |
| 9.2 With co‐interventions | 2 | 187 | Mean Difference (IV, Random, 95% CI) | ‐3.92 [‐6.76, ‐1.08] |
| 10 Adverse events: subgroup analysis (co‐interventions) Show forest plot | 7 | 540 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.54, 2.36] |
| 10.1 Without co‐interventions | 4 | 191 | Risk Ratio (M‐H, Random, 95% CI) | 2.09 [0.33, 13.30] |
| 10.2 With co‐interventions | 3 | 349 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.34, 3.79] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Intraprostatic | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐25.80 [‐30.15, ‐21.45] |
| 1.2 Pelvic floor muscles | 1 | 29 | Mean Difference (IV, Random, 95% CI) | ‐2.6 [‐5.59, 0.39] |
| 2 Prostatitis symptoms: pain subscore Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Intraprostatic | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐14.63 [‐16.76, ‐12.50] |
| 2.2 Pelvic floor muscles | 1 | 29 | Mean Difference (IV, Random, 95% CI) | ‐1.70 [‐3.23, ‐0.17] |
| 3 Prostatitis symptoms: urinary subscore Show forest plot | 2 | 89 | Mean Difference (IV, Random, 95% CI) | ‐2.82 [‐7.40, 1.76] |
| 3.1 Intraprostatic | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐5.17 [‐6.72, ‐3.62] |
| 3.2 Pelvic floor muscles | 1 | 29 | Mean Difference (IV, Random, 95% CI) | ‐0.5 [‐1.85, 0.85] |
| 4 Prostatitis symptoms: quality of life Show forest plot | 2 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4.1 Intraprostatic | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Pelvic floor muscles | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse events Show forest plot | 2 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.25, 99.95] |
| 5.1 Intraprostatic | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 5.00 [0.25, 99.95] |
| 5.2 Pelvic floor muscle | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 6 Urinary symptoms Show forest plot | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐9.67 [‐13.97, ‐5.37] |
| 6.1 Intraprostatic | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐9.67 [‐13.97, ‐5.37] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Prostatitis symptoms: pain subscore Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Prostatitis symptoms: urinary subscore Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Prostatitis symptoms Show forest plot | 7 | 835 | Mean Difference (IV, Random, 95% CI) | ‐3.13 [‐4.99, ‐1.28] |
| 1.1 Prostant versus placebo | 2 | 164 | Mean Difference (IV, Random, 95% CI) | ‐3.93 [‐7.76, ‐0.10] |
| 1.2 Qianlieping (add‐on) versus medical treatment alone | 1 | 154 | Mean Difference (IV, Random, 95% CI) | ‐3.40 [‐5.03, ‐1.77] |
| 1.3 Qianlieantong (add‐on) versus medical treatment alone | 1 | 115 | Mean Difference (IV, Random, 95% CI) | ‐6.67 [‐8.14, ‐5.20] |
| 1.4 Yuleshu (add‐on) versus medical care alone | 1 | 88 | Mean Difference (IV, Random, 95% CI) | ‐1.41 [‐2.57, ‐0.25] |
| 1.5 Aike Mixture versus placebo | 1 | 74 | Mean Difference (IV, Random, 95% CI) | ‐3.59 [‐8.24, 1.06] |
| 1.6 Bazhengsan versus placebo | 2 | 162 | Mean Difference (IV, Random, 95% CI) | ‐1.53 [‐9.28, 6.23] |
| 1.7 Prostatitis Decoction versus placebo | 1 | 78 | Mean Difference (IV, Random, 95% CI) | 1.07 [‐3.78, 5.92] |
| 2 Prostatitis symptoms: pain subscore Show forest plot | 5 | 585 | Mean Difference (IV, Random, 95% CI) | ‐1.49 [‐2.44, ‐0.53] |
| 2.1 Prostant versus placebo | 2 | 164 | Mean Difference (IV, Random, 95% CI) | ‐1.42 [‐2.51, ‐0.34] |
| 2.2 Qianlieantong versus medical treatment alone | 1 | 115 | Mean Difference (IV, Random, 95% CI) | ‐2.54 [‐3.24, ‐1.84] |
| 2.3 Yuleshu versus medical care | 1 | 88 | Mean Difference (IV, Random, 95% CI) | ‐2.52 [‐3.64, ‐1.40] |
| 2.4 Aike Mixture versus placebo | 1 | 74 | Mean Difference (IV, Random, 95% CI) | ‐2.21 [‐4.60, 0.18] |
| 2.5 Bazhengsan versus placebo | 1 | 66 | Mean Difference (IV, Random, 95% CI) | 1.35 [‐1.22, 3.92] |
| 2.6 Prostatitis Decoction versus placebo | 1 | 78 | Mean Difference (IV, Random, 95% CI) | 1.02 [‐1.52, 3.56] |
| 3 Prostatitis symptoms: urinary subscore Show forest plot | 4 | 497 | Mean Difference (IV, Random, 95% CI) | ‐0.78 [‐1.50, ‐0.06] |
| 3.1 Prostant versus placebo | 2 | 164 | Mean Difference (IV, Random, 95% CI) | ‐1.62 [‐3.37, 0.13] |
| 3.2 Qianlieantong versus medical treatment alone | 1 | 115 | Mean Difference (IV, Random, 95% CI) | ‐0.39 [‐1.02, 0.24] |
| 3.3 Aike Mixture versus placebo | 1 | 74 | Mean Difference (IV, Random, 95% CI) | ‐0.84 [‐2.31, 0.63] |
| 3.4 Bazhengsan versus placebo | 1 | 66 | Mean Difference (IV, Random, 95% CI) | 0.25 [‐1.35, 1.85] |
| 3.5 Prostatitis Decoction versus placebo | 1 | 78 | Mean Difference (IV, Random, 95% CI) | ‐0.26 [‐1.73, 1.21] |
| 4 Prostatitis symptoms: quality of life Show forest plot | 2 | 306 | Mean Difference (IV, Random, 95% CI) | ‐0.97 [‐2.14, 0.20] |
| 4.1 Prostant versus placebo | 1 | 88 | Mean Difference (IV, Random, 95% CI) | ‐1.91 [‐1.00, ‐0.82] |
| 4.2 Aike Mixture versus placebo | 1 | 74 | Mean Difference (IV, Random, 95% CI) | ‐1.10 [‐3.36, 1.16] |
| 4.3 Bazhengsan versus placebo | 1 | 66 | Mean Difference (IV, Random, 95% CI) | 0.66 [‐1.65, 2.97] |
| 4.4 Prostatitis Decoction versus placebo | 1 | 78 | Mean Difference (IV, Random, 95% CI) | ‐0.34 [‐2.59, 1.91] |
| 5 Adverse events Show forest plot | 4 | 584 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.22, 8.02] |
| 5.1 Prostant versus placebo | 2 | 212 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.03, 19.50] |
| 5.2 Qianlieping versus medical treatment alone | 1 | 154 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.3 Aike Mixture versus placebo | 1 | 74 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.4 Bazhengsan versus placebo | 1 | 66 | Risk Ratio (M‐H, Random, 95% CI) | 4.33 [0.26, 72.96] |
| 5.5 Prostatitis Decoction versus placebo | 1 | 78 | Risk Ratio (M‐H, Random, 95% CI) | 2.43 [0.14, 42.93] |
| 6 Sexual dysfunction Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 6.1 Yuleshu versus medical care | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Anxiety and depression: anxiety Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 7.1 Yuleshu versus medical care | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Anxiety and depression: depression Show forest plot | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8.1 Yuleshu versus medical care | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |