Аспирин (одна доза) при боли в промежности в раннем послеродовом периоде
Abstract
Background
Perineal trauma (due to spontaneous tears, surgical incision (episiotomy) or in association with operative vaginal birth) is common after vaginal birth, and is often associated with postpartum perineal pain. Birth over an intact perineum may also lead to perineal pain. There are adverse health consequences associated with perineal pain for the women and their babies in the short‐ and long‐term, and the pain may interfere with newborn care and the establishment of breastfeeding. Aspirin has been used in the management of postpartum perineal pain and its effectiveness and safety should be assessed.
Objectives
To determine the efficacy of a single dose of aspirin (acetylsalicylic acid), including at different doses, in the relief of acute postpartum perineal pain.
Search methods
We searched Cochrane Pregnancy and Childbirth's Trials Register (30 August 2016), ClinicalTrials.gov, the WHO International Clinical Trials Registry Platform (ICTRP) (31 May 2016) and reference lists of retrieved studies.
Selection criteria
Randomised controlled trials (RCTs) assessing single dose aspirin compared with placebo, no treatment, a different dose of aspirin, or single dose paracetamol/acetaminophen for women with perineal pain in the early postpartum period. We planned to include cluster‐RCTs but none were identified. Quasi‐RCTs and cross‐over studies were not eligible for inclusion in this review.
Data collection and analysis
Two review authors independently assessed study eligibility, extracted data and assessed the risk of bias of the included RCTs. Data were checked for accuracy. The quality of the evidence for the main comparison (aspirin versus placebo) was assessed using the GRADE approach.
Main results
We included 17 RCTs, with 16 involving 1132 women randomised to aspirin or placebo (one RCT did not report numbers of women). Two RCTs (of 16) did not contribute data to review meta‐analyses. All women had perineal pain post‐episiotomy, and were not breastfeeding. Studies were published between 1967 and 1997, and the risk of bias was often unclear due to poor reporting.
We included four comparisons: aspirin versus placebo (data from 15 RCTs); 300 mg versus 600 mg aspirin (1 RCT); 600 mg versus 1200 mg aspirin (2 RCTs); and 300 mg versus 1200 mg aspirin (1 RCT).
Primary outcomes
Aspirin versus placebo
More women who received aspirin experienced adequate pain relief compared with women who received placebo over four to eight hours after administration (risk ratio (RR) 2.03, 95% confidence intervals (CI) 1.69 to 2.42; 13 RCTs, 1001 women; low‐quality evidence). Women who received aspirin were less likely to need additional pain relief over four to eight hours after administration (RR 0.25, 95% CI 0.17 to 0.37; 10 RCTs, 744 women; very low‐quality evidence). There was no difference in maternal adverse effects over four to eight hours post‐administration (RR 1.08, 95% CI 0.57 to 2.06; 14 RCTs, 1067 women; very low‐quality evidence). Subgroup analyses based on dose did not reveal any clear subgroup differences.
There was no clear difference over four hours after administration between 300 mg and 600 mg aspirin for adequate pain relief (RR 0.82, 95% CI 0.36 to 1.86; 1 RCT, 81 women) or need for additional pain relief (RR 0.68, 95% CI 0.12 to 3.88; 1 RCT, 81 women). There were no maternal adverse effects in either aspirin group.
There was no clear difference over four to eight hours after administration between 600 mg and 1200 mg aspirin for adequate pain relief (RR 0.85, 95% CI 0.52 to 1.39; 2 RCTs, 121 women), need for additional pain relief (RR 1.32, 95% CI 0.30 to 5.68; 2 RCTs, 121 women), or maternal adverse effects (RR 3.00, 95% CI 0.13 to 69.52; 2 RCTs, 121 women).
There was no clear difference over four hours after administration between 300 mg and 1200 mg aspirin for adequate pain relief (RR 0.62, 95% CI 0.29 to 1.32; 1 RCT, 80 women) or need for additional pain relief (RR 2.00, 95% CI 0.19 to 21.18; 1 RCT, 80 women). There were no maternal adverse effects in either aspirin group.
None of the included RCTs reported on neonatal adverse effects.
Secondary outcomes
No studies reported on secondary review outcomes: prolonged hospitalisation due to perineal pain; re‐hospitalisation due to perineal pain; fully breastfeeding at discharge; mixed feeding at discharge; fully breastfeeding at six weeks; mixed feeding at six weeks; perineal pain at six weeks; maternal views; maternal postpartum depression.
Authors' conclusions
We found low‐quality evidence to suggest that single dose aspirin compared with placebo can increase pain relief in women with perineal pain post‐episiotomy. Very low‐quality evidence also suggested that aspirin can reduce the need for additional analgesia, without increasing maternal adverse effects. Evidence was downgraded based on study limitations (risk of bias), imprecision, and publication bias or both. RCTs excluded breastfeeding women so there is no evidence to assess the effects of aspirin on neonatal adverse effects or breastfeeding.
With international guidance recommending mothers initiate breastfeeding within one hour of birth, and exclusively breastfeed for the first six months, the evidence from this review is not applicable to current recommended best practice. Aspirin may be considered for use in non‐breastfeeding women with post‐episiotomy perineal pain. Although formal assessment was beyond the remit of this review, current guidance suggests that other analgesic drugs (including paracetamol) should be considered first for postpartum perineal pain. Such agents are the focus of other reviews in this series on drugs for perineal pain in the early postpartum period. It is considered most likely that if RCTs are conducted in the future they could compare aspirin with other pain relievers. Future RCTs should be designed to ensure high methodological quality, and address gaps in the evidence, such as the secondary outcomes established for this review. Current research has focused on women with post‐episiotomy pain, future RCTs could be extended to women with perineal pain associated with spontaneous tears or operative birth.
PICOs
Резюме на простом языке
Аспирин (одна доза) для облегчения боли в промежности после родов
В чем суть проблемы?
Может ли аспирин облегчить боль у женщин, испытывающих боль в промежности после родов, не вызывая побочных эффектов, как у самих женщин, так у их детей?
Почему это важно?
Многие женщины испытывают боль в промежности (область между влагалищем и задним проходом) после родов. Во время родов промежность может быть травмирована или разорвана, или разрезана для облегчения рождения ребенка (эпизиотомия). После родов боль в промежности может повлиять на способность женщин заботиться о своих новорожденных и установление грудного вскармливания. Если боль в промежности полностью не купирована, могут возникнуть долгосрочные проблемы, включая болезненный половой акт, проблемы с тазовым дном, приводящие к недержанию мочи, опущению или хронической боли в области промежности. Аспирин может быть назначен женщинам, испытывающим боли в промежности после родов, однако его эффективность и безопасность не оценивали в систематическом обзоре. Это часть серии обзоров, в которых изучали лекарства для облегчения боли в промежности в первые несколько недель после родов.
Какие доказательства мы обнаружили?
31 мая 2016 года мы провели поиск доказательств и нашли 17 исследований, опубликованных в период с 1967 по 1997 гг., в которых приняли участие 1 132 женщины. Все женщины испытывали боли в промежности после эпизиотомии (как правило, в течение 48 часов после родов) и не кормили грудью. Женщины принимали внутрь либо аспирин (в дозах от 300 мг до 1200 мг), либо таблетку‐пустышку (плацебо). Качество доказательств в исследованиях было оценено как низкое или очень низкое. Два испытания не предоставили каких‐либо данных для анализа.
У большего числа женщин было адекватное облегчение боли через четыре‐восемь часов после приема аспирина, по сравнению с женщинами, получавшими плацебо (доказательства низкого качества). Женщины реже нуждались в дополнительном облегчении боли через четыре‐восемь часов после приема аспирина (доказательства очень низкого качества). Различий в неблагоприятных эффектах у женщин через четыре‐восемь часов после приема лекарства не наблюдалось (доказательства очень низкого качества).
Мы не обнаружили четких различий в эффекте у женщин, получавших 300 мг в сравнении с 600 мг аспирина (1 испытание), 600 мг в сравнении с 1200 мг аспирина (2 испытания) или 300 мг по сравнению с 1200 мг аспирина (1 испытание), в отношении адекватного облегчения боли, потребности в дополнительном облегчении боли или неблагоприятных эффектов у матери.
Ни в одном исследовании не сообщали о неблагоприятных эффектах аспирина у ребенка, или о других исходах, которые мы планировали оценить: продление госпитализации или повторная госпитализация по поводу боли в промежности; боль в промежности через шесть недель после родов, мнения женщин или послеродовая депрессия.
Что это значит?
Одна доза аспирина может облегчить боль в промежности после эпизиотомии у женщин, не кормящих грудью, на период от четырех до восьми часов после приема. Грудное молоко признано лучшим питанием для младенцев, поэтому рекомендуется по возможности начать грудное вскармливание в течение одного часа после рождения, и продолжать его в течение первых шести месяцев жизни ребенка. Мы не нашли информации для оценки эффектов аспирина у женщин, кормящих грудью, но известно, что аспирин может проникать в грудное молоко.
Authors' conclusions
Summary of findings
| Aspirin compared with placebo for perineal pain in the early postpartum period | ||||||
| Patient or population: women with perineal pain in the early postpartum period Settings: 17 RCTs published from 1967 to 1997 (11 RCTs conducted in USA, 3 in Venezuela, 1 each in Belgium, Canada and India) Intervention: aspirin (single dose) Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Aspirin | |||||
| Adequate pain relief as reported by the woman (4 to 8 hours) | Study population | RR 2.03 (1.69, 2.42) | 1001 (13 RCTs) | ⊕⊕⊝⊝ | ||
| 253 per 1000 | 513 per 1000 (427 to 612) | |||||
| Need for additional pain relief (4 to 8 hours) | Study population | RR 0.25 (0.17, 0.37) | 744 (10 RCTs) | ⊕⊝⊝⊝ | ||
| 267 per 1000 | 67 per 1000 (45 to 99) | |||||
| Maternal adverse effects (4 to 8 hours) | Study population | RR 1.08 (0.57, 2.06) | 1067 (14 RCTs) | ⊕⊝⊝⊝ | ||
| 27 per 1000 | 29 per 1000 (15 to 55) | |||||
| Neonatal adverse effects | (0 RCTs) | Not reported by any of the included RCTs | ||||
| Perineal pain at six weeks postpartum | (0 RCTs) | Not reported by any of the included RCTs | ||||
| *The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Study limitations: downgraded two levels due to the serious risk of bias 2Publication bias: downgraded by one level based on visual inspection of funnel plot which indicates likely publication bias 3Imprecision: downgraded one level due to few events and wide 95% CI around the pooled estimate which includes no effect | ||||||
Background
Description of the condition
Perineal trauma may result from naturally occurring tears, surgical incisions such as episiotomy (cutting of the perineum to enlarge the vaginal opening during the second stage of labour), or in association with operative vaginal births (vacuum or forceps assisted births); and is frequently associated with acute perineal pain in the immediate postpartum period (Chou 2009). Birth over an intact perineum is also often associated with acute postpartum perineal pain. Perineal trauma is common, for example, in high‐income countries such as Australia, where perineal trauma may occur in up to 68% of vaginal births, in association with episiotomy (15.6%), naturally occurring tears (first to fourth degree lacerations) (49.8%), and laceration plus episiotomy (3.0%) (Li 2013).
Short‐term morbidities for the mother arising from perineal trauma may include bleeding, infection, haematoma, and acute postpartum perineal pain, which may also interfere with newborn care and the establishment of breastfeeding (Chou 2009; East 2012a). In the longer‐term, women are at an increased risk of dyspareunia (painful sexual intercourse), pelvic floor problems, and chronic perineal pain (Chou 2009; East 2012a).
Various practices can impact on the extent of perineal trauma sustained during birth, and so can influence the degree of perineal pain experienced by the woman in the immediate postpartum period. Cochrane systematic reviews have shown that antenatal digital perineal massage (Beckmann 2013) and the use of warm compresses on the perineum during the second stage of labour (Aasheim 2011) to be effective in preventing perineal trauma and associated pain.
A variety of practices and agents have also been assessed for the relief of perineal pain in the immediate postpartum period. Cochrane reviews have reported finding limited evidence to support routine use of local cooling (such as with ice packs or cold gel packs) of the perineum (East 2012b), or the application of topical local anaesthetics to the perineum for postpartum perineal pain relief (Hedayati 2005). A Cochrane review found some support for the use of paracetamol to reduce postpartum perineal pain, and decrease the need for additional pain relief. However, the overall quality of included evidence was assessed as unclear, and adverse effects were not assessed (Chou 2013). Other practices and agents that have been systematically reviewed and shown to have varied effectiveness in relieving postpartum perineal pain include: methods or materials or both for suturing perineal tears or episiotomies, therapeutic ultrasound, and rectal analgesia (East 2012a; Hay‐Smith 1998; Hedayati 2003; Kettle 2012). For example, in regard to perineal suturing after childbirth, a Cochrane review showed that continuous suturing techniques for perineal closure, compared with interrupted methods, are associated with less short‐term pain; if the continuous techniques are used for all layers (vagina, perineal muscles and skin), the reduction of pain has been reported to be even greater (Kettle 2012).
Description of the intervention
The history of aspirin began thousands of years ago, with early uses of extracts from plants and herbs containing salicylates (Vane 2003). In the 1870s, it was demonstrated that salicin and salicylic acid from white willow bark could reduce fever, pain and inflammation in people with rheumatic fever (Maclagan 1879). The success of salicylic acid prompted the German pharmaceutical manufacturer, Bayer, to search for a derivative that was equally or more effective. Felix Hoffman, a young chemist at Bayer, motivated by his father's inability to take salicylic acid for his arthritis due to its adverse effects (particularly vomiting), found a way to acetylate the hydroxyl group on the benzene ring of salicylic acid to form acetylated salicylic acid (Vane 2003).
In the first decades of the 1900s, acetylsalicylic acid, or 'aspirin' was considered the supreme analgesic (pain reliever); for three quarters of the 20th century, its use was solely as an analgesic and antipyretic (fever reducing) agent (Vane 2003). In the 1970s and 1980s, as part of his Nobel Prize‐winning work, Sir John Vane demonstrated that aspirin could inhibit the formation of prostaglandins, associated with pain, fever, and inflammation, providing a physiological rationale for the effectiveness of one of the world's most widely used medications. As part of this work, Vane also discovered prostacyclin, an important prostaglandin that plays a vital role in the process of blood coagulation. The potential for aspirin to prevent a range of serious, life‐threatening conditions, including heart attacks and stroke, was recognised following this discovery (Smith 2014).
Aspirin is now considered to be one of the most effective and versatile medications in the world. It is commonly recommended to be taken in the lowest effective dose to avoid adverse effects secondary to higher doses. For example, low‐dose aspirin (75 mg to 150 mg daily) has been shown to provide substantial benefit for preventing serious cardiovascular events (heart attacks, stroke and vascular death) in people with pre‐existing cardiovascular disease or with a history of events (secondary prevention) (ATT Collaboration 2002); in primary prevention, for people without histories of events or previous disease, the value of low and high‐dose aspirin (75 mg to 500 mg daily) remains uncertain (ATT Collaboration 2009). Furthermore, there is increasing evidence that aspirin may reduce the risk of some cancers, and certain pregnancy complications. Long‐term low‐dose aspirin (at least 75 mg daily) has been shown to reduce colorectal cancer incidence and mortality (Rothwell 2010); and low‐dose aspirin is reported to have moderate benefits in preventing pre‐eclampsia and its consequences (Duley 2007), with 75 mg daily recommended for pregnant women a high risk of developing the condition (WHO 2011).
How the intervention might work
Perineal pain is transmitted primarily through the pudendal nerve, a somatic sensory and motor nerve that innervates the external genitalia, as well as bladder and rectum sphincters (Cunningham 2005). Although a detailed description of the mechanism of action and pharmacology of aspirin is beyond the scope of this review, basic concepts are outlined.
The mechanisms by which aspirin exerts its analgesic, anti‐inflammatory, and antipyretic effects were discovered in the 1970s. Aspirin inhibits the activity of the cyclo‐oxygenase (COX) enzymes (irreversible inhibition of COX‐1 and modification of COX‐2), which play important roles in inflammation and nociceptive processes (the encoding and processing in the nervous system of noxious stimuli), such as through the formation of prostaglandins and thromboxanes (Vane 2003).
Through inhibiting these key enzymes, it has also been demonstrated that aspirin can prevent the production of physiologically important prostaglandins and thromboxanes, including those that protect the stomach mucosa from damage by hydrochloric acid, and those that aggregate platelets when required (Vane 2003). It is through these mechanisms that aspirin has been shown to cause adverse effects such as gastrointestinal irritation and occult (hidden) blood loss (Derry 2012). The availability of alternative agents with improved tolerability has reduced the use of aspirin for pain relief over recent years, however in many parts of the world, where alternatives are not available or are more expensive, aspirin is still the most commonly used analgesic for many different pain conditions (Derry 2012; Vane 2003).
A Cochrane systematic review that included 67 trials (involving 5743 adults) that were assessed at moderate to high quality overall, confirmed single‐dose aspirin (300 mg to 1200 mg) to be an effective analgesic for acute postoperative moderate to severe intensity pain (Derry 2012). Higher doses (900 mg to 1000 mg) were shown to be more effective, however, these doses were associated with increased adverse effects, including gastric irritation, nausea, vomiting, drowsiness and dizziness. The pain relief achieved with aspirin was very similar to paracetamol given at the same dose (Derry 2012). Derry 2012 excluded trials where pain was due to trauma, such as is often the case for women with acute perineal pain in the immediate postpartum period. It is considered plausible that aspirin may also be effective in relieving acute perineal pain in the early postpartum period after birth, and so it was important to review the efficacy of single‐dose aspirin when used for this indication.
Why it is important to do this review
Perineal trauma is common after vaginal birth, and frequently associated with acute postpartum perineal pain; birth over an intact perineum is also often associated with perineal pain. Perineal pain may be associated with adverse health consequences for the mother and her baby in the short and long term, such as dyspareunia, pelvic floor problems, and chronic perineal pain, and may also interfere with newborn care, including the establishment of breastfeeding (Chou 2009; East 2012a).
There is currently a dearth of evidence on effective interventions to reduce acute perineal pain in the immediate postpartum period. Previous Cochrane reviews have assessed practices and agents including therapeutic ultrasound (Hay‐Smith 1998), rectal analgesia (Hedayati 2003), local cooling (East 2012b), topical anaesthetics (Hedayati 2005), paracetamol (Chou 2013), and most recently non‐steroidal anti‐inflammatory agents (Wuytack 2016), for the relief of perineal pain in the postpartum period. These reviews have reported mixed results. It is therefore important to establish if aspirin may be effective in relieving perineal pain and improving health outcomes for mother and their babies. Because it is known that salicylate and salicylate metabolites, including aspirin, are excreted in breast milk (NIH 2015), there is potential for effects on breastfeeding babies. Adverse effects or harms for both mothers and their babies therefore also need to be assessed.
We assessed the clinical effectiveness and adverse effects of aspirin given to relieve perineal pain in the early postpartum period after childbirth. This review is one of a series of reviews of drugs for perineal pain in the early postpartum period, all based on the same generic protocol (Chou 2009). This protocol is published in theCochrane Library and describes the methods that shaped the production of all the reviews on drugs for perineal pain. It is available for consultation for prospective reviews undertaken on future drugs that may be introduced for this population and indication.
Objectives
To determine the effects of a single dose of aspirin (acetylsalicylic acid), including at different doses, in the relief of acute postpartum perineal pain.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials and planned to include cluster‐randomised controlled trials. Quasi‐randomised controlled trials and cross‐over trials were excluded. We planned to include studies published as abstracts only, as well as studies published in full‐text form.
Types of participants
All women with acute perineal pain in the early postpartum period after childbirth; defined as the first four weeks after giving birth or as defined by the authors of the studies.
Types of interventions
Single administration of aspirin used to treat perineal pain due to spontaneous lacerations, episiotomy or birth over an intact perineum in the early postpartum period. We included studies in which aspirin was compared with a placebo or no treatment, and where different doses of aspirin (e.g. 75 mg, 300 mg, etc) administered as a single dose, were compared. We also planned to include studies where aspirin was compared with a single dose of paracetamol/acetaminophen for perineal pain in the early postpartum period.
Types of outcome measures
Primary outcomes
-
Adequate pain relief as reported by the woman.*
-
Need for additional pain relief in the first 48 hours for perineal pain.
-
Maternal adverse effects, composite of any of the following: nausea, vomiting, sedation, constipation, diarrhoea, drowsiness, sleepiness, gastric discomfort, psychological impact.
-
Neonatal adverse effects, composite of any of the following: vomiting, sedation, constipation, diarrhoea, drowsiness, sleepiness.
* As determined by more than 50% relief of pain, as stated by the woman or calculated using a formula; see Data collection and analysis for details.
Secondary outcomes
-
Prolonged hospitalisation due to perineal pain.
-
Rehospitalisation due to perineal pain.
-
Fully breastfeeding at discharge.
-
Mixed feeding at discharge.
-
Fully breastfeeding at six weeks.
-
Mixed feeding at six weeks.
-
Perineal pain at six weeks.
-
Maternal views (using a validated questionnaire).
-
Maternal postpartum depression.
Search methods for identification of studies
The methods section of this review is based on a standard template used by Cochrane Pregnancy and Childbirth.
Electronic searches
We searched Cochrane Pregnancy and Childbirth’s Trials Register by contacting their Information Specialist (30 August 2016).
The Register is a database containing over 22,000 reports of controlled trials in the field of pregnancy and childbirth. For full search methods used to populate Pregnancy and Childbirth’s Trials Register including the detailed search strategies for CENTRAL, MEDLINE, Embase and CINAHL; the list of handsearched journals and conference proceedings, and the list of journals reviewed via the current awareness service, please follow this link to the editorial information about Cochrane Pregnancy and Childbirth in the Cochrane Library and select the ‘Specialized Register’ section from the options on the left side of the screen.
Briefly, Cochrane Pregnancy and Childbirth’s Trials Register is maintained by their Information Specialist and contains trials identified from:
-
monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL);
-
weekly searches of MEDLINE (Ovid);
-
weekly searches of Embase (Ovid);
-
monthly searches of CINAHL (EBSCO);
-
handsearches of 30 journals and the proceedings of major conferences; and
-
weekly current awareness alerts for a further 44 journals plus monthly BioMed Central email alerts.
Search results are screened by two people and the full text of all relevant trial reports identified through the searching activities described above is reviewed. Based on the intervention described, each trial report is assigned a number that corresponds to a specific Pregnancy and Childbirth review topic (or topics), and is then added to the Register. The Information Specialist searches the Register for each review using this topic number rather than keywords. This results in a more specific search set which has been fully accounted for in the relevant review sections (Included studies; Excluded studies; Studies awaiting classification).
We also searched ClinicalTrials.gov and the WHO International Clinical Trials Registry Platform (ICTRP) for unpublished, planned and ongoing trial reports using the terms given in Appendix 1 on 31 May 2016.
Searching other resources
We searched for further studies in the reference lists of the studies identified.
We did not apply language or date restrictions.
Data collection and analysis
Assessment of pain
The number of participants achieving adequate pain relief was defined as one of the following.
-
The number of participants reporting "good" or "excellent" pain relief when asked about their level of pain relief four to six hours after receiving their allocated treatment (the data were extracted as dichotomous data).
-
The number of women who reported 50% pain relief, or greater.
-
The number of participants who achieved 50% pain relief, or greater, as calculated by using derived pain relief scores (TOTPAR (total pain relief) or SPID (summed pain intensity differences)) over four to six hours.
It is common to use categorical or visual analogue scales for pain intensity and to calculate the results for each participant, over periods of four or six hours, as SPID or TOTPAR (Moore 1996). From these categorical scales, it was possible to convert results into dichotomous data (the proportion of participants achieving at the least 50%, or greater, max TOTPAR) using standard formulae (Moore 1996; Moore 1997b). Converting data in this way enabled these data to be used in a meta‐analysis (Moore 1997a; Moore 1997b). The following equations were used to estimate the proportions of participants achieving at least 50% of maximum TOTPAR.
Proportion with greater than 50% maxTOTPAR = (1.33 x mean %maxTOTPAR – 11.5)
With %maxTOTPAR = mean TOTPAR x 100/(maximum score x number of hours)
Proportion with greater than 50% maxTOTPAR = (1.36 x mean %maxSPID – 2.3)
With %maxSPID = mean SPID x 100/(maximum score x number of hours)
The number of participants achieving at least 50% maxTOTPAR was then calculated by multiplying the proportions of participants with at least 50% maxTOTPAR by the total number of participants in the treatment groups. The number of participants with at least 50% maxTOTPAR was then used to calculate the relative benefit and number needed to treat to benefit.
Where studies used more than one method of calculating adequate pain relief, preference for analyses and reporting purposes, in order of decreasing preference, was: i) the proportion with at least 50% maxTOTPAR calculated using SPID; ii) the proportion with at least 50% maxTOTPAR calculated using TOTPAR; and iii) the number of participants reporting 'good' or 'excellent' pain relief/number of participants reporting at least 50% pain relief. We also assessed the number of participants who re‐medicated in the period of four to eight hours, as well as the median time to re‐medication, if data were available.
Selection of studies
Two review authors independently assessed for inclusion all the potential studies we identified as a result of the search strategy. We resolved any disagreement through discussion or, if required, we consulted a third review author.
We created a study flow diagram to illustrate numbers of records identified, included and excluded (Figure 1).

Study flow diagram
Data extraction and management
We designed a form to extract data. At least two review authors extracted data using the agreed form for eligible studies. We resolved discrepancies through discussion or, if required, consultation with another member of the review author team. We entered data into Review Manager software (RevMan 2014) and checked for accuracy. When information regarding any steps was unclear, we attempted to contact authors of the original reports to provide further details.
Assessment of risk of bias in included studies
Two review authors independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved any disagreement by discussion or by involving a third assessor.
(1) Random sequence generation (checking for possible selection bias)
We described for each included study the method used to generate the allocation sequence in sufficient detail to allow an assessment of whether it should produce comparable groups.
We assessed the method as:
-
low risk of bias (any truly random process, e.g. random number table; computer random number generator);
-
high risk of bias (any non‐random process, e.g. odd or even date of birth; hospital or clinic record number);
-
unclear risk of bias.
(2) Allocation concealment (checking for possible selection bias)
We described for each included study the method used to conceal allocation to interventions prior to assignment and assessed whether intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment.
We assessed the methods as:
-
low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes);
-
high risk of bias (open random allocation; unsealed or non‐opaque envelopes, alternation; date of birth);
-
unclear risk of bias.
(3.1) Blinding of participants and personnel (checking for possible performance bias)
We described for each included study the methods used, if any, to blind study participants and personnel from knowledge of which intervention a participant received. We considered that studies were at low risk of bias if they were blinded, or if we judged that the lack of blinding would be unlikely to affect results.
We assessed the methods as:
-
low, high or unclear risk of bias for participants;
-
low, high or unclear risk of bias for personnel.
(3.2) Blinding of outcome assessment (checking for possible detection bias)
We described for each included study the methods used, if any, to blind outcome assessors from knowledge of which intervention a participant received.
We assessed methods used to blind outcome assessment as:
-
low, high or unclear risk of bias.
(4) Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data)
We described for each included study, and for each outcome or class of outcomes, the completeness of data including attrition and exclusions from the analysis. We stated whether attrition and exclusions were reported and the numbers included in the analysis at each stage (compared with the total randomised participants), reasons for attrition or exclusion where reported, and whether missing data were balanced across groups or were related to outcomes. Where sufficient information was reported, or could be supplied by the trial authors, we planned to re‐include missing data in the analyses.
We assessed methods as:
-
low risk of bias (e.g. no missing outcome data; missing outcome data balanced across groups);
-
high risk of bias (e.g. numbers or reasons for missing data imbalanced across groups; ‘as treated’ analysis done with substantial departure of intervention received from that assigned at randomisation);
-
unclear risk of bias.
(5) Selective reporting (checking for reporting bias)
We described for each included study how we investigated the possibility of selective outcome reporting bias and what we found.
We assessed the methods as:
-
low risk of bias (where it was clear that all of the study’s pre‐specified outcomes and all expected outcomes of interest to the review were reported);
-
high risk of bias (where not all the study’s pre‐specified outcomes were reported; one or more reported primary outcomes were not pre‐specified; outcomes of interest were reported incompletely and so could not be used; study failed to include results of a key outcome that would have been expected to have been reported);
-
unclear risk of bias.
(6) Other bias (checking for bias due to problems not covered by points (1) to (5))
We described for each included study any important concerns we had about other possible sources of bias including: was the trial stopped early due to some data‐dependent process? Was there extreme baseline imbalance? Has the study been claimed to be fraudulent?
We assessed whether each study was free of other problems that could put it at risk of bias:
-
low risk of other bias;
-
high risk of other bias;
-
unclear whether there was risk of other bias.
(7) Overall risk of bias
We made explicit judgements about whether studies were at high risk of bias, according to the criteria given in the Handbook (Higgins 2011). With reference to points (1) to (6), we assessed the likely magnitude and direction of the bias and whether we considered it is likely to impact on the findings. We planned to assess the impact of the level of bias through undertaking sensitivity analyses ‐ seeSensitivity analysis.
Assessing the quality of the evidence using the GRADE approach
We planned to assess the quality of the evidence using the GRADE approach as outlined in the GRADE handbook in order to assess the quality of the body of evidence relating to the following outcomes for the main comparison: aspirin versus placebo.
-
Adequate pain relief as reported by the woman.
-
Need for additional pain relief in the first 48 hours for perineal pain.
-
Maternal adverse effects, composite of any of the following: nausea, vomiting, sedation, constipation, diarrhoea, drowsiness, sleepiness, gastric discomfort, psychological impact.
-
Neonatal adverse effects, composite of any of the following: vomiting, sedation, constipation, diarrhoea, drowsiness, sleepiness.
-
Perineal pain at six weeks postpartum.
We could however only assess the quality of the evidence for the first three outcomes, as for outcomes 4 and 5, we had no data from the included trials.
We created a 'Summary of findings' table for our main comparison in Review Manager 5.3 (RevMan 2014). A summary of the intervention effect and a measure of quality for each of the outcomes was produced using the GRADE approach. The GRADE approach uses five considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of the body of evidence for each outcome. The evidence can be downgraded from 'high quality' by one level for serious (or by two levels for very serious) limitations, depending on assessments for risk of bias, indirectness of evidence, serious inconsistency, imprecision of effect estimates or potential publication bias.
Measures of treatment effect
Dichotomous data
We presented results as summary risk ratio with 95% confidence intervals for dichotomous data.
Continuous data
We planned to use the mean difference if outcomes were measured in the same way between trials. We planned to use the standardised mean difference to combine trials that measured the same outcome, but used different methods.
Unit of analysis issues
Cluster‐randomised trials
We planned to include cluster‐randomised trials in the analyses along with individually‐randomised trials. If we include cluster‐randomised trials in future updates, we will adjust their sample sizes using the methods described in theHandbook (Higgins 2011) using an estimate of the intra cluster correlation co‐efficient (ICC) derived from the trial (if possible), from a similar trial or from a study of a similar population. If we use ICCs from other sources, we will report this and conduct sensitivity analyses to investigate the effect of variation in the ICC. If we identify both cluster‐randomised trials and individually‐randomised trials, we plan to synthesise the relevant information. We will consider it reasonable to combine the results from both if there is little heterogeneity between the study designs and the interaction between the effect of intervention and the choice of randomisation unit is considered to be unlikely.
We will also acknowledge heterogeneity in the randomisation unit and perform a sensitivity analysis to investigate the effects of the randomisation unit.
Cross‐over trials
We considered cross‐over trials to be inappropriate for this research question and these were not included.
Multi‐armed trials
We included all the relevant intervention groups (aspirin) and control groups (placebo) from multi‐arm trials. We excluded other arms that were not relevant to this review.
Dealing with missing data
We noted levels of attrition for the included studies. We planned to explore the impact of including studies with high levels of missing data in the overall assessment of treatment effect by conducting sensitivity analyses.
We carried out analyses, as far as possible, on an intention‐to‐treat basis for all outcomes. That is, we attempted to include all participants randomised to each group in the analyses, and all participants were analysed in the group to which they were allocated, regardless of whether or not they received the allocated intervention. The denominator for each outcome in each trial was the number randomised minus any participants whose outcomes were known to be missing.
Assessment of heterogeneity
We assessed statistical heterogeneity in meta‐analyses using the T², I² and Chi² statistics. We regarded heterogeneity as substantial if an I² was greater than 30% and either a T² was greater than zero, or there was a low P value (P < 0.10) in the Chi² test for heterogeneity.
Assessment of reporting biases
Because there were 10 or more studies included in the meta‐analyses for 'Adequate pain relief as reported by the woman', 'Need for additional pain relief' and 'Maternal adverse effects', we investigated reporting biases (such as publication bias) using funnel plots. We assessed funnel plot asymmetry visually.
Data synthesis
We carried out statistical analysis using Review Manager software (RevMan 2014). We used fixed‐effect meta‐analysis for combining data because it was considered reasonable to assume that studies were estimating the same underlying treatment effect.
In future updates of this review, if there is clinical heterogeneity sufficient to expect that the underlying treatment effects differed between trials, or where substantial statistical heterogeneity is detected, we will use random‐effects meta‐analysis to produce an overall summary. We will treat the random‐effects summary as the average of the range of possible treatment effects and discuss the clinical implications of treatment effects differing among trials. If the average treatment effect is not clinically meaningful, we will not combine trials. If we use random‐effects analyses in future updates, the results will be presented as the average treatment effect with 95% confidence intervals, and the estimates of Tau² and I².
Subgroup analysis and investigation of heterogeneity
If we had identified substantial heterogeneity, we planned to investigate possible sources using subgroup analyses and sensitivity analyses. We planned to consider whether an overall summary was meaningful, and if it was, use random‐effects analysis to produce the effect.
We planned to carry out the following subgroup analyses:
-
primiparous versus multiparous women;
-
women with perineal trauma versus women who gave birth over intact perineum; and
-
dose of aspirin (i.e. low‐dose versus high‐dose).
However due to the absence of relevant data in the included trials, we were able to conduct analyses based on dose only.
Subgroup analysis were restricted to the review's primary outcomes with reported data.
We assessed subgroup differences by interaction tests available in RevMan 2014. We reported the results of subgroup analyses quoting the Chi² statistic and P value, and the interaction test I² value.
Sensitivity analysis
We planned to conduct sensitivity analyses to explore the effects of trial quality on the outcomes. We planned to explore the effects of trial quality assessed by allocation concealment and random sequence generation (considering selection bias), by omitting studies rated as high or unclear risk of bias for these components. However, because all included trials were assessed with an unclear rating for at least one of these two components, we did not conduct sensitivity analyses.
We also planned to investigate the effects of the randomisation unit (individual versus cluster) on the outcomes, and the impact of including studies with high levels of missing data. We planned to explore the effects of fixed‐effect or random‐effects analyses for outcomes with statistical heterogeneity, and the effects of any assumptions made such as the value of the ICC used for cluster‐randomised trials. However, because we did not include any cluster‐randomised trials, trials with high levels of missing data, or identify outcomes with substantial statistical heterogeneity, we did not conduct sensitivity analyses.
We planned to use only primary outcomes in sensitivity analyses.
Results
Description of studies
Results of the search
The search of the Cochrane Pregnancy and Childbirth's Trials Register retrieved 35 reports. The search of additional sources yielded seven records (ClinicalTrials.gov = five, ICTRP = two records), all of which were screened and considered to be ineligible based on title, abstract or both. This resulted in screening 35 full text reports. Of these, 17 studies (22 records) were included, and 10 studies (11 records) were excluded.
Two studies (two records) await classification: the method of allocation was not clearly reported in Bhounsule 1990; and we were unable to locate a copy of Sunshine 1989.
See Characteristics of included studies; Characteristics of excluded studies; Characteristics of studies awaiting classification; Figure 1.
Included studies
Design and setting
We included 17 studies (22 reports) in this review. All were reported to be randomised controlled trials (RCTs).
The included studies were published between 1967 and 1997; one trial was published in the 1960s; six in the 1970s; nine in the 1980s; and one in the 1990s. Most (11 trials) were conducted in the USA (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; Friedrich 1983; Jain 1978a; Jain 1978b; Jain 1985; London 1983a; London 1983b; Okun 1982), three in Venezuela (Olson 1997; Sunshine 1983a; Sunshine 1983b), and one each in Belgium (De Vroey 1978), Canada (Trop 1983) and India (Mukherjee 1980).
Only one trial (Bloomfield 1970b) had two trial arms, comparing aspirin and placebo. Another trial compared only aspirin and placebo, but had four trial arms (London 1983b). The remaining 15 trials had between three and five trial arms, and in addition to aspirin, assessed a number of other agents for perineal pain in the early postpartum period. These agents included chlorphenesin 400 mg, 800 mg and combination aspirin 300 mg and chlorphenesin 400 mg (Bloomfield 1967); flufenisal 300 mg and 600 mg (Bloomfield 1970a); ibuprofen 300 mg and 900 mg (Bloomfield 1974); diflunisal 125 mg, 250 mg and 500 mg (De Vroey 1978), etodolac 25 mg and 100 mg (Friedrich 1983), piroxicam 20 mg and 40 mg (Jain 1978a); combination aspirin 800 mg and caffeine 64 mg (Jain 1978b); indoprofen 50 and 100 mg (Jain 1985); fluproquazone 100 mg and 200 mg (London 1983a); dipyrone 500 mg (Mukherjee 1980); fendosal 100 mg, 200 mg and 400 mg (Okun 1982); potassium 25 mg, 50 mg and 100 mg (Olson 1997); zomepirac and ibuprofen (Sunshine 1983a); flurbiprofen 25 mg 50 mg and 100 mg (Sunshine 1983b); and tiaprofenic acid 200 and 400 mg (Trop 1983). For the purposes of the review, we analysed only the aspirin and placebo arms from the included trials.
Participants and sample sizes
In total, there were 1132 women in the aspirin and placebo arms of 16 of the 17 included trials, with 617 women randomised to receive aspirin, and 515 to a placebo. In three trials, only the numbers analysed (not randomised) were reported (De Vroey 1978; London 1983a; London 1983b), and in one trial (Trop 1983), the numbers of women were not reported at all. The sample sizes of the trials (including only the relevant arms) ranged from 26 (Bloomfield 1970b) to 178 (Mukherjee 1980). We reported the number of women in other arms of the trials not included in analyses in the Characteristics of included studies tables.
All included trials included women with perineal pain in the early postpartum period post‐episiotomy. One trial recruited and randomised women on the first post‐operative morning (Mukherjee 1980), two within 24 hours of birth (Bloomfield 1967; Bloomfield 1970b), one from 16 to 48 hours following induction of anaesthesia (Friedrich 1983), six within 48 hours of birth (Bloomfield 1970a; Bloomfield 1974; De Vroey 1978; Jain 1978a; London 1983a; Okun 1982); and seven trials did not specify a time period following birth (Jain 1978a; Jain 1985; London 1983b; Olson 1997; Sunshine 1983a; Sunshine 1983b; Trop 1983). No trials were identified that assessed perineal pain associated with naturally occurring tears or birth over an intact perineum. The intensity of women's pain following episiotomy varied among the included trials; eight trials included women with moderate or severe pain (Bloomfield 1967; De Vroey 1978; Friedrich 1983; Jain 1978a; London 1983a; London 1983b; Mukherjee 1980; Sunshine 1983b); three included women with moderate to very severe pain (Bloomfield 1970a; Bloomfield 1974; Okun 1982); one included women with mild to severe pain (Bloomfield 1970b); one included women with at least moderate pain (Jain 1985); and three included women with severe pain (Jain 1978b; Olson 1997; Sunshine 1983a). Pain intensity was not specified in one trial (Trop 1983). Most trials clearly specified that breastfeeding was an exclusion criterion, and excluded women with known sensitivity or allergy to aspirin, and women who had recently received analgesia.
Interventions and comparisons
Of the 17 included trials, 15 included additional arms that assessed other agents. However, we included data from aspirin and placebo arms only in this review. Fifteen trials included comparisons of aspirin and placebo only; the single, oral doses of aspirin in these were 500 mg (Mukherjee 1980), 600 mg (Bloomfield 1967; De Vroey 1978; Jain 1985; Sunshine 1983a; Sunshine 1983b), 648 mg (Jain 1978a), 650 mg (Friedrich 1983; Jain 1978b; London 1983a; Okun 1982; Olson 1997), 900 mg (Bloomfield 1974), and 1200 mg (Bloomfield 1970b). Three trials included two or more aspirin arms (in addition to a placebo arm); Bloomfield 1970a and Trop 1983 compared 600 mg and 1200 mg aspirin, and London 1983b compared 300 mg, 600 mg and 1200 mg aspirin. The number of aspirin and placebo tablets (and dose of the tablets) taken varied across the trials.
Outcomes
Some measure of 'Adequate pain relief as reported by the woman' could be extracted four to eight hours after drug administration from 13 trials and meta‐analysed. Data were not presented in a way that enabled inclusion in the meta‐analysis in four trials (Jain 1978a; Jain 1978b; Okun 1982; Trop 1983).
Three trials in the meta‐analysis provided data on adequate pain relief four hours after taking the medication (London 1983b; Olson 1997; Sunshine 1983a); two trials reported this outcome after five hours (Bloomfield 1970b; Jain 1985), seven trials after six hours (Bloomfield 1967; Bloomfield 1974; De Vroey 1978; Friedrich 1983; London 1983a; Mukherjee 1980; Sunshine 1983b) and one trial after eight hours (Bloomfield 1970a). SPID scores were used to calculate the number of women with adequate pain relief for the meta‐analysis in 11 trials; (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; De Vroey 1978; Friedrich 1983; Jain 1985; London 1983b; Olson 1997; Sunshine 1983a; Sunshine 1983b), one used TOTPAR scores (Mukherjee 1980), and one used the number of women reporting pain relief to be good or excellent (London 1983a).
Five trials provided both SPID and TOTPAR scores (Friedrich 1983; Jain 1985; Olson 1997; Sunshine 1983a; Sunshine 1983b); two trials provided both SPID scores, and the number of women reporting pain relief to be good or excellent (Friedrich 1983; Jain 1985); and five reported SPID scores and the number of women with at least 50% pain relief (or similar) (Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; De Vroey 1978; Mukherjee 1980). In these cases, we used SPID data to calculate the number of women with adequate pain relief for inclusion in the meta‐analysis. In some cases, the number of women with adequate pain relief according to these different measures did not match, and the reasons for discrepancies were not entirely clear, particularly in numbers of women with adequate pain relief when calculated using the SPID versus TOTPAR scores.
Data on the need for additional analgesia which could be included in a meta‐analysis were available from 10 trials (Bloomfield 1970a; Bloomfield 1974; De Vroey 1978; Jain 1978a; Jain 1985; London 1983a; London 1983b; Olson 1997; Sunshine 1983a; Sunshine 1983b) and 14 trials reported data on any maternal adverse effects suitable for meta‐analysis (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1974; De Vroey 1978; Friedrich 1983; Jain 1978a; Jain 1978b; Jain 1985; London 1983a; London 1983b; Mukherjee 1980; Olson 1997; Sunshine 1983a; Sunshine 1983b).
Two trials (Okun 1982; Trop 1983) did not provide any data that could be meta‐analysed.
None of the 17 included trials reported on any of the prespecified secondary outcomes.
Funding sources
Two of the trials reported support or funding solely from the National Institutes of Health (Bloomfield 1967), and the United Stated Public Health Service and National Heart Institute (Bloomfield 1970b). Ten of the trials reported at least partial support from pharmaceutical companies/commercial medical research organisations: Merck Sharp and Dohme Research Laboratories (Bloomfield 1970a; De Vroey 1978), the Upjohn Company (Bloomfield 1974; Sunshine 1983b), American Home Products, Ives Laboratories and Wyeth Laboratories (Jain 1978b), Adria Laboratories (Jain 1985), Sandoz Inc. (London 1983a), the Ciba‐Geigy Corporation (Olson 1997), Boots Pharmaceuticals (Sunshine 1983b) and Roussel Canada Inc. (Trop 1983). Five of the trials (Friedrich 1983; Jain 1978a; London 1983b; Mukherjee 1980; Okun 1982) did not report any sources of support or funding.
Declarations of interests
None of the 17 included trials provided specific declarations of interest for the manuscript authors. It was noted that three of the trials had author(s) with affiliations to pharmaceutical companies/commercial medical research organisations: Merck Sharp and Dohme Research Laboratories (De Vroey 1978); Analgesic Development Ltd. (Olson 1997), and Roussel Canada Inc. (Trop 1983).
Excluded studies
We excluded 10 studies (11 records) for the following reasons: five trials (Bruni 1965; Gruber 1979; Moggian 1972; Sunshine 1983c; Sunshine 1985) included mixed populations of women with postpartum pain (such as uterine cramping), and no results were reported separately for women with perineal pain; one was not a randomised trial (Santiago 1959); three assessed combination agents (not aspirin alone) (Gindhart 1971; Prockop 1960; Rubin 1984); and one assessed twice daily aspirin (not single dose aspirin) (Van der Pas 1984).
Risk of bias in included studies

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies

Risk of bias summary: review authors' judgements about each risk of bias item for each included study
Allocation
Only two trials were assessed to have applied adequate sequence generation methods, both used computer‐generated random sequences (Olson 1997; Sunshine 1983b). The remaining 15 trials did not report on methods used for random sequence generation, and simply reported that the women were 'randomised' (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; De Vroey 1978; Friedrich 1983; Jain 1978a; Jain 1978b; Jain 1985; London 1983a; London 1983b; Mukherjee 1980; Okun 1982; Sunshine 1983a; Trop 1983). All included trials were judged at unclear risk of selection bias, with none reporting methods of allocation concealment.
Blinding
Of the 17 trials, 14 were judged at low risk of both performance and detection bias, with blinding of women and study personnel (who were also the outcome assessors) by using identical placebos (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; De Vroey 1978; Jain 1985; London 1983a; London 1983b; Mukherjee 1980; Okun 1982; Olson 1997; Sunshine 1983a; Sunshine 1983b; Trop 1983). Three trials (Friedrich 1983; Jain 1978a; Jain 1978b) were at unclear risk of performance and detection bias, because although the trials were reported to be 'double‐blind', no information was provided on the nature of the placebos used to determine if blinding could have been successfully achieved.
Incomplete outcome data
Only three trials (Bloomfield 1970b; Jain 1985; Mukherjee 1980) were judged to be at low risk of attrition bias, with no losses or exclusions.
Five trials were assessed at unclear risk of attrition bias (Friedrich 1983; Jain 1978a; Jain 1978b; London 1983a; Trop 1983), largely due to unclear reporting regarding any losses and exclusions, and reasons for missing data, or both.
Nine trials (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1974; De Vroey 1978; London 1983b; Okun 1982; Olson 1997; Sunshine 1983a; Sunshine 1983b) were assessed at high risk of attrition bias. In eight, the trial authors imputed data (i.e. for women requesting additional analgesia, trial authors either used women's pre‐treatment pain intensity/relief scores for all subsequent hours, or used the last observation carried forward method for subsequent hours), which may have introduced bias; in one trial (Bloomfield 1967), women who requested additional analgesia were excluded from the analyses, which may have similarly introduced bias.
Selective reporting
We assessed 12 trials (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; Friedrich 1983; Jain 1985; London 1983a; London 1983b; Mukherjee 1980; Okun 1982; Olson 1997; Sunshine 1983b) at unclear risk of reporting bias, with no access to trial protocols or registrations to confidently assess the risk of selective reporting.
Five trials (De Vroey 1978; Jain 1978a; Jain 1978b; Sunshine 1983a; Trop 1983) were judged to be at high risk of reporting bias. In all five, some of outcome data and results were reported incompletely in the text, which meant these data could not have been extracted, such as for inclusion in a meta‐analysis. For example: "The three drugs were much the same for mean onset, duration and time to peak values. The hypothesis that there is no difference among treatments was rejected at the 0.05 level or better for all variables" (Sunshine 1983a).
Very few outcome data were reported in all 17 trials.
Other potential sources of bias
Nine trials (Bloomfield 1967; Bloomfield 1970a; Jain 1978a; Jain 1985; Mukherjee 1980; Okun 1982; Olson 1997; Sunshine 1983a; Sunshine 1983b) were assessed at low risk of other potential sources of bias. Eight trials (Bloomfield 1970b; Bloomfield 1974; De Vroey 1978; Friedrich 1983; Jain 1978b; London 1983a; London 1983b; Trop 1983) were judged to be at unclear risk of other bias. These trials did not report baseline characteristics in a way that enabled assessment of comparability among groups (with no baseline characteristics reported, or lack of detail reported); one trial (Bloomfield 1970a) reported that most baseline characteristics were similar between groups "However, body weight was not similar in all treatment groups".
Effects of interventions
Comparison 1: Aspirin versus placebo for perineal pain
Fifteen of the 17 included trials contributed data to meta‐analyses in this comparison (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; De Vroey 1978; Friedrich 1983; Jain 1978a; Jain 1978b; Jain 1985; London 1983a; London 1983b; Mukherjee 1980; Olson 1997; Sunshine 1983a; Sunshine 1983b). Two trials (Okun 1982; Trop 1983) did not provide any data that could be meta‐analysed.
Primary outcomes
Adequate pain relief as reported by the woman
Over four to eight hours after drug administration, more women who had received aspirin experienced adequate pain relief compared with women who received placebo (risk ratio (RR) 2.03, 95% confidence intervals (CI) 1.69 to 2.42; 13 trials, 1001 women; Analysis 1.1). The quality of the evidence (GRADE) for this outcome was judged to be low, with downgrading based on study limitations (risk of bias) (summary of findings Table for the main comparison). Visual inspection of the funnel plot for this outcome suggested no clear evidence of reporting bias (Figure 4).

Funnel plot of comparison: 1 Aspirin versus placebo for perineal pain, outcome: 1.1 Adequate pain relief as reported by the woman
Data from the trials not included in the meta‐analysis
-
Jain 1978a: "By all measurements of drug effect... aspirin 648 mg [was] significantly (P < 0.01) superior to placebo in [its] overall analgesic effect and also at second, third and fourth hours after dosing".
-
Jain 1978b: "In comparing 650 mg aspirin with placebo, we detected no significant difference at 1, 2, or 3 hr, but at the fourth hour we noted trends toward significant in favour of aspirin (P < 0.10) by each Kruskal‐Wallis analysis for pain analogue, pain intensity, and pain relief scores. The corresponding analysis of covariance at hour 4 showed differences in favour of aspirin for both pain analogue and pain intensity scores (P < 0.02)".
-
Okun 1982: "In patients with either uterine cramp or episiotomy pain, aspirin... provided greater pain relief (lower mean pain intensity scores) than did placebo from the 2nd through the 8th study hour".
-
Trop 1983: "When compared to placebo both patient's self‐rating scale and nurse's impression scale have shown a significant reduction in pain following treatment... with ASA".
Need for additional pain relief in the first 48 hours for perineal pain
Women who received aspirin were less likely to require additional analgesia over four to eight hours (RR 0.25, 95% CI 0.17 to 0.37; 10 trials, 744 women; Analysis 1.2) after drug administration. The quality of the evidence (GRADE) for this outcome was judged to be very low, with downgrading based on study limitations (risk of bias), and possible publication bias (summary of findings Table for the main comparison). Visual inspection of the funnel plot for this outcome indicated possible evidence of reporting bias, which could be due to some smaller trials producing exaggerated intervention effect estimates (Figure 5).
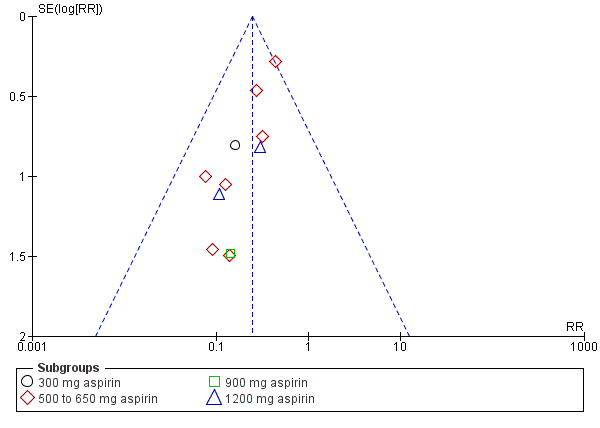
Funnel plot of comparison: 1 Aspirin versus placebo for perineal pain, outcome: 1.2 Need for additional pain relief
Data from the trials not included in the meta‐analysis
-
Bloomfield 1967: not reported; although one woman in the aspirin group was reported to have been "withdrawn owing to distressing pain unrelieved by the study drugs" compared with no women in the placebo group.
-
Okun 1982: "The proportion of patients requiring additional analgesic was significantly different... Approximately 71% of patients in the placebo group needed additional analgesic as compared with... 48% in the aspirin group" (these data related to women with uterine cramp or episiotomy pain).
-
Trop 1983 "None of the patients on… ASA required any additional analgesic during the 4‐hour observation period, but four patients in the placebo group required supplementary medication for pain" (the denominators for each group were not reported).
Maternal adverse effects
There was no clear difference in overall maternal adverse effects over four to eight hours post‐administration (RR 1.08, 95% CI 0.57 to 2.06; 14 trials, 1067 women; Analysis 1.3). The quality of the evidence (GRADE) for this outcome was judged to be very low due to study limitations (risk of bias) and imprecision (summary of findings Table for the main comparison). Visual inspection of the funnel plot for this outcome suggested no clear evidence of reporting bias (Figure 6).

Funnel plot of comparison: 1 Aspirin versus placebo for perineal pain, outcome: 1.3 Maternal adverse effects
Data from the trials not included in the meta‐analysis
-
Okun 1982: "The incidence of side effects was not significantly different among the treatment groups... Three patients each in the placebo group... and 5 patients in the aspirin group reported side effects" (these data related to women with uterine cramp or episiotomy pain).
-
Trop 1983: one woman receiving 1200 mg aspirin (dizziness), no women receiving 600 mg aspirin and two women receiving placebo (nausea) experienced side effects (the denominators for each group were not reported).
Neonatal adverse effects
None of the included trials reported on the primary outcome: neonatal adverse effects.
Subgroup analyses based on dose
We integrated subgroup analyses based on dose into the main analyses, comparing trials using 300 mg, 500 to 650 mg, 900 mg and 1200 mg aspirin. No clear subgroup differences were observed based on dose of aspirin for 'Adequate pain relief as reported by the woman (Test for subgroup differences: Chi² = 0.75, P = 0.86, I² = 0%; Analysis 1.1); 'Need for additional pain relief' (Test for subgroup differences: Chi² = 0.63, P = 0.89, I² = 0%; Analysis 1.2); or 'Maternal adverse effects' (Test for subgroup differences: Chi² = 3.76, df = 2 (P = 0.15), I² = 46.8%; Analysis 1.3).
Secondary outcomes
None of the included trials reported on any of the secondary review outcomes: prolonged hospitalisation due to perineal pain; re‐hospitalisation due to perineal pain; fully breastfeeding at discharge; mixed feeding at discharge; fully breastfeeding at six weeks; mixed feeding at six weeks; perineal pain at six weeks; maternal views (using a validated questionnaire); maternal postpartum depression.
Comparison 2: 300 mg aspirin versus 600 mg aspirin for perineal pain
London 1983b contributed data to this comparison.
Primary outcomes
Adequate pain relief as reported by the woman
There was no clear difference over four hours after drug administration between the 300 mg and 600 mg aspirin groups in the proportion of women who experienced adequate pain relief (RR 0.82, 95% CI 0.36 to 1.86; 1 trial, 81 women; Analysis 2.1).
Need for additional pain relief in the first 48 hours for perineal pain
There was no clear difference over four hours after drug administration between the 300 mg and 600 mg aspirin groups in the proportion of women who required additional pain relief (RR 0.68, 95% CI 0.12 to 3.88; 1 trial, 81 women; Analysis 2.2).
Maternal adverse effects
There were no adverse effects among women in the 300 mg and 600 mg aspirin groups (Analysis 2.3).
Neonatal adverse effects
London 1983b did not report on the primary outcome: neonatal adverse effects.
Secondary outcomes
London 1983b did not report on any of the secondary review outcomes.
Comparison 3: 600 mg aspirin versus 1200 mg aspirin for perineal pain
Bloomfield 1970a and London 1983b contributed data to this comparison.
Primary outcomes
Adequate pain relief as reported by the woman
There was no clear difference over four to eight hours after drug administration between the 600 mg and 1200 mg aspirin groups in the proportion of women who experienced adequate pain relief (RR 0.85, 95% CI 0.52 to 1.39; 2 trials, 121 women; Analysis 3.1).
Need for additional pain relief in the first 48 hours for perineal pain
There was no clear difference over four to eight hours after drug administration between the 600 mg and 1200 mg aspirin groups in the proportion of women who required additional pain relief (RR 1.32, 95% CI 0.30 to 5.68; 2 trials, 121 women; Analysis 3.2).
Maternal adverse effects
There was no clear difference over four to eight hours after drug administration between the 300 mg and 1200 mg aspirin groups in the proportion of women who experienced adverse effects (RR 3.00, 95% CI 0.13 to 69.52; 2 trials, 121 women; Analysis 3.3).
Neonatal adverse effects
Bloomfield 1970a and London 1983b did not report on the primary outcome: neonatal adverse effects.
Secondary outcomes
Bloomfield 1970a and London 1983b did not report on any of the secondary review outcomes.
Comparison 4: 300 mg aspirin versus 1200 mg aspirin for perineal pain
London 1983b contributed data to this comparison.
Primary outcomes
Adequate pain relief as reported by the woman
There was no clear difference over four hours after drug administration between the 300 mg and 1200 mg aspirin groups in the proportion of women who experienced adequate pain relief (RR 0.62, 95% CI 0.29 to 1.32; 1 trial, 80 women; Analysis 4.1).
Need for additional pain relief in the first 48 hours for perineal pain
There was no clear difference over four hours after drug administration between the 300 mg and 1200 mg aspirin groups in the proportion of women who required additional pain relief (RR 2.00, 95% CI 0.19 to 21.18; 1 trial, 80 women; Analysis 4.2).
Maternal adverse effects
There were no adverse effects among women in the 300 mg and 1200 mg aspirin groups (Analysis 4.3).
Neonatal adverse effects
London 1983b did not report on the primary outcome: neonatal adverse effects.
Secondary outcomes
London 1983b did not report on any of the secondary review outcomes.
Discussion
Summary of main results
We included 17 trials, of these, 16 randomised 1132 women to single dose aspirin or placebo for perineal pain in the early postpartum period. Fifteen trials contributed data to four comparisons (aspirin versus placebo; 300 mg versus 600 mg aspirin; 600 mg versus 1200 mg aspirin; 300 mg versus 1200 mg aspirin).
Low‐quality evidence showed that women receiving aspirin (doses ranging from 300 mg to 1200 mg) had a 103% relative increase in adequate pain relief at four to eight hours after administration compared with placebo (from 25% in the placebo group to 47% in the aspirin group) across 13 trials involving 1001 women. Data from four trials (Jain 1978a; Jain 1978b; Okun 1982; Trop 1983) could not be included in the meta‐analysis for this outcome. However, results indicated a benefit from aspirin when compared with placebo. In a subgroup analysis, no clear differences were observed based on dose of aspirin. Neither were clear differences observed between groups for adequate pain relief as reported by the women in comparisons of 300 mg and 600 mg aspirin (1 trial, 81 women), 600 mg and 1200 mg aspirin (2 trials, 121 women), and 300 mg and 1200 mg aspirin (1 trial, 80 women).
Very low‐quality evidence also showed that women receiving aspirin (doses ranging from 300 mg to 1200 mg) had a 75% relative reduction in the need for additional analgesia for perineal pain over four to eight hours after administration (from 27% in the placebo group to 6% in the aspirin group) across 10 trials involving 744 women. Subgroup analysis did not reveal any clear differences based on dose of aspirin. No clear differences were observed between groups for need of additional pain relief in comparisons of 300 mg and 600 mg aspirin (1 trial, 81 women), 600 mg and 1200 mg aspirin (2 trials, 121 women), and 300 mg and 1200 mg aspirin (1 trial, 80 women)
Very low‐quality evidence also showed no clear difference between women receiving aspirin (doses ranging from 300 mg to 1200 mg) versus placebo for maternal adverse effects over four to eight hours after administration (3% in both the aspirin and placebo groups) across 14 trials involving 1067 women. Similarly, there were no subgroup differences based on dose. No clear difference was observed in a comparison of 300 mg and 1200 mg aspirin (2 trials, 121 women).
None of the included trials reported on the review primary outcome ‐ neonatal adverse effects, nor the secondary review outcomes: prolonged hospitalisation due to perineal pain; re‐hospitalisation due to perineal pain; fully breastfeeding at discharge; mixed feeding at discharge; fully breastfeeding at six weeks; mixed feeding at six weeks; perineal pain at six weeks; maternal views (using a validated questionnaire); and maternal postpartum depression.
Overall completeness and applicability of evidence
The included trials enrolled women with perineal pain in the early postpartum period post‐episiotomy. Accordingly, results may not be applicable to other women with perineal pain, such as those with pain following naturally occurring tears or birth over an intact perineum. All included trials compared aspirin with placebo; we were unable to assess the comparative effects of aspirin versus paracetamol as proposed in the protocol for this review.
Most trials recruited women from the USA (11 trials); three trials were conducted in Venezuela, and one in each Belgium, Canada and India. Sixteen trials were published before the 1990s (one in the 1960s, six in the 1970s, and nine in the 1980s). Results may not be applicable to all settings or countries worldwide, nor to current clinical practice.
Although there were more than 1000 women and their babies in the included trials, individually, sample sizes were small, ranging from 26 to 178 women. Most trials reported on the review primary outcomes adequate pain relief as reported by the woman (N = 13), need for additional analgesia (N = 10) and maternal adverse effects (N = 14). Only three (of 13) pre‐specified outcomes were examined in the included trials; there were no data reported for the primary outcome ‐ neonatal adverse effects ‐ or for any of the secondary review outcomes.
Breastfeeding was clearly stated as an exclusion criterion in most trials, and as a result, no data were available to determine any neonatal adverse effects or effects on breastfeeding. Guidance for the management of perineal pain, including in breastfeeding women, recommends that if oral analgesia is required then paracetamol/acetaminophen should be used first unless contraindicated; if paracetamol is not effective, an oral or rectal non‐steroidal anti‐inflammatory (NSAID) agent such as ibuprofen should be considered in the absence of contraindications (Montgomery 2012; NICE 2015; NIH 2015). Although some guidance indicates that low‐dose aspirin may be considered as an antiplatelet drug for use in breastfeeding women (Bell 2011), it is generally recommended to be used cautiously or avoided during breastfeeding because salicylate and salicylate metabolites are excreted in breast milk. Therefore, there is potential for adverse effects in infants. Longer‐term, high‐dose maternal aspirin administration has been associated with a report of infant metabolic acidosis, and aspirin administration to infants with viral infections has been associated with Reye's syndrome (NIH 2015).
It is recognised that breastfeeding is an unequalled way of providing the ideal food for infants. International guidance, including from the World Health Organization, recommends (where possible) initiation of breastfeeding within the first hour after birth, and exclusive breastfeeding for the first six months of life for optimal growth, development and health, followed by age‐appropriate complementary feeding alongside breastfeeding for two years or more (WHO 2001; WHO 2003). The evidence in this review is thus not applicable to current globally recommended best practice.
Quality of the evidence
Many aspects of methodological quality were unclear for several of the included trials (Figure 2; Figure 3). Except for one included trial, all studies were published before the 1990s. We found a lack of methodological detail provided in published reports. Attempts to contact trial authors to obtain additional information have been unsuccessful. Of the 17 included trials, 15 were assessed at unclear risk of selection bias; this judgement was based on study reports not detailing methods for sequence generation. All trials had unclear risk of selection bias; assessment was based on study reports not detailing methods for concealment of allocation. We assessed 14 trials at low risk of performance and detection bias; the risk was judged as unclear for three trials. Most trials were judged to be at unclear or high risk of attrition bias (a number imputing data); and all were at unclear or high risk of reporting bias, with many reporting very limited outcome data, and none had available trial registration or protocols.
We assessed the quality of the evidence using the GRADE approach as outlined in the GRADE Handbook for pre‐specified outcomes analysed in the main comparison (aspirin versus placebo). Our assessment was that evidence quality was low (adequate pain relief as reported by the women) or very low (need for additional pain relief; maternal adverse effects). Our judgements were based on design limitations in the included trials (all outcomes), possible publication bias (need for additional pain relief) and imprecision (maternal adverse effects). See summary of findings Table for the main comparison.
Potential biases in the review process
We took steps to minimise the introduction of bias during the review process. At least two review authors independently assessed trials for inclusion, performed data extraction, and assessed risk of bias for each of the included trials.
A detailed, systematic search process was conducted by the Information Specialist of Cochrane Pregnancy and Childbirth, without language or publication status restrictions to reduce the risk for potential publication bias. We also searched trial registries for unpublished, planned or ongoing trials. It is possible that additional trials assessing aspirin for perineal pain in the early postpartum period have been published but not identified; and that further trials have been conducted but are not yet published; or both. Should any such studies be identified in the future, we will assess these for inclusion in future updates of this review.
We investigated reporting biases (such as publication bias) using funnel plots for our primary outcomes. Although no clear evidence of reporting bias was observed for adequate pain relief as reported by the woman and maternal adverse effects, the funnel plot for need for additional pain relief demonstrated some asymmetry. This could indicate possible reporting bias, with the smaller published trials reporting exaggerated intervention effect estimates, and the possibility of additional small trials (including those reporting smaller effect estimates) remaining unpublished.
Agreements and disagreements with other studies or reviews
Previous Cochrane Reviews have assessed therapeutic ultrasound (Hay‐Smith 1998), rectal analgesia (Hedayati 2003), local cooling (East 2012b) and topical anaesthetics (Hedayati 2005) for the relief of perineal pain in the postpartum period, revealing mixed results. More recently, following publication of a generic protocol (Chou 2009) for a series of reviews of drugs for perineal pain in the early postpartum period, two reviews have assessed paracetamol (Chou 2013), and NSAIDs (Wuytack 2016). Both Chou 2013 (including 10 trials involving 2307 women) and Wuytack 2016 (including 28 trials involving 4181 women) showed benefits for paracetamol and NSAIDs compared with placebo, in terms of an increase in adequate pain relief and reduced need for additional pain relief for women with perineal pain in the early postpartum period. However, like our review, Chou 2013 and Wuytack 2016 found that the risk of bias was unclear for many of the included studies (most of which were also conducted from the 1960s to 1990s), and adverse effects were often not assessed for women, and were not assessed for infants. Breastfeeding women, and thus breastfeeding outcomes, were not included.
Another Cochrane Review (including 37 trials involving 5743 adults) assessed single dose aspirin (doses ranging from 300 mg to 1200 mg) for acute postoperative pain in adults (Derry 2012). Like our review, Derry 2012 found that compared with placebo, aspirin was shown to increase adequate pain relief, and reduced the need for rescue medication. Although Derry 2012 reported that benefits were seen for 600 mg to 650 mg aspirin, 900 mg to 1000 mg aspirin and 1200 mg aspirin, it was reported that lower doses of aspirin (500 mg) were not significantly different from placebo; however, no formal subgroup interaction tests were performed or reported. Derry 2012 reported no difference in adverse effects when 600 mg to 650 mg aspirin was compared with placebo, but reported an increase in adverse effects with 900 mg to 1000 mg aspirin compared with placebo. No formal subgroup interaction test was performed.
No other reviews were identified that assessed single dose aspirin for perineal pain in the early postpartum period.
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies
Risk of bias summary: review authors' judgements about each risk of bias item for each included study
Funnel plot of comparison: 1 Aspirin versus placebo for perineal pain, outcome: 1.1 Adequate pain relief as reported by the woman
Funnel plot of comparison: 1 Aspirin versus placebo for perineal pain, outcome: 1.2 Need for additional pain relief
Funnel plot of comparison: 1 Aspirin versus placebo for perineal pain, outcome: 1.3 Maternal adverse effects
Comparison 1 Aspirin versus placebo for perineal pain, Outcome 1 Adequate pain relief as reported by the woman.
Comparison 1 Aspirin versus placebo for perineal pain, Outcome 2 Need for additional pain relief.

Comparison 1 Aspirin versus placebo for perineal pain, Outcome 3 Maternal adverse effects.

Comparison 2 300 mg aspirin versus 600 mg aspirin for perineal pain, Outcome 1 Adequate pain relief as reported by the woman.
Comparison 2 300 mg aspirin versus 600 mg aspirin for perineal pain, Outcome 2 Need for additional pain relief.
Comparison 2 300 mg aspirin versus 600 mg aspirin for perineal pain, Outcome 3 Maternal adverse effects.

Comparison 3 600 mg aspirin versus 1200 mg aspirin for perineal pain, Outcome 1 Adequate pain relief as reported by the woman.

Comparison 3 600 mg aspirin versus 1200 mg aspirin for perineal pain, Outcome 2 Need for additional pain relief.
Comparison 3 600 mg aspirin versus 1200 mg aspirin for perineal pain, Outcome 3 Maternal adverse effects.
Comparison 4 300 mg aspirin versus 1200 mg aspirin for perineal pain, Outcome 1 Adequate pain relief as reported by the woman.
Comparison 4 300 mg aspirin versus 1200 mg aspirin for perineal pain, Outcome 2 Need for additional pain relief.
Comparison 4 300 mg aspirin versus 1200 mg aspirin for perineal pain, Outcome 3 Maternal adverse effects.
| Aspirin compared with placebo for perineal pain in the early postpartum period | ||||||
| Patient or population: women with perineal pain in the early postpartum period Settings: 17 RCTs published from 1967 to 1997 (11 RCTs conducted in USA, 3 in Venezuela, 1 each in Belgium, Canada and India) Intervention: aspirin (single dose) Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Aspirin | |||||
| Adequate pain relief as reported by the woman (4 to 8 hours) | Study population | RR 2.03 (1.69, 2.42) | 1001 (13 RCTs) | ⊕⊕⊝⊝ | ||
| 253 per 1000 | 513 per 1000 (427 to 612) | |||||
| Need for additional pain relief (4 to 8 hours) | Study population | RR 0.25 (0.17, 0.37) | 744 (10 RCTs) | ⊕⊝⊝⊝ | ||
| 267 per 1000 | 67 per 1000 (45 to 99) | |||||
| Maternal adverse effects (4 to 8 hours) | Study population | RR 1.08 (0.57, 2.06) | 1067 (14 RCTs) | ⊕⊝⊝⊝ | ||
| 27 per 1000 | 29 per 1000 (15 to 55) | |||||
| Neonatal adverse effects | (0 RCTs) | Not reported by any of the included RCTs | ||||
| Perineal pain at six weeks postpartum | (0 RCTs) | Not reported by any of the included RCTs | ||||
| *The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Study limitations: downgraded two levels due to the serious risk of bias 2Publication bias: downgraded by one level based on visual inspection of funnel plot which indicates likely publication bias 3Imprecision: downgraded one level due to few events and wide 95% CI around the pooled estimate which includes no effect | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adequate pain relief as reported by the woman Show forest plot | 13 | 1001 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.03 [1.69, 2.42] |
| 1.1 300 mg aspirin | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.6 [0.36, 18.88] |
| 1.2 500 to 650 mg aspirin | 11 | 800 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.98 [1.64, 2.39] |
| 1.3 900 mg aspirin | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.83 [0.84, 3.99] |
| 1.4 1200 mg aspirin | 3 | 108 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.75 [1.25, 6.06] |
| 2 Need for additional pain relief Show forest plot | 10 | 744 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.25 [0.17, 0.37] |
| 2.1 300 mg aspirin | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.16 [0.03, 0.79] |
| 2.2 500 to 650 mg aspirin | 9 | 569 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.17, 0.41] |
| 2.3 900 mg aspirin | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.60] |
| 2.4 1200 mg aspirin | 2 | 82 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.06, 0.70] |
| 3 Maternal adverse effects Show forest plot | 14 | 1067 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.57, 2.06] |
| 3.1 300 mg aspirin | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 3.2 500 to 650 mg aspirin | 13 | 892 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.51, 2.53] |
| 3.3 900 mg aspirin | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.5 [0.55, 11.41] |
| 3.4 1200 mg aspirin | 2 | 82 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.10 [0.01, 1.80] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adequate pain relief as reported by the woman Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2 Need for additional pain relief Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Maternal adverse effects Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adequate pain relief as reported by the woman Show forest plot | 2 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.52, 1.39] |
| 2 Need for additional pain relief Show forest plot | 2 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.30, 5.68] |
| 3 Maternal adverse effects Show forest plot | 2 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 69.52] |
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Adequate pain relief as reported by the woman Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2 Need for additional pain relief Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3 Maternal adverse effects Show forest plot | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |