نقش آلمتوزوماب برای مالتیپل اسکلروزیس (MS)
چکیده
پیشینه
مالتیپل اسکلروزیس (multiple sclerosis; MS) یک بیماری خود‐ایمنی، وابسته به سلول T، التهابی و دمیلینه کننده (demyelinating) سیستم عصبی مرکزی با یک دوره غیر‐قابل پیشبینی است. در حال حاضر، روشهای درمانی بیماری MS بر درمان تشدید علائم بیماری، پیشگیری از تشدیدهای جدید و اجتناب از پیشرفت ناتوانی تمرکز میکنند. با این حال، در حال حاضر هیچ درمان موثری وجود ندارد که قادر به دستیابی به این اهداف به صورت ایمن و موثر باشد. این امر منجر به توسعه و ارزیابی داروهای جدید شده است. کارآزماییهای بالینی اخیر نشان میدهند که آلمتوزوماب (alemtuzumab)، که یک آنتیبادی مونوکلونال انسانی بر ضد CD52 در سطح سلول است، میتواند یک گزینه امیدوار کننده برای درمان بیماری MS باشد.
اهداف
ارزیابی اثربخشی و بیخطری آلمتوزوماب به تنهایی یا همراه با درمانهای دیگر برای کاهش فعالیت بیماری در بیماران مبتلا به MS از هر نوع.
روشهای جستوجو
پایگاه ثبت کارآزماییهای گروه مالتیپل اسکلروزیس و بیماریهای نادر دستگاه سیستم عصبی مرکزی (CNS) در کاکرین (30 اپریل 2015) را که شامل پایگاه مرکزی ثبت کارآزماییهای بالینی کاکرین (CENTRAL) بود؛ MEDLINE؛ EMBASE؛ CINAHL؛ LILACS و بانک اطلاعاتی پایگاه ثبت کارآزمایی ClinicalTrials.gov و پلتفرم بینالمللی پایگاه ثبت کارآزماییهای بالینی سازمان جهانی بهداشت را جستوجو کردیم. هیچ نوع محدودیتی در منبع، تاریخ یا زبان انتشار وجود نداشت.
معیارهای انتخاب
همه کارآزماییهای بالینی تصادفیسازی شده (randomised clinical trials; RCTs) شامل بزرگسالان مبتلا به هر نوع از بیماری MS بر اساس معیارهای مکدونالد، که به مقایسه آلمتوزوماب به تنهایی یا همراه با داروهای دیگر، در هر دوز و برای هر مدت زمان، در برابر دارونما (placebo) یا هر درمان دارویی فعال دیگر یا آلمتوزوماب در دوز، رژیم یا مدت زمان متفاوت پرداخته باشند. پیامدهای اولیه همزمان شامل بقای بدون عود، پیشرفت پایدار بیماری و تعداد شرکتکنندگان با حداقل یکی از عوارض جانبی (از هر نوعی که باشد)، از جمله عوارض جانبی جدی بود.
گردآوری و تجزیهوتحلیل دادهها
دو نویسنده مرور به طور جداگانه، انتخاب مطالعه، استخراج دادهها و ارزیابی «خطر سوگیری (bias)» را انجام دادند. نویسنده سوم مرور، این فرایند را از نظر دقت بررسی کرد. از ابزار «خطر سوگیری» کاکرین، جهت ارزیابی خطر سوگیری مطالعات وارد شده در این مرور استفاده کردیم. از سیستم درجهبندی توصیه، ارزیابی، توسعه و ارزشیابی (GRADE) برای ارزیابی کیفیت مجموعه شواهد استفاده کردیم. برای اندازهگیری تاثیر درمان بر پیامدهای دو‐حالتی، از خطر نسبی (RR) و برای تاثیر درمان بر پیامدهای پیوسته از تفاوت میانگین (MD) و برای پیامدهای زمان تا یک رویداد از نسبت خطر (HR) استفاده کردیم. 95% فواصل اطمینان (CI) را برای این معیارها محاسبه کردیم. زمانی که هیچ ناهمگونی وجود نداشت از مدل اثر‐ثابت برای ترکیب دادهها استفاده کردیم.
نتایج اصلی
سه RCT (با 1713 شرکتکننده) معیار انتخاب را داشتند و ما آنها را در مرور حاضر وارد کردیم. هر سه کارآزمایی به مقایسه آلمتوزوماب در برابر اینترفرون بتا (interferon beta)‐ 1a زیر‐جلدی برای بیماران مبتلا به MS از نوع عود کننده ‐ فروکش کننده پرداخته بودند. بیماران در مطالعات CARE‐MS و CAMMS223، برای اولین بار بود که تحت درمان قرار میگرفتند (treatment‐naive). مطالعه CARE‐MS II شامل بیمارانی با حداقل یک عود بودند، در حالی که با اینترفرون بتا یا گلاتیرامر استات (glatiramer acetate) درمان میشدند. آلمتوزوماب به مدت 12 یا 24 ماه به بیماران داده شد.برای برخی از پیامدها، دوره پیگیری به 36 ماه رسید. رژیمها شامل موارد زیر بودند: (الف) تجویز داخل‐وریدی 12 میلیگرم یا 24 میلیگرم در روز، یک بار در روز به مدت پنج روز متوالی در ماه 0 و ماه 12 یا (ب) 24 میلیگرم در روز، به صورت داخل‐وریدی، یک بار در روز به مدت سه روز متوالی در ماه 12 و 24. بیماران در بازوی دیگر کارآزماییها، 44 میکروگرم اینترفرون بتا‐a1 به صورت زیر‐جلدی و سه بار در هفته بعد از تیتراسیون (titration) دوز دریافت کردند.
بعد از 24 ماه، آلمتوزوماب 12 میلیگرم با موارد زیر همراه بود: (الف) بقای بدون عود بالاتر (نسبت خطر (HR): 0.50؛ 95% CI؛ 0.41 تا 0.60؛ دو مطالعه؛ 1248 شرکتکننده؛ شواهد با کیفیت متوسط)، (ب) بقای بدون پیشرفت بیماری پایدار بالاتر (HR: 0.62؛ 95% CI؛ 0.44 تا 0.87؛ دو مطالعه؛ 1191 شرکتکننده؛ شواهد با کیفیت متوسط)، (ج) تعداد اندکی بیشتر از شرکتکنندگان با حداقل یک عارضه (HR: 1.04؛ 95% CI؛ 1.01 تا 1.06؛ دو مطالعه؛ 1248 شرکتکننده؛ شواهد با کیفیت متوسط)، (د) تعداد کمتر شرکتکنندگان با ضایعات جدید یا بزرگ T2‐hyperintense در تصویربرداری رزونانس مغناطیسی (magnetic resonance imaging; MRI)؛ (HR: 0.74؛ 95% CI؛ 0.59 تا 0.91؛ دو مطالعه؛ 1238 شرکتکننده؛ I2 = 80%) و (ه) تعداد کمتر انصراف از مطالعه (HR: 0.31؛ 95% CI؛ 0.23 تا 0.41؛ دو مطالعه؛ 1248 شرکتکننده؛ I2 = 29%؛ شواهد با کیفیت پائین).
در 36 ماه، آلمتوزوماب 24 میلیگرم با موارد زیر همراه بود: (الف) بقای بدون عود بالاتر (45 در برابر 17؛ HR: 0.21؛ 95% CI؛ 0.11 تا 0.40؛ یک مطالعه؛ 221 شرکتکننده)، (ب) بقای بدون پیشرفت بیماری پایدار بالاتر (HR: 0.33؛ 95% CI؛ 0.16 تا 0.69؛ یک مطالعه؛ 221 شرکتکننده) و (ج) عدم وجود تفاوت آماری در نرخ شرکتکنندگان با حداقل یک عارضه جانبی. هیچ مطالعهای نیافتیم که هریک از پیامدهای زیر را گزارش داده باشد: نرخ شرکتکنندگان عاری از فعالیتهای بیماری بالینی، کیفیت زندگی، خستگی یا تغییر در تعداد ضایعات ‐MRI T2 و ‐T1 وزندار شده پس از درمان. به دلیل عدم وجود داده، انجام آنالیز زیر‐گروه بر طبق نوع بیماری و ناتوانی درشروع مطالعه امکانپذیر نبود.
نتیجهگیریهای نویسندگان
برای پیامدهای زیر در بیماران مبتلا به MS عود کننده ‐ فروکش کننده، آلمتوزوماب 12 میلیگرم که در 24 ماه ارزیابی شد، بهتر از اینترفرون زیر‐جلدی بتا‐a1 بود: بقای بدون عود، بقای بدون پیشرفت بیماری پایدار، تعداد شرکتکنندگان با حداقل یک عارضه جانبی و تعداد شرکتکنندگان با ضایعات جدید یا بزرگ شده T2‐hyperintense در MRI. کیفیت شواهد برای این پیامدها پائین تا متوسط بود. به نظر میرسد که آلمتوزوماب 24 میلیگرم برای بقای عاری از عود بیماری و بقای بدون پیشرفت بیماری پایدار، در 36 ماه، بهتر از اینترفرون بتا‐a1 زیر‐جلدی باشد.
کارآزماییهای بالینی تصادفیسازی بیشتری به منظور بررسی تاثیرات آلمتوزوماب در فرمهای دیگر بیماری MS و در مقایسه با سایر گزینههای درمانی، مورد نیاز است. این مطالعات جدید باید پیامدهای مرتبط بیشتری را از قبیل نرخ شرکتکنندگان عاری از فعالیتهای بیماری بالینی، کیفیت زندگی، خستگی و عوارض جانبی (نرخهای فردی، عوارض جانبی جدی و عوارض جانبی طولانی‐مدت) ارزیابی کنند. علاوه بر این، این مطالعات جدید باید سایر دوزها و مدت زمانهای مصرف آلمتوزوماب را ارزیابی کنند.
PICOs
خلاصه به زبان ساده
نقش آلمتوزوماب برای مالتیپل اسکلروزیس (MS)
پیشینه
مالتیپل اسکلروزیس (multiple sclerosis; MS) یک بیماری مزمن سیستم عصبی است که افراد جوان و میانسال را تحت تاثیر قرار میدهد. آسیبهای مکرر به غلاف میلین (غشایی است که اعصاب را پوشش داده و از آن محافظت میکند) و دیگر بخشهای اعصاب میتواند منجر به ناتوانی جدی شود. بیماری MS ممکن است با بروز مشکلاتی در سیستم ایمنی بدن مرتبط باشد. آلمتوزوماب (alemtuzumab) یک داروی بیولوژیک (نوعی آنتیبادی) است که در حال حاضر برای بیماریهای دیگر استفاده میشود.
ویژگیهای مطالعه
سه مطالعه (شامل 1713 شرکتکننده) را یافتیم که معیار انتخاب در مرور حاضر را داشتند. تمام مطالعات به مقایسه آلمتوزوماب در برابر اینترفرون بتا (interferon beta)‐a1 زیر‐جلدی در افراد مبتلا به MS از نوع عود کننده ‐ فروکش کننده پرداخته بودند. در دو مطالعه (CARE‐MS و CAMMS223)، شرکتکنندگان برای اولین بار بود که تحت درمان قرار گرفتند (treatment‐naive). مطالعه سوم (CARE‐MS II) شامل شرکتکنندگانی بود که در حالی که با اینترفرون بتا یا گلاتیرامر استات (glatiramer acetate) به مدت حداقل شش ماه تحت درمان بودند، حداقل یک بار عود داشتند.
نتایج کلیدی
مرور این مطالعات مقایسهای نشان داد آلمتوزوماب در مقایسه با اینترفرون بتا‐a1 زیر‐جلدی، خطر عود را کاهش میدهد، عملکرد را بهبود میدهد و به نظر میرسد که خطر کلی عوارض جانبی را افزایش نمیدهد. علاوه بر این، آلمتوزوماب خطر ابتلا را به ضایعات جدید یا بزرگ شده MS که با استفاده از تصویربرداری رزونانس مغناطیسی (magnetic resonance imaging; MRI) شناسایی میشوند، کاهش میدهد. با این حال، در مورد تاثیرات آلمتوزوماب بر چند پیامد مربوط به بیمار مانند (الف) کیفیت زندگی، (ب) نرخ هر یک از عوارض جانبی (به طور جداگانه) و (ج) فراوانی طولانی‐مدت عوارض جانبی و عوارض جانبی جدی، اطلاعات کافی وجود ندارد.
کیفیت شواهد
کیفیت کلی روششناسی مطالعات وارد شده متوسط تا بالا بود. با این حال، به دلیل تعداد کم مطالعات وارد شده و نرخ پائین حوادث، قضاوت ما بر این بود که کیفیت کلی شواهد درباره پیامدهای اصلی از بسیار پائین تا متوسط متغیر است. این بدان معنی است که مطالعات جدید به احتمال زیاد تاثیر مهمی بر اعتماد ما در تخمین این تاثیر خواهند داشت و ممکن است این تخمین را تغییر دهند یا به این معناست که ما در مورد این تخمین بسیار نامطمئن هستیم.
Authors' conclusions
Summary of findings
| Alemtuzumab 12 mg compared to interferon beta‐1a for multiple sclerosis | ||||||
| Patient or population: patients with multiple sclerosis | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Interferon beta‐1a | Alemtuzumab 12 mg | |||||
| Relapse‐free survival | Not estimated | Not estimated | HR 0.50 (0.41 to 0.60) | 1248 | ⊕⊕⊕⊝ | — |
| Sustained disease progression‐free survival | Not estimated | Not estimated | HR 0.62 (0.44 to 0.87) | 1191 | ⊕⊕⊕⊝ | — |
| Number of participants with at least one adverse event | Study population | RR 1.04 | 1248 | ⊕⊕⊕⊝ | — | |
| 94 per 100 | 98 per 100 | |||||
| Moderate | ||||||
| 94 per 100 | 98 per 100 | |||||
| Change in EDSS score | — | The mean change in EDSS score in the intervention groups was | — | 1199 | ⊕⊝⊝⊝ | — |
| Number of participants with new or enlarging T2‐hyperintense lesions | 69 per 100 | 51 per 100 | RR 0.74 | 1238 | ⊕⊕⊕⊝ | — |
| Dropouts | Study population | RR 0.31 | 1248 | ⊕⊕⊝⊝ | — | |
| 24 per 100 | 8 per 100 | |||||
| Moderate | ||||||
| 24 per 100 | 7 per 100 | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Participants and personnel were not blinded and this outcome could be affected by this fact. | ||||||
Background
Description of the condition
Multiple sclerosis (MS) is an autoimmune, inflammatory, demyelinating disease of the central nervous system (brain and spinal cord), the causes of which remain unknown (Coles 1999a; Gray 2004). It is the most common cause of non‐traumatic neurological disability in young adults (Noseworthy 2000). Almost two million people in the world are affected by this condition, which can substantially impair patients' quality of life and is associated with high costs for patients, their families and society in general (Multiple Sclerosis International Federation 2010).
Four types of MS have been identified: relapsing‐remitting (RR), secondary‐progressive (SP), primary‐progressive (PP) and progressive‐relapsing (PR). The disease course is unpredictable; while some individuals are minimally affected, others show rapid progression of the disease, reaching total physical incapacity (Lublin 1996). In the first form, MS is characterised by relapses and remissions (RR), but given time sequelae from relapses may cause increased disability (Hawkins 1999). In some patients, the disease is progressive from its onset (PP); others experience periods of progression followed by relapses and remissions (SP) (Lublin 1996). In other cases, MS shows progression from onset but with clear relapses (PR).
Description of the intervention
Therapeutic strategies for MS aim to treat exacerbations, prevent new exacerbations and avoid progression of disability (Filippini 2013). Current disease‐modifying treatments decrease the frequency of relapse and modestly reduce the accumulation of disability (Coles 2006; Rieckmann 2009). Consequently, new agents that effectively control the disease are needed.
Alemtuzumab (Lemtrada, previously known as Campath‐1H) is a humanised monoclonal antibody against cell surface CD52, which can be found in a variety of cell populations, including B and T lymphocytes, thymocytes and monocytes but not in haematological precursors or plasma cells (Gilleece 1993). However, the exact function of CD52 is still unknown (Xia 1991).
In 2001, alemtuzumab was approved for fludarabine‐resistant B‐cell chronic lymphocytic leukaemia (FDA 2001; Keating 2002). Since that time, it has been used for several other diseases (licensed or off‐label use), including immune thrombocytopenic purpura, aplastic anaemia, autoimmune haemolytic anaemia, vasculitis, hematopoietic stem cell transplants (as a conditioning regimen) and organ transplants (as an induction agent) (Gomez‐Almaguer 2012; Lockwood 2003; Waldmann 2005; Weissenbacher 2010).
A study published in 1999, including 36 participants with progressive MS, reported that daily intravenous infusions of alemtuzumab (20 mg over four hours for five days) were associated with a reduction in gadolinium‐enhanced magnetic resonance imaging (MRI) lesions and a reduction in relapses, with no clinical improvement in disability (Coles 1999b). Open studies involving participants with relapsing‐remitting MS reported that the drug reduced relapse rates and disability (Coles 2006; Hirst 2008).
Alemtuzumab is already approved for MS in the European Union (EMA 2013). The US Food and Drug Administration (FDA) approved alemtuzumab for the treatment of people with RR MS who have had an inadequate response to two or more drugs indicated for the treatment of MS (FDA 2014).
The guidelines from the Association of British Neurologists identified alemtuzumab as having greatest activity in preventing relapses. However, because of safety concerns, the guidelines recommended this drug as a second‐line treatment, or for patients with the rapidly evolving RR form (Scolding 2015).
Alemtuzumab is available for the treatment of MS in 12 mg/1.2 mL single‐dose vials (10 mg/mL). The proposed initial dosage for MS is 12 mg daily for five consecutive days (intravenous infusion), followed by a second treatment course of 12 mg/daily for three consecutive days. The second treatment course is administered 12 months after the first course. Premedication with corticosteroids is recommended immediately before alemtuzumab and during the first three days of any treatment course (FDA 2014). The overall half‐life of the drug is approximately 21 days. Alemtuzumab is available as a liquid to be made up into a solution for infusion (drip) into a vein. An infusion provides 12 mg and lasts around four hours.
Alemtuzumab can produce serious adverse events including other autoimmune syndromes affecting the thyroid and blood cells (thrombocytopenia, haemolytic anaemia, pancytopenia) and nephropathies, and it can increase the risk of thyroid cancer (FDA 2014). At five‐year follow‐up, the cumulative risk of autoimmune disease is approximately 22%, Graves' disease 12%, immune thrombocythaemia purpura 3% and Goodpasture's disease (severe glomerulopathy) 0.4% (Cossburn 2011).
Recently, the FDA updated a general overview of recommendations (Risk Evaluation and Mitigation Strategy Program) about Lemtrada for patients, pharmacies and healthcare providers (FDA 2015).
How the intervention might work
Previous researches have suggested that alemtuzumab depletes the T‐ and B‐cells that may be responsible for cellular damage, while sparing innate immune cells (Rao 2012). Change in the composition of lymphocytes that accompanies lymphocyte reconstitution has also been reported (Hill‐Cawthorne 2012).
Why it is important to do this review
Results of randomised controlled trials (RCTs) of alemtuzumab for MS are promising and a systematic review of all RCTs was warranted to evaluate its effectiveness and safety for MS.
Objectives
To assess the safety and effectiveness of alemtuzumab used alone or associated with other treatments to decrease disease activity in people with any form of MS.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised, double‐blind clinical trials (RCTs). We did not consider cross‐over trials.
Types of participants
We included adults diagnosed with MS according to the McDonald criteria (McDonald 2001; Polman 2011), or Poser criteria (Poser 1983). We considered participants with any form of MS (relapsing‐remitting, primary‐progressive, secondary‐progressive or progressive‐relapsing) for inclusion.
Types of interventions
-
Experimental intervention: alemtuzumab alone or associated with other medications at any dose and for any course duration.
-
Comparator: placebo, any other active drug therapy (i.e. corticosteroids, plasmapheresis, beta interferons, glatiramer acetate, fingolimod, natalizumab, mitoxantrone, teriflunomide or dimethyl fumarate).
Types of outcome measures
Primary outcomes
-
Relapse‐free survival. Relapse was defined as newly developed or recently worsened symptoms of neurological dysfunction, lasting longer than 24 hours and objectively confirmed. However, we considered less stringent criteria and assessed these separately.
-
Sustained disease progression‐free survival, defined as a ≥ 1.0‐point increase in the Expanded Disability Status Scale (EDSS) score (Kurtzke 1983) for participants with a baseline score ≤ 5.0 or a ≥ 0.5‐point increase for participants with a baseline score ≥ 5.5 points confirmed at six months. We considered a one‐point increase in EDSS score confirmed at three months' follow‐up as a surrogate outcome measure of progression.
-
Number of participants with at least one adverse event, including serious adverse events.
All primary outcomes were assessed after 12 and 24 months follow‐up and at the end of the follow‐up period.
Secondary outcomes
-
Number of participants free of clinical disease activity, defined as no relapses and no sustained accumulation of disability. Sustained accumulation disability was defined as an increase of at least 1.5 points on the Expanded Disability Status Scale (EDSS) for patients with a baseline score of 0 and of at least 1.0 point for patients with a baseline score of 1.0 or more.
-
Quality of life as assessed by the Multiple Sclerosis Quality of Life scale (MSQOL)‐54 (Vickrey 1995) or the Multiple Sclerosis Quality of Life Inventory (MSQLI) (Fischer 1999).
-
Change in disability as assessed by the EDSS (Kurtzke 1983).
-
Fatigue as assessed by the Fatigue Severity Scale or the Fatigue Index Scale (Krupp 1989).
-
Number of participants with new or enlarging T2‐hyperintense lesions on magnetic resonance imaging (Li 1999).
-
Number of participants who dropped out.
All secondary outcomes would be assessed after 12 and 24 months and at the end of the follow‐up period.
Search methods for identification of studies
We conducted a systematic search without language restrictions to identify all relevant published randomised controlled trials using the optimally sensitive strategy developed by Cochrane for the identification of RCTs.
Electronic searches
The Trials Search Co‐ordinator searched the Trials Register of the Cochrane Multiple Sclerosis and Rare Diseases of the CNS Group (30 April 2015) which, among other sources, includes trials from:
-
Cochrane Central Register of Controlled Trials (CENTRAL 2015, Issue 4);
-
MEDLINE (PubMed) (1966 to 30 April 2015);
-
EMBASE (Embase.com) (1974 to 30 April 2015);
-
Cumulative Index to Nursing and Allied Health Literature (CINAHL) (EBSCO host) (1981 to 30 April 2015);
-
Latin American and Caribbean Health Science Information Database (LILACS) (Bireme) (1982 to 30 April 2015);
-
ClinicalTrials.gov (www.clinicaltrials.gov);
-
World Health Organization (WHO) International Clinical Trials Registry Platform (apps.who.int/trialsearch).
Information on the Trials Register of the Review Group and details of search strategies used to identify trials can be found in the 'Specialised Register' section within the Cochrane Multiple Sclerosis and Rare Diseases of the CNS Group module.
The keywords used to search for trials for this review are listed in Appendix 1.
Searching other resources
In addition, we used the following methods.
-
We screened the bibliographic references of identified studies to identify additional studies.
-
We contacted pharmaceutical companies (Genzyme‐Sanofi and Bayer) for information on any unpublished trials.
-
We contacted the main authors of studies if data reported in the original articles were incomplete and we asked experts in this field about additional unpublished or ongoing studies.
Data collection and analysis
Selection of studies
Two review authors (RR and GJMP) independently screened the titles and abstracts of all records retrieved by the search in order to identify potentially relevant studies. We retrieved the full‐text reports/publications of those deemed eligible for inclusion and three review authors (RR, GP and MRT) independently read the full texts to identify studies that met the selection criteria. The review authors recorded the reasons for exclusion of rejected studies. Disagreements between two review authors (RR and GJMP) were discussed until a consensus was reached with the consultation of a third review author (MRT) if needed.
Data extraction and management
We used a data collection form to report information on study characteristics and outcome data. Three review authors (RR, GP and MRT) extracted the following information from the primary studies included in the review.
-
Publication details (i.e. year, country, authors).
-
Study design and methods: inclusion/exclusion criteria, randomisation method, allocation concealment, blinding.
-
Setting.
-
Population data (i.e. age, severity of disease, type of MS).
-
Details of intervention (i.e. dose, regimen, duration).
-
Outcome measures (including effectiveness and adverse effects).
-
Number of dropouts.
-
Length of follow‐up.
-
Types of data analyses (e.g. intention‐to‐treat, modified intention‐to‐treat).
-
Any other potential risk of bias.
We discussed disagreements until consensus was reached, with the involvement of a third review author (MRT) if needed. One review author (RR) inserted data into the Review Manager 5.3 software (RevMan 2015). We double‐checked that data were entered correctly into the form.
Assessment of risk of bias in included studies
Three review authors (RR, GJMP and MRT) independently assessed the methodological quality of included clinical trials using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We assessed the risk of bias according to the following domains.
-
Sequence generation: Was the allocation sequence adequately generated?
-
Allocation concealment: Was allocation adequately concealed?
-
Blinding of participants, personnel and outcome assessors: Was knowledge of the allocated interventions adequately prevented during the study?
-
Incomplete outcome data: Were outcome data adequately assessed and accounted for? (We considered a loss to follow‐up rate greater than 15% as high risk).
-
Selective outcome reporting: Were the reports of the study free of any suggestion of selective outcome reporting?
-
Other potential threats to validity: Was the study apparently free from other problems that could put it at risk of bias?
We graded each potential source of bias as high, low or unclear and provided a quote from the study report together with a justification for our judgement in the 'Risk of bias' table.
We considered the overall quality of the studies good if the sequence generation, allocation concealment and blinding (patients and personnel, and assessors) domains were all at low risk of bias. We considered the overall quality of the studies moderate if one of these domains was categorised as being at unclear risk. Finally, we considered the overall quality high if at least one of these domains was categorised as high risk of bias.
Measures of treatment effect
For each outcome, we calculated a summarised estimate of treatment effect (with 95% confidence interval (CI)) for each comparison. We reported dichotomous outcomes as risk ratios (RRs). We used the mean difference (MD) for continuous outcomes and the hazard ratio (HR) for time‐to‐event outcomes.
Unit of analysis issues
The unit of analysis was the individual participant.
Dealing with missing data
In cases where there were missing or unavailable data, we contacted the primary authors for further information. We performed a search for protocols or additional articles related to the included trials (or both). If relevant data were unavailable, we presented and discuss the results in the main text of the review.
Assessment of heterogeneity
We investigated heterogeneity using the Chi2 test and the I2 statistic, which indicates the degree of variation across studies that is due to heterogeneity rather than due to chance. We considered an I2 value greater than 50% as substantial heterogeneity (Higgins 2011). We checked clinical and methodological differences as potential causes of heterogeneity.
Assessment of reporting biases
As it was not possible to pool more than 10 studies, we did not use funnel plots to explore possible publication bias.
Data synthesis
We summarised data using the Review Manager 5 software (RevMan 2015). When significant methodological or clinical heterogeneity existed,we used a random‐effects model; otherwise we used a fixed‐effect model.
Subgroup analysis and investigation of heterogeneity
We planned the following subgroup analyses:
-
Treatment duration (12 or 24 months).
-
Different doses and regimens of alemtuzumab (12 mg or 24 mg).
-
Disease type: relapsing‐remitting, primary‐progressive, secondary‐progressive or progressive‐relapsing.
-
Disability at baseline (EDSS score ≤ 5.0 or ≥ 5.5).
-
Naive or previously treated participants.
However, we did not carry out subgroup analyses to consider disease type and disability at baseline due to lack of available data.
Sensitivity analysis
We planned to perform sensitivity analysis by excluding trials of low or moderate quality (or both) and comparing the results with the overall findings. However, since we included only three trials in the review, we deemed this analysis inappropriate.
'Summary of findings' table
We created a 'Summary of findings' table (summary of findings Table for the main comparison) by using the following outcomes:
-
Relapse‐free survival.
-
Sustained disease progression‐free survival.
-
Number of participants with at least one adverse event, including serious adverse events.
-
Change in disability assessed by the EDSS.
-
Number of participants with new or enlarging T2‐hyperintense lesions.
-
Number of participants who dropped out.
We used the five GRADE parameters (risk of bias, inconsistency, imprecision, indirectness and publication bias) to assess the quality of the body of evidence as it relates to the studies that contributed data to the meta‐analyses for prespecified outcomes. We used the methods and recommendations described in Section 8.5 and Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) using the GRADEpro software (GRADEpro 2008). We justified all decisions to downgrade or upgrade the quality of studies in the footnotes and we made comments to aid readers' understanding of the review when necessary.
Results
Description of studies
Results of the search
The search strategy retrieved 223 references: two in CENTRAL, 128 in MEDLINE, 82 in EMBASE, three in CINAHL, none in PEDro, none in LILACS, four in ClinicalTrials.gov, none in the WHO International Clinical Trials Registry Platform and four from handsearching. We considered a total of 35 references to be potentially eligible. After reading the full text, we included these 35 records. They referred to three RCTs and 32 ancillary reports about these three primary studies. The flow diagram of the process of study identification and selection is presented in Figure 1.

Study flow diagram.
Included studies
The three RCTs included a total of 1713 participants (CAMMS223; CARE‐MS I; CARE‐MS II). All studies were multicentric trials, comparing alemtuzumab versus subcutaneous interferon beta‐1a for patients with relapsing–remitting MS according to the McDonald criteria (McDonald 2001).
Participants were treatment‐naive in the CARE‐MS I and CAMMS223 studies. The CARE‐MS II study included only participants with at least one relapse while being treated with interferon beta or glatiramer for at least six months.
In the CARE‐MS I and CARE‐MS II studies, the interventions were given for 12 months (CARE‐MS I; CARE‐MS II); in the CAMMS223 study, the treatment lasted 24 months (CAMMS223). The following regimens were used in these RCTs:
-
CAMMS223 study, a phase II trial: alemtuzumab (either 12 mg per day or 24 mg per day) was given by intravenous infusion on five consecutive days during the first month and on three consecutive days at months 12 and 24 (CAMMS223).
-
CARE‐MS I (or CAMMS323) study, a phase III trial: alemtuzumab (12 mg per day) was given by intravenous infusion on five consecutive days during the first month and on three consecutive days at month 12 (CARE‐MS I).
-
CARE‐MS II (or CAMMS324) study, a phase III trial: alemtuzumab (either 12 mg per day or 24 mg per day) was given by intravenous infusion on five consecutive days during the first month and on three consecutive days at month 12 (CARE‐MS II).
In all studies, the dose of interferon beta‐1a was 44 μg given subcutaneously three times weekly after dose titration.
Details of these RCTs are available in the table Characteristics of included studies.
Excluded studies
We excluded none of the potentially eligible studies.
Risk of bias in included studies
The risk of bias of each study is detailed in the Characteristics of included studies table. Figure 2 and Figure 3 present the 'Risk of bias' summary along with review authors' judgements about each risk of bias item for each included study. The overall quality of the studies was low since in all of them we categorised at least one of the main domains (generation of allocation sequence, allocation concealment and blinding) as having a high risk of bias.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
We classified all studies as low risk of bias for generation of allocation sequence. The methods were reported in the articles and we judged them to be appropriate.
However, for allocation concealment, we classified one study as having an unclear risk of bias because it did not provide enough information to allow judgement (CAMMS223). We classified the other two studies as having a low risk of bias because they provided an adequate method to ensure allocation concealment (CARE‐MS I; CARE‐MS II).
Blinding
We considered all studies as having a high risk of bias of performance bias (participants and personnel) because both drugs (intervention and comparator) had adverse effects that precluded masking.
We judged the following outcomes separately for detection bias (outcome assessment):
-
EDSS outcome assessment: We classified CARE‐MS I and CARE‐MS II as having a high risk of bias since unmasked raters performed the EDSS assessments. On the other hand, CAMMS223 had a low risk of bias.
-
Efficacy outcomes assessment (except EDSS): We classified all studies as having a low risk of bias.
-
Safety outcomes assessment: CARE‐MS I and CARE‐MS II had a low risk of bias, while CAMMS223 had a high risk of bias.
Incomplete outcome data
One study was at low risk of attrition bias (CAMMS223), one was at high risk because it used an inappropriate modified "intention‐to‐treat" analysis (CARE‐MS I), and one study was at unclear risk of attrition bias (CARE‐MS II)
Selective reporting
We classified two studies as having a low risk of selective reporting bias (CAMMS223; CARE‐MS I). We classified one study as having high risk of bias because the results for some previously planned outcomes were not provided (i.e. quality of life) (CARE‐MS II).
Other potential sources of bias
There were no other known potential sources of bias in the three included trials.
Effects of interventions
Comparison 1: Alemtuzumab 12 mg versus subcutaneous interferon beta‐1a
Primary outcomes
Relapse‐free survival
Alemtuzumab was associated with better relapse‐free survival at 24‐month follow‐up (hazard ratio (HR) 0.50, 95% confidence interval (CI) 0.41 to 0.60; 1248 participants; two studies; moderate quality evidence, I2 = 0%) (Analysis 1.1; Figure 4). This result was consistent when we considered separately naive and previously treated participants.

Forest plot of comparison: 1 Alemtuzumab 12 mg versus interferon beta‐1a, outcome: 1.1 Relapse‐free survival.
Only one study assessed this outcome at 36 months (CAMMS223). This study showed a higher number of participants who relapsed with interferon than with alemtuzumab (45 versus 24; HR 0.31, 95% CI 0.18 to 0.52).
None of the included studies provided data for the 12‐month analysis.
Sustained disease progression‐free survival
Alemtuzumab was associated with a lower number of participants with sustained disease progression‐free survival at both 24‐month (HR 0.62, 95% CI 0.44 to 0.87; 1191 participants; two studies; I2 = 0%) and 36‐month follow‐up (HR 0.25, 95% CI 0.11 to 0.57; 223 participants; one study) (Analysis 1.2; Figure 5). This finding was consistent when we considered a subgroup of previously treated participants. However, for naive participants there was no difference between the interventions.

Forest plot of comparison: 1 Alemtuzumab 12 mg versus interferon beta‐1a, outcome: 1.2 Sustained disease progression‐free survival.
None of the included studies provided data for the 12‐month analysis.
Number of participants with at least one adverse event, including serious adverse events
Alemtuzumab was associated with a higher proportion of participants with at least one adverse event after 24 months (risk ratio (RR) 1.04, 95% CI 1.01 to 1.06; 1248 participants; two studies; I2 = 0%; moderate quality evidence), but not at 36 months (RR 1.00, 95% CI 0.98 to 1.02; 224 participants; one study) (Analysis 1.3; Figure 6).

Forest plot of comparison: 1 Alemtuzumab 12 mg versus interferon beta‐1a, outcome: 1.2 Rate of participants with at least one adverse event.
None of the included studies provided data for the 12‐month analysis.
Secondary outcomes
Number of participants free of clinical disease activity
None of the included studies assessed this outcome.
Quality of life
None of the included studies assessed this outcome.
Change in disability as assessed by the Expanded Disability Status Scale (EDSS)
Alemtuzumab was associated with a significant improvement in EDSS scores after 36 months (mean difference (MD) ‐0.70, 95% CI ‐1.04 to ‐0.36; 223 participants; one study) (CAMMS223). At 24 months, considering both treatment‐naive patients and previously treated patients (who failed after interferon beta or glatiramer treatment), there were no differences in EDSS scores (MD ‐0.20, 95% CI ‐0.60 to 0.20; 1199 participants; two studies; I2 = 88%) (Analysis 1.4). However, when only previously treated patients were assessed, alemtuzumab was associated with better results (MD ‐0.41, 95% CI ‐0.62 to ‐0.20; one study; 628 participants) (CARE‐MS II).
None of the included studies provided data for the 12‐month analysis.
Fatigue as assessed by the Fatigue Severity Scale or the Fatigue Index Scale
None of the included studies assessed this outcome.
Number of participants with new or enlarging T2‐hyperintense lesions on magnetic resonance imaging
Alemtuzumab was associated with a lower rate of participants presenting with new or enlarging lesions, considering both naive and previously treated participants (RR 0.74, 95% CI 0.59 to 0.91; 1238 participants; two studies; I2 = 80%; random‐effects model) (Analysis 1.5).
Number of participants who dropped out
Alemtuzumab was associated with a lower number of dropouts at 24 months (RR 0.31, 95% CI 0.23 to 0.41; 1248 participants; two studies), but not at 36 months (RR 0.81, 95% CI 0.57 to 1.14; 224 participants; one study) (Analysis 1.6).
None of the included studies provided data for the 12‐month analysis.
Comparison 2: Alemtuzumab 24 mg versus subcutaneous interferon beta‐1a
Primary outcomes
Relapse‐free survival
Only one study assessed this outcome at 36 months (CAMMS223). This study showed a higher number of relapses in the interferon group than in the alemtuzumab group (45 versus 17; HR 0.21, 95% CI 0.11 to 0.40) (CAMMS223).
None of the included studies provided data for the 12‐ and 24‐month analyses.
Sustained disease progression‐free survival
Alemtuzumab was associated with a lower number of participants with sustained disease progression‐free survival at 36 months (HR 0.33, 95% CI 0.16 to 0.69; 221 participants; one study) (CAMMS223).
None of the included studies provided data for the 12‐ or 24‐month analyses.
Number of participants with at least one adverse event, including serious adverse events
There were no significant differences between alemtuzumab and subcutaneous interferon beta‐1a at 24 months (RR 1.04, 95% CI 1.00 to 1.07; 391 participants; one study) (CARE‐MS II) or at 36 months (RR 0.99, 95% CI 0.97 to 1.02; 220 participants; one study) (CAMMS223).
None of the included studies provided data for the 12‐month analysis.
Secondary outcomes
Number of participants free of clinical disease activity
None of the included studies assessed this outcome.
Quality of life
None of the included studies assessed this outcome.
Change in disability as assessed by the EDSS
Alemtuzumab was associated with a significant improvement in EDSS scores after 36 months (MD ‐0.83, 95% CI ‐1.17 to ‐0.49; 221 participants; one study) (CAMMS223). None of the included studies provided data for the 12‐ and 24‐month analyses.
Fatigue as assessed by the Fatigue Severity Scale or the Fatigue Index Scale
None of the included studies assessed this outcome.
Number of participants with new or enlarging T2‐hyperintense lesions on magnetic resonance imaging
None of the included studies assessed this outcome.
Number of participants who dropped out
Alemtuzumab was associated with a lower number of dropouts at 24 months (RR 0.27, 95% CI 0.16 to 0.46; 404 participants; one study) (CARE‐MS II), but not at 36 months (RR 0.76, 95% CI 0.53 to 1.09; 221 participants; one study) (CAMMS223).
None of the included studies provided data for the 12‐month analysis.
Discussion
Summary of main results
This systematic review aimed to assess the effects (benefits and harms) of alemtuzumab compared with any other drug treatment for any type of multiple sclerosis (MS).
Based on results of three randomised clinical trials (RCTs), compared to subcutaneous interferon beta‐1a, alemtuzumab 12 mg was associated with:
-
higher relapse‐free survival at 24 months and 36 months;
-
a lower number of participants with sustained disease progression‐free survival;
-
a slightly higher number of participants with at least one adverse event after 24 months;
-
a higher improvement in Expanded Disability Status Scale (EDSS) scores after 36 months;
-
a higher improvement in EDSS scores after 24 months (for patients previously treated with interferon or glatiramer acetate);
-
a lower number of participants with new or enlarging T2‐hyperintense lesions on magnetic resonance imaging;
-
a lower number of dropouts at 24 months, but not at 36 months.
Based on the results of one RCT, compared to subcutaneous interferon beta‐1a, alemtuzumab 24 mg was associated with:
-
higher relapse‐free survival at 36 months;
-
a lower number of participants with sustained disease progression‐free survival at 36 months;
-
no statistical difference in the number of participants with at least one adverse event at 24 and 36 months;
-
a higher improvement in EDSS scores after 36 months;
-
a lower number of dropouts at 24 months, but not at 36 months.
The higher number of participants with at least one adverse event was not associated with a higher dropout rate probably because most of these events were mild or moderate. Data for severe adverse events were not provided separately by any of the included studies.
We included change in EDSS scores as a secondary outcome instead of a primary outcome because short‐term changes in EDSS scores may not be a reliable marker of irreversible change in relapsing–remitting MS (Healy 2013).
Overall completeness and applicability of evidence
We included three RCTs that compared alemtuzumab versus subcutaneous interferon beta‐1a in patients with relapsing–remitting MS. Alemtuzumab was given during 12 or 24 months and the participants had a follow‐up of up to 36 months for some outcomes in one of the included studies. The doses were (a) 12 mg or 24 mg per day intravenously, once a day for five consecutive days at month 0 and 12, or (b) 24 mg per day intravenously, once a day for three consecutive days at month 12 and 24. The control groups received interferon beta‐1a, 44 μg subcutaneously three times weekly after dose titration. Therefore the available evidence is limited to these specific interventions and patients.
There is a lack of evidence for the following outcomes:
-
number of participants free of clinical disease activity;
-
quality of life;
-
fatigue (assessed by the Fatigue Severity Scale or the Fatigue Index Scale, for example).
There are two probable reasons for this lack of evidence: (a) the outcomes were initially proposed in the trial protocols but were not available for this review even after contact with the authors of these studies; (b) the outcomes were not originally planned at the protocol stage of the included RCTs.
We must emphasise that the data at 36‐month follow‐up are based on a small number of participants and this can increase the uncertainty of these findings.
Finally, the three studies only included patients with relapsing–remitting MS and we found no evidence for other forms of the disease.
Quality of the evidence
As presented in summary of findings Table for the main comparison, the quality of the body of evidence obtained for each outcome ranged from very low to moderate.
The overall quality of the RCTs was low since in all of them we categorised at least one of the main domains (generation of allocation sequence, allocation concealment and blinding) as having a high risk of bias. In all studies, the participants and personnel were not blinded because the adverse effects related to each intervention preclude the masking. Additionally, in two studies the assessment of the EDSS scores could also be not blinded. Considering these two facts, we judged separately the risk of bias for EDSS and adverse events.
We noted no statistically significant heterogeneity among the studies for the co‐primary outcomes. The quality of the evidence for dropouts was impaired by the low number of events in the trials.
Potential biases in the review process
To avoid the introduction of bias, we strictly followed all of the recommendations on searching, study selection, data collection, and data analysis from the Cochrane Handbook for Systematic Reviews of Interventions in this review (Higgins 2011).
The strengths of this review include a wide literature search and the use of intention‐to‐treat analyses for dichotomous data.
The limitations of this review include: (a) no assessment of publication bias through funnel plot analysis because there were fewer than 10 studies included in the meta‐analysis and (b) the lack of some outcome data in the included RCTs.
Agreements and disagreements with other studies or reviews
During the conduct of this review a non‐Cochrane systematic review assessing all available treatments for MS was published (CADTH 2013). This review evaluated direct and indirect comparisons between several drugs, including alemtuzumab. The findings are similar to those of our review, including the results of meta‐analysis and the risk of bias of the included RCTs.
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Forest plot of comparison: 1 Alemtuzumab 12 mg versus interferon beta‐1a, outcome: 1.1 Relapse‐free survival.
Forest plot of comparison: 1 Alemtuzumab 12 mg versus interferon beta‐1a, outcome: 1.2 Sustained disease progression‐free survival.
Forest plot of comparison: 1 Alemtuzumab 12 mg versus interferon beta‐1a, outcome: 1.2 Rate of participants with at least one adverse event.
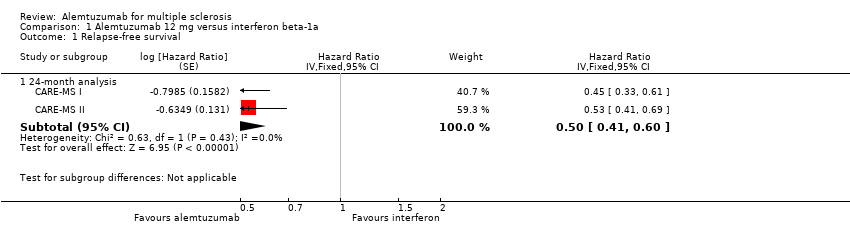
Comparison 1 Alemtuzumab 12 mg versus interferon beta‐1a, Outcome 1 Relapse‐free survival.

Comparison 1 Alemtuzumab 12 mg versus interferon beta‐1a, Outcome 2 Sustained disease progression‐free survival.
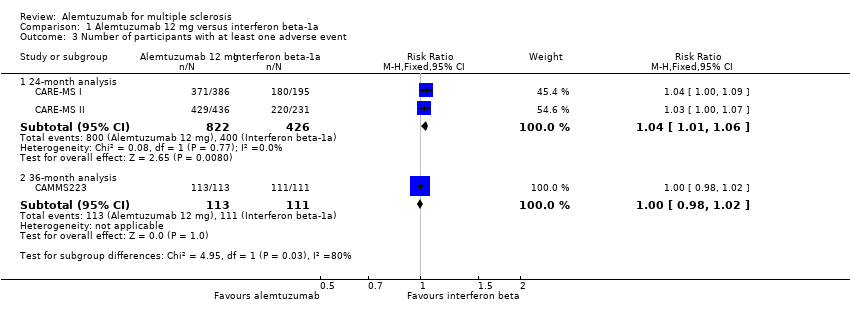
Comparison 1 Alemtuzumab 12 mg versus interferon beta‐1a, Outcome 3 Number of participants with at least one adverse event.
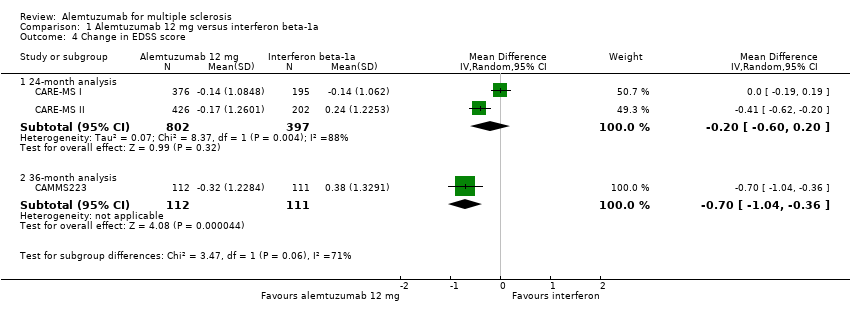
Comparison 1 Alemtuzumab 12 mg versus interferon beta‐1a, Outcome 4 Change in EDSS score.

Comparison 1 Alemtuzumab 12 mg versus interferon beta‐1a, Outcome 5 Number of participants with new or enlarging T2‐hyperintense lesions.
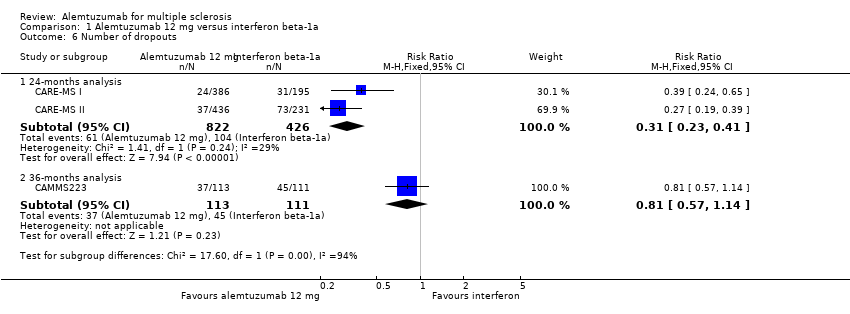
Comparison 1 Alemtuzumab 12 mg versus interferon beta‐1a, Outcome 6 Number of dropouts.
| Alemtuzumab 12 mg compared to interferon beta‐1a for multiple sclerosis | ||||||
| Patient or population: patients with multiple sclerosis | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect | No of participants | Quality of the evidence | Comments | |
| Assumed risk | Corresponding risk | |||||
| Interferon beta‐1a | Alemtuzumab 12 mg | |||||
| Relapse‐free survival | Not estimated | Not estimated | HR 0.50 (0.41 to 0.60) | 1248 | ⊕⊕⊕⊝ | — |
| Sustained disease progression‐free survival | Not estimated | Not estimated | HR 0.62 (0.44 to 0.87) | 1191 | ⊕⊕⊕⊝ | — |
| Number of participants with at least one adverse event | Study population | RR 1.04 | 1248 | ⊕⊕⊕⊝ | — | |
| 94 per 100 | 98 per 100 | |||||
| Moderate | ||||||
| 94 per 100 | 98 per 100 | |||||
| Change in EDSS score | — | The mean change in EDSS score in the intervention groups was | — | 1199 | ⊕⊝⊝⊝ | — |
| Number of participants with new or enlarging T2‐hyperintense lesions | 69 per 100 | 51 per 100 | RR 0.74 | 1238 | ⊕⊕⊕⊝ | — |
| Dropouts | Study population | RR 0.31 | 1248 | ⊕⊕⊝⊝ | — | |
| 24 per 100 | 8 per 100 | |||||
| Moderate | ||||||
| 24 per 100 | 7 per 100 | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). | ||||||
| GRADE Working Group grades of evidence | ||||||
| 1Participants and personnel were not blinded and this outcome could be affected by this fact. | ||||||
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
| 1 Relapse‐free survival Show forest plot | 2 | Hazard Ratio (Fixed, 95% CI) | Subtotals only | |
| 1.1 24‐month analysis | 2 | Hazard Ratio (Fixed, 95% CI) | 0.50 [0.41, 0.60] | |
| 2 Sustained disease progression‐free survival Show forest plot | 3 | Hazard Ratio (Fixed, 95% CI) | Subtotals only | |
| 2.1 24‐month analysis | 2 | Hazard Ratio (Fixed, 95% CI) | 0.62 [0.44, 0.87] | |
| 2.2 36‐month analysis | 1 | Hazard Ratio (Fixed, 95% CI) | 0.25 [0.11, 0.57] | |
| 3 Number of participants with at least one adverse event Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 24‐month analysis | 2 | 1248 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [1.01, 1.06] |
| 3.2 36‐month analysis | 1 | 224 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.98, 1.02] |
| 4 Change in EDSS score Show forest plot | 3 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 24‐month analysis | 2 | 1199 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.60, 0.20] |
| 4.2 36‐month analysis | 1 | 223 | Mean Difference (IV, Random, 95% CI) | ‐0.7 [‐1.04, ‐0.36] |
| 5 Number of participants with new or enlarging T2‐hyperintense lesions Show forest plot | 2 | 1238 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.59, 0.91] |
| 6 Number of dropouts Show forest plot | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 24‐months analysis | 2 | 1248 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.23, 0.41] |
| 6.2 36‐months analysis | 1 | 224 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.57, 1.14] |