Monoterapia con fármacos antiepilépticos para la epilepsia: un metanálisis en red de los datos de los participantes individuales
Resumen
Antecedentes
La epilepsia es una afección neurológica frecuente, con una prevalencia global de alrededor del 1%. Aproximadamente, del 60% al 70% de los pacientes con epilepsia logrará una remisión de las crisis convulsivas a más largo plazo, y la mayoría logra esa remisión poco después del comienzo de la farmacoterapia antiepiléptica. La mayoría de los pacientes con epilepsia son tratados con un único fármaco antiepiléptico (monoterapia) y las guías actuales del National Institute for Health and Care Excellence (NICE) en el Reino Unido para adultos y niños recomiendan la carbamazepina o la lamotrigina como tratamiento de primera línea para las crisis convulsivas de inicio parcial y el valproato de sodio para las crisis convulsivas de inicio generalizado; sin embargo, se dispone de una serie de otros tratamientos con fármacos antiepilépticos (FAE) y se necesita evidencia sobre su efectividad comparativa para informar las opciones de tratamiento.
Objetivos
Comparar el tiempo hasta la interrupción del tratamiento asignado, la remisión y la primera crisis convulsiva de 10 FAE (carbamazepina, fenitoína, valproato de sodio, fenobarbitona, oxcarbazepina, lamotrigina, gabapentina, topiramato, levetiracetam, zonisamida) que se utilizan actualmente como monoterapia en niños y adultos con crisis convulsivas de inicio parcial (parcial sencilla, parcial compleja o secundaria generalizada) o crisis convulsivas tónico‐clónicas generalizadas con o sin otros tipos de crisis convulsivas generalizadas (ausencia, mioclonía).
Métodos de búsqueda
Se buscó en las bases de datos siguientes: registro especializado del Grupo Cochrane de Epilepsia (Cochrane Epilepsy's Specialised Register), CENTRAL, MEDLINE, SCOPUS y en dos registros de ensayos clínicos. Se hicieron búsquedas manuales en revistas relevantes y se estableció contacto con compañías farmacéuticas, investigadores de ensayos originales y expertos en el tema. La fecha de la búsqueda más reciente fue el 27 de julio de 2016.
Criterios de selección
Se incluyeron los ensayos controlados aleatorizados con diseño de monoterapia en adultos o niños con crisis convulsivas de inicio parcial o crisis convulsivas tónico‐clónicas de inicio generalizado (con o sin otros tipos de crisis convulsivas generalizadas).
Obtención y análisis de los datos
Esta fue una revisión de datos de participantes individuales (DPI) y metanálisis en red. El resultado primario fue "tiempo hasta la interrupción del tratamiento asignado", y los resultados secundarios fueron "tiempo hasta lograr la remisión por 12 meses", "tiempo hasta lograr la remisión por seis meses", "tiempo hasta la primera convulsión posterior a la asignación al azar" y "ocurrencia de eventos adversos". Todos los resultados de tiempo hasta el evento se presentan como cocientes de riesgos instantáneos (CRI) proporcionales de Cox con intervalos de confianza (IC) del 95%. Se realizó el metanálisis pareado de las comparaciones directas entre los fármacos dentro de los ensayos para obtener las estimaciones "directas" del efecto del tratamiento y se realizó el metanálisis en red frecuentista para combinar la evidencia directa con la evidencia indirecta a través de la red de tratamiento de diez fármacos. La inconsistencia entre las estimaciones directas y el metanálisis en red se investigó mediante la separación de nodos. Debido a la variabilidad en los métodos y los detalles para informar los eventos adversos, no se realizó el análisis. Se proporcionó un resumen narrativo de los eventos adversos que se informaron con mayor frecuencia.
Resultados principales
Se proporcionaron los DPI de al menos un resultado de esta revisión para 12 391 de un total de 17 961 participantes elegibles (69% de los datos totales) de 36 de los 77 ensayos elegibles (47% de los ensayos totales). No fue posible incluir los DPI de los 41 ensayos restantes en el análisis por diversos motivos, como la imposibilidad de establecer contacto con un autor o un patrocinador para solicitar los datos, que los datos estaban perdidos o ya no estaban disponibles, que los costos y los recursos requeridos para prepararlos son prohibitivos, o por limitaciones de las autoridades locales o específicas de los países.
Fue posible calcular las estimaciones directas del efecto del tratamiento de entre la mitad y dos tercios de las comparaciones entre los resultados de la revisión; sin embargo, en muchas de las comparaciones, los datos provinieron solo de un único ensayo o de un escaso número de participantes, por lo que los intervalos de confianza de las estimaciones fueron amplios.
El metanálisis de la red mostró que para el resultado primario "Tiempo hasta la retirada del tratamiento asignado", en el caso de los individuos con convulsiones parciales; el levetiracetam tuvo un rendimiento (estadísticamente) significativamente mejor que el tratamiento actual de primera línea carbamazepina y el otro tratamiento actual de primera línea lamotrigina tuvo un rendimiento mejor que todos los demás tratamientos (aparte del levetiracetam); la carbamazepina tuvo un rendimiento significativamente mejor que la gabapentina y la fenobarbitona (evidencia de alta calidad). En los pacientes con crisis convulsivas de inicio generalizado, el tratamiento de primera línea valproato de sodio funcionó significativamente mejor que la carbamazepina, el topiramato y la fenobarbitona (evidencia de calidad moderada a alta). Además, para las crisis convulsivas de inicio parcial y generalizado, el tratamiento autorizado más antiguo, la fenobarbitona, parece funcionar peor que todos los otros tratamientos (evidencia de calidad moderada a alta).
El metanálisis en red también mostró que para los resultados secundarios "Tiempo hasta la remisión por 12 meses de las crisis convulsivas" y "Tiempo hasta la remisión por seis meses de las crisis convulsivas", hubo pocas diferencias notables en los tipos de crisis convulsivas parciales o generalizadas (evidencia de calidad moderada a alta). Para el resultado secundario "Tiempo para la primera convulsión", para los individuos con convulsiones parciales; la fenobarbitona tuvo un rendimiento significativamente mejor que los tratamientos actuales de primera línea carbamazepina y lamotrigina; la carbamazepina tuvo un rendimiento significativamente mejor que el valproato de sodio, la gabapentina y la lamotrigina. La fenitoína también funcionó significativamente mejor que la lamotrigina (evidencia de alta calidad). En general, los tratamientos autorizados más antiguos (fenitoína y fenobarbitona) funcionaron mejor que los otros tratamientos para ambos tipos de crisis convulsiva (evidencia de calidad moderada a alta).
En general, la evidencia directa y las estimaciones del metanálisis en red (evidencia directa más indirecta) fueron numéricamente similares y consistentes con los intervalos de confianza de los tamaños de los efectos superpuestos.
Los eventos adversos informados con mayor frecuencia con todos los fármacos fueron somnolencia/fatiga, cefalea o migraña, trastornos gastrointestinales, mareo/debilidad y erupción o trastornos cutáneos.
Conclusiones de los autores
En general, la evidencia de alta calidad proporcionada por esta revisión apoya la orientación actual (por ejemplo, NICE) de que la carbamazepina y la lamotrigina son tratamientos de primera línea adecuados para los individuos con convulsiones de comienzo parcial y también demuestran que el levetiracetam puede ser una alternativa adecuada. La evidencia de alta calidad de esta revisión también apoya la administración de valproato de sodio como tratamiento de primera línea en los pacientes con crisis convulsivas generalizadas tónico‐clónicas (con o sin otros tipos de crisis convulsivas generalizadas) y también demuestra que la lamotrigina y el levetiracetam serían opciones apropiadas para cualquiera de estos tratamientos de primera línea, en particular, en las pacientes en edad fértil, en las que el valproato de sodio puede no ser una opción apropiada de tratamiento debido a la teratogenicidad.
PICOs
Resumen en términos sencillos
Monoterapia con fármacos antiepilépticos (tratamiento con un único fármaco) para la epilepsia
Antecedentes
La epilepsia es un trastorno neurológico frecuente en el que las descargas eléctricas anormales en el cerebro provocan convulsiones recurrentes. En esta revisión, se estudiaron dos tipos de crisis epilépticas: las crisis parciales que comienzan en un área del cerebro y las crisis tónico‐clónicas de inicio generalizado que comienzan en ambos hemisferios cerebrales simultáneamente.
En alrededor del 70% de los pacientes con epilepsia, las crisis se pueden controlar y, en la mayoría, las crisis convulsivas se controlan con un único fármaco antiepiléptico. Actualmente, en el Reino Unido, las guías del National Institute for Health and Care Excellence (NICE) para adultos y niños recomiendan la carbamazepina o la lamotrigina como las primeras opciones de tratamiento a probar para los pacientes con convulsiones parciales recién diagnosticados y el valproato de sodio para los pacientes con convulsiones tónico‐clónicas generalizadas recién diagnosticadas; sin embargo, se dispone de una gama de otros tratamientos con medicamentos antiepilépticos.
La elección del primer fármaco antiepiléptico para un paciente con crisis convulsivas de diagnóstico reciente es de gran importancia y se debe realizar teniendo en cuenta la evidencia de alta calidad de cuán efectivos son los fármacos para controlar las crisis convulsivas y si se asocian con efectos secundarios. También es importante que los fármacos apropiados para diferentes tipos de crisis convulsivas se comparen entre sí.
Métodos de la revisión
Los fármacos antiepilépticos de interés para esta revisión fueron carbamazepina, fenitoína, valproato de sodio, fenobarbitona, oxcarbazepina, lamotrigina, gabapentina, topiramato, levetiracetam y zonisamida. En esta revisión, se evaluó la evidencia de 77 ensayos clínicos controlados aleatorizados que compararon dos o más de los fármacos de interés sobre la base de cuán efectivos son para controlar las crisis convulsivas (es decir, si los pacientes tuvieron recurrencias de las crisis convulsivas o tuvieron períodos prolongados de falta de crisis convulsivas [remisión]) y cuán tolerable fue cualquier efecto secundario de los fármacos. Se pudieron combinar los datos de 12 391 pacientes de 36 de los 77 ensayos; para las 5570 pacientes restantes de 41 ensayos, no se dispuso de datos para utilizarlos en esta revisión.
En esta revisión, se realizaron dos tipos de análisis; en primer lugar, se combinaron los datos disponibles de pares de fármacos que se habían comparado directamente en ensayos clínicos, y en segundo lugar, se realizó un análisis para combinar toda la información de los ensayos clínicos en una "red" de diez fármacos. Este análisis permitió comparar fármacos en la red que no se habían comparado previamente entre sí en los ensayos clínicos.
Resultados clave
De 45 comparaciones pareadas posibles de los diez fármacos de interés en la revisión, estuvieron disponibles datos de ensayos clínicos para poco más de la mitad de estas comparaciones, pero en muchas, solamente un único ensayo había hecho una comparación de los dos fármacos y la comparación no incluyó a muchos pacientes.
El análisis en "red" mostró que los fármacos más antiguos en la red (fenobarbitona y fenitoína) fueron mejores opciones en cuanto al control de las crisis convulsivas que los otros fármacos, pero que estos fármacos más antiguos fueron los peores en cuanto a la retención a largo plazo (interrupción del tratamiento) en comparación con los fármacos más modernos como lamotrigina y levetiracetam.
Los efectos secundarios informados con mayor frecuencia con todos los fármacos fueron somnolencia o fatiga, cefalea o migraña, trastornos gastrointestinales (molestias estomacales), mareo o debilidad y erupción o trastornos cutáneos.
Calidad de la evidencia
Esta revisión proporciona evidencia de alta calidad para pacientes con crisis convulsivas parciales y evidencia de calidad moderada a alta para pacientes con crisis convulsivas tónico‐clónicas generalizadas, y hay menos información disponible de algunos de los fármacos de interés para los pacientes con este tipo de crisis convulsiva.
Conclusiones
Los resultados de la presente revisión apoyan las guías NICE de que la carbamazepina y la lamotrigina son opciones apropiadas como primer tratamiento para los pacientes con crisis convulsivas de inicio parcial y también muestran que el levetiracetam también sería un tratamiento apropiado. Los resultados de la presente revisión también apoyan la administración del valproato de sodio como tratamiento de primera línea en los pacientes con crisis convulsivas tónico‐clónicas generalizadas y también muestran que la lamotrigina y el levetiracetam serían opciones apropiadas, en particular, en las embarazadas o las pacientes que consideran la posibilidad de quedar embarazadas, en las que el valproato de sodio puede no ser una opción apropiada de tratamiento.
¿Cuál es el grado de actualización de esta revisión?
Los autores de la revisión buscaron estudios que se habían publicado hasta el 27 julio 2016.
Authors' conclusions
Summary of findings
| Antiepileptic drug monotherapy for epilepsy: time to withdrawal of allocated treatment for individuals with partial seizures | ||||||
| Patient or population: adults and children with partial seizures Settings: outpatients Intervention: phenobarbitone, phenytoin, sodium valproate, lamotrigine, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: carbamazepine | ||||||
| Intervention (experimental treatment)a,b | Comparison (reference treatment) | No of participants | Relative effect Direct evidence Heterogeneity: I2 | Relative effect Direct plus indirect evidence | Proportion of | Quality of the evidence |
| Phenobarbitone | Carbamazepine | 520 (4 studies) | 1.57 (1.16 to 2.13) I2 = 0% | 1.55 (1.18 to 2.04) | 52.5% | ⊕⊕⊕⊕ |
| Phenytoin | Carbamazepine | 428 (3 studies) | 1.03 (0.74 to 1.42) I2 = 63.6% | 1.13 (0.92 to 1.38) | 12.8% | ⊕⊕⊕⊕ |
| Sodium Valproate | Carbamazepine | 814 (5 studies) | 0.94 (0.73 to 1.19) I2 = 0% | 1.04 (0.86 to 1.25) | 40.1% | ⊕⊕⊕⊕ |
| Lamotrigine | Carbamazepine | 2268 (9 studies) | 0.76 (0.61 to 0.95) I2 = 39.3% | 0.75 (0.65 to 0.86) | 28.9% | ⊕⊕⊕⊕ |
| Oxcarbazepine | Carbamazepine | 562 (2 studies) | 4.62 (0.95 to 22.4) I2 = 0% | 1.09 (0.84 to 1.42) | 5.7% | ⊕⊕⊕⊕ |
| Topiramate | Carbamazepine | 937 (2 studies) | 1.04 (0.52 to 2.07) I2 = 0% | 1.18 (0.98 to 1.43) | 7.4% | ⊕⊕⊕⊕ |
| Gabapentin | Carbamazepine | 954 (2 studies) | 1.14 (0.84 to 1.55) I2 = 0% | 1.20 (1.00 to 1.43) | 87.1% | ⊕⊕⊕⊕ |
| Levetiracetam | Carbamazepine | 1567 (3 studies) | 0.70 (0.52 to 0.94) I2 = 0% | 0.82 (0.69 to 0.97) | 37.9% | ⊕⊕⊕⊕ |
| Zonisamide | Carbamazepine | 583 (1 study) | 1.08 (0.81 to 1.44) I2 = NA) | 1.08 (0.79 to 1.48) | 100% | ⊕⊕⊕⊕ |
| Abbreviations: CI: confidence interval; HR: hazard ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence | ||||||
| aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Antiepileptic drug monotherapy for epilepsy: time to withdrawal of allocated treatment for individuals with partial seizures | ||||||
| Patient or population: adults and children with partial seizures Settings: outpatients Intervention: carbamazepine, phenobarbitone, phenytoin, sodium valproate, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: lamotrigine | ||||||
| Intervention (experimental treatment)a,b | Comparison (reference | No of participants | Relative effect Direct evidence Heterogeneity: I2 | Relative effect Direct plus | Proportion of | Quality of the evidence |
| Carbamazepine | Lamotrigine | 2268 (9 studies) | 1.31 (1.05 to 1.64) I2 = 39.3% | 1.34 (1.17 to 1.53) | 28.9% | ⊕⊕⊕⊕ |
| Phenobarbitone | Lamotrigine | No direct evidence | No direct evidence I2: NA | 2.08 (1.52 to 2.86) | 0% | ⊕⊕⊕⊕ |
| Phenytoin | Lamotrigine | 90 (1 study) | 0.91 (0.47 to 1.76) I2: NA | 1.52 (1.18 to 1.92) | 11.6% | ⊕⊕⊕⊕ |
| Sodium Valproate | Lamotrigine | 221 (3 studies) | 0.71 (0.51 to 1.00) I2 = 45.1% | 1.39 (1.11 to 1.72) | 5.1% | ⊕⊕⊕⊝ |
| Oxcarbazepine | Lamotrigine | 506 (1 study) | 0.69 (0.12 to 4.14) I2: NA | 1.46 (1.11 to 1.92) | 4.4% | ⊕⊕⊕⊕ |
| Topiramate | Lamotrigine | 648 (1 study) | 1.18 (0.86 to 1.62) I2: NA | 1.59 (1.29 to 1.95) | 20.9% | ⊕⊕⊕⊕ |
| Gabapentin | Lamotrigine | 659 (1 study) | 0.62 (0.06 to 6.01) I2: NA | 1.60 (1.31 to 1.96) | 1% | ⊕⊕⊕⊕ |
| Levetiracetam | Lamotrigine | 240 (1 study) | 0.86 (0.58 to 1.28) I2: NA | 1.10 (0.89 to 1.35) | 23.7% | ⊕⊕⊕⊕ |
| Zonisamide | Lamotrigine | No direct evidence | No direct evidence I2: NA | 1.45 (1.03 to 2.04) | 0% | ⊕⊕⊕⊕ |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence | ||||||
| aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Antiepileptic drug monotherapy for epilepsy: time to withdrawal of allocated treatment for individuals with generalised seizures | ||||||
| Patient or population: adults and children with generalised seizures* Settings: outpatients Intervention: carbamazepine, phenobarbitone, phenytoin, lamotrigine, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide. Comparison: sodium valproate | ||||||
| Intervention (experimental treatment)a,b | Comparison (reference | No of | Relative effect Direct evidence Heterogeneity: I2 | Relative effect Direct plus | Proportion of | Quality of the evidence |
| Carbamazepine | Sodium Valproate | 405 (4 studies) | 0.79 (0.45 to 1.37) I2 = 6.6% | 1.42 (1.09 to 1.85) | 27.3% | ⊕⊕⊕⊕ |
| Phenobarbitone | Sodium Valproate | 94 (2 studies) | 1.79 (0.65 to 5.00) I2 = 0% | 2.09 (1.17 to 3.75) | 19.4% | ⊕⊕⊕⊝ |
| Phenytoin | Sodium Valproate | 326 (3 studies) | 1.52 (0.68 to 3.33) I2 = 22.6% | 1.30 (0.79 to 2.15) | 19.3% | ⊕⊕⊕⊕ |
| Lamotrigine | Sodium Valproate | 387 (3 studies) | 0.46 (0.22 to 0.97) I2 = 0% | 0.90 (0.60 to 1.35) | 14.8% | ⊕⊕⊕⊕ |
| Oxcarbazepine | Sodium Valproate | No direct evidence | No direct evidence I2: NA | 1.42 (0.29 to 6.92) | 0% | ⊕⊕⊕⊝ |
| Topiramate | Sodium Valproate | 443 (2 studies) | 1.04 (0.52 to 2.07) I2 = 48.5% | 1.76 (1.22 to 2.53) | 22.4% | ⊕⊕⊕⊝ |
| Gabapentin | Sodium Valproate | No direct evidence | No direct evidence I2: NA | 1.28 (0.16 to 10.5) | 0% | ⊕⊕⊕⊝ |
| Levetiracetam | Sodium Valproate | 512 (1 study) | 0.68 (0.30 to 1.59) I2: NA) | 1.05 (0.58 to 1.90) | 18.6% | ⊕⊕⊕⊕ |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence | ||||||
| *Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Antiepileptic drug monotherapy for epilepsy: time to 12‐month remission for individuals with partial seizures | ||||||
| Patient or population: adults and children with partial seizures Settings: outpatients Intervention: phenobarbitone, phenytoin, sodium valproate, lamotrigine, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: carbamazepine | ||||||
| Intervention (experimental treatment)a,b | Comparison (reference treatment) | No of participants | Relative effect Direct evidence Heterogeneity: I2 | Relative effect Direct plus | Proportion of | Quality of the evidence |
| Phenobarbitone | Carbamazepine | 525 (4 studies) | 1.41 (1.04 to 1.91) I2 = 0% | 1.02 (0.76 to 1.35) | 56.1% | ⊕⊕⊕⊕ |
| Phenytoin | Carbamazepine | 430 (3 studies) | 1.00 (0.76 to 1.32) I2 = 54.8% | 1.03 (0.85 to 1.25) | 18.6% | ⊕⊕⊕⊕ |
| Sodium Valproate | Carbamazepine | 816 (5 studies) | 1.03 (0.85 to 1.25) I2 = 46.4% | 1.05 (0.89 to 1.25) | 27.6% | ⊕⊕⊕⊕ |
| Lamotrigine | Carbamazepine | 891 (2 studies) | 1.02 (0.69 to 1.50) I2 = 0% | 1.16 (0.98 to 1.37) | 17.5% | ⊕⊕⊕⊕ |
| Oxcarbazepine | Carbamazepine | 555 (2 studies) | 1.13 (0.62 to 2.05) I2 = 0% | 0.98 (0.78 to 1.25) | 21% | ⊕⊕⊕⊕ |
| Topiramate | Carbamazepine | 925 (2 studies) | 0.94 (0.48 to 1.83) I2 = 0% | 1.08 (0.92 to 1.27) | 7.2% | ⊕⊕⊕⊕ |
| Gabapentin | Carbamazepine | 651 (1 study) | 0.61 (0.06 to 5.82) I2: NA | 1.20 (0.99 to 1.47) | 10.5% | ⊕⊕⊕⊕ |
| Levetiracetam | Carbamazepine | 1567 (3 studies) | 1.08 (0.81 to 1.42) I2 = 60.8% | 1.35 (1.09 to 1.69) | 14.2% | ⊕⊕⊕⊕ |
| Zonisamide | Carbamazepine | 582 (1 study) | 1.05 (0.85 to 1.30) I2: NA | 1.05 (0.81 to 1.35) | 100% | ⊕⊕⊕⊕ |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence | ||||||
| aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Antiepileptic drug monotherapy for epilepsy: time to 12‐month remission for individuals with partial seizures | ||||||
| Patient or population: adults and children with partial seizures Settings: outpatients Intervention: carbamazepine, phenobarbitone, phenytoin, sodium valproate, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: lamotrigine | ||||||
| Intervention (experimental treatment)a,b | Comparison (reference treatment) | No of participants | Relative effect Direct evidence Heterogeneity: I2 | Relative effect Direct plus | Proportion of | Quality of the evidence |
| Carbamazepine | Lamotrigine | 891 (2 studies) | 0.98 (0.67 to 1.45) I2 = 0% | 0.86 (0.72 to 1.02) | 17.5% | ⊕⊕⊕⊕ |
| Phenobarbitone | Lamotrigine | No direct evidence | No direct evidence I2: NA | 0.88 (0.62 to 1.22) | 0% | ⊕⊕⊕⊕ |
| Phenytoin | Lamotrigine | No direct evidence | No direct evidence I2: NA | 0.89 (0.68 to 1.13) | 0% | ⊕⊕⊕⊕ |
| Sodium Valproate | Lamotrigine | 221 (3 studies) | 0.72 (0.56 to 0.93) I2 = 0% | 0.91 (0.73 to 1.33) | 39.9% | ⊕⊕⊕⊕ |
| Oxcarbazepine | Lamotrigine | 499 (1 study) | 1.49 (0.33 to 6.67) I2: NA | 0.85 (0.66 to 1.09) | 2.8% | ⊕⊕⊕⊕ |
| Topiramate | Lamotrigine | 636 (1 study) | 0.98 (0.29 to 3.25) I2: NA | 0.93 (0.75 to 1.15) | 2.5% | ⊕⊕⊕⊕ |
| Gabapentin | Lamotrigine | 647 (1 study) | 0.74 (0.08 to 6.58) I2: NA | 1.04 (0.84 to 1.30) | 10.1% | ⊕⊕⊕⊕ |
| Levetiracetam | Lamotrigine | 240 (1 study) | 1.02 (0.70 to 1.49) I2: NA | 1.16 (0.93 to 1.47) | 26.6% | ⊕⊕⊕⊕ |
| Zonisamide | Lamotrigine | No direct evidence | No direct evidence I2: NA | 0.91 (0.67 to 1.22) | 0% | ⊕⊕⊕⊕ |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence | ||||||
| aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Antiepileptic drug monotherapy for epilepsy: time to withdrawal of allocated treatment for individuals with generalised seizures | ||||||
| Patient or population: adults and children with generalised seizures* Settings: outpatients Intervention: carbamazepine, phenobarbitone, phenytoin, lamotrigine, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: sodium valproate | ||||||
| Intervention (experimental treatment)a,b | Comparison (reference treatment) | No of participants | Relative effect Direct evidence | Relative effect Direct plus | Proportion of | Quality of the evidence |
| Carbamazepine | Sodium Valproate | 412 (4 studies) | 0.99 (0.69 to 1.39) I2 = 0% | 1.06 (0.88 to 1.27) | 51.1% | ⊕⊕⊕⊕ |
| Phenobarbitone | Sodium Valproate | 98 (2 studies) | 0.86 (0.40 to 1.89) I2 = 42.3% | 1.33 (0.87 to 2.04) | 13% | ⊕⊕⊕⊕ |
| Phenytoin | Sodium Valproate | 269 (4 studies) | 1.15 (0.71 to 1.82) I2 = 0% | 0.91 (0.67 to 1.25) | 44.9% | ⊕⊕⊕⊕ |
| Lamotrigine | Sodium Valproate | 387 (3 studies) | 0.77 (0.38 to 1.56) I2 = 0% | 1.35 (0.57 to 3.13) | 35.7% | ⊕⊕⊕⊕ |
| Oxcarbazepine | Sodium Valproate | No direct evidence | No direct evidence I2: NA | 1.82 (0.50 to 6.67) | 0% | ⊕⊕⊕⊝ |
| Topiramate | Sodium Valproate | 441 (2 studies) | 0.52 (0.26 to 1.04) I2 = 58.5% | 1.12 (0.83 to 1.52) | 10.6% | ⊕⊕⊕⊕ |
| Gabapentin | Sodium Valproate | No direct evidence | No direct evidence I2: NA | 0.79 (0.10 to 6.25) | 0% | ⊕⊕⊕⊝ |
| Levetiracetam | Sodium Valproate | 512 (1 study) | 0.91 (0.49 to 1.70) I2: NA | 1.41 (0.83 to 2.44) | 55.2% | ⊕⊕⊕⊕ |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence | ||||||
| *Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity. aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
Background
Description of the condition
Epilepsy is a common neurological condition in which recurrent, unprovoked seizures occur due to abnormal electrical discharges in the brain, with an estimated incidence of 33 to 57 per 100,000 person‐years worldwide (Annegers 1999; Hirtz 2007; MacDonald 2000; Olafsson 2005; Sander 1996), accounting for approximately 1% of the global burden of disease (WHO 1994). The lifetime risk of epilepsy onset is estimated to be 1300 to 4000 per 100,000 person years (Hauser 1993; Juul‐Jenson 1983), and the lifetime prevalence could be as large as 70 million people world‐wide (Ngugi 2010). It is believed that with effective drug treatment, up to 70% of individuals with active epilepsy have the potential to become seizure‐free and go into long‐term remission shortly after starting drug therapy (Cockerell 1995; Hauser 1993; Sander 2004), and that around 70% of individuals can achieve seizure freedom using a single AED (AED) in monotherapy (Cockerell 1995). The remaining 30% of individuals experience refractory or drug‐resistant seizures, which often require treatment with combinations of AEDs or alternative treatments such as epilepsy surgery (Kwan 2000).
Epilepsy is not a single condition, but is in fact a heterogeneous group of conditions ranging from those with a purely genetic cause to those that are symptomatic of a brain injury (e.g. stroke) or other abnormality (e.g. tumour). We also recognise a range of differing seizure types, and epilepsy syndromes that have been classified by the International League Against Epilespy (ILAE), a classification that continues to be revised as our understanding of the genetics and basic biology of epilepsy improves (Berg 2010; Commission 1981; Commission 1989)
The simplest dichotomy in epilepsy is between partial onset (or focal) and generalised onset seizures. Partial onset seizures originate in one part of the brain and include simple partial, complex partial and secondary generalised seizures (Berg 2010). Generalised seizures originate in both cerebral hemispheres simultaneously and include generalised tonic‐clonic seizures, absence seizures and myoclonic seizures. In this review we focus on this dichotomy rather than specific epilepsy syndromes.
Description of the intervention
For the treatment of partial and generalised onset seizures we included in our evidence base the following 10 AEDs, which at the time of publication of the protocol of this review (December 2014) were licensed and used in clinical practice for use as monotherapy in at least one country (eMC 2014; FDA 2014):
-
carbamazepine;
-
phenobarbitone;
-
phenytoin;
-
sodium valproate;
-
oxcarbazepine;
-
lamotrigine;
-
gabapentin;
-
topiramate;
-
levetiracetam;
-
zonisamide.
Carbamazepine, sodium valproate, phenytoin and phenobarbitone are among the earliest drugs licensed for treating epileptic seizures. Carbamazepine and sodium valproate have been commonly used as monotherapy for partial onset and generalised onset seizures for over 30 years (Shakir 1980), while phenytoin and phenobarbitone have been used in monotherapy for over 50 years (Gruber 1962).
These traditionally used drugs have all been recommended as first‐line treatments due to their effects across a range of seizure types, however they are also associated with a number of adverse effects. Phenytoin and phenobarbitone are no longer considered as first‐line agents in the USA and much of Europe due to worries over adverse events (Wallace 1997; Wilder 1995). Both drugs have been shown to be teratogenic (associated with malformations of an embryo or fetus) and are associated with low folic acid levels and megaloblastic anaemia (a blood disorder marked by the appearance of very large red blood cells (Carl 1992; Gladstone 1992; Meador 2008; Morrow 2006; Nulman 1997)). Phenytoin is particularly associated with fetal hydantoin syndrome, the name given to a group of birth defects associated with exposure to phenytoin (Scheinfeld 2003), and phenobarbitone has been associated with behavioural disturbances, particularly in children (de Silva 1996; Trimble 1988). These agents are however still used as first‐line drugs in low‐ to middle‐income countries (Ogunrin 2005; Pal 1998).
Carbamazepine and sodium valproate are also associated with congenital abnormalities (Canger 1999; Gladstone 1992; Morrow 2006; Nulman 1997; Tomson 2011). Systematic reviews have shown sodium valproate to have the highest incidence of congenital malformations of traditional first‐line AEDs (Meador 2008; Weston 2016), particularly spina bifida, as well as cardiac, craniofacial, skeletal and limb defects known as 'valproate syndrome' (Ornoy 2009). A recent study has shown an increased prevalence of neurodevelopmental disorders following prenatal sodium valproate exposure (Bromley 2013). A recently published Cochrane Review found that levetiracetam and lamotrigine exposure carried the lowest risk of overall congenital malformation, however information regarding specific malformations was lacking (Weston 2016).
In the last 20 years, a second‐generation of AEDs including oxcarbazepine, lamotrigine, gabapentin, topiramate and, most recently, levetiracetam and zonisamide, have been licensed as monotherapy following demonstrations of efficacy, or non‐inferiority within the European Union, compared to the traditional AEDs (for example, Baulac 2012; Bill 1997; Brodie 1995a; Brodie 1999; Brodie 2007; Chadwick 1998; Christe 1997; Dam 1989; Guerreiro 1997; SANAD A 2007, SANAD B 2007; Privitera 2003; Reunanen 1996; Rowan 2005; Steiner 1999; Trinka 2013). Comparative studies have also shown the newer AEDs to be generally well tolerated as monotherapy in both adults and children and related to fewer adverse events, fewer serious adverse events, fewer teratogenic effects and fewer drug interactions with concomitant AEDs and other concomitant medications than the traditional first‐line AEDs (French 2004; French 2007).
Current guidelines from the National Institute for Health and Care Excellence (NICE) for adults and children recommend carbamazepine or lamotrigine as first‐line treatment for partial onset seizures and sodium valproate for generalised onset seizures, on the condition that women and girls of childbearing age are made aware of the potential teratogenic effects of the drug (NICE 2012).
How the intervention might work
AEDs suppress seizures by reducing neuronal excitability, hence reducing the probability that a seizure will occur. Different AEDs have different mechanisms of action; therefore certain AEDs are more effective at treating different seizure types. For example, there are reports of efficacy for sodium valproate in generalised epilepsy syndromes such as juvenile myoclonic epilepsy and absence epilepsy (Bourgeois 1987; Delgado‐Escueta 1984; Grünewald 1993; Jeavons 1977; Penry 1989), while carbamazepine, on the other hand, is reported to exacerbate some generalised seizure types such as myoclonic and absence seizures (Liporace 1994; Shields 1983; Snead 1985).
The majority of traditional AEDs are thought to have multiple mechanisms of action such as blocking ion channels, binding with neurotransmitter receptors or inhibiting the metabolism or reuptake of neurotransmitters. However the precise mechanism of action is not known for all AEDs, particularly sodium valproate. It is thought that one of the mechanisms of action of phenytoin, sodium valproate, carbamazepine, oxcarbazepine and lamotrigine is via blocking of sodium channels (Brodie 1996; Faigle 1990; Granger 1995; Grant 1992; Lees 1993; McLean 1986; Pinder 1977; Ragsdale 1991; Willow 1985), while phenobarbitone binds with gamma‐aminobutyric acid (GABA) A receptors (Rho 1996).
Zonisamide is thought to have multiple mechanisms of action (Endoh 1994; Kawai 1994; Okada 1998; Sackellares 2004; Schauf 1987; Suzuki 1992; Zhu 1999), while the mechanism of actions of gabapentin and topiramate are not fully understood (Brodie 1996; Coulter 1993; Hill 1993; McClean 1995; McLean 1999; White 1997). Levetiracatam has a novel mode of action which is different from that of other AEDs (Cho 2011); it is thought to exhibit its antiepileptic effect by binding to synaptic vesicle protein 2A (encoded within the SV2A gene), influencing excitatory neurotransmitter release (Gillard 2006; Lynch 2004).
Why it is important to do this review
Given that up to 70% of individuals with a new epilepsy diagnosis enter a long‐term remission of seizures shortly after starting drug therapy (Cockerell 1995; Hauser 1993; Sander 2004), the correct choice of first‐line antiepileptic therapy for individuals with newly diagnosed seizures is of great importance. There are currently over 50 AEDs available worldwide for the treatment of all epilepsy syndromes (Epilepsy Foundation of America 2013), and therefore it is important that the choice of first AEDs is based on the highest‐quality evidence regarding potential benefits and harms of various treatments.
We have published a series of Cochrane systematic Reviews investigating pairwise monotherapy comparisons using individual participant data (Marson 2000; Nevitt 2016; Nolan 2013b; Nolan 2013c; Nolan 2015; Nolan 2016a; Nolan 2016b; Nolan 2016d). Each Cochrane Review and meta‐analysis provides high‐quality evidence for each pair of drugs but does not inform a choice among the range of drugs available. Furthermore, direct evidence from randomised controlled trials (RCTs) is not available for some drug comparisons such as between oxcarbazepine and phenobarbitone; therefore it is not possible to make pairwise comparisons of treatment effects between all 10 drugs included in this review. Also, pairwise comparisons between certain drugs are unlikely to be made in the future, such as comparisons with phenobarbitone, which is no longer considered to be a first‐line treatment, so it is unlikely that a RCT will be designed in the future to compare oxcarbazepine with phenobarbitone (Tudur Smith 2007). However, it is possible to estimate an indirect treatment effect size between oxcarbazepine and phenobarbitone using existing evidence comparing oxcarbazepine with phenytoin and phenytoin with phenobarbitone (Nolan 2013b; Nolan 2016d). By similar methodology, an indirect pairwise comparison is possible for all 10 drugs in our treatment network. Indirect comparisons are also valuable in the case that a limited amount of data are available to inform a direct comparison or in the case that evidence informing a direct comparison is of poor methodological quality. The power and precision of a treatment effect estimate can be increased by 'borrowing strength' from the indirect evidence in the network of treatments (Bucher 1997). Eight of the AEDs included in this review have been included in an IPD network meta‐analysis of epilepsy monotherapy drugs (Tudur Smith 2007). We wish to update the information in this network meta‐analysis with new evidence from trials published since 2007 and including evidence for two drugs, which were licensed for use as monotherapy after 2007.
As noted in the series of Cochrane Reviews investigating pairwise monotherapy comparisons, the important efficacy outcomes in epilepsy monotherapy trials often require analysis of time‐to‐event data (for example, time to first seizure after randomisation or time to withdrawal of allocated treatment). Although methods have been developed to synthesise time‐to‐event data using summary information (Parmar 1998; Williamson 2002), the appropriate statistics are not commonly reported in published epilepsy trials (Altman 1995; Nolan 2013a).
Furthermore, although seizure data have been collected in most epilepsy monotherapy trials, we have seen little uniformity in the definition and reporting of outcomes. For example, trials may report time to 12‐month remission but not time to first seizure or vice versa, or some trials may define time to first seizure from the date of randomisation but others use date of achieving maintenance dose. Trial investigators have also adopted differing approaches to the analysis, particularly with respect to the censoring of time‐to‐event data. For these reasons, we performed the pairwise meta‐analyses using IPD, which helps to overcome these problems and is considered to be the 'gold standard' approach to synthesis of censored data (Parmar 1998). We therefore also performed the network meta‐analysis of epilepsy monotherapy drugs as an IPD analysis.
Objectives
To compare the time to withdrawal of allocated treatment, remission and first seizure of 10 AEDs (carbamazepine, phenytoin, sodium valproate, phenobarbitone, oxcarbazepine, lamotrigine, gabapentin, topiramate, levetiracetam, zonisamide) currently used as monotherapy in children and adults with partial onset seizures (simple partial, complex partial or secondary generalised) or generalised tonic‐clonic seizures with or without other generalised seizure types (absence, myoclonus).
Methods
Criteria for considering studies for this review
Types of studies
We included RCTs using either:
-
an adequate method of allocation concealment (e.g. sealed, opaque envelopes);
-
a quasi method of randomisation (e.g. allocation by date of birth).
Trials may be double‐blind, single‐blind or unblinded. We included only trials of a monotherapy design; in other words, all participants are randomised to treatment with a single drug. We excluded trials with an add‐on (polytherapy), or withdrawal to monotherapy designs.
We included trials of parallel designs. We excluded trials of a cross‐over design, as this design is not appropriate for assessing treatment decisions at the time of epilepsy diagnosis and the cross‐over design is also inappropriate for measuring our primary time‐to‐event outcome 'time to withdrawal of allocated treatment', as a withdrawal of allocated treatment in the first treatment period would mean than the participant could not cross into the second treatment period, potentially leading to a large amount of incomplete outcome data and therefore a reduction in statistical power. Furthermore, the use of cross‐over designs is no longer recommended in epilepsy due to concerns over trial duration, large proportions of dropouts, unblinding of masked treatments as participants cross into the second period, and potential carryover effects; a particular concern in trials of a monotherapy design that aim to assess the effect of a single treatment (Engel 2008; Wyllie 2006).
Types of participants
Children or adults with partial onset seizures (simple partial, complex partial, or secondarily generalised tonic‐clonic seizures) or generalised onset tonic‐clonic seizures (with or without other generalised seizure types). We did not include participants with other generalised seizure types alone (for example absence seizures alone without generalised tonic‐clonic seizures) as guidelines for the first‐line treatment of other generalised seizure types are different from the guidelines for generalised tonic‐clonic seizures (NICE 2012), and due to documented evidence that certain drugs of interest in our review may exacerbate some generalised seizure types (How the interventions might work). We also considered individuals with a new diagnosis of epilepsy, or who had had a relapse following antiepileptic monotherapy withdrawal.
We excluded trials that considered AEDs as treatment for conditions other than epilepsy.
Types of interventions
We included the 10 AEDs currently licensed and commonly used as monotherapy in our network of treatments: carbamazepine, phenytoin, sodium valproate, phenobarbitone, oxcarbazepine, lamotrigine, gabapentin, topiramate, levetiracetam, zonisamide.
Included trials had to make at least one pairwise comparison between at least two of the 10 AEDs included in our network. For trials with three treatment arms or more, we included treatment arms only of the 10 AEDs included in our network; treatment arms of drugs not included in our network were excluded from analysis. We did not make pairwise comparisons (direct or indirect) between any AEDs not specified above. We made pairwise comparisons (based on direct or indirect evidence, or both) between all 10 drugs (Data synthesis).
We included trials with multiple arms of the same drug as long as at least one arm of another drug from our network was included (e.g. multiple doses of gabapentin compared to carbamazepine in Chadwick 1998). We pooled multiple dose arms of the same drug in our analysis; dose comparisons are outside the scope of this review.
Types of outcome measures
We investigated the following outcomes in this review (Primary outcomes; Secondary outcomes). Reporting of these outcomes in the original trial report was not an eligibility requirement for inclusion in this review.
Primary outcomes
Time to withdrawal of allocated treatment (retention time). This is a combined outcome reflecting both efficacy and tolerability, as treatment may be withdrawn due to continued seizures, adverse effects or a combination of both. This is an outcome to which the participant makes a contribution, and is the primary effectiveness outcome measure recommended by the Commission on Antiepileptic Drugs of the International League Against Epilepsy (Glauser 2006; ILAE 1998).
Secondary outcomes
-
Time to achieve 12‐month seizure‐free period (remission) after randomisation
-
Time to achieve six‐month seizure‐free period (remission) after randomisation
-
Time to first seizure post randomisation
-
Occurrence of adverse events (to be reported narratively) (Data synthesis)
Search methods for identification of studies
We searched the following databases with no language restrictions:
-
the Cochrane Epilepsy Specialised Register (26 July 2016) using the search strategy outlined in Appendix 1;
-
the Cochrane Central Register of Controlled Trials (CENTRAL; 2016, issue 7) via the Cochrane Register of Studies Online (CRSO, 26 July 2016) using the search strategy outlined in Appendix 2;
-
MEDLINE (Ovid, 1946 to 26 July 2016) using the search strategy outlined in Appendix 3;
-
SCOPUS (1823 to 09 September 2014) using the search strategy outlined in Appendix 4;
-
ClinicalTrials.gov searched on 26 July 2016) using the search strategy outlined in Appendix 5;
-
World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) search portal searched on 26 July 2016), using the search strategy outlined in Appendix 6.
We had originally searched SCOPUS as an alternative to Embase, but this is no longer necessary, because randomised and quasi‐randomised controlled trials in Embase are now included in CENTRAL. We have not, therefore, updated the SCOPUS search.
We also reviewed reference lists of retrieved trials to search for additional reports of relevant trials, reviewed relevant conference proceedings and contacted experts in the field for details of any ongoing or unpublished trials.
Data collection and analysis
Selection of studies
One author (SJN) screened all titles and abstracts of all records identified by the electronic searches as described in Search methods for identification of reviews, according to the inclusion criteria specified above (Types of studies; Types of participants; Types of interventions). Subsequently, two authors (SJN and AGM) independently assessed full‐text publications according to the same inclusion criteria specified above. We resolved disagreements by discussion or by consulting a third author (CT) where necessary. We recorded the reasons for exclusion of trials at both stages of screening. We contacted trial authors for clarification if the eligibility of a trial was unclear from the published information.
Data extraction and management
Requesting individual participant data
For all trials meeting our inclusion criteria, two authors (SJN and AGM) sent a data‐request form to the first or corresponding author, or both, of the trial or to the trial sponsor where appropriate (referred to as data providers in this review).
Our data‐request form asked data providers if the following information was available (tick yes or no).
-
Trial methods:
-
method of generation of random list;
-
method of concealment of randomisation;
-
stratification factors;
-
blinding methods.
-
-
Participant covariates:
-
sex;
-
age;
-
seizure types;
-
epilepsy status (newly diagnosed/relapsed seizures following drug withdrawal);
-
time between first seizure and randomisation;
-
number of seizures prior to randomisation (with dates);
-
presence of neurological signs;
-
electroencephalography (EEG) results;
-
computed tomography (CT) or magnetic resonance imaging (MRI) results;
-
aetiology of seizures (if known).
-
-
Follow‐up data:
-
treatment allocation;
-
date of randomisation;
-
dates of follow‐up;
-
dates of seizures post randomisation or seizure frequency data between follow‐up visits;
-
dates of treatment withdrawal and reason(s) for treatment withdrawal;
-
starting dose of treatment;
-
dates of dose changes;
-
adverse events reported.
-
We also requested any available, related documents such as case report forms, trial protocols, clinical summaries etc. from data providers.
In the event of no response to our IPD request, we sent a follow‐up email to the original data provider contacted. If we still received no response for a particular trial, we attempted to contact another trial author or sponsor where appropriate. If a data provider was unable to make IPD available to us, we recorded the quoted reason why IPD could not be made available and we requested any aggregate data related to our outcome not reported in the publication.
If data could not be obtained (no response to any requests or IPD was not available), two independent authors (SJN and MS) assessed whether any relevant and appropriate aggregate level data was reported in the trial publication or could be indirectly estimated via the methods described in Parmar 1998 and Williamson 2002. We resolved any disagreements on extracted aggregate data by discussion or by consulting a third author (CT) if necessary.
Management of individual participant data
We stored all obtained data on a secure, dedicated network drive accessible only to the statisticians performing analysis (SJN, MS, CT). We checked all provided data for consistency and prepared them for analysis according to a pre‐specified procedure prepared by one author (SJN) (available on request) and piloted by two authors (SJN and MS). For each trial where IPD were supplied, we reproduced results from trial findings where possible and we performed the following consistency checks:
-
trial details cross‐checked against any published report of the trial; original trial authors to be contacted if missing data, errors or inconsistencies were found;
-
review of the chronological randomisation sequence by checking the balance of prognostic factors, taking account of factors stratified for in randomisation procedure.
We discussed any inconsistencies in the provided data with the corresponding data providers. If large or major inconsistencies were present, which could not be resolved by data providers, we did not include the data in any analyses. If minor inconsistencies were present, we analysed the data and conducted sensitivity analyses to test the robustness of results (Sensitivity analysis).
Following consistency checking and data cleaning, we prepared datasets for analysis and calculated outcomes for this review according to the methodology summarised below. We followed a 'standard operating procedure' for the data cleaning and preparation of data for analysis for all datasets to ensure a standardised and consistent approach to analysis throughout this review. Further details of this procedure can be obtained from the corresponding author on request.
Preparation of individual participant data for analysis
For the analysis of time to withdrawal of allocated treatment as a time‐to‐event outcome, we defined an 'event' as either the withdrawal of the allocated treatment due to poor seizure control or adverse events, or both. We also classed non‐compliance with the treatment regimen or the addition of another AED as 'events'. We censored the outcome if treatment was withdrawn because the individual achieved a period of remission, if a participant withdrew from allocated treatment for reasons not related to the treatment (such as loss to follow‐up) or if the individual was still on allocated treatment at the end of follow‐up. Two authors (SJN and AG) independently reviewed reasons for treatment withdrawal for classification as events or censored observations, and we resolved any disagreements by mutual discussion or by involving a third author (CT).
If seizure data were provided or recorded in terms of the number of seizures recorded between clinic visits rather than specific dates of seizures, to enable the calculation of time‐to‐event outcomes, we applied linear interpolation to estimate dates of seizures between follow‐up visits. For example, if the trial recorded four seizures between two visits that occurred on 1 March 2010 and 1 May 2010 (interval of 61 days), then the date of the first seizure would be approximately 13 March 2010. This allowed the computation of an estimate of the time to six‐month remission, 12‐month remission, and first seizure.
We calculated time to six‐month and 12‐month remission from the date of randomisation to the date (or estimated date) the individual had first been free of seizures for six or 12 months respectively. If the person had one or more seizures in the titration period, a six‐month or 12‐month seizure‐free period could also occur between the estimated date of the last seizure in the titration period and the estimated date of the first seizure in the maintenance period
We calculated time to first seizure from the date of randomisation to the date that their first seizure was estimated to have occurred. If seizure data were missing for a particular visit, these outcomes were censored at the previous visit. These outcomes were also censored if the individual died or if follow‐up ceased prior to the occurrence of the event of interest.
Two trials were designed in strata based on whether recommended treatment would be carbamazepine or sodium valproate (Privitera 2003; Trinka 2013). Within the two strata, participants were randomised to topiramate (Privitera 2003) or levetiracetam (Trinka 2013) compared to the recommended treatment of carbamazepine or sodium valproate depending on the strata. To ensure that randomised comparisons were made, we analysed data for these two trials according to the separate strata in this review (i.e. treated as two trials Privitera 2003 carbamazepine branch and Privitera 2003 sodium valproate branch).
Assessment of risk of bias in included studies
Two authors (SJN, JW) independently assessed risk of bias in all included trials using the Cochrane tool for assessing risk of bias (Higgins 2011). The following methodological criteria are assessed according to this tool:
-
selection bias (sequence generation and allocation concealment);
-
performance bias (blinding of participants and personnel);
-
detection bias (blinding of outcome assessment);
-
attrition bias (incomplete outcome data);
-
reporting bias (selective outcome reporting).
We resolved any disagreements by discussion. In theory, a review using IPD should overcome issues of reporting biases as unpublished data can be provided and unpublished outcomes calculated. Any selective reporting bias detected could be assessed with the Outcome Reporting Bias in Trials (ORBIT) classification system (Kirkham 2010). As specified in Data extraction and management, we asked the data providers to provide trial methods such as randomisation and blinding methods, and we discussed any missing data and or inconsistencies, or both with them.
Measures of treatment effect
We summarised all time‐to‐event outcomes using the hazard ratio (HR) as the measure of treatment effect. We calculated outcomes from IPD provided where possible or extracted summary statistics from published trials. We did not attempt to analyse or synthesise adverse event data; a large range of different adverse events are thought to be associated with the 10 different drugs and such data were collected and presented in different ways across trials. For these reasons, we believe a synthesis of adverse event data would present only selective, and potentially misleading information, while a narrative description of adverse event data from IPD or extracted from published trials would be the most informative way of presenting these data.
Unit of analysis issues
We did not encounter any unit of analysis issues. For inclusion in the review, the unit of allocation had to be the individual. Trials of a repeated‐measures (longitudinal) nature or of a cross‐over design were not eligible for inclusion.
Dealing with missing data
For all included trials, we conducted an assessment of the proportion of missing outcome, demographic and covariate data and made a judgement regarding the extent and nature of missing data (e.g. missing at random, missing not at random). We attempted to contact all trial authors in order to request relevant data; we included any information regarding missing data in such requests (Data extraction and management). If further information regarding missing data could not be provided and we judged that an important proportion of data (particularly outcome data) were missing, we conducted sensitivity analyses to investigate the potential impact of the missing data (for example, best case scenario or worst case scenario analyses, assuming those with missing outcome data all had a favourable or unfavourable outcome, respectively).
Assessment of heterogeneity
We used a fixed‐effect model for all pairwise and network meta‐analyses in the first instance as we anticipated that our specific inclusion criteria would result in eligible studies of a similar design and populations and our use of IPD to standardise definitions of outcomes. Also, our previous reviews of this topic have not showed any important heterogeneity (Marson 2000; Nevitt 2016; Nolan 2013b; Nolan 2013c; Nolan 2015; Nolan 2016a; Nolan 2016b; Nolan 2016d); see Data synthesis for further details of pairwise and network meta‐analysis.
For each pairwise comparison, we assessed the presence of heterogeneity statistically using the Q test (P value less than 0.10 for significance) and the I2 statistic with the following interpretation (Higgins 2003):
-
0% to 40%: might not be important;
-
30% to 60%: may represent moderate heterogeneity;
-
50% to 90%: may represent substantial heterogeneity;
-
75% to 100%: considerable heterogeneity.
We also assessed the presence of heterogeneity by visually inspecting forest plots, particularly in terms of the magnitude and direction of effects. If substantial or considerable heterogeneity (i.e. I2 of 50% or over) was found to be present, which we were not able to explain by differences in characteristics of the trials and participants, we planned to perform network meta‐analysis with a random‐effects model.
It was not possible to directly calculate an I2 statistic for the network meta‐analysis due to the between‐study covariance structure required for the network meta‐analysis model (see Data synthesis). However, for this model, we were able to estimate an R statistic, which compares the impact of heterogeneity in the fixed‐effect and random‐effects models (Jackson 2012) and it has been previously shown that R can be used to calculate I2 as follows: I2 = (R2 ‐ 1)/R2 (Higgins 2002)
Therefore we estimated an I2 statistic for the whole treatment network for each analysis and interpreted as above. We also presented an estimate of Tau2 (an estimate of the between‐study variance in random‐effects meta‐analysis) for each analysis and we have taken both statistics into account when interpreting the presence of any important heterogeneity in the treatment network.
Assessment of reporting biases
Two authors (SJN and JW) undertook a full 'Risk of bias' assessment for each eligible trial, including risk of reporting biases. In theory, a review using IPD should overcome issues of reporting biases, as unpublished data can be provided and unpublished outcomes calculated. As specified in Data extraction and management, we asked the data providers for trial methods, such as randomisation and blinding methods, and we discussed any missing data and inconsistencies with them.
If we suspected selective reporting bias in the review, we intended to assess the magnitude and impact of this selective reporting bias using the ORBIT classification system (Kirkham 2010), however we did not have any major concerns about selective reporting bias in this review. The approach to this review (re‐analysis of IPD) helps to overcome issues of reporting bias, as unpublished data can be provided and unpublished outcomes calculated.
Data synthesis
Figure 1 and Figure 2 visually present the network of 45 pairwise comparisons from the 10 antiepileptic treatments of interest to this review.

Network plot of pairwise comparisons in all included studies, studies providing individual participant data (IPD) and studies without IPD
Note that the size of the node indicates the number of studies the drug is included in and the thickness of the edges corresponds to the number of participants contributing to the comparison (i.e. larger node = more studies, thicker edge = more participants).
CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.

Network plot of pairwise comparisons for all included participants (total 17,961 participants), participants with partial seizures and participants with generalised tonic‐clonic seizures with or without other seizure types (shortened to 'generalised seizures' for brevity).
11978 participants were classified as experiencing partial seizures (66.7% of total), 4407 participants were classified as experiencing generalised seizures (24.5% of total) and 1576 had an unclassified or missing seizure type (8.8% of total).
Note that the size of the node indicates the number of studies the drug is included in and the thickness of the edges corresponds to the number of participants contributing to the comparison (i.e. larger node = more studies, thicker edge = more participants).
CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Pairwise and Network meta‐analysis
We used the statistical software package SAS (version 9.3) (SAS 2011) to perform all data cleaning, consistency checking and data preparation (see Data extraction and management) and Stata version 14 (StataCorp 2015) to perform all synthesis of direct and indirect evidence .
We requested data for one trial, Biton 2001, via data sharing portal ClinicalStudyDataRequest.com and the data were provided to us via a remote secure data access system that allowed analysis in SAS‐based statistical software and export of analysis results. We were unable to combine this dataset with the other datasets to perform the analyses described below in Stata version 14, therefore we treated the results exported from the data access system as aggregate data in sensitivity analysis (see Sensitivity analysis).
We took an intention‐to‐treat approach (as far as possible) to analysis; in other words, we analysed participants in the group to which they had been randomised in an individual trial, irrespective of which treatment they had actually received. Therefore, for time‐to‐event outcomes, 'time to six‐month remission', 'time to 12‐month remission' and 'time to first seizure post randomisation', participants were not censored if treatment was withdrawn. For the primary outcome, time to withdrawal of allocated treatment, we considered withdrawals due to lack of efficacy (i.e. recurrent seizures), poor tolerability (i.e. adverse events) or a combination of both poor efficacy and tolerability. Other withdrawals such as losses to follow‐up, non treatment‐related deaths, administrative trial reasons etc. were censored at the time of withdrawal.
For all time‐to‐event outcomes, we investigated the relationship between the time to the event and treatment effect of the AEDs. We fitted a Cox proportional hazards regression model, stratified by trial to preserve the within‐trial randomisation, to the entire individual participant dataset. We fitted this model via the 'mvmeta_make' command in Stata version 14 to produce a dataset in the correct format to perform network meta‐analysis with the 'mvmeta' command (White 2009); in other words, a dataset with trial‐specific estimates of treatment effect (log HR), the associated variance of the treatment effect and covariances where applicable (i.e. correlation between treatment effects for trials with more than two treatment arms).
The Cox proportional hazards model assumes that ratio of hazards (risks) between the two treatment groups is constant over time. To assess the validity of this assumption, we tested the statistical significance of time‐varying covariates for all covariates in the primary model. If we had reason to believe that the proportional hazards assumption had been violated in the primary model, in sensitivity analysis we fitted a parametric, accelerated failure‐time model, stratified by trial, to the entire individual participant dataset via the 'mvmeta_make' command and compared these results to those of the primary analysis (White 2009). An accelerated failure‐time model assumes that treatment effect accelerates or decelerates over time, rather than remains constant as assumed by the Cox proportional hazards model.
We calculated direct pairwise treatment effect estimates (where possible) using the 'metan' command (Palmer 2016) in Stata version 14 to pool trial‐specific log hazard ratios from the Cox proportional hazards model as described above.
We performed network meta‐analysis via the 'mvmeta' command in Stata version 14 assuming equal heterogeneity for all comparisons (i.e. a between‐study covariance structure (variance‐covariance matrix) proportional to unknown parameter Tau2) (White 2009). It was necessary to make an assumption regarding the between‐study covariance structure for a network without pairwise comparisons between all treatments of interest. However, due to this assumption regarding heterogeneity, we could not calculate an I2 statistic directly from the model and had to estimated it (see Assessment of heterogeneity). Network meta‐analysis provided treatment effect estimates combining direct and indirect evidence.
We performed pairwise and network meta‐analyses with a treatment by epilepsy type interaction (see Subgroup analysis and investigation of heterogeneity for further details).
For clinical interest and relevance, we have presented HR estimates from the network model (direct and indirect evidence combined) for each AED in the network compared to the current recommended first‐line treatments (carbamazepine or lamotrigine for partial onset seizures and sodium valproate for generalised onset seizures) and for all comparisons by epilepsy type in the main results of this review via forest plots.
Often rankings of treatments (i.e. the probability that each treatment in the network is the best) are presented for network meta‐analysis; however due to the treatment by epilepsy type interaction in this model, we could not calculate rankings by epilepsy type. Instead, we informally 'ranked' treatments by ordering according to their treatment effect sizes compared to the reference treatment (e.g. better or worse than carbamazepine) on the forest plots presented.
Investigation of consistency in network meta‐analysis
A key assumption made in network meta‐analysis is that treatment effect is 'exchangeable' across all included trials; in other words, the indirect comparison made between two treatments is a feasible comparison to make (known as the transitivity assumption) and that the indirect evidence is consistent with the direct evidence where a comparison exists (known as the consistency assumption).
Transitivity requires that all treatments are "jointly randomisable"; in other words, all 10 AEDs could feasibly be randomised in the same trial and those that are not treatment arms in any given trial are "missing at random" (Lu 2006). This assumption cannot be formally tested statistically; transitivity must be judged by careful consideration of trial settings and characteristics, treatment mechanisms and participant demographics to investigate if any differences would be expected to modify relative treatment effects. Given that all of the 10 drugs within this network are licenced as monotherapy treatments for individuals with newly diagnosed partial onset seizures or generalised onset tonic‐clonic seizures (with or without other generalised seizure types) and have all been used within trials of similar designs, we have no concerns over this transitivity assumption in this network.
The consistency assumption can be evaluated statistically comparing the difference between the direct treatment effect estimate and the indirect estimate for each loop of evidence. Given the complexity of the network model fitted (with treatment by epilepsy type interaction) and the number of multi‐arm trials included in analysis, we performed node splitting in Stata version 14 via the command 'network sidesplit' (Dias 2010; White 2015) to formally estimate differences between direct and indirect evidence for each comparison. In order to examine any clinical inconsistency (i.e. important differences in numerical results between direct, indirect and network results), we have presented HR estimates for direct evidence, indirect evidence (from the node splitting model) and direct plus indirect evidence from the network models for each pairwise comparison via forest plots and discuss the potential origins and implications of any apparent inconsistency. Secondly, we fitted a ‘design‐by‐treatment’ inconsistency model in Stata version 14 via mvmeta (White 2009); this method evaluates both loop and design inconsistencies, particularly within multi‐arm trials (Higgins 2012).
Adverse events
Due to the wide range of events reported in the trials and the different methods of recording and reporting of adverse events, we have not analysed adverse event data in meta‐analysis but have provided a narrative report according to the definition of the events within the data provided to us or in the published paper.
Subgroup analysis and investigation of heterogeneity
There are strong clinical beliefs that certain AEDs are more effective in certain seizure types than others, for example carbamazepine is more effective in partial onset seizures and sodium valproate is more effective in generalised onset seizures (Marson 2000), suggesting that there is a treatment‐by‐seizure type (partial or generalised) interaction. Without taking account of this potential interaction in our analysis, we believe that the key assumption of an exchangeable treatment effect across all included trials would be violated.
To account for this, we conducted all analyses separately by epilepsy type (partial onset or generalised onset) according to the classification of main seizure type at baseline and performed all network meta‐analysis with a treatment‐by‐epilepsy‐type interaction. We classified partial seizures (simple or complex) and partial secondarily generalised seizures as partial epilepsy. We classified primarily generalised seizures as generalised epilepsy. We then judged exchangeability of treatment effect separately by analyses of seizure type.
We also performed an analysis adjusted for age at entry into the trial (an interaction between treatment and age (centred) added to initial Cox proportional hazards model described in Data synthesis) and we compared results to primary analysis with adjustment only for seizure type.
We would have liked to explore other participant covariates specified in Data extraction and management as potential modifiers of treatment effect and as potential sources of heterogeneity or inconsistency, or both, such as seizure frequency before randomisation (time since first ever seizure and/or number of seizures before randomisation) and aetiology of seizures (if known according to pre‐treatment investigations such as EEG, CT and/or MRI scan); however, due to large proportions of missing data for most of these covariates and variability in the definitions of data provided to us for these covariates (see Included studies), an additional adjusted analysis was not appropriate. We will consider other options to explore these covariates for an update of this review.
Sensitivity analysis
As described in Data synthesis, we applied a fixed‐effect model principally to pairwise and network meta‐analysis, and fitted a random‐effects model to both pairwise and network meta‐analysis models in sensitivity analysis, and compared the results.
Also as described in Data synthesis, we applied a Cox proportional hazards model principally to pairwise and network meta‐analysis. We fitted an accelerated failure‐time model, which does not make the assumption of constant treatment effect over time, to both pairwise and network meta‐analysis models in sensitivity analysis and compared the results.
As specified in Data extraction and management, we discussed any inconsistencies in the provided data with the corresponding data providers and performed sensitivity analyses to investigate the impact of any missing data (see Dealing with missing data). If large or major inconsistencies were present, which could not be resolved by the data providers, we would not include the data in any analyses. If minor inconsistencies were present, we included the data in analyses and pursued sensitivity analyses to test the robustness of results included in these data. We performed the following sensitivity analyses due to inconsistencies in IPD provided and compared the results of sensitivity analyses to those of the primary analysis:
-
In Stephen 2007 there were minor inconsistencies between rates of seizure recurrence and reasons for withdrawal between the data provided and the published paper, which the trial authors could not resolve. Therefore we performed sensitivity analysis excluding Stephen 2007 from all analyses.
-
In Reunanen 1996, participants were considered to have completed the trial and hence treatment was withdrawn if they experienced a seizure after week six. This does not correspond with the treatment withdrawal definition used in this review (see Primary outcomes and Data extraction and management). Therefore, we performed sensitivity analysis excluding Reunanen 1996 for the analysis of 'Time to withdrawal of allocated treatment.'
-
In Banu 2007, there were minor inconsistencies between rates of seizure recurrence between the data provided and the published paper, which the authors could not resolve. Therefore we performed sensitivity analysis excluding Banu 2007 from analysis of 'Time to first seizure.' (Data for first seizure recurrence only were available, so this trial did not contribute to outcomes of time to six‐month remission and time to 12‐month remission).
-
Nieto‐Barrera 2001 did not include seizures that occurred during the first four weeks of the trial in efficacy analyses, and dates of seizures before week four were not supplied to us. Therefore, we calculated seizure outcomes as the time to first seizure and time to six‐month remission after week four rather than after randomisation. We performed sensitivity analysis excluding seizure data for Nieto‐Barrera 2001 from analysis of 'Time to first seizure' (this trial was 24 weeks' duration so did not contribute to outcomes of time to six‐month remission and time to 12‐month remission).
-
In Placencia 1993, there were minor inconsistencies between reasons for withdrawal between the data provided and the published paper. We compared reasons for withdrawal in the data provided with reasons reported in the publication and performed a sensitivity analysis for the analysis of 'Time to withdrawal of allocated treatment', with withdrawals reclassified according to definitions from the published paper (this sensitivity analysis was also performed in a previously published Cochrane Review, see Nolan 2016b for further details).
Given that misclassification of seizure type is a recognised problem in epilepsy (whereby some individuals with generalised seizures have been mistakenly classed as having partial onset seizures and vice versa) and such misclassification did impact upon the results of a review in our series of pairwise reviews for monotherapy in epilepsy comparing phenytoin and sodium valproate in which nearly 50% of participants analysed may have had their seizure type misclassified (Nolan 2016d), we investigated the potential impact of misclassification on results in a sensitivity analysis. Given clinical evidence that individuals with generalised onset seizures are unlikely to have an 'age of onset' greater than 25 to 30 years (Malafosse 1994), we examined the distribution of age at onset for individuals with generalised seizures. We identified 1164 participants classified as experiencing generalised seizures and estimated age of onset as greater than 30 years (age of first seizure provided directly in IPD or estimated to be within one year of age of entry into trial for newly diagnosed participants). We performed two sensitivity analyses to investigate misclassification:
-
re‐classification of all individuals with generalised seizures and age of onset greater than 30 years as having partial onset seizures. We then repeated network meta‐analysis with the interaction term of treatment by seizure type with the reclassified seizure type.
-
re‐classification of all individuals with generalised seizure types and age at onset greater than 30 years and those with missing seizure type into an 'unclassified seizure type' group. We then repeated network meta‐analysis with the interaction term of treatment by seizure type, where seizure type is partial epilepsy compared to generalised or unclassified epilepsy.
We were unable to perform network meta‐analysis with a 'three‐way' interaction (i.e. partial epilepsy compared to generalised epilepsy compared to unclassified epilepsy) due to small numbers of participants with unclassified epilepsy on some of the treatments.
Where possible, if IPD were not available for analysis, we attempted to extract aggregate data. Where aggregate hazard ratios and standard errors or confidence intervals could be extracted or estimated from trial publications by seizure type for our outcomes of interest, we incorporated these estimates into network meta‐analysis and compared the results of these sensitivity analyses to those of the primary analysis. As described in Data synthesis, we were provided with IPD for one trial (Biton 2001), in a remote data access system therefore we could not combine this dataset with the other datasets to perform IPD analysis. We also treated our exported results for this trial as aggregate data in sensitivity analysis.
'Summary of findings' table and quality of the evidence
We have presented six 'Summary of findings' tables for our primary outcome and first secondary outcome by epilepsy type and by reference treatment (see Data synthesis for further information);
-
Time to withdrawal of allocated treatment for individuals with partial seizures (reference treatment carbamazepine) (see summary of findings Table for the main comparison)
-
Time to withdrawal of allocated treatment for individuals with partial seizures (reference treatment lamotrigine) (see summary of findings Table 2)
-
Time to withdrawal of allocated treatment for individuals with generalised seizures (reference treatment sodium valproate) (see summary of findings Table 3)
-
Time to 12‐month remission for individuals with partial seizures (reference treatment carbamazepine) (see summary of findings Table 4)
-
Time to 12‐month remission for individuals with partial seizures (reference treatment lamotrigine) (see summary of findings Table 5)
-
Time to 12‐month remission for individuals with generalised seizures (reference treatment sodium valproate) (see summary of findings Table 6)
We have presented the tables following the approach of Salanti 2014 as far as possible ‐ for pairwise comparisons, we have presented the relative effect from direct evidence from pairwise meta‐analysis, number of studies and participants contributing to direct evidence, the relative effect from direct plus indirect evidence from network meta‐analysis, proportion of direct evidence, and quality of the evidence.
We determined quality of the evidence using the GRADE approach (GRADE 2008), whereby we downgraded evidence in the presence of high risk of bias, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results or high probability of publication bias. We downgraded evidence by one level if we considered the limitation to be serious and two levels if we considered it to be very serious. In this context of network meta‐analysis we also considered the proportion of direct evidence and inconsistency of direct and indirect evidence when determining quality of the evidence.
Results
Description of studies
Results of the search
We identified 6762 records from the databases and search strategies outlined in Electronic searches. We found three further records by handsearching and checking reference lists of included studies. We removed 3032 duplicate records and screened 3733 records (title and abstract) for inclusion in the review. We excluded 3591 records based on title and abstract and assessed 142 full‐text articles for inclusion in the review. We excluded 31 studies (described in 32 full‐text articles) from the review (see Excluded studies below) and included 77 trials in the review, which were reported in 95 full‐text articles (see Included studies below). We identified seven studies as ongoing (ACTRN12615000556549; ACTRN12615000639527; ACTRN12615000640505; ACTRN12615000641594; ACTRN12615000643572; NCT01891890; NCT02201251) and seven studies (described in eight records) as awaiting classification (translation: Chen 2013; Korean Zonisamide Study 1999; Park 2001; Rysz 1994; Xu 2012) or further information: IRCT201202068943N1; NCT00154076). See Figure 3 for PRISMA study flow diagram (Moher 2009).

Study flow diagram
Included studies
We included 77 trials in the review (Aikia 1992; Banu 2007; Baulac 2012; Bidabadi 2009; Bill 1997; Biton 2001; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2002; Brodie 2007; Callaghan 1985; Capone 2008; Castriota 2008; Chadwick 1998; Chen 1996; Cho 2011; Christe 1997; Consoli 2012; Cossu 1984; Craig 1994; Czapinski 1997; Dam 1989; de Silva 1996; Dizdarer 2000; Donati 2007; Eun 2012; Feksi 1991; Forsythe 1991; Fritz 2006; Gilad 2007; Guerreiro 1997; Heller 1995; Jung 2015; Kalviainen 2002; Kopp 2007; Korean Lamotrigine Study Group 2008; Kwan 2009; Lee 2011; Lukic 2005; Mattson 1985; Mattson 1992; Mitchell 1987; Miura 1990; Motamedi 2013; NCT01498822; NCT01954121; Nieto‐Barrera 2001; Ogunrin 2005; Pal 1998; Placencia 1993; Privitera 2003; Pulliainen 1994; Ramsey 1983; Ramsey 1992; Ramsey 2007; Ramsey 2010; Rastogi 1991; Ravi Sudhir 1995; Resendiz 2004; Reunanen 1996; Richens 1994; Rowan 2005; Saetre 2007; SANAD A 2007; SANAD B 2007; Shakir 1981; So 1992; Steiner 1999; Steinhoff 2005; Stephen 2007; Suresh 2015; Thilothammal 1996; Trinka 2013; Turnbull 1985; Verity 1995; Werhahn 2015).
Seven trials were available in abstract form only (Bidabadi 2009; Czapinski 1997; Fritz 2006; Kalviainen 2002; Kopp 2007; Lukic 2005; Ramsey 2007), one was available in English only as a clinical trial summary report (Korean Lamotrigine Study Group 2008) and two trials were available only as an online summary (NCT01498822; NCT01954121). Three trials were published in Italian (Capone 2008; Castriota 2008; Cossu 1984) and one in Spanish (Resendiz 2004) and were translated into English. One of the published reports contained results on two separate RCTs run on very similar protocols; although the two trials were reported within the same publication we treated them as separate trials within this systematic review (Brodie 1995a; Brodie 1995b).
Characteristics of included trials
Twenty‐five trials were designed to recruit individuals with partial seizures only (Baulac 2012; Bidabadi 2009; Castriota 2008; Chadwick 1998; Cho 2011; Cossu 1984; Czapinski 1997; Dizdarer 2000; Donati 2007; Eun 2012; Gilad 2007; Jung 2015; Lee 2011; Mattson 1985; Mattson 1992; Mitchell 1987; NCT01498822; NCT01954121; Nieto‐Barrera 2001; Ramsey 2007; Resendiz 2004; SANAD A 2007; So 1992; Suresh 2015; Werhahn 2015). Three trials were designed to recruit individuals with generalised tonic‐clonic seizures with or without other generalised seizure types or unclassified seizure types only (Ramsey 1992; SANAD B 2007; Thilothammal 1996). The remaining 49 trials recruited individuals with partial or generalised tonic‐clonic seizures with or without other generalised seizure types (Aikia 1992; Banu 2007; Bill 1997; Biton 2001; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2002; Brodie 2007; Callaghan 1985; Capone 2008; Chen 1996; Christe 1997; Consoli 2012; Craig 1994; Dam 1989; de Silva 1996; Feksi 1991; Forsythe 1991; Fritz 2006; Guerreiro 1997; Heller 1995; Kalviainen 2002; Kopp 2007; Korean Lamotrigine Study Group 2008; Kwan 2009; Lukic 2005; Miura 1990; Motamedi 2013; Ogunrin 2005; Pal 1998; Placencia 1993; Privitera 2003; Pulliainen 1994; Ramsey 1983; Ramsey 2010; Rastogi 1991; Ravi Sudhir 1995; Reunanen 1996; Richens 1994; Rowan 2005; Saetre 2007; Shakir 1981; Steiner 1999; Steinhoff 2005; Stephen 2007; Trinka 2013; Turnbull 1985; Verity 1995). However five trials did not describe the number of participants with each seizure type recruited (Capone 2008; Dam 1989; Forsythe 1991; Fritz 2006; Saetre 2007).
Forty‐seven trials recruited only individuals with new onset seizures and no previous AED treatment (Aikia 1992; Baulac 2012; Bill 1997; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2007; Castriota 2008; Chen 1996; Cho 2011; Christe 1997; Cossu 1984; Craig 1994; Czapinski 1997; Dam 1989; de Silva 1996; Donati 2007; Eun 2012; Forsythe 1991; Guerreiro 1997; Heller 1995; Jung 2015; Kalviainen 2002; Kopp 2007; Lukic 2005; Mitchell 1987; Miura 1990; Motamedi 2013; NCT01498822; NCT01954121; Ogunrin 2005; Pal 1998; Placencia 1993; Privitera 2003; Pulliainen 1994; Ramsey 1983; Ramsey 1992; Ravi Sudhir 1995; Resendiz 2004; Saetre 2007; Steiner 1999; Steinhoff 2005; Stephen 2007; Suresh 2015; Thilothammal 1996; Turnbull 1985; Werhahn 2015). Three trials recruited individuals with new onset post‐stroke seizures (Consoli 2012; Capone 2008; Gilad 2007), seven trials recruited individuals with new onset or long‐term untreated seizures (Banu 2007; Callaghan 1985; Feksi 1991; Lee 2011; Korean Lamotrigine Study Group 2008; Nieto‐Barrera 2001; Trinka 2013), six trials recruited individuals with new onset, untreated or under‐treated seizures (Mattson 1985; Mattson 1992; Ramsey 2007; Ramsey 2010; Rowan 2005; So 1992), five trials recruited individuals with new onset or relapsed seizures following a period of remission (Chadwick 1998; Kwan 2009; Reunanen 1996; Richens 1994; Verity 1995), three trials recruited individuals with new onset, relapsed seizures following a period of remission or individuals whose previous treatment with an AED had failed (SANAD A 2007; SANAD B 2007; Shakir 1981) and six trials did not state if individuals had received previous AED treatment (Biton 2001; Brodie 2002; Bidabadi 2009; Dizdarer 2000; Fritz 2006; Rastogi 1991).
Twenty‐eight trials were single‐centre and conducted in Bangladesh (Banu 2007) Iran (Bidabadi 2009; Motamedi 2013), Ireland (Callaghan 1985), Italy (Capone 2008; Castriota 2008; Cossu 1984), Taiwan (Chen 1996), Republic of Korea (Cho 2011), the UK (Craig 1994; Forsythe 1991; Stephen 2007; Turnbull 1985), Turkey (Dizdarer 2000), Kenya (Feksi 1991), Israel (Gilad 2007), Germany (Kopp 2007), Serbia and Montenegro (Lukic 2005), the USA (Mitchell 1987), Japan (Miura 1990), Nigeria (Ogunrin 2005), India (Pal 1998; Rastogi 1991; Ravi Sudhir 1995; Suresh 2015; Thilothammal 1996), Ecuador (Placencia 1993) and Finland (Pulliainen 1994).
Forty‐five trials were multicentre, conducted in centres across the USA (Biton 2001; Mattson 1985; Mattson 1992; Ramsey 1983; Ramsey 1992; Ramsey 2007; Ramsey 2010; Rowan 2005), the UK (Brodie 1995a; Brodie 1995b; Brodie 1999; de Silva 1996; Heller 1995; Richens 1994; SANAD A 2007; SANAD B 2007; Steiner 1999; Verity 1995), the UK and New Zealand (Shakir 1981), Europe (Consoli 2012; Dam 1989; Donati 2007; Kalviainen 2002; Saetre 2007; Steinhoff 2005; Werhahn 2015), Europe and Australia (Brodie 2002; Reunanen 1996; Trinka 2013), Europe and South Africa (Brodie 2007), Europe and Mexico (Nieto‐Barrera 2001), Europe, South America and South Africa (Christe 1997), South America and South Africa (Bill 1997; Guerreiro 1997), Republic of Korea (Eun 2012; Jung 2015; Korean Lamotrigine Study Group 2008; Lee 2011; NCT01498822), China (NCT01954121), Hong Kong (Kwan 2009), Mexico (Resendiz 2004), Asia, Australia and Europe (Baulac 2012), Europe, Australia, Canada and South Africa (Chadwick 1998), the USA, Canada, Europe and South America (Privitera 2003).
Four trials did not state whether they were single‐ or multicentre; these trials were conducted in Finland (Aikia 1992), Poland (Czapinski 1997), Germany (Fritz 2006) and the USA (So 1992).
Twenty trials recruited adults and children (Biton 2001; Brodie 1995a; Brodie 1995b; Callaghan 1985; Chadwick 1998; Cho 2011; Feksi 1991; Korean Lamotrigine Study Group 2008; Nieto‐Barrera 2001; Placencia 1993; Privitera 2003; Ramsey 1992; Ramsey 2010; Rastogi 1991; Reunanen 1996; SANAD A 2007; SANAD B 2007; Shakir 1981; Steinhoff 2005; Stephen 2007).
Fifteen trials recruited children; four trials recruited children under the age of 12 years (Bidabadi 2009; Eun 2012; Mitchell 1987; Thilothammal 1996), one trial recruited children under 14 years (Forsythe 1991), three trials recruited children under 15 years (Banu 2007; Chen 1996; Dizdarer 2000), three trials recruited children under 16 years (de Silva 1996; Jung 2015; Verity 1995), one trial recruited children under 17 years (Donati 2007) and three trials recruited children under 18 years (Guerreiro 1997; Pal 1998; Resendiz 2004).
Thirty‐nine trials recruited adults; two trials defined adults as over the age of 13 years (Heller 1995; So 1992), four trials defined adults as over the age of 14 years (Ogunrin 2005; Ravi Sudhir 1995; Steiner 1999; Turnbull 1985); four trials defined adults as over the age of 15 years (Cossu 1984; Dam 1989; Fritz 2006; Pulliainen 1994), nine trials defined adults as over the age of 16 years (Bill 1997; Brodie 2002; Brodie 2007; Christe 1997; Lee 2011; NCT01498822; NCT01954121; Richens 1994; Trinka 2013), nine trials defined adults as over the age of 18 (Baulac 2012; Consoli 2012; Czapinski 1997; Kwan 2009; Lukic 2005; Mattson 1985; Mattson 1992; Ramsey 1983; Suresh 2015) and four trials did not state the minimum age of an ‘adult’ in the trial (Aikia 1992; Capone 2008; Castriota 2008; Gilad 2007). Seven trials recruited elderly participants; two trials recruited participants over the age of 65 years (Brodie 1999; Saetre 2007) and five trials recruited individuals over the age of 60 years (Craig 1994; Motamedi 2013; Ramsey 2007; Rowan 2005; Werhahn 2015).
Three trials did not state the age ranges of eligible participants (Kalviainen 2002; Kopp 2007; Miura 1990).
Table 1 shows the number of participants randomised to each of the 10 drugs, split according to the trials for which individual participant data were available and not available:
| Trial\Drug | CBZ | PHB | PHT | VPS | LTG | OXC | LEV | TPM | GBP | ZNS | Total | Total randomiseda |
| Trials providing individual participant data | ||||||||||||
| 54 | 54 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 108 | 108 | |
| 301 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 282 | 583 | 583 | |
| 0 | 0 | 144 | 0 | 0 | 143 | 0 | 0 | 0 | 0 | 287 | 287 | |
| 0 | 0 | 0 | 69 | 66 | 0 | 0 | 0 | 0 | 0 | 135 | 136 | |
| 66 | 0 | 0 | 0 | 70 | 0 | 0 | 0 | 0 | 0 | 136 | 136 | |
| 63 | 0 | 0 | 0 | 61 | 0 | 0 | 0 | 0 | 0 | 124 | 124 | |
| 48 | 0 | 0 | 0 | 102 | 0 | 0 | 0 | 0 | 0 | 150 | 150 | |
| 291 | 0 | 0 | 0 | 0 | 0 | 288 | 0 | 0 | 0 | 579 | 579 | |
| 74 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 218 | 0 | 292 | 292 | |
| 0 | 0 | 81 | 85 | 0 | 0 | 0 | 0 | 0 | 0 | 166 | 166 | |
| 54 | 10 | 54 | 49 | 0 | 0 | 0 | 0 | 0 | 0 | 167 | 173 | |
| 26 | 0 | 0 | 0 | 0 | 26 | 0 | 0 | 0 | 0 | 52 | 52 | |
| 41 | 0 | 0 | 0 | 43 | 0 | 0 | 0 | 0 | 0 | 84 | 84 | |
| 0 | 0 | 94 | 0 | 0 | 99 | 0 | 0 | 0 | 0 | 193 | 193 | |
| 61 | 58 | 63 | 61 | 0 | 0 | 0 | 0 | 0 | 0 | 243 | 243 | |
| 0 | 0 | 0 | 44 | 37 | 0 | 0 | 0 | 0 | 0 | 81 | 81 | |
| 53 | 0 | 0 | 0 | 57 | 0 | 0 | 0 | 0 | 0 | 110 | 110 | |
| 155 | 155 | 165 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 475 | 475 | |
| 236 | 0 | 0 | 244 | 0 | 0 | 0 | 0 | 0 | 0 | 480 | 480 | |
| 202 | 0 | 0 | 0 | 420 | 0 | 0 | 0 | 0 | 0 | 622 | 622 | |
| 19 | 18 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 55 | 55 | |
| 0 | 47 | 47 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 94 | 94 | |
| 95 | 97 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 192 | 192 | |
| Privitera 2003 (CBZ branch)b | 129 | 0 | 0 | 0 | 0 | 0 | 0 | 266 | 0 | 0 | 395 | 395 |
| Privitera 2003 (VPS branch)b | 0 | 0 | 0 | 78 | 0 | 0 | 0 | 147 | 0 | 0 | 225 | 225 |
| 0 | 0 | 50 | 86 | 0 | 0 | 0 | 0 | 0 | 0 | 136 | 136 | |
| 0 | 0 | 128 | 0 | 0 | 0 | 0 | 133 | 0 | 0 | 261 | 261 | |
| 121 | 0 | 0 | 0 | 230 | 0 | 0 | 0 | 0 | 0 | 351 | 351 | |
| 151 | 0 | 0 | 149 | 0 | 0 | 0 | 0 | 0 | 0 | 300 | 300 | |
| 378 | 0 | 0 | 0 | 378 | 210 | 0 | 378 | 377 | 0 | 1721 | 1721 | |
| 0 | 0 | 0 | 238 | 239 | 0 | 0 | 239 | 0 | 0 | 716 | 716 | |
| 0 | 0 | 95 | 0 | 86 | 0 | 0 | 0 | 0 | 0 | 181 | 181 | |
| 0 | 0 | 0 | 109 | 117 | 0 | 0 | 0 | 0 | 0 | 226 | 227 | |
| Trinka 2013 (CBZ branch)b | 503 | 0 | 0 | 0 | 0 | 0 | 493 | 0 | 0 | 0 | 996 | 999 |
| Trinka 2013 (VPS branch)b | 0 | 0 | 0 | 353 | 0 | 0 | 350 | 0 | 0 | 0 | 703 | 703 |
| 0 | 0 | 70 | 70 | 0 | 0 | 0 | 0 | 0 | 0 | 140 | 140 | |
| 130 | 0 | 0 | 130 | 0 | 0 | 0 | 0 | 0 | 0 | 260 | 260 | |
| 121 | 0 | 0 | 0 | 118 | 0 | 122 | 0 | 0 | 0 | 361 | 361 | |
| Total | 3372 | 439 | 1009 | 1765 | 2024 | 478 | 1253 | 1163 | 595 | 282 | 12,380 | 12,391 |
| Trials not providing individual participant data | ||||||||||||
| Trial\Drug | CBZ | PHB | PHT | VPS | LTG | OXC | LEV | TPM | GBP | ZNS | Total | Total randomiseda |
| 0 | 0 | 18 | 0 | 0 | 19 | 0 | 0 | 0 | 0 | 37 | 37 | |
| 36 | 35 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 71 | 71 | |
| 0 | 0 | 0 | 0 | 151 | 0 | 0 | 0 | 158 | 0 | 309 | 309 | |
| 59 | 0 | 58 | 64 | 0 | 0 | 0 | 0 | 0 | 0 | 181 | 181 | |
| 17 | 0 | 0 | 0 | 0 | 0 | 18 | 0 | 0 | 0 | 35 | 35 | |
| 14 | 0 | 0 | 0 | 0 | 0 | 13 | 0 | 0 | 0 | 27 | 27 | |
| 26 | 25 | 0 | 25 | 0 | 0 | 0 | 0 | 0 | 0 | 76 | 76 | |
| 15 | 0 | 0 | 0 | 0 | 0 | 16 | 0 | 0 | 0 | 31 | 31 | |
| 0 | 0 | 0 | 121 | 0 | 128 | 0 | 0 | 0 | 0 | 249 | 249 | |
| 66 | 0 | 0 | 0 | 0 | 0 | 62 | 0 | 0 | 0 | 128 | 128 | |
| 6 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 12 | 12 | |
| 30 | 30 | 30 | 30 | 0 | 0 | 0 | 0 | 0 | 0 | 120 | 120 | |
| 100 | 0 | 0 | 0 | 0 | 94 | 0 | 0 | 0 | 0 | 194 | 194 | |
| 28 | 0 | 0 | 29 | 0 | 55 | 0 | 0 | 0 | 0 | 112 | 112 | |
| 152 | 150 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 302 | 302 | |
| 23 | 0 | 20 | 21 | 0 | 0 | 0 | 0 | 0 | 0 | 64 | 64 | |
| 0 | 0 | 0 | 0 | 21 | 27 | 0 | 0 | 0 | 0 | 48 | 48 | |
| 32 | 0 | 0 | 0 | 32 | 0 | 0 | 0 | 0 | 0 | 64 | 64 | |
| 64 | 0 | 0 | 0 | 0 | 0 | 57 | 0 | 0 | 0 | 121 | 121 | |
| 70 | 0 | 0 | 0 | 73 | 0 | 0 | 0 | 0 | 0 | 143 | 143 | |
| 6 | 0 | 0 | 3 | 0 | 0 | 6 | 0 | 0 | 0 | 15 | 15 | |
| 129 | 0 | 0 | 0 | 264 | 0 | 0 | 0 | 0 | 0 | 393 | 393 | |
| 0 | 0 | 0 | 38 | 35 | 0 | 0 | 0 | 0 | 0 | 73 | 73 | |
| 15 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 33 | 33 | |
| 66 | 0 | 51 | 46 | 0 | 0 | 0 | 0 | 0 | 0 | 163 | 163 | |
| 0 | 0 | 0 | 0 | 50 | 0 | 50 | 0 | 0 | 0 | 100 | 100 | |
| 0 | 0 | 0 | 0 | 0 | 178 | 175 | 0 | 0 | 0 | 353 | 353 | |
| 215 | 0 | 0 | 0 | 0 | 0 | 218 | 0 | 0 | 0 | 433 | 433 | |
| 23 | 0 | 20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 43 | 43 | |
| 42 | 0 | 45 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 87 | 87 | |
| ? | 0 | 0 | 0 | 0 | 0 | ? | 0 | 0 | 0 | 37 | 37 | |
| 0 | 0 | 45 | 49 | 0 | 0 | 0 | 0 | 0 | 0 | 94 | 94 | |
| 20 | 0 | 20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 40 | 40 | |
| 42 | 0 | 0 | 0 | 0 | 0 | 0 | 46 | 0 | 0 | 88 | 88 | |
| 198 | 0 | 0 | 0 | 200 | 0 | 0 | 0 | 195 | 0 | 593 | 593 | |
| 92 | 0 | 0 | 0 | 93 | 0 | 0 | 0 | 0 | 0 | 185 | 185 | |
| 0 | 0 | 15 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 33 | 33 | |
| 17 | 0 | 0 | 16 | 0 | 0 | 0 | 0 | 0 | 0 | 33 | 33 | |
| 30 | 0 | 0 | 0 | 0 | 0 | 30 | 0 | 0 | 0 | 60 | 60 | |
| 88 | 0 | 0 | 30 | 121 | 0 | 0 | 0 | 0 | 0 | 239 | 239 | |
| 0 | 51 | 52 | 48 | 0 | 0 | 0 | 0 | 0 | 0 | 151 | 151 | |
| Totalc | 1721 | 315 | 374 | 538 | 1040 | 501 | 645 | 46 | 353 | 0 | 5570 | 5570 |
| Grand totalc | 5093 | 754 | 1383 | 2303 | 3064 | 979 | 1898 | 1209 | 948 | 282 | 17,950 | 17,961 |
CBZ: carbamazepine; GBP: gabapentin; IPD: individual participant data; ITT: intention to treat; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aDrug allocated missing for 11 participants in the IPD provided.
bTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review.
cOne trial provided the total number randomised but not the numbers randomised to each group. The 37 participants randomised are counted in the overall totals.
-
5093 participants were randomised to carbamazepine and we were provided with 66% of IPD
-
3064 participants were randomised to lamotrigine and we were provided with 66% of IPD
-
2303 participants were randomised to sodium valproate and we were provided with 77% of IPD
-
1898 participants were randomised to levetiracetam and we were provided with 66% of IPD
-
1383 participants were randomised to phenytoin and we were provided with 73% of IPD
-
1209 participants were randomised to topiramate and we were provided with 96% of IPD
-
979 participants were randomised to oxcarbazepine and we were provided with 49% of IPD
-
948 participants were randomised to gabapentin and we were provided with 63% of IPD
-
754 participants were randomised to phenobarbitone and we were provided with 58% of IPD
-
282 participants were randomised to zonisamide and we were provided with 100% of IPD
-
One trial with 37 participants (Ramsey 2010, IPD not provided) randomised individuals to carbamazepine or levetiracetam but did not state how many individuals were randomised to each drug and for 11 individuals the randomised drug was missing from the IPD
In total, we were provided with data for 12,391 out of a total of 17,961 eligible participants (69% of total data) from 36 out of the 77 eligible trials (47%).
Trials with individual participant data
Individual participant data were available for 36 trials recruiting 12,391 participants (Banu 2007; Baulac 2012; Bill 1997; Biton 2001; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2007; Chadwick 1998; Craig 1994; de Silva 1996; Dizdarer 2000; Eun 2012; Guerreiro 1997; Heller 1995; Kwan 2009; Lee 2011; Mattson 1985; Mattson 1992; Nieto‐Barrera 2001; Ogunrin 2005; Pal 1998; Placencia 1993; Privitera 2003; Ramsey 1992; Ramsey 2010; Reunanen 1996; Richens 1994; SANAD A 2007; SANAD B 2007; Steiner 1999; Stephen 2007; Trinka 2013; Turnbull 1985; Verity 1995; Werhahn 2015).
Table 2; Table 3 and Table 4 show the participant characteristics from the trials providing IPD. Data were available for the following participant characteristics (percentage of 12,391 participants with data available): sex (99.5%, data missing for 75 participants), seizure type (96%, data missing for 555 participants), drug randomised (99.9%, data missing for 11 participants), age at randomisation (99%, data missing for 98 participants), number of seizures in six months prior to randomisation (83%, data missing for 2135 participants), and time since first seizure to randomisation (37%, data missing for 7820 participants).
| Trial | Gender | Epilepsy type | Epilepsy type reclassifiedc | ||||||
| Male | Female | Missing | Genb | Partial | Missing | Genb | Partial | Unclassifiedd | |
| 61 (56%) | 47 (44%) | 0 (0%) | 49 (45%) | 59 (55%) | 0 (0%) | 49 (45%) | 59 (55%) | 0 (0%) | |
| 347 (60%) | 236 (40%) | 0 (0%) | 0 (0%) | 583 (100%) | 0 (0%) | 0 (0%) | 583 (100%) | 0 (0%) | |
| 174 (61%) | 113 (39%) | 0 (0%) | 105 (37%) | 182 (63%) | 0 (0%) | 75 (26%) | 182 (63%) | 30 (10%) | |
| 60 (44%) | 75 (55%) | 1 (1%) | 46 (34%) | 82 (60%) | 8 (6%) | 33 (24%) | 82 (60%) | 21 (15%) | |
| 56 (41%) | 80 (59%) | 0 (0%) | 54 (40%) | 82 (60%) | 0 (0%) | 34 (25%) | 82 (60%) | 20 (15%) | |
| 56 (45%) | 68 (55%) | 0 (0%) | 62 (50%) | 62 (50%) | 0 (0%) | 39 (31%) | 62 (50%) | 23 (19%) | |
| 83 (55%) | 67 (45%) | 0 (0%) | 45 (30%) | 105 (70%) | 0 (0%) | 0 (0%) | 105 (70%) | 45 (30%) | |
| 319 (55%) | 260 (45%) | 0 (0%) | 113 (20%) | 466 (80%) | 0 (0%) | 50 (9%) | 466 (80%) | 63 (11%) | |
| 157 (54%) | 135 (46%) | 0 (0%) | 0 (0%) | 292 (100%) | 0 (0%) | 0 (0%) | 292 (100%) | 0 (0%) | |
| 71 (43%) | 92 (55%) | 3 (2%) | 86 (52%) | 80 (48%) | 0 (0%) | 2 (1%) | 80 (48%) | 84 (51%) | |
| 86 (50%) | 81 (47%) | 6 (3%) | 84 (49%) | 89 (51%) | 0 (0%) | 84 (49%) | 89 (51%) | 0 (0%) | |
| 21 (40%) | 31 (60%) | 0 (0%) | 0 (0%) | 52 (100%) | 0 (0%) | 0 (0%) | 52 (100%) | 0 (0%) | |
| 48 (57%) | 36 (43%) | 0 (0%) | 0 (0%) | 84 (100%) | 0 (0%) | 0 (0%) | 84 (100%) | 0 (0%) | |
| 100 (52%) | 93 (48%) | 0 (0%) | 50 (26%) | 143 (74%) | 0 (0%) | 45 (23%) | 143 (74%) | 5 (3%) | |
| 117 (48%) | 126 (52%) | 0 (0%) | 141 (58%) | 102 (42%) | 0 (0%) | 82 (34%) | 102 (42%) | 59 (24%) | |
| 40 (49%) | 41 (51%) | 0 (0%) | 48 (59%) | 29 (36%) | 4 (5%) | 25 (31%) | 29 (36%) | 27 (33%) | |
| 57 (52%) | 53 (48%) | 0 (0%) | 15 (14%) | 95 (86%) | 0 (0%) | 6 (5%) | 95 (86%) | 9 (8%) | |
| 413 (87%) | 58 (12%) | 4 (1%) | 1 (0%) | 474 (100%) | 0 (0%) | 1 (0%) | 474 (100%) | 0 (0%) | |
| 445 (93%) | 35 (7%) | 0 (0%) | 0 (0%) | 480 (100%) | 0 (0%) | 0 (0%) | 480 (100%) | 0 (0%) | |
| 329 (53%) | 293 (47%) | 0 (0%) | 3 (1%) | 619 (99%) | 0 (0%) | 1 (0%) | 619 (100%) | 2 (0%) | |
| 34 (62%) | 21 (38%) | 0 (0%) | 45 (82%) | 10 (18%) | 0 (0%) | 26 (47%) | 10 (18%) | 19 (35%) | |
| 47 (50%) | 45 (48%) | 2 (2%) | 34 (36%) | 60 (64%) | 0 (0%) | 34 (36%) | 60 (64%) | 0 (0%) | |
| 67 (35%) | 125 (65%) | 0 (0%) | 59 (31%) | 133 (69%) | 0 (0%) | 35 (18%) | 133 (69%) | 24 (13%) | |
| (CBZ branch)a | 215 (54%) | 180 (46%) | 0 (0%) | 88 (22%) | 285 (72%) | 22 (6%) | 51 (13%) | 285 (72%) | 59 (15%) |
| (VPS branch)a | 112 (50%) | 113 (50%) | 0 (0%) | 131 (58%) | 78 (35%) | 16 (7%) | 86 (38%) | 78 (35%) | 61 (27%) |
| 73 (54%) | 63 (46%) | 0 (0%) | 136 (100%) | 0 (0%) | 0 (0%) | 110 (81%) | 0 (0%) | 26 (19%) | |
| 126 (48%) | 135 (52%) | 0 (0%) | 150 (57%) | 53 (20%) | 58 (22%) | 80 (31%) | 53 (20%) | 128 (49%) | |
| 188 (54%) | 163 (46%) | 0 (0%) | 114 (32%) | 237 (68%) | 0 (0%) | 71 (20%) | 237 (68%) | 43 (12%) | |
| 153 (51%) | 147 (49%) | 0 (0%) | 154 (51%) | 146 (49%) | 0 (0%) | 87 (29%) | 146 (49%) | 67 (22%) | |
| 922 (54%) | 755 (44%) | 44 (3%) | 25 (1%) | 1491 (87%) | 205 (12%) | 16 (1%) | 1491 (87%) | 214 (12%) | |
| 420 (59%) | 282 (39%) | 14 (2%) | 463 (65%) | 52 (7%) | 201 (28%) | 397 (55%) | 52 (7%) | 267 (37%) | |
| 101 (56%) | 80 (44%) | 0 (0%) | 91 (50%) | 90 (50%) | 0 (0%) | 55 (30%) | 90 (50%) | 36 (20%) | |
| 114 (50%) | 112 (49%) | 1 (0%) | 32 (14%) | 154 (68%) | 41 (18%) | 29 (13%) | 154 (68%) | 44 (19%) | |
| (CBZ branch)a | 551 (55%) | 448 (45%) | 0 (0%) | 141 (14%) | 858 (86%) | 0 (0%) | 48 (5%) | 858 (86%) | 93 (9%) |
| (VPS branch)a | 398 (57%) | 305 (43%) | 0 (0%) | 513 (73%) | 190 (27%) | 0 (0%) | 285 (41%) | 190 (27%) | 228 (32%) |
| 73 (52%) | 67 (48%) | 0 (0%) | 77 (55%) | 63 (45%) | 0 (0%) | 42 (30%) | 63 (45%) | 35 (25%) | |
| 122 (47%) | 138 (53%) | 0 (0%) | 152 (58%) | 108 (42%) | 0 (0%) | 152 (58%) | 108 (42%) | 0 (0%) | |
| 215 (60%) | 146 (40%) | 0 (0%) | 0 (0%) | 361 (100%) | 0 (0%) | 0 (0%) | 361 (100%) | 0 (0%) | |
| Total | 6971(56%) | 5345 (43%) | 75 (1%) | 3307 (27%) | 8529 (69%) | 555 (4%) | 2130 (17%) | 8529 (69%) | 1732 (14%) |
aTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review.
bGen: Generalised tonic‐clonic seizures with or without other seizure types
cSee Sensitivity analysis for further details of misclassification of epilepsy type
dUnclassified seizures defined as missing seizure type or generalised onset seizures and age of onset of seizures over the age of 30 years (see Sensitivity analysis for further details)
| Trial | Age (years) | Epilepsy duration (years) | Number of seizures in the last 6 months | |||||||
| Mean | SD | Range | Missing | Median | Range | Missing | Median | Range | Missing | |
| 5.7 | 3.5 | 1 to 15 | 0 | 1.2 | 0 to 11.5 | 0 | 24 | 1 to 7200 | 5 | |
| 36.4 | 15.9 | 18 to 75 | 0 | 0.2 | 0 to 17.7 | 30 | 2 | 1 to 30 | 1 | |
| 26.8 | 10.7 | 15 to 91 | 1 | 0.4 | 0 to 25 | 0 | 3 | 0 to 252 | 0 | |
| 32 | 14.5 | 12 to 76 | 0 | 1 | 0 to 53 | 27 | 2 | 0 to 100 | 2 | |
| 34 | 15.8 | 14 to 71 | 0 | 1 | 0 to 18 | 0 | 4 | 1 to 960 | 0 | |
| 30 | 14.1 | 14 to 81 | 0 | 0.5 | 0 to 19.4 | 0 | 3 | 1 to 1020 | 0 | |
| 76.9 | 6 | 65 to 94 | 0 | NA | NA | 150 | 3 | 0 to 163 | 0 | |
| 39 | 16.2 | 15 to 82 | 0 | NA | NA | 579 | 3 | 1 to 1410 | 4 | |
| 35 | 16.6 | 12 to 86 | 0 | 0.5 | 0 to 7.7 | 5 | 4 | 1 to 146 | 6 | |
| 78.2 | 7.1 | 61 to 95 | 3 | NA | NA | 166 | 3 | 0 to 99 | 3 | |
| 9.9 | 3.6 | 3 to 16 | 6 | 0.5 | 0 to 13.7 | 6 | 3 | 1 to 900 | 6 | |
| 10.8 | 2.3 | 4 to 15 | 0 | NA | NA | 52 | 3 | 1 to 60 | 0 | |
| 8.8 | 2.1 | 5 to 13 | 0 | 0.4 | 0 to 4.5 | 0 | 3 | 2 to 11 | 0 | |
| 18.6 | 9.7 | 5 to 53 | 1 | 0.4 | 0 to 20 | 0 | 2 | 0 to 157 | 0 | |
| 32.3 | 14.8 | 13 to 77 | 3 | 1 | 0 to 40 | 4 | 2 | 1 to 579 | 3 | |
| 33.9 | 10.9 | 16 to 56 | 0 | NA | NA | 81 | 1 | 0 to 540 | 0 | |
| 35.8 | 12.2 | 16 to 60 | 0 | NA | NA | 110 | 2 | 0 to 200 | 0 | |
| 41 | 15.5 | 18 to 82 | 4 | 2 | 0.5 to 59 | 5 | 1 | 1 to 100 | 7 | |
| 47.1 | 16.1 | 18 to 83 | 0 | 3 | 1 to 68 | 19 | 12 | 1 to 2248 | 38 | |
| 27.2 | 21.4 | 2 to 84 | 1 | NA | NA | 622 | 3 | 1 to 9000 | 0 | |
| 27.5 | 8.5 | 14 to 55 | 0 | 7 | 3 to 11.5 | 18 | 12 | 6 to 42 | 0 | |
| 11.4 | 5 | 2 to 18 | 0 | 2.5 | 0.5 to 17 | 2 | NA | NA | 94 | |
| 29 | 17.6 | 2 to 68 | 0 | 5 | 0.5 to 44 | 0 | 2 | 0 to 100 | 0 | |
| (CBZ branch)a | 34.4 | 18.4 | 6 to 80 | 0 | NA | NA | 395 | 4 | 0 to 2400 | 0 |
| (VPS branch)a | 32.8 | 19.4 | 6 to 84 | 0 | NA | NA | 225 | 4 | 0 to 20000 | 0 |
| 20.9 | 14.2 | 3 to 64 | 0 | 0 | 0 to 3 | 0 | NA | NA | 136 | |
| 34.1 | 14.8 | 12 to 78 | 0 | NA | NA | 261 | 4 | 0 to 570 | 0 | |
| 32.1 | 14.2 | 12 to 72 | 2 | 0.7 | 0 to 27 | 3 | 3 | 1 to 145 | 1 | |
| 33 | 14.9 | 16 to 79 | 2 | NA | NA | 300 | 4 | 2 to 101 | 5 | |
| 38.4 | 18.3 | 5 to 86 | 44 | NA | NA | 1721 | 4 | 0 to 1185 | 49 | |
| 22.5 | 14.1 | 5 to 77 | 14 | NA | NA | 716 | 3 | 0 to 2813 | 17 | |
| 34.1 | 16.7 | 13 to 75 | 1 | 1.3 | 0 to 28.5 | 1 | 3 | 1 to 600 | 0 | |
| 36 | 16.9 | 13 to 80 | 2 | NA | NA | 227 | 18 | 6 to 1080 | 37 | |
| (CBZ branch)1 | 42.8 | 17.2 | 16 to 89 | 0 | NA | NA | 999 | NA | NA | 999 |
| (VPS branch)1 | 36.5 | 17.8 | 16 to 85 | 1 | NA | NA | 703 | NA | NA | 703 |
| 35.2 | 16.1 | 14 to 70 | 0 | 0.75 | 0.1 to 30 | 0 | 2 | 0 to 60 | 0 | |
| 10.1 | 2.9 | 5 to 16 | 13 | 0.3 | 0 to 5.9 | 32 | 3 | 1 to 104 | 12 | |
| 71.5 | 7.2 | 60 to 95 | 0 | NA | NA | 361 | 2 | 1 to 96 | 7 | |
| Total (missing) | 98 | 7820 | 2135 | |||||||
Abbreviations: SD: Standard deviation
aTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review.
| Trial | Electroencephalographic (EEG) | Computerised Tomography (CT) /Magnetic Resonance Imaging (MRI) | Neurological exams | ||||||
| Normal | Abnormal | Missing | Normal | Abnormal | Missing | Normal | Abnormal | Missing | |
| 49 (45%) | 54 (50%) | 5 (5%) | 21 (19%) | 5 (5%) | 82 (76%) | 0 (0%) | 0 (0%) | 108 (100%) | |
| 0 (0%) | 0 (0%) | 583 (100%) | 0 (0%) | 0 (0%) | 583 (100%) | 478 (82%) | 103 (18%) | 2 (0%) | |
| 126 (44%) | 152 (53%) | 9 (3%) | 173 (60%) | 69 (24%) | 45 (16%) | 0 (0%) | 0 (0%) | 287 (100%) | |
| 0 (0%) | 0 (0%) | 136 (100%) | 0 (0%) | 0 (0%) | 136 (100%) | 89 (65%) | 46 (34%) | 1 (1%) | |
| 62 (46%) | 72 (53%) | 2 (1%) | 82 (60%) | 12 (9%) | 42 (31%) | 123 (90%) | 13 (10%) | 0 (0%) | |
| 76 (61%) | 42 (34%) | 6 (5%) | 72 (58%) | 20 (16%) | 32 (26%) | 108 (87%) | 16 (13%) | 0 (0%) | |
| 0 (0%) | 0 (0%) | 150 (100%) | 62 (41%) | 87 (58%) | 1 (1%) | 90 (60%) | 60 (40%) | 0 (0%) | |
| 0 (0%) | 0 (0%) | 579 (100%) | 0 (0%) | 0 (0%) | 579 (100%) | 493 (85%) | 86 (15%) | 0 (0%) | |
| 107 (37%) | 179 (61%) | 6 (2%) | 0 (0%) | 0 (0%) | 292 (100%) | 0 (0%) | 0 (0%) | 292 (100%) | |
| 28 (17%) | 74 (45%) | 64 (39%) | 0 (0%) | 0 (0%) | 166 (100%) | 0 (0%) | 0 (0%) | 166 (100%) | |
| 0 (0%) | 0 (0%) | 173 (100%) | 0 (0%) | 0 (0%) | 173 (100%) | 152 (88%) | 15 (9%) | 6 (3%) | |
| 18 (35%) | 34 (65%) | 0 (0%) | 50 (96%) | 2 (4%) | 0 (0%) | 0 (0%) | 0 (0%) | 52 (100%) | |
| 6 (7%) | 78 (93%) | 0 (0%) | 75 (89%) | 9 (11%) | 0 (0%) | 83 (99%) | 1 (1%) | 0 (0%) | |
| 92 (48%) | 99 (51%) | 2 (1%) | 126 (65%) | 12 (6%) | 55 (28%) | 0 (0%) | 0 (0%) | 193 (100%) | |
| 0 (0%) | 0 (0%) | 243 (100%) | 0 (0%) | 0 (0%) | 243 (100%) | 222 (91%) | 19 (8%) | 2 (1%) | |
| 0 (0%) | 0 (0%) | 81 (100%) | 0 (0%) | 0 (0%) | 81 (100%) | 0 (0%) | 0 (0%) | 81 (100%) | |
| 58 (53%) | 52 (47%) | 0 (0%) | 74 (67%) | 36 (33%) | 0 (0%) | 110 (100%) | 0 (0%) | 0 (0%) | |
| 126 (27%) | 343 (72%) | 6 (1%) | 308 (65%) | 119 (25%) | 48 (10%) | 0 (0%) | 0 (0%) | 475 (100%) | |
| 0 (0%) | 0 (0%) | 480 (100%) | 0 (0%) | 0 (0%) | 480 (100%) | 0 (0%) | 0 (0%) | 480 (100%) | |
| 0 (0%) | 0 (0%) | 622 (100%) | 0 (0%) | 0 (0%) | 622 (100%) | 0 (0%) | 0 (0%) | 622 (100%) | |
| 0 (0%) | 0 (0%) | 55 (100%) | 37 (67%) | 0 (0%) | 18 (33%) | 55 (100%) | 0 (0%) | 0 (0%) | |
| 0 (0%) | 0 (0%) | 94 (100%) | 0 (0%) | 0 (0%) | 94 (100%) | 24 (26%) | 70 (74%) | 0 (0%) | |
| 180 (94%) | 12 (6%) | 0 (0%) | 0 (0%) | 0 (0%) | 192 (100%) | 0 (0%) | 0 (0%) | 192 (100%) | |
| (CBZ branch)a | 0 (0%) | 0 (0%) | 395 (100%) | 0 (0%) | 0 (0%) | 395 (100%) | 0 (0%) | 0 (0%) | 395 (100%) |
| (VPS branch)a | 0 (0%) | 0 (0%) | 225 (100%) | 0 (0%) | 0 (0%) | 225 (100%) | 0 (0%) | 0 (0%) | 225 (100%) |
| 0 (0%) | 0 (0%) | 136 (100%) | 0 (0%) | 0 (0%) | 136 (100%) | 0 (0%) | 0 (0%) | 136 (100%) | |
| 0 (0%) | 0 (0%) | 261 (100%) | 0 (0%) | 0 (0%) | 261 (100%) | 0 (0%) | 0 (0%) | 261 (100%) | |
| 13 (4%) | 13 (4%) | 325 (93%) | 16 (5%) | 5 (1%) | 330 (94%) | 305 (87%) | 46 (13%) | 0 (0%) | |
| 0 (0%) | 0 (0%) | 300 (100%) | 0 (0%) | 0 (0%) | 300 (100%) | 0 (0%) | 0 (0%) | 300 (100%) | |
| 0 (0%) | 0 (0%) | 1721 (100%) | 0 (0%) | 0 (0%) | 1721 (100%) | 1267 (74%) | 410 (24%) | 44 (3%) | |
| 0 (0%) | 0 (0%) | 716 (100%) | 0 (0%) | 0 (0%) | 716 (100%) | 595 (83%) | 107 (15%) | 14 (2%) | |
| 103 (57%) | 71 (39%) | 7 (4%) | 111 (61%) | 33 (18%) | 37 (20%) | 165 (91%) | 16 (9%) | 0 (0%) | |
| 51 (22%) | 121 (53%) | 55 (24%) | 0 (0%) | 0 (0%) | 227 (100%) | 0 (0%) | 0 (0%) | 227 (100%) | |
| (CBZ branch)1 | 0 (0%) | 0 (0%) | 999 (100%) | 0 (0%) | 0 (0%) | 999 (100%) | 0 (0%) | 0 (0%) | 999 (100%) |
| (VPS branch)1 | 0 (0%) | 0 (0%) | 703 (100%) | 0 (0%) | 0 (0%) | 703 (100%) | 0 (0%) | 0 (0%) | 703 (100%) |
| 70 (50%) | 70 (50%) | 0 (0%) | 17 (12%) | 10 (7%) | 113 (81%) | 0 (0%) | 0 (0%) | 140 (100%) | |
| 0 (0%) | 0 (0%) | 260 (100%) | 0 (0%) | 0 (0%) | 260 (100%) | 0 (0%) | 0 (0%) | 260 (100%) | |
| 117 (32%) | 242 (67%) | 2 (1%) | 78 (22%) | 282 (78%) | 1 (0%) | 0 (0%) | 0 (0%) | 361 (100%) | |
| Total | 1282 (10%) | 1708 (14%) | 9401 (75%) | 1302 (11%) | 701 (6%) | 10,388 (83%) | 4359 (36%) | 1008 (8%) | 7024 (56%) |
aTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review.
Thirteen trials (Baulac 2012; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2007; de Silva 1996; Eun 2012; Heller 1995; Lee 2011; Ogunrin 2005; Pal 1998; Reunanen 1996; Steiner 1999) provided the results of neurological examinations for 5367 participants (43%). Seventeen trials (Banu 2007; Bill 1997; Brodie 1995a; Brodie 1995b; Chadwick 1998; Craig 1994; Dizdarer 2000; Eun 2012; Guerreiro 1997; Lee 2011; Mattson 1985; Placencia 1993; Reunanen 1996; Steiner 1999; Stephen 2007; Turnbull 1985; Werhahn 2015) provided electroencephalographic (EEG) results for 2990 participants (24%). Fifteen trials (Banu 2007; Bill 1997; Brodie 1995a; Brodie 1995b; Brodie 1999; Dizdarer 2000; Eun 2012; Guerreiro 1997; Lee 2011; Mattson 1985; Ogunrin 2005; Reunanen 1996; Steiner 1999; Turnbull 1985; Werhahn 2015) provided computerised tomography/magnetic resonance imaging (CT/MRI) results for 2083 participants (16%).
Trials without individual participant data
The remaining 41 trials recruiting 5570 participants did not provide IPD for the review (Aikia 1992; Bidabadi 2009; Brodie 2002; Callaghan 1985; Capone 2008; Castriota 2008; Chen 1996; Cho 2011; Christe 1997; Consoli 2012; Cossu 1984; Czapinski 1997; Dam 1989; Donati 2007; Feksi 1991; Forsythe 1991; Fritz 2006; Gilad 2007; Jung 2015; Kalviainen 2002; Kopp 2007; Korean Lamotrigine Study Group 2008; Lukic 2005; Mitchell 1987; Miura 1990; Motamedi 2013; NCT01498822; NCT01954121; Pulliainen 1994; Ramsey 1983; Ramsey 2007; Rastogi 1991; Ravi Sudhir 1995; Resendiz 2004; Rowan 2005; Saetre 2007; Shakir 1981; So 1992; Steinhoff 2005; Suresh 2015; Thilothammal 1996).
In response to our direct requests for IPD, trial authors or government sponsors of nine trials confirmed that data were no longer available (Callaghan 1985; Capone 2008; Consoli 2012; Forsythe 1991; Pulliainen 1994; Ramsey 1983; Shakir 1981; So 1992; Thilothammal 1996).
Data could not be provided for three pharmaceutical trials where data were requested via ClinicalStudyDataRequest.Com, due to the cost and resource of locating and preparing data (Kalviainen 2002; Saetre 2007) and due to country‐specific restrictions regarding anonymisation of data (Steinhoff 2005). For three further pharmaceutical company‐sponsored trials, data were not available could not be provided due to time elapsed since the trial was completed (Brodie 2002; Christe 1997; Donati 2007).
The authors of three trials confirmed that the data we required had not been collected (Chen 1996; Lukic 2005; Mitchell 1987) and the authors of two trials stated that data could not be provided due to local authority/ethical restrictions (Cho 2011; Jung 2015).
We were unable to make contact with the authors or sponsors of 14 trials to request data (Aikia 1992; Bidabadi 2009; Castriota 2008; Cossu 1984; Dam 1989; Fritz 2006; Kopp 2007; Miura 1990; Motamedi 2013; Ramsey 2007; Rastogi 1991; Ravi Sudhir 1995; Resendiz 2004; Suresh 2015).
We received an initially positive response from the authors or government sponsors of three trials but no data were provided (Czapinski 1997; Gilad 2007; Rowan 2005) and for two pharmaceutical trials, data could not be made available until a final manuscript had been published for the trials (NCT01498822; NCT01954121). Our IPD request to the sponsor of one trial is still ongoing (Korean Lamotrigine Study Group 2008); if data are provided at a later date for these trials, they will be included in an update of this review.
An author of Feksi 1991 provided access to an IPD dataset, but this was not the final dataset used for the analysis published by the original trial authors. The pharmaceutical company that sponsored the trial, Ciba‐Geigy, who at that time held the product licence for carbamazepine, held the final dataset. Since the trial was undertaken, there have been a number of mergers and restructures within the industry, and the current owners of the data are Novartis. Unfortunately, Novartis were unable to locate the data for this trial. The dataset that we had for this trial contained a number of problems and inconsistencies, and we therefore decided not to include this trial in the meta‐analysis. This was the only trial with major inconsistencies that prevented the inclusion of this IPD in analysis; for details of minor inconsistencies between IPD and published results, see Sensitivity analysis and Other potential sources of bias.
Two trials (Forsythe 1991; Shakir 1981) presented times at which the allocated drug was withdrawn and the reason for withdrawal in the trial publication for each individual. However only Shakir 1981 provided this information according to seizure type, so only results for Shakir 1981 could be incorporated into the analysis of 'Time to withdrawal of allocated treatment' (see Sensitivity analysis). Shakir 1981 presented 'Time on trial drug' in months for each participant, therefore to calculate 'Time to withdrawal of allocated treatment', we assumed that if 'Time spent on trial drug' was five months, the individual spent five full months (152 full days) on the trial drug before withdrawal.
Three trials presented sufficient detail to extract individual withdrawal (Gilad 2007; Steinhoff 2005) or seizure times (Gilad 2007; Consoli 2012) from survival curves, however this information was not separated by seizure type for Consoli 2012 so we could not include the results in analysis for this trial.
A further four trials reported summary statistics or graphical data for one of more outcomes of interest of the review; however none of these trials presented information by seizure type so we could not include the results in analysis (Brodie 2002; Christe 1997; Rowan 2005; Saetre 2007).
The remaining 31 trials did not report any published results relevant to this review (Aikia 1992; Bidabadi 2009; Callaghan 1985; Capone 2008; Castriota 2008; Chen 1996; Cossu 1984; Czapinski 1997; Dam 1989; Donati 2007; Feksi 1991; Fritz 2006; Jung 2015; Kalviainen 2002; Kopp 2007; Korean Lamotrigine Study Group 2008; Lukic 2005; Mitchell 1987; Miura 1990; Motamedi 2013; NCT01498822; NCT01954121; Pulliainen 1994; Ramsey 1983; Ramsey 2007; Rastogi 1991; Ravi Sudhir 1995; Resendiz 2004; So 1992; Suresh 2015; Thilothammal 1996). Details of outcomes considered and a summary of results of each trial for which IPD were not available to us can be found in Table 5.
| Trial | Summary of resultsb |
| 1. MANOVA revealed no significant interaction effect of group and time 2. MANOVA revealed no significant interaction effect of group and time 3. MANOVA revealed no significant interaction effect of group and time 4. MANOVA revealed no significant interaction effect of group and time | |
| 1. CBZ: 64%, PHB: 63% 2. No statistically significant difference between groups 3. No statistically significant difference between groups 4. Mean seizure frequency: CBZ: 0.66, PHB: 0.8 5. Mean duration (seconds): CBZ: 12.63; PHB: 15 | |
| 1. Median time to exit: GBP: 69 days, LTG: 48 days HR: 1.043 (95% confidence interval 0.602 to 1.809) 2. Proportion of evaluable population completing the study – GBP: 71.6%, LTG: 67.1% No difference between groups for time to withdrawal for any reason 3. No difference between groups for time to first seizure 4. GBP: 76.1%, LTG: 76.8% (ITT population) 5. Withdrawals during titration: GBP: 7, LTG: 10 Withdrawals after titration: GBP: 10, LTG: 13 | |
| 1a. PHT: 67%; CBZ: 37%; VPS: 53% 2. PHT: 10%; CBZ: 8%; VPS:11% | |
| 1. CBZ: 76%, LEV: 76% 2. Proportion with AEs: CBZ: 65%, LEV: 50% 3. CBZ: 2 discontinuations due to failure to control seizures and interactions with other medications LEV: 3 discontinuations – 1 death from stroke and 2 due to AEs | |
| 1. No significant difference between groups 2. No significant difference between groups | |
| 1. No significant difference between groups 3. 2 children from PHB group, 1 child from CBZ group and no children from VPS group withdrew from the study because of allergic reactions 4. No significant difference between groups | |
| 1. Overall effect on sleep parameters was comparable between groups. LEV group PSG significant increase post treatment compared to baseline in sleep efficiency (P = 0.039) and in decrease of wake time after sleep onset (P = 0.047), no significant change in other sleep parameters. CBZ group post treatment compared to baseline significant increases in the percentage of slow wave sleep (P = 0.038), no significant change in other sleep parameters 2. No significant difference between baseline and post‐treatment between the 2 groups | |
| 1. OXC 56.6% ; VPS 53.8% 2. No significant difference between groups 3. OXC 40.6% ; VPS 33.9% 4. Efficacy no significant difference between groups Tolerability no significant difference between groups Therapeutic effect no significant difference between groups 5. Proportion of participants experiencing at least 1 AE regardless of relationship to trial drug OXC 89.8%; VPS 87.6% 6. Seizure frequency per week OXC (n = 106) mean 0.17 median 0, VPS (n = 106) mean 0.40, median 0 | |
| 1. No significant difference between groups 2. Completed study LEV 52/62, CBZ 54/66, withdrawals: 8 poor compliance (LEV 4, CBZ 4); 7 severe adverse effect (LEV 3, CBZ 4); 7 unknown cause (LEV 3, CBZ 4) 3. Attention deficit on digital span end of follow up greater in CBZ group than LEV (P = 0.03) Stroop test worse in CBZ than LEV (P = 0.02) No significant difference between groups for other scales. Impairment of activities of daily living greater CBZ than LEV (P = 0.05) 4. 4 participants (LEV 2, CBZ 2) had abnormal EEG at baseline, normal at end of treatment. Drug dose reduction (LEV 4, CBZ 2). Remaining participants unmodified versus baseline 5. No significant difference between groups | |
| 1. Significant decrease in visual‐verbal memory for CBZ and acoustic memory for PHB. No significant differences for other tests | |
| 1. PHB: 60%, PHT: 59%; CBZ: 62%; VPS: 64% | |
| 1. Baseline OXC mean 2.9 (SD 7.0), median 1, range 0‐60 CBZ mean 5.8 (SD 14.7) median 1, range 0‐99 Maintenance phases OXC mean 0.4 (SD 3.0) median 0, range 0‐27 CBZ mean 0.3 (SD 1.4) median 0, range 0‐12 2. Severe side effects CBZ 25, OXC 13, statistically significant difference favouring OXC (P = 0.04) Participants without any side effects CBZ 25, OXC 29 no significant difference between groups (P = 0.22) 3. Global efficacy no significant difference between groups (P = 0.77); global tolerability (P = 0.11) Participants very good/good CBZ 69 (73%), OXC 76 (84%) Participants poor/very poor CBZ 26 (27%), OXC 15 (16%) 4. Nature of side effects same between groups, included tiredness, headache, dizziness, ataxia. Participants withdrawn due to severe side effects CBZ 16, OXC 9 5. Clinically relevant changes observed in 2 participants only, both CBZ group, both stopped treatment | |
| 1. Comparison of cognitive results no significant difference between treatment groups (P = 0.195) No significant difference between treatment groups for secondary variables (psychomotor speed, alertness, memory and learning, attention, intelligence scores) 2. OXC 58%; CBZ 46%; VPS 54% 3. Most common (> 10% reported) side effects OXC fatigue and headache; CBZ fatigue and rash VPS headache, increased appetite, alopecia 4. Good/very good: OXC investigators 84%, participants 82%, parents/carers 86%; Combined CBZ/VPS investigators 77%, participants 73%, parents/carers 80% | |
| 1. Minor adverse effects reported in PHB: 58 participants (39%) reported 86 AEs, CBZ: 46 participants (30%) reported 68 AEs 2. All withdrawals: PHB: 18%, CBZ: 17% Withdrawals due to side‐effects: PHB: 5%, CBZ: 3% 3. Seizure‐free: PHB: 54%, CBZ: 52% > 50% reduction of seizures: PHB: 23%, CBZ: 29% 50% reduction‐50% increase in seizures: PHB: 15%, CBZ: 13% > 50% increase in seizures: PHB: 8%, CBZ: 6% | |
| 1. Significant difference favouring VPS test of speed of information processing No significant differences between treatment groups for any other cognitive tests 2. PHT: 30%; CBZ: 39%; VPS:33% | |
| 1. Seizure freedom: LTG: 38%, OXC: 44% < 50% seizure reduction: LTG: 48%, OXC: 55% 2. Both groups showed improvement in verbal learning and in 1/4 measures of attention. In addition, participants under OXC improved in word fluency. Improved mood was reported with OXC only. | |
| 1. Number of participants experiencing early seizures as first event: LTG 2/32, CBZ 3/32 Number of participants remaining seizure‐free in the follow‐up period: LTG 23/32 (72%), CBZ 14/32 (44%) P = 0.05 2. Incidence of side effects: LTG 2/32 (6.25%), CBZ 12/32 (37.5%) P = 0.05 3. Withdrawals from study due to side effects LTG 1/32 (3%), CBZ 10/32 (31%), P = 0.02 | |
| 1. No difference between groups in terms of social competence; school competence; internalising behaviour problems; externalising behaviour problems; total behaviour problems and anxiety. Significant decrease in depression in LEV group compared to CBZ group (P = 0.027) 2. LEV 95.7% , CBZ 97.1% , P = 0.686 3. LEV 66.7%, CBZ 57.8% , P = 0.317 4. LEV 33.3%, CBZ 46.9%. Number of AEs not significantly different between groups | |
| 1.CBZ: 53% LTG: 56% 2. No significant difference between groups in overall cognitive score. In terms of individual assessments, only Stroop test B showed a statistically significant advantage for LTG. | |
| 1. No significant difference between groups 2. No significant difference between groups | |
| 1. LTG: 65% CBZ: 70% 2. Total seizure‐free rate LTG: 62% CBZ: 63% Time to first seizure: mean (SD): weeks LTG: 10 (5.09), CBZ: 10.82 (6.44) | |
| 1. LTG: 54%, VPS: 55 %, no difference by seizure type 2. LTG: 69%, VPS:68 % | |
| 1. No significant differences between treatment groups 2. Compliance: trend towards better compliance in CBZ group (not significant) Randomised participants only: trend towards higher rate withdrawal from treatment in PHB group (not significant). More mild systemic side‐effects in CBZ group (significant). 3 children switched from CBZ to PHB and 1 from PHB to CB following adverse reactions 3. 6 months: excellent/good: PHB = 15, CBZ = 13 12 months: excellent/good: PHB = 13, CBZ = 9 | |
| 1. Partial seizures ‐ PHT: 32%; CBZ: 40%; VPS : 41% Generalised seizures ‐ PHT :35%; CBZ: 15%; VPS: 7% 1. Partial seizures ‐ PHT: 24%; CBZ: 24%; VPS : 25% Generalised seizures ‐ PHT :13%; CBZ: 0%; VPS: 0% | |
| 1. Seizure recurrence at 2 weeks ‐ LTG: 43% LEV: 35%, p=0.42 Seizure recurrence at 4 weeks ‐ LTG: 39% LEV: 33%, p=0.53 Seizure recurrence at 8 weeks ‐ LTG: 35% LEV: 28%, p=0.50 Seizure recurrence at 12 weeks ‐ LTG: 33% LEV: 24%, p=0.35 Seizure recurrence at 20 weeks ‐ LTG: 31% LEV: 13%, p=0.03 2. No significant difference between groups 3. Proportion with AEs ‐ LTG: 53%, LEV: 67% | |
| 1. LEV: 12.7%, OXC: 23.4% 2. Median months: LEV: 7.6, OXC: NA (fewer than 50% of participants in the OXC group had seizure recurrence) 3. LEV: 53.8%, OXC: 58.5% 4. LEV: 34.7%, OXC: 40.9% | |
| 1. LEV: 47.3%, CBZ: 68.4% 2. LEV: 48.4%, CBZ: 70.2% 3. Number of events: LEV: 88, CBZ: 45 4. Number of events: LEV: 87, CBZ: 39 5. Number of events: LEV: 97, CBZ: 57 | |
| 1. Compared to CBZ, participants on PHT became slower (motor speed of the hand) and their visual memory decreased. There was an equal decrease in negative mood (helplessness, irritability, depression) on PHT and CBZ 2. 3 participants taking PHT complained of tiredness, and 1 participant taking CBZ complained of facial skin problems, another tiredness and memory problems | |
| 1. Incidence of major side effects (proportion of analysed participants): PHT 23%; CBZ: 23% Minor side effects: cognitive impairment and sedation twice as likely on CBZ compared to PHT. Other minor side effects similar between groups. 2. Treatment failures among analysed participants: Seizure control (among analysed participants with no major side effects): PHT: 86%; CBZ: 82% 3. Significantly lower mean LDH level at 24 weeks in CBZ participants than PHT participants. Other laboratory results similar across treatment groups | |
| 1. 8 discontinuations; due to generalised rash (n = 1), excessive tiredness (n = 1), withdrew consent (n = 2), renal transplant (n = 1), lost to follow‐up (n = 2), died (n = 1) 2. 6 participants reported treatment‐emergent side effects. 3. No participants withdrew due to lack of seizure control | |
| 1(a). PHT: 51%, VPS: 49% 1(b). PHT : 24%, VPS: 35% 1(c). PHT: 18%, VPS: 10% 1(d). PHT: 2%, VPS: 6% 2. All reported AEs were minor and similar rates between groups | |
| 1. No significant differences between any tests of cognitive function taken before treatment and after 10‐12 weeks for both treatment groups | |
| 1. Six months of seizure freedom: CBZ: 81%, TPM: 91% 50% reduction of seizures: CBZ: 84% TPM: 97% The average number of seizures was significantly less in the TPM group compared to the CBZ group at 6 and 9 months 2. AEs were mild and similar between groups 3. No significant differences between groups | |
| 1. Significant difference between 3 treatment groups (P = 0.00022) CBZ more early terminators than GBP (P = 0.008) or LTG (P < 0.0001) 2. LTG 51.4%, GBP 47.4%, CBZ 64.3% no significant difference between groups P = 0.09 3. No difference between groups for time to first, second, fifth and tenth seizure (P values = 0.18, 0.13, 0.74, 0.95 respectively) 4. More systemic toxicities on GBP than CBZ or LTG No significant differences in neuro‐toxicities between treatment groups over 12 months 5. Mean serum levels: 6 weeks GBP 8.67 ± 4.83; µg/mL, CBZ 6.79 ± 2.92 µg/mL and LTG 2.87 ± 1.60 µg/mL 52 weeks GBP 8.54 ± 5.57 µg/mL, CBZ 6.48 ± 3.72 µg/mL and LTG 3.46 ± 1.68 µg/mL Overall medical compliance 89% without significant group differences 6. 3 months LTG 49.7%, GBP 43.3%, CBZ 36.0% significant difference between groups P = 0.02 6 months LTG 37.2%, GBP 33.0%, CBZ 28.9% no significant difference between groups P = 0.22 12 months LTG 28.6%, GBP 23.2%, CBZ 22.8% no significant difference between groups P = 0.33 | |
| 1. LTG 68 (73%), CBZ 61 (67%), no significant difference between groups 2. LTG 59 (63%), CBZ 69 (76%), not significant difference P = 0.068 ITT analysis 3. LTG 71 (76%), CBZ 81 (89%), significant difference, P = 0.0234 ITT analysis 4.Hazard ratio (lamotrigine/carbamazepine) 1.50, 95% CI 0.94–2.40, p value 0.092 5. During treatment period LTG 82 (88%) reported 378 AEs, CBZ 79 (86%) reported 310 AEs. No significant differences between groups for any AEs except for immune system Withdrew due to AE LTG 13 (14%), CBZ 23 (25%), P = 0.078 6. No difference between groups even when changes over time corrected for age, gender and baseline score | |
| 1. PHT: 33%; VPS: 39% 2. All reported AEs were minor and similar rates between groups | |
| 1. VPS 7/11 (64%), CBZ 9/14 (64%) 2. At least one AE reported VPS 15/16 (94%), CBZ 16/17 (94%) | |
| 1. FE CBZ group 83/88 (94.3%), LTG group 78/88 (88.6%) no significant difference between groups GE VPS group 25/30 (83.3%) LTG group 20/33 (60.6%) no significant difference between groups 2. FE CBZ group 81%, LTG group 91%, not a significant difference between groups GE VPS group 97%, LTG group 88%, not stated as significant or non‐significant difference 3. At least 1 AE FE CBZ 81 participants (91%), LTG 68 participants (77.3%) GE VPS 25 participants (83.3%), LTG 24 participants (72.7%) Serious AEs FE CBZ 8 participants (9%), LTG 6 participants (7%) GE VPS 1 participant (3%), LTG 5 participants (15%) AEs considered related to study drug FE CBZ 65 participants (74%), LTG 38 participants (43%) GE VPS 16 participants (53%), LTG 15 participants (45.5%) | |
| 1. Mean quality‐of‐life score at baseline CBZ group 31.14 ± 1.83, LEV group 29.76 ± 1.71 (P value = 0.5861) Mean quality of life score after 26 weeks of treatment CBZ group 58.41 ± 1.89, LEV 64.58 ± 2.02 (P value = 0.0302) 2.28 participants in CBZ group, 28 in LEV group Seizure freedom 4 weeks CBZ group 85.72%, LEV group 85.72% (P value = 1); 12 weeks CBZ group 89.29%, LEV group 93.34% (P value = 0.4595); 26 weeks CBZ group 96.43%, LEV group 100% (P value = 0.1212); 6 months CBZ group 71.42% (20 participants), LEV group 78.57% (22 participants) (P value = 0.2529) 3. Participants experiencing at least 1 AE, CBZ group 36.66%, LEV group 40% (P value = 0.77) | |
| 1. PHB: 31%, PHT: 27%, VPS: 21% 2. PHB: 33%, PHT: 63%, VPS: 31% |
AE: adverse event; CBZ: carbamazepine; EEG: electroencephalogram; FE: focal epilepsies; GBP: gabapentin; GE: generalised epilepsies; ITT: intention to treat; LDH: lactic acid dehydrogenase; LEV: levetiracetam; LTG: lamotrigine; MANOVA: repeated measures analysis of variance; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; SD: standard deviation; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aFor further details of adverse events see Table 16 and Table 17.
bSee Table 1 for details of treatment arms in each trial and number of participants randomised to each arm.
cResults not split by treatment arm for Ramsey 2007.
Excluded studies
We excluded 31 studies from the review; three were cross‐over trials (Cereghino 1974; Gruber 1962; Loiseau 1984), three studies were terminated early with no results available (EUCTR2004‐004053‐26‐SE; EUCTR2010‐018284‐42‐NL; ISRCTN73223855), two were not fully randomised (Baxter 1998; Kaminow 2003), one did not recruit participants with epilepsy (Taragano 2003) and the other 22 did not have a monotherapy design (Albani 2006; Alsaadi 2002; Alsaadi 2005; ; Ben‐Menachem 2003; Beydoun 1997; Beydoun 1998; Beydoun 2000; Bittencourt 1993; Canadian Group 1999; Chung 2012; DeToledo 2000; Fakhoury 2004; French 2012; Gilliam 1998; Hakami 2012; Kerr 1999; Kerr 2001; Reinikainen 1984; Reinikainen 1987; Rosenow 2012; Simonsen 1975a; Simonsen 1975b). See Characteristics of excluded studies for further information.
Risk of bias in included studies
For further details, see the Characteristics of included studies and Figure 4.

Risk of bias summary: review authors' judgements about each risk of bias item for each included trial
Allocation
Trials for which we received IPD (information reported in published papers or provided with IPD)
One trial used alternate allocation (quasi randomisation) which we judged to be at high risk of selection bias (Dizdarer 2000). One trial described an adequate method of randomisation, use of a random number list, but reported that allocation was concealed by sealed, opaque envelopes, although this method was not used for all participants in the trial (Placencia 1993), so we also judged this trial to be at high risk of selection bias.
Twenty trials described adequate methods of generation of random sequence and allocation concealment and we judged them to be at low risk of bias. One trial used a random number list and central allocation (Ogunrin 2005). Four trials used block randomisation, of which three concealed allocation with sealed, opaque envelopes (de Silva 1996; Heller 1995; Mattson 1992) and one used central pharmacy allocation (Chadwick 1998). Ten trials used a computer‐generated random sequence. Of these, seven concealed allocation with sealed, opaque envelopes (Bill 1997; Brodie 1995a; Brodie 1995b; Brodie 1999; Guerreiro 1997; Nieto‐Barrera 2001; Reunanen 1996), two used a telephone interactive voice‐response system (Baulac 2012; Brodie 2007) and one used central pharmacy allocation (Werhahn 2015). Five trials used a computer‐generated minimisation programme: four used central telephone allocation (Richens 1994; SANAD A 2007; SANAD B 2007; Verity 1995) and one used central pharmacy allocation (Craig 1994).
Two trials were described as randomised but gave no information about the generation of the random list (unclear risk of bias for generation of random sequence). One of these trials concealed allocation with sealed, opaque envelopes (Banu 2007) and one used a telephone interactive voice‐response system (Trinka 2013) (both low risk of bias for allocation concealment). Five trials gave no information about allocation concealment (unclear risk of bias). Of these, three used a computer‐generated random sequence (Biton 2001; Eun 2012; Privitera 2003) and two used random number tables (Pal 1998; Ramsey 1992) (all low risk of bias for generation of random sequence).
The remaining seven trials were described as randomised but gave no details of methods of generation of random sequence and allocation concealment and we judged them to be at unclear risk of bias (Kwan 2009; Lee 2011; Mattson 1985; Ramsey 2010; Steiner 1999; Stephen 2007; Turnbull 1985).
Trials for which no IPD were available (information reported in published papers only)
We judged two trials to be at high risk of selection bias: one trial reported a method of quota allocation and did not report how allocation was concealed (Forsythe 1991) and the other reported a method of randomisation and allocation concealment based on two Latin squares which seemed to take into account the drug preference of participants (the “drug of first preference” was selected from the randomisation list on a sequential basis) (Callaghan 1985).
Five trials described adequate methods of generation of random sequence and allocation concealment and we judged them to be at low risk of bias. Of these, one trial used a random number list and sealed, opaque envelopes (Feksi 1991) and four trials used a computer‐generated random sequence, including three trials that used central telephone randomisation (Donati 2007; Rowan 2005; Shakir 1981) and one trial that used central pharmacy allocation (Jung 2015).
Six trials gave no information about allocation concealment (unclear risk of bias).Of these, two used block randomisation (Brodie 2002; Chen 1996), one used random number tables (Resendiz 2004) and three used a computer‐generated random sequence (Consoli 2012; Motamedi 2013; Thilothammal 1996) (all low risk of bias for generation of random sequence).
The remaining 28 trials were described as randomised but gave no details of methods of generation of random sequence and allocation concealment and we judged them to be at unclear risk of bias: six were published as abstracts only (Bidabadi 2009; Czapinski 1997; Fritz 2006; Kalviainen 2002; Kopp 2007; Lukic 2005); three were published only as only summary results (Korean Lamotrigine Study Group 2008; NCT01498822; NCT01954121); and nineteen were published as full‐text articles (Aikia 1992; Capone 2008; Castriota 2008; Cho 2011, Christe 1997; Cossu 1984; Dam 1989; Gilad 2007; Mitchell 1987; Miura 1990; Pulliainen 1994; Ramsey 1983; Ramsey 2007; Rastogi 1991; Ravi Sudhir 1995; Saetre 2007; So 1992; Steinhoff 2005; Suresh 2015).
Blinding
Trials for which we received IPD (information reported in published papers or provided with IPD)
Five trials reported that participants, personnel and outcome assessors were blinded via the use of matching placebo tablets (Baulac 2012; Biton 2001; Ogunrin 2005; Ramsey 2010; Steiner 1999). Eleven trials reported that participants and personnel were double‐blinded but gave no information about blinding of outcome assessors (Banu 2007; Bill 1997; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2007; Guerreiro 1997; Mattson 1985; Mattson 1992; Privitera 2003; Werhahn 2015). We judged all of these trials to be at low risk of performance bias but unclear risk of detection bias.
Two trials reported that outcome assessors were blinded but that participants and personnel were not blinded (Craig 1994; Pal 1998) and two trials gave no information about blinding so we judged them to be at unclear risk of performance and detection bias (Placencia 1993; Turnbull 1985).
Fifteen trials were of an open‐label design and judged to be at high risk of performance and detection bias (de Silva 1996; Dizdarer 2000; Eun 2012; Heller 1995; Kwan 2009; Lee 2011; Nieto‐Barrera 2001; Ramsey 1992; Reunanen 1996; Richens 1994; SANAD A 2007; SANAD B 2007; Stephen 2007; Trinka 2013; Verity 1995) and one trial could not blind participants and personnel by design but did not state whether outcome assessors were blinded (Chadwick 1998).
Trials for which no IPD were available (information reported in published papers only)
Five trials reported that outcome assessors were blinded. Of these, three did not state whether participants and personnel were blinded (Chen 1996; Cho 2011; Pulliainen 1994) and in the other two trials participants and personnel were not blinded (Forsythe 1991; Jung 2015). Eleven trials reported that participants and personnel were double‐blinded but gave no information about blinding of outcome assessors (Aikia 1992; Brodie 2002; Christe 1997; Cossu 1984; Dam 1989; Motamedi 2013; Ramsey 1983; Ramsey 2007; Rowan 2005; Saetre 2007; So 1992). We judged all of these trials to be at low risk of performance bias but unclear risk of detection bias.
Twelve trials were of an open‐label design and we judged them to be at high risk of performance and detection bias (Castriota 2008; Consoli 2012; Donati 2007; Gilad 2007; Korean Lamotrigine Study Group 2008; Lukic 2005; Mitchell 1987; NCT01498822; NCT01954121; Resendiz 2004; Steinhoff 2005, Suresh 2015)
Thirteen trials gave no information about blinding so we judged them to be at unclear risk of performance and detection bias. Of these, five were published as abstracts only (Bidabadi 2009; Czapinski 1997; Fritz 2006; Kalviainen 2002; Kopp 2007) and eight were published as full‐text articles (Callaghan 1985; Capone 2008; Feksi 1991; Miura 1990; Rastogi 1991; Ravi Sudhir 1995; Shakir 1981; Thilothammal 1996).
Incomplete outcome data
Trials for which we received individual participant data (information reported in published papers or provided with IPD)
In theory, a review using IPD should overcome issues of attrition bias, as unpublished data can be provided, unpublished outcomes calculated, and all randomised participants can be analysed by an intention‐to‐treat approach. All 36 trials (Banu 2007; Baulac 2012; Bill 1997; Biton 2001; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2007; Chadwick 1998; Craig 1994; de Silva 1996; Dizdarer 2000; Eun 2012; Guerreiro 1997; Heller 1995; Kwan 2009; Lee 2011; Mattson 1985; Mattson 1992; Nieto‐Barrera 2001; Ogunrin 2005; Pal 1998; Placencia 1993; Privitera 2003; Ramsey 1992; Ramsey 2010; Reunanen 1996; Richens 1994; SANAD A 2007; SANAD B 2007; Steiner 1999; Stephen 2007; Trinka 2013; Turnbull 1985; Verity 1995; Werhahn 2015) provided individual participant data for all randomised individuals and reported the extent of follow‐up for each individual. We queried any missing data with the original trial authors. From the information provided by the trial authors, we deemed the small amount of missing data present (see Included studies) to be missing at random and not affecting our analysis so we judged them to be at low risk of bias.
Trials for which no IPD were available (information reported in published papers only)
Seven trials, which were published as abstracts only, did not give enough information to assess selective reporting so we judged them to have unclear risk of bias (Bidabadi 2009; Czapinski 1997; Fritz 2006; Kalviainen 2002; Kopp 2007; Lukic 2005; Ramsey 2007). Three trials excluded the small proportion of participants who withdrew from the trial from analysis but it is unclear whether this would have influenced analysis (Castriota 2008; Chen 1996; Suresh 2015) and two trials did not clearly report whether participants had withdrawn from the trial (Cho 2011; Rastogi 1991) so we also judged these trials to be at unclear risk of bias.
Twelve trials reported attrition rates and used an intention‐to‐treat approach to analysis so we judged them to be at low risk of attrition bias (Brodie 2002; Callaghan 1985; Capone 2008; Cossu 1984; Forsythe 1991; Gilad 2007; Mitchell 1987; Miura 1990; Rowan 2005; Saetre 2007; Shakir 1981; Thilothammal 1996). The remaining 17 trials excluded participants from analysis and did not use an intention‐to‐treat approach to analysis and we judged them to be at high risk of attrition bias (Aikia 1992; Christe 1997; Consoli 2012; Dam 1989; Donati 2007; Feksi 1991; Jung 2015; Korean Lamotrigine Study Group 2008; Motamedi 2013; NCT01498822; NCT01954121; Pulliainen 1994; Ramsey 1983; Ravi Sudhir 1995; Resendiz 2004; So 1992; Steinhoff 2005).
Selective reporting
Trials for which we received IPD (information reported in published papers or provided with IPD)
We requested trial protocols in all IPD requests and protocols were provided for 20 out of the 36 trials providing IPD (Baulac 2012; Bill 1997; Biton 2001; Brodie 1995a; Brodie 1995b; Brodie 1999; de Silva 1996; Guerreiro 1997; Heller 1995; Mattson 1985; Mattson 1992; Nieto‐Barrera 2001; Ogunrin 2005; Reunanen 1996; Richens 1994; SANAD A 2007; SANAD B 2007; Steiner 1999; Verity 1995; Werhahn 2015).
In theory, a review using IPD should overcome issues of reporting biases, as unpublished data can be provided and unpublished outcomes calculated, so we judged all trials providing IPD to be at low risk of bias. We received sufficient IPD to calculate the four outcomes ('Time to withdrawal of allocated treatment', 'Time to six‐month remission, 'Time to 12‐month remission', and 'Time to first seizure') for 20 of the 36 trials (Baulac 2012; Bill 1997; Brodie 2007; de Silva 1996; Dizdarer 2000; Guerreiro 1997; Heller 1995; Kwan 2009; Mattson 1985; Mattson 1992; Placencia 1993; Privitera 2003; Richens 1994; SANAD A 2007; SANAD B 2007Stephen 2007; Trinka 2013; Turnbull 1985; Verity 1995; Werhahn 2015)
We could not calculate 'Time to 12‐month remission' for nine trials as the duration of the trial was less than 12 months (Biton 2001; Brodie 1995a; Brodie 1995b; Chadwick 1998; Eun 2012; Lee 2011; Ramsey 1992; Reunanen 1996; Steiner 1999) and we could not calculate 'Time to 12‐month remission' or 'Time to six‐month remission' for three trials as the duration of the trial was less than six months (Brodie 1999; Nieto‐Barrera 2001; Ramsey 2010).
Withdrawal information was not available for two trials so we could not calculate 'Time to withdrawal of allocated treatment' (Craig 1994; Pal 1998). For two trials we could only calculate 'Time to first seizure': the trial duration of Ogunrin 2005 was 12 weeks, and all randomised participants completed the trial without withdrawing; and Banu 2007 did not record the dates of all seizures after randomisation and dates of withdrawal for allocated treatment for all participants.
Trials for which no IPD were available (information reported in published papers only)
Protocols were not available for any of the 41 trials without IPD available, so we made a judgement of the risk of bias based on the information included in the publications or from the IPD we received (see the Characteristics of included studies tables for more information).
We judged two trials to be at high risk of reporting bias; one trial reported results for outcomes that were not defined in the methods section (Suresh 2015) and one trial did not provide online results for all listed outcomes (NCT01954121).
In 25 trials, expected efficacy and tolerability outcomes were well reported in the methods and results therefore we judged these trials to be at low risk of selective reporting bias (Aikia 1992; Brodie 2002; Callaghan 1985; Chen 1996; Cho 2011, Christe 1997; Consoli 2012; Dam 1989; Donati 2007; Feksi 1991; Gilad 2007; Jung 2015; Korean Lamotrigine Study Group 2008; Mitchell 1987; Motamedi 2013; NCT01498822; Ramsey 1983; Rastogi 1991; Resendiz 2004; Rowan 2005; Saetre 2007; Shakir 1981; So 1992; Steinhoff 2005; Thilothammal 1996).
Seven trials that were published as abstracts only (Bidabadi 2009; Czapinski 1997; Fritz 2006; Kalviainen 2002; Kopp 2007; Lukic 2005; Ramsey 2007) and one trial with a very brief description of methods (Capone 2008) did not give enough information to assess selective reporting so we judged them to have unclear risk of bias. Six trials reported only cognitive outcomes rather than expected efficacy or tolerability outcomes and it was unclear if such outcomes were planned a priori, therefore we also judged these trials to have unclear risk of bias (Castriota 2008; Cossu 1984; Forsythe 1991; Miura 1990; Pulliainen 1994; Ravi Sudhir 1995).
Other potential sources of bias
We detected another source of bias in eight trials.
Following consistency checks of IPD for Placencia 1993; Stephen 2007 and Banu 2007, we found some inconsistencies between the data provided and the results in the publications in terms of withdrawal and seizure recurrences, respectively, which the trial authors could not resolve. We performed sensitivity analysis to investigate the impact of the inconsistent data on our outcomes (see Sensitivity analysis). Furthermore, we received IPD for another trial (Feksi 1991), but too many inconsistencies were present for this data to be usable (see Included studies for further details).
We included one trial with very small participant numbers (six participants randomised to each drug) and very short‐term follow‐up (three weeks) (Cossu 1984), and one trial that terminated early with only 20% of target sample size recruited (Consoli 2012). It is unlikely that either of these trials were adequately powered and of sufficient duration to detect differences. Another trial had several other potential sources of bias (Mitchell 1987); the trial was likely underpowered to detect differences between the treatments, one of the tools for outcome assessment was not fully validated, and non‐randomised children from a related pilot study were included in analysis for some of the outcomes. In one trial, it was unclear if all participants were receiving AED monotherapy treatment (‘total number of AEDs’ described in Table 1 of the publication), so we judged this trial to be at unclear risk of bias (Gilad 2007).
No other sources of bias were identified in the remaining 69 trials.
Effects of interventions
See: Summary of findings for the main comparison Summary of findings ‐ Time to withdrawal of allocated treatment for individuals with partial seizures; Summary of findings 2 Summary of findings ‐ Time to withdrawal of allocated treatment for individuals with partial seizures; Summary of findings 3 Summary of findings ‐ Time to withdrawal of allocated treatment for individuals with generalised seizures; Summary of findings 4 Summary of findings ‐ Time to 12‐month remission for individuals with partial seizures; Summary of findings 5 Summary of findings ‐ Time to 12‐month remission for individuals with partial seizures; Summary of findings 6 Summary of findings ‐ Time to 12‐month remission for individuals with generalised seizures
For brevity throughout the results section, we refer to participants with generalised tonic‐clonic seizures with or without other generalised seizure types as 'participants with generalised seizures.'
Figure 1 and Figure 2 visually present the network of 45 pairwise comparisons from the 10 antiepileptic treatments. Figure 1 also demonstrates the network of the trials with and without IPD provided for analysis and Figure 2 also presents the network of evidence for participants with partial seizures and with generalised seizures. We note that zonisamide has only been used in a single trial recruiting individuals with partial onset seizures only (Baulac 2012), therefore zonisamide does not feature in the network of evidence for generalised seizures and there are 36 pairwise comparisons in this network.
Table 6 shows the total number of participants contributing to each analysis (Table 7 shows the reported reasons for withdrawal from treatment across all studies) and Table 8; Table 9; Table 10; Table 11; Table 12; Table 13; Table 14; Table 15 and Figure 5; Figure 6; Figure 7; Figure 8; Figure 9; Figure 10; and Figure 11 show the results for each of the outcomes below. Results highlighted in bold in the tables indicate statistically significant results and HR less than 1 indicates an advantage to the second drug in the comparison. All results presented were calculated with a fixed‐effect analysis.
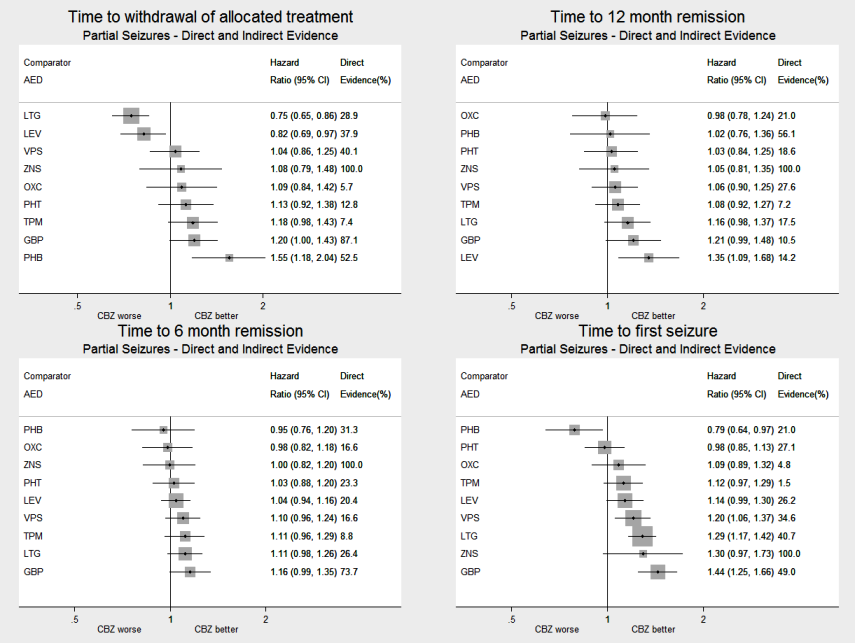
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with partial seizures, all drugs compared to carbamazepine (CBZ)
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.

AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with partial seizures, all drugs compared to lamotrigine (LTG)
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.

AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with generalised seizures, all drugs compared to sodium valproate (VPS)
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
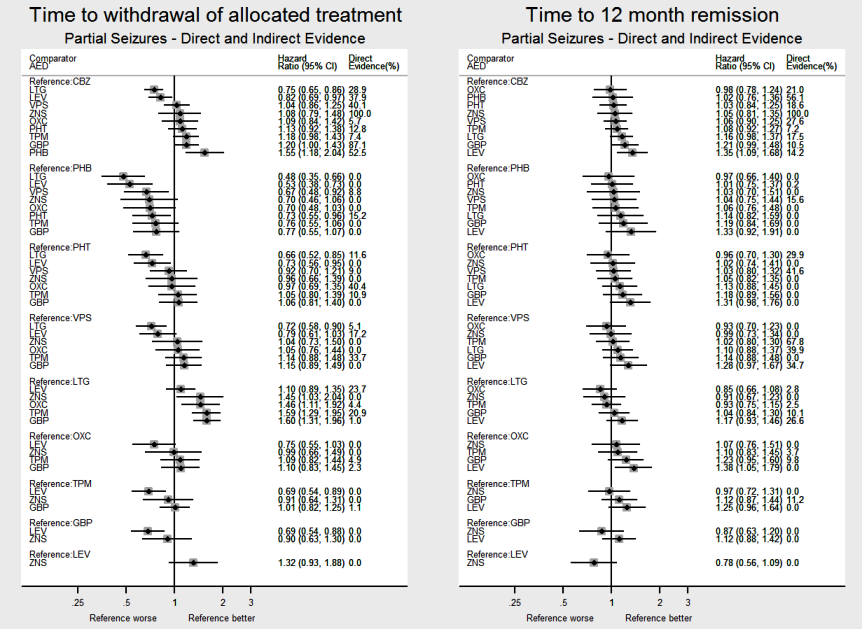
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with partial seizures, all pairwise comparisons for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.

AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with generalised seizures, all pairwise comparisons for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
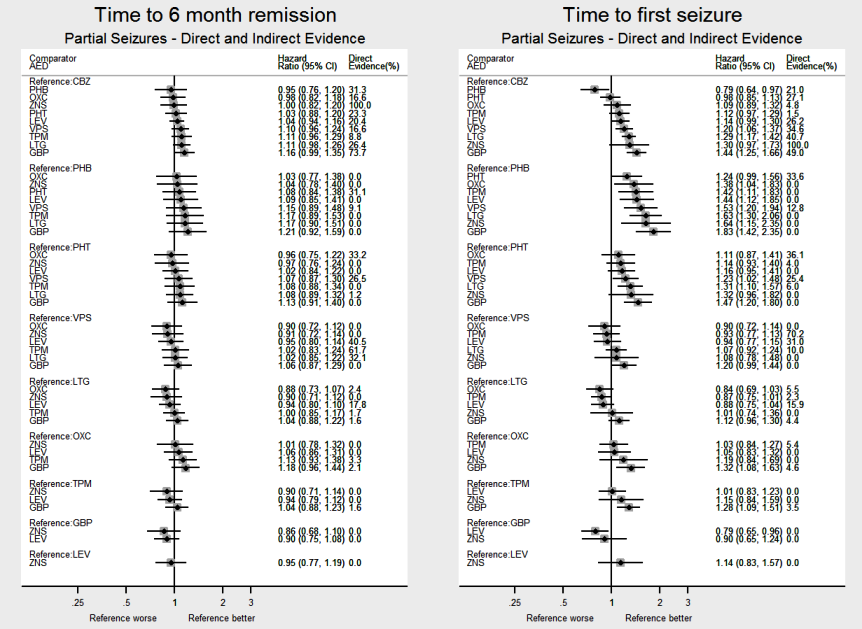
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with partial seizures, all pairwise comparisons for time to six‐month remission and time to first seizure.
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.

AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with generalised seizures, all pairwise comparisons for time to six‐month remission and time to first seizure.
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
| Trial | Time to withdrawal from allocated treatmentc | Time to first seizure | Time to 12‐month remissiond | Time to six‐month remissiond | ||||||||||||
| Cens | Event | Total | Missing | Cens | Event | Total | Missing | Cens | Event | Total | Missing | Cens | Event | Total | Missing | |
| 0 | 0 | 0 | 108 | 39 | 69 | 108 | 0 | 0 | 0 | 0 | 108 | 0 | 0 | 0 | 108 | |
| 392 | 191 | 583 | 0 | 388 | 186 | 574 | 9 | 251 | 323 | 574 | 9 | 194 | 380 | 574 | 9 | |
| 232 | 55 | 287 | 0 | 137 | 145 | 282 | 5 | 190 | 92 | 282 | 5 | 136 | 146 | 282 | 5 | |
| 99 | 36 | 135 | 1 | 64 | 71 | 135 | 1 | 0 | 0 | 0 | 136 | 90 | 45 | 135 | 1 | |
| 78 | 58 | 136 | 0 | 69 | 67 | 136 | 0 | 0 | 0 | 0 | 136 | 122 | 14 | 136 | 0 | |
| 79 | 45 | 124 | 0 | 52 | 72 | 124 | 0 | 0 | 0 | 0 | 124 | 96 | 28 | 124 | 0 | |
| 95 | 55 | 150 | 0 | 70 | 80 | 150 | 0 | 0 | 0 | 0 | 150 | 106 | 44 | 150 | 0 | |
| 323 | 256 | 579 | 0 | 350 | 229 | 579 | 0 | 260 | 319 | 579 | 0 | 177 | 402 | 579 | 0 | |
| 69 | 223 | 292 | 0 | 102 | 190 | 292 | 0 | 0 | 0 | 0 | 292 | 193 | 99 | 292 | 0 | |
| 0 | 0 | 0 | 166 | 68 | 81 | 149 | 17 | 117 | 30 | 147 | 19 | 58 | 89 | 147 | 19 | |
| 100 | 67 | 167 | 6 | 18 | 149 | 167 | 6 | 22 | 145 | 167 | 6 | 19 | 148 | 167 | 6 | |
| 44 | 8 | 52 | 0 | 40 | 12 | 52 | 0 | 11 | 41 | 52 | 0 | 8 | 44 | 52 | 0 | |
| 75 | 9 | 84 | 0 | 52 | 32 | 84 | 0 | 0 | 0 | 0 | 84 | 35 | 49 | 84 | 0 | |
| 151 | 42 | 193 | 0 | 106 | 84 | 190 | 3 | 112 | 78 | 190 | 3 | 84 | 106 | 190 | 3 | |
| 166 | 77 | 243 | 0 | 66 | 177 | 243 | 0 | 78 | 165 | 243 | 0 | 49 | 194 | 243 | 0 | |
| 60 | 21 | 81 | 0 | 38 | 29 | 67 | 14 | 68 | 13 | 81 | 0 | 30 | 50 | 80 | 1 | |
| 73 | 37 | 110 | 0 | 79 | 31 | 110 | 0 | 0 | 0 | 0 | 110 | 39 | 71 | 110 | 0 | |
| 267 | 208 | 475 | 0 | 226 | 238 | 464 | 11 | 325 | 149 | 474 | 1 | 281 | 193 | 474 | 1 | |
| 308 | 172 | 480 | 0 | 165 | 303 | 468 | 12 | 334 | 133 | 467 | 13 | 242 | 225 | 467 | 13 | |
| 511 | 111 | 622 | 0 | 310 | 312 | 622 | 0 | 0 | 0 | 0 | 622 | 431 | 191 | 622 | 0 | |
| 0 | 0 | 0 | 55 | 29 | 26 | 55 | 0 | 0 | 0 | 0 | 55 | 0 | 0 | 0 | 55 | |
| 0 | 0 | 0 | 94 | 41 | 49 | 90 | 4 | 82 | 8 | 90 | 4 | 63 | 27 | 90 | 4 | |
| 158 | 32 | 190 | 2 | 121 | 71 | 192 | 0 | 132 | 60 | 192 | 0 | 69 | 123 | 192 | 0 | |
| (CBZ branch)b | 221 | 174 | 395 | 0 | 208 | 187 | 395 | 0 | 316 | 79 | 395 | 0 | 194 | 201 | 395 | 0 |
| (VPS branch)b | 111 | 114 | 225 | 0 | 119 | 106 | 225 | 0 | 180 | 45 | 225 | 0 | 106 | 119 | 225 | 0 |
| 113 | 23 | 136 | 0 | 81 | 44 | 125 | 11 | 0 | 0 | 0 | 136 | 78 | 47 | 125 | 11 | |
| 192 | 69 | 261 | 0 | 197 | 64 | 261 | 0 | 0 | 0 | 0 | 261 | 0 | 0 | 0 | 261 | |
| 288 | 63 | 351 | 0 | 216 | 135 | 351 | 0 | 0 | 0 | 0 | 351 | 328 | 23 | 351 | 0 | |
| 210 | 76 | 286 | 14 | 91 | 199 | 290 | 10 | 92 | 198 | 290 | 10 | 77 | 213 | 290 | 10 | |
| 857 | 815 | 1672 | 49 | 383 | 1261 | 1644 | 77 | 577 | 1067 | 1644 | 77 | 355 | 1326 | 1681 | 40 | |
| 400 | 299 | 699 | 17 | 182 | 511 | 693 | 23 | 167 | 526 | 693 | 23 | 96 | 610 | 706 | 10 | |
| 108 | 73 | 181 | 0 | 100 | 81 | 181 | 0 | 0 | 0 | 0 | 181 | 157 | 24 | 181 | 0 | |
| 160 | 67 | 227 | 0 | 81 | 140 | 221 | 6 | 172 | 55 | 227 | 0 | 137 | 90 | 227 | 0 | |
| (CBZ branch)b | 760 | 239 | 999 | 0 | 572 | 427 | 999 | 0 | 780 | 219 | 999 | 0 | 336 | 663 | 999 | 0 |
| (VPS branch)b | 583 | 120 | 703 | 0 | 456 | 247 | 703 | 0 | 484 | 219 | 703 | 0 | 191 | 512 | 703 | 0 |
| 91 | 49 | 140 | 0 | 75 | 65 | 140 | 0 | 47 | 93 | 140 | 0 | 36 | 104 | 140 | 0 | |
| 187 | 59 | 246 | 14 | 59 | 187 | 246 | 14 | 84 | 162 | 246 | 14 | 19 | 227 | 246 | 14 | |
| 195 | 166 | 361 | 0 | 249 | 96 | 345 | 16 | 211 | 150 | 361 | 0 | 178 | 183 | 361 | 0 | |
| Total | 7756 | 4109 | 11,865 | 526 | 5699 | 6453 | 12,152 | 239 | 5092 | 4369 | 9461 | 2930 | 4810 | 7010 | 11,820 | 571 |
Abbreviation: cens = censored
aFor two studies we could only calculate 'Time to first seizure'; the study duration of Ogunrin 2005 was 12 weeks, and all randomised participants completed the study without withdrawing; and Banu 2007 did not record the dates of all seizures after randomisation and dates of withdrawal for allocated treatment for all participants.
bTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review.
cWithdrawal information was not available for two trials so we could not calculate 'Time to withdrawal of allocated treatment' (Craig 1994; Pal 1998).
dWe could not calculate 'Time to 12‐month remission' for nine trials as the duration of the study was less than 12 months (Biton 2001; Brodie 1995a; Brodie 1995b; Chadwick 1998; Eun 2012; Lee 2011; Ramsey 1992; Reunanen 1996; Steiner 1999) and we could not calculate 'Time to 12‐month remission' or 'Time to six‐month remission' for three trials as the duration of the study was less than six months (Brodie 1999; Nieto‐Barrera 2001; Ramsey 2010).
| Reason for withdrawal | Classification for analysis | Randomised drug4b | ||||||||||
| CBZ | PHB | PHT | VPS | LTG | OXC | TPM | GBP | LEV | ZNS | Total | ||
| Adverse events | Event | 505 (45%) | 24 (20%) | 93 (35%) | 132 (28%) | 235 (41%) | 56 (41%) | 259 (48%) | 73 (20%) | 134 (39%) | 31 (32%) | 1542 (38%) |
| Inadequate response | Event | 232 (20%) | 20 (16%) | 46 (17%) | 140 (29%) | 144 (26%) | 36 (26%) | 148 (27%) | 223 (62%) | 89 (26%) | 23 (24%) | 1101 (27%) |
| Both adverse events and inadequate response | Event | 148 (13%) | 51 (41%) | 54 (20%) | 107 (22%) | 32 (6%) | 11 (8%) | 46 (8%) | 32 (9%) | 0 (0%) | 0 (0%) | 481 (12%) |
| Protocol violation/non compliance | Event | 102 (9%) | 15 (12%) | 41 (15%) | 11 (2%) | 68 (12%) | 27 (20%) | 0 (0%) | 21 (6%) | 21 (6%) | 3 (3%) | 309 (8%) |
| Withdrew consent | Event | 121 (11%) | 13 (11%) | 25 (9%) | 64 (13%) | 65 (11%) | 2 (1%) | 55 (10%) | 4 (1%) | 68 (20%) | 35 (36%) | 452 (11%) |
| Othera | Event | 29 (3%) | 0 (0%) | 7 (3%) | 24 (5%) | 26 (5%) | 5 (4%) | 37 (7%) | 9 (2%) | 32 (9%) | 4 (4%) | 173 (4%) |
| Total eventsb | 1137 (35%) | 123 (38%) | 266 (31%) | 478 (28%) | 570 (29%) | 137 (29%) | 545 (47%) | 362 (61%) | 344 (27%) | 96 (34%) | 4058 (34%) | |
| Illness or death | Censored | 34 (2%) | 10 (5%) | 17 (3%) | 7 (1%) | 20 (1%) | 1 (0%) | 10 (2%) | 9 (4%) | 0 (0%) | 0 (0%) | 108 (1%) |
| Remission of seizures | Censored | 49 (2%) | 4 (2%) | 38 (6%) | 75 (6%) | 40 (3%) | 12 (4%) | 44 (7%) | 21 (9%) | 0 (0%) | 0 (0%) | 283 (4%) |
| Lost to follow‐up | Censored | 81 (4%) | 31 (16%) | 51 (9%) | 63 (5%) | 33 (3%) | 24 (7%) | 18 (3%) | 0 (0%) | 41 (5%) | 0 (0%) | 342 (4%) |
| Otherc | Censored | 104 (5%) | 6 (3%) | 22 (4%) | 82 (7%) | 31 (2%) | 5 (2%) | 26 (4%) | 26 (12%) | 0 (0%) | 25 (13%) | 327 (4%) |
| Completed study | Censored | 1829 (87%) | 139 (73%) | 468 (79%) | 949 (81%) | 1272 (91%) | 291 (87%) | 501 (84%) | 166 (75%) | 868 (95%) | 161 (87%) | 6644 (86%) |
| Total censoredb | 2097 (65%) | 190 (62%) | 596 (69%) | 1176 (72%) | 1396 (71%) | 333 (71%) | 599 (53%) | 222 (39%) | 909 (73%) | 186 (66%) | 7704 (66%) | |
| Missingd | 24 | 7 | 1 | 26 | 12 | 8 | 14 | 11 | 0 | 0 | 103 | |
| Totale | 3258 | 320 | 863 | 1680 | 1978 | 478 | 1158 | 595 | 1253 | 282 | 11,865 | |
CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aOther treatment‐related reasons included: physician's decision, drug‐related death, pregnancy or perceived remission, or non specific (drug‐related) reason.
bProportions for specific reasons indicate proportion of total events or total censored. Proportion for total events and total censored indicate the proportion of total participants.
cOther non treatment‐related reasons included: epilepsy diagnosis changed, participants developed other medical disorders including neurological and psychiatric disorders or non specific (non drug‐related) reason.
dWe treated those with missing reasons for withdrawal as censored in analysis and performed a sensitivity analysis treating these individuals as having withdrawal 'events.' Results of sensitivity analysis were practically identical and conclusions unchanged so we have presented the results treating these individuals as censored.
eFour studies did not contribute to analysis of time to withdrawal of allocated treatment (Banu 2007; Craig 1994; Ogunrin 2005; Pal 1998).
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number of | Number of | HR (95% CI)b,c | I² statisticd | Direct evidence (%)e | HR (95% CI)b,c | |
| CBZ vs PHB | 4 | 520 | 1.57 (1.16 to 2.13) | 0% | 52.5% | 1.55 (1.18 to 2.04) |
| CBZ vs PHT | 3 | 428 | 1.03 (0.74 to 1.42) | 63.6% | 12.8% | 1.13 (0.92 to 1.38) |
| CBZ vs VPS | 5 | 814 | 0.94 (0.73 to 1.19) | 0% | 40.1% | 1.04 (0.86 to 1.25) |
| CBZ vs LTG | 9 | 2268 | 0.76 (0.61 to 0.95) | 39.3% | 28.9% | 0.75 (0.65 to 0.86) |
| CBZ vs OXC | 2 | 562 | 4.62 (0.95 to 22.4) | 0% | 5.7% | 1.09 (0.84 to 1.42) |
| CBZ vs TPM | 2 | 937 | 1.04 (0.52 to 2.07) | 0% | 7.4% | 1.18 (0.98 to 1.43) |
| CBZ vs GBP | 2 | 954 | 1.14 (0.84 to 1.55) | 0% | 87.1% | 1.20 (1.00 to 1.43) |
| CBZ vs LEV | 3 | 1567 | 0.70 (0.52 to 0.94) | 0% | 37.9% | 0.82 (0.69 to 0.97) |
| CBZ vs ZNS | 1 | 583 | 1.08 (0.81 to 1.44) | NA | 100% | 1.08 (0.79 to 1.48) |
| PHB vs PHT | 3 | 404 | 0.67 (0.50 to 0.91) | 65% | 15.2% | 0.73 (0.55 to 0.96) |
| PHB vs VPS | 2 | 75 | 0.68 (0.34 to 1.36) | 23% | 8.8% | 0.67 (0.48 to 0.92) |
| PHB vs LTG | No direct evidence | 0% | 0.48 (0.35 to 0.66) | |||
| PHB vs OXC | No direct evidence | 0% | 0.70 (0.48 to 1.03) | |||
| PHB vs TPM | No direct evidence | 0% | 0.76 (0.55 to 1.06) | |||
| PHB vs GBP | No direct evidence | 0% | 0.77 (0.55 to 1.07) | |||
| PHB vs LEV | No direct evidence | 0% | 0.53 (0.38 to 0.73) | |||
| PHB vs ZNS | No direct evidence | 0% | 0.70 (0.46 to 1.06) | |||
| PHT vs VPS | 4 | 168 | 1.00 (0.60 to 1.64) | 58.5% | 9% | 0.92 (0.70 to 1.21) |
| PHT vs LTG | 1 | 90 | 1.10 (0.57 to 2.14) | NA | 11.6% | 0.66 (0.52 to 0.85) |
| PHT vs OXC | 2 | 325 | 0.65 (0.32 to 1.32) | 0% | 40.4% | 0.97 (0.69 to 1.35) |
| PHT vs TPM | 1 | 53 | 0.77 (0.38 to 1.57) | NA | 10.9% | 1.05 (0.80 to 1.39) |
| PHT vs GBP | No direct evidence | 0% | 1.06 (0.81 to 1.40) | |||
| PHT vs LEV | No direct evidence | 0% | 0.73 (0.56 to 0.95) | |||
| PHT vs ZNS | No direct evidence | 0% | 0.96 (0.66 to 1.39) | |||
| VPS vs LTG* | 3 | 221 | 1.40 (1.00 to 1.96) | 45.1% | 5.1% | 0.72 (0.58 to 0.90) |
| VPS vs OXC | No direct evidence | 0% | 1.05 (0.76 to 1.44) | |||
| VPS vs TPM | 2 | 111 | 1.66 (1.24 to 2.23) | 48.1% | 33.7% | 1.14 (0.88 to 1.48) |
| VPS vs GBP | No direct evidence | 0% | 1.15 (0.89 to 1.49) | |||
| VPS vs LEV | 1 | 190 | 1.14 (0.73 to 1.75) | NA | 17.2% | 0.79 (0.61 to 1.03) |
| VPS vs ZNS | No direct evidence | 0% | 1.04 (0.73 to 1.50) | |||
| LTG vs OXC | 1 | 506 | 0.69 (0.12 to 4.14) | NA | 4.4% | 1.46 (1.11 to 1.92) |
| LTG vs TPM | 1 | 648 | 1.18 (0.86 to 1.62) | NA | 20.9% | 1.59 (1.29 to 1.95) |
| LTG vs GBP | 1 | 659 | 0.62 (0.06 to 6.01) | NA | 1% | 1.60 (1.31 to 1.96) |
| LTG vs LEV | 1 | 240 | 0.86 (0.58 to 1.28) | NA | 23.7% | 1.10 (0.89 to 1.35) |
| LTG vs ZNS | No direct evidence | 0% | 1.45 (1.03 to 2.04) | |||
| OXC vs TPM | 1 | 496 | 0.87 (0.16 to 4.73) | NA | 4.9% | 1.09 (0.82 to 1.44) |
| OXC vs GBP | 1 | 507 | 0.90 (0.08 to 9.96) | NA | 2.3% | 1.10 (0.83 to 1.45) |
| OXC vs LEV | No direct evidence | 0% | 0.75 (0.55 to 1.03) | |||
| OXC vs ZNS | No direct evidence | 0% | 0.99 (0.66 to 1.49) | |||
| TPM vs GBP | 1 | 649 | 1.04 (0.12 to 9.33) | NA | 1.1% | 1.01 (0.82 to 1.25) |
| TPM vs LEV | No direct evidence | 0% | 0.69 (0.54 to 0.89) | |||
| TPM vs ZNS | No direct evidence | 0% | 0.91 (0.64 to 1.31) | |||
| GBP vs LEV | No direct evidence | 0% | 0.69 (0.54 to 0.88) | |||
| GBP vs ZNS | No direct evidence | 0% | 0.90 (0.63 to 1.30) | |||
| LEV vs ZNS | No direct evidence | 0% | 1.32 (0.93 to 1.88) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first).
bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details.
cNote that HR < 1 indicates an advantage to the second drug in the comparison; results highlighted in bold are statistically significant.
dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence.
eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
For comparisons marked with a *, confidence intervals of direct evidence and network meta‐analysis do not overlap indicating that inconsistency may be present in the results.
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number of | Number of | HR (95% CI)b,c | I² statisticd | Direct | HR (95% CI)b,c | |
| CBZ vs PHB | 3 | 156 | 1.21 (0.51 to 2.86) | 11.8% | 27.3% | 1.47 (0.83 to 2.61) |
| CBZ vs PHT | 2 | 118 | 2.68 (0.95 to 7.57) | 0% | 11.3% | 0.92 (0.59 to 1.42) |
| CBZ vs VPS | 4 | 405 | 1.26 (0.73 to 2.20) | 6.6% | 27.3% | 0.70 (0.54 to 0.92) |
| CBZ vs LTG | 7 | 302 | 1.23 (0.72 to 2.10) | 0% | 39.2% | 0.63 (0.45 to 0.89) |
| CBZ vs OXC | 1 | 9 | 0.39 (0.03 to 4.35) | NA | 3.9% | 1.00 (0.21 to 4.81) |
| CBZ vs TPM | 2 | 101 | 1.10 (0.51 to 2.36) | 0% | 23.2% | 1.24 (0.90 to 1.71) |
| CBZ vs GBP | 1 | 6 | 0.49 (0.03 to 7.90) | NA | 8.5% | 0.90 (0.11 to 7.29) |
| CBZ vs LEV | 2 | 251 | 1.22 (0.74 to 2.02) | 0% | 57% | 0.74 (0.44 to 1.23) |
| PHB vs PHT | 2 | 95 | 1.56 (0.49 to 4.99) | 0% | 16.1% | 0.62 (0.32 to 1.24) |
| PHB vs VPS | 2 | 94 | 0.56 (0.20 to 1.54) | 0% | 19.4% | 0.48 (0.27 to 0.86) |
| PHB vs LTG | No direct evidence | 0% | 0.43 (0.22 to 0.83) | |||
| PHB vs OXC | No direct evidence | 0% | 0.68 (0.13 to 3.60) | |||
| PHB vs TPM | No direct evidence | 0% | 0.84 (0.44 to 1.60) | |||
| PHB vs GBP | No direct evidence | 0% | 0.61 (0.07 to 5.34) | |||
| PHB vs LEV | No direct evidence | 0% | 0.50 (0.23 to 1.09) | |||
| PHT vs VPS | 3 | 326 | 0.66 (0.30 to 1.45) | 22.6% | 19.3% | 0.77 (0.46 to 1.27) |
| PHT vs LTG | 1 | 91 | 1.11 (0.42 to 2.94) | NA | 14.9% | 0.69 (0.39 to 1.20) |
| PHT vs OXC | 2 | 155 | 1.05 (0.44 to 2.52) | 0% | 37.9% | 1.09 (0.21 to 5.56) |
| PHT vs TPM | 1 | 150 | 1.68 (0.49 to 5.69) | NA | 11.2% | 1.35 (0.79 to 2.30) |
| PHT vs GBP | No direct evidence | 0% | 0.98 (0.12 to 8.30) | |||
| PHT vs LEV | No direct evidence | 0% | 0.80 (0.42 to 1.55) | |||
| VPS vs LTG | 3 | 387 | 0.46 (0.22 to 0.97) | 0% | 14.8% | 0.90 (0.60 to 1.35) |
| VPS vs OXC | No direct evidence | 0% | 1.42 (0.29 to 6.92) | |||
| VPS vs TPM* | 2 | 443 | 0.53 (0.27 to 1.07) | 48.5% | 22.4% | 1.76 (1.22 to 2.53) |
| VPS vs GBP | No direct evidence | 0% | 1.28 (0.16 to 10.5) | |||
| VPS vs LEV | 1 | 512 | 0.68 (0.30 to 1.59) | NA | 18.6% | 1.05 (0.58 to 1.90) |
| LTG vs OXC | 1 | 10 | 2.09 (0.34 to 12.8) | NA | 7.6% | 1.58 (0.33 to 7.67) |
| LTG vs TPM | 1 | 14 | 1.10 (0.42 to 2.89) | NA | 7.3% | 1.96 (1.25 to 3.08) |
| LTG vs GBP | 1 | 7 | 2.63 (0.27 to 25.7) | NA | 13.8% | 1.42 (0.17 to 11.6) |
| LTG vs LEV | No direct evidence | 0% | 1.17 (0.63 to 2.19) | |||
| OXC vs TPM | 1 | 14 | 1.31 (0.24 to 7.32) | NA | 9% | 1.24 (0.26 to 5.94) |
| OXC vs GBP | 1 | 7 | 1.26 (0.11 to 14.1) | NA | 12.7% | 0.90 (0.08 to 9.96) |
| OXC vs LEV | No direct evidence | 0% | 0.74 (0.14 to 3.86) | |||
| TPM vs GBP | 1 | 11 | 0.96 (0.11 to 8.67) | NA | 14.6% | 0.73 (0.09 to 5.89) |
| TPM vs LEV | No direct evidence | 0% | 0.60 (0.33 to 1.09) | |||
| GBP vs LEV | No direct evidence | 0% | 0.82 (0.10 to 7.10) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first).
bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details
cNote that HR < 1 indicates an advantage to the second drug in the comparison; results highlighted in bold are statistically significant.
dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence.
eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
For comparisons marked with a *, confidence intervals of direct evidence and network meta‐analysis do not overlap indicating that inconsistency may be present in the results
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number of | Number of | HR (95% CI)b,c | I² statisticd | Direct | HR (95% CI)b,c | |
| CBZ vs PHB | 4 | 525 | 1.41 (1.04 to 1.91) | 0% | 56.1% | 1.02 (0.76 to 1.35) |
| CBZ vs PHT | 3 | 430 | 1.00 (0.76 to 1.32) | 54.8% | 18.6% | 1.03 (0.85 to 1.25) |
| CBZ vs VPS | 5 | 816 | 1.03 (0.85 to 1.25) | 46.4% | 27.6% | 1.05 (0.89 to 1.25) |
| CBZ vs LTG | 2 | 891 | 1.02 (0.69 to 1.50) | 0% | 17.5% | 1.16 (0.98 to 1.37) |
| CBZ vs OXC | 2 | 555 | 1.13 (0.62 to 2.05) | 0% | 21% | 0.98 (0.78 to 1.25) |
| CBZ vs TPM | 2 | 925 | 0.94 (0.48 to 1.83) | 0% | 7.2% | 1.08 (0.92 to 1.27) |
| CBZ vs GBP | 1 | 651 | 0.61 (0.06 to 5.82) | NA | 10.5% | 1.20 (0.99 to 1.47) |
| CBZ vs LEV | 3 | 1567 | 1.08 (0.81 to 1.42) | 60.8% | 14.2% | 1.35 (1.09 to 1.69) |
| CBZ vs ZNS | 1 | 582 | 1.05 (0.85 to 1.30) | NA | 100% | 1.05 (0.81 to 1.35) |
| PHB vs PHT | 4 | 465 | 0.80 (0.59 to 1.10) | 0% | 0.2% | 1.01 (0.75 to 1.37) |
| PHB vs VPS | 2 | 80 | 0.85 (0.51 to 1.40) | 4.4% | 15.6% | 1.04 (0.75 to 1.43) |
| PHB vs LTG | No direct evidence | 0% | 1.14 (0.82 to 1.59) | |||
| PHB vs OXC | No direct evidence | 0% | 0.96 (0.67 to 1.41) | |||
| PHB vs TPM | No direct evidence | 0% | 1.06 (0.76 to 1.47) | |||
| PHB vs GBP | No direct evidence | 0% | 1.19 (0.83 to 1.69) | |||
| PHB vs LEV | No direct evidence | 0% | 1.33 (0.93 to 1.92) | |||
| PHB vs ZNS | No direct evidence | 0% | 1.03 (0.70 to 1.52) | |||
| PHT vs VPS | 4 | 245 | 1.04 (0.78 to 1.40) | 0% | 41.6% | 1.03 (0.80 to 1.32) |
| PHT vs LTG | No direct evidence | 0% | 1.12 (0.88 to 1.45) | |||
| PHT vs OXC | 2 | 318 | 1.21 (0.73 to 2.03) | 0% | 29.9% | 0.95 (0.70 to 1.30) |
| PHT vs TPM | No direct evidence | 0% | 1.05 (0.81 to 1.35) | |||
| PHT vs GBP | No direct evidence | 0% | 1.18 (0.88 to 1.56) | |||
| PHT vs LEV | No direct evidence | 0% | 1.32 (0.98 to 1.75) | |||
| PHT vs ZNS | No direct evidence | 0% | 1.02 (0.74 to 1.41) | |||
| VPS vs LTG | 3 | 221 | 1.37 (1.07 to 1.77) | 0% | 39.9% | 1.10 (0.88 to 1.37) |
| VPS vs OXC | No direct evidence | 0% | 0.93 (0.70 to 1.23) | |||
| VPS vs TPM | 2 | 111 | 1.11 (0.87 to 1.40) | 0% | 67.8% | 1.02 (0.80 to 1.30) |
| VPS vs GBP | No direct evidence | 0% | 1.14 (0.88 to 1.47) | |||
| VPS vs LEV | 1 | 190 | 1.14 (0.84 to 1.55) | NA | 34.7% | 1.28 (0.97 to 1.67) |
| VPS vs ZNS | No direct evidence | 0% | 0.99 (0.74 to 1.35) | |||
| LTG vs OXC | 1 | 499 | 1.49 (0.33 to 6.67) | NA | 2.8% | 0.85 (0.66 to 1.09) |
| LTG vs TPM | 1 | 636 | 0.98 (0.29 to 3.25) | NA | 2.5% | 0.93 (0.75 to 1.15) |
| LTG vs GBP | 1 | 647 | 0.74 (0.08 to 6.58) | NA | 10.1% | 1.04 (0.84 to 1.30) |
| LTG vs LEV | 1 | 240 | 1.02 (0.70 to 1.49) | NA | 26.6% | 1.16 (0.93 to 1.47) |
| LTG vs ZNS | No direct evidence | 0% | 0.91 (0.67 to 1.22) | |||
| OXC vs TPM | 1 | 487 | 0.66 (0.17 to 2.47) | NA | 3.7% | 1.10 (0.83 to 1.45) |
| OXC vs GBP | 1 | 498 | 0.49 (0.05 to 4.74) | NA | 9.8% | 1.23 (0.95 to 1.59) |
| OXC vs LEV | No direct evidence | 0% | 1.37 (1.05 to 1.79) | |||
| OXC vs ZNS | No direct evidence | 0% | 1.06 (0.76 to 1.52) | |||
| TPM vs GBP | 1 | 635 | 0.75 (0.09 to 6.00) | NA | 11.2% | 1.12 (0.87 to 1.45) |
| TPM vs LEV | No direct evidence | 0% | 1.25 (0.96 to 1.64) | |||
| TPM vs ZNS | No direct evidence | 0% | 0.97 (0.72 to 1.32) | |||
| GBP vs LEV | No direct evidence | 0% | 1.12 (0.88 to 1.43) | |||
| GBP vs ZNS | No direct evidence | 0% | 0.87 (0.63 to 1.20) | |||
| LEV vs ZNS | No direct evidence | 0% | 0.78 (0.56 to 1.09) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first).
bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details.
cNote that HR < 1 indicates an advantage to the second drug in the comparison; results highlighted in bold are statistically significant.
dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence.
eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number of | Number of | HR (95% CI)b,c | I² statisticd | Direct | HR (95% CI)b,c | |
| CBZ vs PHB | 3 | 158 | 0.53 (0.28 to 1.00) | 0% | 42.6% | 1.25 (0.83 to 1.89) |
| CBZ vs PHT | 2 | 121 | 1.11 (0.61 to 2.02) | 64.5% | 9.3% | 0.86 (0.65 to 1.16) |
| CBZ vs VPS | 4 | 412 | 1.01 (0.72 to 1.43) | 0% | 51.1% | 0.94 (0.79 to 1.14) |
| CBZ vs LTG | 1 | 9 | 1.33 (0.29 to 6.03) | NA | 7% | 1.28 (0.54 to 3.03) |
| CBZ vs OXC | 1 | 9 | 0.77 (0.15 to 3.89) | NA | 5.6% | 1.72 (0.47 to 6.25) |
| CBZ vs TPM | 2 | 101 | 1.15 (0.52 to 2.53) | 0% | 27.2% | 1.06 (0.78 to 1.45) |
| CBZ vs GBP | 1 | 6 | 2.19 (0.23 to 21.2) | NA | 10.9% | 0.75 (0.10 to 5.88) |
| CBZ vs LEV | 2 | 251 | 1.02 (0.65 to 1.59) | 77.4% | 16.6% | 1.33 (0.81 to 2.22) |
| PHB vs PHT | 3 | 130 | 1.30 (0.61 to 2.78) | 53% | 10.9% | 0.68 (0.44 to 1.08) |
| PHB vs VPS | 2 | 98 | 1.15 (0.53 to 2.49) | 42.3% | 13% | 0.75 (0.49 to 1.15) |
| PHB vs LTG | No direct evidence | 0% | 1.01 (0.40 to 2.63) | |||
| PHB vs OXC | No direct evidence | 0% | 1.37 (0.35 to 5.26) | |||
| PHB vs TPM | No direct evidence | 0% | 0.85 (0.51 to 1.41) | |||
| PHB vs GBP | No direct evidence | 0% | 0.60 (0.07 to 5.00) | |||
| PHB vs LEV | No direct evidence | 0% | 1.06 (0.56 to 2.04) | |||
| PHT vs VPS | 4 | 269 | 0.87 (0.55 to 1.40) | 0% | 44.9% | 1.10 (0.80 to 1.49) |
| PHT vs LTG | No direct evidence | 0% | 1.47 (0.60 to 3.57) | |||
| PHT vs OXC | 2 | 154 | 0.77 (0.41 to 1.47) | 0% | 41.2% | 2.00 (0.53 to 7.69) |
| PHT vs TPM | No direct evidence | 0% | 1.23 (0.81 to 1.85) | |||
| PHT vs GBP | No direct evidence | 0% | 0.87 (0.11 to 7.14) | |||
| PHT vs LEV | No direct evidence | 0% | 1.56 (0.87 to 2.78) | |||
| VPS vs LTG | 3 | 387 | 0.77 (0.38 to 1.56) | 0% | 35.7% | 1.35 (0.57 to 3.13) |
| VPS vs OXC | No direct evidence | 0% | 1.82 (0.50 to 6.67) | |||
| VPS vs TPM | 2 | 441 | 0.52 (0.26 to 1.04) | 58.5% | 10.6% | 1.12 (0.83 to 1.52) |
| VPS vs GBP | No direct evidence | 0% | 0.79 (0.10 to 6.25) | |||
| VPS vs LEV | 1 | 512 | 0.91 (0.49 to 1.70) | NA | 55.2% | 1.41 (0.83 to 2.44) |
| LTG vs OXC | 1 | 10 | 0.58 (0.13 to 2.64) | NA | 9.2% | 1.35 (0.33 to 5.56) |
| LTG vs TPM | 1 | 14 | 1.13 (0.33 to 3.82) | NA | 15.1% | 0.83 (0.35 to 2.00) |
| LTG vs GBP | 1 | 7 | 1.64 (0.18 to 14.8) | NA | 12.5% | 0.59 (0.07 to 5.00) |
| LTG vs LEV | No direct evidence | 0% | 1.05 (0.40 to 2.78) | |||
| OXC vs TPM | 1 | 14 | 1.95 (0.51 to 7.50) | NA | 11.4% | 0.62 (0.17 to 2.27) |
| OXC vs GBP | 1 | 7 | 2.83 (0.29 to 27.6) | NA | 10.9% | 0.44 (0.04 to 4.35) |
| OXC vs LEV | No direct evidence | 0% | 0.78 (0.20 to 3.13) | |||
| TPM vs GBP | 1 | 11 | 1.45 (0.18 to 11.7) | NA | 15.9% | 0.71 (0.09 to 5.56) |
| TPM vs LEV | No direct evidence | 0% | 1.27 (0.68 to 2.33) | |||
| GBP vs LEV | No direct evidence | 0% | 1.79 (0.21 to 14.3) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first).
bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details.
cNote that HR < 1 indicates an advantage to the second drug in the comparison; results in highlighted in bold are statistically significant.
dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence.
eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number of | Number of | HR (95% CI)b,c | I² statisticd | Direct | HR (95% CI)b,c | |
| CBZ vs PHB | 4 | 525 | 1.24 (0.95 to 1.61) | 0% | 31.3% | 0.95 (0.76 to 1.19) |
| CBZ vs PHT | 3 | 430 | 0.85 (0.66 to 1.09) | 4.2% | 23.3% | 1.03 (0.88 to 1.20) |
| CBZ vs VPS | 5 | 816 | 1.06 (0.90 to 1.25) | 56.5% | 16.6% | 1.10 (0.96 to 1.25) |
| CBZ vs LTG | 7 | 1535 | 1.15 (0.89 to 1.48) | 0% | 26.4% | 1.11 (0.98 to 1.27) |
| CBZ vs OXC | 2 | 555 | 1.15 (0.65 to 2.04) | 0% | 16.6% | 0.98 (0.82 to 1.18) |
| CBZ vs TPM | 2 | 925 | 1.05 (0.64 to 1.72) | 0% | 8.8% | 1.11 (0.96 to 1.28) |
| CBZ vs GBP | 2 | 943 | 0.81 (0.52 to 1.27) | 0% | 73.7% | 1.16 (0.99 to 1.35) |
| CBZ vs LEV | 3 | 1567 | 1.06 (0.84 to 1.33) | 37.9% | 20.4% | 1.04 (0.93 to 1.16) |
| CBZ vs ZNS | 1 | 582 | 1.00 (0.82 to 1.20) | NA | 100% | 1.00 (0.83 to 1.20) |
| PHB vs PHT | 4 | 465 | 0.79 (0.60 to 1.05) | 0% | 31.1% | 1.08 (0.85 to 1.37) |
| PHB vs VPS | 2 | 80 | 0.67 (0.42 to 1.08) | 0% | 9.1% | 1.15 (0.89 to 1.49) |
| PHB vs LTG | No direct evidence | 0% | 1.16 (0.90 to 1.52) | |||
| PHB vs OXC | No direct evidence | 0% | 1.03 (0.77 to 1.39) | |||
| PHB vs TPM | No direct evidence | 0% | 1.16 (0.89 to 1.54) | |||
| PHB vs GBP | No direct evidence | 0% | 1.22 (0.93 to 1.59) | |||
| PHB vs LEV | No direct evidence | 0% | 1.10 (0.85 to 1.41) | |||
| PHB vs ZNS | No direct evidence | 0% | 1.04 (0.78 to 1.41) | |||
| PHT vs VPS | 5 | 245 | 0.90 (0.70 to 1.15) | 0% | 26.5% | 1.06 (0.88 to 1.30) |
| PHT vs LTG | 1 | 90 | 0.88 (0.25 to 3.03) | NA | 1.20% | 1.09 (0.88 to 1.32) |
| PHT vs OXC | 2 | 318 | 1.21 (0.79 to 1.87) | 0% | 33.2% | 0.95 (0.75 to 1.22) |
| PHT vs TPM | No direct evidence | 0% | 1.09 (0.88 to 1.33) | |||
| PHT vs GBP | No direct evidence | 0% | 1.12 (0.91 to 1.39) | |||
| PHT vs LEV | No direct evidence | 0% | 1.02 (0.84 to 1.22) | |||
| PHT vs ZNS | No direct evidence | 0% | 0.97 (0.76 to 1.23) | |||
| VPS vs LTG | 3 | 221 | 1.22 (0.97 to 1.52) | 0% | 32.1% | 1.02 (0.85 to 1.22) |
| VPS vs OXC | No direct evidence | 0% | 0.90 (0.72 to 1.12) | |||
| VPS vs TPM | 2 | 111 | 1.08 (0.87 to 1.34) | 0% | 61.7% | 1.02 (0.83 to 1.23) |
| VPS vs GBP | No direct evidence | 0% | 1.05 (0.87 to 1.28) | |||
| VPS vs LEV | 1 | 190 | 1.09 (0.88 to 1.33) | NA | 40.5% | 0.95 (0.79 to 1.14) |
| VPS vs ZNS | No direct evidence | 0% | 0.91 (0.72 to 1.14) | |||
| LTG vs OXC | 1 | 499 | 1.08 (0.27 to 4.32) | NA | 2.4% | 0.88 (0.73 to 1.08) |
| LTG vs TPM | 1 | 636 | 0.89 (0.70 to 1.13) | NA | 1.7% | 1.00 (0.85 to 1.18) |
| LTG vs GBP | 1 | 647 | 1.46 (0.16 to 13.0) | NA | 1.6% | 1.04 (0.88 to 1.22) |
| LTG vs LEV | 1 | 240 | 0.83 (0.59 to 1.17) | NA | 17.8% | 0.93 (0.80 to 1.10) |
| LTG vs ZNS | No direct evidence | 0% | 0.89 (0.71 to 1.12) | |||
| OXC vs TPM | 1 | 487 | 0.86 (0.26 to 2.86) | NA | 3.3% | 1.14 (0.93 to 1.37) |
| OXC vs GBP | 1 | 498 | 1.35 (0.15 to 12.1) | NA | 2.1% | 1.18 (0.96 to 1.43) |
| OXC vs LEV | No direct evidence | 0% | 1.06 (0.86 to 1.32) | |||
| OXC vs ZNS | No direct evidence | 0% | 1.01 (0.78 to 1.32) | |||
| TPM vs GBP | 1 | 635 | 1.56 (0.2 to 12.5) | NA | 1.6% | 1.04 (0.88 to 1.23) |
| TPM vs LEV | No direct evidence | 0% | 0.93 (0.79 to 1.12) | |||
| TPM vs ZNS | No direct evidence | 0% | 0.89 (0.70 to 1.14) | |||
| GBP vs LEV | No direct evidence | 0% | 0.90 (0.75 to 1.09) | |||
| GBP vs ZNS | No direct evidence | 0% | 0.86 (0.68 to 1.10) | |||
| LEV vs ZNS | No direct evidence | 0% | 0.95 (0.77 to 1.19) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first).
bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details.
cNote that HR < 1 indicates an advantage to the second drug in the comparison; results in highlighted in bold are statistically significant.
dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence.
eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number | Number | HR (95% CI)b,c | I² statisticd | Direct | HR (95% CI)b,c | |
| CBZ vs PHB | 3 | 158 | 0.56 (0.33 to 0.96) | 13.2% | 28.2% | 1.28 (0.92 to 1.79) |
| CBZ vs PHT | 2 | 121 | 1.44 (0.82 to 2.55) | 31.4% | 13% | 0.87 (0.68 to 1.10) |
| CBZ vs VPS | 4 | 412 | 1.11 (0.81 to 1.53) | 29.9% | 30.7% | 0.95 (0.84 to 1.09) |
| CBZ vs LTG | 5 | 254 | 0.58 (0.25 to 1.32) | 0% | 0.1% | 1.20 (0.99 to 1.49) |
| CBZ vs OXC | 1 | 9 | 0.79 (0.17 to 3.56) | NA | 4.6% | 1.30 (0.42 to 4.00) |
| CBZ vs TPM | 2 | 101 | 1.00 (0.55 to 1.79) | 0% | 32.8% | 1.11 (0.78 to 1.59) |
| CBZ vs GBP | 1 | 6 | 0.71 (0.07 to 6.90) | NA | 10% | 1.75 (0.23 to 12.5) |
| CBZ vs LEV | 2 | 251 | 1.00 (0.72 to 1.37) | 57.9% | 26.7% | 1.14 (0.85 to 1.52) |
| PHB vs PHT | 3 | 130 | 1.31 (0.67 to 2.53) | 0% | 22.7% | 0.68 (0.47 to 0.98) |
| PHB vs VPS | 2 | 98 | 1.50 (0.72 to 3.11) | 7.5% | 15.3% | 0.75 (0.53 to 1.05) |
| PHB vs LTG | No direct evidence | 0% | 0.94 (0.64 to 1.39) | |||
| PHB vs OXC | No direct evidence | 0% | 1.01 (0.31 to 3.23) | |||
| PHB vs TPM | No direct evidence | 0% | 0.87 (0.53 to 1.41) | |||
| PHB vs GBP | No direct evidence | 0% | 1.37 (0.17 to 11.1) | |||
| PHB vs LEV | No direct evidence | 0% | 0.88 (0.57 to 1.37) | |||
| PHT vs VPS | 4 | 394 | 1.03 (0.68 to 1.54) | 0% | 36.8% | 1.10 (0.85 to 1.43) |
| PHT vs LTG | 1 | 91 | 1.96 (0.37 to 10.2) | NA | 4.4% | 1.39 (1.03 to 1.89) |
| PHT vs OXC | 2 | 154 | 0.71 (0.42 to 1.21) | 0% | 45.1% | 1.49 (0.48 to 4.76) |
| PHT vs TPM | No direct evidence | 0% | 1.28 (0.84 to 1.96) | |||
| PHT vs GBP | No direct evidence | 0% | 2.00 (0.26 to 16.7) | |||
| PHT vs LEV | No direct evidence | 0% | 1.32 (0.89 to 1.92) | |||
| VPS vs LTG | 3 | 387 | 0.84 (0.48 to 1.47) | 0% | 43.5% | 1.27 (1.03 to 1.56) |
| VPS vs OXC | No direct evidence | 0% | 1.35 (0.44 to 4.17) | |||
| VPS vs TPM | 2 | 441 | 0.67 (0.38 to 1.19) | 58.7% | 12.9% | 1.16 (0.81 to 1.67) |
| VPS vs GBP | No direct evidence | 0% | 1.82 (0.24 to 14.3) | |||
| VPS vs LEV | 1 | 512 | 0.88 (0.60 to 1.30) | NA | 48.6% | 1.19 (0.86 to 1.64) |
| LTG vs OXC | 1 | 10 | 0.80 (0.20 to 3.26) | NA | 7.6% | 1.08 (0.35 to 3.33) |
| LTG vs TPM | 1 | 14 | 0.59 (0.30 to 1.16) | NA | 10% | 0.92 (0.62 to 1.37) |
| LTG vs GBP | 1 | 7 | 0.73 (0.08 to 6.57) | NA | 11% | 1.45 (0.19 to 11.1) |
| LTG vs LEV | No direct evidence | 0% | 0.93 (0.65 to 1.33) | |||
| OXC vs TPM | 1 | 14 | 1.34 (0.40 to 4.54) | NA | 9.4% | 0.86 (0.28 to 2.63) |
| OXC vs GBP | 1 | 7 | 0.91 (0.10 to 8.20) | NA | 10.7% | 1.35 (0.15 to 12.5) |
| OXC vs LEV | No direct evidence | 0% | 0.88 (0.27 to 2.78) | |||
| TPM vs GBP | 1 | 11 | 0.68 (0.08 to 5.45) | NA | 13.9% | 1.56 (0.21 to 12.5) |
| TPM vs LEV | No direct evidence | 0% | 1.02 (0.65 to 1.61) | |||
| GBP vs LEV | No direct evidence | 0% | 0.65 (0.08 to 5.00) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first).
bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details.
cNote that HR < 1 indicates an advantage to the second drug in the comparison; results highlighted in bold are statistically significant.
dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence.
eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number | Number | HR (95% CI)b,c | I² statisticd | Direct | HR (95% CI)b,c | |
| CBZ vs PHB | 6 | 581 | 0.99 (0.78 to 1.26) | 54.3% | 21% | 0.79 (0.64 to 0.97) |
| CBZ vs PHT | 4 | 432 | 0.91 (0.72 to 1.16) | 16.1% | 27.1% | 0.98 (0.85 to 1.13) |
| CBZ vs VPS | 5 | 813 | 1.01 (0.86 to 1.19) | 32% | 34.6% | 1.20 (1.06 to 1.37) |
| CBZ vs LTG | 9 | 2252 | 0.98 (0.75 to 1.27) | 0% | 40.7% | 1.29 (1.17 to 1.42) |
| CBZ vs OXC | 2 | 555 | 1.47 (0.57 to 3.81) | 57.3% | 4.8% | 1.09 (0.89 to 1.32) |
| CBZ vs TPM | 2 | 925 | 1.03 (0.51 to 2.08) | 69.3% | 1.5% | 1.12 (0.97 to 1.29) |
| CBZ vs GBP | 2 | 943 | 1.64 (1.14 to 2.36) | 17.7% | 49% | 1.44 (1.25 to 1.66) |
| CBZ vs LEV | 3 | 1552 | 1.18 (0.85 to 1.65) | 0% | 26.2% | 1.14 (0.99 to 1.30) |
| CBZ vs ZNS | 1 | 581 | 1.30 (0.97 to 1.73) | NA | 100% | 1.30 (0.97 to 1.73) |
| PHB vs PHT | 5 | 463 | 1.07 (0.83 to 1.37) | 27.7% | 33.6% | 1.24 (0.99 to 1.56) |
| PHB vs VPS* | 2 | 80 | 0.71 (0.43 to 1.17) | 9.1% | 12.8% | 1.53 (1.20 to 1.94) |
| PHB vs LTG | No direct evidence | 0% | 1.63 (1.30 to 2.06) | |||
| PHB vs OXC | No direct evidence | 0% | 1.38 (1.04 to 1.83) | |||
| PHB vs TPM | No direct evidence | 0% | 1.42 (1.11 to 1.83) | |||
| PHB vs GBP | No direct evidence | 0% | 1.83 (1.42 to 2.35) | |||
| PHB vs LEV | No direct evidence | 0% | 1.44 (1.12 to 1.85) | |||
| PHB vs ZNS | No direct evidence | 0% | 1.64 (1.15 to 2.35) | |||
| PHT vs VPS | 5 | 245 | 0.96 (0.72 to 1.29) | 0% | 25.4% | 1.23 (1.02 to 1.48) |
| PHT vs LTG | 1 | 90 | 0.77 (0.38 to 1.54) | NA | 6% | 1.31 (1.10 to 1.57) |
| PHT vs OXC | 2 | 318 | 1.46 (0.88 to 2.44) | 23.9% | 36.1% | 1.11 (0.87 to 1.41) |
| PHT vs TPM | 1 | 53 | 2.32 (0.95 to 5.70) | NA | 4% | 1.14 (0.93 to 1.40) |
| PHT vs GBP | No direct evidence | 0% | 1.47 (1.20 to 1.80) | |||
| PHT vs LEV | No direct evidence | 0% | 1.16 (0.95 to 1.41) | |||
| PHT vs ZNS | No direct evidence | 0% | 1.32 (0.96 to 1.82) | |||
| VPS vs LTG | 3 | 215 | 1.57 (1.23 to 2.00) | 39.4% | 10% | 1.07 (0.92 to 1.24) |
| VPS vs OXC | No direct evidence | 0% | 0.90 (0.72 to 1.14) | |||
| VPS vs TPM | 2 | 111 | 1.18 (0.93 to 1.50) | 0% | 70.2% | 0.93 (0.77 to 1.13) |
| VPS vs GBP | No direct evidence | 0% | 1.20 (0.99 to 1.44) | |||
| VPS vs LEV | 1 | 190 | 1.27 (0.94 to 1.72) | NA | 31% | 0.94 (0.77 to 1.15) |
| VPS vs ZNS | No direct evidence | 0% | 1.08 (0.78 to 1.48) | |||
| LTG vs OXC | 1 | 499 | 0.87 (0.23 to 3.25) | NA | 5.5% | 0.84 (0.69 to 1.03) |
| LTG vs TPM | 1 | 636 | 0.73 (0.57 to 0.93) | NA | 2.3% | 0.87 (0.75 to 1.01) |
| LTG vs GBP | 1 | 647 | 0.63 (0.07 to 5.42) | NA | 4.4% | 1.12 (0.96 to 1.30) |
| LTG vs LEV | 1 | 229 | 0.84 (0.53 to 1.35) | NA | 15.9% | 0.88 (0.75 to 1.04) |
| LTG vs ZNS | No direct evidence | 0% | 1.01 (0.74 to 1.36) | |||
| OXC vs TPM | 1 | 487 | 0.55 (0.15 to 2.06) | NA | 5.4% | 1.03 (0.84 to 1.27) |
| OXC vs GBP | 1 | 498 | 0.73 (0.08 to 6.49) | NA | 4.6% | 1.32 (1.08 to 1.63) |
| OXC vs LEV | No direct evidence | 0% | 1.05 (0.83 to 1.32) | |||
| OXC vs ZNS | No direct evidence | 0% | 1.19 (0.84 to 1.69) | |||
| TPM vs GBP | 1 | 635 | 1.31 (0.15 to 11.2) | NA | 3.5% | 1.28 (1.09 to 1.51) |
| TPM vs LEV | No direct evidence | 0% | 1.01 (0.83 to 1.23) | |||
| TPM vs ZNS | No direct evidence | 0% | 1.15 (0.84 to 1.59) | |||
| GBP vs LEV | No direct evidence | 0% | 0.79 (0.65 to 0.96) | |||
| GBP vs ZNS | No direct evidence | 0% | 0.90 (0.65 to 1.24) | |||
| LEV vs ZNS | No direct evidence | 0% | 1.14 (0.83 to 1.57) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; zNS: Zonisamide
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first).
bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details.
cNote that HR < 1 indicates an advantage to the second drug in the comparison; results highlighted in bold are statistically significant
dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence.
eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
For comparisons marked with a *, confidence intervals of direct evidence and network meta‐analysis do not overlap indicating that inconsistency may be present in the results.
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number | Number | HR (95% CI)2,3 | I² statistic4 | Direct | HR (95% CI)2,3 | |
| CBZ vs PHB | 5 | 237 | 0.55 (0.33 to 0.92) | 50.4% | 35.5% | 1.10 (0.80 to 1.51) |
| CBZ vs PHT | 3 | 150 | 0.88 (0.51 to 1.54) | 0% | 26.6% | 0.76 (0.59 to 0.98) |
| CBZ vs VPS | 4 | 411 | 1.37 (0.98 to 1.92) | 84.1% | 10.4% | 0.88 (0.76 to 1.03) |
| CBZ vs LTG | 7 | 302 | 1.49 (0.94 to 2.35) | 0% | 0.3% | 0.98 (0.70 to 1.37) |
| CBZ vs OXC | 1 | 9 | 1.55 (0.38 to 6.31) | NA | 9% | 1.09 (0.36 to 3.36) |
| CBZ vs TPM | 2 | 101 | 1.19 (0.56 to 2.50) | 62% | 9% | 1.15 (0.89 to 1.48) |
| CBZ vs GBP | 1 | 6 | 2.83 (0.31 to 25.5) | NA | 10.7% | 0.79 (0.10 to 6.08) |
| CBZ vs LEV | 2 | 251 | 1.04 (0.65 to 1.64) | 0% | 44.9% | 1.19 (0.78 to 1.83) |
| PHB vs PHT | 4 | 161 | 1.41 (0.76 to 2.62) | 46.9% | 20.3% | 0.69 (0.48 to 1.00) |
| PHB vs VPS | 2 | 98 | 1.87 (0.87 to 4.00) | 69.8% | 6.5% | 0.80 (0.57 to 1.12) |
| PHB vs LTG | No direct evidence | 0% | 0.89 (0.56 to 1.42) | |||
| PHB vs OXC | No direct evidence | 0% | 1.00 (0.31 to 3.20) | |||
| PHB vs TPM | No direct evidence | 0% | 1.05 (0.70 to 1.56) | |||
| PHB vs GBP | No direct evidence | 0% | 0.72 (0.09 to 5.68) | |||
| PHB vs LEV | No direct evidence | 0% | 1.09 (0.64 to 1.85) | |||
| PHT vs VPS | 4 | 394 | 1.11 (0.71 to 1.74) | 0% | 36.4% | 1.16 (0.88 to 1.53) |
| PHT vs LTG | 1 | 91 | 1.00 (0.40 to 2.46) | NA | 16.2% | 1.29 (0.85 to 1.97) |
| PHT vs OXC | 2 | 154 | 0.60 (0.33 to 1.10) | 49.7% | 25.2% | 1.44 (0.46 to 4.56) |
| PHT vs TPM | 1 | 150 | 0.63 (0.18 to 2.26) | NA | 9.8% | 1.51 (1.06 to 2.15) |
| PHT vs GBP | No direct evidence | 0% | 1.05 (0.13 to 8.14) | |||
| PHT vs LEV | No direct evidence | 0% | 1.57 (0.96 to 2.58) | |||
| VPS vs LTG | 3 | 377 | 0.64 (0.37 to 1.11) | 23.2% | 31.3% | 1.11 (0.77 to 1.60) |
| VPS vs OXC | No direct evidence | 0% | 1.24 (0.40 to 3.84) | |||
| VPS vs TPM* | 2 | 441 | 0.42 (0.23 to 0.80) | 46.4% | 21% | 1.30 (1.01 to 1.68) |
| VPS vs GBP | No direct evidence | 0% | 0.90 (0.12 to 6.92) | |||
| VPS vs LEV | 1 | 512 | 0.82 (0.48 to 1.40) | NA | 34% | 1.35 (0.86 to 2.13) |
| LTG vs OXC | 1 | 10 | 0.94 (0.25 to 3.57) | NA | 12.2% | 1.12 (0.36 to 3.48) |
| LTG vs TPM | 1 | 14 | 0.61 (0.28 to 1.30) | NA | 13.1% | 1.17 (0.78 to 1.77) |
| LTG vs GBP | 1 | 7 | 1.72 (0.20 to 14.9) | NA | 11.9% | 0.81 (0.11 to 6.25) |
| LTG vs LEV | No direct evidence | 0% | 1.22 (0.71 to 2.10) | |||
| OXC vs TPM | 1 | 14 | 1.90 (0.50 to 7.19) | NA | 13.6% | 1.05 (0.34 to 3.24) |
| OXC vs GBP | 1 | 7 | 1.83 (0.20 to 16.5) | NA | 13.3% | 0.73 (0.08 to 6.49) |
| OXC vs LEV | No direct evidence | 0% | 1.09 (0.33 to 3.62) | |||
| TPM vs GBP | 1 | 11 | 0.96 (0.11 to 8.29) | NA | 13.2% | 0.69 (0.09 to 5.32) |
| TPM vs LEV | No direct evidence | 0% | 1.04 (0.63 to 1.71) | |||
| GBP vs LEV | No direct evidence | 0% | 1.50 (0.19 to 12.0) | |||
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity
aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first).
bHRs and 95% CIs are calculated from fixed‐effect analyses (pairwise and network meta‐analysis); where substantial heterogeneity was present (I2 > 50%), random‐effects meta‐analysis was also conducted, see Effects of interventions for further details.
cNote that HR < 1 indicates an advantage to the second drug in the comparison; results in highlighted in bold are statistically significant.
dNA ‐ heterogeneity is not applicable as only one study contributed direct evidence.
eDirect evidence (%) ‐ proportion of the estimate contributed by direct evidence.
For comparisons marked with a *, confidence intervals of direct evidence and network meta‐analysis do not overlap indicating that inconsistency may be present in the results
All tables and figures of results indicate the proportion of the treatment effect estimate that is contributed by direct evidence (ranging from 0% where no direct comparison exists to 100% for the carbamazepine vs zonisamide comparison, which is disconnected from the rest of the network ‐ see Figure 1). We note that due to the limited amount of evidence for individuals with generalised seizures for some comparisons in the network; some confidence intervals of treatment effect sizes are very wide.
We investigated inconsistency of the direct and network meta‐analysis estimates via node splitting (Dias 2010) and via ‘design‐by treatment’ inconsistency models (Higgins 2012) – see Data synthesis for further detail. Figure 12; Figure 13; Figure 14; Figure 15; Figure 16 and Figure 17 display investigations of inconsistency graphically. Figures show direct evidence, indirect evidence and network meta‐analysis results (direct plus indirect evidence) for all treatments compared to first‐line treatments carbamazepine and lamotrigine for individuals with partial seizures and sodium valproate for individuals with generalised seizures. Numerical results from investigations of inconsistency for all pairwise comparisons are available from the corresponding author on request.

CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with partial seizures compared to carbamazepine (CBZ) for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.

CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with partial seizures compared to lamotrigine (LTG) for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.

CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: Direct, Indirect and Network estimates for individuals with generalised seizures compared to sodium valproate (VPS) for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.

CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with partial seizures compared to carbamazepine (CBZ) for time to six‐month remission and time to first seizure.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
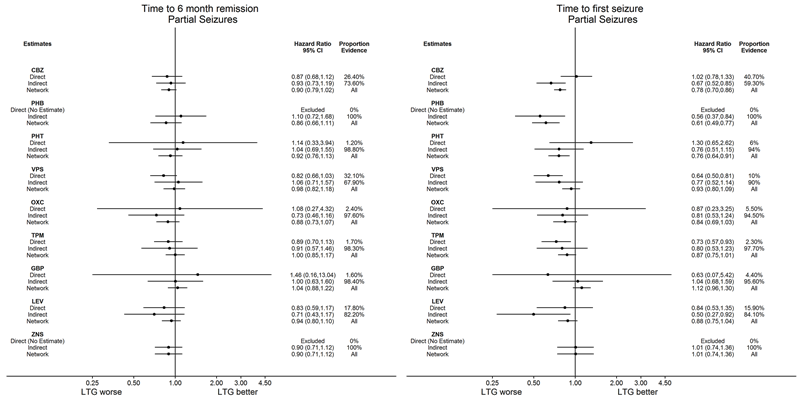
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with partial seizures compared to lamotrigine (LTG) for time to six‐month remission and time to first seizure.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.

CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with generalised seizures compared to sodium valproate (VPS) for time to six‐month remission and time to first seizure.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
We note for the interpretation of these plots that direct evidence comes from the trials that compared the drugs head‐to‐head, indirect evidence comes from the trials that did not compare the drugs head‐to‐head, and direct plus indirect evidence comes from the whole network (head‐to‐head comparisons and indirect comparisons for all drugs).
We examined the numerical results, particularly overlap of confidence intervals of the direct evidence, indirect evidence and network meta‐analysis results. We anticipate that numerical results for the network meta‐analysis will be the most precise. We note potentially important clinical inconsistency to be present where confidence intervals of results from direct evidence and direct plus indirect evidence do not overlap and we consider possible reasons and origins of this inconsistency. Our main concern is statistically significant differences between direct evidence and network meta‐analysis results; however we also note where confidence intervals of results from indirect evidence do not overlap with the confidence intervals of the other estimates.
We conducted a number of sensitivity analyses for each outcome (see Sensitivity analysis for further information). For brevity, we only summarise the conclusions of the sensitivity analyses below rather than presenting full numerical results but these can be made available on request from the corresponding review author.
Time to withdrawal of allocated treatment
The number of participants that contributed to analysis of our primary outcome was 11,865 out of 12,391 participants (96%).
Table 7 shows the reported reasons for withdrawal from treatment across all studies and how we treated each of these reasons in analysis. We note that in some trials, participants many have withdrawn from treatment for a combination of reasons; for the purpose of analysis we have made a judgement regarding the primary reason for withdrawal.
Out of the 11,865 participants who contributed data, 4058 (34%) of individuals prematurely withdrew; fewest participants withdrew from levetiracetam (27%) and sodium valproate (28%) and the most participants withdrew from gabapentin (47%) and phenobarbitone (38%).
The most commonly reported reason for withdrawal from treatment was due to adverse events (38% of all withdrawal 'events'); fewest participants withdrew from gabapentin (20%) and phenobarbitone (20%) due to adverse events and the most participants withdrew from carbamazepine (45%) and topiramate (48%) due to adverse events. Inadequate response (i.e. lack of seizure control) was reported as the reason for withdrawal for 27% of participants ranging from 16% of participants on phenobarbitone to 62% of participants on gabapentin.
We censored 7704 participants out of 11,865 (66%) in the analysis. The majority of censored participants were still taking their allocated treatment at last follow‐up; ranging by drug from 73% (phenobarbitone) to 95% (levetiracetam) of censored participants. Very few participants were lost to follow‐up in the trials (ranging from 0% (gabapentin and zonisamide) to 16% (phenobarbitone)).
For 103 participants, reason for withdrawal was missing (ranging by drug from 0 participants (levetiracetam and zonisamide) to 26 participants (sodium valproate)). We treated those with missing reason for withdrawal as censored in analysis and performed a sensitivity analysis treating these individuals as having withdrawal 'events.' Results of sensitivity analysis were practically identical and conclusions unchanged so we present the results treating these individuals as censored.
We also note that information reported in Table 7 does not take account of randomisation within trials and should be interpreted as exploratory.
Direct evidence
Table 8 (individuals with partial seizures) and Table 9 (individuals with generalised seizures) show the number of trials and participants contributing direct evidence for each of the pairwise comparisons in the network. Twenty out of 45 comparisons had no direct evidence for individuals with partial seizures. Thirteen out of 36 comparisons had no direct evidence for individuals with generalised seizures and eight comparisons for individuals with generalised seizures had fewer than 20 individuals contributing direct evidence resulting in wide confidence intervals around the treatment effect estimate for these comparisons.
The comparisons with the most participants contributing to analysis were carbamazepine vs lamotrigine and carbamazepine vs levetiracetam for individuals with partial seizures and sodium valproate vs levetiracetam and sodium valproate vs topiramate for individuals with generalised seizures.
Table 8 and Table 9 also show estimates for heterogeneity in the direct treatment effects. No substantial heterogeneity was present (I2 greater than 50%) for any comparison for individuals with generalised seizures.
For three comparisons for individuals with partial seizures, substantial heterogeneity was present (I2 greater than 50%). The heterogeneity in these comparisons seemed to originate from difference in trial designs contributing to the pooled result; that is, pooling of trials recruiting children only, adults only or elderly participants only and pooling of double‐blind and open‐label trials (see Nolan 2016b for further discussion of the importance of blinding to the outcome of time‐to‐treatment withdrawal). Repeating analysis with random‐effects did not change conclusions for two of the comparisons (carbamazepine vs phenytoin and phenytoin vs sodium valproate); but for one comparison (phenobarbitone vs phenytoin), when repeating analysis with random‐effects there was no longer a statistically significant advantage to phenytoin: HR 0.42 (0.16 to 1.06)
Network meta‐analysis results (direct plus indirect evidence)
Figure 5 shows how each treatment performed compared to first‐line treatment carbamazepine for individuals with partial seizures (ordered by treatment effect estimate); lamotrigine and levetiracetam are significantly better than carbamazepine, and carbamazepine is significantly better than gabapentin and phenobarbitone.
Figure 6 shows how each treatment performs compared to first‐line treatment lamotrigine for individuals with partial seizures (ordered by treatment effect estimate); lamotrigine is significantly better than all treatments except for levetiracetam.
Figure 7 shows how each treatment performs compared to first‐line treatment sodium valproate for individuals with generalised seizures (ordered by treatment effect estimate); sodium valproate is significantly better than carbamazepine, topiramate and phenobarbitone.
Table 8 and Figure 8 (individuals with partial seizures) and Table 9 and Figure 9 (individuals with generalised seizures) show treatment effect estimates for all pairwise comparisons in the network combining direct with indirect evidence.
In addition to the results described above; for individuals with partial seizures, levetiracetam seems to perform better than most other drugs and for individuals with generalised seizures, lamotrigine seems to perform better than most other drugs. For both individuals with partial seizures and individuals with generalised seizures, phenobarbitone seems to perform worse than most other drugs.
As described further in Assessment of heterogeneity, we could not directly calculate an I2 statistic for the network meta‐analysis but the estimated I2 statistic was 11.7%. When repeating network meta‐analysis with random‐effects the Tau2 statistic was 0.0037, numerical results for treatment effects were very similar (the same to one or two decimal places) and conclusions remained unchanged.
Investigation of inconsistency (node‐splitting)
We fitted the ‘design‐by‐treatment’ inconsistency model to 17 variables and regressed it on 23 designs, five of which were multi‐arm trials (up to five treatment arms). Accounting for the multi‐arm trials, this resulted in an overall test for inconsistency with 36 degrees of freedom, which was not significant (Chi2 statistic (36) = 45.6, P value = 0.1312, heterogeneity (Tau) = 5.65 x 10‐10). Furthermore, there was no significant evidence of inconsistency within any of the 23 designs.
Table 8 (individuals with partial seizures) and Table 9 (individuals with generalised seizures) show treatment effect estimates from direct evidence and from direct plus indirect evidence, Figure 12 and Figure 13 show treatment effect estimates for direct, indirect, and direct plus indirect evidence for individuals with partial seizures compared to carbamazepine and lamotrigine respectively and Figure 14 for individuals with generalised seizures compared to sodium valproate.
We note that for most pairwise comparisons, numerical results of direct evidence and network meta‐analysis are similar, mostly in the same direction and confidence intervals of estimates overlap. For all pairwise comparisons, results from network meta‐analysis are more precise than results from direct evidence (in some cases much more precise where limited direct evidence exists, for example see carbamazepine compared to oxcarbazepine, Figure 12).
For the following comparisons, conclusions drawn from direct evidence and from network meta‐analysis are different (see Table 8 and Table 9).
-
Direct evidence shows a significant advantage to one of the drugs and the network meta‐analysis results show no significant difference between the drugs: sodium valproate vs topiramate (partial seizures).
-
Direct evidence shows no significant difference between the drugs and network meta‐analysis shows a significant advantage for one of the drugs: carbamazepine vs gabapentin, lamotrigine vs oxcarbazepine, lamotrigine vs topiramate, lamotrigine vs gabapentin (all partial seizures); carbamazepine vs sodium valproate, carbamazepine vs lamotrigine, phenobarbitone vs sodium valproate, sodium valproate vs topiramate, lamotrigine vs topiramate (all generalised seizures).
-
No direct evidence exists between the drugs while network meta‐analysis shows a significant advantage for one of the drugs: phenobarbitone vs lamotrigine, phenobarbitone vs levetiracetam, lamotrigine vs zonisamide, topiramate vs levetiracetam, gabapentin vs levetiracetam (all partial seizures); phenobarbitone vs lamotrigine (generalised seizures).
For the following comparisons, confidence intervals for the results from indirect evidence do not overlap with:
-
direct evidence: carbamazepine vs phenytoin (generalised seizures), phenobarbitone vs phenytoin (generalised seizures);
-
network meta‐analysis results: lamotrigine vs phenytoin (partial seizures), carbamazepine vs phenytoin (generalised seizures), lamotrigine vs phenytoin (generalised seizures).
For the following comparisons, confidence intervals for the results from direct evidence and from network meta‐analysis do not overlap which indicates potential inconsistency is present (see Table 7, Table 8, Figure 12; Figure 13 and Figure 14): sodium valproate vs lamotrigine (partial seizures), sodium valproate vs topiramate (generalised seizures).
For the comparison of sodium valproate vs lamotrigine for individuals with partial seizures, from direct evidence only, there is a statistically significant advantage to sodium valproate (HR 1.40 (1.00 to 1.96), however from the network meta‐analysis results, the direction of effect changes to a statistically significant advantage to lamotrigine (HR 0.72 (0.58 to 0.90)). However, for this comparison, only 5.1% of the network estimate is contributed from direct evidence and a moderate amount of heterogeneity is present in this estimate (I2 = 45%), likely due to variability in the trial design of the three trials contributing to this estimate (for example, one trial (SANAD B 2007) was designed to only recruit individuals with generalised or unclassified seizures but did recruit a small number of individuals with partial seizures who contribute to this outcome).
For the comparison of sodium valproate vs topiramate for individuals with generalised seizures, from direct evidence, there is no significant difference between the drugs (HR 0.53 (0.27 to 1.07)), however from the network meta‐analysis results, a statistically significant advantage is shown for sodium valproate (HR 1.76 (1.22 to 2.53)). As above, for this comparison, only 22.4% of the network estimate is contributed from direct evidence and a moderate amount of heterogeneity is present in this estimate (I2 = 48.5%). Again, this heterogeneity is likely due to difference in trial design of the two trials contributing direct evidence (see characteristics of Privitera 2003 for details of stratification).
Furthermore, the 'design‐by treatment' inconsistency model does not show any significant evidence of inconsistency within the network. Therefore, we are not concerned about any impact of this observed inconsistency of numerical results on the conclusions of the review.
Subgroup and sensitivity analysis
See Sensitivity analysis for full details and rationale of all sensitivity analyses conducted.
We performed an additional analysis adjusted for age (as well as epilepsy type ‐ see Subgroup analysis and investigation of heterogeneity). Numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We were able to incorporate aggregate or extracted individual‐level data for 471 participants for four additional trials (Biton 2001; Gilad 2007; Steinhoff 2005; Shakir 1981). Numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We performed two sensitivity analyses to investigate the possibility of generalised seizures being misclassified; in the first analysis we reclassified those with generalised seizures and age of onset greater than 30 years as having partial onset seizures and in the second analysis we reclassified generalised seizure types and age at onset greater than 30 years and those with missing seizure type into an 'unclassified seizure type' group.
For the first analysis; numerical results for individuals with generalised seizures were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions. However, for individuals with partial seizures, most numerical results were similar but the most notable change was that phenytoin was now significantly better than all other treatments.
There was a large amount of heterogeneity present in this sensitivity analysis; the estimated I2 statistic was 98% and when repeating network meta‐analysis with random‐effects, Tau2 was 7.074 and confidence intervals of all treatment effect estimates were very wide so that no significant differences were present between any effect sizes. We are unsure why this sensitivity analysis has introduced a large amount of heterogeneity into analysis for this outcome but not for the other outcomes (as described below). Due to this uncertainty, we do not encourage interpretation of this sensitivity analysis.
For the second analysis of seizure type classification, numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We assessed the validity of the proportional hazards assumption of the Cox model used in the network meta‐analysis (see Data synthesis for further details); numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
We excluded one trial (Stephen 2007) from all analyses due to inconsistencies in provided data. Numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
Another trial (Reunanen 1996) was excluded from analysis due to the definition of withdrawal from allocated treatment. Again, numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
For one trial (Placencia 1993), we performed an additional analysis with different definitions of withdrawal from allocated treatment. Again, numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
Time to achieve 12‐month seizure‐free period (remission) after randomisation
The number of participants that contributed to analysis of our secondary outcome, 'Time to achieve 12‐month seizure‐free period' was 9461 out of 12,391 participants (76%).
Direct evidence
Table 10 (individuals with partial seizures) and Table 11 (individuals with generalised seizures) show the number of trials and participants contributing direct evidence for each of the pairwise comparisons in the network. Twenty‐two out of 45 comparisons had no direct evidence for individuals with partial seizures. Fifteen out of 36 comparisons had no direct evidence for individuals with generalised seizures and nine comparisons for individuals with generalised seizures had fewer than 20 individuals contributing direct evidence resulting in wide confidence intervals around the treatment effect estimate for these comparisons.
The comparisons with the most participants contributing to analysis were carbamazepine vs levetiracetam and carbamazepine vs topiramate for individuals with partial seizures and sodium valproate vs levetiracetam and sodium valproate vs topiramate for individuals with generalised seizures.
Table 10 and Table 11 also show estimates of heterogeneity in the direct treatment effects. For three comparisons for individuals with partial seizures and for four comparisons for individuals with generalised seizures, substantial heterogeneity was present (I2 greater than 50%).
The heterogeneity in these comparisons seemed to originate from differences in trial designs contributing to the pooled result; that is, pooling of trials recruiting children only, adults only or elderly participants only and pooling trials with or without treatment strata (see Data extraction and management for further details). None of the treatment effects with substantial heterogeneity present were statistically significant so conclusions would not change for these treatment effects if random‐effects were applied.
Network meta‐analysis results (direct plus indirect evidence)
Figure 5 shows how each treatment performs compared to first‐line treatment carbamazepine for individuals with partial seizures (ordered by treatment effect estimate); carbamazepine is significantly better than levetiracetam.
Figure 6 shows how each treatment performs compared to first‐line treatment lamotrigine for individuals with partial seizures (ordered by treatment effect estimate); there is no significant difference between lamotrigine and the other treatments.
Figure 7 shows how each treatment performs compared to first‐line treatment sodium valproate for individuals with generalised seizures (ordered by treatment effect estimate); there is no significant difference between sodium valproate and the other treatments.
Table 10 and Figure 8 (individuals with partial seizures) and Table 11 and Figure 9 (individuals with generalised seizures) show treatment effect estimates for all pairwise comparisons in the network combining direct with indirect evidence. In addition to the results described above; there are few notable differences between any of the treatments for either individuals with partial seizures or individuals with generalised seizures.
As described further in Assessment of heterogeneity, we could not directly calculate an I2 statistic for the network meta‐analysis but the estimated I2 statistic was 17.3%. When repeating network meta‐analysis with random‐effects, the Tau2 statistic was 0.005, numerical results for treatment effects were very similar (the same to one or two decimal places) and conclusions remained unchanged.
Investigation of inconsistency (node‐splitting)
We fitted the ‘design‐by‐treatment’ inconsistency model was fitted to 17 variables and regressed it on 18 designs, five of which were multi‐arm trials (up to five treatment arms). Accounting for the multi‐arm trials, this resulted in an overall test for inconsistency with 29 degrees of freedom, which was not significant (Chi2 statistic (29) = 14.3, P value = 0.990, heterogeneity (Tau) = 0.154). Furthermore, there was no significant evidence of inconsistency within any of the 18 designs.
Table 10 (individuals with partial seizures) and Table 11 (individuals with generalised seizures) show treatment effect estimates from direct evidence, and from direct plus indirect evidence, Figure 12 and Figure 13 show treatment effect estimates for direct, indirect, and direct plus indirect evidence for individuals with partial seizures compared to carbamazepine and lamotrigine respectively and Figure 14 for individuals with generalised seizures compared to sodium valproate.
We note that for most pairwise comparisons, numerical results of direct evidence and network meta‐analysis are similar, mostly in the same direction and confidence intervals of estimates overlap. For all pairwise comparisons, results from network meta‐analysis are more precise than results from direct evidence (in some cases much more precise where limited direct evidence exists, for example see carbamazepine compared to gabapentin, Figure 12).
For the following comparisons, conclusions drawn from direct evidence and from network meta‐analysis are different (see Table 10 and Table 11).
-
Direct evidence shows a significant advantage to one of the drugs and the network meta‐analysis results show no significant difference between the drugs: carbamazepine vs phenobarbitone (for both partial seizures and generalised seizures).
-
Direct evidence shows no significant difference between the drugs and network meta‐analysis shows a significant advantage for one of the drugs: carbamazepine vs levetiracetam, sodium valproate vs lamotrigine (all partial seizures).
-
No direct evidence exists between the drugs while network meta‐analysis shows a significant advantage for one of the drugs: oxcarbazepine vs levetiracetam (partial seizures).
For the following comparisons, confidence intervals for the results from indirect evidence do not overlap with:
-
direct evidence: sodium valproate vs topiramate (generalised seizures);
-
network meta‐analysis results: none.
Confidence intervals overlap for the results from direct evidence and from network meta‐analysis for all comparisons, therefore there is no indication that inconsistency is present in the results (see Table 10, Table 11, Figure 12; Figure 13 and Figure 14).
Subgroup and sensitivity analysis
See Sensitivity analysis for full details and rationale of all sensitivity analyses conducted.
We performed an additional analysis adjusted for age (as well as epilepsy type ‐ see Subgroup analysis and investigation of heterogeneity). Numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
No trials reported aggregate or summary data for this outcome, therefore we did not perform any sensitivity analysis incorporating aggregate data.
We performed two sensitivity analyses to investigate the possibility of generalised seizures being misclassified; in the first analysis we reclassified those with generalised seizures and age of onset greater than 30 years as having partial onset seizures and in the second analysis we reclassified those with generalised seizure types and age at onset greater than 30 years, and those with missing seizure type into an 'unclassified seizure type' group. For both analyses, numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We assessed the validity of the proportional hazards assumption of the Cox model used in the network meta‐analysis (see Data synthesis for further details); there was no evidence the assumption was violated for any of the covariates in the network meta‐analysis, so we did not perform any sensitivity analysis.
We excluded one trial (Stephen 2007) from all analyses due to inconsistencies in provided data. Numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
Time to achieve six‐month seizure‐free period (remission) after randomisation
The number of participants that contributed to analysis of our secondary outcome, 'Time to achieve six‐month seizure‐free period' was 11,820 out of 12,391 participants (95%).
Direct evidence
Table 12 (individuals with partial seizures) and Table 13 (individuals with generalised seizures) show the number of trials and participants contributing direct evidence for each of the pairwise comparisons in the network. Twenty‐one out of 45 comparisons had no direct evidence for individuals with partial seizures. Fourteen out of 36 comparisons had no direct evidence for individuals with generalised seizures and eight comparisons for individuals with generalised seizures had fewer than 20 individuals contributing direct evidence resulting in wide confidence intervals around the treatment effect estimate for these comparisons.
The comparisons with the most participants contributing to analysis were carbamazepine vs levetiracetam and carbamazepine vs topiramate for individuals with partial seizures and sodium valproate vs levetiracetam and sodium valproate vs topiramate for individuals with generalised seizures.
Table 12 and Table 13 also show estimates of heterogeneity in the direct treatment effects. For one comparison for individuals with partial seizures and for two comparisons for individuals with generalised seizures, substantial heterogeneity was present (I2 greater than 50%).
The heterogeneity in these comparisons seemed to originate from differences in trial designs contributing to the pooled result; that is, pooling of trials recruiting children only, adults only or elderly participants only and pooling trials with or without treatment strata (see Data extraction and management for further details). None of the treatment effects with substantial heterogeneity present were statistically significant so conclusions would not change for these treatment effects if random‐effects were applied.
Network meta‐analysis results (direct plus indirect evidence)
Figure 5 shows how each treatment performs compared to first‐line treatment carbamazepine for individuals with partial seizures (ordered by treatment effect estimate); there is no significant difference between carbamazepine and the other treatments.
Figure 6 shows how each treatment performs compared to first‐line treatment lamotrigine for individuals with partial seizures (ordered by treatment effect estimate); there is no significant difference between lamotrigine and the other treatments.
Figure 7 shows how each treatment performs compared to first‐line treatment sodium valproate for individuals with generalised seizures (ordered by treatment effect estimate); sodium valproate is significantly better than lamotrigine.
Table 12 and Figure 10 (individuals with partial seizures) and Table 13 and Figure 11 (individuals with generalised seizures) show treatment effect estimates for all pairwise comparisons in the network combining direct with indirect evidence. In addition to the results described above; there are few notable differences between any of the treatments for either individuals with partial seizures or individuals with generalised seizures.
As described further in Assessment of heterogeneity, we could not directly calculate an I2 statistic for the network meta‐analysis but the estimated I2 statistic was 0%. When repeating network meta‐analysis with random‐effects, Tau2 was 7 x 10‐22. As no heterogeneity was present and Tau2 was negligible, numerical results for treatment effects and conclusions were identical.
Investigation of inconsistency (node‐splitting)
We fitted the ‘design‐by‐treatment’ inconsistency model to 17 variables and regressed it on 23 designs, five of which were multi‐arm trials (up to five treatment arms). Accounting for the multi‐arm trials, this resulted in an overall test for inconsistency with 37 degrees of freedom which was not significant (Chi2 statistic (37) = 36.2, P value = 0.508, heterogeneity (Tau) = 8.09 x 10‐12). Furthermore, there was no significant evidence of inconsistency within any of the 23 designs.
Table 12 (individuals with partial seizures) and Table 13 (individuals with generalised seizures) show treatment effect estimates from direct evidence, and from direct plus indirect evidence, Figure 15 and Figure 16 show treatment effect estimates for direct, indirect, and direct plus indirect evidence for individuals with partial seizures compared to carbamazepine and lamotrigine respectively and Figure 17 for individuals with generalised seizures compared to sodium valproate.
We note that for most pairwise comparisons, numerical results of direct evidence and network meta‐analysis are similar, mostly in the same direction and confidence intervals of estimates overlap. For all pairwise comparisons, results from network meta‐analysis are more precise than results from direct evidence (in some cases much more precise where limited direct evidence exists, for example see lamotrigine compared to gabapentin, Figure 16).
For the following comparisons, conclusions drawn from direct evidence and from network meta‐analysis are different (see Table 12 and Table 13).
-
Direct evidence shows a significant advantage to one of the drugs and the network meta‐analysis results show no significant difference between the drugs: carbamazepine vs phenobarbitone (generalised seizures).
-
Direct evidence shows no significant difference between the drugs and network meta‐analysis shows a significant advantage for one of the drugs: sodium valproate vs lamotrigine (generalised seizures).
-
No direct evidence exists between the drugs while network meta‐analysis shows a significant advantage for one of the drugs: none.
For the following comparisons, confidence intervals for the results from indirect evidence do not overlap with:
-
direct evidence: carbamazepine vs phenobarbitone (generalised seizures);
-
network meta‐analysis results: none.
Confidence intervals overlap for the results from direct evidence and from network meta‐analysis for all comparisons, therefore there is no indication that inconsistency is present in the results (see Table 12, Table 13, Figure 15; Figure 16 and Figure 17).
Subgroup and sensitivity analysis
See Sensitivity analysis for full details and rationale of all sensitivity analyses conducted.
We performed an additional analysis adjusted for age (as well as epilepsy type ‐ see Subgroup analysis and investigation of heterogeneity). Numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We were able to incorporate aggregate or extracted individual‐level data for 135 participants for one additional trial (Biton 2001). Numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
We performed two sensitivity analyses to investigate the possibility of generalised seizures being misclassified; in the first analysis we reclassified those with generalised seizures and age of onset greater than 30 years as having partial onset seizures and in the second analysis we reclassified generalised seizure types and age at onset greater than 30 years, and those with missing seizure type into an 'unclassified seizure type' group. For both analyses, numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We assessed the validity of the proportional hazards assumption of the Cox model used in the network meta‐analysis (see Data synthesis for further details); there was no evidence the assumption was violated for any of the covariates in the network meta‐analysis so we did not perform any sensitivity analysis.
We excluded one trial (Stephen 2007) from all analyses due to inconsistencies in provided data. Numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
Time to first seizure post randomisation
The number of participants that contributed to analysis of our secondary outcome, 'Time to first seizure post randomisation' was 12,152 out of 12,391 participants (98%).
Direct evidence
Table 14 (individuals with partial seizures) and Table 15 (individuals with generalised seizures) show the number of trials and participants contributing direct evidence for each of the pairwise comparisons in the network. Twenty out of 45 comparisons had no direct evidence for individuals with partial seizures. Thirteen out of 36 comparisons had no direct evidence for individuals with generalised seizures and eight comparisons for individuals with generalised seizures had fewer than 20 individuals contributing direct evidence resulting in wide confidence intervals around the treatment effect estimate for these comparisons.
The comparisons with the most participants contributing to analysis were carbamazepine vs lamotrigine and carbamazepine vs levetiracetam for individuals with partial seizures and sodium valproate vs levetiracetam and sodium valproate vs topiramate for individuals with generalised seizures.
Table 14 and Table 15 also show estimates of heterogeneity in the direct treatment effects. For three comparisons for individuals with partial seizures and for four comparisons for individuals with generalised seizures, substantial heterogeneity was present (I2 greater than 50%). The heterogeneity in these comparisons seemed to originate from differences in trial designs contributing to the pooled result; that is, pooling of trials recruiting children only, adults only or elderly participants only and pooling trials with or without treatment strata (see Data extraction and management for further details). For the comparisons for individuals with partial seizures, none of the treatment effects with substantial heterogeneity present were statistically significant so conclusions would not change for these treatment effects if random‐effects were applied. For the comparisons for individuals with generalised seizures, repeating analysis with random‐effects did not change conclusions for two of the comparisons (carbamazepine vs sodium valproate and phenytoin vs sodium valproate); but for one comparison (carbamazepine vs phenobarbitone), when repeating analysis with random‐effects there was no longer a statistically significant advantage to phenobarbitone: HR 0.59 (0.27 to 1.26)
Network meta‐analysis results (direct plus indirect evidence)
Figure 5 shows how each treatment performs compared to first‐line treatment carbamazepine for individuals with partial seizures (ordered by treatment effect estimate); phenobarbitone is significantly better than carbamazepine and carbamazepine is significantly better than sodium valproate, lamotrigine and gabapentin.
Figure 6 shows how each treatment performs compared to first‐line treatment lamotrigine for individuals with partial seizures (ordered by treatment effect estimate); phenobarbitone, phenytoin and carbamazepine are significantly better than lamotrigine.
Figure 7 shows how each treatment performs compared to first‐line treatment sodium valproate for individuals with generalised seizures (ordered by treatment effect estimate); sodium valproate is significantly better than topiramate.
Table 14 and Figure 10 (individuals with partial seizures) and Table 15 and Figure 11 (individuals with generalised seizures) show treatment effect estimates for all pairwise comparisons in the network combining direct with indirect evidence. In addition to the results described above; for individuals with partial seizures, phenobarbitone and phenytoin seems to perform better than most other drugs and for individuals with generalised seizures, phenytoin seems to perform better than most other drugs. There were few notable differences between the newer drugs (oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide) for either individuals with partial seizures or individuals with generalised seizures.
As described further in Assessment of heterogeneity, we could not directly calculate an I2 statistic for the network meta‐analysis the estimated I2 statistic was 0%. When repeating network meta‐analysis with random‐effects, Tau2 was 9 x 10‐21. As no heterogeneity was present and Tau2 was negligible, numerical results for treatment effects and conclusions were identical.
Investigation of inconsistency (node‐splitting)
We fitted the ‘design‐by‐treatment’ inconsistency model to 17 variables and regressed it on 23 designs, seven of which were multi‐arm trials (up to five treatment arms). Accounting for the multi‐arm trials, this resulted in an overall test for inconsistency with 43 degrees of freedom, which was not significant (Chi2 statistic (43) = 38.2, P value = 0.680, heterogeneity (Tau) = 0.094). Furthermore, there was no significant evidence of inconsistency within any of the 23 designs.
Table 14 (individuals with partial seizures) and Table 15 (individuals with generalised seizures) show treatment effect estimates from direct evidence, and from direct plus indirect evidence, Figure 15 and Figure 16 show treatment effect estimates for direct, indirect, and direct plus indirect evidence for individuals with partial seizures compared to carbamazepine and lamotrigine respectively and Figure 17 for individuals with generalised seizures compared to sodium valproate.
We note that for most pairwise comparisons, numerical results of direct evidence and network meta‐analysis are similar, mostly in the same direction and confidence intervals of estimates overlap. For all pairwise comparisons, results from network meta‐analysis are more precise than results from direct evidence (in some cases much more precise where limited direct evidence exists, for example see lamotrigine compared to gabapentin, Figure 15).
For the following comparisons; conclusions drawn from direct evidence and from network meta‐analysis are different (see Table 14 and Table 15).
-
Direct evidence shows a significant advantage to one of the drugs and the network meta‐analysis results show no significant difference between the drugs: sodium valproate vs lamotrigine (partial seizures); carbamazepine vs phenobarbitone (generalised seizures).
-
Direct evidence shows no significant difference between the drugs and network meta‐analysis shows a significant advantage for one of the drugs: carbamazepine vs phenobarbitone, carbamazepine vs sodium valproate, carbamazepine vs lamotrigine, phenobarbitone vs sodium valproate, phenytoin vs sodium valproate, phenytoin vs lamotrigine, oxcarbazepine vs gabapentin (all partial seizures), carbamazepine vs phenytoin, phenobarbitone vs phenytoin (generalised seizures).
-
No direct evidence exists between the drugs while network meta‐analysis shows a significant advantage for one of the drugs: phenobarbitone vs lamotrigine, phenobarbitone vs oxcarbazepine, phenobarbitone vs topiramate, phenobarbitone vs gabapentin, phenobarbitone vs levetiracetam, phenobarbitone vs zonisamide, phenytoin vs gabapentin, gabapentin vs levetiracetam (all partial seizures).
Confidence intervals for the results from indirect evidence overlapped with the confidence intervals from direct evidence and from network meta‐analysis for all comparisons.
For the following comparisons, confidence intervals for the results from direct evidence and from network meta‐analysis do not overlap, which indicates potential inconsistency is present (see Table 14, Table 15, Figure 15; Figure 16 and Figure 17): phenobarbitone vs sodium valproate (partial seizures), sodium valproate vs topiramate (generalised seizures).
For the comparison of phenobarbitone vs sodium valproate for individuals with partial seizures, from direct evidence, there is no significant difference between the drugs (HR 0.71 (0.43 to 1.17)), however from the network meta‐analysis results, a statistically significant advantage is shown for phenobarbitone (HR 1.53 (1.20 to 1.94)). For this comparison, only 12.8% of the network estimate is contributed from direct evidence and only 80 individuals contribute to this estimate. This small sample size and imprecision for the direct evidence is likely because sodium valproate is not considered to be a first‐line treatment for partial seizures and although phenobarbitone is a broad spectrum agent for the treatment of many seizure types, it is no longer used as a first‐line treatment (see NICE 2012 and Description of the intervention).
For the comparison of sodium valproate vs topiramate for individuals with generalised seizures, from direct evidence only, there is a statistically significant advantage to topiramate (HR 0.42 (0.23 to 0.80)), however from the network meta‐analysis results, the direction of effect changes to a statistically significant advantage to sodium valproate (HR 1.30 (1.01 to 1.68)). Furthermore, for this comparison, only 21% of the network estimate is contributed from direct evidence and a moderate amount of heterogeneity is present in this estimate (I2 = 46%). The same two trials contribute evidence to this outcome as ‘Time to withdrawal of allocated treatment;’ see above for discussion of the differences in design of these trials.
Furthermore, the 'design‐by treatment' inconsistency model does not show any significant evidence of inconsistency within the network. Therefore, we are not concerned about any impact of this observed inconsistency of numerical results on the conclusions of the review.
Subgroup and sensitivity analysis
See Sensitivity analysis for full details and rationale of all sensitivity analyses conducted.
We performed an additional analysis adjusted for age (as well as epilepsy type ‐ see Subgroup analysis and investigation of heterogeneity). Numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We were able to incorporate aggregate or extracted individual‐level data for 199 participants from two additional trials (Biton 2001; Gilad 2007). Numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with generalised seizures) and conclusions remained unchanged.
We performed two sensitivity analyses to investigate the possibility of generalised seizures being misclassified; in the first analysis we reclassified those with generalised seizures and age of onset greater than 30 years as having partial onset seizures, and in the second analysis we reclassified generalised seizure types and age at onset greater than 30 years, and those with missing seizure type into an 'unclassified seizure type' group. For both analyses, numerical results of this sensitivity analysis were similar; there were some changes in direction of effect size and some changes in the order or 'rank' of treatments compared to the reference treatment but no change in statistical significance for any estimate and no change to conclusions.
We assessed the validity of the proportional hazards assumption of the Cox model used in the network meta‐analysis (see Data synthesis for further details); most numerical results of this sensitivity analysis were similar, however there were a few changes in conclusions from those above, most notably that lamotrigine became significantly better than gabapentin, and that sodium valproate was no longer significantly better than topiramate (or any other treatment).
We excluded one trial (Stephen 2007) from all analyses due to inconsistencies in provided data. Numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with partial seizures) and conclusions remained unchanged.
We excluded another trial (Banu 2007) from analysis due to inconsistencies in provided data. Again, numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with partial seizures) and conclusions remained unchanged.
We excluded one trial (Nieto‐Barrera 2001) from analysis as we were not provided with seizure dates in the first four weeks of the trial. Again, numerical results of this sensitivity analysis were very similar (the same to two decimal places for individuals with partial seizures and one or two decimal places for individuals with partial seizures) and conclusions remained unchanged.
Occurence of adverse events
We were provided with individual participant data for adverse events experienced during the trial for 23 trials (Banu 2007; Baulac 2012; Biton 2001; Brodie 1995a; Brodie 1995b; Brodie 1999; Brodie 2007; Chadwick 1998; Dizdarer 2000; Eun 2012; Kwan 2009; Lee 2011; Nieto‐Barrera 2001; Ogunrin 2005; Privitera 2003; Ramsey 2010; Reunanen 1996; SANAD A 2007; SANAD B 2007; Steiner 1999; Stephen 2007; Trinka 2013; Werhahn 2015). The remaining 13 trials providing IPD, did not provide detailed IPD for adverse events, so we extracted information regarding adverse events from the trial publications (Bill 1997; Craig 1994; de Silva 1996; Guerreiro 1997; Heller 1995; Mattson 1985; Mattson 1992; Pal 1998; Placencia 1993; Ramsey 1992; Richens 1994; Turnbull 1985; Verity 1995). No adverse events data was reported in three of these publications (de Silva 1996; Heller 1995; Turnbull 1985).
We were also able to extract a summary of adverse event data from 26 trials not providing IPD ((Brodie 2002; Callaghan 1985; Capone 2008; Christe 1997; Consoli 2012; Dam 1989; Donati 2007; Feksi 1991; Gilad 2007; Jung 2015; Kalviainen 2002; Korean Lamotrigine Study Group 2008; Motamedi 2013; NCT01498822; NCT01954121; Pulliainen 1994; Ramsey 1983; Rastogi 1991; Resendiz 2004; Rowan 2005; Saetre 2007; Shakir 1981; So 1992; Steinhoff 2005; Suresh 2015; Thilothammal 1996).
No adverse event data was reported in 15 publications (Aikia 1992; Bidabadi 2009; Castriota 2008; Chen 1996; Cho 2011; Cossu 1984; Czapinski 1997; Forsythe 1991; Fritz 2006; Kopp 2007; Lukic 2005; Mitchell 1987; Miura 1990; Ramsey 2007; Ravi Sudhir 1995)
Due to the wide range of events reported in the trials and the different methods of recording and reporting of adverse events, we have not analysed adverse event data in meta‐analysis and provide a narrative report. We took the following approach to the negative synthesis of adverse events. One review author (SJN) grouped verbatim or reported terms extracted from publications or provided in IPD under higher level definitions and discussed any uncertainties in definition with the senior clinical author (AGM). We took the definitions used in this review from a previous review in our series of IPD monotherapy reviews (Nolan 2016a), with further definitions added as appropriate when reviewing the reported terms.
Table 16 describes the number of adverse events and the number of participants experiencing adverse events respectively by drug. Table 17 describes the frequency of some of the most commonly associated side effects of AEDs by drug.
| Drug | Number of participants | Number of participants | Number of events |
| CBZ | 5134 | 3023 | 9769 |
| PHB | 754 | 271 | 181 |
| PHT | 1384 | 614 | 1513 |
| VPS | 2303 | 1294 | 3599 |
| LTG | 3107 | 1608 | 6296 |
| OXC | 978 | 623 | 1000 |
| TPM | 1898 | 920 | 6316 |
| GBP | 1209 | 506 | 2580 |
| LEV | 948 | 1441 | 4258 |
| ZNS | 282 | 182 | 606 |
| Total | 18,045 | 10,482 | 36,118 |
CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aAdverse event data were provided as detailed individual participant data for 23 trials and we extracted summary adverse event information from 36 trial publications. No adverse event data were reported in 18 trial publications.
bSome trial publications reported only on the “most common” adverse events, the totals and frequencies are likely to be an underestimation of the true number of events and number of individuals experiencing events. Furthermore, detailed information was provided in the more recent trial publications and individual participant data requests of more recent trials, often involving newer antiepileptic drugs, such as LTG, LEV and TPM; which may indicate that these newer drugs are associated with more adverse events than older drugs such as PHB and PHT, for which less detailed information was available.
| Event (general description)a,b,c | CBZ | PHB | PHT | VPS | LTG | OXC | TPM | GBP | LEV | ZNS | Total |
| Accidental injury | 100 | 0 | 100 | 28 | 110 | 5 | 95 | 36 | 58 | 8 | 540 |
| Anorexia or weight loss | 126 | 0 | 126 | 24 | 116 | 6 | 394 | 58 | 62 | 25 | 937 |
| Anxiety/depression | 203 | 0 | 203 | 59 | 171 | 32 | 309 | 82 | 163 | 16 | 1238 |
| Aphasia | 59 | 7 | 66 | 11 | 26 | 4 | 106 | 22 | 11 | 2 | 314 |
| Asthenia | 59 | 1 | 60 | 26 | 41 | 1 | 31 | 33 | 37 | 10 | 299 |
| Ataxia | 172 | 37 | 209 | 32 | 55 | 17 | 61 | 40 | 32 | 8 | 663 |
| Cognitive (memory, concentration, confusion etc.) | 321 | 41 | 362 | 100 | 204 | 44 | 439 | 127 | 73 | 19 | 1730 |
| Dental problems | 93 | 0 | 93 | 28 | 62 | 5 | 61 | 24 | 70 | 7 | 443 |
| Dizziness/faintness | 617 | 0 | 617 | 171 | 348 | 140 | 269 | 160 | 394 | 23 | 2739 |
| Drowsiness/fatigue | 1270 | 1 | 1271 | 422 | 539 | 233 | 628 | 326 | 477 | 33 | 5200 |
| Fever or viral infection | 379 | 0 | 379 | 68 | 172 | 24 | 84 | 58 | 338 | 37 | 1539 |
| Gastrointestinal disturbances | 683 | 20 | 703 | 246 | 394 | 33 | 236 | 142 | 284 | 42 | 2783 |
| Hair loss | 47 | 0 | 47 | 130 | 22 | 15 | 39 | 8 | 16 | 3 | 327 |
| Headache or migraine | 843 | 0 | 843 | 264 | 556 | 137 | 315 | 171 | 596 | 47 | 3772 |
| Impotence | 90 | 24 | 114 | 13 | 17 | 0 | 27 | 32 | 11 | 3 | 331 |
| Increased/worsened seizures | 151 | 0 | 151 | 31 | 164 | 6 | 58 | 48 | 140 | 6 | 755 |
| Infection | 121 | 0 | 121 | 19 | 90 | 4 | 56 | 27 | 63 | 5 | 506 |
| Laboratory results abnormal | 367 | 0 | 367 | 103 | 117 | 8 | 47 | 19 | 90 | 32 | 1150 |
| Menstrual problems | 110 | 0 | 110 | 28 | 31 | 1 | 22 | 18 | 39 | 4 | 363 |
| Mood or behavioural change | 279 | 41 | 320 | 128 | 163 | 25 | 415 | 121 | 121 | 15 | 1628 |
| Nausea/vomiting | 413 | 1 | 414 | 167 | 233 | 53 | 132 | 92 | 142 | 20 | 1667 |
| Pain | 345 | 1 | 346 | 65 | 250 | 6 | 154 | 48 | 251 | 25 | 1491 |
| Paraesthesia or tingling | 56 | 0 | 56 | 22 | 33 | 2 | 708 | 34 | 28 | 7 | 946 |
| Problems sleeping/nightmares | 108 | 1 | 109 | 46 | 197 | 16 | 147 | 31 | 101 | 14 | 770 |
| Rash or skin disorder | 701 | 17 | 718 | 46 | 420 | 73 | 163 | 113 | 125 | 31 | 2407 |
| Renal/urinary disorder | 152 | 0 | 152 | 27 | 78 | 2 | 92 | 57 | 93 | 21 | 674 |
| Respiratory disorder | 233 | 0 | 233 | 53 | 124 | 4 | 190 | 23 | 131 | 17 | 1008 |
| Tremor or twitch | 171 | 1 | 172 | 258 | 219 | 19 | 56 | 23 | 51 | 2 | 972 |
| Visual disturbance/nystagmus | 199 | 0 | 199 | 53 | 96 | 33 | 86 | 59 | 33 | 8 | 766 |
| Weight gain | 259 | 0 | 259 | 347 | 167 | 22 | 71 | 258 | 70 | 1 | 1454 |
CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
aVerbatim or reported terms extracted from publications or provided in individual participant data were grouped under the definitions by one review author (SJN) and any uncertainties in definition were discussed with the senior clinical author (AGM).
bAdverse event data were provided as detailed individual participant data for 23 trials and we extracted summary adverse event information from 36 trial publications. No adverse event data were reported in 18 trial publications.
cSome trial publications reported only on the “most common” adverse events, the totals and frequencies are likely to be an underestimation of the true number of events and number of individuals experiencing events. Furthermore, detailed information was provided in the more recent trial publications and individual participant data requests of more recent trials, often involving newer antiepileptic drugs, such as LTG, LEV and TPM; which may indicate that these newer drugs are associated with more adverse events than older drugs such as PHB and PHT ,for which less detailed information was available.
The most commonly occurring adverse events across all drugs were drowsiness/fatigue, headache or migraine, gastrointestinal disturbances, dizziness/faintness and rash or skin disorders.
Drowsiness/fatigue was the most commonly reported adverse event of carbamazepine, phenytoin, sodium valproate, oxcarbazepine and gabapentin. Headache or migraine was the most commonly reported adverse event of lamotrigine, levetiracetam and zonisamide, Paraesthesia (tingling or 'pins and needles') was the most commonly reported adverse event of topiramate and cognitive disorders (memory or concentration difficulties, confusion etc.) and mood or behaviour changes (including aggression) were the most commonly reported adverse event of phenobarbitone.
We note that as some trial publications reported only on the “most common” adverse events, the totals and frequencies are likely to be an underestimation of the true number of events and number of individuals experiencing events. Furthermore in general, more detailed information was provided in the more recent trial publications and IPD requests of more recent trials often involving newer AEDs such as lamotrigine, levetiracetam and topiramate; which may indicate that these newer drugs are associated with more adverse events than older drugs such as phenobarbitone and phenytoin, for which less detailed information was available.
Such limitations must be taken into account when interpreting Table 16 and Table 17 as well as the definitions of adverse events in the review, which were defined by the review authors rather than according to dictionary terminology (such as MedDRA®); we encourage only general comparison of the relative frequencies of different adverse events experienced by participants on different drugs and we do not encourage direct interpretation of numerical frequencies of adverse events.
Discussion
Summary of main results
For brevity throughout the results section, we refer to participants with generalised tonic‐clonic seizures with or without other generalised seizure types as 'participants with generalised seizures.'
Individual participant data were provided for at least one outcome of this review for 12,391 participants with partial seizures or generalised seizures randomised to carbamazepine, phenytoin, sodium valproate, phenobarbitone, oxcarbazepine (oxcarbazepine), lamotrigine, gabapentin, topiramate, levetiracetam or zonisamide (zonisamide) in 36 trials.
We calculated ‘direct estimates’ via meta‐analysis of the head‐to‐head comparisons of the drugs within the trials and performed network meta‐analysis to combine this direct evidence with indirect evidence across the network of 10treatments. Network meta‐analysis provided a total of 45 pairwise comparisons for individuals with partial seizures and 36 pairwise comparisons for individuals with generalised seizures (no participants with generalised onset seizures were randomised to zonisamide).
Direct estimates could be calculated for between half and two thirds of comparisons across the outcomes of the review, however for many of the comparisons, data were contributed by only a single trial or by a small number of participants, or both. Where pooling of head‐to‐head data was possible, direct evidence was generally quite consistent, and where substantial heterogeneity was present between trials (I2 greater than 50%), it is likely that the heterogeneity originated from variability in design of the pooled trials such as pooling of trials recruiting different age groups, pooling of double‐blind and open‐label trials and pooling of trials with and without treatment stratification.
Network meta‐analysis showed that for our primary outcome, ‘Time to withdrawal of allocated treatment,’ for individuals with partial seizures; lamotrigine and levetiracetam were significantly better than first‐line treatment carbamazepine, which was significantly better than gabapentin and phenobarbitone. Lamotrigine was significantly better than all treatments except levetiracetam. For individuals with generalised onset seizures, first‐line treatment sodium valproate performed significantly better than carbamazepine, topiramate and phenobarbitone.
For ‘Time to 12‐month remission of seizures’ and ‘Time to six‐month remission of seizures,’ few notable differences were shown for either seizure type; only that carbamazepine was significantly better than levetiracetam for individuals with partial seizures (12‐month remission) and sodium valproate was significantly better than lamotrigine for individuals with generalised seizures (six‐month remission). Network meta‐analysis also showed that for ‘Time to first seizure,’ for individuals with partial seizures; phenobarbitone was significantly better than both first‐line treatments carbamazepine and lamotrigine; first‐line treatment carbamazepine performed significantly better than sodium valproate, gabapentin and first‐line treatment lamotrigine and phenytoin also performed significantly better than lamotrigine. In general, the earliest licenced treatments (phenytoin and phenobarbitone) performed better than the other treatments for both seizure types.
Results from network meta‐analysis were more precise than results from head‐to‐head comparisons, often much more precise for comparisons where there was limited direct evidence, reflecting the added precision of network meta‐analysis over pairwise meta‐analysis. Across outcomes for the majority of pairwise comparisons, numerical results of direct evidence and network meta‐analysis were similar, mostly in the same direction, confidence intervals of estimates overlapped and there was little indication of inconsistency between direct and network meta‐analysis results. For the few pairwise comparisons where confidence intervals of direct estimates and network meta‐analysis estimates did not overlap, generally direct evidence was limited and contributed only a small proportion of evidence to the network meta‐analysis estimates.
Adverse event data were recorded and reported variably in individual participant datasets and trial publications, therefore we have not attempted to analyse these data and have provided only a narrative report of commonly reported adverse events. The most commonly reported adverse events across all drugs were drowsiness/fatigue, headache or migraine, gastrointestinal disturbances, dizziness/faintness and rash or skin disorders, with some drug‐specific variations (e.g. paraesthesia (tingling or 'pins and needles') was the most commonly reported adverse event of topiramate, and cognitive disorders (memory or concentration difficulties, confusion etc.) and mood or behaviour changes (including aggression) were the most commonly reported adverse event of phenobarbitone).
Overall completeness and applicability of evidence
We have gratefully received IPD for 12,391 out of a total of 17,961 eligible participants (69% of total data) from 36 out of the 77 eligible trials (47%) randomising participants to one of 10 AEDs. We received between 49% and 100% of participant data across the 10 drugs.
Data from the remaining 41 trials could not be provided for a variety of reasons reported by trial authors or sponsors, including data lost or no longer available, cost and resources required to prepare data was prohibitive, local authority‐ or country‐specific restrictions. Furthermore for 15 of these trials, at the time of writing, we have been unable to make contact with an author or sponsor to request data and two trials are currently available only as ClinicalTrials.gov summaries. If data can be made available for any of these additional trials at a later date, they will be included in an update of this review.
Figure 1 shows network plots of pairwise comparisons in all included trials, trials providing IPD and trials without IPD. Visually, the plot of the trials providing IPD is very similar to the plot of all included trials; therefore it is likely that the 69% of participant data we received is a representative sample of all eligible participants and that the 31% of missing participant data can generally be treated as ‘missing at random.’
Specifically, we were provided with IPD for all direct pairwise comparisons in the total network except for oxcarbazepine compared to sodium valproate and oxcarbazepine compared to levetiracetam. In fact, out of all drugs included in the network, we received the lowest proportion of data for oxcarbazepine (49%). The lack of data for these comparisons may have contributed to imprecision of some effect sizes relating to oxcarbazepine (see Figure 9 and Figure 11), therefore we encourage caution when interpreting results relating to oxcarbazepine from this review. We note that the 51% of IPD missing for oxcarbazepine mostly comes from trials for which we could not establish contact with an author or sponsor to request IPD. If additional data can be included in an update for oxcarbazepine, we expect the precision of these estimates to improve.
Figure 2 shows network plots of pairwise comparisons in all eligible participants, from participants with partial seizures and from participants with generalised seizures. The majority of participants recruited into the trials were classified as experiencing partial seizures (66.7% of participants in all trials and 67.5% of participants with IPD provided); this majority is demonstrated in the visual similarity of the network plot for individuals with partial seizures compared to the plot of all participants and reflected in the relative precision of the results of this review for partial seizures compared to generalised seizures.
While a majority of partial seizures compared to generalised seizures is reflective of clinical practice (around 60% of individuals with epilepsy experience partial seizures, NINDS 2015), the proportion of individuals with partial seizures recruited to the trials in this review is even greater. This likely in part reflects the challenges of undertaking trials in children, highlights the need for more large and high‐quality trials.
The remaining participants were classified as experiencing generalised seizures (24.5% of participants in all trials and 26.5% of participants with IPD provided) or unclassified/missing seizure type (8.8% of participants in all trials and 6% of participants with IPD provided). Misclassification of seizure type is a recognised problem in epilepsy (whereby some individuals with generalised seizures have been mistakenly classed as having partial onset seizures and vice versa). The potential impact of this misclassification on results has been shown in our series of Cochrane IPD reviews of monotherapy for epilepsy (Nolan 2016d) whereby up to 50% of individuals classified as experiencing generalised seizures may have had their seizure type misclassified, as an age of seizure onset of over 30 years is unlikely for generalised seizures (Malafosse 1994). Investigation of misclassification within this review (reclassification of 1164 participants with generalised seizures and age of onset of over 30 years, 36% of individuals originally classified at experiencing generalised seizures) did not show any important changes to treatment effect sizes and no changes to conclusions.
This does not, however, indicate that misclassification of seizure type has not occurred in these trials; rather that the primary analysis results are robust to any misclassification. Trials included in this review were published between 1981 and 2015 and a proportion of trials classified generalised and partial onset seizures according to the International League Against Epilepsy (ILAE) classification of 1981 (Commission 1981), rather than the revised ILAE classification in 1989 (Commission 1989) or recently revised terminology (Berg 2010), which may have led to misclassification. Furthermore, several trials were conducted in low‐income countries in Africa, Asia and Central or South America, without access to the same facilities such as EEGs or MRI scanners as trials conducted in the USA and Europe. Within these trials, it is likely that seizure type would have been classified clinically, which may have further contributed to misclassification in these trials
In reality, it is likely that fewer than 20% of participants recruited into all of these trials (17% of participants included in IPD analysis were classified as having generalised seizures following reclassification in sensitivity analysis) experienced generalised seizures which is a lower proportion than would be expected in clinical practice (NINDS 2015). For this reason, treatment effect sizes for generalised seizures, particularly those that are imprecise, should be treated as less applicable than the treatment effect sizes for partial seizures.
In order to provide more precise evidence, applicable to individuals with generalised seizures, it is important both to ensure accurate seizure classification (as far as possible) and to increase the proportion of individuals with generalised seizures recruited into trials of AEDs to better reflect the ‘real world’ ratio of partial to generalised seizures. Increased recruitment of participants may not be straightforward, particularly as those with new onset generalised seizures are expected to be children and adolescents, and recruitment of children into clinical trials comes with difficulties (Joseph 2015); however, if targeted recruitment strategies could be implemented and the evidence base for individuals with generalised seizures increased, this may better inform treatment decisions for this population, particularly for those of childbearing potential, for whom first‐line treatment sodium valproate may not be appropriate (NICE 2012).
Quality of the evidence
This review provides mostly high‐quality evidence for the relative effectiveness of 10 commonly used anti‐epileptic drugs for the treatment of partial seizures and generalised tonic‐clonic seizures. Where limited data were available for a comparison and confidence intervals around treatment effect size results were wide (mostly for individuals with generalised seizures) or where potential inconsistency existed between direct estimates and network meta‐analysis estimates, we judged the quality of the evidence to be moderate and additional data from future trials may impact on these treatment effect estimates (see summary of findings Table for the main comparison; summary of findings Table 2; summary of findings Table 3; summary of findings Table 4; summary of findings Table 5; summary of findings Table 6).
Direct estimates and network meta‐analysis estimates were generally consistent and despite some methodological concerns in several trials contributing to analyses, which may have introduced bias into analyses, or inconsistencies present within individual participant data, (see Risk of bias in included studies); numerous sensitivity analyses were performed to test the robustness of the results in the presence of these biases (see Sensitivity analysis for full details); results of sensitivity analyses were numerically similar and did not lead to any changes to conclusions, therefore it is unlikely that any methodological inadequacies of individual trials has influenced the overall pooled network meta‐analysis results
Potential biases in the review process
The search strategies for this review were extensive and we are confident that we have identified all relevant evidence for this review including ongoing trials.
We have taken an IPD approach to analysis, which has many advantages, such as allowing the standardisation of definitions of outcomes across trials, and reducing attrition and reporting biases, as we can perform additional analyses and calculate additional outcomes from unpublished data. For the outcomes we used in this review that are of a time‐to‐event nature, an IPD approach is considered to be the 'gold standard' approach to analysis (Parmar 1998). Furthermore, the use of IPD in this analysis has allowed us to consider the relationship between treatment effect and seizure type via an interaction term in the network meta‐analysis model and present results separately according to seizure type in the context of the recommended first‐line treatment of the seizure type; an approach which would not have been possible without the use of IPD.
Despite the advantages of an IPD approach, for reasons out of our control, we were not able to obtain IPD for 5570 participants from 41 eligible trials, and for the majority of these trials, no aggregate data were available for our outcomes of interest in trial publications. It is inevitable that the exclusion of 31% of eligible participants may be a source of bias in our analyses, however as discussed in more detail above in Overall completeness and applicability of evidence, we believe that the 69% of participants we were able to include in IPD analyses were a representative sample of the total participants included in all eligible trials and that the benefits of an IPD approach outweigh the limitations.
The majority of IPD requested were provided to us directly but for one trial (Biton 2001) we requested data via data sharing portal ClinicalStudyDataRequest.com and data were provided to us via a remote secure data access system, which allowed analysis in SAS‐based statistical software and export of analysis results. We were unable to combine this dataset with the other datasets to perform the analyses described in Data synthesis, therefore we treated the results exported from the data access system as aggregate data in sensitivity analysis (see Sensitivity analysis). As described above, numerical results were similar and conclusions unchanged following the addition of aggregate data to the IPD analyses, therefore the restricted access format of this single trial does not seem to have impacted on the results of the review; however, we are concerned for updates of this review in particular and for future meta‐analyses of IPD in general, that the provision of data in different formats and the increased use of remote access systems may restrict the analyses that it is possible to perform across all eligible datasets and subsequently impact on meta‐analytic results and the scope of clinical questions that are able to be addressed.
Agreements and disagreements with other studies or reviews
In 2007 our group published a network meta‐analysis (NMA) including IPD for over 6418 participants from 20 trials (also included in the current review) comparing direct and indirect evidence from carbamazepine, phenobarbitone, phenytoin, sodium valproate, lamotrigine, oxcarbazepine, topiramate and gabapentin (Tudur Smith 2007). Results of this NMA showed for partial onset seizures that lamotrigine performed better than all other drugs in terms of treatment withdrawal but may not perform better than carbamazepine in terms of seizure control. Phenobarbitone performed better than other drugs in terms of seizure control but at the expense of increased treatment failure. Overall for individuals with partial seizures, lamotrigine, carbamazepine and oxcarbazepine seemed to provide the best balance of seizure control and treatment failure. As in the current review, data for individuals with generalised seizures were limited and results suggested that sodium valproate or phenytoin may provide the best combination of seizure control and treatment failure.
The present review was designed to update the information in the previous NMA with new evidence from trials published since 2007 and including evidence for two drugs, which were licensed for use as monotherapy after 2007 (levetiracetam and zonisamide).
The results of this review generally agree with the results of the NMA, in addition to providing evidence of the comparative effectiveness of the two new drugs within the spectrum of commonly used anti‐epileptic drugs, and further highlight that nearly 10 years on, data for individuals with generalised seizures are still limited.
Network plot of pairwise comparisons in all included studies, studies providing individual participant data (IPD) and studies without IPD
Note that the size of the node indicates the number of studies the drug is included in and the thickness of the edges corresponds to the number of participants contributing to the comparison (i.e. larger node = more studies, thicker edge = more participants).
CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Network plot of pairwise comparisons for all included participants (total 17,961 participants), participants with partial seizures and participants with generalised tonic‐clonic seizures with or without other seizure types (shortened to 'generalised seizures' for brevity).
11978 participants were classified as experiencing partial seizures (66.7% of total), 4407 participants were classified as experiencing generalised seizures (24.5% of total) and 1576 had an unclassified or missing seizure type (8.8% of total).
Note that the size of the node indicates the number of studies the drug is included in and the thickness of the edges corresponds to the number of participants contributing to the comparison (i.e. larger node = more studies, thicker edge = more participants).
CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
Risk of bias summary: review authors' judgements about each risk of bias item for each included trial
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with partial seizures, all drugs compared to carbamazepine (CBZ)
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with partial seizures, all drugs compared to lamotrigine (LTG)
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with generalised seizures, all drugs compared to sodium valproate (VPS)
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with partial seizures, all pairwise comparisons for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with generalised seizures, all pairwise comparisons for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with partial seizures, all pairwise comparisons for time to six‐month remission and time to first seizure.
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
AED: antiepileptic drug; CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Network meta‐analysis results (direct and indirect evidence combined) for individuals with generalised seizures, all pairwise comparisons for time to six‐month remission and time to first seizure.
Note: direct evidence (%) is the proportion of the estimate contributed by direct evidence and the box size is proportional to the number of participants contributing direct evidence.
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with partial seizures compared to carbamazepine (CBZ) for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with partial seizures compared to lamotrigine (LTG) for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: Direct, Indirect and Network estimates for individuals with generalised seizures compared to sodium valproate (VPS) for time to withdrawal of allocated treatment and time to 12‐month remission.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with partial seizures compared to carbamazepine (CBZ) for time to six‐month remission and time to first seizure.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with partial seizures compared to lamotrigine (LTG) for time to six‐month remission and time to first seizure.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide
Consistency: direct, indirect and network estimates for individuals with generalised seizures compared to sodium valproate (VPS) for time to six‐month remission and time to first seizure.
Note: direct evidence comes from studies that compared the drugs (head‐to‐head comparisons), indirect evidence comes from studies that did not compare the drugs (indirect comparisons) and network evidence comes from the whole network (head‐to‐head and indirect comparisons for all drugs).
Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity.
To see a magnified version of this figure, please see https://epilepsy.cochrane.org/network‐meta‐analysis‐figures.
| Antiepileptic drug monotherapy for epilepsy: time to withdrawal of allocated treatment for individuals with partial seizures | ||||||
| Patient or population: adults and children with partial seizures Settings: outpatients Intervention: phenobarbitone, phenytoin, sodium valproate, lamotrigine, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: carbamazepine | ||||||
| Intervention (experimental treatment)a,b | Comparison (reference treatment) | No of participants | Relative effect Direct evidence Heterogeneity: I2 | Relative effect Direct plus indirect evidence | Proportion of | Quality of the evidence |
| Phenobarbitone | Carbamazepine | 520 (4 studies) | 1.57 (1.16 to 2.13) I2 = 0% | 1.55 (1.18 to 2.04) | 52.5% | ⊕⊕⊕⊕ |
| Phenytoin | Carbamazepine | 428 (3 studies) | 1.03 (0.74 to 1.42) I2 = 63.6% | 1.13 (0.92 to 1.38) | 12.8% | ⊕⊕⊕⊕ |
| Sodium Valproate | Carbamazepine | 814 (5 studies) | 0.94 (0.73 to 1.19) I2 = 0% | 1.04 (0.86 to 1.25) | 40.1% | ⊕⊕⊕⊕ |
| Lamotrigine | Carbamazepine | 2268 (9 studies) | 0.76 (0.61 to 0.95) I2 = 39.3% | 0.75 (0.65 to 0.86) | 28.9% | ⊕⊕⊕⊕ |
| Oxcarbazepine | Carbamazepine | 562 (2 studies) | 4.62 (0.95 to 22.4) I2 = 0% | 1.09 (0.84 to 1.42) | 5.7% | ⊕⊕⊕⊕ |
| Topiramate | Carbamazepine | 937 (2 studies) | 1.04 (0.52 to 2.07) I2 = 0% | 1.18 (0.98 to 1.43) | 7.4% | ⊕⊕⊕⊕ |
| Gabapentin | Carbamazepine | 954 (2 studies) | 1.14 (0.84 to 1.55) I2 = 0% | 1.20 (1.00 to 1.43) | 87.1% | ⊕⊕⊕⊕ |
| Levetiracetam | Carbamazepine | 1567 (3 studies) | 0.70 (0.52 to 0.94) I2 = 0% | 0.82 (0.69 to 0.97) | 37.9% | ⊕⊕⊕⊕ |
| Zonisamide | Carbamazepine | 583 (1 study) | 1.08 (0.81 to 1.44) I2 = NA) | 1.08 (0.79 to 1.48) | 100% | ⊕⊕⊕⊕ |
| Abbreviations: CI: confidence interval; HR: hazard ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence | ||||||
| aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Antiepileptic drug monotherapy for epilepsy: time to withdrawal of allocated treatment for individuals with partial seizures | ||||||
| Patient or population: adults and children with partial seizures Settings: outpatients Intervention: carbamazepine, phenobarbitone, phenytoin, sodium valproate, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: lamotrigine | ||||||
| Intervention (experimental treatment)a,b | Comparison (reference | No of participants | Relative effect Direct evidence Heterogeneity: I2 | Relative effect Direct plus | Proportion of | Quality of the evidence |
| Carbamazepine | Lamotrigine | 2268 (9 studies) | 1.31 (1.05 to 1.64) I2 = 39.3% | 1.34 (1.17 to 1.53) | 28.9% | ⊕⊕⊕⊕ |
| Phenobarbitone | Lamotrigine | No direct evidence | No direct evidence I2: NA | 2.08 (1.52 to 2.86) | 0% | ⊕⊕⊕⊕ |
| Phenytoin | Lamotrigine | 90 (1 study) | 0.91 (0.47 to 1.76) I2: NA | 1.52 (1.18 to 1.92) | 11.6% | ⊕⊕⊕⊕ |
| Sodium Valproate | Lamotrigine | 221 (3 studies) | 0.71 (0.51 to 1.00) I2 = 45.1% | 1.39 (1.11 to 1.72) | 5.1% | ⊕⊕⊕⊝ |
| Oxcarbazepine | Lamotrigine | 506 (1 study) | 0.69 (0.12 to 4.14) I2: NA | 1.46 (1.11 to 1.92) | 4.4% | ⊕⊕⊕⊕ |
| Topiramate | Lamotrigine | 648 (1 study) | 1.18 (0.86 to 1.62) I2: NA | 1.59 (1.29 to 1.95) | 20.9% | ⊕⊕⊕⊕ |
| Gabapentin | Lamotrigine | 659 (1 study) | 0.62 (0.06 to 6.01) I2: NA | 1.60 (1.31 to 1.96) | 1% | ⊕⊕⊕⊕ |
| Levetiracetam | Lamotrigine | 240 (1 study) | 0.86 (0.58 to 1.28) I2: NA | 1.10 (0.89 to 1.35) | 23.7% | ⊕⊕⊕⊕ |
| Zonisamide | Lamotrigine | No direct evidence | No direct evidence I2: NA | 1.45 (1.03 to 2.04) | 0% | ⊕⊕⊕⊕ |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence | ||||||
| aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Antiepileptic drug monotherapy for epilepsy: time to withdrawal of allocated treatment for individuals with generalised seizures | ||||||
| Patient or population: adults and children with generalised seizures* Settings: outpatients Intervention: carbamazepine, phenobarbitone, phenytoin, lamotrigine, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide. Comparison: sodium valproate | ||||||
| Intervention (experimental treatment)a,b | Comparison (reference | No of | Relative effect Direct evidence Heterogeneity: I2 | Relative effect Direct plus | Proportion of | Quality of the evidence |
| Carbamazepine | Sodium Valproate | 405 (4 studies) | 0.79 (0.45 to 1.37) I2 = 6.6% | 1.42 (1.09 to 1.85) | 27.3% | ⊕⊕⊕⊕ |
| Phenobarbitone | Sodium Valproate | 94 (2 studies) | 1.79 (0.65 to 5.00) I2 = 0% | 2.09 (1.17 to 3.75) | 19.4% | ⊕⊕⊕⊝ |
| Phenytoin | Sodium Valproate | 326 (3 studies) | 1.52 (0.68 to 3.33) I2 = 22.6% | 1.30 (0.79 to 2.15) | 19.3% | ⊕⊕⊕⊕ |
| Lamotrigine | Sodium Valproate | 387 (3 studies) | 0.46 (0.22 to 0.97) I2 = 0% | 0.90 (0.60 to 1.35) | 14.8% | ⊕⊕⊕⊕ |
| Oxcarbazepine | Sodium Valproate | No direct evidence | No direct evidence I2: NA | 1.42 (0.29 to 6.92) | 0% | ⊕⊕⊕⊝ |
| Topiramate | Sodium Valproate | 443 (2 studies) | 1.04 (0.52 to 2.07) I2 = 48.5% | 1.76 (1.22 to 2.53) | 22.4% | ⊕⊕⊕⊝ |
| Gabapentin | Sodium Valproate | No direct evidence | No direct evidence I2: NA | 1.28 (0.16 to 10.5) | 0% | ⊕⊕⊕⊝ |
| Levetiracetam | Sodium Valproate | 512 (1 study) | 0.68 (0.30 to 1.59) I2: NA) | 1.05 (0.58 to 1.90) | 18.6% | ⊕⊕⊕⊕ |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence | ||||||
| *Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Antiepileptic drug monotherapy for epilepsy: time to 12‐month remission for individuals with partial seizures | ||||||
| Patient or population: adults and children with partial seizures Settings: outpatients Intervention: phenobarbitone, phenytoin, sodium valproate, lamotrigine, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: carbamazepine | ||||||
| Intervention (experimental treatment)a,b | Comparison (reference treatment) | No of participants | Relative effect Direct evidence Heterogeneity: I2 | Relative effect Direct plus | Proportion of | Quality of the evidence |
| Phenobarbitone | Carbamazepine | 525 (4 studies) | 1.41 (1.04 to 1.91) I2 = 0% | 1.02 (0.76 to 1.35) | 56.1% | ⊕⊕⊕⊕ |
| Phenytoin | Carbamazepine | 430 (3 studies) | 1.00 (0.76 to 1.32) I2 = 54.8% | 1.03 (0.85 to 1.25) | 18.6% | ⊕⊕⊕⊕ |
| Sodium Valproate | Carbamazepine | 816 (5 studies) | 1.03 (0.85 to 1.25) I2 = 46.4% | 1.05 (0.89 to 1.25) | 27.6% | ⊕⊕⊕⊕ |
| Lamotrigine | Carbamazepine | 891 (2 studies) | 1.02 (0.69 to 1.50) I2 = 0% | 1.16 (0.98 to 1.37) | 17.5% | ⊕⊕⊕⊕ |
| Oxcarbazepine | Carbamazepine | 555 (2 studies) | 1.13 (0.62 to 2.05) I2 = 0% | 0.98 (0.78 to 1.25) | 21% | ⊕⊕⊕⊕ |
| Topiramate | Carbamazepine | 925 (2 studies) | 0.94 (0.48 to 1.83) I2 = 0% | 1.08 (0.92 to 1.27) | 7.2% | ⊕⊕⊕⊕ |
| Gabapentin | Carbamazepine | 651 (1 study) | 0.61 (0.06 to 5.82) I2: NA | 1.20 (0.99 to 1.47) | 10.5% | ⊕⊕⊕⊕ |
| Levetiracetam | Carbamazepine | 1567 (3 studies) | 1.08 (0.81 to 1.42) I2 = 60.8% | 1.35 (1.09 to 1.69) | 14.2% | ⊕⊕⊕⊕ |
| Zonisamide | Carbamazepine | 582 (1 study) | 1.05 (0.85 to 1.30) I2: NA | 1.05 (0.81 to 1.35) | 100% | ⊕⊕⊕⊕ |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence | ||||||
| aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Antiepileptic drug monotherapy for epilepsy: time to 12‐month remission for individuals with partial seizures | ||||||
| Patient or population: adults and children with partial seizures Settings: outpatients Intervention: carbamazepine, phenobarbitone, phenytoin, sodium valproate, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: lamotrigine | ||||||
| Intervention (experimental treatment)a,b | Comparison (reference treatment) | No of participants | Relative effect Direct evidence Heterogeneity: I2 | Relative effect Direct plus | Proportion of | Quality of the evidence |
| Carbamazepine | Lamotrigine | 891 (2 studies) | 0.98 (0.67 to 1.45) I2 = 0% | 0.86 (0.72 to 1.02) | 17.5% | ⊕⊕⊕⊕ |
| Phenobarbitone | Lamotrigine | No direct evidence | No direct evidence I2: NA | 0.88 (0.62 to 1.22) | 0% | ⊕⊕⊕⊕ |
| Phenytoin | Lamotrigine | No direct evidence | No direct evidence I2: NA | 0.89 (0.68 to 1.13) | 0% | ⊕⊕⊕⊕ |
| Sodium Valproate | Lamotrigine | 221 (3 studies) | 0.72 (0.56 to 0.93) I2 = 0% | 0.91 (0.73 to 1.33) | 39.9% | ⊕⊕⊕⊕ |
| Oxcarbazepine | Lamotrigine | 499 (1 study) | 1.49 (0.33 to 6.67) I2: NA | 0.85 (0.66 to 1.09) | 2.8% | ⊕⊕⊕⊕ |
| Topiramate | Lamotrigine | 636 (1 study) | 0.98 (0.29 to 3.25) I2: NA | 0.93 (0.75 to 1.15) | 2.5% | ⊕⊕⊕⊕ |
| Gabapentin | Lamotrigine | 647 (1 study) | 0.74 (0.08 to 6.58) I2: NA | 1.04 (0.84 to 1.30) | 10.1% | ⊕⊕⊕⊕ |
| Levetiracetam | Lamotrigine | 240 (1 study) | 1.02 (0.70 to 1.49) I2: NA | 1.16 (0.93 to 1.47) | 26.6% | ⊕⊕⊕⊕ |
| Zonisamide | Lamotrigine | No direct evidence | No direct evidence I2: NA | 0.91 (0.67 to 1.22) | 0% | ⊕⊕⊕⊕ |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence | ||||||
| aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Antiepileptic drug monotherapy for epilepsy: time to withdrawal of allocated treatment for individuals with generalised seizures | ||||||
| Patient or population: adults and children with generalised seizures* Settings: outpatients Intervention: carbamazepine, phenobarbitone, phenytoin, lamotrigine, oxcarbazepine, topiramate, gabapentin, levetiracetam and zonisamide Comparison: sodium valproate | ||||||
| Intervention (experimental treatment)a,b | Comparison (reference treatment) | No of participants | Relative effect Direct evidence | Relative effect Direct plus | Proportion of | Quality of the evidence |
| Carbamazepine | Sodium Valproate | 412 (4 studies) | 0.99 (0.69 to 1.39) I2 = 0% | 1.06 (0.88 to 1.27) | 51.1% | ⊕⊕⊕⊕ |
| Phenobarbitone | Sodium Valproate | 98 (2 studies) | 0.86 (0.40 to 1.89) I2 = 42.3% | 1.33 (0.87 to 2.04) | 13% | ⊕⊕⊕⊕ |
| Phenytoin | Sodium Valproate | 269 (4 studies) | 1.15 (0.71 to 1.82) I2 = 0% | 0.91 (0.67 to 1.25) | 44.9% | ⊕⊕⊕⊕ |
| Lamotrigine | Sodium Valproate | 387 (3 studies) | 0.77 (0.38 to 1.56) I2 = 0% | 1.35 (0.57 to 3.13) | 35.7% | ⊕⊕⊕⊕ |
| Oxcarbazepine | Sodium Valproate | No direct evidence | No direct evidence I2: NA | 1.82 (0.50 to 6.67) | 0% | ⊕⊕⊕⊝ |
| Topiramate | Sodium Valproate | 441 (2 studies) | 0.52 (0.26 to 1.04) I2 = 58.5% | 1.12 (0.83 to 1.52) | 10.6% | ⊕⊕⊕⊕ |
| Gabapentin | Sodium Valproate | No direct evidence | No direct evidence I2: NA | 0.79 (0.10 to 6.25) | 0% | ⊕⊕⊕⊝ |
| Levetiracetam | Sodium Valproate | 512 (1 study) | 0.91 (0.49 to 1.70) I2: NA | 1.41 (0.83 to 2.44) | 55.2% | ⊕⊕⊕⊕ |
| Abbreviations: CI: confidence interval; HR: hazard Ratio; NA: not applicable | ||||||
| GRADE Working Group grades of evidence | ||||||
| *Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity. aOrder of drugs in the table: drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Trial\Drug | CBZ | PHB | PHT | VPS | LTG | OXC | LEV | TPM | GBP | ZNS | Total | Total randomiseda |
| Trials providing individual participant data | ||||||||||||
| 54 | 54 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 108 | 108 | |
| 301 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 282 | 583 | 583 | |
| 0 | 0 | 144 | 0 | 0 | 143 | 0 | 0 | 0 | 0 | 287 | 287 | |
| 0 | 0 | 0 | 69 | 66 | 0 | 0 | 0 | 0 | 0 | 135 | 136 | |
| 66 | 0 | 0 | 0 | 70 | 0 | 0 | 0 | 0 | 0 | 136 | 136 | |
| 63 | 0 | 0 | 0 | 61 | 0 | 0 | 0 | 0 | 0 | 124 | 124 | |
| 48 | 0 | 0 | 0 | 102 | 0 | 0 | 0 | 0 | 0 | 150 | 150 | |
| 291 | 0 | 0 | 0 | 0 | 0 | 288 | 0 | 0 | 0 | 579 | 579 | |
| 74 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 218 | 0 | 292 | 292 | |
| 0 | 0 | 81 | 85 | 0 | 0 | 0 | 0 | 0 | 0 | 166 | 166 | |
| 54 | 10 | 54 | 49 | 0 | 0 | 0 | 0 | 0 | 0 | 167 | 173 | |
| 26 | 0 | 0 | 0 | 0 | 26 | 0 | 0 | 0 | 0 | 52 | 52 | |
| 41 | 0 | 0 | 0 | 43 | 0 | 0 | 0 | 0 | 0 | 84 | 84 | |
| 0 | 0 | 94 | 0 | 0 | 99 | 0 | 0 | 0 | 0 | 193 | 193 | |
| 61 | 58 | 63 | 61 | 0 | 0 | 0 | 0 | 0 | 0 | 243 | 243 | |
| 0 | 0 | 0 | 44 | 37 | 0 | 0 | 0 | 0 | 0 | 81 | 81 | |
| 53 | 0 | 0 | 0 | 57 | 0 | 0 | 0 | 0 | 0 | 110 | 110 | |
| 155 | 155 | 165 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 475 | 475 | |
| 236 | 0 | 0 | 244 | 0 | 0 | 0 | 0 | 0 | 0 | 480 | 480 | |
| 202 | 0 | 0 | 0 | 420 | 0 | 0 | 0 | 0 | 0 | 622 | 622 | |
| 19 | 18 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 55 | 55 | |
| 0 | 47 | 47 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 94 | 94 | |
| 95 | 97 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 192 | 192 | |
| Privitera 2003 (CBZ branch)b | 129 | 0 | 0 | 0 | 0 | 0 | 0 | 266 | 0 | 0 | 395 | 395 |
| Privitera 2003 (VPS branch)b | 0 | 0 | 0 | 78 | 0 | 0 | 0 | 147 | 0 | 0 | 225 | 225 |
| 0 | 0 | 50 | 86 | 0 | 0 | 0 | 0 | 0 | 0 | 136 | 136 | |
| 0 | 0 | 128 | 0 | 0 | 0 | 0 | 133 | 0 | 0 | 261 | 261 | |
| 121 | 0 | 0 | 0 | 230 | 0 | 0 | 0 | 0 | 0 | 351 | 351 | |
| 151 | 0 | 0 | 149 | 0 | 0 | 0 | 0 | 0 | 0 | 300 | 300 | |
| 378 | 0 | 0 | 0 | 378 | 210 | 0 | 378 | 377 | 0 | 1721 | 1721 | |
| 0 | 0 | 0 | 238 | 239 | 0 | 0 | 239 | 0 | 0 | 716 | 716 | |
| 0 | 0 | 95 | 0 | 86 | 0 | 0 | 0 | 0 | 0 | 181 | 181 | |
| 0 | 0 | 0 | 109 | 117 | 0 | 0 | 0 | 0 | 0 | 226 | 227 | |
| Trinka 2013 (CBZ branch)b | 503 | 0 | 0 | 0 | 0 | 0 | 493 | 0 | 0 | 0 | 996 | 999 |
| Trinka 2013 (VPS branch)b | 0 | 0 | 0 | 353 | 0 | 0 | 350 | 0 | 0 | 0 | 703 | 703 |
| 0 | 0 | 70 | 70 | 0 | 0 | 0 | 0 | 0 | 0 | 140 | 140 | |
| 130 | 0 | 0 | 130 | 0 | 0 | 0 | 0 | 0 | 0 | 260 | 260 | |
| 121 | 0 | 0 | 0 | 118 | 0 | 122 | 0 | 0 | 0 | 361 | 361 | |
| Total | 3372 | 439 | 1009 | 1765 | 2024 | 478 | 1253 | 1163 | 595 | 282 | 12,380 | 12,391 |
| Trials not providing individual participant data | ||||||||||||
| Trial\Drug | CBZ | PHB | PHT | VPS | LTG | OXC | LEV | TPM | GBP | ZNS | Total | Total randomiseda |
| 0 | 0 | 18 | 0 | 0 | 19 | 0 | 0 | 0 | 0 | 37 | 37 | |
| 36 | 35 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 71 | 71 | |
| 0 | 0 | 0 | 0 | 151 | 0 | 0 | 0 | 158 | 0 | 309 | 309 | |
| 59 | 0 | 58 | 64 | 0 | 0 | 0 | 0 | 0 | 0 | 181 | 181 | |
| 17 | 0 | 0 | 0 | 0 | 0 | 18 | 0 | 0 | 0 | 35 | 35 | |
| 14 | 0 | 0 | 0 | 0 | 0 | 13 | 0 | 0 | 0 | 27 | 27 | |
| 26 | 25 | 0 | 25 | 0 | 0 | 0 | 0 | 0 | 0 | 76 | 76 | |
| 15 | 0 | 0 | 0 | 0 | 0 | 16 | 0 | 0 | 0 | 31 | 31 | |
| 0 | 0 | 0 | 121 | 0 | 128 | 0 | 0 | 0 | 0 | 249 | 249 | |
| 66 | 0 | 0 | 0 | 0 | 0 | 62 | 0 | 0 | 0 | 128 | 128 | |
| 6 | 6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 12 | 12 | |
| 30 | 30 | 30 | 30 | 0 | 0 | 0 | 0 | 0 | 0 | 120 | 120 | |
| 100 | 0 | 0 | 0 | 0 | 94 | 0 | 0 | 0 | 0 | 194 | 194 | |
| 28 | 0 | 0 | 29 | 0 | 55 | 0 | 0 | 0 | 0 | 112 | 112 | |
| 152 | 150 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 302 | 302 | |
| 23 | 0 | 20 | 21 | 0 | 0 | 0 | 0 | 0 | 0 | 64 | 64 | |
| 0 | 0 | 0 | 0 | 21 | 27 | 0 | 0 | 0 | 0 | 48 | 48 | |
| 32 | 0 | 0 | 0 | 32 | 0 | 0 | 0 | 0 | 0 | 64 | 64 | |
| 64 | 0 | 0 | 0 | 0 | 0 | 57 | 0 | 0 | 0 | 121 | 121 | |
| 70 | 0 | 0 | 0 | 73 | 0 | 0 | 0 | 0 | 0 | 143 | 143 | |
| 6 | 0 | 0 | 3 | 0 | 0 | 6 | 0 | 0 | 0 | 15 | 15 | |
| 129 | 0 | 0 | 0 | 264 | 0 | 0 | 0 | 0 | 0 | 393 | 393 | |
| 0 | 0 | 0 | 38 | 35 | 0 | 0 | 0 | 0 | 0 | 73 | 73 | |
| 15 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 33 | 33 | |
| 66 | 0 | 51 | 46 | 0 | 0 | 0 | 0 | 0 | 0 | 163 | 163 | |
| 0 | 0 | 0 | 0 | 50 | 0 | 50 | 0 | 0 | 0 | 100 | 100 | |
| 0 | 0 | 0 | 0 | 0 | 178 | 175 | 0 | 0 | 0 | 353 | 353 | |
| 215 | 0 | 0 | 0 | 0 | 0 | 218 | 0 | 0 | 0 | 433 | 433 | |
| 23 | 0 | 20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 43 | 43 | |
| 42 | 0 | 45 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 87 | 87 | |
| ? | 0 | 0 | 0 | 0 | 0 | ? | 0 | 0 | 0 | 37 | 37 | |
| 0 | 0 | 45 | 49 | 0 | 0 | 0 | 0 | 0 | 0 | 94 | 94 | |
| 20 | 0 | 20 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 40 | 40 | |
| 42 | 0 | 0 | 0 | 0 | 0 | 0 | 46 | 0 | 0 | 88 | 88 | |
| 198 | 0 | 0 | 0 | 200 | 0 | 0 | 0 | 195 | 0 | 593 | 593 | |
| 92 | 0 | 0 | 0 | 93 | 0 | 0 | 0 | 0 | 0 | 185 | 185 | |
| 0 | 0 | 15 | 18 | 0 | 0 | 0 | 0 | 0 | 0 | 33 | 33 | |
| 17 | 0 | 0 | 16 | 0 | 0 | 0 | 0 | 0 | 0 | 33 | 33 | |
| 30 | 0 | 0 | 0 | 0 | 0 | 30 | 0 | 0 | 0 | 60 | 60 | |
| 88 | 0 | 0 | 30 | 121 | 0 | 0 | 0 | 0 | 0 | 239 | 239 | |
| 0 | 51 | 52 | 48 | 0 | 0 | 0 | 0 | 0 | 0 | 151 | 151 | |
| Totalc | 1721 | 315 | 374 | 538 | 1040 | 501 | 645 | 46 | 353 | 0 | 5570 | 5570 |
| Grand totalc | 5093 | 754 | 1383 | 2303 | 3064 | 979 | 1898 | 1209 | 948 | 282 | 17,950 | 17,961 |
| CBZ: carbamazepine; GBP: gabapentin; IPD: individual participant data; ITT: intention to treat; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide aDrug allocated missing for 11 participants in the IPD provided. | ||||||||||||
| Trial | Gender | Epilepsy type | Epilepsy type reclassifiedc | ||||||
| Male | Female | Missing | Genb | Partial | Missing | Genb | Partial | Unclassifiedd | |
| 61 (56%) | 47 (44%) | 0 (0%) | 49 (45%) | 59 (55%) | 0 (0%) | 49 (45%) | 59 (55%) | 0 (0%) | |
| 347 (60%) | 236 (40%) | 0 (0%) | 0 (0%) | 583 (100%) | 0 (0%) | 0 (0%) | 583 (100%) | 0 (0%) | |
| 174 (61%) | 113 (39%) | 0 (0%) | 105 (37%) | 182 (63%) | 0 (0%) | 75 (26%) | 182 (63%) | 30 (10%) | |
| 60 (44%) | 75 (55%) | 1 (1%) | 46 (34%) | 82 (60%) | 8 (6%) | 33 (24%) | 82 (60%) | 21 (15%) | |
| 56 (41%) | 80 (59%) | 0 (0%) | 54 (40%) | 82 (60%) | 0 (0%) | 34 (25%) | 82 (60%) | 20 (15%) | |
| 56 (45%) | 68 (55%) | 0 (0%) | 62 (50%) | 62 (50%) | 0 (0%) | 39 (31%) | 62 (50%) | 23 (19%) | |
| 83 (55%) | 67 (45%) | 0 (0%) | 45 (30%) | 105 (70%) | 0 (0%) | 0 (0%) | 105 (70%) | 45 (30%) | |
| 319 (55%) | 260 (45%) | 0 (0%) | 113 (20%) | 466 (80%) | 0 (0%) | 50 (9%) | 466 (80%) | 63 (11%) | |
| 157 (54%) | 135 (46%) | 0 (0%) | 0 (0%) | 292 (100%) | 0 (0%) | 0 (0%) | 292 (100%) | 0 (0%) | |
| 71 (43%) | 92 (55%) | 3 (2%) | 86 (52%) | 80 (48%) | 0 (0%) | 2 (1%) | 80 (48%) | 84 (51%) | |
| 86 (50%) | 81 (47%) | 6 (3%) | 84 (49%) | 89 (51%) | 0 (0%) | 84 (49%) | 89 (51%) | 0 (0%) | |
| 21 (40%) | 31 (60%) | 0 (0%) | 0 (0%) | 52 (100%) | 0 (0%) | 0 (0%) | 52 (100%) | 0 (0%) | |
| 48 (57%) | 36 (43%) | 0 (0%) | 0 (0%) | 84 (100%) | 0 (0%) | 0 (0%) | 84 (100%) | 0 (0%) | |
| 100 (52%) | 93 (48%) | 0 (0%) | 50 (26%) | 143 (74%) | 0 (0%) | 45 (23%) | 143 (74%) | 5 (3%) | |
| 117 (48%) | 126 (52%) | 0 (0%) | 141 (58%) | 102 (42%) | 0 (0%) | 82 (34%) | 102 (42%) | 59 (24%) | |
| 40 (49%) | 41 (51%) | 0 (0%) | 48 (59%) | 29 (36%) | 4 (5%) | 25 (31%) | 29 (36%) | 27 (33%) | |
| 57 (52%) | 53 (48%) | 0 (0%) | 15 (14%) | 95 (86%) | 0 (0%) | 6 (5%) | 95 (86%) | 9 (8%) | |
| 413 (87%) | 58 (12%) | 4 (1%) | 1 (0%) | 474 (100%) | 0 (0%) | 1 (0%) | 474 (100%) | 0 (0%) | |
| 445 (93%) | 35 (7%) | 0 (0%) | 0 (0%) | 480 (100%) | 0 (0%) | 0 (0%) | 480 (100%) | 0 (0%) | |
| 329 (53%) | 293 (47%) | 0 (0%) | 3 (1%) | 619 (99%) | 0 (0%) | 1 (0%) | 619 (100%) | 2 (0%) | |
| 34 (62%) | 21 (38%) | 0 (0%) | 45 (82%) | 10 (18%) | 0 (0%) | 26 (47%) | 10 (18%) | 19 (35%) | |
| 47 (50%) | 45 (48%) | 2 (2%) | 34 (36%) | 60 (64%) | 0 (0%) | 34 (36%) | 60 (64%) | 0 (0%) | |
| 67 (35%) | 125 (65%) | 0 (0%) | 59 (31%) | 133 (69%) | 0 (0%) | 35 (18%) | 133 (69%) | 24 (13%) | |
| (CBZ branch)a | 215 (54%) | 180 (46%) | 0 (0%) | 88 (22%) | 285 (72%) | 22 (6%) | 51 (13%) | 285 (72%) | 59 (15%) |
| (VPS branch)a | 112 (50%) | 113 (50%) | 0 (0%) | 131 (58%) | 78 (35%) | 16 (7%) | 86 (38%) | 78 (35%) | 61 (27%) |
| 73 (54%) | 63 (46%) | 0 (0%) | 136 (100%) | 0 (0%) | 0 (0%) | 110 (81%) | 0 (0%) | 26 (19%) | |
| 126 (48%) | 135 (52%) | 0 (0%) | 150 (57%) | 53 (20%) | 58 (22%) | 80 (31%) | 53 (20%) | 128 (49%) | |
| 188 (54%) | 163 (46%) | 0 (0%) | 114 (32%) | 237 (68%) | 0 (0%) | 71 (20%) | 237 (68%) | 43 (12%) | |
| 153 (51%) | 147 (49%) | 0 (0%) | 154 (51%) | 146 (49%) | 0 (0%) | 87 (29%) | 146 (49%) | 67 (22%) | |
| 922 (54%) | 755 (44%) | 44 (3%) | 25 (1%) | 1491 (87%) | 205 (12%) | 16 (1%) | 1491 (87%) | 214 (12%) | |
| 420 (59%) | 282 (39%) | 14 (2%) | 463 (65%) | 52 (7%) | 201 (28%) | 397 (55%) | 52 (7%) | 267 (37%) | |
| 101 (56%) | 80 (44%) | 0 (0%) | 91 (50%) | 90 (50%) | 0 (0%) | 55 (30%) | 90 (50%) | 36 (20%) | |
| 114 (50%) | 112 (49%) | 1 (0%) | 32 (14%) | 154 (68%) | 41 (18%) | 29 (13%) | 154 (68%) | 44 (19%) | |
| (CBZ branch)a | 551 (55%) | 448 (45%) | 0 (0%) | 141 (14%) | 858 (86%) | 0 (0%) | 48 (5%) | 858 (86%) | 93 (9%) |
| (VPS branch)a | 398 (57%) | 305 (43%) | 0 (0%) | 513 (73%) | 190 (27%) | 0 (0%) | 285 (41%) | 190 (27%) | 228 (32%) |
| 73 (52%) | 67 (48%) | 0 (0%) | 77 (55%) | 63 (45%) | 0 (0%) | 42 (30%) | 63 (45%) | 35 (25%) | |
| 122 (47%) | 138 (53%) | 0 (0%) | 152 (58%) | 108 (42%) | 0 (0%) | 152 (58%) | 108 (42%) | 0 (0%) | |
| 215 (60%) | 146 (40%) | 0 (0%) | 0 (0%) | 361 (100%) | 0 (0%) | 0 (0%) | 361 (100%) | 0 (0%) | |
| Total | 6971(56%) | 5345 (43%) | 75 (1%) | 3307 (27%) | 8529 (69%) | 555 (4%) | 2130 (17%) | 8529 (69%) | 1732 (14%) |
| aTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review. | |||||||||
| Trial | Age (years) | Epilepsy duration (years) | Number of seizures in the last 6 months | |||||||
| Mean | SD | Range | Missing | Median | Range | Missing | Median | Range | Missing | |
| 5.7 | 3.5 | 1 to 15 | 0 | 1.2 | 0 to 11.5 | 0 | 24 | 1 to 7200 | 5 | |
| 36.4 | 15.9 | 18 to 75 | 0 | 0.2 | 0 to 17.7 | 30 | 2 | 1 to 30 | 1 | |
| 26.8 | 10.7 | 15 to 91 | 1 | 0.4 | 0 to 25 | 0 | 3 | 0 to 252 | 0 | |
| 32 | 14.5 | 12 to 76 | 0 | 1 | 0 to 53 | 27 | 2 | 0 to 100 | 2 | |
| 34 | 15.8 | 14 to 71 | 0 | 1 | 0 to 18 | 0 | 4 | 1 to 960 | 0 | |
| 30 | 14.1 | 14 to 81 | 0 | 0.5 | 0 to 19.4 | 0 | 3 | 1 to 1020 | 0 | |
| 76.9 | 6 | 65 to 94 | 0 | NA | NA | 150 | 3 | 0 to 163 | 0 | |
| 39 | 16.2 | 15 to 82 | 0 | NA | NA | 579 | 3 | 1 to 1410 | 4 | |
| 35 | 16.6 | 12 to 86 | 0 | 0.5 | 0 to 7.7 | 5 | 4 | 1 to 146 | 6 | |
| 78.2 | 7.1 | 61 to 95 | 3 | NA | NA | 166 | 3 | 0 to 99 | 3 | |
| 9.9 | 3.6 | 3 to 16 | 6 | 0.5 | 0 to 13.7 | 6 | 3 | 1 to 900 | 6 | |
| 10.8 | 2.3 | 4 to 15 | 0 | NA | NA | 52 | 3 | 1 to 60 | 0 | |
| 8.8 | 2.1 | 5 to 13 | 0 | 0.4 | 0 to 4.5 | 0 | 3 | 2 to 11 | 0 | |
| 18.6 | 9.7 | 5 to 53 | 1 | 0.4 | 0 to 20 | 0 | 2 | 0 to 157 | 0 | |
| 32.3 | 14.8 | 13 to 77 | 3 | 1 | 0 to 40 | 4 | 2 | 1 to 579 | 3 | |
| 33.9 | 10.9 | 16 to 56 | 0 | NA | NA | 81 | 1 | 0 to 540 | 0 | |
| 35.8 | 12.2 | 16 to 60 | 0 | NA | NA | 110 | 2 | 0 to 200 | 0 | |
| 41 | 15.5 | 18 to 82 | 4 | 2 | 0.5 to 59 | 5 | 1 | 1 to 100 | 7 | |
| 47.1 | 16.1 | 18 to 83 | 0 | 3 | 1 to 68 | 19 | 12 | 1 to 2248 | 38 | |
| 27.2 | 21.4 | 2 to 84 | 1 | NA | NA | 622 | 3 | 1 to 9000 | 0 | |
| 27.5 | 8.5 | 14 to 55 | 0 | 7 | 3 to 11.5 | 18 | 12 | 6 to 42 | 0 | |
| 11.4 | 5 | 2 to 18 | 0 | 2.5 | 0.5 to 17 | 2 | NA | NA | 94 | |
| 29 | 17.6 | 2 to 68 | 0 | 5 | 0.5 to 44 | 0 | 2 | 0 to 100 | 0 | |
| (CBZ branch)a | 34.4 | 18.4 | 6 to 80 | 0 | NA | NA | 395 | 4 | 0 to 2400 | 0 |
| (VPS branch)a | 32.8 | 19.4 | 6 to 84 | 0 | NA | NA | 225 | 4 | 0 to 20000 | 0 |
| 20.9 | 14.2 | 3 to 64 | 0 | 0 | 0 to 3 | 0 | NA | NA | 136 | |
| 34.1 | 14.8 | 12 to 78 | 0 | NA | NA | 261 | 4 | 0 to 570 | 0 | |
| 32.1 | 14.2 | 12 to 72 | 2 | 0.7 | 0 to 27 | 3 | 3 | 1 to 145 | 1 | |
| 33 | 14.9 | 16 to 79 | 2 | NA | NA | 300 | 4 | 2 to 101 | 5 | |
| 38.4 | 18.3 | 5 to 86 | 44 | NA | NA | 1721 | 4 | 0 to 1185 | 49 | |
| 22.5 | 14.1 | 5 to 77 | 14 | NA | NA | 716 | 3 | 0 to 2813 | 17 | |
| 34.1 | 16.7 | 13 to 75 | 1 | 1.3 | 0 to 28.5 | 1 | 3 | 1 to 600 | 0 | |
| 36 | 16.9 | 13 to 80 | 2 | NA | NA | 227 | 18 | 6 to 1080 | 37 | |
| (CBZ branch)1 | 42.8 | 17.2 | 16 to 89 | 0 | NA | NA | 999 | NA | NA | 999 |
| (VPS branch)1 | 36.5 | 17.8 | 16 to 85 | 1 | NA | NA | 703 | NA | NA | 703 |
| 35.2 | 16.1 | 14 to 70 | 0 | 0.75 | 0.1 to 30 | 0 | 2 | 0 to 60 | 0 | |
| 10.1 | 2.9 | 5 to 16 | 13 | 0.3 | 0 to 5.9 | 32 | 3 | 1 to 104 | 12 | |
| 71.5 | 7.2 | 60 to 95 | 0 | NA | NA | 361 | 2 | 1 to 96 | 7 | |
| Total (missing) | 98 | 7820 | 2135 | |||||||
| Abbreviations: SD: Standard deviation aTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review. | ||||||||||
| Trial | Electroencephalographic (EEG) | Computerised Tomography (CT) /Magnetic Resonance Imaging (MRI) | Neurological exams | ||||||
| Normal | Abnormal | Missing | Normal | Abnormal | Missing | Normal | Abnormal | Missing | |
| 49 (45%) | 54 (50%) | 5 (5%) | 21 (19%) | 5 (5%) | 82 (76%) | 0 (0%) | 0 (0%) | 108 (100%) | |
| 0 (0%) | 0 (0%) | 583 (100%) | 0 (0%) | 0 (0%) | 583 (100%) | 478 (82%) | 103 (18%) | 2 (0%) | |
| 126 (44%) | 152 (53%) | 9 (3%) | 173 (60%) | 69 (24%) | 45 (16%) | 0 (0%) | 0 (0%) | 287 (100%) | |
| 0 (0%) | 0 (0%) | 136 (100%) | 0 (0%) | 0 (0%) | 136 (100%) | 89 (65%) | 46 (34%) | 1 (1%) | |
| 62 (46%) | 72 (53%) | 2 (1%) | 82 (60%) | 12 (9%) | 42 (31%) | 123 (90%) | 13 (10%) | 0 (0%) | |
| 76 (61%) | 42 (34%) | 6 (5%) | 72 (58%) | 20 (16%) | 32 (26%) | 108 (87%) | 16 (13%) | 0 (0%) | |
| 0 (0%) | 0 (0%) | 150 (100%) | 62 (41%) | 87 (58%) | 1 (1%) | 90 (60%) | 60 (40%) | 0 (0%) | |
| 0 (0%) | 0 (0%) | 579 (100%) | 0 (0%) | 0 (0%) | 579 (100%) | 493 (85%) | 86 (15%) | 0 (0%) | |
| 107 (37%) | 179 (61%) | 6 (2%) | 0 (0%) | 0 (0%) | 292 (100%) | 0 (0%) | 0 (0%) | 292 (100%) | |
| 28 (17%) | 74 (45%) | 64 (39%) | 0 (0%) | 0 (0%) | 166 (100%) | 0 (0%) | 0 (0%) | 166 (100%) | |
| 0 (0%) | 0 (0%) | 173 (100%) | 0 (0%) | 0 (0%) | 173 (100%) | 152 (88%) | 15 (9%) | 6 (3%) | |
| 18 (35%) | 34 (65%) | 0 (0%) | 50 (96%) | 2 (4%) | 0 (0%) | 0 (0%) | 0 (0%) | 52 (100%) | |
| 6 (7%) | 78 (93%) | 0 (0%) | 75 (89%) | 9 (11%) | 0 (0%) | 83 (99%) | 1 (1%) | 0 (0%) | |
| 92 (48%) | 99 (51%) | 2 (1%) | 126 (65%) | 12 (6%) | 55 (28%) | 0 (0%) | 0 (0%) | 193 (100%) | |
| 0 (0%) | 0 (0%) | 243 (100%) | 0 (0%) | 0 (0%) | 243 (100%) | 222 (91%) | 19 (8%) | 2 (1%) | |
| 0 (0%) | 0 (0%) | 81 (100%) | 0 (0%) | 0 (0%) | 81 (100%) | 0 (0%) | 0 (0%) | 81 (100%) | |
| 58 (53%) | 52 (47%) | 0 (0%) | 74 (67%) | 36 (33%) | 0 (0%) | 110 (100%) | 0 (0%) | 0 (0%) | |
| 126 (27%) | 343 (72%) | 6 (1%) | 308 (65%) | 119 (25%) | 48 (10%) | 0 (0%) | 0 (0%) | 475 (100%) | |
| 0 (0%) | 0 (0%) | 480 (100%) | 0 (0%) | 0 (0%) | 480 (100%) | 0 (0%) | 0 (0%) | 480 (100%) | |
| 0 (0%) | 0 (0%) | 622 (100%) | 0 (0%) | 0 (0%) | 622 (100%) | 0 (0%) | 0 (0%) | 622 (100%) | |
| 0 (0%) | 0 (0%) | 55 (100%) | 37 (67%) | 0 (0%) | 18 (33%) | 55 (100%) | 0 (0%) | 0 (0%) | |
| 0 (0%) | 0 (0%) | 94 (100%) | 0 (0%) | 0 (0%) | 94 (100%) | 24 (26%) | 70 (74%) | 0 (0%) | |
| 180 (94%) | 12 (6%) | 0 (0%) | 0 (0%) | 0 (0%) | 192 (100%) | 0 (0%) | 0 (0%) | 192 (100%) | |
| (CBZ branch)a | 0 (0%) | 0 (0%) | 395 (100%) | 0 (0%) | 0 (0%) | 395 (100%) | 0 (0%) | 0 (0%) | 395 (100%) |
| (VPS branch)a | 0 (0%) | 0 (0%) | 225 (100%) | 0 (0%) | 0 (0%) | 225 (100%) | 0 (0%) | 0 (0%) | 225 (100%) |
| 0 (0%) | 0 (0%) | 136 (100%) | 0 (0%) | 0 (0%) | 136 (100%) | 0 (0%) | 0 (0%) | 136 (100%) | |
| 0 (0%) | 0 (0%) | 261 (100%) | 0 (0%) | 0 (0%) | 261 (100%) | 0 (0%) | 0 (0%) | 261 (100%) | |
| 13 (4%) | 13 (4%) | 325 (93%) | 16 (5%) | 5 (1%) | 330 (94%) | 305 (87%) | 46 (13%) | 0 (0%) | |
| 0 (0%) | 0 (0%) | 300 (100%) | 0 (0%) | 0 (0%) | 300 (100%) | 0 (0%) | 0 (0%) | 300 (100%) | |
| 0 (0%) | 0 (0%) | 1721 (100%) | 0 (0%) | 0 (0%) | 1721 (100%) | 1267 (74%) | 410 (24%) | 44 (3%) | |
| 0 (0%) | 0 (0%) | 716 (100%) | 0 (0%) | 0 (0%) | 716 (100%) | 595 (83%) | 107 (15%) | 14 (2%) | |
| 103 (57%) | 71 (39%) | 7 (4%) | 111 (61%) | 33 (18%) | 37 (20%) | 165 (91%) | 16 (9%) | 0 (0%) | |
| 51 (22%) | 121 (53%) | 55 (24%) | 0 (0%) | 0 (0%) | 227 (100%) | 0 (0%) | 0 (0%) | 227 (100%) | |
| (CBZ branch)1 | 0 (0%) | 0 (0%) | 999 (100%) | 0 (0%) | 0 (0%) | 999 (100%) | 0 (0%) | 0 (0%) | 999 (100%) |
| (VPS branch)1 | 0 (0%) | 0 (0%) | 703 (100%) | 0 (0%) | 0 (0%) | 703 (100%) | 0 (0%) | 0 (0%) | 703 (100%) |
| 70 (50%) | 70 (50%) | 0 (0%) | 17 (12%) | 10 (7%) | 113 (81%) | 0 (0%) | 0 (0%) | 140 (100%) | |
| 0 (0%) | 0 (0%) | 260 (100%) | 0 (0%) | 0 (0%) | 260 (100%) | 0 (0%) | 0 (0%) | 260 (100%) | |
| 117 (32%) | 242 (67%) | 2 (1%) | 78 (22%) | 282 (78%) | 1 (0%) | 0 (0%) | 0 (0%) | 361 (100%) | |
| Total | 1282 (10%) | 1708 (14%) | 9401 (75%) | 1302 (11%) | 701 (6%) | 10,388 (83%) | 4359 (36%) | 1008 (8%) | 7024 (56%) |
| aTrials designed in two strata based on whether recommended treatment would be CBZ or VPS. Within the two strata, participants were randomised to TPM in Privitera 2003/LEV in Trinka 2013 or CBZ/VPS depending on the strata. Data analysed according to the separate strata (CBZ branch or VPS branch) in this review. | |||||||||
| Trial | Summary of resultsb |
| 1. MANOVA revealed no significant interaction effect of group and time 2. MANOVA revealed no significant interaction effect of group and time 3. MANOVA revealed no significant interaction effect of group and time 4. MANOVA revealed no significant interaction effect of group and time | |
| 1. CBZ: 64%, PHB: 63% 2. No statistically significant difference between groups 3. No statistically significant difference between groups 4. Mean seizure frequency: CBZ: 0.66, PHB: 0.8 5. Mean duration (seconds): CBZ: 12.63; PHB: 15 | |
| 1. Median time to exit: GBP: 69 days, LTG: 48 days HR: 1.043 (95% confidence interval 0.602 to 1.809) 2. Proportion of evaluable population completing the study – GBP: 71.6%, LTG: 67.1% No difference between groups for time to withdrawal for any reason 3. No difference between groups for time to first seizure 4. GBP: 76.1%, LTG: 76.8% (ITT population) 5. Withdrawals during titration: GBP: 7, LTG: 10 Withdrawals after titration: GBP: 10, LTG: 13 | |
| 1a. PHT: 67%; CBZ: 37%; VPS: 53% 2. PHT: 10%; CBZ: 8%; VPS:11% | |
| 1. CBZ: 76%, LEV: 76% 2. Proportion with AEs: CBZ: 65%, LEV: 50% 3. CBZ: 2 discontinuations due to failure to control seizures and interactions with other medications LEV: 3 discontinuations – 1 death from stroke and 2 due to AEs | |
| 1. No significant difference between groups 2. No significant difference between groups | |
| 1. No significant difference between groups 3. 2 children from PHB group, 1 child from CBZ group and no children from VPS group withdrew from the study because of allergic reactions 4. No significant difference between groups | |
| 1. Overall effect on sleep parameters was comparable between groups. LEV group PSG significant increase post treatment compared to baseline in sleep efficiency (P = 0.039) and in decrease of wake time after sleep onset (P = 0.047), no significant change in other sleep parameters. CBZ group post treatment compared to baseline significant increases in the percentage of slow wave sleep (P = 0.038), no significant change in other sleep parameters 2. No significant difference between baseline and post‐treatment between the 2 groups | |
| 1. OXC 56.6% ; VPS 53.8% 2. No significant difference between groups 3. OXC 40.6% ; VPS 33.9% 4. Efficacy no significant difference between groups Tolerability no significant difference between groups Therapeutic effect no significant difference between groups 5. Proportion of participants experiencing at least 1 AE regardless of relationship to trial drug OXC 89.8%; VPS 87.6% 6. Seizure frequency per week OXC (n = 106) mean 0.17 median 0, VPS (n = 106) mean 0.40, median 0 | |
| 1. No significant difference between groups 2. Completed study LEV 52/62, CBZ 54/66, withdrawals: 8 poor compliance (LEV 4, CBZ 4); 7 severe adverse effect (LEV 3, CBZ 4); 7 unknown cause (LEV 3, CBZ 4) 3. Attention deficit on digital span end of follow up greater in CBZ group than LEV (P = 0.03) Stroop test worse in CBZ than LEV (P = 0.02) No significant difference between groups for other scales. Impairment of activities of daily living greater CBZ than LEV (P = 0.05) 4. 4 participants (LEV 2, CBZ 2) had abnormal EEG at baseline, normal at end of treatment. Drug dose reduction (LEV 4, CBZ 2). Remaining participants unmodified versus baseline 5. No significant difference between groups | |
| 1. Significant decrease in visual‐verbal memory for CBZ and acoustic memory for PHB. No significant differences for other tests | |
| 1. PHB: 60%, PHT: 59%; CBZ: 62%; VPS: 64% | |
| 1. Baseline OXC mean 2.9 (SD 7.0), median 1, range 0‐60 CBZ mean 5.8 (SD 14.7) median 1, range 0‐99 Maintenance phases OXC mean 0.4 (SD 3.0) median 0, range 0‐27 CBZ mean 0.3 (SD 1.4) median 0, range 0‐12 2. Severe side effects CBZ 25, OXC 13, statistically significant difference favouring OXC (P = 0.04) Participants without any side effects CBZ 25, OXC 29 no significant difference between groups (P = 0.22) 3. Global efficacy no significant difference between groups (P = 0.77); global tolerability (P = 0.11) Participants very good/good CBZ 69 (73%), OXC 76 (84%) Participants poor/very poor CBZ 26 (27%), OXC 15 (16%) 4. Nature of side effects same between groups, included tiredness, headache, dizziness, ataxia. Participants withdrawn due to severe side effects CBZ 16, OXC 9 5. Clinically relevant changes observed in 2 participants only, both CBZ group, both stopped treatment | |
| 1. Comparison of cognitive results no significant difference between treatment groups (P = 0.195) No significant difference between treatment groups for secondary variables (psychomotor speed, alertness, memory and learning, attention, intelligence scores) 2. OXC 58%; CBZ 46%; VPS 54% 3. Most common (> 10% reported) side effects OXC fatigue and headache; CBZ fatigue and rash VPS headache, increased appetite, alopecia 4. Good/very good: OXC investigators 84%, participants 82%, parents/carers 86%; Combined CBZ/VPS investigators 77%, participants 73%, parents/carers 80% | |
| 1. Minor adverse effects reported in PHB: 58 participants (39%) reported 86 AEs, CBZ: 46 participants (30%) reported 68 AEs 2. All withdrawals: PHB: 18%, CBZ: 17% Withdrawals due to side‐effects: PHB: 5%, CBZ: 3% 3. Seizure‐free: PHB: 54%, CBZ: 52% > 50% reduction of seizures: PHB: 23%, CBZ: 29% 50% reduction‐50% increase in seizures: PHB: 15%, CBZ: 13% > 50% increase in seizures: PHB: 8%, CBZ: 6% | |
| 1. Significant difference favouring VPS test of speed of information processing No significant differences between treatment groups for any other cognitive tests 2. PHT: 30%; CBZ: 39%; VPS:33% | |
| 1. Seizure freedom: LTG: 38%, OXC: 44% < 50% seizure reduction: LTG: 48%, OXC: 55% 2. Both groups showed improvement in verbal learning and in 1/4 measures of attention. In addition, participants under OXC improved in word fluency. Improved mood was reported with OXC only. | |
| 1. Number of participants experiencing early seizures as first event: LTG 2/32, CBZ 3/32 Number of participants remaining seizure‐free in the follow‐up period: LTG 23/32 (72%), CBZ 14/32 (44%) P = 0.05 2. Incidence of side effects: LTG 2/32 (6.25%), CBZ 12/32 (37.5%) P = 0.05 3. Withdrawals from study due to side effects LTG 1/32 (3%), CBZ 10/32 (31%), P = 0.02 | |
| 1. No difference between groups in terms of social competence; school competence; internalising behaviour problems; externalising behaviour problems; total behaviour problems and anxiety. Significant decrease in depression in LEV group compared to CBZ group (P = 0.027) 2. LEV 95.7% , CBZ 97.1% , P = 0.686 3. LEV 66.7%, CBZ 57.8% , P = 0.317 4. LEV 33.3%, CBZ 46.9%. Number of AEs not significantly different between groups | |
| 1.CBZ: 53% LTG: 56% 2. No significant difference between groups in overall cognitive score. In terms of individual assessments, only Stroop test B showed a statistically significant advantage for LTG. | |
| 1. No significant difference between groups 2. No significant difference between groups | |
| 1. LTG: 65% CBZ: 70% 2. Total seizure‐free rate LTG: 62% CBZ: 63% Time to first seizure: mean (SD): weeks LTG: 10 (5.09), CBZ: 10.82 (6.44) | |
| 1. LTG: 54%, VPS: 55 %, no difference by seizure type 2. LTG: 69%, VPS:68 % | |
| 1. No significant differences between treatment groups 2. Compliance: trend towards better compliance in CBZ group (not significant) Randomised participants only: trend towards higher rate withdrawal from treatment in PHB group (not significant). More mild systemic side‐effects in CBZ group (significant). 3 children switched from CBZ to PHB and 1 from PHB to CB following adverse reactions 3. 6 months: excellent/good: PHB = 15, CBZ = 13 12 months: excellent/good: PHB = 13, CBZ = 9 | |
| 1. Partial seizures ‐ PHT: 32%; CBZ: 40%; VPS : 41% Generalised seizures ‐ PHT :35%; CBZ: 15%; VPS: 7% 1. Partial seizures ‐ PHT: 24%; CBZ: 24%; VPS : 25% Generalised seizures ‐ PHT :13%; CBZ: 0%; VPS: 0% | |
| 1. Seizure recurrence at 2 weeks ‐ LTG: 43% LEV: 35%, p=0.42 Seizure recurrence at 4 weeks ‐ LTG: 39% LEV: 33%, p=0.53 Seizure recurrence at 8 weeks ‐ LTG: 35% LEV: 28%, p=0.50 Seizure recurrence at 12 weeks ‐ LTG: 33% LEV: 24%, p=0.35 Seizure recurrence at 20 weeks ‐ LTG: 31% LEV: 13%, p=0.03 2. No significant difference between groups 3. Proportion with AEs ‐ LTG: 53%, LEV: 67% | |
| 1. LEV: 12.7%, OXC: 23.4% 2. Median months: LEV: 7.6, OXC: NA (fewer than 50% of participants in the OXC group had seizure recurrence) 3. LEV: 53.8%, OXC: 58.5% 4. LEV: 34.7%, OXC: 40.9% | |
| 1. LEV: 47.3%, CBZ: 68.4% 2. LEV: 48.4%, CBZ: 70.2% 3. Number of events: LEV: 88, CBZ: 45 4. Number of events: LEV: 87, CBZ: 39 5. Number of events: LEV: 97, CBZ: 57 | |
| 1. Compared to CBZ, participants on PHT became slower (motor speed of the hand) and their visual memory decreased. There was an equal decrease in negative mood (helplessness, irritability, depression) on PHT and CBZ 2. 3 participants taking PHT complained of tiredness, and 1 participant taking CBZ complained of facial skin problems, another tiredness and memory problems | |
| 1. Incidence of major side effects (proportion of analysed participants): PHT 23%; CBZ: 23% Minor side effects: cognitive impairment and sedation twice as likely on CBZ compared to PHT. Other minor side effects similar between groups. 2. Treatment failures among analysed participants: Seizure control (among analysed participants with no major side effects): PHT: 86%; CBZ: 82% 3. Significantly lower mean LDH level at 24 weeks in CBZ participants than PHT participants. Other laboratory results similar across treatment groups | |
| 1. 8 discontinuations; due to generalised rash (n = 1), excessive tiredness (n = 1), withdrew consent (n = 2), renal transplant (n = 1), lost to follow‐up (n = 2), died (n = 1) 2. 6 participants reported treatment‐emergent side effects. 3. No participants withdrew due to lack of seizure control | |
| 1(a). PHT: 51%, VPS: 49% 1(b). PHT : 24%, VPS: 35% 1(c). PHT: 18%, VPS: 10% 1(d). PHT: 2%, VPS: 6% 2. All reported AEs were minor and similar rates between groups | |
| 1. No significant differences between any tests of cognitive function taken before treatment and after 10‐12 weeks for both treatment groups | |
| 1. Six months of seizure freedom: CBZ: 81%, TPM: 91% 50% reduction of seizures: CBZ: 84% TPM: 97% The average number of seizures was significantly less in the TPM group compared to the CBZ group at 6 and 9 months 2. AEs were mild and similar between groups 3. No significant differences between groups | |
| 1. Significant difference between 3 treatment groups (P = 0.00022) CBZ more early terminators than GBP (P = 0.008) or LTG (P < 0.0001) 2. LTG 51.4%, GBP 47.4%, CBZ 64.3% no significant difference between groups P = 0.09 3. No difference between groups for time to first, second, fifth and tenth seizure (P values = 0.18, 0.13, 0.74, 0.95 respectively) 4. More systemic toxicities on GBP than CBZ or LTG No significant differences in neuro‐toxicities between treatment groups over 12 months 5. Mean serum levels: 6 weeks GBP 8.67 ± 4.83; µg/mL, CBZ 6.79 ± 2.92 µg/mL and LTG 2.87 ± 1.60 µg/mL 52 weeks GBP 8.54 ± 5.57 µg/mL, CBZ 6.48 ± 3.72 µg/mL and LTG 3.46 ± 1.68 µg/mL Overall medical compliance 89% without significant group differences 6. 3 months LTG 49.7%, GBP 43.3%, CBZ 36.0% significant difference between groups P = 0.02 6 months LTG 37.2%, GBP 33.0%, CBZ 28.9% no significant difference between groups P = 0.22 12 months LTG 28.6%, GBP 23.2%, CBZ 22.8% no significant difference between groups P = 0.33 | |
| 1. LTG 68 (73%), CBZ 61 (67%), no significant difference between groups 2. LTG 59 (63%), CBZ 69 (76%), not significant difference P = 0.068 ITT analysis 3. LTG 71 (76%), CBZ 81 (89%), significant difference, P = 0.0234 ITT analysis 4.Hazard ratio (lamotrigine/carbamazepine) 1.50, 95% CI 0.94–2.40, p value 0.092 5. During treatment period LTG 82 (88%) reported 378 AEs, CBZ 79 (86%) reported 310 AEs. No significant differences between groups for any AEs except for immune system Withdrew due to AE LTG 13 (14%), CBZ 23 (25%), P = 0.078 6. No difference between groups even when changes over time corrected for age, gender and baseline score | |
| 1. PHT: 33%; VPS: 39% 2. All reported AEs were minor and similar rates between groups | |
| 1. VPS 7/11 (64%), CBZ 9/14 (64%) 2. At least one AE reported VPS 15/16 (94%), CBZ 16/17 (94%) | |
| 1. FE CBZ group 83/88 (94.3%), LTG group 78/88 (88.6%) no significant difference between groups GE VPS group 25/30 (83.3%) LTG group 20/33 (60.6%) no significant difference between groups 2. FE CBZ group 81%, LTG group 91%, not a significant difference between groups GE VPS group 97%, LTG group 88%, not stated as significant or non‐significant difference 3. At least 1 AE FE CBZ 81 participants (91%), LTG 68 participants (77.3%) GE VPS 25 participants (83.3%), LTG 24 participants (72.7%) Serious AEs FE CBZ 8 participants (9%), LTG 6 participants (7%) GE VPS 1 participant (3%), LTG 5 participants (15%) AEs considered related to study drug FE CBZ 65 participants (74%), LTG 38 participants (43%) GE VPS 16 participants (53%), LTG 15 participants (45.5%) | |
| 1. Mean quality‐of‐life score at baseline CBZ group 31.14 ± 1.83, LEV group 29.76 ± 1.71 (P value = 0.5861) Mean quality of life score after 26 weeks of treatment CBZ group 58.41 ± 1.89, LEV 64.58 ± 2.02 (P value = 0.0302) 2.28 participants in CBZ group, 28 in LEV group Seizure freedom 4 weeks CBZ group 85.72%, LEV group 85.72% (P value = 1); 12 weeks CBZ group 89.29%, LEV group 93.34% (P value = 0.4595); 26 weeks CBZ group 96.43%, LEV group 100% (P value = 0.1212); 6 months CBZ group 71.42% (20 participants), LEV group 78.57% (22 participants) (P value = 0.2529) 3. Participants experiencing at least 1 AE, CBZ group 36.66%, LEV group 40% (P value = 0.77) | |
| 1. PHB: 31%, PHT: 27%, VPS: 21% 2. PHB: 33%, PHT: 63%, VPS: 31% | |
| AE: adverse event; CBZ: carbamazepine; EEG: electroencephalogram; FE: focal epilepsies; GBP: gabapentin; GE: generalised epilepsies; ITT: intention to treat; LDH: lactic acid dehydrogenase; LEV: levetiracetam; LTG: lamotrigine; MANOVA: repeated measures analysis of variance; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; SD: standard deviation; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide aFor further details of adverse events see Table 16 and Table 17. | |
| Trial | Time to withdrawal from allocated treatmentc | Time to first seizure | Time to 12‐month remissiond | Time to six‐month remissiond | ||||||||||||
| Cens | Event | Total | Missing | Cens | Event | Total | Missing | Cens | Event | Total | Missing | Cens | Event | Total | Missing | |
| 0 | 0 | 0 | 108 | 39 | 69 | 108 | 0 | 0 | 0 | 0 | 108 | 0 | 0 | 0 | 108 | |
| 392 | 191 | 583 | 0 | 388 | 186 | 574 | 9 | 251 | 323 | 574 | 9 | 194 | 380 | 574 | 9 | |
| 232 | 55 | 287 | 0 | 137 | 145 | 282 | 5 | 190 | 92 | 282 | 5 | 136 | 146 | 282 | 5 | |
| 99 | 36 | 135 | 1 | 64 | 71 | 135 | 1 | 0 | 0 | 0 | 136 | 90 | 45 | 135 | 1 | |
| 78 | 58 | 136 | 0 | 69 | 67 | 136 | 0 | 0 | 0 | 0 | 136 | 122 | 14 | 136 | 0 | |
| 79 | 45 | 124 | 0 | 52 | 72 | 124 | 0 | 0 | 0 | 0 | 124 | 96 | 28 | 124 | 0 | |
| 95 | 55 | 150 | 0 | 70 | 80 | 150 | 0 | 0 | 0 | 0 | 150 | 106 | 44 | 150 | 0 | |
| 323 | 256 | 579 | 0 | 350 | 229 | 579 | 0 | 260 | 319 | 579 | 0 | 177 | 402 | 579 | 0 | |
| 69 | 223 | 292 | 0 | 102 | 190 | 292 | 0 | 0 | 0 | 0 | 292 | 193 | 99 | 292 | 0 | |
| 0 | 0 | 0 | 166 | 68 | 81 | 149 | 17 | 117 | 30 | 147 | 19 | 58 | 89 | 147 | 19 | |
| 100 | 67 | 167 | 6 | 18 | 149 | 167 | 6 | 22 | 145 | 167 | 6 | 19 | 148 | 167 | 6 | |
| 44 | 8 | 52 | 0 | 40 | 12 | 52 | 0 | 11 | 41 | 52 | 0 | 8 | 44 | 52 | 0 | |
| 75 | 9 | 84 | 0 | 52 | 32 | 84 | 0 | 0 | 0 | 0 | 84 | 35 | 49 | 84 | 0 | |
| 151 | 42 | 193 | 0 | 106 | 84 | 190 | 3 | 112 | 78 | 190 | 3 | 84 | 106 | 190 | 3 | |
| 166 | 77 | 243 | 0 | 66 | 177 | 243 | 0 | 78 | 165 | 243 | 0 | 49 | 194 | 243 | 0 | |
| 60 | 21 | 81 | 0 | 38 | 29 | 67 | 14 | 68 | 13 | 81 | 0 | 30 | 50 | 80 | 1 | |
| 73 | 37 | 110 | 0 | 79 | 31 | 110 | 0 | 0 | 0 | 0 | 110 | 39 | 71 | 110 | 0 | |
| 267 | 208 | 475 | 0 | 226 | 238 | 464 | 11 | 325 | 149 | 474 | 1 | 281 | 193 | 474 | 1 | |
| 308 | 172 | 480 | 0 | 165 | 303 | 468 | 12 | 334 | 133 | 467 | 13 | 242 | 225 | 467 | 13 | |
| 511 | 111 | 622 | 0 | 310 | 312 | 622 | 0 | 0 | 0 | 0 | 622 | 431 | 191 | 622 | 0 | |
| 0 | 0 | 0 | 55 | 29 | 26 | 55 | 0 | 0 | 0 | 0 | 55 | 0 | 0 | 0 | 55 | |
| 0 | 0 | 0 | 94 | 41 | 49 | 90 | 4 | 82 | 8 | 90 | 4 | 63 | 27 | 90 | 4 | |
| 158 | 32 | 190 | 2 | 121 | 71 | 192 | 0 | 132 | 60 | 192 | 0 | 69 | 123 | 192 | 0 | |
| (CBZ branch)b | 221 | 174 | 395 | 0 | 208 | 187 | 395 | 0 | 316 | 79 | 395 | 0 | 194 | 201 | 395 | 0 |
| (VPS branch)b | 111 | 114 | 225 | 0 | 119 | 106 | 225 | 0 | 180 | 45 | 225 | 0 | 106 | 119 | 225 | 0 |
| 113 | 23 | 136 | 0 | 81 | 44 | 125 | 11 | 0 | 0 | 0 | 136 | 78 | 47 | 125 | 11 | |
| 192 | 69 | 261 | 0 | 197 | 64 | 261 | 0 | 0 | 0 | 0 | 261 | 0 | 0 | 0 | 261 | |
| 288 | 63 | 351 | 0 | 216 | 135 | 351 | 0 | 0 | 0 | 0 | 351 | 328 | 23 | 351 | 0 | |
| 210 | 76 | 286 | 14 | 91 | 199 | 290 | 10 | 92 | 198 | 290 | 10 | 77 | 213 | 290 | 10 | |
| 857 | 815 | 1672 | 49 | 383 | 1261 | 1644 | 77 | 577 | 1067 | 1644 | 77 | 355 | 1326 | 1681 | 40 | |
| 400 | 299 | 699 | 17 | 182 | 511 | 693 | 23 | 167 | 526 | 693 | 23 | 96 | 610 | 706 | 10 | |
| 108 | 73 | 181 | 0 | 100 | 81 | 181 | 0 | 0 | 0 | 0 | 181 | 157 | 24 | 181 | 0 | |
| 160 | 67 | 227 | 0 | 81 | 140 | 221 | 6 | 172 | 55 | 227 | 0 | 137 | 90 | 227 | 0 | |
| (CBZ branch)b | 760 | 239 | 999 | 0 | 572 | 427 | 999 | 0 | 780 | 219 | 999 | 0 | 336 | 663 | 999 | 0 |
| (VPS branch)b | 583 | 120 | 703 | 0 | 456 | 247 | 703 | 0 | 484 | 219 | 703 | 0 | 191 | 512 | 703 | 0 |
| 91 | 49 | 140 | 0 | 75 | 65 | 140 | 0 | 47 | 93 | 140 | 0 | 36 | 104 | 140 | 0 | |
| 187 | 59 | 246 | 14 | 59 | 187 | 246 | 14 | 84 | 162 | 246 | 14 | 19 | 227 | 246 | 14 | |
| 195 | 166 | 361 | 0 | 249 | 96 | 345 | 16 | 211 | 150 | 361 | 0 | 178 | 183 | 361 | 0 | |
| Total | 7756 | 4109 | 11,865 | 526 | 5699 | 6453 | 12,152 | 239 | 5092 | 4369 | 9461 | 2930 | 4810 | 7010 | 11,820 | 571 |
| Abbreviation: cens = censored aFor two studies we could only calculate 'Time to first seizure'; the study duration of Ogunrin 2005 was 12 weeks, and all randomised participants completed the study without withdrawing; and Banu 2007 did not record the dates of all seizures after randomisation and dates of withdrawal for allocated treatment for all participants. | ||||||||||||||||
| Reason for withdrawal | Classification for analysis | Randomised drug4b | ||||||||||
| CBZ | PHB | PHT | VPS | LTG | OXC | TPM | GBP | LEV | ZNS | Total | ||
| Adverse events | Event | 505 (45%) | 24 (20%) | 93 (35%) | 132 (28%) | 235 (41%) | 56 (41%) | 259 (48%) | 73 (20%) | 134 (39%) | 31 (32%) | 1542 (38%) |
| Inadequate response | Event | 232 (20%) | 20 (16%) | 46 (17%) | 140 (29%) | 144 (26%) | 36 (26%) | 148 (27%) | 223 (62%) | 89 (26%) | 23 (24%) | 1101 (27%) |
| Both adverse events and inadequate response | Event | 148 (13%) | 51 (41%) | 54 (20%) | 107 (22%) | 32 (6%) | 11 (8%) | 46 (8%) | 32 (9%) | 0 (0%) | 0 (0%) | 481 (12%) |
| Protocol violation/non compliance | Event | 102 (9%) | 15 (12%) | 41 (15%) | 11 (2%) | 68 (12%) | 27 (20%) | 0 (0%) | 21 (6%) | 21 (6%) | 3 (3%) | 309 (8%) |
| Withdrew consent | Event | 121 (11%) | 13 (11%) | 25 (9%) | 64 (13%) | 65 (11%) | 2 (1%) | 55 (10%) | 4 (1%) | 68 (20%) | 35 (36%) | 452 (11%) |
| Othera | Event | 29 (3%) | 0 (0%) | 7 (3%) | 24 (5%) | 26 (5%) | 5 (4%) | 37 (7%) | 9 (2%) | 32 (9%) | 4 (4%) | 173 (4%) |
| Total eventsb | 1137 (35%) | 123 (38%) | 266 (31%) | 478 (28%) | 570 (29%) | 137 (29%) | 545 (47%) | 362 (61%) | 344 (27%) | 96 (34%) | 4058 (34%) | |
| Illness or death | Censored | 34 (2%) | 10 (5%) | 17 (3%) | 7 (1%) | 20 (1%) | 1 (0%) | 10 (2%) | 9 (4%) | 0 (0%) | 0 (0%) | 108 (1%) |
| Remission of seizures | Censored | 49 (2%) | 4 (2%) | 38 (6%) | 75 (6%) | 40 (3%) | 12 (4%) | 44 (7%) | 21 (9%) | 0 (0%) | 0 (0%) | 283 (4%) |
| Lost to follow‐up | Censored | 81 (4%) | 31 (16%) | 51 (9%) | 63 (5%) | 33 (3%) | 24 (7%) | 18 (3%) | 0 (0%) | 41 (5%) | 0 (0%) | 342 (4%) |
| Otherc | Censored | 104 (5%) | 6 (3%) | 22 (4%) | 82 (7%) | 31 (2%) | 5 (2%) | 26 (4%) | 26 (12%) | 0 (0%) | 25 (13%) | 327 (4%) |
| Completed study | Censored | 1829 (87%) | 139 (73%) | 468 (79%) | 949 (81%) | 1272 (91%) | 291 (87%) | 501 (84%) | 166 (75%) | 868 (95%) | 161 (87%) | 6644 (86%) |
| Total censoredb | 2097 (65%) | 190 (62%) | 596 (69%) | 1176 (72%) | 1396 (71%) | 333 (71%) | 599 (53%) | 222 (39%) | 909 (73%) | 186 (66%) | 7704 (66%) | |
| Missingd | 24 | 7 | 1 | 26 | 12 | 8 | 14 | 11 | 0 | 0 | 103 | |
| Totale | 3258 | 320 | 863 | 1680 | 1978 | 478 | 1158 | 595 | 1253 | 282 | 11,865 | |
| CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide aOther treatment‐related reasons included: physician's decision, drug‐related death, pregnancy or perceived remission, or non specific (drug‐related) reason. | ||||||||||||
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number of | Number of | HR (95% CI)b,c | I² statisticd | Direct evidence (%)e | HR (95% CI)b,c | |
| CBZ vs PHB | 4 | 520 | 1.57 (1.16 to 2.13) | 0% | 52.5% | 1.55 (1.18 to 2.04) |
| CBZ vs PHT | 3 | 428 | 1.03 (0.74 to 1.42) | 63.6% | 12.8% | 1.13 (0.92 to 1.38) |
| CBZ vs VPS | 5 | 814 | 0.94 (0.73 to 1.19) | 0% | 40.1% | 1.04 (0.86 to 1.25) |
| CBZ vs LTG | 9 | 2268 | 0.76 (0.61 to 0.95) | 39.3% | 28.9% | 0.75 (0.65 to 0.86) |
| CBZ vs OXC | 2 | 562 | 4.62 (0.95 to 22.4) | 0% | 5.7% | 1.09 (0.84 to 1.42) |
| CBZ vs TPM | 2 | 937 | 1.04 (0.52 to 2.07) | 0% | 7.4% | 1.18 (0.98 to 1.43) |
| CBZ vs GBP | 2 | 954 | 1.14 (0.84 to 1.55) | 0% | 87.1% | 1.20 (1.00 to 1.43) |
| CBZ vs LEV | 3 | 1567 | 0.70 (0.52 to 0.94) | 0% | 37.9% | 0.82 (0.69 to 0.97) |
| CBZ vs ZNS | 1 | 583 | 1.08 (0.81 to 1.44) | NA | 100% | 1.08 (0.79 to 1.48) |
| PHB vs PHT | 3 | 404 | 0.67 (0.50 to 0.91) | 65% | 15.2% | 0.73 (0.55 to 0.96) |
| PHB vs VPS | 2 | 75 | 0.68 (0.34 to 1.36) | 23% | 8.8% | 0.67 (0.48 to 0.92) |
| PHB vs LTG | No direct evidence | 0% | 0.48 (0.35 to 0.66) | |||
| PHB vs OXC | No direct evidence | 0% | 0.70 (0.48 to 1.03) | |||
| PHB vs TPM | No direct evidence | 0% | 0.76 (0.55 to 1.06) | |||
| PHB vs GBP | No direct evidence | 0% | 0.77 (0.55 to 1.07) | |||
| PHB vs LEV | No direct evidence | 0% | 0.53 (0.38 to 0.73) | |||
| PHB vs ZNS | No direct evidence | 0% | 0.70 (0.46 to 1.06) | |||
| PHT vs VPS | 4 | 168 | 1.00 (0.60 to 1.64) | 58.5% | 9% | 0.92 (0.70 to 1.21) |
| PHT vs LTG | 1 | 90 | 1.10 (0.57 to 2.14) | NA | 11.6% | 0.66 (0.52 to 0.85) |
| PHT vs OXC | 2 | 325 | 0.65 (0.32 to 1.32) | 0% | 40.4% | 0.97 (0.69 to 1.35) |
| PHT vs TPM | 1 | 53 | 0.77 (0.38 to 1.57) | NA | 10.9% | 1.05 (0.80 to 1.39) |
| PHT vs GBP | No direct evidence | 0% | 1.06 (0.81 to 1.40) | |||
| PHT vs LEV | No direct evidence | 0% | 0.73 (0.56 to 0.95) | |||
| PHT vs ZNS | No direct evidence | 0% | 0.96 (0.66 to 1.39) | |||
| VPS vs LTG* | 3 | 221 | 1.40 (1.00 to 1.96) | 45.1% | 5.1% | 0.72 (0.58 to 0.90) |
| VPS vs OXC | No direct evidence | 0% | 1.05 (0.76 to 1.44) | |||
| VPS vs TPM | 2 | 111 | 1.66 (1.24 to 2.23) | 48.1% | 33.7% | 1.14 (0.88 to 1.48) |
| VPS vs GBP | No direct evidence | 0% | 1.15 (0.89 to 1.49) | |||
| VPS vs LEV | 1 | 190 | 1.14 (0.73 to 1.75) | NA | 17.2% | 0.79 (0.61 to 1.03) |
| VPS vs ZNS | No direct evidence | 0% | 1.04 (0.73 to 1.50) | |||
| LTG vs OXC | 1 | 506 | 0.69 (0.12 to 4.14) | NA | 4.4% | 1.46 (1.11 to 1.92) |
| LTG vs TPM | 1 | 648 | 1.18 (0.86 to 1.62) | NA | 20.9% | 1.59 (1.29 to 1.95) |
| LTG vs GBP | 1 | 659 | 0.62 (0.06 to 6.01) | NA | 1% | 1.60 (1.31 to 1.96) |
| LTG vs LEV | 1 | 240 | 0.86 (0.58 to 1.28) | NA | 23.7% | 1.10 (0.89 to 1.35) |
| LTG vs ZNS | No direct evidence | 0% | 1.45 (1.03 to 2.04) | |||
| OXC vs TPM | 1 | 496 | 0.87 (0.16 to 4.73) | NA | 4.9% | 1.09 (0.82 to 1.44) |
| OXC vs GBP | 1 | 507 | 0.90 (0.08 to 9.96) | NA | 2.3% | 1.10 (0.83 to 1.45) |
| OXC vs LEV | No direct evidence | 0% | 0.75 (0.55 to 1.03) | |||
| OXC vs ZNS | No direct evidence | 0% | 0.99 (0.66 to 1.49) | |||
| TPM vs GBP | 1 | 649 | 1.04 (0.12 to 9.33) | NA | 1.1% | 1.01 (0.82 to 1.25) |
| TPM vs LEV | No direct evidence | 0% | 0.69 (0.54 to 0.89) | |||
| TPM vs ZNS | No direct evidence | 0% | 0.91 (0.64 to 1.31) | |||
| GBP vs LEV | No direct evidence | 0% | 0.69 (0.54 to 0.88) | |||
| GBP vs ZNS | No direct evidence | 0% | 0.90 (0.63 to 1.30) | |||
| LEV vs ZNS | No direct evidence | 0% | 1.32 (0.93 to 1.88) | |||
| CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). For comparisons marked with a *, confidence intervals of direct evidence and network meta‐analysis do not overlap indicating that inconsistency may be present in the results. | ||||||
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number of | Number of | HR (95% CI)b,c | I² statisticd | Direct | HR (95% CI)b,c | |
| CBZ vs PHB | 3 | 156 | 1.21 (0.51 to 2.86) | 11.8% | 27.3% | 1.47 (0.83 to 2.61) |
| CBZ vs PHT | 2 | 118 | 2.68 (0.95 to 7.57) | 0% | 11.3% | 0.92 (0.59 to 1.42) |
| CBZ vs VPS | 4 | 405 | 1.26 (0.73 to 2.20) | 6.6% | 27.3% | 0.70 (0.54 to 0.92) |
| CBZ vs LTG | 7 | 302 | 1.23 (0.72 to 2.10) | 0% | 39.2% | 0.63 (0.45 to 0.89) |
| CBZ vs OXC | 1 | 9 | 0.39 (0.03 to 4.35) | NA | 3.9% | 1.00 (0.21 to 4.81) |
| CBZ vs TPM | 2 | 101 | 1.10 (0.51 to 2.36) | 0% | 23.2% | 1.24 (0.90 to 1.71) |
| CBZ vs GBP | 1 | 6 | 0.49 (0.03 to 7.90) | NA | 8.5% | 0.90 (0.11 to 7.29) |
| CBZ vs LEV | 2 | 251 | 1.22 (0.74 to 2.02) | 0% | 57% | 0.74 (0.44 to 1.23) |
| PHB vs PHT | 2 | 95 | 1.56 (0.49 to 4.99) | 0% | 16.1% | 0.62 (0.32 to 1.24) |
| PHB vs VPS | 2 | 94 | 0.56 (0.20 to 1.54) | 0% | 19.4% | 0.48 (0.27 to 0.86) |
| PHB vs LTG | No direct evidence | 0% | 0.43 (0.22 to 0.83) | |||
| PHB vs OXC | No direct evidence | 0% | 0.68 (0.13 to 3.60) | |||
| PHB vs TPM | No direct evidence | 0% | 0.84 (0.44 to 1.60) | |||
| PHB vs GBP | No direct evidence | 0% | 0.61 (0.07 to 5.34) | |||
| PHB vs LEV | No direct evidence | 0% | 0.50 (0.23 to 1.09) | |||
| PHT vs VPS | 3 | 326 | 0.66 (0.30 to 1.45) | 22.6% | 19.3% | 0.77 (0.46 to 1.27) |
| PHT vs LTG | 1 | 91 | 1.11 (0.42 to 2.94) | NA | 14.9% | 0.69 (0.39 to 1.20) |
| PHT vs OXC | 2 | 155 | 1.05 (0.44 to 2.52) | 0% | 37.9% | 1.09 (0.21 to 5.56) |
| PHT vs TPM | 1 | 150 | 1.68 (0.49 to 5.69) | NA | 11.2% | 1.35 (0.79 to 2.30) |
| PHT vs GBP | No direct evidence | 0% | 0.98 (0.12 to 8.30) | |||
| PHT vs LEV | No direct evidence | 0% | 0.80 (0.42 to 1.55) | |||
| VPS vs LTG | 3 | 387 | 0.46 (0.22 to 0.97) | 0% | 14.8% | 0.90 (0.60 to 1.35) |
| VPS vs OXC | No direct evidence | 0% | 1.42 (0.29 to 6.92) | |||
| VPS vs TPM* | 2 | 443 | 0.53 (0.27 to 1.07) | 48.5% | 22.4% | 1.76 (1.22 to 2.53) |
| VPS vs GBP | No direct evidence | 0% | 1.28 (0.16 to 10.5) | |||
| VPS vs LEV | 1 | 512 | 0.68 (0.30 to 1.59) | NA | 18.6% | 1.05 (0.58 to 1.90) |
| LTG vs OXC | 1 | 10 | 2.09 (0.34 to 12.8) | NA | 7.6% | 1.58 (0.33 to 7.67) |
| LTG vs TPM | 1 | 14 | 1.10 (0.42 to 2.89) | NA | 7.3% | 1.96 (1.25 to 3.08) |
| LTG vs GBP | 1 | 7 | 2.63 (0.27 to 25.7) | NA | 13.8% | 1.42 (0.17 to 11.6) |
| LTG vs LEV | No direct evidence | 0% | 1.17 (0.63 to 2.19) | |||
| OXC vs TPM | 1 | 14 | 1.31 (0.24 to 7.32) | NA | 9% | 1.24 (0.26 to 5.94) |
| OXC vs GBP | 1 | 7 | 1.26 (0.11 to 14.1) | NA | 12.7% | 0.90 (0.08 to 9.96) |
| OXC vs LEV | No direct evidence | 0% | 0.74 (0.14 to 3.86) | |||
| TPM vs GBP | 1 | 11 | 0.96 (0.11 to 8.67) | NA | 14.6% | 0.73 (0.09 to 5.89) |
| TPM vs LEV | No direct evidence | 0% | 0.60 (0.33 to 1.09) | |||
| GBP vs LEV | No direct evidence | 0% | 0.82 (0.10 to 7.10) | |||
| CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). For comparisons marked with a *, confidence intervals of direct evidence and network meta‐analysis do not overlap indicating that inconsistency may be present in the results | ||||||
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number of | Number of | HR (95% CI)b,c | I² statisticd | Direct | HR (95% CI)b,c | |
| CBZ vs PHB | 4 | 525 | 1.41 (1.04 to 1.91) | 0% | 56.1% | 1.02 (0.76 to 1.35) |
| CBZ vs PHT | 3 | 430 | 1.00 (0.76 to 1.32) | 54.8% | 18.6% | 1.03 (0.85 to 1.25) |
| CBZ vs VPS | 5 | 816 | 1.03 (0.85 to 1.25) | 46.4% | 27.6% | 1.05 (0.89 to 1.25) |
| CBZ vs LTG | 2 | 891 | 1.02 (0.69 to 1.50) | 0% | 17.5% | 1.16 (0.98 to 1.37) |
| CBZ vs OXC | 2 | 555 | 1.13 (0.62 to 2.05) | 0% | 21% | 0.98 (0.78 to 1.25) |
| CBZ vs TPM | 2 | 925 | 0.94 (0.48 to 1.83) | 0% | 7.2% | 1.08 (0.92 to 1.27) |
| CBZ vs GBP | 1 | 651 | 0.61 (0.06 to 5.82) | NA | 10.5% | 1.20 (0.99 to 1.47) |
| CBZ vs LEV | 3 | 1567 | 1.08 (0.81 to 1.42) | 60.8% | 14.2% | 1.35 (1.09 to 1.69) |
| CBZ vs ZNS | 1 | 582 | 1.05 (0.85 to 1.30) | NA | 100% | 1.05 (0.81 to 1.35) |
| PHB vs PHT | 4 | 465 | 0.80 (0.59 to 1.10) | 0% | 0.2% | 1.01 (0.75 to 1.37) |
| PHB vs VPS | 2 | 80 | 0.85 (0.51 to 1.40) | 4.4% | 15.6% | 1.04 (0.75 to 1.43) |
| PHB vs LTG | No direct evidence | 0% | 1.14 (0.82 to 1.59) | |||
| PHB vs OXC | No direct evidence | 0% | 0.96 (0.67 to 1.41) | |||
| PHB vs TPM | No direct evidence | 0% | 1.06 (0.76 to 1.47) | |||
| PHB vs GBP | No direct evidence | 0% | 1.19 (0.83 to 1.69) | |||
| PHB vs LEV | No direct evidence | 0% | 1.33 (0.93 to 1.92) | |||
| PHB vs ZNS | No direct evidence | 0% | 1.03 (0.70 to 1.52) | |||
| PHT vs VPS | 4 | 245 | 1.04 (0.78 to 1.40) | 0% | 41.6% | 1.03 (0.80 to 1.32) |
| PHT vs LTG | No direct evidence | 0% | 1.12 (0.88 to 1.45) | |||
| PHT vs OXC | 2 | 318 | 1.21 (0.73 to 2.03) | 0% | 29.9% | 0.95 (0.70 to 1.30) |
| PHT vs TPM | No direct evidence | 0% | 1.05 (0.81 to 1.35) | |||
| PHT vs GBP | No direct evidence | 0% | 1.18 (0.88 to 1.56) | |||
| PHT vs LEV | No direct evidence | 0% | 1.32 (0.98 to 1.75) | |||
| PHT vs ZNS | No direct evidence | 0% | 1.02 (0.74 to 1.41) | |||
| VPS vs LTG | 3 | 221 | 1.37 (1.07 to 1.77) | 0% | 39.9% | 1.10 (0.88 to 1.37) |
| VPS vs OXC | No direct evidence | 0% | 0.93 (0.70 to 1.23) | |||
| VPS vs TPM | 2 | 111 | 1.11 (0.87 to 1.40) | 0% | 67.8% | 1.02 (0.80 to 1.30) |
| VPS vs GBP | No direct evidence | 0% | 1.14 (0.88 to 1.47) | |||
| VPS vs LEV | 1 | 190 | 1.14 (0.84 to 1.55) | NA | 34.7% | 1.28 (0.97 to 1.67) |
| VPS vs ZNS | No direct evidence | 0% | 0.99 (0.74 to 1.35) | |||
| LTG vs OXC | 1 | 499 | 1.49 (0.33 to 6.67) | NA | 2.8% | 0.85 (0.66 to 1.09) |
| LTG vs TPM | 1 | 636 | 0.98 (0.29 to 3.25) | NA | 2.5% | 0.93 (0.75 to 1.15) |
| LTG vs GBP | 1 | 647 | 0.74 (0.08 to 6.58) | NA | 10.1% | 1.04 (0.84 to 1.30) |
| LTG vs LEV | 1 | 240 | 1.02 (0.70 to 1.49) | NA | 26.6% | 1.16 (0.93 to 1.47) |
| LTG vs ZNS | No direct evidence | 0% | 0.91 (0.67 to 1.22) | |||
| OXC vs TPM | 1 | 487 | 0.66 (0.17 to 2.47) | NA | 3.7% | 1.10 (0.83 to 1.45) |
| OXC vs GBP | 1 | 498 | 0.49 (0.05 to 4.74) | NA | 9.8% | 1.23 (0.95 to 1.59) |
| OXC vs LEV | No direct evidence | 0% | 1.37 (1.05 to 1.79) | |||
| OXC vs ZNS | No direct evidence | 0% | 1.06 (0.76 to 1.52) | |||
| TPM vs GBP | 1 | 635 | 0.75 (0.09 to 6.00) | NA | 11.2% | 1.12 (0.87 to 1.45) |
| TPM vs LEV | No direct evidence | 0% | 1.25 (0.96 to 1.64) | |||
| TPM vs ZNS | No direct evidence | 0% | 0.97 (0.72 to 1.32) | |||
| GBP vs LEV | No direct evidence | 0% | 1.12 (0.88 to 1.43) | |||
| GBP vs ZNS | No direct evidence | 0% | 0.87 (0.63 to 1.20) | |||
| LEV vs ZNS | No direct evidence | 0% | 0.78 (0.56 to 1.09) | |||
| CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number of | Number of | HR (95% CI)b,c | I² statisticd | Direct | HR (95% CI)b,c | |
| CBZ vs PHB | 3 | 158 | 0.53 (0.28 to 1.00) | 0% | 42.6% | 1.25 (0.83 to 1.89) |
| CBZ vs PHT | 2 | 121 | 1.11 (0.61 to 2.02) | 64.5% | 9.3% | 0.86 (0.65 to 1.16) |
| CBZ vs VPS | 4 | 412 | 1.01 (0.72 to 1.43) | 0% | 51.1% | 0.94 (0.79 to 1.14) |
| CBZ vs LTG | 1 | 9 | 1.33 (0.29 to 6.03) | NA | 7% | 1.28 (0.54 to 3.03) |
| CBZ vs OXC | 1 | 9 | 0.77 (0.15 to 3.89) | NA | 5.6% | 1.72 (0.47 to 6.25) |
| CBZ vs TPM | 2 | 101 | 1.15 (0.52 to 2.53) | 0% | 27.2% | 1.06 (0.78 to 1.45) |
| CBZ vs GBP | 1 | 6 | 2.19 (0.23 to 21.2) | NA | 10.9% | 0.75 (0.10 to 5.88) |
| CBZ vs LEV | 2 | 251 | 1.02 (0.65 to 1.59) | 77.4% | 16.6% | 1.33 (0.81 to 2.22) |
| PHB vs PHT | 3 | 130 | 1.30 (0.61 to 2.78) | 53% | 10.9% | 0.68 (0.44 to 1.08) |
| PHB vs VPS | 2 | 98 | 1.15 (0.53 to 2.49) | 42.3% | 13% | 0.75 (0.49 to 1.15) |
| PHB vs LTG | No direct evidence | 0% | 1.01 (0.40 to 2.63) | |||
| PHB vs OXC | No direct evidence | 0% | 1.37 (0.35 to 5.26) | |||
| PHB vs TPM | No direct evidence | 0% | 0.85 (0.51 to 1.41) | |||
| PHB vs GBP | No direct evidence | 0% | 0.60 (0.07 to 5.00) | |||
| PHB vs LEV | No direct evidence | 0% | 1.06 (0.56 to 2.04) | |||
| PHT vs VPS | 4 | 269 | 0.87 (0.55 to 1.40) | 0% | 44.9% | 1.10 (0.80 to 1.49) |
| PHT vs LTG | No direct evidence | 0% | 1.47 (0.60 to 3.57) | |||
| PHT vs OXC | 2 | 154 | 0.77 (0.41 to 1.47) | 0% | 41.2% | 2.00 (0.53 to 7.69) |
| PHT vs TPM | No direct evidence | 0% | 1.23 (0.81 to 1.85) | |||
| PHT vs GBP | No direct evidence | 0% | 0.87 (0.11 to 7.14) | |||
| PHT vs LEV | No direct evidence | 0% | 1.56 (0.87 to 2.78) | |||
| VPS vs LTG | 3 | 387 | 0.77 (0.38 to 1.56) | 0% | 35.7% | 1.35 (0.57 to 3.13) |
| VPS vs OXC | No direct evidence | 0% | 1.82 (0.50 to 6.67) | |||
| VPS vs TPM | 2 | 441 | 0.52 (0.26 to 1.04) | 58.5% | 10.6% | 1.12 (0.83 to 1.52) |
| VPS vs GBP | No direct evidence | 0% | 0.79 (0.10 to 6.25) | |||
| VPS vs LEV | 1 | 512 | 0.91 (0.49 to 1.70) | NA | 55.2% | 1.41 (0.83 to 2.44) |
| LTG vs OXC | 1 | 10 | 0.58 (0.13 to 2.64) | NA | 9.2% | 1.35 (0.33 to 5.56) |
| LTG vs TPM | 1 | 14 | 1.13 (0.33 to 3.82) | NA | 15.1% | 0.83 (0.35 to 2.00) |
| LTG vs GBP | 1 | 7 | 1.64 (0.18 to 14.8) | NA | 12.5% | 0.59 (0.07 to 5.00) |
| LTG vs LEV | No direct evidence | 0% | 1.05 (0.40 to 2.78) | |||
| OXC vs TPM | 1 | 14 | 1.95 (0.51 to 7.50) | NA | 11.4% | 0.62 (0.17 to 2.27) |
| OXC vs GBP | 1 | 7 | 2.83 (0.29 to 27.6) | NA | 10.9% | 0.44 (0.04 to 4.35) |
| OXC vs LEV | No direct evidence | 0% | 0.78 (0.20 to 3.13) | |||
| TPM vs GBP | 1 | 11 | 1.45 (0.18 to 11.7) | NA | 15.9% | 0.71 (0.09 to 5.56) |
| TPM vs LEV | No direct evidence | 0% | 1.27 (0.68 to 2.33) | |||
| GBP vs LEV | No direct evidence | 0% | 1.79 (0.21 to 14.3) | |||
| CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number of | Number of | HR (95% CI)b,c | I² statisticd | Direct | HR (95% CI)b,c | |
| CBZ vs PHB | 4 | 525 | 1.24 (0.95 to 1.61) | 0% | 31.3% | 0.95 (0.76 to 1.19) |
| CBZ vs PHT | 3 | 430 | 0.85 (0.66 to 1.09) | 4.2% | 23.3% | 1.03 (0.88 to 1.20) |
| CBZ vs VPS | 5 | 816 | 1.06 (0.90 to 1.25) | 56.5% | 16.6% | 1.10 (0.96 to 1.25) |
| CBZ vs LTG | 7 | 1535 | 1.15 (0.89 to 1.48) | 0% | 26.4% | 1.11 (0.98 to 1.27) |
| CBZ vs OXC | 2 | 555 | 1.15 (0.65 to 2.04) | 0% | 16.6% | 0.98 (0.82 to 1.18) |
| CBZ vs TPM | 2 | 925 | 1.05 (0.64 to 1.72) | 0% | 8.8% | 1.11 (0.96 to 1.28) |
| CBZ vs GBP | 2 | 943 | 0.81 (0.52 to 1.27) | 0% | 73.7% | 1.16 (0.99 to 1.35) |
| CBZ vs LEV | 3 | 1567 | 1.06 (0.84 to 1.33) | 37.9% | 20.4% | 1.04 (0.93 to 1.16) |
| CBZ vs ZNS | 1 | 582 | 1.00 (0.82 to 1.20) | NA | 100% | 1.00 (0.83 to 1.20) |
| PHB vs PHT | 4 | 465 | 0.79 (0.60 to 1.05) | 0% | 31.1% | 1.08 (0.85 to 1.37) |
| PHB vs VPS | 2 | 80 | 0.67 (0.42 to 1.08) | 0% | 9.1% | 1.15 (0.89 to 1.49) |
| PHB vs LTG | No direct evidence | 0% | 1.16 (0.90 to 1.52) | |||
| PHB vs OXC | No direct evidence | 0% | 1.03 (0.77 to 1.39) | |||
| PHB vs TPM | No direct evidence | 0% | 1.16 (0.89 to 1.54) | |||
| PHB vs GBP | No direct evidence | 0% | 1.22 (0.93 to 1.59) | |||
| PHB vs LEV | No direct evidence | 0% | 1.10 (0.85 to 1.41) | |||
| PHB vs ZNS | No direct evidence | 0% | 1.04 (0.78 to 1.41) | |||
| PHT vs VPS | 5 | 245 | 0.90 (0.70 to 1.15) | 0% | 26.5% | 1.06 (0.88 to 1.30) |
| PHT vs LTG | 1 | 90 | 0.88 (0.25 to 3.03) | NA | 1.20% | 1.09 (0.88 to 1.32) |
| PHT vs OXC | 2 | 318 | 1.21 (0.79 to 1.87) | 0% | 33.2% | 0.95 (0.75 to 1.22) |
| PHT vs TPM | No direct evidence | 0% | 1.09 (0.88 to 1.33) | |||
| PHT vs GBP | No direct evidence | 0% | 1.12 (0.91 to 1.39) | |||
| PHT vs LEV | No direct evidence | 0% | 1.02 (0.84 to 1.22) | |||
| PHT vs ZNS | No direct evidence | 0% | 0.97 (0.76 to 1.23) | |||
| VPS vs LTG | 3 | 221 | 1.22 (0.97 to 1.52) | 0% | 32.1% | 1.02 (0.85 to 1.22) |
| VPS vs OXC | No direct evidence | 0% | 0.90 (0.72 to 1.12) | |||
| VPS vs TPM | 2 | 111 | 1.08 (0.87 to 1.34) | 0% | 61.7% | 1.02 (0.83 to 1.23) |
| VPS vs GBP | No direct evidence | 0% | 1.05 (0.87 to 1.28) | |||
| VPS vs LEV | 1 | 190 | 1.09 (0.88 to 1.33) | NA | 40.5% | 0.95 (0.79 to 1.14) |
| VPS vs ZNS | No direct evidence | 0% | 0.91 (0.72 to 1.14) | |||
| LTG vs OXC | 1 | 499 | 1.08 (0.27 to 4.32) | NA | 2.4% | 0.88 (0.73 to 1.08) |
| LTG vs TPM | 1 | 636 | 0.89 (0.70 to 1.13) | NA | 1.7% | 1.00 (0.85 to 1.18) |
| LTG vs GBP | 1 | 647 | 1.46 (0.16 to 13.0) | NA | 1.6% | 1.04 (0.88 to 1.22) |
| LTG vs LEV | 1 | 240 | 0.83 (0.59 to 1.17) | NA | 17.8% | 0.93 (0.80 to 1.10) |
| LTG vs ZNS | No direct evidence | 0% | 0.89 (0.71 to 1.12) | |||
| OXC vs TPM | 1 | 487 | 0.86 (0.26 to 2.86) | NA | 3.3% | 1.14 (0.93 to 1.37) |
| OXC vs GBP | 1 | 498 | 1.35 (0.15 to 12.1) | NA | 2.1% | 1.18 (0.96 to 1.43) |
| OXC vs LEV | No direct evidence | 0% | 1.06 (0.86 to 1.32) | |||
| OXC vs ZNS | No direct evidence | 0% | 1.01 (0.78 to 1.32) | |||
| TPM vs GBP | 1 | 635 | 1.56 (0.2 to 12.5) | NA | 1.6% | 1.04 (0.88 to 1.23) |
| TPM vs LEV | No direct evidence | 0% | 0.93 (0.79 to 1.12) | |||
| TPM vs ZNS | No direct evidence | 0% | 0.89 (0.70 to 1.14) | |||
| GBP vs LEV | No direct evidence | 0% | 0.90 (0.75 to 1.09) | |||
| GBP vs ZNS | No direct evidence | 0% | 0.86 (0.68 to 1.10) | |||
| LEV vs ZNS | No direct evidence | 0% | 0.95 (0.77 to 1.19) | |||
| CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number | Number | HR (95% CI)b,c | I² statisticd | Direct | HR (95% CI)b,c | |
| CBZ vs PHB | 3 | 158 | 0.56 (0.33 to 0.96) | 13.2% | 28.2% | 1.28 (0.92 to 1.79) |
| CBZ vs PHT | 2 | 121 | 1.44 (0.82 to 2.55) | 31.4% | 13% | 0.87 (0.68 to 1.10) |
| CBZ vs VPS | 4 | 412 | 1.11 (0.81 to 1.53) | 29.9% | 30.7% | 0.95 (0.84 to 1.09) |
| CBZ vs LTG | 5 | 254 | 0.58 (0.25 to 1.32) | 0% | 0.1% | 1.20 (0.99 to 1.49) |
| CBZ vs OXC | 1 | 9 | 0.79 (0.17 to 3.56) | NA | 4.6% | 1.30 (0.42 to 4.00) |
| CBZ vs TPM | 2 | 101 | 1.00 (0.55 to 1.79) | 0% | 32.8% | 1.11 (0.78 to 1.59) |
| CBZ vs GBP | 1 | 6 | 0.71 (0.07 to 6.90) | NA | 10% | 1.75 (0.23 to 12.5) |
| CBZ vs LEV | 2 | 251 | 1.00 (0.72 to 1.37) | 57.9% | 26.7% | 1.14 (0.85 to 1.52) |
| PHB vs PHT | 3 | 130 | 1.31 (0.67 to 2.53) | 0% | 22.7% | 0.68 (0.47 to 0.98) |
| PHB vs VPS | 2 | 98 | 1.50 (0.72 to 3.11) | 7.5% | 15.3% | 0.75 (0.53 to 1.05) |
| PHB vs LTG | No direct evidence | 0% | 0.94 (0.64 to 1.39) | |||
| PHB vs OXC | No direct evidence | 0% | 1.01 (0.31 to 3.23) | |||
| PHB vs TPM | No direct evidence | 0% | 0.87 (0.53 to 1.41) | |||
| PHB vs GBP | No direct evidence | 0% | 1.37 (0.17 to 11.1) | |||
| PHB vs LEV | No direct evidence | 0% | 0.88 (0.57 to 1.37) | |||
| PHT vs VPS | 4 | 394 | 1.03 (0.68 to 1.54) | 0% | 36.8% | 1.10 (0.85 to 1.43) |
| PHT vs LTG | 1 | 91 | 1.96 (0.37 to 10.2) | NA | 4.4% | 1.39 (1.03 to 1.89) |
| PHT vs OXC | 2 | 154 | 0.71 (0.42 to 1.21) | 0% | 45.1% | 1.49 (0.48 to 4.76) |
| PHT vs TPM | No direct evidence | 0% | 1.28 (0.84 to 1.96) | |||
| PHT vs GBP | No direct evidence | 0% | 2.00 (0.26 to 16.7) | |||
| PHT vs LEV | No direct evidence | 0% | 1.32 (0.89 to 1.92) | |||
| VPS vs LTG | 3 | 387 | 0.84 (0.48 to 1.47) | 0% | 43.5% | 1.27 (1.03 to 1.56) |
| VPS vs OXC | No direct evidence | 0% | 1.35 (0.44 to 4.17) | |||
| VPS vs TPM | 2 | 441 | 0.67 (0.38 to 1.19) | 58.7% | 12.9% | 1.16 (0.81 to 1.67) |
| VPS vs GBP | No direct evidence | 0% | 1.82 (0.24 to 14.3) | |||
| VPS vs LEV | 1 | 512 | 0.88 (0.60 to 1.30) | NA | 48.6% | 1.19 (0.86 to 1.64) |
| LTG vs OXC | 1 | 10 | 0.80 (0.20 to 3.26) | NA | 7.6% | 1.08 (0.35 to 3.33) |
| LTG vs TPM | 1 | 14 | 0.59 (0.30 to 1.16) | NA | 10% | 0.92 (0.62 to 1.37) |
| LTG vs GBP | 1 | 7 | 0.73 (0.08 to 6.57) | NA | 11% | 1.45 (0.19 to 11.1) |
| LTG vs LEV | No direct evidence | 0% | 0.93 (0.65 to 1.33) | |||
| OXC vs TPM | 1 | 14 | 1.34 (0.40 to 4.54) | NA | 9.4% | 0.86 (0.28 to 2.63) |
| OXC vs GBP | 1 | 7 | 0.91 (0.10 to 8.20) | NA | 10.7% | 1.35 (0.15 to 12.5) |
| OXC vs LEV | No direct evidence | 0% | 0.88 (0.27 to 2.78) | |||
| TPM vs GBP | 1 | 11 | 0.68 (0.08 to 5.45) | NA | 13.9% | 1.56 (0.21 to 12.5) |
| TPM vs LEV | No direct evidence | 0% | 1.02 (0.65 to 1.61) | |||
| GBP vs LEV | No direct evidence | 0% | 0.65 (0.08 to 5.00) | |||
| CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). | ||||||
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number | Number | HR (95% CI)b,c | I² statisticd | Direct | HR (95% CI)b,c | |
| CBZ vs PHB | 6 | 581 | 0.99 (0.78 to 1.26) | 54.3% | 21% | 0.79 (0.64 to 0.97) |
| CBZ vs PHT | 4 | 432 | 0.91 (0.72 to 1.16) | 16.1% | 27.1% | 0.98 (0.85 to 1.13) |
| CBZ vs VPS | 5 | 813 | 1.01 (0.86 to 1.19) | 32% | 34.6% | 1.20 (1.06 to 1.37) |
| CBZ vs LTG | 9 | 2252 | 0.98 (0.75 to 1.27) | 0% | 40.7% | 1.29 (1.17 to 1.42) |
| CBZ vs OXC | 2 | 555 | 1.47 (0.57 to 3.81) | 57.3% | 4.8% | 1.09 (0.89 to 1.32) |
| CBZ vs TPM | 2 | 925 | 1.03 (0.51 to 2.08) | 69.3% | 1.5% | 1.12 (0.97 to 1.29) |
| CBZ vs GBP | 2 | 943 | 1.64 (1.14 to 2.36) | 17.7% | 49% | 1.44 (1.25 to 1.66) |
| CBZ vs LEV | 3 | 1552 | 1.18 (0.85 to 1.65) | 0% | 26.2% | 1.14 (0.99 to 1.30) |
| CBZ vs ZNS | 1 | 581 | 1.30 (0.97 to 1.73) | NA | 100% | 1.30 (0.97 to 1.73) |
| PHB vs PHT | 5 | 463 | 1.07 (0.83 to 1.37) | 27.7% | 33.6% | 1.24 (0.99 to 1.56) |
| PHB vs VPS* | 2 | 80 | 0.71 (0.43 to 1.17) | 9.1% | 12.8% | 1.53 (1.20 to 1.94) |
| PHB vs LTG | No direct evidence | 0% | 1.63 (1.30 to 2.06) | |||
| PHB vs OXC | No direct evidence | 0% | 1.38 (1.04 to 1.83) | |||
| PHB vs TPM | No direct evidence | 0% | 1.42 (1.11 to 1.83) | |||
| PHB vs GBP | No direct evidence | 0% | 1.83 (1.42 to 2.35) | |||
| PHB vs LEV | No direct evidence | 0% | 1.44 (1.12 to 1.85) | |||
| PHB vs ZNS | No direct evidence | 0% | 1.64 (1.15 to 2.35) | |||
| PHT vs VPS | 5 | 245 | 0.96 (0.72 to 1.29) | 0% | 25.4% | 1.23 (1.02 to 1.48) |
| PHT vs LTG | 1 | 90 | 0.77 (0.38 to 1.54) | NA | 6% | 1.31 (1.10 to 1.57) |
| PHT vs OXC | 2 | 318 | 1.46 (0.88 to 2.44) | 23.9% | 36.1% | 1.11 (0.87 to 1.41) |
| PHT vs TPM | 1 | 53 | 2.32 (0.95 to 5.70) | NA | 4% | 1.14 (0.93 to 1.40) |
| PHT vs GBP | No direct evidence | 0% | 1.47 (1.20 to 1.80) | |||
| PHT vs LEV | No direct evidence | 0% | 1.16 (0.95 to 1.41) | |||
| PHT vs ZNS | No direct evidence | 0% | 1.32 (0.96 to 1.82) | |||
| VPS vs LTG | 3 | 215 | 1.57 (1.23 to 2.00) | 39.4% | 10% | 1.07 (0.92 to 1.24) |
| VPS vs OXC | No direct evidence | 0% | 0.90 (0.72 to 1.14) | |||
| VPS vs TPM | 2 | 111 | 1.18 (0.93 to 1.50) | 0% | 70.2% | 0.93 (0.77 to 1.13) |
| VPS vs GBP | No direct evidence | 0% | 1.20 (0.99 to 1.44) | |||
| VPS vs LEV | 1 | 190 | 1.27 (0.94 to 1.72) | NA | 31% | 0.94 (0.77 to 1.15) |
| VPS vs ZNS | No direct evidence | 0% | 1.08 (0.78 to 1.48) | |||
| LTG vs OXC | 1 | 499 | 0.87 (0.23 to 3.25) | NA | 5.5% | 0.84 (0.69 to 1.03) |
| LTG vs TPM | 1 | 636 | 0.73 (0.57 to 0.93) | NA | 2.3% | 0.87 (0.75 to 1.01) |
| LTG vs GBP | 1 | 647 | 0.63 (0.07 to 5.42) | NA | 4.4% | 1.12 (0.96 to 1.30) |
| LTG vs LEV | 1 | 229 | 0.84 (0.53 to 1.35) | NA | 15.9% | 0.88 (0.75 to 1.04) |
| LTG vs ZNS | No direct evidence | 0% | 1.01 (0.74 to 1.36) | |||
| OXC vs TPM | 1 | 487 | 0.55 (0.15 to 2.06) | NA | 5.4% | 1.03 (0.84 to 1.27) |
| OXC vs GBP | 1 | 498 | 0.73 (0.08 to 6.49) | NA | 4.6% | 1.32 (1.08 to 1.63) |
| OXC vs LEV | No direct evidence | 0% | 1.05 (0.83 to 1.32) | |||
| OXC vs ZNS | No direct evidence | 0% | 1.19 (0.84 to 1.69) | |||
| TPM vs GBP | 1 | 635 | 1.31 (0.15 to 11.2) | NA | 3.5% | 1.28 (1.09 to 1.51) |
| TPM vs LEV | No direct evidence | 0% | 1.01 (0.83 to 1.23) | |||
| TPM vs ZNS | No direct evidence | 0% | 1.15 (0.84 to 1.59) | |||
| GBP vs LEV | No direct evidence | 0% | 0.79 (0.65 to 0.96) | |||
| GBP vs ZNS | No direct evidence | 0% | 0.90 (0.65 to 1.24) | |||
| LEV vs ZNS | No direct evidence | 0% | 1.14 (0.83 to 1.57) | |||
| CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; zNS: Zonisamide aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). For comparisons marked with a *, confidence intervals of direct evidence and network meta‐analysis do not overlap indicating that inconsistency may be present in the results. | ||||||
| Comparisiona | Direct evidence (pairwise meta‐analysis) | Direct plus indirect evidence | ||||
| Number | Number | HR (95% CI)2,3 | I² statistic4 | Direct | HR (95% CI)2,3 | |
| CBZ vs PHB | 5 | 237 | 0.55 (0.33 to 0.92) | 50.4% | 35.5% | 1.10 (0.80 to 1.51) |
| CBZ vs PHT | 3 | 150 | 0.88 (0.51 to 1.54) | 0% | 26.6% | 0.76 (0.59 to 0.98) |
| CBZ vs VPS | 4 | 411 | 1.37 (0.98 to 1.92) | 84.1% | 10.4% | 0.88 (0.76 to 1.03) |
| CBZ vs LTG | 7 | 302 | 1.49 (0.94 to 2.35) | 0% | 0.3% | 0.98 (0.70 to 1.37) |
| CBZ vs OXC | 1 | 9 | 1.55 (0.38 to 6.31) | NA | 9% | 1.09 (0.36 to 3.36) |
| CBZ vs TPM | 2 | 101 | 1.19 (0.56 to 2.50) | 62% | 9% | 1.15 (0.89 to 1.48) |
| CBZ vs GBP | 1 | 6 | 2.83 (0.31 to 25.5) | NA | 10.7% | 0.79 (0.10 to 6.08) |
| CBZ vs LEV | 2 | 251 | 1.04 (0.65 to 1.64) | 0% | 44.9% | 1.19 (0.78 to 1.83) |
| PHB vs PHT | 4 | 161 | 1.41 (0.76 to 2.62) | 46.9% | 20.3% | 0.69 (0.48 to 1.00) |
| PHB vs VPS | 2 | 98 | 1.87 (0.87 to 4.00) | 69.8% | 6.5% | 0.80 (0.57 to 1.12) |
| PHB vs LTG | No direct evidence | 0% | 0.89 (0.56 to 1.42) | |||
| PHB vs OXC | No direct evidence | 0% | 1.00 (0.31 to 3.20) | |||
| PHB vs TPM | No direct evidence | 0% | 1.05 (0.70 to 1.56) | |||
| PHB vs GBP | No direct evidence | 0% | 0.72 (0.09 to 5.68) | |||
| PHB vs LEV | No direct evidence | 0% | 1.09 (0.64 to 1.85) | |||
| PHT vs VPS | 4 | 394 | 1.11 (0.71 to 1.74) | 0% | 36.4% | 1.16 (0.88 to 1.53) |
| PHT vs LTG | 1 | 91 | 1.00 (0.40 to 2.46) | NA | 16.2% | 1.29 (0.85 to 1.97) |
| PHT vs OXC | 2 | 154 | 0.60 (0.33 to 1.10) | 49.7% | 25.2% | 1.44 (0.46 to 4.56) |
| PHT vs TPM | 1 | 150 | 0.63 (0.18 to 2.26) | NA | 9.8% | 1.51 (1.06 to 2.15) |
| PHT vs GBP | No direct evidence | 0% | 1.05 (0.13 to 8.14) | |||
| PHT vs LEV | No direct evidence | 0% | 1.57 (0.96 to 2.58) | |||
| VPS vs LTG | 3 | 377 | 0.64 (0.37 to 1.11) | 23.2% | 31.3% | 1.11 (0.77 to 1.60) |
| VPS vs OXC | No direct evidence | 0% | 1.24 (0.40 to 3.84) | |||
| VPS vs TPM* | 2 | 441 | 0.42 (0.23 to 0.80) | 46.4% | 21% | 1.30 (1.01 to 1.68) |
| VPS vs GBP | No direct evidence | 0% | 0.90 (0.12 to 6.92) | |||
| VPS vs LEV | 1 | 512 | 0.82 (0.48 to 1.40) | NA | 34% | 1.35 (0.86 to 2.13) |
| LTG vs OXC | 1 | 10 | 0.94 (0.25 to 3.57) | NA | 12.2% | 1.12 (0.36 to 3.48) |
| LTG vs TPM | 1 | 14 | 0.61 (0.28 to 1.30) | NA | 13.1% | 1.17 (0.78 to 1.77) |
| LTG vs GBP | 1 | 7 | 1.72 (0.20 to 14.9) | NA | 11.9% | 0.81 (0.11 to 6.25) |
| LTG vs LEV | No direct evidence | 0% | 1.22 (0.71 to 2.10) | |||
| OXC vs TPM | 1 | 14 | 1.90 (0.50 to 7.19) | NA | 13.6% | 1.05 (0.34 to 3.24) |
| OXC vs GBP | 1 | 7 | 1.83 (0.20 to 16.5) | NA | 13.3% | 0.73 (0.08 to 6.49) |
| OXC vs LEV | No direct evidence | 0% | 1.09 (0.33 to 3.62) | |||
| TPM vs GBP | 1 | 11 | 0.96 (0.11 to 8.29) | NA | 13.2% | 0.69 (0.09 to 5.32) |
| TPM vs LEV | No direct evidence | 0% | 1.04 (0.63 to 1.71) | |||
| GBP vs LEV | No direct evidence | 0% | 1.50 (0.19 to 12.0) | |||
| CBZ: carbamazepine; CI: confidence interval; GBP: gabapentin; HR: hazard ratio; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide Generalised tonic‐clonic seizures with or without other seizure types is shortened to 'Generalised seizures' for brevity aOrder of drugs in the table: most commonly used drug first (carbamazepine), then drugs are ordered approximately by the date they were licenced as a monotherapy treatment (oldest first). For comparisons marked with a *, confidence intervals of direct evidence and network meta‐analysis do not overlap indicating that inconsistency may be present in the results | ||||||
| Drug | Number of participants | Number of participants | Number of events |
| CBZ | 5134 | 3023 | 9769 |
| PHB | 754 | 271 | 181 |
| PHT | 1384 | 614 | 1513 |
| VPS | 2303 | 1294 | 3599 |
| LTG | 3107 | 1608 | 6296 |
| OXC | 978 | 623 | 1000 |
| TPM | 1898 | 920 | 6316 |
| GBP | 1209 | 506 | 2580 |
| LEV | 948 | 1441 | 4258 |
| ZNS | 282 | 182 | 606 |
| Total | 18,045 | 10,482 | 36,118 |
| CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide aAdverse event data were provided as detailed individual participant data for 23 trials and we extracted summary adverse event information from 36 trial publications. No adverse event data were reported in 18 trial publications. | |||
| Event (general description)a,b,c | CBZ | PHB | PHT | VPS | LTG | OXC | TPM | GBP | LEV | ZNS | Total |
| Accidental injury | 100 | 0 | 100 | 28 | 110 | 5 | 95 | 36 | 58 | 8 | 540 |
| Anorexia or weight loss | 126 | 0 | 126 | 24 | 116 | 6 | 394 | 58 | 62 | 25 | 937 |
| Anxiety/depression | 203 | 0 | 203 | 59 | 171 | 32 | 309 | 82 | 163 | 16 | 1238 |
| Aphasia | 59 | 7 | 66 | 11 | 26 | 4 | 106 | 22 | 11 | 2 | 314 |
| Asthenia | 59 | 1 | 60 | 26 | 41 | 1 | 31 | 33 | 37 | 10 | 299 |
| Ataxia | 172 | 37 | 209 | 32 | 55 | 17 | 61 | 40 | 32 | 8 | 663 |
| Cognitive (memory, concentration, confusion etc.) | 321 | 41 | 362 | 100 | 204 | 44 | 439 | 127 | 73 | 19 | 1730 |
| Dental problems | 93 | 0 | 93 | 28 | 62 | 5 | 61 | 24 | 70 | 7 | 443 |
| Dizziness/faintness | 617 | 0 | 617 | 171 | 348 | 140 | 269 | 160 | 394 | 23 | 2739 |
| Drowsiness/fatigue | 1270 | 1 | 1271 | 422 | 539 | 233 | 628 | 326 | 477 | 33 | 5200 |
| Fever or viral infection | 379 | 0 | 379 | 68 | 172 | 24 | 84 | 58 | 338 | 37 | 1539 |
| Gastrointestinal disturbances | 683 | 20 | 703 | 246 | 394 | 33 | 236 | 142 | 284 | 42 | 2783 |
| Hair loss | 47 | 0 | 47 | 130 | 22 | 15 | 39 | 8 | 16 | 3 | 327 |
| Headache or migraine | 843 | 0 | 843 | 264 | 556 | 137 | 315 | 171 | 596 | 47 | 3772 |
| Impotence | 90 | 24 | 114 | 13 | 17 | 0 | 27 | 32 | 11 | 3 | 331 |
| Increased/worsened seizures | 151 | 0 | 151 | 31 | 164 | 6 | 58 | 48 | 140 | 6 | 755 |
| Infection | 121 | 0 | 121 | 19 | 90 | 4 | 56 | 27 | 63 | 5 | 506 |
| Laboratory results abnormal | 367 | 0 | 367 | 103 | 117 | 8 | 47 | 19 | 90 | 32 | 1150 |
| Menstrual problems | 110 | 0 | 110 | 28 | 31 | 1 | 22 | 18 | 39 | 4 | 363 |
| Mood or behavioural change | 279 | 41 | 320 | 128 | 163 | 25 | 415 | 121 | 121 | 15 | 1628 |
| Nausea/vomiting | 413 | 1 | 414 | 167 | 233 | 53 | 132 | 92 | 142 | 20 | 1667 |
| Pain | 345 | 1 | 346 | 65 | 250 | 6 | 154 | 48 | 251 | 25 | 1491 |
| Paraesthesia or tingling | 56 | 0 | 56 | 22 | 33 | 2 | 708 | 34 | 28 | 7 | 946 |
| Problems sleeping/nightmares | 108 | 1 | 109 | 46 | 197 | 16 | 147 | 31 | 101 | 14 | 770 |
| Rash or skin disorder | 701 | 17 | 718 | 46 | 420 | 73 | 163 | 113 | 125 | 31 | 2407 |
| Renal/urinary disorder | 152 | 0 | 152 | 27 | 78 | 2 | 92 | 57 | 93 | 21 | 674 |
| Respiratory disorder | 233 | 0 | 233 | 53 | 124 | 4 | 190 | 23 | 131 | 17 | 1008 |
| Tremor or twitch | 171 | 1 | 172 | 258 | 219 | 19 | 56 | 23 | 51 | 2 | 972 |
| Visual disturbance/nystagmus | 199 | 0 | 199 | 53 | 96 | 33 | 86 | 59 | 33 | 8 | 766 |
| Weight gain | 259 | 0 | 259 | 347 | 167 | 22 | 71 | 258 | 70 | 1 | 1454 |
| CBZ: carbamazepine; GBP: gabapentin; LEV: levetiracetam; LTG: lamotrigine; OXC: oxcarbazepine; PHB: phenobarbitone; PHT: phenytoin; TPM: topiramate; VPS: sodium valproate; ZNS: zonisamide aVerbatim or reported terms extracted from publications or provided in individual participant data were grouped under the definitions by one review author (SJN) and any uncertainties in definition were discussed with the senior clinical author (AGM). bAdverse event data were provided as detailed individual participant data for 23 trials and we extracted summary adverse event information from 36 trial publications. No adverse event data were reported in 18 trial publications. cSome trial publications reported only on the “most common” adverse events, the totals and frequencies are likely to be an underestimation of the true number of events and number of individuals experiencing events. Furthermore, detailed information was provided in the more recent trial publications and individual participant data requests of more recent trials, often involving newer antiepileptic drugs, such as LTG, LEV and TPM; which may indicate that these newer drugs are associated with more adverse events than older drugs such as PHB and PHT ,for which less detailed information was available. | |||||||||||