Fotoféresis extracorpórea versus tratamiento alternativo para la enfermedad injerto contra huésped crónica posterior al trasplante de células madre hematopoyéticas en pacientes pediátricos
Resumen
Antecedentes
La enfermedad injerto contra huésped (EIcH) crónica es una causa importante de morbilidad y mortalidad después del trasplante de células madre hematopoyéticas, que ocurre en el 6% al 65% de los receptores. Actualmente, la base terapéutica para la EIcH crónica son los corticosteroides, que se combinan con frecuencia con otros agentes inmunosupresores en pacientes con manifestaciones de que no responden a los esteroides. No hay un tratamiento estándar establecido para la EIcH crónica que no responde a los esteroides. Las opciones terapéuticas para estos pacientes incluyen la fotoféresis extracorpórea (FEC), un tratamiento inmunorregulador que incluye la obtención ex vivo de células mononucleares de la sangre periférica, la exposición al agente fotoactivo 8‐metoxipsoraleno, la radiación ultravioleta y la readministración del producto celular procesado. Los mecanismos de acción de la FEC no se comprenden por completo. Ésta es una actualización de una revisión Cochrane publicada por primera vez en 2014.
Objetivos
Evaluar la efectividad y la seguridad de la FEC para el tratamiento de la EIcH crónica en niños y adolescentes después del trasplante de células madre hematopoyéticas.
Métodos de búsqueda
Se hicieron búsquedas en las bases de datos Registro Cochrane Central de Ensayos Controlados (Cochrane Central Register of Controlled Trials; CENTRAL) (número 9, 2015), MEDLINE y EMBASE, desde su inicio hasta el 23 de septiembre de 2015. Se hicieron búsquedas en las listas de referencias de los estudios potencialmente relevantes sin ninguna restricción de idioma. Se realizaron búsquedas en ocho registros de ensayos y cinco actas de congresos el 29 de septiembre de 2015.
Criterios de selección
Ensayos controlados aleatorizados (ECA) que compararan FEC con o sin tratamiento alternativo versus tratamiento alternativo solo en pacientes pediátricos con EIcH crónica después del trasplante de células madre hematopoyéticas.
Obtención y análisis de los datos
Dos autores de la revisión realizaron de forma independiente la selección de los estudios. Los desacuerdos en la selección de los ensayos se resolvieron mediante consulta con un tercer autor de la revisión.
Resultados principales
No se identificaron estudios adicionales en esta actualización de la revisión de 2015, por lo que no hubo estudios que cumplieran los criterios de inclusión para esta revisión.
Conclusiones de los autores
No es posible evaluar, sobre la base de ECA, la eficacia de la FEC para el tratamiento de la EIcH crónica en pacientes pediátricos después del trasplante de células madre hematopoyéticas, ya quela versión original de esta revisión y la primera actualización de la misma no hallaron ningún ECA. Las recomendaciones actuales se basan solamente en estudios retrospectivos u observacionales. Por lo tanto, idealmente la FEC se debe aplicar solamente en el contexto de ensayos controlados. Sin embargo, la realización de ECA en esta población de pacientes será un desafío debido al número limitado de pacientes, la presentación variable de la enfermedad y la falta de criterios de respuesta bien definida. Se necesitarán colaboración internacional, ensayos multicéntricos y financiamiento apropiado para dichos ensayos. Si se toman decisiones de tratamiento según los datos clínicos se realizan a favor de la FEC, los pacientes se deben monitorizar cuidadosamente con respecto a los efectos beneficiosos y perjudiciales. Además, se deben hacer esfuerzos para compartir esta información con otros médicos, por ejemplo, al establecer registros para los pacientes pediátricos que son tratados con FEC.
PICOs
Resumen en términos sencillos
Fotoféresis extracorpórea para la enfermedad injerto contra huésped crónica posterior al trasplante de células madre hematopoyéticas en pacientes pediátricos
Antecedentes
La enfermedad de injerto contra huésped crónica es una complicación frecuente después del trasplante de células madre hematopoyéticas (TCMH; trasplante de células madre que forman sangre). Las células inmunitarias (leucocitos) del donante reconocen las células del paciente como extrañas ("no propias"). Por lo tanto, las células inmunes trasplantadas atacan las células del paciente. Los principales órganos afectados son la piel, el hígado y los intestinos, entre otros. Estas reacciones inmunológicas pueden causar una inflamación aguda (hinchazón repentina) seguida de cambios crónicos (a largo plazo) en órganos (p.ej. fibrosis, cicatrización de los pulmones). El tratamiento de primera línea suele consistir en fármacos inmunosupresores (que reducen la potencia del sistema inmunológico del cuerpo) en forma de corticoides, en combinación con otros agentes inmunosupresores en los casos refractarios (en los que la enfermedad es resistente al tratamiento). Estos fármacos se supone que suprimen el ataque mediado por la reacción inmune de las células del paciente. La eficacia limitada y los efectos secundarios graves de estos fármacos han provocado la aplicación de varios enfoques alternativos.
La fotoféresis extracorpórea (FEC) es un tratamiento inmunorregulador que incluye la obtención de células inmunitarias de sangre periférica fuera del cuerpo del paciente. Estas células inmunitarias se exponen a un agente fotoactivo (una sustancia química que responde a la exposición a la luz; p.ej. el 8‐metoxipsoraleno), con la subsiguiente radiación ultravioleta‐A, y luego se infunden nuevamente. Los efectos inmunorreguladores de este procedimiento no se han dilucidado por completo. Varias recomendaciones para la práctica clínica actual indican considerar la FEC en pacientes pediátricos con enfermedad injerto contra huésped crónica.
Características de los estudios
Se buscaron ensayos controlados aleatorizados (estudios clínicos en los que los pacientes se asignan al azar a uno de dos o más grupos de tratamiento) en las bases de datos científicas diseñados para evaluar la efectividad y la seguridad de la FEC en el tratamiento de la enfermedad injerto contra huésped crónica en niños y adolescentes (menores de 18 años de edad) después del TCMH.
Resultados
En la versión original de esta revisión y en esta actualización de 2015 no se encontraron ECA que analizaran la eficacia de la FEC para los pacientes pediátricos con enfermedad crónica de injerto contra huésped después de TCMH. Las recomendaciones actuales se basan solamente en estudios retrospectivos (estudios en los que los resultados ocurrieron en los pacientes antes del comienzo del estudio) u observacionales (estudios en los que los investigadores no intervienen y sencillamente observan el curso de los eventos). Por lo tanto, idealmente la FEC se debe aplicar en pacientes pediátricos solamente en el contexto de ECA. La FEC se puede considerar en pacientes con EIcH crónica que no responde a los esteroides y recordar que este tratamiento no está apoyado por evidencia de alto nivel. Si se toman decisiones de tratamiento según los datos clínicos a favor de la FEC, los pacientes se deben monitorizar cuidadosamente con respecto a los efectos beneficiosos y perjudiciales y se deben hacer esfuerzos para compartir esta información con otros médicos, por ejemplo, al establecer registros para los pacientes pediátricos que son tratados con FEC.
Authors' conclusions
Background
Description of the condition
Haematopoietic stem cell transplantation (HSCT) is a curative treatment option for children with haematological malignancies, haemoglobinopathies, immune deficiencies and inborn errors of metabolism (Diaconescu 2005; Gaziev 2010; Kennedy‐Nasser 2006; Peters 2003; Rappeport 2011; Walters 2000; Walters 2005). Chronic graft‐versus‐host disease (cGvHD) is considered one of the major complications following HSCT, limiting its wider application (Billingham 1966; Ferrara 2004; Sullivan 2004). cGvHD was traditionally defined by manifestation after 100 days following HSCT (Gilman 2000). Several advances in the practice of HSCT (including different haematopoietic stem cell sources, intensity of conditioning regimen, immunosuppression and donor lymphocyte infusions) resulted in a more time variable presentation of cGvHD (Bunin 2008; Eapen 2004). Presently, cGvHD is described according to its pattern of presentation: following HSCT as a progression from acute graft‐versus‐host disease (aGvHD) (progressive form); as a recurrence after a disease‐free interval (quiescent form); and without a history of aGvHD (de novo form) (Shulman 1988). The US National Institutes of Health (NIH) developed a diagnosis and scoring system defining cGvHD based on specific clinical signs and histopathology rather than time of onset after HSCT (Filipovich 2005; Jagasia 2009). The NIH global severity score uses a numerical scoring system for individual organs to calculate a summary scale according to the number and severity of organs involved. This provides a more clinically informative and discriminating severity measure for use in clinical trials and is an indicator for the necessity of systemic immunosuppressive treatment (Arai 2010).
The incidence of cGvHD varies between 6% and 65%, depending on the transplant procedure and disease‐specific variables (Meisel 2007; Rocha 2000; Zecca 2002). Well‐known risk factors for developing cGvHD are precedent aGvHD, stem cell donor, the preparative regimen, prophylactic procedures and the underlying disease (Flowers 2011; Rocha 2000; Zecca 2002). The exact pathogenesis of cGvHD remains unclear. Several studies demonstrated the role of T cells in the development of cGvHD (De Bueger 1993; Higman 2004; Mutis 1999). Increased levels of non‐specific antibodies in people with cGvHD and response to B‐cell‐depleting antibodies suggest that B cells also play a role (Allan 2007; Zhang 2006). Moreover, a co‐ordinated T‐cell B‐cell interaction to minor histocompatibility alloantigens seems to account for the manifestation of cGvHD (Miklos 2005). Soluble inflammation‐associated factors are also involved in the pathogenesis of cGvHD (Fujii 2008; Lin 2003). Other pathogenetic suggestions include a defective negative selection of autoreactive T cells due to thymic damage, aberrant production of transforming growth factor‐beta and activation of the platelet‐derived growth factor receptor, as well as deficiency of CD8+ cells (Martin 2008; Toubai 2008). Clinical manifestations of cGvHD are separated into rather inflammatory, acute type features (erythematous rash, mucositis, diarrhoea, transaminitis and pulmonary infiltrates) as opposed to more fibrotic, chronic characteristics (sclerotic and lichen planus‐like skin changes, fasciitis, sicca syndrome, oesophageal strictures and bronchiolitis obliterans) (Flowers 2002; Higman 2004). The most commonly affected organs, isolated or in combination, are the skin and oral cavity, with over 70% involvement for both, followed by eyes, liver, lungs, gastrointestinal tract and musculoskeletal system (Atkinson 1990; Beredjiklian 1998; Chosidow 1992; Cooke 2009; Dudek 2003; Filipovich 2005; Janin 1994; Lee 2003; Melin‐Aldana 2007; Pavletic 2005; Sullivan 1981; Treister 2005).
Both cGvHD and treatment of cGvHD lead to significant morbidity and mortality in children; however, outcome reports for cGvHD are mostly based on adult retrospective studies with a five‐year overall survival rate of 60% to 90% in the low‐risk range, 40% to 75% in the intermediate‐risk range, and 10% to 60% in the high‐risk range, using the previous grading and scoring system (Akpek 2003; Cahn 2005; Couriel 2006; Lee 2003; Shimada 2005). cGvHD accounts for 54% of deaths in adults two years after transplantation among the non‐relapse‐related deaths (Higman 2004; Horowitz 2004; Wingard 2002). The only available study with a larger cohort in children reports an overall six‐year disease‐free survival of 68% in the presence of cGvHD (Zecca 2002). According to reports in adults, immunosuppressive therapy is still required in 50% of people five years after the diagnosis of cGvHD (Stewart 2004).
Management options
Since aGvHD also has a significant impact on the pathogenesis of cGvHD, prophylaxis to prevent graft‐versus‐host disease (GvHD) plays a major role (Ram 2009). Prevention is based on T‐cell inhibition including the following strategies: inhibition of T‐cell activation and function (calcineurin inhibitor), inhibition of T‐cell proliferation (methotrexate, mycophenolate mofetil) and elimination of T cells (alemtuzumab, anti‐thymoglobulin) (Shah 2007; Storb 1986). The majority of children undergoing HSCT receive GvHD prophylaxis, starting before transplantation and typically continuing for up to six months after transplantation (Barrett 2008).
The clinical management of people with cGvHD is complicated due to the variability of disease manifestation, clinical course, infectious complications and treatment‐related toxicity (Flowers 2002). Therefore, treatment of cGvHD in children is highly variable, and, due to lack of paediatric studies, mostly based on the experience in adults. In people with mild cGvHD and high‐risk malignancies where a graft‐versus‐leukaemia effect gains importance, topical therapy should be considered (Filipovich 2005). Drug therapy for mild cGvHD mostly includes locally applied immunosuppressive agents (steroids and calcineurin inhibitors) for the affected organ (skin, eyes, oral cavity and lungs) (Jacobsohn 2010). In contrast, moderate‐to‐severe cGvHD is mostly treated with systemic steroids (Filipovich 2005; Salmasian 2010). Prednisone is usually tapered (alternate day) for the next two to three months (Sullivan 1988a). Some reports suggest better response rates and fewer corticosteroid complications when adding a calcineurin inhibitor (ciclosporin, tacrolimus) to systemic steroids (Koc 2002; Sullivan 1988b; Vogelsang 2001). Regarding the effect of corticosteroids on the growth and development of children and the mean duration of therapy of three years for people with cGvHD, the management of a daily calcineurin inhibitor with alternate day prednisone is considered standard for the treatment of cGvHD (Koc 2002; Sullivan 1988a). This therapeutic regimen yielded an objective response in more than 50% of people (Sullivan 1988a; Sullivan 1988b).
In cases where there is no response to therapy within four weeks, second‐line treatment is mostly considered (Jacobsohn 2010). There are various salvage therapies including general immunosuppressants (mycophenolate mofetil, methotrexate, sirolimus, thalidomide, pentostatin and hydroxychloroquine) and monoclonal antibodies (rituximab) (Akpek 2001; Bolanos‐Meade 2008; Busca 2000; Cutler 2006; Fraser 2007; Gilman 2006; Goldberg 2003; Jacobsohn 2009; Johnston 2005; Lopez 2005; Martin 2009; Mookerjee 1999; Vogelsang 1992).
Description of the intervention
Extracorporeal photopheresis (ECP), an immunomodulatory therapy, may play a role in the treatment of cGvHD. During ECP, peripheral mononuclear cells are collected ex vivo by leukapheresis, incubated with the photoactive and photosensitising drug 8‐methoxypsoralen (8‐MOP), exposed to ultraviolet‐A (UV‐A) light and then re‐infused into the patient without any adverse effects to other organs (Bethea 1999; Girardi 2002; Heald 1989). Psoralen occurs naturally in the seeds of the furocoumarin family of plants and its exposure to UV‐A light (wavelength 200 to 350 nm) facilitates the intercalation of psoralen with deoxyribonucleic acid (DNA), leading to the formation of both monofunctional and bifunctional adducts, which results in programmed cell death (apoptosis) of the majority of cells (Yoo 1996). Initially, the patients received psoralen orally prior to the leukapheresis (Bethea 1999). However, oral application results in an inconstant absorption of the drug and considerable gastrointestinal adverse effects (Brickl 1984). The now generally used ex vivo method, with the incubation of the collected cells in a bag, significantly reduces the exposure of the patient to 8‐MOP (Schooneman 2003).
ECP has been successfully applied in the treatment of cutaneous T‐cell lymphoma since the 1980s (Edelson 1987). Following this observation, the method has been implemented for a wider spectrum of immunologically mediated diseases such as systemic scleroderma, autoimmune disorders, solid organ rejection, aGvHD and cGvHD (Szodoray 2010). In the paediatric setting, many authors report a response rate of 33% to 93% to ECP as second‐line treatment in steroid‐resistant cGvHD (Foss 2005; Messina 2003; Smith 1998; Sniecinski 1994). People with steroid‐refractory cGvHD that respond to ECP have a higher five‐year survival rate compared with non‐responders (96% with responders versus 85% with non‐responders) (Perotti 2010). Adverse reactions are uncommon (less than 0.003%), transient and mild (nausea, hypotension, dizziness, cytopenia, skin infection at site of venous access and abnormal clotting to heparin) (Kanold 2003). Moreover, ECP is not associated with increased risk of systemic infections and relapse of malignant disease (Dall'Amico 2002; Hackstein 2009; Scarisbrick 2008).
How the intervention might work
The mechanisms of action of ECP are not completely understood. It has been shown that the procedure induces apoptosis in mononuclear white blood cells (Voss 2010). However, only a small percentage of the peripheral mononuclear cells is treated and, therefore, an immunomodulatory effect of the apoptotic cells is hypothesised (Heshmati 2003). One suspected mechanism is that apoptotic T‐cell fragments presented by dendritic cells induce an anti‐idiotypic T‐suppressor activity, or downregulate a pre‐existing T‐cell response, and in this way generate a tolerogenic response and modulate cytokine production (Bladon 2006; Legitimo 2007; Xia 2009). In summary, the postulated mechanisms involved include: reduced stimulation of effector T cells, deletion of effector T cells, induction of regulatory T cells, increase of anti‐inflammatory cytokines (i.e. tumour necrosis factor‐beta, interleukin‐10), and reduction of proinflammatory cytokines (interleukin‐1beta, interleukin‐6, and tumour necrosis factor‐alpha) (Fimiani 2004). On the basis of this hypothesis, photopheresis seems to downregulate the T‐cell alloreactivity that plays the central role in the pathogenesis of GvHD after HSCT (Lamioni 2005; Maeda 2005).
Why it is important to do this review
cGvHD remains one of the major challenges for transplant‐related morbidity and mortality after stem cell transplantation in paediatric patients. All conventional therapies, including established first‐line therapy, have considerable adverse effects and probably increase the risk of infections and relapse of malignant disease. Therefore, it is essential to develop new therapeutic approaches for the selective immune control of cGvHD without generalised immunosuppression‐related complications (infections and pharmacological toxicity issues) (Wolff 2011). ECP seems to be an effective immunomodulatory therapy with very mild, if any, adverse effects and, therefore, may be a promising alternative for improving morbidity and mortality in children with cGvHD.
The current review is the first update of the original review and protocol (Weitz 2012; Weitz 2014).
Objectives
To evaluate the effectiveness and safety of ECP for the management of cGvHD in children and adolescents after HSCT.
Methods
Criteria for considering studies for this review
Types of studies
We intended to consider randomised controlled trials (RCTs) for this review if they assessed any clinical outcome as described in the Types of outcome measures section. For medical reasons (e.g. we did not consider cGvHD a stable condition), we aimed to include only trials with a parallel group design. We excluded studies restricted to adults (≥18 years of age). For studies including both children and adults, we planned to include studies if children represented more than 50% of participants in the study.
Types of participants
We planned to include children and adolescents under 18 years of age who underwent HSCT therapy with presence of cGvHD, independent of the underlying disease and donor source. We planned to consider all stages and grades of cGvHD, independent of the type of organ involvement.
Types of interventions
For the purpose of this review, we considered systemic steroids with or without calcineurin inhibitors as standard treatment as first‐line therapy and considered all other treatment as second‐line therapy of cGvHD.
The following comparisons for ECP for cGvHD after HSCT were conceivable.
-
ECP versus standard treatment in paediatric patients with cGvHD as first‐line treatment.
-
ECP plus standard treatment versus standard treatment alone in paediatric patients with cGvHD as first‐line treatment.
-
ECP versus standard treatment in paediatric patients with steroid‐/calcineurin‐inhibitor‐refractory cGvHD (second‐line treatment).
-
ECP plus standard treatment versus standard treatment alone in paediatric patients with steroid‐/calcineurin‐inhibitor‐refractory cGvHD (second‐line treatment).
These comparisons constituted four separate groups and we anticipated analysing them separately.
Types of outcome measures
Primary outcomes
-
Response to ECP treatment (defined as either classical response rates (i.e. number of people in complete or partial remission) or percentage of achieved reduction in either NIH score (Filipovich 2005; Jagasia 2009), scales of Akpek and Lee (Akpek 2001; Lee 2003), or steroid‐tapering under therapy with ECP (defined as number of people with at least 25% reduction in steroid dose).
Secondary outcomes
-
Overall survival (defined as the time to death from any cause, starting at the day of HSCT).
-
Failure‐free survival (defined as progression of GvHD, expressed as change in NIH score or intensification of treatment, or both).
-
Adverse events.
-
Quality of life.
-
Cost of intervention per month including length of hospital stay in days, number of outpatient attendances, direct medical resource use, direct medical costs, indirect medical resource use or costs and patient out‐of‐pocket expenses.
Search methods for identification of studies
We applied no language restrictions.
Electronic searches
The search for the original version of this review was done on 12 September 2012 in the Cochrane Central Register of Controlled Trials (CENTRAL) (Issue 9, 2012); MEDLINE/PubMed (1945 to 12 September 2012) and EMBASE (Ovid) (1980 to 12 September). The search strategies for the different electronic databases (using a combination of controlled vocabulary and text words) are shown in Appendix 1; Appendix 2; and Appendix 3.
For the 2015 search update, we applied the same strategy without changes, from the date of the last search to 23 September 2015.
Searching other resources
Conference proceedings
We carried out an electronic search for the original version of this review using the regular search with keyword terms ("extracorporeal"; "photopheresis"; "photochemotherapy"; "psoralen"; "graft‐versus‐host") for the conference proceedings of the following societies on 12 September 2012.
-
International Society for Paediatric Oncology (SIOP) (2007 to 2012).
-
American Society of Clinical Oncology (ASCO): Journal of Clinical Oncology (1995 to 2012).
-
American Society of Hematology (ASH): Blood (2001 to 2012).
-
American Society of Blood and Marrow Transplantation: Biology of Blood and Marrow Transplantation (1990 to 2012).
-
European Group for Blood and Marrow Transplantation (EBMT): Bone Marrow Transplantation (2000 to 2012).
We searched the reference lists of relevant articles and review articles.
We carried out a search update on 29 September 2015 without modifications, to cover conference proceedings from after the initial search date.
Electronic search in databases of ongoing trials
For the original version of this review, we searched the following clinical trials registries for ongoing or recently completed trials, and for locating potential links to other related databases and resources on 12 September 2012. Regular search with keywords ("extracorporeal"; "photopheresis"; "photochemotherapy"; "psoralen"; "graft‐versus‐host") was used where applicable, no time restrictions applied and the search not limited to trials involving children.
-
ISRCTN registry (controlled‐trials.com/).
-
ClinicalTrials.gov (ClinicalTrials.gov).
-
World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch).
-
Trials Central (www.trialscentral.org/) (search via "condition": "graft vs host disease").
-
Internet Portal of the German Clinical Trials Register (DRKS) (www.drks.de/).
-
NCIC Clinical Trials Group (www.ctg.queensu.ca/).
-
National Cancer Institute (www.cancer.gov) (under heading "childhood cancers" and "cancers in childhood and adolescents" using "research" button).
-
Australian New Zealand Clinical Trials Registry (ANZCTR) (www.anzctr.org.au/trialSearch.aspx).
For the 2015 update of this review, we repeated the initial search without modifications from the date of the last search to 29 September 2015.
Data collection and analysis
Selection of studies
The original review was undertaken by four review authors (MW, BS, FM, DB). One review author (MW) screened all titles and abstracts of the references identified by the search strategies for relevance. We only excluded citations that were clearly irrelevant at this stage. We considered citations as irrelevant that included only adults, were animal studies, did not describe cGvHD and that used stem cell sources other than haematopoietic. Two review authors (MW, DB) independently screened the remaining titles, excluded all irrelevant publications and recorded details of the studies together with the reasons for exclusion. We resolved any disagreement on the eligibility of studies through discussion and consensus. We obtained full‐text versions of all potentially relevant papers. Two review authors (MW, DB) independently screened these manuscripts, identified potentially relevant studies and assessed eligibility of studies for inclusion. We resolved any disagreements on the eligibility of studies through discussion and consensus. For this 2015 update, an additional author (MS) screened titles, abstracts and potentially relevant studies.
Data extraction and management
-
We planned to extract data using a data extraction form developed by the review authors, and one of the review authors (MW) would transcribe the data into Review Manager 5 (RevMan 2014). Another review author (JM) was to verify all data entry for discrepancies. We planned to resolve any disagreements on data extraction and management issues through discussion and consensus, or if necessary through a third review author (DB or BS). We intended to request missing data from the original investigators.
-
Two review authors (MW, JM) would have completed the 'Characteristics of included studies' table. Study characteristics would have included place of publication, date of publication, population characteristics, setting, detailed nature of intervention, detailed nature of comparator and detailed nature of outcomes. A key purpose of these data was to define unexpected clinical heterogeneity in included studies independently from analysis of results.
-
Two review authors (MW, JM) intended to carefully record reasons why an included study did not contribute data on a particular outcome and to consider the possibility of selective reporting of results on particular outcomes.
Assessment of risk of bias in included studies
Two review authors (MW, DB) planned to assess independently each included study for risk of bias using the definitions for the different risk of bias items as stated in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011): random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting and any other potential threats to validity (Higgins 2011; Kjaergard 2001; Moher 1998; Schulz 1995). We also planned to assess the risk of bias for blinding of outcome assessors and incomplete outcome data separately for each outcome. We aimed to consider a trial as having a low risk of bias if we assessed all domains as adequate. We planned to consider a trial as having a high risk of bias if we assessed one or more domain as inadequate or unclear. We wanted to report the 'Risk of bias' table as part of the 'Characteristics of included studies' table and present a 'Risk of bias' summary figure that would detail all of the judgements made for all included studies in the review (Higgins 2011). For each included study, we planned to assess selective reporting bias by comparing the methods and results section of the individual studies. We intended to resolve any disagreements on the assessment of risk of bias through discussion and consensus, or if necessary through a third review author (JM or BS) and to explore the impact of the level of bias through undertaking sensitivity analyses (see Sensitivity analysis).
Measures of treatment effect
We planned to analyse extracted data using Review Manager 5 (RevMan 2014).
We planned to extract hazard ratios (HR) with their 95% confidence intervals (CI) for time‐to‐event outcomes, such as mortality. If HRs were not provided, we intended to use the indirect estimation methods described in Parmar 1998 and Williamson 2002 to calculate them. As an alternative, we intended to use the proportions of participants with the respective outcomes measured at certain time points to calculate risk ratios (RR).
We planned to express results for binary outcomes as RRs with 95% CIs as measures of uncertainty. For continuous outcomes, we planned to express the results as mean differences (MD), with 95% CIs as measures of uncertainty.
Unit of analysis issues
Since, for medical reasons, we only aimed to include parallel group randomised trials, unit of analysis issues related to cross‐over and cluster randomised trials were not relevant for this systematic review. In the context of ECP, "body‐part randomisation" and "body part analyses" did not make sense, so related issues do not need to be discussed here. In cases of parallel group designs with three or more treatment groups, we planned to divide the control group into several parts, so that the total number added up to the original size of the group.
Dealing with missing data
We planned to contact original investigators for missing data regarding study selection, data extraction and risk of bias assessment. To optimise the strategy for dealing with missing data, we intended to conduct an intention‐to‐treat analysis, which would have included all participants who did not receive the assigned intervention according to the protocol, as well as those who were lost to follow‐up. If unsuccessful, we wanted to address the potential impact of missing data on the findings of the review in the 'Discussion' section.
Assessment of heterogeneity
We planned to assess statistical heterogeneity using the I2 statistic (Higgins 2002; Higgins 2003). This measure describes the percentage of total variation across studies that is caused by heterogeneity rather than by chance (Higgins 2003). The values of I2 lie between 0% and 100%. We planned to use a simplified categorisation of heterogeneity with the following categories: low (I2 less than 30%), moderate (I2 between 30% and 60%) and high (I2 more than 60%) (Deeks 2011).
If moderate or high heterogeneity had been detected, we intended to explore clinical heterogeneity by examining differences between groups as detailed below (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
We minimised the likelihood of publication bias by using a comprehensive search strategy without language restrictions and we also searched trial registries. In addition to the evaluation of reporting bias, as described in the Assessment of risk of bias in included studies section, we planned to assess reporting bias by constructing a funnel plot where a sufficient number of studies had been identified (i.e. at least 10 studies included in a meta‐analysis). If there were fewer studies, the power of these tests would be too low to distinguish chance from real asymmetry (Sterne 2011).
Data synthesis
For future updates, we will conduct meta‐analyses of pooled data from all contributing studies using Review Manager 5 (RevMan 2014). We intended to use a fixed‐effect model as primary analysis. If we had found high clinical, methodological or statistical heterogeneity (I2 more than 50%), we would, as a secondary analysis, have used a random‐effects model and reported results from both models. We planned to summarise studies in cases where pooling of results was not possible.
Subgroup analysis and investigation of heterogeneity
We planned to assess clinical heterogeneity by examining differences due to:
-
underlying disease;
-
type of haematopoietic stem cell source;
-
age of children at HSCT;
-
age at start of ECP;
-
type of conditioning regimen;
-
type of GvHD prophylaxis regimen; and
-
degree of human leukocyte antigen (HLA) compatibility (matched sibling donor versus matched unrelated donor versus mismatched unrelated donor).
Sensitivity analysis
We planned to investigate the robustness of our results through a sensitivity analysis on the basis of risk of bias of the included studies by defining the following groups:
-
low risk of bias (adequate sequence generation and allocation concealment; successful blinding of all participants, care providers and outcome assessors; incomplete outcome data for less than 20% of participants; no selective reporting or other sources of bias);
-
high risk of bias (no adequate sequence generation and allocation concealment; no adequate blinding of all participants, care providers and outcome assessors; incomplete outcome data for more than 20% of participants; selective reporting or other sources of bias);
-
unclear risk of bias (rating of unclear risk of bias in at least one of these seven categories).
We intended to perform sensitivity analyses for each risk of bias item separately.
Results
Description of studies
We found no RCTs meeting the inclusion criteria for the original version of this review and its 2015 update (see: Characteristics of excluded studies table; Characteristics of ongoing studies table).
Results of the search
We performed the initial search on 12 September 2012. The search yielded 68 articles including three duplicates in CENTRAL, MEDLINE/PubMed and EMBASE (Ovid) (Figure 1). After title and abstract screening, we excluded 53 of the unique articles, as the intervention was not ECP.
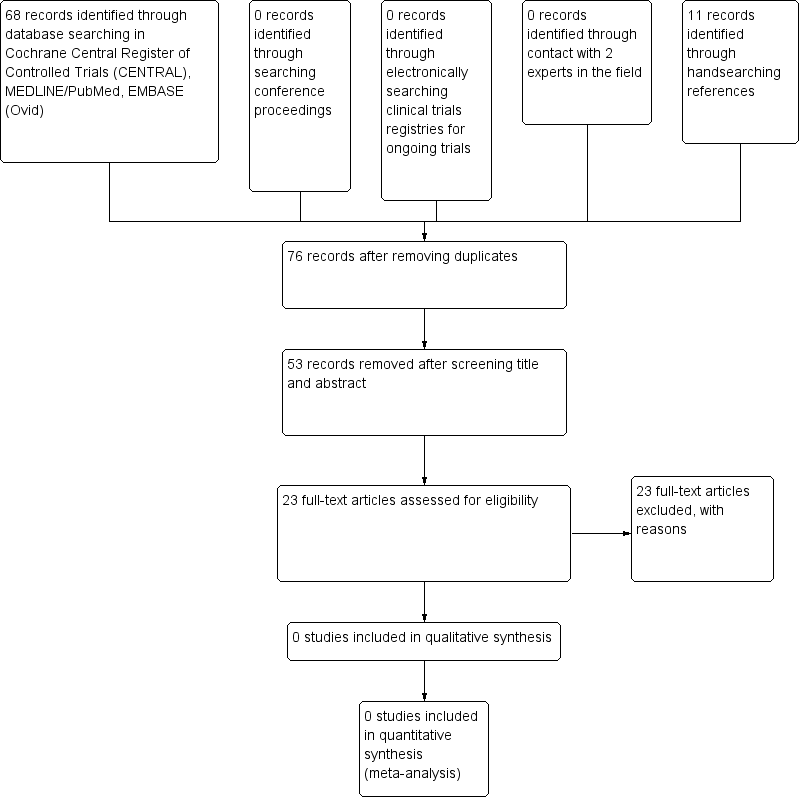
Identification of potentially eligible studies.
We screened 12 full‐text articles but we found only one RCT. However, this RCT was not eligible for inclusion in the review (see Excluded studies). We found another 11 studies by searching the references of the full‐texts articles. In total, the 23 articles were not eligible and we excluded them following full‐text screening. Reasons for exclusion were as follows:
-
13 were case report/case series;
-
four were reviews reporting case reports/case series;
-
four were prospective, not randomised, not controlled clinical trials;
-
one was a retrospective case series;
-
one was an RCT including less than 50% of children.
The search of the trial registers and conference proceedings identified no eligible studies. We contacted two experts in the field, which did not yield any studies.
We performed the latest search on 23 September 2015, yielding 20 new articles in CENTRAL, MEDLINE/PubMed, EMBASE (Ovid) and conference proceedings (Figure 2). After title and abstract screening, we excluded 15 of the unique articles for the following reasons:
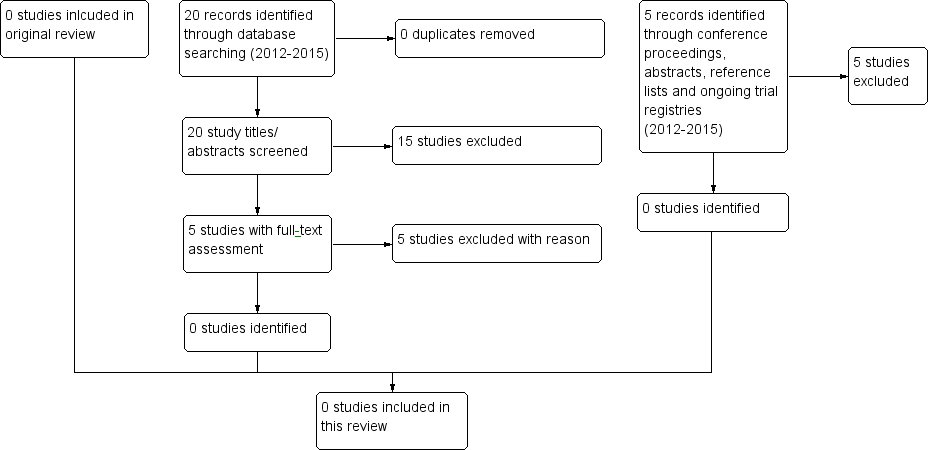
Identification of potentially eligible studies for 2015 update.
-
two were animal studies (mouse model);
-
six were interventions other than ECP;
-
one was a survey;
-
one did not concern cGvHD;
-
five were conference abstracts without references to published studies.
We screened five full‐text articles but we found no RCTs to include in this review. We found a further five studies while searching the references of the full‐text articles. These additional articles were also not eligible RCTs and we excluded them after abstract screening. Reasons for exclusion were as follows:
-
four were case series;
-
one was an RCT including no children.
The search of the trial registers (run 29 September 2015) identified no eligible studies.
Included studies
We found no RCTs in this 2015 review update meeting the inclusion criteria for this review.
Excluded studies
For this 2015 review update, we excluded additional data from two case series and three reviews.
See Characteristics of excluded studies table.
Risk of bias in included studies
We found no studies meeting the inclusion criteria for the original version of this review and its 2015 update. For that reason, the assessment of risk of bias was not applicable.
Effects of interventions
We found no studies meeting the inclusion criteria for this 2015 review update. For that reason the effectiveness and safety of ECP for the management of cGvHD in children and adolescents after HSCT remains unclear.
Discussion
cGvHD remains one of the major challenges for transplant‐related morbidity and mortality after HSCT in paediatric patients. ECP represents an alternative second‐line treatment option in paediatric patients with cGvHD after HSCT. However, there were no data from RCTs available in the original version of this review and in the 2015 update to support or refute treatment with ECP in children with cGvHD after HSCT. Therefore, this systematic review cannot establish whether such a treatment is effective in paediatric patients with cGvHD after HSCT.
We limited the search strategy to children and adolescents under 18 years of age. However, we found one RCT during the initial 2014 review that assessed the efficacy of ECP in cGvHD in mainly adults (greater than 50%). This randomised controlled, single‐blinded trial investigated the effect of ECP on the cutaneous manifestation of cGvHD in people with failed corticosteroid treatment following HSCT (Flowers 2008). People were eligible in case of: corticosteroid refractoriness defined as lack of response or disease progression after administration of a least 1 mg/kg of methylprednisolone equivalent, corticosteroid‐dependency after more than 10 mg of methylprednisolone equivalent to control skin manifestation, and corticosteroid intolerance due to intolerable adverse effects. Steroids had to be applied in a stable dose for at least two weeks prior to randomisation and had to be maintained on the same dose level for the first six study weeks (with the exception of reduction due to adverse effects). Immunosuppressants such as tacrolimus, mycophenolate and ciclosporin A were accepted as concomitant medication if they had been introduced at least four weeks before randomisation. Discontinuation of these agents during the study period was only permitted for safety reasons. The primary objective of the study was the median percentage change of total skin score (TSS) at week 12 compared with the pretreatment value. Secondary objectives included: the proportion of participants with at least 25% improvement in TSS, 50% or greater reduction in daily steroid dose compared with the baseline dose, or improvement of at least 25% in TSS in conjunction with steroid‐sparing at weeks 12 and 24. Skin assessments were performed on alternate weeks up to study week 12 and afterwards on alternate weeks up to study week 24. Participants were randomised with a block scheme in a 1 : 1 ratio to the conventional therapy arm (49 participants) or to the conventional therapy arm combined with ECP (50 participants). Conventional therapy consisted of corticosteroids and other immunosuppressive agents in refractory participants. ECP was administered three times during week one, and then twice weekly on consecutive days during weeks two to 12. Responding participants could continue ECP treatment until week 24 following the schedule of two treatments every week. Ten participants (four in the study group, six in the control group) withdrew consent prior to study week 12. Four participants died due to multiple organ failure, cardiac failure and infection. The changes in TSS from baseline until week 12 between the ECP group (‐14.5%) and the control group (‐8.5%) were not statistically different. However, ECP‐treated participants had a 50% or greater reduction in the total daily dose of corticosteroids. At week 12, the percentage of participants experiencing both a 50% or greater reduction in daily corticosteroid dose and a 25% or greater improvement in the TSS was higher in the ECP group (8%) than the control group (0%). Regarding TSS, 40% of the participants in the ECP group had a complete or partial skin response compared with 10% in the control group. Current clinical practice guidelines already recommend consideration of ECP as second‐line therapy option in adults and paediatric patients with cGvHD based on the available and partially limited evidence (Wolff 2011). This 2015 review update found no further RCTs on ECP treatment in cGvHD patients.
Apart from this RCT, which enrolled mainly adults, the search strategy applied for the initial 2014 review identified 21 non‐randomised, paediatric studies including several case reports, case series and observational studies that suggest a benefit of ECP in paediatric patients with cGvHD. We identified one major non‐randomised, non‐controlled prospective study including children with aGvHD (33 participants) and cGvHD (44 participants) resistant to conventional immunosuppressive therapy (Messina 2003). Depending on the original disorder and haematopoietic stem cell source, immunosuppressive prophylaxis consisted either of ciclosporin A alone, short‐term methotrexate plus rabbit anti‐thymoglobulin (ATG) or ciclosporin A combined with steroids. People with cGvHD and skin involvement were assessed according to the extent of skin surface involved (per cent ESS) and skin severity score (SSS) in monthly intervals. The clinical stage in people with organ involvement was graded for each organ and then combined to an overall grade following published criteria (Ferrara 1991; Przepiorka 1995; Sullivan 1991). Resistance to conventional immunosuppressive therapy after complete remission was defined as lack of clinical stabilisation or improvement after treatment with prednisolone at a dosage of 2.5 mg/kg for at least seven days and no response to at least two lines of alternative immunosuppressive treatment options (such as ciclosporin A or tacrolimus). The median Lansky/Karnofsky performance score at the start of the ECP was 60% (range 30% to 90%). Photopheresis was carried out on two consecutive days weekly for the first month, every two weeks for the second and third month, followed by monthly intervals for at least three months. Clinical evaluation of the participants was done at months one, two, three and six after initiation of ECP. Complete response was defined as clinical stage 0 or I, partial response as improvement greater than 50% and no response as stable or progressive disease or improvement less than 50%. For the participants with cGvHD, the median Lansky/Karnofsky score improved from 60% to 90% (range 60% to 100%). About 59% of the participants had an overall improvement evaluated according to organ involvement. The median improvement in ESS was 26% and SSS scores was 8 points. At the end of ECP treatment, 44% of participants had a complete response; 29% a partial response; 5% no response; and 10 participants died due to complication of GvHD, infection or relapse of the primary disease. Participants with cGvHD responding to ECP had a significantly better five‐year overall survival of 96.1% (95% CI 88.7% to 100%) compared with non‐responders with 58.4% (95% CI 34.5% to 82.4%). The five studies identified during the 2015 review update were non‐randomised and retrospective or summarised studies that did not meet the search criteria.
The presented studies provided only limited evidence for the efficacy of ECP in paediatric patients with cGvHD after HSCT and, therefore, should not solely be used to establish recommendations in paediatric patients. It is questionable if insights gained from data of the RCT performed in a predominantly adult population can be transferred to children (Flowers 2008). Further evaluation in controlled trials, preferably in RCTs, including cost‐effectiveness analyses is urgently needed. However, performing RCTs in this age group will be challenging due to the limited number of children, the variable disease presentation and the lack of well‐defined response criteria. International multicentre trials with appropriate funding will be needed to study the efficacy of ECP in cGvHD in children after HSCT. As of this 2015 review update, there is still no ongoing prospective RCT investigating the efficacy of ECP in people with cGvHD after HSCT to help to define the role of ECP in treatment of cGvHD in children and adolescents.
Identification of potentially eligible studies.
Identification of potentially eligible studies for 2015 update.