Transfusiones de plasma antes de las punciones lumbares y la colocación de catéteres epidurales para pacientes con coagulación anormal
Resumen
Antecedentes
La inserción de una aguja de punción lumbar o de un catéter epidural puede asociarse con hemorragia antes y después de la intervención. Los pacientes que requieren dicha intervención pueden presentar trastornos de la coagulación como resultado de la enfermedad subyacente, de las comorbilidades o de los efectos del tratamiento. En algunos centros de atención, la práctica clínica implica mitigar el riesgo de hemorragia en estos pacientes mediante la transfusión de plasma de forma profiláctica para corregir las deficiencias de los factores de coagulación antes de la intervención. Sin embargo, la transfusión de plasma no está exenta de riesgos, y aún no es posible precisar si esta intervención se asocia con tasas reducidas de hemorragia u otros resultados clínicamente significativos.
Objetivos
Evaluar el efecto de diferentes regímenes de transfusión de plasma profilácticos antes de la inserción de una aguja de punción lumbar o un catéter epidural en pacientes con coagulación anormal.
Métodos de búsqueda
Se realizaron búsquedas de ensayos controlados aleatorios (ECA), ensayos controlados no aleatorios (ECNA) y estudios controlados de antes y después (CAD) en CENTRAL (la Biblioteca Cochrane 2016; Número 11), MEDLINE (desde 1946), Embase (desde 1974), CINAHL (desde 1937), la Transfusion Evidence Library (desde 1950) y otras cinco bases de datos electrónicas así como ClinicalTrials.gov y la World Health Organization International Clinical Trials Registry Platform (ICTRP) para obtener ensayos en curso hasta el 9 de enero de 2017.
Criterios de selección
Se planificó incluir ECA, ECNA y CAD sobre las transfusiones de plasma administradas para prevenir las hemorragias en pacientes de cualquier edad con una coagulopatía que requerían la inserción de una aguja de punción lumbar o un catéter epidural. Si se hubiesen identificado, se habrían excluido los estudios no controlados, los estudios transversales y los estudios de casos y controles. Sólo se habrían incluido los ECA con asignación al azar por grupos, los ensayos no aleatorios por grupos, y los CAD con al menos dos sitios de intervención y dos sitios de control. En los estudios con sólo una intervención o un sitio de control, la intervención (o comparación) se ve completamente afectada por factores de confusión relacionados con el sitio de estudio, lo cual dificulta atribuir cualquier diferencia observada a la intervención en lugar de a otras variables propias del sitio.
Se planificó excluir a los pacientes con hemofilia debido a que deben ser tratados con el concentrado del factor apropiado. También se planificó excluir a los pacientes que recibían warfarina debido a que las guías recomiendan la administración del concentrado del complejo de protrombina para la reversión de urgencia de la warfarina.
Obtención y análisis de los datos
Se utilizaron los procedimientos metodológicos estándar previstos por la Colaboración Cochrane.
Resultados principales
No se identificaron ECA, ECNA o CAD completados ni en curso.
Conclusiones de los autores
No existe evidencia de ECA, ECNA y CAD para determinar si se requieren transfusiones de plasma antes de la inserción de una aguja de punción lumbar o un catéter epidural y, si se necesitan transfusiones de plasma, cuál es el grado de coagulopatía al cual deben administrarse. Se necesitaría diseñar un estudio con al menos 47 030 participantes para poder detectar un aumento en el número de pacientes que presentan hemorragia después de la punción lumbar o el anestésico epidural de uno en 1000 a dos en 1000.
PICO
Resumen en términos sencillos
Transfusiones de plasma antes de la inserción de agujas de punción lumbar en pacientes con coagulación anormal
Pregunta de la revisión
Se evaluó la evidencia acerca de si los pacientes con coagulación anormal (coagulación deficiente) requieren una transfusión de plasma antes de la inserción de una aguja de punción lumbar o un catéter epidural, y si es así, cuál es el grado de coagulación anormal al que se necesita una transfusión de plasma.
Antecedentes
Los pacientes con coagulación anormal pueden requerir una punción lumbar o anestesia epidural. La punción lumbar se realiza generalmente mediante la inserción de una aguja entre los huesos (vértebras) de la parte baja de la columna vertebral en el líquido que rodea la médula espinal (el grupo de nervios que recorre la columna vertebral y conecta el cerebro con el cuerpo). Las punciones lumbares se realizan para obtener una muestra de dicho líquido o para administrar el tratamiento en el líquido (quimioterapia o un anestésico). La aguja de punción lumbar se retira inmediatamente después de que se haya tomado la muestra de líquido o de que se haya administrado el tratamiento. La epidural incluye la inserción de una aguja de diámetro más grande que una aguja de punción lumbar. La aguja epidural atraviesa los mismos tejidos que la aguja de punción lumbar, aunque se detiene apenas penetra el saco de líquido alrededor de la médula espinal. En este caso, el tratamiento se inyecta en el espacio inmediatamente fuera del saco de líquido (llamado espacio epidural). A menudo se pasa un tubo pequeño (un catéter epidural) a través de la aguja epidural y se deja en posición para poder administrar medicación anestésica local adicional. La práctica actual en muchos países es administrar transfusiones de plasma para prevenir hemorragias graves debidas a la intervención cuando los análisis de sangre que evalúan la coagulación son anormales. Aunque el riesgo de hemorragia parece ser muy bajo, si se presentan hemorragias, pueden ser muy graves. La corrección de las anomalías en la coagulación con transfusiones de plasma no está exenta de riesgos, y no puede precisarse si esta práctica es beneficiosa o perjudicial. Los pacientes pueden estar expuestos a los riesgos de una transfusión plaquetaria sin evidentes efectos clínicos beneficiosos. El riesgo de que una transfusión de plasma pueda causar efectos perjudiciales graves, como la transmisión de una infección o problemas respiratorios graves, es muy bajo.
Características de los estudios
Se efectuaron búsquedas en las bases de datos científicas para obtener estudios clínicos (ensayos controlados aleatorios y estudios no aleatorios bien diseñados) de pacientes de cualquier edad con coagulación anormal que requerían una punción lumbar o anestesia epidural. La evidencia está actualizada hasta el 9 enero de 2017. En esta revisión, no se encontraron estudios relevantes.
Resultados clave
No hay resultados porque no se encontraron estudios relevantes. El riesgo de hemorragia después de una punción lumbar o epidural es muy bajo y se necesitaría un estudio muy amplio (con aproximadamente 50 000 pacientes) para saber si es útil la transfusión de plasma antes de estas intervenciones.
Calidad de la evidencia
No se evaluó la calidad de la evidencia porque no se incluyeron estudios.
Authors' conclusions
Background
Description of the condition
Abnormal coagulation refers to the condition in which the blood's ability to clot is impaired (Hunt 2014). People requiring insertion of a lumbar puncture (LP) needle or an epidural catheter often develop abnormal coagulation as a consequence of their underlying illness, co‐morbidities or the effects of treatment.
People requiring LPs and epidurals can have a variety of conditions and include people with liver failure, people who are critically ill and people requiring chemotherapy (Doherty 2014).
An LP is usually performed by inserting a needle into the lower back (underneath the spinal L4 bony process) (Williams 2008). A diagnostic LP is an invasive procedure to obtain samples of cerebrospinal fluid (CSF) (Doherty 2014). CSF is the fluid that bathes and protects the brain and spinal cord. The CSF obtained can then be used for the investigation of haematological malignancies (Vavricka 2003), subarachnoid haemorrhages, meningitis (Riordan 2002), or neurological disorders. LPs are performed by doctors or specially trained nurses. Therapeutic LPs administer drugs into the CSF. This can be for the administration of therapeutics such as chemotherapy or antibiotics, or administration of local anaesthetic to the nerves of the lower spine (spinal anaesthetic) (Doherty 2014). This usually involves inserting a fine needle into the lower back, administration of the therapeutic agent and then removal of the needle (Ng 2004).
An epidural catheter is inserted to administer anaesthetic. Epidural anaesthesia typically involves inserting a larger diameter needle than a spinal needle. The epidural needle passes through the same tissues as a spinal needle but stops short of penetrating the dura (tissue sac that contains CSF). An epidural catheter is often passed through the needle and left in position so that additional local anaesthetic medications can be administered (Ng 2004).The most common indication for epidural anaesthesia is in pregnant women to aid pain relief during labour (Venn 2015). However, epidural anaesthesia can also be used in postoperative pain management especially for people with lower limb ischaemia (narrowing or blockage of the arteries, which markedly reduces blood flow to the legs and feet) (Venn 2015), and people undergoing thoracic surgery (Mendola 2009), as alternatives to general anaesthesia.
In the general population, the risk of a spinal haematoma is very low 0.85 per 100,000 (95% confidence interval (CI) 0 to 1.8 per 100,000) (Cook 2009). The risk varies depending upon the type of person undergoing the procedure (1 in 200,000 epidural anaesthetic procedures during labour to 1 in 3600 epidural anaesthetic procedures in older women having knee surgery) (Li 2010; Moen 2004; Ruppen 2006; Vandermeulen 1994). Risk factors for major bleeding are multifactorial and include: increasing age (the procedure is more difficult in older people due to changes to the spine that occur with age), low platelet count, abnormal coagulation (including anticoagulant medication) and traumatic needle or catheter insertion (Erbay 2014; Li 2010; Moen 2004; Vandermeulen 1994). Performing an LP or administration of epidural anaesthesia is a relative contraindication in people with abnormal coagulation due to this perceived higher risk of complications (AAGBI 2013). However, overall, there are no current reliable estimates of the risks of adverse effects such as spinal haematomas in people with abnormal coagulation (AAGBI 2013; Cook 2009).
A large national study of fresh frozen plasma (FFP) use in critical illness reported that 30% of people admitted to the intensive care unit (ICU) developed an abnormality of coagulation (Walsh 2010). The aetiology of coagulopathy in critical illness is complex and multi‐factorial; sepsis, haemodilution, haemorrhage, disseminated intravascular coagulation, hepatic and renal disease and anti‐coagulant medication are all implicated (Hunt 2014). The causes of abnormal coagulation in people who are not critically ill are similarly broad.
Description of the intervention
Current practice in many countries is to correct abnormal coagulation tests ((prolonged prothrombin time (PT) or elevated international normalised ratio (INR)) with transfusion of plasma prior to insertion of an LP needle or epidural catheter, in order to mitigate the risk of serious peri‐ or post‐procedural bleeding (Moiz 2006; NICE 2015; Vlaar 2009; Yaddanapudi 2014).
Plasma is the liquid component of blood (Benjamin 2012). FFP refers to plasma that is frozen within eight hours of removal to ‐30°C, whereas frozen plasma (F24) is that which is frozen within 24 hours. Both contain concentrations of clotting factors equivalent to those found in in vivo blood, although the levels of factor V and VIII fall rapidly on thawing (Stanworth 2007). Current recommendations regarding the correction of coagulopathy prior to invasive procedures reflect expert opinion rather than high‐quality evidence from randomised controlled trials (RCTs) (AAGBI 2013; NICE 2015). An INR greater than or equal to 1.5 is frequently advocated as the threshold above which patients should undergo correction of coagulopathy prior to insertion of an LP needle or epidural catheter (Hunt 2014; NICE 2015). Whilst the use of standard laboratory tests of coagulation to assess bleeding has been criticised, an INR over 1.5 demarcates the level above which the activity of some coagulation factors falls to less than 50% (Hall 2014). An alternative approach to transfusing based on an INR threshold (which only detects low coagulation factor levels) is to use a test such as rotational thromboelastometry (ROTEM) or thromboelastography (TEG) that assesses how well a blood clot forms in whole blood (haemostasis) (Kinnaird 2013). ROTEM and TEG not only assess coagulation factor function, but also platelet function, strength of the clot and whether the clot is rapidly broken down (Whiting 2014).
Recent studies report that 15% to 26% of non‐bleeding critically ill patients receive prophylactic FFP transfusions prior to an invasive procedure (Dara 2005; Stanworth 2010; Stanworth 2011). However, there remains substantial heterogeneity in clinicians' views about the effectiveness of this intervention, with doubts over its effectiveness and the balance of the risk‐benefit ratio (Watson 2011).
How the intervention might work
Plasma transfusion is administered to people with abnormal coagulation in order to correct multiple clotting factor deficiencies and therefore reduce the incidence of bleeding. A dose of at least 10 mL to 15 mL/kg is required to significantly improve the INR (O'Shaughnessy 2004). However, clinical studies indicate that the INR is often minimally reduced following administration of FFP, especially when only modestly increased pre‐transfusion (Abdel‐Wahab 2006; Stanworth 2011). It remains unclear whether plasma transfusion in people with abnormal coagulation, despite improving standard laboratory tests of coagulation, reduces the incidence of clinically important bleeding or improves other meaningful patient‐oriented outcomes such as mortality.
Risks associated with the intervention
If plasma transfusions are ineffective, people are exposed to the risks associated with plasma transfusion unnecessarily. These include transfusion‐associated lung injury (Khan 2007; Rana 2006), transfusion‐associated circulatory overload (Narick 2011), multi‐organ failure (Watson 2009), and sepsis (Sarani 2008).
The requirement to administer plasma to correct coagulopathy prior to insertion of an LP needle or epidural catheter may additionally delay the start of a treatment. This could lead to unnecessary delays and cancellations of procedures, which may be time‐critical in an emergency situation. Delays in initiating treatment may lead to poorer patient outcomes (increased morbidity and mortality). It may also mean that a person does not receive regional anaesthesia, but instead receives a general anaesthetic that may place them at greater risk of complications (Amini 2015; Sanford 2015).
Why it is important to do this review
It is uncertain whether plasma transfusions are effective at preventing bleeding in patients with abnormal coagulation undergoing an invasive procedure (Desborough 2012; Hunt 2014; Segal 2005; Stanworth 2007). If effective, the INR threshold above which plasma transfusions are clinically effective is also uncertain. Wide variation in the use of FFP prior to invasive procedures exists, indicating significant clinician uncertainty and potentially exposing patients to varying risks (Watson 2011).
Previous systematic reviews have either only assessed the evidence from RCTs (Stanworth 2004; Yang 2012); only assessed the evidence associated with one or two outcomes (all‐cause mortality and multi‐organ failure) (Murad 2010); or were performed more than 10 years ago (Segal 2005; Stanworth 2004). In these previous systematic reviews, there was no RCT evidence for the use of plasma transfusions prior to insertion of an LP needle or epidural catheter (Murad 2010; Segal 2005; Stanworth 2004; Yang 2012). This review addresses an important question for clinicians and the best available evidence needs to be summarised. This review will therefore summarise the evidence from a broad range of studies and include a broad range of outcomes.
Objectives
To assess the effect of different prophylactic plasma transfusion regimens prior to insertion of a lumbar puncture needle or epidural catheter in people with abnormal coagulation.
Methods
Criteria for considering studies for this review
Types of studies
We planned to include randomised controlled trials (RCTs), non‐randomised controlled trials (non‐RCTs) and controlled before‐after studies (CBAs), irrespective of language or publication status. If identified, we planned to exclude uncontrolled studies, cross‐sectional studies and case‐control studies.
We would only have included cluster‐RCTs, non‐randomised cluster trials, and CBAs with at least two intervention sites and two control sites. This was because in studies with only one intervention or control site, the intervention (or comparison) is completely confounded by study site making it difficult to attribute any observed differences to the intervention rather than to other site‐specific variables.
Types of participants
People, of any age, with abnormal coagulation (as defined by the included studies) requiring insertion of a lumbar puncture needle or epidural catheter.
If identified, we would not have included studies involving people with haemophilia as they should be treated with the appropriate factor concentrate (WFH 2012). Similarly, we would not have included studies involving people on warfarin as guidelines recommend the use of prothrombin complex concentrate for emergency reversal of warfarin (Keeling 2011; Tran 2013).
Types of interventions
Comparison 1: Plasma transfusion (for example, when an international normalised ratio (INR) is 1.5 or above; INR 2 or above; INR 3 or above; or other study‐specified INR or prothrombin time (PT) ratio threshold; or thromboelastography (TEG)‐guided) versus no plasma transfusion.
Comparison 2: Plasma transfusion when an INR is at a higher threshold (for example, INR 2 or above or INR > 3 or TEG‐guided) versus plasma transfusion when INR is at a lower threshold (for example INR 1.5 or above).
Types of outcome measures
Primary outcomes
-
Major procedure‐related bleeding within 24 hours of the procedure (as defined by 24 hours after removal of lumbar puncture needle or catheter in the case of epidural anaesthesia). For example: spinal haematoma; intraventricular, intracerebral or subarachnoid haemorrhage; or major bleeding (not further defined), as reported by individual studies.
-
Serious adverse events:
-
transfusion‐related complications within 24 hours of the procedure (including transfusion‐related acute lung injury (TRALI), transfusion‐transmitted infection, transfusion‐associated circulatory overload (TACO), transfusion‐associated dyspnoea (TAD), acute transfusion reactions);
-
venous and arterial thromboembolism (including deep vein thrombosis; pulmonary embolism; stroke; myocardial infarction) (up to 30 days);
-
lumbar puncture (LP)‐related or epidural anaesthetic‐related complications within seven days of the procedure (infection, headache, cerebral herniation, neurological symptoms such as radicular pain or numbness, back pain).
-
Secondary outcomes
-
All‐cause mortality (up to 24 hours and up to 30 days).
-
Minor LP‐related or epidural anaesthetic‐related bleeding within 24 hours of the procedure (defined as prolonged bleeding at the insertion site that only required treatment with a pressure bandage) or minor bleeding (not further defined) as reported by individual studies.
-
Total number of days in hospital.
-
Proportion of patients receiving plasma transfusions within 24 hours of the procedure.
-
Change in baseline coagulation test abnormalities PT ratio, INR or as defined by the study within 24 hours after the plasma transfusion.
-
Quality of life, as defined by the individual studies.
Search methods for identification of studies
The Systematic Review Initiative’s Information Specialist (CD) formulated the search strategies in collaboration with the Cochrane Haematological Malignancies Group.
Electronic searches
We searched the following databases.
-
Cochrane Central Register of Controlled Trials (CENTRAL, the Cochrane Library, 2016, Issue 11) (http://www.cochranelibrary.com/) (Appendix 1).
-
MEDLINE (OvidSP, Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations, Ovid MEDLINE 1946 to 9th January 2017) (Appendix 2).
-
Embase (OvidSP, 1974 to 9th January 2017) (Appendix 3).
-
CINAHL (EBSCOHost, 1937 to 9th January 2017) (Appendix 4).
-
PubMed (e‐publications ahead of print only) (http://www.ncbi.nlm.nih.gov/pubmed) (Appendix 5).
-
Transfusion Evidence Library (1950 to 9th January 2017) (www.transfusionevidencelibrary.com), this includes a search of grey literature (Appendix 6).
-
LILACS (1980 to 9th January 2017) (http://lilacs.bvsalud.org/en/) (Appendix 7).
-
IndMed (1986 to 9th January 2017) (http://indmed.nic.in/indmed.html) (Appendix 8).
-
KoreaMed (1958 to 9th January 2017) (http://koreamed.org/) (Appendix 9).
-
PakMediNet (1995 to 9th January 2017) (http://www.pakmedinet.com/) (Appendix 10).
-
Web of Science: Conference Proceedings Citation Index‐Science (CPCI‐S) (Thomson Reuters, 1990 to 9th January 2017) (Appendix 11).
-
Transfusion Evidence Library (1950 to 9th January 2017) (www.transfusionevidencelibrary.com), this includes a search of grey literature (Appendix 6).
-
LILACS (1980 to 9th January 2017) (http://lilacs.bvsalud.org/en/) (Appendix 7).
-
IndMed (1986 to 9th January 2017) (http://indmed.nic.in/indmed.html) (Appendix 8).
-
KoreaMed (1958 to 9th January 2017) (http://koreamed.org/) (Appendix 9).
-
PakMediNet (1995 to 9th January 2017) (http://www.pakmedinet.com/) (Appendix 10).
-
Web of Science: Conference Proceedings Citation Index‐Science (CPCI‐S) (Thomson Reuters, 1990 to 9th January 2017) (Appendix 11).
We searched for ongoing trials in the following clinical trial registers to 9th January 2017.
-
ClinicalTrials.gov (https://www.clinicaltrials.gov/) (Appendix 12).
-
World Health Organization International Clinical Trials Registry Platform (ICTRP) (http://apps.who.int/trialsearch/) (Appendix 13).
We combined searches in MEDLINE and Embase with the recommended Cochrane RCT search filters (Lefebvre 2011) and with systematic review and observational studies filters based on those of the Scottish Intercollegiate Guidelines Network (SIGN) (www.sign.ac.uk/methodology/filters.html). Searches in CINAHL were combined with SIGN systematic review, observational studies and RCT filters. We did not limit searches by language, year of publication or publication type.
If we had identified studies for inclusion we had planned to search MEDLINE (Ovid) for errata or retraction statements for the reports of these studies.
Searching other resources
We conducted handsearching of the reference lists of any relevant systematic reviews to identify further relevant studies. For future iterations of this review, we will make contact with lead authors of relevant studies to identify any unpublished material, missing data or information regarding ongoing studies.
Data collection and analysis
We summarised data in accordance with standard Cochrane methodologies.
Selection of studies
We selected studies with reference to the methods outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). The Systematic Review Initiative’s Information Specialist (CD) initially screened all search hits for relevance against the eligibility criteria and discarded all those that were clearly irrelevant. Thereafter, two review authors (LE, MD) independently screened all the remaining references for relevance against the full eligibility criteria.
Full‐text papers were retrieved for all references for which a decision on eligibility could not be made from title and abstract alone. We did not request additional information from study authors because it was not necessary to assess the eligibility for inclusion of individual studies. The two review authors discussed the results of study selection and resolved any discrepancies between themselves without the need for a third review author.
The results of study selection were reported using a PRISMA flow diagram (Moher 2009). We recorded the reasons for excluding studies based on full‐text assessment and added those to the Characteristics of excluded studies table.
We planned to collate multiple reports of one study, so that the study, and not the report, was the unit of analysis.
Data extraction and management
As recommended in the Cochrane Handbook for Systematic Reviews of Interventions, two review authors (LE, MD) planned to independently extract data onto standardised forms and perform a cross‐check (Higgins 2011a). However, no completed or ongoing study was included in this review.
We planned to extract the following information for each study.
-
Source: Study ID; report ID; review author ID; date of extraction; ID of author checking extracted data; citation of paper; contact authors details.
-
General study information: Publication type; study objectives; funding source; conflict of interest declared; other relevant study publication reviewed.
-
Study details and methods: Location; country; setting; number of centres; total study duration; recruitment dates; length of follow‐up; power calculation; primary analysis (and definition); stopping rules; method of sequence generation; allocation concealment; blinding (of clinicians, participants and outcome assessors); any other concerns regarding bias; inclusion and exclusion criteria.
-
Characteristics of interventions: Number of study arms; description of experimental arm; description of control arm;and other relevant information.
-
Characteristics of participants: Age; gender; primary diagnosis; subgroup classification of primary disease type where appropriate, severity of primary disease, where appropriate, prognostic classification of primary disease where appropriate; additional therapy received; risk of alloimmunisation; baseline haematology laboratory parameters; confounders reported.
-
Participant flow: Total number screened for inclusion; total number recruited; total number excluded; total number allocated to each study arm; total number analysed (for review outcomes); number of allocated patients who received planned treatment; number of dropouts with reasons (percentage in each arm); protocol violations; missing data.
-
Outcomes: Major procedure‐related bleeding within 24 hours of the procedure; serious adverse events (transfusion‐related complications within 24 hours of the procedure; venous and arterial thromboembolism; LP‐related or epidural anaesthetic‐related complications within seven days of the procedure); all‐cause mortality (up to 24 hours and up to 30 days); minor LP‐related or epidural anaesthetic‐related bleeding within 24 hours of the procedure; total number of days in hospital; proportion of patients receiving plasma transfusions within 24 hours of the procedure; venous and arterial thromboembolism; change in baseline coagulation test abnormalities; quality of life, as defined by the individual studies.
-
For interventional cohort and pre‐post single arm or multiple arms studies, we also planned to collect data if available on: confounding factors, the comparability of groups on confounding factors; methods used to control for confounding and on multiple effect estimates (both unadjusted and adjusted estimates) as recommended in chapter 13 of theCochrane Handbook of Systematic Reviews of Interventions (Reeves 2011).
Assessment of risk of bias in included studies
Randomised controlled trials (RCTs)
We planned to assess the risk of bias for all included RCTs using the Cochrane 'Risk of bias' tool according to chapter eight of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). However, no completed study was included in this review.
In future updates of this review, if there are included RCTs, two review authors will work independently to assess each element of potential bias listed below as 'high', 'low' or 'unclear' risk of bias. We will report a brief description of the judgement statements upon which the authors have assessed potential bias in the Characteristics of included studies table. We will ensure that a consensus on the degree of risk of bias is met through comparison of the review authors’ statements and where necessary, through consultation with a third review author (SS). We will use Cochrane's tool for assessing risk of bias, that will include the following domains.
Selection bias
We will describe for each included study if and how the allocation sequence was generated and if allocation was adequately concealed prior to assignment. We will also describe the method used to conceal the allocation sequence in detail and determine if intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment.
Performance bias
We will describe for each included study, where possible, if the study participants and personnel were adequately blinded from knowledge of which intervention a participant received. We will judge studies as low risk of bias if they were blinded, or if we judge that lack of blinding could not have affected the results.
Detection bias:
Was blinding of the outcome assessors effective in preventing systematic differences in the way in which the outcomes were determined?
Attrition bias
We will describe for each included study the attrition bias due to amount, nature or handling of incomplete outcome data. We will also try to evaluate whether intention‐to‐treat (ITT) analysis was performed or could be performed from published information.
Reporting bias
We will describe for each included study the possibility of selective outcome reporting bias.
Other issues
Was the study apparently free of other problems that could put it at risk of bias?
We will summarise the risk of bias for each key outcome for each included study. We will judge studies with at least one domain of high risk at high risk of bias overall etc.
Non‐randomised studies
We planned to use ROBINS‐I tool (formerly known as ACROBAT‐NRSI) to rate the quality of non‐randomised controlled trials (non‐RCTs) and controlled before‐after studies (CBAs) studies (Sterne 2014). This tool is based on the Cochrane 'Risk of bias' tool for rating the quality of RCTs (Higgins 2011c). The tool covers seven domains and the quality of evidence is rated as low, moderate, serious, critical or no information (see Appendix 14 for a copy of the tool), and uses signalling questions for the assessment of:
-
Bias due to confounding;
-
Bias in the selection of participants;
-
Bias in measurement of interventions;
-
Bias due to departure from intended interventions;
-
Bias due to missing data;
-
Bias in measurement of outcomes;
-
Bias in the selection of the reported result.
However, no completed study was included in this review.
We planned to resolve disagreements on the assessment of quality of an included study by discussion until we reach consensus or failing that by consulting a third review author.
We pre‐specified the following main potential confounding factors.
-
Primary diagnosis of patient (e.g. liver disease; critical illness; pregnancy).
-
Age: variability in the age of patients included, e.g. paediatric (less than 16 years) versus adult (> 16 years) versus older adult (> 60 years).
-
Gender: male to female ratio.
-
Previous severe bleeding (e.g. World Health Organization (WHO) grade 3 or 4 or equivalent).
Measures of treatment effect
We did not perform any of the planned analyses because no completed study was included in this review.
In future updates of this review we will perform the following.
-
Randomised controlled trials (RCTs)
-
For continuous outcomes, we will record the mean, standard deviation (SD) and total number of participants in both the treatment and control groups. For dichotomous outcomes, we will record the number of events and the total number of participants in both the treatment and control groups.
-
For continuous outcomes using the same scale, we will perform analyses using the mean difference (MD) with 95% confidence intervals (CIs). If continuous outcomes are reported using different scales we will use standardised mean difference (SMD).
-
If available, we will extract and report hazard ratios (HRs) for time‐to‐event‐data (mortality or time in hospital) data. If HRs are not available, we will make every effort to estimate as accurately as possible the HR using the available data and a purpose‐built method based on the Parmar and Tierney approach (Parmar 1998; Tierney 2007). If sufficient studies provide HRs, we will use HRs in favour of risk ratios (RRs) or MDs in a meta‐analysis, but for completeness, we will also perform a separate meta‐analysis of data from studies providing only RRs or MDs for the same outcome.
-
For dichotomous outcomes, we will report the pooled RR with a 95% CI. (Deeks 2011). Where the number of observed events is small (< 5% of sample per group), and where trials have balanced treatment groups, we will report the Peto’s Odds Ratio (OR) with 95% CI (Deeks 2011).
-
For cluster‐randomised trials, we will extract and report direct estimates of the effect measure (e.g. RR with a 95% CI) from an analysis that accounts for the clustered design. We will obtain statistical advice (MT) to ensure the analysis is appropriate. If appropriate analyses are not available, we will make every effort to approximate the analysis following the recommendations in Chapter 16 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c).
-
If data allow, we will undertake quantitative assessments using Review Manager 5 (RevMan 2014).
-
-
Non‐randomised studies
-
For dichotomous outcomes, if available we will extract and report the RR with a 95% CI from statistical analyses adjusting for baseline differences (such as Poisson regressions or logistic regressions) or the ratio of RRs (i.e. the RR post intervention/RR pre intervention).
-
For continuous variables, if available we will extract and report the absolute change from a statistical analysis adjusting for baseline differences (such as regression models, mixed models or hierarchical models), or the relative change adjusted for baseline differences in the outcome measures (i.e. the absolute post‐intervention difference between the intervention and control groups, as well as the absolute pre‐intervention difference between the intervention and control groups/the post‐intervention level in the control group) (EPOC 2015).
-
If data allow, we will undertake quantitative assessments using Review Manager 5 (RevMan 2014).
-
-
All studies
-
Where appropriate, we will report the number needed to treat for an additional beneficial outcome (NNTB) and the number needed to treat for an additional harmful outcome (NNTH) with 95% CIs.
-
If we cannot report the available data in any of the formats described above, we will perform a narrative report, and if appropriate, we will present the data in tables.
-
Unit of analysis issues
We planned to treat any unit of analysis issues in accordance with the advice given in Chapter 16 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c). However, no completed study was identified in this review and there were therefore no unit of analysis issues.
Dealing with missing data
We did not need to contact any study authors directly to enable us to make a decision on whether a study should be excluded.
Assessment of heterogeneity
We did not perform any of the planned analyses because no completed study was included in this review.
In future updates of this review we will:
-
Combine the data to perform a meta‐analysis, if the clinical and methodological characteristics of individual studies are sufficiently homogeneous. We will analyse the data in RCTs, non‐RCTs, and CBA studies separately;
-
Evaluate the extent of heterogeneity by visual inspection of forest plots as well as by utilising statistical methods;
-
Assess statistical heterogeneity of treatment effects between studies using a Chi2 test with a significance level at P < 0.1. We will use the I2 statistic to quantify the degree of potential heterogeneity and classify it as low if the I2 is ≤ 50%, moderate if the I2 is 50% to 80% or considerable if the I2 is > 80%. We will use the random‐effects model for low to moderate heterogeneity. If statistical heterogeneity is considerable, and we cannot identify a cause for the heterogeneity, the overall summary statistic will not be reported. Potential causes of heterogeneity will be assessed by sensitivity and subgroup analyses (Deeks 2011).
Assessment of reporting biases
We were unable to perform a formal assessment of potential publication bias (small‐trial bias) by generating a funnel plot and statistically test using a linear regression test (Sterne 2011), because there were no completed trials within this review.
Data synthesis
We planned to perform analyses according to the recommendations of Chapter 9 of the Cochrane Handbook for Systematic Reviews of Interventions, using aggregated data for analysis (Deeks 2011). We did not perform any of the planned analyses because no completed study was included in this review.
In future updates of this review we will perform the following:
-
If studies are sufficiently homogenous in their study design, we will conduct meta‐analyses according to Cochrane recommendations (Deeks 2011). We will not conduct meta‐analyses that include both RCTs and non‐RCTs. We will conduct separate meta‐analyses for each comparison. Different thresholds within the comparisons will only be grouped together if they are considered to be clinically similar.
-
Randomised controlled trials (RCTs)
-
For RCTs where meta‐analysis is feasible, we will use the random‐effects model for pooling the data. For binary outcomes, we will base the estimation of the between‐study variance using the Mantel‐Haenszel method. We will use the inverse‐variance method for continuous outcomes, outcomes that include data from cluster‐RCTs, or outcomes where HRs are available. If heterogeneity is found to be above 80%, and we identify a cause for the heterogeneity, we will explore this with subgroup analyses. If we cannot find a cause for the heterogeneity then we will not perform a meta‐analysis, but comment on the results as a narrative with the results from all studies presented in tables.
-
-
Non‐randomised studies
-
If meta‐analysis is feasible for non‐RCTs or CBA studies, we will analyse non‐RCTs and CBA studies separately. We will only analyse outcomes with adjusted effect estimates if these are adjusted for the same factors using the inverse‐variance method as recommended in chapter 13 of the Cochrane Handbook of Systematic Reviews of Interventions (Reeves 2011).
-
-
All studies
-
We will use the random‐effects model for all analyses as we anticipate that true effects will be related but will not be the same for included studies. If we cannot perform a meta‐analysis, we will comment on the results as a narrative with the results from all studies presented in tables.
-
Summary of Findings
We planned to use the GRADE tool (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of evidence for each outcome. We planned to present a 'Summary of 'findings' table as suggested in Chapters 11 and 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Schunemann 2011a; Schunemann 2011b). The outcomes we planned to include are listed below in order of most relevant endpoints for patients.
-
Major procedure‐related bleeding.
-
Serious adverse events ‐ transfusion‐related.
-
Serious adverse events ‐ venous and arterial thromboembolism.
-
Serious adverse events ‐ LP or epidural‐related.
-
All‐cause mortality.
-
Total number of days in hospital.
-
Quality of life, as defined by the individual studies.
Subgroup analysis and investigation of heterogeneity
If adequate data were available, we planned to perform subgroup analyses for each of the following outcomes in order to assess the effect on heterogeneity.
-
Type of participant (such as obstetric, intensive care, liver disease, etc.).
-
Age of participants grouped as infant (nought to one year); paediatric (one to 16 years) adult (17 years to 60 years) elderly adult (greater than 60 years).
-
Underlying bleeding tendencies (e.g. associated thrombocytopenia, platelet dysfunction).
-
Type of procedure (diagnostic LP; therapeutic LP; epidural catheter).
-
Type of plasma component.
-
Dose of plasma component.
However, no completed study was identified in this review and therefore we could not perform any subgroup analyses.
Sensitivity analysis
We planned to assess the robustness of our findings by performing the following sensitivity analyses according to the recommendations
of Cochrane (Deeks 2011) where appropriate:
-
including only those studies with a ‘low risk of bias’ (e.g. RCTs with methods assessed as low risk for random sequence generation and concealment of treatment allocation);
-
including only those studies with less than a 20% dropout rate.
However, no completed study was identified in this review and therefore we could not perform any sensitivity analyses.
Results
Description of studies
See Characteristics of excluded studies.
Results of the search
The search (conducted 9th January 2017) identified a total of 2173 potentially‐relevant records. There were 1377 records after duplicates were removed. Two review authors (LE; MD) excluded 1373 records on the basis of the abstract. Four full‐text articles were retrieved for assessment by the same two review authors. All four studies were excluded (Figure 1).
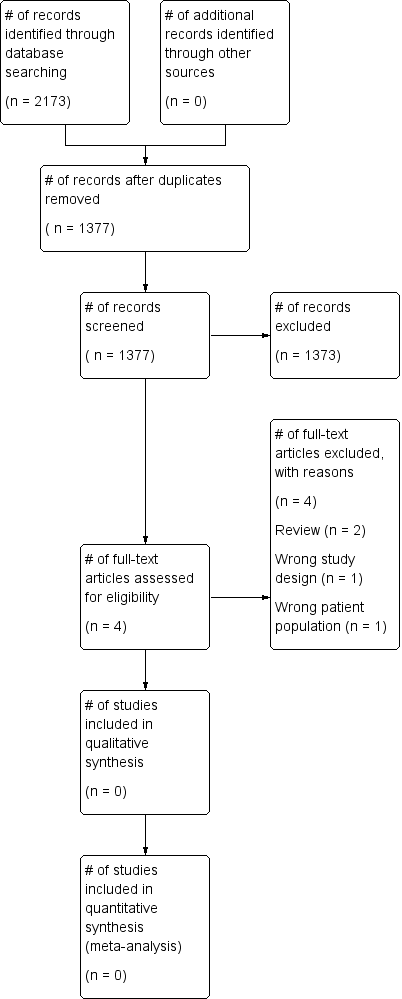
Study flow diagram.
Included studies
No completed or ongoing trials were included in this review. No studies that fitted the criteria were found.
Excluded studies
See Characteristics of excluded studies for further details.
• Two studies were reviews (Bellini 2014; Tryba 1989).
• One study did not include people with abnormal coagulation prior to insertion of the epidural catheter (Lim 2006).
• One study was a single‐centre retrospective observational study (Friedman 1989).
Risk of bias in included studies
No trials were identified for inclusion in the review.
Effects of interventions
No trials were identified for inclusion in the review.
Discussion
Summary of main results
There were no completed or ongoing studies that were relevant to this review.
Overall completeness and applicability of evidence
This review did not identify any completed randomised controlled trials (RCTs), non‐randomised controlled trials (non‐RCTs) or controlled before‐after studies (CBAs) eligible for inclusion and therefore there is no evidence that can be assessed.
Any future study would need to be very large to detect a difference in the risk of bleeding. For example, if we assumed that major bleeding occurred in 1 out of 1000 people who had an LP when their INR was 1.5 or below, and that the risk of major bleeding doubled to 2 out of 1000 when their INR was 3, we would need to design a study with at least 47,030 participants to be able to detect this difference with 80% power and 5% significance (calculated using a power calculator at Sealed Envelope).
Quality of the evidence
This review did not identify any completed studies and therefore there is no evidence that could be assessed.
Potential biases in the review process
To our knowledge, our review process was free from bias. We conducted a comprehensive search, searching data sources (including multiple databases and clinical trial registries) to ensure that all relevant studies would be captured. The relevance of each paper identified was carefully assessed and all screening and data extractions were performed in duplicate.
We prespecified all outcomes and subgroups prior to analysis.
Agreements and disagreements with other studies or reviews
Of the four systematic reviews that addressed the use of plasma transfusion prior to a procedure (Murad 2010; Segal 2005; Stanworth 2004; Yang 2012), only one contained a study that assessed the use of plasma transfusions in people who required a lumbar puncture (Segal 2005). This study was the single‐centre retrospective study excluded from this review because it was the wrong study design (Friedman 1989). No other studies were identified.
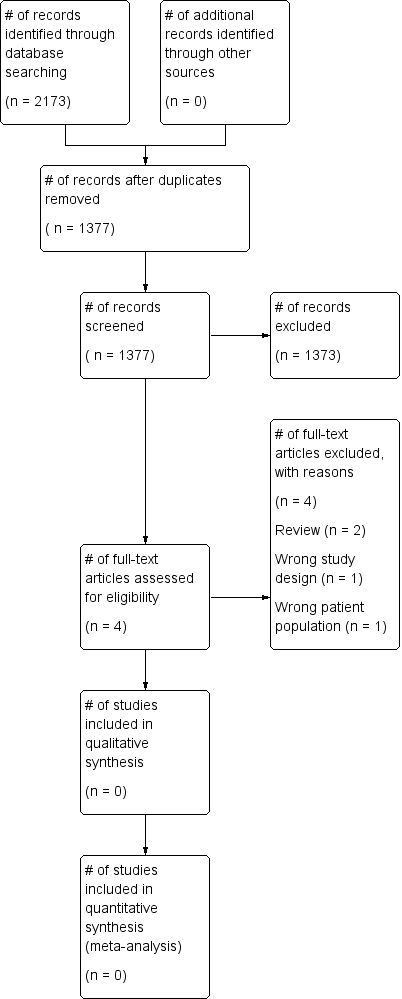
Study flow diagram.