Retuksimab za liječenje inhibitora u osoba s naslijeđenom teškom hemofilijom
Abstract
Background
Hemophilia A and B are inherited coagulation disorders characterized by a reduced or absent level of factor VIII or factor IX respectively. The severe form is characterized by a factor level less than 0.01 international units (IU) per milliliter. The development of inhibitors in hemophilia is the main complication of treatment, because the presence of these antibodies, reduces or even nullifies the efficacy of replacement therapy, making it very difficult to control the bleeding. People with inhibitors continue to have significantly higher risks of morbidity and mortality, with considerable treatment costs. Given the wide 'off‐label' use of rituximab for treating people with hemophilia and inhibitors, its efficacy and safety need to be evaluated. This is an update of a previously published Cochrane Review.
Objectives
To assess the efficacy and safety of rituximab for treating inhibitors in people with inherited severe hemophilia A or B.
Search methods
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group's Coagulopathies Trials Register, complied from electronic database searches and handsearching of journals and conference abstract books. We searched the reference lists of relevant articles and reviews and also searched for ongoing or unpublished studies. We also undertook further searches of other bibliographic databases and trial registries.
Date of last search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Coagulopathies Trials Register: 16 February 2017.
Selection criteria
Randomized controlled trials and controlled clinical trials investigating the efficacy and safety of rituximab for treating inhibitors in people with hemophilia.
Data collection and analysis
No randomized controlled trials matching the selection criteria were eligible for inclusion.
Main results
No randomized controlled trials on rituximab for treating inhibitors in people with hemophilia were identified.
Authors' conclusions
We were unable to identify any relevant trials on the efficacy and safety of rituximab for treating inhibitors in people with hemophilia. The research evidence available is from case reports and case series. Randomized controlled trials are needed to evaluate the efficacy and safety of rituximab for this condition. However, prior to the publication of any possible future randomized controlled trials, meta‐analysis of case reports and case series may provide some evidence.
PICOs
Laički sažetak
Retuksimab za liječenje inhibitora faktora zgrušavanja u osoba s naslijeđenom teškom hemofilijom
Istraživačko pitanje
U ovom Cochrane sustavnom pregledu literature analizirali smo dostupne dokaze iz istraživanja u kojima je ispitano je li retuksimab učinkovit i siguran u liječenju inhibitora faktora zgrušavanja u osoba s teškom hemofillijom. Ovaj pregled je obnovljena verzija ranije objavljenog Cochrane sustavnog pregleda.
Dosadašnje spoznaje
Hemofilija A i B su nasljedna stanja sa smanjenom razinom ( ili je uopće nema) faktora VIII (hemofilija A) ili faktora IX (hemofilija B) u krvi. U teškim oblicima koncentracije ovih faktora su nemjerljive (manje od 0.01 internacionalnih jedinica (IU) po mililitru). Ljudi sa hemofilijom imaju opasnost od krvarenja koja se mogu javiti spontano, nakon traume ili invazivnog medicinskog postupka. Stoga ih treba liječiti koncentratima faktora zgrušavanja, ili nakon što se ti događaje dogode ili preventivno. Nažalost, oko 30% osoba s teškom hemofilijom A i 1% do 6% osoba sa teškom hemofilijom B mogu razviti antitijela (inhibitore) na faktor VIII ili faktor IX, jer imunološki sustav ne prepoznaje faktore. Razvijanje inhibitora predstavlja glavnu komplikaciju u liječenju hemofilije, jer njihova prisutnost smanjuje ili poništava učinkovitost nadomjesne terapije, čime se otežava kontrola krvarenja. Štoviše, kada su inhibitori prisutni nemoguće je započeti preventivno liječenje koncentracijama faktora VIII ili faktora IX. Stoga je važno ukloniti inhibitore i omogućiti uspješan nastavak liječenja. Korišenje retuksimaba u ovoj situaciji predstavlja liječenje izvan odobrenih indikacija (engl. off‐label use), ali se u nekim studijama taj lijek pokazao učinkovit za uklanjanje inhibitora kod osoba sa hemofilijom. Stoga smo željeli vidjeti je li retuksimab bolji od liječenja standardnom terapijom ili drugim terapijama bez retuksimaba, i da li je to sigurno i može li spasiti ljude od po život opasnih krvarenja i ogromnih financijskih troškova.
Datum pretraživanja
Dokazi se temelje na literaturi objavljenoj do: 16. veljače 2016.
Ključni rezultati
Nismo pronašli niti jedan randomizirani kontrolirani pokus koji procjenjuje retuksimab u ljudi s teškom hemofilijom. Potrebne su dobro ustrojene kontrolirane studije kako bi se procjenili koristi i rizici upotrebe retuksimab‐a kod ljudi sa teškom hemofilijom. Dok se ne objave kontrolirane studije, samo ograničeni dokazi niske razine, temeljeni na individualnim slučajevima, mogu liječnicima biti vodič u donošenju kliničkih odluka.
Authors' conclusions
Background
For a glossary of terms used in this review please refer to the appendices (Appendix 1).
Description of the condition
Hemophilia is a rare, inherited, X‐linked, recessive disorder in which the blood does not clot normally (NHLBI 2011). The severe form is characterized by a factor level less than 0.01 international units (IU) per milliliter (mL). Treatment consists of administering factor VIII (FVIII) concentrate (for hemophilia A) or factor IX (FIX) concentrate (for hemophilia B) on demand when bleeding occurs, or prophylactically to prevent bleeding (Coppola 2012; Iorio 2011). Inhibitors, neutralizing antibodies toward FVIII or FIX, can occur when the body's immune system does not recognize clotting factor concentrates as self protein (WFH 2013). These inhibitors reduce or even nullify the efficacy of factor concentrates.There are several known risk factors for the occurrence of inhibitors; either genetic risk factors (nature of gene defect, ethnicity); or non‐genetic risk factors (intensive factor exposure at the time of surgery, and prophylactic or on‐demand treatment regimens) (Iorio 2010a). The source of FVIII used for replacement therapy may also have an effect on inhibitor development. Studies have shown that for previously untreated patients (PUPs) with severe hemophilia A, high‐dose intensive FVIII treatment increases the risk for inhibitor development, but low‐dose prophylactic treatment decreases it, especially in those with low‐inhibitor‐risk FVIII genotype mutations (Gouw 2007a; Gouw 2013a). Although much debated, recent studies have suggested no significant difference in the risk of inhibitor development in PUPs between recombinant and plasma‐derived FVIII concentrate therapy (Gouw 2007b; Gouw 2013b). A clinical trial randomizing PUPs to either plasma‐derived or recombinant FVIII concentrates is ongoing (SIPPET trial) and is expected to provide further high‐quality evidence to clarify the issue (NCT01064284).
Blood screening can show the presence of a bleeding disorder. The activated partial thromboplastin time (APTT) assay often suggests the presence of inhibitors, when it is not corrected (either at all or partially) by mixing with normal plasma. The Nijmegen assay is performed to confirm diagnosis, which can determine the titer of inhibitors (Verbruggen 1995). The amount of inhibitors in the blood is measured in Bethesda Units (BU), where a level of more than five BU is referred to as 'high titer' and less than five BU as 'low titer' (WFH 2012a). Inhibitors usually occur between the first 10 to 20 exposure days and therefore most often develop in childhood. Approximately 30% of children with hemophilia A (Gouw 2013b), and 1% to 6% of individuals with hemophilia B (WFH 2012b), develop inhibitors. In people with hemophilia, sometimes the bleeding is spontaneous; surgical operations and strenuous exercise can also be precipitating factors for acute bleeding.
People with hemophilia and inhibitors commonly receive bypassing agents to treat acute bleeding episodes and immune tolerance induction (ITI) to eradicate the inhibitors. Bypassing agents, such as recombinant‐activated FVII concentrate (rFVIIa) or activated prothrombin complex concentrate (aPCC), are insensitive to inhibitors (Iorio 2010b). As an alternative, and where resources are not a limitation, bypassing agents can be used prophylactically (Leissinger 2011). In hemophilia A, the standard treatment, ITI, consists of the regular infusion of FVIII concentrate in an attempt to achieve immunologic tolerance. Success is no different between using a high‐ or low‐dose ITI regimen, although a high‐dose regimen may achieve success more rapidly (Hay 2012). People with hemophilia B and high‐titer FIX inhibitors may also be treated with ITI, although this approach is less frequently successful than for people with hemophilia A (DiMichele 2007). However, ITI is expensive and not always effective for everyone; it requires specialized medical expertise over a long period of time.
People with inhibitors continue to have significantly high risk for morbidity and mortality, with considerable treatment costs. The ultimate eradication of inhibitors remains the most challenging treatment issue in people with severe hemophilia.
Description of the intervention
Many immunomodulatory approaches have been studied in the search for more effective techniques to eradicate clotting factor inhibitors. The chimeric monoclonal antibody, rituximab, seems to be a particularly promising agent. In 1997, the USA's Food and Drug Administration (FDA) approved rituximab, also known as MabThera® (F. Hoffmann‐La Roche Ltd., Pharmaceuticals Division, Basel, Switzerland) and Rituxan® (IDEC Pharmaceuticals, San Diego, CA, and Genentech, Inc, San Francisco, CA), for use in adult CD20⁺ B‐cell lymphomas (Kavcic 2013). To date, this treatment is licensed for the following indications in adults: non‐Hodgkin's lymphoma; chronic lymphocytic leukemia; rheumatoid arthritis; granulomatosis with polyangiitis (Wegener's granulomatosis); and microscopic polyangiitis (FDA 2013). In pediatric hematology‐oncology departments, rituximab is widely used 'off‐label' (Yadav 2012) to treat such diseases as chronic immune thrombocytopenia (Grace 2012), systemic lupus erythematosus (Nwobi 2008) and autoimmune hemolytic anemia (Kuzmanovic 2012). Given its 'off‐label' use, studies in the hemophilia population are limited. The standard single dose of rituximab for treating inhibitors is 375 mg/m², administered intravenously on a weekly schedule for four weeks. Rituximab is currently available in a 10 mg/mL concentrate of either 10 mL (100 mg) or 50 mL (500 mg) (Pescovitz 2006). The price in Italy is currently the equivalent of USD 575 (100 mg) and USD 1445 (500 mg), while in the USA it is USD 568 (100 mg) and USD 2840 (500 mg). It is an expensive drug and not covered by many insurance plans or by government funding.
How the intervention might work
Rituximab is a chimeric mouse‐human monoclonal immunoglobulin G1 antibody against the CD20 antigen on the surface of B lymphocytes (Borker 2011). The binding of rituximab to CD20, which is a B‐cell differentiation maker, may cause B‐cell death in three ways, including complement‐dependent cytotoxicity, stimulation of apoptosis, or antibody‐dependent cell‐mediated cytotoxicity (Selewski 2010). Rituximab targets the CD20⁺ B‐cells, with a rapid and sustained elimination. Generally, rituximab is used as second‐line treatment for inhibitors, in those resistant to standard ITI. It is hypothesized that rituximab, eliminating B cells which produce inhibitors, can reduce the titer facilitating ITI in resistant cases. A national cohort in the UK has shown the efficacy of this treatment (Collins 2009). Rituximab has been used for treating people with hemophilia and inhibitors with a response rate up to 63% (Borker 2011).
Why it is important to do this review
Two non‐Cochrane systematic reviews have examined the literature on rituximab for treating children with hemophilia who express clotting factor inhibitors. One review, published in 2008, was based on studies including only people with congenital hemophilia (Franchini 2008). Another review, published in 2007, included eight children with hemophilia (Giulino 2007). These two reviews included only case reports and case series involving a relatively small number of participants, making the reviews potentially biased and imprecise. The two reviews were unable to draw definitive conclusions or make recommendations about using rituximab to manage hemophilia in people with inhibitors.
Although rituximab has become widely available as a therapy option, its high price and the lack of definitive evidence about its efficacy and safety remain barriers to its acceptance by patients and physicians. In an attempt to guide both groups as they decide on the most appropriate hemophilia treatment, we undertook a Cochrane systematic review to evaluate the efficacy and safety of rituximab to eradicate inhibitors complicating congenital hemophilia treatment. This is an update of a previously published Cochrane Review (Liu 2015).
Objectives
To assess the efficacy and safety of rituximab in the treatment of inhibitors in people with severe hemophilia A or B.
Methods
Criteria for considering studies for this review
Types of studies
Randomized and quasi‐randomized controlled trials (RCTs; quasi‐RCTs). Controlled clinical trials would be considered if no RCTs or quasi‐RCTs were found.
Types of participants
People of any age with confirmed congenital hemophilia A or B and inhibitors.
A positive inhibitor result is any result above the cut‐off, measured by an assay that can quantify the level of the inhibitor (e.g. Nijmegen or Bethesda assay).
Types of interventions
Intervention group: intravenous rituximab, irrespective of dose or duration and interval of administration.
Control group: placebo, no treatment, intervention not involving rituximab or a different dose of intravenous rituximab.
Any adjunct therapy, if recommended or prescribed, should be given to both groups.
Types of outcome measures
Primary outcomes
-
Remission or no response (Hay 2012)
-
complete remission: negative inhibitor titer, FVIII recovery over 66% of expected, and FVIII recovery lasting longer than six hours
-
partial remission: negative inhibitor titer, but persistently abnormal recovery or half‐life; responding clinically to FVIII replacement without an anamnestic increase in inhibitor titer
-
failure: failure of the inhibitor to decline by 20% over any six‐month period; or failure to achieve tolerance or partial response; or withdrawal from the trial for any reason before tolerance was achieved
-
relapse: inhibitor recurrence during the 12‐month follow‐up period on prophylaxis after tolerance was achieved, as evidenced by recurrent positive Bethesda titer or a decline in FVIII recovery or half‐life below trial limits
-
Secondary outcomes
-
Months to complete remission (time‐to‐event data)
-
Duration of remission (months)
-
Relapse rate following remission
-
Cost of care
-
Quality of life (e.g. as reported within trials or specifically using validated questionnaires)
-
All‐cause mortality
-
Adverse drug reactions
Search methods for identification of studies
There were no restrictions regarding language or publication status.
Electronic searches
We searched relevant studies from the Cystic Fibrosis and Genetic Disorders Group's Coagulopathies Trials Register using the terms: haemophilia* AND rituximab.
The Coagulopathies Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of the Cochrane Library) and weekly searches of MEDLINE and the prospective handsearching of one journal ‐ Haemophilia. Unpublished work is identified by searching the abstract books of major conferences: the European Haematology Association conference; the American Society of Hematology conference; the British Society for Haematology Annual Scientific Meeting; the Congress of the World Federation of Hemophilia; the European Association for Haemophilia and Allied Disorders, the American Society of Gene and Cell Therapy and the International Society on Thrombosis and Haemostasis. For full details of all searching activities for the register, please see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group's website.
Date of the most recent search of the Cystic Fibrosis and Genetic Disorders Group's Coagulopathies Trials Regsiter: 16 February 2017.
-
Bibliographic databases: we retrieved articles from the following databases: PubMed; Embase; the Chinese National Knowledge Infrastructure (CNKI) database; the Chinese Biomedical Literature Database (CBM); VIP; WANFANG; and LILACS. We searched all databases from their earliest records to 15 May 2017. Detailed search strategies are given in the appendices (Appendix 2; Appendix 3).
-
Trials registries: we searched OpenGrey (www.greynet.org/), Clinicaltrials.gov (https://clinicaltrials.gov/), and the WHO International Clinical Trials Registry Platform (ICTRP) (www.who.int/ictrp/en/) (searched using the terms: 'hemophilia' and 'rituximab')
-
We analyzed the websites of the following organizations to identify grey literature: the Canadian Hemophilia Society; the National Hemophilia Foundation; the National Heart, Lung and Blood Institute (USA); the Hemophilia Alliance.
Searching other resources
-
Researchers: we contacted the major researchers in this field to gather information on unpublished or ongoing trials.
-
Pharmaceutical companies: we contacted the pharmaceutical companies: F. Hoffmann‐La Roche Inc.; IDEC Pharmaceuticals; and Genentech, Inc. for information on unpublished or ongoing trials.
-
Reference lists: we manually searched the reference lists of relevant reviews, systematic reviews and original research articles.
Data collection and analysis
Given that we did not identify any eligible trials, we did not apply the process below; however, if we include any trials in a future update, the following methods will be applied.
Selection of studies
Two authors will independently scan the titles and abstracts of all reports identified from the literature searches. The complete articles will be obtained if we need more information beyond what is in the title and abstract. Two authors will independently apply the inclusion criteria to identified articles in order to determine whether we should include these in the review or not. We will resolve disagreements by discussion among co‐authors and, when necessary, with the Cochrane Cystic Fibrosis and Genetic Disorders Group. We will use reference management software to merge search results and remove duplicate records. We will also scrutinize each trial to ensure that no trial is reported in multiple publications. We will document reasons for excluding trials.
Data extraction and management
Two authors will independently extract data using pre‐designed forms. These data will include characteristics of the trial design, participants, interventions and outcomes. The authors will test and optimize the data extraction form before extraction after a preliminary test. If data for a trial are incomplete, the review authors will contact the primary trial author for further information and clarification. The authors aim to detect and resolve any discrepancies by discussion and cross‐checking.
If any included trials involve children and adults we will attempt to extract data separately for children. If any trials examine hemophilia A or B with inhibitors in addition to other hematological diseases, we will include these and attempt to extract the relevant data.
Assessment of risk of bias in included studies
Two authors will independently assess the risk of bias of included trials using a standardized assessment form. The authors will assess bias in RCTs across the following six domains according to the domain‐based evaluation recommended in the Cochrane Handbook for Systematic reviews of Interventions (Higgins 2011a): sequence generation; allocation concealment; blinding of participants and personnel; blinding to outcome assessment; incomplete outcome data; and selective outcome reporting. If we are unable to obtain important information from the published trials, we will contact the primary author of the relevant trial. We will aim to resolve any disagreements by discussion.
Measures of treatment effect
To measure treatment effects, we will use the rate ratio (RR) for dichotomous data; we will also use the hazard ratios (HR) to measure time‐to event data. We will use the mean difference (MD) for measuring continuous data. If trials report the same outcome on different scales which cannot be converted to the same unit, we will use the standardized mean difference (SMD). We will report all of these measurements with their corresponding 95% confidence intervals (CI).
Unit of analysis issues
We will include cluster‐randomized trials and cross‐over trials along with individual‐randomized trials. In this systematic review, we will treat each group or cluster in cluster‐randomized trials as the unit of analysis; we will use the intracluster correlation coefficient (ICC) to estimate the relative variability within and between clusters, according to the relevant section of the Cochrane Handbook of Systematic Reviews of Interventions (Higgins 2011b). For cross‐over trials with binary outcomes, we will adopt the Becker‐Balagtas approach to combine cross‐over trials with parallel trials for meta‐analysis (Stedman 2011). For cross‐over trials with continuous outcomes, each individual will act as the unit of analysis and we will conduct approximate paired analyses (Higgins 2011b).
Dealing with missing data
If data on individuals are missing, such as when randomized participants are excluded from the analysis, we will conduct intention‐to‐treat analyses. We will follow the principal options and adopt four general recommendations for dealing with missing data, as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). These are:
-
whenever possible, we will contact the original investigators to request missing data;
-
we will make explicit the assumptions of any methods used to cope with missing data: for example, that the data are assumed missing at random, or that missing values were assumed to have a particular value such as a poor outcome;
-
we will perform sensitivity analyses to assess how sensitive results are to reasonable changes in the assumptions that are made;
-
we will address the potential impact of missing data on the findings of the review in the discussion section.
If necessary, and possible, the authors plan to impute any missing standard deviations (SDs), following advice from chapter 16 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b).
Assessment of heterogeneity
The authors will identify any statistical heterogeneity among the included studies based on the Chi² test ( χ² or Chi²) included in the forest plots. A P value of less than or equal to 0.1 will be taken as evidence of heterogeneity in treatment effects, and the I² statistic will be used to quantify the heterogeneity. We will interpret the level of heterogeneity according to the Cochrane Handbook for Systematic reviews of Interventions (Higgins 2011a):
-
0% to 40%, may not be important;
-
30% to 60%, may represent moderate heterogeneity;
-
50% to 90%, may represent substantial heterogeneity;
-
75% to 100%, represents considerable heterogeneity.
The authors will also refer to the forest plots to see if the CIs overlap in order to further identify any possible heterogeneity.
Assessment of reporting biases
If we include 10 or more trials, the authors will use a funnel plot to detect small‐study effects, including publication bias. We will use a linear regression approach proposed by Egger to test for funnel plot asymmetry (Egger 1997).
Data synthesis
The authors will use Review Manager software to analyze data (RevMan 2014). If the trials show homogeneity, we will carry out a meta‐analysis of pooled research outcomes using a fixed‐effects model. If the trials show substantial heterogeneity, we will use a random‐effects model or describe the results narratively. If we are unable to undertake any meta‐analyses, we will summarize the trial results in tables. We will describe the time to response and response duration with medians, minimum and maximum values. If we are able to include sufficient data, we will group outcome data for analysis at six months, one year and annually thereafter. However, if outcome data were recorded at other time periods, then we will consider examining these as well.
If rituximab is given as an adjunct therapy to ITI, these trials will be analysed separately from those in which rituximab is given alone.
Subgroup analysis and investigation of heterogeneity
If 10 or more trials are included, and if we identify heterogeneity (I² equals more than 50%), we plan to investigate this by performing the following subgroup analyses:
-
hemophilia groups: hemophilia A; hemophilia B;
-
treatment groups: rituximab alone; rituximab plus other treatment.
-
participants: PUPs; previously treated patients.
Sensitivity analysis
If the authors are able to include at least 10 trials in the review, we will perform a sensitivity analysis to determine whether the conclusions are robust to decisions made during the review process. We therefore plan to perform sensitivity analyses by re‐analyzing the data statistically, such as using a fixed‐effects model first and then a random‐effects model, and vice versa.
Results
Description of studies
No trials were found which were eligible for inclusion in the review.
Results of the search
No trials were found which were eligible for inclusion in the review (Figure 1).
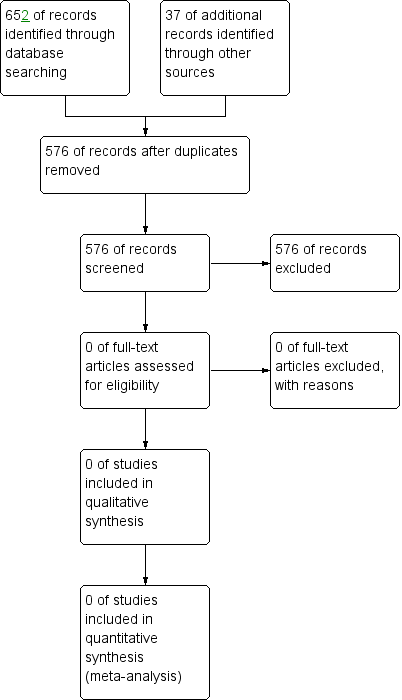
Study flow diagram.
Included studies
No trials were found which were eligible for inclusion in the review.
Excluded studies
No trials were found which were eligible for inclusion in the review.
Risk of bias in included studies
No trials were found which were eligible for inclusion in the review.
Effects of interventions
No trials were found which were eligible for inclusion in the review.
Discussion
Summary of main results
No controlled trials were identified which were eligible for inclusion in this review.
Agreements and disagreements with other studies or reviews
As described in the Background section, we are aware of two systematic review about rituximab for treatment of people with hemophilia and inhibitors (Franchini 2008; Giulino 2007). However, neither of them included any controlled trials. A total of 29 studies, with information on 49 cases were included in the Franchini review (Franchini 2008). The individuals included had congenital hemophilia A and B with inhibitors, were resistant to previous immune tolerance induction (ITI) regimens and were treated with rituximab (Franchini 2008). The dose of rituximab was 375 mg/m² per week, with the total number of doses varying from 4 to 11 according to the individuals condition (Franchini 2008), although the four‐dose schedule was the most common one (Collins 2009). Treatment with rituximab provided a durable remission in 26 out of 49 cases (53.1%). However, the follow‐up (three to 34 months) was not long enough and there is potential to overestimate the efficacy, as individuals who relapsed would be missed. The degree of disease severity was shown to be the primary influencing factor for the effect of rituximab, with those with mild and moderate disease showing a higher rate of response than those with severe hemophilia. In the Giulino review, eight children with congenital hemophilia were treated with rituximab for inhibitors. Five of these children (63%) had an initial response, but two of them relapsed at 11 and eight months, respectively; only one of them responded to a second course of rituximab (Giulino 2007). For this update, we identified a new case report which reported on a successful treatment of an individual with hemophilia B with inhibitors using ITI with rituximab, who reported that the CD20‐positive lymphocyte count was a useful marker to guide ITI therapy (Kobayashi 2015).
Regarding safety issues, rituximab‐related adverse reactions have been reported to include the following: sinusitis (Carcao 2006); nausea (Collins 2009; Fox 2006); dyspnea (Mateo 2006; Moschovi 2006); hypotension (Mateo 2006); urinary infection (Fox 2006); fever (Moschovi 2006); abdominal pain (Cooper 2006); and headache (Collins 2009). However, no severe adverse reactions have been reported for this disease (Collins 2009; Franchini 2008). First infusion‐related side effects were common and mild (Franchini 2008; Giulino 2007).
In the absence of evidence from randomized controlled trials (or controlled trials), clinicians are only able to use information from observational studies of lower quality taking into account the limitations of doing so.