Infiltración con bupivacaína liposomal en el sitio quirúrgico para el tratamiento del dolor posoperatorio
Resumen
Antecedentes
A pesar de las técnicas analgésicas de modalidades múltiples, el dolor posoperatorio agudo sigue siendo una necesidad de salud no atendida; hasta un 75% de los pacientes sometidos a cirugía informan dolor significativo. La bupivacaína liposomal es un analgésico que consiste en clorhidrato de bupivacaína encapsulado en múltiples bicapas lipídicas no concéntricas, que ofrecen un método nuevo de liberación sostenida.
Objetivos
Evaluar la eficacia analgésica y los efectos adversos de la infiltración con bupivacaína liposomal en el sitio quirúrgico para el tratamiento del dolor posoperatorio.
Métodos de búsqueda
El 13 enero 2016, se hicieron búsquedas en CENTRAL, MEDLINE, MEDLINE In‐Process, Embase, ISI Web of Science y en listas de referencias de artículos recuperados. Se obtuvieron informes de ensayos clínicos y sinopsis de estudios publicados y no publicados en fuentes de Internet, y se buscaron ensayos en curso en bases de datos de ensayos clínicos.
Criterios de selección
Se incluyeron los ensayos clínicos aleatorios doble ciego, controlados con placebo o con control activo en pacientes a partir de los 18 años de edad sometidos a cirugía electiva, en cualquier sitio quirúrgico, cuando comparaban la infiltración con bupivacaína liposomal en el sitio quirúrgico con placebo u otro tipo de analgesia.
Obtención y análisis de los datos
Dos autores de la revisión consideraron de forma independiente los ensayos para la inclusión, evaluaron el riesgo de sesgo y extrajeron los datos. Se realizó el análisis de datos utilizando técnicas estadísticas estándar, tal como se describe en el Manual Cochrane para Revisiones Sistemáticas de Intervenciones (Cochrane Handbook for Systematic Reviews of Interventions), utilizando Review Manager 5.3. Se planificó realizar un metanálisis y producir una tabla de “Resumen de los hallazgos” para cada comparación, sin embargo, hubo datos insuficientes para asegurar una respuesta clínicamente significativa. Por lo tanto, se han producido dos tablas de “Resumen de los hallazgos” en un formato narrativo. Cuando fue posible, se evaluó la calidad de las pruebas mediante GRADE.
Resultados principales
Se identificaron nueve estudios (10 informes, 1377 participantes) que cumplieron los criterios de inclusión. Cuatro ensayos de fase II de aumento y disminución de la dosis, diseñados para evaluar y demostrar la eficacia y la seguridad, presentaron datos agrupados que no pudieron utilizarse. De los cinco estudios de brazos paralelos restantes (965 participantes), dos fueron controlados con placebo y tres utilizaron la infiltración del anestésico local clorhidrato de bupivacaína como control. Con la herramienta Cochrane, la mayoría de los estudios se consideró, en general, en riesgo de sesgo poco claro; sin embargo, dos estudios estuvieron en riesgo alto de sesgo de informe selectivo y cuatro estudios estuvieron en riesgo alto de sesgo debido al tamaño (menos de 50 participantes por brazo de tratamiento).
Tres estudios (551 participantes) informaron el resultado primario de la intensidad del dolor acumulativo durante 72 horas después de la cirugía. En comparación con placebo, la bupivacaína liposomal se asoció con una puntuación de dolor acumulativo inferior entre el final de la cirugía (0 horas) y las 72 horas (un estudio, muy baja calidad). En comparación con el clorhidrato de bupivacaína, dos estudios no mostraron ninguna diferencia para este resultado (pruebas de muy baja calidad); sin embargo, no se realizó un metanálisis por las diferencias en la población y el procedimiento quirúrgicos (aumento de mamas versus artroplastia de la rodilla).
No se informó ningún evento adverso grave asociado con la administración de bupivacaína liposomal, y ninguno de los cinco estudios informó las pérdidas debido a los eventos adversos relacionados con los fármacos (pruebas de calidad moderada).
Un estudio informó una puntuación media inferior del dolor a las 12 horas asociada con la bupivacaína liposomal en comparación con clorhidrato de bupivacaína, pero no a las 24; a las 48 ni a las 72 horas después de la cirugía (pruebas de muy baja calidad).
Dos estudios (382 participantes) informaron un tiempo más largo hasta la primera dosis de opiáceos posoperatorios en comparación con placebo (pruebas de baja calidad).
Dos estudios (325 participantes) informaron el uso total de opiáceos posoperatorios durante las primeras 72 horas: un estudio informó un uso de opiáceos acumulativo inferior para la bupivacaína liposomal comparada con placebo (pruebas de muy baja calidad); un estudio no informó ninguna diferencia en comparación con clorhidrato de bupivacaína (pruebas de muy baja calidad).
Tres estudios (492 participantes) informaron el porcentaje de participantes que no requirieron opiáceos posoperatorios durante las 72 horas iniciales después de la intervención quirúrgica. Uno de los dos estudios que comparaban bupivacaína liposomal con placebo demostró que un número mayor de participantes que recibieron bupivacaína liposomal no requirieron opiáceos posoperatorios (pruebas de muy baja calidad). Los otros dos estudios, uno versus placebo y otro versus clorhidrato de bupivacaína, no encontraron diferencias en la necesidad de opiáceos (pruebas de muy baja calidad). No se agruparon los resultados por la heterogeneidad significativa entre los estudios (I2 = 92%).
Todos los estudios incluidos informaron los eventos adversos en el plazo de 30 días de la intervención quirúrgica; los más frecuentes fueron náuseas, estreñimiento y vómitos. De los cinco estudios de brazos paralelos, ninguno realizó ni informó las evaluaciones económicas sanitarias ni los resultados informados por los pacientes diferentes del dolor.
Utilizando GRADE, la calidad de las pruebas varió de moderada a muy baja. La limitación principal fue la escasez de datos para los resultados de interés. Además, el riesgo de sesgo en varios estudios fue alto; este hecho dio lugar a una disminución adicional en la calidad.
Conclusiones de los autores
La bupivacaína liposomal en el sitio quirúrgico parece aliviar el dolor posoperatorio en comparación con placebo; sin embargo, en la actualidad, las pruebas limitadas no demuestran que sea superior al clorhidrato de bupivacaína. No se informó ningún evento adverso grave relacionado con los fármacos ni pérdidas del estudio debido a los eventos adversos relacionados con los fármacos. En general, debido a la baja calidad y al volumen reducido de pruebas la confianza en el cálculo del efecto es limitada y el efecto verdadero puede ser significativamente diferente de este cálculo.
PICO
Resumen en términos sencillos
Bupivacaína liposomal en el sitio de la cirugía para el tratamiento del dolor
Conclusión
La bupivacaína liposomal administrada en el sitio de la cirugía parece aliviar el dolor posoperatorio en comparación con placebo (agua salada). Actualmente hay pruebas limitadas sobre su efectividad en comparación con otros analgésicos, como el clorhidrato de bupivacaína. Se necesitan estudios grandes adicionales para constatar si la bupivacaína liposomal tiene una función en esta área.
Antecedentes
A pesar de los analgésicos, tres de cuatro pacientes informan dolor luego de la cirugía. Un método para tratar el dolor incluye la inyección de un analgésico por parte del cirujano en el sitio de la cirugía con objeto de bloquear los nervios que envían las señales de dolor al cerebro. Se ha desarrollado un nuevo fármaco denominado bupivacaína liposomal diseñado para liberar el analgésico durante un tiempo mucho mayor y proporcionar un alivio del dolor prolongado. Esta revisión se ha diseñado para considerar la efectividad de la bupivacaína liposomal inyectada en el sitio de la cirugía para tratar el dolor y también para considerar si hay algún riesgo asociado con su uso.
Características de los estudios y resultados clave
En enero de 2016, se encontraron nueve estudios (10 informes) con 1377 pacientes que evaluaron la bupivacaína liposomal después de cinco tipos diferentes de cirugía: reemplazo total de rodilla; hemorroidectomía; reparación de la hernia inguinal; bunionectomía y aumento de mamas. Los resultados indicaron que, en comparación con placebo (agua salada), la bupivacaína liposomal fue mejor para aliviar el dolor cuando se la inyectó en el sitio de la cirugía y también redujo tanto la necesidad general de analgésicos opiáceos adicionales (potentes) como el tiempo antes de dicha necesidad. Sin embargo, las pruebas limitadas no indicaron que la bupivacaína liposomal fuese mejor que el analgésico clorhidrato de bupivacaína utilizado actualmente. En general, en ninguno de los estudios incluidos hubo abandonos por los efectos secundarios relacionados con los fármacos.
Calidad de la evidencia
Debido al escaso número de estudios y a algunas limitaciones en la calidad de estos ensayos, la calidad de las pruebas se clasificó de moderada a muy baja. Se necesita más investigación para evaluar la función de la infiltración con bupivacaína liposomal en el sitio quirúrgico para tratar el dolor después de la intervención quirúrgica.
Authors' conclusions
Summary of findings
| Liposomal bupivacaine infiltration at the surgical site compared with placebo for the management of postoperative pain | |||
| Patient or population: aged 18 years and older undergoing elective surgery at any surgical site Settings: inpatient Intervention: surgical site infiltration of liposomal bupivacaine Comparison: surgical site infiltration of placebo | |||
| Outcomes | Impact | Number of participants | Quality of the evidence |
| Cumulative pain score from the end of operation (0 hours) to 72 hours (NRS 0 to 10) | A reduction in cumulative pain score associated with the use of liposomal bupivacaine was reported in one study. The mean cumulative pain score from the end of operation to 72 hours (NRS 0 to 10) in the placebo control group was 202.5 points with the mean cumulative pain score from the end of operation to 72 hours in the liposomal bupivacaine intervention group being 60.7 points lower (90.4 lower to 31.1 lower). | 189 participants (1 study) | ⊕⊝⊝⊝ |
| Serious adverse events | No reported drug‐related serious adverse events, no study withdrawals due to drug‐related adverse events | 382 participants (2 studies) | ⊕⊕⊝⊝ |
| Mean pain score at 12, 24, 48, 72 and 96 hours following surgery (NRS 0 to 10) | No data reported | No studies | |
| Time to first postoperative opioid dose over initial 72 hours | A longer time to first postoperative opioid dose associated with the use of liposomal bupivacaine was reported in two studies. In the placebo control group the time to first postoperative opioid was 4.3 and 1.2 hours compared to 7.2 and 14.3 hours in the liposomal bupivacaine groups respectively. The distribution of data was not reported. | 382 participants (2 studies) | ⊕⊕⊝⊝ |
| Total postoperative opioid consumption over first 72 hours | A reduction in total postoperative opioid consumption over first 72 hours associated with the use of liposomal bupivacaine was reported in one study. In the placebo control group the mean cumulative parenteral morphine equivalent dose over the first 72 hours was 29.1 mg and was 6.8 mg lower (12.8 mg lower to 0.9 mg lower) in the liposomal bupivacaine intervention group. | 189 participants (1 study) | ⊕⊝⊝⊝ |
| Percentage of participants not requiring postoperative opioids over initial 72 hours | One study reported a higher proportion of participants not requiring postoperative opioids over initial 72 hours associated with the use of liposomal bupivacaine (RR 0.82; 95% CI 0.72 to 0.94), and one study found no difference (RR 0.99; 95% CI 0.95 to 1.03). | 382 participants (2 studies) | ⊕⊝⊝⊝ |
| Incidence of adverse events within 30 days of surgery | The incidence of cardiac events and wound complications within 30 days of surgery were not reported in any study Adverse events within 30 days of surgery were reported in all studies with nausea, constipation and vomiting being the most common. | 382 participants (2 studies) | ⊕⊕⊝⊝ |
| CI: confidence interval; NRS: numeric rating scale; RR: risk ratio | |||
| GRADE Working Group grades of evidence | |||
| aWe downgraded the quality of this evidence due to the sparseness of data (‐1), indirectness (‐1) and risk of bias (‐1) due to the unclear risk of bias due to the sample size (50‐199). | |||
| Liposomal bupivacaine infiltration at the surgical site compared with bupivacaine hydrochloride for the management of postoperative pain | |||
| Patient or population: aged 18 years and older undergoing elective surgery at any surgical site Settings: inpatient Intervention: surgical site infiltration of liposomal bupivacaine Comparison: surgical site infiltration of bupivacaine hydrochloride | |||
| Outcomes | Impact | Number of participants | Quality of the evidence |
| Cumulative pain score from the end of operation (0 hours) to 72 hours (NRS 0 to 10) | No difference in cumulative pain score was reported in two studies. In one study the mean cumulative pain score from the end of operation to 72 hours (NRS 0 to 10) in the active control group was 335.0 points and 24.0 points higher (5.7 lower to 53.7 higher) in the liposomal bupivacaine intervention group. In the other study the mean cumulative pain score from the end of operation to 72 hours (NRS 0 to 10) in the active control group was 468.2 points and 26.7 points lower (91.3 lower to 37.9 higher) in the liposomal bupivacaine intervention group. Data were not pooled as differences in outcomes were expected due to differences in surgical interventions between studies. | 379 participants (2 studies) | ⊕⊝⊝⊝ |
| Serious adverse events | No reported drug‐related serious adverse events, no study withdrawals due to drug‐related adverse events | 583 participants (3 studies) | ⊕⊕⊕⊝ |
| Mean pain score at 12, 24, 48, 72 and 96 hours following surgery (NRS 0 to 10) | A reduction in mean pain score at 12 hours, but not 24, 48 or 72 hours, associated with the use of liposomal bupivacaine was reported in one study. Mean pain score at these time points were not reported in other studies. In the study that reported mean pain score (NRS 0 to 10) at 12 hours in the active control group it was 6.9 points and 1.3 points lower (2.4 lower to 0.2 lower) in the liposomal bupivacaine intervention group at this time point. | 134 participants (1 study) | ⊕⊝⊝⊝ |
| Time to first postoperative opioid dose over initial 72 hours | No data reported | No studies | |
| Total postoperative opioid consumption over first 72 hours | No difference in cumulative parenteral morphine equivalent dose over first 72 hours was reported in one study though no estimate of variance was provided and as such estimates of effect could not be calculated. | 134 participants (1 study) | ⊕⊝⊝⊝ |
| Percentage of participants not requiring postoperative opioids over initial 72 hours | No difference in the percentage of participants not requiring postoperative opioids over initial 72 hours was reported in one study (RR 0.95; 95% CI 0.86 to 1.05). | 134 participants (1 study) | ⊕⊝⊝⊝ |
| Incidence of adverse events within 30 days of surgery | The incidence of cardiac events and wound complications within 30 days of surgery were not reported in any study Adverse events within 30 days of surgery were reported in all studies with nausea, constipation and vomiting being the most common. | 583 participants (3 studies) | ⊕⊕⊕⊝ |
| CI: confidence interval; NRS: numeric rating scale; RR: risk ratio | |||
| GRADE Working Group grades of evidence | |||
| aWe downgraded the quality of this evidence one level due to the sparseness of data, a further level because Smoot 2012 was subject to a high risk of bias due to the risk of performance bias and attrition bias due to early termination of the study (as well as the unclear risk of bias due to the sample size (50‐199)), and a further level due to inconsistency. We did not pool of results as we predicted that participant characteristics, as well as nature of postoperative pain, would be different following breast augmentation and knee replacement. As such we expected there to be heterogeneity of the results due to population characteristics, not due to intervention characteristics. | |||
Background
Description of the condition
The treatment of acute postoperative pain remains an unmet health need. Despite the development of guidelines to assist clinicians and allied health professionals to recognise and treat the so‐called ‘fifth vital sign’, it has been reported that up to three quarters of surgical patients receive inadequate pain relief (Apfelbaum 2003; Gan 2014; Lorentzen 2012; Nimmaanrat 2007). Optimising postoperative pain management, and reducing the requirement for systemic analgesia, in particular opiates, through the use of multi‐modal analgesia has many benefits. These include patient benefits, such as reduced morbidity and mortality, as well as benefits to the healthcare system through enhanced patient satisfaction and reduced healthcare‐associated costs including a reduced postoperative length of stay. Furthermore, there is increasing evidence that optimising perioperative and postoperative analgesia reduces the incidence of chronic post‐surgical pain as well as enhancing long‐term patient‐reported functional outcomes (Kehlet 2006).
Description of the intervention
The concept of multi‐modal analgesia was introduced over 20 years ago and its use has expanded to many surgical specialties (Kehlet 1993). Multi‐modal analgesia employs a range of techniques all aiming to inhibit the multiple pathways of nociceptive stimuli along their path, from the site of surgical injury, passing through the peripheral nervous system to the central nervous system. Using paracetamol, non‐steroidal anti‐inflammatory drugs (NSAIDs), gabapentinoids, as well as local and regional anaesthetic techniques, the need for oral or parenteral opioids in the postoperative period, and as a consequence their side effects, is reduced. Local anaesthetic incisional infiltration, where local anaesthetic is infiltrated at the site of the surgical incision at the time of surgery, and local anaesthetic peripheral nerve blocks are commonly used as part of a multi‐modal regime with the view that modification of pain stimuli at their origin will reduce the transmission of nociceptive stimuli, thereby reducing downstream organ dysfunction and pain and stress responses, including centrally mediated changes in the spinal cord or cerebral cortex (Kehlet 2006). The use of liposomal bupivacaine for peripheral nerve blockade will be the subject of a separate review (Hamilton 2016).
Local anaesthetic incisional infiltration is used in a wide range of operations. The local anaesthetic can be administered prior to wound incision as pre‐emptive analgesia, during surgery, or immediately following wound closure. Bupivacaine hydrochloride is the most commonly used local anaesthetic for local infiltration, however its duration of action is a major limiting factor. Despite the addition of drugs such as epinephrine and clonidine to enhance the duration of action many people report significant rebound pain when the effect of the local anaesthetic wears off (Apfelbaum 2003). As such, there has been a great deal of interest in sustained‐release local anaesthetics such as liposomal bupivacaine, which are administered in the same manner but have been reported to have an effect that lasts significantly longer than currently used drugs (Grant 2004).
The adverse effects of bupivacaine and liposomal bupivacaine administered at the surgical site are typical of those associated with other amide‐type local anaesthetics. A major cause of adverse reactions to these drugs is high plasma levels, which may be due to overdosage, rapid absorption from the injection site, diminished tolerance, accidental intravascular injection or slow metabolic degradation. Side effects that require immediate treatment are related to neurological and cardiovascular toxicity, which can cause fits and cardiac arrest resistant to standard treatment. These reactions are generally dose‐related and due to excessively high plasma levels. Other side effects include gastrointestinal symptoms (nausea, vomiting, constipation), nervous system side effects (peri‐oral tingling, dizziness, headache, syncope, somnolence), skin side effects (pruritus), fungal infections and pyrexia. In addition, for liposomal bupivacaine the potential exists for local adverse effects due to the liposomal component, which is known to undergo slow lipid degradation and clearance at the injection site.
How the intervention might work
Liposomal bupivacaine consists of bupivacaine hydrochloride encapsulated within multiple, non‐concentric lipid bi‐layers. This encapsulation technique produces vesicles of a diameter of 10 to 20 micrometres that contain the active drug, which offers a novel method of sustained release (Spector 1996). Release of the active drug from these multi‐vesicular liposomes is via three mechanisms, membrane breakdown, membrane reorganisation and diffusion (Mantripragada 2002). The relative importance of each mechanism is not known.
Following its release from the liposome vesicles, the active component bupivacaine hydrochloride, an amide local anaesthetic, binds to the intracellular portion of voltage‐gated sodium channels thereby preventing depolarisation of the nerve cell and thus conduction of nociceptive stimuli. Bupivacaine hydrochloride is subsequently metabolised, primarily in the liver via a microsomal cytochrome P450 3A4 mediated pathway to pipecoloxylidide, with 5% undergoing renal excretion and around 15% being excreted unchanged (Gantenbein 2000). The multi‐vesicular liposome component of liposome bupivacaine undergoes a slow process of lipid degradation and clearance; studies have demonstrated that a significant proportion of the liposome component is detectable at the injection site at periods exceeding 21 days following administration (Mantripragada 2002).
Why it is important to do this review
Regional anaesthetic techniques using local anaesthetics have an established role as part of a multi‐modal technique across a wide range of surgical specialties. Currently their duration of action is a major limiting factor with patients reporting rebound pain. Liposomal bupivacaine is a new therapy utilising a novel mechanism to provide sustained release of local anaesthetic at the origin of pain, which has the potential to address this limitation. At present there are a limited number of trials evaluating liposomal bupivacaine for the management of postoperative pain. This independent review has been designed to critically appraise the current literature on liposomal bupivacaine administered at the surgical site in people aged 18 years and over undergoing elective surgery to evaluate its clinical and cost effectiveness in managing postoperative pain.
Objectives
To assess the analgesic efficacy and adverse effects of liposomal bupivacaine infiltration at the surgical site for the management of postoperative pain.
Methods
Criteria for considering studies for this review
Types of studies
We included prospective randomised and quasi‐randomised controlled trials (including cluster‐randomised trials) that compared liposomal bupivacaine infiltration against placebo or other types of analgesia. We included data from clinical trials registries and clinical trial records in the review. We included studies irrespective of publication status or language.
Types of participants
We included all trials with participants aged 18 years and older undergoing elective surgery at any surgical site, without restriction on any co‐morbidities.
Types of interventions
We included all double‐blind randomised controlled trials (RCTs) that compared the effects of a single dose of liposomal bupivacaine infiltrated at the surgical site against placebo or other types of analgesia delivered systemically, via local infiltration, perineural injection, or epidural or subarachnoid (spinal) routes. We considered studies reporting on pre‐emptive, intraoperative and postoperative wound infiltration eligible for inclusion provided the drug was administered not earlier than 30 minutes prior to the procedure or later than 30 minutes after wound closure.
Types of outcome measures
We included patient‐reported outcome measures of pain, use of supplementary opiate analgesia (incidence of supplementary analgesia, time to supplementary analgesia, mean and total opiate consumption, opiate or other analgesia‐related adverse events) and measures of cost effectiveness. We included withdrawals from the trials and adverse events.
Primary outcomes
-
Cumulative pain intensity assessed on a 100 mm visual analogue scale (VAS) over the initial 72 hours following surgery, at rest or with activity. However, we considered all types of pain scales with standardisation of pain intensity data described by other means than a 100 mm VAS, where possible.
-
Serious adverse events, specifically incidence of cardiac events and incidence of wound complications within 30 days of surgery.
Secondary outcomes
-
Mean pain score, at rest or with activity, assessed on a 100 mm VAS at 12, 24, 48, 72 and 96 hours following surgery. We considered all types of pain scales with standardisation of pain intensity data described by other means than a 100 mm VAS, where possible.
-
Time to first postoperative opioid dose over initial 72 hours.
-
Total postoperative opioid consumption over first 72 hours.
-
Percentage of participants not requiring postoperative opioids over initial 72 hours.
-
Health economics assessed using a recognised health economic technique.
-
Incidence of adverse events within 30 days of surgery.
-
Patient‐reported outcomes, using validated outcome scores, at any time point following surgery.
Search methods for identification of studies
Electronic searches
We searched the following electronic databases:
-
CENTRAL (in the Cochrane Library) Issue 1, 2016;
-
MEDLINE (Ovid) 1946 to 13 January 2016;
-
Embase (Ovid) 1974 to 13 January 2016;
-
Web of Science (ISI Web of Knowledge) 1945 to 13 January 2016;
We used MeSH or equivalent and text word terms with no language restrictions. We tailored searches to individual databases. The search strategies used are shown in Appendix 1.
Searching other resources
We searched the metaRegister of controlled trials (mRCT) (www.isrctn.com/page/mrct) (4 January 2016), clinicaltrials.gov (www.clinicaltrials.gov) (4 January 2016) and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) (http://apps.who.int/trialsearch/) (4 January 2016) for ongoing trials. In addition, we searched reference lists of reviews and retrieved articles for additional studies and citation searches performed on key articles. We contacted study authors where necessary for additional information.
Data collection and analysis
Selection of studies
We assessed studies independently and in duplicate for eligibility (TWH, VA). In the first instance, we selected studies from the title and abstract. For those deemed relevant, we obtained the full text. Different pairs of authors (TWH, VA, LHS) assessed the full text according to the eligibility criteria. We resolved disagreement by consensus with input from the senior author (HP). We have presented a summary of the search strategy yield and study selection as a PRISMA flowchart (Liberati 2009). We retrieved the full texts of eligible studies and collated data where there were multiple publications of individual studies.
Data extraction and management
Two authors (TWH, VA) extracted data independently and in duplicate and recorded them onto a pre‐tested, standardised, electronic data collection form. We resolved inconsistency in data collection by discussion with input of a third author (LHS). Where additional information was required we contacted the study authors and study sponsors.
Assessment of risk of bias in included studies
We used the Oxford Quality Score (Jadad 1996) as the basis for inclusion, limiting inclusion to studies that were randomised and double blind as a minimum.
Two authors (TWH, VA) also independently assessed risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a) and adapted from those used by Cochrane Pregnancy and Childbirth, with any disagreements resolved by discussion. We assessed the following for each study.
-
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (any truly random process, e.g. random number table; computer random number generator); unclear risk of bias (method used to generate sequence not clearly stated). We excluded studies using a non‐random process (e.g. odd or even date of birth; hospital or clinic record number).
-
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions prior to assignment determines whether intervention allocation could have been foreseen in advance of or during recruitment, or changed after assignment. We assessed the methods as: low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes); unclear risk of bias (method not clearly stated). We excluded studies that did not conceal allocation (e.g. open list).
-
Blinding of participants and personnel (checking for possible performance bias). We assessed the methods used to blind study participants and personnel from knowledge of which intervention a participant received. We assessed methods as: low risk of bias (study states that it was blinded and describes the method used to achieve blinding, such as identical tablets matched in appearance or smell, or a double‐dummy technique); unclear risk of bias (study states that it was blinded but does not provide an adequate description of how it was achieved). We excluded studies that were not double blind.
-
Blinding of outcome assessment (checking for possible detection bias). We assessed the methods used to blind study participants and outcome assessors from knowledge of which intervention a participant received. We assessed the methods as: low risk of bias (study has a clear statement that outcome assessors were unaware of treatment allocation, and ideally describes how this was achieved); unclear risk of bias (study states that outcome assessors were blind to treatment allocation but lacks a clear statement on how it was achieved). Studies where outcome assessment was not blinded were excluded.
-
Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data). We assessed the methods used to deal with incomplete data as: low risk (less than 10% of participants did not complete the study or used ‘baseline observation carried forward’ analysis, or both); unclear risk of bias (used 'last observation carried forward' analysis); high risk of bias (used 'completer' analysis).
-
Selective outcome reporting. We compared outcomes of interest published in the protocol, clinical trials registry entry and methods section against those published in the study report. Where all outcomes of interest were reported then we considered these studies as at low risk of bias. Where there was incomplete outcome data reporting we considered these studies as at high risk of bias.
-
Size of study (checking for possible biases confounded by small size). We assessed studies as being at: low risk of bias (200 participants or more per treatment arm); unclear risk of bias (50 to 199 participants per treatment arm); high risk of bias (fewer than 50 participants per treatment arm).
Measures of treatment effect
A lack of data prevented a quantitative assessment of the efficacy of liposomal bupivacaine infiltration at the surgical site for the management of postoperative pain. For dichotomous data we planned to calculate the risk ratio (RR) and for continuous data the standardised mean difference (SMD), along with 95% confidence intervals (95% CI) (RevMan 2014). We planned, where possible, for efficacy outcomes, to calculate the numbers needed to treat for a beneficial outcome (NNTB) and harmful outcome (NNTH) for adverse events.
Unit of analysis issues
We assessed outcomes at the patient level and proposed to analyse studies involving multiple treatment arms by dividing the sample size of the control group into the appropriate number of groups depending on the number of arms of the trial.
Dealing with missing data
We contacted study authors and sponsors to request further information in the event of missing data. We did not attempt data imputation because of the controversies associated with imputing data from multiple scoring schemes, especially due to possible small sample sizes per scoring scale (Sterne 2009).
Assessment of heterogeneity
We examined the heterogeneity of included studies, where possible, using the I2 statistic (Higgins 2003) as described in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011). Where there was substantial heterogeneity (that is I2 greater than 85%) we did not attempt pooled analysis. Had it been possible to perform meta‐analysis, as we expected a degree of variability among the eligible studies in terms of the measurement scale used and the subjectivity of the outcome, we planned to use a random‐effects model.
Assessment of reporting biases
We assessed for publication bias, due to non‐reporting of negative studies, by contacting the principal investigators of unpublished trials registered as completed on trial registries. As there were fewer than 10 studies included we did not explore publication bias by means of a funnel plot.
Data synthesis
A lack of data prevented a quantitative assessment of the efficacy of liposomal bupivacaine infiltration at the surgical site for the management of postoperative pain and as such we did not perform meta‐analysis. In future updates of this review, where outcome data are found to be of sufficient quality, and participants, interventions, comparisons and outcomes judged to be sufficiently similar to ensure an answer that is clinically meaningful, then we will perform a meta‐analysis.
Quality of the Evidence
We planned to assess the quality of the evidence for each of the primary and secondary outcomes independently in duplicate (TH, LS) using the GRADE system for all of the primary and secondary outcomes assessed. However, this was not possible for all outcomes due to the lack of data available.
The GRADE approach uses five considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of the body of evidence for each outcome.
The GRADE system uses the following criteria for assigning grade of evidence.
-
High: we are very confident that the true effect lies close to that of the estimate of the effect.
-
Moderate: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different.
-
Low: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect.
-
Very low: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect.
The grade is decreased if:
-
serious (‐1) or very serious (‐2) limitation to study quality;
-
important inconsistency (‐1);
-
some (‐1) or major (‐2) uncertainty about directness;
-
imprecise or sparse data (‐1);
-
high probability of reporting bias (‐1).
Further information on the use of the GRADE System and GRADEprofiler Guideline Development Tool software can be found in Chapter 12.2 of the Cochrane Handbook for Systematic Reviews of Interventions (GRADEPro GDT 2015; Schünemann 2011).
'Summary of findings' table
Due to the lack of data we have produced two 'Summary of findings' tables as a narrative to present the main findings in a transparent and simple tabular format. In future updates, we will update these depending on data availability.
Subgroup analysis and investigation of heterogeneity
We did not perform subgroup analysis due to lack of sufficient data. In future updates of this review we will carry out subgroup analysis for different doses (based on the licensed recommendations for dosage) of liposomal bupivacaine administered and different surgical sites. The indications for these subgroup analyses are that in basic science studies it has been demonstrated that a dose response curve is seen and, as such, the dose of liposomal bupivacaine may have an effect on outcome. Furthermore, different surgeries will have different pain profiles and, in addition, the release pattern of bupivacaine hydrochloride from liposomal bupivacaine may be altered by the local environment and therefore different efficacies may be observed at different surgical sites.
Sensitivity analysis
We planned to perform sensitivity analysis based on the following domains from the Cochrane tool for assessing risk of bias: blinding of outcome assessment and incomplete outcome data. Due to lack of sufficient data, we did not perform sensitivity analysis.
Results
Description of studies
Results of the search
Using electronic searches we identified 179 possible studies for inclusion. We identified an additional 43 possible studies, 40 by searches of clinical trials registers and three by searching reference lists of included studies. After removal of duplicates, we screened the titles of 127 records and excluded 59 studies as these were irrelevant. We explored the full text of 68 studies. We excluded 21 studies (see Excluded studies) and identified 37 ongoing studies, leaving nine studies (10 reports) for inclusion in the review (see Characteristics of included studies). For a flowchart of the study selection process, please see Figure 1.

Study flow diagram
Included studies
We identified nine studies (10 reports) involving 1377 participants that met inclusion criteria, with 780 participants randomised to receive liposomal bupivacaine infiltration at the surgical site. All studies were conducted in the inpatient setting. In the control group, two studies used placebo (0.9% sodium chloride) at the surgical site (one haemorrhoidectomy Gorfine 2011 (results of this study also reported by Schmidt 2012 (secondary reference of Gorfine 2011)), one bunionectomy (Golf 2011)) and seven studies used bupivacaine hydrochloride at the surgical site (Bramlett 2012; Haas 2012; Langford 2008; NCT 00744848; NCT 00745290; Smoot 2012 (long‐term follow‐up of this study reported by Minkowitz 2012 (secondary reference of Smoot 2012); White 2009).
Studies were conducted across five surgical sites including:
-
total knee replacement; two studies, 383 participants (Bramlett 2012; NCT 00745290);
-
haemorrhoidectomy; three studies, 493 participants (Gorfine 2011; Haas 2012; NCT 00744848);
-
inguinal hernia repair; two studies, 174 participants (Langford 2008; White 2009);
-
bunionectomy; one study, 193 participants (Golf 2011);
-
breast augmentation, one study, 134 participants (Smoot 2012).
The dose of liposomal bupivacaine in included studies ranged from 66 mg to 532 mg. The timing of administration of liposomal bupivacaine or control varied between studies. In one study it was administered in a staged fashion, starting after dissection (Bramlett 2012), in three studies it was administered intra‐operatively but the timing was not specified (Golf 2011; NCT 00745290; White 2009) and in five studies it was administered at the end of surgery (Gorfine 2011; Haas 2012; Langford 2008; NCT 00744848; Smoot 2012). We identified five simultaneous parallel‐arm studies (Golf 2011; Gorfine 2011; NCT 00744848; NCT 00745290; Smoot 2012) and four Phase II adaptive trials, where the dose was escalating/de‐escalating (Bramlett 2012; Haas 2012; Langford 2008; White 2009). The adaptive trials randomised sequential cohorts of participants to control or intervention arms, with the dose of liposomal bupivacaine in the intervention arm increased or decreased conditional on the efficacy and safety of the previous cohort. We have discussed the results of the simultaneous parallel‐arm and adaptive‐design studies separately.
We have given details of randomisation schedule and interventions, together with details of all eligible studies, in the Characteristics of included studies tables. Outcomes of interest were not investigated in all studies or not reported (reporting bias), or reported in an idiosyncratic manner in adaptive trial designs. As such we were not able to include data from every study in all analyses.
Excluded studies
We excluded 21 studies as:
-
nine were not RCTs;
-
six were review papers;
-
two trials, both assessing liposomal bupivacaine for bilateral breast augmentation, used the patient as own control preventing inclusion of these data;
-
two trials were not appropriately blinded, with the outcome assessors or participants, or both, not blinded to treatment allocation;
-
one trial did not assess liposomal bupivacaine at the surgical site;
-
one included bupivacaine hydrochloride, at different doses, in both the control and intervention arm.
The study that included bupivacaine hydrochloride, at different doses in both the control and intervention arm, was an RCT evaluating the efficacy of liposomal bupivacaine at the surgical site for the management of pain following total knee replacement. This trial compared 266 mg liposomal bupivacaine mixed with 75 mg bupivacaine hydrochloride against an active control arm of 150 mg bupivacaine hydrochloride. At the time of writing the trial protocol it was not advised to mix liposomal bupivacaine with other drugs, in particular bupivacaine, due the risk of premature de‐encapsulation of liposomal bupivacaine. As such we excluded this study from the analysis. However, it must be noted that in December 2015, an amendment to the FDA‐licensed indication was made which approved admixing liposomal bupivacaine with bupivacaine, including co‐administration in the same syringe, as it has been proposed that admixing with bupivacaine hydrochloride enhances early postoperative analgesia. Whilst excluded from this analysis, in future updates of this review we will include studies evaluating liposomal bupivacaine admixed with bupivacaine hydrochloride. We have given details of excluded studies in the Characteristics of excluded studies tables.
We also identified 37 ongoing studies, details of which are given in the Characteristics of ongoing studies tables.
Risk of bias in included studies
A summary of the risk of bias assessment using the Cochrane tool can be seen in Figure 2 and Figure 3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study
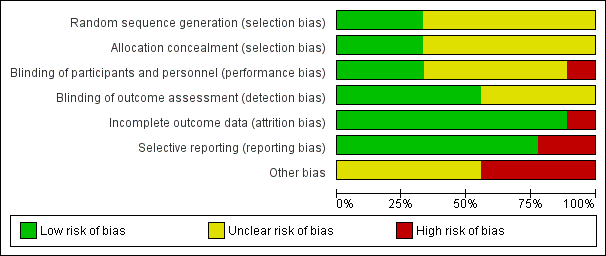
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies
On the five‐point Oxford Scale (Jadad 1996) addressing randomisation, blinding, and withdrawals, five studies scored three points (Langford 2008; NCT 00744848; NCT 00745290; Smoot 2012; White 2009), three studies scored four points (Golf 2011; Gorfine 2011; Haas 2012), and one study scored five points (Bramlett 2012), with studies scoring three or more points considered unlikely to be subject to major systematic bias (Khan 1996).
Allocation
Three studies clearly described the method of randomisation (computer‐generated randomisation) and allocation concealment (central‐allocation) and we assigned them a low risk of bias for selection bias (Bramlett 2012; Golf 2011; Smoot 2012).
Six studies (two conference abstracts (Langford 2008; White 2009), two clinical trials registry entries (NCT 00744848; NCT 00745290) and two complete manuscripts (Gorfine 2011; Haas 2012)), did not describe the method of random sequence generation or allocation concealment and as such presented an unclear risk of selection bias.
Blinding
Blinding of participants and personnel (performance bias)
Liposomal bupivacaine is cloudy and has a different visual appearance to both normal saline and bupivacaine hydrochloride. Furthermore liposomal bupivacaine is more viscous than both normal saline and bupivacaine hydrochloride. As such there is a risk of performance bias by the surgeon who administers drug at the time of surgery. To reduce the risk of performance bias the injection technique was standardised in three trials (Bramlett 2012; Gorfine 2011; Haas 2012) and we regarded these as presenting a low risk of performance bias.
Five studies (two conference abstracts (Langford 2008; White 2009), two clinical trials registry entries (NCT 00744848; NCT 00745290) and one manuscript (Golf 2011), did not state whether the injection technique was standardised, presenting an unclear risk of performance bias.
One study (Smoot 2012), left the injection technique at the discretion of the operating surgeon, presenting a high risk of performance bias. Both the participant and outcome assessor remained blinded and as such we considered this study double‐blind and included it in this review.
Blinding of outcome assessment (detection bias)
Five studies blinded participants and staff involved in assessment of outcome measures to the treatment allocation (Bramlett 2012; Golf 2011; Gorfine 2011; Haas 2012; Smoot 2012), presenting a low risk of detection bias.
Four studies (two conference abstracts (Langford 2008; White 2009) and two clinical trials registry entries (NCT 00744848; NCT 00745290), did not state whether the participants and staff were blinded, presenting an unclear risk of detection bias.
Incomplete outcome data
We assessed one study as having a high risk of attrition bias (Smoot 2012), as it was terminated early by the study sponsor for "administrative reasons". We assessed all other studies as having a low risk of attrition bias with greater than 95% follow‐up of randomised participants.
Selective reporting
We assessed seven of the nine studies as having a low risk of reporting bias. As each included study investigated a number of outcome measures, often at multiple time points, outcome measures were commonly reported as being non‐significant without other measures of variance being reported. We assessed two studies as having a high risk of reporting bias (Bramlett 2012; Haas 2012), as cumulative pain scores other than that of the primary endpoint were not reported.
Other potential sources of bias
As the majority of trials were drug development trials, that is, Phase II and Phase III, the sample size of the treatment and control groups was small. We considered four of the included trials at high risk of bias due to having fewer than 50 participants per treatment arm (Bramlett 2012; Haas 2012; Langford 2008; White 2009), with the remainder considered at unclear risk of bias due to sample sizes of between 50 and 199 participants per treatment arm (Golf 2011; Gorfine 2011; NCT 00744848; NCT 00745290; Smoot 2012). All studies were commissioned, funded or published by Pacira Pharmaceuticals Incorporated, manufacturer of liposomal bupivacaine, presenting an unclear risk of bias.
Effects of interventions
See: Summary of findings for the main comparison Summary of findings: liposomal bupivacaine vs placebo; Summary of findings 2 Summary of findings: liposomal bupivacaine vs bupivacaine hydrochloride
Results of simultaneous parallel‐arm studies
The five simultaneous parallel‐arm studies reported the following outcomes (Golf 2011; Gorfine 2011; NCT 00744848; NCT 00745290; Smoot 2012).
Cumulative pain intensity over 72 hours following surgery
-
Three studies with 551 participants assessed cumulative pain intensity over 72 hours following surgery (Gorfine 2011; NCT 00745290; Smoot 2012). One study assessing liposomal bupivacaine 266 mg in participants undergoing excisional haemorrhoidectomy, was placebo controlled (Gorfine 2011), and two studies using liposomal bupivacaine 532 mg, in total knee replacement and breast augmentation respectively, used bupivacaine hydrochloride 200 mg as a control (NCT 00745290; Smoot 2012). They recorded pain scores on an 11‐point Numeric Rating Scale (NRS; 0 to 10) with cumulative pain intensity over 72 hours calculated using a windowed worst observation carried forward, plus last observation carried forward method in two studies (Gorfine 2011; Smoot 2012) (Appendix 2). One study did not specify the method of calculation (NCT 00745290).
-
We have reported the results of these studies in Analysis 1.1 (Figure 4).
-
One placebo‐controlled study demonstrated a significant reduction in cumulative pain score over 72 hours associated with the use of liposomal bupivacaine (Gorfine 2011). Using GRADE, we downgraded the quality of this evidence one level due to the sparseness of data, a further level due to indirectness and concerns about the generalisability of limited data presented to the population undergoing elective surgery, and one level due to the unclear risk of bias presented by the sample size of the included study (summary of findings Table for the main comparison). Overall we judged the evidence to be of very low quality, meaning that we have very little confidence in the effect estimate and that the true effect is likely to be substantially different from the estimate of effect.
-
The two studies that used bupivacaine hydrochloride as a control did not demonstrate a difference in cumulative pain scores from 0 to 72 hours associated with the use of liposomal bupivacaine (NCT 00745290; Smoot 2012). Whilst both studies did not demonstrate a difference in cumulative pain score, we decided not to pool the data as we predicted that participant characteristics, as well as the nature of postoperative pain, would be different following breast augmentation and knee replacement, and as such a pooled analysis would not be appropriate. Using GRADE, we downgraded the quality of this evidence one level due to the sparseness of data, a further level due to Smoot 2012 being subject to a high risk of performance bias and attrition bias due to early termination of the study (as well as an unclear risk of bias due to the sample size), and by a further level due to indirectness and concerns about the generalisability of limited data presented to the population undergoing elective surgery (summary of findings Table 2). Overall we judged the evidence to be of very low quality, meaning that we have very little confidence in the effect estimate and that the true effect is likely to be substantially different from the estimate of effect.
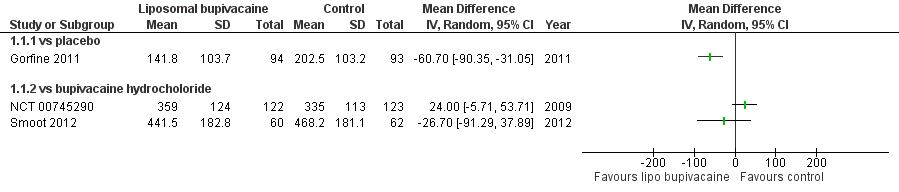
Forest plot of comparison: 1 Liposomal bupivacaine vs control, outcome: 1.1 Cumulative pain score 0 to 72 hours
Serious adverse events
-
There were no serious adverse events reported to be associated with the use of liposomal bupivacaine and none of the five studies (Golf 2011; Gorfine 2011; NCT 00744848; NCT 00745290; Smoot 2012), 964 participants, reported withdrawals due to drug‐related adverse events.
-
Compared to placebo, 2 studies, 382 participants, using GRADE, we downgraded the quality of this evidence one level due sparseness of the data and one level due to Golf 2011 being subject to a high risk of performance bias and both studies presenting an unclear risk of bias due to their sample size (50‐199). Overall we judged the evidence to be of low quality, meaning our confidence in the effect estimate is limited and that the true effect may be substantially different from the estimate of the effect.
-
Compared to bupivacaine hydrochloride, 3 studies, 583 participants, using GRADE, we downgraded the quality of this evidence one level due to Smoot 2012 being subject to a high risk of performance bias and attrition bias due to early termination of the study. Overall we judged the evidence to be of moderate quality, meaning we were moderately confident in the effect estimate and that the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different.
Mean pain score at 12, 24, 48, 72 and 96 hours following surgery
-
The mean pain score using an 11‐point NRS (0 to 10) at 12, 24, 48 and 72 hours following breast augmentation surgery was reported by Smoot 2012, 136 participants, who found a significantly lower pain score at 12 hours in those participants receiving liposomal bupivacaine 532 mg compared to bupivacaine hydrochloride 200 mg (P = 0.014; Figure 5) with no difference in mean pain score (mean NRS not reported) found at 24, 48 or 72 hours.
-
The other simultaneous parallel‐arm studies did not report mean pain score at 12, 24, 48, 72 or 96 hours following surgery.
-
Using GRADE, we downgraded the quality of this evidence one level due to the sparseness of data; a further level due to Smoot 2012 being subject to a high risk of performance bias and attrition bias due to early termination of the study; and by a further level due to indirectness and the limitations in interpreting data from a single study. Overall we judged the evidence to be of very low quality, meaning we had very little confidence in the effect estimate and that the true effect is likely to be substantially different from the estimate of effect.

Table of results for included simultaneous parallel‐arm trials
Time to first postoperative opioid dose over initial 72 hours
-
Two studies, 382 participants, reported the median time to first opioid dose following bunionectomy and haemorrhoidectomy respectively with both studies finding the time to first postoperative opioid dose to be significantly longer (P < 0.0001) in those participants receiving liposomal bupivacaine (106 mg and 266 mg respectively) compared to placebo (Golf 2011; Gorfine 2011). We did not pool the data as they were not normally distributed and in addition participant characteristics, as well as the nature of postoperative pain, would be different following bunionectomy and haemorrhoidectomy; as such, a pooled analysis would not be appropriate.
-
The other simultaneous parallel‐arm studies did not report time to first postoperative opioid dose.
-
Using GRADE, we downgraded the quality of this evidence one level due to the sparseness of data and a further level due to Golf 2011 being subject to a high risk of performance bias. Overall we judged the evidence to be of low quality, meaning that our confidence in the effect estimate was limited and the true effect may be substantially different from our estimate of effect.
Total postoperative opioid consumption over first 72 hours
-
Two studies with 325 participants reported the total postoperative opioid consumption over the first 72 hours (Gorfine 2011; Smoot 2012).
-
One study compared liposomal bupivacaine 266 mg with placebo (Gorfine 2011) in participants undergoing haemorrhoidectomy and reported a reduction in cumulative parenteral morphine equivalent dose of 6.8 mg (95% CI (‐12.8 mg to ‐0.9 mg) for the liposomal bupivacaine arm. Using GRADE, we downgraded the quality of this evidence one level due to the sparseness of data, a further level due to indirectness due to the limitations in interpreting data from a single study, and a further level due to the unclear risk of bias due the sample size of the included study (50‐199). Overall we judged the evidence to be of very low quality, meaning we had very little confidence in the effect estimate and that the true effect is likely to be substantially different from the estimate of effect.
-
One trial compared liposomal bupivacaine 532 mg with bupivacaine hydrochloride 200 mg (Smoot 2012) in participants undergoing breast augmentation and found no difference in cumulative parenteral morphine equivalent dose (Figure 5). Using GRADE, we downgraded the quality of this evidence one level due to the sparseness of data, a further level due to indirectness due to the limitations in interpreting data from a single study and a further level due to Smoot 2012 being subject to a high risk of performance bias and attrition bias due to early termination of the study. Overall we judged the evidence to be of very low quality, meaning we had very little confidence in the effect estimate and that the true effect is likely to be substantially different from the estimate of effect.
Percentage of participants not requiring postoperative opioids over initial 72 hours
-
Three studies with 492 participants reported the percentage of participants not requiring postoperative opioids over initial 72 hours following surgery (Golf 2011; Gorfine 2011; Smoot 2012). Two studies were placebo controlled (Gorfine 2011; Golf 2011), and one study used bupivacaine hydrochloride as a control (Smoot 2012). One of the two studies comparing liposomal bupivacaine with placebo demonstrated a higher number of participants receiving liposomal bupivacaine did not require postoperative opioids. The other two studies, one versus placebo, one versus bupivacaine hydrochloride, found no difference in opioid requirement. Due to significant heterogeneity between the studies (I2 = 92%) we have not shown the pooled result (Figure 6). Possible reasons for heterogeneity could be due to differences in pain response after different surgical procedures or differences in pain response between patient groups undergoing specific surgeries.
-
Compared to placebo, 2 studies, 382 participants, using GRADE, we downgraded the quality of this evidence one level due to the sparseness of data, one level due to inconsistency of data, and a further level due to Golf 2011 being subject to a high risk of performance bias and unclear risk of bias due to the sample size (50‐199). Overall we judged the evidence to be of very low quality, meaning we had very little confidence in the effect estimate and that the true effect is likely to be substantially different from the estimate of effect.
-
Compared to bupivacaine hydrochloride, 1 study, 134 participants, using GRADE, we downgraded the quality of this evidence one level due to the sparseness of data, a further level due to Smoot 2012 being subject to a high risk of performance bias and attrition bias due to early termination of the study and by a further level due to indirectness and the limitations in interpreting data from a single study. Overall we judged the evidence to be of very low quality, meaning we had very little confidence in the effect estimate and that the true effect is likely to be substantially different from the estimate of effect.

Forest plot of comparison: 1 Liposomal bupivacaine vs control, outcome: 1.2 Participants not requiring postoperative opioids
Health economics assessment
-
None of the included studies presented a health economic assessment.
Incidence of adverse events within 30 days of surgery
-
None of the included studies reported the incidence of cardiac events and wound complications within 30 days of surgery.
-
All the included studies reported adverse events within 30 days of surgery, with nausea, constipation and vomiting being the most common.
-
Nausea was reported in 38% of participants receiving placebo (Golf 2011), 31% to 58% of those receiving bupivacaine hydrochloride (NCT 00744848; NCT 00745290; Smoot 2012) and 44% to 62% of those participants receiving liposomal bupivacaine (Golf 2011; NCT 00744848; NCT 00745290; Smoot 2012).
-
Constipation was reported in between 9% and 38% of participants receiving bupivacaine hydrochloride (NCT 00744848; NCT 00745290; Smoot 2012) and between 16% and 46% of participants receiving liposomal bupivacaine (NCT 00744848; NCT 00745290; Smoot 2012).
-
Vomiting was reported in 18% of participants receiving placebo (Golf 2011), 20% to 34% of those receiving bupivacaine hydrochloride (NCT 00745290; Smoot 2012) and 15% to 31% of participants receiving liposomal bupivacaine (Golf 2011; NCT 00745290; Smoot 2012).
-
We decided not to pool the data as we predicted that participant characteristics, as well as the nature of adverse events, would be different following different surgical procedures and, as such, a pooled analysis would not be appropriate.
-
Compared to placebo, 2 studies, 382 participants, using GRADE, we downgraded the quality of this evidence one level due sparseness of the data and one level due to Golf 2011 being subject to a high risk of performance bias and both studies presenting an unclear risk of bias due to their sample size (50‐199). Overall we judged the evidence to be of low quality, meaning our confidence in the effect estimate is limited and that the true effect may be substantially different from the estimate of the effect.
-
Compared to bupivacaine hydrochloride, 3 studies, 583 participants, using GRADE, we downgraded the quality of this evidence one level due to Smoot 2012 being subject to a high risk of performance bias and attrition bias due to early termination of the study. Overall we judged the evidence to be of moderate quality, meaning we were moderately confident in the effect estimate and that the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different.
Patient‐reported outcomes
-
None of the included studies reported patient‐reported outcomes (outside of pain), using validated outcome scores, at any time point following surgery.
Results of adaptive‐design trials
We identified four dose‐escalating/de‐escalating studies (Bramlett 2012; Haas 2012; Langford 2008; White 2009). Dose‐escalating/de‐escalating studies are designed to evaluate efficacy and safety. An illustrative example of a typical adaptive‐design trial is shown in Figure 7. In the four Phase II adaptive‐design studies (Bramlett 2012; Haas 2012; Langford 2008; White 2009), the results from the control groups of all dose‐escalating steps in the randomisation process were reported collectively as a single population. Data from adaptive‐design trials cannot be included in meta‐analysis for a number of reasons: a) the decision to escalate or de‐escalate a dose is conditional on the failure of the previous dose on either the efficacy, or safety, or cost‐effectiveness of the intervention, introducing bias in any pooled analysis, and b) the randomisation ratio is altered with each escalation/de‐escalation while the control group population is typically reported cumulatively for all dose levels. We therefore have decided to report the information from these studies as a narrative and we have included it in Figure 8. In hindsight, due to the role of adaptive‐design trials in identifying an efficient and safe dose for further exploration of the intervention in larger scale trials, and the limitations imposed in including such data in meta‐analyses, we may consider excluding trials of this design from the definition in Types of studies in future updates of this review.

Illustrative example of an adaptive‐design trial. The decision to escalate, or de‐escalate a dose is conditional on the failure of the previous dose on the efficacy, or safety, or cost‐effectiveness of the intervention, introducing bias in any pooled analysis. The randomisation ratio is altered with each escalation/de‐escalation while the control group population is typically reported cumulatively for all dose levels

Table of results for adaptive‐design trials
Discussion
Summary of main results
We identified nine studies that met inclusion criteria for this review. Four Phase II dose‐escalating/de‐escalating studies (Bramlett 2012; Haas 2012; Langford 2008; White 2009), designed to evaluate and demonstrate efficacy and safety, presented pooled data which could not be used in this analysis. Of the remaining five studies two were placebo controlled (Golf 2011; Gorfine 2011) and three used bupivacaine hydrochloride as a control (NCT 00744848; NCT 00745290; Smoot 2012).
Compared to placebo one study (Gorfine 2011) reported a lower cumulative pain score 0 to 72 hours after surgery, two studies reported a longer time to first postoperative opioid (Golf 2011; Gorfine 2011), and one study reported a lower cumulative opioid consumption 0 to 72 hours after surgery associated with the used of liposomal bupivacaine (Gorfine 2011). Compared to bupivacaine hydrochloride two studies found no difference in the cumulative pain score 0 to 72 hours after surgery associated with the use of liposomal bupivacaine (NCT 00745290; Smoot 2012), and one study reported a lower mean pain score at 12 hours, but not at 24, 48 or 72 hours postoperatively (Smoot 2012). Three studies reported the number of participants not requiring postoperative opioids (Golf 2011; Gorfine 2011; Smoot 2012), however significant heterogeneity (I2 = 92%) was observed, limiting further analysis. Data comparing liposomal bupivacaine with femoral nerve block were not available for inclusion in the analysis.
Of the five parallel‐arm studies which did not have an adaptive design assessing liposomal bupivacaine against either placebo or bupivacaine hydrochloride, no studies reported health economic assessments or patient‐reported outcomes other than pain (Golf 2011; Gorfine 2011; NCT 00744848; NCT 00745290; Smoot 2012). Nausea, constipation and vomiting were the most commonly reported adverse events. Data regarding cardiac events and wound complications were not reported. No withdrawals were reported to be due to drug‐related adverse events.
Using GRADE we considered the quality of evidence to be very low to moderate with further research considered very likely to have an important impact on our confidence in the estimate of effect. This assessment of quality was predominantly due to sparseness of data as well as a high risk of bias in some of the included studies.
Liposomal bupivacaine does appear to have efficacy in reducing postoperative pain compared to placebo when infiltrated at the surgical site, but, at present the limited evidence does not demonstrate superiority to bupivacaine hydrochloride. Due to the low quality and volume of evidence our confidence in the effect estimate is limited and the true effect may be substantially different from our estimate.
Overall completeness and applicability of evidence
The main limitations of this review are the small number of studies, incomplete outcome data reporting and significant heterogeneity observed between studies. The use of adaptive‐design dose‐escalating/de‐escalating Phase II studies is necessary to evaluate and demonstrate efficacy and safety of a novel drug, however, when the decision to escalate/de‐escalate is conducted in a conditional manner this can lead to the introduction of bias as well as leading to imbalance in the randomisation ratio (where the control group is reported cumulatively). This review found four of the nine included studies to be of an adaptive design, and we decided to exclude these from the analysis. Assessing the five remaining studies, these were conducted across four surgical sites (bunionectomy, haemorrhoidectomy, knee replacement and breast augmentation) with differences in pain profiles, as well as differences in the way that people report pain at these surgical sites being a possible explanation for the heterogeneity seen in this review.
Quality of the evidence
The quality of evidence ranged from moderate to very low across the different outcomes. The major limitation in quality was the sparseness of data for the outcomes of interest. In addition, we assessed a number of included studies as at high risk of bias resulting in further downgrading of the quality assessment. As such our confidence in the effect estimate was limited and the true effect may be substantially different from our estimates of effect.
Potential biases in the review process
Liposomal bupivacaine is a relatively new drug and has been recently licensed. As such many studies investigating its efficacy and safety are currently underway. Our review identified 37 ongoing studies (See Characteristics of ongoing studies) which will report over the next few years. Whilst every effort has been made to minimise bias in this review, new evidence will continue to emerge in this field that may impact on the conclusions drawn. Methods used to minimise the possibility of bias in this review included the use of a comprehensive broad search strategy based on previous Cochrane Reviews for RCTs in postoperative pain, such that we could identify all relevant studies. Additionally, we searched reference lists of potentially relevant studies and reviews, and searched trials registries.
Agreements and disagreements with other studies or reviews
The results of this meta‐analysis disagree with previous meta‐analyses performed by Bergese 2012a and Dasta 2012 which reported the use of liposomal bupivacaine to be associated with lower cumulative pain scores between 0 and 72 hours after the end of the operation, longer time to first opioid and lower cumulative postoperative opioid usage between 0 and 72 hours postoperatively compared to bupivacaine hydrochloride control. There are several reasons for the disagreement seen between ours and the previous meta‐analyses. Firstly, previous meta‐analyses included the results of adaptive‐design studies, which, as discussed, we do not believe is valid. Secondly, previous meta‐analyses pooled data from a range of different surgical procedures in which patient demographics and postoperative pain profiles would be expected to be different; and finally both previous meta‐analyses were performed in collaboration with Pacira Pharmaceuticals Inc. who manufacture the drug and funded the drug development studies, and as such had privileged access to the complete set of data for each of the studies.

Study flow diagram

Risk of bias summary: review authors' judgements about each risk of bias item for each included study

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies

Forest plot of comparison: 1 Liposomal bupivacaine vs control, outcome: 1.1 Cumulative pain score 0 to 72 hours

Table of results for included simultaneous parallel‐arm trials

Forest plot of comparison: 1 Liposomal bupivacaine vs control, outcome: 1.2 Participants not requiring postoperative opioids

Illustrative example of an adaptive‐design trial. The decision to escalate, or de‐escalate a dose is conditional on the failure of the previous dose on the efficacy, or safety, or cost‐effectiveness of the intervention, introducing bias in any pooled analysis. The randomisation ratio is altered with each escalation/de‐escalation while the control group population is typically reported cumulatively for all dose levels

Table of results for adaptive‐design trials

Comparison 1 Liposomal bupivacaine vs control, Outcome 1 Cumulative pain score 0 to 72 hours.

Comparison 1 Liposomal bupivacaine vs control, Outcome 2 Participants not requiring postoperative opioids.
| Liposomal bupivacaine infiltration at the surgical site compared with placebo for the management of postoperative pain | |||
| Patient or population: aged 18 years and older undergoing elective surgery at any surgical site Settings: inpatient Intervention: surgical site infiltration of liposomal bupivacaine Comparison: surgical site infiltration of placebo | |||
| Outcomes | Impact | Number of participants | Quality of the evidence |
| Cumulative pain score from the end of operation (0 hours) to 72 hours (NRS 0 to 10) | A reduction in cumulative pain score associated with the use of liposomal bupivacaine was reported in one study. The mean cumulative pain score from the end of operation to 72 hours (NRS 0 to 10) in the placebo control group was 202.5 points with the mean cumulative pain score from the end of operation to 72 hours in the liposomal bupivacaine intervention group being 60.7 points lower (90.4 lower to 31.1 lower). | 189 participants (1 study) | ⊕⊝⊝⊝ |
| Serious adverse events | No reported drug‐related serious adverse events, no study withdrawals due to drug‐related adverse events | 382 participants (2 studies) | ⊕⊕⊝⊝ |
| Mean pain score at 12, 24, 48, 72 and 96 hours following surgery (NRS 0 to 10) | No data reported | No studies | |
| Time to first postoperative opioid dose over initial 72 hours | A longer time to first postoperative opioid dose associated with the use of liposomal bupivacaine was reported in two studies. In the placebo control group the time to first postoperative opioid was 4.3 and 1.2 hours compared to 7.2 and 14.3 hours in the liposomal bupivacaine groups respectively. The distribution of data was not reported. | 382 participants (2 studies) | ⊕⊕⊝⊝ |
| Total postoperative opioid consumption over first 72 hours | A reduction in total postoperative opioid consumption over first 72 hours associated with the use of liposomal bupivacaine was reported in one study. In the placebo control group the mean cumulative parenteral morphine equivalent dose over the first 72 hours was 29.1 mg and was 6.8 mg lower (12.8 mg lower to 0.9 mg lower) in the liposomal bupivacaine intervention group. | 189 participants (1 study) | ⊕⊝⊝⊝ |
| Percentage of participants not requiring postoperative opioids over initial 72 hours | One study reported a higher proportion of participants not requiring postoperative opioids over initial 72 hours associated with the use of liposomal bupivacaine (RR 0.82; 95% CI 0.72 to 0.94), and one study found no difference (RR 0.99; 95% CI 0.95 to 1.03). | 382 participants (2 studies) | ⊕⊝⊝⊝ |
| Incidence of adverse events within 30 days of surgery | The incidence of cardiac events and wound complications within 30 days of surgery were not reported in any study Adverse events within 30 days of surgery were reported in all studies with nausea, constipation and vomiting being the most common. | 382 participants (2 studies) | ⊕⊕⊝⊝ |
| CI: confidence interval; NRS: numeric rating scale; RR: risk ratio | |||
| GRADE Working Group grades of evidence | |||
| aWe downgraded the quality of this evidence due to the sparseness of data (‐1), indirectness (‐1) and risk of bias (‐1) due to the unclear risk of bias due to the sample size (50‐199). | |||
| Liposomal bupivacaine infiltration at the surgical site compared with bupivacaine hydrochloride for the management of postoperative pain | |||
| Patient or population: aged 18 years and older undergoing elective surgery at any surgical site Settings: inpatient Intervention: surgical site infiltration of liposomal bupivacaine Comparison: surgical site infiltration of bupivacaine hydrochloride | |||
| Outcomes | Impact | Number of participants | Quality of the evidence |
| Cumulative pain score from the end of operation (0 hours) to 72 hours (NRS 0 to 10) | No difference in cumulative pain score was reported in two studies. In one study the mean cumulative pain score from the end of operation to 72 hours (NRS 0 to 10) in the active control group was 335.0 points and 24.0 points higher (5.7 lower to 53.7 higher) in the liposomal bupivacaine intervention group. In the other study the mean cumulative pain score from the end of operation to 72 hours (NRS 0 to 10) in the active control group was 468.2 points and 26.7 points lower (91.3 lower to 37.9 higher) in the liposomal bupivacaine intervention group. Data were not pooled as differences in outcomes were expected due to differences in surgical interventions between studies. | 379 participants (2 studies) | ⊕⊝⊝⊝ |
| Serious adverse events | No reported drug‐related serious adverse events, no study withdrawals due to drug‐related adverse events | 583 participants (3 studies) | ⊕⊕⊕⊝ |
| Mean pain score at 12, 24, 48, 72 and 96 hours following surgery (NRS 0 to 10) | A reduction in mean pain score at 12 hours, but not 24, 48 or 72 hours, associated with the use of liposomal bupivacaine was reported in one study. Mean pain score at these time points were not reported in other studies. In the study that reported mean pain score (NRS 0 to 10) at 12 hours in the active control group it was 6.9 points and 1.3 points lower (2.4 lower to 0.2 lower) in the liposomal bupivacaine intervention group at this time point. | 134 participants (1 study) | ⊕⊝⊝⊝ |
| Time to first postoperative opioid dose over initial 72 hours | No data reported | No studies | |
| Total postoperative opioid consumption over first 72 hours | No difference in cumulative parenteral morphine equivalent dose over first 72 hours was reported in one study though no estimate of variance was provided and as such estimates of effect could not be calculated. | 134 participants (1 study) | ⊕⊝⊝⊝ |
| Percentage of participants not requiring postoperative opioids over initial 72 hours | No difference in the percentage of participants not requiring postoperative opioids over initial 72 hours was reported in one study (RR 0.95; 95% CI 0.86 to 1.05). | 134 participants (1 study) | ⊕⊝⊝⊝ |
| Incidence of adverse events within 30 days of surgery | The incidence of cardiac events and wound complications within 30 days of surgery were not reported in any study Adverse events within 30 days of surgery were reported in all studies with nausea, constipation and vomiting being the most common. | 583 participants (3 studies) | ⊕⊕⊕⊝ |
| CI: confidence interval; NRS: numeric rating scale; RR: risk ratio | |||
| GRADE Working Group grades of evidence | |||
| aWe downgraded the quality of this evidence one level due to the sparseness of data, a further level because Smoot 2012 was subject to a high risk of bias due to the risk of performance bias and attrition bias due to early termination of the study (as well as the unclear risk of bias due to the sample size (50‐199)), and a further level due to inconsistency. We did not pool of results as we predicted that participant characteristics, as well as nature of postoperative pain, would be different following breast augmentation and knee replacement. As such we expected there to be heterogeneity of the results due to population characteristics, not due to intervention characteristics. | |||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Cumulative pain score 0 to 72 hours Show forest plot | 3 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 1.1 vs placebo | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 vs bupivacaine hydrocholoride | 2 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participants not requiring postoperative opioids Show forest plot | 3 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 vs placebo | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 vs bupivacaine hydrochloride | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |